Note: Descriptions are shown in the official language in which they were submitted.
1
Title: A process for enzymatic synthesis of amides from amines and carboxylic
acids or esters
Field of the invention
The present invention relates to a process for enzymatic synthesis of amides
from amines and
carboxylic acids or esters using a lipase.
Background of the invention and prior art
Amide linkage is important in development of numerous compounds, such a
pharmaceutical
drugs and polymers. Several processes for direct catalytic amidation have been
developed
over the years.
In thermal amidation, no catalyst may be used. This process is performed at
high temperature
(> 140 C) and the yield is dependent on the temperature used, the
concentration of the
substrate, the solvent used and other parameters.
Metal-based amidations have been done using boron-based catalysts or palladium-
based
catalyst. Although higher yield can be obtained compared to thermal amidation,
the processes
are expensive and time consuming. Recycling of catalysts and solvents is
challenging.
Neither thermal nor metal based amidations are environmentally friendly
processes. Several
attempts have been made to improve the efficiencies of the processes and
reduce the costs
and carbon food-print.
In amidation processes, water must be removed to improve the yield of the
processes. Most
of amidation processes are therefore performed under reduced pressure. This
increases costs
and thus increases difficulties for large scale amidation. Molecular sieves
may be used as well,
but these are still expensive for use at large scale. A Dean-Stark apparatus
may be used as well
to remove water from an amidation process.
Enzymatic amidation has been developed over the years using different kind of
enzymes like
lipases. These so-called biocatalysts can be used at lower temperature and
show good
selectivity. However, current technologies show very limited substrate scope
and often
require long reaction times (days). Combining enzymatic amidation with a
palladium catalyst
may result in a yield of about 70% as shown by Palo-Nieto et al., ACS Catal.,
2016, 6, 3932-
3940.
Another drawback of biocatalysts is costs. To reduce the costs and improve
efficiency of the
amidation process, the enzymes can be immobilized e.g. on beads during the
reaction. This
allows recirculation of the enzyme. The use of flow reactors has further
improved the
biocatalytic amidation process. However, recirculation of the lipase is both
time- and cost-
ineffective.
Up to date, there is no environmentally friendly catalytic amidation process
that is sufficiently
efficient and cost effective to be used for large scale production. This is a
top priority of the
American Chemical Society Green Chemistry Pharmaceutical Roundtable
(https://www.acsgcipr.org). Today, most methods utilize stoichiometric amounts
of toxic
activating reagents, Dunetz et. al. Org. Process. Res. Dev. 2016, 20, 140.
Thus, there is still a
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
2
need for a greener and more cost effective amidation process that can be used
at a larger
scale.
Capsaicinoids are commonly used in food environmentally friendly products.
Capsaicin is also
widely used in the pharmaceutical industry. Capsaicin is for example used as
an analgesic in
topical ointments and dermal patches to relieve minor aches and pains of
muscles and joints
associated with arthritis, backache, strains and sprains, or to reduce the
symptoms of
peripheral neuropathy.
Capsaicinoids can be isolated from natural sources (e.g. Capsicum spp pepper
fruits), but this
gives predominantly capsaicin and dihydrocapsaicin, since many of the other
capsaicinoids are
present only in trace amounts. Chemical synthesis is thus useful to obtain the
more
uncommon capsaicinoids, such as nonivamide, and for making none-natural
capsaicinoids.
Capsaicinoids can be prepared from vanillin by first reducing vanillin oxime
using a mixture of
an excess of metal (Zn) and ammonium formate in methanol under reflux to
obtain
vanillylamine. Alternatively, the amide bond-formation can be accomplished by
an enzyme-
catalyzed transformation between vanillylamine and different fatty acid
derivatives.
W02015/144902A1 discloses a multi-catalytic cascade relay sequence involving
an enzyme
cascade system that when integrated with other catalytic systems, such as
heterogeneous
metal catalysts and organic catalysts, converts an alcohol to an amine and
amide in sequence
or in one-pot.
US2017081277A1 discloses an amidation using dialkyl-amines as substrates.
Novozym 435()
immobilized on beads are used. A Dean Stark apparatus may be used to remove
ethanol from
the reaction mixture. The reactions are performed under reduced pressure. For
large scale
manufacturing, beads are not suitable because it is costly and time consuming
to separate the
beads from the reaction mixture. Further, for large scale production, reduced
pressure is
preferably avoided to reduce cost and time of the overall process.
US6022718 discloses a process for preparation of capsaicin analogues using
hydrolysis and
capsaicin as starting materials.
Pithani S., Using spinchem rotation bed reactor technology for immobilized
enzymatic
reactions: a case study, Org. Process Res. Dev., 2019, vol.23, pages 1926-
1931, discloses
advantages of using rotary bed immobilized lipase. An acylation reaction is
used to
demonstrate that lipase (Novozym 435-) can be used in a rotating bed reactor.
The loading
was limited to 10 wt% due to high costs. A loading of 5 to 10 wt% was deemed
sufficient to
achieve a conversion of 45-50% within 6 hours. The overall yield after
upscaling was 39%.
Although Pithani shows that a rotation bed reactor is useful for acylations,
it also shows that
it is costly and results in a conversion of 45-50% with an overall yield of
39%. The results
disclosed in Pithani are disconcerting for large scale production using a
rotation bed reactor.
There is an increasing need for large scale production of amide compounds like
capsaicinoids.
Such processes are preferably efficient and effective having improved yields
compared to
known processes. Such amidation processes are preferably environmentally
friendly and
especially cost effective.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
3
Summary of the invention
It is an object of the present invention to at least partly overcome the above-
mentioned
problems, and to provide an improved process for the synthesis of amides from
amines and
carboxylic acids or esters.
This object is achieved by a process as defined in the claims.
One aspect relates to a process for enzymatic synthesis of amides of formula
Ill from amines
of formula I and compounds of formula II,
1 3 2 Lipase 1 2
R -NH2 R O-R-C(0)-R R -
N(H)-C(0)-R
I II III
wherein R1 is selected from the group comprising or consisting of Ca_nalkyl-,
12a I kynyl-, C112a I koxy-, C112al kyl-O-C1_12a lkyl-, C1_22a I kyl-OC(0)-
C1_12a lkyl-,
12a I kyl-, Ci_i2a I kyl-N HC(0)-Ci_na I kyl-, C3_12cycloa lkyl-, C3_12cycloa
Ike nyl-, C5_12aryl-, C3-
12cycloa I kyl-C1_6a I kyl-, Ca_12cycloa I kenyl-Ci_6a I kyl- and C5_12a ryl-
C1_6a I kyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, Ci
6hydroxyalkyl-,
C1_6arnineoxyalkyl-, Ci_6annideyalkyl-, Ca_6carboxyalkyl-, Ci
6su1fu ralkyl-, CF6sulfidealkyl- and CF6alkoxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
wherein R2 is selected from the group comprising or consisting of hydrogen,
C1_30alkyl-, Ci
ma Ike nyl-, C1_30a I kynyl-, C13oa I koxy-, C1_3ca lky1-0-Ci_12a I kyl-,
Ci_30alkyl-NHC(0)-Ci_22alkyl-, CI_12cycloalkyl-, CO_12cycloalkenyl- and C5-
12aryl-, C342cycloalkyl-Ci_6alkyl-, C3_i2cycloalkenyl-Ci_salkyl- and Cs_naryl-
Ci_6alkyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amide,
Ci_6hydroxyalkyl-
, Ci_Ghaloya lkyl-, C1_6ca rboxya I kyl-, C1_6s ulfidea I kyl- and C1_6a I
koxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_balkyl-,
6a1keny1-, C16alkynyl-,
kyl-NH-
Ci_6alkyl-,
C3_12cycloalkyl-, C3_12cycloalkenyl- and C542aryl-, C3-
ncycloa I kyl-C1_6a I kyl-, C342cycloa I kenyl-Ci_6a I kyl- and C512a ryl-
Ci_6a I kyl-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, Ci
6hydr0xya1ky1-, C1_6haloyalkyl-, Ci_6amineoxyalkyl-, C16amideyalkyl-,
C1_6carboxyalkyl-, C1_
65u1fura1ky1-, C1_6sulfidealkyl- and Ci_6alkoxy-, and
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
4
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S. and
wherein R is a bond or C1_6alkyl-,
characterized in that
the lipase is immobilized on a rotary bed reactor or on a spin-fixed-bed
reactor and a Dean-
Stark apparatus is used for dehydration.
In some aspects, lipase is immobilized on a rotary bed reactor and a Dean-
Stark apparatus is
used for dehydration.
In some aspects, a lipase immobilized on beads is disclaimed.
In the processes of the invention, as defined anywhere in here, a combination
of enzyme
catalysis and azeotropic dehydration is used for direct catalytic amide
synthesis. The enzyme,
lipase, is immobilized on a rotary bed reactor or on a spin-fixed-bed reactor.
Compared to
immobilizing lipase on beads or using sieves, the lipase in the process of the
invention can
easily be recirculated. This allows the process to be performed in a time- and
cost-effective
manner, especially at large scale.
The unique combination of a rotary bed reactor or a spin-fixed-bed reactor and
a Dean-Stark
apparatus improves the yield (>90, or 99%) as well as the conversion rate (>90
or 99%). The
unique combination allows the use of wet raw material. The process can be
performed at
atmospheric pressure and at temperatures below 100 C (60 ¨ 90 C). The process
is
environmentally friendly. The process is suitable for large scale production
of amides.
An easy workup and purification process allow the process to be used at a
large scale. The
enzymes and the solvents used, if any, are easy to recycle, which in turn
makes large scale
production feasible. The unique combination of an immobilized enzyme on a
rotary bed
reactor or on a spin-fixed-bed reactor and a Dean Trap apparatus allows the
process to be
extended to the synthesis of other amides and esters.
In one aspect, the process is performed under neat conditions. The process can
be performed
without any solvent. This may improve the efficiency and effective and
environmentally
friendliness of the process. It also reduces costs for performing the process.
A neat process
further reduces costs for the process on a large scale.
Compared to known processes, the direct amidation process of the invention has
an improved
conversation rate as well as an improved yield. Less process steps are needed
for the
amidation, which reduces time and costs. The mass flow is improved in the
process of the
invention. Because the enzyme is immobilized/fixed, the reaction products can
easily be
filtered off and purified. The process of the invention has an improved
reaction rate.
The process allows for effective and efficient large-scale production of amide
compounds like
capsaicinoids. The process has improved yields compared to known processes.
The amidation
processes are environmentally friendly and especially cost-effective.
The combined use of the rotary bed reactor and the Dean-Stark apparatus allows
for control
of the moisture content during the process. A low moisture content improves
conversion rate
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
and yield. The results in Cycle 2 of Table 1 in example 14, show that even raw
material having
a moisture content of 23wt% can be used. This improves the flexibility of the
process. This also
improves the feasibility for large scale use of the process.
In some aspects, the option for R2 to be a (dialkyl)-amine is disclaimed.
5 According to some aspect of the invention, is
selected from the group comprising or
consisting of C1-12alkyl-, C1_12alkenyl-,
C342cycloalkyl-, C3 ucycloalkenyl-, C5_12aryl-, C342cycloalkyl-C16alkyl-,
C3_12cycloa I kenyl-C1_6alkyl- and
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C1_6hydroxyalkyl-, C1_
6haloyalkyl-, Cl_6carboxyalkyl-, = C1_6sulfidealkyl- and
Ci_6alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
C1_30alkyl-, Ci
30a Ike nyl-, Ci_30a I kynyl-, Ci_302 I koxy-, C1_3c2 lky1-0-Ci_i2a I kyl-,
Ci_30a lkyl-OC(0)-C1_nalkyl-, C3_
12cycloalkyl-, C3_12cycloalkenyl- and C5_12aryl-,
C3_12cycloalkenyl-C1_
6alkyl- and Cs_12aryl-Ci_Balkyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C1_6hydroxyalkyl-, Ci
shaloyalkyl-, Cl_6carboxyalkyl-, = C1_6sulfidealkyl- and
Ci_6a1koxy-, and
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_6alkyl-, Ci_
6a I kenyl-, C1_6a I kynyl-, C1_6a I koxy-, C1_6a I
kyl-OC (0)-Ci_6a I kyl-, C3-
12cyc10a1ky1-, C3_12cycloalkenyl- and Cs_naryl-, C3_12cycloa lkyl-Ci_Ga I kyl-
, C3_12cycloa Ikenyl-
salkyl- and C5_naryl-Ca_6a1ky1-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C1_6hydroxyalkyl-,
6haloyalkyl-, Ci_6carboxyalkyl-, = Ci_6sulfidealkyl- and Ci_6alkoxy-, and
wherein R is a bond or Ci_Galkyl-.
According to some aspect of the invention, RI- is selected from the group
comprising or
consisting of C1_12alkyl-, C1_12alkenyl-,
C1-
3.2aI kyl-OC(0)-Ci_na I kyl-, C342cycloalkyl-, C3_12cycloa Ike nyl-, C5_12aryl-
, C342cycloa I kyl-Ci_6alkyl-,
C3_12cycloa I kenyl-C1_6alkyl- and C542aryl-Ci_ealkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, and Ci_6a1k0xy-, and
wherein R2 is selected from the group comprising or consisting of Ca_30alkyl-,
C1_30alkenyl-,
wherein R3 is selected from the group comprising or consisting of hydrogen,
C1_6alkyl-, and
wherein R is a bond or Ci_6alkyl-.
According to some aspect of the invention, R1 is selected from the group
comprising or
consisting of C1_6alkyl-,
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
6
OC(0)-Ci_6a I kyl
C3_6cycl oa I kyl-, C3_6cyclo2 I kenyl-, C6a ryl-, C3_6cycloa I kyl-
CJ._62 I kyl-, C3_
6cycloal kenyl-Ci_6a lkyl- and C6aryl-C1_3alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, and Ci_6alkoxy-, and
wherein R2 is selected from the group comprising or consisting of C-i_malkyl-,
C-msalkenyl-,
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_3alkyl-, and
wherein R is a bond or C1_3alkyl-.
According to some aspect of the invention, RI- is selected from the group
comprising or
consisting of Cl-Gal kyl-,
C3_6cycloalkyl-, C3-
6cycloalkenyl-, C6_7aryl-, C3_6cyc102lkyl-C1_3alkyl-, C3_6cycloalkenyl-
Ci_3alkyl- and C5_7aryl-C1_
3alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C1_3hydroxyalkyl-, Ci
3haloyalkyl-, and C1_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
C548alkyl-, Cs-
isalke nyl-, C5_isalkoxy-, Csalkyl-O-Ci_Ba I kyl-, and Cs_isalkyl-OC(0)-Ci_6al
kyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen and carboxy,
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_3alkyl-, Ci
3a1koxy- and Ci_3alkyl-O-Ci_3alkyl-, and
wherein R is a bond or Ci_3alkyl-.
According to some aspect of the invention, 11' is selected from the group
comprising or
consisting of hydrogen, C6_7ary1- and Cs_7ary1-C1_3a lkyl-,
which [0-may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and C1_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_isalkyl- and C5
15a Ike nyl-,
wherein R3 is selected from the group comprising or consisting of hydrogen,
CF3alkyl- and
wherein R is a bond or Ci_3alkyl-.
According to some aspect of the invention, R1 is C5_7aryl-Ci_3alkyl-,
which RI-may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy and C13alkoxy-,
wherein R2 is selected from the group comprising C5_16alkyl- and Cs_isalkenyl-
, and
wherein R3 is hydrogen, methyl or ethyl, and
wherein R is a bond.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
7
The process with these compounds results in improved yields and conversion
rates, which is
especially important for large scale production.
According to some aspect of the invention, 111 is C5_7aryl-C1_3alkyl-,
which RI-may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and Ci_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
C546alkyl- and C5_
isa Ike nyl-,
wherein R3 is hydrogen, methyl or ethyl, and
wherein R is a bond or Ci.2alkyl-.
According to some aspect of the invention, Rl is C6aryl-Ci_2alkyl-,
which RI-may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and C1_2alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
C7_102 lkyl- and C7-
loalkenyl-, and
wherein R3 is hydrogen, methyl, or ethyl
wherein R is a bond or Ci_2alkyl-.
According to some aspect of the invention, R1- is CGaryl-, or CGaryl-Ci_2a1ky1-
, optionally be
substituted with one or more substituent selected from the group comprising or
consisting of
hydrogen, hydroxy, oxy, and methoxy-.
According to some aspect of the invention, R2 is hydrogen, methanyl, ethanyl,
heptanyl,
octanyl, 8-methyl-nonanyl, octadecanyl or 8-methyl-nonenyl.
The process with these compounds results in improved yields and conversion
rates, which is
especially important for large scale production.
One aspect relates to a process for enzymatic synthesis of amides of formula
Ill from amines
of formula land compounds of formula Ila,
1 3 2 Lipase 1 2
R-NH2 R0-C(0)-R R -N(H
)-C( 0 )- R
ha Ill
wherein R1 is selected from the group comprising or consisting of Ca_nalkyl-,
12a I kynyl-, Ci_12a I koxy-,
C1_i2a I kyl-OC(0)-C1_12alkyl-, C1_122 lkyl-N H-C1_
12a I kyl-, C1_12a I kyl-N HC(0)-Ci_12al kyl-, C3_12cycloa lkyl-,
C3_12cycloa Ike nyl-, C5_12aryl-, C3-
12cyc10alkyl-C1_6alkyl-, C342cycloa I kenyl-Ci_6a I kyl- and C5_12a ryl-Ci_6a
I kyl-,
which RI-may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, Ci
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
8
Ghydroxyalkyl-, = Ci_Gaminexyalkyl-,
= Ci_Gcarboxyalkyl-, C1_
6sulfu ralkyl-, Ci_6sulfidealkyl- and Ci_6alkoxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
wherein R2 is selected from the group comprising or consisting of hydrogen,
Ci_30alkyl-, Ci
30a Ike nyl-, C1-3oa IlkynY1-, C1-30alkoxy-,
lkyl-, C1-30alkyl-OC(0)-C1_12alkyl-, C1-
C1_30a1kyl-NHC(0)-Ci_i2alkyl-, C342cycloalkyl-, C342cycloalkenyl- and C5_
12aryl-, = C3_12cyc1oalkenyl-C1_6alkyl- and
C5_12aryl-C1_salkyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, C1_
6hydroxyalkyl-, =
Ci_6carboxyalkyl-,
6sulfu ralkyl-, C16sulfidealkyl- and Ci_6alkoxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_olkyl-, C1-
Ga I ken yl-, Cl_Ga I kyl-OC(0)-Ci_Ga
I kyl-,
C342cycloalkyl-, C3_12cycloalkenyl- and Cs_izaryl-, C3_
12CYCI0a I kyl-C1_6a I kyl C342cycloa I kenyl-Ci_6a I kyl- and Cs-12a ryl-
C1_6a I kyl-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, Ci
Ghydroxyalkyl-, = Ci_Gaminexyalkyl-,
= Ci_Gcarboxyalkyl-, C1-
6sulfu ralkyl-, C1_6sulfidealkyl- and C1_6a1k0xy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S
characterized in that
the lipase is immobilized on a rotary bed reactor or on a spin-fixed-bed
reactor and a Dean-
Stark apparatus is used for dehydration.
According to some aspect of the invention,
is selected from the group comprising or
consisting of Ci_salkyl-, C1-6a
C3_6cycloalkyl-, C3-
6cycloa I kenyl-, C6_7aryl-, C3_6cycloalkyl-C1_3alkyl-, C3_6cycloa Ike nyl-
Ci_3a1 kyl- and C5_7a
3a lkyl-,
which RI-may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C1_3hydroxyalkyl-, Ci
3haloyalkyl-, and Ci_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_isalkyl-, Cs_
isalke nyl-, Cs_isalkoxy-, Cs_i5alky1-0-Ci_6a I kyl-, and C5 asalkyl-OC(0)-
Ci_Gal kyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen and carboxy, and
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
9
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_3alkyl-, Ci
3a1k0xy- and C13alkyl-O-C1_3alkyl-.
According to some aspect of the invention, 111- is C5_7aryl-C1_3alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and Ci_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_nalkyl- and Cs_
isalkenyl-, and
wherein R3 is hydrogen, methyl or ethyl.
According to some aspect of the invention, 1:11- is C6aryl-Cl_2alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and Ci_2a1k0xy-, and
wherein R2 is selected from the group comprising or consisting of C7_1oalkyl-
and CTioalkenyl-,
and wherein R3 is hydrogen, methyl or ethyl.
The process allows for effective and efficient large-scale production of amide
compounds like
capsaicinoids and derivatives thereof. The process has improved yields
compared to known
processes. The amidation processes are environmentally friendly and especially
cost effective.
