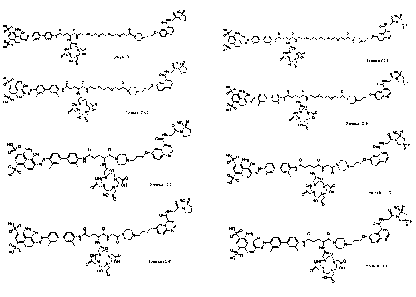Note: Descriptions are shown in the official language in which they were submitted.
3,210,863
TRUNCATED EVANS BLUE MODIFIED FIBROBLAST ACTIVATION PROTEIN
INHIBITOR, PREPARATION METHOD AND APPLICATION THEREOF
TECHNICAL FIELD
The present disclosure relates to the fields of nuclear medicine and molecular
imaging, and specifically
relates to a truncated Evans Blue modified fibroblast activation protein
inhibitor, preparation and labelling thereof
and application thereof.
BACKGROUND
A fibroblast activation protein (FAP) is a membrane serine peptidase that is
expressed on the surface
of a tumor stroma activated fibroblast and plays an important role in
generation and development processes
of tumors. Previous studies show that the FAP is generally not expressed in
normal human tissues, but
selectively highly expressed on surfaces of stromal fibroblasts of more than
90% of epithelial malignant
tumors, including breast cancer, ovarian cancer, lung cancer, colorectal
cancer, gastric cancer and
pancreatic cancer. In view of widespread expression and important role in
tumors, the FAP has become an
important target for imaging and therapy of tumors.
At present, a radionuclide labeled fibroblast activation protein inhibitor
(FAPI), represented by a
quinolinic acid derivative, has made important progress in the field of
accurate imaging of tumors. For
example, PET/CT imaging agents such as PHEAPI-02 and PHEAPi-04 have realized
specific imaging of
more than 30 different types of tumors. Compared with FDG imaging, FAPI
imaging has lower background
in brain, liver and oropharyngeal mucosa and higher detection rate of tumor
lesions. According to current
reports, the FAPI is rapidly cleared in blood circulation and rapidly eluted
at a tumor site. Such metabolic
characteristics are favorable for imaging because clean background can be
provided. However, the
metabolic characteristics are unfavorable for therapy because rapid metabolism
and elution lead to low
effective dose and short retention time at a tumor site, high dose or a more
frequent administration method
is required to meet therapeutic needs, and the possibility of adverse
reactions is increased.
For example, FAPI-02 is completely cleared in blood circulation within one
hour, and 24 hours later,
the retained dose at a tumor site is decreased by about 75%. Although a non-
pharmacophore part of the
structure of the FAPI has been optimized in recent research work, the dose
uptake of the FAPI in tumors
and the retention time are improved to an extremely limited extent, and the
needs of therapeutic use cannot
be met. Persons of ordinary skill in the field know that when a small molecule
medicine has too short
circulation time in blood vessels or is quickly cleared by the body, binding
of the medicine to a target will
be insufficient. Therefore, during preparation of an FAPI probe, it is
possible to increase the dose uptake
and prolong the retention time of the probe at a target site when the half-
life of the probe in blood
circulation is properly prolonged.
Therefore, a new strategy is required to prolong the half-life of the FAPI
probe in blood circulation, so
that the FAPI probe can have appropriate metabolic dynamics, higher dose
uptake in tumors and longer
retention time in tumors to meet requirements of nuclide therapy and imaging.
Date Regue/Date Received 2023-12-12
3,210,863
SUMMARY
Based on the above background, a primary purpose of the present disclosure is
to develop a kind of
conjugates of truncated Evans Blue (tEB) and a fibroblast activation protein
inhibitor (FAPI). The
conjugate is characterized in that through effective binding of the truncated
Evans Blue to serum albumin,
the albumin is used as a delivery carrier of the FAPI, so that the half-life
of the FAPI in peripheral blood is
prolonged, the uptake and accumulation in tumors are increased, and the
retention time is prolonged.
According to the tEB-FAPI conjugates developed by the present disclosure, the
defects of too fast
metabolism of the small molecule FAPI and too short retention time in a target
organ can be overcome,
nuclide therapy and imaging effects of targeting FAP are improved, and the
potential for clinical
application and popularization is achieved.
Another purpose of the present disclosure is to provide a radiolabeled
fibroblast activation protein
inhibitor modified by truncated Evans Blue (tEB-FAPI) with a long half-life in
blood circulation.
Another purpose of the present disclosure is to provide a preparation method
of a radiolabeled
tEB-FAPI complex.
Another purpose of the present disclosure is to provide application of the
complex in nuclide imaging
and therapy by targeting FAP tumors.
Technical solutions for realizing the above primary purpose of the present
disclosure include the
following two aspects: synthesis of ligands and radiolabeling of the ligands.
In a first aspect, the present disclosure provides a truncated Evans Blue
(tEB) modified fibroblast
activation protein inhibitor (FAPI). The compound has the following structure
shown in Formula (I), and is
denoted as "tEB-FAPI":
HO r,
NH2
0--"S
OH
0õs, 114
'0 R2
HO Formula (I)
where in
L1 is a lysine or glutamic acid structure, or a derivative compound structure
containing a lysine or
glutamic acid structure;
L2 is -(CH2)11-, wherein n is an integer from 0 to 30, wherein each CH2 can be
individually substituted
or unsubstituted with -0-, -NH-, -(CO)-, -NH(C0)-, or -(C0)-NH-, provided that
no two adjacent CH2
groups are substituted;
L3 is -(C112).-, wherein m is an integer from 0 to 30, wherein each CH2 can be
individually substituted
or unsubstituted with -0- or -(CO)-, provided that no two adjacent CH2 groups
are substituted;
L4 is -(CH2)p-, wherein p is an integer from 0 to 30, wherein each CH2 can be
individually substituted
or unsubstituted with -0-, -NH-, -(CO)-, -NH(C0)-, or -(C0)-NH-, provided that
no two adjacent CH2
groups are substituted;
X is selected from N, C, 0, S. or any one of the following structures:
2
Date Regue/Date Received 2023-12-12
3,210,863
0
NNH
N
/ H H , and 0 =
RI is the following structure of a fibroblast activation protein inhibitor:
0
R4
HN--/ R3
0
1-0
R2 is a nuclide chelating group, and is selected from any one of the following
structures:
o
0
0HrN N -=\0
OH HOOC, 0
0 HOr_J HO e HOOC-\ 1 II
HO-
0 , 0 -N-NH , and HOOC \-COOH
0
OH N 0
0N N OH
akh
NCS
OH
and R3-R4 are the same or different, and are independently selected from H or
F.
In a preferred solution of the present disclosure, the L2 in Formula (I) is -
(CH2).-; n is an integer from
0 to 16, is more preferably an integer from 0 to 12, and is further preferably
0, 3, or 10; and each -CH2-
may be individually substituted or unsubstituted with -0-, -NH-, or -(C0)-,
provided that no two adjacent
-CH2- groups are substituted.
In a preferred solution of the present disclosure, the L3 in Formula (I) is -
(C112).-; m is an integer
from 0 to 20, is more preferably an integer from 1 to 6, and is further
preferably 2 or 3; and each -CH2-
may be individually substituted or unsubstituted with -0-, provided that no
two adjacent -CH2- groups are
substituted.
In a preferred solution of the present disclosure, the L4 in Formula (I) is -
(CH2),-; p is an integer from
0 to 20, is more preferably an integer from 0 to 12, is further preferably 3,
4, 9, or 12, and is most
preferably 3; and each -CH2- may be individually substituted or unsubstituted
with -0-, -NH-, -(CO)-,
-NH(C0)-, or -(C0)-NH-, provided that no two adjacent -CH2- groups are
substituted.
