Note: Descriptions are shown in the official language in which they were submitted.
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
DETECTION OF LOW ABUNDANCE VIRUSES
CROSS-REFERENCE
[0001] This patent application claims the benefit of U.S. Provisional Patent
Application No.
63/002,969, filed on March 31, 2020 which is incorporated herein by reference
in its entirety.
BACKGROUND
[0002] Viral infection represents an ongoing threat to both human health and
the global
economy. For example, the coronavirus COVID-19 is predicted to result in
millions of deaths
globally. Testing for infection is a primary method of identifying infected
patients, as well as a
tool for allocating essential medical resources to infected populations.
However, state of the art
testing methods may suffer from extended workflows, safety issues, or low
accuracy. There
exists a need for improved testing methods to address these challenges.
BRIEF SUMMARY
[0003] Provided herein are compositions, methods, and systems for detecting
low-abundance
viruses. Provided herein are methods of detecting nucleic acids comprising: a)
providing a
sample from a source, wherein the sample comprises at least one viral
ribonucleic acid; b)
heating the sample; c) reverse transcribing the at least one viral ribonucleic
acid to generate at
least one cDNA, wherein the at least one viral ribonucleic acid is not
subjected to a purification
step prior to reverse transcribing; and d) detecting the at least one cDNA.
Further provided
herein are methods wherein in the purification step comprises binding the at
least one viral
ribonucleic acid to a solid support. Further provided herein are methods
wherein the purification
step comprises precipitating the least one viral ribonucleic acid or use of
ion-exchange
chromatography. Further provided herein are methods wherein the purification
step comprises
hybridizing the least one viral ribonucleic acid to an array. Further provided
herein are methods
wherein reverse transcribing comprises use of a reverse transcriptase. Further
provided herein
are methods wherein the method further comprises amplification of the at least
one cDNA.
Further provided herein are methods wherein the at least one viral ribonucleic
acid is obtained
from a respiratory virus. Further provided herein are methods wherein the
respiratory virus is a
coronavirus. Further provided herein are methods wherein the coronavirus is
selected from
Covid-19, SARS, MERS, bovine coronaviruses, norovirus, orthoreoviruses
(reoviruses), human
rotaviruses, human coronaviruses, or adenoviruses. Further provided herein are
methods wherein
the at least one viral ribonucleic acid encodes for a viral nucleocapsid.
Further provided herein
are methods wherein the at least one viral ribonucleic acid is an Ni gene, an
N2 gene, or an N3
gene. Further provided herein are methods wherein detecting comprises binding
the at least one
1
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
cDNA with at least one probe. Further provided herein are methods wherein the
probe comprises
a reporter moiety. Further provided herein are methods wherein detection
comprises RT-PCR,
RT-LAMP, RT-PTA, or RT-RPA. Further provided herein are methods wherein the
method
further comprises contacting the sample with a lysis buffer prior to step (c).
Further provided
herein are methods wherein the lysis buffer comprises a proteinase. Further
provided herein are
methods wherein the source is selected from nasopharyngeal or oropharyngeal
swabs, sputum,
lower respiratory tract aspirates, bronchoalveolar lavage, nasopharyngeal
wash/aspirate, or nasal
aspirate. Further provided herein are methods wherein heating the sample
comprises: heating the
sample at a first temperature for a first length of time; and heating the
sample at a second
temperature for a second length of time. Further provided herein are methods
wherein the first
temperature is 30-45 degrees C. Further provided herein are methods wherein
the second
temperature is 80-90 degrees C. Further provided herein are methods wherein
the first time is
10-30 min. Further provided herein are methods wherein the second time is 10-
30 min.
[0004] Provided herein are methods of detecting a virus comprising: a)
providing a sample
from a source, wherein the sample comprises at least one viral genome copy; b)
heating the
sample; c) amplifying the at least one viral genome copy to generate an
amplified viral genome;
d) detecting the amplified viral genome, wherein the at least one viral genome
copy is not
subjected to a purification step prior to detecting. Further provided herein
are methods wherein
the sample comprises 1000-10,000 viral genome copies. Further provided herein
are methods
wherein the sample comprises 10-100 viral genome copies. Further provided
herein are methods
wherein amplifying comprises subjecting the sample to fewer than 30 PCR
cycles. Further
provided herein are methods wherein amplifying comprises subjecting the sample
to fewer than
40 PCR cycles. Further provided herein are methods wherein the viral amplified
genome is
detected in less than 3 hours. Further provided herein are methods wherein the
viral amplified
genome is detected in less than 2 hours. Further provided herein are methods
wherein the
purification step comprises binding the at least one viral genome copy to a
solid support.
Further provided herein are methods wherein the purification step comprises
precipitating the
least one viral genome copy or use of ion-exchange chromatography. Further
provided herein
are methods wherein the purification step comprises hybridizing the least one
viral genome copy
to an array. Further provided herein are methods wherein the at least one
viral genome copy is
obtained from a respiratory virus. Further provided herein are methods wherein
the respiratory
virus is a coronavirus. Further provided herein are methods wherein the
coronavirus is selected
from SARS, MERS, Covid-19, bovine, norovirus, orthoreoviruses (reoviruses),
human
rotaviruses, human coronaviruses, or adenoviruses. Further provided herein are
methods wherein
the source is selected from nasopharyngeal or oropharyngeal swabs, sputum,
lower respiratory
2
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
tract aspirates, bronchoalveolar lavage, nasopharyngeal wash/aspirate, or
nasal aspirate. Further
provided herein are methods wherein heating the sample comprises: heating the
sample at a first
temperature for a first length of time; and heating the sample at a second
temperature for a
second length of time. Further provided herein are methods wherein the first
temperature is 30-
45 degrees C. Further provided herein are methods wherein the second
temperature is 80-90
degrees C. Further provided herein are methods wherein the first time is 10-30
min. Further
provided herein are methods wherein the second time is 10-30 min. Further
provided herein are
methods wherein detection comprises RT-PCR, RT-LAMP, RT-PTA, or RT-RPA.
[0005] Provided herein are methods of detecting a virus comprising: a)
providing at least 48
samples, wherein at least some of the at least 48 samples comprises at least
one viral genome
copy; b) heating the at least 48 samples; c) amplifying the at least one viral
genome copy to
generate an amplified viral genome; d) determining the presence or absence of
the amplified
viral genome for each sample, wherein the at least one viral genome copy is
not subjected to a
purification step prior to determining, and wherein the at least 48 samples
are analyzed in
parallel. Further provided herein are methods comprising providing at least 90
samples. Further
provided herein are methods comprising providing at least 300 samples. Further
provided herein
are methods wherein determining the presence or absence of the viral amplified
genome occurs
in less than 3 hours. Further provided herein are methods wherein determining
the presence or
absence of the viral amplified genome occurs in less than 2 hours. Further
provided herein are
methods wherein the rate of determining the presence or absence of the
amplified viral genome
is at least 2 samples per minute. Further provided herein are methods wherein
the rate of
determining the presence or absence of the amplified viral genome is at least
3 samples per
minute. Further provided herein are methods wherein the rate of determining
the presence or
absence of the amplified viral genome is at least 5 samples per minute.
Further provided herein
are methods wherein the method comprises at least 190 samples, and wherein
determining the
presence or absence of the amplified viral genome for all of the at least 48
samples occurs in no
more than 90 min. Further provided herein are methods wherein the method
comprises at least
384 samples, and wherein determining the presence or absence of the amplified
viral genome for
all of the at least 48 samples occurs in no more than 60 min. Further provided
herein are
methods wherein the purification step comprises binding the at least one viral
genome copy to a
solid support. Further provided herein are methods wherein the purification
step comprises
precipitating the least one viral genome copy or use of ion-exchange
chromatography. Further
provided herein are methods wherein the purification step comprises
hybridizing the least one
viral genome copy to an array. Further provided herein are methods wherein the
at least one
viral genome copy is obtained from a respiratory virus. Further provided
herein are methods
3
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
wherein the respiratory virus is a coronavirus. Further provided herein are
methods wherein the
coronavirus is selected from SARS, MERS, Covid-19, bovine, norovirus,
orthoreoviruses
(reoviruses), human rotaviruses, human coronaviruses, or adenoviruses. Further
provided herein
are methods wherein heating the at least 48 samples comprises: heating the at
least 48 samples at
a first temperature for a first length of time; and heating the at least 48
samples at a second
temperature for a second length of time. Further provided herein are methods
wherein the first
temperature is 30-45 degrees C. Further provided herein are methods wherein
the second
temperature is 80-90 degrees C. Further provided herein are methods wherein
the first time is
10-30 min. Further provided herein are methods wherein the second time is 10-
30 min. Further
provided herein are methods wherein the at least one viral genome copy
comprises DNA.
Further provided herein are methods wherein the at least one viral genome copy
comprises
RNA. Further provided herein are methods wherein determining comprises RT-PCR,
RT-
LAMP, RT-PTA, or RT-RPA.
INCORPORATION BY REFERENCE
[0006] All publications, patents, and patent applications mentioned in this
specification are
herein incorporated by reference to the same extent as if each individual
publication, patent, or
patent application was specifically and individually indicated to be
incorporated by reference.
BRIEF DESCRIPTION OF THE DRAWINGS
[0007] The novel features of the invention are set forth with particularity in
the appended
claims. A better understanding of the features and advantages of the present
invention will be
obtained by reference to the following detailed description that sets forth
illustrative
embodiments, in which the principles of the invention are utilized, and the
accompanying
drawings of which:
[0008] Figure 1A illustrates a standard process for viral detection comprising
the steps of
sample acquisition, sample extraction, sample assay, and reporting. In some
instances, the
sample extraction step leads to reduced sensitivity and slower workflow speed.
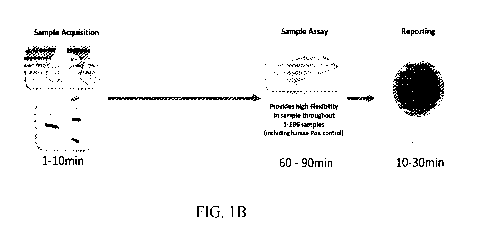
[0009] Figure 1B illustrates a process described herein for viral detection
comprising the steps
of sample acquisition, sample assay, and reporting without a sample extraction
step. Processing
times and sample throughput are shown for example purposes only.
[0010] Figure 2 illustrates a workflow for viral detection without a sample
extraction step.
[0011] Figure 3 illustrates a plot of a normalized reporter value (ARn) vs.
PCR cycles for a
viral detection experiment. Signals obtained from various plasmid (DNA)
concentrations
(number of genome copies, cp) of Covid-19 control/N1 nucleic acid standards
are show.
4
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
Processing times are shown for example purposes only; methods described herein
in some
instances result in faster or slower processing times for various steps.
[0012] Figure 4 illustrates a plot and plate layout for a viral (RNA)
detection experiment. The
plot illustrates analytical sensitive of a normalized reporter value (ARn) vs.
PCR cycles.
Processing times are shown for example purposes only.
[0013] Figure 5 illustrates a workflow for viral detection using lyophilized
beads, without a
sample extraction or purification step. The final detection step in some
instances comprises
qRT-PCR, LAMP, RT-PTA, or RT ¨ Recombinase Polymerase Amplification (RPA). The
plate
size is shown as an example only; other plate sizes are also compatible with
the methods
described herein.
DETAILED DESCRIPTION OF THE INVENTION
[0014] There is a need to develop new scalable, accurate and efficient methods
for viral
detection.
which would overcome limitations in the current methods by increasing
accuracy, safety, and
sensitivity. Provided herein are compositions and methods for viral detection
which do no
comprise one or more sample extraction steps. Such methods in some instances
reduce
workflow times while maintaining high sensitivity and accuracy. Further
provided herein are
methods of viral detection which limit exposure of method operators to
potentially infectious
pathology (e.g., live virus).
Definitions
[0015] Unless defined otherwise, all technical and scientific terms used
herein have the same
meaning as is commonly understood by one of ordinary skill in the art to which
these inventions
belong.
[0016] Throughout this disclosure, numerical features are presented in a range
format. It
should be understood that the description in range format is merely for
convenience and brevity
and should not be construed as an inflexible limitation on the scope of any
embodiments.
Accordingly, the description of a range should be considered to have
specifically disclosed all
the possible subranges as well as individual numerical values within that
range to the tenth of
the unit of the lower limit unless the context clearly dictates otherwise. For
example, description
of a range such as from 1 to 6 should be considered to have specifically
disclosed subranges
such as from 1 to 3, from 1 to 4, from 1 to 5, from 2 to 4, from 2 to 6, from
3 to 6 etc., as well as
individual values within that range, for example, 1.1, 2, 2.3, 5, and 5.9.
This applies regardless
of the breadth of the range. The upper and lower limits of these intervening
ranges may
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
independently be included in the smaller ranges, and are also encompassed
within the invention,
subject to any specifically excluded limit in the stated range. Where the
stated range includes
one or both of the limits, ranges excluding either or both of those included
limits are also
included in the invention, unless the context clearly dictates otherwise.
[0017] The terminology used herein is for the purpose of describing particular
embodiments
only and is not intended to be limiting of any embodiment. As used herein, the
singular forms
"a," "an" and "the" are intended to include the plural forms as well, unless
the context clearly
indicates otherwise. It will be further understood that the terms "comprises"
and/or
"comprising," when used in this specification, specify the presence of stated
features, integers,
steps, operations, elements, and/or components, but do not preclude the
presence or addition of
one or more other features, integers, steps, operations, elements, components,
and/or groups
thereof. As used herein, the term "and/or" includes any and all combinations
of one or more of
the associated listed items.
[0018] Unless specifically stated or obvious from context, as used herein, the
term "about" in
reference to a number or range of numbers is understood to mean the stated
number and
numbers +/- 10% thereof, or 10% below the lower listed limit and 10% above the
higher listed
limit for the values listed for a range.
[0019] The terms "subject" or "patient" or "individual", as used herein, refer
to animals,
including mammals, such as, e.g., humans, veterinary animals (e.g., cats,
dogs, cows, horses,
sheep, pigs, etc.) and experimental animal models of diseases (e.g., mice,
rats). In accordance
with the present invention there may be employed conventional molecular
biology,
microbiology, and recombinant DNA techniques within the skill of the art. Such
techniques are
explained fully in the literature. See, e.g., Sambrook, Fritsch & Maniatis,
Molecular Cloning: A
Laboratory Manual, Second Edition (1989) Cold Spring Harbor Laboratory Press,
Cold Spring
Harbor, New York (herein "Sambrook et al., 1989"); DNA Cloning: A practical
Approach,
Volumes I and II (D.N. Glover ed. 1985); Oligonucleotide Synthesis (MJ. Gait
ed. 1984);
Nucleic Acid Hybridization (B.D. Hames & S.J. Higgins eds. (1985);
Transcription and
Translation (B.D. Hames & S.J. Higgins, eds. (1984); Animal Cell Culture (R.I.
Freshney, ed.
(1986); Immobilized Cells and Enzymes IRL Press, (1986); B. Perbal, A
practical Guide To
Molecular Cloning (1984); F.M. Ausubel et al. (eds.), Current Protocols in
Molecular Biology,
John Wiley & Sons, Inc. (1994); among others.
[0020] The term "nucleic acid" encompasses multi-stranded, as well as single-
stranded
molecules. In double- or triple-stranded nucleic acids, the nucleic acid
strands need not be
coextensive (i.e., a double- stranded nucleic acid need not be double-stranded
along the entire
length of both strands). Nucleic acid templates described herein may be any
size depending on
6
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
the sample (from small cell-free DNA fragments to entire genomes), including
but not limited to
50-300 bases, 100-2000 bases, 100-750 bases, 170-500 bases, 100-5000 bases, 50-
10,000 bases,
or 50-2000 bases in length. In some instances, templates are at least 50, 100,
200, 500, 1000,
2000, 5000, 10,000, 20,000 50,000, 100,000, 200,000, 500,000, 1,000,000 or
more than
1,000,000 bases in length. Methods described herein provide for the
amplification of nucleic
acid acids, such as nucleic acid templates. Methods described herein
additionally provide for the
generation of isolated and at least partially purified nucleic acids and
libraries of nucleic acids.
Nucleic acids include but are not limited to those comprising DNA, RNA,
circular RNA,
mtDNA (mitochondrial DNA), cfDNA (cell free DNA), cfRNA (cell free RNA), siRNA
(small
interfering RNA), cffDNA (cell free fetal DNA), mRNA, tRNA, rRNA, miRNA
(microRNA),
synthetic polynucleotides, polynucleotide analogues, viral DNA, viral RNA, any
other nucleic
acid consistent with the specification, or any combinations thereof. The
length of
polynucleotides, when provided, are described as the number of bases and
abbreviated, such as
nt (nucleotides), bp (bases), kb (kilobases), or Gb (gigabases).
[0021] The term "droplet" as used herein refers to a volume of liquid on a
droplet actuator.
Droplets in some instances, for example, be aqueous or non-aqueous or may be
mixtures or
emulsions including aqueous and non-aqueous components. For non-limiting
examples of
droplet fluids that may be subjected to droplet operations, see, e.g., Int.
Pat. Appl. Pub. No.