According to some aspect of the invention, compounds of formula III are
compounds of
formula IV
0
R6 II
tii=
n N R2
R50
OR4 IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising or consisting of C3_30alkyl-,
C3_30alkenyl-, C3_
aoalkynyl-, C342cycloalkyl-, C342cycloalkenyl- and C5_12aryl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amide,
Ci_6hydroxyalkyl-
, Ci_6carboxyalkyl-, C1_6sulfidealkyl- and
6alkoxy- and Cs_naryl-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
wherein re or R5 is independently selected from the group comprising or
consisting of
hydrogen, Ci_6a I kyl-, C2_6a1 kenyl-, C2_6alkynyl-, C340cycloalkyl-,
C3_1ocycloa I ke nyl- and C6_12aryl-,
which R4 or R5 may optionally be independently substituted with one or more
substituent
selected from the group comprising or consisting of hydroxy, oxy, halogen,
carboxy, amine,
amide, C1_6hydroxyalkyl-,
C1_6haloyalkyl-, C1_6a mineoxyalkyl-, C1-
6carboxyal kyl-, C1_6su Ifidealkyl- and Ci_olkoxy-, and
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
wherein R6 is selected from the group comprising or consisting of hydrogen,
hydroxy, oxy,
halogen, carboxy, amine, amide, Ci_ioalkyl-, C2_10alkenyl-, C2_10alkynyl-,
C3_12cycloalkyl-, C3_
5 licycloalkenyl- and C5_12ary1-,
which R6 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide,
shYdroxyalkyl-, Cl_6amineoxyalkyl-,
Ca_scarboxyalkyl-, Ci
6su1fu ralkyl-, C1_6su1f1dea1ky1- and C1_6alkoxy-, and
10 wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
The process with these corn pounds results in improved yields and conversion
rates, which is
especially important for large scale production.
In some aspects, wherein compounds of formula III are compounds of formula IV
n is 1 or 2,
R2 is selected from the group comprising or consisting of C3_30alkyl-,
C3_30alkenyl-,
R4 or R5 is independently selected from the group comprising or consisting of
hydrogen, Ci
3a1ky1, and
R6 is hydrogen.
In some aspects, wherein compounds of formula III are compounds of formula IV
n is 1 or 2,
R2 is selected from the group comprising C2_isalkyl- and C3_18alkenyl-,
R4 or R5 is independently selected from the group comprising hydrogen,
Ci_salkyl-, and
R6 is hydrogen.
In some aspects, wherein compounds of formula III are compounds of formula IV
n is 1 or 2,
R2 is selected from the group comprising Cb_ibalkyl- and Cs_isalkenyl-,
R4 or R5 is independently selected from the group comprising hydrogen,
Ci_3alkyl-, and
R6 is hydrogen.
In some aspects, wherein compounds of formula III are compounds of formula IV,
R2 is
methanyl, ethanyl, heptanyl, octanyl, 8-methyl-nonanyl or octadecanyl or 8-
methyl-nonenyl.
The process with these compounds results in improved yields and conversion
rates, which is
especially important for large scale production.
According to some aspect of the invention, no solvent is used.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
11
According to some aspect of the invention, the solvent is an organic solvent
selected from the
group comprising or consisting of methyl tert-butyl ether, diisopropylether,
&alkyl ethers, hexane and other C540alkanes, cyclohexane and other
Cs_locycloalkanes,
benzene, toluene, xylene, tert-butanol, tert amyl alcohol, other bulky
secondary or tertiary Cs
-
10 alcohols and any esters thereof. In some aspects, the organic solvent is
selected from the
group comprising or consisting of diisopropylether, cyclohexane, toluene and
tert-butanol, or
mixtures thereof. In some aspects, the solvent is cyclohexane, toluene or
diisopropylether
(DIPE). In some aspects, the solvent is diisopropylether (DIPE). In some
aspects, the solvent is
cyclohexane. In some aspects, the solvent is toluene. In some aspects, the
solvent is tert-
According to some aspect of the invention, when R3 is not hydrogen, the
solvent is selected
from the group comprising or consisting of methyl tert-butyl ether,
diisopropylether, Ci_balkyl-
O-C1_6alkyl ethers, hexane and other Cs_ioalkanes, cyclohexane and other
Cs_locycloalkanes,
benzene, toluene, xylene, tert-butanol, tert amyl alcohol, other bulky
secondary or tertiary C5-
10 alcohols and their esters. In some aspects, the organic solvent is selected
from the group
comprising or consisting of diisopropylether, cyclohexane, toluene and tert-
butanol, or
mixtures thereof. In some aspects, the solvent is cyclohexane, toluene or
diisopropylether
(DIPE). In some aspects, the solvent is diisopropylether (DIPE). In some
aspects, the solvent is
cyclohexane. In some aspects, the solvent is toluene. In some aspects, the
solvent is tea-
butanol. In some aspects, the solvent is recyclable. In some aspects, the
solvent is recycled. In
some aspects, the solvent is recycled for at least 70% or 80% or 90%.
Recycling the solvent
reduces the overall costs for the process and also reduced the carbon
footprint of the process.
According to some aspect of the invention, the lipase is selected from the
group comprising
or consisting of Candida antarctica lipase A, Candida antarctica lipase B,
cross-linked Substilisin
A protease, Porcine pancreas lipase, Candida cylindracea lipase, Rhizopus a
rrhizus, Penicillum
cyclopium, Mucor miehei, Thermomyces lanuginosus lipase, Candida rugosa lipase
and
Pseudomonas lipoprotein lipase. In one aspect, the lipase is selected from the
group
comprising or consisting of Candida antarctica lipase A and Candida antarctica
lipase B. In one
aspect, the lipase is Candida antarctica lipase . In one aspect, the lipase is
Candida antarctica
lipase B (Novozym 4351. The immobilized enzymes, like Candida antarctica
Lipase B or C.
antarctica lipase A, are commercially and easily available under tradenames
like Novozym
435¨. The availability at relative low cost is important for a cost-effective
process, especially
for large scale processes.
According to some aspect of the invention, the process temperature is between
15 C and
150 C, or between 15 C and 115 C, or between 50 and 90, or 70 -80 C. The
relative low
temperature is important for a cost-effective process, especially for large
scale processes.
According to some aspect of the invention, the process is performed at a
pressure between
0.900 and 0.200 MPa, or at atmospheric pressure (about 0.1 MPa). Performing
the process at
atmospheric pressure is important for a cost-effective process, especially for
large scale
processes.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
12
According to some aspect of the invention, the rotary bed reactor is loaded
for 10 to 75wt%
with the lipase. According to some aspect of the invention, the rotary bed
reactor is loaded
for 11 to 60wt% with the lipase. According to some aspect of the invention,
the rotary bed
reactor is loaded for 15 to 50w1% with the lipase. The unique combination of
an immobilized
enzyme on a rotary bed reactor or on a spin-fixed-bed reactor and a Dean Trap
apparatus
improves the conversion rate and yield of the process. Because the process is
both time- and
cost-effective, a possible additional cost for loading of the lipase with more
than 10 wt%
loading becomes affordable.
According to some aspect of the invention, the rate of agitation is 150 to 600
rpm or 200 to
500 rpm, or 200 to 450 rpm.
The invention also relates to a process for synthesis of compounds of formula
II, wherein R2 is
a C6_isalkyl or C6_isalkenyl_ According to some aspect, compounds of formula
II, wherein R2 is
a CG_isalkyl or C6_1galkenyl, which may be straight or branched, are prepared
comprising the
steps of
ppii3 ___________________________________
HO R.--Br step A HO R2 ¨PPh3 Br
base 0 0
iso-butyraldehyde HO isomerization
3' HO.R2
solvent
step B
hydrogenation
0
HO
step A-1, wherein the reaction is performed without solvent or with an organic
solvent,
step B-1, wherein a solvent is an aprotic organic solvent, and
step B-1, wherein a base is a sodium or potassium alkoxides ,
optionally isomerization step C-1, wherein a catalyst is selected from the
group comprising or
consisting of HNO2, HNO3 and combinations of NaNO2/HNO3, NaNO2/NaNO3/H2504,
that can
generate HNO2 or HNO3, and
hydrogenation step D-1, wherein a catalyst is a heterogeneous hydrogenation
catalyst and a
hydrogen source is hydrogen gas.
In some aspects, R2 is a C6_ioalkyl.
In some aspects, the organic solvent in step A-1 is ethyl acetate,
wherein the aprotic organic solvent in step B-1 is selected from the group
comprising or
consisting of 2-methyl tetra hydrofuran, tetrahydrofuran and toluene,
wherein the sodium or potassium alkoxide base in step B-1 is selected from the
group
comprising or consisting of NaH, KH, t-BuOK, t-BuONa, and
CA 03215699 2023- 10- 16
WO 2022/229314 PCT/EP2022/061321
1.3
wherein the heterogeneous hydrogenation catalyst in hydrogenation step D-1 is
selected from
the group comprising or consisting of Pd/C and Pd/A1203.
In some aspects, the organic solvent in step A-1 is ethyl acetate, the aprotic
organic solvent in
step B-1 is 2-methyl tetrahydrofuran, the sodium or potassium alkoxide base in
step B-1 is t-
BuOK, and the heterogeneous hydrogenation catalyst in hydrogenation step D-1
is Pd/C.
In the production of 8-methyl-6-nonenoic acid, using 2-MeTHF as recyclable
solvent for the
key Wittig reaction between (6-Carboxyhexyl)triphenylphosphonium bromide and
iso-
butyraldehyde improves conversion rate and yield. The synthesis is time- and
cost-effective
with high yields and conversion rates. This is especially important for large
scale process.
Further solvents may be used in the process steps. Extraction and filtration
may be performed
between the steps.
The process may be performed at room temperature. The process can be performed
at
atmospheric pressure (approximately 1 atm or 0.1 MPa).
The invention also relates to a process for a new synthetic route to 8-methyl-
6-nonanoic acid,
which is used for the direct production of dihydro-capsaicin. The process
starts from
cyclohexanone and iso-butyraldehyde as raw materials, with aldol condensation,
Baeyer-
Villiger oxidation and hydrogenation as key steps.
According to some aspect of the invention, compounds of formula II, wherein R2
is 8-methyl-
nonanyl, are prepared comprising the steps of
0 OH 0
0 + catalyst &IT,. catalyst
step A step B
0 0
C step
ep step D
0 0
HO =
step E step F HO
step A-2, wherein the reaction is performed without solvent or with any
organic solvent and
a catalyst is selected from the group comprising or consisting of amines and
inorganic bases,
step B-2, wherein the reaction is performed without solvent or with an organic
solvent, and a
catalyst is an acid,
step C-2, wherein a catalyst is a heterogeneous hydrogenation catalyst, and a
hydrogen source
is hydrogen gas,
step D-2, wherein an oxidant is a peroxide, and a catalyst is a lipase, and
step E-2, wherein a reaction medium is an acidic media, and
Step F-2, wherein a catalyst is a heterogeneous hydrogenation catalyst, and a
hydrogen source
is hydrogen gas.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
14
According to some aspects, the organic solvent in step A-2 is selected from
the group
comprising or consisting of toluene and aromatic solvents, THF and ethers,
dichloromethane
and halogenated solvents, and the catalyst is selected from the group
comprising or consisting
of pyrrolidine and corresponding salts, NaOH and KOH,
wherein the organic solvent in step B-2 is selected from the group comprising
or consisting of
toluene, and the acid is selected from the group comprising or consisting of p-
Ts0H, sulfuric
acid and Amberlyst-15,
wherein the catalyst in step C-2 is selected from the group comprising or
consisting of Pd/C,
Pd/A1203,
wherein the oxidant in step 0-2 is selected from the group comprising or
consisting of aqueous
H202 and peroxy acids and the lipase is selected from the group comprising or
consisting of
Candida antarctica lipase A, Candida antarctica lipase B, cross-linked
Substilisin A protease,
Porcine pancreas lipase, Candida cylindracea lipase, Rhizopus arrhizus,
Penicillum cyclopium,
Mucor miehei, Thermomyces lanuginosus lipase, Candida rugosa lipase and
Pseudomonas
lipoprotein lipase, and
wherein the reaction medium in step E-2 is selected from the group comprising
or consisting
of aqueous sulfuric acid solution, and
wherein the catalyst in step F-2 is selected from the group comprising or
consisting of Pd/C,
Pd/A1203, Pd/ molecular sieves, Pt/C, Pt/A1203, and Pt/molecular sieves.
A Dean Stark trap may be used in step B-2.
According to some aspects, the organic solvent in step A-2 is toluene, and the
catalyst is
pyrrolidine, the organic solvent in step B-2 is toluene and the acid is p-
Ts0H, the catalyst in
step C-2 is Pd/C, the oxidant in step D-2 is aqueous H202 and the lipase is
Candida antarctica
lipase B, the reaction medium in step E-2 is aqueous sulfuric acid solution,
and the catalyst in
step F-2 is Pd/C.
The synthesis is time and cost effective with high yields and conversion
rates. This is especially
important for large scale process.
Further solvents may be used in the process steps. Extraction and filtration
may be performed
between the steps.
The process may be performed at room temperature. The process can be performed
at
atmospheric pressure (approximately 1 atm or 0.1 MPa).
The process as defined anywhere herein are useful for large scale production
of compounds
of formula III. In some aspects, the process is used for large scale
production (>0.5 or > 1 kg)
of compounds of formula Ill.
Brief description of the drawings
The invention will now be explained more closely by the description of
different embodiments
of the invention and with reference to the appended figures.
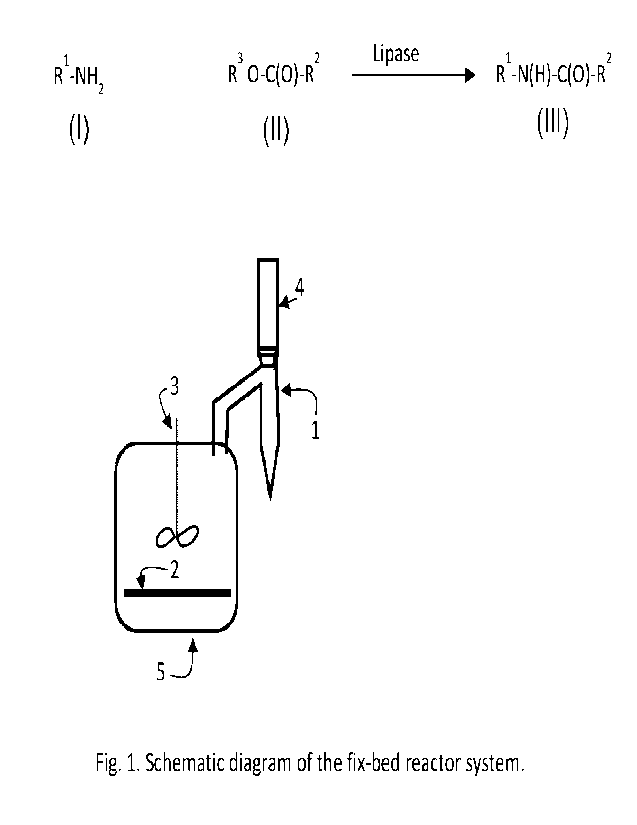
Fig. 1 shows a system for performing the process of the invention.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
Detailed description of various embodiments of the invention
Definitions
Room temperature is a temperature between 15 and 25 C.
Et0Ac is ethyl acetate.
5 DIPE is diisopropylether.
KOtBu is potassium tert-butoxide.
2-MeTHF is 2-methyltetrahydrofuran.
ET20 is diethyl ether.
AcOH is acetic acid.
10 p-Ts0H is p-toluenesulfonic acid or tosylic acid.
tBuOH is tert-butyl alcohol.
equiv. is equivalent. equivalent
As used herein, the term "wt%" or "w/w%" or "w%" means weight percentage,
which is a
percentage of the total weight.
15 As used herein, the term "optional" or "optionally" means that the
subsequently described
event or circumstance may but need not occur, and that the description
includes instances
where the event or circumstance occurs and instances where it does not.
As used herein, the terms "C, used alone or as a suffix or prefix, is intended
to include
hydrocarbon-containing groups; n is an integer from 1 to 30.
As used herein, the term "halogen" or "halo", used alone or as suffix or
prefix, is intended to
include bromine, chlorine, fluorine, and iodine.
As used herein, the term ''hetero'', used alone or as a suffix or prefix, is
intended to include
alkyl, cycloalkyl and aryl groups in which one or more of the carbon atoms
(and certain
associated hydrogen atoms) are independently replaced with the same or
different hetero
atoms (5, 0 or N) or heteroatomic groups. Examples of heteroatomic groups
include, but are
not limited to, 0 , S, 00, SS, OS, NR, =N¨N=, ¨N=N¨, ¨N=N¨NR¨, ¨PR¨,
¨P(0)2¨, ¨POR¨, ¨0¨P(0)2¨, ¨SO¨, ¨Sn(R)2¨, and the like.
As used herein, the term "Ci_30-al kyr, used alone or as a suffix or prefix,
is intended to include
both branched and straight chain saturated aliphatic hydrocarbon groups having
from 1 to 30
carbon atoms. Examples of C1_4.-alkyl include methyl, ethyl, n-propyl, i-
propyl, n-butyl, i-butyl,
sec-butyl, and tert-butyl.
The term "alkenyl" refers to a monovalent straight or branched chain
hydrocarbon radical
having at least one carbon-carbon double bond and comprising at least 2 up to
about 30
carbon atoms. The double bond of an alkenyl can be unconjugated or conjugated
to another
unsaturated group. Suitable alkenyl groups include, but are not limited to
C2_6alkenyl groups,
such as vinyl, allyl, butenyl, pentenyl, hexenyl, butadienyl, pentadienyl,
hexadienyl, 2-
CA 03215699 2023- 10- 16
WO 2022/229314 PCT/EP2022/061321
16
ethylhexenyl, 2-propy1-2-butenyl, 4-(2-methyl-3-butene)-pentenyl. An alkenyl
can be
unsubstituted or substituted with one or two suitable substituents.
The term "alkynyl" refers to a monovalent straight or branched chain
hydrocarbon radical
having at least one carbon-carbon triple bond and comprising at least 2 and up
to about 12
carbon atoms. The triple bond of an alkynyl can be unconjugated or conjugated
to another
unsaturated group. Suitable alkynyl groups include, but are not limited to
C2_6alkynyl groups,
such as acetylenyl, methylacetylenyl, butynyl, pentynyl, hexynyl. An alkynyl
can be
unsubstituted or substituted with one or two suitable substituents.
As used herein, the term "C1-6-alkoxy", used alone or as a suffix och prefix,
refers to a C1-6-alkyl
radical, which is attached to the remainder of the molecule through an oxygen
atom. Examples
of C1-4-alkoxy include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, i-
butoxy, sec-butoxy
and tert-butoxy.
As used herein, the term "cycloalkyl", and "cycloalkenyl" used alone or as a
suffix or prefix, is
intended to include saturated or partially unsaturated cyclic alkyl radical.
Where a specific
level of saturation is intended, the nomenclature cycloakanyl or cycloalkenyl
is used. Examples
of cycloalkyl groups include, but is not limited to, groups derived from
cyclopropane,
cyclobutene, cyclopentane, cyclohexane and the like.
As used herein, the term "aryl" refers to either a monocyclic aromatic ring
having 5 or 12 ring
members or a multiple ring system having at least one carbocyclic aromatic
ring fused to at
least one carbocyclic aromatic ring, cycloalkyl ring or a heterocycloalkyl
ring. For example, aryl
includes a phenyl ring fused to a 5- to 7- membered heterocycloalkyl ring
containing one or
more heteroatoms independently selected from N, 0, and S.
As used herein, the term "C.5_12-aryl-C1-6-alkyl" refers to a phenyl group
that is attached
through a C1_6-alkyl radical. Examples of C6-aryl -C1_3-alkyl include
phenylmethyl (benzyl), 1-
phenylethyl and 2-phenylethyl.
Figure 1 shows a system for performing the process. In a reactor 5, the lipase
is immobilized
on a rotary fix bed 2. A motor 3 is used for rotation of the fixed bed 2. The
reactor 5 is
connected to a Dean Stark apparatus 1, which is connected to a condenser 4.
In the present invention, the process is performed using the lipase, which is
immobilized on a
rotary bed reactor together with a Dean-Stark apparatus for dehydration.
This process may be used for the preparation of capsaicinoids, but also for
the amidation of
numerous of other amines with carboxylic acids or esters.
