Note: Descriptions are shown in the official language in which they were submitted.
CYSTE1NE ENGINEERED ANTIBODIES AND CONJUGATES
[0001] REFERENCE TO RELATED APPLICATIONS
I 00021 'fhis non-provisional application filed under 37 CFR
1.53(b), claims the
benefit under 35 USC 119(e) of U.S. Provisional Application Serial No.
61/352,728 filed on
8 June 2010.
[0003] FIELD OF THE INVENTION
[00041 The invention relates generally to antibodies engineered
with reactive cysteine
residues and more specifically to antibodies with therapeutic or diagnostic
applications. The
cysteine engineered antibodies may be conjugated with chemotherapeutic drugs,
toxins,
affinity ligands such as biotin, and detection labels such as fluorophores.
The invention also
relates to methods of using antibodies and antibody-drug conjugate compounds
for in vitro, in
situ, and in vivo diagnosis or treatment of mammalian cells, or associated
pathological
conditions.
[0005] BACKGROUND OF THE INVENTION
[00061 Antibody drug conjugates (ADC) are attractive targeted chemo-
therapeutic
molecules as they combine ideal properties of both antibodies and cytotoxic
drugs by
targeting potent cytotoxic drugs to the antigen-expressing tumor cells,
thereby enhancing
their anti-tumor activity. The successful ADC development for a given target
antigen depends
on optimization of antibody selection, linker stability, cytotoxic drug
potency and mode of
linker-drug conjugation to the antibody.
100071 Conventional means of attaching, i.e. linking through
covalent bonds, a drug
moiety to an antibody generally leads to a heterogeneous mixture of molecules
where the
drug moieties are attached at a number of sites on the antibody. For example,
cytotoxic drugs
have typically been conjugated to antibodies through the often-numerous lysine
residues of
an antibody, generating a heterogeneous antibody-drug conjugate mixture.
Depending on
reaction conditions, the heterogeneous mixture typically contains a
distribution of antibodies
with from 0 to about 8, or more, attached drug moieties. In addition, within
each subgroup of
conjugates with a particular integer ratio of drug moieties to antibody, is a
potentially
heterogeneous mixture where the drug moiety is attached at various sites on
the antibody.
Analytical and preparative methods are inadequate to separate and characterize
the antibody-
1
Date Recue/Date Received 2023-11-15
drug conjugate species molecules within the heterogeneous mixture resulting
from a
conjugation reaction. Antibodies are large, complex and structurally diverse
biomolecules,
often with many reactive functional groups. Their reactivities with linker
reagents and drug-
linker intermediates arc dependent on factors such as pH, concentration, salt
concentration,
and co-solvents. Furthermore, the multistep conjugation process may be
nonreproduciblc due
to difficulties in controlling the reaction conditions and characterizing
reactants and
intermediates.
[0008] Cysteine thiols are reactive at neutral pH, unlike most amines
which are
protonated and less nucl cophi I ic near pH 7. Since free thiol (RSFI,
sulfhydryl) groups are
relatively reactive, proteins with cysteine residues often exist in their
oxidized form as
disulfide-linked oligomers or have internally bridged disulfide groups.
Antibody cysteine
thiol groups are generally more reactive, i.e. more nucleophilic, towards
electrophilic
conjugation reagents than antibody amine or hydroxyl groups. Engineering in
cysteine thiol
groups by the mutation of various amino acid residues of a protein to cysteine
amino acids is
potentially problematic, particularly in the case of unpaired (free Cys)
residues or those
which are relatively accessible for reaction or oxidation. In concentrated
solutions of the
protein, whether in the periplasm of E. coli, culture supernatants, or
partially or completely
purified protein, unpaired Cys residues on the surface of the protein can pair
and oxidize to
form intermolecular disulfides, and hence protein dimers or multimers.
Disulfide dimer
formation renders the new Cys unreactive for conjugation to a drug, ligand, or
other label.
Furthermore, if the protein oxidatively forms an intramolecular disulfide bond
between the
newly engineered Cys and an existing Cys residue, both Cys groups are
unavailable for active
site participation and interactions. Furthermore, the protein may be rendered
inactive or non-
specific, by misfolding or loss of tertiary structure (Zhang et al (2002)
Anal. Biochem. 311:1-
9).
[0009] Antibodies with cysteine substitutions (ThioMabs) at sites
where the
engineered cysteines arc available for conjugation but do not perturb
immunoglobulin folding
and assembly or alter antigen binding and effector functions (Junutula, et
al., 2008b Nature
Biotech., 26(8):925-932; Doman et at (2009) Blood 114(13):2721-2729; US
7521541; US
7723485; W02009/052249). These ThioMabs can then be conjugated to cytotoxic
drugs
through the engineered cysteine thiol groups to obtain ThioMab drug conjugates
(TDC) with
uniform stoichiometry (-2 drugs per antibody). Studies with multiple
antibodies against
different antigens have shown that TDC are as efficacious as conventional ADC
in xenograft
models and are tolerated at higher doses in relevant preclinical models.
ThioMab drug
2
Date Recue/Date Received 2023-11-15
conjugates have been engineered with drug attachment at different parts of the
antibody (light
chain-Fab, heavy chain-Fab and heavy chain-Fe). The in vitro & in vivo
stability, efficacy
and PK properties of TDC provide a unique advantage over conventional ADC due
to their
homogeneity and site-specific conjugation to cytotoxic drugs.
[0010] SUMMARY
[0011] The invention includes an isolated cysteine engineered antibody
comprising a
free cysteine amino acid in the heavy chain or light chain.
[0012] An aspect of the invention is a process to prepare the isolated
cysteine
engineered antibody by mutagenizing a nucleic acid sequence of a parent
antibody by
replacing one or more amino acid residues by cysteine to encode the cysteine
engineered
antibody; expressing the cysteine engineered antibody; and isolating the
cysteine engineered
antibody.
100131 Another aspect of the invention is a conjugate of the isolated
cysteine
engineered antibody wherein the antibody is covalently attached to a capture
label, a
detection label, a drug moiety, or a solid support.
[0014] BRIEF DESCRIPTION OF THE DRAWINGS
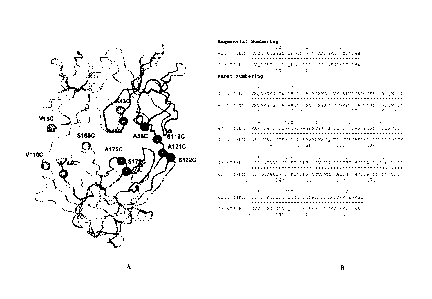
[0015] Figure lA shows a three-dimensional representation of the
hu4D5Fabv7
antibody fragment derived by X-ray crystal coordinates. The structure
positions of the
exemplary engineered Cys residues of the heavy and light chains are numbered
(according to
a sequential numbering system).
[0016] Figure 1B shows a sequential numbering scheme (top row),
starting at the N-
terminus in comparison with the Kabat numbering scheme (bottom row) for
4D5v7fabH.
Kabat numbering insertions are noted by a,b,c.
[0017] Figures 2A and 2B show binding measurements with detection of
absorbance
at 450nm of hu4D5Fabv8 and hu4D5Fabv8 Cys mutant (ThioFab) phage variants: (A)
non-
biotinylated phage-hu4D5Fabv8 and (B) biotinylated phage-hu4D5Fabv8 (B) by the
PHESELECTOR assay for interactions with BSA (open bar), HER2 (striped bar) or
streptavidin (solid bar).
[0018] Figures 3A and 3B show binding measurements with detection of
absorbance
at 450nm of hu4D5Fabv8 (left) and hu4D5Fabv8 Cys mutant (ThioFab) variants:
(A) non-
biotinylated phage-hu4D5Fabv8 and (B) biotinylated phage-hu4D5Fabv8 by the
PHESELECTOR assay for interactions with: BSA (open bar), FIER2 (striped bar)
and
3
Date Recue/Date Received 2023-11-15
streptavidin (solid bar). Light chain variants are on the left side and heavy
chain variants are
on the right side. Thiol reactivity = 013450 for streptavidin binding 013450
mm for HER2
(antibody) binding
[0019] Figure 4A shows Fractional Surface Accessibility values of
residues on wild
type hu4D5Fabv8. Light chain sites are on the left side and heavy chain sites
are on the right
side.
100201 Figure 4B shows binding measurements with detection of
absorbance at
450nm of biotinylated hu4D5Fabv8 (left) and hu4D5Fabv8 Cys mutant (ThioFab)
variants
for interactions with HER2 (day 2), streptavidin (SA) (day 2), HER2 (day 4),
and SA (day 4).
Phage-hu4D5Fabv8 Cys variants were isolated and stored at 4 C. Biotin
conjugation was
carried out either at day 2 or day 4 followed by PHESELECTOR analyses to
monitor their
interaction with Her2 and streptavidin as described in Example 2, and probe
the stability of
reactive thiol groups on engineered ThioFab variants.
[0021] Figure 5 shows binding measurements with detection of
absorbance at 450nm
of biotin-maleimide conjugated-hu4D5Fabv8 (A121C) and non-biotinylated wild
type
hu4D5Fabv8 for binding to streptavidin and HER2. Each Fab was tested at 2 ng
and 20 ng.
100221 Figure 6 shows ELISA analysis with detection of absorbance at
450nm of
biotinylated ABP-hu4D5Fabv8 wild type (wt), and ABP-hu4D5Fabv8 cysteine
mutants
V110C and A121C for binding with rabbit albumin, streptavidin (SA), and HER2.
100231 Figure 7 shows ELISA analysis with detection of absorbance at
450nm of
biotinylated ABP-hu4D5Fabv8 cysteine mutants (ThioFab variants): (left to
right) single Cys
variants ABP-V1 10C, ABP-A121C, and double Cys variants ABP-V110C-A88C and ABP-
V110C-A121C for binding with rabbit albumin, HER2 and streptavidin (SA), and
probing
with Fab-HRP or SA-HRP.
[0024] Figure 8 shows binding of biotinylated ThioFab phage and an
anti -phage HRP
antibody to HER2 (top) and Streptavidin (bottom).
[0025] Figure 9A shows a cartoon depiction of biotinylated antibody
binding to
immobilized HER2 with binding of HRP labeled secondary antibody for absorbance
detection.
[0026] Figure 9B shows binding measurements with detection of
absorbance at
450nm of biotin-maleimide conjugated thio-trastuzumab variants and non-
biotinylated wild
type trastuzumab in binding to immobilized HER2. From left to right: V1 10C
(single cys),
A121C (single cys), V110C/A121C (double cys), and trastuzumab. Each thio IgG
variant
4
Date Recue/Date Received 2023-11-15
and trastuzumab was tested at 1, 10, and 100 ng.
[0027] Figure 10A shows a cartoon depiction of biotinylated antibody
binding to
immobilized HER2 with binding of biotin to anti-IgG-HRP for absorbance
detection.
[0028] Figure 10B shows binding measurements with detection of
absorbance at
450nm of biotin-mal eimi de conjugated-thio trastuzumab variants and non-
biotinylated wild
type trastuzumab in binding to immobilized streptavidin. From left to right:
V11 0C (single
cys), A121C (single cys), V110C/A121C (double cys), and trastuzumab. Each thio
IgG
variant and trastuzumab was tested at 1, 10, and 100 ng.
[0029] Figure 11 shows the general process to prepare a cysteine
engineered antibody
(ThioMab) expressed from cell culture for conjugation.
[0030] DETAILED DESCRIPTION OF EXEMPLARY EMBODIMENTS
[0031] Reference will now be made in detail to certain embodiments of
the invention,
examples of which are illustrated in the accompanying structures and formulas.
While the
invention will be described in conjunction with the enumerated embodiments, it
will be
understood that they are not intended to limit the invention to those
embodiments. On the
contrary, the invention is intended to cover all alternatives, modifications,
and equivalents,
which may be included within the scope of the present invention as defined by
the claims.
[0032] One skilled in the art will recognize many methods and
materials similar or
equivalent to those described herein, which could be used in the practice of
the present
invention. The present invention is in no way limited to the methods and
materials described.
[0033] Unless defined otherwise, technical and scientific terms used
herein have the
same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs, and are consistent with: Singleton et al (1994) Dictionary
of Microbiology
and Molecular Biology, 2nd Ed., J. Wiley & Sons, New York, NY; and Janeway,
C., Travers,
P., Walport, M., Shlomchik (2001) Immunobiology, 5th Ed., Garland Publishing,
New York.
[0034] DEFINITIONS
[0035] Unless stated otherwise, the following terms and phrases as
used herein are
intended to have the following meanings:
[0036] When trade names are used herein, applicants intend to
independently include
the trade name product formulation, the generic drug, and the active
pharmaceutical
ingredient(s) of the trade name product.
[0037] The term "antibody" herein is used in the broadest sense and
specifically
Date Recue/Date Received 2023-11-15
covers monoclonal antibodies, polyclonal antibodies, dimers, multimers,
multispecific
antibodies (e.g., bispecific antibodies), and antibody fragments, so long as
they exhibit the
desired biological activity (Miller et al (2003) Jour. of Immunology 170:4854-
4861).
Antibodies may be murine, human, humanized, chimeric, or derived from other
species. An
antibody is a protein generated by the immune system that is capable of
recognizing and
binding to a specific antigen. (Janeway, C., Travers, P., Walport, M.,
Shlomchik (2001)
Immuno Biology, 5th Ed., Garland Publishing, New York). A target antigen
generally has
numerous binding sites, also called epitopes, recognized by CDRs on multiple
antibodies.
Each antibody that specifically binds to a different epitope has a different
structure. Thus,
one antigen may have more than one corresponding antibody. An antibody
includes a full-
length immunoglobulin molecule or an immunologically active portion of a full-
length
immunoglobulin molecule, i.e., a molecule that contains an antigen binding
site that
immunospecifically binds an antigen of a target of interest or part thereof,
such targets
including but not limited to, cancer cell or cells that produce autoimmune
antibodies
associated with an autoimmune disease. The immunoglobulin disclosed herein can
be of any
type (e.g., IgG, IgE, IgM, IgD, and IgA), class (e.g., IgGI, IgG2, lgG3, IgG4,
IgAl and
IgA2) or subclass of immunoglobulin molecule. The immunoglobulins can be
derived from
any species. In one aspect, however, the immunoglobulin is of human, murine,
or rabbit
origin.
[0038] "Antibody fragments" comprise a portion of a full length
antibody, generally
the antigen binding or variable region thereof. Examples of antibody fragments
include Fab,
Fab', F(a1302, and Fv fragments; diabodies; linear antibodies; minibodies
(Olafsen et al (2004)
Protein Eng. Design & Sel. 17(4):315-323), fragments produced by a Fab
expression library,
anti-idiotypie (anti-1d) antibodies, CDR (complementary determining region),
and epitope-
binding fragments of any of the above which immunospecifically bind to cancer
cell antigens,
viral antigens or microbial antigens, single-chain antibody molecules; and
multispecific
antibodies formed from antibody fragments.
[0039] The term "monoclonal antibody" as used herein refers to an
antibody obtained
from a population of substantially homogeneous antibodies, i.e., the
individual antibodies
comprising the population are identical except for possible naturally
occurring mutations that
may be present in minor amounts. Monoclonal antibodies arc highly specific,
being directed
against a single antigenic site. Furthermore, in contrast to polyclonal
antibody preparations
which include different antibodies directed against different determinants
(epitopes), each
monoclonal antibody is directed against a single determinant on the antigen.
In addition to
6
Date Recue/Date Received 2023-11-15
their specificity, the monoclonal antibodies are advantageous in that they may
be synthesized
uncontaminated by other antibodies. The modifier "monoclonal" indicates the
character of
the antibody as being obtained from a substantially homogeneous population of
antibodies,
and is not to be construed as requiring production of the antibody by any
particular method.
For example, the monoclonal antibodies to be used in accordance with the
present invention
may be made by the hybridoma method first described by Kohler et at (1975)
Nature
256:495, or may be made by recombinant DNA methods (see for example: US
4816567; US
5807715). The monoclonal antibodies may also be isolated from phage antibody
libraries
using the techniques described in Clackson et at (1991) Nature, 352:624-628;
Marks et al
(1991) J. Mol. Biol., 222:581-597; for example.
[0040] The monoclonal antibodies herein specifically include
"chimeric" antibodies
in which a portion of the heavy and/or light chain is identical with or
homologous to
corresponding sequences in antibodies derived from a particular species or
belonging to a
particular antibody class or subclass, while the remainder of the chain(s) is
identical with or
homologous to corresponding sequences in antibodies derived from another
species or
belonging to another antibody class or subclass, as well as fragments of such
antibodies, so
long as they exhibit the desired biological activity (US 4816567; and Morrison
et at (1984)
Proc. Natl. Acad. Sci. USA, 81:6851-6855). Chimeric antibodies of interest
herein include
"primatized" antibodies comprising variable domain antigen-binding sequences
derived from
a non-human primate (e.g., Old World Monkey, Ape etc) and human constant
region
sequences.
[0041] An "intact antibody" herein is one comprising a VL and VH
domains, as well
as a light chain constant domain (CL) and heavy chain constant domains, CH1,
CH2 and
CH3. The constant domains may be native sequence constant domains (e.g., human
native
sequence constant domains) or amino acid sequence variant thereof. The intact
antibody may
have one or more "effector functions" which refer to those biological
activities attributable to
the Fc constant region (a native sequence Fc region or amino acid sequence
variant Fc
region) of an antibody. Examples of antibody effector functions include Clq
binding;
complement dependent cytotoxicity; Fc receptor binding; antibody-dependent
cell-mediated
cytotoxicity (ADCC); phagocytosis; and down regulation of cell surface
receptors such as B
cell receptor and BCR.
[0042] Depending on the amino acid sequence of the constant domain of
their heavy
chains, intact antibodies can be assigned to different "classes." There are
five major classes
of intact immunoglobulin antibodies: IgA, IgD, IgE, IgG, and IgM, and several
of these may
7
Date Recue/Date Received 2023-11-15
be further divided into "subclasses" (isotypes), e.g., IgGI, IgG2, IgG3, IgG4,
IgA, and IgA2.
The heavy-chain constant domains that correspond to the different classes of
antibodies are
called a, 6, E, y, and , respectively. The subunit structures and three-
dimensional
configurations of different classes of immunoglobulins are well known. Ig
forms include
hinge-modifications or hingeless forms (Roux et al (1998) J. Immunol. 161:4083-
4090; Lund
et al (2000) Eur. J. Biochem. 267:7246-7256; US 2005/0048572; US
2004/0229310).
[0043] An "ErbB receptor" is a receptor protein tyrosine kinase which
belongs to the
ErbB receptor family whose members are important mediators of cell growth,
differentiation
and survival. The ErbB receptor family includes four distinct members
including epidermal
growth factor receptor (EGFR, ErbB I, HER1), HER2 (ErbB2 or p185neu), HER3
(ErbB3)
and HER4 (ErbB4 or tyro2). A panel of anti-ErbB2 antibodies has been
characterized using
the human breast tumor cell line SKBR3 aludziak et al (1989) Mol. Cell. Biol.
9(3):1165-
1172. Maximum inhibition was obtained with the antibody called 4D5 which
inhibited
cellular proliferation by 56%. Other antibodies in the panel reduced cellular
proliferation to a
lesser extent in this assay. The antibody 4D5 was further found to sensitize
ErbB2-
overexpressing breast tumor cell lines to the cytotoxic effects of TNF-a (US
5677171). The
anti-ErbB2 antibodies discussed in Hudziak et al. are further characterized in
Fendly et al
(1990) Cancer Research 50:1550-1558; Kotts et al. (1990) In Vitro 26(3):59A;
Sarup et al.
(1991) Growth Regulation 1:72-82; Shepard et al. J. (1991) Clin. lmmunol.
11(3):117-127;
Kumar et al. (1991) Mol. Cell. Biol. 11(2):979-986; Lewis et al. (1993) Cancer
Immunol.
Immunother. 37:255-263; Pietras et al. (1994) Oncogene 9:1829-1838; Vitetta et
al. (1994)
Cancer Research 54:5301-5309; Sliwkowski et al. (1994) J. Biol. Chem.
269(20):14661-
14665; Scott et al. (1991) J. Biol. Chem. 266:14300-5; D'souza et al. Proc.
Natl. Acad. Sci.
(1994) 91:7202-7206; Lewis et al. (1996) Cancer Research 56:1457-1465; and
Schaefer et al.
(1997) Oncogene 15:1385-1394.
[0044] The ErbB receptor will generally comprise an extracellular
domain, which
may bind an ErbB ligand; a lipophilic transmembrane domain; a conserved
intracellular
tyrosine kinase domain; and a carboxyl-terminal signaling domain harboring
several tyrosine
residues which can be phosphorylated. The ErbB receptor may be a "native
sequence" ErbB
receptor or an "amino acid sequence variant" thereof. Preferably, the ErbB
receptor is native
sequence human ErbB receptor. Accordingly, a "member of the ErbB receptor
family"
includes EGFR (ErbB1), ErbB2, ErbB3, ErbB4.
[0045] The term "amino acid sequence variant" refers to polypeptides
having amino
acid sequences that differ to some extent from a native sequence polypeptide.
Ordinarily,
8
Date Recue/Date Received 2023-11-15
amino acid sequence variants will possess at least about 70% sequence identity
with at least
one receptor binding domain of a native ErbB ligand or with at least one
ligand binding
domain of a native ErbB receptor, and preferably, they will be at least about
80%, more
preferably, at least about 90% homologous by sequence with such receptor or
ligand binding
domains. The amino acid sequence variants possess substitutions, deletions,
and/or insertions
at certain positions within the amino acid sequence of the native amino acid
sequence.
Amino acids are designated by the conventional names, one-letter and three-
letter codes.
[0046] "Sequence identity" is defined as the percentage of residues in
the amino acid
sequence variant that are identical after aligning the sequences and
introducing gaps, if
necessary, to achieve the maximum percent sequence identity. Methods and
computer
programs for the alignment are well known in the art. One such computer
program is "Align
2," authored by Genentech, Inc., which was filed with user documentation in
the United
States Copyright Office, Washington, DC 20559, on December 10, 1991.
100471 "Native antibodies" are usually heterotetrameric glycoproteins
of about
150,000 daltons, composed of two identical light (L) chains and two identical
heavy (H)
chains. Each light chain is linked to a heavy chain by one covalent disulfide
bond, while the
number of disulfide linkages varies among the heavy chains of different immuno
globulin
isotypes. Each heavy and light chain also has regularly spaced intrachain
disulfide bridges.
Each heavy chain has at one end a variable domain (VH) followed by a number of
constant
domains. Each light chain has a variable domain at one end (VI) and a constant
domain at its
other end. The constant domain of the light chain is aligned with the first
constant domain of
the heavy chain, and the light-chain variable domain is aligned with the
variable domain of
the heavy chain. Particular amino acid residues are believed to form an
interface between the
light chain and heavy chain variable domains.
[0048] The term "variable" refers to the fact that certain portions of
the variable
domains differ extensively in sequence among antibodies and are used in the
binding and
specificity of each particular antibody for its particular antigen. However,
the variability is
not evenly distributed throughout the variable domains of antibodies. It is
concentrated in
three segments called hypervariable regions both in the light chain and the
heavy chain
variable domains. The more highly conserved portions of variable domains are
called the
framework regions (FRs). The variable domains of native heavy and light chains
each
comprise four FRs, largely adopting a I3-sheet configuration, connected by
three
hypervariable regions, which form loops connecting, and in some cases forming
part of, the
13-sheet structure. The hypervariable regions in each chain are held together
in close
9
Date Recue/Date Received 2023-11-15
proximity by the FRs and, with the hypervariable regions from the other chain,
contribute to
the formation of the antigen-binding site of antibodies (see Kabat et at
(1991) Sequences of
Proteins of Immunological Interest, 5th Ed. Public Health Service, National
Institutes of
Health, Bethesda, MD). The constant domains are not involved directly in
binding an
antibody to an antigen, but exhibit various effector functions, such as
participation of the
antibody in antibody dependent cellular cytotoxicity (ADCC).
[0049] The term "hypervariable region" when used herein refers to the
amino acid
residues of an antibody which are responsible for antigen-binding. The
hypervariable region
generally comprises amino acid residues from a "complementarity determining
region" or
"CDR" (e.g., residues 24-34 (L1), 50-56 (L2) and 89-97 (L3) in the light chain
variable
domain and 31-35 (H1), 50-65 (H2) and 95-102 (H3) in the heavy chain variable
domain;
Kabat et al supra) and/or those residues from a "hypervariable loop" (e.g.,
residues 26-32
(L1), 50-52 (L2) and 91-96 (L3) in the light chain variable domain and 26-32
(H1), 53-55
(H2) and 96-101 (H3) in the heavy chain variable domain; Chothia and Lesk
(1987) J. Mol.
Biol., 196:901-917). "Framework Region" or "FR" residues are those variable
domain
residues other than the hypervariable region residues as herein defined.