W02007/120241. Any suitable system for forming and manipulating droplets can
be used in the
embodiments presented herein. For example, in some instances a droplet
actuator is used. For
non-limiting examples of droplet actuators which can be used, see, e.g., U.S.
Pat. No. 6,911,132,
6,977,033, 6,773,566, 6,565,727, 7,163,612, 7,052,244, 7,328,979, 7,547,380,
7,641,779, U.S.
Pat. Appl. Pub. Nos. U520060194331, U520030205632, U520060164490,
U520070023292,
U520060039823, U520080124252, U520090283407, U520090192044, U520050179746,
U520090321262, U520100096266, U520110048951, Int. Pat. Appl. Pub. No.
W02007/120241.
In some instances, beads are provided in a droplet, in a droplet operations
gap, or on a droplet
operations surface. In some instances, beads are provided in a reservoir that
is external to a
droplet operations gap or situated apart from a droplet operations surface,
and the reservoir may
be associated with a flow path that permits a droplet including the beads to
be brought into a
droplet operations gap or into contact with a droplet operations surface. Non-
limiting examples
of droplet actuator techniques for immobilizing magnetically responsive beads
and/or non-
magnetically responsive beads and/or conducting droplet operations protocols
using beads are
described in U.S. Pat. Appl. Pub. No. U520080053205, Int. Pat. Appl. Pub. No.
W02008/098236, W02008/134153, W02008/116221, W02007/120241. Bead
characteristics
may be employed in the multiplexing embodiments of the methods described
herein. Examples
7
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
of beads having characteristics suitable for multiplexing, as well as methods
of detecting and
analyzing signals emitted from such beads, may be found in U.S. Pat. Appl.
Pub. No.
US20080305481, US20080151240, US20070207513, US20070064990, US20060159962,
US20050277197, US20050118574.
[0022] As used herein, the term "unique molecular identifier (UMI)" refers to
a unique nucleic
acid sequence that is attached to each of a plurality of nucleic acid
molecules. When
incorporated into a nucleic acid molecule, an UMI in some instances is used to
correct for
subsequent amplification bias by directly counting UMIs that are sequenced
after amplification.
The design, incorporation and application of UMIs is described, for example,
in Int. Pat. Appl.
Pub. No. WO 2012/142213, Islam et al. Nat. Methods (2014) 11:163-166, and
Kivioj a, T. et al.
Nat. Methods (2012) 9: 72-74.
[0023] As used herein, the term "barcode" refers to a nucleic acid tag that
can be used to
identify a sample or source of the nucleic acid material. Thus, where nucleic
acid samples are
derived from multiple sources, the nucleic acids in each nucleic acid sample
are in some
instances tagged with different nucleic acid tags such that the source of the
sample can be
identified. Barcodes, also commonly referred to indexes, tags, and the like,
are well known to
those of skill in the art. Any suitable barcode or set of barcodes can be
used. See, e.g., non-
limiting examples provided in U.S. Pat. No. 8,053,192 and Int. Pat. Appl. Pub.
No.
W02005/068656. Barcoding of single cells can be performed as described, for
example, in U.S.
Pat. Appl. Pub. No. 2013/0274117.
[0024] The terms "solid surface," "solid support" and other grammatical
equivalents herein
refer to any material that is appropriate for or can be modified to be
appropriate for the
attachment of the primers, barcodes and sequences described herein. Exemplary
substrates
include, but are not limited to, glass and modified or functionalized glass,
plastics (including
acrylics, polystyrene and copolymers of styrene and other materials,
polypropylene,
polyethylene, polybutylene, polyurethanes, TeflonTm, etc.), polysaccharides,
nylon,
nitrocellulose, ceramics, resins, silica, silica-based materials (e.g.,
silicon or modified silicon),
carbon, metals, inorganic glasses, plastics, optical fiber bundles, and a
variety of other polymers.
In some embodiments, the solid support comprises a patterned surface suitable
for
immobilization of primers, barcodes and sequences in an ordered pattern.
[0025] As used herein, the term "biological sample" includes, but is not
limited to, tissues,
cells, biological fluids and isolates thereof. Cells or other samples used in
the methods described
herein are in some instances isolated from human patients, animals, plants,
soil or other samples
comprising microbes such as bacteria, fungi, protozoa, etc. In some instances,
the biological
sample is of human origin. In some instances, the biological is of non-human
origin. The cells in
8
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
some instances undergo PTA methods described herein and sequencing. Variants
detected
throughout the genome or at specific locations can be compared with all other
cells isolated from
that subject to trace the history of a cell lineage for research or diagnostic
purposes.
[00026] The term "identity" or "homology" refer to the percentage of amino
acid residues in
the candidate sequence that are identical with the residue of a corresponding
sequence to which
it is compared, after aligning the sequences and introducing gaps, if
necessary to achieve the
maximum percent identity for the entire sequence, and not considering any
conservative
substitutions as part of the sequence identity. Conservative substitutions in
some instances
involve substitution of one amino acid of similar shape (e.g., tyrosine for
phenylalanine) or
charge (glutamic acid for aspartic acid) for another. A polynucleotide or
polynucleotide region
(or a peptide or peptide region) comprises a certain percentage (for example,
80%, 85%, 90%, or
95%) of "sequence identity" or "homology" to another sequence means that, when
aligned, that
percentage of bases (or amino acids) are the same in comparing the two
sequences. Neither N-
or C-terminal extensions nor insertions shall be construed as reducing
identity or homology.
Alignment and the percent homology or sequence identity in some instances are
determined
using software programs known by those skilled the art. In some instances,
default parameters
are used for alignment. An exemplary alignment program is BLAST, using default
parameters.
In particular, programs are BLASTN and BLASTP, using the following default
parameters:
Genetic code=standard; filter=none; strand= both; cutoff=60; expect= 10;
Matrix=BLOSUM62;
Descriptions=50 sequences; sort by=HIGH SCORE; Databases=non-redundant,
GenBank+EMBL+DDBJ+PDB+GenBank CDS translations+SwissProtein+SPupdate+PIR.
Similarity, or percent similarity in some instances of two sequences is the
sum of both identical
and similar matches (residues that have undergone conservative substitution).
In some instances,
similarity is measured using the program BLAST "Positives."
[0027] Methods of Viral Detection
[0028] Described herein are methods of detecting viruses. In some instances,
such methods
comprise one or more steps in a workflow. A standard method of viral detection
is shown in
FIG. 1A. In a first step, a sample (e.g., biological sample) is acquired from
a source. In some
instances, a source is a patient, surface, or other source. In a second step,
the sample is extracted
to isolate nucleic acids. In a third step, the extracted nucleic acids are
assayed or identified to
establish if they comprise nucleic acids of a virus. In a fourth step, results
of the assay are
reported to a healthcare provider, patient, electronic display, or electronic
database.
[0029] Described herein are methods for viral detection which may eliminate
one or more
sample extraction steps (FIG. 1B). Such methods in some instances comprise at
least the steps
9
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
of sample acquisition, and sample assay. In some instances, methods described
herein comprise
at least the steps of sample acquisition, sample assay, and reporting. In some
instances, methods
described herein are capable of multiplexing.
[0030] Samples may be acquired from any source which may contain nucleic
acids. Such
samples in some instances are utilized during a sample acquisition step. In
some instances, a
source includes but is not limited to a fluid (e.g., water source, bodily
fluid), gas (air sample), or
solid (medical surface, mask). In some instances, a source is a fluid. In some
instances, the fluid
is obtained from an animal. In some instances, the animal is a mammal. In some
instances, the
mammal is a human. Samples in some instances are obtained from blood, serum,
plasma, bone
marrow, urine, saliva, mucus, cerebrospinal fluid, pleural fluid, pericardial
fluid, ascites, or
aqueous humor. In some instances, samples are obtained from upper or lower
respiratory
sources. In some instances, sources include but are not limited to
nasopharyngeal or
oropharyngeal swabs, sputum, lower respiratory tract aspirates,
bronchoalveolar lavage,
nasopharyngeal wash/aspirate, or nasal aspirate. In some instances, samples
comprise indwelling
medical devices, such as but not limited to, intravenous catheters, urethral
catheters,
cerebrospinal shunts, prosthetic valves, artificial joints, or endotracheal
tubes. In some instances
samples are obtained from swabs of a surface. In some instances, a surface
comprises the
respiratory tract, nose, ear, throat, lung, or esophagus. Acquisition of
samples in some instances
is completed in about 10 sec, 20 sec, 30 sec, 45 sec, 1 min, 2 min, 5 min, 8
min, 10 min, or about
15 min. Acquisition of samples in some instances is completed in no more than
10 sec, 20 sec,
30 sec, 45 sec, 1 min, 2 min, 5 min, 8 min, 10 min, or no more than 15 min.
Acquisition of
samples in some instances is completed in 10 sec-15 min, 10 sec-10 min, 10 sec-
5 min, 10 sec-1
min, 30 sec-5 min, 30 sec-2 min, 1 min-12 min, 1 min-10 min, 1 min-5 min, 2
min-15 min, 5
min-15 min, or 5 min-10 min.
[0031] Extraction steps may be used to purify nucleic acids prior to a sample
assay step. In
some instances, methods described herein to not comprise an extraction step.
In some instances,
methods described herein comprise no more than 4, 3, 2, or 1 extraction steps.
In some
instances, a method described herein does not comprise an extraction step. In
some instances, a
method described herein does not comprise binding nucleic acids to a solid
support,
precipitating nucleic acids, or ion-exchange chromatography. In some instance,
extraction steps
include cell lysis, nucleic acid binding, washing bound nucleic acids, drying
bound nucleic
acids, and eluting bound nucleic acids. In some instances, extraction steps
comprise binding a
nucleic acid to a solid support. In some instances, extract steps comprise
precipitating a nucleic
acid. In some instances, extraction steps comprise hybridizing a nucleic acid
to an array. In some
instances In some instances, extraction comprises binding nucleic acids to a
solid support. In
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
some instances, extraction comprises use of beads (e.g., SPRI beads). In some
instances,
extraction comprises use of ion-exchange chromatography. In some instances, a
workflow is
limited to extracting 5-10, 5-100, 12-96, 24-64, 8-96, 48-96, or 48-72 samples
in a single batch.
In some instances, one or more extraction steps is completed in 10-240 min, 10-
180 min, 10-
120 min, 90-180 min, 120-180 min, 60-180 min, or 120-300 min. In some
instances, one or
more extraction steps is completed in at least 10 min, 20 min, 30 min, 45 min,
60 min, 90 min,
120 min, 180 min, or at least 240 min.
[0032] One or more additional steps may precede a sample assay step. In some
instances,
methods described herein comprise treatment of a sample with a lysis buffer
prior to an assay
step. In some instances, a lysis buffer comprises a proteinase. In some
instances, the proteinase
is proteinase K or Pk. In some instances, the lysis buffer is stored as a
lyophilized powder. In
some instances, the lyophilized powder comprises a stabilizer. In some
instances, the stabilizer
is a sugar. In some instances, the sugar is selected from maltose, trehalose,
cellobiose, sucralose,
isomaltose, raffinose, or isomaltulose. In some instances, a stabilizer is
present at about 1%, 2%,
5%, 10%, 15%, 20%, 30%, 50%, or about 75% (w/w). In some instances, a
stabilizer is present
at 1-5%, 1-20%, 5-20%, 10-50%, 20-50%, or 15-30% (w/w). In some instances, a
lysis buffer
comprises a reducing agent. In some instances, the reducing agent is DTT or
beta-
mercaptoethanol. In some instances, the lysis buffer comprises a surfactant.
In some instances, a
sample is heat treated prior to an assay step. In some instances, a sample is
heated at one or more
temperatures, each for a period of time. In some instances, heating the sample
deactivates one or
more enzymes in the sample, such as RNAses. In some instances, a sample is
heated to a first
temperature for a first time, and then heated at a second temperature for a
second time. In some
instances, the first temperature is 25-75 deg C, 25-60 deg C, 25-50 deg C, 25-
40 deg C, 30-45
deg C, 35-45 deg C, 35-50 deg C, or 30-60 deg C. In some instances, the first
temperature is
about 25 deg C, 30 deg C, 32 deg C, 35 deg C, 37 deg C, 39 deg C, 40 deg C, 42
deg C, 45 deg
C, 50 deg C, 55 deg C, or about 60 deg C. In some instances, the second
temperature is 65-95
deg C, 65-90 deg C, 65-85 deg C, 65- 80 deg C, 70-95 deg C, 75-90 deg C, 78-84
deg C, or 80-
90 deg C. In some instances, the first temperature is about 60 deg C, 65 deg
C, 70 deg C, 75 deg
C, 80 deg C, 85 deg C, 90 deg C, 95 deg C, about 98 deg C. In some instances,
the first time is
5-30 min, 10-20 min, 5-20 min, 8-13 min, or 15-30 min. In some instances, the
first time is
about 5 min, 8 min, 10 min, 12 min, 15 min, 17 min, 20 min, 30 min, or 45 min.
In some
instances, the second time is 5-30 min, 10-20 min, 5-20 min, 8-13 min, or 15-
30 min. In some
instances, the second time is about 5 min, 8 min, 10 min, 12 min, 15 min, 17
min, 20 min, 30
min, or 45 min. In some instances, the first temperature is 25-75 deg C, 25-60
deg C, 25-50 deg
C, 25-40 deg C, 30-45 deg C, 35-45 deg C, 35-50 deg C, or 30-60 deg C and the
first time is 10-
11
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
20 min. In some instances, the first temperature is about 25 deg C, 30 deg C,
32 deg C, 35 deg
C, 37 deg C, 39 deg C, 40 deg C, 42 deg C, 45 deg C, 50 deg C, 55 deg C, or
about 60 deg C and
the first time is about 15 min. In some instances, the second temperature is
65-95 deg C, 65-90
deg C, 65-85 deg C, 65- 80 deg C, 70-95 deg C, 75-90 deg C, 78-84 deg C, or 80-
90 deg C and
the first time is 10-20 min. In some instances, the first temperature is about
60 deg C, 65 deg C,
70 deg C, 75 deg C, 80 deg C, 85 deg C, 90 deg C, 95 deg C, about 98 deg C and
the first time is
about 15 min.
[0033] Sample assays may be used to detect the presence of viral particles.
Various viral
particles may be detected using a sample assay. In some instances, viral
particles comprise
nucleic acids. In some instances, nucleic acids comprise DNA or RNA. In some
instances,
nucleic acids comprise RNA. In some instances, an assay step comprises
analysis of a positive
control. In some instances, a positive control comprises nucleic acids
associated with a virus. In
some instances, a positive control comprises RNA. In some instances, a
positive control
comprises DNA. In some instances, a positive control comprises a plasmid. In
some instances, a
positive control is generated in-situ. In some instances, an assay step
comprises a negative
control (no template control). In some instances, a negative control does not
comprise viral
nucleic acids. In some instances, an assay step comprises analysis of a
positive control and a
negative control. Positive controls in some instances are specific to a
specific type of virus. In
some instances, a positive control is a COVID-19 plasmid. In some instances, a
positive control
comprises an RNA copy of a viral gene. In some instances, viral genes include
but are not
limited to Ni, N2, and/or N3. In some instances, a control targeting human
RNaseP is used to
establish a sample comprises at least some nucleic acids for testing,
regardless of whether it
comprises viral nucleic acids. In some instances, a negative sample control
(without sample) is
used to establish if any cross-contamination has occurred between samples. In
some instances, a
virus is detected by the presence of one or more different nucleic acids. In
some instances, a
sample assay is completed in about 10 min, 20 min, 30 min, 45 min, 60 min, 90
min, 120 min, or
about 180 min. In some instances, a sample assay is completed in no more than
10 min, 20 min,
30 min, 45 min, 60 min, 90 min, 120 min, or no more than 180 min. In some
instances, a sample
assay is completed in 10 min-180 min 10-120 min, 10-60 min, 10-30 min, 30-180
min, 30-120
min, 60-120 min, 60-90 min, 90-120 min, or 45-100 min.
[0034] Sample assays may comprise one or more reporter assays to quantify
viral nucleic
acids. In some instances, sample assays comprise a probe comprising a
recognition moiety and a
reporter moiety. In some instances, a recognition moiety binds to a viral
component, such as a
viral nucleic acid (or fragment thereof). In some instances, a reporter moiety
generates a signal
which indicates the presence of a viral nucleic acid. In some instances,
signals include but are
12
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
not limited to fluorescence, phosphorescence, chemiluminescence,
antibody/antigen binding,
radioactivity, mass tags, next generation sequencing, or other detectable
signal. In some
instances, sample assays comprise use of a polymerase chain reaction. In some
instances, sample
assays comprise a reverse transcriptase. In some instances, sample assays
comprise a
polymerase. In some instances, sample assays comprise quantitative polymerase
chain reactions
(qPCR), or real-time PCR. In some instances, sample assays comprise
quantitative reverse-
transcriptase polymerase chain reactions (qRT-PCR). In some instances, a
sample assay step
comprises use of one or more primers, such as a forward primer and a reverse
primer. In some
instances, the amount of nucleic acid in a sample is quantified after one or
more PCR cycles. In
some instances, a sample assay step comprises about 1, 2, 5, 10, 12, 15, 18,
20, 25, 30, 35, 40, or
about 45 PCR cycles. In some instances, a sample assay step comprises no more
than 1, 2, 5, 10,
12, 15, 18, 20, 25, 30, 35, 40, or no more than 45 PCR cycles. In some
instances, sample assays
comprise reverse transcription of RNA into cDNA. In some instances, sample
assay steps
comprise binding of a reporter moiety to a target nucleic acid (e.g., viral
nucleic acids). In some
instance, a probe comprises a quenching moiety. In some instances, a probe
comprises a nucleic
acid complementary to a viral nucleic acid (Table 1). Any number of probes are
in some
instances used during a sample assay step. In some instances, probes target
Covid-19 nucleic
acids. In some instances a sample assay comprises at least two probes. In some
instances a first
probe is configured to bind a viral nucleic acid, and a second probe is
configured to bind a
control (non-viral) nucleic acid. In some instances, a control nucleic acid is
a human gene or
fragment thereof.