The process may be used for synthesis of amides of formula III from amines of
formula I and
compounds of formula II or Ila as shown below,
1 3 2 3 2 Lipase 1
2
R -NH 2 R0-R-C(0)-R or 1 0-C(0)-R R - N(H)-
C(0)-R
11 ha Ill
or
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
17
1-NH2 3 2 Lipase 1 2
R R 0-R-C(0)-R __________ R -N(H)-R-C(0)-R
I II III
may be selected from the group comprising Ci_nalkyl-,
12a I koxy-, C112a I kyl-O-Ci_12a lkyl-, C1_12a I kyl-OC(0)-Ci_na I kyl-,
C1_12a I kyl-N lkyl-, Ci
12alkyl-NHC(0)-Ci_ualkyl-, C3_12cycloalkyl-, C3_12cycloalkenyl-, C5_12aryl-,
C3_12cycloalkyl-Ci_
fialkyl-, C1_12cycloalkenyl-Cl_6alkyl- and C5_12a ryl-Ci_6alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
C1_6hydroxyalkyl-, Ci
shaloyalkyl-, C1_6aminexyalkyl-, C1_6amideya1ky1-, Ci_6carboxyalkyl-,
Ci_6su1fura1ky1-, C1-
6sulfidealkyl- and Ca_6a1k0xy-.
R1 may be selected from the group comprising Ci_olkyl-,
C3_6cycloalkyl-, C3_6cycloalkenyl-, C6_2aryl-, Ca_6cycloalkyl-CiAalkyl-,
C3_6cycloalkenyl-
Ci_3alkyl- and C5_2aryl-Ci_3alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, Ci_Awdroxyalkyl-,
C1_2haloyalkyl-, and
Ci_3a I koxy-.
Or R1 may be C5_7aryl-Ci_3alkyl-, or C6_7aryl-Ci_2alkyl-, or C6aryl-C1_3alkyl-
, optionally substituted
with hydrogen, hydroxy and/or methoxy.
R2 may be selected from the group comprising hydrogen, Ci_30alkyl-,
CF3oalkynyl-
, Ci_30a I koxy-, Ci_30a I kyl-O-Ci_12a1 kyl-, Ciioa lkyl-OC(0)-Ci_12a I kyl-,
Ci_30a I kyl-N H-C1_i2a I kyl-,
malkyl-NHC(0)-C1-12alkyl-, C3-12cycloalkyl-, C3-12cycloalkenyl- and Cs-12a ryl-
,
C3_12cycloa I kenyl-Ci_6a I kyl- and Cs_12a
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
Cl_6hydroxyalkyl-,
6ha1oya1ky1-, C1_6aminexyalkyl-, C1_6amideya1ky1-, Cl_6carboxyalkyl-,
C1-
6sulfidealkyl- and Ci_6alkoxy-.
R2 may be selected from the group comprising hydrogen, Cl_oalkyl-,
C3_12cycloalkyl-, C3_12cycloalkenyl- and C5_12ary1-.
R2 may be selected from the group comprising hydrogen, Cs_isalkyl-,
Cs_isalkoxy-
, Cs_isalky1-0-Ci_Ga I kyl-, and Cs_isa I kyl-OC(0)-C1_Ga I kyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen and carboxy.
Or R2 may be selected from the group comprising hydrogen, Cs_Thalkyl- and
Cs_Thalkenyl- or C7-
17a1ky1- and C7_16alkenyl-, or C7_1oalkyl- and C7_10alkenyl-.
R3 is selected from the group comprising hydrogen, C1_6alkyl-,
C1_
6a Ikoxy-,
C16a lkyl-O-Cisa I kyl-, Ca&a I kyl-OC(0)-C15a I kyl-, C16a I kyl- N H-C16a I
kyl-, Cisa I kyl-
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
1.8
N HC(0)-C1_62 I kyl-, C3_12cyc10a1ky1-, C3_12cycloa Ike nyl- and C5_122 ryl-,
C3_12cycloa I kyl-Ci_Galkyl-, C3_
12cycloalkenyl-Cl_6a lkyl- and C5_12aryl-C1_6alkyl-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
C1_6hydroxyalkyl-,
6haloyalkyl-, Cl_6aminexyalkyl-, C1_6amideyalkyl-, Cl6carboxyalkyl-,
Cl6sulfuralkyl-, C1-
6sulfidealkyl- and Ci_6alkoxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S
R3 may be selected from the group comprising hydrogen, Cr3alkyl-, Ci_3alkoxy-
and Ci_3alkyl-
0-C1_3a1 kyl-.
Or R3 may be hydrogen, methyl, ethyl. R3 may be hydrogen.
R may be a bond. R may be C1_3alkyl-, or methyl or ethyl.
The compounds of formula III may be represented the structure of IV
0
R6
n NAR2
R50
OR4 IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising C3_2Dalkyl-, C3_20alkenyl-,
C3_20alkynyl-, C3-
12cycloalkyl-, C342cyc10a1keny1- and Cs_12aryl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
C1_6hydroxyalkyl-,
6haloyalkyl-, Ci_6a minexya I kyl-,
Ci_6carboxyalkyl-, Ca_6sulfura lkyl-, C1-
ssulfidealkyl- and Cr6alkoxy- and C542aryl-,
wherein R4 or R5 is selected from the group comprising hydrogen, C1_6alkyl-,
C2_6alkenyl-, C2-
6alkynyl-, C3_10cycloa I kyl-, C3_1ocycloal kenyl- and CS_12aryl-,
wherein R6 is selected from the group comprising hydrogen, hydroxy, oxy,
halogen, carboxy,
amine, amide, C1_6alkyl-, C2_6a I ke nyl-, C2_6alkynyl-, C3_6cyc10a I kyl-,
C3_6cycloa Ike nyl- and C5_6aryl-
'
which R6 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
Ci_6hydroxyalkyl-, Ci
6haloyalkyl-, C1_6aminexyalkyl-, C1_6amideyalkyl-, C16carboxyalkyl-,
C16sulfuralkyl-,
6sulfidealkyl- and Ci_6alkoxy-, and.
The compounds of formula III may be represented the structure of IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising or consisting of C5_18alkyl-,
Cs_asalkenyl-, Cs-
isalkoxy-, C5_18alkyl-O-Ci_6alkyl-, and Cs_isalkyl-OC(0)-Ci_6alkyl-,
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
19
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydroxy, oxy, halogen and carboxy,
wherein R4 or R5 is selected from the group comprising or consisting of
hydrogen and C1_3alkyl-
, and
R6 may be selected from the group comprising or consisting of hydrogen,
hydroxy and oxy.
The compounds of formula III may be represented the structure of IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising C6_12a lkyl- and C6_12alkenyl-
, or CTioalkyl- and
C7_10alkenyl-,
wherein R4 or R5 is selected from the group comprising hydrogen, methyl or
ethyl, and
R6 is hydrogen.
Prior art processes for preparation of capsaicinoids
Ester as acyl donor: need very dry amine (<3wt% water content), otherwise
water will cover
the amine on the bottom and retard the reaction. It is difficult to reach full
conversion of the
ester without addition of excess amine.
HO 161 NH2
S 0 Clz
1.1
OMe
HO
HO
Et20 CI
OMe
Expensive anhydrous solvent and toxic SOCl2 were required in this process.
Comparing with
the enzymatic process, the yield was much lower and the resulting appearance
of the product
was much worse. The product was sticky with brownish-yellow color. See example
25.
Enzymatic process
HO
NH2 enzyme
molecular sieves ri
HO solvent
OMe HO
OMe
This process requires large amounts of molecular sieves, which require bigger
equipment
when scaling up. Filtration and purification is necessary, which is time- and
cost-consuming.
See example 23.
25 Both prior art processes are time consuming, expensive with yields that
are too low to be used
for large scale production in an economically feasible manner.
The process of the invention may be used for the preparation of capsaicinoids
according to
the scheme below.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
o
HO
OMe
capsaicin
0
11
HO =
NH2 noxoxym 435
reflux with =
HO
Dean-Stark distillation
OMe OMe
dihydrocapsaicia
(1110 l'111)1
HO
OMe nonivainide
In the process of the invention, no solvent may be used.
If a solvent is used, the solvent may be an organic solvent selected from the
group comprising
or consisting of methyl tert-butyl ether, diisopropylether, CF6alkyl-0-
C3_6alkyl ethers, hexane
5 and other Cs_loalkanes, cyclohexane and other Cs_locycloalkanes, benzene,
toluene, xylene,
tert-butanol, tert amyl alcohol, other bulky secondary or tertiary C5-10
alcohols and any esters
thereof. The solvent may be toluene, diisopropylether or cyclohexane.
When R3is not hydrogen, the solvent may be selected from the group comprising
or consisting
of methyl tert-butyl ether, diisopropylether, CF6alkyl-O-Ci_salkyl ethers,
hexane and other C5-
10 ioalka nes, cyclohexane and other Cs_mcycloalkanes, benzene, toluene,
xylene, tert-butanol,
tert amyl alcohol, other bulky secondary or tertiary C5-20 alcohols and their
esters.
The solvent may be toluene, diisopropylether or cyclohexane.
The lipase may be selected from the group comprising or consisting of Candida
antarctica
lipase A, Candida antarctica lipase B, cross-linked Substilisin A protease,
Porcine pancreas
15 lipase, Candida cylindracea lipase, Rhizopus arrhizus, Penicillum
cyclopium, Mucor miehei,
Thermomyces lanuginosus lipase, Candida rugosa lipase and Pseudomonas
lipoprotein lipase.
The process may be performed at a temperature between room temperature and 150
C, or
between room temperature and 115 C.
The process may be performed at a pressure between 0.900 and 0.200 MPa, or
about 0.1 MPa.
20 The compounds of formula II may be prepared comprising or consisting of
the steps of
0 0
+PPh3 _____________________________________________ e
HO R2¨Br step A HO R--PPh3 Br
base
)((s 0
iso-butyraldehyde somerization
solvent HO i R2-
step B
hydrogenation
0
HO
CA 03215699 2023- 10- 16
WO 2022/229314 PCT/EP2022/061321
21
wherein R2 is a CG_isalkyl or CG_isalkenyl, which may be straight or branched,
step A-1, the reaction is performed without solvent or with any organic
solvent, such as Et0Ac,
step B-1, a solvent is selected from the group comprising or consisting of 2-
methyl
tetrahydrofuran, tetra hydrofuran, toluene and any other aprotic organic
solvent,
step B-1, a base is selected from the group comprising or consisting of NaH,
KH, t-BuOK, t-
BuONa and another sodium or potassium al koxides,
isomerization step C-1, a catalyst is selected from the group comprising or
consisting of HNO2,
HNO3 and any other combination that can generate HNO2 or HNO3, and
hydrogenation step D-1, a catalyst is selected from the group comprising or
consisting of Pd/C,
Pd/A1203 and any other heterogeneous hydrogenation catalyst, a hydrogen source
is hydrogen
gas.
The compounds of formula II may be prepared comprising or consisting of the
steps of
0 OH 0
+ catalystarty_ catalyst
step A step B
0 0
step C step D
0 0
=
step E HO step F HO
step A-2, the reaction is performed without solvent or with any organic
solvents, such as
toluene, a catalyst is selected from the group comprising or consisting of
pyrrolidine, other
amines and corresponding salts, NaOH, KOH, and other inorganic bases,
step B-2, the reaction is performed without solvent or with an organic
solvent, such as toluene,
a catalyst is selected from the group c comprising or consisting of p-Ts0H,
sulfuric acid,
Amberlyst-15, and other acids,
step C-2 and step F-2, a hydrogen source is hydrogen gas, a catalyst is
selected from the group
comprising or consisting of Pd/C, Pd/A1203 and another heterogeneous
hydrogenation catalyst,
step D-2, an oxidants is selected from the group comprising or consisting of
aqueous H202,
peroxyacids and another peroxides, a catalyst is selected from the group
comprising or
consisting of Candida antarctica lipase A, Candida antarctica lipase B, cross-
linked Substilisin
A protease, Porcine pancreas lipase, Candida cylindracea lipase, Rhizopus a
rrhizus, Penicillum
cyclopium, Mucor miehei, Thermomyces la nuginosus lipase, Candida rugosa
lipase and
Pseudomonas lipoprotein lipase, and
step E-2, a reaction medium is selected from the group comprising or
consisting of aqueous
sulfuric acid solution or another strong acidic media, and
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
22
Step F-2, a catalyst is selected from the group comprising or consisting of
Pd/C, Pd/A1203, Pd/
molecular sieves, Pt/C, Pt/A1203, and Pt/molecular sieves, a hydrogen source
is hydrogen gas.
The processes for the preparation of compounds of formula II may be performed
at a
temperature is between room temperature and 150 C, or between room temperature
and
115 C. These processes may be performed at a pressure between 0.900 and 0.200
MPa, or
about 0.1 MPa.
Experimental sections
Preparation of vanillylanni ne
Vanillylamine was prepared from its hydrochloride salt. The HCI salt was
purchased from
commercial suppliers or prepared according to literature procedures
(ChemBioChem 2009,
10, 823; J. Med. Chem. 2018, 61, 8225.).
0 NH2=HC1 Si NH2
NaOH
HO water HO
OMe OMe
Example 1:50.00 g of vanillylamine HCI salt was dissolved in 500 mL of cold
water (-5 C), and
cooled with an ice-bath, 1 equivalent of 3 M NaOH (87.9 mL) was portion-wise
added in 10
min while keeping vigorous stirring. The internal temperature kept about 5 C.
After addition
of all bases, the milky solution was stirred for further 5 min, then filtered.
The white product
in the funnel was washed twice with cold water (5 C, 100 mLx2), then dried
under vacuum
until the weight remains the same. 37.32 g (92.4% yield) of product was
obtained.
Example 2: 500.0g of vanillylamine HCI salt was dissolved in 5 L of water (10 -
15 C), 1
equivalent of 3 M NaOH was portion-wise added in 20 min while keeping vigorous
stirring.
After addition of all bases, the milky solution was stirred for further 10
min, then filtered. The
white product in the funnel was washed twice with cold water (1 Lx2), then
dried in a vacuum
chamber at SO C for 24 hours. 478.0 g of off-white product was obtained with
19.5wt%
moisture content (determined with Kern DBS 60-3 moisture analyser).
Preparation of fatty acids
Preparation of fatty acids with Wittig reaction as key step
3
0 0
HO..--11.õ....-----..õ..^....õ..-Br + PPh3
solvent HO) PPh BreL--
--------'¨'.
t-BuOK \....--"' . 0
0
iso-butyraldehyde isomerization
______________________ 7...
2-MeTHF HO
hydrogenation
0
HO
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
23
Example 3: Preparation of (6-Carboxyhexyl)triphenylphosphonium bromide.
In a 1 L round-bottom flask, 97.53 g 6-bromohexanoic acid and 131.15 g (1.0
equiv)
triphenylphosphine (PPh3) were dissolved in 500 mL Et0Ac. The mixture was
heated at 75-80
C and stirred for 7 days. After filtration, the collected product was washed
with Et0Ac (50
mLx2) and then dried under vacuum to give 221.80 g white powder (97.0% yield).
The
combined filtrate was recycled as solvent for more batches.
Example 4: Preparation of (Z)-8-methyl-6-nonenoic acid.
In a 1 L two-necked round-bottom flask 100.00 g (6-
Carboxyhexyl)triphenylphosphoniurn
(Ph3P0) bromide and 49.07 g (2.0 equiv) KOtBu were dissolved in 300 mL 2-MeTHF
under
protection of nitrogen atmosphere and cooled using an ice water bath. The
reaction mixture
turned bright orange color while the compounds were dissolved. A solution of
18.92 g (1.2
equiv) isobutyraldehyde in 200 mL 2-MeTHF was slowly added to the cold
reaction mixture,
which quickly turned white. After the addition was completed, the reaction
mixture was
warmed to room temperature and stirred for 6 h. The reaction was quenched by
addition of
500 mL H20. The MeTHF solvent was recovered by distillation. After cooling to
room
temperature, most of Ph3P0 was precipitated and collected as white powder by
filtration. The
filtrate was acidified with concentrated HCI to pH 2, the formed organic layer
was collected,
and the water phase was extracted with Et20 (100 mLx2). The organic phases
were combined,
dried with anhydrous Na2SO4, and concentrated to give 56.6 g crude product.
This crude
product was then distilled under reduced pressure to give (Z)-8-methyl-6-
nonenoic acid (32.76
g, 88% yield, ZIE 11:1 by NMR analysis) as colorless oil product.
Example 5: Preparation of 8-methyl nonanoic acid.
32 g of (Z)-8-methyl-6-nonenoic acid was dissolved in 150 mL of diisopropyl
ether. 0.5 mol%
of Pd/C powder was then dispersed in this solution. The mixture was
hydrogenated with H2
balloon at room temperature overnight. The catalyst was recovered by
filtration. The solvent
was recovered by distillation. 8-methyl nonanoic acid was obtained as
colorless oil with >99%
yield.
Example 6: Preparation of (E)-8-methyl-6-nonenoic acid.
In a 1 L two-necked round-bottom flask 100.00 g (6-
Carboxyhexyl)triphenylphosphonium
bromide and 49.07 g (2.0 equiv) KOtBu were dissolved in 300 mL 2-MeTHF under
protection
of nitrogen atmosphere and cooled using an ice water bath. The reaction
mixture turned
bright orange color while the compounds were dissolved. A solution of 18.92 g
(1.2 equiv)
isobutyraldehyde in 200 mL 2-MeTHF was slowly added to the cold reaction
mixture, which
quickly turned white. After the addition was completed, the reaction mixture
was warmed to
room temperature and stirred for 6 h. The reaction was quenched by addition of
500 mL H20.
The MeTHF solvent was recovered by distillation. After cooling to room
temperature, most of
Ph3P0 was precipitated and collected as white powder by filtration. The
filtrate was acidified
with concentrated HCI to pH 2, the formed organic layer was collected, and the
water phase
was extracted with DI PE (100 mLx2). The organic phases were combined and
concentrated.
This crude intermediate was then treated with concentrated HNO3 (0.03 equiv)
at 85 C under
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
24
protection of nitrogen atmosphere for 24 hours. After cooling, the mixture was
washed with
water (50 mLx2). The aqueous phases were combined and extracted with DIPE (50
mLx2). The
organic phases were combined, dried with anhydrous Na2504, and concentrated to
give 53.1
g crude product. This crude product was then distilled under reduced pressure
to give (E)-8-
methyl-6-nonenoic acid (30.28, 81% yield, EIZ 86:14 by NMR analysis) as
colorless oil product.
Preparation of 8-methyl nonanoic acid starting from cyclohexanone and
isobutyraldehyde
0 AcOH 0 OH 0
0 pyrrolidine p-Ts0H
______________________________________________________________ L:rr
+
83% yield
0o for 2 steps
Pd/C,H novozym 435 0
full conversion
30w% H202
91% conversion
0 0
SOW% H2S 04 Pd/C, H,
=
HO HO
37% yield for 4 steps
Example 7: Preparation of 2-(2-methylpropylidene)cyclohexan-1-one
0 1. AcOH 0
0 pyrrolidine
Q.y.õ
2. p-Ts0H
50 g isobutyraldehyde, 102 g cyclohexanone (1.5 equiv), 5 mol% of pyrrolidine
and 5 mol%
AcOH were heated and stirred at 40 C for 12 h. After cooling to room
temperature, the
mixture was dispersed in 200 mL of water and 100 mL of toluene and organic
phase was
separated. The aqueous phase was extracted with toluene (50 mL x 2). The
organic phases
were combined and treated with 4 mol% of p-Ts0H=H20 catalyst at refluxing
condition for 2
hours with a Dean-Stark trap to collect the generated water. After cooling
again to room
temperature, the acid was removed by washing with 30 mL of aqueous 1 M NaOH
solution.
Toluene and excess cyclohexanone were recovered by distillation. The enone
product (87.6 g,
83% yield, light yellow) was then distilled out under reduced pressure.
Example 8: Preparation of 2-isobutylcyclohexanone
5 g of 2-(2-methylpropylidene)cyclohexan-1-one was dissolved in 10 mL of
Et0Ac. 0.2 mol%
of Pd/C was added, and the hydrogenation was conducted at room temperature
with H2
balloon for 4 hours. Full conversion was achieved based on NMR analysis. The
catalyst was
recovered by filtration, and the filtrate was directly used in the following
oxidation.
Example 9: Preparation of 7-isobutyloxepan-2-one
To the solution of 2-isobutylcyclohexa none in Et0Ac, were added Noyozyrn
435(tm) (250 mg)
and 30% aqueous H202(3 equiv). The mixture was stirred and heated at 50 C.
After 24 h, 91%
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
conversion was achieved based on NMR analysis. After cooling to room
temperature, the
lipase catalyst was recovered by filtration. The filtrate was washed with 5%
aqueous Na25203
solution and brine to remove excess peroxide. The organic phase was
concentrated to give
the crude lactone.