In a preferred embodiment of the present disclosure, the X in Formula (I) is
the L3 is
-(CH2)3-, the L4 is -(CH2)0-, and the R2 is DOTA. That is to say, a preferred
compound tEB-FAPI of the
present disclosure has the following structure shown in Formula II:
3
Date Regue/Date Received 2023-12-12
3,210,863
Nc,>LFI
HN I -/ R4
0 R3
HO 0
= NH2 * N/1
N
HO,= '0 N
0 N OH
HO- \.?
0 Formula (II)
wherein R3 and R4 are both H or both F, L1 is a glutamic acid or lysine
structure, and L2 is -(CH2)0,
-NH-CH2-(C0)-, -NH-CH2-(CH2OCH2)2-CH2-(C0)-, -NH-CH2-(CH2OCH2)4-CH2(C0)-, -
(C0)-CH2-(C0)-,
-(C0)-(C112)2-(C0)-, -(C0)-CH2-(CH2OCH2)2-CH2(C0)-, or -(C0)-CH2-(CH2OCH2)4-
CH2(C0)-.
In a more preferred embodiment of the present disclosure, the X in Formula (I)
is \---/ , the Li
is a glutamic acid structure, the L2 is -(CH2)0-, -NH-CH2-(C0)-, -NH-CH2-
(CH2OCH2)2-C112-(C0)-, or
-NH-CH2-(C1120C112)4-CH2(C0)-, the L3 is -(CH2)3-, the L4 is -(CH2)0-, the R2
is DOTA, and the R3 and
R4 are both H or both F.
/-11
In another more preferred embodiment of the present disclosure, the X in
Formula (I) is \ ,
the LI is a lysine structure, the L2 is -(C0)-CH2-(C0)-, -(C0)-(C112)2-(C0)-,
-(C0)-C112-(C1120C112)2-CH2(C0)-, or -(C0)-C112-(CH20C112)4-CH2(C0)-, the L3
is - (C H2)3 -, the L4 is
-(CH2)0-, the R2 is DOTA, and the R3 and R4 are both H or both F.
In a further preferred solution of the present disclosure, the compound tEB-
FAPI has any one of the
following structures shown in Formula (II-1) to Formula (11-1 6):
o Na H
0
I=K)
0 0
j--P NI
4/41 Nri 1-14 H
=
CA5sj 0
L,N1
Formula (II-1),
H
NC
0 y,
HN-j-NJ
HO 0
'64. NH2 0 0 0
N 46 N'
Nt w HN "\-21
0,
""\r0 Olt= N
=
Formula (11-2),
4
Date Regue/Date Received 2023-12-12
3,210,863
Ns H
HN-1- Na
0
HO )0
0 0
0- 0
%,
Fici '0
OHFN N -"\O
0 N
) OH
L,
HO Formula (II-3),
0 NCõ õH
HN-).- N
0
¨
HcOi ,eo NH2 0 0 /
N
N NK'YL N'ThrNN
HN 0
0,..s ,
HO, ' 0
0Hr N
k N i 1
0
OH
L.,yN
H0-
0 Formula (II-4),
t H
0\\ NC
HN-T-N4 F
0 F
HO ,0 ¨
NH2 0 0 0
0' OH N . . N),õõ.."yky"..õõØ.õõ...Ø^.,,,O,,,...Ø---õKi---- \ _
j_,P * N/
N" HN " NJ''
-= 0 ?0
0
.kN ) 61
t.....,N
HO--
Formula (II-5),
0 NC, H
HNi. --Ni4
0 F
Hq AD F
= NH 0 0 0
OH 1100 /
H
N' HN L../
0,
HO
Z' 5 \gH
0 1. ,...."N
Fia-,
Formula (II-6),
NOõ H
0
HN--, ______________________________________________
0 F
F
¨
HO
NH2 0
0 0 i
N
OH
H
41 N A A
O 41 ise HN 0
'_0
H0 '0
0HrN/1 N ' \
.=\ ,,N i ' OH
HO 4
0 Formula (II-7),
5
Date Regue/Date Received 2023-12-12
3,210,863
o No
HN, H
\---N__
j ' F
HO F 0 _
NH 2 0 0
0' OH 0 /
N
H
NI' HN \--/
0,
'S, 0
HO 'C> 7Th
OHFN N.'\O
0.),,LN) 1 OH
Formula (II-8),
HO-
0
N0 H
0 HNj\--No
0
_
HO /
NH2
0 N
Ise H HN 0 0
CI'-'s, 0
H0
0Hr-N N-\0
0
.),NIJI ) 1 OH
L
HO4
o Formula (II-9),
O,
No, H
HN--Y-Na
0
HO
s -0 ¨
S' 2 0 0
0* OH
NH /
r--\ __./..._ jo *
N
N N-kr----"'"'il N N
FIN
0,
Sõ 0
HO '' NfTh
011- N-\rO
O
0 NH
HO-
o Formula (II-10),
0 Noõ,,H
0
HN----,--
0
Ho 0 _
s= NH2 o o
,N CI hi
Al' HN \¨/-
0, =;(2,
HO /:3 01-rN \N-Nr0
kN ) O
o N H
HO--
Formula (II-11),
6
Date Regue/Date Received 2023-12-12
3,210,863
0 NC H
HN .,,,
N
-1-
0
HO
, 0 -_
o'S'''' NH2 0 0
H 0 /
OH
4141 0 N' H HN 0
....../"=xy"....-'oN/M-",/k, M j---/
,s,
HO '13 N/Th
oi-r N" \A
OH
HO
Formula (II-12),
o NC, H
HN F
0 F
_
HO /
0 NH2
0 0 * N
,N KritY"'"N'irThrN\____ JH
N' H HN 0 0
0.
HO/
OFirN=Th N--\0
_) 1
0 N OH
HO4
0 Formula (II-13),
0
NC H
4,
HN-j-N3c-F
0 F
HO 0 --
\S"' NH2 0 0
01' OH
1\1)-'11.1-HC.1----\ _7---jo * =
N
0õs ,
<,' 0
Hd "0 =Th
01141 NO
OH
HO4
0 Formula (II-
14),
o NCõ 1-1
HN--*Ns--F
0 F
HO 0
S N''' 1-12 0 0
OH ¨
/
N
N N
tr H MN 0 1 --_.=
0,
0
Hd 7----- A
OWN 0
0 A1 OH L.....
HO--
Formula (II-15),
or
7
Date Regue/Date Received 2023-12-12
3,210,863
0NC H
HNJ1-0 \_4-FF
*0
N142 0 0
H N = ai
11 HN
h7-1:2µ
0+0
Formula (II-16).
On the above basis, the present disclosure further provides a method for
preparing the compound
tEB-FAPI shown in Formula (II-1). The method includes the following steps:
()reacting 6-hydroxy-4-quinolinecarboxylic acid with tert-butyl glycinate by
amide condensation,
followed by reactions with 1-bromo-3-chloropropane and tert-butyl 1-
piperazinecarboxylate in sequence;
then, removing Boc and tert-butyl protective groups under the action of TFA,
and introducing a Boc
protective group to amino, followed by an amide condensation reaction with (S)-
pyrrolidene-2-carbonitrile
hydrochloride; then, removing the Boc protective group using p-toluenesulfonic
acid, followed by a
condensation reaction with 5,8,11,14-tetraoxa-2-azaheptadecanedioic acid- 1-
tert-buty1 ester; and removing
the Boc protective group again under the action of p-toluenesulfonic acid to
obtain an intermediate
compound A;
introducing a Boc protective group to one end of 4,4'-diamino-3,3'-dimethyl
biphenyl, followed by
a reaction with monosodium 1-amino-8-naphthol-2,4-disulfonate to prepare a
truncated Evans Blue
derivative; removing the Boc protective group, followed by an amide
condensation reaction with
N-tert-butyloxycarbonyl-L-glutamic acid- 1-tert-buty 1 ester; then, removing
Boc and tert-butyl protective
groups under the action of TFA; and then carrying out a reaction with di-tert-
butyl dicarbonate, and
introducing a Boc protective group to amino to obtain an intermediate compound
B; and
reacting the intermediate compound A with the intermediate compound B by amide
condensation;
then removing the Boc protective group using p-toluenesulfonic acid; and
finally, carrying out a reaction
with DOTA-NHS to obtain the compound shown in Formula (II-1).
