Note: Descriptions are shown in the official language in which they were submitted.
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
METHODS FOR TREATING PANCREATIC CANCER AND OTHER SOLID
TUMORS
[0001] The invention relates to methods and medicaments useful for treating
solid tumors,
e.g., pancreatic adenocarcinoma and other solid tumors with a combination of
CEND-1
peptide and anti-cancer therapies, e.g., standard-of-care anti-cancer
therapies.
BACKGROUND
[0002] The National Cancer Institute estimates that in 2018 approximately
1,735,350 new
cases of cancer will be diagnosed in the United States and 609,640 people will
die from the
disease. Despite advances in the treatment of certain forms of cancer through
surgery,
radiotherapy, chemotherapy, and most recently immunotherapy, most types of
solid tumors
are essentially incurable. Even when an effective treatment is available for a
particular cancer,
the side effects from the treatment can have a significant adverse impact on a
patient's quality
of life.
[0003] Pancreatic cancer is an especially serious cancer and a life-
threatening condition.
In most cases, early stages of the disease are asymptomatic and less than 20%
of pancreatic
cancers are amenable to surgery. Moreover, invasive and metastatic pancreatic
cancers
respond poorly to existing treatments in chemotherapy and radiotherapy, with
response rates
typically less than 30%. The National Cancer Institute (NCI) estimate that
survival rate for
cancer of the exocrine pancreas is less than 5% and the median survival time
after diagnosis
is less than a year. The continuing poor prognosis and lack of effective
treatments for
pancreatic cancer highlight an unmet medical need to develop less toxic and
more efficient
treatment strategies that improve the clinical management and prognosis of
patients afflicted
with pancreatic cancer.
[0004] An important reason for why most anti-cancer agents have toxicity
and limited
efficacy for solid tumors is the fact that anti-cancer drugs only penetrate 3-
5 cell diameters
deep from the blood vessels, leaving some areas of the tumor exposed to an
ineffective
concentration of the drug or to no drug at all. As an example, studies have
suggested that less
than 1% of the administered nabpaclitaxel may be able to penetrate / enter the
pancreatic
ductal adenocarcinoma tissue.
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
SUMMARY.
Improved Penetration of Chemotherapeutics with CEND-1
[0005] The results from both in vivo and in vitro pharmacology and
mechanistic studies
indicate that combining the invention CEND-1/iRGD-analog (Fig. 2) with
chemotherapeutics
significantly increases the tumor penetration of these drugs and improves
their efficacy.
Although the invention methods are applicable to a broad class of cancers
and/or solid tumors,
the initial indication for this investigational drug is pancreatic ductal
adenocarcinoma (PDAC)
because, in addition to its poor prognosis, it is characterized by a dense
extracellular matrix
stroma, which acts as a physical barrier to drug entry. Since the tumor homing
and the
transport process initiated by CEND-1 have been shown to be active in the PDAC
stroma and
preclinical studies have shown increased drug penetration and efficacy in
different kinds of
PDAC models, CEND-1 appears particularly well suited to target PDAC.
[0006] Provided herein are methods for treating, inhibiting, or reducing
the volume of a
tumor of a cancer in a subject or patient in need thereof, wherein the method
comprises
administering CEND-1, or a pharmaceutically acceptable salt thereof, in a
combination with
simultaneous, separate or sequential administration of at least one anti-
cancer agent or
therapy. In certain embodiments, the tumor is a malignant solid tumor
characterized by dense
tumor stroma. In other embodiments, the tumor is a solid tumor of a cancer
selected from the
group consisting of: breast cancer, squamous cell cancer, small-cell lung
cancer, non-small
cell lung cancer, gastrointestinal cancer, pancreatic cancer, glioblastoma,
cervical cancer,
ovarian cancer, liver cancer, bladder cancer, hepatoma, colon cancer,
colorectal cancer,
endometrial carcinoma, salivary gland carcinoma, kidney cancer, liver cancer,
prostate cancer,
vulval cancer, thyroid cancer, hepatic carcinoma, and head and neck cancer. In
another
embodiment, the pancreatic cancer is selected from the group consisting of:
primary
pancreatic cancer, metastatic pancreatic cancer, refractory pancreatic cancer,
cancer drug
resistant pancreatic cancer and adenocarcinoma. In a particular embodiment,
the cancer is
ductal adenocarcinoma (such as Stage 0-IV, and the like.
[0007] In particular embodiments, the anti-cancer agent or therapy is
selected from the
group consisting of: a chemotherapeutic agent, small molecule, antibody,
antibody drug
conjugate, nanoparticle, cell therapy, polypeptides, peptides,
peptidomimetics, nucleic acid-
molecules, ribozymes, antisense oligonucleotides, and nucleic acid molecules
encoding
- 2 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
transgenes, viruses, cytokines, cytotoxic polypeptides; pro-apoptotic
polypeptides, anti-
angiogenic polypeptides. cytotoxic cells such as cytotoxic T cells, and/or
vaccines (mRNA or
DNA).
[0008] In other embodiments, the chemotherapeutic agent is selected from
one or more of
the group consisting of: taxane, docetaxel, paclitaxel, nab-paclitaxel, a
nucleoside,
gemcitabine, an anthracycline, doxorubicin, an alkylating agent, a vinca
alkaloid, an anti-
metabolite, a platinum agent, cisplatin, carboplatin, a steroid, methotrexate,
an antibiotic,
adriamycin, an isofamide, a selective estrogen receptor modulator, a
maytansinoid,
mertansine, emtansine, an antibody such, trastuzumab, an anti-epidermal growth
factor
receptor 2 (HER2) antibody, trastuzumab, a caspase, caspase-8; diphtheria
toxin A chain,
Pseudomonas exotoxin A, cholera toxin, ligand fusion toxins, DAB389EGF,
Ricinus
communis toxin (ricin); chimeric antigen receptor T cells (CAR-T), chimeric
antigen receptor
macrophages (CAR-M), chimeric antigen receptor natural killer cells (CAR-K),
and tumor-
infiltrating lymphocytes (TIL), anti-PD-1 antibodies, nivolumab,
pembrolizumab,
atezolizumab, avelumab, durvalumab; anti-CTLA-4 antibodies. ipilimumab;
bispecific
antibodies, catumaxomab, Moderna' s mRNA-4157 and/or BioNTech' s BNT122.