5 Example 10: Preparation of 8-methyl-6-nonanoic acid
The above crude lactone was dispersed in 5 M H2504 (40 mL) and heated at 110 C
oil bath.
After 20 h, the mixture was cooled to room temperature, extracted with DIPE
(20 nIL x 3). The
organic phases were washed with brine, dried over anhydrous Na2SO4 and
filtered. To the
filtrate, 0.5 mol% Pd/C powder was added, and the hydrogenation was conducted
at room
10 temperature with Hz balloon for 24 hours. After filtration to recover
the Pd catalyst, the filtrate
was concentrated and purified by flash chromatography on silica gel to give
the 8-methyl-6-
nonanoic acid (2.1 g, 37% yield from 5 g of the enone product of Example 7).
Preparation of capsaicinoids
0
0 NI-12
HO)LR Novozym 435 N.AR
HO
reflux with
1-10
Dean-Stark distillation
OMe OMe
15 Example 11: Preparation of capsaicin with excess amine in D1PE. At
normal atmospheric
pressure (approximately 1 atm), 8-methyl-6-nonenoic acid (3.65 g), vanillyl
amine (1.1 equiv.),
Novozym 435(tm) on beads (498.8 mg, 14 w/w% E/S) were refluxed (about 69 C)
in diisopropyl
ether (45 mL) with a Dean-Stark trap to collect the generated water. After
stirring at about
300 rpm overnight (19 h), the mixture was filtered to recover the lipase
catalyst, and the
20 filtrate was washed with 0.5 M aqueous HC1 solution (10 mL). The aqueous
phase was
extracted with Et20 (10 mLx2) and the organic phase were combined, dried over
anhydrous
Na2SO4 and concentrated to give 6.53 g product (99.7% yield, very pale yellow
color).
Example 12: Preparation of nonivamide with excess fatty acid in toluene. At
normal
atmospheric pressure (approximately 1 atm), Vanillyla mine (4.89 g, 2.00 wt%
water),
25 nonanoic acid (1.01 equiv.) and Novozym 435(tm) on beads (1 g, 20 w/w%
E/S) were refluxed
(about 110 C) in toluene (50 mL) with a Dean-Stark trap to collect the
generated water. After
stirring at about 300 rpm overnight (16 h), the conversion of nonanoic acid
was >99%. After
filtration to recover enzyme catalyst, the mixture was concentrated to give
9.14 g of product
(99.6% yield, white color).
Example 13: Preparation of nonivamide with excess fatty acid in cyclohexane.
At normal
atmospheric pressure (approximately 1 atm), Vanillyl amine (4.89 g, 2.00 wt%
water),
nonanoic acid (1.01 equiv.) and Novozym 435(") on beads (1 g, 20 w/w% E/S)
were refluxed
(about 81 C) in cyclohexane (50 mL) with a Dean-Stark trap to collect the
generated water.
After stirring at about 300 rpm overnight (16 h), the conversion of nonanoic
acid was >99%.
After filtration to recover enzyme catalyst, the mixture was concentrated to
give 9.10 g of
product (99.1% yield, pale yellow color).
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
26
Because the use of lipase on beads is not feasible for large scale production
due to costs for
work-up, like filtering the lipase, etc., next experiment was performed using
lipase
immobilized on a rotary bed reactor.
Example 14: Preparation of dihydrocapsaicin in fix-bed reactor. At normal
atmospheric
pressure (approximately 1 atm), Ina1L reactor equipped with a rotating fix-bed
filled with 12
g of Novozym 435(') (45-60 w/w% E/S), vanillylarnine, slightly excess 8-methyl
nonanoic acid
(1.01 equiv), and diisopropyl ether (600 mL) were refluxed (about 69 C) with
a Dean-Stark
trap to collect the generated water (Figure 1). During reaction, the rpm was
fixed at about
300 rpm.
After reaction, the hot solution was released out and cooled to room
temperature. The
dihydrocapsaicin product crystallized and was collected by filtration. The
filtrate was directly
recycled as solvent for more batches. The results are shown in Table 1. The
yield of
dihydrocapsaicin was 99.7% in average.
Table 1. Preparation of dihydrocapsaicin infix-bed reactor
so N.2 0
+ Novozym 435 (1101
HO HO reflux with HO
OMe Dean-Stark distillation
OMe
1.5 in fix-bed reactor
Amine moisture content of amine Reaction time Conversion
Yield
Cycle
(g) (wt%) (h) (%)
(%)
1 29.41 10.18 24 >99
2 34.34 23.08 24 >99
99.8
3 31.80 16.95 24 >99
100
4 31.80 16.95 24 >99
5 31.80 16.95 24 >99
6 24.89 16.95 16 >99
The results show good conversion rates and yields even after 6 cycles.
Example 15: Preparation of capsaicin in fix-bed reactor (45w/w% E/S). The
reactor system of
Example 14 was cleaned by refluxing with DIPE solvent to remove residual
dihydrocapsaicin.
In this reactor, at normal atmospheric pressure (approximately 1 atm),
vanillylamine, slightly
excess 8-methyl-6-nonenoic acid (1.01 equiv), and diisopropyl ether (600 mL)
were refluxed
(about 69 C) with a Dean-Stark trap to collect the generated water. During
reaction, the rpm
was fixed at about 300 rpm. After reaction, the hot solution was released out
and cooled to
room temperature. The capsaicin product crystallized and was collected by
filtration. The
filtrate was directly recycled as solvent for more batches. The results are
shown in Table 2.
The yield of capsaicin was 99.4% in average.
Table 2. Preparation of capsaicin in fix-bed reactor.
CA 03215699 2023- 10- 16
WO 2022/229314 PCT/EP2022/061321
27
o
o ----
0 NH2 Novozym 435 0 II ---"" =
HO
HO reflux with HO
OMe Dean-Stark distillation
OMe
in fix-bed reactor
Amine moisture content of amine Reaction time Conversion
Yield
Cycle
(g) (wt%) (h) (%)
(%)
1 32.18 16.95 24 >99
2 32.18 16.95 24 >99 99.5
3 32.18 16.95 24 >99 99.9
4 27.83 3.90 20 >99
27.83 3.90 20 >99
The results show good conversion rates and yields even after 5 cycles.
Example 15a: Preparation of nonivamide in fix-bed reactor (15-21 w/w% EN. The
reactor
system of Example 15 was cleaned by refluxing with DIF'E solvent to remove
residual capsaicin.
5 In
this reactor, at normal atmospheric pressure (approximately 1 atm),
vanillylamine, slightly
excess nonanoic acid (1.01 equiv), and diisopropyl ether (600 mL) were
refluxed (about 69 C)
with a Dean-Stark trap to collect the generated water. During reaction, the
rpm was fixed at
about 300 rpm. After reaction, the hot solution was released out and cooled to
room
temperature. The nonivamide product crystallized and was collected by
filtration. The filtrate
was directly recycled as solvent for more batches. The results are shown in
Table 3a. The yield
of nonivamide was 99.8% in average.
Table 3a. Preparation of nonivamide in fix-bed reactor.
o
o
0 NH2
+ Novozym 435 40 il
HO a
HO reflux with HO
OMe Dean-Stark distillation
OMe
in fix-bed reactor
Cycle Amine (g) moisture content of amine (wt%) Reaction time (h) Conversion
(%)
1 39.89 3.90 24 >99
2 39.89 3.90 24 >99
3 44.88 3.90 24 >99
4 45.48 5.16 24 >99
5 45.48 5.16 24 >99
6 45.48 5.16 24 >99
7 50.53 5.16 24 >99
8 48.90 2.00 24 >99
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
28
9 58.68 2.00 24 >99
58.68 2.00 24 >99
11 78.24 2.00 24 >99
The results show good conversion rates and yields even after 11 cycles.
Example 1613: Preparation of nonivamide in 100 L fix-bed reactor. At normal
atmospheric
pressure (approximately 1 atm), in a 100 L reactor equipped with a rotating
fix-bed filled with
1 kg of Novozym 435") (50-100 w/w% US), vanillylamine, excess 8-methyl
nonanoic acid
5
(1.03 equiv), and diisopropyl ether (90 L) were refluxed (about 69 C) with a
Dean-Stark trap
to collect the generated water. During reaction, the rpm was fixed at about
250 rpm. After
reaction, the hot solution was released out and cooled to 15 C. The
nonivamide product
crystallized and was collected by filtration. The filtrate was directly
recycled as solvent for
more batches. The results are shown in Table 3b. The results show that the
process of the
10 invention can be used for
large scale production of amides.
Table 3b. Preparation of nonivamide in 100 L fix-bed reactor
Cycle Amine (g) moisture content of amine (wt%) Reaction time (h) yield (%)
1 1014 5.51 15
96.4
2 1992 2.66 24
95.6
3 1936 <0.5 24
95.1
The results show good conversion rates and yields even after 3 cycles when the
process is
used at large scale.
Comparative examples:
Preparation of capsaicin from fatty acid with drying agents
o
HO 10
NI-12 Novozym 435
molecular sieves
________________________________________________________ tb-
1 IN1
HO solvent
OMe HO
OMe
Example 17: To a 200 mL reactor, were added in toluene (150 mL), 4 A molecular
sieves (10 g),
immobilized enzyme (Novozym 435"), 0.59 g), 8-methyl-6-nonenoic acid (2.02 g),
and
va nillyla mine (2 equiv). The reaction was conducted at 80 C and monitored
by NM R. After 6
hours, the conversion of acid was >99%. After filtration, the organic filtrate
was cooled,
successively washed with 1 M HCI (20 mLx2), water (20 mLx2), and brine (20
mL). After drying
with anhydrous Na2SO4, the solvent was removed under vacuum. 3.07 g (84.7%
yield) of
capsaicin was obtained.
Example 18: To a 25 mL flask, were added in t-BuOH (8 mL), 4 A molecular
sieves (600 mg),
immobilized enzyme (Novozym 435"), 75 mg), 8-methy1-6-nonenoic acid (341 g),
and
va nillyla mine (1.06 equiv)_ The reaction was conducted at 80 C and monitored
by NM R. After
10 hours, the conversion of acid was about 95%.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
29
Preparation of capsaicin with ester as acyl donor:
I.N112
HO
OMe
NR
0 0 H2SO4 enzyme a
HOR Me0H solvent HO
or ElOH OMe
R' = Me, Et
Example 19: Preparation of methyl ester. 4.73 g of 8-methyl-6-nonenoic acid
was dissolved in
30 mL of Me0H. To this solution, 5 drops of concentrated H2SO4 was added as
catalyst. The
resulting solution was refluxed overnight. After cooling to room temperature,
most of Me0H
was removed by rotary evaporator and the residue was dissolved in Et20 (30 mL)
and washed
with 5% Na2CO3 solution (10 mLx2). The organic phase was dried with anhydrous
Na2SO4 and
concentrated to give the ester product with >99% yield.
Example 20: Preparation of capsaicin with ester in fix-bed reactor. In a 1 L
reactor equipped
with a rotating fix-bed filled with 6 g of Novozym 435(') (10-20 w/w% E/5),
ethyl ester of 8-
methyl 6-nonenoic acid, excess vanillylamine (1.1 equiv), and diisopropyl
ether (600 mL) were
refluxed. After reaction, the hot solution was released out and cooled to room
temperature.
The solution was successively washed with 0.5 M HC1 (60 mL), water (60 mL),
and brine (20
mL). After drying with anhydrous Na2SO4, the solvent was recovered by rotary
evaporation.
The capsaicin product was obtained with light yellow color. The results are
shown in Table 4.
Table 4. Preparation of capsaicin with ester infix-bed reactor.
Cycle Ethyl ester (g) Time (h) Conversion (%) Yield (%)
1 30 22 >99 92
2 30 16 >99 93
3 60 40 90 80
4 30 42 >99 93
Example 21: Preparation of capsaicin from ester with distillation apparatus.
To a flask
equipped with short-path distillation apparatus, 923 mg of methyl ester of 8-
methyl-6-
nonenoic acid, 200 mg of Novozym 435(tm) on beads and 1.1 equiv. of
vanillylamine were
heated at 80 C in 20 mL of t-BuOH. After 20 hours, the conversion of ester
was >99%
according to NMR analysis.
The results show that using esters is possible, but that the yield is lower
compared to earlier
examples 14, 15 and 16. An extra process step is needed because the methyl
ester is prepared
from the corresponding acid. Further, the work-up is tedious when up-scaling.
Besides,
recycling of the solvent is difficult, which is important for reducing costs
and environmental
impact of the process at large scale production.
Example 22: Preparation of capsaicinoids in neat condition (no solvent).
Experimental results
are shown in Table 5.
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
Table 5_ Preparation of capsaicinoids in neat condition.
Lipase
Vanillylamine Acyl donor Temp Time
Yield
Entry (g/g product
(equiv) (equiv) ( C) (h)
CYO
amine)
nonanoic acid
1* 1 0.2 100 18 nonivamide
37
(3 equiv)
ethyl 8-methyl-
2 1 6-nonenoate 0.2 80 16 capsaicin
91
(2 equiv)
8-methyl-6-
3 2 nonenoic acid 150 16 capsaicin
37
(1 equiv)
methyl 8-
methyl-6-
4 1.2 nonenoate 150 16 capsaicin
10
(1 equiv)
under vacuum.
The results show that the yield of the process is decreased using reduced
pressure (Entry 1).
Example 23: Preparation of capsaicin with lipase catalyst and without
dehydration.
5
Vanillylamine (1 mmol), 8-methy1-6-nonenoic acid (1 mmol), and Novozym 435(")
(45 mg)
were stirred in toluene (4 mL) and heated at 80 C for 48 h. 72% conversion
was achieved
based on N MR analysis.
Example 24: Preparation of capsaicin without lipase catalyst and with Dean-
Stark distillation.
With a Dean-Stark trap to collect generated water, vanillylamine (1 mmol) and
8-methyl-6-
10
acid (1 mmol) were refluxed in toluene (4 mL) at 115 C oil bath for 20 h. 19%
conversion was achieved based on N MR analysis.
Example 25: Preparation of capsaicin with acid chloride as acyl donor
/01 NH2 0
SOC1, HO
0 0 ONk H
Et20
_____________________________________________________________ HO
ome
In one 1 L flask, 47.17 g of 8-methyl-6-nonenoic acid was dissolved in 400 mL
of anhydrous
15
Et20. 30.1 mL of S0Cl2 (1.5 equiv) was dissolved in 100 mL of anhydrous Et20,
and slowly added
to the acid solution. The resulting solution was refluxing for 3 hours, and
then the excess SOC12
and solvent were removed under reduced pressure. The resulting acid chloride
was then
dissolved in 200 mL of anhydrous Et20. To a slurry of 84.7 g vanillylamine (2
equiv) in 400 mL
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
31
of anhydrous Et20, was added slowly the acid chloride solution in 2 hours.
After addition, the
refluxing was continued for 2 hours. The mixture was cooled with ice water
bath, the
precipitate was filtered. The organic filtrate was successively washed with 1
M HCI (50 mLx2),
water (50 mLx2), and brine (50 mL). After drying with anhydrous Na2SO4, the
solvent was
removed under vacuum. 54.3 g (64.2% yield) of capsaicin was obtained.
Preparation of miscellaneous amides by the combination of enzymatic catalysis
and Dean-
Stark distillation.
Preparation of (R)-2-methoxy-N-(1-phenylethyl)acetamide
NH2 0 0
0
CALB
DIPE "- HN
7 OA
reflux with
Dean-Stark distillation
Example 26. With a Dean-Stark trap to collect generated water, 1-phenylethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435(') on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 10 h. After work
up, (R)-2-methoxy-
N-(1-phenylethypacetamide was obtained with 85% yield and 8% ee.
Example 27. With a Dean-Stark trap to collect generated water, 1-phenylethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435(tm) on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 3 h. After work
up, (R)-2-methoxy-
N-(1-phenylethyl)acetamide was obtained with 75% yield and 55% ee.
Example 28. With a Dean-Stark trap to collect generated water, 1-phenylethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435(trn) on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 1 h. After work
up, (R)-2-methoxy-
N-(1-phenylethyl)acetamide was obtained with 48% yield and 97% ee.
Example 29. Preparation of (R)-2-methoxy-N-(1-(4-methoxyphenyl)ethypacetamide.
NH2 0 0
Et0)0 CALB 0
DIPE
reflux with p-Me0Ph
Dean-Stark distillation
With a Dean-Stark trap to collect generated water, 1-(4-methoxyphenyl)ethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435(lm) on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 0.83 h. After work
up, (R)-2-methoxy-
N-(1-(4-methoxyphenyl)ethypacetamide was obtained with 44% yield and 97% ee.
Example 30. Preparation of N-phenethylnonanamide.
0
0 CALB
HNA,...(CHACH3
Ph
HO2)6CH3
reflux with
Dean-Stark distillation
CA 03215699 2023- 10- 16
WO 2022/229314
PCT/EP2022/061321
32
With a Dean-Stark trap to collect generated water, 2-phenylethan-1-amine (1
mmol),
nonanoic acid (1.05 mmol), and Novozym 435(t1) on beads (45 mg) were refluxed
in
diisopropryl ether (30 mL) at 90 C oil bath for 10 h. After work up, N-
phenethylnonanamide
was obtained with 98% yield.
Example 31. Preparation of N-phenethylstearamide.
0
0 CALB
HN2)15CF13
HOH2)15CH3 DI
Ph N H2 + PE
reflux with
Dean-Stark distillation
With a Dean-Stark trap to collect generated water, 2-phenylethan-1-amine (1
mmol), stearic
acid (1.05 mmol), and Novozym 435(") on beads (45 mg) were refluxed in
diisopropryl ether
(30 mL) at 90 C oil bath for 10 h. After work up, N-phenethylstearamide was
obtained with
98% yield.
The present invention is not limited to the embodiments disclosed but may be
varied and
modified within the scope of the following claims.
CA 03215699 2023- 10- 16
PCT/EP 2022/061 321 - 27.01.2023
1
Title: A process for enzymatic synthesis of amides from amines and carboxylic
acids or esters.
Field of the invention
The present invention relates to a process for enzymatic synthesis of amides
from amines and
carboxylic acids or esters using a lipase.
Background of the invention and prior art
Amide linkage is important in development of numerous compounds, such a
pharmaceutical
drugs and polymers. Several processes for direct catalytic amidation have been
developed
over the years.
In thermal amidation, no catalyst may be used. This process is performed at
high temperature
(> 140 C) and the yield is dependent on the temperature used, the
concentration of the
substrate, the solvent used and other parameters.
Metal-based amidations have been done using boron-based catalysts or palladium-
based
catalyst. Although higher yield can be obtained compared to thermal amidation,
the processes
are expensive and time consuming. Recycling of catalysts and solvents is
challenging.
Neither thermal nor metal based amidations are environmentally friendly
processes. Several
attempts have been made to improve the efficiencies of the processes and
reduce the costs
and carbon food-print.
In amidation processes, water must be removed to improve the yield of the
processes. Most
of amidation processes are therefore performed under reduced pressure. This
increases costs
and thus increases difficulties for large scale amidation. Molecular sieves
may be used as well,
but these are still expensive for use at large scale. A Dean-Stark apparatus
may be used as well
to remove water from an amidation process.
Enzymatic amidation has been developed over the years using different kind of
enzymes like
lipases. These so-called biocatalysts can be used at lower temperature and
show good
selectivity. However, current technologies show very limited substrate scope
and often
require long reaction times (days). Combining enzymatic amidation with a
palladium catalyst
may result in a yield of about 70% as shown by Palo-Nieto et al., ACS Catal.,
2016, 6, 3932-
3940.
Another drawback of biocatalysts is costs. To reduce the costs and improve
efficiency of the
amidation process, the enzymes can be immobilized e.g. on beads during the
reaction. This
allows recirculation of the enzyme. The use of flow reactors has further
improved the
biocatalytic amidation process. However, recirculation of the lipase is both
time- and cost-
ineffective.
Up to date, there is no environmentally friendly catalytic amidation process
that is sufficiently
efficient and cost effective to be used for large scale production. This is a
top priority of the
American Chemical Society Green Chemistry Pharmaceutical Roundtable
(https:fiwww.acsgcipr.org). Today, most methods utilize stoichiometric amounts
of toxic
activating reagents, Dunetz et. al. Org. Process. Res. Dev. 2016, 20, 140.
Thus, there is still a
CA 03215699 2023- 10- 16
AMENDED SHEET
1
Title: A process for enzymatic synthesis of amides from amines and carboxylic
acids or esters
Field of the invention
The present invention relates to a process for enzymatic synthesis of amides
from amines and
carboxylic acids or esters using a lipase.