[0035] Table 1: Primers and Probes for Viral Detection using qRT-PCR
SEQ Description Sequence
ID
NO
1 nCoV_Nl_ForwardPrimer GACCCCAAAATCAGCGAAAT
2 nCoV_Nl_ReversePrimer TCTGGTTACTGCCAGTTGAATCTG
3 nCoV_Nl_Probe FAM-ACCCCGCATTACGTTTGGTGGACC-
BHQ 1
4 nCoV_N2_ForwardPrimer TTACAAACATTGGCCGCAAA
R nCoV_N2ReversePrimer GCGCGACATTCCGAAGAA
6 nCoV_N2_Probe FAM-ACAATTTGC CCCCAGCGCTTCAG-
BHQ 1
13
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
7 nCoV_N3 _ForwardPrimer GGGAGCCTTGAATACACCAAAA
8 nCoV_N3 _ReversePrimer TGTAGCACGATTGCAGCATTG
9 nCoV_N3 _Probe FAM-AYCACATTGGCACCCGCAATCCTG-
BHQ1
RNAseP_Forward_Primer GAGCGGCTGTCTCCACAAGT
11 RNAseP_Reverse_Primer AGATTTGGACCTGCGAGCG
12 RNAseP_Probe FAM¨TTCTGACCTGAAGGCTCTGCGCG¨
BHQ- 1
13 SARS_polyprotein_la_Forward_Primer GCCgTAgTgTCAgTATCATCACC
14 SARS_polyprotein_la_Reverse_Primer AATAggACCAATCTCTgTAA,gAgCC
SARS_polyprotein_la_Probe FAM-TCACTTCgTCATC A AAgAC ATC [T_5 -
TAMRA] gAggA gCp
16 SARS_polyprotein_lab_Forward_Primer TITTgrfgrITCAACTggATACCAT
17 SARS_polyprotein_lab_Reverse_Primer GAAACTgAgACgCgAgCTATgT
18 SARS_polyprotein_lab_Probe FAM-
CATCCTgATTATgTACgACTCCTAAC[T_5-
TAMRA]CACgAAp
19 SAR_Surface_Spike_Glycoprotein_Forward_Primer gAggTCTTTTATTgAggACTTgCTC
SAR_Surface_Spike_Glycoprotein_Reverse_Primer gCATTCgCCATATTgerIcAT
21 SAR_Surface_Spike_Glycoprotein_Probe FAM-
gCCAgC ATCAgCgAgTgICACCITA [T_5-
TAMRA]p
[0036] FAM = 6-carboxyfluorescein; BHQ-1= Black Hole Quencher 1; lower case
indicates
ribonucleotides; [T 5-TAMRA] = 5-carboxytetramethylrhodamine attached to 5-
ethylamino-
dThymidin; p = phosphate
[0037] Sample assays may comprise loop-mediated isothermal amplification
(LAMP). Sample
assays in some instances comprise reverse-transcriptase loop-mediated
isothermal amplification
(RT-LAMP). In some instances, a sample assay comprises use of an isothermal
polymerase. In
some instances, a sample assay comprises use of an isothermal polymerase and a
reverse
transcriptase. In some instances, each PCR cycle during LAMP is held at a
relatively constant
temperature, for example 45-50 deg C, 50-55 deg C, 55-60 deg C, 60-65 deg C,
65-70 deg C, or
70-75 deg C. In some instances, primers used in sample assays comprise loop
primers (primers
comprising intramolecular loops). In some instances the assay readout includes
colorimetric
detection.
[0038] Sample assays may comprise reverse transcriptase PTA (RT-PTA). Sample
assays in
some instances comprise an RT-PCR reaction to generate cDNA, followed by use
of the PTA
14
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
method to amplify the cDNA library. Such libraries are then sequenced, for
example, using Next
Generation Sequencing to detect the presence of viral nucleic acids.
[0039] Sample assays may comprise reverse transcriptase RPA (RT-RPA). Sample
assays in
some instances comprise an RT-RPA reaction to generate cDNA, followed by use
of the RPA
method to amplify the cDNA library and detect the viral genome using a primer
and probe. In
some instances, RPA comprises use of a recombinase, a single stranded DNA
binding protein
(SSB), and a strand-displacing enzyme. In some instances, each PCR cycle in
RPA is held at a
relatively constant temperature, for example 30-50 deg C, 30-55 deg C, 35-45
deg C, 30-40 deg
C, 35-40 deg C, or 37-42 deg C.
[0040] Methods described herein may be used to detect viruses, virus
particles, or other viral
component or sub-component of a virus. In some instances, viruses comprise
respiratory viruses.
Virus include but are not limited to influenza or a coronavirus. In some
instances, virus
possesses hemagglutinin activity. In some instances, the virus is capable of
infecting mammalian
cells. In some instances, the virus is capable of infecting erythrocytes. In
some instances, the
coronavirus comprises SARS, MERS, Covid-19, bovine, norovirus, orthoreoviruses
(reoviruses), human rotaviruses, human coronaviruses, adenoviruses,
filoviruses, or other
coronavirus. In some instances, the coronavirus is SARS. In some instances,
the coronavirus is
Covid-19. In some instances, the coronavirus is a bovine coronavirus. In some
instances, the
coronavirus is norovirus. In some instances, the coronavirus is an
orthoreoviruses (e.g.,
reoviruses). In some instances, the coronavirus is a human rotaviruses, In
some instances, the
coronavirus is a human coronaviruses. In some instances, the coronavirus is an
adenovirus. In
some instances, the influenza is selected from avian flu, swine flu, or other
flu.
[0041] In some instances, the virus is Abelson leukemia virus; Abelson murine
leukemia
virus; Abelson's virus; Acute laryngotracheobronchitis virus; Adelaide River
virus; Adeno
associated virus group; Adenovirus; African horse sickness virus; African
swine fever virus;
AIDS virus; Aleutian mink disease parvovirus; Alpharetrovirus; Alphavirus; ALV
related virus;
Amapari virus; Aphthovirus; Aquareovirus; Arbovirus; Arbovirus C; arbovirus
group A;
arbovirus group B; Arenavirus group; Argentine hemorrhagic fever virus;
Argentine
hemorrhagic fever virus; Arterivirus; Astrovirus; Ateline herpesvirus group;
Aujezky's disease
virus; Aura virus; Ausduk disease virus; Australian bat lyssavirus;
Aviadenovirus; avian
erythroblastosis virus; avian infectious bronchitis virus; avian leukemia
virus; avian leukosis
virus; avian lymphomatosis virus; avian myeloblastosis virus; avian
paramyxovirus; avian
pneumoencephalitis virus; avian reticuloendotheliosis virus; avian sarcoma
virus; avian type C
retrovirus group; Avihepadnavirus; or Avipoxvirus; B virus.
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
[0042] In some instances, the virus is B19 virus; Babanki virus; baboon
herpesvirus;
baculovirus; Barmah Forest virus; Bebaru virus; Berrimah virus;
Betaretrovirus; Birnavirus;
Bittner virus; BK virus; Black Creek Canal virus; bluetongue virus; Bolivian
hemorrhagic fever
virus; Boma disease virus; border disease of sheep virus; borna virus; bovine
alphaherpesvirus 1;
bovine alphaherpesvirus 2; bovine coronavirus; bovine ephemeral fever virus;
bovine
immunodeficiency virus; bovine leukemia virus; bovine leukosis virus; bovine
mammillitis
virus; bovine papillomavirus; bovine papular stomatitis virus; bovine
parvovirus; bovine
syncytial virus; bovine type C oncovirus; bovine viral diarrhea virus; Buggy
Creek virus; bullet
shaped virus group; Bunyamwera virus supergroup; Bunyavirus; Burkitt's
lymphoma virus; or
Bwamba Fever.
[0043] In some instances, the virus is CA virus; Calicivirus; California
encephalitis virus;
camelpox virus; canarypox virus; canid herpesvirus; canine coronavirus; canine
distemper virus;
canine herpesvirus; canine minute virus; canine parvovirus; Cano Delgadito
virus; caprine
arthritis virus; caprine encephalitis virus; Caprine Herpes Virus; Capripox
virus; Cardiovirus;
caviid herpesvirus 1; Cercopithecid herpesvirus 1; cercopithecine herpesvirus
1; Cercopithecine
herpesvirus 2; Chandipura virus; Changuinola virus; channel catfish virus;
Charleville virus;
chickenpox virus; Chikungunya virus; chimpanzee herpesvirus; chub reovirus;
chum salmon
virus; Cocal virus; Coho salmon reovirus; coital exanthema virus; Colorado
tick fever virus;
Coltivirus; Columbia SK virus; common cold virus; contagious eethyma virus;
contagious
pustular dermatitis virus; Coronavirus; Corriparta virus; coryza virus; covid-
19 (Wuhan virus);
cowpox virus; coxsackie virus; CPV (cytoplasmic polyhedrosis virus); cricket
paralysis virus;
Crimean-Congo hemorrhagic fever virus; croup associated virus; Cryptovirus;
Cypovirus;
Cytomeg Finalovirus; cytomegalovirus group; or cytoplasmic polyhedrosis virus.
[0044] In some instances, the virus is deer papillomavirus; deltaretrovirus;
dengue virus;
Densovirus; Dependovirus; Dhori virus; diploma virus; Drosophila C virus; duck
hepatitis B
virus; duck hepatitis virus 1; duck hepatitis virus 2; duovirus; Duvenhage
virus; or Deformed
wing virus DWV.
[0045] In some instances, the virus is eastern equine encephalitis virus;
eastern equine
encephalomyelitis virus; EB virus; Ebola virus; Ebola-like virus; echo virus;
echovirus;
echovirus 10; echovirus 28; echovirus 9; ectromelia virus; EEE virus; ETA
virus; ETA virus;
encephalitis virus; encephalomyocarditis group virus; encephalomyocarditis
virus; Enterovirus;
enzyme elevating virus; enzyme elevating virus (LDH); epidemic hemorrhagic
fever virus;
epizootic hemorrhagic disease virus; Epstein-Barr virus; equid
alphaherpesvirus 1; equid
alphaherpesvirus 4; equid herpesvirus 2; equine abortion virus; equine
arteritis virus; equine
encephalosis virus; equine infectious anemia virus; equine morbillivirus;
equine
16
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
rhinopneumonitis virus; equine rhinovirus; Eubenangu virus; European elk
papillomavirus;
European swine fever virus; Everglades virus; or Eyach virus.
[0046] In some instances, the virus is felid herpesvirus 1; feline
calicivirus; feline
fibrosarcoma virus; feline herpesvirus; feline immunodeficiency virus; feline
infectious
peritonitis virus; feline leukemia/sarcoma virus; feline leukemia virus;
feline panleukopenia
virus; feline parvovirus; feline sarcoma virus; feline syncytial virus;
Filovirus; Flanders virus;
Flavivirus; foot and mouth disease virus; Fort Morgan virus; Four Corners
hantavirus; fowl
adenovirus 1; fowlpox virus; Friend virus; Gammaretrovirus; GB hepatitis
virus; GB virus;
German measles virus; Getah virus; gibbon ape leukemia virus; glandular fever
virus; goatpox
virus; golden shinner virus; Gonometa virus; goose parvovirus; granulosis
virus; Gross' virus;
ground squirrel hepatitis B virus; group A arbovirus; Guanarito virus; guinea
pig
cytomegalovirus; or guinea pig type C virus.
[0047] Hantaan virus; Hantavirus; hard clam reovirus; hare fibroma virus; HCMV
(human
cytomegalovirus); hemadsorption virus 2; hemagglutinating virus of Japan;
hemorrhagic fever
virus; hendra virus; Henipaviruses; Hepadnavirus; hepatitis A virus; hepatitis
B virus group;
hepatitis C virus; hepatitis D virus; hepatitis delta virus; hepatitis E
virus; hepatitis F virus;
hepatitis G virus; hepatitis nonA nonB virus; hepatitis virus; hepatitis virus
(nonhuman);
hepatoencephalomyelitis reovirus 3; Hepatovirus; heron hepatitis B virus;
herpes B virus; herpes
simplex virus; herpes simplex virus 1; herpes simplex virus 2; herpesvirus;
herpesvirus 7;
Herpesvirus ateles; Herpesvirus hominis; Herpesvirus infection; Herpesvirus
saimiri;
Herpesvirus suis; Herpesvirus varicellae; Highlands J virus; Hirame
rhabdovirus; hog cholera
virus; human adenovirus 2; human alphaherpesvirus 1; human alphaherpesvirus 2;
human
alphaherpesvirus 3; human B lymphotropic virus; human betaherpesvirus 5; human
coronavirus;
human cytomegalovirus group; human foamy virus; human gammaherpesvirus 4;
human
gammaherpesvirus 6; human hepatitis A virus; human herpesvirus 1 group; human
herpesvirus 2
group; human herpesvirus 3 group; human herpesvirus 4 group; human herpesvirus
6; human
herpesvirus 8; human immunodeficiency virus; human immunodeficiency virus 1;
human
immunodeficiency virus 2; human papillomavirus; human T cell leukemia virus;
human T cell
leukemia virus I; human T cell leukemia virus II; human T cell leukemia virus
III; human T cell
lymphoma virus I; human T cell lymphoma virus II; human T cell lymphotropic
virus type 1;
human T cell lymphotropic virus type 2; human T lymphotropic virus I; human T
lymphotropic
virus II; human T lymphotropic virus III; Ichnovirus; infantile
gastroenteritis virus; infectious
bovine rhinotracheitis virus; infectious haematopoietic necrosis virus;
infectious pancreatic
necrosis virus; influenza virus A; influenza virus B; influenza virus C;
influenza virus D;
17
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
influenza virus pr8; insect iridescent virus; insect virus; iridovirus;
Japanese B virus; Japanese
encephalitis virus; JC virus; Junin virus;
[0048] In some instances, the virus is Kaposi's sarcoma-associated
herpesvirus; Kemerovo
virus; Kilham's rat virus; Klamath virus; Kolongo virus; Korean hemorrhagic
fever virus; kumba
virus; Kysanur forest disease virus; Kyzylagach virus; La Crosse virus; lactic
dehydrogenase
elevating virus; lactic dehydrogenase virus; Lagos bat virus; Langur virus;
lapine parvovirus;
Lassa fever virus; Lassa virus; latent rat virus; LCM virus; Leaky virus;
Lentivirus;
Leporipoxvirus; leukemia virus; leukovirus; lumpy skin disease virus;
lymphadenopathy
associated virus; Lymphocryptovirus; lymphocytic choriomeningitis virus; or
lymphoproliferative virus group.
[0049] In some instances, the virus is Machupo virus; mad itch virus;
mammalian type B
oncovirus group; mammalian type B retroviruses; mammalian type C retrovirus
group;
mammalian type D retroviruses; mammary tumor virus; Mapuera virus; Marburg
virus;
Marburg-like virus; Mason Pfizer monkey virus; Mastadenovirus; Mayaro virus;
ME virus;
measles virus; Menangle virus; Mengo virus; Mengovirus; Middelburg virus;
milkers nodule
virus; mink enteritis virus; minute virus of mice; MLV related virus; MM
virus; Mokola virus;
Molluscipoxvirus; Molluscum contagiosum virus; monkey B virus; monkeypox
virus;
Mononegavirales; Morbillivirus; Mount Elgon bat virus; mouse cytomegalovirus;
mouse
encephalomyelitis virus; mouse hepatitis virus; mouse K virus; mouse leukemia
virus; mouse
mammary tumor virus; mouse minute virus; mouse pneumonia virus; mouse
poliomyelitis virus;
mouse polyomavirus; mouse sarcoma virus; mousepox virus; Mozambique virus;
Mucambo
virus; mucosal disease virus; mumps virus; murid betaherpesvirus 1; murid
cytomegalovirus 2;
murine cytomegalovirus group; murine encephalomyelitis virus; murine hepatitis
virus; murine
leukemia virus; murine nodule inducing virus; murine polyomavirus; murine
sarcoma virus;
Muromegalovirus; Murray Valley encephalitis virus; myxoma virus; Myxovirus;
Myxovirus
multiforme; or Myxovirus parotitidis.