[0050] Papain digestion of antibodies produces two identical antigen-
binding
fragments, called "Fab" fragments, each with a single antigen-binding site,
and a residual
"Fc" fragment, whose name reflects its ability to crystallize readily. Pepsin
treatment yields
an F(ab')2 fragment that has two antigen-binding sites and is still capable of
cross-linking
antigen.
[0051] "Fv" is the minimum antibody fragment which contains a complete
antigen-
recognition and antigen-binding site. This region consists of a dimer of one
heavy chain and
one light chain variable domain in tight, non-covalent association. It is in
this configuration
that the three hypervariable regions of each variable domain interact to
define an antigen-
binding site on the surface of the VH-VL dimer. Collectively, the six
hypervariable regions
confer antigen-binding specificity to the antibody. However, even a single
variable domain
(or half of an Fv comprising only three hypervariable regions specific for an
antigen) has the
ability to recognize and bind antigen, although at a lower affinity than the
entire binding site.
[0052] The Fab fragment also contains the constant domain of the light
chain and the
first constant domain (CH1) of the heavy chain. Fab' fragments differ from Fab
fragments by
the addition of a few residues at the carboxy terminus of the heavy chain CH1
domain
including one or more cysteines from the antibody hinge region. Fab'-SH is the
designation
herein for Fab' in which the cysteine residue(s) of the constant domains bear
at least one free
Date Recue/Date Received 2023-11-15
thiol group. F(ab')2 antibody fragments originally were produced as pairs of
Fab' fragments
which have hinge cysteincs between them. Other chemical couplings of antibody
fragments
are also known.
[0053] The "light chains" of antibodies from any vertebrate species
can be assigned to
one of two clearly distinct types, called kappa (K) and lambda (k), based on
the amino acid
sequences of their constant domains.
[0054] "Single-chain Fv" or "scFv" antibody fragments comprise the VH
and VL
domains of antibody, wherein these domains are present in a single polypeptide
chain.
Preferably, the Fv polypeptide further comprises a polypeptide linker between
the VII and
VL domains which enables the scFv to form the desired structure for antigen
binding. For a
review of scFv, see Pliickthun in The Pharmacology of Monoclonal Antibodies,
vol. 113,
Rosenburg and Moore eds., Springer-Verlag, New York, pp. 269-315 (1994). Anti-
ErbB2
antibody scFv fragments are described in WO 93/16185; US Patent Nos. 5571894;
and
5587458.
100551 "Humanized" forms of non-human (e.g., rodent) antibodies are
chimeric
antibodies that contain minimal sequence derived from non-human
immunoglobulin.
Humanization is a method to transfer the murinc antigen binding information to
a non-
immunogenic human antibody acceptor, and has resulted in many therapeutically
useful
drugs. The method of humanization generally begins by transferring all six
murine
complementarity determining regions (CDRs) onto a human antibody framework
(Jones et al,
(1986) Nature 321:522-525). These CDR-grafted antibodies generally do not
retain their
original affinity for antigen binding, and in fact, affinity is often severely
impaired. Besides
the CDRs, select non-human antibody framework residues must also be
incorporated to
maintain proper CDR conformation (Chothia et al (1989) Nature 342:877). The
transfer of
key mouse framework residues to the human acceptor in order to support the
structural
conformation of the grafted CDRs has been shown to restore antigen binding and
affinity
(Riechmann et al (1992) J. Mol. Biol. 224, 487-499; Foote and Winter, (1992)
J. Mol. Biol.
224:487-499; Presta et al (1993) J. Immunol. 151, 2623-2632; Werther et al
(1996) J.
Immunol. Methods 157:4986-4995; and Presta et al (2001) Thromb. Haemost.
85:379-389).
For the most part, humanized antibodies are human immunoglobulins (recipient
antibody) in
which residues from a hypervariable region of the recipient arc replaced by
residues from a
hypervariable region of a non-human species (donor antibody) such as mouse,
rat, rabbit or
nonhuman primate having the desired specificity, affinity, and capacity. In
some instances,
framework region (FR) residues of the human immunoglobulin are replaced by
11
Date Recue/Date Received 2023-11-15
corresponding non-human residues. Furthermore, humanized antibodies may
comprise
residues that are not found in the recipient antibody or in the donor
antibody. These
modifications are made to further refine antibody performance. In general, the
humanized
antibody will comprise substantially all of at least one, and typically two,
variable domains,
in which all or substantially all of the hypervariable loops correspond to
those of a non-
human immunoglobulin and all or substantially all of the FRs are those of a
human
immunoglobulin sequence. The humanized antibody optionally also will comprise
at least a
portion of an immunoglobulin constant region (Fc), typically that of a human
immunoglobulin. For further details, see US 6407213; Jones et al (1986)
Nature, 321:522-
525; Riechmann et at (1988) Nature 332:323-329; and Presta, (1992) Curr. Op.
Struct. Biol.,
2:593-596.
[0056] A "free cysteine amino acid" refers to a cysteine amino acid
residue which has
been engineered into a parent antibody, has a thiol functional group (-SH),
and is not paired
as an intramolecular or intermolecular disulfide bridge.
[0057] The term "thiol reactivity value" is a quantitative
characterization of the
reactivity of free cysteine amino acids. The thiol reactivity value is the
percentage of a free
cysteine amino acid in a cysteine engineered antibody which reacts with a
thiol-reactivc
reagent, and converted to a maximum value of 1. For example, a free cysteine
amino acid on
a cysteine engineered antibody which reacts in 100% yield with a thiol-
reactive reagent, such
as a biotin-maleimide reagent, to form a biotin-labelled antibody has a thiol
reactivity value
of 1Ø Another cysteine amino acid engineered into the same or different
parent antibody
which reacts in 80% yield with a thiol-reactive reagent has a thiol reactivity
value of about
0.8. Another cysteine amino acid engineered into the same or different parent
antibody
which fails totally to react with a thiol-reactive reagent has a thiol
reactivity value of O.
Determination of the thiol reactivity value of a particular cysteine may be
conducted by
ELISA assay, mass spectroscopy, liquid chromatography, autoradiography, or
other
quantitative analytical tests.
[0058] A "parent antibody" is an antibody comprising an amino acid
sequence from
which one or more amino acid residues are replaced by one or more cysteine
residues. The
parent antibody may comprise a native or wild type sequence. The parent
antibody may have
pre-existing amino acid sequence modifications (such as additions, deletions
and/or
substitutions) relative to other native, wild type, or modified forms of an
antibody. A parent
antibody may be directed against a target antigen of interest, e.g. a
biologically important
polypeptide. Antibodies directed against nonpolypeptide antigens (such as
tumor-associated
12
Date Recue/Date Received 2023-11-15
glycolipid antigens; see US 5091178) are also contemplated.
100591 Exemplary parent antibodies include antibodies having affinity
and selectivity
for cell surface and transmembrane receptors and tumor-associated antigens
(TAA).
[0060] An "isolated" antibody is one which has been identified and
separated and/or
recovered from a component of its natural environment. Contaminant components
of its
natural environment are materials which would interfere with diagnostic or
therapeutic uses
for the antibody, and may include enzymes, hormones, and other proteinaceous
or
nonproteinaceous solutes. In preferred embodiments, the antibody will be
purified (1) to
greater than 95% by weight of antibody as determined by the Lowry method, and
most
preferably more than 99% by weight, (2) to a degree sufficient to obtain at
least 15 residues
of N-terminal or internal amino acid sequence by use of a spinning cup
sequenator, or (3) to
homogeneity by SDS-PAGE under reducing or nonreducing conditions using
Coomassie blue
or, preferably, silver stain. Isolated antibody includes the antibody in situ
within recombinant
cells since at least one component of the antibody's natural environment will
not be present.
Ordinarily, however, isolated antibody will be prepared by at least one
purification step.
100611 An antibody "which binds" a molecular target or an antigen of
interest, e.g.,
ErbB2 antigen, is one capable of binding that antigen with sufficient affinity
such that the
antibody is useful in targeting a cell expressing the antigen. Where the
antibody is one which
binds ErbB2, it will usually preferentially bind ErbB2 as opposed to other
ErbB receptors,
and may be one which does not significantly cross-react with other proteins
such as EGFR,
ErbB3 or ErbB4. In such embodiments, the extent of binding of the antibody to
these non-
ErbB2 proteins (e.g., cell surface binding to endogenous receptor) will be
less than 10% as
determined by fluorescence activated cell sorting (FACS) analysis or
radioimmunoprecipitation (RIA). Sometimes, the anti-ErbB2 antibody will not
significantly
cross-react with the rat neu protein, e.g., as described in Schecter et al.
(1984) Nature 312:513
and Drebin et al (1984) Nature 312:545-548.
[0062] Molecular targets for antibodies encompassed by the present
invention include
CD proteins and their ligands, such as, but not limited to: (i) CD3, CD4, CD8,
CD19, CD20,
CD22, CD34, CD40, CD79a (CD79a), and CD79f3 (CD79b); (ii) members of the ErbB
receptor family such as the EGF receptor, HER2, HER3 or HER4 receptor; (iii)
cell adhesion
molecules such as LFA-1, Macl, p150,95, VLA-4, ICAM-1, VCAM and av/133
integrin,
including either alpha or beta subunits thereof (e.g. anti-CD1 1 a, anti-CD18
or anti-CD11b
antibodies); (iv) growth factors such as VEGF; IgE; blood group antigens;
flk2/flt3 receptor;
13
Date Recue/Date Received 2023-11-15
obesity (OB) receptor; mpl receptor; CTLA-4; protein C, BR3, c-met, tissue
factor, 37 etc;
and (v) cell surface and transmembrane tumor-associated antigens (TAA).
[0063] Unless indicated otherwise, the term "monoclonal antibody 4D5"
refers to an
antibody that has antigen binding residues of, or derived from, the murine 4D5
antibody
(ATCC CRL 10463). For example, the monoclonal antibody 4D5 may be murine
monoclonal antibody 4D5 or a variant thereof, such as a humanized 4D5.
Exemplary
humanized 4D5 antibodies include huMAb4D5-1, huMAb4D5-2, huMAb4D5-3,
huMAb4D5-4, huMAb4D5-5, huMAb4D5-6, huMAb4D5-7 and huMAb4D5-8 (trastuzumab,
HERCEPTINO) as in US Patent No. 5821337.
[0064] The terms "treat" and "treatment" refer to both therapeutic
treatment and
prophylactic or preventative measures, wherein the object is to prevent or
slow down (lessen)
an undesired physiological change or disorder, such as the development or
spread of cancer.
For purposes of this invention, beneficial or desired clinical results
include, but are not
limited to, alleviation of symptoms, diminishment of extent of disease,
stabilized (i.e., not
worsening) state of disease, delay or slowing of disease progression,
amelioration or
palliation of the disease state, and remission (whether partial or total),
whether detectable or
undetectable. "Treatment" can also mean prolonging survival as compared to
expected
survival if not receiving treatment. Those in need of treatment include those
already with the
condition or disorder as well as those prone to have the condition or disorder
or those in
which the condition or disorder is to be prevented.
[0065] The term "therapeutically effective amount" refers to an amount
of a drug
effective to treat a disease or disorder in a mammal. In the case of cancer,
the therapeutically
effective amount of the drug may reduce the number of cancer cells; reduce the
tumor size;
inhibit (i.e., slow to some extent and preferably stop) cancer cell
infiltration into peripheral
organs; inhibit (i.e., slow to some extent and preferably stop) tumor
metastasis; inhibit, to
some extent, tumor growth; and/or relieve to some extent one or more of the
symptoms
associated with the cancer. To the extent the drug may prevent growth and/or
kill existing
cancer cells, it may be cytostatic and/or cytotoxic. For cancer therapy,
efficacy can, for
example, be measured by assessing the time to disease progression (TTP) and/or
determining
the response rate (RR).
[0066] The terms "cancer" and "cancerous" refer to or describe the
physiological
condition in mammals that is typically characterized by unregulated cell
growth. A "tumor"
comprises one or more cancerous cells. Examples of cancer include, but are not
limited to,
14
Date Recue/Date Received 2023-11-15
carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies.
More
particular examples of such cancers include squamous cell cancer (e.g.,
epithelial squamous
cell cancer), lung cancer including small- cell lung cancer, non-small cell
lung cancer
("NSCLC"), adenocarcinoma of the lung and squamous carcinoma of the lung,
cancer of the
peritoneum, hepatocellular cancer, gastric or stomach cancer including
gastrointestinal
cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer,
liver cancer, bladder
cancer, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal
cancer, endometrial or
uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate
cancer, vulval
cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma,
as well as head
and neck cancer.
[0067] An "ErbB-expressing cancer" is one comprising cells which have
ErbB
protein present at their cell surface. An "ErbB2-expressing cancer" is one
which produces
sufficient levels of ErbB2 at the surface of cells thereof, such that an anti-
ErbB2 antibody can
bind thereto and have a therapeutic effect with respect to the cancer.
[0068] A cancer which "overexpresses" an antigenic receptor is one
which has
significantly higher levels of the receptor, such as ErbB2, at the cell
surface thereof,
compared to a noncancerous cell of the same tissue type. Such overcxpression
may be
caused by gene amplification or by increased transcription or translation.
Receptor
overexpression may be determined in a diagnostic or prognostic assay by
evaluating
increased levels of the receptor protein present on the surface of a cell
(e.g., via an
immunohistochemistry assay; IHC). Alternatively, or additionally, one may
measure levels
of receptor-encoding nucleic acid in the cell, e.g., via fluorescent in situ
hybridization (FISH;
see WO 98/45479), southern blotting, or polymerase chain reaction (PCR)
techniques, such
as real time quantitative PCR (RT-PCR).
[0069] The term "cytotoxic agent" as used herein refers to a substance
that inhibits or
prevents the function of cells and/or causes destruction of cells. The term is
intended to
include radioactive isotopes (e.g., 211 At,1311, 1251, 90y, 186- e,
R 188Re, 153sm, 212Bi, 32/3,
u and
radioactive isotopes of Lu), chemotherapeutic agents, and toxins such as small
molecule
toxins or enzymatically active toxins of bacterial, fungal, plant or animal
origin, including
synthetic analogs and derivatives thereof.
[0070] "Phagc display" is a technique by which variant polypcptidcs
arc displayed as
fusion proteins to a coat protein on the surface of phage, e.g., filamentous
phage, particles.
One utility of phage display lies in the fact that large libraries of
randomized protein variants
can be rapidly and efficiently sorted for those sequences that bind to a
target molecule with
Date Recue/Date Received 2023-11-15
high affinity. Display of peptide and protein libraries on phage has been used
for screening
millions of polypeptides for ones with specific binding properties. Polyvalent
phage display
methods have been used for displaying small random peptides and small
proteins, typically
through fusions to either pill or pVIII of filamentous phage (Wells and
Lowman, (1992)
Curr. Opin. Struct. Biol., 3:355-362, and references cited therein). In
monovalent phage
display, a protein or peptide library is fused to a phage coat protein or a
portion thereof, and
expressed at low levels in the presence of wild type protein. Avidity effects
are reduced
relative to polyvalent phage so that sorting is on the basis of intrinsic
ligand affinity, and
phagemid vectors are used, which simplify DNA manipulations. Lowman and Wells,
Methods: A companion to Methods in Enzymology, 3:205-0216 (1991). Phage
display
includes techniques for producing antibody-like molecules (Janeway, C.,
Travers, P.,
Walport, M., Shlomchik (2001) Immunobiology, 5th Ed., Garland Publishing, New
York,
p627-628; Lee et al).
100711 A "phagemid" is a plasmid vector having a bacterial origin of
replication, e.g.,
Co 1E1, and a copy of an intergenic region of a bacteriophage. The phagemid
may be used on
any known bacteriophage, including filamentous bacteriophage and lambdoid
bacteriophage.
The plasmid will also generally contain a selectable marker for antibiotic
resistance.
Segments of DNA cloned into these vectors can be propagated as plasmids. When
cells
harboring these vectors are provided with all genes necessary for the
production of phage
particles, the mode of replication of the plasmid changes to rolling circle
replication to
generate copies of one strand of the plasmid DNA and package phage particles.
The
phagemid may form infectious or non-infectious phage particles. This term
includes
phagemids which contain a phage coat protein gene or fragment thereof linked
to a
heterologous polypeptide gene as a gene fusion such that the heterologous
polypeptide is
displayed on the surface of the phage particle.
100721 "Linker", "Linker Unit", or "link" means a chemical moiety
comprising a
covalent bond or a chain of atoms that covalently attaches an antibody to a
drug moiety. In
various embodiments, a linker is specified as L. Linkers include a divalent
radical such as an
alkyldiyl, an arylene, a heteroarylene, moieties such as: ¨(CR2).0(CR2)õ¨,
repeating units of
alkyloxy (e.g. polyethylenoxy, PEG, polymethyleneoxy) and alkylamino (e.g.
polyethyleneamino, JeffamineTm); and diacid ester and amides including
succinate,
succinamide, diglycolate, malonate, and caproamide.
100731 The term "label" means any moiety which can be covalently
attached to an
16
Date Recue/Date Received 2023-11-15
antibody and that functions to: (i) provide a detectable signal; (ii) interact
with a second label
to modify the detectable signal provided by the first or second label, e.g.
FRET (fluorescence
resonance energy transfer); (iii) stabilize interactions or increase affinity
of binding, with
antigen or ligand; (iv) affect mobility, e.g. electrophoretic mobility, or
cell-permeability, by
charge, hydrophobicity, shape, or other physical parameters, or (v) provide a
capture moiety,
to modulate ligand affinity, antibody/antigen binding, or ionic complexation.
[0074] Stereochemical definitions and conventions used herein
generally follow S. P.
Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book
Company, New York; and Eliel, E. and Wilen, S., Stereochemistty of Organic
Compounds
(1994) John Wiley & Sons, Inc., New York. Many organic compounds exist in
optically
active forms, i.e., they have the ability to rotate the plane of plane-
polarized light. In
describing an optically active compound, the prefixes D and L, or R and S. are
used to denote
the absolute configuration of the molecule about its chiral center(s). The
prefixes d and 1 or
(+) and (-) are employed to designate the sign of rotation of plane-polarized
light by the
compound, with (-) or 1 meaning that the compound is levorotatory. A compound
prefixed
with (+) or d is dextrorotatory. For a given chemical structure, these
stereoisomers are
identical except that they are mirror images of one another. A specific
stereoisomer may also
be referred to as an enantiomer, and a mixture of such isomers is often called
an enantiomeric
mixture. A 50:50 mixture of enantiomers is referred to as a racemic mixture or
a racemate,
which may occur where there has been no stereoselection or stereospecificity
in a chemical
reaction or process. The terms "racemic mixture" and "racemate" refer to an
equimolar
mixture of two enantiomeric species, devoid of optical activity.
[0075] The phrase "pharmaceutically acceptable salt," as used herein,
refers to
pharmaceutically acceptable organic or inorganic salts of an ADC. Exemplary
salts include,
but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide,
iodide, nitrate,
bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid
citrate, tartrate,
oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate,
gentisinate, fumarate,
gluconate, glucuronate, saccharate, formate, benzoate, glutamate,
methanesulfonate,
ethanesulfonate, benzenesulfonate, p-toluenesulfonate, and pamoate (i.e., 1,1'
-methylene-bis
-(2-hydroxy-3- naphthoate)) salts. A pharmaceutically acceptable salt may
involve the
inclusion of another molecule such as an acetate ion, a succinatc ion or other
countcrion. The
counterion may be any organic or inorganic moiety that stabilizes the charge
on the parent
compound. Furthermore, a pharmaceutically acceptable salt may have more than
one
charged atom in its structure. Instances where multiple charged atoms are part
of the
17
Date Recue/Date Received 2023-11-15
pharmaceutically acceptable salt can have multiple counter ions. Hence, a
pharmaceutically
acceptable salt can have one or more charged atoms and/or one or more
counterion.
[0076] "Pharmaceutically acceptable solvate" refers to an association
of one or more
solvent molecules and an ADC. Examples of solvents that form pharmaceutically
acceptable
solvates include, but are not limited to, water, isopropanol, ethanol,
methanol, DMSO, ethyl
acetate, acetic acid, and ethanolamine.
[0077] CYSTEINE ENGINEERED ANTIBODIES
[0078] The compounds of the invention include cysteine engineered
antibodies where
one or more amino acids of a wild-type or parent antibody are replaced with a
cysteine amino
acid. Any form of antibody may be so engineered, i.e. mutated. For example, a
parent Fab
antibody fragment may be engineered to form a cysteine engineered Fab,
referred to herein as
"Thiaab." Similarly, a parent monoclonal antibody may be engineered to form a
"ThioMab." It should be noted that a single site mutation yields a single
engineered cysteine
residue in a ThioFab, while a single site mutation yields two engineered
cysteine residues in a
ThioMab, due to the dimeric nature of the IgG antibody. Mutants with replaced
("engineered") cysteine (Cys) residues are evaluated for the reactivity of the
newly
introduced, engineered cysteine thiol groups. The thiol reactivity value is a
relative,
numerical term in the range of 0 to 1.0 and can be measured for any cysteine
engineered
antibody. Thiol reactivity values of cysteine engineered antibodies of the
invention are in the
ranges of 0.6 to 1.0; 0.7 to 1.0; or 0.8 to 1Ø
[0079] The design, selection, and preparation methods of the invention
enable
cysteine engineered antibodies which are reactive with electrophilic
fimctionality. These
methods further enable antibody conjugate compounds such as antibody-drug
conjugate
(ADC) compounds with drug molecules at designated, designed, selective sites.
Reactive
cysteine residues on an antibody surface allow specifically conjugating a drug
moiety through
a thiol reactive group such as maleimide or haloacetyl. The nucleophilic
reactivity of the
thiol functionality of a Cys residue to a maleimide group is about 1000 times
higher
compared to any other amino acid functionality in a protein, such as amino
group of lysine
residues or the N-terminal amino group. Thiol specific functionality in
iodoacetyl and
maleimide reagents may react with amine groups, but higher pH (>9.0) and
longer reaction
times are required (Garman, 1997, Non-Radioactive Labelling: A Practical
Approach,
Academic Press, London).
[0080] Cysteine engineered antibodies of the invention preferably
retain the antigen
18
Date Recue/Date Received 2023-11-15
binding capability of their wild type, parent antibody counterparts. Thus,
cysteine engineered
antibodies are capable of binding, preferably specifically, to antigens. Such
antigens include,
for example, tumor-associated antigens (TAA), cell surface receptor proteins
and other cell
surface molecules, transmembrane proteins, signalling proteins, cell survival
regulatory
factors, cell proliferation regulatory factors, molecules associated with (for
e.g., known or
suspected to contribute functionally to) tissue development or
differentiation, lymphokines,
cytokines, molecules involved in cell cycle regulation, molecules involved in
vasculogenesis
and molecules associated with (for e.g., known or suspected to contribute
functionally to)
angiogenesis. The tumor-associated antigen may be a cluster differentiation
factor (i.e., a CD
protein). An antigen to which a cysteine engineered antibody is capable of
binding may be a
member of a subset of one of the above-mentioned categories, wherein the other
subset(s) of
said category comprise other molecules/antigens that have a distinct
characteristic (with
respect to the antigen of interest).
[0081] The parent antibody may also be a humanized antibody
selected from
huMAb4D5-1, huMAb4D5-2, huMAb4D5-3, huMAb4D5-4, huMAb4D5-5, huMAb4D5-6,
huMAb4D5-7 and liuMAb4D5-8 (Trastuzumab, HERCEPTINC)) as described in Table 3
of
US 5821337,
humanized 520C9 (WO 93/21319)
and humanized 2C4 antibodies as described herein.
[0082] Cysteine engineered antibodies of the invention may bc site-
specifically and
efficiently coupled with a thiol-reactive reagent. The thiol-reactive reagent
may be a
multifunctional linker reagent, a capture, i.e. affinity, label reagent (e.g.
a biotin-linker
reagent), a detection label (e.g. a fluorophore reagent), a solid phase
immobilization reagent
(e.g. SEPHAROSETM, polystyrene, or glass), or a drug-linker intermediate. One
example of
a thiol-reactive reagent is N-ethyl maleimide (NEM). In an exemplary
embodiment, reaction
of a ThioFab with a biotin-linker reagent provides a biotinylated ThioFab by
which the
presence and reactivity of the engineered cysteine residue may be detected and
measured.
Reaction of a ThioFab with a multifunctional linker reagent provides a ThioFab
with a
functionalized linker which may be further reacted with a drug moiety reagent
or other label.
Reaction of a ThioFab with a drug-linker intermediate provides a ThioFab drug
conjugate.
[0083] The exemplary methods described here may be applied
generally to the
identification and production of antibodies, and more generally, to other
proteins through
application of the design and screening steps described herein.