Background of the invention and prior art
Amide linkage is important in development of numerous compounds, such a
pharmaceutical
drugs and polymers. Several processes for direct catalytic amidation have been
developed
over the years.
In thermal amidation, no catalyst may be used. This process is performed at
high temperature
(> 140 C) and the yield is dependent on the temperature used, the
concentration of the
substrate, the solvent used and other parameters.
Metal-based amidations have been done using boron-based catalysts or palladium-
based
catalyst. Although higher yield can be obtained compared to thermal amidation,
the processes
are expensive and time consuming. Recycling of catalysts and solvents is
challenging.
Neither thermal nor metal based amidations are environmentally friendly
processes. Several
attempts have been made to improve the efficiencies of the processes and
reduce the costs
and carbon food-print.
In amidation processes, water must be removed to improve the yield of the
processes. Most
of amidation processes are therefore performed under reduced pressure. This
increases costs
and thus increases difficulties for large scale amidation. Molecular sieves
may be used as well,
but these are still expensive for use at large scale. A Dean-Stark apparatus
may be used as well
to remove water from an amidation process.
Enzymatic amidation has been developed over the years using different kind of
enzymes like
lipases. These so-called biocatalysts can be used at lower temperature and
show good
selectivity. However, current technologies show very limited substrate scope
and often
require long reaction times (days). Combining enzymatic amidation with a
palladium catalyst
may result in a yield of about 70% as shown by Palo-Nieto et al., ACS Catal.,
2016, 6, 3932-
3940.
Another drawback of biocatalysts is costs. To reduce the costs and improve
efficiency of the
amidation process, the enzymes can be immobilized e.g. on beads during the
reaction. This
allows recirculation of the enzyme. The use of flow reactors has further
improved the
biocatalytic amidation process. However, recirculation of the lipase is both
time- and cost-
ineffective.
Up to date, there is no environmentally friendly catalytic amidation process
that is sufficiently
efficient and cost effective to be used for large scale production. This is a
top priority of the
American Chemical Society Green Chemistry Pharmaceutical Roundtable
(https://www.acsgcipr.org). Today, most methods utilize stoichiometric amounts
of toxic
activating reagents, Dunetz et. al. Org. Process. Res. Dev. 2016, 20, 140.
Thus, there is still a
CA 03215699 2023- 10- 16
PCT/EP 2022/061 321 - 27.01.2023
2
need for a greener and more cost effective amidation process that can be used
at a larger
scale.
Capsaicinoids are commonly used in food environmentally friendly products.
Capsaicin is also
widely used in the pharmaceutical industry. Capsaicin is for example used as
an analgesic in
topical ointments and dermal patches to relieve minor aches and pains of
muscles and joints
associated with arthritis, backache, strains and sprains, or to reduce the
symptoms of
peripheral neuropathy.
Capsaicinoids can be isolated from natural sources (e.g. Capsicum spp pepper
fruits), but this
gives predominantly capsaicin and dihydrocapsaicin, since many of the other
capsaicinoids are
present only in trace amounts. Chemical synthesis is thus useful to obtain the
more
uncommon capsaicinoids, such as nonivamide, and for making none-natural
capsaicinoids.
Capsaicinoids can be prepared from vanillin by first reducing vanillin oxime
using a mixture of
an excess of metal (Zn) and ammonium formate in methanol under reflux to
obtain
vanillylamine. Alternatively, the amide bond-formation can be accomplished by
an enzyme-
catalyzed transformation between vanillylamine and different fatty acid
derivatives.
W02015/144902A1 discloses a multi-catalytic cascade relay sequence involving
an enzyme
cascade system that when integrated with other catalytic systems, such as
heterogeneous
metal catalysts and organic catalysts, converts an alcohol to an amine and
amide in sequence
or in one-pot.
US2017081277A1 discloses an amidation using dialkyl-amines as substrates.
Novozym 435(-)
immobilized on beads are used. A Dean Stark apparatus may be used to remove
ethanol from
the reaction mixture. The reactions are performed under reduced pressure. For
large scale
manufacturing, beads are not suitable because it is costly and time consuming
to separate the
beads from the reaction mixture. Further, for large scale production, reduced
pressure is
preferably avoided to reduce cost and time of the overall process.
U56022718 discloses a process for preparation of capsaicin analogues using
hydrolysis and
capsaicin as starting materials.
Pithani S., Using spinchem rotation bed reactor technology for immobilized
enzymatic
reactions: a case study, Org. Process Res. Dev., 2019, vol.23, pages 1926-
1931, discloses
advantages of using rotary bed immobilized lipase. An acylation reaction is
used to
demonstrate that lipase (Novozym 435-) can be used in a rotating bed reactor.
The loading
was limited to 10 wt% due to high costs. A loading of 5 to 10 wt% was deemed
sufficient to
achieve a conversion of 45-50% within 6 hours. The overall yield after
upscaling was 39%.
Although Pithani shows that a rotation bed reactor is useful for acylations,
it also shows that
it is costly and results in a conversion of 45-50% with an overall yield of
39%. The results
disclosed in Pithani are disconcerting for large scale production using a
rotation bed reactor.
There is an increasing need for large scale production of amide compounds like
capsaicinoids.
Such processes are preferably efficient and effective having improved yields
compared to
known processes. Such amidation processes are preferably environmentally
friendly and
especially cost effective.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
3
Summary of the invention
It is an object of the present invention to at least partly overcome the above-
mentioned
problems, and to provide an improved process for the synthesis of amides from
amines and
carboxylic acids or esters.
This object is achieved by a process as defined in the claims.
One aspect relates to a process for enzymatic synthesis of amides of formula
Ill from amines
of formula I and compounds of formula II,
1 3 2 Lipase 1
R -NH2 + RO-R-C(0)-R R -N(H)-C(0)-R2
i II III
wherein R1 is selected from the group comprising or consisting of Ci_nalkyl-,
Ci_nalkenyl-, Ci_
12a lkynyl-, C1_12a I koxy-, CI...12a I kyl-O-C3.42a I kyl-, C1_12a I kyl-
OC(0)-Ci12a lkyl-, C1_12a I kyl-N H-Ci_
12a lkyl-, Ci-12a I kyl-N HC(0)-C142a I kyl-, C3-12cycloalkyl-, C342cycloa Ike
nyl-, C5_12aryl-, C3-
12CYCI0a I kyl-Ci_6a I kyl-, C3_12cyc10a I kenyl-Ci..6a I kyl- and CsAla ryl-
Ci_6a I kyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, Ci_
6 hyd roxya I kyl-, C1-6 haloya lkyl-, Ci_6amineoxyalkyl-, Ci_6a mideya I kyl-
, Ci_6ca rboxya I kyl-, Cl-
65U Ifuralkyl-, Ci_6s ulfidea lkyl- and Ci_6a I koxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or 5,
wherein R2 is selected from the group comprising or consisting of hydrogen,
Ci_30a1ky1-, Ci_
30a Ikenyl-, Ci_30a I kynyl-, Ci_30a I koxy-, Ci_30a I kyl-O-CiAla lkyl-,
Ci_30a lkyl-OC(0)-CiA2a I kyl-, Cl-
30a lkyl-N H-Ci_12a I kyl-, Ci_30 I kyl-NHC(0)-Ci_12a I kyl-, C342cycloalkyl-,
C342cycloa I ke nyl- and Cs_
12a ryl-, C342cyc10a lkyl-Ci_6alkyl-, C3_12cyc10a I kenyl-Ci_6a I kyl- and Cs-
12a ryl-C1-6a I kyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amide,
Ci_6hydroxya lkyl-
, Ci_6ha loya I kyl-, Ci_6ca rboxya I kyl-, C1_6sulfuralkyl-, Ci_6su lfidea I
kyl- and C1_6a I koxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S,
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_6alkyl-, Ci_
6a I kenyl-, Ci_6a I kynyl-, C1_6a I koxy-, Ci_6a I kyl-O-Ci_6a lkyl-,
Ci_6alkyl-OC(0)-Ci_6alkyl-, Ci_6a lkyl-N H-
C1..6alkyl-, C3_6alkyl-NHC(0)-Ci_6a1ky1-, C342cycloalkyl-, C342cycloalkenyl-
and C6_12aryl-, C3-
12CYCI0a I kyl-Ci_6a I kyl-, C3-12cycloa I kenyl-Ci-6a I kyl- and Cs-i2a ryl-
Ci-6a I kyl-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, Ci-
6 hyd roxya I kyk, C1-6 haloya lkyl-, C1_6amineoxyalkyl-, Ci_6a mideya I kyl-,
Ci_6ca rboxya I kyl-, Ci_
CA 032156993523- 106Sil1fura1ky1-, Ci_6sulfidealkyl- and Ci_6a I koxy-, and
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
4
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S. and
wherein R is a bond or Ci.6alkyl-,
characterized in that
the lipase is immobilized on a rotary bed reactor or on a spin-fixed-bed
reactor and a Dean-
Stark apparatus is used for dehydration.
In some aspects, lipase is immobilized on a rotary bed reactor and a Dean-
Stark apparatus is
used for dehydration.
In some aspects, a lipase immobilized on beads is disclaimed.
In the processes of the invention, as defined anywhere in here, a combination
of enzyme
catalysis and azeotropic dehydration is used for direct catalytic amide
synthesis. The enzyme,
lipase, is immobilized on a rotary bed reactor or on a spin-fixed-bed reactor.
Compared to
immobilizing lipase on beads or using sieves, the lipase in the process of the
invention can
easily be recirculated. This allows the process to be performed in a time- and
cost-effective
manner, especially at large scale.
The unique combination of a rotary bed reactor or a spin-fixed-bed reactor and
a Dean-Stark
apparatus improves the yield (>90, or 99%) as well as the conversion rate (>90
or 99%). The
unique combination allows the use of wet raw material. The process can be
performed at
atmospheric pressure and at temperatures below 100 C (60 ¨ 90 C). The process
is
environmentally friendly. The process is suitable for large scale production
of amides.
An easy workup and purification process allow the process to be used at a
large scale. The
enzymes and the solvents used, if any, are easy to recycle, which in turn
makes large scale
production feasible. The unique combination of an immobilized enzyme on a
rotary bed
reactor or on a spin-fixed-bed reactor and a Dean Trap apparatus allows the
process to be
extended to the synthesis of other amides and esters.
In one aspect, the process is performed under neat conditions. The process can
be performed
without any solvent. This may improve the efficiency and effective and
environmentally
friendliness of the process. It also reduces costs for performing the process.
A neat process
further reduces costs for the process on a large scale.
Compared to known processes, the direct amidation process of the invention has
an improved
conversation rate as well as an improved yield. Less process steps are needed
for the
amidation, which reduces time and costs. The mass flow is improved in the
process of the
invention. Because the enzyme is immobilized/fixed, the reaction products can
easily be
filtered off and purified. The process of the invention has an improved
reaction rate.
The process allows for effective and efficient large-scale production of amide
compounds like
capsaicinoids. The process has improved yields compared to known processes.
The amidation
processes are environmentally friendly and especially cost-effective.
The combined use of the rotary bed reactor and the Dean-Stark apparatus allows
for control
CA 03215699 2023- 100i the moisture content during the process. A low moisture
content improves conversion rate
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
and yield. The results in Cycle 2 of Table 1 in example 14, show that even raw
material having
a moisture content of 23wt% can be used. This improves the flexibility of the
process. This also
improves the feasibility for large scale use of the process.
In some aspects, the option for R2 to be a (dialkyl)-amine is disclaimed.
5 According to some aspect of the invention, R1 is selected from the group
comprising or
consisting of C1_12alkyl-, C1_12a1kenyl-, C1_22alkynyl-,
12a lkyl-OC(0)-C2_22a I kyl-, C3_12cycloalkyl-, C342cyc10alkenyl-, C5_22ary1-,
C342cycloa I kyl-C1_6a I kyl-,
C3_22cycloa I ke nyl-C1-6a lkyl- and Cs-12a ryl-C1-6a I kyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C2_6hydroxyalkyl-, C1_
6 ha loya lkyl-, C1_6carboxyalkyl-, C1_65u lfu ra I kyl-, C1_6s ulfidea lkyl-
and C2_6a1koxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Ci_soalkyl-,
30a lkenyl-, Ci_30a I kynyl-, C1_30a I koxy-, C1_30a I kyl-O-C1_12alkyl-,
C2_30a lkyl-OC(0)-C1_12a I kyl-, C3-
ucycloalkyl-, C3-12cycloalkenyl- and Cs-12aryl-, C3-12cycloalkyl-C1-6a1kyl-,
C3-12cycloalkenyl-C1-
6a I kyl- and Cs_12ary1-C1_6a1ky1-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C1_6hydroxyalkyl-,
6 ha loya lkyl-, C1_6carboxyalkyl-, C1_6su lfu ra I kyl-, C1_6sulfidealkyl-
and C2_6a I koxy-, and
wherein R3 is selected from the group comprising or consisting of hydrogen,
C1_6alkyl-, C1_
6a I kenyl-, C1_6a I kynyl-, C1_6a I koxy-, C1_6a I kyl-O-C1_6a I kyl-, C1-6a
I kyl-OC(0)-C1-6a I kyl-, C3-
ucycloa I kyl-, C3_22cycloalkenyl- and Cs_12aryl-, C3_12cyc1oa lkyl-C2_6a I
kyl-, C342cycloa I ke
6a I kyl- and Cs-12a ryl-C1-6a lkyl-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
C1_6hydroxyalkyl-, C1_
6 ha loya lkyl-, C1_6carboxyalkyl-, C1_65u lfu ra I kyl-, C1_6sulfidealkyl-
and C1_6a1koxy-, and
wherein R is a bond or C1_6a1ky1-.
According to some aspect of the invention, R1 is selected from the group
comprising or
consisting of C1_12alkyl-, C1_12a1kenyl-, C1_22a1kyny1-, C1_12alkoxy-,
C1_12alkyl-O-C1_12alkyl-,
12a lkyl-OC(0)-C1-12a I kyl-, C3-12cycloalkyl-, Cs-ncycloalkenyl-, CS-12a ryl-
, C3-12cycloa I kyl-C1-6a I kyl-,
C3_22cycloa I ke nyl-C1_6a lkyl- and Cs_12a ryl-C1_6a I kyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, and C1_6a1koxy-, and
wherein R2 is selected from the group comprising or consisting of C1_30a1ky1-,
C1_30alkenyl-,
wherein R3 is selected from the group comprising or consisting of hydrogen,
C1_6a1ky1-, and
wherein R is a bond or C1_6a1ky1-.
According to some aspect of the invention, R1 is selected from the group
comprising or
consisting of Clzalkyl-, C1-6a1kenyl-, C1_6alkynyl-, C1_6alkoxy-,
C1_6alkyl-
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
6
C3_6cyc1oalkyl-, C3_6cyc1oa1keny1-, C6aryl-, C3_6cyc1oalkyl-C1.6a1ky1-, C3.
6cycloalkenyl-Ci_6a1ky1- and C6aryl-Ci-3alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, and C1_6a1koxy-, and
wherein R2 is selected from the group comprising or consisting of Ci_malkyl-,
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_3a1ky1-, and
wherein R is a bond or C1_3a1ky1-.
According to some aspect of the invention, R1 is selected from the group
comprising or
consisting of Ci_6alkyl-,
C3_6cyc1oa1kyl-, C3-
6cyc10a I kenyl-, C6-7aryl-, C3-6cycloa I kyl-Ci_3a I kyl-, C3_6cycloa I
kyl- and Cs-7a ryl-C7.-
3alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
Ci_3hydroxyalkyl-,
3haloyalkyl-, and C1_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_isalkyl-, C5.
18a Ikenyl-, Cs-isalkoxy-, CsAsalkyl-O-C1_6a1kyl-, and C5-15a
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen and carboxy,
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_3a1ky1-,
3a1k0xy- and Ci_3alkyl-O-Ci_3a1ky1-, and
wherein R is a bond or Ci_3a1ky1-.
According to some aspect of the invention, R1 is selected from the group
comprising or
consisting of hydrogen, C6_7ary1- and C5_7ary1-Ci_3a1ky1-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and Ci_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_isalkyl- and Cs_
wherein R3 is selected from the group comprising or consisting of hydrogen,
Ci_3a1ky1- and
wherein R is a bond or Ci_3a1ky1-.
According to some aspect of the invention, R1 is Cs_7aryl-C1_3a1ky1-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy and Ci_3alkoxy-,
wherein R2 is selected from the group comprising Cs_malkyl- and Cs_isalkenyl-,
and
wherein R3 is hydrogen, methyl or ethyl, and
wherein R is a bond.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
7
The process with these compounds results in improved yields and conversion
rates, which is
especially important for large scale production.
According to some aspect of the invention, R1 is Cs_7aryl-C1_3a1ky1-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and C1_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_malkyl- and Css-
na lkenyl-,
wherein R3 is hydrogen, methyl or ethyl, and
wherein R is a bond or Ci..2a1ky1-.
According to some aspect of the invention, R1 is C6aryl-Ci_2a1ky1-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and C1_2alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
C7oalkyl- and C7-
ioa lkenyl-, and
wherein R3 is hydrogen, methyl, or ethyl
wherein R is a bond or C1_2alkyl-.
According to some aspect of the invention, R1 is C6aryl-, or C6aryl-Ci_2alkyl-
, optionally be
substituted with one or more substituent selected from the group comprising or
consisting of
hydrogen, hydroxy, oxy, and methoxy-.
According to some aspect of the invention, R2 is hydrogen, methanyl, ethanyl,
heptanyl,
octanyl, 8-methyl-nonanyl, octadecanyl or 8-methyl-nonenyl.
The process with these compounds results in improved yields and conversion
rates, which is
especially important for large scale production.
One aspect relates to a process for enzymatic synthesis of amides of formula
Ill from amines
of formula I and compounds of formula Ila,
Lipase
Ris-NH2 + R3 0-C(0)-R2
_________________________________________________________ 0. R1-N(H)-C(0)-R2
I Ha III
wherein R1 is selected from the group comprising or consisting of Ci_nalkyl-,
Ci_nalkenyl-, Ci_
12a I kynyl-, CiAla I kOXY-, C1-12a lky1-0-C142alkyl-, Ci_12a1kyl-OC(0)-
Ci_12a1ky1-, Ci_nalkyl-N H-C2_
22a I kyl-, C1-12alkyl-NHC(0)-Ci_12a1kyl-, C342cycloalkyl-, Ca_ncycloalkenyl-,
Cs_naryl-, C3-
ncycloa I kyl-C1_6a I kyl-, C342cycloa I kenyl-Ci_6a I kyl- and Cs_na ryl-
Ci_6a I kyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, Ci_
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
8
6 hydroxyalkyl-, C3_6a mi nexya I kyl-, C3_6ca
rboxya I kyl-,
ssu Ifura lkyl-, C3_6sulfidealkyl- and C3-6a I koxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S,
wherein R2 is selected from the group comprising or consisting of hydrogen,
C3_30alkyl-,
30a Ikenyl-, Ci_30a I kynyl-, C3_30a I koxy-, Ci_30a I kyl-O-C342a lkyl-,
Ci_30a lkyl-OC(0)-Ci_37a I kyl-, Cl-
30a lkyl-NH-C342alkyl-, C3_30a I kyl-NHC(0)-C3_32a I kyl-, C342cyc10alkyl-,
C342cycloa I ke nyl- and C5-
12a ryl-, C3-37cyc10a C3-37cyc10a I kenyl-C3_6a I kyl- and C5-
17a ryl-C3-6a I kyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, C1_
6hydroxyalkyl-, C1_6haloyalkyl-, C3_6aminexyalkyl-, C1_6amideyalkyl-,
C1_6carboxyalkyl-, C1-
65ulfuralkyl-, C3_6sulfidealkyl- and C3_6a I koxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or 5,
wherein R3 is selected from the group comprising or consisting of hydrogen,
CI.6a1ky1-,
6a I kenyl-, C3_6a I kynyl-, C3_6a I koxy-, C3_6a I kyl-O-C3_6a lkyl-,
C3_6alkyl-OC(0)-C3_6a1ky1-,
C3_6alkyl-NHC(0)-C3_6a1ky1-, C347cycloalkyl-, C347cycloalkenyl- and Cs_12aryl-
, C3-
12CYCI0a I kyl-Ci_6a I kyl-, C342cycloa I kenyl-C3_6a I kyl- and Cs_37a ryl-
C3_6a I kyl-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, C1-
6 hydroxyalkyl-, C3_6ha1oya1ky1-, C3_6a mi nexya I kyl-, C3_6a m ideya I kyl-,
C3_6ca rboxya I kyl-, C1..
su Ifura lkyl-, C3_6su1fidea1ky1- and C3-6a I koxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S
characterized in that
the lipase is immobilized on a rotary bed reactor or on a spin-fixed-bed
reactor and a Dean-
Stark apparatus is used for dehydration.