[0050] In some instances, the virus is Nairobi sheep disease virus;
Nairovirus; Nanirnavirus;
Nariva virus; Ndumo virus; Neethling virus; Nelson Bay virus; neurotropic
virus; New World
Arenavirus; newborn pneumonitis virus; Newcastle disease virus; Nipah virus;
noncytopathogenic virus; Norwalk virus; nuclear polyhedrosis virus (NPV);
nipple neck virus;
O'nyong'nyong virus; Ockelbo virus; oncogenic virus; oncogenic viruslike
particle;
oncornavirus; Orbivirus; Orf virus; Oropouche virus; Orthohepadnavirus;
Orthomyxovirus;
Orthopoxvirus; Orthoreovirus; Orungo; ovine papillomavirus; ovine catarrhal
fever virus; or owl
monkey herpesvirus.
18
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
[0051] In some instances, the virus is Palyam virus; Papillomavirus;
Papillomavirus sylvilagi;
Papovavirus; parainfluenza virus; parainfluenza virus type 1; parainfluenza
virus type 2;
parainfluenza virus type 3; parainfluenza virus type 4; Paramyxovirus;
Parapoxvirus;
paravaccinia virus; Parvovirus; Parvovirus B19; parvovirus group; Pestivirus;
Phlebovirus;
phocine distemper virus; Picodnavirus; Picornavirus; pig cytomegalovirus-
pigeonpox virus; Piry
virus; Pixuna virus; pneumonia virus of mice; Pneumovirus; poliomyelitis
virus; poliovirus;
Polydnavirus; polyhedral virus; polyoma virus; Polyomavirus; Polyomavirus
bovis;
Polyomavirus cercopitheci; Polyomavirus hominis 2; Polyomavirus maccacae 1;
Polyomavirus
muris 1; Polyomavirus muris 2; Polyomavirus papionis 1; Polyomavirus papionis
2;
Polyomavirus sylvilagi; Pongine herpesvirus 1; porcine epidemic diarrhea
virus; porcine
hemagglutinating encephalomyelitis virus; porcine parvovirus; porcine
transmissible
gastroenteritis virus; porcine type C virus; pox virus; poxvirus; poxvirus
variolae; Prospect Hill
virus; Provirus; pseudocowpox virus; pseudorabies virus; psittacinepox virus;
or quailpox virus.
[0052] In some instances, the virus is rabbit fibroma virus; rabbit kidney
vaculolating virus;
rabbit papillomavirus; rabies virus; raccoon parvovirus; raccoonpox virus;
Ranikhet virus; rat
cytomegalovirus; rat parvovirus; rat virus; Rauscher's virus; recombinant
vaccinia virus;
recombinant virus; reovirus; reovirus 1; reovirus 2; reovirus 3; reptilian
type C virus; respiratory
infection virus; respiratory syncytial virus; respiratory virus;
reticuloendotheliosis virus;
Rhabdovirus; Rhabdovirus carpia; Rhadinovirus; Rhinovirus; Rhizidiovirus; Rift
Valley fever
virus; Riley's virus; rinderpest virus; RNA tumor virus; Ross River virus;
Rotavirus; rougeole
virus; Rous sarcoma virus; rubella virus; rubeola virus; Rubivirus; or Russian
autumn
encephalitis virus.
[0053] In some instances, the virus is SA 11 simian virus; SA2 virus; Sabia
virus; Sagiyama
virus; Saimirine herpesvirus 1; salivary gland virus; sandfly fever virus
group; Sandjimba virus;
SARS virus; SDAV (sialodacryoadenitis virus); sealpox virus; Semliki Forest
Virus; Seoul
virus; sheeppox virus; Shope fibroma virus; Shope papilloma virus; simian
foamy virus; simian
hepatitis A virus; simian human immunodeficiency virus; simian
immunodeficiency virus;
simian parainfluenza virus; simian T cell lymphotrophic virus; simian virus;
simian virus 40;
Simplexvirus; Sin Nombre virus; Sindbis virus; smallpox virus; South American
hemorrhagic
fever viruses; sparrowpox virus; Spumavirus; squirrel fibroma virus; squirrel
monkey retrovirus;
SSV 1 virus group; STLV (simian T lymphotropic virus) type I; STLV (simian T
lymphotropic
virus) type II; STLV (simian T lymphotropic virus) type III; stomatitis
papulosa virus;
submaxillary virus; suid alphaherpesvirus 1; suid herpesvirus 2; Suipoxvirus;
swamp fever virus;
swinepox virus; or Swiss mouse leukemia virus.
19
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
[0054] In some instances, the virus is TAC virus; Tacaribe complex virus;
Tacaribe virus;
Tanapox virus; Taterapox virus; Tench reovirus; Theiler's encephalomyelitis
virus; Theiler's
virus; Thogoto virus; Thottapalayam virus; Tick borne encephalitis virus;
Tioman virus;
Togavirus; Torovirus; tumor virus; Tupaia virus; turkey rhinotracheitis virus;
turkeypox virus;
type C retroviruses; type D oncovirus; type D retrovirus group; ulcerative
disease rhabdovirus;
Una virus; Uukuniemi virus group; vaccinia virus; vacuolating virus; varicella
zoster virus;
Varicellovirus; Varicola virus; variola major virus; variola virus; Vasin
Gishu disease virus;
VEE virus; Venezuelan equine encephalitis virus; Venezuelan equine
encephalomyelitis virus;
Venezuelan hemorrhagic fever virus; vesicular stomatitis virus; Vesiculovirus;
Vilyuisk virus;
viper retrovirus; viral haemorrhagic septicemia virus; Visna Maedi virus;
Visna virus; volepox
virus; VSV (vesicular stomatitis virus); Wallal virus; Warrego virus; wart
virus; WEE virus;
West Nile virus; western equine encephalitis virus; western equine
encephalomyelitis virus;
Whataroa virus; Winter Vomiting Virus; woodchuck hepatitis B virus; woolly
monkey sarcoma
virus; wound tumor virus; WRSV virus; Yaba monkey tumor virus; Yaba virus;
Yatapoxvirus;
yellow fever virus; or the Yug Bogdanovac virus.
[0055] Methods described herein may detect low concentrations or amounts of
viruses in a
sample. In some instances, the amount of a virus is represented in terms of
the number of
genome copies (cp). In some instance, a method described herein detects about
1, 2, 5, 10, 15,
20, 25, 50, 100, 200, 500, 1000, 5000, 10,000, 50,000, 100,000, 500,000, or
about 1,000,000 cp
of a virus in a sample. In some instance, a method described herein detects no
more than 1, 2, 5,
10, 15, 20, 25, 50, 100, 200, 500, 1000, 5000, 10,000, 50,000, 100,000,
500,000, or no more
than 1,000,000 cp of a virus in a sample. In some instance, a method described
herein detects at
least 1, 2, 5, 10, 15, 20, 25, 50, 100, 200, 500, 1000, 5000, 10,000, 50,000,
100,000, 500,000, or
at least 1,000,000 cp of a virus in a sample. In some instance, a method
described herein detects
1-10, 1-100, 1-500, 1-1000, 1-5000, 1-10,000, 10-1000, 10-5000, 10-100,000,
100-10,000, 100-
100,000, 100-1,000,000, 1000-5000, 1000-10,000, 1000-50,000, or 50,000-
1,000,000 cp of a
virus in a sample.
[0056] Detection may be defined as a measured signal greater than a background
or control
signal. In some instances, detection is defined as a normalized reporter value
(ARn). In some
instances, the reporter value is obtained from a fluorescent signal. In some
instances, the
normalized reporter value is calculated as the experimental signal value minus
the background
signal. In some instances, the normalized reporter value is calculated as the
experimental signal
value minus the control signal. In some instances, a method described herein
produces a
normalized reporter value of at least 0.01, 0.02, 0.03, 0.04, 0.05, 0.06 or at
least 0.07. In some
instances, a method described herein produces a normalized reporter value of
at least 0.01, 0.02,
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
0.03, 0.04, 0.05, 0.06 or at least 0.07, for a sample comprising at least 1
cp. In some instances, a
method described herein produces a normalized reporter value of at least 0.01,
0.02, 0.03, 0.04,
0.05, 0.06 or at least 0.07, for a sample comprising at least 2 cp. In some
instances, a method
described herein produces a normalized reporter value of at least 0.01, 0.02,
0.03, 0.04, 0.05,
0.06 or at least 0.07, for a sample comprising at least 5 cp. In some
instances, a method
described herein produces a normalized reporter value of at least 0.01, 0.02,
0.03, 0.04, 0.05,
0.06 or at least 0.07, for a sample comprising at least 8 cp. In some
instances, a method
described herein produces a normalized reporter value of at least 0.01, 0.02,
0.03, 0.04, 0.05,
0.06 or at least 0.07, for a sample comprising at least 10 cp. In some
instances, a method
described herein produces a normalized reporter value of at least 0.01, 0.02,
0.03, 0.04, 0.05,
0.06 or at least 0.07, for a sample comprising at least 20 cp. In some
instances, a method
described herein produces a normalized reporter value of at least 0.01, 0.02,
0.03, 0.04, 0.05,
0.06 or at least 0.07, for a sample comprising at least 1 cp and subjected to
no more than 45 PCR
cycles. In some instances, a method described herein produces a normalized
reporter value of at
least 0.01, 0.02, 0.03, 0.04, 0.05, 0.06 or at least 0.07, for a sample
comprising at least 2 cp and
subjected to no more than 40 PCR cycles. In some instances, a method described
herein
produces a normalized reporter value of at least 0.01, 0.02, 0.03, 0.04, 0.05,
0.06 or at least 0.07,
for a sample comprising at least 5 cp and subjected to no more than 38 PCR
cycles. In some
instances, a method described herein produces a normalized reporter value of
at least 0.01, 0.02,
0.03, 0.04, 0.05, 0.06 or at least 0.07, for a sample comprising at least 8 cp
and subjected to no
more than 36 PCR cycles. In some instances, a method described herein produces
a normalized
reporter value of at least 0.01, 0.02, 0.03, 0.04, 0.05, 0.06 or at least
0.07, for a sample
comprising at least 10 cp and subjected to no more than 34 PCR cycles. In some
instances, a
method described herein produces a normalized reporter value of at least 0.01,
0.02, 0.03, 0.04,
0.05, 0.06 or at least 0.07, for a sample comprising at least 20 cp and
subjected to no more than
32 PCR cycles.
[0057] Described herein are methods and compositions for analysis of single
viral particles.
Such methods may comprise "Primary Template-Directed Amplification" (PTA) to
obtain
libraries of nucleic acids for sequencing. In some instances, PTA is combined
with additional
steps or methods such as RT-PCR or proteome/protein quantification techniques
(e.g., mass
spectrometry, antibody staining, etc.). In some instances, various components
of a cell are
physically or spatially separated from each other during individual analysis
steps. For example, a
workflow in some instances comprises the general steps of labeling proteins,
generating mRNA,
generating RT-PCR libraries, isolating genomic DNA, subjecting the genomic DNA
to PTA,
generating a gDNA library, and sequencing the two libraries. Proteins are
first labeled with
21
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
antibodies and sorted based on fluorescent markers. After RT-PCR, first strand
mRNA products
are generated and then removed for analysis. Libraries are then generated from
RT-PCR
products and barcodes present on protein-specific antibodies, which are
subsequently sequenced.
In parallel, genomic DNA from the same cell is subjected to PTA, a library
generated, and
sequenced. Sequencing results from the genome, proteome, and transcriptome are
in some
instances pooled using bioinformatics methods. Methods described herein in
some instances
comprise any combination of labeling, cell sorting, affinity
separation/purification, lysing of
specific cell components (e.g., outer membrane, nucleus, etc.), RNA
amplification, DNA
amplification (e.g., PTA), or other step associated with protein, RNA, or DNA
isolation or
analysis.
[0058] Described herein are methods of sample analysis comprising analysis of
RNA and
DNA from a sample source comprising a putative virus. In some instances, the
method
comprises isolation of single cells, lysis of single cells, and reverse
transcription (RT). In some
instances, reverse transcription is carried out with template switching
oligonucleotides (TS0s).
In some instances, TSOs comprise a molecular TAG such as biotin, which allows
subsequent
pull-down of cDNA RT products, and PCR amplification of RT products to
generate a cDNA
library. Alternatively or in combination, centrifugation is used to separate
RNA in the
supernatant from cDNA in the cell pellet. Remaining cDNA is in some instances
fragmented and
removed with UDG (uracil DNA glycosylase), and alkaline lysis is used to
degrade RNA and
denature the genome. After neutralization, addition of primers and PTA,
amplification products
are in some instances purified on SPRI (solid phase reversible immobilization)
beads, and
ligated to adapters to generate a gDNA library. In some instances, a pull-down
purification step
is not required.
[0059] Methods described herein (e.g., PTA) may be used as a replacement for
any number of
other known methods in the art which are used for single cell sequencing
(multiomics or the
like). PTA may substitute genomic DNA sequencing methods such as MBA,
PicoPlex, DOP-
PCR, MALBAC, or target-specific amplifications. In some instances, PTA
replaces the standard
genomic DNA sequencing method in a multiomics method including DR-seq (Dey et
al., 2015),
G&T seq (MacAulay et al., 2015), scMT-seq (Hu et al., 2016), sc-GEM (Cheow et
al., 2016),
scTrio-seq (Hou et al., 2016), simultaneous multiplexed measurement of RNA and
proteins
(Darmanis et al., 2016), scCOOL-seq (Guo et al., 2017), CITE-seq (Stoeckius et
al., 2017),
REAP-seq (Peterson et al., 2017), scNMT-seq (Clark et al., 2018), or SIDR-seq
(Han et al.,
2018). In some instances, a method described herein comprises PTA and a method
of
polyadenylated mRNA transcripts. In some instances, a method described herein
comprises PTA
and a method of non-polyadenylated mRNA transcripts. In some instances, a
method described
22
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
herein comprises PTA and a method of total (polyadenylated and non-
polyadenylated) mRNA
transcripts.
[0060] In some instances, PTA is combined with a standard RNA sequencing
method to
obtain genome and transcriptome data. In some instances, a multiomics method
described herein
comprises PTA and one of the following: Drop-seq (Macosko, et al. 2015), mRNA-
seq (Tang et
al., 2009), InDrop (Klein et al., 2015), MARS-seq (Jaitin et al., 2014), Smart-
seq2
(Hashimshony, et al., 2012; Fish et al., 2016), CEL-seq (Jaitin et al., 2014),
STRT-seq (Islam, et
al., 2011), Quartz-seq (Sasagawa et al., 2013), CEL-seq2 (Hashimshony, et al.
2016), cytoSeq
(Fan et al., 2015), SuPeR-seq (Fan et al., 2011), RamDA-seq (Hayashi, et al.
2018), MATQ-seq
(Sheng et al., 2017), or SMARTer (Verboom et al., 2019).
[0061] RT reactions may be used to reverse transcribe RNA (e.g., viral RNA).
Various
reaction conditions and mixes are in some instances used for generating cDNA
libraries for
transcriptome analysis of virus-containing samples, wherein the cDNA libraries
are analyzed by
methods such as LAMP or PTA. In some instances, an RT reaction mix is used to
generate a
cDNA library. In some instances, the RT reaction mixture comprises a crowding
reagent, at least
one primer, a template switching oligonucleotide (TSO), a reverse
transcriptase, and a dNTP
mix. In some instances, an RT reaction mix comprises an RNAse inhibitor. In
some instances an
RT reaction mix comprises one or more surfactants. In some instances an RT
reaction mix
comprises Tween-20 and/or Triton-X. In some instances an RT reaction mix
comprises Betaine.
In some instances an RT reaction mix comprises one or more salts. In some
instances an RT
reaction mix comprises a magnesium salt (e.g., magnesium chloride) and/or
tetramethylammonium chloride. In some instances an RT reaction mix comprises
gelatin. In
some instances an RT reaction mix comprises PEG (PEG1000, PEG2000, PEG4000,
PEG6000,
PEG8000, or PEG of other length). In some instances an RT reaction mix
contains gelatin or
bovine serum albumin.
Primary Template-Directed Amplification
[0062] Described herein are nucleic acid amplification methods, such as
"Primary Template-
Directed Amplification (PTA)." Such methods in some instances are combined
with reverse
transcription. In some instance, PTA is used to detect low amounts of viral
cDNA. With the
PTA method, amplicons are preferentially generated from the primary template
("direct copies")
using a polymerase (e.g., a strand displacing polymerase). Consequently,
errors are propagated
at a lower rate from daughter amplicons during subsequent amplifications
compared to MDA.
The result is an easily executed method that, unlike existing WGA protocols,
can amplify low
DNA input including the genomes of single cells with high coverage breadth and
uniformity in
an accurate and reproducible manner. Moreover, the terminated amplification
products can
23
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
undergo direction ligation after removal of the terminators, allowing for the
attachment of a cell
barcode to the amplification primers so that products from all cells can be
pooled after
undergoing parallel amplification reactions. In some instances, terminator
removal is not
required prior to amplification and/or adapter ligation.
[0063] Described herein are methods employing nucleic acid polymerases with
strand
displacement activity for amplification. In some instances, such polymerases
comprise strand
displacement activity and low error rate. In some instances, such polymerases
comprise strand
displacement activity and proofreading exonuclease activity, such as 3'->5'
proofreading
activity. In some instances, nucleic acid polymerases are used in conjunction
with other
components such as reversible or irreversible terminators, or additional
strand displacement
factors. In some instances, the polymerase has strand displacement activity,
but does not have
exonuclease proofreading activity. For example, in some instances such
polymerases include
bacteriophage phi29 (129) polymerase, which also has very low error rate that
is the result of
the 3'->5' proofreading exonuclease activity (see, e.g., U.S. Pat. Nos.
