Note: Descriptions are shown in the official language in which they were submitted.
- 1 -
FIVE-MEMBERED RING DERIVATIVE AND MEDICAL USE THEREOF
Technical Field
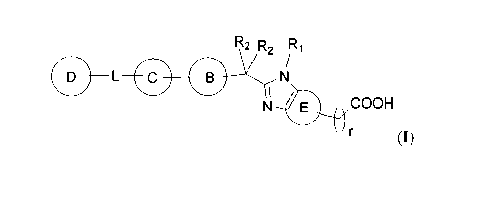
[0001] The present invention relates to a compound represented by general
formula
(I) or a stereoisomer, tautomer, deuterated substance, solvate, prodrug,
metabolite,
pharmaceutically acceptable salt or co-crystal thereof, an intermediate
thereof, and a
preparation method therefor, as well as an application in the preparation of a
drug for
treating diabetes.
Background Art
[0002] Diabetes is a group of metabolic diseases characterized
by hyperglycemia.
Hyperglycemia is caused by defective insulin secretion or impaired insulin
biological
action, or both. The long-term high blood sugar in diabetes leads to chronic
damage and
dysfunction of various tissues, especially eyes, kidneys, heart, blood
vessels, and nerves.
Diabetes is mainly divided into two types. Type 1 diabetes: destruction of
islet B cells
leads to absolute insulin deficiency. Type 2 diabetes: insulin resistance is
dominant with
insulin relative deficiency or impaired insulin secretion is dominant with
insulin resistance.
[0003] Drugs for type 2 diabetes can be divided into six major
classes (insulin, insulin
secretagogues, biguanides, glucosidase inhibitors, thiazolidinediones, SGLT2
inhibitors),
each of which works through a different primary mechanism. However, with the
exception
of GLP-1 receptor agonists and SGLT2 inhibitors, these drugs have limited
efficacy and
cannot address the most important problems, namely decreased cellular function
and
associated obesity.
[0004] GLP-1 is a 30-amino acid long intestinal insulin stimulating hormone
secreted
by L cells in the intestine. GLP-1 stimulates insulin secretion, reduces
glucagon secretion,
inhibits gastric emptying, reduces appetite, and stimulates 13-cell
proliferation in a
physiological and glucose-dependent manner. In nonclinical experiments, GLP-1
promotes
the sustainability of 13 cells by stimulating the transcription of genes
important for glucose-
dependent insulin secretion and by promoting (3 cell regeneration. In healthy
individuals,
GLP- 1 plays an important role in the regulation of postprandial blood,
leading to increased
CA 03203276 2023- 6- 22
- 2 -
peripheral glucose uptake by stimulating glucose-dependent insulin secretion
from the
pancreas. GLP-1 also inhibits glucagon secretion, resulting in decreased
hepatic glucose
output. In addition, GLP-1 delays gastric emptying, slows small bowel
motility, and delays
food absorption.
[0005] GLP-1 receptor agonists, such as GLP-1, liraglutide and exendin-4,
are
polypeptide drugs and are mostly used for injection. Small molecule GLP-1
receptor
agonists have become a hot spot in drug development in recent years due to
their high oral
bi availability potential.
Summary of the Invention
[0006] The object of the present invention is to provide a
compound capable of
stimulating a GLP-1 receptor or a stereoisomer, tautomer, deuterated
substance, solvate,
prodrug, metabolite, pharmaceutically acceptable salt or co-crystal thereof,
and an
intermediate and a preparation method thereof, as well as an application in
the preparation
of a drug for treating diabetes.
[0007] The compound of the present invention has good activity
of stimulating a
GLP-1 receptor, good pharmacokinetic performance and bioavailability, oral
performance
and good safety.
[0008] The present invention provides a compound or a
stereoisomer, tautomer,
deuterated substance, solvate, prodrug, metabolite, pharmaceutically
acceptable salt or co-
crystal thereof, and the compound is selected from a compound represented by
general
formula (I), wherein,
R2 R2 iR1
DI ______________________________ Le
/
N ' COOH
(I)
[0009] In some embodiments, ring B is selected from a 4- to 12-
membered
heterocyclic ring, C5-12 carbocyclic ring, C6-10 aromatic ring or a 5- to 12-
membered
heteroaromatic ring. The carbocyclic ring, heterocyclic ring, aromatic ring or
heteroaromatic ring is optionally further substituted with 0 to 4 (for
example, 0, 1, 2, 3 or
CA 03203276 2023- 6- 22
- 3 -
4) RB, the heterocyclyl or heteroaromatic ring contains 1 to 3 (for example 1,
2 or 3)
heteroatoms selected from 0, S and N.
[0010]
In some embodiments, ring B is selected from a benzene ring, a 4- to 8-
membered heterocyclyl, C5-8 carbocyclyl or a 5- to 6-membered heteroaromatic
ring. The
benzene ring, carbocyclyl, heterocyclyl or heteroaromatic ring is optionally
further
substituted with 0 to 4 (for example 0, 1, 2, 3 or 4) RB, the heterocyclic
ring contains 1 to 3
(for example, 1, 2 or 3) heteroatoms selected from 0, S and N.
[0011]
In some embodiments, ring B is selected from one of the following
substituted
or unsubstituted groups: cyclohexyl, cyclohexenyl, azacyclohexenyl,
piperidinyl, phenyl,
pyrazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl or triazinyl, which,
when
substituted, is optionally further substituted with 0 to 4 (for example, 0, 1,
2, 3 or 4) RB.
[0012]
In some embodiments, ring B is selected from one of the following
substituted
or unsubstituted groups: pyrimidin-4(3H)-one (that is, the ortho position of
the pyrimidine
N atom is substituted with a hydroxyl group, and pyrimidin-4(3H)-one is formed
through
tautomerism), pyridazine-3(2H)-one (that is, the ortho position of the
pyridazine N atom is
substituted with a hydroxyl group, and pyridazine-3(2H)-one is formed through
tautomerism), pyridine-2(/H)-one (that is, the ortho position of the pyridine
N atom is
substituted with a hydroxyl group, and pyridin-2(/H)-one is formed through
tautomerism).
[0013]
In some embodiments, ring B is selected from one of the following
substituted
or unsubstituted groups: piperazinyl, tetrahydropyrrolyl or 1,4-diazepanyl,
which, when
substituted, is optionally further substituted with 0 to 4 (for example, 0, 1,
2, 3 or 4) RB.
[0014]
In some embodiments, ring B is selected from one of the following
substituted
r')C
A4lNj ANI
N
or unsubstituted groups:
0 0
C)I\JC /\)c
N)
?NO'k -css:N/k ?Nk nN
`?-=
, the left side of which is directly
CA 03203276 2023- 6- 22
- 4 -
connected to ring C, and which, when substituted, is optionally further
substituted with 1, 2
or 3 Rs.
[0015]
In some embodiments, ring B is selected from one of the following
substituted
0
C)I\JC
µ7õN '7õNJ
or unsubstituted groups:
0
N
or
the left side of which is directly connected to ring C, and which,
when substituted, is optionally further substituted with 1, 2 or 3 RB.
[0016]
In some embodiments, ring B is selected from one of the following
substituted
1-/Nrµ
or unsubstituted groups: -z C or
, the left side of which is
directly connected to ring C, and which, when substituted, is optionally
further substituted
with 1, 2 or 3 RB.
[0017]
In some embodiments, ring B is selected from one of the following
substituted
0
NC ,0)?
or unsubstituted groups: '1/2-)
or
, the left side of which is directly connected to ring C, and which,
when substituted, is optionally further substituted with 1, 2 or 3 R.
[0018] In some
embodiments, ring B is selected from one of the following substituted
0 0
N
or unsubstituted groups: or
, the left side of which is directly
connected to ring C, and which, when substituted, is optionally further
substituted with 1, 2
or 3 Rs.
CA 03203276 2023- 6- 22
-5-
1100191
In some embodiments, ring B is selected from one of the following
substituted
0 0 0
N-\ N-\ Ni
N
or unsubstituted groups:
NN
, N
or
, the left side of which is directly
connected to ring C, and which, when substituted, is optionally further
substituted with 1, 2
or 3 Rs.
R2 R2
[0020] In some embodiments, is selected from
or
N
, the left side of which is directly connected to ring C, and which, when the
6-membered ring in the spiro ring is substituted, is optionally further
substituted with 1, 2
or 3 Rs.
[0021] In some
embodiments, ring B is selected from one of the following substituted
N
or unsubstituted groups: V--) , or -
, the left side
of which is directly connected to ring C, and which, when substituted, is
optionally further
substituted with 1, 2 or 3 RB.
[0022]
In some embodiments, ring C is selected from C6_io carbocyclic ring, a 5-
to
10-membered heterocyclic ring, C6-10 aromatic ring or a 5- to 10-membered
heteroaromatic
ring. The carbocyclic ring, heterocyclic ring, aromatic ring or heteroaromatic
ring is
optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) Rc,
the heterocyclic
ring or heteroaromatic ring contains 1 to 5 (for example, 1, 2, 3, 4 or 5)
heteroatoms
selected from 0, S and N.
[0023] In some
embodiments, ring C is selected from one of the following substituted
or unsubstituted groups: A benzene ring, a pyrrole ring, a pyrazole ring, a
pyridine ring, a
furan ring, a thiophene ring, an imidazole ring, a thiazole ring, a oxazole
ring, an
isothiazole ring, an isoxazole ring, a triazole ring, a tetrazole ring, a
oxadiazole ring, a
CA 03203276 2023- 6- 22
- 6 -
thiadiazole ring, a pyridazine ring, a pyrimidine ring, a pyrazine ring or a
triazine ring,
which, when substituted, is optionally further substituted with 0 to 4 (for
example, 0, 1, 2,
3 or 4) Rc.
[0024]
In some embodiments, ring C is selected from a benzene ring, a 5- to 6-
memberedmonocyclic heteroaromatic ring, a 5-membered fused 5-membered
heteroaromatic ring, a 5-membered fused 6-membered heteroaromatic ring or a 6-
membered fused 6-membered heteroaromatic ring. The benzene ring or
heteroaromatic
ring is optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3 or
4) Rc, the
heteroaromatic ring contains 1 to 5 (for example, 1, 2, 3, 4 or 5) heteroatoms
selected from
0, S and N.
[0025]
In some embodiments, ring C is selected from one of the following
substituted
Nk !\lk
Nk
or unsubstituted groups: a benzene ring,
¨
N77"Nk N
N N
N N or
, which = , when substituted, is optionally further
substituted with 1, 2 or 3 Rc, and the left side of which is connected to L.
[0026]
In some embodiments, ring C is selected from one of the following
substituted
\
or unsubstituted groups: or
, which, when substituted, is optionally
further substituted with 1, 2 or 3 Rc, and the left side of which is connected
to L.
0
L
[0027] In some embodiments, is selected from
, the right
side of which is connected to ring B, and in which the benzene ring is
optionally further
substituted with 1, 2 or 3 Rc.
CA 03203276 2023- 6- 22
-7-
1100281 In some embodiments, L is selected from a bond, 0, S, -
NRL-, -C(R02-, -
C(RL)2-C(RL)2-, -Y-C(RL)2-, -C(RL)2-Y-, -Y-C(RL)2-C(RL)2-, -C(RL)2-C(RL)2.-Y-
or -
C(RL)2-Y-C(R02-.
[0029] In some embodiments, L is selected from a bond, 0, S, -
NRL-, -CHRL-, -
CHRL-CHRL-, -Y-CHRL-, -CHRL-Y-, -Y-CHRL-CHRL-, -CHRL-CHRL-Y- or -CHRL-Y-
CHRL-.
[0030] In some embodiments, L is selected from -Y-CHRL-, -CHRL-Y-
, -CHRL-
CURL-, -CHRL-CHRL-Y- or -Y-CHRL-CHRL-.
[0031] In some embodiments, Y is selected from 0, S or -NRL-.
[0032] In some embodiments, L is selected from -CH20-, -OCH2-, -CH2NH-, -
NIICH2-, -CII2C112-, -OCH2C112-, -CH2C1120-, -NIICH2C112- Or -CII2C112N11-,
and the
right side of L is connected with ring C.
[0033] In some embodiments, L is selected from -CH20-, and the
right side of L is
connected to ring C.
[0034] In some embodiments, ring D is selected from a 6- to 10-membered
aromatic
ring or a 5- to 12-membered heteroaromatic ring. The aromatic ring or
heteroaromatic ring
is optionally further substituted with 0 to 5 RD. The heteroaromatic ring
contains 1 to 5 (for
example, 1, 2, 3, 4 or 5) heteroatoms selected from 0, S and N.
[0035] In some embodiments, ring D is selected from a benzene
ring, a naphthalene
ring, a 5- to 6-membered monocyclic heteroaromatic ring, a 5-membered fused 5-
membered heteroaromatic ring, a 5-membered fused 6-membered heteroaromatic
ring or a
6-membered fused 6-membered heteroaromatic ring. The benzene ring, naphthalene
ring or
heteroaromatic ring is optionally further substituted with 0 to 4 (for
example, 0, 1, 2, 3 or
4) RD, the heteroaromatic ring contains 1 to 5 (for example, 1, 2, 3, 4 or 5)
heteroatoms
selected from 0, S and N.
[0036] In some embodiments, ring D is selected from a benzene
ring, a naphthalene
ring, a pyrrole ring, a pyrazole ring, a pyridine ring, a furan ring, a
thiophene ring, an
imidazole ring, a thiazole ring, a oxazole ring, an isothiazole ring, an
isoxazole ring, a
triazole ring, a oxadiazole ring, a thiadiazole ring, a pyridazine ring, a
pyrimidine ring, a
pyrazine ring or a triazine ring, which, when substituted, is optionally
further substituted
with 0 to 4 (for example, 0, 1, 2, 3 or 4) RD.
CA 03203276 2023- 6- 22
-8-
1100371 In some embodiments, ring D is selected from a benzene
ring or a pyridine
ring, and the benzene ring or pyridine ring is optionally further substituted
with 1, 2 or 3
RD.
[0038] In some embodiments, ring D is selected from a
substituted or unsubstituted
benzene ring, and when substituted, is optionally further substituted with 1,
2 or 3 RD.
[0039] In some embodiments, ring E is selected from a 5-membered
heterocyclic
ring, and the heterocyclic ring is optionally further substituted with 0 or 1
substituent
selected from H, halogen, CN, OH, NH2, C1-6 alkyl, C1-6 alkoxy or C3-6
cycloalkyl. The
heterocyclic ring contains 1 to 2 heteroatoms selected from 0, S and N.
[00401 In some embodiments, r is selected from 0 or 1.
N ' N-'
[0041] In some embodiments, r=1, f is selected from N
N ' N
[0042] In some embodiments, r=0, l: is selected from
N N N N
N
-Th N
N
0
,)1, S H 0 , S
, , , or .
N N
/ / S
N N
[0043] In some embodiments, 1=0, I' is selected from ,
S , or S .
[0044] In some embodiments, ring E is selected from a 5-membered
non-aromatic
heterocyclic ring or a 5-membered heteroaromatic ring, and the heterocyclic
ring or
CA 03203276 2023- 6- 22
- 9 -
heteroaromatic ring is optionally further substituted with 0 or 1 substituent
selected from
H, halogen, CN, OH, NH2, C1-4 alkyl, C14 alkoxy or C3-6 cycloalkyl, the
heterocyclic ring
or heteroaromatic ring contains 1 to 2 heteroatoms selected from 0, S and N.
[0045] In some embodiments, R1 is selected from H, halogen, OH, -
SH, CF3, CN, Ci-
6 alkyl, C1-6 alkoxy, -Ci_3 alkylene-Z-00_3 alkylene-Ria, -00-4 alkylene-Ria.
The alkyl,
alkoxy, and alkylene are optionally further substituted with 0 to 4 (such as
0, 1 , 2, 3 or 4)
substituents selected from H, halogen, =0, CN, OH, -N(R1b)2, C1-6 alkyl,
halogen-
substituted C1-6 alkyl, hydroxy-substituted C1-6 alkyl, cyano-substituted C1-6
alkyl, C1-6
alkoxy, C3-6 cycloalkyl, a 3- to 6 -membered heterocycloalkyl, a 5- to 10-
membered
heteroaryl. The heterocycloalkyl or heteroaryl contains 1 to 3 (for example,
1, 2 or 3)
heteroatoms selected from 0, S and N.
[0046] In some embodiments, R1 is selected from H, halogen, OH, -
SH, CF3, CN, Ci-
4 alkyl, C14 alkoxy, -Ci_2 alkylene-Z-00_2 alkylene-Ria, -00_4 alkylene-Ria.
The alkyl,
alkoxy, and alkylene are optionally further substituted with 0 to 4 (such as
0, 1 , 2, 3 or 4)
substituents selected from H, halogen, =0, CN, OH, -N(R1b)2, C1_4 alkyl,
halogen-
substituted C1_4 alkyl, hydroxy-substituted Ci_4 alkyl, cyano-substituted C1_4
alkyl, C1-4
alkoxy, C3-6 cycloalkyl, a 3- to 6 -membered heterocycloalkyl, a 5- to 6-
membered
heteroaryl. The heterocycloalkyl or heteroaryl contains 1 to 3 (for example,
1, 2 or 3)
heteroatoms selected from 0, S and N.
[0047] In some embodiments, R1 is selected from H, F, Cl, OH, CN, CF3,
methyl,
ethyl, propyl, methoxy, ethoxy, propoxy, isopropoxy, -ethylene-Z-methylene-Ri
a, -
ethylene-Z-ethylene-Ri a, -ethylene-Z-Ri a, -R1 a, -methylene-Ri a, -ethylene-
Ri a. The methyl,
ethyl, propyl, methoxy, ethoxy, methylene, ethylene are optionally further
substituted with
0 to 3 (for example 0, 1, 2 or 3) substituents selected from H, F, Cl, =0, CN,
01-1, -N(Rib)2,
methyl, ethyl, propyl, CF3, -CH2F, -CHF2, 1 to 3 F-substituted ethyl,
hydroxymethyl,
hydroxyethyl, cyano-substituted methyl, cyano-substituted ethyl, methoxy,
ethoxy,
isopropoxy, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, oxiranyl,
aziridinyl,
oxetanyl, azetidinyl, tetrahydrofuranyl, tetrahydro-2H-pyranyl, dioxolanyl,
dioxanyl,
pyrrolidinyl, piperidinyl, imidazolidinyl, oxazolidinyl, oxazinanyl,
morpholinyl,
hexahydropyrimidinyl, piperazinyl, pyrrolyl, pyridyl, pyrazolyl, triazolyl,
tetrazolyl,
CA 03203276 2023- 6- 22
- 10 -
imidazolyl, thiazolyl, thienyl, furyl, oxazolyl, isoxazolyl, oxadiazolyl,
isothiazolyl,
pyrimidinyl, pyrazinyl or pyridazinyl.
[0048] In some embodiments, Ri is selected from H, CN, CF3,
CHF2, CH2F, -CH2OH,
-CH(OH)CH3, methyl, ethyl, methoxy, ethoxy, propoxy, isopropoxy,
methoxymethyl,
methoxyethyl, ethoxym ethyl , i sopropoxym ethyl , -ethyl en e-Z-m ethyl en e-
Ri a, -ethyl en e-Z-
ethylene-Ri a, -ethylene-Z-Ri a, Ria- or -methylene-R1 a-
o
[0049] In some embodiments, Ri is selected from -CH7CH7OCH3,
</N
0
(crk.k. 0 N 0 0
or C
CC-kk ga\____µ, 0
[0050] In some embodiments, Ri is selected from
or
0
[0051] In some embodiments, Z is selected from a bond, N(Rib), 0
or S.
[0052] In some embodiments, Z is selected from NH, N(CH3) or 0.
[0053] In some embodiments, Ria is selected from C1_6 alkyl, C1-
6 alkoxy, Cs
cycloalkyl, a 3- to 8-membered heterocycloalkyl, a 6- to 10-membered aryl or a
5- to 12-
membered heteroaryl. The cycloalkyl, heterocycloalkyl, aryl or heteroaryl is
optionally
further substituted with 0 to 4 (for example 0, 1, 2, 3 or 4) substituents
selected from H,
halogen, =0, OH, CN, C1-6 alkyl, C1_6 alkoxy or -N(R1b)2. The alkyl or alkoxy
is optionally
further substituted with 0 to 4 (such as 0, 1, 2, 3 or 4) substituents
selected from H,
halogen, =0, CN, OH, -N(R1b)2, C1-6 alkyl, C1_6 alkoxy or C3-6 cycloalkyl. The
heterocycloalkyl or heteroaryl contains 1 to 3 (for example, 1, 2 or 3)
heteroatoms selected
from 0, S and N.
[0054] In some embodiments, Ria is selected from C1-4 alkyl, C1-
4 alkoxy, C3-6
cycloalkyl, a 3- to 6-membered heterocycloalkyl, phenyl or a 5- to 6-membered
heteroaryl.
The cycloalkyl, heterocycloalkyl, phenyl or heteroaryl is optionally further
substituted with
0 to 4 (for example, 0, 1, 2, 3 or 4) substituents selected from H, halogen,
=0, OH, CN, CI_
CA 03203276 2023- 6- 22
- 11 -
4 alkyl, Ci_4 alkoxy or -N(R1b)2. The alkyl or alkoxy is optionally further
substituted with 0
to 4 (such as 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, CN,
01-1, -N(Rib)2,
C14 alkyl, C14 alkoxy or C3-6 cycloalkyl. The heterocycloalkyl or heteroaryl
contains 1 to 3
(for example, 1, 2 or 3) heteroatoms selected from 0, S and N.
[0055] In some embodiments, Ria is selected from methyl, ethyl, isopropyl,
propyl,
methoxy, ethoxy, propoxy, isopropoxy, cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl,
oxiranyl, aziridinyl, oxetanyl, azetidinyl, tetrahydrofuranyl, tetrahydro-2H-
pyranyl,
dioxolanyl, dioxanyl, dioxanyl, pyrrolidinyl, piperidinyl, imidazolidinyl,
oxazolidinyl,
oxazinanyl, morpholinyl, hexahydropyrimidinyl, piperazinyl, pyrrolyl, pyridyl,
pyrazolyl,
triazolyl, tetrazolyl, imidazolyl, thiazolyl, thienyl, furyl, oxazolyl,
isoxazolyl, oxadiazolyl,
isothiazolyl, pyrimidinyl, pyrazinyl or phenyl. The Ria is optionally further
substituted with
0 to 3 substituents selected from H, F, =0, OH, CN, methyl, ethyl, propyl,
CF3, -CH2F, -
CHF2, 1 to 3 F-substituted ethyl, hydroxymethyl, hydroxyethyl, cyano-
substituted methyl,
cyano-substituted ethyl, methoxy, ethoxy or -N(Rib)2.
[0056] In some embodiments, Ria is selected from methyl, ethyl, isopropyl,
propyl,
methoxy, ethoxy, propoxy, isopropoxy, cyclopropyl, cyclobutyl, cyclopentyl,
cyclohexyl,
oxiranyl, oxetanyl, azetidinyl, tetrahydrofuranyl, tetrahydro-2H-pyranyl,
pyrrolidinyl,
piperidinyl, pyrazolyl, 1,2,3-triazolyl, 1,2,4-triazolyl, imidazolyl,
thiazolyl, thienyl, furyl,
oxazolyl, isoxazolyl, 1,2,4-oxadiazolyl, isothiazolyl, pyrimidyl, pyridine or
phenyl. The
Ria is optionally further substituted with 0 to 3 (such as 0, 1, 2 or 3 )
substituents selected
from H, F, CN, methyl, ethyl, propyl, CF3, methoxy or ethoxy.
[0057] In some embodiments, each Rib is independently selected
from H or C1-6 alkyl;
the alkyl is optionally further substituted with 0 to 3 (such as 0, 1, 2 or 3)
substituents
selected from H, halogen, =0, CN, OH, NH2, Ci-6alkyl, C1-6 alkoxy or C3-6
cycloalkyl.
[0058] In some embodiments, each R11) is independently selected from H or
C1_4 alkyl
which alkyl is optionally further substituted with 0 to 3 (such as 0, 1, 2 or
3) substituents
selected from H, halogen, =0, CN, OH, NH2, C1_4 alkyl, C1_4 alkoxy or C3-6
cycloalkyl.
[0059] In some embodiments, each Rib is independently selected
from H, methyl or
ethyl.
[0060] In some embodiments, each RL is independently selected from H,
halogen, -
SH, CN, OH, CF3, NH2, C1-6 alkyl, C1-6 alkoxy, C3-6 cycloalkyl. The alkyl,
cycloalkyl or
CA 03203276 2023- 6- 22
- 12 -
alkoxy is optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3
or 4) substituents
selected from H, halogen, =0, OH, CN, NH2, C1-6 alkyl, C3-6 cycloalkyl or C1-6
alkoxy.
[0061] In some embodiments, each RL is independently selected
from H, halogen,
CN, OH, -SH, CF3, NH2, C1_4 alkyl, C1_4 alkoxy, C3-6 cycloalkyl. The alkyl,
cycloalkyl or
alkoxy is optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3
or 4) substituents
selected from H, halogen, =0, OH, CN, NH2, C14 alkyl, C36 cycloalkyl or C14
alkoxy.
[0062] In some embodiments, each RL is independently selected
from H, F, Cl, CN,
CF3, OH, NH?, methyl, ethyl, propyl, CF3, -CH2F, -CHF2, 1 to 3 F-substituted
ethyl,
hydroxymethyl, hydroxyethyl, cyano-substituted methyl, cyano-substituted
ethyl, methoxy,
ethoxy, cyclopropyl, cyclobutyl or cyclopentyl.
[0063] In some embodiments, each RL is independently selected
from H, F, methyl,
ethyl or propyl.
[0064] In some embodiments, each RL is independently selected
from H or methyl.
[0065] In some embodiments, R2 is selected from H, halogen or
C1_6alkyl.
[0066] In some embodiments, R2 is selected from H, halogen or C1_4alkyl.
[0067] In some embodiments, R2 is selected from H.
[0068] In some embodiments, R2 is selected from H, F, methyl or
ethyl.
[0069] In some embodiments, each RB, Rc or RD is independently
selected from H,
halogen, =0, CN, CF3, OH, -SH, NH2, C1-6 alkyl, C3-6 cycloalkyl or C1-6
alkoxy. The alkyl,
cycloalkyl or alkoxy is optionally further substituted with 0 to 4 (for
example, 0, 1, 2, 3 or
4) substituents selected from H, halogen, =0, OH, CN, NH2, Ci_6 alkyl, C3_6
cycloalkyl or
C1-6 alkoxy.
[0070] In some embodiments, each RB, Rc or RD is independently
selected from H,
halogen, =0, CN, CF3, OH, -SH, NH2, C14 alkyl, C3-6 cycloalkyl or C1_4 alkoxy.
The alkyl,
cycloalkyl or alkoxy is optionally further substituted with 0 to 4 (for
example, 0, 1, 2, 3 or
4) substituents selected from H, halogen, =0, OH, CN, NH2, C1_4 alkyl, C3-6
cycloalkyl or
C14 alkoxy.
[0071] In some embodiments, each RB, Rc or RD is independently
selected from H,
halogen, =0, CN, OH, -SH, NH2, methyl, ethyl, propyl, isopropyl, methoxy,
ethoxy,
propoxy, isopropoxy, tert-butyloxy, butoxy, cyclopropyl or cyclobutyl. The
methyl, ethyl,
propyl, isopropyl, methoxy, ethoxy, propoxy, isopropoxy, tert-butyloxy,
butoxy,
CA 03203276 2023- 6- 22
- 13 -
cyclopropyl or cyclobutyl is optionally further substituted with 0 to 4 (for
example 0, 1, 2,
3 or 4) substituents selected from H, halogen, =0, OH, CN, NH2, C1_4 alkyl, C3-
6
cycloalkyl or C1_4 alkoxy substituents.
[0072] In some embodiments, each RB, Rc or RD is independently
selected from H, F,
Cl, =0, CN, CF3, OH, NH2, methyl, ethyl, propyl, -CH2F, -CHF2, 1 to 3 F-
substituted
ethyl, hydroxymethyl, hydroxyethyl, cyano-substituted methyl, cyano-
substituted ethyl,
methoxy, ethoxy, propoxy, isopropoxy, tert-butyloxy, trifluoromethoxy,
cyclopropyl,
cyclobutyl or cyclopentyl.
[0073] In some embodiments, each RE is independently selected
from H, =0, F, OH,
CN, CF3, methyl, ethyl, propyl, cyclopropyl, cyclobutyl, cyclopentyl, methoxy
or ethoxy.
[0074] In some embodiments, each Rc is independently selected
from H, F, Cl, OH,
CN, NH2, CF3, methyl, ethyl, propyl, cyclopropyl, cyclobutyl, cyclopentyl,
methoxy or
ethoxy.
[0075] In some embodiments, each RE is independently selected
from H, =0, F, CN,
CF3, methyl, ethyl or cyclopropyl.
[0076] In some embodiments, each RE is independently selected
from H, F, =0 or
methyl.
[0077] In some embodiments, each Rc is independently selected
from H, F, CF3 or
methyl.
[0078] In some embodiments, each Rc is independently selected from H, F,
Cl, CN,
CF3, methyl, ethyl, methoxy or ethoxy.
[0079] In some embodiments, each RD is independently selected
from H, F, Cl, CN,
CF3, CHF2, methyl, ethyl, methoxy, ethoxy, tert-butyloxy or trifluoromethoxy.
[0080] In some embodiments, each RE is independently selected
from H, =0, F, CN,
CF3, methyl, ethyl or cyclopropyl.
[0081] In some embodiments, each Rc is independently selected
from H, F, Cl, CN,
CF3, methyl, ethyl, methoxy or ethoxy.
[0082] In some embodiments, each RD is independently selected
from H, F, Cl, CN,
CF3, CHF2, methyl or ethyl.
[0083] In some embodiments, two RB, two Rc, two RD, Rc and RL, or RD and RL
and
the atoms attached thereto together form a C3_iocarbocyclic ring or a 3- to 10-
membered
CA 03203276 2023- 6- 22
- 14 -
heterocyclic ring. The carbocyclic ring or heterocyclic ring is optionally
further substituted
with 0 to 4 (for example, 0, 1, 2, 3 or 4) substituents selected from H,
halogen, =0, OH,
CN, NH2, C1-6 alkyl, C3-6 cycloalkyl or C1-6 alkoxy. The alkyl, cycloalkyl or
alkoxy is
optionally further substituted with 0 to 4 (for example 0, 1, 2, 3 or 4)
substituents selected
from H, halogen, =0, OH, CN, NH2, C1-6 alkyl, C3-6 cycloalkyl or C1-6 alkoxy.
The
heterocyclic ring contains 1 to 3 (for example, 1, 2 or 3) heteroatoms
selected from 0, S
and N.
[0084] Tn some embodiments, two RB, two Rc, two RD, Rc and RL,
or RB and R2 and
the atoms attached thereto together form a C3_7carbocyclic ring or a 4- to 7-
membered
heterocyclic ring. The carbocyclic ring or heterocyclic ring is optionally
further substituted
with 0 to 4 (for example, 0, 1, 2, 3 or 4) substituents selected from H,
halogen, =0, OH,
CN, NH2, C1_4 alkyl, C3-6 cycloalkyl or C1-4 alkoxy. The alkyl, cycloalkyl or
alkoxy is
optionally further substituted with 0 to 4 (for example 0, 1, 2, 3 or 4)
substituents selected
from H, halogen, =0, OH, CN, NH2, C1_4 alkyl, C3-6 cycloalkyl or C1_4 alkoxy.
The
heterocyclic ring contains 1 to 3 (for example, 1, 2 or 3) heteroatoms
selected from 0, S
and N.
[0085] In some embodiments, Rc and RL together with the atoms
attached thereto
form a C3-6 carbocyclic ring or a 4- to 7-membered heterocyclic ring. The
carbocyclic ring
or heterocyclic ring is further substituted with 0 to 4 (for example, 0, 1, 2,
3 or 4)
substituents selected from H, halogen, =0, OH, CN, NH2, C1_4 alkyl, C3-6
cycloalkyl or C1-4
alkoxy. The alkyl, cycloalkyl or alkoxy is optionally further substituted with
0 to 4 (for
example 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, OH, CN,
NH2, C1-4
alkyl, C3-6 cycloalkyl or C1-4 alkoxy. The heterocyclic ring contains 1 to 3
(for example, 1,
2 or 3) heteroatoms selected from 0, S and N.
[0086] In some embodiments, two RB, or RL and Rc are directly connected
(between
two RB, or RL and Rc) to form C4_6carbocyclic ring or a 4- to 7-membered
heterocyclic
ring. The carbocyclic ring or heterocyclic ring is optionally further
substituted with 0 to 4
(for example, 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, 01-
1, CN, NH2, C1-4
alkyl, C3-6 cycloalkyl or C1-4 alkoxy. The alkyl, cycloalkyl or alkoxy is
optionally further
substituted with 0 to 4 (for example 0, 1, 2, 3 or 4) substituents selected
from H, halogen,
CA 03203276 2023- 6- 22
- 15 -
=0, OH, CN, NH2, C1-4 alkyl, C3-6 cycloalkyl or C1-4 alkoxy. The heterocyclic
ring contains
1 to 3 (for example, 1, 2 or 3) heteroatoms selected from 0, S and N.