[0009] In particular embodiments, the CEND-1 (e.gõ the iRGD-analog set
forth in Figure
2) is administered in an amount selected from the group consisting of: about
0.2 to 20 mg/kg
body weight/per dose of cancer therapy, about 0.3 to 17 mg/kg body weight/per
dose of cancer
therapy, about 0.4 to 14 mg/kg body weight/per dose of cancer therapy, about
0.5 to 11 mg/kg
body weight/per dose of cancer therapy, about 0.6 to 8 mg/kg body weight/per
dose of cancer
therapy, about 0.7 to 5 mg/kg body weight/per dose of cancer therapy, about
0.8 to 3.2 mg/kg
body weight/per dose of cancer therapy. In a particular embodiment, CEND-1 is
administered
in an amount corresponding to 3.2 mg/kg body weight/per dose of cancer
therapy.
[0010] In certain embodiments, CEND-1 is administered before or during the
administration of anti-cancer therapy, wherein the cancer therapy is at a
dosing regimen
selected from the group consisting of: 4 times/day, 3 times/day, twice daily,
once daily, once
every other day, once every 2nd day, once every 3rd day, once every 4th day,
once every 5th
day, once every 6th day, once weekly, once every 8th day, once every 9th day,
once every
10th day, once every 1 lth day, once every 12th day, once every 13th day, once
every 2 weeks,
once every 3 weeks, and/or once per month. In one embodiment, CEND-1 is
present in a dry
formulation or suspended in a biocompatible medium.
- 3 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
[0011] In particular embodiments, the biocompatible media is selected from
the group
consisting of: water, buffered aqueous media, saline, buffered saline,
optionally buffered
solutions of amino acids, optionally buffered solutions of proteins,
optionally buffered
solutions of sugars, optionally buffered solutions of vitamins, optionally
buffered solutions of
synthetic polymers, and lipid-containing emulsions. In a particular
embodiment, CEND-1 is
administered intravenously.
[0012] Also provided herein is a method of treating pancreatic cancer in a
patient in need
thereof, comprising administering to the patient an effective amount of CEND-
1, in
combination with gemcitabine and/or nab-paclitaxel, or pharmaceutically
acceptable salts
thereof In certain embodiments, the pancreatic cancer is selected from the
group consisting
of: primary pancreatic cancer, metastatic pancreatic cancer, refractory
pancreatic cancer,
cancer drug resistant pancreatic cancer and adenocarcinoma. In a particular
embodiment, the
cancer is ductal adenocarcinoma (Stage 0-IV).
[0013] In certain ebodiments, CEND-1 is administered in an amount selected
from the
group consisting of: about 0.2 to 20 mg/kg body weight/per dose of cancer
therapy, about 0.3
to 17 mg/kg body weight/ per dose of cancer therapy, about 0.4 to 14 mg/kg
body weight/per
dose of cancer therapy, about 0.5 to 11 mg/kg body weight/per dose of cancer
therapy, about
0.6 to 8 mg/kg body weight/per dose of cancer therapy, about 0.7 to 5 mg/kg
body weight/per
dose of cancer therapy, about 0.8 to 3.2 mg/kg body weight/per dose of cancer
therapy. In
once embodiment, CEND-1 is administered in an amount corresponding to 3.2
mg/kg body
weight/per dose of cancer therapy.
[0014] In particular embodiments, CEND-1 is administered before or during
the
administration of anti-cancer therapy, wherein the cancer therapy is at a
dosing regimen
selected from the group consisting of: 4 times/day, 3 times/day, twice daily,
once daily, once
every other day, once every 2nd day, once every 3rd day, once every 4th day,
once every 5th
day, once every 6th day, once weekly, once every 8th day, once every 9th day,
once every
10th day, once every 11th day, once every 12th day, once every 13th day, once
every 2 weeks,
once every 3 weeks, and/or once per month. In a particular embodiment,
CEND-1 is administered in a range amount selected from: 0.01-100, 0.02-90,
0.03-80, 0.04-
70, 0.05-60, 0.06-50, 0.07-40, 0.08-30, 0.09-30, 0.1-25, 0.11-20, 0.12-15,
0.13-10, 0.14-9,
- 4 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
0.15-8, 0.16-7, 0.17-6, 0.18-5, 0.19-4, or 0.2-3.2 mg/kg body weight/day or
per dose of
chemotherapy;
nab-paclitaxel is administered in a range amount selected from: 1-500, 10-450,
20-400, 30-
350, 40-300, 50-250, 60-200, 70-175, 80-160, 90-150, 100-140, 110-140, 115-135
or 120-130
mg/m2; and
gemcitabine is administered in a range amount selected from: 1-5000, 100-4500,
200-4000,
300-3500, 400-3000, 500-2500, 550-2000, 600-1750, 650-1500, 700-1400, 750-
1300, 800-
1200, or 900-1100 mg/m2.
[0015] In yet another embodiment, CEND-1 is administered in a range of 0.2-
3.2 mg/kg
body weight/day or per dose of chemotherapy; nab-paclitaxel is administered at
125 mg/m2;
and gemcitabine is administered at 1000 mg/m2.
[0016] In certain embodiments of the invention methods provided herein,
efficacy or
clinical activity of the method is measured by determining: Overall Response
Rate (ORR),
Progression Free Survival (PFS) and/or Overall Survival (OS). In yet further
embodiments,
efficacy or clinical activity of the method is measured by determining one or
more of: an
Overall Response Rate (ORR) selected from greater than 25%, 30%, 35%, 40%,
45%, 50%,
55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or greater that 95%; a Progression
Free Survival
(PFS) selected from greater than 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%,
70%,
75%, 80%, 85%, 90%, or greater that 95%; and/or an Overall Survival (OS)
selected from
greater than 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%,
90%,
or greater that 95%.
[0017] Also provided herein are pharmaceutical composition comprising: a
CEND-
1/iRGD-analog and a pharmaceutically acceptable excipient. In a particular
embodiment, the
invention composition corresponds to the iRGD-analog is set forth as the
structure in Figure
2 (CEND-1/iRGD-analog). The invention iRGD-analog differs from the prior art
iRGD
peptides in the specific moieties used to block the amino and carboxy termini,
which has
resulted in significant advantages over prior art cyclic iRGD peptides. For
example, the
invention CEND-1/iRGD-analog (set forth in Figure 2) has the following
Molecular formula
C37 H60 N14 014 S2; a MW 989.1; and the recent CAS Registry#: 2580154-02-3.