5,198,543 and 5,001,050).
In some instances, non-limiting examples of strand displacing nucleic acid
polymerases include,
e.g., genetically modified phi29 (129) DNA polymerase, Klenow Fragment of DNA
polymerase
I (Jacobsen et al., Eur. J. Biochem. 45:623-627 (1974)), phage M2 DNA
polymerase
(Matsumoto et al., Gene 84:247 (1989)), phage phiPRD1 DNA polymerase (Jung et
al., Proc.
Natl. Acad. Sci. USA 84:8287 (1987); Zhu and Ito, Biochim. Biophys. Acta.
1219:267-276
(1994)), Bst DNA polymerase (e.g., Bst large fragment DNA polymerase (Exo(-)
Bst; Aliotta et
al., Genet. Anal. (Netherlands) 12:185-195 (1996)), exo(-)Bca DNA polymerase
(Walker and
Linn, Clinical Chemistry 42:1604-1608 (1996)), Bsu DNA polymerase, VentR DNA
polymerase
including VentR (exo-) DNA polymerase (Kong et al., J. Biol. Chem. 268:1965-
1975 (1993)),
Deep Vent DNA polymerase including Deep Vent (exo-) DNA polymerase, IsoPol DNA
polymerase, DNA polymerase I, Therminator DNA polymerase, T5 DNA polymerase
(Chatterjee et al., Gene 97:13-19 (1991)), Sequenase (U.S. Biochemicals), T7
DNA polymerase,
T7-Sequenase, T7 gp5 DNA polymerase, PRDI DNA polymerase, T4 DNA polymerase
(Kaboord and Benkovic, Curr. Biol. 5:149-157 (1995)). Additional strand
displacing nucleic
acid polymerases are also compatible with the methods described herein. The
ability of a given
polymerase to carry out strand displacement replication can be determined, for
example, by
using the polymerase in a strand displacement replication assay (e.g., as
disclosed in U.S. Pat.
No. 6,977,148). Such assays in some instances are performed at a temperature
suitable for
optimal activity for the enzyme being used, for example, 32 C for phi29 DNA
polymerase, from
46 C to 64 C for exo(-) Bst DNA polymerase, or from about 60 C to 70 C for an
enzyme from a
hyperthermophylic organism. Another useful assay for selecting a polymerase is
the primer-
24
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
block assay described in Kong et al., J. Biol. Chem. 268:1965-1975 (1993). The
assay consists
of a primer extension assay using an M13 ssDNA template in the presence or
absence of an
oligonucleotide that is hybridized upstream of the extending primer to block
its progress. Other
enzymes capable of displacement the blocking primer in this assay are in some
instances useful
for the disclosed method. In some instances, polymerases incorporate dNTPs and
terminators at
approximately equal rates. In some instances, the ratio of rates of
incorporation for dNTPs and
terminators for a polymerase described herein are about 1:1, about 1.5:1,
about 2:1, about 3:1
about 4:1 about 5:1, about 10:1, about 20:1 about 50:1, about 100:1, about
200:1, about 500:1,
or about 1000:1. In some instances, the ratio of rates of incorporation for
dNTPs and terminators
for a polymerase described herein are 1:1 to 1000:1,2:1 to 500:1, 5:1 to
100:1, 10:1 to 1000:1,
100:1 to 1000:1, 500:1 to 2000:1, 50:1 to 1500:1, or 25:1 to 1000:1.
[0064] Described herein are methods of amplification wherein strand
displacement can be
facilitated through the use of a strand displacement factor, such as, e.g.,
helicase. Such factors
are in some instances used in conjunction with additional amplification
components, such as
polymerases, terminators, or other component. In some instances, a strand
displacement factor is
used with a polymerase that does not have strand displacement activity. In
some instances, a
strand displacement factor is used with a polymerase having strand
displacement activity.
Without being bound by theory, strand displacement factors may increase the
rate that smaller,
double stranded amplicons are reprimed. In some instances, any DNA polymerase
that can
perform strand displacement replication in the presence of a strand
displacement factor is
suitable for use in the PTA method, even if the DNA polymerase does not
perform strand
displacement replication in the absence of such a factor. Strand displacement
factors useful in
strand displacement replication in some instances include (but are not limited
to) BMRF1
polymerase accessory subunit (Tsurumi et al., J. Virology 67(12):7648-7653
(1993)), adenovirus
DNA-binding protein (Zijderveld and van der Vliet, J. Virology 68(2): 1158-
1164 (1994)),
herpes simplex viral protein ICP8 (Boehmer and Lehman, J. Virology 67(2):711-
715 (1993);
Skaliter and Lehman, Proc. Natl. Acad. Sci. USA 91(22):10665-10669 (1994));
single-stranded
DNA binding proteins (SSB; Rigler and Romano, J. Biol. Chem. 270:8910-8919
(1995)); phage
T4 gene 32 protein (Villemain and Giedroc, Biochemistry 35:14395-14404
(1996);T7 helicase-
primase; T7 gp2.5 SSB protein; Tte-UvrD (from Thermoanaerobacter
tengcongensis), calf
thymus helicase (Siegel et al., J. Biol. Chem. 267:13629-13635 (1992));
bacterial SSB (e.g., E.
coil SSB), Replication Protein A (RPA) in eukaryotes, human mitochondrial SSB
(mtSSB), and
recombinases, (e.g., Recombinase A (RecA) family proteins, T4 UvsX, 5ak4 of
Phage HK620,
Rad51, Dmcl, or Radb). Combinations of factors that facilitate strand
displacement and priming
are also consistent with the methods described herein. For example, a helicase
is used in
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
conjunction with a polymerase. In some instances, the PTA method comprises use
of a single-
strand DNA binding protein (SSB, T4 gp32, or other single stranded DNA binding
protein), a
helicase, and a polymerase (e.g., SauDNA polymerase, Bsu polymerase, Bst2.0,
GspM,
GspM2.0, GspSSD, or other suitable polymerase). In some instances, reverse
transcriptases are
used in conjunction with the strand displacement factors described herein.
[0065] Described herein are amplification methods comprising use of terminator
nucleotides,
polymerases, and additional factors or conditions. For example, such factors
are used in some
instances to fragment the nucleic acid template(s) or amplicons during
amplification. In some
instances, such factors comprise endonucleases. In some instances, factors
comprise
transposases. In some instances, mechanical shearing is used to fragment
nucleic acids during
amplification. In some instances, nucleotides are added during amplification
that may be
fragmented through the addition of additional proteins or conditions. For
example, uracil is
incorporated into amplicons; treatment with uracil D-glycosylase fragments
nucleic acids at
uracil-containing positions. Additional systems for selective nucleic acid
fragmentation are also
in some instances employed, for example an engineered DNA glycosylase that
cleaves modified
cytosine-pyrene base pairs. (Kwon, et al. Chem Biol. 2003, 10(4), 351).
[0066] Described herein are amplification methods comprising use of terminator
nucleotides,
which terminate nucleic acid replication thus decreasing the size of the
amplification products.
Such terminators are in some instances used in conjunction with polymerases,
strand
displacement factors, or other amplification components described herein. In
some instances,
terminator nucleotides reduce or lower the efficiency of nucleic acid
replication. Such
terminators in some instances reduce extension rates by at least 99.9%, 99%,
98%, 95%, 90%,
85%, 80%, 75%, 70%, or at least 65%. Such terminators in some instances reduce
extension
rates by 50%-90%, 60%-80%, 65%-90%, 70%-85%, 60%-90%, 70%-99%, 80%-99%, or 50%-
80%. In some instances terminators reduce the average amplicon product length
by at least
99.9%, 99%, 98%, 95%, 90%, 85%, 80%, 75%, 70%, or at least 65%. Terminators in
some
instances reduce the average amplicon length by 50%-90%, 60%-80%, 65%-90%, 70%-
85%,
60%-90%, 70%-99%, 80%-99%, or 50%-80%. In some instances, amplicons comprising
terminator nucleotides form loops or hairpins which reduce a polymerase's
ability to use such
amplicons as templates. Use of terminators in some instances slows the rate of
amplification at
initial amplification sites through the incorporation of terminator
nucleotides (e.g.,
dideoxynucleotides that have been modified to make them exonuclease-resistant
to terminate
DNA extension), resulting in smaller amplification products. By producing
smaller
amplification products than the currently used methods (e.g., average length
of 50-2000
nucleotides in length for PTA methods as compared to an average product length
of >10,000
26
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
nucleotides for MDA methods) PTA amplification products in some instances
undergo direct
ligation of adapters without the need for fragmentation, allowing for
efficient incorporation of
cell barcodes and unique molecular identifiers (UMI).
[0067] Terminator nucleotides are present at various concentrations depending
on factors such
as polymerase, template, or other factors. For example, the amount of
terminator nucleotides in
some instances is expressed as a ratio of non-terminator nucleotides to
terminator nucleotides in
a method described herein. Such concentrations in some instances allow control
of amplicon
lengths. In some instances, the ratio of non-terminator to terminator
nucleotides is about 2:1,
5:1,7:1, 10:1, 20:1, 50:1, 100:1, 200:1, 500:1, 1000:1, 2000:1, or 5000:1. In
some instances the
ratio of non-terminator to terminator nucleotides is 2:1-10:1, 5:1-20:1, 10:1-
100:1,20:1-200:1,
50:1-1000:1, 50:1-500:1, 75:1-150:1, or 100:1-500:1. In some instances, at
least one of the
nucleotides present during amplification using a method described herein is a
terminator
nucleotide. Each terminator need not be present at approximately the same
concentration; in
some instances, ratios of each terminator present in a method described herein
are optimized for
a particular set of reaction conditions, sample type, or polymerase. Without
being bound by
theory, each terminator may possess a different efficiency for incorporation
into the growing
polynucleotide chain of an amplicon, in response to pairing with the
corresponding nucleotide
on the template strand. For example, in some instances, a terminator pairing
with cytosine is
present at about 3%, 5%, 10%, 15%, 20%, 25%, or 50% higher concentration than
the average
terminator concentration. In some instances, a terminator pairing with thymine
is present at
about 3%, 5%, 10%, 15%, 20%, 25%, or 50% higher concentration than the average
terminator
concentration. In some instances, a terminator pairing with guanine is present
at about 3%, 5%,
10%, 15%, 20%, 25%, or 50% higher concentration than the average terminator
concentration.
In some instances, a terminator pairing with adenine is present at about 3%,
5%, 10%, 15%,
20%, 25%, or 50% higher concentration than the average terminator
concentration. In some
instances, a terminator pairing with uracil is present at about 3%, 5%, 10%,
15%, 20%, 25%, or
50% higher concentration than the average terminator concentration. Any
nucleotide capable of
terminating nucleic acid extension by a nucleic acid polymerase in some
instances is used as a
terminator nucleotide in the methods described herein. In some instances, a
reversible terminator
is used to terminate nucleic acid replication. In some instances, a non-
reversible terminator is
used to terminate nucleic acid replication. In some instances, non-limited
examples of
terminators include reversible and non-reversible nucleic acids and nucleic
acid analogs, such
as, e.g., 3' blocked reversible terminator comprising nucleotides, 3'
unblocked reversible
terminator comprising nucleotides, terminators comprising 2' modifications of
deoxynucleotides, terminators comprising modifications to the nitrogenous base
of
27
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
deoxynucleotides, or any combination thereof. In one embodiment, terminator
nucleotides are
dideoxynucleotides. Other nucleotide modifications that terminate nucleic acid
replication and
may be suitable for practicing the invention include, without limitation, any
modifications of the
r group of the 3' carbon of the deoxyribose such as inverted
dideoxynucleotides, 3' biotinylated
nucleotides, 3' amino nucleotides, 3'-phosphorylated nucleotides, 3'-0-methyl
nucleotides, 3'
carbon spacer nucleotides including 3' C3 spacer nucleotides, 3' C18
nucleotides, 3' Hexanediol
spacer nucleotides, acyclonucleotides, and combinations thereof In some
instances, terminators
are polynucleotides comprising 1, 2, 3, 4, or more bases in length. In some
instances, terminators
do not comprise a detectable moiety or tag (e.g., mass tag, fluorescent tag,
dye, radioactive
atom, or other detectable moiety). In some instances, terminators do not
comprise a chemical
moiety allowing for attachment of a detectable moiety or tag (e.g., "click"
azide/alkyne,
conjugate addition partner, or other chemical handle for attachment of a tag).
In some instances,
all terminator nucleotides comprise the same modification that reduces
amplification to at region
(e.g., the sugar moiety, base moiety, or phosphate moiety) of the nucleotide.
In some instances,
at least one terminator has a different modification that reduces
amplification. In some instances,
all terminators have a substantially similar fluorescent excitation or
emission wavelengths. In
some instances, terminators without modification to the phosphate group are
used with
polymerases that do not have exonuclease proofreading activity. Terminators,
when used with
polymerases which have 3'->5' proofreading exonuclease activity (such as,
e.g., phi29) that can
remove the terminator nucleotide, are in some instances further modified to
make them
exonuclease-resistant. For example, dideoxynucleotides are modified with an
alpha-thio group
that creates a phosphorothioate linkage which makes these nucleotides
resistant to the 3'->5'
proofreading exonuclease activity of nucleic acid polymerases. Such
modifications in some
instances reduce the exonuclease proofreading activity of polymerases by at
least 99.5%, 99%,
98%, 95%, 90%, or at least 85%. Non-limiting examples of other terminator
nucleotide
modifications providing resistance to the 3'->5' exonuclease activity include
in some instances:
nucleotides with modification to the alpha group, such as alpha-thio
dideoxynucleotides creating
a phosphorothioate bond, C3 spacer nucleotides, locked nucleic acids (LNA),
inverted nucleic
acids, 2' Fluoro bases, 3' phosphorylation, 2'-0-Methyl modifications (or
other 2'-0-alkyl
modification), propyne-modified bases (e.g., deoxycytosine, deoxyuridine), L-
DNA nucleotides,
L-RNA nucleotides, nucleotides with inverted linkages (e.g., 5'-5' or 3'-3'),
5' inverted bases
(e.g., 5' inverted 2',3'-dideoxy dT), methylphosphonate backbones, and trans
nucleic acids. In
some instances, nucleotides with modification include base-modified nucleic
acids comprising
free 3' OH groups (e.g., 2-nitrobenzyl alkylated HOMedU triphosphates, bases
comprising
modification with large chemical groups, such as solid supports or other large
moiety). In some
28
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
instances, a polymerase with strand displacement activity but without 3'-
>5'exonuclease
proofreading activity is used with terminator nucleotides with or without
modifications to make
them exonuclease resistant. Such nucleic acid polymerases include, without
limitation, Bst DNA
polymerase, Bsu DNA polymerase, Deep Vent (exo-) DNA polymerase, Klenow
Fragment
(exo-) DNA polymerase, Therminator DNA polymerase, and VentR (exo-).
Primers and Amplicon Libraries
[0068] Described herein are amplicon libraries resulting from amplification of
at least one
target nucleic acid molecule (e.g., viral nucleic acid). Such libraries are in
some instances
generated using the methods described herein, such as those using terminators.
Such methods
comprise use of strand displacement polymerases or factors, terminator
nucleotides (reversible
or irreversible), or other features and embodiments described herein. In some
instances,
amplicon libraries generated by use of terminators described herein are
further amplified in a
subsequent amplification reaction (e.g., PCR). In some instances, subsequent
amplification
reactions do not comprise terminators. In some instances, amplicon libraries
comprise
polynucleotides, wherein at least 50%, 60%, 70%, 80%, 90%, 95%, or at least
98% of the
polynucleotides comprise at least one terminator nucleotide. In some
instances, the amplicon
library comprises the target nucleic acid molecule from which the amplicon
library was derived.
The amplicon library comprises a plurality of polynucleotides, wherein at
least some of the
polynucleotides are direct copies (e.g., replicated directly from a target
nucleic acid molecule,
such as genomic DNA, RNA, or other target nucleic acid). For example, at least
0.5%, 1%, 2%,
3%, 4%, 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or more than 95%
of the
amplicon polynucleotides are direct copies of the at least one target nucleic
acid molecule. In
some instances, at least 5% of the amplicon polynucleotides are direct copies
of the at least one
target nucleic acid molecule. In some instances, at least 10% of the amplicon
polynucleotides
are direct copies of the at least one target nucleic acid molecule. In some
instances, at least 15%
of the amplicon polynucleotides are direct copies of the at least one target
nucleic acid molecule.