According to some aspect of the invention, R1 is selected from the group
comprising or
consisting of C3_6a I kyl-, C3_6a I kenyl-, C3_6a I koxy-, C3_6a I kyl-O-C3_6a
I kyl-, C3_6cycloa I kyl-, C3-
6cyc10a I kenyl-, C6-7aryl-, C3-6cycloa I kyl-C3_3a I kyl-, C3_6cycloa Ikenyl-
C3_3a I kyl- and Cs-7a ryl-Ci-
3a I kyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy,
CIahydroxyalkyl-,
3ha1oya1ky1-, and C3_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_isalkyl-, Cs_
isa Ikenyl-, Cs-isa I koxy-, Cs_isa lky1-0-146a I kyl-, and C5-15a I kyl-OC(0)-
C3.6a I kyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen and carboxy, and
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
9
wherein R3 is selected from the group comprising or consisting of hydrogen,
C2_3a1ky1-, Ci_
3a1koxy- and Cl-3alkyl-O-C23alkyl-.
According to some aspect of the invention, R1 is Cs_2ary1-C2_3a1ky1-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and C2_3alkoxy-, and
wherein R2 is selected from the group comprising or consisting of hydrogen,
Cs_nalkyl- and Cs-
isalkenyl-, and
wherein R3 is hydrogen, methyl or ethyl.
According to some aspect of the invention, R1 is C6aryl-C2_2a1ky1-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy and C2_2a1k0xy-, and
wherein R2 is selected from the group comprising or consisting of Cmoalkyl-
and C7oalkenyl-,
and wherein R3 is hydrogen, methyl or ethyl.
The process allows for effective and efficient large-scale production of amide
compounds like
capsaicinoids and derivatives thereof. The process has improved yields
compared to known
processes. The amidation processes are environmentally friendly and especially
cost effective.
According to some aspect of the invention, compounds of formula III are
compounds of
formula IV
0
R6
40/ - n NAR2
H
R60
OR4 IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising or consisting of C3_30alkyl-,
C3_30alkenyl-, C3-
30a lkynyl-, C342cyc10a1kyl-, C342cycloalkenyl- and Cs_22aryl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amide,
C2_6hydroxya I kyl-
, C3-6ha loya I kyl-, C2-6a m ideya I kyl-, C2_6carboxyalkyl-, C2_65u Ifu ra I
kyl-, C3_6sulfidealkyl- and Ci_
6a1koxy- and C6_32ary1-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or 5,
wherein R4 or Rs is independently selected from the group comprising or
consisting of
hydrogen, C3_6alkyl-, C2_6a1keny1-, C2_6a1kynyl-, C3_20cycloalkyl-,
C340cycloalkenyl- and C5_22aryl-,
which R4 or RS may optionally be independently substituted with one or more
substituent
selected from the group comprising or consisting of hydroxy, oxy, halogen,
carboxy, amine,
amide, C3_6hyd roxya I kyl-, C2_6ha loya I kyl-, C3_6amineoxyalkyl-,
C2_6amideyalkyl-, CI.-
CA 03215699 2023 6ca rboxyalkyl-, C2_6sulfuralkyl-, C2_6sulfidealkyl- and
C1_6alkoxy-, and
- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
wherein R6 is selected from the group comprising or consisting of hydrogen,
hydroxy, oxy,
halogen, carboxy, amine, amide, Ci_ioalkyl-, C240alkenyl-, Cmoalkynyl-,
C342cycloalkyl-, C3-
5 32cycloalkenyl- and Cs-varyl-,
which R6 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydrogen, hydroxy, oxy, halogen, carboxy, amine,
amide, C1-
6hydroxyalkyl-, C1-6ha1oya1ky1-, C1_6amineoxyalkyl-, C1_6amideyalkyl-,
C1_6carb0xya1kyl-, C1-
6sulfuralkyl-, Ci_6su1fidea1ky1- and C1_6alkoxy-, and
10 wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S.
The process with these compounds results in improved yields and conversion
rates, which is
especially important for large scale production.
In some aspects, wherein compounds of formula III are compounds of formula IV
n is 1 or 2,
R2 is selected from the group comprising or consisting of C3_30alkyl-,
C3_30alkenyl-,
R4 or R5 is independently selected from the group comprising or consisting of
hydrogen, C1_
3a1ky1, and
R6 is hydrogen.
In some aspects, wherein compounds of formula III are compounds of formula IV
n is 1 or 2,
R2 is selected from the group comprising C348alkyl- and C348alkenyl-,
R4 or R5 is independently selected from the group comprising hydrogen,
C1_6alkyl-, and
R6 is hydrogen.
In some aspects, wherein compounds of formula III are compounds of formula IV
n is 1 or 2,
R2 is selected from the group comprising Cs_malkyl- and Cs_isalkenyl-,
R4 or R5 is independently selected from the group comprising hydrogen,
Ci_3a1ky1-, and
R6 is hydrogen.
In some aspects, wherein compounds of formula III are compounds of formula IV,
R2 is
methanyl, ethanyl, heptanyl, octanyl, 8-methyl-nonanyl or octadecanyl or 8-
methyl-nonenyl.
The process with these compounds results in improved yields and conversion
rates, which is
especially important for large scale production.
According to some aspect of the invention, no solvent is used.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
11
According to some aspect of the invention, the solvent is an organic solvent
selected from the
group comprising or consisting of methyl tert-butyl ether, diisopropylether,
Ci_6a1ky1-0-
6a1ky1 ethers, hexane and other Cs_walkanes, cyclohexane and other
C540cycloalkanes,
benzene, toluene, xylene, tert-butanol, tert amyl alcohol, other bulky
secondary or tertiary Cs-
10 alcohols and any esters thereof. In some aspects, the organic solvent is
selected from the
group comprising or consisting of diisopropylether, cyclohexane, toluene and
tert-butanol, or
mixtures thereof. In some aspects, the solvent is cyclohexane, toluene or
diisopropylether
(DIPE). In some aspects, the solvent is diisopropylether (DIPE). In some
aspects, the solvent is
cyclohexane. In some aspects, the solvent is toluene. In some aspects, the
solvent is tert-
According to some aspect of the invention, when R3 is not hydrogen, the
solvent is selected
from the group comprising or consisting of methyl tert-butyl ether,
diisopropylether, Ci_6a1ky1-
O-C1.6a1ky1 ethers, hexane and other Cs_ioalka nes, cyclohexane and other
Cs_iocycloalka nes,
benzene, toluene, xylene, tert-butanol, tert amyl alcohol, other bulky
secondary or tertiary Cs.
10 alcohols and their esters. In some aspects, the organic solvent is selected
from the group
comprising or consisting of diisopropylether, cyclohexane, toluene and tert-
butanol, or
mixtures thereof. In some aspects, the solvent is cyclohexane, toluene or
diisopropylether
(DIPE). In some aspects, the solvent is diisopropylether (DIPE). In some
aspects, the solvent is
cyclohexane. In some aspects, the solvent is toluene. In some aspects, the
solvent is tert-
butanol. In some aspects, the solvent is recyclable. In some aspects, the
solvent is recycled. In
some aspects, the solvent is recycled for at least 70% or 80% or 90%.
Recycling the solvent
reduces the overall costs for the process and also reduced the carbon
footprint of the process.
According to some aspect of the invention, the lipase is selected from the
group comprising
or consisting of Candida antarctica lipase A, Candida antarctica lipase B,
cross-linked Substilisin
A protease, Porcine pancreas lipase, Candida cylindracea lipase, Rhizopus
arrhizus, Penicillum
cyclopium, Mucor miehei, Thermomyces lanuginosus lipase, Candida rugosa lipase
and
Pseudomonas lipoprotein lipase. In one aspect, the lipase is selected from the
group
comprising or consisting of Candida antarctica lipase A and Candida antarctica
lipase B. In one
aspect, the lipase is Candida antarctica lipase . In one aspect, the lipase is
Candida antarctica
lipase B (Novozym 4351. The immobilized enzymes, like Candida antarctica
Lipase B or C.
antarctica lipase A, are commercially and easily available under tradenames
like Novozym
435-. The availability at relative low cost is important for a cost-effective
process, especially
for large scale processes.
According to some aspect of the invention, the process temperature is between
15 C and
150 C, or between 15 C and 115 C, or between 50 and 90, or 70 -80 C. The
relative low
temperature is important for a cost-effective process, especially for large
scale processes.
According to some aspect of the invention, the process is performed at a
pressure between
0.090 and 0.200 MPa, or at atmospheric pressure (about 0.1 MPa). Performing
the process at
atmospheric pressure is important for a cost-effective process, especially for
large scale
processes.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
12
According to some aspect of the invention, the rotary bed reactor is loaded
for 10 to 75wt%
with the lipase. According to some aspect of the invention, the rotary bed
reactor is loaded
for 11 to 60wt% with the lipase. According to some aspect of the invention,
the rotary bed
reactor is loaded for 15 to 50wt% with the lipase. The unique combination of
an immobilized
enzyme on a rotary bed reactor or on a spin-fixed-bed reactor and a Dean Trap
apparatus
improves the conversion rate and yield of the process. Because the process is
both time- and
cost-effective, a possible additional cost for loading of the lipase with more
than 10 wt%
loading becomes affordable.
According to some aspect of the invention, the rate of agitation is 150 to 600
rpm or 200 to
500 rpm, or 200 to 450 rpm.
The invention also relates to a process for synthesis of compounds of formula
II, wherein R2 is
a C648alkyl or C648alkenyl. According to some aspect, compounds of formula II,
wherein R2 is
a C6_18a1ky1 or C6_18a1keny1, which may be straight or branched, are prepared
comprising the
steps of
0 0
ppii3 e
HO R2¨Br step A HO R2¨PPh3 Br
base 0 0
isomerization iso-butyraldehyde
solvent 110)L-' -RD-r H0)11R.2*)y
step B
ihydrogenation
0
HO R2(
step A-1, wherein the reaction is performed without solvent or with an organic
solvent,
step B-1, wherein a solvent is an aprotic organic solvent, and
step B-1, wherein a base is a sodium or potassium alkoxides ,
optionally isomerization step C-1, wherein a catalyst is selected from the
group comprising or
consisting of HNO2, HNO3 and combinations of NaNO2/HNO3, NaNO2/NaNO3/H2504,
that can
generate HNO2 or HNO3, and
hydrogenation step 0-1, wherein a catalyst is a heterogeneous hydrogenation
catalyst and a
hydrogen source is hydrogen gas.
In some aspects, R2 is a C6-walkyl.
In some aspects, the organic solvent in step A-1 is ethyl acetate,
wherein the aprotic organic solvent in step B-1 is selected from the group
comprising or
consisting of 2-methyl tetra hydrofuran, tetrahydrofuran and toluene,
wherein the sodium or potassium alkoxide base in step B-1 is selected from the
group
comprising or consisting of NaH, KH, t-BuOK, t-BuONa, and
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
13
wherein the heterogeneous hydrogenation catalyst in hydrogenation step D-1 is
selected from
the group comprising or consisting of Pd/C and Pd/A1203.
In some aspects, the organic solvent in step A-1 is ethyl acetate, the aprotic
organic solvent in
step B-1 is 2-methyl tetrahydrofuran, the sodium or potassium alkoxide base in
step B-1 is t-
BuOK, and the heterogeneous hydrogenation catalyst in hydrogenation step 0-1
is Pd/C.
In the production of 8-methyl-6-nonenoic acid, using 2-MeTHF as recyclable
solvent for the
key Wittig reaction between (6-Carboxyhexyl)triphenylphosphonium bromide and
iso-
butyraldehyde improves conversion rate and yield. The synthesis is time- and
cost-effective
with high yields and conversion rates. This is especially important for large
scale process.
Further solvents may be used in the process steps. Extraction and filtration
may be performed
between the steps.
The process may be performed at room temperature. The process can be performed
at
atmospheric pressure (approximately 1 atm or 0.1 MPa).
The invention also relates to a process for a new synthetic route to 8-methyl-
6-nonanoic acid,
which is used for the direct production of dihydro-capsaicin. The process
starts from
cyclohexanone and iso-butyraldehyde as raw materials, with aldol condensation,
Baeyer-
Villiger oxidation and hydrogenation as key steps.
According to some aspect of the invention, compounds of formula II, wherein R2
is 8-methyl-
nonanyl, are prepared comprising the steps of
o 0 OH 0
0 (yy, catalyst ,
catalyst
step A * step B I:j,kr' 'sre
0 0
._...Ø)...)_
step C step D
0 0
step E ' HOy step g fity'lL"."
step A-2, wherein the reaction is performed without solvent or with any
organic solvent and
a catalyst is selected from the group comprising or consisting of amines and
inorganic bases,
step B-2, wherein the reaction is performed without solvent or with an organic
solvent, and a
catalyst is an acid,
step C-2, wherein a catalyst is a heterogeneous hydrogenation catalyst, and a
hydrogen source
is hydrogen gas,
step 0-2, wherein an oxidant is a peroxide, and a catalyst is a lipase, and
step E-2, wherein a reaction medium is an acidic media, and
Step F-2, wherein a catalyst is a heterogeneous hydrogenation catalyst, and a
hydrogen source
is hydrogen gas.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
14
According to some aspects, the organic solvent in step A-2 is selected from
the group
comprising or consisting of toluene and aromatic solvents, THF and ethers,
dichloromethane
and halogenated solvents, and the catalyst is selected from the group
comprising or consisting
of pyrrolidine and corresponding salts, NaOH and KOH,
wherein the organic solvent in step 8-2 is selected from the group comprising
or consisting of
toluene, and the acid is selected from the group comprising or consisting of p-
Ts0H, sulfuric
acid and Amberlyst-15,
wherein the catalyst in step C-2 is selected from the group comprising or
consisting of Pd/C,
Pd/A1203,
wherein the oxidant in step D-2 is selected from the group comprising or
consisting of aqueous
H202 and peroxy acids and the lipase is selected from the group comprising or
consisting of
Candida antarctica lipase A, Candida antarctica lipase B, cross-linked
Substilisin A protease,
Porcine pancreas lipase, Candida cylindracea lipase, Rhizopus arrhizus,
Penicillum cyclopium,
Mucor miehei, Thermomyces lanuginosus lipase, Candida rugosa lipase and
Pseudomonas
lipoprotein lipase, and
wherein the reaction medium in step E-2 is selected from the group comprising
or consisting
of aqueous sulfuric acid solution, and
wherein the catalyst in step F-2 is selected from the group comprising or
consisting of Pd/C,
Pd/A1203, Pd/ molecular sieves, Pt/C, Pt/A1203, and Pt/molecular sieves.
A Dean Stark trap may be used in step B-2.
According to some aspects, the organic solvent in step A-2 is toluene, and the
catalyst is
pyrrolidine, the organic solvent in step 8-2 is toluene and the acid is p-
Ts0H, the catalyst in
step C-2 is Pd/C, the oxidant in step D-2 is aqueous H202 and the lipase is
Candida antarctica
lipase 13, the reaction medium in step E-2 is aqueous sulfuric acid solution,
and the catalyst in
step F-2 is Pd/C.
The synthesis is time and cost effective with high yields and conversion
rates. This is especially
important for large scale process.
Further solvents may be used in the process steps. Extraction and filtration
may be performed
between the steps.
The process may be performed at room temperature. The process can be performed
at
atmospheric pressure (approximately 1 atm or 0.1 MPa).
The process as defined anywhere herein are useful for large scale production
of compounds
of formula III. In some aspects, the process is used for large scale
production (>0.5 or > 1 kg)
of compounds of formula III.
Brief description of the drawings
The invention will now be explained more closely by the description of
different embodiments
of the invention and with reference to the appended figures.
F*. CA 03215699 2023- 1 shows a system for performing the process of
the invention.
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
Detailed description of various embodiments of the invention
Definitions
Room temperature is a temperature between 15 and 25 C.
Et0Ac is ethyl acetate.
5 DIPE is diisopropylether.
KOtBu is potassium tert-butoxide.
2-MeTHF is 2-methyltetrahydrofuran.
ET20 is diethyl ether.
AcOH is acetic acid.
10 p-Ts0H is p-toluenesulfonic acid or tosylic acid.
tBuOH is tert-butyl alcohol.
equiv. is equivalent. equivalent
As used herein, the term "wt%" or "w/w%" or "w%" means weight percentage,
which is a
percentage of the total weight.
15 As used herein, the term "optional" or "optionally" means that the
subsequently described
event or circumstance may but need not occur, and that the description
includes instances
where the event or circumstance occurs and instances where it does not.
As used herein, the terms "Cr,", used alone or as a suffix or prefix, is
intended to include
hydrocarbon-containing groups; n is an integer from 1 to 30.
As used herein, the term "halogen" or "halo", used alone or as suffix or
prefix, is intended to
include bromine, chlorine, fluorine, and iodine.
As used herein, the term "hetero", used alone or as a suffix or prefix, is
intended to include
alkyl, cycloalkyl and aryl groups in which one or more of the carbon atoms
(and certain
associated hydrogen atoms) are independently replaced with the same or
different hetero
atoms (5, 0 or N) or heteroatomic groups. Examples of heteroatomic groups
include, but are
not limited to, ¨0¨, ¨S¨, ¨0-0¨, ¨5-5¨, ¨0¨S¨, NR, =N¨N=, ¨N=N¨, ¨N=N¨NR¨,
¨PR¨,
¨P(0)2¨, ¨POR¨, ¨0¨P(0)2¨, ¨SO¨, ¨502¨, ¨Sn(R)2¨, and the like.
As used herein, the term "C1_30-alkyl", used alone or as a suffix or prefix,
is intended to include
both branched and straight chain saturated aliphatic hydrocarbon groups having
from 1 to 30
carbon atoms. Examples of CI...I-alkyl include methyl, ethyl, n-propyl, i-
propyl, n-butyl, i-butyl,
sec-butyl, and tert-butyl.
The term "alkenyl" refers to a monovalent straight or branched chain
hydrocarbon radical
having at least one carbon-carbon double bond and comprising at least 2 up to
about 30
carbon atoms. The double bond of an alkenyl can be unconjugated or conjugated
to another
unsaturated group. Suitable alkenyl groups include, but are not limited to
C2_6alkenyl groups,
such as vinyl, ally!, butenyl, pentenyl, hexenyl, butadienyl, pentadienyl,
hexadienyl, 2-
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
16
ethylhexenyl, 2-propy1-2-butenyl, 4-(2-methyl-3-butene)-pentenyl. An alkenyl
can be
unsubstituted or substituted with one or two suitable substituents.
The term "alkynyl" refers to a monovalent straight or branched chain
hydrocarbon radical
having at least one carbon-carbon triple bond and comprising at least 2 and up
to about 12
carbon atoms. The triple bond of an alkynyl can be unconjugated or conjugated
to another
unsaturated group. Suitable alkynyl groups include, but are not limited to
C2_6alkynyl groups,
such as acetylenyl, methylacetylenyl, butynyl, pentynyl, hexynyl. An alkynyl
can be
unsubstituted or substituted with one or two suitable substituents.
As used herein, the term "C2_6-alkoxy", used alone or as a suffix och prefix,
refers to a Ci_6-alkyl
radical, which is attached to the remainder of the molecule through an oxygen
atom. Examples
of C2_4-alkoxy include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, i-
butoxy, sec-butoxy
and tert-butoxy.
As used herein, the term "cycloalkyl", and "cycloalkenyl" used alone or as a
suffix or prefix, is
intended to include saturated or partially unsaturated cyclic alkyl radical.
Where a specific
level of saturation is intended, the nomenclature cycloakanyl or cycloalkenyl
is used. Examples
of cycloalkyl groups include, but is not limited to, groups derived from
cyclopropane,
cyclobutene, cyclopentane, cyclohexane and the like.
As used herein, the term "aryl" refers to either a monocyclic aromatic ring
having 5 or 12 ring
members or a multiple ring system having at least one carbocyclic aromatic
ring fused to at
least one carbocyclic aromatic ring, cycloalkyl ring or a heterocycloalkyl
ring. For example, aryl
includes a phenyl ring fused to a 5- to 7- membered heterocycloalkyl ring
containing one or
more heteroatoms independently selected from N, 0, and S.
As used herein, the term "Cs.12-aryl-C2_6-alkyl" refers to a phenyl group that
is attached
through a Ci_6-alkyl radical. Examples of C6-aryl -C2_3-alkyl include
phenylmethyl (benzyl), 1-
phenylethyl and 2-phenylethyl.