Whereas
one prior art iRGD with at least one inferior therapeutic property corresponds
to an iRGD
- 5 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
having the Molecular Formula: C35H57N13014S2; a Molecular Weight of 948.04;
and CAS
Registry No. 1392278-76-0.
[0018] Advantages of the invention iRGD-analog (Figure 2; C37 H60 N14 014
S2; MW
989.1), relative to prior art CAS Registry No. 1392278-76-0 cyclic peptide and
other known
iRGD molecules, while maintaining favorable in vitro/in vivo potency and
efficacy include
one or more of the following:
Favorable pharmacokinetic properties;
Improved stability in plasma/serum;
Improved stability in formulated solution;
Improved stability in storage; and/or
Improved protection from proteases such as aminopeptidases and
carboxypeptidases.
[0019] In certain embodiments, favorable and/or improved pharmacokinetic
properties
are selected from one or more of absorption, distribution, metabolism, and/or
excretion.
[0020] Also provided herein is a kit or composition comprising an iRGD-
analog (CEND-
1); and an anti-cancer agent. In a particular embodiment, the iRGD-analog is
set forth as the
structure in Figure 2.
BRIEF DESCRIPTION OF THE DRAWINGS
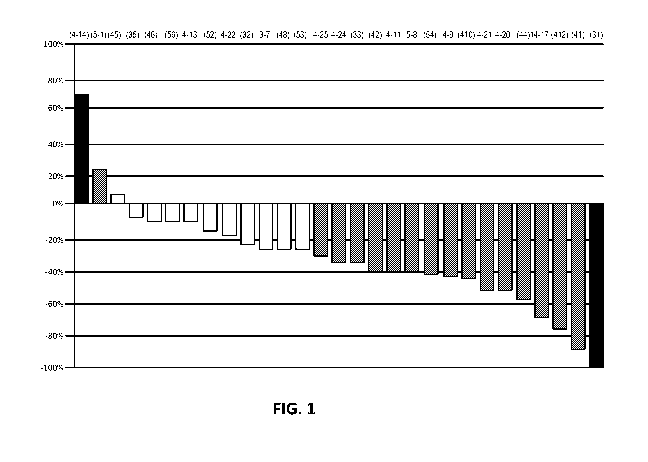
[0021] FIG. 1 shows a waterfall plot described in the Examples.
[0022] FIG. 2 shows the chemical structure of the invention CEND-1 or CEND-
1/iRGD-
analog cyclic peptide having the Molecular formula C37 H60 N14 014 S2; a MW of
989.1;
and the CAS Registry#: 2580154-02-3. It has all natural amino acids and can
also be
represented as follows: Ac-Cys-Arg-Gly-Asp-Lys-Gly-Pro-Asp-Cys-NH2 (Cys & Cys
Bridge). It can also br represented as follows: L-cysteinyl-L-arginylglycyl-L-
.alpha.-aspartyl-
L-lysylglycyl-L-prolyl-L-.alpha.-aspartyl-, cyclic (1.fwdarw.9)-disulfide,
with N-terminal
amino group blocked by an acetyl group and the C-terminal carbonyl group by a
carboxyamide
group.
- 6 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
DETAILED DESCRIPTION
Provided herein are methods for treating, inhibiting, or reducing the volume
of a tumor
of a cancer in a subject or patient in need thereof, wherein the method
comprises administering
CEND-1, or a pharmaceutically acceptable salt thereof, in a combination with
simultaneous,
separate or sequential administration of at least one anti-cancer agent or
therapy. The
invention provides improved methods and medicaments for more effectively
treating solid
tumors with anti-cancer therapies. CEND-1 is a tumor-penetrating peptide (also
known as
iRGD and internalizing arginylglycylaspartic acid cyclic peptide), with a
cyclizing (S-S bond
through the cysteine side chains) structure containing nine amino acids. In a
particular
embodiment, an invention CEND-1/iRGD-analog corresponds to the invention iRGD-
analog
peptide sequence corresponding to the specific cyclic peptide chemical
structure set forth in
Figure 2, set forth as Ac-Cys-Arg-Gly-Asp-Lys-Gly-Pro-Asp-Cys-NH2 and having
CAS
Registry #2580154-02-3. The pharmacological effect of CEND-1 is restricted to
tumors via
the primary RGD tumor homing motif interaction with av-integrins (highly
expressed in
growing tumors but not in healthy tissues). The secondary CendR' ¨ motif
modulates the
tumor microenvironment via NRP-1. Based on experimental models, the
interaction with
neuropilin-1 leads to transformation of the solid tumor microenvironment into
a temporary
drug conduit, allowing an efficient tumor access of anti-cancer therapies
given in combination
with CEND-1. Studies have demonstrated that CEND-1 increases, via the above-
mentioned
tumor microenvironment modulation mechanism, accumulation and penetration of
anticancer
drugs into tumors, but not into normal tissues. As a result, anti-tumor
activity is enhanced,
while the therapeutic margins/safety profile is potentially improved. In
addition to the
invention CEND-1/iRGD-analog (Figure 2); other iRGD peptides and anglogs known
in the
art, such as those described hereinabove, can be used in the invention
methods, in view of the
data, dosages and results provided herein.
[0023] In certain embodiments, the tumor is a malignant solid tumor
characterized by
dense tumor stroma. In other embodiments, the tumor is a solid tumor of a
cancer selected
from the group consisting of: breast cancer, squamous cell cancer, small-cell
lung cancer, non-
small cell lung cancer, gastrointestinal cancer, pancreatic cancer,
glioblastoma, cervical
cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, colon cancer,
colorectal
cancer, endometrial carcinoma, salivary gland carcinoma, kidney cancer, liver
cancer, prostate
cancer, vulval cancer, thyroid cancer, hepatic carcinoma, and head and neck
cancer. In
- 7 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
another embodiment, the pancreatic cancer is selected from the group
consisting of: primary
pancreatic cancer, metastatic pancreatic cancer, refractory pancreatic cancer,
cancer drug
resistant pancreatic cancer and adenocarcinoma. In a particular embodiment,
the cancer is
ductal adenocarcinoma (such as Stage 0-Iv, and the like.
[0024] As used herein the phrase "solid tumor" refers to essentially solid
neoplasmic
growth, with low liquid content that is other than a cyst or tumor metastasis
(i.e. at its
metastatic stage of disease).
[0025] As used herein, the phrase "in a combination" refers to
administering more that
one therapeutic agent to a respective patient in need thereof. In particular
embodiments,
CEND-1 is administered with at least one other anti-cancer therapeutic agent.