In some instances, at least 20% of the amplicon polynucleotides are direct
copies of the at least
one target nucleic acid molecule. In some instances, at least 50% of the
amplicon
polynucleotides are direct copies of the at least one target nucleic acid
molecule. In some
instances, 1.5-50%, 1.5-10%, 1.5-30%, 3-50%, 3%-5%, 3-10%, 5%-10%, 10%-20%,
20%-30%,
30%-40%, 5%-30%, 10%-50%, or 15%-75% of the amplicon polynucleotides are
direct copies
of the at least one target nucleic acid molecule. In some instances, at least
some of the
polynucleotides are direct copies of the target nucleic acid molecule, or
daughter (a first copy of
the target nucleic acid) progeny. For example, at least 0.1%, 1%, 2%, 3%, 4%,
5%, 10%, 20%,
30%, 40%, 50%, 60%, 70%, 80%, 90%, 95% or more than 95% of the amplicon
polynucleotides
29
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
are direct copies of the at least one target nucleic acid molecule or daughter
progeny. In some
instances, at least 1.5% of the amplicon polynucleotides are direct copies of
the at least one
target nucleic acid molecule or daughter progeny. In some instances, at least
3% of the amplicon
polynucleotides are direct copies of the at least one target nucleic acid
molecule or daughter
progeny. In some instances, at least 5% of the amplicon polynucleotides are
direct copies of the
at least one target nucleic acid molecule or daughter progeny. In some
instances, at least 10% of
the amplicon polynucleotides are direct copies of the at least one target
nucleic acid molecule or
daughter progeny. In some instances, at least 20% of the amplicon
polynucleotides are direct
copies of the at least one target nucleic acid molecule or daughter progeny.
In some instances, at
least 30% of the amplicon polynucleotides are direct copies of the at least
one target nucleic acid
molecule or daughter progeny. In some instances, 1.5-50%, 1.5-10%, 1.5-30%, 3%-
5%, 3%-
10%, 5%-10%, 10%-20%, 20%-30%, 30%-40%, 5%-30%, 10%-50%, or 15%-75% of the
amplicon polynucleotides are direct copies of the at least one target nucleic
acid molecule or
daughter progeny. In some instances, direct copies of the target nucleic acid
are 50-2500, 75-
2000, 50-2000, 25-1000, 50-1000, 500-2000, or 50-2000 bases in length. In some
instances,
daughter progeny are 1000-5000, 2000-5000, 1000-10,000, 2000-5000, 1500-5000,
3000-7000,
or 2000-7000 bases in length. In some instances, the average length of PTA
amplification
products is 25-3000 nucleotides in length, 50-2500, 75-2000, 50-2000, 25-1000,
50-1000, 500-
2000, or 50-2000 bases in length. In some instance, amplicons generated from
PTA are no more
than 5000, 4000, 3000, 2000, 1700, 1500, 1200, 1000, 700, 500, or no more than
300 bases in
length. In some instance, amplicons generated from PTA are 1000-5000, 1000-
3000, 200-2000,
200-4000, 500-2000, 750-2500, or 1000-2000 bases in length. Amplicon libraries
generated
using the methods described herein in some instances comprise at least 1000,
2000, 5000,
10,000, 100,000, 200,000, 500,000 or more than 500,000 amplicons comprising
unique
sequences. In some instances, the library comprises at least 100, 200, 300,
400, 500, 600, 700,
800, 900, 1000, 1100, 1200, 1300, 1400, 1500, 2000, 2500, 3000, or at least
3500 amplicons. In
some instances, at least 0.5%, 1%, 1.5%, 2%, 3%, 4%, 5%, 10%, 15%, 20%, 25%,
30% or more
than 30% of amplicon polynucleotides having a length of less than 1000 bases
are direct copies
of the at least one target nucleic acid molecule. In some instances, at least
0.5%, 1%, 1.5%, 2%,
3%, 4%, 5%, 10%, 15%, 20%, 25%, 30% or more than 30% of amplicon
polynucleotides having
a length of no more than 2000 bases are direct copies of the at least one
target nucleic acid
molecule. In some instances, at least 0.5%, 1%, 1.5%, 2%, 3%, 4%, 5%, 10%,
15%, 20%, 25%,
30% or more than 30% of amplicon polynucleotides having a length of 3000-5000
bases are
direct copies of the at least one target nucleic acid molecule. In some
instances, the ratio of
direct copy amplicons to target nucleic acid molecules is at least 10:1,
100:1, 1000:1, 10,000:1,
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
100,000:1, 1,000,000:1, 10,000,000:1, or more than 10,000,000:1. In some
instances, the ratio of
direct copy amplicons to target nucleic acid molecules is at least 10:1,
100:1, 1000:1, 10,000:1,
100,000:1, 1,000,000:1, 10,000,000:1, or more than 10,000,000:1, wherein the
direct copy
amplicons are no more than 700-1200 bases in length. In some instances, the
ratio of direct copy
amplicons and daughter amplicons to target nucleic acid molecules is at least
10:1, 100:1,
1000:1, 10,000:1, 100,000:1, 1,000,000:1, 10,000,000:1, or more than
10,000,000:1. In some
instances, the ratio of direct copy amplicons and daughter amplicons to target
nucleic acid
molecules is at least 10:1, 100:1, 1000:1, 10,000:1, 100,000:1, 1,000,000:1,
10,000,000:1, or
more than 10,000,000:1, wherein the direct copy amplicons are 700-1200 bases
in length, and
the daughter amplicons are 2500-6000 bases in length. In some instances, the
library comprises
about 50-10,000, about 50-5,000, about 50-2500, about 50-1000, about 150-2000,
about 250-
3000, about 50-2000, about 500-2000, or about 500-1500 amplicons which are
direct copies of
the target nucleic acid molecule. In some instances, the library comprises
about 50-10,000, about
50-5,000, about 50-2500, about 50-1000, about 150-2000, about 250-3000, about
50-2000,
about 500-2000, or about 500-1500 amplicons which are direct copies of the
target nucleic acid
molecule or daughter amplicons. The number of direct copies may be controlled
in some
instances by the number of PCR amplification cycles. In some instances, no
more than 30, 25,
20, 15, 13, 11, 10, 9, 8, 7, 6, 5, 4, or 3 PCR cycles are used to generate
copies of the target
nucleic acid molecule. In some instances, about 30, 25, 20, 15, 13, 11, 10, 9,
8, 7, 6, 5, 4, or
about 3 PCR cycles are used to generate copies of the target nucleic acid
molecule. In some
instances, 3, 4, 5, 6, 7, or 8 PCR cycles are used to generate copies of the
target nucleic acid
molecule. In some instances, 2-4, 2-5, 2-7, 2-8, 2-10, 2-15, 3-5, 3-10, 3-15,
4-10, 4-15, 5-10 or
5-15 PCR cycles are used to generate copies of the target nucleic acid
molecule. Amplicon
libraries generated using the methods described herein are in some instances
subjected to
additional steps, such as adapter ligation and further PCR amplification. In
some instances, such
additional steps precede a sequencing step. In some instances, the method
comprises
amplification of a genomic or fragment thereof in the presence of at least one
terminator
nucleotide, wherein the number of amplification cycles is less than 12, 10, 9,
8, 7, 6, 5, 4, or less
than 3 cycles. In some instances, the average length of amplification products
is 100-1000, 200-
500, 200-700, 300-700, 400-1000, or 500-1200 bases in length. In some
instances, the method
comprises amplification of a genomic or fragment thereof in the presence of at
least one
terminator nucleotide, wherein the number of amplification cycles is no more
than 6 cycles. In
some instances, the at least one terminator nucleotide does comprise a
detectable label or tag. In
some instances, the amplification comprises 2, 3, or 4 terminator nucleotides.
In some instances,
at least two of the terminator nucleotides comprise a different base. In some
instances, at least
31
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
three of the terminator nucleotides comprise a different base. In some
instances, four terminator
nucleotides each comprise a different base. The number of direct copies may be
controlled in
some instances by the number of amplification cycles. In some instances, no
more than 30, 25,
20, 15, 13, 11, 10, 9, 8, 7, 6, 5, 4, or 3 cycles are used to generate copies
of the target nucleic
acid molecule. In some instances, about 30, 25, 20, 15, 13, 11, 10, 9, 8, 7,
6, 5, 4, or about 3
cycles are used to generate copies of the target nucleic acid molecule. In
some instances, 3, 4, 5,
6, 7, or 8 cycles are used to generate copies of the target nucleic acid
molecule. In some
instances, 2-4, 2-5, 2-7, 2-8, 2-10, 2-15, 3-5, 3-10, 3-15, 4-10, 4-15, 5-10
or 5-15 cycles are used
to generate copies of the target nucleic acid molecule. Amplicon libraries
generated using the
methods described herein are in some instances subjected to additional steps,
such as adapter
ligation and further amplification. In some instances, such additional steps
precede a sequencing
step. In some instances, the cycles are PCR cycles. In some instances, the
cycles represent
annealing, extension, and denaturation. In some instances, the cycles
represent annealing,
extension, and denaturation which occur under isothermal or essentially
isothermal conditions.
[0069] Amplicon libraries of polynucleotides generated from the PTA methods
and
compositions (terminators, polymerases, etc.) described herein in some
instances have increased
uniformity. Uniformity, in some instances, is described using a Lorenz curve
or other such
method. Such increases in some instances lead to lower sequencing reads needed
for the desired
coverage of a target nucleic acid molecule (e.g., genomic DNA, RNA, or other
target nucleic
acid molecule). For example, no more than 50% of a cumulative fraction of
polynucleotides
comprises sequences of at least 80% of a cumulative fraction of sequences of
the target nucleic
acid molecule. In some instances, no more than 50% of a cumulative fraction of
polynucleotides
comprises sequences of at least 60% of a cumulative fraction of sequences of
the target nucleic
acid molecule. In some instances, no more than 50% of a cumulative fraction of
polynucleotides
comprises sequences of at least 70% of a cumulative fraction of sequences of
the target nucleic
acid molecule. In some instances, no more than 50% of a cumulative fraction of
polynucleotides
comprises sequences of at least 90% of a cumulative fraction of sequences of
the target nucleic
acid molecule. In some instances, uniformity is described using a Gini index
(wherein an index
of 0 represents perfect equality of the library and an index of 1 represents
perfect inequality). In
some instances, amplicon libraries described herein have a Gini index of no
more than 0.55,
0.50, 0.45, 0.40, or 0.30. In some instances, amplicon libraries described
herein have a Gini
index of no more than 0.50. In some instances, amplicon libraries described
herein have a Gini
index of no more than 0.40. Such uniformity metrics in some instances are
dependent on the
number of reads obtained. For example no more than 100 million, 200 million,
300 million, 400
million, or no more than 500 million reads are obtained. In some instances,
the read length is
32
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
about 50,75, 100, 125, 150, 175, 200, 225, or about 250 bases in length. In
some instances,
uniformity metrics are dependent on the depth of coverage of a target nucleic
acid. For example,
the average depth of coverage is about 10X, 15X, 20X, 25X, or about 30X. In
some instances,
the average depth of coverage is 10-30X, 20-50X, 5-40X, 20-60X, 5-20X, or 10-
20X. In some
instances, amplicon libraries described herein have a Gini index of no more
than 0.55, wherein
about 300 million reads was obtained. In some instances, amplicon libraries
described herein
have a Gini index of no more than 0.50, wherein about 300 million reads was
obtained. In some
instances, amplicon libraries described herein have a Gini index of no more
than 0.45, wherein
about 300 million reads was obtained. In some instances, amplicon libraries
described herein
have a Gini index of no more than 0.55, wherein no more than 300 million reads
was obtained.
In some instances, amplicon libraries described herein have a Gini index of no
more than 0.50,
wherein no more than 300 million reads was obtained. In some instances,
amplicon libraries
described herein have a Gini index of no more than 0.45, wherein no more than
300 million
reads was obtained. In some instances, amplicon libraries described herein
have a Gini index of
no more than 0.55, wherein the average depth of sequencing coverage is about
15X. In some
instances, amplicon libraries described herein have a Gini index of no more
than 0.50, wherein
the average depth of sequencing coverage is about 15X. In some instances,
amplicon libraries
described herein have a Gini index of no more than 0.45, wherein the average
depth of
sequencing coverage is about 15X. In some instances, amplicon libraries
described herein have a
Gini index of no more than 0.55, wherein the average depth of sequencing
coverage is at least
15X. In some instances, amplicon libraries described herein have a Gini index
of no more than
0.50, wherein the average depth of sequencing coverage is at least 15X. In
some instances,
amplicon libraries described herein have a Gini index of no more than 0.45,
wherein the average
depth of sequencing coverage is at least 15X. In some instances, amplicon
libraries described
herein have a Gini index of no more than 0.55, wherein the average depth of
sequencing
coverage is no more than 15X. In some instances, amplicon libraries described
herein have a
Gini index of no more than 0.50, wherein the average depth of sequencing
coverage is no more
than 15X. In some instances, amplicon libraries described herein have a Gini
index of no more
than 0.45, wherein the average depth of sequencing coverage is no more than
15X. Uniform
amplicon libraries generated using the methods described herein are in some
instances subjected
to additional steps, such as adapter ligation and further PCR amplification.
In some instances,
such additional steps precede a sequencing step.
[0070] Primers comprise nucleic acids used for priming the amplification
reactions described
herein. Such primers in some instances include, without limitation, random
deoxynucleotides of
any length with or without modifications to make them exonuclease resistant,
random
33
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
ribonucleotides of any length with or without modifications to make them
exonuclease resistant,
modified nucleic acids such as locked nucleic acids, DNA or RNA primers that
are targeted to a
specific genomic region, and reactions that are primed with enzymes such as
primase. In the
case of whole genome PTA, it is preferred that a set of primers having random
or partially
random nucleotide sequences be used. In a nucleic acid sample of significant
complexity,
specific nucleic acid sequences present in the sample need not be known and
the primers need
not be designed to be complementary to any particular sequence. Rather, the
complexity of the
nucleic acid sample results in a large number of different hybridization
target sequences in the
sample, which will be complementary to various primers of random or partially
random
sequence. The complementary portion of primers for use in PTA are in some
instances fully
randomized, comprise only a portion that is randomized, or be otherwise
selectively randomized.
The number of random base positions in the complementary portion of primers in
some
instances, for example, is from 20% to 100% of the total number of nucleotides
in the
complementary portion of the primers. In some instances, the number of random
base positions
in the complementary portion of primers is 10% to 90%, 15-95%, 20%-100%, 30%-
100%, 50%-
100%, 75-100% or 90-95% of the total number of nucleotides in the
complementary portion of
the primers. In some instances, the number of random base positions in the
complementary
portion of primers is at least 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, or at
least 90% of the
total number of nucleotides in the complementary portion of the primers. Sets
of primers having
random or partially random sequences are in some instances synthesized using
standard
techniques by allowing the addition of any nucleotide at each position to be
randomized. In
some instances, sets of primers are composed of primers of similar length
and/or hybridization
characteristics. In some instances, the term "random primer" refers to a
primer which can exhibit
four-fold degeneracy at each position. In some instances, the term "random
primer" refers to a
primer which can exhibit three-fold degeneracy at each position. Random
primers used in the
methods described herein in some instances comprise a random sequence that is
3, 4, 5, 6, 7, 8,
10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, or more bases in length. In some
instances, primers
comprise random sequences that are 3-20, 5-15, 5-20, 6-12, or 4-10 bases in
length. Primers may
also comprise non-extendable elements that limit subsequent amplification of
amplicons
generated thereof. For example, primers with non-extendable elements in some
instances
comprise terminators. In some instances, primers comprise terminator
nucleotides, such as 1, 2,
3, 4, 5, 10, or more than 10 terminator nucleotides. Primers need not be
limited to components
which are added externally to an amplification reaction. In some instances,
primers are
generated in-situ through the addition of nucleotides and proteins which
promote priming. For
example, primase-like enzymes in combination with nucleotides is in some
instances used to
34
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
generate random primers for the methods described herein. Primase-like enzymes
in some
instances are members of the DnaG or AEP enzyme superfamily. In some
instances, a primase-
like enzyme is TthPrimPol. In some instances, a primase-like enzyme is T7 gp4
helicase-
primase. Such primases are in some instances used with the polymerases or
strand displacement
factors described herein. In some instances, primases initiate priming with
deoxyribonucleotides.
In some instances, primases initiate priming with ribonucleotides.
[0071] The PTA amplification can be followed by selection for a specific
subset of amplicons.
Such selections are in some instances dependent on size, affinity, activity,
hybridization to
probes, or other known selection factor in the art. In some instances,
selections precede or
follow additional steps described herein, such as adapter ligation and/or
library amplification. In
some instances, selections are based on size (length) of the amplicons. In
some instances,
smaller amplicons are selected that are less likely to have undergone
exponential amplification,
which enriches for products that were derived from the primary template while
further
converting the amplification from an exponential into a quasi-linear
amplification process. In
some instances, amplicons comprising 50-2000, 25-5000, 40-3000, 50-1000, 200-
1000, 300-
1000, 400-1000, 400-600, 600-2000, or 800-1000 bases in length are selected.
Size selection in
some instances occurs with the use of protocols, e.g., utilizing solid-phase
reversible
immobilization (SPRI) on carboxylated paramagnetic beads to enrich for nucleic
acid fragments
of specific sizes, or other protocol known by those skilled in the art.
Optionally or in
combination, selection occurs through preferential amplification of smaller
fragments during
PCR while preparing sequencing libraries, as well as a result of the
preferential formation of
clusters from smaller sequencing library fragments during Illumina sequencing.