A preferred method for preparing the compound tEB-FAPI shown in Formula (II-1)
of the present
disclosure specifically includes the following steps:
dissolving 6-hydroxy-4-quinolinecarboxylic acid (compound 1) and tert-butyl
glycinate in
N,N-dimethylformamide, and adding HATU to obtain a compound 2; dissolving the
compound 2 in
N,N-dimethylformamide, adding 1-bromo-3-chloropropane and potassium carbonate,
and heating the
reaction system to 60 C for a certain period of time to obtain a compound 3;
dissolving the compound 3
in N,N-dimethylformamide, and adding tert-butyl 1-piperazinecarboxylate and
potassium iodide for a
reaction to obtain a compound 4; dissolving the compound 4 in a
trifluoroacetic acid solution for removing
8
Date Regue/Date Received 2023-12-12
3,210,863
protective groups to obtain a compound 5; dissolving the compound 5 in N,N-
dimethylformamide, and
adding di-tert-butyl dicarbonate and an acid binding agent to obtain a
compound 6; the compound 6
reacting with (S)-pyrrolidene-2-carbonitrile hydrochloride under the action of
HATU and DIPEA to make a
condensation to obtain a compound 7; removing a protective group of the
compound 7 under the action of
p-toluenesulfonic acid to obtain a compound 8; the compound 8 reacting with
5,8,11,14-tetraoxa-2-azaheptadecanedioic acid-1 -tert-butyl ester under the
action of HATU and DIPEA to
make a condensation to obtain a compound 9; and removing a protective group of
the compound 9 under
the action of p-toluenesulfonic acid to obtain a compound 10 (namely, the
intermediate compound A);
reacting 4,4'-diamino-3,3'-dimethyl biphenyl (compound 11) with di-tert-butyl
dicarbonate to obtain a
compound 12; reacting the compound 12 with monosodium 1-amino-8-naphthol-2,4-
disulfonate and
sodium nitrite to prepare a truncated Evans Blue derivative (compound 13);
removing a Boc protective
group of the compound 13 to obtain a compound 14; reacting the compound 14
with
N-tert-butyloxycarbonyl-L-glutamic acid-l-tert-butyl ester under the action of
HATU and DIPEA to make
a condensation to obtain a compound 15; dissolving the compound 15 in a
trifluoroacetic acid solution for
removing a protective group to obtain a compound 16; and dissolving the
compound 16 in
N,N-dimethylformamide, and adding di-tert-butyl dicarbonate and an acid
binding agent to obtain a
compound 17 (namely, the intermediate compound B); and
reacting the compound 17 with the compound 10 under the action of HATU and
DIPEA to make a
condensation to obtain a compound 18; then removing a protective group of the
compound 18 under the
action of p-toluenesulfonic acid to obtain a compound 19; and reacting the
compound 19 with DOTA-NHS
to obtain the final compound 20 shown in Formula (11-1).
A synthesis route in the above specific steps is as follows:
juk
0,1 L.,õ,NBoc 13'in 0
NH
0 NH
0 OH HATU
4
1 HN
2 3
TFA
BocNa 0 9N
?in')
'1,1 0 NH (C4-1 "'")
HN Be 0 HN
NH (Boc)20
F1)%
HATU 0(5
7
6
5
Ts0H
9
Date Regue/Date Received 2023-12-12
3,210,863
Lit:is3, O.
J'iLN7.5
HNC) ri NCI 3 ENcaq`-'40H
-1
C51"--NO
õ---,-------
N MOH
.N.õ,..J
__________________________ N... ,1 0 NH 1.- Oe.,,,,,,) ti I ON
HATU 0 5
0 0) 5
LL
ic BocHNfj H,NO
B 9 10
HO, ,0
0
OH
HO
0,S - ,s,,0 NH,
(Bcc)20 n O, DIPEA +Na-d - OH
WA
NHEoc
H2N 1-12 ---11' H,N __________________ NHBoc õ
___=,.
1%0
NaNO, H0
11 12 13
0
Hq 0
cr NH, HOYLO)C HR,s-P
, NH, 0__/--\ ¨CI¨ TFA
tia = H N
NH, NHBcc 0
I.- HI NHBoc
NH NH
N'4 N4.4
HATU
0,
Hd HO ¨
S,,
HO0
14 15 16
NC,5(Boc),0, DIFEA
H
0 HCt _c, H
.--- ' NH,
HO,,0 IV 0' H NHBoc
0.,,S' NH, 10, HATU
NH
4 ____________________________________________________
N'4
P N
H
NHBoc HO/S,0
0, -
'.
Hd '. 18 17
NC
,i,
Hd-N3
Hq _
NH, 0 0
H 0 NI'
Nr- \ ..../.__/
J4 NH, \--7
(:= -
/%0
HO -
NNC.:
19 DOTA-
NHS H N-7-
W
FIC1 "5) NH,
OH N ii ai riLyt
¨ ¨...........--õ,.....XN/--Lf_i
\--_/
0 Flo
HO' %C)
OXierN ri
14.
0
25 Preparation methods of other tEB-FAPI compounds in solutions of the
present disclosure are similar
to the preparation method of the compound 20, and preparation can be carried
out basically based on an
existing conventional means with reference to the synthesis route of the
compound 20.
In another aspect, the present disclosure further provides a radiolabeled tEB-
FAPI complex. The
complex is obtained by using the compound shown in Formula (I) of the present
disclosure as a ligand and
labeling the ligand with a radionuclide. The radiolabeled complex can be used
as a novel radioactive
Date Regue/Date Received 2023-12-12
3,210,863
diagnostic and therapeutic probe for tumors, namely, a radionuclide diagnostic
probe or a radionuclide
therapeutic probe. The radionuclide may be selected from any one of 177Lu,
90y, 18F, 64cu, 68Ga, 62cu, 67cu,
86y, 89zr, 99mTc, 89sr, 153sm, 149Tb, 161Tb, 186Re, 188Re, 212pb, 213Bi,
223Ra, 225Ac, 226Th, 227Th, 1311, 211m, Or
"In, and is preferably 68Ga, '77Lu, or "Y.
The complex of the present disclosure preferably has the following structure
shown in Formula (IV):
N,
0
HN R4
HO
s .0
N. H2
O'S
4
OH
N-Li-L2-X-L3-0 141 N' HN
(")'-',8$120
HO -
0 N-Nr.0
LtN
0 Formula (IV)
where in
L1 is a lysine or glutamic acid structure, or a derivative compound structure
containing a lysine or
glutamic acid structure;
L2 is -(CH2),,, wherein n is an integer from 0 to 30, wherein each CH2 may be
individually substituted
or unsubstituted with -0-, -NH-, -(CO)-, -NH(C0)-, or -(C0)-NH-, provided that
no two adjacent CH2
groups are substituted;
L3 is -(CH2),11-, wherein m is an integer from 0 to 30, wherein each CH2 may
be individually
substituted or unsubstituted with -0- or -(C0)-, provided that no two adjacent
CH2 groups are substituted;
x is selected from N, C, 0, S. or the following structures:
0
egsr
N-1
/NH H H N', and 0 =
R3 and R4 are the same or different, and are independently selected from H or
F;
and M is a radionuclide selected from any one of "Ga, 177Lu, or "Y.
In a preferred solution of the complex of the present disclosure, the L2 in
Formula (IV) is -(CH2)u-; n
is an integer from 0 to 16, is more preferably an integer from 0 to 12, and is
further preferably 0, 3, or 10;
wherein each -CH2- may be individually substituted or unsubstituted with -0-, -
NH-, or -(CO)-, provided
that no two adjacent -CH2- groups are substituted. More preferably, the L2 is -
(CH2)o, -NH-CH2-(C0)-,
-NII-C112-(CH20C112)2-CH2-(C0)-,
-NH-C112-(CH2OCH2)4-CH2(C0)-, -(C0)-CH2-(C0)-,
-(C0)-(CH2)24C0)-, -(C0)-CH2-(CH2OCH2)2-C1-12(C0)-, or -(C0)-CH2-(CH2OCH2).4-
CH2(C0)-.