[0026] As used herein, the phrase "simultaneous, separate or sequential
administration"
refers to administering CEND-1 at the same time as the one or more other
cancer therapeutic
agents; or either before or after administration with the co-administered anti-
cancer agents;
such that the co-administration can be from separate pharmaceutical
compositions
administered with either the same or different dosing regimens. In certain
embodiments,
CEND-1 is administered before the subsequent and sequential administration of
the one or
more anti-cancer agents.
[0027] As used herein, the term "malignant" refers to a tumor or cancer in
which abnormal
cells divide without control and can invade nearby tissues. Malignant cancer
cells can also
spread to other parts of the body through the blood and lymph systems.
[0028] Based on the novel drug conduit mechanism discovered by the present
inventors,
the methods and medicaments of the present invention are suitable for using
CEND-1 (e.g.,
an iRGD-analog) to enhance the therapeutic effects of any anticancer agent
used to treat solid
tumors. The methods and medicaments of the present invention can thus contain
combinations
of CEND-1/iRGD-analog with any anticancer agent used to treat solid tumors,
such as at least
one of a taxane such as docetaxel or paclitaxel (including nab-paclitaxel), a
nucleoside such
as gemcitabine, an anthracyclin such as doxorubicin, an alkylating agent, a
vinca alkaloid, an
anti-metabolite, a platinum agent such as cisplatin or carboplatin, a steroid
such as
methotrexate, an antibiotic such as adriamycin, an isofamide, a selective
estrogen receptor
modulator, or an antibody such as trastuzumab.
- 8 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
[0029] An anticancer agent whose effects can be enhanced by CEND-1 can be
an antibody
such as a humanized monoclonal antibody. As an example, the anti-epidermal
growth factor
receptor 2 (HER2) antibody, trastuzumab (Herceptin: Genentech, South San
Francisco, Calif)
is a therapeutic agent useful in a conjugate for treating HER2/neu
overexpressing breast
cancers (White et al., Annu. Rev. Med. 52:125-141 (2001)).
[0030] Anticancer agents whose effects can be enhanced by CEND-1 also can
be
cytotoxic agents, which, as used herein, can be any molecule that directly or
indirectly
promotes cell death. Useful cytotoxic agents include, without limitation,
small molecules,
polypeptides, peptides, peptidomimetics, nucleic acid-molecules, cells and
viruses. As non-
limiting examples, useful cytotoxic agents include cytotoxic small molecules
such as
doxorubicin, docetaxel or trastuzumab, antimicrobial peptides such as those
described further
below; pro-apoptotic polypeptides such as caspases and toxins, for example,
caspase-8;
diphtheria toxin A chain, Pseudomonas exotoxin A, cholera toxin, ligand fusion
toxins such
as DAB389EGF, Ricinus communis toxin (ricin); and cytotoxic cells such as
cytotoxic T cells.
See, for example, Martin et al., Cancer Res. 60:3218-3224 (2000); Kreitman and
Pastan,
Blood 90:252-259 (1997); Allam et al., Cancer Res. 57:2615-2618 (1997); and
Osborne and
Coro nado-Heinsohn, Cancer J. Sci. Am. 2: 175 (1996). One skilled in the art
understands that
these and additional cytotoxic agents described herein or known in the art can
be combined
with CEND-1 in the disclosed methods and medicaments.
[0031] In one embodiment, an anticancer agent whose effects can be enhanced
by CEND-
1 can be a therapeutic polypeptide. As used herein, a therapeutic polypeptide
can be any
polypeptide with a biologically useful function. Useful therapeutic
polypeptides encompass,
without limitation, cytokines, antibodies, cytotoxic polypeptides; pro-
apoptotic polypeptides;
and anti-angiogenic polypeptides. An anticancer agent whose effects can be
enhanced by
CEND-1 can be an anti-angiogenic agent. As used herein, the term "anti-
angiogenic agent'
means a molecule that reduces or prevents angiogenesis, which is the growth
and development
of blood vessels. The combination of CEND-1 with anti-angiogenic agents can be
used to treat
cancer associated with angiogenesis. A variety of anti-angiogenic agents can
be prepared by
routine methods. Such anti-angiogenic agents include, without limitation,
small molecules;
proteins such as dominant negative forms of angiogenic factors, transcription
factors and
antibodies; peptides; and nucleic acid molecules including ribozymes,
antisense
oligonucleotides, and nucleic acid molecules encoding, for example, dominant
negative forms
- 9 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
of angiogenic factors and receptors, transcription factors, and antibodies and
anti gen-binding
fragments thereof. See, for example, Hagedorn and Bikfalvi, Crit. Rev. Oncol.
Hematol.
34:89-110 (2000), and Kirsch et al., J. Neurooncol. 50:149-163 (2000).
[0032] In particular embodiments, the anti-cancer agent or therapy is
selected from the
group consisting of: a chemotherapeutic agent, small molecule, antibody,
antibody drug
conjugate, nanoparticle, cell therapy, polypeptides, peptides,
peptidomimetics, nucleic acid-
molecules, ribozymes, antisense oligonucleotides, and nucleic acid molecules
encoding
transgenes, viruses, cytokines, cytotoxic polypeptides; pro-apoptotic
polypeptides, anti-
angiogenic polypeptides. cytotoxic cells such as cytotoxic T cells, and/or
vaccines (mRNA or
DNA).
[0033] In other embodiments, the chemotherapeutic agent is selected from
one or more of
the group consisting of: taxane, docetaxel, paclitaxel, nab-paclitaxel, a
nucleoside,
gemcitabine, an anthracycline, doxorubicin, an alkylating agent, a vinca
alkaloid, an anti-
metabolite, a platinum agent, cisplatin, carboplatin, a steroid, methotrexate,
an antibiotic,
adriamycin, an isofamide, a selective estrogen receptor modulator, a
maytansinoid,
mertansine, emtansine, an auristatin, monomethyl auristatin E (MMAE) and F
(MMAF), a
natural antimitotic drug, an antibody, trastuzumab, an anti-epidermal growth
factor receptor 2
(HER2) antibody, trastuzumab, a caspase, caspase-8; diphtheria toxin A chain,
Pseudomonas
exotoxin A, cholera toxin, ligand fusion toxins, DAB389EGF, Ricinus communis
toxin
(ricin); chimeric antigen receptor T cells (CAR-T), chimeric antigen receptor
macrophages
(CAR-M), chimeric antigen receptor natural killer cells (CAR-K), and tumor-
infiltrating
lymphocytes (TIL), anti-PD-1 antibodies, nivolumab, pembrolizumab,
atezolizumab,
avelumab, durvalumab; anti-CTLA-4 antibodies. ipilimumab; bispecific
antibodies,
catumaxomab, anti-CD47 antibodies, enfortumab, sacituzumab, antibody-drug
conjugates.