Other strategies
to select for smaller fragments are also consistent with the methods described
herein and
include, without limitation, isolating nucleic acid fragments of specific
sizes after gel
electrophoresis, the use of silica columns that bind nucleic acid fragments of
specific sizes, and
the use of other PCR strategies that more strongly enrich for smaller
fragments. Any number of
library preparation protocols may be used with the PTA methods described
herein. Amplicons
generated by PTA are in some instances ligated to adapters (optionally with
removal of
terminator nucleotides). In some instances, amplicons generated by PTA
comprise regions of
homology generated from transposase-based fragmentation which are used as
priming sites.
[0072] The non-complementary portion of a primer used in PTA can include
sequences which
can be used to further manipulate and/or analyze amplified sequences. An
example of such a
sequence is a "detection tag". Detection tags have sequences complementary to
detection probes
and are detected using their cognate detection probes. There may be one, two,
three, four, or
more than four detection tags on a primer. There is no fundamental limit to
the number of
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
detection tags that can be present on a primer except the size of the primer.
In some instances,
there is a single detection tag on a primer. In some instances, there are two
detection tags on a
primer. When there are multiple detection tags, they may have the same
sequence or they may
have different sequences, with each different sequence complementary to a
different detection
probe. In some instances, multiple detection tags have the same sequence. In
some instances,
multiple detection tags have a different sequence.
[0073] Another example of a sequence that can be included in the non-
complementary portion
of a primer is an "address tag" that can encode other details of the
amplicons, such as the
location in a tissue section. In some instances, a cell barcode comprises an
address tag. An
address tag has a sequence complementary to an address probe. Address tags
become
incorporated at the ends of amplified strands. If present, there may be one,
or more than one,
address tag on a primer. There is no fundamental limit to the number of
address tags that can be
present on a primer except the size of the primer. When there are multiple
address tags, they
may have the same sequence or they may have different sequences, with each
different sequence
complementary to a different address probe. The address tag portion can be any
length that
supports specific and stable hybridization between the address tag and the
address probe. In
some instances, nucleic acids from more than one source can incorporate a
variable tag
sequence. This tag sequence can be up to 100 nucleotides in length, preferably
1 to 10
nucleotides in length, most preferably 4, 5 or 6 nucleotides in length and
comprises
combinations of nucleotides. In some instances, a tag sequence is 1-20, 2-15,
3-13, 4-12, 5-12,
or 1-10 nucleotides in length For example, if six base-pairs are chosen to
form the tag and a
permutation of four different nucleotides is used, then a total of 4096
nucleic acid anchors (e.g.
hairpins), each with a unique 6 base tag can be made.
[0074] Primers described herein may be present in solution or immobilized on a
solid support.
In some instances, primers bearing sample barcodes and/or UMI sequences can be
immobilized
on a solid support. The solid support can be, for example, one or more beads.
In some instances,
individual cells are contacted with one or more beads having a unique set of
sample barcodes
and/or UMI sequences in order to identify the individual cell. In some
instances, lysates from
individual cells are contacted with one or more beads having a unique set of
sample barcodes
and/or UMI sequences in order to identify the individual cell lysates. In some
instances, purified
nucleic acid from individual cells are contacted with one or more beads having
a unique set of
sample barcodes and/or UMI sequences in order to identify the purified nucleic
acid from the
individual cell. The beads can be manipulated in any suitable manner as is
known in the art, for
example, using droplet actuators as described herein. The beads may be any
suitable size,
including for example, microbeads, microparticles, nanobeads and
nanoparticles. In some
36
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
embodiments, beads are magnetically responsive; in other embodiments beads are
not
significantly magnetically responsive. Non-limiting examples of suitable beads
include flow
cytometry microbeads, polystyrene microparti des and nanoparticles,
functionalized polystyrene
microparticles and nanoparticles, coated polystyrene microparticles and
nanoparticles, silica
microbeads, fluorescent microspheres and nanospheres, functionalized
fluorescent microspheres
and nanospheres, coated fluorescent microspheres and nanospheres, color dyed
microparticles
and nanoparticles, magnetic microparticles and nanoparticles,
superparamagnetic microparticles
and nanoparticles (e.g., DYNABEADS available from Invitrogen Group, Carlsbad,
CA),
fluorescent microparticles and nanoparticles, coated magnetic microparticles
and nanoparticles,
ferromagnetic microparticles and nanoparticles, coated ferromagnetic
microparticles and
nanoparticles, and those described in U.S. Pat. Appl. Pub. No. US20050260686,
US20030132538, US20050118574, 20050277197, 20060159962. Beads may be pre-
coupled
with an antibody, protein or antigen, DNA/RNA probe or any other molecule with
an affinity for
a desired target. In some embodiments, primers bearing sample barcodes and/or
UMI sequences
can be in solution. In certain embodiments, a plurality of droplets can be
presented, wherein
each droplet in the plurality bears a sample barcode which is unique to a
droplet and the UMI
which is unique to a molecule such that the UMI are repeated many times within
a collection of
droplets. In some embodiments, individual cells are contacted with a droplet
having a unique set
of sample barcodes and/or UMI sequences in order to identify the individual
cell. In some
embodiments, lysates from individual cells are contacted with a droplet having
a unique set of
sample barcodes and/or UMI sequences in order to identify the individual cell
lysates. In some
embodiments, purified nucleic acid from individual cells are contacted with a
droplet having a
unique set of sample barcodes and/or UMI sequences in order to identify the
purified nucleic
acid from the individual cell. Various microfluidics platforms may be used for
analysis of single
cells. Cells in some instances are manipulated through hydrodynamics (droplet
microfluidics,
inertial microfluidics, vortexing, microvalves, microstructures (e.g.,
microwells, microtraps)),
electrical methods (dielectrophoresis (DEP), electroosmosis), optical methods
(optical tweezers,
optically induced dielectrophoresis (ODEP), opto-thermocapillary), acoustic
methods, or
magnetic methods. In some instances, the microfluidics platform comprises
microwells. In some
instances, the microfluidics platform comprises a PDMS (Polydimethylsiloxane)-
based device.
Non-limited examples of single cell analysis platforms compatible with the
methods described
herein are: ddSEQ Single-Cell Isolator, (Bio-Rad, Hercules, CA, USA, and
Illumina, San Diego,
CA, USA)); Chromium (10x Genomics, Pleasanton, CA, USA)); Rhapsody Single-Cell
Analysis
System (BD, Franklin Lakes, NJ, USA); Tapestri Platform (MissionBio, San
Francisco, CA,
USA)), Nadia Innovate (Dolomite Bio, Royston, UK); Cl and Polaris (Fluidigm,
South San
37
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
Francisco, CA, USA); ICELL8 Single-Cell System (Takara); MSND (Wafergen);
Puncher
platform (Vycap); CellRaft AIR System (CellMicrosystems); DEPArray NxT and
DEPArray
System (Menarini Silicon Biosystems); AVISO CellCelector (ALS); and InDrop
System
(1CellBio).
[0075] PTA primers may comprise a sequence-specific or random primer, an
address tag, a
cell barcode and/or a unique molecular identifier (UMI). In some instances, a
primer comprises
a sequence-specific primer. In some instances, a primer comprises a random
primer. In some
instances, a primer comprises a cell barcode. In some instances, a primer
comprises a sample
barcode. In some instances, a primer comprises a unique molecular identifier.
In some instances,
primers comprise two or more cell barcodes. Such barcodes in some instances
identify a unique
sample source, or unique workflow. Such barcodes or UMIs are in some instances
5, 6, 7, 8, 9,
10, 11, 12, 15, 20, 25, 30, or more than 30 bases in length. Primers in some
instances comprise
at least 1000, 10,000, 50,000, 100,000, 250,000, 500,000, 106, 107, 108, 109,
or at least 1010
unique barcodes or UMIs. In some instances primers comprise at least 8, 16,
96, or 384 unique
barcodes or UMIs. In some instances a standard adapter is then ligated onto
the amplification
products prior to sequencing; after sequencing, reads are first assigned to a
specific cell based on
the cell barcode. Suitable adapters that may be utilized with the PTA method
include, e.g.,
xGeng Dual Index UMI adapters available from Integrated DNA Technologies
(IDT). Reads
from each cell is then grouped using the UMI, and reads with the same UMI may
be collapsed
into a consensus read. The use of a cell barcode allows all cells to be pooled
prior to library
preparation, as they can later be identified by the cell barcode. The use of
the UMI to form a
consensus read in some instances corrects for PCR bias, improving the copy
number variation
(CNV) detection. In addition, sequencing errors may be corrected by requiring
that a fixed
percentage of reads from the same molecule have the same base change detected
at each
position. This approach has been utilized to improve CNV detection and correct
sequencing
errors in bulk samples. In some instances, UMIs are used with the methods
described herein, for
example, U.S Pat. No. 8,835,358 discloses the principle of digital counting
after attaching a
random amplifiable barcode. Schmitt. et al and Fan et al. disclose similar
methods of correcting
sequencing errors.
[0076] The methods described herein may further comprise additional steps,
including steps
performed on the sample or template. Such samples or templates in some
instance are subjected
to one or more steps prior to PTA. In some instances, samples comprising cells
are subjected to
a pre-treatment step. For example, cells undergo lysis and proteolysis to
increase chromatin
accessibility using a combination of freeze-thawing, Triton X-100, Tween 20,
and Proteinase K.
Other lysis strategies are also be suitable for practicing the methods
described herein. Such
38
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
strategies include, without limitation, lysis using other combinations of
detergent and/or
lysozyme and/or protease treatment and/or physical disruption of cells such as
sonication and/or
alkaline lysis and/or hypotonic lysis. In some instances, cells are lysed with
mechanical (e.g.,
high pressure homogenizer, bead milling) or non-mechanical (physical,
chemical, or biological).
In some instances, physical lysis methods comprise heating, osmotic shock,
and/or cavitation. In
some instances, chemical lysis comprises alkali and/or detergents. In some
instances, biological
lysis comprises use of enzymes. Combinations of lysis methods are also
compatible with the
methods described herein. Non-limited examples of lysis enzymes include
recombinant
lysozyme, serine proteases, and bacterial lysins. In some instances, lysis
with enzymes
comprises use of lysozyme, lysostaphin, zymolase, cellulose, protease or
glycanase. In some
instances, the primary template or target molecule(s) is subjected to a pre-
treatment step. In
some instances, the primary template (or target) is denatured using sodium
hydroxide, followed
by neutralization of the solution. Other denaturing strategies may also be
suitable for practicing
the methods described herein. Such strategies may include, without limitation,
combinations of
alkaline lysis with other basic solutions, increasing the temperature of the
sample and/or altering
the salt concentration in the sample, addition of additives such as solvents
or oils, other
modification, or any combination thereof. In some instances, additional steps
include sorting,
filtering, or isolating samples, templates, or amplicons by size. For example,
after amplification
with the methods described herein, amplicon libraries are enriched for
amplicons having a
desired length. In some instances, amplicon libraries are enriched for
amplicons having a length
of 50-2000, 25-1000, 50-1000, 75-2000, 100-3000, 150-500, 75-250, 170-500, 100-
500, or 75-
2000 bases. In some instances, amplicon libraries are enriched for amplicons
having a length no
more than 75, 100, 150, 200, 500, 750, 1000, 2000, 5000, or no more than
10,000 bases. In some
instances, amplicon libraries are enriched for amplicons having a length of at
least 25, 50, 75,
100, 150, 200, 500, 750, 1000, or at least 2000 bases.
[0077] Methods and compositions described herein may comprise buffers or other
formulations. Such buffers in some instances comprise surfactants/detergent or
denaturing
agents (Tween-20, DMSO, D1VIF, pegylated polymers comprising a hydrophobic
group, or other
surfactant), salts (potassium or sodium phosphate (monobasic or dibasic),
sodium chloride,
potassium chloride, TrisHC1, magnesium chloride or sulfate, Ammonium salts
such as
phosphate, nitrate, or sulfate, EDTA), reducing agents (DTT, THP, DTE, beta-
mercaptoethanol,
TCEP, or other reducing agent) or other components (glycerol, hydrophilic
polymers such as
PEG). In some instances, buffers are used in conjunction with components such
as polymerases,
strand displacement factors, terminators, or other reaction component
described herein. Buffers
may comprise one or more crowding agents. In some instances, crowding reagents
include
39
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
polymers. In some instances, crowding reagents comprise polymers such as
polyols. In some
instances, crowding reagents comprise polyethylene glycol polymers (PEG). In
some instances,
crowding reagents comprise polysaccarides. Without limitation, examples of
crowding reagents
include ficoll (e.g., ficoll PM 400, ficoll PM 70, or other molecular weight
ficoll), PEG (e.g.,
PEG1000, PEG 2000, PEG4000, PEG6000, PEG8000, or other molecular weight PEG),
dextran
(dextran 6, dextran 10, dextran 40, dextran 70, dextran 6000, dextran 138k, or
other molecular
weight dextran).
[0078] The nucleic acid molecules amplified according to the methods described
herein may
be sequenced and analyzed using methods known to those of skill in the art.
Non-limiting
examples of the sequencing methods which in some instances are used include,
e.g., sequencing
by hybridization (SBH), sequencing by ligation (SBL) (Shendure et al. (2005)
Science
309:1728), quantitative incremental fluorescent nucleotide addition sequencing
(QIFNAS),
stepwise ligation and cleavage, fluorescence resonance energy transfer (FRET),
molecular
beacons, TaqMan reporter probe digestion, pyrosequencing, fluorescent in situ
sequencing
(FISSEQ), FISSEQ beads (U.S. Pat. No. 7,425,431), wobble sequencing (Int. Pat.
Appl. Pub.
No. W02006/073504), multiplex sequencing (U.S. Pat. Appl. Pub. No.
U52008/0269068;
Porreca et al., 2007, Nat. Methods 4:931), polymerized colony (POLONY)
sequencing (U.S.
Patent Nos. 6,432,360, 6,485,944 and 6,511,803, and Int. Pat. Appl. Pub. No.
W02005/082098),
nanogrid rolling circle sequencing (ROLONY) (U.S. Pat. No. 9,624,538), allele-
specific oligo
ligation assays (e.g., oligo ligation assay (OLA), single template molecule
OLA using a ligated
linear probe and a rolling circle amplification (RCA) readout, ligated padlock
probes, and/or
single template molecule OLA using a ligated circular padlock probe and a
rolling circle
amplification (RCA) readout), high-throughput sequencing methods such as,
e.g., methods using
Roche 454, Illumina Solexa, AB-SOLiD, Helicos, Polonator platforms and the
like, and light-
based sequencing technologies (Landegren et al. (1998) Genome Res. 8:769-76;
Kwok (2000)
Pharmacogenomics 1:95-100; and Shi (2001) Clin. Chem.47:164-172). In some
instances, the
amplified nucleic acid molecules are shotgun sequenced.
Kits
[0079] Described herein are kits for the detection of viral nucleic acids from
samples. In some
instances, a kit described herein comprises one or more of a sampling device,
one or more
positive control nucleic acids, negative control, primers, probes, reverse
transcriptase,
polymerase, sample plates, sample tubes, pipets, or lysis buffer. In some
instances, a lysis buffer
comprises a reducing agent. In some instances, a lysis buffer comprises
proteinase K or
proteinase pk. In some instances, a kit described herein comprises an qRT-PCR
master mix. In
some instances, a master mix comprises a polymerase (e.g., TaqMan, or other
polymerase),
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
uracil-N-glycosylase, dNTPs with dUTP, passive reference dyes (e.g., ROX dye),
and other
buffers. In some instances, the plate is a 96 or 386 well plate. In some
instances, the primers and
probes are configured to detect a virus (e.g., Covid-19, SARS, or MERS). In
some instances, the
master mix is attached to a bead. In some instances, kits further comprise
reagents for RT-
LAMP or RT-PTA methods.
[0080] Described herein are kits facilitating the practice of the PTA method
with RT-PCR to
detect viral nucleic acids. Various combinations of the components set forth
above in regard to
exemplary reaction mixtures and reaction methods can be provided in a kit
form. A kit may
include individual components that are separated from each other, for example,
being carried in
separate vessels or packages. A kit in some instances includes one or more sub-
combinations of
the components set forth herein, the one or more sub-combinations being
separated from other
components of the kit. The sub-combinations in some instances are combinable
to create a
reaction mixture set forth herein (or combined to perform a reaction set forth
herein). In
particular embodiments, a sub-combination of components that is present in an
individual vessel
or package is insufficient to perform a reaction set forth herein. However,
the kit as a whole in
some instances includes a collection of vessels or packages the contents of
which can be
combined to perform a reaction set forth herein.
[0081] A kit can include a suitable packaging material to house the contents
of the kit. The
packaging material in some instances is constructed by well-known methods,
preferably to
provide a sterile, contaminant-free environment. The packaging materials
employed herein
include, for example, those customarily utilized in commercial kits sold for
use with nucleic acid
sequencing systems. Exemplary packaging materials include, without limitation,
glass, plastic,
paper, foil, and the like, capable of holding within fixed limits a component
set forth herein. The
packaging material can include a label which indicates a particular use for
the components. The
use for the kit that is indicated by the label in some in instances is one or
more of the methods
set forth herein as appropriate for the particular combination of components
present in the kit.
For example, a label in some instances indicates that the kit is useful for a
method of detecting
mutations in a nucleic acid sample using the PTA method. Instructions for use
of the packaged
reagents or components can also be included in a kit. The instructions will
typically include a
tangible expression describing reaction parameters, such as the relative
amounts
of kit components and sample to be admixed, maintenance time periods for
reagent/sample
admixtures, temperature, buffer conditions, and the like. It will be
understood that not all
components necessary for a particular reaction need be present in a particular
kit. Rather one or
more additional components in some instances are provided from other sources.