[0087] In some embodiments, RL and Rc are directly connected to
form a 4, 5, 6 or 7-
membered heterocyclic ring. The heterocyclic ring is optionally further
substituted with 0
to 4 (for example 0, 1, 2, 3 or 4) substituents selected from H, F, =0, OH,
CN, NH2,
methyl, ethyl, isopropyl, cyclopropyl, cyclobutyl, cyclopentyl or methoxy. The
heterocyclic ring contains 1 to 2 heteroatoms selected from 0, S and N.
[0088] Tn some embodiments, Rc and RL together with the atoms
attached thereto
form a 4- to 7-membered heterocyclic ring. The heterocyclic ring is further
substituted with
0, 1, 2, 3 or 4 substituents selected from H, F, methyl or ethyl. The
heterocyclic ring
contains 1 to 2 heteroatoms selected from 0, S and N.
[0089] In some embodiments, RB and R2 together with the atoms
attached thereto
form a C3-6 carbocyclic ring or a 4- to 7-membered heterocyclic ring. The
carbocyclic ring
or heterocyclic ring is further substituted with 0 to 4 (for example, 0, 1, 2,
3 or 4)
substituents selected from H, halogen, =0, OH, CN, NI-12, C1_4 alkyl, C3-6
cycloalkyl or C14
alkoxy. The alkyl, cycloalkyl or alkoxy is optionally further substituted with
0 to 4 (for
example 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, OH, CN,
NH2, C1-4
alkyl, C3-6 cycloalkyl or C1-4 alkoxy. The heterocyclic ring contains 1 to 3
(for example, 1,
2 or 3) heteroatoms selected from 0, S and N.
[0090] As the first embodiment of the present invention, the compound
represented
by the above general formula (I) or a stereoisomer, tautomer, deuterated
substance, solvate,
prodrug, metabolite, pharmaceutically acceptable salt or co-crystal, wherein,
[0091] Ring B is selected from a 4- to 12-membered heterocyclic
ring, C5-12
carbocyclic ring, C6-10 aromatic ring or a 5- to 12-membered heteroaromatic
ring. The
carbocyclic ring, heterocyclic ring, aromatic ring or heteroaromatic ring is
optionally
further substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) R. The
heterocyclyl or
heteroaromatic ring contains 1 to 3 (for example 1, 2 or 3) heteroatoms
selected from 0, S
and N.
[0092] Ring C is selected from C6-10 carbocyclic ring, a 5- to
10-membered
heterocyclic ring, C6_10 aromatic ring or a 5- to 10-membered heteroaromatic
ring. The
carbocyclic ring, heterocyclic ring, aromatic ring or heteroaromatic ring is
optionally
CA 03203276 2023- 6- 22
- 16 -
further substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) Rc. The
heterocyclic ring or
heteroaromatic ring contains 1 to 5 (for example, 1, 2, 3, 4 or 5) heteroatoms
selected from
0, S and N.
[0093] L is selected from a bond, 0, S, -NRL-, -C(RL)2-, -C(RL)2-
C(RL)2-, -Y-C(RL)2-,
-C(RL)2-Y-, -Y-C(RL)2-C(RL)2-, -C(RL)2-C(RL)2.-Y- or -C(RL)2-Y-C(RL)2.-.
[0094] Y is selected from 0, S or -NRL-.
[0095] Ring D is selected from a 6- to 10-membered aromatic ring
or a 5- to 12-
membered heteroaromatic ring. The aromatic ring or heteroaromatic ring is
optionally
further substituted with 0 to 5 RD. The heteroaromatic ring contains 1 to 5
(for example, 1,
2, 3, 4 or 5) heteroatoms selected from 0, S and N.
[0096] Ring E is selected from a 5-membered heterocyclic ring,
and the heterocyclic
ring is optionally further substituted with 0 or 1 substituent selected from
H, halogen, CN,
OH, NI-12, C1_6 alkyl, C1_6 alkoxy or C3-6 cycloalkyl. The heterocyclic ring
contains 1 to 2
heteroatoms selected from 0, S and N.
[0097] R1 is selected from H, halogen, OH, -SH, CF3, CN, C1_6 alkyl, C1_6
alkoxy, -
C1-3 alkylene-Z-Co_3 alkylene-Ri a, -00_4 alkylene-Ria or -N(R1b)2. The alkyl,
alkoxy, and
alkylene are optionally further substituted with 0 to 4 (such as 0, 1 , 2, 3
or 4) substituents
selected from H, halogen, =0, CN, OH, -N(R16)2, C1-6 alkyl, halogen-
substituted C1-6 alkyl,
hydroxy-substituted C1_6 alkyl, cyano-substituted C1_6 alkyl, C1_6 alkoxy,
C3_6 cycloalkyl, a
3- to 6 -membered heterocycloalkyl, a 5- to 10-membered heteroaryl. The
heterocycloalkyl
or heteroaryl contains 1 to 3 (for example, 1, 2 or 3) heteroatoms selected
from 0, S and N.
[0098] Z is selected from a bond, N(Rib), 0 or S.
[0099] R a is selected from C1_6 alkyl, C1_6 alkoxy, C3_8
cycloalkyl, a 3- to 8-
membered heterocycloalkyl, a 6- to 10-membered aryl or a 5- to 12-membered
heteroaryl.
The cycloalkyl, heterocycloalkyl, aryl or heteroaryl is optionally further
substituted with 0
to 4 (for example 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0,
OH, CN, C1-6
alkyl, C1_6 alkoxy or -N(R16)2. The alkyl or alkoxy is optionally further
substituted with 0
to 4 (such as 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, CN,
OH, -N(R1b)2,
C1-6 alkyl, Ci-6alkoxy or C3-6 cycloalkyl. The heterocycloalkyl or heteroaryl
contains 1 to 3
(for example, 1, 2 or 3) heteroatoms selected from 0, S and N.
CA 03203276 2023- 6- 22
- 17 -
[0100] Each R11) is independently selected from H or C1-6 alkyl;
the alkyl is optionally
further substituted with 0 to 3 substituents selected from H, halogen, =0, CN,
OH, NI-12,
C1-6 alkyl, C1-6 alkoxy or C3-6 cycloalkyl.
[0101] R2 is selected from H, halogen or Ci_6alkyl.
[0102] Each RL is independently selected from H, halogen, -SH, CN, OH, CF3,
NH2,
C1 6 alkyl, Ci 6 alkoxy, C36 cycloalkyl. The alkyl, cycloalkyl or alkoxy is
optionally further
substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) substituents selected
from H, halogen,
=0, OH, CN, NH2, C1-6 alkyl, C3-6 cycloalkyl or C1-6 alkoxy.
[0103] Each RB, Rc or RD is independently selected from H,
halogen, =0, CN, CF3,
OH, -SH, NH2, Ci _6 alkyl, C3_6 cycloalkyl or Ci _6 alkoxy. The alkyl,
cycloalkyl or alkoxy is
optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4)
substituents selected
from H, halogen, =0, OH, CN, NH2, C1-6 alkyl, C3-6 cycloalkyl or C1-6 alkoxy.
[0104] Alternatively, two RB, two Rc, two RD, Rc and RL, RD and
RL, or RB and R2
and the atoms attached thereto together form a C3_iocarbocyclic ring or a 3-
to 10-
membered heterocyclic ring. The carbocyclic ring or heterocyclic ring is
optionally further
substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) substituents selected
from H, halogen,
=0, OH, CN, NH2, C1-6 alkyl, C3-6 cycloalkyl or Ci_6 alkoxy. The alkyl,
cycloalkyl or
alkoxy is optionally further substituted with 0 to 4 (for example 0, 1, 2, 3
or 4) substituents
selected from H, halogen, =0, OH, CN, NH2, Ci-6alkyl, C3-6 cycloalkyl or C1-6
alkoxy. The
said heterocyclic ring contains 1 to 3 (for example, 1, 2 or 3) heteroatoms
selected from 0,
S and N.
[0105] r is selected from 0 or 1.
[0106] As the second embodiment of the present invention, the
compound
represented by the above general formula (I) or a stereoisomer, tautomer,
deuterated
substance, solvate, prodrug, metabolite, pharmaceutically acceptable salt or
co-crystal,
wherein,
[0107] Ri is selected from H, halogen, OH, -SH, CF3, CN, C14
alkyl, C14 alkoxy, -
C1-2 alkylene-Z-Co_2 alkylene-Ria, -Co-4 alkylene-Ria. The alkyl, alkoxy, and
alkylene are
optionally further substituted with 0 to 4 (such as 0, 1 , 2, 3 or 4)
substituents selected from
H, halogen, =0, CN, OH, -N(Rib)2, Ci_4 alkyl, halogen-substituted C14 alkyl,
hydroxy-
substituted C14 alkyl, cyano-substituted C14 alkyl, C14 alkoxy, C3-6
cycloalkyl, a 3- to 6 -
CA 03203276 2023- 6- 22
- 18 -
membered heterocycloalkyl, a 5- to 6-membered heteroaryl. The heterocycloalkyl
or
heteroaryl contains 1 to 3 (for example, 1, 2 or 3) heteroatoms selected from
0, S and N.
[0108] Ri a is selected from C14 alkyl, Ci_4 alkoxy, C3-6
cycloalkyl, a 3- to 6-
membered heterocycloalkyl, phenyl or a 5- to 6-membered heteroaryl. The
cycloalkyl,
heterocycloalkyl, phenyl or heteroaryl is optionally further substituted with
0 to 4 (for
example, 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, OH, CN,
Ci4 alkyl, Cl
4 alkoxy or -N(Rib)2. The alkyl or alkoxy is optionally further substituted
with 0 to 4 (such
as 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, CN, OH, -
N(Rib)2, C14 alkyl,
C14 alkoxy or C3-6 cycloalkyl. The heterocycloalkyl or heteroaryl contains 1
to 3 (for
1() example, 1, 2 or 3) heteroatoms selected from 0, S and N.
[0109] Each Rib is independently selected from H or C1_4 alkyl;
the alkyl is optionally
further substituted with 0 to 3 substituents selected from H, halogen, =0, CN,
OH, NH2,
C14 alkyl, C1_4 alkoxy or C3-6 cycloalkyl.
[0110] Each RL is independently selected from H, halogen, CN,
OH, -SH, CF3, NH2,
Ci_4 alkyl, C1_4 alkoxy, C3-6 cycloalkyl. The alkyl, cycloalkyl or alkoxy is
optionally further
substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) substituents selected
from H, halogen,
=0, OH, CN, NH2, C1_4 alkyl, C3-6 cycloalkyl or C1_4 alkoxy.
[0111] Each RB, Rc or RD is independently selected from H,
halogen, =0, CN, CF3,
OH, -SH, NH2, C1_4 alkyl, C3_6 cycloalkyl or C14 alkoxy. The alkyl, cycloalkyl
or alkoxy is
optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4)
substituents selected
from H, halogen, =0, OH, CN, NH2, C1-4 alkyl, C3-6 cycloalkyl or Ci _4 alkoxy.
[0112] Alternatively, two RB, two Rc, two RD, Rc and RL or RB
and R2 and the atoms
attached thereto together form a C3_7carbocyclic ring or a 4- to 7-membered
heterocyclic
ring. The carbocyclic ring or heterocyclic ring is optionally further
substituted with 0 to 4
(for example, 0, 1, 2, 3 or 4) substituents selected from H, halogen, =0, OH,
CN, NH2, C14
alkyl, C3-6 cycloalkyl or C14 alkoxy. The alkyl, cycloalkyl or alkoxy is
optionally further
substituted with 0 to 4 (for example 0, 1, 2, 3 or 4) substituents selected
from H, halogen,
=0, OH, CN, NH2, C14 alkyl, C3-6 cycloalkyl or C14 alkoxy. The heterocyclic
ring contains
1 to 3 (for example, 1, 2 or 3) heteroatoms selected from 0, S and N.
[0113] The definitions of other groups are the same as those in the first
embodiment.
CA 03203276 2023- 6- 22
-19-
1101141 As the third embodiment of the present invention, the
compound represented
by the above general formula (I) or a stereoisomer, tautomer, deuterated
substance, solvate,
prodrug, metabolite, pharmaceutically acceptable salt or co-crystal, wherein,
[0115] Ring B is selected from a benzene ring, a 4- to 8-
membered heterocyclyl, C5-8
carbocyclyl or a 5- to 6-membered heteroaromatic ring. The benzene ring,
carbocyclyl,
heterocyclyl or heteroaromatic ring is optionally further substituted with 0
to 4 (for
example 0, 1, 2, 3 or 4) RB, the heterocyclic ring contains 1 to 3 (for
example, 1, 2 or 3)
heteroatoms selected from 0, S and N.
[0116] Ring C is selected from a benzene ring, a 5- to 6-
membered monocyclic
heteroaromatic ring, a 5-membered fused 5-membered heteroaromatic ring, a 5-
membered
fused 6-membered heteroaromatic ring or a 6-membered fused 6-membered
heteroaromatic ring. The benzene ring or heteroaromatic ring is optionally
further
substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) Rc, the heteroaromatic
ring contains 1
to 5 (for example, 1, 2, 3, 4 or 5) heteroatoms selected from 0, S and N.
[0117] Ring D is selected from a benzene ring, a naphthalene ring, a 5- to
6-
membered monocyclic heteroaromatic ring, a 5-membered fused 5-membered
heteroaromatic ring, a 5-membered fused 6-membered heteroaromatic ring or a 6-
membered fused 6-membered heteroaromatic ring. The benzene ring, naphthalene
ring or
heteroaromatic ring is optionally further substituted with 0 to 4 (for
example, 0, 1, 2, 3 or
4) RD, the heteroaromatic ring contains 1 to 5 (for example, 1, 2, 3, 4 or 5)
heteroatoms
selected from 0, S and N.
[0118] Ring E is selected from a 5-membered non-aromatic
heterocyclic ring or a 5-
membered heteroaromatic ring, and the heterocyclic ring or heteroaromatic ring
is
optionally further substituted with 0 or 1 substituent selected from H,
halogen, CN, OH,
NH2, C14 alkyl, C14 alkoxy or C3-6 cycloalkyl, the heterocyclic ring or
heteroaromatic ring
contains 1 to 2 heteroatoms selected from 0, S and N.
[0119] R1 is selected from H, F, Cl, OH, CN, CF3, methyl, ethyl,
propyl, methoxy,
ethoxy, propoxy, isopropoxy, -ethylene-Z-methylene-Ri a, -ethylene-Z-ethylene-
Ri a, -
ethylene-Z-Ri a, -RI a, -methylene-R 1 a, -ethylene-RI a. The methyl, ethyl,
propyl, methoxy,
ethoxy, methylene, ethylene are optionally further substituted with 0 to 3
substituents
selected from H, F, Cl, =0, CN, OH, -N(R1b)2, methyl, ethyl, propyl, CF3, -
CH2F, -CHF2, 1
CA 03203276 2023- 6- 22
- 20 -
to 3 F-substituted ethyl, hydroxymethyl, hydroxyethyl, cyano-substituted
methyl, cyano-
substituted ethyl, methoxy, ethoxy, isopropoxy, cyclopropyl, cyclobutyl,
cyclopentyl,
cyclohexyl, oxiranyl, aziridinyl, oxetanyl, azetidinyl, tetrahydrofuranyl,
tetrahydro-2H-
pyranyl, dioxolanyl, dioxanyl, pyrrolidinyl, piperidinyl, imidazolidinyl,
oxazolidinyl,
oxazinanyl, morpholinyl, hexahydropyrimidinyl, piperazinyl, pyrrolyl, pyridyl,
pyrazolyl,
triazolyl, tetrazolyl, imidazolyl, thiazolyl, thienyl, furyl, oxazolyl,
isoxazolyl, oxadiazolyl,
isothiazolyl, pyri midi nyl , pyrazinyl or pyridazinyl
[0120] Z is selected from 0, S or N(R 10-
[0121] Ri a is selected from methyl, ethyl, isopropyl, propyl,
methoxy, ethoxy,
propoxy, isopropoxy, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
oxiranyl,
aziridinyl, oxetanyl, azetidinyl, tetrahydrofuranyl, tetrahydro-2H-pyranyl,
dioxolanyl,
dioxanyl, dioxanyl, pyrrolidinyl, piperidinyl, imidazolidinyl, oxazolidinyl,
oxazinanyl,
morpholinyl, hexahydropyrimidinyl, piperazinyl, pyrrolyl, pyridyl, pyrazolyl,
triazolyl,
tetrazolyl, imidazolyl, thiazolyl, thienyl, furyl, oxazolyl, isoxazolyl,
oxadiazolyl,
isothiazolyl, pyrimidinyl, pyrazinyl or phenyl. The Ri a is optionally further
substituted with
0 to 3 substituents selected from H, F, =0, OH, CN, methyl, ethyl, propyl,
CF3, -CH2F, -
CHF2, 1 to 3 F-substituted ethyl, hydroxymethyl, hydroxyethyl, cyano-
substituted methyl,
cyano-substituted ethyl, methoxy, ethoxy or -N(Rib)2.
[0122] Each Rib is independently selected from H, methyl or
ethyl.
[0123] R2 is selected from H, F, methyl or ethyl.
[0124] Each RL is independently selected from H, F, Cl, CN, CF3,
OH, NH2, methyl,
ethyl, propyl, CF3, -CH2F, -CHF2, 1 to 3 F-substituted ethyl, hydroxymethyl,
hydroxyethyl,
cyano-substituted methyl, cyano-substituted ethyl, methoxy, ethoxy,
cyclopropyl,
cyclobutyl or cyclopentyl.
[0125] Each RB, Rc or RD is independently selected from H, F, Cl, =0, CN,
CF3, OH,
NH2, methyl, ethyl, propyl, -CH2F, -CHF2, 1 to 3 F-substituted ethyl,
hydroxymethyl,
hydroxyethyl, cyano-substituted methyl, cyano-substituted ethyl, methoxy,
ethoxy,
propoxy, isopropoxy, tert-butyloxy, trifluoromethoxy, cyclopropyl, cyclobutyl
or
cyclopentyl.
[0126] Alternatively, RB and R2 together with the atoms attached thereto
form a C3-6
carbocyclic ring (for example, C3, C4, C5 or C6 carbocyclic ring) or a 4- to 7-
membered
CA 03203276 2023- 6- 22
- 21 -
(for example, 4, 5, 6 or 7-membered) heterocyclic ring. The carbocyclic ring
or
heterocyclic ring is further substituted with 0 to 4 (for example, 0, 1, 2, 3
or 4) substituents
selected from H, halogen, =0, OH, CN, NH2, C1_4 alkyl, C3-6 cycloalkyl or C1_4
alkoxy.
The alkyl, cycloalkyl or alkoxy is optionally further substituted with 0 to 4
(for example 0,
1, 2, 3 or 4) substituents selected from H, halogen, =0, OH, CN, NH), C1_4
alkyl, C3-6
cycloalkyl or C14 alkoxy. The heterocyclic ring contains 1 to 3 (for example,
1, 2 or 3)
heteroatoms selected from 0, S and N.
[0127] Alternatively, Rc and RL together with the atoms attached
thereto form a C3-6
carbocyclic ring (for example, C3, C4, C5 or C6 carbocyclic ring) or a 4- to 7-
membered
(for example, 4, 5, 6 or 7-membered) heterocyclic ring. The carbocyclic ring
or
heterocyclic ring is further substituted with 0 to 4 (for example, 0, 1, 2, 3
or 4) substituents
selected from H, halogen, =0, OH, CN, NH2, C1_4 alkyl, C3-6 cycloalkyl or Ci_4
alkoxy.
The alkyl, cycloalkyl or alkoxy is optionally further substituted with 0 to 4
(for example 0,
1, 2, 3 or 4) substituents selected from H, halogen, =0, OH, CN, NH), C1_4
alkyl, C3-6
cycloalkyl or C14 alkoxy. The heterocyclic ring contains 1 to 3 (for example,
1, 2 or 3)
heteroatoms selected from 0, S and N.
[0128] The definitions of other groups are the same as those in
any one of the first
and second embodiments.
[0129] As the fourth embodiment of the present invention, the
compound represented
by the above general formula (I) or a stereoisomer, tautomer, deuterated
substance, solvate,
prodrug, metabolite, pharmaceutically acceptable salt or co-crystal, wherein,
[0130] L is selected from a bond, 0, S, -NRL-, -CHRL-, -CHRL-
CHRL-, -Y-CHRL-, -
CHRL-Y-, -Y-CHRL-CHRL-, -CHRL-CHRL-Y- or -CHRL-Y-CHRL-.
[0131] Each RL is independently selected from H, F, methyl,
ethyl or propyl.
[0132] Y is selected from 0, S or -NRL-.
[0133] Ring B is selected from one of the following substituted
or unsubstituted
groups: cyclohexyl, cyclohexenyl, azacyclohexenyl, piperidinyl, phenyl,
pyrazolyl,
pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl or triazinyl, which, when
substituted, is
optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) RB.
CA 03203276 2023- 6- 22
- 22 -
[0134] Or ring B is selected from one of the following
substituted or unsubstituted
groups: piperazinyl, tetrahydropyrrolyl or 1,4-diazepanyl, which, when
substituted, is
optionally further substituted with 0 to 4 (for example, 0, 1, 2, 3 or 4) RE.
[0135] Ring C is selected from one of the following substituted
or unsubstituted
groups: A benzene ring, a pyrrole ring, a pyrazole ring, a pyridine ring, a
furan ring, a
thiophene ring, an imidazole ring, a thiazole ring, a oxazole ring, an
isothiazole ring, an
isoxazole ring, a triazole ring, a tetrazole ring, a oxadiazole ring, a
thiadiazole ring, a
pyridazine ring, a pyrimidine ring, a pyrazine ring or a triazine ring, which,
when
substituted, is optionally further substituted with 0 to 4 (for example, 0, 1,
2, 3 or 4) Rc.
[0136] Ring D is selected from one of the following substituted or
unsubstituted
groups: A benzene ring, a naphthalene ring, a pyrrole ring, a pyrazole ring, a
pyridine ring,
a furan ring, a thiophene ring, an imidazole ring, a thiazole ring, a oxazole
ring, an
isothiazole ring, an isoxazole ring, a triazole ring, a oxadiazole ring, a
thiadiazole ring, a
pyridazine ring, a pyrimidine ring, a pyrazine ring or a triazine ring, which,
when
substituted, is optionally further substituted with 0 to 4 (for example, 0, 1,
2, 3 or 4) RD.
N
11-
N /
110137] When r=1, ( is selected from N
N N N
[0138] When r=0, ( is selected from
,
`1"--
N N N
, , NI-1?'= H 0
, ,
,
--
N N N
N \ N N
S jl ,,i,1 H 0 S
, , or .
[0139] The definitions of other groups are the same as those in any one of
the first,
second and third embodiments.
CA 03203276 2023- 6- 22
- 23 -
[0140]
As the fifth embodiment of the present invention, the compound represented
by the above general formula (I) or a stereoisomer, tautomer, deuterated
substance, solvate,
prodrug, metabolite, pharmaceutically acceptable salt or co-crystal, wherein,
[0141]
L is selected from -Y-CHRL-, -CHRL-Y-, -CHRL-CHRL-, -CHRL-CHRL-Y- or
-Y-CHRL-CHRL-.
[0142] Y is selected from 0, S or -NRL-.
[0143] Each RL is independently selected from H or methyl.
[0144]
Ring B is selected from one of the following substituted or unsubstituted
0
N AN N
groups: µV-) , , , , ,
or
, the left side of which is directly connected to ring C, and which, when
substituted, is optionally further substituted with 1, 2 or 3 RB.
[0145]
Or ring B is selected from one of the following substituted or
unsubstituted
'?,õN_, ) 1--N¨e_
/ i¨N
groups: , C \.,,s_v j
or
, the left side of which is directly
--
connected to ring C, and which, when substituted, is optionally further
substituted with 1, 2
or 3 RB.
[0146]
Or ring B is selected from one of the following substituted or
unsubstituted
0 0 0
-LI\J)c N
1
N_,c,
I\ j
-- N
groups: N
, , , , ,
,
I I
,-
, or ''l
, the left side of which is directly connected to ring
C, and which, when substituted, is optionally further substituted with 1, 2 or
3 RB.
CA 03203276 2023- 6- 22
- R R2 24-
2
[0147] Or 0 is selected from or kN
, the
left side of which is directly connected to ring C, and which, when the 6-
membered ring in
the spiro ring is substituted, is optionally further substituted with 1, 2 or
3 RB.
[0148]
Ring C is selected from one of the following substituted or unsubstituted
-1`1=Nik
(x`1\1)4- eCNA- NO$-
groups: a benzene ring, ¨/ , \--/
N
or N
, which, when substituted, is optionally further substituted with 1, 2 or 3
Rc,
and the left side of which is connected to L.
[0149]
Ring D is selected from a benzene ring or a pyridine ring, and the benzene
ring or pyridine ring is optionally further substituted with 1, 2 or 3 R.
[0150]
Each RB is independently selected from H, =0, F, OH, CN, CF3, methyl,
ethyl,
propyl, cyclopropyl, cyclobutyl, cyclopentyl, methoxy or ethoxy.
[0151]
Each Rc is independently selected from H, F, Cl, OH, CN, NH2, CF3, methyl,
ethyl, propyl, cyclopropyl, cyclobutyl, cyclopentyl, methoxy or ethoxy.
[0152] Each RD is
independently selected from H, F, Cl, CN, CF3, CHF2, methyl,
ethyl, methoxy, ethoxy, tert-butyloxy or trifluoromethoxy.
[0153]
Alternatively, Rc and RL together with the atoms attached thereto form a 4-
to
7-membered heterocyclic ring. The heterocyclic ring is further substituted
with 0, 1, 2, 3 or
4 substituents selected from H, F, methyl or ethyl. The heterocyclic ring
contains 1 to 2
heteroatoins selected from 0, S and N.
[0154]
The definitions of other groups are the same as those in any one of the
first,
second, third and fourth embodiments.
CA 03203276 2023- 6- 22
- 25 -
[0155]
As the sixth embodiment of the present invention, the compound represented
by the above general formula (I) or a stereoisomer, tautomer, deuterated
substance, solvate,
prodrug, metabolite, pharmaceutically acceptable salt or co-crystal, wherein,
[0156] L is selected from -CH20-, and the right side of L is
connected to ring C.
[0157] R1 is
selected from H, CN, CF3, CHF2, CH2F, -CH2OH, -CH(OH)CH3, methyl,
ethyl, methoxy, ethoxy, propoxy, isopropoxy, methoxymethyl, methoxyethyl,
ethoxymethyl, isopropoxymethyl, -ethylene-Z-methylene-R I a, -ethylene-Z-
ethylene-R I a, -
ethylene-Z-R 1 a, -R1 a or -methylene-R1 a-
[0158] Z is selected from NH, N(C113) or 0.
[01591 Ri a is
selected from methyl, ethyl, isopropyl, propyl, methoxy, ethoxy,
propoxy, isopropoxy, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
oxiranyl, oxetanyl,
azetidinyl, tetrahydrofuranyl, tetrahydro-2H-pyranyl, pyrrolidinyl,
piperidinyl, pyrazolyl,
1,2,3-triazolyl,
imidazolyl, thiazolyl, thienyl, furyl, oxazolyl, isoxazolyl,
1,2,4-oxadiazolyl, isothiazolyl, pyrimidyl, pyridine or phenyl. The Ri a is
optionally further
substituted with 0, 1, 2 or 3 substituents selected from F, CN,
methyl, ethyl, propyl,
CF3, methoxy or ethoxy.
[0160]
Ring C is selected from one of the following substituted or unsubstituted
N \
groups: or
, which, when substituted, is optionally further substituted
with 1, 2 or 3 Rc, and the left side of which is connected to L.
[0161] Ring D is
selected from a benzene ring or pyridine, and the benzene ring or
pyridine is optionally further substituted with 1, 2 or 3 RD.
0
L
[0162] Or is selected from
, the right side of which is
connected to ring B, and in which the benzene ring is optionally further
substituted with 1,
2 or 3 Rc.
[0163] Each RB is
independently selected from H, =0, F, CN, CF3, methyl, ethyl or
cyclopropyl.
CA 03203276 2023- 6- 22
- 26 -
[0164] Each Rc is independently selected from H, F, Cl, CN, CF3,
methyl, ethyl,
methoxy or ethoxy.
[0165] Each RD is independently selected from H, F, Cl, CN, CF3,
CHF?, methyl or
ethyl.
[0166] The definitions of other groups are the same as those in any one of
the first,
second, third, fourth and fifth embodiments.
[0167] As the seventh embodiment of the present invention, the
compound
represented by the above general formula (1) or a stereoisomer, tautomer,
deuterated
substance, solvate, prodrug, metabolite, pharmaceutically acceptable salt or
co-crystal,
wherein,
-kk \..k 0
[0168] R1 is selected from (c gi or
[0169] R2 is selected from H;
[0170] Ring B is selected from one of the following substituted
or unsubstituted
A 5-
r-N )r( is+j
L1/2N.
groups: or
, the left side of which is
directly connected to ring C, and which, when substituted, is optionally
further substituted
with 1, 2 or 3 RB;
R2 ,R2
B
[0171] Or is selected from
, the left side of which is
directly connected to ring C, and which, when the 6-membered ring in the Spiro
ring is
substituted, is optionally further substituted with 1, 2 or 3 RB;
N N N
/ II / S II /
N
[0172] r=0, is selected from
or
N
[0173] The definitions of other groups are the same as those in
any one of the first,
second, third, fourth, fifth and sixth embodiments.
CA 03203276 2023- 6- 22
- 27 -
[0174] The present invention provides the following compound or
a stereoisomer,
tautomer, deuterated substance, solvate, prodrug, metabolite, pharmaceutically
acceptable
salt or co-crystal thereof,
C)--
NC. ,--,N,---,,N NC, --,
,,N
,)..._s
I [I, \ _, N õCOON
F y
(:) N) N---{ 1
.- OH
II
F 0
O,-- 0--
NC NC--
,0õN -,, ) 14-4 N(/ OH
F
-, =Ns----H.,i 0 H
F ,--,-.- S f
0 0
('=),
NC 'Isl-')--N \. NC N.
N
'I 1 ,,Z) N ,N) \ ---c-N
1 '
11, /'-\'_
7 I s'y OH õ,
1 I ----- --1 OH
F F N--
0 0
0----- 0-7---
.F ,N F N,
NC- --;(- \ \, ir--S NC 7
,.0_1:7N-- N--=( 0 \ 7reN-Z N---'k . -
0
0 OH t_z
OH
0///-/ 0-/
,F iz---,f1õ,,,.Nh NC, . r
,0 , ._- ON õ -i J N /\s ', '1 1 OH
7 OH F J. 0
C)-- 0-
F F
0 N NC F
N
N M-1 /
-_,
NC N =-''") OH
N
S \S--
0 OH
CA 03203276 2023- 6- 22
- 28 -
o
cr:1 oTh
F
NC, F )___N
0 -N N
/
-1- 0 N, y r IV-------li N 11 OH
N
S
- ---
s-- -r0 NC
01 0
-I
F
0-- 0--
,
C1,,,_
N T\
CI NII*1/
1
(:),INI. ) N--- \ OH
N
F 1
OH
1 S
1 t
F0 , 2----, 0
0--
0-----
NN r
'14-'
_
NC z--- 7F 1 f----11_1-11. 1 j N-j<
- N ¨ \S )21 NC 1-
s OH
If
11
) 7) OH F -04N 10
0
----...=
0 --\
F
F Cci-
/
NC F
-N
N )1-
M-1
0 N
N '-''''j NOH
r
NC
S
0
OH
I 1
F
NC),_ F OTh
NC _ F /
N
1 11 )---
---
N---< -
K C:$
L
S"---)
S
r r
O OH H
0-- 0-- CI F
NC- F
N
N
0 N'll / 1 0 N
0 N
1 0
N / \
S
0 0
S
OH
OH
(:)-- NC F
NC Ai F
1\1 N
N 0 0 N N
\-\'' /
S
0 1 0
S
OH
OH
CA 03203276 2023- 6- 22
- 29 -
CI F
0--- CI F
0--
--õ, ---- /LO r'il''N
' 0
N- 1 0 0 N
'S S
---, .-.:"- OH
OH
0-- CI F
0---) CI N
-Nil- 1;
N N 0
OH OH
CI F Cr,- 1:1J--1,
I\10õ ,.-------....-
Nz
0 rN7C N 1 1 r N,,) \
0 N
OH
S F 0
OH
0--a ON
/
NC,, ''N" _ N, NC, .-
7 0 N, ,-, ) IV----=c'
OH r',i /
'
1 11 'I S ; :r
s iOH
F
0 0
0--J\
I NC, F /
NC
r N--;-N,,,_
1 J 1:1 -..<'
J. ,0 N õN T ---`-,10 N,,, -
J N /
\ OH T s If'
'S 0
F
0
OH
I
NCF (c-aN F 0--
\
i NC F
..-- ---_-- :Is /
11 /µ>-----, A 0 N -L 3
0 ) N-Z( 11 - ---- --.Tr 7.