[0084] Such an approach may be applied to the conjugation of other
thiol-reactive
agents in which the reactive group is, for example, a maleimide, an
iodoacetamide, a pyridyl
19
Date Recue/Date Received 2023-11-15
disulfide, or other thiol-reactive conjugation partner (Haugland, 2003,
Molecular Probes
Handbook of Fluorescent Probes and Research Chemicals, Molecular Probes, Inc.;
Brinkley,
1992, Bioconjugate Chem. 3:2; Garman, 1997, Non-Radioactive Labelling: A
Practical
Approach, Academic Press, London; Means (1990) Bioconjugate Chem. 1:2;
Hermanson, G.
in Bioconjugate Techniques (1996) Academic Press, San Diego, pp. 40-55, 643-
671). The
partner may be a cytotoxic agent (e.g. a toxin such as doxorubicin or
pertussis toxin), a
fluorophore such as a fluorescent dye like fluorescein or rhodamine, a
chelating agent for an
imaging or radiotherapeutic metal, a peptidyl or non-peptidyl label or
detection tag, or a
clearance-modifying agent such as various isomers of polyethylene glycol, a
peptide that
binds to a third component, or another carbohydrate or lipophilic agent.
100851 The sites identified on the exemplary antibody fragment,
hu4D5Fabv8, herein
are primarily in the constant domain of an antibody which is well conserved
across all species
of antibodies. These sites should be broadly applicable to other antibodies,
without further
need of structural design or knowledge of specific antibody structures, and
without
interference in the antigen binding properties inherent to the variable
domains of the
antibody.
100861 Cysteine engineered antibodies which may be useful in the
treatment of cancer
include, but are not limited to, antibodies against cell surface receptors and
tumor-associated
antigens (TAA). Such antibodies may be used as naked antibodies (unconjugated
to a drug or
label moiety) or as Formula I antibody-drug conjugates (ADC). Tumor-associated
antigens
are known in the art, and can prepared for use in generating antibodies using
methods and
information which are well known in the art. In attempts to discover effective
cellular targets
for cancer diagnosis and therapy, researchers have sought to identify
transmembrane or
otherwise tumor-associated polypeptides that are specifically expressed on the
surface of one
or more particular type(s) of cancer cell as compared to on one or more normal
non-
cancerous cell(s). Often, such tumor-associated polypeptides are more
abundantly expressed
on the surface of the cancer cells as compared to on the surface of the non-
cancerous cells.
The identification of such tumor-associated cell surface antigen polypeptides
has given rise to
the ability to specifically target cancer cells for destruction via antibody-
based therapies.
100871 Examples of TAA include, but are not limited to, TAA (1)-(36)
listed below.
For convenience, information relating to these antigens, all of which arc
known in the art, is
listed below and includes names, alternative names, Genbank accession numbers
and primary
reference(s), following nucleic acid and protein sequence identification
conventions of the
National Center for Biotechnology Information (NCBI). Nucleic acid and protein
sequences
Date Recue/Date Received 2023-11-15
corresponding to TAA (1)-(36) are available in public databases such as
GenBank. Tumor-
associated antigens targeted by antibodies include all amino acid sequence
variants and
isoforms possessing at least about 70%, 80%, 85%, 90%, or 95% sequence
identity relative to
the sequences identified in the cited references, or which exhibit
substantially the same
biological properties or characteristics as a TAA having a sequence found in
the cited
references. For example, a TAA having a variant sequence generally is able to
bind
specifically to an antibody that binds specifically to the TAA with the
corresponding
sequence listed.
[0088] TUMOR-ASSOCIATED ANTIGENS (1)-(36):
[0089] (1) BMPR1B (bone morphogenetic protein receptor-type I13,
Genbank
accession no. NM 001203) ten Dijke,P., et al Science 264 (5155):101-104
(1994), Oncogene
14 (11):1377-1382 (1997)); W02004063362 (Claim 2); W02003042661 (Claim 12);
US2003134790-Al (Page 38-39); W02002102235 (Claim 13; Page 296); W02003055443
(Page 91-92); W0200299122 (Example 2; Page 528-530); W02003029421 (Claim 6);
W02003024392 (Claim 2; Fig 112); W0200298358 (Claim 1; Page 183); W0200254940
(Page 100-101); W0200259377(Page 349-350); W0200230268 (Claim 27; Page 376);
W0200148204 (Example; Fig 4); NP_001194 bone morphogenetic protein receptor,
type TB
/pic1=NP_001194.1. Cross-references: MIM:603248; NP_001194.1; AY065994
[0090] (2) E16 (LAT1, SLC7A5, Genbank accession no. NM_003486)
Biochem.
Biophys. Res, Commun. 255 (2), 283-288 (1999), Nature 395 (6699):288-291
(1998),
Gaugitsch, H.W., et al (1992) J. Biol. Chem. 267 (16):11267-11273);
W02004048938
(Example 2); W02004032842 (Example IV); W02003042661 (Claim 12); W02003016475
(Claim 1); W0200278524 (Example 2); W0200299074 (Claim 19; Page 127-129);
W0200286443 (Claim 27; Pages 222, 393); W02003003906 (Claim 10; Page 293);
W0200264798 (Claim 33; Page 93-95); W0200014228 (Claim 5; Page 133-136);
US2003224454 (Fig 3); W02003025138 (Claim 12; Page 150); NP_003477 solute
carrier
family 7 (cationic amino acid transporter, y+system), member 5 /pid=-
NP_003477.3 - Homo
sapiens; Cross-references: MIM:600182; NP_003477.3; NM_015923; NM_003486_1
[0091] (3) STEAP1 (six transmembrane epithelial antigen of prostate,
Genbank
accession no. NM 012449); Cancer Res. 61(15), 5857-5860 (2001), Hubert, R.S.,
et al
(1999) Proc. Natl. Acad. Sci. U.S.A. 96 (25):14523-14528); W02004065577 (Claim
6);
W02004027049 (Fig 1L); EP1394274 (Example 11); W02004016225 (Claim 2);
21
Date Recue/Date Received 2023-11-15
W02003042661 (Claim 12); US2003157089 (Example 5); US2003185830 (Example 5);
US2003064397 (Fig 2); W0200289747 (Example 5; Page 618-619); W02003022995
(Example 9; Fig 13A, Example 53; Page 173, Example 2; Fig 2A); NP_036581 six
transmembrane epithelial antigen of the prostate
Cross-references: MIM:604415; NP_036581.1; NM_012449_1
[0092] (4) 0772P (CA125, MUC16, Genbank accession no. AF361486); J.
Biol.
Chem. 276 (29):27371-27375 (2001)); W02004045553 (Claim 14); W0200292836
(Claim
6; Fig 12); W0200283866 (Claim 15; Page 116-121); US2003124140 (Example 16);
Cross-
references: GI:34501467; AAK74120.3; AF361486_1
[0093] (5) MPF (MPF, MSLN, SMR, megakaryocyte potentiating factor,
mesothelin,
Genbank accession no. NM 005823) Yamaguchi, N., et al Biol. Chem. 269 (2), 805-
808
(1994), Proc. Natl. Acad. Sci. U.S.A. 96 (20):11531-11536 (1999), Proc. Natl.
Acad. Sci.
U.S.A. 93 (1):136-140 (1996), J. Biol. Chem. 270 (37):21984-21990 (1995));
W02003101283 (Claim 14); (W02002102235 (Claim 13; Page 287-288); W02002101075
(Claim 4; Page 308-309); W0200271928 (Page 320-321); W09410312 (Page 52-57);
Cross-
references: MIM:601051; NP 005814.2; NM 005823 1
[0094] (6) Napi3b (NAPI-3B, NPTIIb, SLC34A2, solute carrier family 34
(sodium
phosphate), member 2, type 11 sodium-dependent phosphate transporter 3b,
Genbank
accession no. NM 006424) J. Biol. Chem. 277 (22):19665-19672 (2002), Genomics
62
(2):281-284 (1999), Feild, J.A., et al (1999) Biochem. Biophys. Res. Commun.
258 (3):578-
582); W02004022778 (Claim 2); EP1394274 (Example 11); W02002102235 (Claim 13;
Page 326); EP875569 (Claim 1; Page 17-19); W0200157188 (Claim 20; Page 329);
W02004032842 (Example IV); W0200175177 (Claim 24; Page 139-140); Cross-
references:
MIM:604217; NP 006415.1; NM_006424_1
[0095] (7) Sema 5b (FLJ10372, KIAA1445, Mm.42015, SEMA5B, SEMAG,
Semaphorin 5b Hlog, sema domain, seven thrombospondin repeats (type 1 and type
1-like),
transmembrane domain (TM) and short cytoplasmic domain, (semaphorin) 5B,
Genbank
accession no. AB040878); Nagase T., et al (2000) DNA Res. 7 (2):143-150);
W02004000997 (Claim 1); W02003003984 (Claim 1); W0200206339 (Claim 1; Page
50);
W0200188133 (Claim 1; Page 41-43, 48-58); W02003054152 (Claim 20);
W02003101400
(Claim 11); Accession: Q9P283; EMBL; AB040878; BAA95969.1. Genew; HGNC:10737
[0096] (8) PSCA hlg (2700050C12Rik, C530008016Rik, R1KEN cDNA
2700050C12, RIKEN cDNA 2700050C12 gene, Genbank accession no. AY358628); Ross
et
al (2002) Cancer Res. 62:2546-2553; US2003129192 (Claim 2); US2004044180
(Claim 12);
22
Date Recue/Date Received 2023-11-15
US2004044179 (Claim 11); US2003096961 (Claim 11); US2003232056 (Example 5);
W02003105758 (Claim 12); US2003206918 (Example 5); EP1347046 (Claim 1);
W02003025148 (Claim 20); Cross-references: GI:37182378; AAQ88991.1; AY358628_1
[0097] (9) ETBR (Endothelin type B receptor, Genbank accession no.
AY275463);
Nakamuta M., et al Biochem. Biophys. Res. Commun. 177, 34-39, 1991; Ogawa Y.,
et al
Biochem. Biophys. Res. Commun. 178, 248-255, 1991; Arai H., et al Jpn. Circ.
J. 56, 1303-
1307, 1992; Arai H., et al J. Biol. Chem. 268, 3463-3470, 1993; Sakamoto A.,
Yanagisawa
M., et al Biochem. Biophys. Res. Commun. 178, 656-663, 1991; Elshourbagy N.A.,
et al J.
Biol. Chem. 268, 3873-3879, 1993; Haendler B., et al J. Cardiovasc. Pharmacol.
20, sl-S4,
1992; Tsutsumi M., et al Gene 228, 43-49, 1999; Strausberg R.L., et al Proc.
Natl. Acad. Sci.
U.S.A. 99, 16899-16903, 2002; Bourgeois C., et al J. Clin. Endocrinol. Metab.
82, 3116-
3123, 1997; Okamoto Y., et al Biol. Chem. 272, 21589-21596, 1997; Verheij
J.B., et al Am.
J. Med. Genet. 108, 223-225, 2002; Hofstra R.M.W., et al Eur. J. Hum. Genet.
5, 180-185,
1997; Puffenberger E.G., et al Cell 79, 1257-1266, 1994; Attie T., et al, Hum.
Mol. Genet. 4,
2407-2409, 1995; Auricchio A., et al Hum. Mol. Genet. 5:351-354, 1996; Amiel
J., et al
Hum. Mol. Genet. 5, 355-357, 1996; Hofstra R.M.W., et al Nat. Genet. 12, 445-
447, 1996;
Svensson P.J., et al Hum. Genet. 103, 145-148, 1998; Fuchs S., et al Mol. Mcd.
7, 115-124,
2001; Pingault V., et al (2002) Hum. Genet. 111, 198-206; W02004045516 (Claim
1);
W02004048938 (Example 2); W02004040000 (Claim 151); W02003087768 (Claim 1);
W02003016475 (Claim 1); W02003016475 (Claim 1); W0200261087 (Fig 1);
W02003016494 (Fig 6); W02003025138 (Claim 12; Page 144); W0200198351 (Claim 1;
Page 124-125); EP522868 (Claim 8; Fig 2); W0200177172 (Claim 1; Page 297-299);
US2003109676; US6518404 (Fig 3); US5773223 (Claim la; Col 31-34); W02004001004
[0098] (10) MSG783 (RNF124, hypothetical protein FLJ20315, Genbank
accession
no. NM 017763); W02003104275 (Claim 1); W02004046342 (Example 2);
W02003042661 (Claim 12); W02003083074 (Claim 14; Page 61); W02003018621 (Claim
1); W02003024392 (Claim 2; Fig 93); W0200166689 (Example 6); Cross-references:
LocusID:54894; NP 060233.2; NM_017763_1
[0099] (11) STEAP2 (HGNC_8639, IPCA-1, PCANAP1, STAMP1, STEAP2,
STMP, prostate cancer associated gene 1, prostate cancer associated protein 1,
six
transmembrane epithelial antigen of prostate 2, six transmembranc prostate
protein, Genbank
accession no. AF455138); Lab. Invest. 82 (11):1573-1582 (2002)); W02003087306;
US2003064397 (Claim 1; Fig 1); W0200272596 (Claim 13; Page 54-55); W0200172962
(Claim 1; Fig 4B); W02003104270 (Claim 11); W02003104270 (Claim 16);
US2004005598
23
Date Recue/Date Received 2023-11-15
(Claim 22); W02003042661 (Claim 12); US2003060612 (Claim 12; Fig 10);
W0200226822
(Claim 23; Fig 2); W0200216429 (Claim 12; Fig 10); Cross-references:
GI:22655488;
AAN04080.1; AF455138_1
[00100] (12) TrpM4 (BR22450, FLJ20041, TRPM4, TRPM4B, transient
receptor
potential cation channel, subfamily M, member 4, Genbank accession no.
NM_017636); Xu,
X.Z., et al Proc. Natl. Acad. Sci. U.S.A. 98 (19):10692-10697 (2001), Cell 109
(3):397-407
(2002), J. Biol. Chem. 278 (33):30813-30820 (2003)); US2003143557 (Claim 4);
W0200040614 (Claim 14; Page 100-103); W0200210382 (Claim 1; Fig 9A);
W02003042661 (Claim 12); W0200230268 (Claim 27; Page 391); US2003219806 (Claim
4); W0200162794 (Claim 14; Fig 1A-D); Cross-references: MIM:606936; NP
060106.2;
NM 017636 1
[00101] (13) CRIPTO (CR, CR1, CRGF, CRIPTO, TDGF1, teratocarcinoma-
derived
growth factor, Genbank accession no. NP 003203 or NM_003212); Ciccodicola, A.,
et al
EMBO J. 8 (7):1987-1991 (1989), Am. J. Hum. Genet. 49 (3):555-565 (1991));
US2003224411 (Claim 1); W02003083041 (Example 1); W02003034984 (Claim 12);
W0200288170 (Claim 2; Page 52-53); W02003024392 (Claim 2; Fig 58); W0200216413
(Claim 1; Page 94-95, 105); W0200222808 (Claim 2; Fig 1); US5854399 (Example
2; Col
17-18); US5792616 (Fig 2); Cross-references: MIM:187395; NP_003203.1;
NM_003212_1
[00102] (14) CD21 (CR2 (Complement receptor 2) or C3DR (C3d/Epstein
Barr virus
receptor) or Hs.73792 Genbank accession no. M26004); Fujisaku et al (1989) J.
Biol. Chem.
264 (4):2118-2125); Weis J.J., et al J. Exp. Med. 167, 1047-1066, 1988; Moore
M., et al
Proc. Natl. Acad. Sci. U.S.A. 84, 9194-9198, 1987; Barel M., et al Mol.
Immunol. 35, 1025-
1031, 1998; Weis J.J., et al Proc. Natl. Acad. Sci. U.S.A. 83, 5639-5643,
1986; Sinha S.K., et
al (1993) J. Immunol. 150, 5311-5320; W02004045520 (Example 4); US2004005538
(Example 1); W02003062401 (Claim 9); W02004045520 (Example 4); W09102536 (Fig
9.1-9.9); W02004020595 (Claim 1); Accession: P20023; Q13866; Q14212; EMBL;
M26004; AAA35786.1.
[00103] (15) CD79b (CD79B, CD7913, IGb (immunoglobulin-associated
beta), B29,
Genbank accession no. NM 000626 or 11038674); Proc. Natl. Acad. Sci. U.S.A.
(2003) 100
(7):4126-4131, Blood (2002) 100 (9):3068-3076, Muller et al (1992) Eur. J.
Immunol. 22
(6):1621-1625); W02004016225 (claim 2, Fig 140); W02003087768, US2004101874
(claim
1, page 102); W02003062401 (claim 9); W0200278524 (Example 2); US2002150573
(claim
5, page 15); U55644033; W02003048202 (claim 1, pages 306 and 309); WO
99/558658,
24
Date Recue/Date Received 2023-11-15
US6534482 (claim 13, Fig 17A/B); W0200055351 (claim 11, pages 1145-1146);
Cross-
references: MIM:147245; NP_000617.1; NM_000626_1
[00104] (16) FcRH2 (IFGP4,1RTA4, SPAP1A (SH2 domain containing
phosphatase
anchor protein la), SPAP1B, SPAP1C, Genbank accession no. NM 030764,
AY358130);
Genome Res. 13 (10):2265-2270 (2003), lmmunogenetics 54 (2):87-95 (2002),
Blood 99
(8):2662-2669 (2002), Proc. Natl. Acad. Sci. U.S.A. 98 (17):9772-9777 (2001),
Xu, M.J., et
al (2001) Biochem. Biophys. Res. Commun. 280 (3):768-775; W02004016225 (Claim
2);
W02003077836; W0200138490 (Claim 5; Fig 18D-1-18D-2); W02003097803 (Claim 12);
W02003089624 (Claim 25); Cross-references: MIM:606509; NP_110391.2;
NM_030764_1
[00105] (17) HER2 (ErbB2, Genbank accession no. M11730); Coussens L.,
et al
Science (1985) 230(4730):1132-1139); Yamamoto T., et al Nature 319, 230-234,
1986;
Semba K., et al Proc. Natl. Acad. Sci. U.S.A. 82, 6497-6501, 1985; Swiercz
J.M., et al J. Cell
Biol. 165, 869-880, 2004; Kuhns J.J., et al J. Biol. Chem. 274, 36422-36427,
1999; Cho H.-
S., et al Nature 421, 756-760, 2003; Ehsani A., et al (1993) Genomics 15, 426-
429;
W02004048938 (Example 2); W02004027049 (Fig 11); W02004009622; W02003081210;
W02003089904 (Claim 9); W02003016475 (Claim 1); US2003118592; W02003008537
(Claim 1); W02003055439 (Claim 29; Fig 1A-B); W02003025228 (Claim 37; Fig 5C);
W0200222636 (Example 13; Page 95-107); W0200212341 (Claim 68; Fig 7);
W0200213847 (Page 71-74); W0200214503 (Page 114-117); W0200153463 (Claim 2;
Page 41-46); W0200141787 (Page 15); W0200044899 (Claim 52; Fig 7); W0200020579
(Claim 3; Fig 2); US5869445 (Claim 3; Col 31-38); W09630514 (Claim 2; Page 56-
61);
EP1439393 (Claim 7); W02004043361 (Claim 7); W02004022709; W0200100244
(Example 3; Fig 4); Accession: P04626; EMBL; M11767; AAA35808.1. EMBL; M11761;
AAA35808.1
[00106] (18) NCA (CEACAM6, Genbank accession no. M18728); Barnett T.,
et al
Genomics 3, 59-66, 1988; Tawaragi Y., et al Biochem. Biophys. Res. Commun.
150, 89-96,
1988; Strausberg R.L., et al Proc. Natl. Acad. Sci. U.S.A. 99:16899-16903,
2002;
W02004063709; EP1439393 (Claim 7); W02004044178 (Example 4); W02004031238;
W02003042661 (Claim 12); W0200278524 (Example 2); W0200286443 (Claim 27; Page
427); W0200260317 (Claim 2); Accession: P40199; Q14920; EMBL; M29541;
AAA59915.1. EMBL; M18728
[00107] (19) MDP (DPEP1, Genbank accession no. BC017023); Proc. Natl.
Acad. Sci.
U.S.A. 99 (26):16899-16903 (2002)); W02003016475 (Claim 1); W0200264798 (Claim
33;
Page 85-87); JP05003790 (Fig 6-8); W09946284 (Fig 9); Cross-references:
MIM:179780;
Date Recue/Date Received 2023-11-15
AAH17023.1; BC017023_1
[00108] (20) IL20Ra (IL20Ra, ZCYTOR7, Genbank accession no. AF184971);
Clark
H.F., et al Genome Res. 13, 2265-2270, 2003; Mungall A.J., et al Nature 425,
805-811, 2003;
Blumberg H., et al Cell 104, 9-19, 2001; Dumoutier L., et al J. Immunol. 167,
3545-3549,
2001; Parrish-Novak J., et at J. Biol. Chem. 277, 47517-47523, 2002; Pletnev
S., et al (2003)
Biochemistry 42:12617-12624; Sheikh F., et al (2004) J. Immunol. 172, 2006-
2010;
EP1394274 (Example 11); US2004005320 (Example 5); W02003029262 (Page 74-75);
W02003002717 (Claim 2; Page 63); W0200222153 (Page 45-47); US2002042366 (Page
20-
21); W0200146261 (Page 57-59); W0200146232 (Page 63-65); W09837193 (Claim 1;
Page
55-59); Accession: Q9UHF4; Q6UWA9; Q96SH8; EMBL; AF184971; AAF01320.1.
[00109] (21) Brevican (BCAN, BEHAB, Genbank accession no. AF229053);
Gary
S.C., et al Gene 256, 139-147, 2000; Clark H.F., et al Genome Res. 13, 2265-
2270, 2003;
Strausberg R.L., et al Proc. Natl. Acad. Sci. U.S.A. 99, 16899-16903, 2002;
US2003186372
(Claim 11); US2003186373 (Claim 11); US2003119131 (Claim 1; Fig 52);
US2003119122
(Claim 1; Fig 52); US2003119126 (Claim 1); US2003119121 (Claim 1; Fig 52);
US2003119129 (Claim 1); US2003119130 (Claim 1); US2003119128 (Claim 1; Fig
52);
US2003119125 (Claim 1); W02003016475 (Claim 1); W0200202634 (Claim 1)
[00110] (22) EphB2R (DRT, ERK, Hek5, EPHT3, Tyro5, Genbank accession
no.
NM 004442); Chan,J. and Watt, V.M., Oncogene 6 (6), 1057-1061 (1991) Oncogene
10
(5):897-905 (1995), Annu. Rev. Neurosci. 21:309-345 (1998), Int. Rev. Cytol.
196:177-244
(2000)); W02003042661 (Claim 12); W0200053216 (Claim 1; Page 41); W02004065576
(Claim 1); W02004020583 (Claim 9); W02003004529 (Page 128-132); W0200053216
(Claim 1; Page 42); Cross-references: MIM:600997; NP_004433.2; NM_004442_1
[00111] (23) ASLG659 (B7h, Genbank accession no. AX092328);
US20040101899
(Claim 2); W02003104399 (Claim 11); W02004000221 (Fig 3); US2003165504 (Claim
1);
US2003124140 (Example 2); US2003065143 (Fig 60); W02002102235 (Claim 13; Page
299); US2003091580 (Example 2); W0200210187 (Claim 6; Fig 10); W0200194641
(Claim
12; Fig 7b); W0200202624 (Claim 13; Fig 1A-1B); US2002034749 (Claim 54; Page
45-46);
W0200206317 (Example 2; Page 320-321, Claim 34; Page 321-322); W0200271928
(Page
468-469); W0200202587 (Example 1; Fig 1); W0200140269 (Example 3; Pages 190-
192);
W0200036107 (Example 2; Page 205-207); W02004053079 (Claim 12); W02003004989
(Claim 1); W0200271928 (Page 233-234, 452-453); WO 0116318
[00112] (24) PSCA (Prostate stem cell antigen precursor, Genbank
accession no.
26
Date Recue/Date Received 2023-11-15
AJ297436); Reiter R.E., et al Proc. Natl. Acad. Sci. U.S.A. 95, 1735-1740,
1998; Gu Z., et at
Oncogenc 19, 1288-1296, 2000; Biochcm. Biophys. Res. Commun. (2000) 275(3):783-
788;
W02004022709; EP1394274 (Example 11); US2004018553 (Claim 17); W02003008537
(Claim 1); W0200281646 (Claim 1; Page 164); W02003003906 (Claim 10; Page 288);
W0200140309 (Example 1; Fig 17); US2001055751 (Example 1; Fig lb); W0200032752
(Claim 18; Fig 1); W09851805 (Claim 17; Page 97); W09851824 (Claim 10; Page
94);
W09840403 (Claim 2; Fig 1B); Accession: 043653; EMBL; AF043498; AAC39607.1
[00113] (25) GEDA (Genbank accession No. AY260763); AAP14954 lipoma
HMGIC
fusion-partner-like protein /pid=AAP14954.1 - Homo sapiens (human);
W02003054152
(Claim 20); W02003000842 (Claim 1); W02003023013 (Example 3, Claim 20);
US2003194704 (Claim 45); Cross-references: GI:30102449; AAP14954.1; AY260763_1
[00114] (26) BAFF-R (B cell -activating factor receptor, BLyS receptor
3, BR3,
Genbank accession No. AF116456); BAFF receptor /pid=NP_443177.1 - Homo
sapiens:
Thompson, J.S., et al Science 293 (5537), 2108-2111 (2001); W02004058309;
W02004011611; W02003045422 (Example; Page 32-33); W02003014294 (Claim 35; Fig
6B); W02003035846 (Claim 70; Page 615-616); W0200294852 (Col 136-137);
W0200238766 (Claim 3; Page 133); W0200224909 (Example 3; Fig 3); Cross-
references:
M1M:606269; NP 443177.1; NM_052945_1; AF132600
[00115] (27) CD22 (B-cell receptor CD22-B isoform, BL-CAM, Lyb-8, Lyb8,
SIGLEC-2, FLJ22814, Genbank accession No. AK026467); Wilson et al (1991) J.