Moderna' s mRNA-4157 and/or BioNTech's BNT122.
[0034] In particular embodiments, the CEND-1 (e.gõ the iRGD-analog set
forth in Figure
2) is administered in an amount selected from the group consisting of: about
0.2 to 20 mg/kg
body weight/per dose of cancer therapy, about 0.3 to 17 mg/kg body weight/per
dose of cancer
therapy, about 0.4 to 14 mg/kg body weight/per dose of cancer therapy, about
0.5 to 11 mg/kg
body weight/per dose of cancer therapy, about 0.6 to 8 mg/kg body weight/per
dose of cancer
therapy, about 0.7 to 5 mg/kg body weight/per dose of cancer therapy, about
0.8 to 3.2 mg/kg
- 10 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
body weight/per dose of cancer therapy. In a particular embodiment, CEND-1 is
administered
in an amount corresponding to 3.2 mg/kg body weight/per dose of cancer
therapy.
[0035] As used herein, the phrae "per dose of cancer therapy" refers to the
co-
administration of CEND-1 with one or more anti-cancer agents, such that each
time an anti-
cancer therapeutic is administered, CEND-1 is likewise co-administered to
facilitate the
therapeutics penetration into the tumor. The co-administration per dose of
CEND-1 does not
need to be exactly simultaneous with the therapeutic agent(s), and CEND-1 can
be
administered either before or after the administration of the therapeutic
agent.
[0036] In certain embodiments, CEND-1 is administered before or during the
administration of anti-cancer therapy, wherein the cancer therapy is at a
dosing regimen
selected from the group consisting of: 4 times/day, 3 times/day, twice daily,
once daily, once
every other day, once every 2nd day, once every 3rd day, once every 4th day,
once every 5th
day, once every 6th day, once weekly, once every 8th day, once every 9th day,
once every
10th day, once every 11th day, once every 12th day, once every 13th day, once
every 2 weeks,
once every 3 weeks, and/or once per month. In one embodiment, CEND-1 is
present in a dry
formulation or suspended in a biocompatible medium.
[0037] In particular embodiments, the biocompatible media is selected from
the group
consisting of: water, buffered aqueous media, saline, buffered saline,
optionally buffered
solutions of amino acids, optionally buffered solutions of proteins,
optionally buffered
solutions of sugars, optionally buffered solutions of vitamins, optionally
buffered solutions of
synthetic polymers, and lipid-containing emulsions. In a particular
embodiment, CEND-1 is
administered intravenously.
[0038] The method of the present invention is particularly suitable for the
treatment of
pancreatic cancer, which is characterized by a prominent dense tumor stroma,
acting as a
physical barrier to drug entry. Therefore, advanced pancreatic cancer was
chosen as the first
clinical indication for CEND-1. As an example of clinical usefulness, we show
safety and
efficacy results of CEND-1 when given alone or in combination with nab-
paclitaxel and
gemcitabine, including its ability to enhance tumor response.
[0039] Also provided herein is a method of treating pancreatic cancer in a
patient in need
thereof, comprising administering to the patient an effective amount of CEND-
1, in
combination with gemcitabine and/or nab-paclitaxel, or pharmaceutically
acceptable salts
- 11 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
thereof In certain embodiments, the pancreatic cancer is selected from the
group consisting
of: primary pancreatic cancer, metastatic pancreatic cancer, refractory
pancreatic cancer,
cancer drug resistant pancreatic cancer and adenocarcinoma. In a particular
embodiment, the
cancer is ductal adenocarcinoma (Stage 0-IV).
[0040] In another embodiment the afore described CEND-1 for use in the
treatment of
pancreatic cancer can be administered in combination with at least one
additional anti-cancer
drug, which preferably is known to be effective against pancreatic cancer,
such as
gemcitabine. In context of the present invention it was found that using a
CEND-1 can enhance
the clinical activity of other pancreatic cancer drugs such as gemcitabine and
nab-paclitaxel
administered by the intravenous route.
[0041] In certain embodiments, CEND-1 is administered in an amount selected
from the
group consisting of: about 0.2 to 20 mg/kg body weight/per dose of cancer
therapy, about 0.3
to 17 mg/kg body weight/ per dose of cancer therapy, about 0.4 to 14 mg/kg
body weight/per
dose of cancer therapy, about 0.5 to 11 mg/kg body weight/per dose of cancer
therapy, about
0.6 to 8 mg/kg body weight/per dose of cancer therapy, about 0.7 to 5 mg/kg
body weight/per
dose of cancer therapy, about 0.8 to 3.2 mg/kg body weight/per dose of cancer
therapy. In
once embodiment, CEND-1 is administered in an amount corresponding to 3.2
mg/kg body
weight/per dose of cancer therapy.
[0042] In particular embodiments, CEND-1 is administered before or during
the
administration of anti-cancer therapy, wherein the cancer therapy is at a
dosing regimen
selected from the group consisting of: 4 times/day, 3 times/day, twice daily,
once daily, once
every other day, once every 2nd day, once every 3rd day, once every 4th day,
once every 5th
day, once every 6th day, once weekly, once every 8th day, once every 9th day,
once every
10th day, once every 11th day, once every 12th day, once every 13th day, once
every 2 weeks,
once every 3 weeks, and/or once per month. In a particular embodiment,
[0043] CEND-1 is administered in a range amount selected from: 0.01-100,
0.02-90, 0.03-
80, 0.04-70, 0.05-60, 0.06-50, 0.07-40, 0.08-30, 0.09-30, 0.1-25, 0.11-20,
0.12-15, 0.13-10,
0.14-9, 0.15-8, 0.16-7, 0.17-6, 0.18-5, 0.19-4, or 0.2-3.2 mg/kg body
weight/day or per dose
of chemotherapy;
- 12 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
[0044] nab-paclitaxel is administered in a range amount selected from: 1-
500, 10-450, 20-
400, 30-350, 40-300, 50-250, 60-200, 70-175, 80-160, 90-150, 100-140, 110-140,
115-135 or
120-130 mg/m2; and
[0045] gemcitabine is administered in a range amount selected from: 1-5000,
100-4500,
200-4000, 300-3500, 400-3000, 500-2500, 550-2000, 600-1750, 650-1500, 700-
1400, 750-
1300, 800-1200, or 900-1100 mg/m2.