µS"
OH
OH
.-,
I N ' N----,µ
NC ) N
F - N NC / r , , ,,.----' F -
J ---- N
-..N ,---
-i: - ni...'1, . 1
,::: NJ ) N-
I S --1:)
( 1
.
OH ) \S
r
OH
CA 03203276 2023- 6- 22
- 30 -
NN---- (-0
N=--/
\
N / NC _F 7
NC
(--Isr--1-N\
--y..,---
c , li- ' ¨
N -,õ_rk_,0 J N /
I
- _L
0 S- y
r ,
OH
OH
,------
NN' 0 0\ 7
\ ---(
\?
1 , ,-: F i
NC ,F ^ N
NC
JIIIIIIN'ThiNs
r -.- 7 -
N _ ON N-2-
1I
N N,
1 N = ri
1 s
, \S
0
OH
OH
,.. 7-
0\
(
0--)
F
NC 7
NC
N.N __- ,-
r-N1---N
--L 0N N4 \ XI, N
0 - ---, =,--.1.--
- /-- -
s. y0
'S-
I
OH
OH
\,
0¨,/ OTh
CI
NC F F
=NI N
L i 0 N CNII N / I .
ON)N / II
- -..---- -_.- ..õ===õ,.7- --õ,---
0
0
I s
-, --:-
OH
OH
0-a 0¨
NC F /
N
NC , ,,, F
'Y' r
( ICI N
--. N-
I
ji:>'1--.1%1 \-1
il sS
0
j S 1%0
OH F'
OH
I I
Om OTh
NC -_,
c
N
NC F .N ^ N 'r ,r-F
-'rslI .; r -
',--:,
i ON, (J r',i / 11 .ON N---
I
')-'
.--0
S y-
r U -,
F-
OH - "F
OH
I I
0¨
0 \
--\
/ NC F / NC F
. -
'? 11-
\ ,O, N
N-- -
;
OH 1-'
OH
-
CA 03203276 2023- 6- 22
-31-
0
/ '''
NC,. F N--- _______________________________________ 0
i--N---T,-N
0õN_ N.,) N- )---1, NC ,_,,r F /---N, N¨S OH
,10
--..zi0 N.r
,- OH J
,
o
crD-3 \
b2----_,\ ,
N_ _ 0 NC, F ,)-L. , .N
NC F ,-- t
/ , I r N -''''
N N" S OH ' -I o N.
.-- ---,--- - - - -- ----- \
\
-C a r-- 1 'T s----r-
_. y_,N.. 0, ,._
OH
N_J--
0---J)
Oa \ 0
0
i NC F )t
NC .r F )1 . ----N
,, N N
Nil - ')1 I ) /
0õ, NN N \
'--j-N,0 ,N N_____c 3 õ,
1 s 0
, O
OH H
03
CC-J) N
j NC F _, N ..,,---.õ,,,õ N
NC ,- F N ,_,, õN I 11 /
0,,_, N,,,,,,,,.. N N \ 0
- ...0õ ,N,
s_ 0 I s
OH
OH
CrD-N
[-_;\ i
/ NC .,_ , F N.,..
NC ,-,..., F _N..----T-1,:q 'r T \ ---,
-),-- ir
0 NI
N As! 0 0
- = ...---------
r
/ OH -. OH
\
ga,
OA) /
NC,,,,r- F N.. ---
,_ õN
NC F N ,-- ,N 11 r - iS
'r: =----- N' 1- '')''
s;,N N -4. n
---,1- N \ µ-
'' -c-- õ,-- - ------E, -,-, r
OH
OH
j
0 0
----1 )
, _N NC ,c
NC ,
I 0 N, --, - N -----< \ 0
----, -,,, --,_, 1 .7- N µs- --
-t-
0 N. ,,,- N-----C
F ---.--- OH
F OH
CA 03203276 2023- 6- 22
- 32 -
CL , _F
0
0 I Iii____ F
NC , )1- N 0 --
1 Nil 1-1-
1 ,0 N - N N-J, -1 0
\
S COOH ) r N-
F S-1-
NCO2H
CI, 0
CI , F
p...../2 1
0 -CI 6--tejµ I / µ= 1 11 /)--
F 0, ), '..-, , N--_,/ ,Ai
I 'S '0
,--
\S- 'CO2H
OH
CI F F CI ,_ F
I
0 r_00 r
N
;I
S--NCO2H
-
\S- 'CO2H
CI -
1----,-;" -----
I I i 01)
!--0
^-7.N___
/ 0 1
F 0 \ L N--/C), F
1 S
-I
OH OH
CI ,.,-, Oq CI,
01
I /
F N i0
F 0
0, ',.-
U
i
'S -'
OH OH
CI ,,,
01-i NC 011--1,
,
/
FO \ I F 0 N
N \ ' N-__ \
N--7
I S
OH
OH 0
CN
0 ----.
/ \\ I > <, F 1
NC /--N N-1--S OH
( N
õ,j0õN1,,,,N, _I Y,
I I N T
%1 z
0
F
Oa\ 1:1-.N
/ F i
NC ).N NC, N
0 N, N---- I
----- 0 ,r=-0,..,N1,---, N 8,,,\ 17)
I ,
i 1 r
F F OH
OH
CA 03203276 2023- 6- 22
- 33 -
C)-- C)--
F F
NC ,N NC , .., ,I,c r=lz_
- (:) fq,, (:)N,---.j N--_< \ ,õ
F
F '-' OH F OH
F O
i F
NC N NC
1 -------1 1 11
N
,
I ' s
F OH
OH
0-1 F NC y, F
F
rel NC 1., ,?.
1 0
lj ON, N- =< I , N.,.y..- ry
___<, ,
i
=.- r- ,u1 F
S CO2H
OH
J ] 0 IN NC F 0 \ NC F
F F
N N
C:) N N --{ \ ()N
' I sS--- CO2H 5- CO2H
F F F
F
NC
F ,F 0--7 NC 0---
,
N
0 N N ---/0 ,)_
0 N N---c __1
S¨ CO2H ,--- T.
's CO2H
F F F
NC F 0-
CI
0-1)
1
1 Th
,rJN N-0
1
0 N} T N
s co2H s y
õ F ,
OH
CI
CF)-- CI
0--
1 1
N
Nli.-- 0 N N"10 N NMI' /
O\1J N 0 / \ 0 N \
0
S S
OH
OH
CA 03203276 2023 6 22
- 34
o
z 1µ171 z
0 N 0 N
0 0
OH OH
N7fl
N--\
CI O\ ¨
,
/ NC
r
0 rF
J N /
0
0
OH
OH
[0175] The present invention relates to a pharmaceutical composition
comprising the
above-mentioned compound or a stereoisomer, tautomer, deuterated substance,
solvate,
prodrug, metabolite, pharmaceutically acceptable salt or co-crystal thereof,
and a
pharmaceutically acceptable carrier.
[0176] The present invention relates to the above-mentioned compound or a
stereoisomer, tautomer, deuterated substance, solvate, prodrug, metabolite,
pharmaceutically acceptable salt or co-crystal thereof, or an application of
the above-
mentioned pharmaceutical composition in the preparation of a drug for treating
diabetes.
[0177] A compound has a structure as shown below,
on
X N
N
0
0 Rm
[0178] X is selected from H or Br;
[0179] Rm is selected from H or C1_4alky1; the alkyl is optionally further
substituted
with 0 to 4 substituents selected from halogen, C1-4 alkyl, C1-4 alkoxy or C6-
10 carbocyclic
ring.
[0180] Synthetic method I:
CA 03203276 2023- 6- 22
- 35 -
R1
R1111 Ri NH2
Rml HN
E COORm2 __
02N / COO IRm2 __ (1-3)
02N)COORrn2
(I-1) (1-2) (1-4)
R1 R2 R2 R1
V / OL C OH
HN
_____________________________________ 1Rm3
H2N E COORrr2 (1-7) N E
COORm2
(1-5) (1-6)
R2 R2 R1 R2 R2 R1
I / /
COORm2 N E COOH
[0181] The compound of general formula (I-1) is nitrated to
obtain the corresponding
compound of general formula (I-2).
[0182] The compound of general formula (I-2) and the compound of general
formula
(I-3) are subjected to a substitution reaction to obtain the corresponding
compound of
general formula (I-4).
[0183] The compound of general formula (I-4) is reduced to
obtain the corresponding
compound of general formula (I-5).
[0184] The compound of general formula (I-5) is subjected to a ring-closing
reaction
to obtain the corresponding compound of general formula (I-6).
[0185] The compound of general formula (I-6) and the compound of
general formula
(I-7) are subjected to a substitution reaction to obtain the corresponding
compound of
general formula (I-8).
[0186] The compound of the general formula (I-8) is subjected to a
decarboxylation
protecting group reaction to obtain the corresponding compound of the general
formula (I).
[0187] RI and R1113 are selected from F, Cl, Br, I, OTf, etc.
[0188] Rm2 is selected from carboxyl protecting groups,
preferably Ci_6alkyl or
benzyl.
[0189] The definitions of other groups are the same as those in any
embodiment of
the above general formula (I).
[0190] Unless stated to the contrary, the terms used in the
description and claims have
the following meanings.
CA 03203276 2023- 6- 22
- 36 -
[0191] The carbon, hydrogen, oxygen, sulfur, nitrogen or F, Cl,
Br, I involved in the
groups and compounds of the present invention all comprise their isotopes, and
the carbon,
hydrogen, oxygen, sulfur or nitrogen involved in the groups and compounds of
the present
invention is optionally further substituted with one or more of their
corresponding isotopes,
wherein the isotopes of carbon comprise 12C, '3C and 14C, the isotopes of
hydrogen
comprise protium (H), deuterium (D, also known as heavy hydrogen), tritium (T,
also
known as superheavy hydrogen), the isotopes of oxygen comprise 160, 170 and
180, the
isotopes of sulfur comprise 32S, 33S, 34S and 36S, the isotopes of nitrogen
comprise 14N and
15N, the isotopes of fluorine comprise 17F and 19F, the isotopes of chlorine
comprise 35C1
and 37C1, and the isotopes of bromine comprise 79Br and 81Br.
[0192] "Halogen" means F, Cl, Br or I.
[0193] "Halogen-substituted" refers to F, Cl, Br or I
substitution, including but not
limited to a substitution with 1 to 10 substituents selected from F, Cl, Br or
I, a substitution
with 1 to 6 substituents selected from F, Cl, Br or I, a substitution with 1
to 4 substituents
selected from F, Cl, Br or I. "Halogen-substituted" is referred to simply as
"halo".
[0194] "Alkyl" means a substituted or unsubstituted linear or
branched saturated
aliphatic hydrocarbon group, including but not limited to an alkyl group of 1
to 20 carbon
atoms, an alkyl group of 1 to 8 carbon atoms, an alkyl group of 1 to 6 carbon
atoms, or an
alkyl group of 1 to 4 carbon atoms. Non-limiting examples include methyl,
ethyl, n-propyl,
isopropyl, n-butyl, sec-butyl, neobutyl, tert-butyl, n-pentyl, isoamyl,
neopentyl, n-hexyl
and various branched isomers thereof; The definition of the alkyl described
herein is
consistent with this definition. Alkyl group can be monovalent, divalent,
trivalent or
tetravalent.
[0195] "Heteroalkyl" refers to a substituted or unsubstituted
alkyl group in which one
or more (including but not limited to 2, 3, 4, 5 or 6) carbon atoms are
replaced by
heteroatoms (including but not limited to N, 0 or S). Non-limiting examples
include -
X(CH2)v-X(CH2)v-X(CH2)v-H (v is an integer from 1 to 5, X is independently
selected
from bonds or heteroatoms, heteroatoms include but not limited to N, 0 or S,
and at least
one X is selected from heteroatoms, and N or S in heteroatoms can be oxidized
to various
oxidation states). Heteroalkyl group can be monovalent, divalent, trivalent or
tetravalent.
CA 03203276 2023- 6- 22
- 37 -
[0196] An "alkylene" means a substituted or unsubstituted
straight or branched chain
divalent saturated hydrocarbon group, including -(CH2),- (v is an integer from
1 to 10),
and examples of alkylene include, but are not limited to, methylene, ethylene,
propylene,
butylene, etc.
[0197] "Heteroalkylene" means a substituted or unsubstituted alkylene group
in
which one or more (including but not limited to 2, 3, 4, 5 or 6) carbon atoms
are replaced
by heteroatoms (including but not limited to N, 0 or S). Non-limiting examples
include -
X(CH2)v-X(CH2)v-X(CH9)v-, wherein v is an integer from 1 to 5, each X is
independently
selected from a bond, N, 0 or S, and at least one X is selected from N, 0 or
S.
[0198] "Cycloalkyl" means a substituted or unsubstituted saturated
carbocyclic
hydrocarbon group, usually having from 3 to 10 carbon atoms, and non-limiting
examples
include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, etc.
The
"cycloalkyl" herein is as defined above. Cycloalkyl group can be monovalent,
divalent,
trivalent or tetravalent.
[0199] "Heterocycloalkyl" means a substituted or unsubstituted saturated
heteroatom-
containing cyclic hydrocarbon group including but not limited to 3 to 10
atoms, 3 to 8
atoms, and 1 to 3 atoms selected from N, 0 or S. N and S selectively
substituted in
heterocycloalkyl ring can be oxidized to various oxidation states. The
heterocycloalkyl
group can be connected to a heteroatom or a carbon atom, the heterocycloalkyl
group can
be connected to an aromatic ring or a non-aromatic ring, and the
heterocycloalkyl group
can be connected to a bridged ring or a spiro ring. Non-limiting examples
include oxiranyl,
aziridinyl, oxetanyl, azetidinyl, tetrahydrofuranyl, tetrahydro-2H-pyranyl,
dioxolanyl,
dioxanyl, pyrrolidinyl, piperidinyl, imidazolidinyl, oxazolidinyl, oxazinanyl,
morpholinyl,
hexahydropyrimidinyl or piperazinyl. Heterocycloalkyl group can be monovalent,
divalent,
trivalent or tetravalent.
[0200] "Alkenyl" means a substituted or unsubstituted straight
and branched
unsaturated hydrocarbon group having at least 1, usually 1, 2 or 3 carbon-
carbon double
bonds, with a main chain including but not limited to 2 to 10, 2 to 6, or 2 to
4 carbon
atoms. Examples of alkenyl include, but are not limited to vinyl, allyl, 1-
propenyl, 2-
propenyl, 1-butenyl, 2-butenyl, 3-butenyl, 1-pentenyl, 2-pentenyl, 3-pentenyl,
4-pentenyl,
1-methyl-1-butenyl, 2-methyl-1-butenyl, 2-methyl-3-butenyl, 1-hexenyl, 2-
hexenyl, 3-
CA 03203276 2023- 6- 22
- 38 -
hexenyl, 4-hexenyl, 5-hexenyl, 1-methyl-1 -pentenyl, 2-methyl-1 -pentenyl, 1-
heptenyl, 2-
heptenyl, 3-heptenyl, 4-heptenyl, 1-octenyl, 3-octenyl, 1-nonenyl, 3-nonenyl,
1-decenyl, 4-
decenyl, 1,3-butadiene, 1,3-pentadiene, 1,4-pentadiene, 1,4-hexadiene, etc.
The definition
of the alkenyl described herein is consistent with this definition. Alkenyl
group can be
monovalent, divalent, trivalent or tetravalent.
[0201]
"Alkynyl" means a substituted or unsubstituted straight and branched
monovalent unsaturated hydrocarbon group having at least 1, usually 1, 2 or 3
carbon-
carbon triple bonds, with a main chain including but not limited to 2 to 10
carbon atoms, 2
to 6 carbon atoms or 2 to 4 carbon atoms. Examples of alkynyl include but are
not limited
to ethynyl, propargyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-
butynyl, 1-pentynyl,
2-pentynyl, 3-pentynyl, 4-pentynyl, 1-methyl-1-butynyl, 2-methyl-1-butynyl, 2-
methy1-3-
butynyl, 1-hexynyl, 2-hexynyl, 3-hexynyl, 4-hexynyl, 5- hexynyl, 1-methyl- 1 -
pentynyl, 2-
methyl-1 -pentynyl, 1-heptynyl, 2-heptynyl, 3-heptynyl, 4-heptynyl, 1-octynyl,
3-octynyl,
1-nonynyl, 3-nonynyl, 1-decynyl, 4-decynyl, etc. Alkynyl group can be
monovalent,
divalent, trivalent or tetravalent.
[0202]
"Alkoxy" means a substituted or unsubstituted -0-alkyl group. Non-limiting
examples include methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, sec-butoxy,
tert-
butoxy, n-pentoxy, n-hexyloxy, cyclopropoxy and cyclobutoxy.
[0203]
"Carbocycly1" or "carbocyclic ring" means a substituted or unsubstituted
saturated or unsaturated aromatic ring or non-aromatic ring, and the aromatic
ring or non-
aromatic ring can be 3- to 8-membered monocyclic ring, 4- to 12-membered
bicyclic ring
or 10- to 15-membered tricyclic ring system. The carbocyclyl can be connected
to an
aromatic ring or a nonaromatic ring, and the aromatic ring or nonaromatic ring
is
optionally a monocyclic ring, a bridged ring or a spirocyclic ring. Non-
limiting examples
include cyclopropane, cyclobutane, cyclopentane, cyclohexane, cycloheptane, 1-
cyclopentyl-1-enyl , 1-cyclopenty1-2-enyl, 1-cyclopenty1-3-enyl, cyclohexyl, 1-
cyclohexyl-
d2-enyl, 1-cyclohexy1-3-enyl, cyclohexenyl, benzene ring, naphthalene ringõ
Cl>,
or
. A "carbocycly1" or "carbocyclic ring" can be
,
monovalent, divalent, trivalent or tetravalent.
CA 03203276 2023- 6- 22
- 39 -
[0204]
"Heterocycly1" or "heterocyclic ring" refers to a substituted or
unsubstituted
saturated or unsaturated aromatic ring or non-aromatic ring, and the aromatic
ring or non-
aromatic ring can be 3- to 8-membered monocyclic ring, 4- to 12-membered
bicyclic ring
or 10- to 15-membered tricyclic ring system, and contains one or more
(including but not
limited to 2, 3, 4 or 5) heteroatoms selected from N, 0 or S, and the
optionally substituted
N or S in the heterocyclyl ring can be oxidized into various oxidation states.
Heterocyclyl
can be connected to other groups through its heteroatoms or carbon atoms,
heterocyclyl
can be connected to other groups through its aromatic ring or nonaromatic
ring, and
heterocyclyl can be connected to a bridged ring or a Spiro ring (when the ring
of the
heterocyclyl is a bridged ring or a spiro ring, the site where the
heterocyclyl is connected to
other groups is on the bridged ring or Spiro ring). Non-limiting examples
include oxiranyl,
azacyclopropyl, oxetanyl, azetidinyl, 1,3-dioxolane, 1,4-dioxolane, 1,3-
dioxane,
azacycloheptyl, pyridyl, furanyl, thienyl, pyranyl, N-alkylpyrrolyl,
pyrimidinyl, pyrazinyl,
pyridazinyl, imidazolyl, piperidyl, morpholinyl, thiomorpholinyl, 1,3-
dithianyl,
dihydrofuranyl, dihydropyranyl, dithiolanyl, tetrahydrofuranyl,
tetrahydropyrrolyl,
tetrahydroimidazolyl, tetrahydrothiazolyl, tetrahydropyranyl,
benzimidazolyl,
benzopyridyl, pyrrolopyridyl, benzodihydrofuranyl, pyrrolyl, pyrazolyl,
thiazolyl,
oxazolyl, pyrazinyl, indazolyl, benzothiophenyl, benzofuranyl, benzopyrrolyl,
benzimidazolyl, benzothiazolyl, benzoxazolyl, benzopyridyl, benzopyrimidinyl,
benzopyrazinyl, piperazinyl,
azabicyclo[3.2.11octyl, azabicyclo [5 .2.0]nonalkyl ,
0
oxatricyclo[5.3.1.11dodecyl, azaadamantyl, oxaspiro[3.31heptyl,
0 0,7 FC1
,
Nr>
N .ccN
----- (ON,N 0 N
N N or
. A "heterocyclyl" or
"heterocyclic ring" can be monovalent, divalent, trivalent or tetravalent.
[0205] "Spiro
ring" or "Spiro ring group" means a polycyclic group that shares one
atom (called Spiro atom) between substituted or unsubstituted inonocyclic
rings. The
number of ring atoms in the Spiro ring system includes but is not limited to 5
to 20. 6 to 14,
CA 03203276 2023- 6- 22
-40-
6 to 12, or 6 to 10, wherein one or more rings may contain 0 or more
(including but not
limited to 1, 2, 3 or 4) double bonds, and can optionally contain 0 to 5
heteroatoms
selected from N, 0 or S(=0),
/---\\_.,;
i--I X/ X x ,0 xr3 x
\I
\ __ /
.,
NH
/ ________________________________________________________ ' -0 ,
/---- , ( Y > ><
'
\/ ----, s-
,--- '---
9
¨0\ \
/ x ----il I
---- / \ ---- v
9,
C / \ 7---- NH , 1 - , / ' \
i L) /NH / j / __ \
\
\ \ \ /X\ HN7\>('
X \ NH
\ / / \ /NH
HN ,NH
\/ - - .
[0206] "Spiro ring" or "spiro ring group" can be monovalent, divalent,
trivalent or
tetravalent.
[0207] "Fused ring" or "fused ring group" refers to a polycyclic
group in which each
ring in the system shares an adjacent pair of atoms with other rings in the
system, wherein
one or more rings may contain 0 or more (including but not limited to 1, 2, 3
or 4) double
bonds, and may be substituted or unsubstituted, and each ring in the fused
ring system may
contain 0 to 5 heteroatoms or groups containing heteroatoms (including but not
limited to
N, S(=0)n or 0, n is 0, 1 or 2). The number of the ring atoms in the fused
ring system
includes but is not limited to 5 to 20, 5 to 14, 5 to 12, or 5 to 10. Non-
limiting examples
HN-----\NH HN/--------- NH
i I nclude: CL7N1H , ------./ \---------/ .
,0-, 0
0, <__>, i\C>, -"'"-
---II , N--- (
\
N ,
( ':- -------, ---\
-------- S , ----- , <-1-> , ail , C- ''> \
_ _ ,-- ,s¨
(1___);, o- -----\ o ;------\
----/.,
--< I
) _______________________________ / L __ NH 1 /
------ -----N1 , '------ N
, ---
H __s or
=
[0208] "Fused ring" or "fused ring group" may be monovalent,
divalent, trivalent or
tetravalent.
[0209] "Bridged ring" or "bridged ring group" means a
substituted or unsubstituted
polycyclic group containing any two atoms that are not directly connected, and
may
CA 03203276 2023- 6- 22
- 41 -
contain 0 or more double bonds. Any ring in the bridged ring system may
contain 0 to 5
groups selected from heteroatoms or groups containing heteroatoms (including
but not
limited to N, S(=0)n or 0, wherein n is 0, 1 or 2). The number of ring atoms
includes, but
is not limited to, 5 to 20, 5 to 14, 5 to 12 or 5 to 10. Non-limiting examples
include
zr-Ity
0 HO N-
0
0
o'
HN/ 0 0
/ FIN
/0
H N/ 0 0 H H H
HN
, cub ane or
adatnantane. "Bridged ring" or "bridged ring group" may be monovalent,
divalent,
trivalent or tetravalent.
[0210]
"Carbospiro ring", "Spiro ring carbocyclyl", "spirocarbocyclyr or
"carbospiro
ring group" means a "Spiro ring" with a ring system consisting only of carbon
atoms. The
definitions of "carbospiro ring", "Spiro ring carbocycly1", "spirocarbocyclyr
or
"carbospiro ring group" used herein is consistent with that of spiro ring.
[0211]
"Carbo-fused ring", "fused ring carbocycly1", "fused carbocyclyr or "carbo-
fused ring group" means a "fused ring" with a ring system consisting only of
carbon
atoms. The definition of "carbo-fused ring", "fused ring carbocycly1", "fused
carbocyclyr
or "carbo-fused ring group" used herein is consistent with that of fused ring.
[0212]
"Carbo-bridged ring", "bridged ring carbocycly1", "bridged carbocyclyr or
"carbo-bridged ring group" means a "bridged ring" with a ring system
consisting only of
carbon atoms. The definitions of "carbo-bridged ring", "bridged ring
carbocycly1",
"bridged carbocyclyr or "carbo-bridged ring group" used herein is consistent
with that of
bridged ring.
[0213]
"Heteromonocyclic ring", "monocyclic heterocyclyr or "heteromonocycly1"
means "heterocycly1" or "heterocyclic ring" with a monocyclic system. The
definition of
the heterocyclyl, "monocyclic heterocycly1" or "heteromonocycly1" used herein
is
consistent with that of heterocyclic ring.
CA 03203276 2023- 6- 22
-42 -
[0214]
"Hetero-fused ring", "hetero-fused ring group", "fused ring heterocycly1"
or
"hetero-fused ring group" means a "fused ring" containing a heteroatom. The
definition of
hetero-fused ring, "hetero-fused ring group", "fused ring heterocycly1" or
"hetero-fused
ring group" used herein is consistent with that of fused ring.
[0215] "Hetero-
spiro ring", "hetero-spiro ring group", "spiro ring heterocycly1" or
"hetero-spiro ring group" means a "Spiro ring" containing a heteroatom. The
definition of
hetero-spiro ring, "hetero-spiro ring group", "Spiro ring heterocycly1" or
"hetero-spiro ring
group" used herein is consistent with that of Spiro ring.
[0216]
"Hetero-bridged ring", "hetero-bridged ring group", "bridged ring
heterocycly1" or "hetero-bridged ring group" means a "bridged ring" containing
a
heteroatom. The definition of hetero-bridged ring, "hetero-bridged ring
group", "bridged
ring heterocycly1" or "hetero-bridged ring group" used herein is consistent
with that of
bridged ring.
[0217]
"Aryl" or "aromatic ring" means a substituted or unsubstituted aromatic
hydrocarbon group with a single ring or a fused ring, and the number of ring
atoms in the
aromatic ring includes but not limited to 6 to 18, 6 to 12 or 6 to 10 carbon
atoms. The aryl
ring may be fused to a saturated or unsaturated carbocyclic ring or
heterocyclic ring,
wherein the ring connected to the parent structure is an aryl ring. Non-
limiting examples
include a benzene ring, a naphthalene ring
Or
-0
. "Aryl" or "aromatic ring"
may be monovalent, divalent, trivalent or tetravalent. When divalent,
trivalent or
tetravalent, the point of connection is on the aryl ring.
[0218]
"Heteroaryl" or "heteroaromatic ring" means a substituted or unsubstituted
aromatic hydrocarbon group containing 1 to 5 heteroatoms or groups containing
heteroatoms (including but not limited to N, 0 or S(=0 )n, n is 0, 1 or 2),
and the number
of ring atoms in the heteroarotnatic ring includes but not limited to 5-15, 5-
10 or 5-6. Non-
limiting examples of heteroaryl include, but are not limited to pyridyl,
furanyl, thienyl,
pyridinyl, pyranyl, N-alkylpyrrolyl, pyrimidinyl, pyrazinyl, pyridazinyl,
imidazolyl,
benzopyrazole, benzimidazolyl, benzopyridine, pyrrolopyridine and the like.
The
heteroaryl ring may be fused to a saturated or unsaturated carbocyclic ring or
heterocyclic
CA 03203276 2023- 6- 22
-43 -
ring, wherein the ring connected to the parent structure is an heteroaryl
ring. Non-limiting
examples
include
N0 N0 N
fN-s N 0
>
N N N. N.
and
. The definition of the heteroaryl used herein is consistent with this
definition.
Heteroaryl group can be monovalent, divalent, trivalent or tetravalent. When
divalent,
trivalent or tetravalent, the point of connection is on the heteroaryl ring.
[0219]
"5-membered ring fused 5-membered heteroaromatic ring" means a 5 fused 5-
membered fused heteroaromatic ring, wherein at least one of the two fused
rings contains
more than one heteroatom (including but not limited to 0, S or N), and the
entire group is
aromatic. Non-limiting examples include pyrrolopyrrole ring, pyrazolopyrrole
ring,
pyrazolopyrazole ring, pyrrolofuran ring, pyrazolofuran ring, pyrrolothiophene
ring and
pyrazolothiophene ring.
[0220]
"5 fused 6-membered heteroaromatic ring" means a 5 fused 6-membered fused
heteroaromatic ring, at least one of the two fused rings contains more than
one heteroatom
(including but not limited to 0, S or N), and the entire group is aromatic.
Non-limiting
examples include benzo 5-membered heteroaryl and 6-membered heteroaromatic
ring
fused 5-membered heteroaromatic ring.
[0221]
"Substitution" or "substituted" means a substitution with 1 or more
(including
but not limited to 2, 3, 4 or 5) substituents including but not limited to H,
F, Cl, Br, I, alkyl,
cycloalkyl, alkoxy, haloalkyl, mercaptan, hydroxyl, nitro, sulfhydryl, amino,
cyano,
isocyano, aryl, heteroaryl, heterocyclyl, bridged ring group, Spiro ring
group, fused ring
group, hydroxyalkyl, =0, carbonyl, aldehyde, carboxylic acid, formate, -(CH2).-
C(=0)-Ra,
-0-(CH2)m-C(=0)-R', -(CH2)m-C(=0)-NRbRc, -(CH2)mS(=0)nRa, -(CH2)m-alkenyl-Ra,
ORE'
or -(CH2)m-alkynyl-Ra (wherein m and n are 0, 1 or 2), arylthio, thiocarbonyl,
silyl or -
NRbRc and the like, wherein Rb and RC are independently selected from H,
hydroxyl,
amino, carbonyl, alkyl, alkoxy, cycloalkyl, heterocyclyl, aryl, heteroaryl,
sulfonyl,
trifluoromethylsulfonyl. Alternatively, Rb and RC may form a five- or six-
membered
cycloalkyl or heterocyclyl. Ra and Rd are each independently selected from
aryl, heteroaryl,
CA 03203276 2023- 6- 22
- 44 -
alkyl, alkoxy, cycloalkyl, heterocyclyl, carbonyl, ester group, bridged ring
group, spiro
ring group or bicyclic ring group.
[0222]
"Containing 1 to 5 heteroatoms selected from 0, S or N" means containing 1,
2, 3, 4 or 5 heteroatoms selected from 0, S or N.
[0223]
"Substituted with 0 to X substituents" refers to substituted with 0, 1, 2, 3
... X
substituents, wherein X is selected from any integer between 1 and 10. For
example,
"substituted with 0 to 4 substituents" refers to substituted with 0, 1, 2, 3
or 4 substituents.
For example, "substituted with 0 to 5 substituents" refers to substituted with
0, 1, 2, 3, 4 or
5 substituents. For example, "bridged-heterocyclic ring is optionally further
substituted
with 0 to 4 substituents selected from H or F" means that the bridged-
heterocyclic ring is
optionally further substituted with 0, 1, 2, 3 or 4 substituents selected from
H or F.
[0224]
An X- to Y-membered ring (X is selected from an integer less than Y and
greater than or equal to 3, and Y is selected from any integer between 4 and
12) includes
X-, X+1-, X+2-, X+3-, X+4-,
Y-membered rings. Rings include a heterocyclic ring, a
carbocyclic ring, an aromatic ring, aryl, heteroaryl, cycloalkyl, a mono-
heterocyclic ring, a
fused heterocyclic ring, a spiro-heterocyclic ring or a bridged-heterocyclic
ring. For
example, "a 4- to 7-membered mono-heterocyclic ring" refers to a 4-membered, 5-
membered, 6-membered or 7-membered mono-heterocyclic ring, and "a 5- to 10-
membered fused heterocyclic ring" refers to a 5-membered, 6-membered, 7-
membered, 8-
membered, 9-membered or 10-membered fused heterocyclic ring.
[0225]
"Pharmaceutically acceptable salt" or "pharmaceutically acceptable salt
thereof" refers to a salt of the compound of the present invention maintaining
the
biological effectiveness and characteristics of the free acid or free base,
and obtained by
reacting the free acid with a non- toxic inorganic base or organic base,
reacting the free
base with a non- toxic inorganic acid or organic acid.
[0226]
"Pharmaceutical composition" refers to a mixture of one or more compounds
of the present invention, or stereoisomers, tautomers, deuterated substances,
solvates,
prodrugs, metabolites, pharmaceutically acceptable salts or co-crystals
thereof and other
chemical components, wherein "other chemical components" refer to
pharmaceutically
acceptable carriers, excipients and/or one or more other therapeutic agents.
CA 03203276 2023- 6- 22
-45 -
[0227] "Carrier" refers to a material that does not cause
significant irritation to the
organism and does not eliminate the biological activity and characteristics of
the
administered compound.