Exp. Med.
173:137-146; W02003072036 (Claim 1; Fig 1); Cross-references: MIM:107266;
NP 001762.1; NM 001771 1
[00116] (28) CD79a (CD79A, CD79a, immunoglobulin-associated alpha, a B
cell-
specific protein that covalently interacts with 1g beta (CD79B) and forms a
complex on the
surface with Ig M molecules, transduces a signal involved in B-cell
differentiation), pI: 4.84,
MW: 25028 TM: 2 [P] Gene Chromosome: 19q13.2, Genbank accession No.
NP 001774.10); W02003088808, US20030228319; W02003062401 (claim 9);
US2002150573 (claim 4, pages 13-14); W09958658 (claim 13, Fig 16); W09207574
(Fig
1); US5644033; Ha et al (1992) J. Immunol. 148(5):1526-1531; Mueller et al
(1992) Eur. J.
Biochem. 22:1621-1625; Hashimoto et al (1994) Immunogenetics 40(4):287-295;
Preud'homme et al (1992) Clin. Exp. Immunol. 90(1):141-146; Yu et al (1992) J.
Immunol.
148(2) 633-637; Sakaguchi et al (1988) EMBO J. 7(11):3457-3464
[00117] (29) CXCR5 (Burkitt's lymphoma receptor 1, a G protein-coupled
receptor
27
Date Recue/Date Received 2023-11-15
that is activated by the CXCL13 chemokine, functions in lymphocyte migration
and humoral
defense, plays a role in HIV-2 infection and perhaps development of AIDS,
lymphoma,
myeloma, and leukemia); 372 aa, pl: 8.54 MW: 41959 TM: 7 [P] Gene Chromosome:
11q23.3, Genbank accession No. NP_001707.1); W02004040000; W02004015426;
US2003105292 (Example 2); US6555339 (Example 2); W0200261087 (Fig 1);
W0200157188 (Claim 20, page 269); W0200172830 (pages 12-13); W0200022129
(Example 1, pages 152-153, Example 2, pages 254-256); W09928468 (claim 1, page
38);
US5440021 (Example 2, col 49-52); W09428931 (pages 56-58); W09217497 (claim 7,
Fig
5); Dobner et al (1992) Eur. J. Immunol. 22:2795-2799; Barella et al (1995)
Biochem. J.
309:773-779
[00118] (30) HLA-DOB (Beta subunit of MUG class II molecule (Ia
antigen) that
binds peptides and presents them to CD4+ T lymphocytes); 273 aa, pl: 6.56, MW:
30820.TM: 1 [P] Gene Chromosome: 6p21.3, Genbank accession No. NP_002111.1);
Tonnelle et al (1985) EMBO J. 4(11):2839-2847; Jonsson et al (1989)
Immunogenetics
29(6):411-413; Beck et al (1992) J. Mol. Biol. 228:433-441; Strausberg eta!
(2002) Proc.
Natl. Acad. Sci USA 99:16899-16903; Servenius eta! (1987) J. Biol. Chem.
262:8759-8766;
Beck et al (1996) J. Mol. Biol. 255:1-13; Narusc et al (2002) Tissue Antigens
59:512-519;
W09958658 (claim 13, Fig 15); US6153408 (Col 35-38); US5976551 (col 168-170);
US6011146 (col 145-146); Kasahara et al (1989) Immunogenetics 30(1):66-68;
Larhammar
et al (1985) J. Biol. Chem. 260(26):14111-14119
[00119] (31) P2X5 (Purinergic receptor P2X ligand-gated ion channel 5,
an ion
channel gated by extracellular ATP, may be involved in synaptic transmission
and
neurogenesis, deficiency may contribute to the pathophysiology of idiopathic
detrusor
instability); 422 aa), pl: 7.63, MW: 47206 TM: 1 [P] Gene Chromosome: 17p13.3,
Genbank
accession No. NP 002552.2); Le eta! (1997) FEBS Lett. 418(1-2):195-199;
W02004047749; W02003072035 (claim 10); Touchman et al (2000) Genome Res.
10:165-
173; W0200222660 (claim 20); W02003093444 (claim 1); W02003087768 (claim 1);
W02003029277 (page 82)
[00120] (32) CD72 (B-cell differentiation antigen CD72, Lyb-2); 359 aa,
pl: 8.66,
MW: 40225, TM: 1 [P] Gene Chromosome: 9p13.3, Genbank accession No.
NP_001773.1);
W02004042346 (claim 65); W02003026493 (pages 51-52, 57-58); W0200075655 (pages
105-106); Von Hoegen et al (1990) J. lmmunol. 144(12):4870-4877; Strausberg et
al (2002)
Proc. Natl. Acad. Sci USA 99:16899-16903.
[00121] (33) LY64 (Lymphocyte antigen 64 (RP105), type I membrane
protein of the
28
Date Recue/Date Received 2023-11-15
leucine rich repeat (LRR) family, regulates B-cell activation and apoptosis,
loss of function is
associated with increased disease activity in patients with systemic lupus
erythematosis); 661
aa, p1: 6.20, MW: 74147 TM: 1 [P] Gene Chromosome: 5q12, Genbank accession No.
NP 005573.1); US2002193567; W09707198 (claim 11, pages 39-42); Miura et al
(1996)
Genomics 38(3):299-304; Miura et al (1998) Blood 92:2815-2822; W02003083047;
W09744452 (claim 8, pages 57-61); W0200012130 (pages 24-26)
[00122] (34) FcRH1 (Fe receptor-like protein 1, a putative receptor for
the
immunoglobulin Fe domain that contains C2 type Ig-like and ITAM domains, may
have a
role in B-lymphocyte differentiation); 429 aa, pI: 5.28, MW: 46925 TM: 1 [P]
Gene
Chromosome: 1q21-1q22, Genbank accession No. NP 443170.1); W02003077836;
W0200138490 (claim 6, Fig 18E-1-18-E-2); Davis et al (2001) Proc. Natl. Acad.
Sci USA
98(17):9772-9777; W02003089624 (claim 8); EP1347046 (claim 1); W02003089624
(claim
7)
[00123] (35) IRTA2 (Immunoglobulin superfamily receptor translocation
associated 2,
a putative immunoreceptor with possible roles in B cell development and
lymphomagenesis;
deregulation of the gene by translocation occurs in some B cell malignancies);
977 aa, pI:
6.88, MW: 106468, TM: 1 [P] Gene Chromosome: 1q21, Genbank accession No.
Human:AF343662, AF343663, AF343664, AF343665, AF369794, AF397453, AK090423,
AK090475, AL834187, AY358085; Mouse:AK089756, AY158090, AY506558;
NP 112571.1; W02003024392 (claim 2, Fig 97); Nakayama et al (2000) Biochem.
Biophys.
Res. Commun. 277(1):124-127; W02003077836; W0200138490 (claim 3, Fig 18B-1-18B-
2)
[00124] (36) TENB2 (TMEFF2, tomoregulin, TPEF, 1-IPP1, TR, putative
transmembrane proteoglycan, related to the EGF/heregulin family of growth
factors and
follistatin); 374 aa, NCBI Accession: AAD55776, AAF91397, AAG49451, NCBI
RefSeq:
NP 057276; NCBI Gene: 23671; OMIM: 605734; SwissProt Q9UIK5; Genbank accession
No. AF179274; AY358907, CAF85723, CQ782436; W02004074320; JP2004113151;
W02003042661; W02003009814; EP1295944 (pages 69-70); W0200230268 (page 329);
W0200190304; US2004249130; US2004022727; W02004063355; US2004197325;
U52003232350; U52004005563; US2003124579; Hone et al (2000) Genomics 67:146-
152;
Uchida ct al (1999) Biochcm. Biophys. Res. Commun. 266:593-602; Liang et al
(2000)
Cancer Res. 60:4907-12; Glynne-Jones et al (2001) Int J Cancer. Oct 15;
94(2):178-84.
[00125] The parent antibody may also be a fusion protein comprising an
albumin-
binding peptide (ABP) sequence (Dennis et al. (2002) "Albumin Binding As A
General
29
Date Recue/Date Received 2023-11-15
Strategy For Improving The Pharmacokinetics Of Proteins" J Biol Chem.
277:35035-35043;
WO 01/45746). Antibodies of the invention include fusion proteins with ABP
sequences
taught by: (i) Dennis et al (2002) J Biol Chem. 277:35035-35043 at Tables III
and IV, page
35038; (ii) US 20040001827 at [0076]; and (iii) WO 01/45746 at pages 12-13,
[90126] MUTACIENESIS
[00127] DNA encoding an amino acid sequence variant of the starting
polypeptide is
prepared by a variety of methods known in the art. These methods include, but
are not
limited to, preparation by site-directed (or oligonucleotide-mediated)
mutagenesis, PCR
mutagenesis, and cassette mutagenesis of an earlier prepared DNA encoding the
polypeptide.
Variants of recombinant antibodies may be constructed also by restriction
fragment
manipulation or by overlap extension PCR with synthetic oligonucleotides.
Mutagenic
primers encode the cysteine codon replacement(s). Standard mutagenesis
techniques can be
employed to generate DNA encoding such mutant cysteine engineered antibodies.
General
guidance can be found in Sambrook et al Molecular Cloning, A Laboratory
Manual, Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989; and Ausubel et
al Current
Protocols in Molecular Biology, Greene Publishing and Wiley-Interscience, New
York, N.Y.,
1993.
[00128] Site-directed mutagenesis is one method for preparing
substitution variants,
i.e. mutant proteins. This technique is well known in the art (see for
example, Carter (1985)
et al Nucleic Acids Res. 13:4431-4443; Ho et al (1989) Gene (Amst.) 77:51-59;
and Kunkel
et at (1987) Proc. Natl. Acad. Sci. USA 82:488). Briefly, in carrying out site-
directed
mutagenesis of DNA, the starting DNA is altered by first hybridizing an
oligonucleotide
encoding the desired mutation to a single strand of such starting DNA. After
hybridization, a
DNA polymerase is used to synthesize an entire second strand, using the
hybridized
oligonucleotide as a primer, and using the single strand of the starting DNA
as a template.
Thus, the oligonucleotide encoding the desired mutation is incorporated in the
resulting
double-stranded DNA. Site-directed mutagenesis may be carried out within the
gene
expressing the protein to be mutagenized in an expression plasmid and the
resulting plasmid
may be sequenced to confirm the introduction of the desired cysteine
replacement mutations
(Liu et al (1998) J. Biol. Chem. 273:20252-20260). Site-directed of protocols
and formats,
including those commercially available, e.g. QuikChange Multi Site-Directed
Mutagenesis
Kit (Stratagene, La Jolla, CA).
Date Recue/Date Received 2023-11-15
[00129] PCR mutagenesis is also suitable for making amino acid sequence
variants of
the starting polypeptide. See Higuchi, (1990) in PCR Protocols, pp.177-183,
Academic
Press; Ito et al (1991) Gene 102:67-70; Bernhard et al (1994) Bioconjugate
Chem. 5:126-132;
and Vallette et al (1989) Nuc. Acids Res. 17:723-733. Briefly, when small
amounts of
template DNA are used as starting material in a PCR, primers that differ
slightly in sequence
from the corresponding region in a template DNA can be used to generate
relatively large
quantities of a specific DNA fragment that differs from the template sequence
only at the
positions where the primers differ from the template.
[00130] Another method for preparing variants, cassette mutagenesis, is
based on the
technique described by Wells et at (1985) Gene 34:315-323. The starting
material is the
plasmid (or other vector) comprising the starting polypeptide DNA to be
mutated. The
codon(s) in the starting DNA to be mutated are identified. There must be a
unique restriction
endonuclease site on each side of the identified mutation site(s). If no such
restriction sites
exist, they may be generated using the above described oligonucleotide-
mediated
mutagenesis method to introduce them at appropriate locations in the starting
polypeptide
DNA. The plasmid DNA is cut at these sites to linearize it. A double-stranded
oligonucleotide encoding the sequence of the DNA between the restriction sites
but
containing the desired mutation(s) is synthesized using standard procedures,
wherein the two
strands of the oligonucleotide are synthesized separately and then hybridized
together using
standard techniques. Oligonucleotides are prepared by the phosphoramidite
synthesis method
(US 4415732; US 4458066; Beaucage, S. and Iyer, R. (1992) "Advances in the
synthesis of
oligonucleotides by the phosphoramidite approach", Tetrahedron 48:2223-2311).
This
double-stranded oligonucleotide is referred to as the cassette. This cassette
is designed to
have 5' and 3' ends that are compatible with the ends of the linearized
plasmid, such that it
can be directly ligated to the plasmid. This plasmid now contains the mutated
DNA
sequence. Mutant DNA containing the encoded cysteine replacements can be
confirmed by
DNA sequencing.
[00131] Single mutations are also generated by oligonucleotide directed
mutagenesis
using double stranded plasmid DNA as template by PCR based mutagenesis
(Sambrook and
Russel, (2001) Molecular Cloning: A Laboratory Manual, 3rd edition; Zoller et
al (1983)
Methods Enzymol. 100:468-500; Zoller, M.J. and Smith, M. (1982) Nucl. Acids
Res.
10:6487-6500).
[00132] In the present invention, hu4D5Fabv8 displayed on M13 phage
(Gerstner et al
(2002) "Sequence Plasticity In The Antigen-Binding Site Of A Therapeutic Anti-
HER2
31
Date Recue/Date Received 2023-11-15
Antibody", J Mol Biol. 321:851-62) was used for experiments as a model system.
Cysteine
mutations were introduced in hu4D5Fabv8-phage, hu4D5Fabv8, and ABP-hu4D5Fabv8
constructs. The hu4D5-ThioFab-Phage preps were carried out using the
polyethylene glycol
(PEG) precipitation method as described earlier (Lowman, Henry B. (1998)
Methods in
Molecular Biology (Totowa, New Jersey) 87 (Combinatorial Peptide Library
Protocols) 249-
264).
[00133] PHESELECTOR ASSAY
[00134] The PHESELECTOR (Phage ELISA for Selection of Reactive Thiols)
assay
allows for detection of reactive cysteine groups in antibodies in an ELISA
phage format. The
process of coating the protein (e.g. antibody) of interest on well surfaces,
followed incubation
with phage particles and then HRP labeled secondary antibody with absorbance
detection is
detailed in Example 2. Mutant proteins displayed on phage may be screened in a
rapid,
robust, and high-throughput manner. Libraries of cysteine engineered
antibodies can be
produced and subjected to binding selection using the same approach to
identify
appropriately reactive sites of free Cys incorporation from random protein-
phage libraries of
antibodies or other proteins. This technique includes reacting cysteine mutant
proteins
displayed on phage with an affinity reagent or reporter group which is also
thiol-reactive.
Figure 8 illustrates the PHESELECTOR Assay by a schematic representation
depicting the
binding of Fab or ThioFab to HER2 (top) and biotinylated ThioFab to
streptavidin (bottom).
[00135] PROTEIN EXPRESSION AND PURIFICATION
[00136] DNA encoding the cysteine engineered antibodies is readily
isolated and
sequenced using conventional procedures (e.g., by using oligonucleotide probes
that are
capable of binding specifically to genes encoding the heavy and light chains
of murine
antibodies). The hybridoma cells serve as a source of such DNA. Once isolated,
the DNA
may be placed into expression vectors, which are then transfected into host
cells such as E.
coli cells, simian COS cells, Chinese Hamster Ovary (CHO) cells, or other
mammalian host
cells, such as myeloma cells (US 5807715; US 2005/0048572; US 2004/0229310)
that do not
otherwise produce the antibody protein, to obtain the synthesis of monoclonal
antibodies in
the recombinant host cells. The yields of hu4D5Fabv8 cysteine engineered
antibodies were
similar to wild type hu4D5Fabv8. Review articles on recombinant expression in
bacteria of
DNA encoding the antibody include Skerra et al (1993) Curr. Opinion in
Immunol. 5:256-
262 and Pliickthun (1992) Immunol. Revs. 130:151-188.
32
Date Recue/Date Received 2023-11-15
[00137] After design and selection, cysteine engineered antibodies,
e.g. ThioFabs, with
highly reactive unpaired Cys residues, may be produced by: (i) expression in a
bacterial, e.g.
E. coli, system or a mammalian cell culture system (WO 01/00245), e.g. Chinese
Hamster
Ovary cells (CHO); and (ii) purification using common protein purification
techniques
(Lowman et al (1991) J. Biol. Chem. 266(17):10982-10988).
[00138] ThioFabs were expressed upon induction in 34B8, a non-
suppressor E. coil
strain (Baca et al (1997) Journal Biological Chemistry 272(16):10678-84). See
Example 3a.
The harvested cell pellet was resuspended in PBS (phosphate buffered saline),
total cell lysis
was performed by passing through a microfluidizer and the ThioFabs were
purified by
affinity chromatography with protein G SEPHAROSETM (Amersham). ThioFabs were
conjugated with biotin-PEO-maleimide as described above and the biotinylated-
ThioFabs
were further purified by Superdex-200TM (Amersham) gel filtration
chromatography, which
eliminated the free biotin-PEO-maleimide and the oligomeric fraction of
ThioFabs.
[00139] MASS SPECTROSCOPY ANALYSIS
[00140] Liquid chromatography electrospray ionization mass
spectrometric (LC-ESI-
MS) analysis was employed for the accurate molecular weight determination of
biotin
conjugated Fab (Cole, R.B. Electro Spray Ionization Mass Spectrometry:
Fundamentals,
Instrumentation And Applications. (1997) Wiley, New York). The amino acid
sequence of
biotinylated hu4D5Fabv8 (A121C) peptide was determined by tryptic digestion
followed by
LC-ESI-Tandem MS analysis (Table 4, Example 3b).
[00141] The antibody Fab fragment hu4D5Fabv8 contains about 445 amino
acid
residues, including 10 Cys residues (five on the light and five on the heavy
chain). The high-
resolution structure of the humanized 4D5 variable fragment (Fv4D5) has been
established,
see: Eigenbrot et al "X-Ray Structures Of The Antigen-Binding Domains From
Three
Variants Of Humanized Anti-P185her2 Antibody 4D5 And Comparison With Molecular
Modeling" (1993) J Mol Biol. 229:969-995). All the Cys residues are present in
the form of
disulfide bonds, therefore these residues do not have any reactive thiol
groups to conjugate
with drug-maleimide (unless treated with a reducing agent). Hence, the newly
engineered Cys
residue, can remain unpaired, and able to react with, i.e. conjugate to, an
electrophilic linker
reagent or drug-linker intermediate, such as a drug-maleimide. Figure lA shows
a three-
dimensional representation of the hu4D5Fabv8 antibody fragment derived by X-
ray crystal
coordinates. The structure positions of the engineered Cys residues of the
heavy and light
chains arc numbered according to a sequential numbering system. This
sequential numbering
33
Date Recue/Date Received 2023-11-15
system is correlated to the Kabat numbering system (Kabat et al., (1991)
Sequences of
Proteins of Immunological Interest, 5th Ed. Public Health Service, National
Institutes of
Health, Bethesda, MD) for the 4d5v7fabH variant of trastuzumab according to
Figure 1B
which shows the sequential numbering scheme (top row), starting at the N-
terminus, differs
from the Kabat numbering scheme (bottom row) by insertions noted by a,b,c.
Using the
Kabat numbering system, the actual linear amino acid sequence may contain
fewer or
additional amino acids corresponding to a shortening of, or insertion into, a
FR or CDR of the
variable domain. The cysteine engineered heavy chain variant sites are
identified by the
sequential numbering and Kabat numbering schemes in the following chart:
4D5Fab Heavy chain variants Sequential Numbering Kabat Numbering
A40C Ala-40 Ala-40
A88C Ala-88 Ala-84
S119C Ser-119 Ser-112
S120C Ser-120 Ser-113
A121C Ala-121 Ala-114
S122C Ser-122 Ser-1 15
A175C Ala-175 Ala-168
1001421 M13 phagemid-Cys mutant Fabs (Figures 3A and 3B) can be rapidly
screened
compared to Fab proteins. Phagemid-ThioFab binding to antigen and to
streptavidin can be
tested by coating HER2 and streptavidin, respectively, onto ELISA plates
followed by
probing with anti-Fab-HRP (Horse radish peroxidase) as described in Example 2
and
depicted in Figure 8. This method allowed simultaneous monitoring of the
effect on the
antigen binding and the reactivity of the thiol group by the engineered Cys
residue/conjugated biotin molecule. Also, the method can be applied to screen
the reactive
thiol groups for any protein displayed on M13 phage. Conjugated or
unconjugated
phagemid-ThioFabs are purified by simple PEG precipitation.
1001431 The antigen-binding fragment of humanized 4D5 (hu4D5Fab) is
well
expressed in E. Coli and has been displayed on bacteriophage (Garrard et al
(1993) Gene
128:103-109). The antibody Fab fragment hu4D5Fabv8 was displayed on M13 phage
as a
model system in the ELISA based assay to probe thiol reactivity. Figure 8 is a
graphical
representation of the PHESELECTOR assay, depicting binding of a biotinylated
ThioFab
phage and an anti-phage HRP antibody to HER2 (top) and Streptavidin (bottom).
Five amino
acid residues (L-Ala43, H-Ala40, H-Ser119, H-Ala121 and H-Ser122) were
initially selected
from crystal structure information as remote from the antigen binding surface
(Eigenbrot et
34
Date Recue/Date Received 2023-11-15
al. (1993) J Mol Biol. 229:969-995). The Protein Database X-ray crystal
structure was
designated as 1FVC. Cys residues were engineered at these positions by site
directed
mutagenesis. ThioFab-phage preparations were isolated and reacted with the
biotinylation
reagent.
[00144] Biotin conjugated and unconjugated variants were tested for
HER2 and
streptavidin binding using an ELISA based PHESELECTOR assay (Figure 8, Example
2)
with an HRP (horseradish peroxidase)-conjugated anti-phage antibody. The
interaction of
non-biotinylated phage-hu4D5Fabv8 (Figure 2A) and biotinylated phage-
hu4D5Fabv8
(Figure 2B) with BSA (open box), HER2 (grey box) or streptavidin (solid box)
were
monitored through anti-M13-horseradish peroxidase (HRP) antibody by developing
a
standard HRP reaction and measuring absorbance at 450 nm. The absorbance
produced by
turnover of a colorimetric substrate was measured at 450 nm. The reactivity of
ThioFab with
HER2 measures antigen binding. The reactivity of ThioFab with streptavidin
measures the
extent of biotinylation. The reactivity of ThioFab with BSA is a negative
control for
nonspecific interaction. As seen in Figure 2A, all the ThioFab-phage variants
have similar
binding to HER2 compared to that of wild type hu4D5Fabv8-phage. Furthermore,
conjugation with biotin did not interfere in the ThioFab binding to HER2
(Figure 2B).
[001451 Surprisingly and unexpectedly, the ThioFabs-phage samples
showed varying
levels of streptavidin binding activity. From all the tested phage-ThioFabs,
the A121C
cysteine engineered antibody exhibited maximal thiol reactivity. Even though
wild type
hu4D5Fabv8-phage was incubated with the same amounts of biotin-maleimide,
these phage
had little streptavidin binding indicating that preexisting cysteine residues
(involved in
disulfide bond formation) from the hu4D5Fabv8 and M13 phage coat proteins did
not
interfere with the site-specific conjugation of biotin-maleimide. These
results demonstrate
that the phage ELISA assay can be used successfully to screen reactive thiol
groups on the
Fab surface.
[00146] The PHESELECTOR assay allows screening of reactive thiol groups
in
antibodies. Identification of the A121C variant by this method is exemplary.