Figure 1 shows a system for performing the process. In a reactor 5, the lipase
is immobilized
on a rotary fix bed 2. A motor 3 is used for rotation of the fixed bed 2. The
reactor 5 is
connected to a Dean Stark apparatus 1, which is connected to a condenser 4.
In the present invention, the process is performed using the lipase, which is
immobilized on a
rotary bed reactor together with a Dean-Stark apparatus for dehydration.
This process may be used for the preparation of capsaicinoids, but also for
the amidation of
numerous of other amines with carboxylic acids or esters.
The process may be used for synthesis of amides of formula III from amines of
formula I and
compounds of formula II or Ila as shown below,
3 2 3 2 Lipase 1
2
R1-NH2 + RO-R-C(0)-R or 1O-C(0)-R -IP' R -N(H)-C(0)-R
I II ha
III
or
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
17
1 3 2 Lipase 1 2
R-NH 2 + R O-R-C(0)-R _________ ii, R-N(H)-R-C(0)-R
i II 111
R1 may be selected from the group comprising C142alkyl-, C1-12alkenyl-,
C142alkynyl-, Ci-
12a lkoxy-, C142a I kyl-O-CiAla I kyl-, Ci_22a I kyl-OC(0)-Ci_12a lkyl-,
C2_12a I kyl-N H-Ci_12a I kyl-, Ci_
12a lkyl-N HC(0)-C142a I kyl-, C3-22cycloa I kyl-, C342cyc10a I kenyl-, Cs-12a
ryl-, C3-22cyc10a I kyl-Ci-
6a I kyl-, C342cycloa Ikenyl-C2_6a I kyl- and C542aryl-C2_6alkyl-,
which R1 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
Ci_6hydroxyalkyl-, Ci_
6 ha loya lkyl-, Ci_6a m i nexya I kyl-, C1_6a mideya I kyl-, C1_6ca rboxya I
kyl-, C2_6su Ifura I kyl-, CI._
6sulfidealkyl- and C1_6alkoxy-.
R1 may be selected from the group comprising Ci_6alkyl-, Ci_6alkenyl-,
Ci_6alkoxy-, Ci_6alkyl-O-
Ci_6alkyl-, C3_6cycloa lkyl-, C3_6cycloa I ke nyl-, C6_2ary1-, C3_6cyc1oa lkyl-
Ci_3a I kyl-, C3_6cyc1oa I ke nyl-
C1-3a lkyl- and Cs_2a ryl-Ci-3a I kyl-,
which RIL may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, C1_3hydroxyalkyl-,
Ci_3ha1oya1ky1-, and
Ci_3alkoxy-.
Or R1 may be Cs_2aryl-Ci_3alkyl-, or C6_2ary1-Ci_2a1ky1-, or C6aryl-Ci_3a1ky1-
, optionally substituted
with hydrogen, hydroxy and/or methoxy.
R2 may be selected from the group comprising hydrogen, Ci_30alkyl-,
Ci_30alkenyl-, C1_30alkynyl-
, Ci_30a I koxy-, Ci_30a lkyl-O-C142a I kyl-, Ci_30a lkyl-OC(0)-C142a I kyl-,
C1-30a I kyl-N H-C142a I kyl-, Ci_
30alkyl-NHC(0)-C142a1ky1-, C3_12cyc1oa1ky1-, C342cycloalkenyl- and C5_22aryl-,
C342cycloalkyl-Ci_
6a1ky1-, C342cycloalkenyl-C2_6a1kyl- and Cs42aryl-C1-6alkyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
C1_6hydroxyalkyl-, C1-
6 ha loya lkyl-, Ci_6a m i nexya I kyl-, C1_6a mideya I kyl-, C1_6ca rboxya I
kyl-, C2_65u If ura I kyl-, Ci_
6su1fidea1ky1- and C1_6a1k0xy-.
R2 may be selected from the group comprising hydrogen, C3_30alkyl-,
C3_30a1keny1-, C3_30alkynyl-
, C342cycloalkyl-, C3t2cycloalkenyl- and C5-12a ryl-.
R2 may be selected from the group comprising hydrogen, Cs_malkyl-, Cstolkenyl-
, Cstsalkoxy-
, Cs-18a I kyl-O-Ci_6a I kyl-, and Cs_28a I kyl-OC(0)-Ci_6a I kyl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen and carboxy.
Or R2 may be selected from the group comprising hydrogen, C6_16a1ky1- and
Cs_26a1keny1- or C7-
12a lkyl- and C2-16a I kenyl-, or C2-wa I kyl- and C2-10a Ike nyl-.
R3 is selected from the group comprising hydrogen, Ci_6a1ky1-, Ci-6a1keny1-,
Ci-6a1kyny1-, Ci-
CA 032156993523- 3.06alkoxy-, Ci_6a lky1-0-Ci_6a I kyl-, Ci_6a I kyl-OC(0)-
Ci_6a I kyl-, C2_6a I kyl-N H-C2_6a I kyl-, Ci_6a I kyl-
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
18
N HC(0)-Ci_6a I kyl-, C3_12cyc1oa1ky1-, C342cycloa I kenyl- and Cs_naryl-,
C342cyc1oa I lkyl-, C3.
12CYCI0a I ke nyl-C1_6a I kyl- and Cs-na ryl-C1-6a I kyl-,
which R3 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
Ci_6hydroxyalkyl-,
shaloyalkyl-, Ci_6a m i nexya I kyl-, C1-6a mideya I kyl-, C1-6ca rboxya I kyl-
, Ci_6su lfura I kyl-,
6su1fidea1ky1- and Ci_6a1koxy-, and
wherein one or more carbon in a cycloalkyl, cycloalkenyl or aryl may be
substituted with one
or more heteroatoms selected from 0, N or S
R3 may be selected from the group comprising hydrogen, Ci_salkyl-, Ciaalkoxy-
and C1_3a1ky1-
0-Ci_3a I kyl-.
Or R3 may be hydrogen, methyl, ethyl. R3 may be hydrogen.
R may be a bond. R may be Ci_3a1ky1-, or methyl or ethyl.
The compounds of formula III may be represented the structure of IV
0
R6
(1101 = n NAR2
R50
IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising C3_20a1ky1-, C3_20a1keny1-,
C3_20a1kyny1-, C3-
12CYCIOa I kyl-, C3-12cyc10a1keny1- and Cs-naryl-,
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
Ci_6hydroxyalkyl-,
6 ha loya lkyl-, Ci_6a m i nexya I kyl-, C1_6a mideya I kyl-, C1_6ca rboxya I
kyl-, Ci_6su lfura I kyl-,
6su Ifidea lkyl- and Ci_6a1k0xy- and Cs_naryl-,
wherein R4 or RS is selected from the group comprising hydrogen, Ci_6a1ky1-,
C2_6a1keny1-, C2-
6a I kynyl-, C340cycloa I kyl-, C3-iocycloa I kenyl- and Cs-12a ryl-,
wherein R6 is selected from the group comprising hydrogen, hydroxy, oxy,
halogen, carboxy,
amine, amide, Ci_6a1ky1-, C2_6a1keny1-, C2_6a1kyny1-, C3_6cycloalkyl-,
C3_6cycloalkenyl- and Cs_6ary1-
,
which R6 may optionally be substituted with one or more substituent selected
from the group
comprising hydrogen, hydroxy, oxy, halogen, carboxy, amine, amide,
Ci_6hydroxyalkyl-,
6 ha loya lkyl-, Ci_6a m i nexya I kyl-, Ci_6a mideya I kyl-, Ci_6ca rboxya I
kyl-, Ci_6su lfura I kyl-,
6su1fidea1ky1- and C1_6a1koxy-, and.
The compounds of formula III may be represented the structure of IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising or consisting of Cs_nalkyl-,
Cs_nalkenyl-, Cs..
CA 03215699 2023- lolle lkoxy-, Cs_18alkyl-O-Ci_6a1ky1-, and Cs_i8alkyl-OC(0)-
Ci_6a I kyl-,
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
19
which R2 may optionally be substituted with one or more substituent selected
from the group
comprising or consisting of hydroxy, oxy, halogen and carboxy,
wherein R4 or R5 is selected from the group comprising or consisting of
hydrogen and Ci_3a1ky1-
, and
S R6 may be selected from the group comprising or consisting of hydrogen,
hydroxy and oxy.
The compounds of formula III may be represented the structure of IV
wherein n is 1 or 2,
wherein R2 is selected from the group comprising C642alkyl- and C6_22alkenyl-,
or Cmoalkyl- and
C240alkenyl-,
wherein R4 or R5 is selected from the group comprising hydrogen, methyl or
ethyl, and
R6 is hydrogen.
Prior art processes for preparation of capsaidnoids
Ester as acyl donor: need very dry amine (<3wt% water content), otherwise
water will cover
the amine on the bottom and retard the reaction. It is difficult to reach full
conversion of the
ester without addition of excess amine.
ao NH2 0
He
0 0 OM. SOC12 * njl'Y
Et20 a HO
Me
Expensive anhydrous solvent and toxic SOCl2 were required in this process.
Comparing with
the enzymatic process, the yield was much lower and the resulting appearance
of the product
was much worse. The product was sticky with brownish-yellow color. See example
25.
Enzymatic process
* NH2 enzyme
molecular sieves
HO
HO solvent
OMe HO
= Me
This process requires large amounts of molecular sieves, which require bigger
equipment
when scaling up. Filtration and purification is necessary, which is time- and
cost-consuming.
See example 23.
Both prior art processes are time consuming, expensive with yields that are
too low to be used
for large scale production in an economically feasible manner.
The process of the invention may be used for the preparation of capsaicinoids
according to
the scheme below.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
O
111)
HO
OMe
eapsaicin
0
0
* NH2 novozym 435
01 11.1(1 ream with
HO HO
Dean-Stark distillation
OMe OMe
diltydrocapsaicin
0 0
HO)WW
HO
OMe nonivamide
In the process of the invention, no solvent may be used.
If a solvent is used, the solvent may be an organic solvent selected from the
group comprising
or consisting of methyl tert-butyl ether, diisopropylether, Ci_6alkyl-O-
Ci_Ealkyl ethers, hexane
5 and other Cs_walkanes, cyclohexane and other Cs_iocycloalkanes, benzene,
toluene, xylene,
tert-butanol, tert amyl alcohol, other bulky secondary or tertiary Cs_io
alcohols and any esters
thereof. The solvent may be toluene, diisopropylether or cyclohexane.
When R3 is not hydrogen, the solvent may be selected from the group comprising
or consisting
of methyl tert-butyl ether, diisopropylether, Ci_6alkyl-O-Ci_6alkyl ethers,
hexane and other Cs_
10 ioalkanes, cyclohexane and other Cs_iocycloalkanes, benzene, toluene,
xylene, tert-butanol,
tert amyl alcohol, other bulky secondary or tertiary Cs_io alcohols and their
esters.
The solvent may be toluene, diisopropylether or cyclohexane.
The lipase may be selected from the group comprising or consisting of Candida
antarctica
lipase A, Candida antarctica lipase B, cross-linked Substilisin A protease,
Porcine pancreas
15 lipase, Candida cylindracea lipase, Rhizopus arrhizus, Penicillum
cyclopium, Mucor miehei,
Thermomyces lanuginosus lipase, Candida rugosa lipase and Pseudomonas
lipoprotein lipase.
The process may be performed at a temperature between room temperature and 150
C, or
between room temperature and 115 C.
The process may be performed at a pressure between 0.090 and 0.200 MPa, or
about 0.1 MPa.
20 The compounds of formula II may be prepared comprising or consisting of
the steps of
0 0
+ PPh3 _______________________________________ A 2 e
HO R2¨Br step A HO R--PP113 Br
base 0 0
iso-butyraldehyde isomerization
________________________ DI*
solvent HO)L .RX HOAR2
step B
Ihrhogenation
0
HO R2y
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
21
wherein R2 is a C648a1ky1 or C648a1keny1, which may be straight or branched,
step A-1, the reaction is performed without solvent or with any organic
solvent, such as Et0Ac,
step B-1, a solvent is selected from the group comprising or consisting of 2-
methyl
tetrahydrofuran, tetrahydrofuran, toluene and any other aprotic organic
solvent,
step B-1, a base is selected from the group comprising or consisting of NaH,
KH, t-BuOK, t-
BuONa and another sodium or potassium alkoxides,
isomerization step C-1, a catalyst is selected from the group comprising or
consisting of HNO2,
HNO3 and any other combination that can generate HNO2 or HNO3, and
hydrogenation step D-1, a catalyst is selected from the group comprising or
consisting of Pd/C,
Pd/A1203 and any other heterogeneous hydrogenation catalyst, a hydrogen source
is hydrogen
gas.
The compounds of formula II may be prepared comprising or consisting of the
steps of
o 0 OH 0
0 iyy, catalyst
Ily catalyst a...-õr
4. a A ..
step step B
0 0
step C - step D
0 0
stop 13 _____________ HOA----------- step F
step A-2, the reaction is performed without solvent or with any organic
solvents, such as
toluene, a catalyst is selected from the group comprising or consisting of
pyrrolidine, other
amines and corresponding salts, NaOH, KOH, and other inorganic bases,
step B-2, the reaction is performed without solvent or with an organic
solvent, such as toluene,
a catalyst is selected from the group c comprising or consisting of p-Ts0H,
sulfuric acid,
Amberlyst-15, and other acids,
step C-2 and step F-2, a hydrogen source is hydrogen gas, a catalyst is
selected from the group
comprising or consisting of Pd/C, Pd/A1203 and another heterogeneous
hydrogenation catalyst,
step D-2, an oxidants is selected from the group comprising or consisting of
aqueous H202,
peroxyacids and another peroxides, a catalyst is selected from the group
comprising or
consisting of Candida antarctica lipase A, Candida antarctica lipase B, cross-
linked Substilisin
A protease, Porcine pancreas lipase, Candida cylindracea lipase, Rhizopus
arrhizus, Penicillum
cyclopium, Mucor miehei, Thermomyces lanuginosus lipase, Candida rugosa lipase
and
Pseudomonas lipoprotein lipase, and
step E-2, a reaction medium is selected from the group comprising or
consisting of aqueous
sulfuric acid solution or another strong acidic media, and
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
22
Step F-2, a catalyst is selected from the group comprising or consisting of
Pd/C, Pd/A1203, Pd/
molecular sieves, PVC, Pt/A1203, and Pt/molecular sieves, a hydrogen source is
hydrogen gas.
The processes for the preparation of compounds of formula II may be performed
at a
temperature is between room temperature and 150 C, or between room temperature
and
115 C. These processes may be performed at a pressure between 0.090 and 0.200
MPa, or
about 0.1 MPa.
Experimental sections
Preparation of va ni Ilvlami ne
Vanillylamine was prepared from its hydrochloride salt. The HCI salt was
purchased from
commercial suppliers or prepared according to literature procedures
(ChemBioChem 2009,
10,823; J. Med. Chem. 2018, 61, 8225.).
10 NH2 =HC1 * NH2
NaOH
__),...
HO water HO
OMe OMe
Example 1: 50.00 g of vanillylamine HCl salt was dissolved in 500 mL of cold
water (-5 C), and
cooled with an ice-bath, 1 equivalent of 3 M NaOH (87.9 mL) was portion-wise
added in 10
min while keeping vigorous stirring. The internal temperature kept about 5 C.
After addition
of all bases, the milky solution was stirred for further 5 min, then filtered.
The white product
in the funnel was washed twice with cold water (5 C, 100 mLx2), then dried
under vacuum
until the weight remains the same. 37.32 g (92.4% yield) of product was
obtained.
Example 2: 500.0 g of vanillylamine HCI salt was dissolved in 5 L of water (10
¨ 15 C), 1
equivalent of 3 M NaOH was portion-wise added in 20 min while keeping vigorous
stirring.
After addition of all bases, the milky solution was stirred for further 10
min, then filtered. The
white product in the funnel was washed twice with cold water (1 Lx2), then
dried in a vacuum
chamber at 50 C for 24 hours. 478.0 g of off-white product was obtained with
19.5wt%
moisture content (determined with Kern DBS 60-3 moisture analyser).
Preparation of fatty acids
Preparation of fatty acids with Wittig reaction as key step
I.....................5)PPh3 BP
0
110...1.-s,..........Br + Pph3 -).-
solvent HO
t-BuOK
0 0
iso-butyraldehyde rization
_____________________ Yo- ../". isome
2-MeTHF HO HO
1 hydrogenation
0
H0)(""?....."'""*--'''''"
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
23
Example 3: Preparation of (6-Carboxyhexyl)triphenylphosphonium bromide.
In a 1 L round-bottom flask, 97.53 g 6-bromohexanoic acid and 131.15 g (1.0
equiv)
triphenylphosphine (PPh3) were dissolved in 500 mL Et0Ac. The mixture was
heated at 75-80
C and stirred for 7 days. After filtration, the collected product was washed
with Et0Ac (50
mLx2) and then dried under vacuum to give 221.80 g white powder (97.0% yield).
The
combined filtrate was recycled as solvent for more batches.
Example 4: Preparation of (Z)-8-methyl-6-nonenoic acid.
In a 1 L two-necked round-bottom flask 100.00 g (6-
Carboxyhexyl)triphenylphosphonium
(Ph3P0) bromide and 49.07 g (2.0 equiv) KOtBu were dissolved in 300 mL 2-MeTHF
under
protection of nitrogen atmosphere and cooled using an ice water bath. The
reaction mixture
turned bright orange color while the compounds were dissolved. A solution of
18.92 g (1.2
equiv) isobutyraldehyde in 200 ml 2-MeTHF was slowly added to the cold
reaction mixture,
which quickly turned white. After the addition was completed, the reaction
mixture was
warmed to room temperature and stirred for 6 h. The reaction was quenched by
addition of
500 mL H20. The MeTHF solvent was recovered by distillation. After cooling to
room
temperature, most of Ph3P0 was precipitated and collected as white powder by
filtration. The
filtrate was acidified with concentrated HCI to pH 2, the formed organic layer
was collected,
and the water phase was extracted with Et20 (100 mLx2). The organic phases
were combined,
dried with anhydrous Na2SO4, and concentrated to give 56.6 g crude product.
This crude
product was then distilled under reduced pressure to give (Z)-8-methyl-6-
nonenoic acid (32.76
g, 88% yield, ZJE 11:1 by NMR analysis) as colorless oil product.
Example 5: Preparation of 8-methyl nonanoic acid.
32 g of (Z)-8-methyl-6-nonenoic acid was dissolved in 150 mL of diisopropyl
ether. 0.5 mol%
of Pd/C powder was then dispersed in this solution. The mixture was
hydrogenated with H2
balloon at room temperature overnight. The catalyst was recovered by
filtration. The solvent
was recovered by distillation. 8-methyl nonanoic acid was obtained as
colorless oil with >99%
yield.
Example 6: Preparation of (E)-8-methyl-6-nonenoic acid.
In a 1 L two-necked round-bottom flask 100.00 g (6-
Carboxyhexyl)triphenylphosphonium
bromide and 49.07 g (2.0 equiv) KOtBu were dissolved in 300 mL 2-MeTHF under
protection
of nitrogen atmosphere and cooled using an ice water bath. The reaction
mixture turned
bright orange color while the compounds were dissolved. A solution of 18.92 g
(1.2 equiv)
isobutyraldehyde in 200 mL 2-MeTHF was slowly added to the cold reaction
mixture, which
quickly turned white. After the addition was completed, the reaction mixture
was warmed to
room temperature and stirred for 6 h. The reaction was quenched by addition of
500 mL H20.
The MeTHF solvent was recovered by distillation. After cooling to room
temperature, most of
Ph3P0 was precipitated and collected as white powder by filtration. The
filtrate was acidified
with concentrated HCI to pH 2, the formed organic layer was collected, and the
water phase
was extracted with DIPE (100 mLx2). The organic phases were combined and
concentrated.
This crude intermediate was then treated with concentrated HNO3 (0.03 equiv)
at 85 C under
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
24
protection of nitrogen atmosphere for 24 hours. After cooling, the mixture was
washed with
water (50 mLx2). The aqueous phases were combined and extracted with DIPE (50
mLx2). The
organic phases were combined, dried with anhydrous Na2SO4, and concentrated to
give 53.1
g crude product. This crude product was then distilled under reduced pressure
to give (E)-8-
methyl-6-nonenoic acid (30.2 g, 81% yield, Eg 86:14 by NMR analysis) as
colorless oil product.