[0046] In yet another embodiment: CEND-1 is administered in a range of 0.2-
3.2 mg/kg
body weight/day or per dose of chemotherapy; nab-paclitaxel is administered at
125 mg/m2;
and gemcitabine is administered at 1000 mg/m2.
[0047] In certain embodiments of the invention methods provided herein,
efficacy or
clinical activity of the method is measured by determining: Overall Response
Rate (ORR),
Progression Free Survival (PFS) and/or Overall Survival (OS). In yet further
embodiments,
efficacy or clinical activity of the method is measured by determining one or
more of: an
Overall Response Rate (ORR) selected from greater than 25%, 30%, 35%, 40%,
45%, 50%,
55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, or greater that 95%; a Progression
Free Survival
(PFS) selected from greater than 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%,
70%,
75%, 80%, 85%, 90%, or greater that 95%; and/or an Overall Survival (OS)
selected from
greater than 25%, 30%, 35%, 40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%,
90%,
or greater that 95%.
[0048] Also provided herein are pharmaceutical composition comprising: a
CEND-
1/iRGD-analog and a pharmaceutically acceptable excipient. Pharmaceutically
acceptable
excipients are well-known in the art. The CEND-1 compositions can be
administered to an
individual (such as human) via a bolus injection or an infusion, via various
routes, including,
for example, intravenous, intra-arterial, intraperitoneal, intrapulmonary,
oral and inhalation,
subcutaneous. In some embodiments, the composition is administered
intravenously.
[0049] The formulations can be presented in unit-dose or multi-dose sealed
containers,
such as ampules and vials, and can be stored in a freeze-dried (lyophilized)
condition requiring
only the addition of the sterile liquid excipient, for example, saline, for
injections, immediately
prior to us.
- 13 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
[0050] In a particular embodiments, CEND-1 for Injection is a sterile,
white, lyophilized
powder supplied as 100 mg per vial of active ingredient dose strength for
intravenous
administration. CEND-1 Injection consists of CEND-1 drug substance with sodium
acetate
trihydrate and mannitol as excipients.
[0051] In a particular embodiment, the invention composition corresponds to
the iRGD-
analog is set forth as the structure in Figure 2 (CEND-1/iRGD-analog). The
invention iRGD-
analog differs from the prior art iRGD peptides in the specific moieties used
to block the
amino and carboxy termini, which has resulted in significant advantages over
prior art cyclic
iRGD peptides. For example, the invention CEND-1/iRGD-analog (set forth in
Figure 2) has
the following Molecular formula C37 H60 N14 014 S2; a MW 989.1; and the recent
CAS
Registry#: 2580154-02-3. Whereas one prior art iRGD with at least one inferior
therapeutic
property corresponds to an "academic" iRGD having the Molecular Formula:
C35H57N13014S2;
a Molecular Weight of 948.04; and CAS Registry No. 1392278-76-0.
[0052] Advantages of the invention iRGD-analog (Figure 2; C37 H60 N14 014
S2; MW
989.1), relative to prior art CAS Registry No. 1392278-76-0 cyclic peptide and
other known
iRGD molecules, while maintaining favorable in vitro/in vivo potency and/or
efficacy, include
one or more of the following:
Favorable pharmacokinetic properties;
Improved stability in plasma/serum;
Improved stability in formulated solution;
Improved stability in storage; and/or
Improved protection from proteases such as aminopeptidases and
carboxypeptidases.
In certain embodiments, favorable and/or improved pharmacokinetic properties
are selected
from one or more of absorption, distribution, metabolism, and/or excretion.
[0053] As used herein, the phrase "while maintaining favorable in vitro/in
vivo potency
and/or efficacy" refers to the continued affect of CEND-1 on the respective
therapeutic agents,
such that the efficacy and/or potency is not diminished by CEND-1.
- 14 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
[0054] Also provided herein is a kit or composition comprising an iRGD-
analog (CEND-
1); and an anti-cancer agent. The kit of claim 26, wherein the iRGD-analog is
set forth as the
structure in Figure 2.
EXAMPLES
Example 1: Phase I trial (referred to as CEND-001 trial) of CEND-1 in
combination with
gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer.
[0055] This example demonstrates that CEND-1 was well tolerated in
combination with
gemcitabine and nab-paclitaxel and provided clinical benefit in patients with
advanced
pancreatic cancer. When compared to benchmark trials, the response rates are
more than
doubled. CEND-1 is also referred to herein as the iRGD-analog or CEND-1/iRGD-
analogFfiguel corresponding to the chemical structure set forth in Figure 2
and CAS Registry
# 2580154-02-3.
Materials:
[0056] CEND-1 drug product is a synthetic peptide manufactured using solid
phase
peptide synthetic techniques with high chemical purity. CEND-1 for Injection
is a sterile,
white, lyophilized powder supplied as 100 mg per vial of active ingredient
dose strength for
intravenous administration. CEND-1 Injection consists of CEND-1 drug substance
with
sodium acetate trihydrate and mannitol as excipients.
[0057] Methods: The open-label, dose escalation, multicenter (3 active
sites in Australia)
trial involved a run-in phase with ascending doses of CEND-1 monotherapy (1-7
days),
followed by the combination of CEND-1 with nab-paclitaxel (125 mg/m2) and
gemcitabine
(1000 mg/m2) on days 1, 8, 15 of a 21-day treatment cycle. Patients will first
receive the
intravenous infusion of nabpaclitaxel (125mg/m2 over 30 minutes ( 3 minutes)).
CEND-1 is
given intravenously at the applicable dose level as a slow IV push over 1
minute ( 30 seconds)
immediately following completion of the post-nabpaclitaxel saline flush. The
intravenous
infusion of gemcitabine (1000mg/m2 over 30 minutes ( 3 minutes)) will be
started as soon as
possible, but at the latest within 10 minutes of CEND-1 administration.