In a preferred solution of the complex of the present disclosure, the L3 in
Formula (IV) is -(CH2).-; m
is an integer from 0 to 20, is more preferably an integer from 1 to 6, and is
further preferably 2 or 3;
wherein each -CH2- may be individually substituted or unsubstituted with -0-,
provided that no two
11
Date Regue/Date Received 2023-12-12
3,210,863
adjacent -CH2- groups are substituted. More preferably, the L3 is -(CH2)3-.
The radiolabeled complex of the present disclosure can be prepared from a
compound containing a
radionuclide and the compound shown in Formula (I) of the present disclosure
by a variety of existing
labeling methods. A labeling method of the present disclosure preferably
includes the following wet
method or freeze-drying method.
A wet labeling solution includes: dissolving an appropriate amount of the
compound shown in
Formula (I) of the present disclosure in a buffer solution or deionized water;
and adding a radionuclide
solution to the obtained solution for a reaction under closed conditions for 5-
40 min to produce a
radionuclide labeled complex.
Alternatively, a freeze-drying labeling solution includes: dissolving an
appropriate amount of the
compound shown in Formula (I) of the present disclosure in a buffer solution
or deionized water; treating
the obtained solution by aseptic filtration, followed by dividing and
separately loading into containers,
freeze-drying and sealing with a stopper to obtain a freeze-dried medicine
box; and then adding an
appropriate amount of an acetic acid solution or a buffer solution to the
freeze-dried medicine box for
dissolution, and adding a corresponding radionuclide solution for a reaction
under closed conditions for
5-40 min to produce a radionuclide labeled complex. The container for loading
is preferably a frozen
storage tube or a controlled antibiotic bottle. An excipient, such as mannitol
and ascorbic acid, can also be
added to the medicine box according to the forming situation of a freeze-dried
powder in the medicine box,
and the medicine box can achieve an optimal forming effect by adjusting the
dose of the compound shown
in Formula (I) of the present disclosure and the excipient.
Products obtained according to the wet labeling solution and the freeze-drying
labeling solution can
be further prepared into injections by conventional treatment (such as
chromatographic separation and
purification, rotary evaporation to remove the solvent, dissolution of
residues with PBS or water or normal
saline, and aseptic filtration).
In a preferred specific embodiment of the present disclosure, with the
compound 20 shown in Formula
(II-1) as a ligand, a preferred preparation method of a radiolabeled compound
20 is a wet labeling method.
The method includes the following steps: dissolving the compound 20 in a
buffer solution or deionized
water; adding a fresh radioactive solution for a reaction under closed
conditions at 37-90 C for 5-40 min,
followed by cooling; adding water for diluting a reaction solution, followed
by separation and purification
with a Sep-Pak C18 chromatographic column; rinsing the chromatographic column
with a buffer solution
or water to remove unreacted radioactive ions; and conducting rinsing with a
hydrochloric acid-ethanol
solution or an ethanol solution, and conducting dilution with normal saline or
PBS, followed by aseptic
filtration to obtain an injection of a radiolabeled complex having the
structure shown in Formula (IV-1),
where a radionuclide M is "Ga, '77Lu, or "Y.
12
Date Regue/Date Received 2023-12-12
3,210,863
NC, H
NaHN
0
HO m
N H2 0 0 0
O'S
OH
HO -C)
0
04
Formula (IV-1)
Another preferred preparation method of a radiolabeled compound 20 of the
present disclosure is a
freeze-drying labeling method. The method includes: dissolving the compound 20
and other necessary
reagents in a buffer solution, and treating the obtained solution by aseptic
filtration, followed by loading
into a frozen storage tube, freeze-drying and sealing to obtain a freeze-dried
medicine box; adding an
appropriate amount of a buffer solution to the freeze-dried medicine box for
dissolution, and adding a
newly prepared radioactive solution for a reaction under closed conditions at
37-120 C for 5-40 min,
followed by cooling; adding water for diluting a reaction solution, followed
by separation and purification
with a Sep-Pak C18 chromatographic column; rinsing the chromatographic column
with a buffer solution
or water to remove unreacted radioactive ions; and conducting rinsing with a
hydrochloric acid-ethanol
solution or an ethanol solution, and conducting dilution with normal saline or
PBS, followed by aseptic
filtration to obtain an injection of a radiolabeled complex having the
structure shown in Formula (IV-1),
where a radionuclide M is 68Ga,177Lu, or NY.
Other chemicals used in the above synthesis steps are commercially available
products.
The buffer solution is a substance for stabilizing the pII value of a reaction
solution, and may be
acetate, lactate, tartrate, malate, maleate, succinate, ascorbate, carbonate,
phosphate and a mixture thereof.
In another aspect, the present disclosure also provides application of the tEB-
FAPI compound shown
in Formula (I) or a pharmacologically acceptable salt thereof in preparation
of medicines in nuclide therapy
or imaging of tumors with high expression of FAP.
The present disclosure also provides application of the radiolabeled tEB-FAPI
complex shown in
Formula (IV) in nuclide therapy and imaging of tumors with high expression of
FAP.
In preferred application of the present disclosure, the complex is formulated
as an injection, and then
intravenously injected into patients with tumors with high expression of FAP.
In the application of the present disclosure, the tumors with high expression
of FAP include, but are
not limited to, breast cancer, ovarian cancer, lung cancer, colorectal cancer,
gastric cancer or pancreatic
cancer.
The present disclosure provides a truncated Evans Blue modified fibroblast
activation protein
inhibitor tEB-FAPI and a radionuclide labeled complex thereof, and also
provides a preparation method
and a labeling method of the compound. Biological test results show that the
inhibitor has the
characteristics of significantly prolonging the half-life in blood
circulation, improving the uptake and
accumulation in tumors and prolonging the retention time. Such novel
properties are not available in other
13
Date Regue/Date Received 2023-12-12
3,210,863
FAPI imaging agents at present, and the inhibitor is suitable for nuclide
therapy and imaging of tumors
with high expression of FAP.
BRIEF DESCRIPTION OF DRAWINGS
FIG. 1 is a diagram showing mass spectrum of compound 2 in Example 1 of the
present disclosure.
FIG. 2 shows nuclear magnetic hydrogen spectrum of compound 2 in Example 1 of
the present
disclosure.
FIG. 3 shows nuclear magnetic carbon spectrum of compound 2 in Example 1 of
the present
disclosure.
FIG. 4 is a diagram showing mass spectrum of compound 3 in Example 1 of the
present disclosure.
FIG. 5 shows nuclear magnetic hydrogen spectrum of compound 3 in Example 1 of
the present
disclosure.
FIG. 6 is a diagram showing mass spectrum of compound 4 in Example 1 of the
present disclosure.
FIG. 7 shows nuclear magnetic hydrogen spectrum of compound 4 in Example 1 of
the present
disclosure.
FIG. 8 shows nuclear magnetic carbon spectrum of compound 4 in Example 1 of
the present
disclosure.
FIG. 9 is a diagram showing mass spectrum of compound 7 in Example 1 of the
present disclosure.
FIG. 10 shows nuclear magnetic hydrogen spectrum of compound 7 in Example 1 of
the present
disclosure.
FIG. 11 shows nuclear magnetic carbon spectrum of compound 7 in Example 1 of
the present
disclosure.
FIG. 12 is a diagram showing mass spectrum of compound 10 in Example 1 of the
present disclosure.
FIG. 13 is a diagram showing mass spectrum of compound 20 in Example 1 of the
present disclosure.
FIG. 14 is a diagram showing mass spectrum of compound in Example 10 of the
present disclosure.