Preparation of 8-methyl nonanoic acid starting from cyclohexanone and
isobutyraldehyde
0 AcOH 0 OH 0
4. a pymplidine p-Ts0H
83% yield
0 0 for 2 steps
Pd/C, H novozym 435
full conversion
30w% H202
91% conversion
0 0
50W% H2SO4 Pd/C H
HO
Hey
37% yield for 4 steps
Example 7: Preparation of 2-(2-methylpropylidene)cyclohexan-1-one
0 1. AcOH 0
0 lidine
P2Yrn3T OH
. p- s
50 g isobutyraldehyde, 102 g cyclohexanone (1.5 equiv), 5 mol% of pyrrolidine
and 5 mol%
AcOH were heated and stirred at 40 C for 12 h. After cooling to room
temperature, the
mixture was dispersed in 200 mL of water and 100 mL of toluene and organic
phase was
separated. The aqueous phase was extracted with toluene (50 mL x 2). The
organic phases
were combined and treated with 4 mol% of p-Ts0H4+120 catalyst at refluxing
condition for 2
hours with a Dean-Stark trap to collect the generated water. After cooling
again to room
temperature, the acid was removed by washing with 30 mL of aqueous 1 M NaOH
solution.
Toluene and excess cyclohexanone were recovered by distillation. The enone
product (87.6 g,
83% yield, light yellow) was then distilled out under reduced pressure.
Example 8: Preparation of 2-isobutylcyclohexanone
5 g of 2-(2-methylpropylidene)cyclohexan-1-one was dissolved in 10 mL of
Et0Ac. 0.2 mol%
of Pd/C was added, and the hydrogenation was conducted at room temperature
with H2
balloon for 4 hours. Full conversion was achieved based on NMR analysis. The
catalyst was
recovered by filtration, and the filtrate was directly used in the following
oxidation.
Example 9: Preparation of 7-isobutyloxepan-2-one
To the solution of 2-isobutylcyclohexanone in Et0Ac, were added Novozym
435(tm) (250 mg)
CA 03215699 2023- loaild 30% aqueous H202 (3 equiv). The mixture was stirred
and heated at 50 C. After 24 h, 91%
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
conversion was achieved based on NMR analysis. After cooling to room
temperature, the
lipase catalyst was recovered by filtration. The filtrate was washed with 5%
aqueous Na2S203
solution and brine to remove excess peroxide. The organic phase was
concentrated to give
the crude lactone.
5 Example 10: Preparation of 8-methyl-6-nonanoic acid
The above crude lactone was dispersed in 5 M H2504 (40 mL) and heated at 110 C
oil bath.
After 20 h, the mixture was cooled to room temperature, extracted with DIPE
(20 mL x 3). The
organic phases were washed with brine, dried over anhydrous Na2SO4 and
filtered. To the
filtrate, 0.5 mol% Pd/C powder was added, and the hydrogenation was conducted
at room
10 temperature with H2 balloon for 24 hours. After filtration to recover
the Pd catalyst, the filtrate
was concentrated and purified by flash chromatography on silica gel to give
the 8-methyl-6-
nonanoic acid (2.1 g, 37% yield from 5 g of the enone product of Example 7).
Preparation of capsaicinoids
0
NH2
0 NR
io
HOAR ______________________________________ Novozym 435 401
reflux with
HO HO
Dean-Stark distillation
OMe OMe
15 Example 11: Preparation of capsaicin with excess amine in DIPE. At
normal atmospheric
pressure (approximately 1 atm), 8-methyl-6-nonenoic acid (3.65 g), vanillyl
amine (1.1 equiv.),
Novozym 435(tm) on beads (498.8 mg, 14 w/w% E/S) were refluxed (about 69 C) in
diisopropyl
ether (45 mL) with a Dean-Stark trap to collect the generated water. After
stirring at about
300 rpm overnight (19 h), the mixture was filtered to recover the lipase
catalyst, and the
20 filtrate was washed with 0.5 M aqueous HCI solution (10 mL). The aqueous
phase was
extracted with Et20 (10 mLx2) and the organic phase were combined, dried over
anhydrous
Na2SO4 and concentrated to give 6.53 g product (99.7% yield, very pale yellow
color).
Example 12: Preparation of nonivamide with excess fatty acid in toluene. At
normal
atmospheric pressure (approximately 1 atm), Vanillylamine (4.89 g, 2.00 wt%
water),
25 nonanoic acid (1.01 equiv.) and Novozym 435(") on beads (1 g, 20 w/w%
E/S) were refluxed
(about 110 C) in toluene (50 mL) with a Dean-Stark trap to collect the
generated water. After
stirring at about 300 rpm overnight (16 h), the conversion of nonanoic acid
was >99%. After
filtration to recover enzyme catalyst, the mixture was concentrated to give
9.14 g of product
(99.6% yield, white color).
Example 13: Preparation of nonivamide with excess fatty acid in cyclohexane.
At normal
atmospheric pressure (approximately 1 atm), Vanillyl amine (4.89 g, 2.00 wt%
water),
nonanoic acid (1.01 equiv.) and Novozym 435(tm) on beads (1 g, 20 w/w% E/S)
were refluxed
(about 81 C) in cyclohexane (50 mL) with a Dean-Stark trap to collect the
generated water.
After stirring at about 300 rpm overnight (16 h), the conversion of nonanoic
acid was >99%.
After filtration to recover enzyme catalyst, the mixture was concentrated to
give 9.10 g of
product (99.1% yield, pale yellow color).
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
26
Because the use of lipase on beads is not feasible for large scale production
due to costs for
work-up, like filtering the lipase, etc., next experiment was performed using
lipase
immobilized on a rotary bed reactor.
Example 14: Preparation of dihydrocapsaicin in fix-bed reactor. At normal
atmospheric
pressure (approximately 1 atm), In a 1 L reactor equipped with a rotating fix-
bed filled with 12
g of Novozym 435(tm) (45-60 ION% E/S), vanillylamine, slightly excess 8-methyl
nonanoic acid
(1.01 equiv), and diisopropyl ether (600 mL) were refluxed (about 69 C) with
a Dean-Stark
trap to collect the generated water (Figure 1). During reaction, the rpm was
fixed at about
300 rpm.
After reaction, the hot solution was released out and cooled to room
temperature. The
dihydrocapsaicin product crystallized and was collected by filtration. The
filtrate was directly
recycled as solvent for more batches. The results are shown in Table 1. The
yield of
dihydrocapsaicin was 99.7% in average.
Table 1. Preparation of dihydrocapsaicin in fix-bed reactor
o
o
* NH2 IP
HO Novozym
+ HOA'"'"y s.
reflux with HO
OMe Dean-Stark
distillation
OMe
in fix-bed reactor
Amine moisture content of amine Reaction time Conversion
Yield
Cycle
(g) (wt%) (h) (%)
(%)
1 29.41 10.18 24 >99
2 34.34 23.08 24 >99 99.8
3 31.80 16.95 24 >99 100
4 31.80 16.95 . 24 >99
5 31.80 16.95 24 >99
6 24.89 16.95 16 >99
The results show good conversion rates and yields even after 6 cycles.
Example 15: Preparation of capsaicin in fix-bed reactor (45w/w% E/S). The
reactor system of
Example 14 was cleaned by refluxing with DIPE solvent to remove residual
dihydrocapsaicin.
In this reactor, at normal atmospheric pressure (approximately 1 atm),
vanillylamine, slightly
excess 8-methyl-6-nonenoic acid (1.01 equiv), and diisopropyl ether (600 mL)
were refluxed
(about 69 C) with a Dean-Stark trap to collect the generated water. During
reaction, the rpm
was fixed at about 300 rpm. After reaction, the hot solution was released out
and cooled to
room temperature. The capsaicin product crystallized and was collected by
filtration. The
filtrate was directly recycled as solvent for more batches. The results are
shown in Table 2.
The yield of capsaicin was 99.4% in average.
Table 2. Preparation of capsaicin in fix-bed reactor.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
27
o
io, NH2 + He r Novozym 4 SO35
lL'' __________________________________________________ a
HO reflux with HO
OMe Dean-Stark distillation
OMe
in fix-bed reactor
Amine moisture content of
amine Reaction time Conversion Yield
Cycle
(g) (wt%) (h) (%)
(%)
1 32.18 16.95 24 >99
2 32.18 16.95 24 >99 99.5
3 32.18 16.95 24 >99 99.9
4 27.83 3.90 20 >99
27.83 3.90 20 >99
The results show good conversion rates and yields even after 5 cycles.
Example 16a: Preparation of nonivamide in fix-bed reactor (15-21 wiw% E/S).
The reactor
system of Example 15 was cleaned by refluxing with DIPE solvent to remove
residual capsaicin.
5 In this reactor, at normal atmospheric pressure (approximately 1 atm),
vanillylamine, slightly
excess nonanoic acid (1.01 equiv), and diisopropyl ether (600 mt.) were
refluxed (about 69 C)
with a Dean-Stark trap to collect the generated water. During reaction, the
rpm was fixed at
about 300 rpm. After reaction, the hot solution was released out and cooled to
room
temperature. The nonivamide product crystallized and was collected by
filtration. The filtrate
was directly recycled as solvent for more batches. The results are shown in
Table 3a. The yield
of nonivamide was 99.8% in average.
Table 3a. Preparation of nonivamide in fix-bed reactor.
o
oN-J1-,...----,-------------,...--
so NH2 401
+ Novozym 435 HeIL,'"'.-'-",....,'
.. a
HO reflux with HO
OMe Dean-Stark distillation
OMe
in fix-bed reactor
Cycle Amine (g) moisture content of amine (wt%) Reaction time (h) Conversion
(%)
1 39.89 3.90 24 >99
.
2 39.89 3.90 24 >99
3 44.88 3.90 24 >99
4 45.48 5.16 24 >99
5 45.48 5.16 24 >99
_
6 45.48 - 5.16 24 - >99
_
7 50.53 5.16 24 >99
8 48.90 2.00 24 >99
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
28
9 58.68 2.00 24 >99
10 58.68 2.00 24 >99
11 78.24 2.00 24 >99
The results show good conversion rates and yields even after 11 cycles.
Example 16b: Preparation of nonivamide in 100 L fix-bed reactor. At normal
atmospheric
pressure (approximately 1 atm), in a 100 L reactor equipped with a rotating
fix-bed filled with
1 kg of Novozym 435(tm) (50-100 w/w% E/S), vanillylamine, excess 8-methyl
nonanoic acid
(1.03 equiv), and diisopropyl ether (90 L) were refluxed (about 69 C) with a
Dean-Stark trap
to collect the generated water. During reaction, the rpm was fixed at about
250 rpm. After
reaction, the hot solution was released out and cooled to 15 C. The
nonivamide product
crystallized and was collected by filtration. The filtrate was directly
recycled as solvent for
more batches. The results are shown in Table 3b. The results show that the
process of the
invention can be used for large scale production of amides.
Table 3b. Preparation of nonivamide in 100 L fix-bed reactor
Cycle Amine (g) moisture content of amine (wt%) Reaction time (h) yield (%)
1 1014 5.51 15
96.4
2 1992 2.66 24
95.6
3 1936 <0.5 24
95.1
The results show good conversion rates and yields even after 3 cycles when the
process is
used at large scale.
Corn pa rative examples:
Preparation of capsaicin from fatty acid with drying agents
o HO +as
ip Nil 2 Novozym 435
molecular sieves
HO solvent
OMe HO
OMe
Example 17: To a 200 mL reactor, were added in toluene (150 mL), 4 A molecular
sieves (10 g),
immobilized enzyme (Novozym 435(tm), 0.59 g), 8-methyl-6-nonenoic acid (2.02
g), and
vanillylamine (2 equiv). The reaction was conducted at 80 C and monitored by
NMR. After 6
hours, the conversion of acid was >99%. After filtration, the organic filtrate
was cooled,
successively washed with 1 M HCI (20 mLx2), water (20 mLx2), and brine (20
mL). After drying
with anhydrous Na2SO4, the solvent was removed under vacuum. 3.07 g (84.7%
yield) of
capsaicin was obtained.
Example 18: To a 25 mL flask, were added in t-BuOH (8 mL), 4 A molecular
sieves (600 mg),
immobilized enzyme (Novozym 435(tm), 75 mg), 8-methyl-6-nonenoic acid (341 g),
and
vanillylamine (1.06 equiv). The reaction was conducted at 80 C and monitored
by NMR. After
10 hours, the conversion of acid was about 95%.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
29
Preparation of capsaicin with ester as acvl donor:
SO NH2
HO
0 0 OMe
H2SO4 enzyme
Me0H 12!0')IW'R solvent HO
or Et0H OMe
R. = Me, Et
Example 19: Preparation of methyl ester. 4.73 g of 8-methyl-6-nonenoic acid
was dissolved in
30 mL of Me0H. To this solution, 5 drops of concentrated H2SO4 was added as
catalyst. The
resulting solution was refluxed overnight. After cooling to room temperature,
most of Me0H
was removed by rotary evaporator and the residue was dissolved in Et20 (30 mL)
and washed
with 5% Na2CO3 solution (10 mLx2). The organic phase was dried with anhydrous
Na2SO4 and
concentrated to give the ester product with >99% yield.
Example 20: Preparation of capsaicin with ester in fix-bed reactor. In a 1 L
reactor equipped
with a rotating fix-bed filled with 6 g of Novozym 435(m) (10-20 w/w% E/S),
ethyl ester of 8-
methyl 6-nonenoic acid, excess vanillylamine (1.1 equiv), and diisopropyl
ether (600 mL) were
refluxed. After reaction, the hot solution was released out and cooled to room
temperature.
The solution was successively washed with 0.5 M HCl (60 mL), water (60 mL),
and brine (20
mL). After drying with anhydrous Na2SO4, the solvent was recovered by rotary
evaporation.
The capsaicin product was obtained with light yellow color. The results are
shown in Table 4.
Table 4. Preparation of capsaicin with ester in fix-bed reactor.
Cycle Ethyl ester (g) Time (h) Conversion (%) Yield (%)
1 30 22 >99 92
2 30 16 >99 93
3 60 40 90 80
4 30 42 >99 93
Example 21: Preparation of capsaicin from ester with distillation apparatus.
To a flask
equipped with short-path distillation apparatus, 923 mg of methyl ester of 8-
methyl-6-
nonenoic acid, 200 mg of Novozym 435( ) on beads and 1.1 equiv. of
vanillylamine were
heated at 80 C in 20 mL of t-BuOH. After 20 hours, the conversion of ester
was >99%
according to NMR analysis.
The results show that using esters is possible, but that the yield is lower
compared to earlier
examples 14, 15 and 16. An extra process step is needed because the methyl
ester is prepared
from the corresponding acid. Further, the work-up is tedious when up-scaling.
Besides,
recycling of the solvent is difficult, which is important for reducing costs
and environmental
impact of the process at large scale production.
Example 22: Preparation of capsaicinoids in neat condition (no solvent).
Experimental results
are shown in Table 5.
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
Table 5. Preparation of capsaicinoids in neat condition.
Lipase
Vanillylamine Acyl donor Temp Time
Yield
Entry (gig product
(equiv) (equiv) ( C) (h)
(%)
amine)
nonanoic acid
1* 1 0.2 100 18
nonivamide 37
(3 equiv)
ethyl 8-methyl-
2 1 6-nonenoate 0.2 80 16
capsaicin 91
(2 equiv)
8-methyl-6-
3 2 nonenoic acid - 150 16
capsaicin 37
(1 equiv)
methyl 8-
methyl-6-
4 1.2 nonenoate - 150 16
capsaicin 10
(1 equiv)
* under vacuum.
The results show that the yield of the process is decreased using reduced
pressure (Entry 1).
Example 23: Preparation of capsaicin with lipase catalyst and without
dehydration.
5 Vanillylamine (1 mmol), 8-methyl-6-nonenoic acid (1 mmol), and Novozym
435(tm) (45 mg)
were stirred in toluene (4 mL) and heated at 80 C for 48 h. 72% conversion
was achieved
based on NMR analysis.
Example 24: Preparation of capsaicin without lipase catalyst and with Dean-
Stark distillation.
With a Dean-Stark trap to collect generated water, vanillylamine (1 mmol) and
8-methyl-6-
10 nonenoic acid (1 mmol) were refluxed in toluene (4 mL) at 115 C oil
bath for 20 h. 19%
conversion was achieved based on NMR analysis.
Example 25: Preparation of capsaicin with acid chloride as acyl donor
AO :2 0
HO
0 0 = Me lb til)
SOCl2
crily __________________________________________________________ . H I
ome
In one 1 L flask, 47.17 g of 8-methyl-6-nonenoic acid was dissolved in 400 ml
of anhydrous
15 Et20. 30,1 mL of SOCl2 (1.5 equiv) was dissolved in 100 mL of anhydrous
Et20, and slowly added
to the acid solution. The resulting solution was refluxing for 3 hours, and
then the excess SOCl2
and solvent were removed under reduced pressure. The resulting acid chloride
was then
dissolved in 200 mL of anhydrous Et20. To a slurry of 84.7 g vanillylamine (2
equiv) in 400 mL
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
31
of anhydrous Et20, was added slowly the acid chloride solution in 2 hours.
After addition, the
refluxing was continued for 2 hours. The mixture was cooled with ice water
bath, the
precipitate was filtered. The organic filtrate was successively washed with 1
M HCI (50 mLx2),
water (50 mLx2), and brine (50 mL). After drying with anhydrous Na2SO4, the
solvent was
removed under vacuum. 54.3 g (64.2% yield) of capsaicin was obtained.
Preparation of miscellaneous amides by the combination of enzymatic catalysis
and Dean-
Stark distillation.
Preparation of (R)-2-methoxy-N-(1-phenylethyl)acetamide
NH2 0 0
CALB
Ph''`. + Et0A'.A.. DIPE ' HN3L0- '"-
S
reflux with Pli.
Dean-Stark distillation
Example 26. With a Dean-Stark trap to collect generated water, 1-phenylethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435( ) on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 10 h. After work
up, (R)-2-methoxy-
N-(1-phenylethyl)acetamide was obtained with 85% yield and 8% ee.
Example 27. With a Dean-Stark trap to collect generated water, 1-phenylethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435(tm) on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 3 h. After work
up, (R)-2-methoxy-
N-(1-phenylethyl)acetamide was obtained with 75% yield and 55% ee.
Example 28. With a Dean-Stark trap to collect generated water, 1-phenylethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435("") on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 1 h. After work
up, (R)-2-methoxy-
N-(1-phenylethyl)acetamide was obtained with 48% yield and 97% ee.
Example 29. Preparation of (R)-2-methoxy-N-(1-(4-
methoxyphenyl)ethyl)acetamide.
NH2 0 0
CALB
,...
p-Me0Ph).'''' + Et0A--"- .'"- DIPE 7.
reflux with p-Me0Ph
Dean-Stark distillation
With a Dean-Stark trap to collect generated water, 1-(4-methoxyphenyl)ethan-1-
amine (1
mmol), ethyl 2-methoxyacetate (2 mmol), and Novozym 435(") on beads (45 mg)
were
refluxed in diisopropryl ether (30 mL) at 90 C oil bath for 0.83 h. After work
up, (R)-2-methoxy-
N-(1-(4-methoxyphenyl)ethyflacetamide was obtained with 44% yield and 97% ee.
Example 30. Preparation of N-phenethylnonanamide.
0
0 CALB
ph,,,-.,,,_,, N H2 +
HO)t.(
CH2)6CH3 DIPE 1... HNA(CH2)6CH3
reflux with Ph
Dean-Stark distillation
CA 03215699 2023- 10- 16
AMENDED SHEET
PCT/EP 2022/061 321 - 27.01.2023
32
With a Dean-Stark trap to collect generated water, 2-phenylethan-1-amine (1
mmol),
nonanoic acid (1.05 mmol), and Novozym 435(tm) on beads (45 mg) were refluxed
in
diisopropryl ether (30 mL) at 90 C oil bath for 10 h. After work up, N-
phenethylnonanamide
was obtained with 98% yield.
Example 31. Preparation of N-phenethylstearamide.
0
0 CALB
ph,".,,,N H2 4.
- 110)L'"-(
C1-12)15C1-13 DIPE I..
HNH2)15CH3
reflux with Ph)
Dean-Stark distillation
With a Dean-Stark trap to collect generated water, 2-phenylethan-1-amine (1
mmol), stearic
acid (1.05 mmol), and Novozym 435(tm) on beads (45 mg) were refluxed in
diisopropryl ether
(30 mL) at 90 C oil bath for 10 h. After work up, N-phenethylstearamide was
obtained with
98% yield.
The present invention is not limited to the embodiments disclosed but may be
varied and
modified within the scope of the following claims.
CA 03215699 2023- 10- 16
AMENDED SHEET