The entire Fab
molecule may be effectively searched to identify more ThioFab variants with
reactive thiol
groups. A parameter, fractional surface accessibility, was employed to
identify and
quantitate the accessibility of solvent to the amino acid residues in a
polypeptide. The
surface accessibility can be expressed as the surface area (A2) that can be
contacted by a
solvent molecule, e.g. water. The occupied space of water is approximated as a
1.4 A radius
sphere. Software is freely available or licensable (Secretary to CCP4,
Daresbury Laboratory,
Date Recue/Date Received 2023-11-15
Warrington, WA4 4AD, United Kingdom, Fax: (+44) 1925 603825, or by interne:
www.ccp4.ac.uk/dist/html/INDEX.html) as the CCP4 Suite of crystallography
programs
which employ algorithms to calculate the surface accessibility of each amino
acid of a protein
with known x-ray crystallography derived coordinates ("The CCP4 Suite:
Programs for
Protein Crystallography" (1994) Acta. Cryst. D50:760-763). Two exemplary
software
modules that perform surface accessibility calculations are "AREAIMOL" and
"SURFACE",
based on the algorithms of B.Lee and F.M.Richards (1971) J.Mol.Biol. 55:379-
400.
AREAIMOL defines the solvent accessible surface of a protein as the locus of
the centre of a
probe sphere (representing a solvent molecule) as it rolls over the Van der
Waals surface of
the protein. AREAIMOL calculates the solvent accessible surface area by
generating surface
points on an extended sphere about each atom (at a distance from the atom
centre equal to the
sum of the atom and probe radii), and eliminating those that lie within
equivalent spheres
associated with neighboring atoms. AREAIMOL finds the solvent accessible area
of atoms
in a PDB coordinate file, and summarizes the accessible area by residue, by
chain and for the
whole molecule. Accessible areas (or area differences) for individual atoms
can be written to
a pseudo-PDB output file. AREAIMOL assumes a single radius for each element,
and only
recognizes a limited number of different elements. Unknown atom types (i.e.
those not in
AREA1MOL's internal database) will be assigned the default radius of 1.8 A.
The list of
recognized atoms is:
Atom Atomic no. Van der Waals rad. (A)
6 1.80
7 1.65
0 8 1.60
Mg 12 1.60
16 1.85
1.90
Cl 17 1.80
Co 27 1.80
1001471 AREAIMOL and SURFACE report absolute accessibilities, i.e. the
number of
square Angstroms (A). Fractional surface accessibility is calculated by
reference to a
standard state relevant for an amino acid within a polypeptide. The reference
state is
trip eptide Gly-X-Gly, where X is the amino acid of interest, and the
reference state should be
an 'extended' conformation, i.e. like those in beta-strands. The extended
conformation
maximizes the accessibility of X. A calculated accessible area is divided by
the accessible
36
Date Recue/Date Received 2023-11-15
area in a Gly-X-Gly tripeptide reference state and reports the quotient, which
is the fractional
accessibility. Percent accessibility is fractional accessibility multiplied by
100.
[00148] Another exemplary algorithm for calculating surface
accessibility is based on
the SOLV module of the program xsae (Broger, C., F. Hoffman-LaRoche, Basel)
which
calculates fractional accessibility of an amino acid residue to a water sphere
based on the X-
ray coordinates of the polypeptide.
[00149] The fractional surface accessibility for every amino acid in
hu4D5Fabv7 was
calculated using the crystal structure information (Eigenbrot et al. (1993) J
Mol Biol.
229:969-995; US 7521541). The following two criteria were applied to identify
the residues
of hu4D5Fabv8 that can be engineered to replace with Cys residues:
[00150] 1. Amino acid residues that are completely buried are
eliminated, i.e. less than
10% fractional surface accessibility. There are 134 (light chain) and 151
(heavy chain)
residues of hu4D5Fabv8 that are more than 10% accessible (fractional surface
accessibility).
The top ten most accessible Ser, Ala and Val residues were selected due to
their close
structural similarity to Cys over other amino acids, introducing only minimal
structural
constraints in the antibody by newly engineered Cys. Other cysteine
replacement sites can
also be screened, and may be useful for conjugation.
[00151] 2. Residues are sorted based on their role in functional and
structural
interactions of Fab. The residues which are not involved in antigen
interactions and distant
from the existing disulfide bonds were further selected. The newly engineered
Cys residues
should be distinct from, and not interfere with, antigen binding nor mispair
with cysteines
involved in disulfide bond formation.
[00152] Thiol reactivity may be generalized to any antibody where
substitution of
amino acids with reactive cysteine amino acids may be made within the ranges
in the light
chain selected from: L-10 to L-20; L-38 to L-48; L-105 to L-115; L-139 to L-
149; L-163 to
L-173; and within the ranges in the heavy chain selected from: H-35 to H-45; H-
83 to H-93;
H-114 to H-127; and H-170 to LI-184, and in the Fe region within the ranges
selected from H-
268 to H-291; H-319 to H-344; H-370 to H-380; and H-395 to H-405.
[00153] Thiol reactivity may also be generalized to certain domains of
an antibody,
such as the light chain constant domain (CL) and heavy chain constant domains,
CHI, CH2
and CH3. Cysteine replacements resulting in thiol reactivity values of about
0.8 and higher
may be made in the heavy chain constant domains a, 6, E, y, and II of intact
antibodies: IgA,
IgD, IgE, IgG, and IgM, respectively, including the IgG subclasses: IgGl,
IgG2, IgG3, IgG4,
IgA, and IgA2.
37
Date Recue/Date Received 2023-11-15
[00154] It is evident from the crystal structure data that the selected
10 Cys mutants are
far away from the antigen-combining site, such as the interface with HER2 in
this case.
These mutants can be tested experimentally for indirect effects on functional
interactions.
The thiol reactivities of all the Cys Fab variants were measured and
calculated as described in
Examples 1 and 2, and presented in Table 1. The residues L-V15C, L-V110C, H-
A88C and
H-A121C have reactive and stable thiol groups (Figures 3A and 3B). Mutants
V15C,
V110C, A144C, S168C are light chain Cys variants. Mutants A88C, A121C, A175C,
S179C
are heavy chain Cys variants. It was surprising and unexpected that the sites
with high
fractional surface accessibility did not have the highest thiol reactivity as
calculated by the
PHESELECTOR assay (Table 1). In other words, fractional surface accessibility
(Figure 1A)
did not correlate with thiol reactivity (Table 1). In fact, the Cys residues
engineered at the
sites with moderate surface accessibility of 20% to 80% (Figure 4A), or
partially exposed
sites, like Ala or Val residues, exhibited better thiol reactivity, i.e. >0.6,
(Figure 3B, Table 1)
than the Cys introduced at Ser residues, thus necessitating the use of
PHESELECTOR assay
in the screening of thiol reactive sites since the crystal structure
information alone is not
sufficient to select these sites (Figure 3B and 4A).
[00155] Thiol reactivity data is shown in Figures 3A and 3B for amino
acid residues of
4D5 ThioFab Cys mutants: (3A) non-biotinylated (control) and (3B) biotinylated
phage-
ThioFabs. Reactive thiol groups on antibody/Fab surface were identified by
PHESELECTOR assay analyses for the interaction of non-biotinylated phage-
hu4D5Fabv8
(3A) and biotinylated phage-hu4D5Fabv8 (3B) with BSA (open box), HER2 (grey
box) or
streptavidin (solid box). The assay was carried out as described in Example 2.
Light chain
variants are on the left side and heavy chain variants are on the right side.
The binding of
non-biotinylated 4D5 ThioFab Cys mutants is low as expected, but strong
binding to HER2 is
retained. The ratio of binding to streptavidin and to HER2 of the biotinylated
4D5 ThioFab
Cys mutants gives the thiol reactivity values in Table 1. Background
absorbance at 450 urn
or small amounts of non-specific protein binding of the biotinylated 4D5
ThioFab Cys
mutants to BSA is also evident in Figure 3B. Fractional Surface Accessibility
values of the
selected amino acid residues that were replaced with a Cys residue are shown
in Figure 4A.
Fractional surface accessibility was calculated from the available hu4D5Fabv7
structure
(Eigenbrot et al. (1993) J Mol Biol. 229:969-995). The conformational
parameters of the
hu4D5Fabv7 and hu4D5Fabv8 structures are highly consistent and allow for
determination of
any correlation between fractional surface accessibility calculations of
hu4D5Fabv7 and thiol
reactivity of hu4D5Fabv8 cysteine mutants. The measured thiol reactivity of
phage ThioFab
38
Date Recue/Date Received 2023-11-15
Cys residues introduced at partially exposed residues (Ala or Val) have better
thiol reactivity
compared to the ones introduced at Scr residues (Table 1). It can be seen from
thc ThioFab
Cys mutants of Table 1 that there is little or no correlation between thio
reactivity values and
fractional surface accessibility.
[00156] Amino acids at positions L-15, L-43, L-110, L-144, L-168, H-40,
H-88, H-
119, H-121, H-122, H-175, and H-179 of an antibody may generally be mutated
(replaced)
with free cysteine amino acids. Ranges within about 5 amino acid residues on
each side of
these positions may also be replaced with free cysteine acids, i.e. L-10 to L-
20; L-38 to L-48;
L-105 to L-115; L-139 to L-149; L-163 to L-173; H-35 to H-45; H-83 to H-93; H-
114 to H-
127; and H-170 to H-184, as well as the ranges in the Fe region selected from
H-268 to H-
291; H-319 to H-344; H-370 to H-380; and H-395 to H-405, to yield the cysteine
engineered
antibodies of the invention.
Table 1. Thiol reactivity of phage-ThioFabs
Phage-ThioFab Thiol Fractional Surface
construct Reactivity* Accessibility (%)
hu4D5Fabv8-wt 0.125
L-V15C 0.934 52.46
L-A43C 0.385 26.80
L-V110C 0.850 44.84
L-A144C 0.373 23.65
L-S168C 0.514 79.68
H-A40C 0.450 21.97
H-A88C 0.914 51.60
H-S119C 0.680 18.88
H-A121C 0.925 33.05
H-S122C 0.720 72.87
H-A175C 0.19 23.80
H-S179C 0.446 99.48
L = ligjht chain, H = heavy chain, A = alanine, S = serine, V = valine, C =
cysteine
Thiol reactivity is measured as the ratio of 0D450. for streptavidin binding
to 0D450. for
HER2 (antibody) binding (Example 2). Thiol reactivity value of 1 indicates
complete
biotinylation of the cysteine thiol.
[00157] Two Cys variants from light chain (L-V15C and L-V110C) and two
from
heavy chain (H-A88C and 1-1-A121C) were selected for further analysis as these
variants
39
Date Recue/Date Received 2023-11-15
showed the highest thiol reactivity (Table 1).
[00158] Unlike phage purification, Fab preparation may require 2-3
days, depending
on the scale of production. During this time, thiol groups may lose reactivity
due to oxidation.
To probe the stability of thiol groups on hu4D5Fabv8-phage, stability of the
thiol reactivity
of phage-thioFabs was measured (Figure 4B). After ThioFab-phage purification,
on day 1,
day 2 and day 4, all the samples were conjugated with biotin-PEO-maleimide and
probed
with phage ELISA assay (PHESELECTOR) to test HER2 and streptavidin binding. L-
V15C,
L-V110C, H-A88C and H-A121C retain significant amounts of thiol reactivity
compared to
other ThioFab variants (Figure 4B).
[00159] LABELLED CYSTEINE ENGINEERED ANTIBODIES
[00160] The cysteine engineered antibodies of the invention may be
conjugated with
any label moiety which can be covalently attached to the antibody through a
reactive cysteine
thiol group (Singh et al (2002) Anal. Biochem. 304:147-15; Harlow E. and Lane,
D. (1999)
Using Antibodies: A Laboratory Manual, Cold Springs Harbor Laboratory Press,
Cold Spring
Harbor, NY; Lundblad R.L. (1991) Chemical Reagents for Protein Modification,
2nd ed.
CRC Press, Boca Raton, FL). The attached label may function to: (i) provide a
detectable
signal; (ii) interact with a second label to modify the detectable signal
provided by the first or
second label, e.g. to give FRET (fluorescence resonance energy transfer);
(iii) stabilize
interactions or increase affinity of binding, with antigen or ligand; (iv)
affect mobility, e.g.
electrophoretic mobility or cell-permeability, by charge, hydrophobicity,
shape, or other
physical parameters, or (v) provide a capture moiety, to modulate ligand
affinity,
antibody/antigen binding, or ionic complexation.
[00161] Labelled cysteine engineered antibodies may be useful in
diagnostic assays,
e.g., for detecting expression of an antigen of interest in specific cells,
tissues, or serum. For
diagnostic applications, the antibody will typically be labeled with a
detectable moiety.
Numerous labels are available which can be generally grouped into the
following categories:
[00162] (a) Radioisotopes (radionuclides), such as 3H, tic, 14C,
18F, 32p, 35s, 154cu,
68Ga, 86y, "Zr, "Tc, 111111 12315124j, 1251, 1311,
I33Xe, I"Lu, 21'At, or 2I3Bi. Radioisotope
labelled antibodies are useful in receptor targeted imaging experiments. The
antibody can be
labeled with ligand reagents that bind, chelate or otherwise complex a
radioisotope metal
where the reagent is reactive with the engineered cysteine thiol of the
antibody, using the
techniques described in Current Protocols in Immunology, (1991) Volumes 1 and
2, Coligen
et al, Ed. Wiley-Interscience, New York, NY, Pubs.. Chelating ligands which
may complex a
Date Recue/Date Received 2023-11-15
metal ion include DOTA, DO __ l'P, DOTMA, DTPA and TETA (Macrocyclics, Dallas,
TX).
Radionuclides can be targetted via complexation with the antibody-drug
conjugates of the
invention (Wu et al (2005) Nature Biotechnology 23(9):1137-1146). DOTA-
maleimide
reagents react with the free cysteine amino acids of the cysteine engineered
antibodies and
provide a metal complexing ligand on the antibody (Lewis et al (1998) Bioconj.
Chem. 9:72-
86). Chelating linker labelling reagents such as DOTA-NHS (1,4,7,10-
tetraazacyclododecane-1,4,7,10-tetraacetic acid mono (N-hydroxysuccinimide
ester) are
commercially available (Macrocyclics, Dallas, TX). Receptor target imaging
with
radionuclide labelled antibodies can provide a marker of pathway activation by
detection and
quantitation of progressive accumulation of antibodies in tumor tissue (Albert
et al (1998)
Bioorg. Med. Chem. Lett. 8:1207-1210).
[00163] Metal-chelate complexes suitable as antibody labels for imaging
experiments
(US 2010/0111856; US 5342606; US 5428155; US 5316757; US 5480990; US 5462725;
US
5428139; US 5385893; US 5739294; US 5750660; US 5834456; Hnatowich et al
(1983) J.
Immunol. Methods 65:147-157; Meares et al (1984) Anal. Biochem. 142:68-78;
Mirzadeh et
al (1990) Bioconjugate Chem. 1:59-65; Meares et at (1990) J. Cancer1990,
Suppl. 10:21-26;
Izard et al (1992) Bioconjugate Chem. 3:346-350; Nikula et al (1995) Nucl.
Med. Biol.
22:387-90; Camera et al (1993) Nucl. Med. Biol. 20:955-62; Kukis et al (1998)
J. Nucl. Med.
39:2105-2110; Verel et al (2003) J. Nucl. Med. 44:1663-1670; Camera et al
(1994) J. Nucl.
Med. 21:640-646; Ruegg et al (1990) Cancer Res. 50:4221-4226; Verel et al
(2003) J. Nucl.
Med. 44:1663-1670; Lee et al (2001) Cancer Res. 61:4474-4482; Mitchell, et at
(2003) J.
Nucl. Med. 44:1105-1112; Kobayashi et al (1999) Bioconjugate Chem. 10:103-111;
Miederer
et al (2004) J. Nucl. Med. 45:129-137; DeNardo et al (1998) Clinical Cancer
Research
4:2483-90; Blend et at (2003) Cancer Biotherapy & Radiopharmaceuticals 18:355-
363;
Nikula et al (1999) J. Nucl. Med. 40:166-76; Kobayashi et al (1998) J. Nucl.
Med. 39:829-36;
Mardirossian et al (1993) Nucl. Med. Biol. 20:65-74; Roselli et al (1999)
Cancer Biotherapy
& Radiopharmaceuticals, 14:209-20).
[00164] (b) Fluorescent labels such as rare earth chelates (europium
chelates),
fluorescein types including FITC, 5-carboxyfluorescein, 6-carboxy fluorescein;
rhodamine
types including TAMRA; dansyl; Lissamine; cyanines; phycoerythrins; Texas Red;
and
analogs thereof. The fluorescent labels can be conjugated to antibodies using
the techniques
disclosed in Current Protocols in Immunology, supra, for example. Fluorescent
dyes and
fluorescent label reagents include those which are commercially available from
Invitrogen/Molecular Probes (Eugene, OR) and Pierce Biotechnology, Inc.
(Rockford, IL).
41
Date Recue/Date Received 2023-11-15
[00165] Detection labels such as fluorescent dyes and chemiluminescent
dyes (Briggs
et al (1997) "Synthesis of Functionalised Fluorescent Dyes and Their Coupling
to Amines
and Amino Acids," J. Chem. Soc., Perkin-Trans. 1:1051-1058) provide a
detectable signal
and are generally applicable for labelling antibodies, preferably with the
following properties:
(i) the labelled antibody should produce a very high signal with low
background so that small
quantities of antibodies can be sensitively detected in both cell-free and
cell-based assays;
and (ii) the labelled antibody should be photostable so that the fluorescent
signal may be
observed, monitored and recorded without significant photo bleaching. For
applications
involving cell surface binding of labelled antibody to membranes or cell
surfaces, especially
live cells, the labels preferably (iii) have good water-solubility to achieve
effective conjugate
concentration and detection sensitivity and (iv) are non-toxic to living cells
so as not to
disrupt the normal metabolic processes of the cells or cause premature cell
death.
[00166] (c) Various enzyme-substrate labels are available or
disclosed (US
4275149). The enzyme generally catalyzes a chemical alteration of a
chromogenic substrate
that can be measured using various techniques. For example, the enzyme may
catalyze a
color change in a substrate, which can be measured spectrophotometrically.
Alternatively, the
enzyme may alter the fluorescence or chcmiluminescence of the substrate.
Techniques for
quantifying a change in fluorescence are described above. The chemiluminescent
substrate
becomes electronically excited by a chemical reaction and may then emit light
which can be
measured (using a chemiluminometer, for example) or donates energy to a
fluorescent
acceptor. Examples of enzymatic labels include luciferases (e.g., firefly
luciferase and
bacterial luciferase; US 4737456), luciferin, 2,3-dihydrophthalazinediones,
malate
dehydrogenase, urease, peroxidase such as horseradish peroxidase (HRP),
alkaline
phosphatase (AP), 0-galactosidase, glucoamylase, lysozyme, saccharide oxidases
(e.g.,
glucose oxidase, galactose oxidase, and glucose-6-phosphate dehydrogenase),
heterocyclic
oxidases (such as unease and xanthine oxidase), lactoperoxidase,
microperoxidase, and the
like. Techniques for conjugating enzymes to antibodies are described in
O'Sullivan et al
(1981) -Methods for the Preparation of Enzyme-Antibody Conjugates for use in
Enzyme
Immunoassay", in Methods in Enzytn. (ed J. Langone & H. Van Vunakis), Academic
Press,
New York, 73:147-166.
[00167] Examples of enzyme-substrate combinations (US 4275149; US
4318980)
include, for example:
[00168] (i) Horseradish peroxidase (HRP) with hydrogen peroxidase as
a
42
Date Recue/Date Received 2023-11-15
substrate, wherein the hydrogen peroxidase oxidizes a dye precursor (e.g.,
orthophenylene
diamine (OPD) or 3,3',5,5'-tetramethylbenzidine hydrochloride (TMB));
[00169] (ii) alkaline phosphatase (AP) with para-nitrophenyl
phosphate as
chromogenic substrate; and
[00170] (iii) P-D-galactosidase (13-D-Gal) with a chromogenic
substrate (e.g., p-
nitropheny1-13-D-galactosidase) or fluorogenic substrate 4-methylumbellifery1-
13-D-
galactosidase.
[00171] A label may be indirectly conjugated with a cysteine engineered
antibody. For
example, the antibody can be conjugated with biotin and any of the three broad
categories of
labels mentioned above can be conjugated with avidin or streptavidin, or vice
versa. Biotin
binds selectively to streptavidin and thus, the label can be conjugated with
the antibody in
this indirect manner. Alternatively, to achieve indirect conjugation of the
label with the
polypeptide variant, the polypeptide variant is conjugated with a small hapten
(e.g., digoxin)
and one of the different types of labels mentioned above is conjugated with an
anti-hapten
polypeptide variant (e.g., anti-digoxin antibody). Thus, indirect conjugation
of the label with
the polypeptide variant can be achieved (Hermanson, G. (1996) in Bioconjugate
Techniques
Academic Press, San Diego).
[00172] The polypeptide variant of the present invention may be
employed in any
known assay method, such as ELISA, competitive binding assays, direct and
indirect
sandwich assays, and immunoprecipitation assays (Zola, (1987) Monoclonal
Antibodies: A
Manual of Techniques, pp.147-158, CRC Press, Inc.).
[00173] A detection label may be useful for localizing, visualizing,
and quantitating a
binding or recognition event. The labelled antibodies of the invention can
detect cell-surface
receptors. Another use for detectably labelled antibodies is a method of bead-
based
immunocapture comprising conjugating a bead with a fluorescent labelled
antibody and
detecting a fluorescence signal upon binding of a ligand. Similar binding
detection
methodologies utilize the surface plasmon resonance (SPR) effect to measure
and detect
antibody-antigen interactions.
[00174] Labelled cysteine engineered antibodies of the invention are
useful as imaging
biomarkers and probes by the various methods and techniques of biomedical and
molecular
imaging such as: (i) MRI (magnetic resonance imaging); (ii) MicroCT
(computerized
tomography); (iii) SPECT (single photon emission computed tomography); (iv)
PET
(positron emission tomography) Tinianow, J. et al (2010) Nuclear Medicine and
Biology,
43
Date Recue/Date Received 2023-11-15
37(3):289-297; Chen et at (2004) Bioconjugate Chem. 15:41-49; US 2010/0111856
(v)
bioluminescence; (vi) fluorescence; and (vii) ultrasound. Immunoscintigraphy
is an imaging
procedure in which antibodies labeled with radioactive substances are
administered to an
animal or human patient and a picture is taken of sites in the body where the
antibody
localizes (US 6528624). Imaging biomarkers may be objectively measured and
evaluated as
an indicator of normal biological processes, pathogenic processes, or
pharmacological
responses to a therapeutic intervention. Biomarkers may be of several types:
Type 0 are
natural history markers of a disease and correlate longitudinally with known
clinical indices,
e.g. MRI assessment of synovial inflammation in rheumatoid arthritis; Type I
markers
capture the effect of an intervention in accordance with a mechanism-of-
action, even though
the mechanism may not be associated with clinical outcome; Type II markers
function as
surrogate endpoints where the change in, or signal from, the biomarker
predicts a clinical
benefit to "validate" the targeted response, such as measured bone erosion in
rheumatoid
arthritis by CT. Imaging biomarkers thus can provide pharmacodynamic (PD)
therapeutic
information about: (i) expression of a target protein, (ii) binding of a
therapeutic to the target
protein, i.e. selectivity, and (iii) clearance and half-life pharmacokinetic
data. Advantages of
in vivo imaging biomarkers relative to lab-based biomarkers include: non-
invasive treatment,
quantifiable, whole body assessment, repetitive dosing and assessment, i.e.
multiple time
points, and potentially transferable effects from preclinical (small animal)
to clinical (human)
results. For some applications, bioimaging supplants or minimizes the number
of animal
experiments in preclinical studies.
[00175] Peptide labelling methods are well known. See Haugland, 2003,
Molecular
Probes Handbook of Fluorescent Probes and Research Chemicals, Molecular
Probes, Inc.;
Brinkley, 1992, Bioconjugate Chem. 3:2; Garman, (1997) Non-Radioactive
Labelling: A
Practical Approach, Academic Press, London; Means (1990) Bioconjugate Chem.
1:2;
Glazer et al (1975) Chemical Modification of Proteins. Laboratory Techniques
in
Biochemistry and Molecular Biology (T. S. Work and E. Work, Eds.) American
Elsevier
Publishing Co., New York; Lundblad, R. L. and Noyes, C. M. (1984) Chemical
Reagents for
Protein Modification,Vols. I and II, CRC Press, New York; Pfleiderer, G.
(1985) "Chemical
Modification of Proteins", Modern Methods in Protein Chemistry, H. Tschesche,
Ed., Walter
DeGryter, Berlin and New York; and Wong (1991) Chemistry of Protein
Conjugation and
Cross-linking, CRC Press, Boca Raton, Fla.); De Leon-Rodriguez et al (2004)
Chem.Eur. J.
10:1149-1155; Lewis et al (2001) Bioconjugate Chem. 12:320-324; Li et al
(2002)
Bioconjugate Chem. 13:110-115; Mier et al (2005) Bioconjugate Chem. 16:240-
237.
44
Date Recue/Date Received 2023-11-15
[00176] Peptides and proteins labelled with two moieties, a fluorescent
reporter and
quencher in sufficient proximity undergo fluorescence resonance energy
transfer (FRET).