[0228] "Excipient" refers to an inert substance added to a
pharmaceutical composition
to facilitate the administration of a compound. Non-limiting examples include
calcium
carbonate, calcium phosphate, sugar, starch, cellulose derivatives (including
microcrystalline cellulose), gelatin, vegetable oils, polyethylene glycols,
diluents,
granulating agents, lubricants, adhesives and disintegrants.
[0229] "Prodrug" refers to a substance that can be converted
into a biologically active
compound of the present invention through in vivo metabolism. The prodrug of
the present
invention is prepared by modifying the amino or carboxyl group in the compound
of the
present invention, and the modification can be removed by conventional
operations or in
vivo to obtain the parent compound. When the prodrug of the present invention
is
administered to a mammalian individual, the prodrug is split to form free
amino or
carboxyl group.
[0230] The term "co-crystal" refers to a crystal formed by the
combination of active
pharmaceutical ingredient (API) and co-crystal former (CCF) under the action
of hydrogen
bonds or other non-covalent bonds. The pure state of API and CCF are both
solid at room
temperature, and there is a fixed stoichiometric ratio between various
components. The co-
crystal is a multi-component crystal, which includes both a binary co-crystal
formed
between two neutral solids and a multi-element co-crystal formed between a
neutral solid
and a salt or solvate.
[0231] "Animal" is meant to include mammals, such as humans,
companion animals,
zoo animals, and domestic animals, preferably humans, horses, or dogs.
[0232] The term "stereoisomer" refers to an isomer produced as a result of
different
spatial arrangement of atoms in molecules, including cis-trans isomers,
enantiomers and
conformational isomers.
[0233] "Tautomer" refers to a functional group isomer produced
by the rapid
movement of an atom in two positions in a molecule, such as keto-enol
isomerization,
amide-imino alcohol isomerization, etc.
CA 03203276 2023- 6- 22
-46 -
[0234] "Optional" or "optionally" or "selective" or
"selectively" refers to that the
events or conditions subsequently described may but not necessarily occur, and
the
description includes the case where the events or conditions occur and do not
occur. For
example, "heterocyclyl optionally substituted with alkyl" refers to that the
alkyl may but
not necessarily exist, and the description includes the case where the
heterocyclyl is
substituted with an alkyl group and the case where the heterocyclyl is not
substituted with
alkyl.
[0235] "TC50" refers to the concentration of a drug or inhibitor
required to inhibit half
of a given biological process (or a component of the process such as an
enzyme, a receptor
1(3 and a cell).
Detailed Description of Embodiments
[0236] The technical solutions of the present invention will be
described in detail by
the following examples, but the scope of protection of the present invention
includes but is
not limited thereto.
[0237] The compounds used in the reactions described herein are
prepared according
to organic synthesis techniques known to those skilled in the art, and
starting from
commercially available chemicals and(or) compounds described in chemical
documents.
"Commercially available chemicals" are obtained from regular commercial
sources, and
suppliers include: Titan Technology Co., Ltd., Energy Chemical Co., Ltd.,
Shanghai Demo
Co., Ltd., Chengdu Kelong Chemical Co., Ltd., Accela ChemBio Co., Ltd.,
PharmaBlock
Sciences (Nanjing), Inc., WuXi Apptec Co., Ltd., J&K Scientific and the like.
[0238] The structures of the compounds are determined by nuclear
magnetic
resonance (NMR) or (and) mass spectrometry (MS). The NMR shift (6) is given in
the unit
of 10-6 (ppm). NMR is measured with (Bruker Avance III 400 and Bruker Avance
300)
NMR instrument, and the solvent for determination is deuterated dimethyl
sulfoxide
(DMSO-d6), deuterated chloroform (CDC13), deuterated methanol (CD30D), and the
internal standard is tetramethylsilane (TMS);
[0239] MS is measured with (Agilent 6120B(ESI) and Agilent
6120B(APCI));
[0240] HPLC is determined with Agilent 1260DAD high pressure liquid
chromatograph (Zorbax SB-C18 100 x 4.6 mm, 3.5 pM);
CA 03203276 2023- 6- 22
-47 -
[0241] Yantai Huanghai HSGF254 or Qingdao GF254 silica gel plate
is used as a thin
layer chromatography silica plate, and the silica gel plate for the thin layer
chromatography
(TLC) is of the specification of 0.15 mm-0.20 mm, and the specification when
separating
and purifying a product by thin layer chromatography is 0.4 mm - 0.5 mm.
[0242] For the column chromatography, Yantai Huanghai silica gel of 200-300
mesh
silica gel is generally used as a carrier.
Example 1
(S)-2-((4-(6-((4-cyano-2-fluoroben7y1)oxy)pyridin-2-yl)piperidin-l-yl)methyl)-
1-(oxetan-
2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 1)
ci--1
NC
N N
0
1 S
F -/-
OH
Compound 1
01\.)NH2 F-A H \----A H
Br) 0 Br__, p lc-1
S 0¨ 02N S ¨
Step 1 Step 2 02N'-s 0-
Step 3 H2Nlz -S ()¨
la lb lc Id
NC, 0 ,
NH
N (5--
NC N, ''NT\
Step 4 CI N S 0¨
Step 5 F I _,
le (=)
if
H
)1\111;N 1g NC N\
N
I].
OH
Compound 1
Step 1: methyl 4-bromo-5-nitrothiophene-2-carboxylate (lc)
Br 0
02N /"----S ¨
lb
[0243] Under ice-salt bath conditions, la (2.2 g, 10.0 mmol) was
added to
concentrated sulfuric acid (10 mL), and fuming nitric acid (945.0 mg, 15.0
mmol) was
dissolved in concentrated sulfuric acid (5 mL) and slowly added dropwise into
the reaction
CA 03203276 2023- 6- 22
-48 -
liquid; after the dropwise addition, the reaction was continued for 30 minutes
in the ice-salt
bath. The reaction liquid was slowly poured into an ice-water solution, and a
large amount
of white solids precipitated out. The filter cake was collected by filtration,
washed with
water (10 mL x 2), and dried to obtain lb (2.6 g, yield 99.3%).
[0244] LCMS m/z = 265.9 [M+1]+.
[0245] 'H NMR (400 MHz, CDC13) 6 7.71 (s, 1H), 3.96 (s, 311).
Step 2: methyl (S)-5-nitro-4-((oxetan-2-ylmethyl)amino)thiophene-2-carboxylate
(1c)
H
0
-
- 2 - S
lc
[0246] At room temperature, lb (L3 g, 5.0 mmol) was dissolved in
acetonitrile (30
mL), then lc-1 (436.0 mg, 5.0 mmol) and potassium carbonate (2A g, 15.0 mmol)
were
added (100 mL) successively, heated to 60 C, and continued to react for 18 h.
The reaction
liquid was cooled to room temperature, filtered, and the filtrate was
collected and
concentrated. The obtained crude product was separated and purified by silica
gel column
chromatography (petroleum ether/ethyl acetate (v/v) = 2:1), and concentrated
to obtain lc
(854.0 mg, yield 62.7%).
[0247] LCMS m/z = 2711 [M+1]+.
[0248] 11-1 NMR (400 MHz, CDCb) 6 8A2 ¨ 8.03 (m, 1H), 7.35 (s,
1H), 5.10 ¨ 5.00
(in, 1H), 4.74 ¨ 4.66 (in, 1H), 4.58 ¨ 4.50 (in, 1H), 192 (s, 3H), 3.71 ¨ 3.57
(in, 2H), 2.80
¨ 2.68 (m, 1H), 2.60 ¨ 2.48 (in, 1H).
Step 3: methyl (S)-5-amino-4-((oxetan-2-ylmethyl)amino)thiophene-2-carboxylate
(1d)
H
0
H2N 0
Id
[0249] At room temperature, lc (854M mg, 3.1 mmol) was dissolved
in methanol (30
mL), then palladium on carbon (170.0 mg, L6 inmol) was added, and the reaction
was
continued for 18 h under hydrogen atmosphere. The reaction liquid was filtered
to remove
palladium on carbon, the filtrate was collected and concentrated to obtain a
crude product,
and the obtained crude product was separated and purified by silica gel column
CA 03203276 2023- 6- 22
-49 -
chromatography (petroleum ether/ethyl acetate (v/v)=1:1), and concentrated to
obtain 1 d
(410.0 mg, yield 53.9%).
[0250] LCMS m/z = 243.1 [M+1] .
[0251] 111 NMR (400 MHz, DMSO - d6) 6 7.15 (s, 111), 5.89 (s,
211), 4.83 -4.74 (m,
1H), 4.55 - 4.47 (m, 1H), 4.47 - 4.39 (m, 1H), 4.23 - 4.15 (m, 1H), 3.66 (s,
3H), 3.25 -
3.11 (m, 214), 2.64- 2.55 (m, 111), 2.46 - 2.36 (m, 111).
Step 4: methyl (S)-2-(chloromethyl)-1-(oxetan-2-ylmethyl)-1H-thieno[2,3-
d]imidazole-5-
carboxylate ( 1 e)
/
CI 0-
1e
[0252] At room temperature, ld (410.0 mg, 1.7 mmol) was dissolved in
acetonitrile
(10 mL), then le-1 (391.0 mg, 2.5 mmol) and p-toluenesulfonic acid (15.0 mg,
0.1 mmol)
were added successively, and finally the reaction liquid was heated to 60 C
for 5 h under
the protection of nitrogen. The reaction liquid was cooled to room
temperature, saturated
sodium bicarbonate solution was added to quench the reaction, extracted with
dichloromethane (25 mL x 3), and the organic phase was combined, dried over
anhydrous
sodium sulfate, filtered and concentrated to obtain a crude product, which was
directly
used in the next reaction without further purification.
[0253] LCMS m/z = 301.0 [M+11+.
Step 5: methyl (S)-24(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-yppiperidin-1-
yl)methyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d]imidazole-5-carboxyl ate (10
NC
N Tr=Lch
if
[0254] At room temperature, if-1 (527.0 mg, 1.7 mmol) was
dissolved in acetonitrile
(15 mL) solution, and then crude product le (508.0 mg, 1.7 mmol) and potassium
carbonate (702.0 mg, 5.1 mmol) were added to the reaction system successively,
and
CA 03203276 2023- 6- 22
- 50 -
heated to 60 C for 18 h. The reaction liquid was cooled to room temperature,
concentrated
to remove the solvent. The crude product was dissolved in water (20 mL), and
then
extracted with ethyl acetate (20 mL x 4). The organic phase was washed with
aqueous
saturated NaC1 solution, dried over anhydrous sodium sulfate, and concentrated
by
filtration. The obtained crude product was separated and purified by silica
gel column
chromatography (petroleum ether/ethyl acetate (v/v)=1:3), and concentrated to
obtain 1 f
(135.0 mg, yield 13.8%).
[0255] LCMS m/7 = 576.2 [M+1] .
Step 6: (S)-2-((4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-yppiperidin-1-
yOmethyl)-1-
1() (oxetan-2-ylmethyl)-1H-thieno [2,3-dlimidazole-5-carboxylic acid
(Compound 1)
om
NC N N
0 N N
0
OH
Compound 1
[0256] At room temperature, if (137.0 mg, 0.24 inmol) was
dissolved in (6 mL) and
water (3 InL) solution, then 1 g (CAS: 5807-14-7) (166.0 mg, L20 mmol) was
added, and
the reaction was continued for 24 h. 1N hydrochloric acid was used to adjust
pH to 6, then
dichloromethaneanethanol = 10:1 (20 tiaL x 3) was used for extraction. The
organic phase
was combined, dried over anhydrous sodium sulfate, filtered and concentrated.
The
obtained crude product was subjected to preparative liquid chromatography to
obtain (S)-
2-((4-(6-((4-cyano-2-fluorobenzypoxy)pyridin-2-yepiperidin-l-yemethyl)-1-
(oxetan-2-
ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (compound 1)
trifluoroacetic acid
salt (60 mg, yield 44.9%). Compound 1 was obtained by preparative thin layer
chromatography (dichloromethanehnethanol (v/v) = 10:1).
[0257] Conditions for preparative liquid chromatography:
[0258] Instrument and preparative column: waters 2767
(preparative liquid phase
chromatographic instrument) was used; the preparative column model was
SunFire@Prep
C18 (19 mm x 250 mm).
[0259] Preparation method: The crude product was dissolved in
DMF and filtered
with a 0.45 in filter membrane to prepare into a sample liquid.
CA 03203276 2023- 6- 22
-51-
1102601 Mobile phase system: acetonitrile/water (containing 1%
TFA). Gradient
elution: acetonitrile content 20-65%, elution flow rate: 12 mL/min, elution
time 18 min.
[0261] Compound 1:
[0262] LCMS m/z = 562.2 [M+1] .
[0263] 1H NMR (400 MHz, CD30D) 6 7.79 (s, 1H), 7.70-7.61 (m, 2H), 7.60-7.53
(m,
2H), 6.90 (d, 111), 6.75 (d, 114), 5.53 (s, 211), 5.26-5.17 (m, 114), 4.74-
4.65 (m, 214), 4.62-
4.55 (ni, 1H), 4.52-4.40 (ni, 3H), 3.67-3.55 (ni, 2H), 3.15-3.03 (ni, 2H),
2.98-2.87 (m, 1H),
2.85 ¨ 2.75 (m, 1H), 2.56-2.45 (m, 1H), 2.11 ¨ 1.99 (m, 411).
Example 2
(S)-2-((64(4-cyano-2-fluorobenzypoxy)-3',6'-dihydro-[2,4'-bipyridin1-1.(21-1)-
yl)methyl)-
1-(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 2)
OTh
NC
0
OH
Compound 2
NC 40
NH HCI
gil 11/11: ONJ
OMe 0 F
0 2a-1
2c-1
H2Nz 0- Step 1 N S 0- Step 2 Br N S 0-
.. Step 3
1d 2a 2b
N N
NC
11,õ,0¨N,XnN-t-11 0 j 1 g
0 N N4 I
NCNTN
=Sy I S)
Step 4
2c OH
Compound 2
Step 1: methyl (S)-2-methy1-1-(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-
carboxylate (2a)
2a
[0264] At room temperature, ld (1.2 g, 5.1 mmol) was added to
glacial acetic acid
(50 mL), 2a-1 (926.0 mg, 7.7 mmol) was added to the reaction liquid, and the
temperature
CA 03203276 2023- 6- 22
- 52 -
was raised to 70 C for 1 hour. The glacial acetic acid was distilled off under
reduced
pressure, and the obtained crude product was separated and purified by silica
gel column
chromatography (petroleum ether/ethyl acetate (v/v)=1:4), and concentrated to
obtain 2a
(680.0 mg, yield 50.1%).
[0265] LCMS m/z = 267.1 [M+1]+.
[0266] 'H NMR (400 MHz, CDC13) 6 7.68 (s, 111), 5.19 ¨ 5.11 (m,
114), 4.66 ¨ 4.58
(in, 1H), 4.36¨ 4.29 (in, 1H), 4.26 (d, 2H), 3.89 (s, 3H), 2.79 ¨ 2.68 (m,
1H), 2.63 (s, 3H),
2.44 ¨ 2.34 (m, 1H).
Step 2: methyl (S)-2-(bromomethyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3 -d]imi
dazol e-5 -
carboxylate (2b)
0
\) ________________________________________________ =/
Br 0-
2b
[0267] At room temperature, 2a (680.0 mg, 2.6 mmol) was
dissolved in 1,2-
dichloroethane (50 'EL) solution, then AIBN (84.0 mg, 0.5 inmol) was added,
and NB S(1.1
g, 6.4 mmol) was added in portions in the process of slowly raising the
temperature to
50 C. After the addition, the reaction liquid was heated to 70 C and continued
to react for
8 h. The reaction liquid was cooled to room temperature, saturated sodium
thiosulfate
solution (30 mL) was added to quench the reaction and extracted with
dichloromethane (30
mL x 3). The organic phase was combined, dried over anhydrous sodium sulfate,
filtered
and concentrated to obtain a crude product. The crude product was separated
and purified
by silica gel column chromatography (petroleum ether/ethyl acetate (v/v)=1:1),
and
concentrated to obtain 2b (440.0 mg, yield 51.5%).
[0268] LCMS in/z = 344.9 [M+1]+.
Step 3: methyl (S)-2-((64(4-cyano-2-fluorobenzypoxy)-3',6'-dihydro-[2,4'-
bipyridin1-
1'(2'H)-yetnethyl)-1-(oxetan-2-ylinethyl)-1H-thieno 112,3 -d] imidazole-5-c
arboxylate (2c)
CA 03203276 2023- 6- 22
NC
N
0 N N
0
0
2c
[0269] At room temperature, 2c-1 (69.0 mg, 0.2 mmol) was
dissolved in acetonitrile
(5 mL) solution, then potassium carbonate (83.0 mg, 0.6 mmol) and 2b (69.0 mgõ
0.2
mmol) were added, and the temperature was raised to 40 C for 4 h. The reaction
liquid was
filtered to remove the inorganic base, the filtrate was collected and
concentrated to obtain a
crude product, and the obtained crude product was separated and purified by
silica gel
column chromatography (dichloromethane/methanol (v/v) = 20:1), and
concentrated to
obtain 2c (75.0 mg, yield 66.0%).
[0270] LCMS in/z = 574.2 [M+1] .
Step 4: (S)-2((64(4-cyano-2-fluorobenzypoxy)-3',6'-dihydro- [2,4'-
bipyridin] - (2'H)-
yOmethyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-carboxylic
acid
(Compound 2)
NC
N
0
OH
Compound 2
[0271] At room temperature, 2c (75.0 mg, 0.13 mmol) was
dissolved in acetonitrile (5
in-L) and water (1 naL), then 1 g (CAS: 5807-14-7) (91.0 mg, 0.66 iirmol) was
added, and
the reaction was continued for 24 h. 1N hydrochloric acid was used to adjust
pH to 6, then
dichloromethane:methanol = 10:1 (20 inL x 3) was used for extraction. The
organic phase
was combined, dried over anhydrous sodium sulfate, filtered and concentrated
to obtain a
crude product. The obtained crude product was subjected to prep-HPLC to obtain
(S)-2-
((4-(6((4-cyano -2-fluorobenzypoxy)pyridin-2-yppiperidin- 1 -yemethyl)-1-
(oxetan-2-
ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (compound 2)
trifluoroacetic acid
salt and then preparative thin-layer chromatography (dichlorotnethane:
methanol
(v/v)=10:1) to obtain the compound 2 (16.0 mg, yield 21.9%).
CA 03203276 2023- 6- 22
- 54 -
[0272] Preparation conditions:
[0273] instrument and preparative column: waters 2767
(preparative liquid phase
chromatographic instrument) was used; the preparative column model was
SunFire@Prep
C18 (19 mm x 250 mm).
[0274] Preparation method: The crude product was dissolved in DMF and
filtered
with a 0.45 !..tm filter membrane to prepare into a sample liquid.
[0275] Mobile phase system: acetonitrile/water (containing 1%
TFA). Gradient
elution: acetonitrile content 20-65%, elution flow rate: 12 mUmin, elution
time 18 min.
[0276] Compound 2:
[0277] LCMS m/z = 560.2 1M+11+.
[0278] 111 NMR (400 MHz, CD30D) 6 7.73 (s, 111), 7.69 ¨ 7.60 (m,
211), 7.59 ¨ 7.51
(m, 2H), 7.07 (d, 1H), 6.73 (d, 1H), 6.70 ¨ 6.65 (m, 1H), 5.53 (s, 2H), 5.22 ¨
5.14 (m, 1H),
4.72 ¨ 4.53 (m, 311), 4.45 ¨4.37 (m, 114), 4.10¨ 3.99 (m, 211), 3.37-3.33 (m,
211), 2.95 ¨
2.87 (m, 2H), 2.77 ¨ 2.69 (m, 1H), 2.66¨ 2.57 (m, 2H),2.49 ¨ 2.42 (m, 1H).
Example 3
2-(((S)-4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-2-methylpiperazin-l-
ypmethyl)-
1-(((S)-oxetan-2-y1)methyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid
(Compound 3)
7
NC
0 )V r\l) N
0
OH
Compound 3
NBoc NfO
NC .F HN.õ.) NC - F NC FBV N S 0¨
.0 ,N_CI 3b 0 m t(j.NBoc r - NH
N,õ,) 2b
Step 1 Step 2 UN
Step 3
3a
3c 3d
rp7 CN:YN 0
NC 1 gH
, NC -F N
k,1õ0õ N
s 0 Step 4
0, OH
3f Compound 3
Step 1: tert-butyl (S)-4-(6-((4-cyano-2-fluorobenzypoxy)pyridin-2-y1)-2-
methylpiperazine-
l-carboxylate (3c)
CA 03203276 2023- 6- 22
- 55 -
NC
NBoc
0 N N
3c
[0279] 3a (524 mg, 2.0 minol), 3b (400 mg, 2.0 minol), Pd2(dba)3
(192 mg, 0.2
mmol), RuPhos (2-bicyclohexylphosphine-2',6'-diisopropoxybiphenyl) (184 mg,
0.4
mmol) and cesium carbonate (L3 g, 4M mmol) were added to 1,4-dioxane (20 mL).
The
reaction flask was placed at 90 C and stirred for 6 hours after nitrogen
replacement for
protection. Then the reaction liquid was cooled to room temperature and
quenched by
pouring into aqueous saturated ammonium chloride solution (100 mL), then
extracted with
ethyl acetate (30 naL x 3), and the organic phase was dried over anhydrous
sodium sulfate,
filtered and spin-dried. The crude product was separated and purified by
silica gel column
chromatography (petroleum ether/ethyl acetate (v/v) = 5:1), and concentrated
to obtain 3c
(700 mg, yield 82%).
[0280] LCMS m/z = 427.3[M+ 1 r.
Step 2: (S)-3-fluoro-4-(46-(3-methylpiperazin-1-yl)pyridin-2-yl)oxy)methyl)
benzonitrile
(3d)
NC
r'NH
0 N
3d
[0281] 3c (213 mg, 0.5 mmol) was added into a mixed solvent of
dichloromethane (6
inL) and trifluoroacetic acid (1 inL), and stirred at 15 C for 1 hour. The
reaction liquid was
directly concentrated under reduced pressure to obtain 3d trifluoroacetic acid
salt (230 mg,
crude product), which was directly used in the next reaction without further
purification.
[0282] LCMS tn/z = 327A [1\4+1] +.
Step 3: methyl
2-(((S)-4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-2-
tnethylpiperazin-1-ypmethyl)-1-4(S)-oxetan-2-ypinethyl)-1H-thieno [2,3-
d]imidazole-5-
carboxylate (3t)
CA 03203276 2023- 6- 22
- 5 6 -
NC
N
0 N N N
0
0
3f
[0283] 3d (40 mg, 0.11 minol) and 2b (3g mg, 0.11 mmol) and
potassium carbonate
(47 mg, 0.33 trump were added into acetonitrile (3 mL), and stirred at 12 C
for 7 hours.
The reaction system was quenched by adding into an aqueous saturated ammonium
chloride solution (20 mL), then extracted with ethyl acetate (30 mL x 3). The
organic
phase was combined, dried over anhydrous sodium sulfate and filtered and the
filtrate was
spin-dried. Separation and purification by silica gel column chromatography
(dichloromethane/methanol (v/v) = 20:1) was performed to give 3f (40 mg, yield
60%).
[0284] LCMS m/z = 591.2 [M+1r.
Step 4: 2-(((S)-4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-y1)-2-
methylpiperazin-l-
yOmethyl)-1-(((S)-oxetan-2-yemethyl)-1H-thieno [2,3 -d] imidazole-5-c arb
oxylic acid
(Compound 3)
NC
0 N N
0
OH
Compound 3
[0285] 3f (40 mg, 0.067 inmol) and 1 g (47 mg, 0.033 mmol) were
added to a mixed
solution of acetonitrile (3 mL) and water (0.6 mL), and stirred at 12 C for 10
hours. The
reaction liquid was then adjusted to pH = 6 with 1N hydrochloric acid, then
extracted with
ethyl acetate (10 mL x 3). The organic phase was combined and dried over
anhydrous
sodium sulfate, filtered and spin-dried, and prep-HPLC was performed to give 2-
((S)-4-(6-
((4-cy ano-2-fluorobenzyl)oxy)pyridin-2-y1)-2-methylpiperazin- 1-yl)methyl)-1-
((S)-oxetan-
2-yenriethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (compound 3)
trifluoroacetate
salt. After separation by silica gel column chromatography
(dichloromethane/methanol
(v/v) = 20:1), compound 3 (10 mg, yield 25.6%) was obtained.
[0286] Preparation conditions:
CA 03203276 2023- 6- 22
- 57 -
[0287] instrument and preparative column: waters 2767
(preparative liquid phase
chromatographic instrument) was used; the preparative column model was
SunFire@Prep
C18 (19 mm x 250 mm).
[0288] Preparation method: The crude product was dissolved in
DMF and filtered
with a 0.45 tm filter membrane to prepare into a sample liquid.
[0289] Mobile phase system: acetonitrile/water (containing 1%
TFA). Gradient
elution: acetonitrile content 20-65%, elution flow rate: 12 mL/min, elution
time 18 min.
[0290] LCMS m/z = 577.2 [M+1] .
[0291] 111 NMR (400 MHz, CD30D) 6 7.80 (s, 111), 7.61 (t, 111),
7.53 (t, 211), 7.44 (t,
1H), 6.27 (d, 1H), 6.13 (d, 1H), 5.43 (s, 2H), 5.27 ¨ 5.21 (m, 1H), 4.75 ¨
4.68 (m, 1H),
4.65 ¨ 4.57 (m, 211), 4.43 ¨ 4.32 (m, 211), 3.85 ¨ 3.79 (m, 111), 3.76¨ 3.70
(m, 111), 3.61 ¨
3.56 (m, 1H), 3.12¨ 3.03 (m, 1H), 2.93 ¨2.85 (m, 1H), 2.80¨ 2.71 (m, 2H), 2.63
¨ 2.57
(m, 1H), 2.51 ¨ 2.43 (m, 1H), 2.40 ¨ 2.32 (m, 1H), 1.18 (d, 311).
Example 4
24(4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo[d][1,3]dioxo1-4-yppiperidin-1-
y1)methyl)-1-(((S)-oxetan-2-y1)methyl)-1H-thieno[2,3-dlimidazole-5-carboxylic
acid
(Compound 4)
ci
J- -0
s
OH
Compound 4
Br BOO
Bac
OH
E C)13 CNBoc
OH 0
4b F
^
Cr Step 3 F Step 0
RP
Step 2 111
4a 4c
4e CI 4164' 4f
=pTSA N
Step 4
Br N S 0¨ 1
0 2b F 9 ,C rjj'I;_rsii?"1
Cirrf1V
õO>S0 Step 5 \S% Step 6 t
0H0
CI
4h
Compound 4
Step 1: 4-Bromo-2-(4-chloro-2-fluoropheny1)-2-methylbenzo[d][1,3]dioxole (4c)
CA 03203276 2023- 6- 22
- 58 -
Br
0
0
CI
4c
[0292] Substrates 4a (4L0 g, 0.2 mol) and 4b (45.0 g, 0.237
mol), and p-
toluenesulfonic acid monohydrate (900 mg, 4.75 mmol) were added to toluene
(200 mL),
subjected to nitrogen replacement for protection, and placed at 140 C for
reflux and water
separation for 60 hours. The reaction liquid was cooled to room temperature,
spin-dried,
and the residue was separated and purified by silica gel column chromatography
to obtain
4c (18.0 g, yield 22%).
Step 2: tert-butyl 4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo [d] [1,3]
dioxo1-4-y1)-3,6-
dihydropyridine-1(2H)-carboxylate (4e)
Boc
0
0
CI
4e
[0293] 4b (2.0 g, 5.82 mime and 4c (1.62 g, 5.24 mime,
Pd(dpp0C12 (470 mg, 0.58
minol) and sodium carbonate (3.08 g, 29.1 ininol) were added to a mixed
solvent of 1,4-
dioxane (30 mL) and water (S mL), subjected to nitrogen replacement for
protection,
stirred at 90 C for 20 hours, cooled to room temperature, and extracted with
ethyl acetate
(80 mL x 3). The organic phase was combined, dried over anhydrous sodium
sulfate,
filtered, and spin-dried, and the residue was separated and purified by silica
gel column
chromatography to obtain 4e (1.6 g, yield 62%).
[0294] LCMS in/z = 390.0[M-55]t
Step 3: tert-butyl
44244 -chloro-2-fluoropheny1)-2-methylbenzo [d] [1,3] dioxo1-4-
yl)piperidine-l-carboxylate (40
CA 03203276 2023- 6- 22
- 59 -
Boc
F 0
0
CI
4f
[0295] 4e (L6 g, 3.59 mmol) and Raney nickel (160 mg) were added
to a mixed
solvent of methanol (15 mL) and tetrahydrofuran (10 mL), subjected to hydrogen
replacement for protection, and stirred at room temperature for 8 hours. The
reaction liquid
was filtered and spin-dried, and the residue was separated and purified by
silica gel column
chromatography to obtain 4f (1.4 g, yield 87%).
[0296] LCMS in/z = 392.2[M-55] .
Step 4: 4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo [d] [1,3] dioxo1-4-
yl)piperidine P-
toluenesulfonate (4g)
.pTSA
F 0
0
CI
4g
[0297] 4f (0.8 g, L79 mmol) and p-toluenesulfonic acid
monohydrate (410 mg, 2J5
mmol) were added to ethyl acetate (20 mL), subjected to nitrogen replacement
for
protection, and stirred at 45 C for 12 hours. The reaction liquid was cooled
to room
temperature, filtered, and washed with ethyl acetate to obtain the p-
toluenesulfonate 4g
(620 mg, yield 99%) as a solid.
[0298] LCMS m/z = 348.0 [M+1]+.
Step 5:
methyl 24(4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo [d] [1, 3] dioxo1-4-
yl)piperidin-l-y1)
methyl)-1-(((S)-oxetan-2-yl)tnethyl)-1H-thieno [2,3 -d] imidazole-5-
carboxylate (41)
CA 03203276 2023- 6- 22
- 60 -
CI
0 1\17
F 0 N 0
4i
[0299] 4g (150 mg, 0.3 mmol) and 2b (103 mg, 0.3 mmol), and
potassium carbonate
(210 mg, 1.5 inmol) were added into acetonitrile (5 mL) and stirred at room
temperature
for 5 hours. The reaction liquid was directly mixed with silica gel, and
separated and
purified by silica gel column chromatography to obtain 4i (200 mg, yield 76%).
[0300] LCMS m/z = 612.3 [M+1]+.
Step 6: 2-44-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo [d] [1,3] dioxo1-4-
yepiperidin-1-
yOmethyl)-1-4(S)-oxetan-2-yl)methyl)-1H-thieno [2,3 -d] imidazole-5-c arb
oxylic acid
(Compound 4)
ci om
0
F 0 N
0
OH
Compound 4
[0301] 4i (200 mg, 0.33 intnol) and lithium hydroxide
monohydrate (140 mg, 3.3
inmol) were added to a mixed solvent of acetonitrile (4 inL) and water (1 mL),
and stirred
at 50 C for 8 hours. The reaction liquid was cooled to room temperature,
adjusted with 1N
hydrochloric acid to pH=6, and extracted with ethyl acetate. The organic phase
was dried
with anhydrous sodium sulfate, filtered and spin-dried. The residue was
separated and
purified by silica gel column chromatography to obtain the product (24(4-(2-(4-
chloro-2-
fluoropheny1)-2-methylbenzo[d] [1,3] dioxo1-4-yepiperidine-1 -yemethyl)-14(S)-
oxetan-2-
yOmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (compound 4) (50 ing,
yield
25%).
[0302] LCMS in/z = 598.1 [M+1]+.
[0303] 11-1 NMR (400 MHz, DMS0-(16) 8 7.73 (s, 1H),7.63 ¨ 7.51
(in, 2H), 7.34 (d,
1H), 6.83¨ 630 (in, 3H), 5.15 ¨ 5.04 (in, 1H), 4.67 ¨ 4.58 (in, 1H), 4.55 ¨
4.43 (in, 2H),
CA 03203276 2023- 6- 22
-61-
4.41 ¨ 4.33(m, 1H), 3.80 (d, 1H), 3.69 (d, 1H), 2.97 (d, 1H), 2.87 (d, 1H),
2.73¨ 2.59 (m,
214), 2.46 ¨ 2.37 (m, 114), 2.23 ¨2.09 (m, 214), 2.02 (s, 311), 1.83¨ 1.61 (m,
411).