- 15 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
Safety / Dose Escalation (Cohort la)
Dose Run-in (7 days) Treatment (28-day cycle)
Level
1 CEND-1 nabpaclitaxel 125 mg/m2
(1-6 pts) 0.2 mg/kg CEND-1 0.2 mg/kg
gemcitabine 1000mg/m2
2 CEND-1 nabpaclitaxel 125 mg/m2
(1-6 pts) 0.8 mg/kg CEND-1 0.8 mg/kg
gemcitabine 1000 mg/m2
3 CEND-1 nabpaclitaxel 125 mg/m2
(3-6 pts) 1.6 mg/kg CEND-1 1.6 mg/kg
gemcitabine 1000 mg/m2
4 CEND-1 nabpaclitaxel 125 mg/m2
(3-6 pts) 3.2 mg/kg CEND-1 3.2 mg/kg
gemcitabine 1000 mg/m2
[0058] Patients (n=31) who had measurable metastatic pancreatic cancer, no
prior
treatments for metastatic disease and an ECOG PS of 0 to 1 were included.
Primary endpoints
are safety and optimal biologic dose, secondary and exploratory endpoints
included response
rates, pharmacokinetics and biomarkers.
[0059] Results: 29 patients completed the first treatment cycle and were
evaluable for
response (data cutoff, 27 April 2020). No dose limiting toxicities were
observed. AEs were
generally consistent with those of nabpaclitaxel and gemcitabine. The only
drug related grade
(gr) 3 - 4 adverse events (AEs) present in >3 patients were neutropenia in 18
(62%) and anemia
in 5 (17%) patients. By investigator assessed RECIST 1.1 criteria, 1 pt had a
complete
response (3.4%), 16 pt. with partial response (55%), 10 pt. with stable
disease (34%), and 2
Pt. with progressive disease (6.9%). Among the patients with elevated CA19-9
with a
postbaseline assessment available, A total of 96% of the patients had a
decrease from baseline
of at least 20%, and 74% had a decrease of at least 90% and/or had the CA19-9
levels
normalized to baseline.
[0060] Conclusions: Administration of CEND-1 in combination with nab-
paclitaxel and
gemcitabine is safe, with no dose-limiting toxicities. The incidence of Grade
3 and 4 Adverse
Event is lower than in similar published trials. The median duration of
treatment was longer
and the response rates were >2 times higher than in the benchmark trials.
- 16 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
BASELINE SUBJECT CHARACTERISTICS
Age
Median 62 ...........
Min-Max 42-80
Distribution ¨ no. (%)
<65 yr 15
65 yr 14
Sex ¨ no. (%)
Female 11
Male 18
Race or ethnic group ¨ no.(%)
Asian 2
White 25
Other 2
ECOG Performance Score
0 10
1 19
[0061] Frequencies below are compared with iMPACT3 trial, data in
parenthesis (Von
Hoff et al., 2013).
EFFICACY RESULTS ¨ RESPONSE RATES
[0062] The overall response rate (ORR) for all evaluable patients (N = 29)
was 55% (vs.
23%). The overall disease control rate for 16 weeks was 76% (vs. 48%).
[0063] Figure 1 corresponds to a waterfall plot of maximum percentage
changes from
baseline in the size of target lesions according to the Response Evaluation
Criteria In Solid
Tumors 1.1. A total of 16 patients exhibited partial response (55%) and 10
patients had stable
disease (34%).
CA19-9
[0064] A total of 24 patients had an elevated baseline CA19-9 (>37 U/L). Of
these, 23
patients had at least one on-treatment CA19-9 measurement. A total of 96% of
the patients
had a decrease from baseline of at least 20% (vs. 61%), and 74% had a decrease
of at least
90% and/or had the CA19-9 levels normalized to baseline.
TREATMENT EXPOSURE
[0065] The median duration of treatment was 6.9+ months (vs. 3.9 months),
with 76%
receiving treatment for at least 6 months (vs. 32%). 86% of the patients had
reductions in the
- 17 -
CA 03182546 2022-11-04
WO 2021/226148
PCT/US2021/030740
nab-paclitaxel dose (vs. 41%) and 76% had reductions in the gemcitabine dose
(vs. 47%). In
total, 53% of all nab-paclitaxel doses administered during the study were at
the full dose of
125 mg per square meter (vs. 71%). The median relative dose intensity (the
proportion of the
administered cumulative dose relative to the planned cumulative dose) in the
nab-paclitaxel¨
gemcitabine group was 78% for nab-paclitaxel and 82% for gemcitabine (vs 81%
and 75%,
respectively).
SAFETY
[0066]
Table 2 below shows frequencies of bone marrow toxicity observed according to
the National Cancer Institute Common Terminology Criteria for Adverse Events
(CTCAE),
version 5. The frequency of grade 3-4 bone marrow toxicity in this material
was 66% for
neutropenia, 14% for leukopenia, 23% for neutropenia, 3% for thrombocytopenia
and 24%
for anaemia. The data from the iMPACT3 [Von Hoff et al. 2013] phase III trial
are shown on
the middle column for comparison.
no./total no. (%)
nab-13arlitaxel plus Gemcitabine N
CEND-1 plus nab-Paclitaxel
421)
plus Gemcitabine (N=29)
Grade n hematologic adverse event
Neutropenia 153/405 (38) 19/29
(66)
Leukopenia 124/405 (31) 4/29 (14)
Thrombocytopenia 52/405 (13) 1/29(3)
Anemia 53/405 (13) 7/29(24)
Grade ?..3 nonhematologic adverse event occurring in >5% of patients
Fatigue 70(17) 1/29 (3)
Peripherai neuropathy / Median time to onset 70 (17) / 140 days
4/29 (14) 216 days
Diarrhea 24(5) 1/29(3)
Table 2
CEND1-001 STUDY
[0067] In
the CEND1-001 trial, as set forth above, CEND-1 was given initially at
escalating doses from 0.2 mg/kg to 3.2 mg/kg during a run-in period of 1 to 7
days, during
which PK and safety of the single agent were assessed.
[0068]
There were 8 patients in Cohort la: 1 patient at dose level 1 (CEND-1 0.2
mg/kg),
1 patient at dose level 2 (0.8 mg/kg), 3 patients at dose level 3 (1.6 mg/kg)
and 3 patients at
dose level 4 (3.2 mg/kg). There were 23 patients in Cohort lb, 11 patients at
dose level 3 (1.6
mg/kg), 11 patients at dose level 4 (3.2 mg/kg), and 1 patient who was
assigned to dose level
- 18 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
4 (3.2 mg/kg) but withdrew from the study following the run-in period and only
received the
run-in dosing with CEND-1 0.2 mg/kg.