Reporter groups are typically fluorescent dyes that are excited by light at a
certain
wavelength and transfer energy to an acceptor, or quencher, group, with the
appropriate
Stokes shift for emission at maximal brightness. Fluorescent dyes include
molecules with
extended aromaticity, such as fluorescein and rhodamine, and their
derivatives. The
fluorescent reporter may be partially or significantly quenched by the
quencher moiety in an
intact peptide. Upon cleavage of the peptide by a peptidase or protease, a
detectable increase
in fluorescence may be measured (Knight, C. (1995) "Fluorimetric Assays of
Proteolytic
Enzymes", Methods in Enzymology, Academic Press, 248:18-34).
[00177] The labelled antibodies of the invention may also be used as an
affinity
purification agent. In this process, the labelled antibody is immobilized on a
solid phase such
a Sephadex resin or filter paper, using methods well known in the art. The
immobilized
antibody is contacted with a sample containing the antigen to be purified, and
thereafter the
support is washed with a suitable solvent that will remove substantially all
the material in the
sample except the antigen to be purified, which is bound to the immobilized
polypeptide
variant. Finally, the support is washed with another suitable solvent, such as
glycine buffer at
pH 5.0 that will release the antigen from the polypeptide variant.
[00178] Labelling reagents typically bear reactive functionality which
may react (i)
directly with a cysteine thiol of a cysteine engineered antibody to form the
labelled antibody,
(ii) with a linker reagent to form a linker-label intermediate, or (iii) with
a linker antibody to
form the labelled antibody. Reactive functionality of labelling reagents
include: maleimide,
haloacetyl, iodoacetamide succinimidyl ester (e.g. NHS, N-hydroxysuccinimide),
isothiocyanate, sulfonyl chloride, 2,6-dichlorotriazinyl, pentafluorophenyl
ester, and
phosphoramidite, although other functional groups can also be used.
[00179] CONJUGATION OF BIOTIN-MALEIMIDE TO THIOFABS
[00180] The above-described ThioFab properties were established in the
presence of
phage because fusion of the Fab to the phage coat protein could potentially
alter Cys thiol
accessibility or reactivity. Therefore, the ThioFab constructs were cloned
into an expression
vector under alkaline phosphatase promoter (Chang et al (1987) Gene 55:189-
196) and the
ThioFab expression was induced by growing E. coli cells in the phosphate-free
medium.
T'hioFabs were purified on a Protein G SEPHAROSETM column and analyzed on
reducing
and non-reducing SDS-PAGE gels. These analyses allow assessment of whether
ThioFabs
Date Recue/Date Received 2023-11-15
retained their reactive thiol group or were rendered inactive by forming
intramolecular or
intermolecular disulfide bonds. ThioFabs L-V15C, L-V110C, H-A88C, and H-A121C
were
expressed and purified by Protein-G SEPHAROSErm column chromatography (see
methods
sections for details). Purified proteins were analyzed on SDS-PAGE gel in
reducing (with
DTT) and non-reducing (without DTT) conditions. Other reducing agents such as
BME
(beta-mercaptoethanol) can used in the gel to cleave interchain disulfide
groups. It is evident
from SDS-PAGE gel analysis that the major (-90%) fraction of ThioFab is in the
monomeric
form, while wild type hu4D5Fabv8 is essentially in the monomeric form (47
kDa).
[00181] ThioFab (A121C) and wild type hu4D5Fabv8 were incubated with
100 fold
excess of biotin-maleimide for 3 hours at room temperature and the
biotinylated Fabs were
loaded onto a Superdex-200TM gel filtration column. This purification step was
useful in
separating monomeric Fab from oligomeric Fab and also from excess free biotin-
maleimide
(or free cytotoxic drug).
[00182] Figure 5 shows validation of the properties of ThioFab variants
in the absence
of the phage context. The proteins without phage fusion, hu4D5Fabv8 and
hu4D5Fabv8-
A121C (ThioFab-A121C), were expressed and purified using protein-G agarose
beads
followed by incubation with 100 fold molar excess of biotin-malcimidc.
Strcptavidin and
HER2 binding of a biotinylated cys engineered ThioFab and a non-biotinylated
wild type Fab
was compared. The extent of biotin conjugation (interaction with streptavidin)
and their
binding ability to HER2 were monitored by ELISA analyses. Each Fab was tested
at 2ng and
2Ong.
[00183] Biotinylated A121C ThioFab retained comparable HER2 binding to
that of
wild type hu4D5Fabv8 (Figure 5). Wild type Fab and A121C-ThioFab were purified
by gel
filtration column chromatography. The two samples were tested for HER2 and
streptavidin
binding by ELISA using goat anti-Fab-HRP as secondary antibody. Both wild type
(open
box) and ThioFab (dotted box) have similar binding to HER2 but only ThioFab
retained
streptavidin binding. Only a background level of interaction with streptavidin
was observed
with non-biotinylated wild type hu4D5Fabv8 (Figure 5). Mass spectral (LC-ESI-
MS)
analysis of biotinylated-ThioFab (A121C) resulted in a major peak with 48294.5
daltons
compared to the wild type hu4D5Fabv8 (47737 daltons). The 537.5 daltons
difference
between the two molecules exactly corresponds to a single biotin-maleimide
conjugated to
the ThioFab. Mass spec protein sequencing (LC-ES1-Tandem mass spec analysis)
results
further confirmed that the conjugated biotin molecule was at the newly
engineered Cys
residue (Table 8, Example 3b).
46
Date Recue/Date Received 2023-11-15
[00184] SITE SPECIFIC CONJUGATION OF 1310TIN-MALEIMIDE TO
ALBUMIN BINDING PEPTIDE (ABP)-THIOFABS
[00185] Plasma-protein binding can be an effective means of
improving the
pharmacokinetic properties of short lived molecules. Albumin is the most
abundant protein in
plasma. Serum albumin binding peptides (ABP) can alter the pharmacodynamics of
fused
active domain proteins, including alteration of tissue uptake, penetration,
and diffusion.
These pharmacodynamic parameters can be modulated by specific selection of the
appropriate serum albumin binding peptide sequence (US 20040001827). A series
of
albumin binding peptides were identified by phage display screening (Dennis et
al. (2002)
"Albumin Binding As A General Strategy For -Improving The Pharmacokinctics Of
Proteins"
J Biol Chem. 277:35035-35043; WO 01/45746). Compounds of the invention include
ABP
sequences taught by: (i) Dennis et al (2002) J Biol Chem. 277:35035-35043 at
Tables III and
IV, page 35038; (ii) US 20040001827 at [0076]; and (iii) WO 01/45746 at pages
12-13
[00186] Albumin Binding (ABP)-Fabs were engineered by fusing an
albumin binding
peptide to the C-terminus of Fab heavy chain in 1:1 stoichiometric ratio (1
ABP / 1 Fab). It
was shown that association of these ABP-Fabs with albumin increased their half
life by more
than 25 fold in rabbits and mice. The above described reactive Cys residues
can therefore be
introduced in these ABP-Fabs and used for site-specific conjugation with
cytotoxic drugs
followed by in vivo animal studies.
[00187] Exemplary albumin binding peptide sequences include, but are
not limited to
the amino acid sequences listed in SEQ ID NOS: 1-5:
CDKTHTGGGSQRLMEDICLPRWGCLWEDDF SEQ ID NO:1
QRLMEDICLPRWGCLWEDDF SEQ ID NO:2
QRL IED I C L PRWGCLWEDDF SEQ ID NO:3
RLIEDICLPRWGCLWEDD SEQ ID NO:4
DI CL PRWGCLW SEQ ID NO:5
[00188] The albumin binding peptide (ABP) sequences bind albumin
from multiple
species (mouse, rat, rabbit, bovine, rhesus, baboon, and human) with Kd
(rabbit) = 0.3 p.M.
The albumin binding peptide does not compete with ligands known to bind
albumin and has a
half life (TV2) in rabbit of 2.3 hr. ABP-ThioFab proteins were purified on BSA-
SEPHAROSETm followed by biotin-maleimide conjugation and purification on
Superdex-
S200 column chromatography as described in previous sections. Purified
biotinylated
47
Date Recue/Date Received 2023-11-15
proteins were homogeneous and devoid of any oligomeric forms (Example 4).
[00189] Figure 6 shows thc properties of Albumin Binding Peptide (ABP)-
ThioFab
variants. ELISA analyses were carried out to test the binding ability of ABP-
hu4D5Fabv8-
wt, ABP-hu4D5Fabv8-V110C and ABP-hu4D5Fabv8-A121C with rabbit albumin,
streptavidin and HER2. Biotinylated ABP-ThioFabs are capable of binding to
albumin and
HER2 with similar affinity to that of wild type ABP-hu4D5Fabv8 as confirmed by
ELISA
(Figure 6) and BIAcore binding kinetics analysis (Table 2). An ELISA plate was
coated with
albumin, HER2 and SA as described. Binding of biotinylated ABP-ThioFabs to
albumin,
HER2 and SA was probed with anti-Fab HRP. Biotinylated ABP-ThioFabs were
capable of
binding to streptavidin compared to non biotinylated control ABP-hu4D5Fabv8-wt
indicating
that ABP-ThioFabs were conjugated with biotin maleimide like ThioFabs in a
site specific
manner as the same Cys mutants were used for both the variants (Figure 6).
Table 2. BIAcore kinetic analysis for HER2 and rabbit albumin binding to
biotinylated
ABP- hu4D5Fabv8 wild type and ThioFabs
Antibody kon (Ms') koff (s-') Kd (nM)
HER2 binding
wild type 4.57 x 105 4.19 x 10-5 0.0917
V110C 4.18x 105 4.05x 10-5 0.097
A121C 3.91 x 105 4.15 x 10-5 0.106
Rabbit albumin binding
wild type 1.66 x 105 0.0206 124
V110C 2.43 x 105 0.0331 136
A121C 1.70 x 105 0.0238 140
ABP = albumin binding peptide
[00190] Alternatively, an albumin-binding peptide may be linked to the
antibody by
covalent attachment through a linker moiety.
[00191] ENGINEERING OF ABP-THIOFABS WITH TWO FREE THIOL GROUPS
PER FAB
[00192] The above results indicate that all four (L-V15C, L-V110C, H-A88C
and H-
A121C) thioFab (eysteine engineered Fab antibodies) variants have reactive
thiol groups that
can be used for site specific conjugation with a label reagent, linker
reagent, or drug-linker
intermediate. L-V15C can be expressed and purified but with relatively low
yields. However
the expression and purification yields of L-V110C, H-A88C and H-A121C variants
were
similar to that of hu4D5Fabv8. Therefore these mutants can be used for further
analysis and
recombined to get more than one thiol group per Fab. Towards this objective,
one thiol group
48
Date Recue/Date Received 2023-11-15
on the light and one on heavy chain were constructed to obtain two thiol
groups per Fab
molecule (L-V110C/H-A88C and L-V110C/H-A121C). These two double Cys variants
were
expressed in an E. coli expression system and purified. The homogeneity of
purified
biotinylated ABP-ThioFabs was found to be similar to that of single Cys
variants.
[00193] The effects of engineering two reactive Cys residues per Fab
was investigated
(Figure 7). The presence of a second biotin was tested by probing the binding
of biotinylated
ABP-ThioFab to SA using streptavidin-HRP (Figure 7). For HER2/Fab analysis, an
ELISA
plate was coated with HER2 and probed with anti-Fab HRP. For SA/Fab analysis,
an ELISA
plate was coated with SA and probed with anti-Fab HRP. For SA/SA analysis, an
ELISA
plate was coated with SA and probed with SA-HRP. Figure 7. ELISA analyses for
the
interaction of biotinylated ABP-hu4D5Fabv8 cys variants with HER2,
streptavidin (SA).
HER2/Fab, SA/Fab and SA/SA indicate that their interactions were monitored by
anti-Fab-
HRP, SA-HRP, respectively. SA/Fab monitors the presence of single biotin per
Fab and
more than one biotin per Fab is monitored by SA/SA analysis. Binding of HER2
with double
cys mutants is similar to that of single Cys variants (Figure 7). However the
extent of
biotinylation on double Cys mutants was higher compared to single Cys variants
due to more
than one free thiol group per Fab molecule (Figure 7).
[00194] ENGINEERING OF TRIO IgG VARIANTS OF TRASTUZUMAB
[00195] Cysteine was introduced into the full-length monoclonal
antibody,
trastuzumab (HERCEPT1N , Genentech Inc.) at certain residues. The single cys
mutants H-
A88C, H-A121C and L-V110C of trastuzumab, and double cys mutants V110C-A121C
and
V110C-A121C of trastuzumab were expressed in CHO (Chinese Hamster Ovary) cells
by
transient fermentation in media containing 1 mM cysteine. The A88C mutant
heavy chain
sequence (450 aa) is SEQ ID NO:6. The A121C mutant heavy chain sequence (450
aa) is
SEQ ID NO:7. The V110C mutant light chain sequence (214 aa) is SEQ ID NO:8.
49
Date Recue/Date Received 2023-11-15
EVQLVE SGGGLVQPGGSLRLSCAASGFN I Kai' Y I HWVRQAPGKGLEWVARI Y P TNGYTRY
ADSVKGRFT I SADT SKNTAYLQMN S LRCEDTAVYYC SRWGGDGFYAMDYWGQGT LVTVS S
AS TKGPSVF PLAP S SKS T SGGTAALGCLVKDYFPEPVTVSWNSGAL T SGVHTF PAVLQSS
GLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGG
PSVFLFPPKPKDTLMI SRT PEVT CVVVDVS HE DPEVKFNWYVDGVEVHNAKTKPREEQYN
S TYRVVSVLTVLHQDWLNGKEYKCKVSNKAL PAP IEKT I SKAKGQ PRE PQVYT L PPSREE
MTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVOKSRW
QQGNVFSCSVMHEALHNHYTQKSL S LS PGK
SEQ ID NO:6
EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPINGYTRY
ADSVKGRFT I SADT S KNTAYLQMN S LRAEDTAVYYC SRWGGDGFYAMDYWGQGT LVTVS S
CS TKGPSVF PLAP S SKS T SGGTAALGCLVKDYFPEPVTVSWNSGAL T SGVHTFPAVLQSS
GLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGG
PSVFLFPPKPKDTLMI SRT PEVT CVVVDVS HE DPEVKFNWYVDGVEVHNAKTKPREEQYN
S T YRVVSVLTVLHQDWLNGKEYKCKVSNKAL PAP IEKT I S KAKGQ PRE PQVYT L PPSREE
MTKNQVSLICLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGS FFLYSKLTVDKSRW
QQGNVFSCSVMHEALHNHYTQKSLSLSPGK
SEQ ID NO:7
DI QMTQS PS SLSASVGDRVT I TCRASQDVNTAVAWYQQKPGKAPKLL IYSASFLYSGVPS
RFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTCAAPSVFIFPP
S DEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQE SVTEQDSKDS TY SLSS TLT
LSKADYEKHKVYACEVTHQGLSS PVTKSFNRGEC
SEQ ID NO:8
[00196] According to one embodiment, the cysteine engineered thio-
trastuzumab
antibodies comprise one or more of the following variable region heavy chain
sequences with
a free cysteine amino acid (SEQ ID NOS: 9-16).
Date Recue/Date Received 2023-11-15
Mutant Sequence SEQ ID NO:
A40C WVRQCPGKGL SEQ ID NO:9
A88C NSLRCEDTAV SEQ ID NO:10
S119C LVTVCSASTKGPS SEQ ID NO:11
S120C LVTVSCASTKGPS SEQ ID NO:12
A121C LVTVSSCSTKGPS SEQ ID NO:13
S122C LVTVSSACTKGPS SEQ ID NO:14
A175C HTFPCVLQSSGLYS SEQ ID NO:15
SI79C HTFPAVLQCSGLYS SEQ ID NO:16
[001971 According to another embodiment, the cysteine engineered thio-
trastuzumab
antibodies comprise one or more of the following variable region light chain
sequences with a
free cysteine amino acid (SEQ ID NOS: 17-27).
Mutant Sequence SEQ ID NO:
V15C SLSASCGDRVT SEQ ID NO:17
A43C QKPGKCPKLLI SEQ ID NO:18
V110C EIKRTCAAPSV SEQ ID NO:19
S114C TCAAPCVFIFPP SEQ ID NO:20
S121C FI FPPCDEQLK SEQ ID NO:21
S127C DEQLKCGTASV SEQ ID NO:22
A144C FY PRECKVQWK SEQ ID NO:23
A153C WKVDNCLQSGN SEQ ID NO:24
N158C ALQSGCSQESV SEQ ID NO:25
S168C VTEQDCKDSTY SEQ ID NO:26
V205C GLSSPCTKSFN SEQ ID NO:27
[00198] The resulting full-length, thio-trastuzumab IgG variants were
assayed for thiol
reactivity and TiF,R2 binding activity. Figure 9A shows a cartoon depiction of
biotinylated
antibody binding to immobilized 1-LER2 and HRP labeled secondary antibody for
absorbance
detection. Figure 9B shows binding measurements to immobilized HER2 with
detection of
absorbance at 450 nm of (left to right): non-biotinylated wild type
trastuzurnab (Wt), biotin-
maleimide conjugated thio-trastuzumab variants V11 0C (single cys), A121C
(single cys), and
V1 I 0C-A121C (double cys). Each thio IgG variant and trasturttmab was tested
at 1, 10, and
100 ng. The measurements show that biotinylated anti-HER2 ThioMabs retain HER2
binding
activity.
[00199] Figure I OA shows a cartoon depiction of a biotinylated
antibody binding to
immobilized HER2 with binding of biotin to anti-IgG-HRP for absorbance
detection. Figure
10B shows binding measurements with detection of absorbance at 450 nm of
biotin-
maleimide conjugated thio-trastuzumab variants and non-biotinylated wild type
trastuzumab
51
Date Recue/Date Received 2023-11-15
in binding to streptavidin. From left to right: V110C (single cys), A121C
(single cys),
V110C/A121C (double cys), and trastuzumab. Each thio IgG trastuzumab variant
and parent
trastuzumab was tested at 1, 10, and 100 ng. The measurements show that the
HER2
ThioMabs have high thiol reactivity.
[00200] Cysteine was introduced into the full-length 21-19 anti-EphB2R
antibody at
certain residues. The single cys mutant H-A121C of 2H9 was expressed in CHO
(Chinese
Hamster Ovary) cells by transient fermentation in media containing 1 mM
cysteine. The
A121C 2H9 mutant heavy chain sequence (450 aa) is SEQ ID NO:28.
EVQLVES GGGLVQPGGSLRLSCAAS GYTFT SYWMHWVRQAPGKGLEWVGFINP S TGYTDY
NQKFKDRFT I SADT S KNTAYLQMN S LRAEDTAVYYC TRRPK I PRHANVFWGQGT LVTVS S
CS TKGPSVFPLAPS SKS T SGGTAALGCLVKDYFPEPVTVSWNSGAL T SGVHTFPAVLQS S
GLYSLSSVVTVPSSSLGTQTY ICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGG
PSVFLFPPKPKDTLMI SRT PEVT CVVVDVS HE D PEVKFNWYVDGVEVHNAKTKPREEQYN
STYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKT I SKAKGQ PRE PQVYTL P PSREE
MTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTT PPVLDSDGSFFLYSKLTVDKSRW
QQGNVFS C SVMHEALHNHYTQKS L S LS PGK
SEQ ID NO:28
[00201] Cysteine engineered thio-2H9 antibodies comprise the following
Fe constant
region heavy chain sequences with a free cysteine amino acid (SEQ ID NOS: 29-
38).
Mutant Sequence SEQ ID NO:
V273C HE DPECKFNWYVDGVEVHNAKTKPR SEQ ID NO:29
V279C HEDPEVKFNWYCDGVEVHNAKTKPR SEQ ID NO:30
V282C HE DPEVKFNWYVDGCEVHNAKTK PR SEQ ID NO:31
V284C HE DPEVKFNWYVDGVECHNAKTKPR SEQ ID NO:32
A287C HE DPEVKFNWYVDGVEVHNCKTKPR SEQ ID NO:33
S324C YKCKVCNKALP SEQ ID NO:34
S337C IEKT ICKAKGQ PR SEQ ID NO:35
A339C IEKT I SKCKGQ PR SEQ ID NO:36
S375C KGFY PC D IAVE SEQ ID NO:37
S400C PPVLDCDGSFF SEQ ID NO:38
[00202] Cysteine was introduced into the full-length 3A5 anti-MUC16
antibody at
certain residues. The single cys mutant H-A121C of 3A5 was expressed in CHO
(Chinese
Hamster Ovary) cells by transient fermentation in media containing 1 mM
cysteine. The
A121C 3A5 mutant heavy chain sequence (446 aa) comprises SEQ ID NO:39.
52
Date Recue/Date Received 2023-11-15
DVOLQESGPGLVNPSQSLSLTCTVTGYS I TNDYAWNWIRQFPGNKLEWMGYINY SGYT TY
NPSLKSRI S I TRDT SKNQFFLHLNSVTTEDTATYYCARWDGGLTYWGQGTLVTVSACS TK
GPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS
LSSVVTVPS SSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVF
LFPPKPKDTLMI SRT PEVTCVVVDVSHE DPEVKFNWYVDGVEVHNAKTKPREEQYNS TYR
VVSVLTVLHQDWLNGKEYKCKVSNKALPAP IEKT I SKAKGQ PRE PQVYTL PPS REEMTKN
QVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGN
VFSCSVMHEALHNHYTQKSLSLSPGK
SEQ ID NO:39
[00203] Cysteine engineered thio-3A5 anti-MUC16 antibodies comprise the
following
variable region heavy chain sequences with a free cysteine amino acid (SEQ ID
NOS: 40-44).
Mutant Sequence SEQ ID NO:
F45C NWIRQCPGNK SEQ ID NO:40 ,
A90C LNSCTTEDTAT SEQ ID NO:41
A121C GQGTLVTVSACSTKGPSVFPL SEQ ID NO:42
A175C HTFPCVLQSSGLYS SEQ ID NO:43
V176C HTFPACLQSSGLYS SEQ ID NO:44
[00204] Cysteine engineered thio-3A5 anti-MUC16 antibodies comprise the
following
variable region light chain sequences with a free cysteine amino acid (SEQ ID
NOS: 45-49).
Mutant Sequence SEQ ID NO:
L15C FL SVSCGGRVT SEQ ID NO:45
A43C QKPGNC PRLL SEQ ID NO:46
V110C EIKRTCAAPSV SEQ ID NO:47
A144C FY PRECKVQWK SEQ ID NO:48
5168C VTEQDCKDS TY SEQ ID NO:49
[00205] ENGINEERING AND THIOL REACTIVITY OF 4D5 ANTI-HER2
THIOFABS
[00206] Cysteine was introduced into each position of the heavy chain
and light chain
of the anti-HER2 hu4D5Fabv8 Fab fragment antibody. All 440 of the heavy chain
mutants
and light chain mutants were prepared according to the methods described
herein. Thiol
reactivity was measured according to the PHESELECTOR assay. Heavy chain
sequences are
53
Date Recue/Date Received 2023-11-15
numbered by the Sequential numbering system. Light chain sequences follow the
Kabat
numbering system. In the light chain, both Kabat and Sequential numbering
denotes same
numbers.
[00207] Heavy chain hu4D5Fabv8 mutants were selected for efficient
binding to
HER2 receptor protein (Figures 2 and 3) and thiol reactivity with the
biotinylation reagent,
Biotin-PEO-maleimide (Examples 1 and 2). Certain heavy chain mutants had
limited or
compromised binding to HER2 ECD because this is an important residue for
antigen binding
(HER2), located in CDRs in the variable region of the antibody-Fab. Some of
the residues
located in the constant domain of the Fabs also resulted in poor HER2 binding
because these
residues may contribute to structure and folding of Fab, thus resulting in
poor 4D5-Fab
display on M13-page (Junutula, J.R. et al. (2008) J. Immunol Methods, 332:41-
52). Heavy
chain hu4D5Fabv8 mutants with poor HER2 ECD binding included cysteine
mutations at
positions 1, 21, 31, 33-36, 38, 48-50, 59, 87, 95, 101, 104, 129, 131, 132,
136, 153, 155, 159,
166, 169, 170, 172, 197, 198, 202, 215, 219. Wild type cysteine variants 22,
96, 147, 203,
223 were measured. Other heavy chain mutants had limited thiol reactivity with
the
biotinylation reagent. The free cysteine amino acid residue is in the center
with flanking
residues in the sequences in the middle column of Table 3. The substituted
amino acid and
position in the heavy chain are designated in the left column. Heavy chain
hu4D5Fabv8
mutants SEQ ID NOS: 50-98 of Table 3 have retained HER2 binding and thiol
reactivity
values of about 0.8 or higher, excluding wild type cysteine variants.
Antibodies with SEQ ID
NOS: 50-98 (Table 3) have demonstrated thiol reactivity and may be useful to
form covalent
attachments with a capture label, a detection label, a drug moiety, or a solid
support. The
heavy chain mutants of Table 3 may be conjugated as ThioFabs or ThioMabs for
example as
antibody-drug conjugates.