Example 5
24(4-(2-(4-cyano-2-fluoropheny1)-2-methylbenzo[d] [1,3] dioxo1-4-yppiperidin-1-
yl)methyl)-1-(((S)-oxetan-2-yl)methyl)-1H-thieno[2,3-d]imidazole-5-carboxylic
acid
(Compound 5)
NC
0 N z
F 0 N 0
OH
Compound 5
(:)D
Boc
Boc
N 0
=pTSA I \
Br N S 0-
jo(0 F 0 F 2b
b Step 1
NC 5g NC 5
Step 2 Step 3
CI-- 4f
NC la NC
0 F õ5.1 011µ1' F a ), N--cs1 0
Step 4 \s 0
0- OH
5j Compound 5
Step 1: tert-butyl 4-(2-(4-cyano-2-fluoropheny1)-2-
methylbenzordi 1-1,31dioxo1-4-
yl)piperidine-l-carboxylate (5g)
Boc
F 0
0
NC
5g
[0304] 4f (600 mg, L34 mmol) and Pd2(dba)3 (65 mg, 0.07 mmol),
dppf (84 mg, 0.15
mmol), Zn(CN)2 (264 mg, 2.25 inmol), and zinc powder (24 mg, 0.37 minol) were
added
to DMA (15 inL), subjected to nitrogen replacement for protection, and stirred
at 120 C
for 20 hours. The reaction liquid was cooled to room temperature, filtered and
spin-dried,
CA 03203276 2023- 6- 22
- 62 -
and the residue was separated and purified by silica gel column chromatography
to obtain
g (150 mg, yield 23%).
[0305] LCMS m/z = 383.0[M-55]t
Step 2: 4-(2-(4-cyano-2-fluoropheny1)-2-methylbenzo[d][1,3]dioxol-4-
ylipiperidine P-
5 toluenesulfonate (5h)
=pTSA
xl
0
NC
5h
[0306] 5g (200 mg, 0.46 mmol) and p-toluenesulfonic acid
monohydrate (130 mg,
0.69 mmol) were added to ethyl acetate (8 tnL), subjected to nitrogen
replacement for
protection, and stirred at 45 C for 12 hours. The reaction liquid was cooled
to room
temperature, filtered, and washed with ethyl acetate to obtain the p-
toluenesulfonate 5h
(250 mg, yield 90%) as a solid.
[0307] LCMS in/z = 339.2 [M+1] .
Step 3: methyl 2-((4-(2-(4-cy ano-2-fluoropheny1)-2-
methylbenzo [d] [1,3] dioxo1-4-
yl)piperidin-l-y1) methyl)-1-(((S)-oxetan-2-yl)tnethyl)-1H-thieno
[2,3 -d] imidazole-5-
carboxylate (5j)
NC
0
F 0 N 0
5j
[0308] 5h (120 mg, 0.2 mmol) and 2b (821ng, 0.22mm01), and
potassium carbonate
(160 mg, 1.1 mmol) were added into acetonitrile (5 mL) and stirred at room
temperature
for 5 hours. The reaction liquid was directly mixed with silica gel, and
separated and
purified by silica gel column chromatography to obtain 5j (100 mg, yield 83%).
[0309] LCMS tn/z = 603.2 [M+1] .
CA 03203276 2023- 6- 22
- 63 -
Step 4: 24(4-(2-(4-cyano-2-fluoropheny1)-2-methylbenzo[d] [1,3] di oxo1-4-
yl)piperi din-1-
yOmethyl)-1-(((S)-oxetan-2-yOmethyl)-1H-thi en o [2,3-dlimidazole-5-carboxylic
acid
(Compound 5)
NC
(c-A
F 0 N
S
OH
Compound 5
[0310] 5j (100ing, 0Ø17 inmol) and lithium hydroxide monohydrate (115
ing, 2.73
mmol) were added to a mixed solvent of acetonitrile (4 inL) and water (1 inL),
and stirred
at 50 C for 8 hours. The reaction liquid was cooled to room temperature,
adjusted with 1N
hydrochloric acid to pH=6, and extracted with ethyl acetate. The organic phase
was dried
with anhydrous sodium sulfate, filtered and spin-dried. The residue was
separated and
purified by silica gel column chromatography to obtain (24(4-(2-(4-cyano-2-
fluoropheny1)-2-methylbenzo[d] [1,3]dioxo1-4-yepiperidine-1-yemethyl)-1-((S)-
oxoalkane-2-y1)methyl)-1H-thieno 112,3 -d] imidazole-5-c arboxylic acid
(compound 5) (50
ing, 48% yield).
[0311] LCMS m/z = 589.2 [M+1] .
[0312] 1H NMR (400 MHz. DMSO-c/o) 6 12.94 (hr, 1H), 7.97 (d, 1H), 7.81 (s,
1H),
7.77 ¨ 7.69 (in, 2H), 6.85¨ 6.70 (in, 3H), 5.14 ¨ 5.05 (in, 1H), 4.69 ¨ 4.61
(in, 1H), 4.57 ¨
4.44 (in, 2H), 4.40 ¨ 4.32(in, 1H), 3.82 (d, 1H), 3.72 (d, 1H), 2.98 (d, 1H),
2.88 (d, 1H),
2.73¨ 2.60 (in, 2H), 2.45¨ 2.36 (in. 1H), 2.24 ¨2.10 (in, 2H), 2.05 (s, 3H),
1.79 ¨ 1.63 (in,
4H).
NCO NC
0 N 0
F 0 N F 0 N
0 0
OH OH
Compound 5-a Compound 5-b
[0313] 2g compound 5 was prepared by SFC and dried to obtain
compound 5-1 (600
ingõ yield 30%) as a white solid with a retention time of 6.441 minutes.
compound 5-2
(500 mg, yield 25%) as a white solid with a retention time of 7.104 minutes.
CA 03203276 2023- 6- 22
- 64 -
[0314] Preparation conditions:
[0315] instrument and preparative column: Waters 150 MGM chiral
liquid phase
chromatographic instrument was used, and the preparative column model was
DAICEL
CHIRALPAK AD, I.D., 5 m, inner diameter x length = 250 x 30 mm.
[0316] Preparation method: The crude product was dissolved in ethanol and
filtered
with a 0.45 !..im filter membrane to prepare into a sample liquid.
[0317] Mobile phase system: carbon dioxide/ethanol (0.1% NH3
H20). Gradient
elution: the ethanol content was 25%, and the elution time was 11 minutes.
[0318] Compound 5-1:
[0319] LCMS m/z = 589.2 [M+11+.
[0320] 1H NMR (400 MHz, DMSO-d6) 6 8.01-7.93 (m, 111), 7.79(s,
1H),7.77-7.70
(m, 2H), 6.84-6.69 (m, 3H), 5.15-5.06 (m, 111), 4.69-4.60 (m, 111), 4.57-4.44
(m, 2H),
4.42-4.34(m, 1H), 3.85-3.78 (m, 1H), 3.76-3.68 (m, 111), 3.02-2.93 (m, 111),
2.92-2.84 (m,
111), 2.74-2.58 (m, 2H), 2.46-2.36 (m, 111), 2.25-2.10(m, 2H), 2.05 (s, 311),
1.81-1.64 (m,
411).
[0321] The structure of compound 5-1 is one of the above
formulas 5-a and 5-b;
[0322] Compound 5-2:
[0323] LCMS m/z = 589.2 [M+1]+.
[0324] 1H NMR (400 MHz, DMSO-d6) 6 8.00-7.93 (m, 1H), 7.77-7.67
(m, 3H), 6.84-
6.72 (m, 3H), 5.13-5.04 (m, 1H), 4.67-4.58 (m, 111), 4.56-4.44 (m, 2H), 4.41-
4.33(m, 1H),
3.83-3.77 (m, 1H), 3.74-3.67 (m, 1H), 3.01-2.93 (m, 1H), 2.91-2.83 (m, 1H),
2.72-2.59 (m,
211), 2.46-2.35 (m, 111), 2.23-2.10 (m, 211), 2.05 (s, 311), 1.79-1.61 (m,
411).
[0325] The structure of compound 5-2 is one of the above
formulas 5-a and 5-b; and
compound 5-2 and compound 5-1 were isomers for each other, that is, when the
structure
of compound 5-1 was the structure of formula 5-a, the structure of compound 5-
2 was the
structure of formula 5-b; when the structure of compound 5-1 was the structure
of formula
5-b, the structure of compound 5-2 was the structure of formula 5-a.
Example 6
(S)-2-(4-(64(4-cyano-2-fluorobenzypoxy)pyridi n-2-y1)-2-fluorobenzy1)-1-
(oxetan-2-
ylmethyl)-111-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 6)
CA 03203276 2023- 6- 22
- 65 -
Fr.------1
NC N
N
0
S
Compound 6
r)o_3
HNjp
Rip 0
B F H2 N - ,L4 - '0- Br 0- \
--t.-I 9
;.N ___________________________________________________________________ -1
.. 40 0
r 1--
Step 1 NH=FICI Step 2 step 4 N
u
Step 3 F(. s IN S U-
rsirsA
6a 6b 6c 6f
6e
0,CN 0, N N Fi ir,o,,
F 6NC , F _ 14--,/
lg H NC
Step 5 n_o N, a- ----Kõ __ Step 6 1---3
o
OH 6h Compound 6
Step 1: ethyl 2-(4-bromo-2-fluorophenypacetimidate hydrochloride (6b)
Br F
0
N H +I CI
6b
[0326] 6a (5.0 g, 26.04 minol) was added to anhydrous ethanol (3.0 mL),
placed at
0 C and fully stirred, and at the same time, dry hydrogen chloride gas was
introduced into
the reaction liquid until it was saturated, and the reaction liquid was
continuously stirred at
this temperature for 10 hours. Diethyl ether (40 inL) was then added to the
reaction liquid,
filtered to obtain a solid that was further washed with diethyl ether (100 mL
x 2 ), and
dried to obtain 6b (5.0 g, yield 72%).
[0327] II-1 NMR (400 MHz, DMSO-do) 6 12.04 (s, 2H), 7.61 (d,
1H), 7.51 ¨ 7A3 (in,
2H), 446 (q, 2H), 4A4 (s, 2H), 1.26 (t, 3H).
[0328] 19F NMR (400 MHz, DMSO-do) 6 111.30.
Step 2: 4-broino-2-fluoro-1-(2,2,2-triethoxyethyl)benzene (6c)
CA 03203276 2023- 6- 22
Br F
0
0,
0
6c
[0329] 6b (5.0 g, 19.30 ininol) was added into anhydrous ethanol
(40 itiL), and stirred
at room temperature (15 C) for 48 hours. Then the reaction liquid was directly
filtered to
obtain a filtrate, and the filtrate was spin-dried to make a slurry with a
mixed solvent of
petroleum ether and ethyl acetate (100 inL) (petroleum ether/ethyl acetate
(v/v) = 10:1) and
filtered, and the filtrate was spin-dried to obtain 6c (4M g, yield 66%).
[0330] 11-1 NMR (400 MHz, CDC13) 7.45 (t, 1H), 7.23 ¨ 7.14 (in,
2H), 3.50 (q, 6H),
3.07 (s, 2H), 1.16 (t, 9H).
Step 3: methyl (S)-2-(4-bromo-2-fluorobenzy1)- 1-(oxetan-2-ylinethyl)- 1H-
thieno [2,3-
d]imidazole-5-carboxylate (6e)
Br ;):_)]
0
N¨S 0-
6e
[0331] 6d (500 mg, 2.06 mmol) and 6c (1.0 g, 3.19 mmol) were
added to glacial
acetic acid (20 inL), and stirred at 60 C for 10 hours. The reaction system
was cooled to
room temperature, then was slowly added to an aqueous saturated sodium
bicarbonate
solution (200 mL) to quench the reaction, and then extracted with ethyl
acetate (50 mL x
3). The organic phase was combined, dried over anhydrous sodium sulfate, and
filtered.
The filtrate was spin-dried, and the residue was separated and purified by
silica gel column
chromatography to obtain 6e (430 mg, yield 47%).
[0332] LCMS m/z = 439.0 [M+1]+.
Step 4: methyl (S)-2-(2-fluoro-4-(4,4,5 ,5-tetramethy1-1,3 ,2-dioxaborolan-2-
yl)benzy1)-1-
(oxetan-2-yltnethyl)-1H-thieno [2,3 -d] imidazole-5-c arboxylate (61)
CA 03203276 2023- 6- 22
- 67
oo
0-B
0-
6f
[0333] 6e (400 mg, 0.98 mmol) and pinacol biborate (262 mg, 1.0
mmol),
Pd(dppf)C12.DCM (81 mg, OA mmol), and potassium acetate (196 mg, 2.0 mmol)
were
added to 1,4-dioxane (20 mL), and the reaction flask was subjected to nitrogen
replacement for protection, and the reaction liquid in the reaction flask was
stirred at
100 C for 5 hours. The reaction liquid was cooled to room temperature, then
water (100
mL) was added and extracted with ethyl acetate (50 mL x 3). The organic phase
was
combined, dried over anhydrous sodium sulfate and filtered, the filtrate was
spin-dried, and
the residue was separated and purified by silica gel column chromatography to
obtain 6f
(340 mg, yield 71%).
[0334] LCMS in/z = 487.2 [M+1]+.
Step 5: methyl (S)-2-(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-2-
fluorobenzy1)-1-
(oxetan-2-ylinethyl)-1H-thieno 112,3 -d] imidazole-5-c arboxylate (6h)
NC
0 N
0
6h
[0335] 6f (340 mg, 030 mmol) and 6g (220 mg, 0.84 mmol), Pd(dppf)C12.DCM
(83
mg, OA minol) and potassium carbonate (290 mg, 2A ininol) were added to a
mixed
solution of 1,4-dioxane (10 mL) and water (2 mL), and the reaction flask was
subjected to
nitrogen replacement for protection, and the reaction liquid in the reaction
flask was stirred
at 100 C for 5 hours. The reaction liquid was cooled to room temperature, then
water (50
inL) was added, and extracted with ethyl acetate (30 mL x 3). The organic
phase was
combined, dried over anhydrous sodium sulfate, and filtered. The filtrate was
spin-dried,
and the residue was separated and purified by silica gel column chromatography
to obtain
6h (200 mg, yield 48%).
CA 03203276 2023- 6- 22
- 68 -
[0336] LCMS m/z = 587.1 [M+1]+.
Step 6: (S)-2-(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-2-fluorobenzy1)-
1-(oxetan-
2-ylmethyl)-1H-thieno[2,3-d]imidazole-5 -carboxylic acid (Compound 6)
,70
NC
,
0 N N I
0
OH
Compound 6
[0337] 6h (100 mg, 0.17 mmol) and 1 g (120 mg, 0.85 inmol) were added to a
mixed
solution of acetonitrile (5 mL) and water (1 inL), and stirred at room
temperature (12 C)
for 10 hours. Then the reaction liquid was adjusted to pH=6 with 1M
hydrochloric acid,
continuously stirred until a white solid precipitated, and filtered and the
solid was washed
with water. The solid was dissolved in dichloromethane, dried over anhydrous
sodium
sulfate and filtered. The organic phase was spin-dried. The residue was
separated by
preparative HPLC to give (S)-2-(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-
2-
fluorobenzy1)-1-(oxoalkane-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-c
arboxylic acid
(compound 6) tritluoroacetate salt, which was separated and purified by silica
gel column
chromatography to obtain compound 6 (40 mg, yield 41%).
[0338] LCMS in/z = 573.1 [M+1] .
[0339] 114 NIVIR (400 MHz, CD30D) 6 12.90 (br, 1H), 7.91 (d,
1H), 7.88 ¨ 7.80 (in,
4H), 7.79 ¨ 7.68 (in, 2H) 7.64 (d, 1H), 7.39 (t, 1H), 6.92 (d, 1H), 5.61 (s,
2H), 5.06 ¨ 4.98
(in, 1H), 4.60(dd, 1H), 4.54 ¨ 4.44 (in, 2H), 4.43 ¨ 4.28 (in, 3H), 2.73 ¨
2.61 (in, 1H),
2.40¨ 2.27 (in, 1H).
Example 7:
(S)-2-(4-(64(4-cy ano-2-fluorobenzypoxy)pyridin-2-yl)benzyl)- 1 -(oxetan-2-
ylmethyl)- 1H-
thieno[2,3-d[imidazole-5-carboxylic acid (Compound 7)
o
NC
OH
Compound 7
CA 03203276 2023- 6- 22
- 69 -
`ci.;.A
OEt r- rCN _________________________ H2N S 0¨ ld
Br N11-1.HCI 13r' 1 0 ___
Step 1 Br' Step 2
7a 7b 7c 0, Step 3 \L)
7d 6
NCyp-
6])
0 N CI
T F Step 4 NC Step -N
3a
__________________________________________________________ NcçON N-tro
F 5
OH
7e Compound 7
Step 1: ethyl 2-(4-bromophenyl)acetimidate hydrochloride (7b)
OEt
NH.HCI
Br
7b
[0340] Under the condition of ice bath, the substrate 7a (19.6
g, 100.0 mmol) was
dissolved in a solution of 33% hydrochloric acid in ethanol (45 mL). After the
addition, the
temperature was naturally raised to room temperature for overnight reaction.
At room
temperature, anhydrous diethyl ether (100 mL) was added to the reaction
system, stirred
for 5 minutes and filtered, and the filter cake was washed with diethyl ether
(20 mL x 2),
collected and dried to obtain the product 7b of interest (22.0 g , yield
79.4%) as a white
solid.
[0341] LCMS m/z = 242.1 [M+11 .
Step 2: methyl (S)-2-(4-bromobenzy1)-1-(ox etan-2-ylmethyl)-1H-thi eno [2,3-
dlimidazole-
5-carboxylate (7c)
on
N
Br 0
7c
[0342] At room temperature, the substrate 7b (1.4 g, 5.0 mmol) was
dissolved in
glacial acetic acid (50 mL), and ld (1.2 g, 5.0 mmol) was added. After the
addition, the
reaction system was heated to 45 C for overnight reaction under nitrogen
protection. The
reaction solvent was distilled off under reduced pressure to obtain a crude
product, which
was separated and purified by silica gel column chromatography to obtain 7c
(740.0 mg,
yield 35.2%) as a white solid.
[0343] LCMS m/z = 421.0 [M+1]+.
CA 03203276 2023- 6- 22
- 70 -
Step 3: methyl (S)-1-(oxetan-2-ylmethyl)-2-(4-(4,4,5,5-tetramethy1-1,3,2-
dioxaborolan-2-
y1) benzy1)-1H-thieno[2,3-dlimidazole-5-carboxylate (7d)
cr)
/
0
7d N
[0344] At room temperature, 7c (740.0 mg. 1.8 mmol) was
dissolved in 1.4-dioxane
(30 inL), and then pinacol biborate (820.0 mg, 2.7 mmol), Pd(dppf)C12 (147.0
mg, 0.2
mmol) and potassium acetate (519.0 mg. 5.3 mmol) were added successively.
After the
addition, the reaction system was heated to 100 C for overnight reaction under
nitrogen
protection. The reaction liquid was cooled to room temperature and filtered.
The filter cake
was washed with dichloromethane (10 mLx2). The filtrate was collected and
concentrated
to obtain a crude product, which was purified by silica gel column
chromatography to
obtain 7d (680.0 mg, yield 82.4%) as a yellow solid.
[0345] LCMS m/z = 469.2 [M+1]+.
Step 4: methyl (S)-2-(4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-yebenzy1)-1-
(oxetan-
2-ylmethyl)-1H-thieno [2,3-d] imidazole-5 -c arboxy late (7e)
o
NC
I17,_N,, = N
7e
[0346] At room temperature, 3a (130.0 mg, 0.5 mmol) was
dissolved in a mixed
solvent of 1,4-dioxane (10 mL) and water (1 mL), and then 7d (193.0 mg, 0.4
mmol) and
Pd(dppf)C12 (34.0 mg, 0.04 mmol) and cesium carbonate (261.0 mg, 0.8 niftiol)
were
added successively. After the addition, the reaction system was heated to 100
C for
overnight reaction under nitrogen protection. The reaction liquid was cooled
to room
temperature and filtered. The filter cake was washed with dichloromethane (10
mL x 2).
The filtrate was collected and concentrated to obtain a crude product, which
was separated
and purified by silica gel column chromatography to obtain 7e (109.0 mg, yield
46.6% ) as
a white solid.
CA 03203276 2023- 6- 22
-71-
1103471 LCMS m/z = 569.3 [M+1]+.
Step 5: (S)-2-(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-
yObenzy1)-1-(oxetan-2-
ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 7)
NC
'
\S¨ o
OH
Compound 7
[0348] At room temperature, 7e (109.0 mg, 0.2 mmol) was dissolved in a
mixed
solvent of acetonitrile (5 mL) and water (1 mL), and then 1 g (CAS: 5807-14-7)
(80.0 mg,
0.6 mmol) was added. After the addition, the reaction was carried out at room
temperature
for 24 hours. Acetonitrile was distilled off under reduced pressure. The crude
product was
dissolved in water (5 mL), adjusted to pH=6 with 1N hydrochloric acid. The
aqueous
phase was extracted with ethyl acetate (15 mL x 3). The organic phase was
combined,
dried over anhydrous sodium sulfate, filtered and concentrated to obtain a
crude product,
which was separated and purified by silica gel column chromatography to obtain
compound 7 (26.0 mg, yield 24.5%) as a white solid.
[0349] Compound 7:
[0350] LCMS m/z = 555.2 [M+1] .
[0351] 111 NMR (400 MHz, DMSO-d6) 6 7.98 (d, 2H), 7.93 ¨ 7.88
(m, 111), 7.82 (t,
1H), 7.78 ¨ 7.68 (m, 3H), 7.57 (d, 1H), 7.38 (d, 2H), 6.87 (d, 1H), 5.60 (s,
2H), 4.94 ¨ 4.86
(m, 1H), 4.54 ¨ 4.30 (m, 6H), 2.65 ¨2.56 (m, 111), 2.33 ¨ 2.24 (m, 111).
Example 8
(S)-2-(4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-y1)-2-fluoro-5-methylbenzy1)-
1-
(oxetan-2-ylmethyl)-1H-thieno[2,3-dlimidazole-5-carboxylic acid (Compound 8)
r/1
NC , N
T
)- )0, N 0
T
OH
Compound 8
CA 03203276 2023- 6- 22
- 72 -
FO F F F F
-1
_-OH _____________________________________ ib OMs ____ 6 'CN __
Br ' Br"'r NI-
1.1-1C1
Step 1 Step 2 Br '
Step 3 Br
Step 4 Br
8a 8 b 8c 8d 8e
r---1
HN 0 NC,,,,,
Q rrC7 --.
HN C' ri)
' F
2 0- F if-P-7
F 0--
I d
_\p-B 0 A--1\s-iro 3a
Step 5 Br
B--Yo Step 6 ))s0 (:) Step 7
,
8f 0 8g
0- r In
F N N F
NC
NI) NC ,-
...,., -T... H
I 1
-,1)L,0
0 N ep 8
St
F OH
0,
8h Compound 8
Step 1: (4-bromo-2-fluoro-5-methylphenyl)methanol (8b)
F
OH
Br
8b
[0352] 8a (10.0 g, 43 mmol) and a 2M solution of borane in
tetrahydrofuran (26 tnL,
51.6 minol) were added to tetrahydrofuran (40 inL) and stirred at room
temperature for 30
hours. Then the reaction liquid was slowly added to 0.5 N hydrochloric acid
(100 mL) to
quench the reaction, and then extracted with ethyl acetate (80 mL x 3). The
organic phase
was dried over anhydrous sodium sulfate and filtered, and the liquid was
concentrated to
dryness. The residue was separated and purified by silica gel column
chromatography to
obtain 8b (9.4 g, yield 99%).
[0353] LCMS m/z = 201.0[M-17]t
Step 2: 4-bromo-2-fluoro-5-methylbenzyl tnethanesulfonate (8c)
F
0Ms
Br
8c
[0354] 8b (2.18 g, 10 minol) and triethylamine (2.0 g, 20
minol) were added to dry
dichloromethane (20 mL) and stirred at 0 C for 10 minutes, and then methyl
sulfonyl
CA 03203276 2023- 6- 22
- 73 -
chloride (1.38 g, 12 mmol), was slowly added to the reaction system and the
reaction was
stirred at 0 C for further 30 minutes. Water (20 mL) was added to the reaction
liquid,
extracted with dichloromethane (50 mL x 3). The organic phase was dried over
anhydrous
sodium sulfate, filtered, and the filtrate was directly concentrated under
reduced pressure to
obtain 8c (3.0 g, yield 99%), which was directly used in the next reaction
without further
purification.
Step 3: 2-(4-bromo-2-fluoro-5-methylphenyl)acetonitrile (8d)
CN
Br
8d
[0355] 8c (3.0 g, 10 inmol) and sodium cyanide (440 mg, 9 inmol)
were added to
DIVIF (15 mL), and stirred at room temperature for 40 hours. Ethyl acetate
(300 inL) was
added to the reaction system, washed with water (50 mL x 3). The aqueous phase
was
added to an aqueous sodium hypochlorite solution (50 mL) and stirred for 5
hours. The
organic phase was combined, dried over anhydrous sodium sulfate and filtered.
The filtrate
was concentrated to dryness and purified by silica gel column chromatography
to give 8d
(L5 g, yield 66%).
Step 4: ethyl 2-(4-bromo-2-fluoro-5-methylphenyl)acetimidate hydrochloride
(8e)
Br NH=FICI
8e
[0356] 8d (1.5g, 6.6 ininol) was added into ethanol (5 inL) and
stirred at room
temperature. Then, dry hydrogen chloride gas was introduced into the reaction
liquid for 1
hour, and then continually stirred for 10 hours. Diethyl ether (10 inL) was
added to the
reaction liquid, stirred until a solid precipitated, and filtered. The filter
cake was washed
with diethyl ether and dried to obtain 8e (L5 g, yield 73%).
Step 5: methyl (S)-2-(4-bromo-2-fluoro-5-methylbenzy1)-1-(oxetan-2-ylmethyl)-
1H-
thieno 112,3 -d] imidazole -5 -c arboxylate (8f)
CA 03203276 2023- 6- 22
Br 0
8f
[0357] 8e (300 mg, Idi.96 mmol) and Id (250 mg, LO mmol) were
added to glacial
acetic acid (10 mL), subjected to nitrogen replacement for protection and then
stirred at
40 C for 10 hours. The reaction liquid was added to a aqueous saturated sodium
bicarbonate solution to quench the reaction, and extracted with ethyl acetate
(30 mL x 3).
The organic phase was dried over anhydrous sodium sulfate and filtered. The
liquid was
concentrated to dryness, and the residue was separated and purified by silica
gel column
chromatography to obtain 8f (100 mg, yield 25%).
[0358] LCMS m/z = 453.0[M+ lr.
Step 6: methyl (S)-2-(2-fluoro-5-methy1-4 -(4,4,5 ,5-tetramethy1-1,3,2-dio
xaborolan-2-y1)
benzy1)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3 -d] imidazole-5-c arboxylat e
(8g)
8g
[0359] 8f (100 mg, 0.22 mmol), pinacol biborate (69 mg, 0.26
mmol), Pd(dppf)C12
(18 mg, 0.022 mmol) and potassium acetate (45 mg, 0.44 mmol) were added to 1,4-
dioxane (8 mL), subjected to nitrogen replacement for protection and then
stirred at 100 C
for 12 hours. Then the reaction liquid was cooled to room temperature,
concentrated to
dryness, and separated and purified by silica gel column chromatography to
obtain 8g (70
mg, yield 63%).
[0360] LCMS tn/z = 501.2[M+ 1 r.
Step 7: methyl (S)-2-(4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-2-fluoro-
5-
methylbenzy1)-1-(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylate
(8h)
CA 03203276 2023- 6- 22
-75 -
õ70
NC
I /
0 N
0
0
8h
[0361] 8g (70 ing, 0.14 minol), 3a (50 mg, 0.2 ininol),
Pd(dppt)C12dichloromethane
complex (18 mg, 0.02inino1) and potassium carbonate (55 mg , 0.4 intnol) were
added to a
mixed solvent of 1,4-dioxane (5 mL) and water (1 mL), subjected to nitrogen
replacement
for protection and then stirred at 100 C for 6 hours. The reaction liquid was
cooled to room
temperature, quenched with an aqueous saturated ammonium chloride solution (20
inL),
and extracted with ethyl acetate (20 mL x 3). The organic phase was dried over
anhydrous
sodium sulfate, filtered, spin-dried, and separated and purified by silica gel
column
chromatography to obtain 8h (60 mg, yield 71%).
[0362] LCMS in/z = 601.3 [M+1[+.
Step 8: (S)-2-(4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-y1)-2-fluoro-5-
methylbenzy1)-
1-(oxetan-2-ylinethyl)-1H-thieno [2, 3-d] imidazole-5-c arboxylic acid
(Compound 8)
,70
NC
/
0 N N
0
OH
Compound 8
[0363] 8h (60 mg, 0.1 mmol) and lg (CAS: 5807-14-7) (70 mg, 0.5
mmol) were
added into a mixed solvent of acetonitrile (5 inL) and water (1.0 inL), and
stirred at 50 C
for 5 hours. The reaction liquid was cooled to room temperature, adjusted to
pH=6 with 1N
hydrochloric acid, then extracted with ethyl acetate (20 mL x 3). The organic
phase was
combined, dried over anhydrous sodium sulfate and filtered and the liquid was
concentrated to dryness under reduced pressure. The residue was separated and
purified by
silica gel plate chromatography to obtain compound 8 (10 mg, yield 17%).
[0364] LCMS in/z = 587.3 [M+1[+.
[0365] 11-1 NMR (400 MHz, DMSO-d6) 6 12.89(s, 1H), 7.95 ¨ 7.80
(in, 3H), 7.77 ¨
7.66 (in, 2H), 7.26 ¨ 7.16 (in, 3H), 6.94(d, 1H), 6.51 (s, 2H), 5.06¨ 4.97
(in, 1H), 4.61 (in,
CA 03203276 2023- 6- 22
- 76 -
1H), 4.54 ¨ 4.45 (m, 2H), 4.39 ¨ 4.22 (m, 3H), 2.74 ¨ 2.65 (m, 1H), 2.38¨ 2.30
(m, 1H),
2.22(s, 3H).
Example 9:
(S)-2-((4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-6-oxopyridazin-1(61-1)-
yOmethyl)-
1-(oxetan-2-ylmethyl)-1H-thienor,3-dlimidazole-5-carboxylic acid (Compound 9)
icr:
o
NC _
-N TI-NI>r____
- 1 0. .1µ1 N N----/ \1
\
I S---COOH
F-. ,---.--
Compound 9
NC
Br 6 INI__ B (:j 0 6--
I1, 0 -- F N I
OH
' u, _ N
, NH 2b 'S -0O2Me e- 1\1-N, 9c F
CI
I N ' CIN N 7 \ F N,)LIN
9a Step 1 9b S CO2Me Step 2 1
9d S CO2Me
Step 3
0 01- 0o
I
NC. Step 4 NC N
0 1 j ¨?1,`----..
N,. ' - NN I
I S'cO2Me F I
F
9h Compound 9
Step 1: methyl (S)-2-((4-chloro-6-oxopyridazin-1(611)-yOmethyl)-1-(oxetan-2-
ylmethyl)-
1H-thienor,3-dlimidazole-5-carboxylate (9b)
(c-A
0
Am N
CI N N 1 1
S co2me
9b
[0366] The raw materials 9a (120 mg, 0.87 mmol) and 2b (300 mg,
0.87 mmol) were
added into a reaction flask and dissolved in acetonitrile (10inL). Anhydrous
potassium
carbonate (250 mg, 1.80 mmol) was added, reacted at 40 C for 4h and cooled to
room
temperature. The solid was filtered, the filtrate was concentrated under
reduced pressure,
and the residue was purified by silica gel column chromatography to obtain
product 9b
(247 mg, yield 71.8%) as a yellow solid.
[0367] LCMS m/z = 395.0 [M+Hr.
CA 03203276 2023- 6- 22
- 77 -
Step 2: methyl (S)-2-((4-(6-fluoropyridin-2-y1)-6-oxopyridazin-1(6H)-
yl)methyl)-1-
(oxetan-2-ylmethyl)-1H-thieno [2,3-d]imidazol e-5-carboxylate (9d)
0
/
N N
S 002me
9d
[0368] 9b (40 mg, 0.10 mmol), 9c (30 mg, 0.20 mmol), anhydrous
potassium
carbonate (40 mg, 0.20 mmol), and Pd(dppt)C12 (15 mg, 0.01 mmol) were added to
a
reaction flask successively, dissolved in a mixed solution of 1,4-dioxane (2
mL)/water (0.4
mL), subjected to nitrogen replacement, stirred at 90 C for 5 h, cooled to
room temperature
and filtered. The filtrate was concentrated under reduced pressure, and the
residue was
purified by silica gel column chromatography to obtain product 9d (22 mg,
yield 48.3%) as
a yellow solid.
[0369] LCMS in/z = 456.1 [M+Hr.
Step 3: methyl (S)-2-((4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-6-
oxopyridazin-
1(6H)-ye methyl)-1-(oxetan-2-ylinethyl)-1H-thieno [2,3 -di imidazole-5-c
arboxylate (9h)
NC
CO2me
9h
[0370] 9d (22 mg, 0.05 mmol) and 9f (15 mg, 0.09 mmol) were added into a
reaction
flask and dissolved in tetrahydrofuran solution (1 mL). Potassium tert-
butoxide (10 mg,
0.07 mmol) was added under nitrogen, and stirred at room temperature for 1 h.