The instructions
provided with a kit in some instances identify the additional component(s)
that are to be
41
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
provided and where they can be obtained. In one embodiment, a kit provides at
least one
amplification primer; at least one nucleic acid polymerase; a mixture of at
least two nucleotides,
wherein the mixture of nucleotides comprises at least one terminator
nucleotide which
terminates nucleic acid replication by the polymerase; and instructions for
use of the kit. In some
instances, the kit provides reagents to perform the methods described herein,
such as PTA. In
some instances, a kit further comprises reagents configured for gene editing
(e.g., Crispricas9 or
other method described herein). In some instances, a kit comprises a variant
polymerase
described herein.
[0082] In a related aspect, the invention provides a kit comprising a reverse
transcriptase, a
nucleic acid polymerase, one or more amplification primers, a mixture of
nucleotides
comprising one or more terminator nucleotides, and optionally instructions for
use. In one
embodiment of the kits of the invention, the nucleic acid polymerase is a
strand displacing DNA
polymerase. In one embodiment of the kits of the invention, the nucleic acid
polymerase is
selected from bacteriophage phi29 (129) polymerase, genetically modified phi29
(129) DNA
polymerase, Klenow Fragment of DNA polymerase I, phage M2 DNA polymerase,
phage
phiPRD1 DNA polymerase, Bst DNA polymerase, Bst large fragment DNA polymerase,
exo(-)
Bst polymerase, exo(-)Bca DNA polymerase, Bsu DNA polymerase, VentR DNA
polymerase,
VentR (exo-) DNA polymerase, Deep Vent DNA polymerase, Deep Vent (exo-) DNA
polymerase, IsoPol DNA polymerase, DNA polymerase I, Therminator DNA
polymerase, T5
DNA polymerase, Sequenase, T7 DNA polymerase, T7-Sequenase, and T4 DNA
polymerase. In
one embodiment of the kits of the invention, the nucleic acid polymerase has
3'->5' exonuclease
activity and the terminator nucleotides inhibit such 3'->5' exonuclease
activity (e.g., nucleotides
with modification to the alpha group [e.g., alpha-thio dideoxynucleotides], C3
spacer
nucleotides, locked nucleic acids (LNA), inverted nucleic acids, 2' fluor
nucleotides, 3'
phosphorylated nucleotides, 2'-0-Methyl modified nucleotides, trans nucleic
acids). In one
embodiment of the kits of the invention, the nucleic acid polymerase does not
have 3'->5'
exonuclease activity (e.g., Bst DNA polymerase, exo(-) Bst polymerase, exo(-)
Bca DNA
polymerase, Bsu DNA polymerase, VentR (exo-) DNA polymerase, Deep Vent (exo-)
DNA
polymerase, Klenow Fragment (exo-) DNA polymerase, Therminator DNA
polymerase). In one
specific embodiment, the terminator nucleotides comprise modifications of the
r group of the 3'
carbon of the deoxyribose. In one specific embodiment, the terminator
nucleotides are selected
from 3' blocked reversible terminator comprising nucleotides, 3' unblocked
reversible
terminator comprising nucleotides, terminators comprising 2' modifications of
deoxynucleotides, terminators comprising modifications to the nitrogenous base
of
deoxynucleotides, and combinations thereof In one specific embodiment, the
terminator
42
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
nucleotides are selected from dideoxynucleotides, inverted dideoxynucleotides,
3' biotinylated
nucleotides, 3' amino nucleotides, 3'-phosphorylated nucleotides, 3'-0-methyl
nucleotides, 3'
carbon spacer nucleotides including 3' C3 spacer nucleotides, 3' C18
nucleotides, 3' hexanediol
spacer nucleotides, acyclonucleotides, and combinations thereof
Numbered Embodiments
[0083] Provided herein are the following numbered embodiments: Embodiment 1. A
method
of detecting nucleic acids comprising: a. providing a sample from a source,
wherein the sample
comprises at least one viral ribonucleic acid; b. heating the sample; c.
reverse transcribing the at
least one viral ribonucleic acid to generate at least one cDNA, wherein the at
least one viral
ribonucleic acid is not subjected to a purification step prior to reverse
transcribing; and d.
detecting the at least one cDNA. Embodiment 2. The method of embodiment 1,
wherein the
purification step comprises binding the at least one viral ribonucleic acid to
a solid support.
Embodiment 3. The method of embodiment 1, wherein the purification step
comprises
precipitating the least one viral ribonucleic acid or use of ion-exchange
chromatography.
Embodiment 4. The method of embodiment 1, wherein the purification step
comprises
hybridizing the least one viral ribonucleic acid to an array. Embodiment 5.
The method of any
one of embodiments 1-4, wherein reverse transcribing comprises use of a
reverse transcriptase.
Embodiment 6. The method of any one of embodiments 1-5, wherein the method
further
comprises amplification of the at least one cDNA. Embodiment 7. The method of
any one of
embodiments 1-6, wherein the at least one viral ribonucleic acid is obtained
from a respiratory
virus. Embodiment 8. The method of embodiment 7, wherein the respiratory virus
is a
coronavirus. Embodiment 9. The method of embodiment 8, wherein the coronavirus
is selected
from Covid-19, SARS, MERS, bovine coronaviruses, norovirus, orthoreoviruses
(reoviruses),
human rotaviruses, human coronaviruses, or adenoviruses. Embodiment 10. The
method of any
one of embodiments 1-9, wherein the at least one viral ribonucleic acid
encodes for a viral
nucleocapsid. Embodiment 11. The method of embodiment 10, wherein the at least
one viral
ribonucleic acid is an Ni gene, an N2 gene, or an N3 gene. Embodiment 12. The
method of any
one of embodiments 1-11, wherein detecting comprises binding the at least one
cDNA with at
least one probe. Embodiment 13. The method of embodiment 12, wherein the probe
comprises a
reporter moiety. Embodiment 14. The method of embodiment 12, wherein detection
comprises
RT-PCR, RT-LAMP, RT-PTA, or RT-RPA. Embodiment 15. The method of any one of
embodiments 1-14, wherein the method further comprises contacting the sample
with a lysis
buffer prior to step (c). Embodiment 16. The method of embodiment 15, wherein
the lysis buffer
comprises a proteinase. Embodiment 17. The method of any one of embodiments 1-
16, wherein
the source is selected from nasopharyngeal or oropharyngeal swabs, sputum,
lower respiratory
43
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
tract aspirates, bronchoalveolar lavage, nasopharyngeal wash/aspirate, or
nasal aspirate.
Embodiment 18. The method of any one of embodiments 1-17, wherein heating the
sample
comprises: a. heating the sample at a first temperature for a first length of
time; and b. heating
the sample at a second temperature for a second length of time. Embodiment 19.
The method of
embodiment 18, wherein the first temperature is 30-45 degrees C. Embodiment
20. The method
of embodiment 18 or 19, wherein the second temperature is 80-90 degrees C.
Embodiment 21.
The method of any one of embodiments 18-20, wherein the first time is 10-30
min. Embodiment
22. The method of any one of embodiments 18-21, wherein the second time is 10-
30 min.
Embodiment 23. A method of detecting a virus comprising: a. providing a sample
from a source,
wherein the sample comprises at least one viral genome copy; b. heating the
sample; c.
amplifying the at least one viral genome copy to generate an amplified viral
genome; d.
detecting the amplified viral genome, wherein the at least one viral genome
copy is not
subjected to a purification step prior to detecting. Embodiment 24. The method
of embodiment
23, wherein the sample comprises 1000-10,000 viral genome copies. Embodiment
25. The
method of embodiment 23, wherein the sample comprises 10-100 viral genome
copies.
Embodiment 26. The method of embodiment 24, wherein amplifying comprises
subjecting the
sample to fewer than 30 PCR cycles. Embodiment 27. The method of embodiment
25, wherein
amplifying comprises subjecting the sample to fewer than 40 PCR cycles.
Embodiment 28. The
method of any one of embodiments 23-27, wherein the viral amplified genome is
detected in
less than 3 hours. Embodiment 29. The method of any one of embodiments 23-28,
wherein the
viral amplified genome is detected in less than 2 hours. Embodiment 30. The
method of any one
of embodiments 23-29, wherein the purification step comprises binding the at
least one viral
genome copy to a solid support. Embodiment 31. The method of any one of
embodiments 23-
29, wherein the purification step comprises precipitating the least one viral
genome copy or use
of ion-exchange chromatography. Embodiment 32. The method of any one of
embodiments 23-
29, wherein the purification step comprises hybridizing the least one viral
genome copy to an
array. Embodiment 33. The method of any one of embodiments 23-32, wherein the
at least one
viral genome copy is obtained from a respiratory virus. Embodiment 34. The
method of
embodiment 33, wherein the respiratory virus is a coronavirus. Embodiment 35.
The method of
embodiment 34, wherein the coronavirus is selected from SARS, MERS, Covid-19,
bovine,
norovirus, orthoreoviruses (reoviruses), human rotaviruses, human
coronaviruses, or
adenoviruses. Embodiment 36. The method of any one of embodiments 23-35,
wherein the
source is selected from nasopharyngeal or oropharyngeal swabs, sputum, lower
respiratory tract
aspirates, bronchoalveolar lavage, nasopharyngeal wash/aspirate, or nasal
aspirate. Embodiment
37. The method of any one of embodiments 23-36, wherein heating the sample
comprises: a.
44
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
heating the sample at a first temperature for a first length of time; and b.
heating the sample at a
second temperature for a second length of time. Embodiment 38. The method of
embodiment
37, wherein the first temperature is 30-45 degrees C. Embodiment 39. The
method of
embodiment 37 or 38, wherein the second temperature is 80-90 degrees C.
Embodiment 40. The
method of any one of embodiments 37-39, wherein the first time is 10-30 min.
Embodiment 41.
The method of any one of embodiments 37-40, wherein the second time is 10-30
min.
Embodiment 42. The method of any one of embodiments 23-41, wherein detection
comprises
RT-PCR, RT-LAMP, RT-PTA, or RT-RPA. Embodiment 43. A method of detecting a
virus
comprising: a. providing at least 48 samples, wherein at least some of the at
least 48 samples
comprises at least one viral genome copy; b. heating the at least 48 samples;
c. amplifying the at
least one viral genome copy to generate an amplified viral genome; d.
determining the presence
or absence of the amplified viral genome for each sample, wherein the at least
one viral genome
copy is not subjected to a purification step prior to determining, and wherein
the at least 48
samples are analyzed in parallel. Embodiment 44. The method of embodiment 43,
comprising
providing at least 90 samples. Embodiment 45. The method of embodiment 43,
comprising
providing at least 300 samples. Embodiment 46. The method of any one of
embodiments 43-45,
wherein determining the presence or absence of the viral amplified genome
occurs in less than 3
hours. Embodiment 47. The method of any one of embodiments 43-45, wherein
determining the
presence or absence of the viral amplified genome occurs in less than 2 hours.
Embodiment 48.
The method of any one of embodiments 43-45, wherein the rate of determining
the presence or
absence of the amplified viral genome is at least 2 samples per minute.
Embodiment 49. The
method of any one of embodiments 43-45, wherein the rate of determining the
presence or
absence of the amplified viral genome is at least 3 samples per minute.
Embodiment 50. The
method of any one of embodiments 43-45, wherein the rate of determining the
presence or
absence of the amplified viral genome is at least 5 samples per minute.
Embodiment 51. The
method of embodiment 43, wherein the method comprises at least 190 samples,
and wherein
determining the presence or absence of the amplified viral genome for all of
the at least 48
samples occurs in no more than 90 min. Embodiment 52. The method of embodiment
43,
wherein the method comprises at least 384 samples, and wherein determining the
presence or
absence of the amplified viral genome for all of the at least 48 samples
occurs in no more than
60 min. Embodiment 53. The method of any one of embodiments 43-52, wherein the
purification step comprises binding the at least one viral genome copy to a
solid support.
Embodiment 54. The method of any one of embodiments 43-52, wherein the
purification step
comprises precipitating the least one viral genome copy or use of ion-exchange
chromatography.
Embodiment 55. The method of any one of embodiments 43-52, wherein the
purification step
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
comprises hybridizing the least one viral genome copy to an array. Embodiment
56. The
method of any one of embodiments 43-55, wherein the at least one viral genome
copy is
obtained from a respiratory virus. Embodiment 57. The method of embodiment 56,
wherein the
respiratory virus is a coronavirus. Embodiment 58. The method of embodiment
57, wherein the
coronavirus is selected from SARS, MERS, Covid-19, bovine, norovirus,
orthoreoviruses
(reoviruses), human rotaviruses, human coronaviruses, or adenoviruses.
Embodiment 59. The
method of any one of embodiments 43-58, wherein heating the at least 48
samples comprises: a.
heating the at least 48 samples at a first temperature for a first length of
time; and b. heating the
at least 48 samples at a second temperature for a second length of time.
Embodiment 60. The
method of embodiment 59, wherein the first temperature is 30-45 degrees C.
Embodiment 61.
The method of embodiment 59 or 60, wherein the second temperature is 80-90
degrees C.
Embodiment 62. The method of any one of embodiments 59-61, wherein the first
time is 10-30
min. Embodiment 63. The method of any one of embodiments 59-62, wherein the
second time is
10-30 min. Embodiment 64. The method of any one of embodiments 43-63, wherein
the at least
one viral genome copy comprises DNA. Embodiment 65. The method of any one of
embodiments 43-63, wherein the at least one viral genome copy comprises RNA.
Embodiment
66. The method of any one of embodiments 43-63, wherein determining comprises
RT-PCR,
RT-LAMP, RT-PTA, or RT-RPA. Embodiment
EXAMPLES
[0084] The following examples are set forth to illustrate more clearly the
principle and
practice of embodiments disclosed herein to those skilled in the art and are
not to be construed
as limiting the scope of any claimed embodiments. Unless otherwise stated, all
parts and
percentages are on a weight basis.
EXAMPLE 1: Sensitivity of Covid-19 assay
[0085] Covid-19 cDNA plasmid or RNA at varying concentrations (genome copy
number, cp)
was added to 20 uL of sample lysis buffer containing proteinase K or
proteinase Pk. The sample
was heated for 15 minutes at 37 degrees C, then heated for 15 minutes at 85
degrees C. Then, 10
uL of the heat-treated sample was added to a 96 or 384 well plate along with
Covid-19 specific
qRT-PCR primers and probes, and Thermo Q7 RT PCR system reagents master mix.
The plate
was then sealed and subjected to PCR cycles. Plots of normalized reporter
values vs. number of
PCR cycles for samples of varying concentrations of Covid-19 (DNA) and Ni
(RNA) are shown
in FIG. 3 and FIG. 4, respectively. The assay showed at least 5 logs of
dynamic range (10-
1,000,000 cp/assay), with single viral particle detection possible. The
overall test had a duration
of 2.5 hours, but could be shortened to 60 min total.
46
CA 03178445 2022-09-29
WO 2021/202344 PCT/US2021/024596
EXAMPLE 2: Covid-10 assay with qRT-PCR in bead format
[0086] The general procedures of Example 1 are followed with modification; the
sample
assay is replaced with a qRT-PCR bead format (FIG. 5). In this format, all
reagents required for
qRT-PCR are contained on the beads. The total time to complete the assay is
approximately 2
hours. Alternative bead-based formats such as RT-LAMP or RT-PTA/sequencing may
also be
used.
EXAMPLE 3: Validation of Human sample testing for Covid-19 Virus
[0087] A contrived clinical study is performed to evaluate the performance of
the Covid-19
RT-PCR following the general procedures described in Example 1 or Example 2. A
total of 100
individual clinical respiratory samples: 50 NP (Nasopharyngeal) swabs and 50
BALs
(bronchoalveolar lavages), are used in this study. 100 negatives and 80
contrived positives are
tested. Negative samples include 50 NP swabs and 50 BALs. Positive samples are
comprised of
40 NP swabs and 40 BALs spiked with quantitated live SARS-CoV-2. 10 samples
each are
spiked at 8x, 4x, 2x, and lx LoD (limit of detection, lowest concentration of
virus that can be
detected 95% of the time). The positive and negative percent agreements
between the COVID-
19 RT-PCR test and the expected results are calculated.
EXAMPLE 4: Human testing for Covid-19 Virus
[0088] Following the general procedures of Example 1 or Example 2, 5,000 BALs
samples
obtained from 2,500 humans suspected of having Covid-19 virus are tested using
the workflow
shown in FIG. 2 or FIG. 5. Individuals identified as infected are notified and
appropriate
isolation or protective measures are taken to prevent spread of the disease.
[0089] While preferred embodiments of the present invention have been shown
and described
herein, it will be obvious to those skilled in the art that such embodiments
are provided by way
of example only. Numerous variations, changes, and substitutions will now
occur to those
skilled in the art without departing from the invention. It should be
understood that various
alternatives to the embodiments of the invention described herein may be
employed in practicing
the invention. It is intended that the following claims define the scope of
the invention and that
methods and structures within the scope of these claims and their equivalents
be covered
thereby.
47