Table 3 Efficient binding, thiol-reactive heavy chain hu4D5Fabv8
mutants
HC-L4C EVQCVESGG SEQ ID NO: 50
HC-G8C QLVESCGGLVQ SEQ ID NO: 51
HC-G100 VESGGCLVQPG SEQ ID NO: 52
HC-L20C GGSLRCSCAAS SEQ ID NO: 53
HC-A23C LRLSCCASGFN SEQ ID NO: 54
HC-G26C SCAASCFNIKD SEQ ID NO: 55
HC-F27C CAASGCNIKDT SEQ ID NO: 56
HC-T32C FNIKDCYIHWV SEQ ID NO: 57
HC-Q39C IHWVRCAPGKG SEQ ID NO: 58
HC-P41C WVRQACGKGLE SEQ ID NO: 59
54
Date Recue/Date Received 2023-11-15
HC-K430 RQAPGCGLEWV SEQ ID NO: 60
HC-G44C QAPGKCLEWVA SEQ ID NO: 61
HC-W47C GKGLECVARIY SEQ ID NO: 62
HC-S 63C TRYADCVKGRF SEQ ID NO: 63
HC-F680 SVKGRCT I SAD SEQ ID NO: 64
HC-D730 FT I SACTSKNT SEQ ID NO: 65
HC-K760 SADTSCNTAYL SEQ ID NO: 66
HC-T780 DT SKNCAYLQM SEQ ID NO: 67
HC-Y80C SKNTACLQMNS SEQ ID NO: 68
HC-L81C KNTAYCQMNSL SEQ ID NO: 69
HC-Q820 NTAYLCMNSLR SEQ ID NO: 70
HC-L86C LQMNSCRAEDT SEQ ID NO: 71
HC-A880 MNSLRCEDTAV SEQ ID NO: 72
HC-D90C SLRAECTAVYY SEQ ID NO: 73
HC-V93C AEDTACYYCSR SEQ ID NO: 74
HC-Y 94C EDTAVCYCSRW SEQ ID NO: 75
HC-R98C VYYCSCWGGDG SEQ ID NO: 76
HC-G1000 YCSRWCGDGFY SEQ ID NO: 77
HC-D1080 GFYAMCYWGQG SEQ ID NO: 78
HC-G113C DYWGQCTLVTV SEQ ID NO: 79
HC-T117C QGTLVCVSSAS SEQ ID NO: 80
HC-A1210 VTVSSCSTKGP SEQ ID NO: 81
HC-G1250 SAS TKCPSVFP SEQ ID NO: 82
HC-G141C KS T SGCTAALG SEQ ID NO: 83
HC-P1540 VKDYFCEPVTV SEQ ID NO: 84
HC-N1620 VTVSWCSGALT SEQ ID NO: 85
HC-S1630 TVSWNCGALTS SEQ ID NO: 86
HC-01640 VSWNSCALTSG SEQ ID NO: 87
HC-S1680 SGALTCGVHTF SEQ ID NO: 88
HC-F1730 SGVHTCPAVLQ SEQ ID NO: 89
HO-T1900 LSSVVCVPSSS SEQ ID NO: 90
HC-S1940 VTVPSCSLGTQ SEQ ID NO: 91
HC-T2000 SLGTQCYICNV SEQ ID NO: 92
HC-V2050 TY I CNCNHKP S SEQ ID NO: 93
HC-N2110 NHKPSCTKVDK SEQ ID NO: 94
HC- T212 C HKPSNCKVDKK SEQ ID NO: 95
HC-V2140 PSNTKCDKKVE SEQ ID NO: 96
HC-K217C TKVDKCVEPKS SEQ ID NO: 97
HC-T2260 KSCDKCH SEQ ID NO: 98
[00208] Light chain hu4D5Fabv8 mutants were selected for efficient
binding to HER2
receptor protein (Figures 2 and 3) and thiol reactivity with the biotinylation
reagent, Biotin-
PEO-maleimide (Examples 1 and 2). Certain light chain mutants had limited or
Date Recue/Date Received 2023-11-15
compromised binding to HER2 because this is an important residue for antigen
binding
(HER2), located in CDRs in the variable region of the antibody-Fab. Some of
the residues
located in constant domain of Fab also resulted in poor HER2 binding because
these residues
may contribute to structure and folding of Fab, thus resulting in poor 4D5-Fab
display on
M13-page (Junutula, J.R. et al. (2008) J. Irnmunol Methods, 332:41-52). Light
chain
hu4D5Fabv8 mutants with poor binding to HER2 included cysteine mutants at
positions 4,
29-32, 35, 36, 50, 82, 86, 89-91, 113, 115, 117, 120, 126, 128, 139, 141, 146,
148, 179, 186,
192, 202. Wild type cysteine variants 23, 134, 194, 214 were measured. Other
light chain
mutants had limited thiol reactivity with the biotinylation reagent. The free
cysteine amino
acid residue is in the center with flanking residues in the sequences in the
middle column of
Table 4. The substituted amino acid and position in the light chain are
designated in the left
column. Light chain hu4D5Fabv8 mutants SEQ ID NOS: 99-147 of Table 4 have
retained
HER2 binding and thiol reactivity values of about 0.8 or higher, excluding
wild type cysteine
variants. Antibodies with SEQ ID NOS: 99-147 (Table 4) have demonstrated thiol
reactivity
and may be useful to form covalent attachments with a capture label, a
detection label, a drug
moiety, or a solid support. The light chain mutants of Table 4 may be
conjugated as
ThioFabs or ThioMabs for example as antibody-drug conjugates.
Table 4 Efficient binding, thiol-reactive light chain hu4D5Fabv8
mutants
LC-S9C MTQSPCSLSAS SEQ ID NO: 99
LC-L46C GKAPKCLIYSA SEQ ID NO: 100
LC-Y49C PKLLICSASFL SEQ ID NO: 101
LC-F53C IYSASCLYSGV SEQ ID NO: 102
LC-T72C SGTDFCLTISS SEQ ID NO: 103
LC-L730 GTDFTCTISSL SEQ ID NO: 104
LC-T740 TDFTLCISSLQ SEQ ID NO: 105
LC-175C DFTLTCSSLQP SEQ ID NO: 106
LC-S770 TLTISCLQPED SEQ ID NO: 107
LC-Q79C TISSLCPEDFA SEQ ID NO: 108
LC-P80C ISSLQCEDFAT SEQ ID NO: 109
LC-Y92C YCQQHCTTPPT SEQ ID NO: 110
LC-P95C QHYTTCPTFGQ SEQ ID NO: 111
LC-G99C TPPTFCQGTKV SEQ ID NO: 112
LC-G101C PTFGQCTKVEI SEQ ID NO: 113
LC-K1030 FGQGTCVEIKR SEQ ID NO: 114
LC-E1050 QGTKVCIKRTV SEQ ID NO: 115
LC-V1100 EIKRTCAAPSV SEQ ID NO: 116
LC-A1120 KRTVACPSVFI SEQ ID NO: 117
56
Date Recue/Date Received 2023-11-15
LC-S114C TVAAPCVFI FP SEQ ID NO: 118
LC-F1160 AAPSVCIFPPS SEQ ID NO: 119
LC-F1180 PSVFICPPSDE SEQ ID NO: 120
LC-S121C FIFPPCDEQLK SEQ ID NO: 121
LC-L125C PSDEQCKSGTA SEQ ID NO: 122
LC-S1270 DEQLKCGTASV SEQ ID NO: 123
LC-T1290 QLKSGCASVVC SEQ ID NO: 124
LC-A1300 LKSGTCSVVCL SEQ ID NO: 125
LC-S1310 KSGTACVVCLL SEQ ID NO: 126
LC-N137C VVCLLCNFYPR SEQ ID NO: 127
LC-N1380 VCLLNCFY PRE SEQ ID NO: 128
LC-Y1400 LLNNFCPREAK SEQ ID NO: 129
LC-R1420 NNFYPCEAKVQ SEQ ID NO: 130
LC-A144C FYPRECKVQWK SEQ ID NO: 131
LC-Q1470 REAKVCWKVDN SEQ ID NO: 132
LC-K1490 AKVQWCVDNAL SEQ ID NO: 133
LC-D1510 VQWKVCNALQS SEQ ID NO: 134
LC-Q155C VDNALCSGNSQ SEQ ID NO: 135
LC-Q1600 QSGNSCESVTE SEQ ID NO: 136
LC-A1840 LTLSKCDYEKH SEQ ID NO: 137
LC-D1850 TLSKACYEKHK SEQ ID NO: 138
LC-K188C KADYECHKVYA SEQ ID NO: 139
LC-T1970 YACEVCHQGLS SEQ ID NO: 140
LC-G2000 EVTHQCLSSPV SEQ ID NO: 141
LC-L2010 VTHQGCSS PVT SEQ ID NO: 142
LC-S2030 HQGLSCPVTKS SEQ ID NO: 143
LC-P2040 QGLSSCVTKSF SEQ ID NO: 144
LC-V205C GLSS PCTKS FN SEQ ID NO: 145
LC-T2060 LS SPVCKSFNR SEQ ID NO: 146
LC-K207C SSPVTCSFNRG SEQ ID NO: 147
[00209] THIOL REACTIVITY OF THIOMABS
[00210] The thiol reactivity offal! length, IgG cysteine engineered
antibodies
(ThioMabs) was measured by biotinylation and streptavidin binding (US
7521541). A
western blot assay was set up to screen the ThioMab that is specifically
conjugated with
biotin-maleimide. In this assay, the antibodies are analyzed on reducing SDS-
PAGE and the
presence of Biotin is specifically probed by incubating with streptavidin-HRP.
As seen from
figure 18, the streptavidin-HRP interaction is either observed in heavy chain
or light chain
depending on which engineered cys variant is being used and no interaction is
seen with wild
type, indicating that ThioMab variants specifically conjugated the biotin at
engineered Cys
57
Date Recue/Date Received 2023-11-15
residue. Figure 18 shows denaturing gel analysis of reduced, biotinylated Thio-
IgG variants
after capture on immobilized anti-IgG-HRP (top gel) and streptavidin-HRP
(bottom gel).
Lane 1: 3A5 H-A121C. Lane 2: 3A5 L-V110C. Lane 3: 2H9 H-A121C. Lane 4: 2H9 L-
V110C. Lane 5: anti-EphB2R 2H9 parent, wild type. Each mutant (lanes 1-4) was
captured
by anti-IgG with HRP detection (top) indicating that selectivity and affinity
were retained.
Capture by immobilized streptavidin with HRP detection (bottom) confirmed the
location of
biotin on heavy and light chains. The location of cysteine mutation on the
cysteine
engineered antibodies in lanes 1 and 3 is the heavy chain. The location of
cysteine mutation
on the cysteine engineered antibodies in lanes 2 and 4 is the light chain. The
cysteine
mutation site undergoes conjugation with the biotin-maleimide reagent.
[00211] Analysis of the ThioMab cysteine engineered antibodies of
Figure 18 and a
2H9 V15C variant by LC/MS gave quantitative indication of thiol reactivity
(Table 5).
Table 5 LC/MS quantitation of biotinylation of ThioMabs - Thiol
reactivity
ThioMab variant number of biotin per ThioMab
2H9 wt 0.0
2H9 L-V15C 0.6
2H9 L-V110C 0.5
2H9 H-A121C 2.0
3A5 L-V110C 1.0
3A5 H-A121C 2.0
[00212] Cysteine engineering was conducted in the constant domain, i.e.
Fc region, of
IgG antibodies. A variety of amino acid sites were converted to cysteine sites
and the
expressed mutants, i.e. cysteine engineered antibodies, were assessed for
their thiol reactivity.
Biotinylated 2H9 ThioMab Fe variants were assessed for thiol reactivity by HRP
quantitation
by capture on immobilized streptavidin in an ELISA assay (Figure 19). An ELISA
assay was
established to rapidly screen the Cys residues with reactive Thiol groups. As
depicted in
Figure 19 schematic diagram, the streptavidin-biotin interaction is monitored
by probing with
anti-IgG-HRP followed by measuring absorbance at 450 nm. These results
confirmed 2H9-
ThioFc variants V282C, A287C, A339C, S375C and S400C had moderate to highest
Thiol
reactivity. The extent of biotin conjugation of 2H9 ThioMab Fe variants was
quantitated by
LS/MS analysis as reported in Table 6. The LS/MS analysis confirmed that the
A282C,
S375C and S400C variants had 100% biotin conjugation and V284C and A339C had
50%
conjugation, indicating the presence of a reactive cysteine thiol group. The
other ThioFc
variants, and the parent, wild type 2H9, had either very little biotinylation
or none.
58
Date Recue/Date Received 2023-11-15
Table 6 LC/MS quantitation of biotinylation of 2H9 Fe ThioMabs
2H9 ThioMab Fc variant % biotinylation
=V273C 0
V279C 31
V282C 100
V284C 50
A287C 0
S324C 71
S337C 0
A339C 54
S375C 100
S400C 100
(wild type 2H9) 0
[00213] THIOL REACTIVITY OF TH10-4D5 FAB LIGHT CHAIN VARIANTS
[00214] Screening of a variety of cysteine engineered light chain
variant Fabs of the
antiErbB2 antibody 4D5 gave a number of variants with a thiol reactivity value
of 0.6 and
higher (Table 7), as measured by the PHESELECTOR assay of Figure 8. The thiol
reactivity
values of Table 7 are normalized to the heavy chain 4D5 ThioFab variant (HC-
A121C) which
is set at 100%, assuming complete biotinylation of HC-A121C variant, and
represented as per
cent values.
Table 7 Thiol reactivity per cent values of 4D5 ThioFab light
chain variants
4D5 ThioFab variant Thiol reactivity value
(%)
V15C 100
V110C 95
S114C 78
S121C 75
S127C 75
A153C 82
N158C 77
V205C 78
(HC-A121C) 100
(4D5 wild type) 25
59
Date Recue/Date Received 2023-11-15
[00215] ANTIBODY-DRUG CONJUGATES
[00216] The cysteine engineered antibodies of the invention may be
conjugated with
any therapeutic agent, i.e. drug moiety, which can be covalently attached to
the antibody
through a reactive cysteine thiol group.
[00217] An exemplary embodiment of an antibody-drug conjugate (ADC)
compound
comprises a cysteine engineered antibody (Ab), and a drug moiety (D) wherein
the antibody
has one or more free cysteine amino acids, and the antibody is attached
through the one or
more free cysteine amino acids by a linker moiety (L) to D; the composition
having Formula
Ab-(L-D)P
[00218] where p is 1, 2, 3, or 4. The number of drug moieties which may
be
conjugated via a thio[ reactive linker moiety to an antibody molecule is
limited by the number
of cysteine residues which are introduced by the methods described herein.
Exemplary ADC
of Formula I therefore comprise antibodies which have 1, 2, 3, or 4 engineered
cysteine
amino acids.
[00219] Another exemplary embodiment of an antibody-drug conjugate
compound
(ADC) comprises a cysteine engineered antibody (Ab), an albumin-binding
peptide (ABP)
and a drug moiety (D) wherein the antibody is attached to the drug moiety by a
linker moiety
(L) and the antibody is attached to the albumin-binding peptide by an amide
bond or a second
linker moiety; the composition having Formula la:
ABP-Ab-(L-D)P Ia
[00220] where p is 1, 2, 3, or 4.
[00221] The ADC compounds of the invention include those with utility
for anticancer
activity. In particular, the compounds include a cysteine-engineered antibody
conjugated, i.e.
covalently attached by a linker, to a drug moiety, i.e. toxin. When the drug
is not conjugated
to an antibody, the drug has a cytotoxic or cytostatic effect. The biological
activity of the
drug moiety is thus modulated by conjugation to an antibody. The antibody-drug
conjugates
(ADC) of the invention selectively deliver an effective dose of a cytotoxic
agent to tumor
tissue whereby greater selectivity, i.e. a lower efficacious dose, may be
achieved.
[00222] DRUG MOIETIES
[00223] The drug moiety (D) of the antibody-drug conjugates (ADC)
includes any
Date Recue/Date Received 2023-11-15
compound, moiety or group which has a cytotoxic or cytostatic effect. Drug
moieties
include: (i) chemotherapeutic agents, which may function as microtubulin
inhibitors, mitosis
inhibitors, topoisomerase inhibitors, or DNA intercalators; (ii) protein
toxins, which may
function enzymatically; and (iii) radioisotopes.
[00224] Exemplary drug moieties include, but are not limited to, a
maytansinoid, an
auristatin, a dolastatin, a trichothecene, CC1065, a calicheamicin and other
enediyne
antibiotics, a taxane, an anthracycline, and stereoisomers, isosteres, analogs
or derivatives
thereof
[00225] Maytansine compounds suitable for use as maytansinoid drug
moieties are
well known in the art, and can be isolated from natural sources according to
known methods,
produced using genetic engineering techniques (see Yu et al (2002) PROC. NAT.
ACAD.
SCI. (USA) 99:7968-7973), or maytansinol and maytansinol analogues prepared
synthetically according to known methods.
[00226] Exemplary maytansinoid drug moieties include those having a
modified
aromatic ring, such as: C-19-dechloro (US 4256746) (prepared by lithium
aluminum hydride
reduction of ansamytocin P2); C-20-hydroxy (or C-20-demethyl) +/-C-19-dechloro
(US Pat.
Nos. 4361650 and 4307016) (prepared by demethylation using Streptomyces or
Actinomyces
or dechlorination using LAH); and C-20-demethoxy, C-20-acyloxy (-000R), +/-
dechloro
(U.S. Pat. No. 4,294,757) (prepared by acylation using acyl chlorides), and
those having
modifications at other positions
[00227] Exemplary maytansinoid drug moieties also include those having
modifications such as: C-9-SH (US 4424219) (prepared by the reaction of
maytansinol with
H2S or P2S5); C-14-alkoxymethyl(demethoxy/CH2 OR)(US 4331598); C-14-
hydroxymethyl
or acyloxymethyl (CH2OH or CH20Ac) (US 4450254) (prepared from Nocardia); C-15-
hydroxy/acyloxy (US 4364866) (prepared by the conversion of maytansinol by
Streptomyces); C-15-methoxy (US Pat. Nos. 4313946 and 4315929) (isolated from
Trewia
nudfflora); C-18-N-demethyl (US Pat. Nos. 4362663 and 4322348) (prepared by
the
demethylation of maytansinol by Streptomyces); and 4,5-deoxy (US 4371533)
(prepared by
the titanium trichloride/LAH reduction of maytansinol). Many positions on
maytansine
compounds are known to be useful as the linkage position, depending upon the
type of link.
For example, for forming an ester linkage, the C-3 position having a hydroxyl
group, the C-
14 position modified with hydroxymethyl, the C-15 position modified with a
hydroxyl group
and the C-20 position having a hydroxyl group are all suitable.
[00228] The drug moiety (D) of the antibody-drug conjugates (ADC) of
Formula I
61
Date Recue/Date Received 2023-11-15
include maytansinoids having the structure:
H3C (CR2),¨S
0
H3C 0 0
CI \NI 0
CH30
0
N/L0
HO I
CH30 H
[00229] where the wavy line indicates the covalent attachment of the
sulfur atom of D
to a linker (L) of an antibody-drug conjugate (ADC). R may independently be H
or a Ci¨C6
alkyl selected from methyl, ethyl, 1-propyl, 2-propyl, 1-butyl, 2-methyl-1-
propyl, 2-butyl, 2-
methy1-2-propyl, 1-pentyl, 2-pentyl, 3-pentyl, 2-methy1-2-butyl, 3-methy1-2-
butyl, 3-methyl-
1-butyl, 2-methyl-I -butyl, 1-hexyl, 2-hexyl, 3-hexyl, 2-methyl-2-pentyl, 3-
methyl-2-pentyl,
4-methy1-2-pentyl, 3-methyl-3-pentyl, 2-methy1-3-pentyl, 2,3-dimethy1-2-butyl,
and 3,3-
dimethy1-2-butyl. The alkylene chain attaching the amide group to the sulfur
atom may be
methanyl, ethanyl, or propyl, i.e. m is 1, 2, or 3.
[00230] Maytansine compounds inhibit cell proliferation by inhibiting
the formation of
microtubules during mitosis through inhibition of polymerization of the
microtubulin protein,
tubulin (Remillard et al (1975) Science 189:1002-1005). Maytansine and
maytansinoids are
highly cytotoxic but their clinical use in cancer therapy has been greatly
limited by their
severe systemic side-effects primarily attributed to their poor selectivity
for tumors. Clinical
trials with maytansine had been discontinued due to serious adverse effects on
the central
nervous system and gastrointestinal system (Issel et al (1978) Can. Treatment.
Rev. 5:199-
207).
[00231] Maytansinoid drug moieties are attractive drug moieties in
antibody-drug
conjugates because they are: (i) relatively accessible to prepare by
fermentation or chemical
modification, derivatization of fermentation products, (ii) amenable to
derivatization with
functional groups suitable for conjugation through the non-disulfide linkers
to antibodies, (iii)
stable in plasma, and (iv) effective against a variety of tumor cell lines (US
2005/0169933;
WO 2005/037992; US 5208020).
[00232] As with other drug moieties, all stereoisomers of the
maytansinoid drug
moiety are contemplated for the compounds of the invention, i.e. any
combination of R and S
62
Date Recue/Date Received 2023-11-15
configurations at the chiral carbons of D. In one embodiment, the maytansinoid
drug moiety
(D) will have the following stercochemistry:
H3S (CR2)m¨S-
0
0
H3C 0 0
CI \N 7 0
õA\
CH30
0
NLO
Ho I
CH30 H
1002331 Exemplary embodiments of maytansinoid drug moieties include:
DM1,
(CR2)m ¨ CH2C1-12; DM3, (CR2)m ¨ CH2CH2CH(C143); and DM4, (CR2)õ, ¨
CH2CH2C(CH3)2,
having the structures:
H3S CH2CH2S-
0
0
H3C 0 0
CI \N 0
DM 1
CH30
0
- NLO
I
CH30HO H
63
Date Recue/Date Received 2023-11-15
CH3
CH2CH2C S ___________________________________________________
H3C\
0
H3C 0 0
0
CI \N 0
CH30 I DM3
0
. N
.1-1(75 I
CH30 H
CH3
H3C CH2CH2C S _______________________________________________
0 \N-4,
0 CH3
H3C 0 0
CI \NJ 0
DM4
CH30
0
EHO I
CH30 H
[00234] The linker may be attached to the maytansinoid molecule at
various positions,
depending on the type of the link. For example, an ester linkage may be formed
by reaction
with a hydroxyl group using conventional coupling techniques. The reaction may
occur at
the C-3 position having a hydroxyl group, the C-14 position modified with
hydroxymethyl,
the C-15 position modified with a hydroxyl group, and the C-20 position having
a hydroxyl
group. In a preferred embodiment, the linkage is formed at the C-3 position of
maytansinol
or a maytansinol analogue.
[00235] The drug moiety (D) of the antibody-drug conjugates (ADC) of
Formula I also
include dolastatins and their peptidic analogs and derivatives, the
auristatins (US Patent Nos.
5635483; 5780588). Dolastatins and auristatins have been shown to interfere
with
microtubulc dynamics, GTP hydrolysis, and nuclear and cellular division (Woykc
et al (2001)
Antimicrob. Agents and Chemothcr. 45(12):3580-3584) and have anticancer (US
5663149)
64
Date Recue/Date Received 2023-11-15
and antifungal activity (Pettit et al (1998) Antimicrob. Agents Chemother.
42:2961-2965).
Various forms of a dolastatin or auristatin drug moiety may be covalently
attached to an
antibody through thc N (amino) terminus or the C (carboxyl) terminus of the
peptidic drug
moiety (WO 02/088172; Doromna et at (2003) Nature Biotechnology 21(7):778-784;
Francisco et al (2003) Blood 102(4):1458-1465).
[002361 Drug moieties include dolastatins, auristatins (US 5635483;
US 5780588; US
5767237; US 6124431), and analogs and derivatives thereof. Dolastatins and
auristatins have
been shown to interfere with microtubule dynamics, GTP hydrolysis, and nuclear
and cellular
division (Woyke et al (2001) Antimicrob. Agents and Chemother. 45(12):3580-
3584) and
have anticancer (US 5663149) and antifungal activity (Pettit et al (1998)
Antimicrob. Agents
Chemother. 42:2961-2965). The dolastatin or auristatin drug moiety may be
attached to the
antibody through the N (amino) terminus or the C (carboxyl) terminus of the
peptidic drug
moiety (WO 02/088172).
[00237] Exemplary auristatin embodiments include the N-terminus
linked
monomethylauristatin drug moieties DE and DF, disclosed in US 7498298 and US
7659241.