The
reaction was quenched with aqueous ammonium chloride solution (1 mL) and
extracted
with ethyl acetate 2 mL x 3. The organic phase was combined, dried over
anhydrous
sodium sulfate, filtered and concentrated, and the residue was separated and
purified by
silica gel column chromatography to obtain 9h (20 mg, yield 71.1 %) as a
yellow solid.
[0371] LCMS m/z = 587.1 1M+Hr.
Step 4: (S)-2-((4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-6-oxopyridazin-
1(611)-
yl)methyl)-1-(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic
acid
(Compound 9)
CA 03203276 2023- 6- 22
- 78 -
O----
0
NC.,õ )N--\ N\
1 ' 1\1, N-J-C1
T s COON
F
Compound 9
[0372] At room temperature, 9h (20 mg, 0.03 mmol) was dissolved
in a solution of
acetonitrile (2 mL) and water (0.5 mL), then 1 g (CAS: 5807-14-7) (40 mg, 0.28
mmol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
1N hydrochloric acid to pH=6, and then extracted with dichloromethane:methanol
= 10:1
(2 mL x 3). The organic phase was combined, dried over anhydrous sodium
sulfate,
filtered and concentrated, and the residue was separated and purified by
silica gel plate
chromatography to obtain compound 9 (5 mg, yield 25.7%).
[0373] LCMS m/z = 573.1 [M+H]t
[0374] 111 NMR (400 MHz, DMSO-d6) 6 13.14¨ 12.78 (m, 111), 8.62 (d, 111),
7.98 ¨
7.68 (m, 6H), 7.57 (d, 1H), 7.08 (d, 1H), 5.68 ¨ 5.50 (m, 4H), 5.04 (d, 1H),
4.70 (dd, 1H),
4.58 (d, 111), 4.48 (dd, 111), 4.33 (dd, 1H), 2.74 ¨ 2.63 (m, 1H), 2.33 (dd,
111).
Example 10:
2-(4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo [d] [1,3]dioxo1-4-y1)-2-
fluorobenzy1)-1-
(((S)-oxetan-2-yl)methyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid
(Compound 10)
1-- F
a ---- I F 0,
/
/3, cl , IT¨Ii",
--- N.--_.
k. )
-s - 'co2H
Compound 10
i:
HN---, 1d
:D_J
I-12N-// )õ,
Br - F
BrF__cN
_ I
--- NH HCI F
OEt Step 2 )\_"1 Step 3
Step 1 CO2Me
10a 10b
Br 10c S
CL F
Br a ah, F
F F
"N -0
Step 5 0 IN. --- \ --\
-- N 1
--\
s CO2Me C)-13 "10d Step 4 6 CO2Me S cO2H
10e glip
Compound 10
CA 03203276 2023- 6- 22
- 79 -
Step 1: ethyl 2-(4-bromo-2-fluorophenyl)acetimidate hydrochloride (10b)
Br
NH HCI
OEt
10b
[0375] 10a (10.0 g, 52.1 mmol) was added into a reaction flask,
and 33% hydrogen
chloride-ethanol solution was introduced under ice bath, and then the
temperature was
gradually raised to room temperature for overnight reaction, and a large
number of white
solids were precipitated. Diethyl ether was added and stirred for 0.5h, the
solid was
filtered, washed with diethyl ether, and dried to obtain 10b (9.3 g, yield
60.6%) as a white
solid.
Step 2: methyl (R)-2- (4-bromo-2-fluorobenzy1)- 1-(oxetan-2-ylmethyl)- 1H-
thieno [2,3-
d]imidazole-5-carboxylate (10c)
Ot]
/ \
s CO2Me
Br 10c
[0376] 10b (500 mg, 2.06 inmol) and ld (620 mg, 2.10 mmol) were
dissolved in
glacial acetic acid (20 mL), subjected to nitrogen replacement, stirred at 45
C for 12 h, and
cooled to room temperature. The reaction liquid was concentrated under reduced
pressure,
and the residue was separated and purified by silica gel column chromatography
to obtain
the product 10c of interest (350 mg, yield 39.9%) as a yellow solid.
[0377] LCMS in/z = 439.0 [M+Hr.
Step 3: methyl 2-(2-fluoro-4- (4,4,5 -trimethyl-1,3 ,2-dioxaborolan-2-
yl)benzy1)-1- (((S )-
oxetan-2-y1) methyl)-1H-thieno[2,3-dlimidazole-5-carboxylate (10d)
0,B N
S CO2Me
10d
[0378] 10c (350 mg, 0.79 inmol) and pinacol diboronate (305 mg,
1.20 inmol),
potassium acetate (235 mg, 2.40 mmol) and Pd(dppt)C12 (65 mg, 0.08 mmol) were
added
CA 03203276 2023- 6- 22
- 80 -
to a reaction flask successively, dissolved in 1,4-dioxane solution (10 mL),
subjected to
nitrogen replacement, stirred at 90 C for 5 h, cooled to room temperature and
filtered. The
filtrate was concentrated under reduced pressure to obtain crude product 10d
as a yellow
oil, which was directly used in the next reaction without further
purification.
Step 4: methyl 2-(4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo[d][1,31dioxo1-4-
y1)-2-
fluorobenzyl)-1-(((S)-oxetan-2-yOmethyl)-1H-thieno[2,3-d]imidazole-5-
carboxylate (10e)
CI
0
J
SCOMe
10e
[0379]
The crude products 10d and 4c (344 mg, 1.01 mmol), anhydrous potassium
carbonate (230 mg, 1.60 mmol) and Pd(dppf)C12 (65 mg, 0.08 mmol) were added to
a
reaction flask successively, dissolved in a mixed solution of 1,4-dioxane (10
mL)/water (2
mL), subjected to nitrogen replacement, stirred at 90 C for 5 h and cooled to
room
temperature. the solid was filtered, the filtrate was concentrated under
reduced pressure,
and purified on a silica gel column to obtain the product 10e (350 mg, yield
71.2% over
two steps) as a yellow solid.
[0380] LCMS m/z = 623.1 [M+H]t
Step 5:
2-(4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzokfl[1,31dioxo1-4-y1)-2-
fluorobenzy1)-1-(((S)-oxetan-2-yOmethyl)-1H-thieno[2,3-d]imidazole-5-
carboxylic acid
(Compound 10)
CIF 0
(N 1
0 N
\_J, I
N--((
Compound 10S-- COOH
[0381] At room
temperature, 10e (350 mg, 0.70 mmol) was dissolved in acetonitrile
(5 mL) and water (1 mL), then lithium hydroxide (126.0 mg, 3.01 mmol) was
added and
the reaction was continued for 24 h. The reaction liquid was adjusted with 1N
hydrochloric
acid to pH=6, and then extracted with a mixed solvent of
dichloromethane:methano1=10:1
(20 mL x 3). The organic phase was combined, dried over anhydrous sodium
sulfate,
concentrated by filtration, and concentrated under reduced pressure, and the
residue was
CA 03203276 2023- 6- 22
- 81 -
separated and purified by silica gel column chromatography to obtain compound
10 (100.0
mg, yield 29.3%) as a yellow solid.
[0382] LCMS m/z = 609.2 [M+H]+.
[0383] 111 NMR (400 MHz, DMSO-d6) 6 7.73 (br, 111), 7.62-7.54
(m, 411), 7.42-7.33
(m, 2H), 7.22-7.18 (m, 2H), 7.01-6.94 (m, 2H), 5.04-4.96 (m, 1H), 4.62-4.53
(m, 1H),
4.52-4.42 (m, 211), 4.40-4.27 (m, 314), 2.73-2.61 (m, 114), 2.39-2.27 (m,
111), 2.09 (s, 311).
Example 11:
2-(4-(2-(4-chloro-2-fluoropheny1)-2-methylben zo [d] [1,3] di oxo1-4-yOben
zy1)-1-4(S)-
oxetan-2-yl)methyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 11)
ci oTh
0 /
F 0 N
0
OH
Compound 11
1(;-
CI-0-\--Ck Br Cl 'n
0 F ) 4c 0
Step 2
CI O
13 -
7d Step 1 F 0,
ha ,0
r
Compound 11 11 OH
Step 1: methyl
2-(4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo[d] [1,3] di oxo1-4-
yl)benzy1)-1-(((S)-oxetan-2-yl)methyl)-1H-thi eno [2,3-d]imidazole-5 -
carboxylate (11a)
cion
0 /
F 0 N
0
lia 0,
[0384] At room temperature, substrate 4c (91.0 mg, 0.26 mmol) was dissolved
in a
mixed solvent of 1,4-dioxane (5 mL) and water (0.5 mL), and then 7d (103.0 mg,
0.22
mmol) and Pd(dppt)C12 (18.0 mg, 0.02 minol) and cesium carbonate (215.0 mg,
0.66
minol) were added successively. After the addition, the reaction system was
heated to
100 C for overnight reaction under nitrogen protection. The reaction liquid
was cooled to
room temperature and filtered. The filter cake was washed with dichloromethane
(10 mL x
2). The filtrate was collected and concentrated to obtain a crude product,
which was
separated and purified by silica gel column chromatography to obtain product
lla of
interest (110.0 mg, yield 82.7% ) as a white solid.
CA 03203276 2023- 6- 22
- 82 -
[0385] LCMS m/z = 605.3 [M+1]+.
Step 2: 2-(4-(2-(4-chloro-2-fluoropheny1)-2-methylbenzo[d][1,3]dioxo1-4-
yObenzy1)-1-
(((S)-oxetan-2-yOmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound
11)
cI
o --
F O. \
S"-yu
OH
Compound 11
[0386] At room temperature, substrate ha (110.0 mg, 0.18 mmol) was
dissolved in a
mixed solvent of acetonitrile (5 mL) and water (1 mL), and then 1 g (CAS: 5807-
14-7)
(4.4.0 ) Dec-5-ene (23.0 mg, 0.54 mmol) was added. After the addition, the
reaction was
carried out at room temperature for 24 hours. Acetonitrile was distilled off
under reduced
pressure. The crude product was dissolved in water (5 mL), adjusted to pH=6
with 1N
hydrochloric acid. The aqueous phase was extracted with ethyl acetate (15 mL x
3). The
organic phase was combined, dried over anhydrous sodium sulfate, filtered and
concentrated to obtain a crude product, which was separated and purified by
silica gel
column chromatography to obtain compound 11(25.0 mg, yield 23.4%) as a white
solid.
[0387] LCMS m/z = 591.2 [M+1] .
[0388] 1H NMR (400 MHz, DMSO-d6) 6 12.90 (brs, 1H), 7.82 (s, 1H), 7.74 -
7.67
(m, 214), 7.62 - 7.52 (m, 214), 7.42 - 7.37 (m, 211), 7.36 - 7.32 (dd, 114),
7.16 - 7.10 (m,
1H), 6.95 (d, 2H), 4.97 -4.87 (m, 1H), 4.60- 4.52 (m, 1H), 4.51 - 4.40 (m,
2H), 4.38 -
4.28 (m, 314), 2.68 - 2.56 (m, 111), 2.35 - 2.23 (m, 111), 2.06 (s, 314).
Example 12
(S)-2-((4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-yppiperidin-1-y1)methyl)-1-
((tetrahydrofuran-2-yl)m ethyl)-1H-thi eno [2,3 -d] imi dazol e-5-carboxylic
acid (Compound
12)
NC
OH
Compound 12
CA 03203276 2023- 6- 22
- 83
NI-12 EN1 0\
BI-N{/(0 12a 1, 0 0 _____
I \ \
0 12b
Step 3 N 0
02N S Step 1 02N S Step 2 H2Iµr
\---S 0-
N S 0-
lb 12c 12d
CNN
Cr\, 0N
C\j
1 f-1
____________________ Br 4\10_40 _______________ NC
Step 4 N
) N--2<sc, Step 6
Step 5
S 0¨
12e 0,
12f
NC-, -N
N, No
`s-
OH
Compound 12
Step 1: methyl
(S)-5-nitro-4-(((tetrahydrofuran-2-yl)methyl)amino)thiophene-2-
carboxylate (12b)
H
0
02N z---S
12 b
[0389] lb (2 g,
7.55 minol), 12a (838 mg, 8.31 mmol), Pd2(dba)3 (346 ing, 0A2
mmol), t-BuBrettphos (366 mg, 036 minol) and cesium carbonate (4.92 g, 15.1
minol)
were added into a round bottom flask, and 1,4-dioxane (10 mL) was added. The
reaction
liquid was stirred at a constant temperature of 100 C for 18 hours after the
reaction flask
was subjected to nitrogen replacement for protection. After the reaction was
completed, the
reaction liquid was cooled to room temperature. 20 mL of water was added, then
extracted
with ethyl acetate (20 mL x 3). The organic phase was washed with aqueous
saturated
sodium chloride solution, then dried over anhydrous sodium sulfate, filtered
and spin-
dried. The residue was separated and purified by silica gel column
chromatography to
obtain 12b (L6 g. yield 74%) as a light yellow solid.
[0390] LCMS in/z = 287.0 [M+1]+.
Step 2: methyl
(S )-5-amino-4-(((tetrahydrofuran-2-yl)methyeamino)thiophene-2-
carboxylate (12c)
CA 03203276 2023- 6- 22
- 84
H
0 N 0
H2N S
12c
[0391] Raney nickel (60 mg) was added to Me0H (2 mL) and THF (2
mL)
containing 12b (200 mg, 0.70 mmol). The reaction liquid was stirred at room
temperature
for 1 h after the reaction flask was subjected to hydrogen replacement. After
the reaction
was completed, the reaction liquid was filtered over celite and spin-dried,
and the obtained
crude product 12c was directly used in the next step.
Step 3: methyl (S)-2-methy1-1-((tetrahydrofuran-2-ypmethyl)-1H-thieno [2, 3-d]
imidazole-
-carboxylate (12d)
/<0
0-
12d
[0392] At room temperature, 12c was added to acetonitrile (5 mL), and
ytterbium
trifluoromethanesulfonate (11 mg, 0.02 mmol) and trimethyl orthoacetate (63
mg, 0.52
mmol) were added to the reaction liquid successively, and the temperature was
raised to
70 C for reaction for 2 hours. The reaction liquid was cooled to room
temperature. Glacial
acetic acid was distilled off under reduced pressure, and the residue was
separated and
purified by silica gel column chromatography to obtain 12d (50 mg, yield 26%
over two
step).
[0393] LCMS tn/z = 281.1 [M+1]+.
Step 4: methyl (S)-2-(bromomethyl)-1-((tetrahydrofuran-2-y1) methyl)-1H-thieno
[2,3-
d]imidazole-5-carboxylate (12e)
Br N
(\ \>
0 ¨
12e
CA 03203276 2023- 6- 22
- 85 -
[0394] At room temperature, 12d (50 mg, 0.18 mmol) was dissolved
in 1,2-
dichloroethane (2 mL) solution, then AIBN (3.2 mg, 0.02 mmol) was added, and
NBS(80.1
g, 0.45 mmol) was added in portions in the process of slowly raising the
temperature to
50 C. After the addition, the reaction liquid was heated to 70 C and continued
to react for
8 h. The reaction liquid was cooled to room temperature, saturated sodium
thiosulfate
solution (5 mL) was added to quench the reaction and extracted with
dichloromethane (5
niL x 3). The organic phase was combined, dried over anhydrous sodium sulfate,
filtered
and concentrated to obtain a crude product. The residue was separated and
purified by
silica gel column chromatography to obtain 12e (26 mg, yield 40%) as a dark
yellow solid.
[0395] LCMS m/z = 359.0 [M+1[+.
Step 5: methyl (S)-24(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-yppiperidin-1-
y1)methyl)-1-((tetrahydrofuran-2-y1)methyl)-1H-thieno[2,3-d]imidazole-5-
carboxylate
(120
NC
/
0 N N
0
121
[0396] At room temperature, 12e (26 ing, 0.07 tntnol) was dissolved in
acetonitrile (1
mL) solution, then potassium carbonate (29.0 mg, .21 mmol) and lf-1 (24.8 mg,
0.08
mmol) were added, stirred at room temperature for 18 h. The reaction liquid
was filtered to
remove the inorganic base, and the filtrate was collected and concentrated to
obtain a crude
product, which was separated and purified by silica gel column chromatography
to obtain
12f (20 mg, yield 49%) as a yellow solid.
[0397] LCMS ink = 590.3 1M+11h.
Step 6: (S)-24(4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-yppiperidin-l-
yOmethyl)-1-
((tetrahydrofuran-2-yemethyl)-1H-thieno[2.3-d]imidazole-5-carboxylic acid
(Compound
12)
CA 03203276 2023- 6- 22
- 86 -
(OD)
NC
0 N
0
OH
Compound 12
[0398] At room temperature, 12f (20ing, 0.03 minol) was
dissolved in a solution of
acetonitrile (1 inL) and water (0.2 inL), then 1 g (CAS: 5807-14-7) (20.9 mg,
0.15 mmol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
1N hydrochloric acid to pH=6, and then extracted with a mixed solvent of
dichloromethane:methano1=10:1 (20 mL x 3). The organic phase was combined,
dried over
anhydrous sodium sulfate, filtered and concentrated. The obtained crude
product was
subjected to prep-HPLC to obtain compound 12 (5.0 mg, yield 29 %) as a white
solid.
[0399] Preparation conditions:
[0400] instrument and preparative column: waters 2767 (preparative liquid
phase
chromatographic instrument) was used; the preparative column model was
SunFire@Prep
C18 (19 mm x 250 mm).
[0401] Preparation method: The crude product was dissolved in
DMF and filtered
with a 0.45 pin filter membrane to prepare into a sample liquid.
[0402] Mobile phase system: acetonitrile/water (containing 1% TFA).
Gradient
elution: acetonitrile content 20-65%, elution flow rate: 12 inUmin, elution
time 18 min.
[0403] LCMS in/z = 576.3 [M+1]+.
[0404] 114 NMR (400 MHz, CD30D) 8 7.87 (s, 1H), 7.68 (t, 2H),
7.57 (t, 2H), 6.93
(d, 1H), 6.78 (d, 1H), 5.54 (s, 2H), 4.76-4.62 (in, 2H), 4.62-4.56 (m, 1H),
4.40-4.33 (in,
1H), 4.24-4.15 (in, 1H), 3.95-3.73 (in, 4H), 3.42-3.36 (in, 1H), 3.09-2.98
(in, 1H), 2.27-
2.09 (in, 5H), 2.03-1.81 (in, 3H), 1.69-1.58 (in, 1H).
Example 13
(S)-2-44-(64(4-cy ano-2-fluorobenzypoxy)pyridin-2-yepiperazin-l-yl)methyl)- 1-
(oxetan-
2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-c arboxylic acid
CA 03203276 2023- 6- 22
NC - 87 -
F 0 N71\1) N OH
0
Compound 13
HINnN
1`3, Boc
rro N CI ____________________________
frO N NON _______________________________________________________ Cy N' -)
NCF3 Step 1 NCF Boc Step 2 NC-F
KNH
13b 13c
C1-i)
BrNjz
2bS On)
NC
Step 3
ouN, \ 0- step 4
F
N \ OH
0
13d
Compound 13 0
Step 1: tert-butyl 4-(6-((4-cyan o-2-fluorob en zyl)oxy)pyri din-2-yl)pi
perazi n e-1-carboxyl ate
(13b)
0
NCF
NBoc
13b
[0405] 3a (1 g, 3.82 mmol), 13a (781 mg, 4.20 mmol), Pd2(dba)3
(174 mg, 0.19
mmol), RuPhos (178 mg, 0.38 mmol) and cesium carbonate (2.48 g, 7.64 mmol)
were
added into a round bottom flask, and 1,4-dioxane (10 mL) was added. The
reaction liquid
was stirred at a constant temperature of 100 C for 18 hours after the reaction
flask was
subjected to nitrogen replacement for protection. After the reaction was
completed, the
reaction liquid was cooled to room temperature. 20 mL of water was added, then
extracted
with ethyl acetate (20 mL x 3). The organic phase was washed with aqueous
saturated
NaC1 solution, then dried over anhydrous sodium sulfate, filtered and spin-
dried. The
obtained crude product was separated and purified by silica gel column
chromatography to
obtain 13b (700 mg, yield 44%) as a light yellow solid.
[0406] LCMS m/z = 357.1 [M-55] .
Step 2: 3-fluoro-4-(((6-(piperazin-1-yl)pyridin-2-ypoxy)methypbenzonitrile
(13c)
CA 03203276 2023- 6- 22
- 88
N
NC H
13c
[0407] 13b (100 mg, 0.24 mmol) was added into a round bottom
flask, then
tritluoroacetic acid (0.5 inL) and dichloromethane (1 inL) were added and
stirred at room
temperature for 5 h. After the reaction was completed, the reaction liquid was
directly
concentrated under reduced pressure to obtain 13c (90 mg, yield 92%) as a
colorless oil. It
was directly used in the next step without further purification.
[0408] LCMS m/z = 313.1 [M+1]+.
Step 3: methyl (S)-24(4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-yepiperazin-l-
yOmethyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-carboxylate
(13d)
NC
11\11\-N
F 0 N,N) N
0
13d
[0409] At room temperature, 13c (50 mg, 0.16 mmol) was dissolved
in acetonitrile (2
mL) solution, then potassium carbonate (66.3 mg, 0.48 mmol) and id-1 (55.0 mg,
0.16
mmol) were added, stirred at room temperature for 2 h. The reaction liquid was
filtered to
remove the inorganic base, and the filtrate was collected and concentrated to
obtain a crude
product, which was separated and purified by silica gel column chromatography
to obtain
13d (50 mg, yield 54%) as a yellow solid.
[0410] LCMS in/z = 577.2 [M+1] .
Step 4: (S)-24(4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-yl)piperazin-l-
yOmethyl)-1-
(oxetan-2-ylinethyl)-1H-thieno [2,3 -d] imidazole-5-c arboxylic acid (Compound
13)
NC
F 0 1µ1,7N1) N
OH
0
Compound 13
CA 03203276 2023- 6- 22
- 89 -
[0411] At room temperature, 13d (50 mg, 0.09 mmol) was dissolved
in a solution of
acetonitrile (1 mL) and water (0.2 mL), then 1 g (CAS: 5807-14-7) (62.6 mg,
0.45 mmol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
1N hydrochloric acid to pH=6, and then extracted with
dichloromethane:methano1=10:1
(20 mL x 3). The organic phase was combined, dried over anhydrous sodium
sulfate,
filtered and concentrated. The obtained crude product was subjected to prep-
HPLC to
obtain compound 13 (30 mg, yield 59 %) as a white solid.
[0412] Preparation conditions:
[0413] instrument and preparative column: waters 2767
(preparative liquid phase
chromatographic instrument) was used; the preparative column model was
SunFire@Prep
C18 (19 mm x 250 mm).
[0414] Preparation method: The crude product was dissolved in
DMF and filtered
with a 0.45 !..tm filter membrane to prepare into a sample liquid.
[0415] Mobile phase system: acetonitrile/water (containing 1%
TFA). Gradient
elution: acetonitrile content 20-65%, elution flow rate: 12 mL/min, elution
time 18 min.
[0416] LCMS m/z = 563.2 [M+11+.
[0417] 111 NMR (400 MHz, DMSO-d6) 6 13.17 (brs, 114), 7.96 (s,
111), 7.88 (d, 111),
7.74-7.65 (m, 2H), 7.56 (t, 1H), 6.45 (d, 1H), 6.23 (d, 1H), 5.42 (s, 2H),
5.07-4.97 (m, 1H),
4.75-4.66 (m, 111), 4.63-4.46 (m, 411), 4.38-4.31 (m, 111), 4.06-3.52 (m,
411), 3.43-3.15 (m,
4H), 2.79-2.64 (m, 1H), 2.37-2.26 (m, 1H).
Example 14
(S)-24(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-1,4-diazepan-1-yOmethyl)-
1-
(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 14)
N-_¨\ 0
/ \\
NCT NS
OH
Compound 14
CA 03203276 2023- 6- 22
- 90-
O
NC
_NBoc
Boc alb NH
NC Br N S 0¨ 2b
' a N CI [11(1 14a I 0 N 2 _____________________
IMF 0INTN, j
3a 14b 14c
N 0 N- 0
/--\\
NC
(¨N N S 0¨
/¨N N S OH
0 N, ,NO
NN
1 j
14d
Compound 14
Step 1: tert-butyl 4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-y1)-1,4-diazepane-
1-
carboxylate (14b)
,Bob
NC
0 N
14b
[0418] At room temperature, substrate 3a (393.0 mg, 1.5 mmol) was dissolved
in 1,4-
dioxane (25 inL), and then 14a (301.0 mg, 1.5 mmol), Pd2(dba)3 (138.0 mg, 0.15
mmol),
Ru-Phos (140.0 mg, 0.3 mmol) and cesium carbonate (1.5 g, 4.5 minol) were
added
successively. After addition, the reaction system was heated to 100 C under
the protection
of nitrogen for overnight reaction. The reaction liquid was cooled to room
temperature,
filtered to remove inorganic salts, and concentrated to obtain a crude
product, which was
separated and purified by silica gel column chromatography to obtain the
compound 14b of
interest (462.0 ing, yield 72.3%).
[0419] LCMS in/z = 427.2 [M+1]+.
Step 2: 4-(((6-(1,4 -Diazep an-l-yppyridin-2-ypoxy)methyl)- 3 -fluorob
enzonitrile (14c)
NC /NH
0 N
14c
[0420] At room temperature, the substrate 14b (462.0 mg, 1.1
mmol) was dissolved in
dichloromethane (20 inL), and then trifluoroacetic acid (4 inL) was added
dropwise to the
reaction system. After the dropwise addition, the reaction was carried out at
room
temperature for 2 hours. The reaction liquid was concentrated to obtain the
crude product
CA 03203276 2023- 6- 22
- 91 -
14c as a brown viscous liquid, which was directly used in the next reaction
without any
purification.
[0421] LCMS m/z = 327.1 [M+1] .
Step 3: methyl (S)-2-((4-(64(4-cyano-2-fluorobenzypoxy)pyridin-2-y1)-1,4-
diazepan-1-
yl)methyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d]imidazole-5-carboxyl ate
(14d)
0
\>
NC N"----S 0-
0 N,
14d
[0422] At room temperature, the substrate 14c (115.0 mg, 0.35
mmol) was dissolved
in acetonitrile (5 inL), then 2b (121.0 mg, 0.35 mmol) and potassium carbonate
(244.0 mg,
1.8 mmol) were added successively. After the addition, the reaction system was
reacted at
room temperature for 4 hours. The reaction liquid was filtered, and the filter
cake was
washed with dichloromethane (10 mL x 2). The filtrate was collected and
concentrated to
obtain a crude product, which was separated and purified by silica gel column
chromatography to obtain 3d (138.0 mg, yield 66.5%) as a white solid.
[0423] LCMS m/z = 591.3 [M+1]+.
Step 4: (S)-2-44-
(6-((4-cy ano-2-fluorobenzypoxy)pyridin-2-y1)-1,4-diazep an-1-
yOmethyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-carboxylic
acid
(Compound 14)
(c>"1
NC
N
K
N -------S OH
0õNõNIJõ
Compound 14
[0424] At room temperature, substrate 14d (138.0 mg, 0.2 mmol)
was dissolved in a
mixed solvent of acetonitrile (5 mL) and water (1 mL), and then 1 g (CAS: 5807-
14-7)
(98.0 mg, 0.7 mmol) was added. After the addition, the reaction was carried
out at room
temperature for 24 hours. Acetonitrile was distilled off under reduced
pressure. The crude
product was dissolved in water (5 mL), adjusted to pH=6 with 1N hydrochloric
acid. The
CA 03203276 2023- 6- 22
- 92 -
aqueous phase was extracted with ethyl acetate (15 mL x 3). The organic phase
was
combined, dried over anhydrous sodium sulfate, filtered and concentrated to
obtain a crude
product, which was separated and purified by silica gel column chromatography
to obtain
compound 14 (66.0 mg, yield 48.9%) as a white solid.
[0425] LCMS m/z = 577.2 [M+1]+.
[0426] 11-1 NMR (400 MHz, DMSO-d6) 6 12.94 (brs, 114), 7.87 ¨
7.80 (m, 111), 7.77
(s, 1H), 7.68¨ 7.63 (m, 1H), 7.62 ¨ 7.56 (m, 1H), 7.43 (t, 1H), 6.16 (d, 1H),
6.05 (d, 1H),
5.36 (s, 2H), 4.99 ¨ 4.92 (m, 1H), 4.53 ¨ 4.40 (m, 2H), 4.38 ¨ 4.28 (m, 2H),
3.87-3.74 (m,
21-1), 3.61 (t, 211), 3.51 (t, 21-1), 2.70¨ 2.62 (m, 211), 2.60¨ 2.52 (m,
311), 2.34 ¨ 2.20 (m,
1H), 1.75¨ 1.66 (m, 2H).
Example 15
(S)-24(4-(24(4-cyano-2-fluorobenzypoxy)pyridin-3-yppiperidin-l-ypmethyl)-1-
(oxetan-
2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 15)
CN
FH
0 N /
N
N OH
0
Compound 15
0 / Boc Boc
Boc-N1BL N N¨\
0
N A 15a-1
1_0\yc N
F \ ___________ 0)0
Br ¨ Step 1 NC N¨ Step 2 Njc__/ 'NI¨
Step 3
15a 15b
15c
N CN
HN¨ Br , CN
¨1
N S \ I
0 0-
2b 0 F "IN
0¨)
) \
NC 41, \N¨ Step 4 Step 5 Nb7C
N4s0H
15d 0
15e 0
Compound 15
Step 1: tert-butyl 24(4-cyano-2-fluorobenzypoxy)-3',6'-dihydro-[3,4'-
bipyridine]-1'(2'14)-
carboxylate (15b)
CA 03203276 2023- 6- 22
- 93 -
Boo,
¨
F
0 /
NC N-
15b
[0427] 15a-1 (504.9 mg, L63 tnmol), 15a (500 mg, 1.63 mmol),
Pd(dppf)C12 (133
mg, 016 mmol) and cesium carbonate (1.06 g, 126 mmol) were added into a round
bottom flask, and 1,4-dioxane (10 mL) was added. The reaction liquid was
stirred at a
constant temperature of 100 C for 18 hours after the reaction flask was
subjected to
nitrogen replacement for protection. After the reaction was completed, the
reaction liquid
was cooled to room temperature. 10 inL of water was added, then extracted with
ethyl
acetate (6 inL x 3). The organic phase was washed with aqueous saturated NaCl
solution,
then dried over anhydrous sodium sulfate and filtered. The filtrate was
concentrated to
dryness. The obtained crude product was separated and purified by silica gel
column
chromatography to obtain 15b (600 mg, yield 90%) as a light yellow solid.
[0428] LCMS tn/z = 410.2 [M+1] .
Step 2: tert-butyl 4-(24(4-cyano-2-fluorobenzypoxy)pyridin-3-yl)piperidine-1-c
arboxylate
(15c)
Boc
0 /
NCN-
15c
[0429] Palladium on carbon (60 mg) was added to tetrahydrofuran
(10 inL)
containing 15b (600 mg, 1.47 mmol). The reaction liquid was stirred at room
temperature
for 18 h after the reaction flask was subjected to hydrogen replacement. After
the reaction
was completed, the reaction liquid was filtered over celite, and the liquid
was concentrated
to dryness. The crude product was separated and purified by silica gel column
chromatography to obtain 15c (450 mg, yield 74%) as a light yellow solid.
[0430] LCMS in/z = 412.1 [M+1]+.
Step 3: 3-fluoro-4-(((3-(piperidin-4-yl)pyridin-2-yl)oxy)methyl)benzonitrile
(15d)
CA 03203276 2023- 6- 22
- 94 -
HN_
0 /
NC N-
15d
[0431] 15c (100 mg, 0.24 mmol) was added into a round bottom
flask, then
tritluoroacetic acid (0.5 inL) and dichloromethane (1 inL) were added and
stirred at room
temperature for 5 h. After the reaction was completed, the reaction liquid was
directly
concentrated under reduced pressure to obtain 15d (90 mg, yield 92%) as a
colorless oil. It
was directly used in the next step without further purification.
[0432] LCMS in/z = 312.1 [M+1]+.
Step 4: methyl (S)-24(4-(24(4-cyano-2-fluorobenzyl)oxy)pyridin-3-yepiperidin-1-
yOmethyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-carboxylate
(15e)
CN
FH
N \ 0
N
0
15e
[0433] At room temperature, 15d (25 mg, 0.08 minol) was
dissolved in acetonitrile (1
inL) solution, then potassium carbonate (33.2 mg, 0.24 mmol) and 2b (27.5 mg,
0.08
inmol) were added, stirred at room temperature for 5 h. The reaction liquid
was filtered to
remove the inorganic base, and the filtrate was collected and concentrated to
obtain a crude
product, which was separated and purified by silica gel column chromatography
to obtain
15e (20 mg, yield 43%) as a brown solid.
[0434] LCMS in/z = 576.2 [M+1]+.