FIG. 15 is a diagram showing the mass spectrum of a compound in Example 11 of
the present
disclosure.
FIG. 16 is an HPLC chromatogram of compound 10 in Example 1 of the present
disclosure.
FIG. 17 is an HPLC chromatogram of compound 17 in Example 1 of the present
disclosure.
FIG. 18 is an HPLC chromatogram of a reaction system of compound 17 and
compound 10 in
Example 1 of the present disclosure.
FIG. 19 is an HPLC chromatogram of compound 19 in Example 1 of the present
disclosure.
FIG. 20 is an HPLC chromatogram of a reaction system of compound 19 and DOTA-
NHS in Example
1 of the present disclosure.
FIG. 21A and FIG. 21B show MicroPET imaging of a 68Ga labeled tEB-FAPI complex
of the present
disclosure and 68Ga labeled FAPI-02 in normal mice.
FIG. 22 shows SPECT imaging of 177Lu-tEB-FAPI prepared in Example 40 of the
present disclosure
14
Date Regue/Date Received 2023-12-12
3,210,863
in normal mice at different time points.
FIG. 23 shows SPECT imaging of "Thu-tEB-FAPI prepared in Example 40 of the
present disclosure
in xenograft model mice with human pancreatic cancer at different time points.
DETAILED DESCRIPTION OF EMBODIMENTS
Technical solutions of the present disclosure are further explained and
described below in conjunction
with specific embodiments and attached drawings.
Example 1: Preparation of a tEB-FAPI conjugatesconnector (compound 20)
Synthesis of compound 2:
Compound 1 (6-hydroxy-4-quinolinecarboxylic acid, 1.89 g, 10.0 mmol), tert-
butyl glycinate (1.89 g,
10.0 mmol), HATU (3.8 g, 10.0 mmol) and N,N-diisopropylethylamine (2.6 g, 20.0
mmol) were
sequentially put into 30 mL of N,N-dimethylformamide in a 100 mL flask. A
reaction mixture was stirred
overnight, and reduced pressure distillation was conducted to remove the
solvent to obtain a crude product.
Then purification was conducted with a silica gel column (a ratio of
dichloromethane to methanol was 30:1)
to obtain a white solid compound 2 with a yield of 87%. FIG. 1 is a diagram
showing the mass spectrum of
compound 2. FIG. 2 shows nuclear magnetic hydrogen spectrum of the compound 2.
FIG. 3 shows the
nuclear magnetic carbon spectrum of compound 2.
Synthesis of compound 3:
Compound 2 (1.51 g, 5.0 mmol), 1-bromo-3-chloropropane (1.55 g, 10.0 mmol) and
potassium
carbonate (1.38 g, 10.0 mmol) were sequentially put into 50 mL of N,N-
dimethylformamide in a 100 mL
flask. The system was heated to 60 C and stirred overnight at 60 C, and
reduced pressure distillation was
conducted to remove the solvent to obtain a crude product. Then purification
was conducted with a silica
gel column (a ratio of dichloromethane to methanol was 50:1) to obtain a white
solid compound 3 with a
yield of 63%. FIG. 4 is a diagram showing the mass spectrum of compound 3.
FIG. 5 shows nuclear
magnetic hydrogen spectrum of compound 3.
Synthesis of compound 4:
Compound 3 (0.76 g, 2.0 mmol), tert-butyl 1-piperazinecarboxylate (0.55 g, 3.0
mmol) and potassium
iodide (0.49 g, 3.0 mmol) were sequentially put into 30 mi., of acetonitrile
in a 100 mL flask. The system
was heated to 60 C and stirred overnight at 60 C, and reduced pressure
distillation was conducted to
remove the solvent to obtain a crude product. Then purification was conducted
with a silica gel column (a
ratio of dichloromethane to methanol was 30:1) to obtain a white solid
compound 4 with a yield of 58%.
MS(ESI)m/z calculated for [C281-140N4061: 528.29; found: 529.10 [M+H]t FIG. 6
is a diagram showing the
mass spectrum of compound 4. FIG. 7 shows nuclear magnetic hydrogen spectrum
of compound 4. FIG. 8
shows the nuclear magnetic carbon spectrum of compound 4.
Synthesis of compound 5:
Compound 4 (0.52 g, 1.0 mmol) was dissolved in 10 mL of a mixed solution of
dichloromethane and
trifluoroacetic acid (at a volume ratio of 9:1) in an ice bath. The system was
heated to room temperature for
Date Regue/Date Received 2023-12-12
3,210,863
a reaction for 2 h, and after the reaction was completed, reduced pressure
distillation was conducted to
remove the solvent. Then the resulting product was dissolved in 10 mL of N,N-
dimethylformamide for
later use.
Synthesis of compound 6:
Di-tert-butyl dicarbonate (0.22 g, LO mmol) and N,N-diisopropylethylamine
(0.39 g, 3M mmol) were
separately added to an N,N-dimethylformamide solution of the compound 5. The
system was stirred
overnight at room temperature, and reduced pressure distillation was conducted
to remove the solvent to
obtain a crude product. Then purification was conducted with a silica gel
column (a ratio of
dichloromethane to methanol was 10:1) to obtain a white solid compound 6 with
a yield of 72%.
Synthesis of compound 7:
Compound 6 (0.47 g, 1.0 mmol), (S)-pyrrolidene-2-carbonitrile hydrochloride
(0.13 g, 10.0 mmol),
HATU (0.38 g, 1.0 mmol) and N,N-diisopropylethylamine (0.26 g, 2.0 mmol) were
sequentially put into 10
mL of N,N-dimethylformamide in a 100 mL flask. A reaction mixture was stirred
at room temperature until
a reaction was completed, and reduced pressure distillation was conducted to
remove the solvent to obtain
a crude product. Then purification was conducted with a silica gel column (a
ratio of dichloromethane to
methanol was 50:1) to obtain a white solid compound 7 with a yield of 85%.
FIG. 9 is a diagram showing
mass spectrum of compound 7. FIG. 10 shows nuclear magnetic hydrogen spectrum
of compound 7. FIG.
11 shows the nuclear magnetic carbon spectrum of compound 7.
Synthesis of compound 8:
Compound 7 (0.55 g, 1.0 mmol) and p-toluenesulfonic acid monohydrate (0.27 g,
1.5 mmol) were
sequentially put into 10 mL of acetonitrile in a 100 mL flask. The reaction
system was heated to 60 C and
stirred until a reaction was completed, and reduced pressure distillation was
conducted to remove the
solvent to obtain a crude product.
Synthesis of a compound 9:
5,8,11,14-tetraoxa-2-azaheptadecanedioic acid-l-tert-butyl ester (0.19 g, 1.0
mmol), HATU (0.38 g,
1.0 mmol), N,N-diisopropylethylamine (0.26 g, 2.0 mmol) and 10 mL of N,N-
dimethylformamide were
separately put into the reaction flask of compound 8. A reaction mixture was
stirred overnight, and reduced
pressure distillation was conducted to remove the solvent to obtain a crude
product. Then purification was
conducted with a silica gel column (ratio of dichloromethane to methanol was
50:1) to obtain a white solid
compound 9 with a yield of 64%.
Synthesis of compound 10:
Compound 9 (0.61 g, 1.0 mmol) and p-toluenesulfonic acid monohydrate (0.27 g,
1.5 mmol) were
sequentially put into 10 mL of acetonitrile in a 100 mL flask. The reaction
system was heated to 60 C and
stirred until a reaction was completed, and reduced pressure distillation was
conducted to remove the
solvent to obtain a crude product. Then purification was conducted with a
silica gel column (ratio of
dichloromethane to methanol was 10:1) to obtain a white solid compound 10 with
a yield of 59%. MS(ESI)
16
Date Regue/Date Received 2023-12-12
3,210,863
m/z calculated for [C351151N7081: 697.38; found: 698.43 [M+H]t FIG. 12 is a
diagram showing the mass
spectrum of the compound 10.