[0069] Of the 31 patients enrolled, 29 were evaluated for efficacy, 31
were evaluated for
PK and 30 were evaluated for PD (N=14 at the 1.6 mg/kg CEND-1 dose and N=14 at
the 3.2
mg/kg CEND-1 dose level, not including the 2 patients in the CEND-1 low dose
group). There
were 10 patient deaths reported during the study, 9 caused by progression of
primary disease
(metastatic pancreatic cancer) and 1 due to a left middle cerebral artery
stroke.
[0070] Confirmed objective responses (OR) occurred in 17/29 (58.6%)
patients (95% CI
= 38.9, 76.5). The objective response rates for patients treated with CEND-1
1.6 mg/kg and
3.2 mg/kg were 50% (7/14) and 61.5% (8/13) (61.5%), respectively. Disease
control was
defined as CR + PR + SD > 16 weeks and the disease control rate (DCR) was
64.3% (9/14) in
the CEND-1 1.6 mg/kg group and 92.3% (12/13) in the CEND-1 3.2 mg/kg group.
Overall,
the number of patients with progression was 16/29 (55.2%) and the median time
to progression
was approximately 9.7 months.
[0071] These response rates (OR) are clearly above and a marked
improvement over what
has been achieved in comparable trials historically, Table 3. In the Phase 3
registration trial
for nab-paclitaxel, the response rate in first-line metastatic pancreatic
cancer patients treated
with the gemcitabine / nab-paclitaxel combo was 23% and the PFS 5.5 months
(Von Hoff et
al., 2013).
Outcomes of Gemcitabine and Nab-Paclitaxel in Metastatic Pancreatic Cancer
Gemcitabine and Nab-Paclitaxel Arm
Study Stage Investigational Phase N ORR mOS mPFS PFS
PFS
Agent (Placebo) ( /0) (months) (months) 6m
lY
CYO CYO
VonHoff IV Gem vs Gem/NP 3 432 23 8.5 5.5 45*
17*
2013-
International
Renouf IV Durvalumab and 2 61 23 8.8 5.4 40*
18*
2020- Tremelimumab
Canada
- 19 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
Van Custem IV PEGPH20 2 165 36 11.5 7.1 52*
23*
2020-
International
Hu 2019- IV Tarextumab 2 88 31.8 7.9 5.5 38*
11*
USA
Karasic III/IV Hydroxychloroquine 2 57 21.1
12.1 6.4 15* 50**
2019-USA
Tempero IV Ibrutinib 3 213 42 10.8 6.0 50*
19*
2019-
International
Table 3
[0072] Because of the trend for an improved outcome with the 3.2 mg/kg
dose level, this
was chosen as the dose for further exploration in future studies.
TUMOR BIOMARKERS
[0073] The number of patients with a >50% reduction in CA19-9 from
Baseline increased
to a high of 20/22 (90.9%) patients at Cycle 5 Day 1.
[0074] Tumor biomarker results of CEND-1 at the dose levels of 1.6 mg/kg
and 3.2 mg/kg
show a trend of decreasing CA values over successive cycles of dosing. This
supports the
further development of CEND-1, in combination with drugs such as Nab-
paclitaxel and
Gemcitabine, in patients with metastatic cancers.
CEND-1 PHAR1VIACOKINETICS
[0075] Overall, the median Tmax for CEND-1 was 0.067 hours over all days
of PK
sampling (minimum was 0.03, maximum 0.55). There was dose proportional
increase in Cmax
without increase with repeat dosing.
[0076] Assessment of plasma CEND-1 parameters demonstrated that exposure
(AUCO-t,
AUCO-6h and AUCO-inf) followed the same pattern described for Cmax with a
trend to
increase with increased dose. Dose normalized PK parameters (AUCO-t/D, AUCO-
6h/D and
AUCO-inf/D) were similar between visits and doses.
[0077] CEND-1 was eliminated with median T1/2 values between 1.6 hours
and 1.8 hours
over all days of PK sampling. CL mean values were between 106.8 mL/h/kg and
266.5
- 20 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
mL/h/kg. The terminal volume of distribution (Vz) mean values were between
220.9 mL/kg
and 277.4 mL/kg over all days of PK sampling
CEND-1 SAFETY
[0078] During the CEND-1 run-in during dose escalation, the following
definition of DLT
was used:
CEND-1 monotherapy:
A DLT in the run-in period was defined as:
-Grade 4 neutropenia lasting > 5 days or Grade 3 or 4 neutropenia with fever
and/or infection
-Grade 4 thrombocytopenia (or Grade 3 with bleeding)
-Grade 3 or 4 treatment-related non-hematological toxicity (Grade 3 nausea,
vomiting or diarrhea that last > 72 hours despite maximal treatment
constitutes a DLT,
insufficient treatment will not constitute an exception to the DLT criteria,
as this would
constitute inadequate conduct of the study)
-Dosing delay greater than 2 weeks due to treatment-emergent AEs or related
severe laboratory abnormalities.
[0079] There were no DLTs or grade 3 or 4 adverse events at any CEND-1 dose
level
during the single agent run-in portion of the study and no clinically
significant adverse events
attributable to CEND-1 were reported.
During the combination portion of the study, the following definition of dose-
limiting
toxicity was used:
-Any side effect that is more severe, longer in duration or more frequent than
side effects expected from the nab-paclitaxel and gemcitabine package insert.
-Any side effect that is not included in the nab-paclitaxel and gemcitabine
package insert that meets the DLT definition of the monotherapy above.
-21 -
CA 03182546 2022-11-04
WO 2021/226148 PCT/US2021/030740
[0080] There were no DLTs reported during the study. The majority of TEAEs
were
CTCAE grade 1 or 2. The number of reported TEAEs at each grade was similar
between
CEND-1 dose levels. Overall, the severity of TEAEs did not increase with CEND-
1 dose. The
most common CTCAE grade 3-4 TEAEs by SOC were blood and lymphatic system
disorders.
Overall, 22 (71.0%) patients reported SAEs. There was no trend of increasing
frequency of
SAEs with increasing CEND- 1 dose. The most common SAEs by SOC were infections
and
infestations. The safety data for CEND-1 suggest a favorable benefit-risk
profile and safety
profile. The absence of any CEND-1 monotherapy related SAEs and low frequency
of CEND-
1 combination therapy related SAEs supports the continued evaluation of this
investigational
therapy for metastatic exocrine pancreatic cancer.
- 22 -