Step 5: (S)-24(4-(24(4-cyano-2-fluorobenzypoxy)pyridin-3-yppiperidin-l-
ypinethyl)-1-
(oxetan-2-ylinethyl)-1H-thieno [2,3 -d] imid azole-5-c arboxylic acid
(Compound 15)
CA 03203276 2023- 6- 22
- 95 -
CN
0 N
N
N OH
I z 0
Compound 15
[0435] At room temperature, 15e (20ing, 0.03 mmol) was dissolved
in a solution of
acetonitrile (1 mL) and water (0.2 naL), then 1 g (CAS: 5807-14-7) (20.9 mg,
0.15 minol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
1N hydrochloric acid to pH=6, and then extracted with a mixed solvent of
dichloromethaneanethano1=10:1 (20 mL x 3). The organic phase was combined,
dried over
anhydrous sodium sulfate, filtered and concentrated. The obtained crude
product was
subjected to prep-HPLC to obtain compound 15 (6.0 mg, yield 32%) as a white
solid.
[0436] Preparation conditions:
[0437] instrument and preparative column: waters 2767 (preparative liquid
phase
chromatographic instrument) was used; the preparative column model was
SunFire@Prep
C18 (19 mm x 250 mm).
[0438] Preparation method: The crude product was dissolved in
DMF and filtered
with a 0.45 [fin filter membrane to prepare into a sample liquid.
[0439] Mobile phase system: acetonitrile/water (containing 1% TFA).
Gradient
elution: acetonitrile content 20-65%, elution flow rate: 12 inL/min, elution
time 18 min.
[0440] Compound 15:
[0441] LCMS in/z = 562.2 [M+1]+.
[0442] 11-1 NMR (400 MHz, DMSO-d6) 8 13.22 (brs, 1H), 8.07-8.04
(in, 1H), 7.98 (s,
1H), 7.91 (d, 1H), 7.74-7.67 (in, 2H), 7.63-7.56 (in, 1H), 7.08-7.02 (in, 1H),
5.53 (s, 2H),
5.06-4.98 (in, 1H), 4.77-4.63 (in, 3H), 4.63-4.56 (in, 1H), 4.54-4.47 (in,
1H), 4.38-4.31 (in,
1H), 3.74-3.64 (in, 2H), 3.36-3.23 (in, 2H), 3.15-3.04 (in, 1H), 2.77-2.66
(in, 1H), 2.37-
2.26 (in, 1H), 2.09-1.99 (in, 2H), 1.96-1.84 (in, 2H).
Example 16
(S)-2-44-(64(4-cy ano-2-fluorobenzypoxy)pyridin-2-yepiperidin- 1-yl)methyl)-3-
(oxetan-
2-ylinethyl)-3H- thieno [2,3-d] imidazole-5-c arboxylic acid
CA 03203276 2023- 6- 22
- 96 -
C;,--
NC N
0
1
F -
OH
Compound 16
CI'C)Me
rOMe
\--\ OMe
02N
16a
01NH2 02N, 0 H2N 0 le-1 , 0 lc-
1
0¨ Step 2 f____("N s o¨
Step 3
CIXSO¨ Step 1
L-0 H 16b Lo H 16c
NC-NH
N S 0 F lf-1 NC,2 1 /¨K Isl-A Io ,
N, N ' ' Step 5
CI N Step 4 --1,f0
16d F -- 0-
16e
0--i
NO IN( ,01'S
--C .--- 0
F 1;
6H
Compound 16
Step 1: methyl (S)-4-nitro-5-((oxetan-2-ylmethyl)amino)thiophene-2-carboxylate
(16b)
02N , o
1 ________________________________________________
F---N---s O¨
H
L----0
16b
[0443] At room temperature, 16a (2 g, 9.0 mmol) was dissolved in
acetonitrile (50
triL) solution, then potassium carbonate (3.73 g, 27.0 inmol) and lc-1 (783
mg, 9.0 mmol)
were added, and the temperature was raised to 70 C for 18 h. The reaction
liquid was
filtered to remove solids, and the filtrate was collected and concentrated to
obtain a crude
product, which was separated and purified by silica gel column chromatography
to obtain
16b (2 g, yield 83%).
[0444] LCMS in/z = 273.1 [M+1]+.
Step 2: methyl (S)-4-amino-5-((oxetan-2-ylmethyl)amino)thiophene-2-carboxylate
(16c)
CA 03203276 2023- 6- 22
- 97 -
H2N 0
0
16c
[0445] Zinc powder (L2 g, 18.4 inmol) was added to acetic acid
(20 inL) containing
16b (1.0 g, 3.68 mmol), and the reaction liquid was stirred at room
temperature for 1 h.
After the reaction was completed, the reaction liquid was filtered over
celite, and the
filtrate was concentrated to dryness. The residue was separated and purified
by silica gel
column chromatography to obtain 16c (700 mg, yield 79%) as a light yellow
solid.
[0446] LCMS in/z = 243.1 [M+1]+.
Step 3: methyl (S)-2-(chloroinethyl)-3 -(oxetan-2-ylinethyl)-3H-thieno [2,3 -
(1] imidazole-5-
carboxylate (16d)
0
CI Nj)_
0¨
16d
[0447] At room temperature, 16c (700 mg, 2.89 ininol) was added
to acetonitrile (20
inL), and le-1 (668.2 ing, 4.34 -mime and p-toluenesulfonic acid (24.9 ing,
0.14 ininol)
were added to the reaction liquid successively, heated to 60 C for 5 hours.
Acetonitrile was
distilled off under reduced pressure, and the obtained crude product was
purified by silica
gel column chromatography to obtain 16d (45 ing, yield 5.2%) as a yellow
solid.
[0448] LCMS in/z = 301.1 [M+1]+.
Step 4: methyl (S)-24(4-(64(4-cyano-2-fluorobenzyl)oxy)pyridin-2-yepiperidin-l-
yl)methyl)-3-(oxetan-2-ylmethyl)-3H-thieno[2,3-(11imidazole-5-carboxylate
(16e)
NC
/ S
--- 0
0¨
16e
[0449] At room temperature, 16d (45 ing, 0.15 minol) was dissolved in
acetonitrile (2
inL) solution, then potassium carbonate (62.1 mg, 0.45 mmol) and if-1 (46.5
mg, 0.15
CA 03203276 2023- 6- 22
- 98 -
mmol) were added, stirred at room temperature for 18 h. The reaction liquid
was filtered to
remove solids, and the filtrate was collected and concentrated to obtain a
crude product,
which was separated and purified by silica gel column chromatography to obtain
16e (50
mg, yield 58%) as a yellow solid.
[0450] LCMS m/z = 576.2 1M+11+.
Step 5: (S)-2-((4-(64(4-cyano-2-fluorobenzypoxy)pyri din-2-y] ipiperi din -1-
yl)methyl)-3-
(oxetan-2-y1 methyl)-3H-thi eno 12,3-dlimidazole-5-carboxylic acid (Compound
16)
o
NC
s
0 N '
---' 0
OH
Compound 16
[0451] At room temperature, 16e (40 mg, 0.07 mmol) was dissolved
in a solution of
acetonitrile (1 inL) and water (0.2 inL), then 1 g (CAS: 5807-14-7) (483 mg,
0.35 mmol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
1N hydrochloric acid to pH=6, and then extracted with a mixed solvent of
dichloromethane:methano1=10:1 (20 mL x 3). The organic phase was combined,
dried over
anhydrous sodium sulfate, filtered and concentrated. The obtained crude
product was
subjected to prep-HPLC to obtain compound 16 (20 mg, yield 51 %) as a white
solid.
[0452] Preparation conditions:
[0453] instrument and preparative column: waters 2767
(preparative liquid phase
chromatographic instrument) was used; the preparative column model was
SunFire@Prep
C18 (19 mm x 250 mm).
[0454] Preparation method: The crude product was dissolved in DMF and
filtered
with a 0.45 [lin filter membrane to prepare into a sample liquid.
[0455] Mobile phase system: acetonitrile/water (containing 1%
TFA). Gradient
elution: acetonitrile content 20-65%, elution flow rate: 12 inUmin, elution
time 18 min
[0456] LCMS in/z = 562.3 [M+1]+.
[0457] 114 NMR (400 MHz, DMSO-d6) 8 13A5 (brs, 1H), 7.89 (d, 1H), 7.86 (s,
1H),
7.76-7.69 (in, 3H), 6.98-6.91 (in, 1H), 6.79 (d, 1H), 5A9 (s, 2H), 5.14-5.06
(in, 1H), 4.69
CA 03203276 2023- 6- 22
- 99 -
(s, 2H), 4.66-4.60 (m, 1H), 4.57-4.49 (m, 2H), 4.40-4.33 (m, 1H), 3.80-3.65
(m, 2H), 3.32-
3.19 (m, 214), 2.96-2.86 (m, 111), 2.80-2.70 (m, 114), 2.42-2.31 (m, 111),
2.12-1.93 (m, 411).
Example 17
(S)-2-((4-(6-((4-cyano-2-fluorobenzyl)oxy)-5-fluoropyridin-2-yppiperidin-1-
yOmethyl)-1-
(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 17)
O¨L7
NC
0 N
0
OH
Compound 17
,N Boc ^N Bac Boc step 3 Boc
__________________________________________ HO
Step 1 Step 2
N N, N, 1_J
F F
17a 17b 17c
17d
o
(5])N
NC
, H Step 5 Step 6 NC
_,N,Tv
___________________ WI 0 N
Step 4
F F F- OH
17e 17f Compound 17
Step 1: 2-(1-(tert-butoxycarbonyl)piperidin-4-y1)-5-fluoropyridine1-oxide
(17b)
Boc
17b
[0458] At room temperature, in-chloroperoxybenzoic acid (3.08 g, 17.82
minol) was
added to a solution of 17a (2.00 g, 7.13 inmol) in dichloromethane (20 ml),
and then stirred
and reacted at room temperature for 14 hours. The insolubles were removed by
filtration,
the liquid was washed with an equal volume of water, dried over anhydrous
sodium sulfate
and concentrated to dryness, and the residue was separated and purified by
silica gel
column chromatography to obtain the product 17b of interest (1.20 g, yield:
56.8%).
Step 2: tert-buty14-(5-fluoro-6-hydroxypyridin-2-yppiperidine-1-carboxylate
(17c)
CA 03203276 2023- 6- 22
- 100 -
N,Boc
HO N
17c
[0459] Under ice-water bath, triethylamine (0.82 g, 8.10 mmol)
was added to a
solution of 17b (L20 g, 4.05 mmol) and trifluoroacetic anhydride (8.51 g, 40.5
mmol) in
tetrahydrofuran (20 ml). After one hour, the temperature was raised to room
temperature to
react for 14 hours. The reaction liquid was concentrated to dryness under
reduced pressure,
and the residue was separated and purified by column chromatography to
obtainl7c (0.8 g,
yield: 66.6%).
[0460] LCMS in/z=241.1 [M-55] +.
Step 3: tert-butyl 4-(6-((4-cyano-2-fluorobenzypoxy)-5-fluoropyridin-2-
yl)piperidine-1-
carboxylate (17d)
NC -1\1,Boc
0 N
,
17d
[0461] At room temperature, tri-n-butylphosphine (513 mg, 2.54
mmol), diisopropyl
azodicarboxylate (513 mg, 2.54 mmol) were added to a solution of 17c (0.5 g,
1.69 minol)
and 3-Fluoro-4-(hydroxymethyebenzonitrile (383 mg, 2.54 mmol) in toluene (5
me. The
temperature was raised to 80 C to react for 14 hours. The reaction liquid was
concentrated
under reduced pressure. The residue was separated and purified by silica gel
column
chromatography to obtain 17d (0.2 g, yield: 27.6%).
[0462] LCMS in/z=374.1[M-55] .
Step 4: 3-fluoro-4-(43-fluoro-6-(piperidin-4-yl)pyridin-2-
yl)oxy)tnethyl)benzonitrile (17e)
NC
NH
F
17e
CA 03203276 2023- 6- 22
- 101 -
[0463] At room temperature, trifluoroacetic acid (2 ml) was
added to a solution of
17d (0.2 g, 0.47 mmol) in dichloromethane (2 ml), and then reacted at room
temperature
for 3 hours. The reaction liquid was concentrated to dryness under reduced
pressure, and
the residue was separated and purified by column chromatography to obtain 17e
(0.1 g,
yield: 64.60%).
Step 5: methyl (S)-24(4-(6-((4-cyano-2-fluorobenzypoxy)-5-fluoropyridin-2-
yl)piperidin-
l-yl)methyl)-1-(oxetan-2-ylmethyl)-1H-thieno[2,3-dlimidazole-5-carboxylate
(170
0 m
NC
0 N N
F 00
17f
[0464] At room temperature, potassium carbonate (82.93 mg, 0.60
mmol) was added
to a solution of 17e (0.1 g, 0.30 mmol) and 2b (103.56 mg, 0.30 mmol) in
acetonitrile (10
ml), and then reacted at room temperature for 4 hours. The solid was removed
by filtration,
and the liquid was concentrated to dryness to obtain 17f. The crude product
yield was
calculated as 100% and the next step was directly carried out.
[0465] LCMS iniz=594.2(M+1) .
Step 6: (S)-24(4-(6-((4-cyano-2-fluorobenzypoxy)-5-fluoropyridin-2-yepiperidin-
l-
yOmethyl)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-carboxylic
acid
(Compound 17)
on
NC
0 N
0
F OH
Compound 17
[0466] At room temperature, lg (CAS: 5807-14-7) (139.2 mg, 1.00
mmol) was added
to a mixed solution of 17f (0.15 g, 0.25 mmol) in acetonitrile (5 ml) and
water (5 ml) and
then reacted at room temperature for 14 hours. The reaction liquid was
concentrated to
obtain a crude product. The crude product was purified by Pre-HPLC (instrument
and
CA 03203276 2023- 6- 22
- 102 -
preparative column: Waters 2767 (preparative liquid phase chromatographic
instrument)
was used, and the preparative column model was Sunfire@PrepC18, 5 [im, inner
diameter
x length = 19 mm x 250 mm). Preparation method: The crude product was
dissolved in
DMF and filtered with a 0.45 [tm filter membrane to prepare into a sample
liquid. Mobile
phase system: acetonitrile/water (containing 0.5% ammonium bicarbonate).
Gradient
elution method: gradient elution with acetonitrile from 5% to 50% (flow rate:
15 mL/min;
elution time 15 min), compound 17 (0.05 g, yield: 34.51%) was obtained.
[0467] LCMS m/7 = 580.2[M+1] .
[0468] 111 NMR (400 MHz,CD30D) 6 7.69 (t, 111), 7.65 (s, 111),
7.63-7.53 (m, 211),
7.45-7.38 (m, 1H), 6.87-6.83(m, 1H), 5.59 (s, 2H), 5.26-5.18 (m, 1H), 4.72-
4.54(m, 3H),
4.47-4.39 (m, 111), 4.12-4.00 (m, 211), 3.28-3.16 (m, 211), 2.82-2.69 (m,
211), 2.59-2.44 (m,
3H), 1.95-1.82 (m, 4H).
Example 18
(S)-2-(4-(644-cyano-2-fluorobenzypoxy)-5-fluoropyridin-2-y1)-2-fluorobenzy1)-1-
(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 18)
OraN
NC
T I0 N
0
F OH
Compound 18
F
NC'C T: OH
002Me
18b NC 0.B1 NICS-
0 10d
F
CI, Br _______________________________________________________ oth
111-P 0 N Br ___________________________________________
Step 1 F Fp' Step 2
18a 18c
18d
Step 3 NC 0 N
F 'T-C)
OH
Compound 18
Step 1: 44(6-bromo-3-fluoropyridin-2-yl)oxy)methyl)-3-fluorobenzonitrile (18c)
CA 03203276 2023- 6- 22
- 103 -
NC
0 N Br
F
18c
[0469] Under ice bath, 18a (278 mg, L32 mmol) was added to a
solution of 18b (200
mg, L32 mmol) and potassium tert-butoxide (22L8 mg, L98 mmol) in
tetrahydrofuran (6
ml) and reacted at room temperature for 3 hours. The insolubles were removed
by
filtration, the liquid was washed once with aqueous saturated ammonium
chloride solution
(6 ml), dried over anhydrous sodium sulfate, concentrated to dryness, and the
residue was
separated and purified by silica gel column chromatography to obtain the
product 18c of
interest (250 mg, yield: 58.5%).
Step 2: methyl (S)-2-(4-(64(4-cy ano -2-fluorobenzyl)oxy)-5-
fluoropyridin-2-y1)-2-
fluorobenzy1)- 1-(oxetan-2-ylmethyl)- 1H-thieno [2,3-d] imidazole-5 -c arboxy
late (18d)
FO
NC
/
0 N
0
18d
[0470] The crude products 10d and 18c (178 mg, 0.55 minol),
anhydrous potassium
carbonate (115 ingõ 0.80 mmol) and Pd(dppf)C12 (32.5 mg, 0.04 mmol) were added
to a
reaction flask successively, dissolved in a mixed solution of 1,4-dioxane
(5mL)/water
(1mL), subjected to nitrogen replacement, stirred at 90 C for 5 h and filtered
to remove
solids. The filtrate was concentrated under reduced pressure, and the residue
was separated
and purified by silica gel column chromatography to obtain the product 18b
(140 mg, yield
42.1% over two steps) as a yellow solid.
[0471] LCMS in/z = 605.2 [M+Hr.
Step 3: (S)-2-(4-(64(4-cyano-2-fluorobenzypoxy)-5-fluoropyridin-2-y1)-2-
f1uorobenzy1)-
1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-c arboxylic acid (Compound
18)
CA 03203276 2023- 6- 22
- 104 -
F
NC
/
0 N N
0
OH
Compound 18
[0472] At room temperature, 18b (80 mg, 0.13 mmol) was dissolved
in a solution of
acetonitrile (3 tnL) and water (0.6 mL), then 1 g (CAS: 5807-14-7) (54.2 mg,
0.39 mmol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
1N hydrochloric acid to pH=6, and then extracted with a mixed solvent of
dichloromethane:methano1=10:1 (20 mL x 3). The organic phase was combined,
dried over
anhydrous sodium sulfate, concentrated by filtration, and concentrated under
reduced
pressure, and the residue was separated and purified by silica gel column
chromatography
to obtain compound 18 (7 mg, yield 9.1%) as a white solid.
[0473] LCMS tn/z = 591.1 [M+Hr.
[0474] 11-1 NMR (400 MHz, DMSO-do) 6 7.96-7.91 (m, 1H), 7.86-
7.72 (m, 6H), 7.70-
7.65 (m, 1H), 7.29 (t, 1H), 5.70 (s, 2H), 5.05-4.97 (m, 1H), 4.64-4.56 (m,
1H), 4.52-4.45
(in, 2H), 4.41-4.28 (in, 3H), 2.70-2.64 (in, 1H), 2.35-2.29 (in, 1H).
Example 19
(S)-2-(4-(6-((4-cyanobenzypoxy)-5-fluoropyridin-2-y1)-2-fluorobenzy1)-1-
(oxetan-2-
ylmethyl)-1H-thieno12,3-dlimidazole-5-carboxylic acid (Compound 19)
FO
NC
/
0 N N
0
OH
Compound 19
CA 03203276 2023- 6- 22
- 105 -
F
NC P-13 S CO2Me
CI Br (r3-
19b --\r--No 10d
N o
.N_Br N
18a Step 1 F'' Step 2
19c 19d
F-1
Step 3 Ne....rni ^ N
N---(s
it) -
F OH
Compound 19
Step 1: 4-(((6-bromo-3-fluoropyridin-2-yl)oxy)methylibenzonitrile (19c)
NC
0 N Br
F
19c
[0475] Under ice bath, 18a (300 mg, L43 mmol) was added to a
solution of 19b
(285.7 mg, 1.43 mmol) and potassium tert-butoxide (240 mg, 2.15 mmol) in
tetrahydrofuran (10 ml) and stirred and reacted at room temperature for 3
hours. The
insolubles were removed by filtration, the liquid was washed with an equal
volume of
water, dried over anhydrous sodium sulfate and concentrated to dryness, and
the residue
was separated and purified by silica gel column chromatography to obtain the
product 19c
of interest (250 mg, yield: 57.1%).
Step 2: methyl(S)-2-(4-(64(4-cyanobenzypoxy)-5-fluoropyridin-2-y1)-2-
fluorobenzy1)-1-
(oxetan-2-ylinethyl)-1H-thieno [2,3 -d] imidazole-5-c arboxylate (19d)
NC
I /
0 N
0
19d
[0476] The crude products 10d and 19c (168.3 ing, 0.55 intnol),
anhydrous potassium
carbonate (115 mg, 0.80 ininol) and Pd(dppf)C12 (32.5 mg, 0.04 inmol) were
added to a
reaction flask successively, dissolved in a mixed solution of 1,4-dioxane (5
mL)/water (1
mL), subjected to nitrogen replacement, stirred at 90 C for 5 h and cooled to
room
CA 03203276 2023- 6- 22
- 106 -
temperature. the solid was filtered, the filtrate was concentrated under
reduced pressure,
and purified on a silica gel column to obtain the product 19d (180 mg, yield
67 % over two
steps) as a yellow solid.
[0477] LCMS m/z = 587.1 [M+H]t
Step 3: (S)-2-(4-(6-((4-cyanobenzyl)oxy)-5-fluoropyridin-2-y1)-2-fluorobenzy1)-
1-(oxetan-
2-ylmethyl)-114-thieno[2,3-d]imidazole-5 -carboxylic acid (Compound 19)
FO
NC
/
0 N N I
0
OH
Compound 19
[0478] At room temperature, 19d (80 mg, 0.14 mmol) was dissolved
in a solution of
acetonitrile (3 inL) and water (0.6 inL), then 1 g (CAS: 5807-14-7) (56.9 mg,
0.41 mmol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
IN hydrochloric acid to pH=6, and then extracted with a mixed solvent of
dichloromethane:methano1=10:1 (20 mL x 3). The organic phase was combined,
dried over
anhydrous sodium sulfate, concentrated by filtration, and concentrated under
reduced
pressure, and the residue was separated and purified by silica gel column
chromatography
to obtain compound 19 (40 mg, yield 50%) as a white solid.
[0479] LCMS in/z = 573.1 [M+Hr.
[0480] 11-1 NIVIR (400 MHz, DMSO-do) 6 7.90-7.77 (in, 6H), 7.71
(d, 2H), 7.68-7.64
(in, 1H). 7.38 (t, 1H), 5.67 (s, 2H), 5.04-4.96 (in, 1H), 4.63-4.55 (in, 1H),
4.53-4.44 (in,
2H), 4.42-4.28 (m, 3H), 2.72-2.62 (in, 1H), 2.38-2.27 (in, 1H).
Example 20
(S)-2-(4-(64(4-cyanobenzypoxy)pyridin-2-y1)-2-fluorobenzy1)-1-(oxetan-2-
ylmethyl)-1H-
thieno [2,3 -di imidazole-5-c arboxylic acid (Compound 20)
CA 03203276 2023- 6- 22
- 107 -
FO
NC
/
0 N N
0
OH
Compound 20
-]
0-B 10 \I--(sCO2Me
CI k ________________________
19b NC. 0 10d NC
CI k CI
rsk 7 A,
Step 1 Step 2
20a 20b
FO
Step 3 N
I
OH
Compound 20
Step 1: 4-(((6-chloropyridin-2-yeoxy)methyl)benzonitrile (20a)
NC
0 N CI
20a
[0481] Under ice bath, 2,6-dichloropyridine (5 g, 33.8 mmol) was added to a
solution
of 19b (4.4 g, 33.8 mmol) and potassium tert-butoxide (7.6 g, 67.6 mmol) in
tetrahydrofuran (100 ml) and stirred and reacted at room temperature for 3
hours. The
insolubles were removed by filtration, the liquid was washed with an equal
volume of
water, dried over anhydrous sodium sulfate and concentrated to dryness, and
the residue
was separated and purified by silica gel column chromatography to obtain the
product 20a
of interest (4.7 g, yield: 57.0%).
Step 2: nriethyl(S)-2-(4-(64(4-cyanobenzypoxy)pyridin-2-y1)-2-fluorobenzy1)-1-
(oxetan-2-
ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylate (20b)
CA 03203276 2023- 6- 22
- 108 -
F
NC
I /
0 N
0
20b
[0482] The crude products 10d and 20a (134.2 mg, 0.55 mmol),
anhydrous potassium
carbonate (115 mg, 0.80 mmol) and Pd(dppf)C12 (32.5 mg, 0.04 inmol) were added
to a
reaction flask successively, dissolved in a mixed solution of 1,4-dioxane (5
mL)/water (1
inL), subjected to nitrogen replacement, stirred at 90 C for 5 h and cooled to
room
temperature, the solid was filtered, the filtrate was concentrated under
reduced pressure,
and purified on a silica gel column to obtain the product 20b (160 mg, yield
61.5% over
two steps) as a yellow solid.
[0483] LCMS in/z = 569.1 [1\4+Hr.
Step 3: (S)-2-(4-(64(4-cy anobenzyl)oxy)pyridin-2-y1)-2-fluorobenzy1)-1-
(oxetan-2-
ylmethyl)-1H-thieno [2, 3-d] itnid azole-5-c arboxylic acid (Compound 20)
FO
NC
/
0 N
0
OH
Compound 20
[0484] At room temperature, 19d (80 mg, 0.14 mmol) was dissolved
in a solution of
acetonitrile (3 InL) and water (0.6 InL), then 1 g (CAS: 5807-14-7) (56.9 mg,
0.41 mmol)
was added, and the reaction was continued for 24 h. The reaction liquid was
adjusted with
1N hydrochloric acid to pH=6, and then extracted with a mixed solvent of
dichloromethane:methano1=10:1 (20 mL x 3). The organic phase was combined,
dried over
anhydrous sodium sulfate, concentrated by filtration, and concentrated under
reduced
pressure. The residue was purified by Pre-HPLC (instrument and preparative
column:
Waters 2767 (preparative liquid phase chromatographic instrument) was used,
and the
preparative column model was Sunfire@PrepC18, 5 [tin, inner diameter x length
= 19 mm
x 250 mm). Preparation method: The crude product was dissolved in DMF and
filtered
with a 0.45 [tin filter membrane to prepare into a sample liquid. Mobile phase
system:
CA 03203276 2023- 6- 22
- 109 -
acetonitrile/water (containing 0.5% ammonium bicarbonate). Gradient elution
method:
acetonitrile content 5-50% (flow rate: 15 mL/min; elution time 15 min),
compound 20 (20
mg, yield: 25.8%) was obtained.
[0485] LCMS m/z=555.1 [M+1]+.
[0486] 1H NMR (400 MHz, DMSO-do) 6 7.88-7.80 (m, 6H), 7.68 (d, 2H), 7.63
(d,
111), 7.38 (t, 114), 6.93 (d, 114), 5.69 (s, 211), 5.05-4.97 (m, 114), 4.63-
4.56(m, 111), 4.52-
4.44 (m, 2H), 4.42-4.29 (in, 3H), 2.72-2.62 (m, 1H), 2.37-2.28 (m, 1H).
Example 21
(S)-2-(4-(6-((4-cyano-2-fluorobenzyl)oxy)pyridin-2-y1)-2,5-difluorobenzy1)-1-
(oxetan-2-
ylmethyl)-1H-thieno[2,3-dlimidazole-5-carboxylic acid (Compound 21)
NC F Cra)
F
N
I /
0 1\1 N \
1 S CO2H
Compound 21
ly_3
HN 0
Br,,---õrõF Br -,,, F Br ¨ F Q
i ---T NH HCI H2N 1d
F-ii ,)--. ,CO2H Step 1 F CONH2 Step 2 F J1. ,--A. CN
Step 3 F'-'-- OEt
Step 4
21a 21b 21c 21d
F (-1- NC., (5])
F NC, F F
F 3a 0N)
CO2Me
)
HO d IR---i, 1 )'
¨ r.--- -NI
,,,, N--/c?-1
Br CO2Me CO2Me 1 I I
S
F Step 5 H6 F Step 6
21e 21 21g
rr1L'i
N, N O
H NC-F
-I" 7 F
1 ---TN__
Step 7 0 N, --, N---t 11
5 CO2H
Compound 21
Step 1: 2-(4-bromo-2,5-difluorophenyl)acetic acid (21b)
Br F
F CON H2
21b
[0487] 21a (7.5 g, 29.88 mmol) was added into thionyl chloride (20.0 mL),
and
stirred at 70 C for 2 hours. Then the reaction liquid was cooled to room
temperature and
CA 03203276 2023- 6- 22
- 110 -
concentrated to dryness and the concentrated residue was added to
dichloromethane (30
mL). The system was placed at 0 C and ammonia water (15 mL) was added, and a
large
amount of white solid was precipitated and filtered. The obtained solid was
further washed
with water (20 mL x 2), and the solid was dried to give 21b (7.3 g, yield
97%).
[0488] LCMS m/z = 250.1 [M+1]+.
Step 2: 2-(4-bromo-2,5-difluorophenypacetonitrile (21c)
Br
CN
21c
[0489] 21b (7.3 g, 29.20 mmol) was added into phosphorus
oxychloride (30.0 mL),
and stirred at 100 C for 2 hours. Then the reaction liquid was cooled to room
temperature,
the system was poured into ice water (100 mL), and then extracted with ethyl
acetate (100
inL x 3). The organic phase was combined, dried over anhydrous sodium sulfate
and
filtered, the filtrate was concentrated to dryness, and the residue was
separated and purified
by silica gel column chromatography to obtain 21c (6.0 g, yield 88%).
[0490] LCMS in/z = 232.1 [M+1] .
Step 3: ethyl 2-(4-bromo-2,5-difluorophenypacetimidate hydrochloride (21d)
Br
NH.HCI
OEt
21d
[0491] 21c (6.0 g, 25.86 mmol) was added to anhydrous ethanol
(3.0 mL) and fully
stirred at 0 C. At the same time, 33% hydrochloric acid-ethanol solution (25
inL) was
added. The temperature was gradually raised to room temperature and the
reaction liquid
was stirred for 10 hours. Diethyl ether (40 inL) was then added, filtered to
obtain a solid
that was further washed with diethyl ether (100 mL x 2 ), and dried to obtain
21d (5.8 g,
yield 71%).
[0492] LCMS inh = 278.1 [M-C1r.
Step 4: methyl (S)-2-(4 -bromo-2,5-difluorobenzy1)- 1-(oxetan-2-ylinethyl)- 1H-
thieno [2,3-
d]imidazole-5-carboxylate (21e)
CA 03203276 2023- 6- 22
- 111
OTh
/
Br
S CO2Me
21e
[0493] 21d (3.0 g, 9.58 mmol) and id (2.5 g, 10.3 inmol) were
added to glacial acetic
acid (50 inL), and stirred at 60 C for 10 hours. The reaction system was
cooled to room
temperature, then was slowly added to an aqueous saturated sodium bicarbonate
solution
(200 inL) to quench the reaction, and then extracted with ethyl acetate (50
inL x 3). The
organic phase was combined, dried over anhydrous sodium sulfate, and filtered.
The
filtrate was spin-dried, and the residue was separated and purified by silica
gel column
chromatography to obtain 21e (1.34 g, yield 30%).
[0494] LCMS in/z = 457.1 [M+1] .
Step 5: (S)-(2,5-difluoro-4((5-(methoxycarbony1)- 1-(oxet an-2-ylmethyl)- 1H-
thieno [2,3 -
d]imidazol-2-yl)methyl)phenyl)boronic acid (210
FO
HO,B
S CO2Me
OH F
21f
[0495] 21e (600 mg, 1.3 mmol) and pinacol biborate (519 mg, 1.9
mmol),
Pd(dpp0C12=DCM (110 mg, 0.1 mmol), and potassium acetate (400 mg, 4.0 mmol)
were
added to 1,4-dioxane (10 inL), and the reaction flask was subjected to
nitrogen
replacement for protection, and the reaction liquid in the reaction flask was
stirred at
100 C for 5 hours. The reaction liquid was cooled to room temperature, then
water (50
inL) was added, and extracted with ethyl acetate (50 inL x 3). The organic
phase was
combined, dried over anhydrous sodium sulfate, and filtered. The filtrate was
concentrated
to dryness to obtain crude compound 21f (438 mg, yield 80%).
[0496] LCMS m/z = 423.1[M+1]+.
Step 6: methyl (S)-2-(4-(64(4-cyano-2-
fluorobenzyl)oxy)pyridin-2-y1)-2,5-
difluorobenzy1)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d] imidazole-5-c
arboxylate (21g)
CA 03203276 2023- 6- 22
- 112
CC-J)
0 N
S CO2Me
F
21g
[0497] 21f (60 mg, 0.14 mmol) and 3a (34 mg, 0.13 mmol),
Pd(dppf)C12.DCM (10
mg, 0.01 mmol) and potassium carbonate (36 mg, 0.26 mmol) were added to a
mixed
solution of 1,4-dioxane (2 mL) and water (0.5 mL), and the reaction flask was
subjected to
nitrogen replacement for protection, and the reaction liquid in the reaction
flask was stirred
at 100 C for 5 hours. The reaction liquid was cooled to room temperature, then
water (2
mL) was added, and extracted with ethyl acetate (10 mL x 3). The organic phase
was
combined, dried over anhydrous sodium sulfate, and filtered. The filtrate was
concentrated
to dryness, and the residue was separated and purified by silica gel column
chromatography to obtain 21g (46 mg, yield 54%).