A synthesis route in the above steps is as follows:
0
At, N., H2N H
= NH
0 AH
HN-Th
---'-] 0 NH
_________________ i _____________ 3, ¨Pr 1µ1==,/
\ A 0 ,
. tw
HATU HO Cl,õ."\--* 0 i
= =H *
Nr 4
1 2 3
TFA
'I
Eoctv---) it CN
14 y Boces) 0
Lj c.,N,1
rA01-1 Illsr''l
1
ilir N-' . _ .4_
.K
HATU % 0 NH (B 020
0
7 * N'
10 ,r
a
5
,If Ts0H
0 0 ii till N3
I? NC 5
N
1-14 AO NC H 139*OH N
r'N'0 ii. -.. Ts0H
0 pc-, 0N,J
HATU jp
o .õ_ o i
BocHN() H2N()
8 9 10
Synthesis of compound 12:
4,4'-Diamino-3,3'-dimethyl biphenyl (compound 11) (2.12 g, 10.0 mmol), di-tert-
butyl dicarbonate
(2.2 g, 10.0 mmol), N,N-diisopropylethylamine (1.3 g, 10.0 mmol) and 20 mL of
dichloromethane were
separately put into a 100 mL flask, and stirred overnight at room temperature.
After monitoring by HPLC
that a reaction was completed (r.t. was 10.13 min), reduced pressure
distillation was conducted to remove
the solvent to obtain a crude product. Then purification was conducted with a
silica gel column (ratio of
petroleum ether to ethyl acetate was 5:1) to obtain a white solid compound 12
with a yield of 59%.
Synthesis of compound 13:
Compound 12 (0.31 g, 1.0 mmol) and 4 mL of acetonitrile were separately put
into a 50 mL flask in an
ice bath, 1.5 mL of 2 M hydrochloric acid was added dropwise to the reaction
flask for a reaction for 15
min, and sodium nitrite (0.068 g, 1.0 mmol) was added to 2 mL of water for
dissolution and then added
dropwise to the reaction flask for reaction for half an hour to obtain a
solution A for later use. Monosodium
1-amino-8-naphthol-2,4-disulfonate (0.33 g, 1.0 mmol), sodium carbonate (0.105
g, 1.0 mmol) and 5 mL of
water were added to another 50 mL reaction flask in an ice bath to obtain a
solution B, and the solution A
was slowly added dropwise to the solution 13 and stirred for reaction for 2 h
in the ice bath. Then
purification was conducted with a reversed phase column, followed by freeze-
drying to obtain pure
compound 13 with a yield of 47%.
17
Date Regue/Date Received 2023-12-12
3,210,863
Synthesis of compound 14:
Compound 13 (0.52 g, 1.0 mmol) was dissolved in trifluoroacetic acid in an ice
bath. The system was
heated to room temperature for a reaction for 2 h, and after the reaction was
completed, reduced pressure
distillation was conducted to remove the solvent to obtain a crude product.
Then purification was
conducted on the crude product with a reversed phase column, followed by
freeze-drying to obtain pure
compound 14 with a yield of 73%.
Synthesis of compound 15:
Compound 14 (0.54 g, 1.0 mmol), N-tert-buty loxycarbonyl-L-glutamic acid- 1-
tert-butyl ester (0.30 g,
1M mmol), HATU (0.38 g, 1.0 mmol), N,N-diisopropylethylamine (0.26 g, 2.0
mmol) and 10 mL of
N,N-dimethylformamide were separately put into a 100 mL flask. A reaction
mixture was stirred until a
reaction was completed, and reduced pressure distillation was conducted to
remove the solvent to obtain a
crude product. Then purification was conducted on the crude product with a
reversed phase column,
followed by freeze-drying to obtain pure compound 15 with a yield of 52%.
Synthesis of compound 16:
Tert-butyl and Boc protective groups were removed using a mixture of
thioanisole, 1,2-ethanedithiol,
anisole and TFA (at a ratio of 5:3:2:90) at room temperature. After a reaction
was completed, the TFA was
removed by an argon flow, and the resulting product was dissolved in 10 mL of
N,N-dimethylformamide
for later use.
Synthesis of compound 17:
Di-tert-butyl dicarbonate (0.22 g, LO mmol) and N,N-diisopropylethylamine
(0.39 g, 3M mmol) were
separately added to an N,N-dimethylformamide solution of the compound 16. The
system was stirred
overnight at room temperature, and a reaction was completed according to
monitoring by HPLC (r.t. was
10.84 min). Reduced pressure distillation was conducted to remove the solvent
to obtain a crude product.
Then purification was conducted on the crude product with a reversed phase
column, followed by
freeze-drying to obtain pure compound 17 with a yield of 43% in two steps.
Synthesis of compound 18:
Compound 17 (0.77 g, 1.0 mmol), compound 10 (0.51 g, LO mmol), HATU (0.38 g,
1.0 mmol),
N,N-diisopropylethylamine (0.26 g, 2.0 mmol) and 10 mL of N,N-
dimethylformamide were separately put
into a 50 mL flask. A reaction mixture was stirred for a reaction, and the
reaction was completed according
to monitoring by HPLC (r.t. was 12.16 min). Reduced pressure distillation was
conducted to remove the
solvent to obtain a crude product. Then purification was conducted on the
crude product with a reversed
phase column, followed by freeze-drying to obtain pure compound 18 with a
yield of 55%.
Synthesis of compound 19:
Compound 15 (0.13 g, 0.1 mmol) and p-toluenesulfonic acid monohydrate (0.05 g,
0.3 mmol) were
sequentially put into 5 mL of acetonitrile in a 25 mL flask. The reaction
system was heated to 60 C and
stirred for reaction, and the process of removing protective groups was
monitored by HPLC until the
18
Date Regue/Date Received 2023-12-12
3,210,863
reaction was completed (r.t. was 10.47 min). Reduced pressure distillation was
conducted to remove the
solvent to obtain a crude product. Then purification was conducted on the
crude product with a reversed
phase column, followed by freeze-drying to obtain pure compound 19 with a
yield of 61%.
Synthesis of compound 20:
Compound 19 (0.12 g, 0.1 mmol), DOTA-NHS (0.05 g, 0.1 mmol) and N,N-
diisopropylethylamine
(0.04 g, 0.3 mmol) were sequentially put into 5 mL of N,N-dimethylformamide in
a 25 mL flask. The
reaction system was stirred for reaction at room temperature, and the process
of removing protective
groups was monitored by HPLC until the reaction was completed (r.t. was 11.35
min). Reduced pressure
distillation was conducted to remove the solvent to obtain a crude product.
Then purification was
conducted on the crude product with a reversed phase column, followed by
freeze-drying to obtain pure
compound 20 with a yield of 53%. MS(ESI)m/z calculated for [C801-
1104N16024S21: 1736.69; found:
1737.743 [M+H]+. FIG. 13 is a diagram showing mass spectrum of compound 20.
A synthesis route in the above steps is as follows:
FIR ,
--- - NH,
0 OH
0. ¨ HQ 0
H,
(Boc),O, DIPEA iNa-u - OH
TFA
H, It __ 1. H,N NHBcc ,4 NH Boc
)
S,
NaN 0, lid
11 12 13
C--/--¨CH
O'S- NH' HO)LYLO)C Hqs,0 H. S% NH,
H NHBoc 0' TFA 0%
NI'l NH NH
14 NV
0. ¨ HATU
a.
Hd 0
)1
Hc:cyst7 I>
14 15 18
H
5\ (Boc),O,
DIFEA
HN
cr, NH,
HR .., NHBoc
10, HATU H
H 1 ________ IP
)P1 H iScb
NHBoc HO
CL, ¨
S.,
Hd 18 17
\sr 51;4µC-6
HN
.¨
Hq
0' H = * NHO
NIS NH,
19 DOTA-
NHS 0 r.i-3
HN-) ---N
Hq "
1)1
)P \ ---24
c'',s----0
HO
c f Xici 1 ) IN. )- - \ic7cH3
Ho_e
19
Date Regue/Date Received 2023-12-12
3,210,863
Examples 2- Examples 16
Compounds in Examples 2- Examples 16 have structures shown in Formula (II-2)
to Formula (II-16)
respectively, and preparation methods of the compounds can refer to the
preparation method in Example 1.