[0498] LCMS m/z = 605.2 [M+1]+.
Step 7: (S)-2-(4-(6-((4-cyano-2-fluorobenzypoxy)pyridin-2-y1)-2,5-
difluorobenzy1)-1-
(oxetan-2-ylmethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid (Compound 21)
NC
0 N
S CO2 H
F
Compound 21
[0499] 21g (46 ing, 0.076 mmol) and 1 g (32 ing, 0.228 mmol) were added to
a mixed
solution of acetonitrile (3 inL) and water (0.5 mL), and stirred at room
temperature for 16
hours. Then the reaction liquid was adjusted to pH=6 with 1N hydrochloric
acid,
continuously stirred until a white solid precipitated, and filtered and the
solid was washed
with water. The solid was dissolved in dichloromethane, dried over anhydrous
sodium
sulfate and filtered. The organic phase was concentrated to dryness, the
residue was
separated and purified by silica gel column chromatography to obtain compound
21 (10
mg, yield 22%).
[0500] LCMS in/z = 591.0 [M+1]+.
CA 03203276 2023- 6- 22
-113-
1105011 1H NMR (400 MHz, DMSO-d6) 6 12.91 (br, 1H), 7.93-7.83 (m,
3H), 7.78-
7.67 (m, 3H), 7.53-7.48 (m, 1H), 7.37-7.30 (m, 1H), 7.01-6.95 (m, 1H), 5.59
(s, 214), 5.08-
4.99 (m, 1H), 4.67-4.58 (m, 1H), 4.55-4.45 (m, 2H), 4.43-4.29 (m, 3H), 2.74-
2.64 (m, 1H),
2.40-2.29 (m, 1H).
Example 22
(S)-2-(4-(6-((4-cyano-2-fluorobenzypoxy)-5-fluoropyridin-2-y1)-2,5-
difluorobenzy1)-1-
(oxetan-2-ylmethyl)-1H-thienor,3-dlimidazole-5-carboxylic acid (Compound 22)
oI
NC F
/
0 N N
S 002H
Compound 22
FO
co2NmCe.: N
n'n
NC mr, HO g N NC F
NB 1114P 0 r OH F 21f
= N,
LT: 02Me Step 2
Step 1 F N_cCO2H
18c 22b Compound
22
Step 1: methyl (S)-2-(4-(64(4-cyano-2-fluorobenzyl)oxy)-5-fluoropyridin-2-y1)-
2,5-
difluorobenzy1)-1-(oxetan-2-ylmethyl)-1H-thienor,3-dlimidazole-5-carboxylate
(22b)
NC
/
0 N N
S CO2Me
F
22b
[0502] 21f (143 ing, 0.34mm01) and 18c (110 ing, 0.34mmo1),
Pd(dppf)C12.DCM (40
mg, 0.04 mmol) and potassium carbonate (140 mg, 1.01 mmol) were added to a
mixed
solution of 1,4-dioxane (5 inL) and water (1 mL), and the reaction flask was
subjected to
nitrogen replacement for protection, and the reaction liquid in the reaction
flask was stirred
at 100 C for 5 hours. The reaction liquid was cooled to room temperature, then
water (5
tuL) was added, and extracted with ethyl acetate (10 tuL x 3). The organic
phase was
combined, dried over anhydrous sodium sulfate, and filtered. The filtrate was
concentrated
to dryness, and the residue was separated and purified by silica gel column
chromatography to obtain 22b (170 nag, yield 80%).
CA 03203276 2023- 6- 22
-114-
1105031 LCMS m/z = 623.1 [M+1]+.
Step 2:
(S)-2-(4-(64(4-cyano-2-fluorobenzyl)oxy)-5-fluoropyridin-2-y1)-2,5-
difluorobenzy1)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d]imidazole-5-carboxylic
acid
(Compound 22)
NC
/
S CO2H
F
Compound 22
[0504] 22b (50 mg, 0.08 mmol) and 1 g (34 mg, 0.24 mmol) were
added to a mixed
solution of acetonitrile (5 inL) and water (1 inL), and stirred at room
temperature for 16
hours. Then the reaction liquid was adjusted to pH=6 with 1N hydrochloric
acid,
continuously stirred until a white solid precipitated, and filtered and the
solid was washed
with water. The solid was dissolved in dichloromethane, dried over anhydrous
sodium
sulfate and filtered. The organic phase was concentrated to dryness, the
residue was
separated by preparative HPLC to obtain compound 22 (10 mg, yield 20%).
[0505] LCMS tn/z = 609.1 [M+1]+.
[0506] 114 NMR (400 MHz, DIVISO-d6) 6 7.95-7.90 (in, 1H), 7.88-
7.82 (in, 2H), 7.80-
7.67 (m, 3H), 7.55-7.50(m, 1H), 7.37-7.31 (m, 1H), 5.68 (s, 2H), 5.06-5.00
(in, 1H), 4.66-
4.59 (m, 1H), 4.54-4.45 (m, 2H), 4.40-4.29 (m, 3H), 2.69-2.64 (m, 1H), 2.37-
2.30 (m, 1H).
Example 23
(S)-2-(4-(64(4-cy anobenzypoxy)pyridin-2-y1)-2,5 -difluorobenzy1)-1-(oxetan-2-
ylmethyl)-
1H-thieno [2,3-d] imidazole-5-carboxylic acid (Compound 23)
NC
() N
N¨_/<
S CO2H
Compound 23
(r)NC ,
NC
N
HO B N r "0õ!11''i NC
N N 0])
1-:õ.11-.. 0 N CI OH F 2if S CO2Me q1113-'' N, 1 g H
J=J
co2Nne Step 2 'IL; S CO2H Step 1
20b 23b Compound
23
CA 03203276 2023- 6- 22
- 115 -
Step 1: methyl (S)-2-(4-(64(4-cyano-2-fluorobenzypoxy)-5-fluoropyridin-2-y1)-
2,5-
difluorobenzy1)-1-(oxetan-2-ylmethyl)-1H-thieno [2,3-d]imidazole-5-carboxylate
(23b)
NC s iCr)
0 N
S CO2Me
F
23b
[0507] 21f (200 mg, 0.49mino1) and 20b (120 mg, 0.49mino1),
Pd(dppf)C12=DCM (40
mg, 0.04 mmol) and potassium carbonate (140 mg, 1.01 mmol) were added to a
mixed
solution of 1,4-dioxane (5 inL) and water (1 inL), and the reaction flask was
subjected to
nitrogen replacement for protection, and the reaction liquid in the reaction
flask was stirred
at 100 C for 5 hours. The reaction liquid was cooled to room temperature, then
water (5
inL) was added, and extracted with ethyl acetate (10 inL x 3). The organic
phase was
combined, dried over anhydrous sodium sulfate, and filtered. The filtrate was
concentrated
to dryness, and the residue was separated and purified by silica gel column
chromatography to obtain 23b (143 mg, yield 50%).
[0508] LCMS m/z = 587.0 [M+1]+.
Step 2: (S)-2-(4-(64(4-cy anobenzyl)oxy)pyridin-2-y1)-2,5-difluorob enzy1)-1-
(oxetan-2-
ylinethyl)-1H-th ieno [2,3-d] im idazole-5-carboxyl ic acid (Compound 23)
NC
/
0 N
S CO2H
F
Compound 23
[0509] 23b (50 mg, 0.08 mmol) and 1 g (34 mg, 0.24 mmol) were
added to a mixed
solution of acetonitrile (5 inL) and water (1 inL), and stirred at room
temperature for 16
hours. Then the reaction liquid was adjusted to pH=6 with 1N hydrochloric
acid,
continuously stirred until a white solid precipitated, and filtered and the
solid was washed
with water. The solid was dissolved in dichloromethane, dried over anhydrous
sodium
sulfate and filtered. The organic phase was concentrated to dryness, the
residue was
separated and purified by silica gel column chromatography to obtain compound
23 (15
mg, yield 30%).
CA 03203276 2023- 6- 22
-116-
1105101 LCMS m/z = 573.1 [M+1]+.
[0511] 111 NMR (400 MHz, DMSO-d6) 6 7.92-7.83 (m, 411), 7.73-
7.64 (m, 311), 7.53-
7.47 (m, 1H), 7.37-7.29 (m, 1H), 7.02-6.96 (m, 1H), 5.57 (s, 2H), 5.08-5.00
(m, 1H), 4.66-
4.59 (m, 111), 4.54-4.45 (m, 211), 4.39-4.29 (m, 311), 2.75-2.64 (m, 111),
2.39-2.29 (m, 111).
Example 24:
2-44-(2-(5-chloropyridin-2-y1)-2-methylbenzo[d] [1,3]dioxo1-4-ylipiperidin-1-
ylimethyl)-
1-4(S)-oxetan-2-ylimethyl)-1H-thieno[2,3-d]imidazole-5-carboxylic acid
0A7
-Th\-
0 NN
0
OH
Compound 24
Boc
Br Boc N.
Bockp-B
I OH Br
N - n 0 \
' OH 24c
I
0 - CI
24a 24b CI 24e
CI
24d
0 CI
ciz4NINIrso 0i)
CI
-c?
le
0 N 0 0 CJI4--1 ______ 0 N-
IP S
OH
CI 0- Compound
24
24f 24h
Step 1: 2-(4-bromo-2-methylbenzo[d][1,3]dioxo1-2-y1)-5-chloropyridine (24b)
Br
0
TrO
24b
[0512] 15a (276 mg, 2.0 mmol) and 3-bromocatechol (378 mg, 2.0
mmol) were
dissolved in tetrahydrofuran (6 mL), and Ru3(C0)12 (128 mg, 0.2 minol) was
added to the
system. The reaction flask was subjected to nitrogen replacement for
protection, and the
reaction liquid in the reaction flask was stirred at 110 C for 10 hours. The
reaction liquid
was cooled to room temperature, then concentrated under reduced pressure and
purified by
silica gel column chromatography to obtain lb as a gray oily liquid (190 mg,
yield 30%).
CA 03203276 2023- 6- 22
-117-
1105131 LCMS m/z = 326.0 [M+1]+.
Step 2: tert-butyl 4-(2-(5-chloropyridin-2-y1)-2-methylbenzo[d][1,31dioxo1-4-
y1)-3,6-
dihydropyridine-1(2H)-carboxylate (24d)
Boc
0
<0
CI N
[0514] lb (190 mg, 0.58 mmol), lc (180 mg, 0Ø58 mmol), cesium carbonate
(945
mg, 2.9 mmol) and Pd(dppf)C12.DCM (47 mg, 0.058 trunol) were dissolved in a
mixed
solvent of 1,4-dioxane (15 inL) and water (3 mL), subjected to nitrogen
replacement and
then heated to 90 C for 3 hours. The reaction liquid was cooled to room
temperature.
Water (30 inL) was added, extracted with ethyl acetate (30 inL x 3). The
organic phase
was combined and washed with saturated brine (30 inL x 1), and the obtained
organic
phase was dried over anhydrous sodium sulfate and filtered. The filtrate was
concentrated
under reduced pressure, and purified by silica gel column chromatography to
obtain ld
(180 mg, yield 72%) as a colorless oily liquid.
[0515] LCMS tn/z = 373.1 [M-55]+.
Step 3: tert-butyl 44245 -chloropyridin-2-y1)-2-methylbenzo [d]
[1,3] dioxo1-4-y1)
piperidine- 1-c arboxylate (24e)
Boc
0
CI N
[0516] ld (170 mg, 0.4 mmol) was dissolved in a mixed solvent of
tetrahydrofuran (3
inL) and methanol (3 inL), and Raney-Ni (20 mg) was added. The reaction was
carried out
at room temperature for 10 h after hydrogen replacement for protection. The
reaction liquid
was filtered, the filter cake (20 inL x 3) was washed with tetrahydrofuran,
the filtrate was
concentrated under reduced pressure, and purified by silica gel column
chromatography to
obtain 24e as a colorless oily liquid (120 mg, yield 70%).
CA 03203276 2023- 6- 22
-118-
1105171 LCMS m/z = 431.1 [M+1].
Step 4: 5-chloro-2-(2-methyl-4-(piperidin-4-yObenzo[d] [1,3] di oxo1-2-yl)pyri
dine p-
toluenesulfonate (240
=pTSA N
0
CI N
[0518] 24e (120 mg, 0.28 mmol) was dissolved in ethyl acetate (5 inL), and
p-
toluenesulfonic acid monohydrate (80 mg, 0.42 minol) was added. The reaction
was
carried out at 50 C for 10 hours after hydrogen replacement for protection.
The reaction
liquid was cooled to room temperature and then concentrated under reduced
pressure to
obtain the p-toluenesulfonate salt of crude product 24f, which was directly
used in the next
reaction without further purification.
[0519] LCMS in/z = 33L2 [M+1] .
Step 5: methyl 2-((4-(2-(5-chloropyridin-2-y1)-2-
methylbenzo [d] [1,3] dioxo1-4-
yl)piperidin-1-yl)methyl)-1-(((S)-oxetan-2-y1)methyl)-1H-thieno [2,3 -d]
imidazole-5 -
carboxylate (24h)
ci
NCN
0 Ns
0-
24h
[0520] The crude product 24f obtained in the previous step was
dissolved in
acetonitrile (3 mL), then potassium carbonate (60 mg, 0.42 inmol) and
intermediate le (42
mg, 0.14 mmol) were added. The reaction was carried out at 60 C for 6 hours
after the
reaction flask was subjected to nitrogen replacement for protection. The
reaction liquid
was cooled to room temperature, then concentrated under reduced pressure and
purified by
silica gel column chromatography to obtain 24h (80 mg, yield 96%) as a light
yellow solid.
[0521] LCMS in/z = 595.2 [M+1] .
CA 03203276 2023- 6- 22
- 119 -
Step 6: 2-((4-(2-(5-chloropyridin-2-y1)-2-methylbenzo [d]
[1,3] di oxo1-4-yl)piperi din-1-
yOmethyl)-1-(((S)-oxetan-2-yOmethyl)-1H-thieno [2,3-dlimidazole-5-carboxylic
acid
(Compound 24)
N 'er\
0 N
S
OH
Compound 24
[0522] Compound 24h (80 mg, 0.134 mmol) and lithium hydroxide (56 ing, 1.34
ininol) were added to a mixed solvent of acetonitrile (3 inL) and water ((16
inL), and
stirred at 50 C for 6 hours. 1N hydrochloric acid was used to adjust pH = 6,
then water (15
inL) was added, extracted with ethyl acetate (20 inL x 3), washed with
saturated brine (20
inL x 1). The organic phase was dried over anhydrous sodium sulfate and
filtered. The
solvent was removed from the filtrate under reduced pressure, and the residue
was purified
by column chromatography (dichloromethane/methano1=15:1) to obtain the
compound 24
of interest (53 ing, yield 70%).
[0523] LCMS in/z = 58L2 [M+Hr.
[0524] 11-1 NMR (400 MHz, CD30D) 8 8.59 (s, 1H), 7.88 (dd, 1H),
7.73 ¨ 7.60 (in,
2H), 6.84 ¨ 6.76 (in, 1H), 6.72 (d, 2H), 5.24 ¨ 5.15 (in, 1H), 4.71 ¨ 4.60
(in, 2H), 4.60 ¨
4.51 (in, 1H), 4.47 ¨ 4.38 (in, 1H), 4.21 (s, 2H), 3.45 ¨ 3.32 (in, 2H), 2.90
¨ 2.66 (in, 4H),
2.55 ¨ 2.42 (in, 1H), 2.01 (s, 3H), 1.99 ¨ 1.89 (in, 4H).
Biological test examples
[0525] 1. HEK293/CRE-luc/GLP-1R cell viability
[0526] Cell: HEK293/CRE-luc/GLP-1R
[0527] Cell culture medium: DMEM + 10% FBS + 400 ILighnl G418 +
100 vg/m1
Hygromycin B
[0528] Frozen stock solution: 90% FBS, 10% (V/V) DMSO
[0529] Detection buffer: DMEM + 1% FBS
[0530] Experimental procedure
[0531] Cells were cultured in DMEM medium + 10% FBS + 400 Kg/m' G418 +
100
Hygromycin B in a CO2 incubator at 37 C, and passaged once every 3-4 days.
CA 03203276 2023- 6- 22
-120-
1105321 Cell plating: Trypsinization adjusted the cell density to
1.67x105 cells/mL; 60
tiL of cells per well (10000 cells/well) was inoculated in a 384-well plate
containing
compounds; A NC well (negative control) and a background well (no cells) were
set.
Incubation was performed for about 18 2 h in the incubator.
[0533] Compounds were serially diluted with the detection buffer, and the
detection
concentration was 0.01 nM-1000 nM.
[0534] The cell culture plate was removed and all supernatant
was aspirated from the
cells. The cells were washed gently 2 times with 1X PBS.
[0535] The diluted compound was added to a 384-well plate (10
[iL/well), and three
replicate wells were set for each concentration. 10 lit. of the detection
buffer was added to
the NC well, sealed and incubated at 37 C for 6 hours.
[0536] The plate was removed, the cells were allowed to
equilibrate to room
temperature (at least 15 min), and then all supernatant was aspirated from the
cells.
[0537] Steady-Glo Reagent was added to the sample well at 10
L/well and
incubated at room temperature for 5 min to lyse the cells.
[0538] The test results were read with a BMG microplate reader.
[0539] Data processing
[0540] The mean background value was calculated.
[0541] The induction factor (Fold of induction, Fl) = (induction
well value -
background value) / (negative control well value - background value) was
calculated.
[0542] The EC50 value of the sample was calculated using
Graphpad Prim 8.0
software using four-parameter fitting analysis.
[0543] Statistic analysis
[0544] Mean and standard error (Mean SEM) were calculated for
all results and
Graphpad Prism software was used for statistical analysis. Specific data were
presented in
the form of graphs. P<0.05 was considered to be statistically different.
[0545] Biological test results:
Compounds EC50 (nM)
Trifluoroacetate salt of compound 1 A
Trifluoroacetate salt of compound 2 A
Trifluoroacetate salt of compound 3 A
CA 03203276 2023- 6- 22
- 121 -
Compound 4 A
Compound 5 A
Compound 5-2 A
Compound 6 A
Compound 7 A
Compound 8 A
Compound 9 A
Compound 10 A
Compound 11 A
Compound 12 A
Compound 13 A
Compound 15 A
Compound 16 A
Compound 17 A
Compound 18 A
Compound 20 A
Compound 21 A
Compound 22 A
Compound 23 A
Compound 24 A
[0546] A < 10 nM, 10 nM < B <50 nM, 50nM < C < 100 nM
[0547] Conclusion: The compound of the present invention has
good agonistic effect
on GLP-1 receptor, and has a EC50 value less than 100 nM, for example the EC50
values of
the trifluoroacetate salt of compound 1, the trifluoroacetate salt of compound
2, the
trifluoroacetate salt of compound 3, Compound 4, Compound 5, Compound 5-2,
Compound 6, Compound 7, Compound 8, Compound 9, Compound 10, Compound 11,
Compound 12, Compound 13, Compound 15, Compound 16, Compound 17, Compound
18, Compound 20, Compound 21, Compound 22, Compound 23, and Compound 24 were
less than 10 nM.
[0548] 2. Effects of the compounds on hERG potassium channels
[0549] Experimental platform: electrophysiological manual patch-
clamp system
CA 03203276 2023- 6- 22
-122-
1105501 Cell line: Chinese hamster ovary (CHO) cells stably
expressing hERG
potassium ion channel
[0551] Experimental method: For CHO (Chinese Hamster Ovary)
cells stably
expressing hERG potassium channel, whole cell patch-clamp technique was used
to
recorded hERG potassium channel current at room temperature in this
experiment. The
glass microelectrode is made of a glass electrode blank (BF150-86-10, Sutter)
by a puller.
The tip resistance after filling the liquid in the electrode is about 2-5 MEI
The glass
microelectrode can be connected to the patch-clamp amplifier by inserting the
glass
microelectrode into an amplifier probe. The clamping voltage and data
recording are
controlled and recorded by the pClamp 10 software through a computer. The
sampling
frequency is 10 kHz, and the filtering frequency is 2 kHz. After the whole
cell records
were obtained, the cells were clamped at -80 mV, and the step voltage that
induced the
hERG potassium current (/ hERG) was depolarized from -80 mV to +20 mV for 2 s,
then
repolarized to -50 mV, and returned to -80 mV after 1 s. This voltage
stimulation was
given every 10 s, and the administration process was started after the hERG
potassium
current was confirmed to be stable (at least 1 minute). The compound was
administered for
at least 1 minute at each test concentration, and at least 2 cells (n>2) were
tested at each
concentration.
[0552] Data processing:
[0553] Data analysis processing was carried out by using pClamp 10,
GraphPad
Prism 5 and Excel software. The inhibition degree of hERG potassium current
(peak value
of hERG tail current induced at -50 mV) at different compound concentrations
was
calculated by the following formula: Inhibition % = [1 ¨ (///o)]x100%.
[0554] Among the formula, Inhibition % represents the percentage
of inhibition of
hERG potassium current by the compound, and / and /o represent the amplitude
of hERG
potassium current after and before dosing, respectively.
[0555] Compound IC50 was calculated using GraphPad Prism 5
software by fitting
according to the following equation:
[0556] Y = Bottom + (Top-Bottom)/(1+10^((LogIC50-X) x
HillSlope))
[0557] Among the equation, X represents the Log value of the tested
concentration of
the test sample, Y represents the inhibition percentage at the corresponding
concentration,
CA 03203276 2023- 6- 22
- 123 -
and Bottom and Top represent the minimum and maximum inhibition percentage,
respectively.
[0558] Biological test results
Compounds Structure ICso
(1.11\4)
O-1)
NC
Compound 1 33.6
ON N¨ ):1
S
OH
N ,N
Compound 2 C 52.2
/s r 0
OH
NC
Compound 5-2 o > 40
F 0 0
OH
0
NC
Compound 6 >40
F
OH
Compound 6
[0559] Conclusion: The compounds of the present invention have
less hERG
inhibitory effect.
[0560] 3. Stability test in mouse liver microsomes
[0561] At 37 C, 1 ILEA/1 of the test compound was incubated with
mouse liver
microsomes (0.5 mg/mL) supplemented with NADPH regeneration system for 5, 10,
20,
30 and 60 minutes, and then LC-MS/MS method was used to detect the
concentration of
the test compound in the generated sample. The half-life (Ti/2) and inherent
clearance rate
(CLint(mic)) of the compound in mouse liver microsome solution were obtained
by
calculating at the corresponding time points and the remaining percentage of
the
compound.
[0562] Biological test results:
CA 03203276 2023- 6- 22
- 124 -
T1/2 CLint(mio
Compounds Structure
(min) (Ill./min/mg)
Compound 2 NC
1419
0.977
\S
OH
NC OrI
Compound 5-2 / 0 N \\ 5588
0.252
F 0, N 0
OH
NC
Compound 6 I 296
4.69
OH
Compound 6
Compound DZ-6
(See WO
2020207474
NC os F1)J.N 130
10.6
ON
for Compound 84)
[0563] Conclusion: The compound of the present invention has
good stability in
mouse liver microsomes. For example, compared with the control compound, the
clearance
rate of compound 6 in mouse liver microsomes was significantly slowed down,
and the
half-life was significantly prolonged. The Tin ratio of compound 6 to compound
DZ-6 was
greater than 2.
[0564] 4. Stability test in
monkey liver microsomes
[0565] At 37 C, 1 iuM of the test compound was incubated with
monkey liver
microsomes (0.5 mg/mL) supplemented with NADPH regeneration system for 5, 15,
30,
45 and 60 minutes, and then LC-MS/MS method was used to detect the
concentration of
the test compound in the generated sample. The half-life (Ti/2) and inherent
clearance rate
(CLint(mic)) of the compound in monkey liver microsome solution were obtained
by
calculating at the corresponding time points and the remaining percentage of
the
compound.
[0566] Biological test results:
CA 03203276 2023- 6- 22
- 125 -
Tin
CLint(mic)
Compounds Structure
(min) (IL/min/mg)
o
Compound 2 NC 842 1.60
N
- s
OH
[0567] Conclusion: The compound of the present invention has
good stability in
monkey liver microsomes.
[0568] 6. Stability test in canine liver microsomes
[0569] At 37 C, 1 ILEM of the test compound was incubated with
canine liver
microsomes (0.5 mg/mL) supplemented with NADPH regeneration system for 5, 15,
30,
45 and 60 minutes, and then LC-MS/MS method was used to detect the
concentration of
the test compound in the generated sample. The half-life (Tin) and inherent
clearance rate
(CLint(mic)) of the compound in canine liver microsome solution were obtained
by
calculating at the corresponding time points and the remaining percentage of
the
compound.
[0570] Biological test results:
T1/2
CLint(mic)
Compounds Structure
(min) (IL/min/mg)
O
Compound 1 NC NMN
6116 0.23
0 N N
OH
[0571] Conclusion: The compound of the present invention has
good stability in
canine liver microsomes.
[0572] 7. Stability test in human liver microsomes
[0573] At 37 C, 1 tiM of the test compound was incubated with human liver
microsomes (0.5 mg/mL) supplemented with NADPH regeneration system for 5, 10,
20,
30 and 60 minutes, and then LC-MS/MS method was used to detect the
concentration of
the test compound in the generated sample. The half-life (Tin) and inherent
clearance rate
(CLint(mic)) of the compound in human liver microsome solution were obtained
by
CA 03203276 2023- 6- 22
- 126 -
calculating at the corresponding time points and the remaining percentage of
the
compound.
[0574] Biological test results:
T112
CLint(mic)
Compounds Structure
(min) (nUmin/mg)
_70
NO
Compound 6 o N 554 2.50
sO
OH
Compound 6
Compound DZ-6
(See WO
1.1)
NC 40 F 173 8.01
2020207474 *
N,
OH
for Compound 84)
[0575] Conclusion: The compound of the present invention has
good stability in
canine liver microsomes. For example, compared with the control compound, the
clearance
rate of compound 6 in monkey liver microsomes was significantly slowed down,
and the
half-life was significantly prolonged. The Tin ratio of compound 6 to compound
DZ-6 was
greater than 3.
[0576] 8. Activity of the compounds on PDE10A1
[0577] In this experiment, the effect of the compounds on the inhibition of
PDE10A1
activity was detected by the method of fluorescence polarization. PDE10A1 kit
was used
for detection (Biomol, BPS-60385). According to the kit instruction, FAM-
Cyclic-3',5'-
AMP (200 nM) dilution was added to wells other than the blank control
well(Blank) at
12.5 l/well; PDE test buffer was added to the blank control well (Blank) at
22.5 [LI/well;
PDE test buffer was added to the Substrate control well at 10 p1/well; a
compound working
solution was added to the corresponding well of the test plate at 2.5 I/well;
PDE test
buffer (10% DMSO) was added to the positive control well (Positive control),
the
Substrate control well, and the blank control (Blank) well at 2.5 [LI/well,
respectively;
PDE10A1 enzyme was diluted to 1 pg/ 1 with PDE test buffer, and the enzyme
solution
was added to the Substrate control well and the positive control well
(Positive control) at
10 l/well; the final concentration of the test compound was 300 [LM-15.24 nM,
and the
CA 03203276 2023- 6- 22
- 127 -
final concentration of the positive reference compound TAK063 was 100 nM-
0.00508 nM;
incubation was performed at room temperature for 1 hour; the Binding Agent was
diluted
in a ratio of 1: 100 with Binding Agent Diluent (cAMP) for later use; after
the incubation,
the Binding Agent dilution was added to the test plate at 50 ill/well, and
incubated at room
temperature for 20 minutes with slow shaking; after the incubation, Envision
was used for
FP detection (Excitation 480 nm, Emission 535 nm); Data were normalized to %
inhibition
and IC50 values were calculated using a Graphpad 4 parameter logistic
equation.
[0578] Conclusion: The compounds of the present invention have a
small inhibitory
effect on PDE10A1, for example, for the inhibitory effect of Compound 2 on
PDE10A1,
IC50 (pM) > 500.
[0579] 8. Pharmacokinetic test
Evaluation of pharmacokinetics in mice
[0580] Male C57 mice (purchased from Hunan SJA Laboratory Animal
Co., Ltd,
license number: SCXK (Xiang) 2019-0004), 20-25g, were fasted overnight. On the
day of
the experiment, 18 male C57 mice were divided into 2 groups, with 9 mice in
each group.
The test compound was administered intravenously and intragastrically at doses
of 5
mg/kg and 20 mg/kg respectively. Before administration and 5 min, 15 min, 30
min, 1 h, 2
h, 4 h, 6 h, 8 h and 24 h after administration, 0.1 ml of blood was collected
from the orbital
venous plexus, placed in an EDTAK2 centrifuge tube, and centrifuged at 6000
rpm at 4 C
for 5 min, and then the plasma was collected. The plasma sample was added to
an
acetonitrile solution containing an internal standard (dexamethasone, 500
ng/mL), vortexed
and centrifuged at 10,000 rpm for 10 min, and the supernatant was taken for LC-
MS/MS
analysis. Pharmacokinetic parameters were calculated using the non-
compartmental model
in WinNonlin Version 8.0 (Pharsight, Mountain View, CA) pharmacokinetic
software.
[0581] Conclusion: The compounds of the present invention have good oral
performance, for example, the bioavailability of compound 6 in C57 mice is
>25%.
Evaluation of pharmacokinetics in cynomolgus monkeys
[0582] Male cynomolgus monkeys (raised in Suzhou LEOLAB Bio
Tech. Co.,Ltd.),
weighing 3-5 kg, were fasted overnight (14-18 h), 3-5 kg, were fasted
overnight. On the
day of the experiment, 6 male cynomolgus monkeys were divided into 2 groups,
with 3
mice in each group. The test compound was administered intravenously and
CA 03203276 2023- 6- 22
- 128 -
intragastrically at doses of 2 mg/kg and 10 mg/kg respectively. Before
administration and
min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h, 8 h 10 h, 12 hand 24 h after
administration, 1 ml
of blood was collected via the limb vein, placed in an EDTAK2 centrifuge tube,
and
centrifuged at 6000 rpm at 4 C for 5 min, and then the plasma was collected.
The plasma
5 sample was added to an acetonitrile solution containing an internal standard
(dexamethasone, 500 ng/mL), vortexed and centrifuged at 10,000 rpm for 10 min,
and the
supernatant was taken for LC-MS/MS analysis. Pharmacokinetic parameters were
calculated using the non-compartmental model in WinNonlin Version 8.0
(Pharsight,
Mountain View, CA) pharmacokinetic software. Conclusion: The compounds of the
present invention have good metabolic properties. For example, compound 6 has
a low
clearance rate and a long half-life in monkeys, which has the advantages of
longer drug
action time and lower frequency of administration.
[0583] 9. Cytochrome P450 isoenzyme inhibition test
[0584] Experimental objective: The inhibitory effect of the test
compound on the
activity of human liver microsomal cytochrome P450 isoenzyme (CYP1A2, CYP2C19,
CYP2D6 and CYP3A4) was determined.
[0585] Experimental operation: The test compound at an
incubation concentration of
0, 0.03, 0.1, 0.3, 1, 3, 10, 30 and 100 [xM was pre-incubated with the human
liver
microsome solutions containing 5 enzyme mixed substrates (liver microsome
concentration: 0.1 mg/ mL) in a water bath at 37 C for 10 min, respectively,
and then a
coenzyme factor (NADPH) was added to start the reaction. After incubation in a
water
bath at 37 C for 10 min, the reaction was terminated by adding glacial
acetonitrile solution
containing an internal standard, shaken for 1 min, centrifuged at 4 C and
10,000 rpm for
10 min, and the supernatant was taken for LC/MS/MS analysis. The amount of
metabolites
produced by each CYP enzyme substrate was detected.
[0586] The activity of each CYP isoenzyme was reflected by the
production rate of
each metabolite. The activity of each isoenzyme in the solvent control
incubation system
without test substances was set as 100%, and the ratio of the production rate
of metabolites
in the case of different concentrations of test substances to the production
rate of
metabolites in the case of a solvent control sample was taken as the
percentage of residual
activity of each isoenzyme. With the residual activity percentage as the
ordinate and the
CA 03203276 2023- 6- 22
- 129 -
inhibitor concentration as the abscissa, Graphpad prism 5.0 was used to plot
the data and
calculate the IC50 value of the test substance for each CYP isoenzyme.
[0587] Conclusion: The compounds of the present invention have
weak inhibitory
activity on cytochrome P450 isoenzymes, for example, for the inhibitory
activity of
compound 6 on CYP2C19, CYP1A2, CYP2D6 and CYP3A4-M, IC50 > 50 M.
CA 03203276 2023- 6- 22