The glutamic acid structure reacting with the compound 14 was substituted with
a lysine structure, or the
5,8,11,14-tetraoxa-2-azaheptadecanedioic acid-l-tert-butyl ester reacting with
the compound 8 was
substituted with 5,8,11-trioxa-2-azatridecanediic acid- 1-tert-
butyl ester, tert-butyl
9-amino-4,7-dioxazononate, tert-butyl glycinate or other suitable compounds,
or the
(S)-pyrrolidene-2-carbonitrile hydrochloride reacting with the compound 6 was
substituted with
3,3-difluoropyrrolidene hydrochloride, or the above compounds were substituted
at the same time to obtain
corresponding structures as follows:
HN-376
=
HO
µ13j)
0'
41
HP0
0,n; N5\e
Formula (11-2),
NS 11
Ni-Na
0
= )0 tot
o Ne_14._r
01.1
%*41 IP HN
'1= 3 tAN
jt)
Formula (II-3),
H
H14_3_Na
0
HO 0 0 0
14' HN 0
0,
Hd
0Hr N. V\
o NN ) OH
\
HO--e
0 Formula (II-4),
NG H
0\\
HN-dr.
= z
HO
. 0
cp
S' * NH2 0 0 0
N \
FN
= '= C) ..i===
Formula (I1-5),
Date Regue/Date Received 2023-12-12
3,210,863
0 Ns H
0 F F
HO
t,0 NH2 0 0 -
0'
46A\" N . = 11)1111 NnN..../-/C3 * /
Hid
OTN 0
k 6H
HO-
Formula (1I-6),
0 NS H
HN--)\--N F
0 F
Ho ,0 ¨
o 0/
(3..= NH2 OH
ii
HO
OHPN'y
OH
0 NIL/N)
140--
Formula (1I-7),
NC, H
0
HN--)\-N.,/e'l-F
0
F
HO\ 0
, 0 -
0
S' NH2
H
0,
0
HO '()
/Th
OH FN N'\O
) [
0 L,,,N OH
Formula (II-8),
HO4
0 N16, H
HNi-- Na
0
_
HO
õ...;0 H2
o = *H 14 A A eY--^-11-irmr-11Th\__7--r-P
lii N' HN 0 0
%
Hd w-N/MN
0/N NJ OH
HO-
Formula (II-9),
21
Date Regue/Date Received 2023-12-12
3,210,863
0 NC H
.s
= HN-)0 -
HO .0 -
' hI2OH *0 /
N * = Nr---/-
\ -/
14' HN \ ¨11
0,
'0
ow
N
HO--.
Formula (II-10),
,H
..._, O Ncy_ra
HN
0
HO n
NH 2 OH 0 0
* /
N
N
0
Hd
0
kN ) ' OH
\ ....../N
HO4
0
Formula (II-11),
is, H
HN C-5-Na
=
HO, ,0
0 0
N/
411\ N
HCS '1)
ar--isl
0),tiN N le
Formula (II-12),
NCt H
HNF
0 F
HoCk' NH2
HN 0 0
0
Hd `c)
dtrni le
L,N
H0 Formula (II-13),
22
Date Regue/Date Received 2023-12-12
3,210,863
0 NS3 H
HN---)N(F
----
0 F
HO 0 --
NH2 0
OH N . W=
i m
0 "
% I
HO
0
N ) OH
Lig
HO-
Formula (II-14),
0 NS. H
FK)0 ¨
' NH2 0 F
0' /
p . 4 wily=-=,/ .r..0-cr-J.0
N' H FPI
0,
i 6-7
o LyN
HO-
Formula (II-15),
or
Ks H
=
HN- 0
HO
o- 0 ) ---I-;
NH2 0 0 ¨
H .
OH ,N 4 4 ,,,K.,(rg Ir,õ0,,,Ø.,,,,O,.."Ø----õ,11,,. /Th ,--P ir N
.
N\ /
.-.C)
HO
7N NiSH
HO-
0
Formula (11-16).
The mass spectrum of compound (II-10) in Example 10 is shown in FIG. 14. The
mass spectrum of
compound (II-11) in Example 11 is shown in FIG. 15.
Examples 17- Examples 38:
With reference to the preparation methods in Examples 1- Examples 16, a tEB-
FAPI compound shown
in the following Formula (I) was prepared.
HO
, .0
- NH2
CYS
OH H
N N¨L1-1-2¨X-1-3-Ri
I
NI'
Fld 'ID R2
Formula (I)
Ex a
mpl X L1 L2 L3 L4 RI R2
e
23
Date Regue/Date Received 2023-12-12
3,210,863
cta-
1-isr\N- 17 h"_) Iti!",IR
\-/ H .
s--T. 0
,I1,-(0-.µ,..0 ,,.
\- 1 tiN.1- HY-N,)\õ,f;
- NI 1 0 õ.51."51
18 1-NCN-1....-11' o
63-*6ns-µ '''.µ 144' i--.,-: '1=-+
L.t. HOe
%..-8
FNH II 0 545l1) 4,:: -
14,õ \r,
19 -N N-
0
µ'IL'ICHr2 '--'4""µ µ11.--V
N HO2filj
_4l_3
20 Z-F`I...P-1 ' \ - --c. \-'11 ''') .- n&-µ =,,,N,;140'
HY ')
.-=nr-\N-1 1" / 11, = OLNY1 %^µ
N
21 \---Fi'.
H 0 '110---4 "---1,-4-.., = ,
Ir KV- H
,=,.
N-I WI 5WN1_F '--1
22 ''- \-\i---ic
0/A,NL), OH
HO-
-
Ote_A
ililL IN1
0 0
23 4111e i)c ,...-11-13^-)-6 µ F10-4
NH Hy
24 i'NI L
H H \ j--cõ.
H 0
,
t/N..., F Z
25 N 5,1 HOOC ) 0
II
'4.-\_:/-c,'
N HOOC-/ \-
COOH
26
0 51..., 0
r,\
r'N-- i '''' /
).-;
N.N= 1 N.A"---' -.'"--Chrµ ,,,.-µ 1-11-1." 4--/ 1- .
-c --4-t=
HO
orl
_A- - r
-NH.-\_=/µ r-,HN P14,
27 \-"Ii-%. \. N.-11-''(cr"-)-6 nrµ ;; '-11-v-41--
/ 744 H
ILOLN-
=1:
0 0
11.......tr'<, HOC
O,1
28 ,,,18',, NH '1/4 -.1t--+'6 '''Y -N10. i -tillL 1 HD
c'''' \ --/
I Hooc-/ \
-COOH
7 0 NC H
= il,,XN3 0
29 N
It, ri5tflaH
N''1D
i\s. . . . . õ \ JA
µ J Nes-
- - - i=-= 0 - f
fj
W '0
ai H
24
Date Regue/Date Received 2023-12-12
3,210,863
0, 0 1)LN .'3-,
30 S
N F101
31 0
1
ilY
-
-r tl,7_,
Foct 0
Hil \_virick r µ,Il Hooc-, ...1N
32 C ---.0,...-0.--r, ./..(0,.4 1.11;,õ,41L!
-41- v How-,
...'\
0 0
-3i
4 ft j
-
1
_ Ho_z
\-}-iCil II 31.)41-3 S
34 v......-õ, _
1 _
-,\
35 HO- NH µ,11----*-4,--...e. v".....---y
-
.
Hq
e.c
0 0 Pi.:ISA
9H/ Q-31' 36 i-04- NH - v----,./ - F
I =-ctl,:õ.1 oi, IV Ni
di
po\
0 0
.\),e. po,7- s
37 ;---0-i NH N,,11......-,,----k-y, \ / -
1
0 0 74,
r- \ \ }ti Cl.raN
Olt,
ti,"`y0
3 8 --tk_r-1 NH \-).-f....1/4 v-------y. -
_
Example 39: Preparation of a radioactive 68Ga labeled tEB-FAPI complex
Wet method: A hydrochloric acid solution of about 18.5-1,850 MBq of 68GaC13
(rinsed from a
germanium-gallium generator) was added to an acetic acid-acetate solution (LO
g/L) containing 0.5 mL of
compound 20 prepared in Example 1 in a centrifuge tube, and the reaction was
carried out at 37 C for 20
min. A small C18 separation column was slowly rinsed with 10 mL of anhydrous
ethanol first, and then
rinsed with 10 mL of water. A resulting labeled solution was diluted with 10
mL of water, and then
sampled to the separation column. Unlabeled 68Ga ions were removed with 10 mL
of water, and rinsing
was conducted with 0.3 mL of a 10 mM ethanol solution of HC1 to obtain 68Ga
labeled tEB-FAPI complex.
The rinsed solution was diluted with normal saline, followed by aseptic
filtration to obtain an injection of
the 68Ga labeled tEB-FAPI complex.
Date Regue/Date Received 2023-12-12
3,210,863
Freeze-drying method: A hydrochloric acid solution of about 18.5-1,850 MBq of
68GaC13 (rinsed with
a germanium-gallium generator) was added to a freeze-dried medicine box
containing the compound 20,
and uniformly mixed for a reaction at 37 C for 20 min. A small C18 separation
column was slowly rinsed
with 10 mL of anhydrous ethanol first, and then rinsed with 10 mL of water. A
resulting labeled solution
was diluted with 10 mL of water, and then sampled to the separation column.
Unlabeled 68Ga ions were
removed with 10 mL of water, and rinsing was conducted with 0.3 mL of a 10 mM
ethanol solution of HC1
to obtain a rinsed solution of a complex. The rinsed solution was diluted with
normal saline, followed by
aseptic filtration to obtain an injection of the 68Ga labeled tEB-FAPI
complex.
Example 40: Preparation of a 177Lu labeled tEB-FAPI complex
Wet method: A sodium acetate solution of about 18.5-1,850 MBq of 177LuC13 was
separately added to
an acetic acid-acetate solution (1.0 g/L) containing 0.5 mL of compound 20 in
Example 1, the compound
(Formula (11-2)) in Example 2 and the compound (Formula (II-3)) in Example 3
in three centrifuge tubes,
and reaction was carried out at 90 V for 20 min. A small C18 separation column
was slowly rinsed with
10 mL of anhydrous ethanol first, and then rinsed with 10 mL of water.
Resulting labeled solution was
diluted with 10 mL of water, and then sampled to the separation column.
Unlabeled 177Lu ions were
removed with 10 mL of water, and rinsing was conducted with 0.3 mL of a 10 mM
ethanol solution of HC1
to obtain three i77Lu labeled tEB-FAPI complexes. The rinsed solutions were
diluted with normal saline,
followed by aseptic filtration to obtain injections of the three 177Lu labeled
tEB-FAPI complexes.
Freeze-drying method: A sodium acetate solution of about 18.5-1,850 MBq of
177LuC13 was separately
added to three freeze-dried medicine boxes containing compound 20 in Example
1, the compound (Formula
(II-2)) in Example 2 and the compound (Formula (II-3)) in Example 3, and
uniformly mixed for reactions
at 90 C for 20 min. A small C18 separation column was taken, slowly rinsed
with 10 mL of anhydrous
ethanol first, and then rinsed with 10 mL of water. Resulting labeled
solutions were diluted with 10 mL of
water, and then sampled to the separation column. Unlabeled F77Lu ions were
removed with 10 mL of water,
and rinsing was conducted with 0.3 mL of a 10 mM ethanol solution of HC1 to
obtain rinsed solutions of
three 177Lu labeled tEB-FAPI complexes. The rinsed solutions were diluted with
normal saline, followed
by aseptic filtration to obtain injections of the three 177Lu labeled tEB-FAPI
complexes.
Experimental Example: Analysis and application effect
1. HPLC analysis and identification
An HPLC system was as follows: SHIMADZUTm LC-20A; and a C18 chromatographic
column
(YMCTm, 3 pm, 4.6*150 mm) was used for analysis. Detection was conducted at a
wavelength of 254 nm
and a flow rate of 1 mL/min according to the following rinsing gradient: at 0-
3 min, 10% of acetonitrile
and 90% of water (50 mM ammonium acetate) were remained unchanged; at 3-16
min, the system was
increased to include 90% of acetonitrile and 10% of water (50 mM ammonium
acetate); at 16-18 min, 90%
of acetonitrile and 10% of water (50 mM ammonium acetate) were remained; at 18-
20 min, the system was
reduced to include 10% of acetonitrile and 90% of water (50 mM ammonium
acetate); and at 20-22 min,
26
Date Regue/Date Received 2023-12-12
3,210,863
10% of acetonitrile and 90% of water (50 mM ammonium acetate) were retained.
Compound 10, compound 17, a reaction system of compound 10 and compound 17,
compound 19 and
a reaction system of compound 19 and DOTA-NIIS in Example 1 were identified
and analyzed according
to the above system. Results obtained are shown in FIG. 16 to FIG. 20.
The two radiolabeled probes prepared in Example 39 and Example 40 were used as
experimental
agents below, and determination of properties of the probes is described as
follows.
2. MicroPET imaging of a 68Ga labeled tEB-FAPI complex in normal mice
68Ga-tEB-FAPI with a purity of greater than 95% was prepared by the method in
Example 39. 3.7
MBq of the 68Ga-tEB-FAPI or 68Ga-FAPI-02 (as a control) was intravenously
injected into tails of normal
FVB mice anesthetized with isoflurane. Then MicroPET imaging was conducted
after administration for
0-120 min. Results are shown in FIG. 21A and FIG. 21B. The results show that
the 68Ga-tEB-FAPI
complex in Example 39 has higher uptake in the cardiac blood pool of the mice
(FIG. 21A), while the
68Ga-FAPI-02 is almost completely cleared in the test period (FIG. 21B),
indicating that the half-life in
blood circulation can be obviously prolonged by introducing truncated Evans
Blue.
3. Uptake experiment of a '77Lu labeled tEB-FAPI complex in tumors in
xenograft model mice with
human pancreatic cancer
177Lu-tEB-FAPI with a purity of greater than 95% was prepared by the method in
Example 40. 1.3
MBq of the '77Lu-tEB-FAPI was intravenously injected into tails of normal mice
and xenograft model mice
with human pancreatic cancer separately. SPECT imaging was conducted at
different time points after
injection. Results are shown in FIG. 22 and FIG. 23. The results show that the
177Lu-tEB-FAPI has good
pharmacokinetics in the normal mice, and can be continuously taken up by tumor
tissues in the xenograft
model mice with human pancreatic cancer and maintained for more than 48 h,
indicating that the tEB-FAPI
has the characteristics of significantly improving the uptake in tumors and
prolonging the retention time,
and can be used as a therapeutic agent and an imaging agent for tumors.
In summary, the truncated Evans Blue modified fibroblast activation protein
inhibitor provided by the
present disclosure can significantly prolong the half-life in blood
circulation, improve the uptake and
accumulation in tumors and prolong the tumor retention. Such novel properties
are not available to other
FAPI imaging agents. According to further preclinical animal level studies and
clinical studies, it is proven
that the inhibitor is expected to be used in radionuclide therapy and imaging
of tumors with high
expression of FAP.
Although the present disclosure has been described in detail by general
descriptions, specific
embodiments and tests above, it is obvious to persons skilled in the field
that some modifications or
improvements can be made on the basis of the present disclosure. Therefore,
all the modifications or
improvements made without departing from the spirit of the present disclosure
shall fall within the
protection scope of the present disclosure.
27
Date Regue/Date Received 2023-12-12