1002381 The drug moiety (D) of the antibody-drug conjugates (ADC) of
Formula I
include the monomethylauristatin drug moieties MMAE and MMAF linked through
the N-
terminus to the antibody, and having the structures:
0 OH
I 0 0
0 0
MMAE
0
ANrrQr.rNH
0
0 0 0,
0 0 OH MMAF
[00239] Typically, peptide-based drug moieties can be prepared by
forming a peptide
bond between two or more amino acids and/or peptide fragments. Such peptide
bonds can be
prepared, for example, according to the liquid phase synthesis method (see E.
Schroder and
K. Liibke, "The Peptides", volume 1, pp 76-136, 1965, Academic Press) that is
well known in
Date Recue/Date Received 2023-11-15
the field of peptide chemistry.
[00240] The drug moiety includes calicheamicin, and analogs and
derivatives thereof.
The calicheamicin family of antibiotics are capable of producing double-
stranded DNA
breaks at sub-picomolar concentrations. For the preparation of conjugates of
the
calicheamicin family, see US 5712374; US 5714586; US 5739116; US 5767285; US
5770701, US 5770710; US 5773001; US 5877296. Structural analogues of
calicheamicin
which may be used include, but are not limited to, yil, az', 1131, N-acetyl-
yil, PSAG and WI
(Hinman et al Cancer Research 53:3336-3342 (1993), Lode et al Cancer Research
58:2925-
2928 (1998).
[00241] Protein toxins include: diphtheria A chain, nonbinding active
fragments of
diphtheria toxin, exotoxin A chain (from Pseudomonas aeruginosa), ricin A
chain (Vitetta et
al (1987) Science, 238:1098), abrin A chain, modeccin A chain, alpha-sarcin,
Aleurites fordii
proteins, dianthin proteins, Phytolaca americana proteins (PAPI, PAPI1, and
PAP-S),
momordica charantia inhibitor, curcin, crotin, sapaonaria officinalis
inhibitor, gelonin,
mitogellin, restrictocin, phenomycin, enomycin, and the tricothecenes (WO
93/21232).
[00242] Therapeutic radioisotopes include: 32p, 33p, 90y, 125j, 1311,
131In, 153Sm, 186Re,
188 211 212 212
Re, At, Bi, Pb, and radioactive isotopes of Lu.
[00243] The radioisotope or other labels may be incorporated in the
conjugate in
known ways (Fraker et al (1978) Biochem. Biophys. Res. Commun. 80: 49-57;
"Monoclonal
Antibodies in Immunoscintigraphy" Chatal, CRC Press 1989). Carbon-14-labeled 1-
isothiocyanatobenzy1-3-methyldiethylene triaminepentaacetic acid (MX-DTPA) is
an
exemplary chelating agent for conjugation of a radionuclide to the antibody
(WO 94/11026).
[00244] LINKERS
[00245] A "Linker" (L) is a bifunctional or multifunctional moiety
which can be used
to link one or more Drug moieties (D) and an antibody unit (Ab) to form
antibody-drug
conjugates (ADC) of Formula I. Antibody-drug conjugates (ADC) can be
conveniently
prepared using a Linker having reactive functionality for binding to the Drug
and to the
Antibody. A cysteine thiol of a cysteine engineered antibody (Ab) can form a
bond with a
functional group of a linker reagent, a drug moiety or drug-linker
intermediate.
[00246] In one aspect, a Linker has a reactive site which has an
electrophilic group that
is reactive to a nucicophilic cysteine present on an antibody. The cystcinc
thiol of the
antibody is reactive with an electrophilic group on a Linker and forms a
covalent bond to a
Linker. Useful electrophilic groups include, but are not limited to, maleimide
and
haloacetamide groups.
66
Date Recue/Date Received 2023-11-15
[00247] Cysteine engineered antibodies react with linker reagents or
drug-linker
intetmediates, with electrophilic functional groups such as maleimide or a-
halo carbonyl,
according to the conjugation method at page 766 of Klussman, et al (2004),
Bioconjugate
Chemistry 15(4):765-773, and according to the protocol of Example 4.
[00248] In yet another embodiment, the reactive group of a linker
reagent or drug-
linker intermediate contains a thiol-reactive functional group that can form a
bond with a free
cysteine thiol of an antibody. Examples of thiol-reaction functional groups
include, but are
not limited to, maleimide, a-haloacetyl, activated esters such as suceinimide
esters,
4-nitrophenyl esters, pentafluorophenyl esters, tetrafluorophenyl esters,
anhydrides, acid
chlorides, sulfonyl chlorides, isocyanates and isothiocyanates.
[00249] In another embodiment, the linker may be a dendritic type
linker for covalent
attachment of more than one drug moiety through a branching, multifunctional
linker moiety
to an antibody (Sun et al (2002) Bioorganic & Medicinal Chemistry Letters
12:2213-2215;
Sun et al (2003) Bioorganic & Medicinal Chemistry 11:1761-1768; King (2002)
Tetrahedron
Letters 43:1987-1990). Dendritic linkers can increase the molar ratio of drug
to antibody, i.e.
loading, which is related to the potency of the ADC. Thus, where a cysteine
engineered
antibody bears only one reactive cysteine thiol group, a multitude of drug
moieties may be
attached through a dendritic linker.
[00250] The linker may comprise amino acid residues which links the
antibody (Ab) to
the drug moiety (D) of the cysteine engineered antibody-drug conjugate (ADC)
of the
invention. The amino acid residues may form a dipeptide, tripeptide,
tetrapeptide,
pentapeptide, hexapeptide, heptapeptide, octapeptide, nonapeptide,
decapeptide,
undecapeptide or dodecapeptide unit. Amino acid residues include those
occurring naturally,
as well as minor amino acids and non-naturally occurring amino acid analogs,
such as
citrulline.
[00251] Useful amino acid residue units can be designed and optimized
in their
selectivity for enzymatic cleavage by a particular enzymes, for example, a
tumor-associated
protease to liberate an active drug moiety. In one embodiment, an amino acid
residue unit,
such as valine-eitrulline (vc or val-cit), is that whose cleavage is catalyzed
by cathepsin B, C
and D, or a plasmin protease.
[00252] A linker unit may be of the self-immolative type such as a p-
aminobenzylcarbamoyl (PAB) unit where the ADC has the exemplary structure:
67
Date Recue/Date Received 2023-11-15
Qm
Ab _______________________ Aa-Ww¨NH-(=1)¨\
_________________________________________________ 0 C D
I /
0
P
[00253] wherein Q is -C1-C8 alkyl, -0-(C1-C8 alkyl), -halogen, -nitro
or -cyano; m is an
integer ranging from 0-4; and p ranges from 1 to 4.
[00254] Other examples of self-immolative spacers include, but are not
limited to,
aromatic compounds that are electronically similar to the PAB group such as 2-
aminoimidazol-5-methanol derivatives (US 7375078; Hay et at. (1999) Bioorg.
Med. Chem.
Lett. 9:2237) and ortho or para-aminobenzylacetals. Spacers can be used that
undergo
cyclization upon amide bond hydrolysis, such as substituted and unsubstituted
4-
aminobutyric acid amides (Rodrigues et al (1995) Chemistry Biology 2:223),
appropriately
substituted bicyclo[2.2.1] and bicyclo[2.2.2] ring systems (Storm et al (1972)
J. Amer. Chem.
Soc. 94:5815) and 2-aminophenylpropionic acid amides (Amsberry, et al (1990)
J. Org.
Chem. 55:5867). Elimination of amine-containing drugs that are substituted at
glycine
(Kingsbury et al (1984) J. Med. Chem. 27:1447) are also examples of self-
immolative spacer
useful in ADCs.
[00255] In another embodiment, linker L may be a dendritic type linker
for covalent
attachment of more than one drug moiety through a branching, multifunctional
linker moiety
to an antibody (Sun et al (2002) Bioorganic & Medicinal Chemistry Letters
12:2213-2215;
Sun et al (2003) Bioorganic & Medicinal Chemistry 11:1761-1768). Dendritic
linkers can
increase the molar ratio of drug to antibody, i.e. loading, which is related
to the potency of
the ADC. Thus, where a cysteine engineered antibody bears only one reactive
cysteine thiol
group, a multitude of drug moieties may be attached through a dendritic linker
(WO
2004/01993; Szalai et at (2003) J. Amer. Chem. Soc. 125:15688-15689; Shamis et
al (2004)
J. Amer. Chem. Soc. 126:1726-1731; Amir et at (2003) Angew. Chem. Int. Ed.
42:4494-
4499).
[00256] Embodiments of the Formula Ia antibody-drug conjugate compounds
include
(val-cit), (MC-val-cit), and (MC-val-cit-PAB):
68
Date Recue/Date Received 2023-11-15
i H 0
Ab \ Aa Niryi.L.)--YY-D)
H Of37. P
HN
0NH2 val-cit
/ 0
o )cry on \
/p
HN
0NH2 MC-val-cit
0
0 "D )
\
0 H 040
Ab4N,,,,,,,,õ,-,.,õ,,,K, YT N,...11,
N - N
0 11 0 ='' I
.1 H P
HN
0` N H2
MC-val-cit-PAB
[00257] Other exemplary embodiments of the Formula Ia antibody-drug
conjugate
compounds include the structures:
0
0 0 0 \
II II
4s N¨X¨Ial¨D 1 Ab __ S CH2C¨Y¨C¨D 1
Ab
0 / p / P
r_3(0
0 \
/ 0 II
i
N¨CH2-0--C¨D
II
Ab _________ , S CH2C¨D ) Ab'--(S
\ 0
P P
XlVd
69
Date Recue/Date Received 2023-11-15
C' k
-- Ab(S¨CH 2 C-ki
[00258] where X is:
¨CH2-0¨ , ¨(CF12)n¨ , (CH2CH20)n-
0
'
RI
0
or ¨(CH2)n¨C¨N¨(CH2)n¨
=
Y is:
/ or ¨N¨(CH2)n-
[00259] and R is independently H or C1¨C6 alkyl; and n is 1 to 12.
[00260] In another embodiment, a Linker has a reactive functional group
which has a
nucleophilic group that is reactive to an electrophilic group present on an
antibody. Useful
electrophilic groups on an antibody include, but are not limited to, aldehyde
and ketone
carbonyl groups. The heteroatom of a nucleophilic group of a Linker can react
with an
electrophilic group on an antibody and form a covalent bond to an antibody
unit. Useful
nucleophilic groups on a Linker include, but are not limited to, hydrazide,
oxime, amino,
hydrazine, thiosemicarbazone, hydrazine carboxylate, and arylhydrazide. The
electrophilic
group on an antibody provides a convenient site for attachment to a Linker.
[00261] Typically, peptide-type Linkers can be prepared by forming a
peptide bond
between two or more amino acids and/or peptide fragments. Such peptide bonds
can be
prepared, for example, according to the liquid phase synthesis method (E.
Schroder and K.
Liibke (1965) "The Peptides", volume 1, pp 76-136, Academic Press) which is
well known in
the field of peptide chemistry.
[00262] In another embodiment, the Linker may be substituted with
groups which
modulated solubility or reactivity. For example, a charged substituent such as
sulfonate (-
S03-) or ammonium, may increase water solubility of the reagent and facilitate
the coupling
reaction of the linker reagent with the antibody or the drug moiety, or
facilitate the coupling
Date Recue/Date Received 2023-11-15
reaction of Ab-L (antibody-linker intermediate) with D, or D-L (drug-linker
intermediate)
with Ab, depending on the synthetic route employed to prepare the ADC.
1002631 The compounds of the invention expressly contemplate, but are
not limited to,
ADC prepared with linker reagents: BMPEO, BMPS, EMCS, GMBS, HBVS, LC-SMCC,
MBS, MPBH, SBAP, SIA, STAB, SMCC, SMPB, SMPH, sulfo-EMCS, sulfo-GMBS, sulfo-
KMUS, sulfo-MBS, sulfo-SIAB, sulfo-SMCC, and sulfo-SMPB, and SVSB
(succinimidyl-
(4-vinylsulfone)benzoate), and including bis-maleimide reagents: DTME, BMB,
BMDB,
BMOE, BM(PEO)3, and BM(PEO)4, which are commercially available from Pierce
Biotechnology, Inc., Customer Service Department, P.O. Box 117, Rockford, IL.
61105
U.S.A, 1-800-874-3723, International +815-968-0747. See pages 467-498, 2003-
2004
Applications Handbook and Catalog. Bis-maleimide reagents allow the attachment
of the
thiol group of a cysteine engineered antibody to a thiol-containing drug
moiety, label, or
linker intermediate, in a sequential or concurrent fashion. Other functional
groups besides
maleimide, which are reactive with a thiol group of a cysteine engineered
antibody, drug
moiety, label, or linker intermediate include iodoacetamide, bromoacetamide,
vinyl pyridine,
disulfide, pyridyl disulfide, isocyanate, and isothiocyanate.
0
0
0 0 0
BM(PEO)2 BM(PEO)3
[00264] Useful linker reagents can also be obtained via other
commercial sources, such
as Molecular Biosciences Inc.(Boulder, CO), or synthesized in accordance with
procedures
described in Toki et al (2002) J. Org. Chem. 67:1866-1872; Walker, M.A. (1995)
J. Org.
Chem. 60:5352-5355; Frisch et at (1996) Bioconjugate Chem. 7:180-186; US
6214345; WO
02/088172; US 2003130189; US2003096743; WO 03/026577; WO 03/043583; and WO
04/032828.
[00265] An exemplary valine-citrulline (val-cit or vc) dipeptide linker
reagent having a
maleimide Stretcher and a para-aminobenzylcarbamoyl (PAB) self-immolative
Spacer has
the structure:
71
Date Recue/Date Received 2023-11-15
0
Qm \\(5.0
H3C-õ,(CH3 0
111JLN NO2
Fmoc-N>-1
H 0
NH
H21\1.-
[00266] where Q is -C1-C8 alkyl, -0-(C1-C8 alkyl), -halogen, -nitro or -
cyano; and m is
an integer ranging from 0-4.
[00267] An exemplary phe-lys(Mtr) dipeptide linker reagent having a
maleimide
Stretcher unit and a p-aminobenzyl self-immolative Spacer unit can be prepared
according to
Dubowchik, et al. (1997) Tetrahedron Letters, 38:5257-60, and has the
structure:
C1"0/--OH
X, \
Ph 0
LFN1JLN
Fmoc-N H
HOTh
HN¨Mtr
[00268] where Mtr is mono-4-methoxytrityl, Q is -CI-C8 alkyl, -0-(C1-C8
alkyl), -
halogen, -nitro or -cyano; and m is an integer ranging from 0-4.
[00269] Exemplary antibody-drug conjugate compounds of the invention
include:
_______ 9 Y Ab S 0
0')L-Ny
0 I 0,, 0
0 0
`= 0 OH /
0
Ab-MC-vc-PAB-MMAF
Ab _____ S __ 0
võ)0L, OH
0õ, 0
Val-Cit¨N 0
0
Ab-MC-vc-PAB-MMAE
72
Date Recue/Date Received 2023-11-15
Ab 0
0 H 0
fN,./=======,..)L---.NryNxiLN.,,,,c11) IN FN.' OH
Ab-MC-MMAE
Ab 0
H 0
I 0 0
0 0 0
0 OH )
Ab-MC-MMAF
[00270] where Val is valine; Cit is citrulline; p is 1, 2, 3, or 4; and
Ab is a cysteine
engineered antibody. Other exemplary antibody drug conjugates where
maytansinoid drug
moiety DM1 is linked through a BMPEO linker to a thiol group of trastuzumab
have the
structure:
0
0
_________________________________________________________ Ab
n 0
r0
H3C, ,CH2CH2S
0 N¨µ
H3C, 0 0
CI N 7 0
õNO
CH30
0
Ho i
CH30 H
[00271] where Ab is a cysteine engineered antibody; n is 0, 1, or 2;
and p is 1, 2, 3, or
4.
[00272] PREPARATION OF ANTIBODY-DRUG CONJUGATES
[00273] The ADC of Formula I may be prepared by several routes,
employing organic
chemistry reactions, conditions, and reagents known to those skilled in the
art, including: (1)
reaction of a cysteine group of a cysteine engineered antibody with a linker
reagent, to form
antibody-linker intermediate Ab-L, via a covalent bond, followed by reaction
with an
activated drug moiety D; and (2) reaction of a nucleophilic group of a drug
moiety with a
linker reagent, to form drug-linker intermediate D-L, via a covalent bond,
followed by
reaction with a cysteine group of a cysteine engineered antibody. Conjugation
methods (1)
and (2) may be employed with a variety of cysteine engineered antibodies, drug
moieties, and
73
Date Recue/Date Received 2023-11-15
linkers to prepare the antibody-drug conjugates of Formula I.
[00274] Antibody cysteine thiol groups are nucleophilic and capable of
reacting to
form covalent bonds with electrophilic groups on linker reagents and drug-
linker
intermediates including: (i) active esters such as NHS esters, HOBt esters,
haloformates, and
acid halides; (ii) alkyl and benzyl halides, such as haloacetamides; (iii)
aldehydes, ketones,
carboxyl, and maleimide groups; and (iv) disulfides, including pyridyl
disulfides, via sulfide
exchange. Nucleophilic groups on a drug moiety include, but are not limited
to: amine, thiol,
hydroxyl, hydrazide, oxime, hydrazine, thiosemicarbazone, hydrazine
carboxylate, and
arylhydrazide groups capable of reacting to form covalent bonds with
electrophilic groups on
linker moieties and linker reagents.
[00275] Maytansine may, for example, be converted to May-SSCH3, which
can be
reduced to the free thiol, May-SH, and reacted with a modified antibody (Chari
et al (1992)
Cancer Research 52:127-131) to generate a maytansinoid-antibody
immunoconjugate with a
disulfide linker. Antibody-maytansinoid conjugates with disulfide linkers have
been reported
(WO 04/016801; US 6884874; US 2004/039176 Al; WO 03/068144; US 2004/001838 Al;
US Patent Nos. 6441163, 5208020, 5416064; WO 01/024763). The disulfide linker
SPP is
constructed with linker reagent N-succinimidyl 4-(2-pyridylthio) pentanoate.
[00276] Under certain conditions, the cysteine engineered antibodies
may be made
reactive for conjugation with linker reagents by treatment with a reducing
agent such as DTT
(Cleland's reagent, dithiothreitol) or TCEP (tris(2-carboxyethyl)phosphine
hydrochloride;
Getz et al (1999) Anal. Biochem. Vol 273:73-80; Soltec Ventures, Beverly, MA).
Full
length, cysteine engineered monoclonal antibodies (ThioMabs) expressed in CHO
cells were
reduced with about a 50 fold excess of TCEP for 3 hrs at 37 C to reduce
disulfide bonds
which may form between the newly introduced cysteine residues and the cysteine
present in
the culture media. The reduced ThioMab was diluted and loaded onto HiTrap S
column in 10
mM sodium acetate, pH 5, and eluted with PBS containing 0.3M sodium chloride.
Disulfide
bonds were reestablished between cysteine residues present in the parent Mob
with dilute
(200 nM) aqueous copper sulfate (CuSO4) at room temperature, overnight. Other
oxidants,
i.e. oxidizing agents, and oxidizing conditions, which are known in the art
may be used.
Ambient air oxidation is also effective. This mild, partial reoxidation step
forms intrachain
disulfides efficiently with high fidelity. An approximate 10 fold excess of
drug-linker
intetinediate, e.g. BM(PEO)4-DM1 was added, mixed, and let stand for about an
hour at room
temperature to effect conjugation and form the ThioMab antibody-drug
conjugate. The
conjugation mixture was gel filtered and loaded and eluted through a HiTrap S
column to
74
Date Recue/Date Received 2023-11-15
remove excess drug-linker intermediate and other impurities.
[00277] Figure 11 shows the general process to prepare a cysteine
engineered antibody
expressed from cell culture for conjugation. Cysteine adducts, presumably
along with
various interchain disulfide bonds, are reductively cleaved to give a reduced
form of the
antibody. The interchain disulfide bonds between paired cysteine residues are
reformed
under partial oxidation conditions, such as exposure to ambient oxygen. The
newly
introduced, engineered, and unpaired cysteine residues remain available for
reaction with
linker reagents or drug-linker intermediates to form the antibody conjugates
of the invention.
The ThioMabs expressed in mammalian cell lines result in externally conjugated
Cys adduct
to an engineered Cys through ¨S-S- bond formation. Hence the purified ThioMabs
have to be
treated with reduction and oxidation procedures as described in Example 11 to
produce
reactive ThioMabs. These ThioMabs are used to conjugate with maleimide
containing
cytotoxic drugs, fluorophores, and other labels.
[00278] A variety of ThioFab and ThioMab antibody-drug conjugates were
prepared
(Examples 4-8). Cysteine mutant hu4D5Fabv8 (V110C) was conjugated with the
maytansinoid drug moiety DM1 with a bis-maleimido linker reagent BMPEO to form
hu4D5Fabv8 (V110C) -BMPEO-DM1 (Example 8).
[00279] IN VITRO CELL PROLIFERATION ASSAYS
[00280] Generally, the cytotoxic or cytostatic activity of an antibody-
drug conjugate
(ADC) is measured by: exposing mammalian cells having receptor proteins, e.g.
HER2, to
the antibody of the ADC in a cell culture medium; culturing the cells for a
period from about
6 hours to about 5 days; and measuring cell viability. Cell-based in vitro
assays were used to
measure viability (proliferation), cytotoxicity, and induction of apoptosis
(easpase activation)
of the ADC of the invention.
[00281] The in vitro potency of antibody-drug conjugates was measured
by a cell
proliferation assay (Figures 10 and 11, Example 9). The CellTiter-Glo
Luminescent Cell
Viability Assay is a commercially available (Promega Corp., Madison, WI),
homogeneous
assay method based on the recombinant expression of Coleoptera luciferase (US
Patent Nos.
5583024; 5674713 and 5700670). This cell proliferation assay determines the
number of
viable cells in culture based on quantitation of the ATP present, an indicator
of metabolically
active cells (Crouch ct al (1993) J. Immunol. Meth. 160:81-88; US 6602677).
The CcllTitcr-
Gb Assay was conducted in 96 well format, making it amenable to automated
high-
throughput screening (HTS) (Cree et al (1995) AntiCancer Drugs 6:398-404). The
homogeneous assay procedure involves adding the single reagent (CellTiter-Glo
Reagent)
Date Recue/Date Received 2023-11-15
directly to cells cultured in serum-supplemented medium. Cell washing, removal
of medium
and multiple pipetting steps are not required. The system detects as few as 15
cells/well in a
384-well format in 10 minutes after adding reagent and mixing. The cells may
be treated
continuously with ADC, or they may be treated and separated from ADC.
Generally, cells
treated briefly, i.e. 3 hours, showed the same potency effects as continuously
treated cells.
[00282] The homogeneous "add-mix-measure" format results in cell lysis
and
generation of a luminescent signal proportional to the amount of ATP present.
The amount of
ATP is directly proportional to the number of cells present in culture. The
CellTiter-Glo
Assay generates a "glow-type" luminescent signal, produced by the luciferase
reaction, which
has a half-life generally greater than five hours, depending on cell type and
medium used.
Viable cells are reflected in relative luminescence units (RLU). The
substrate, Beetle
Luciferin, is oxidatively decarboxylated by recombinant firefly luciferase
with concomitant
conversion of ATP to AMP and generation of photons.
[00283] IN VIVO EFFICACY
[00284] The in vivo efficacy of two albumin binding peptide-DM1
(maytansinoid)-
antibody-drug conjugates (ADC) of the invention is measured by a high
expressing HER2
transgenic explant mouse model (Figure 12, Example 10). An allograft is
propagated from
the Fo5 mmtv transgenic mouse which does not respond to, or responds poorly
to,
HERCEPTIN therapy. Subjects were treated once with ABP-rhuFab4D5-cys(light
chain)-
DM1; ABP-rhuFab4D5-cys(heavy chain)-DM1; and placebo PBS buffer control
(Vehicle)
and monitored over 3 weeks to measure the time to tumor doubling, log cell
kill, and tumor
shrinkage.
[00285] ADMINISTRATION OF ANTIBODY-DRUG CONJUGATES
[00286] The antibody-drug conjugates (ADC) of the invention may be
administered by
any route appropriate to the condition to be treated. The ADC will typically
be administered
parenterally, i.e. infusion, subcutaneous, intramuscular, intravenous,
intradermal, intrathecal
and epidural.
[00287] PHARMACEUTICAL FORMULATIONS
[00288] Pharmaceutical formulations of therapeutic antibody-drug
conjugates (ADC)
of the invention are typically prepared for parenteral administration, i.e.
bolus, intravenous,
intratumor injection with a pharmaceutically acceptable parenteral vehicle and
in a unit
dosage injectable form. An antibody-drug conjugate (ADC) having the desired
degree of
purity is optionally mixed with pharmaceutically acceptable diluents,
carriers, excipients or
76
Date Recue/Date Received 2023-11-15
stabilizers (Remington's Pharmaceutical Sciences (1980) 16th edition, Osol, A.
Ed.), in the
form of a lyophilized formulation or an aqueous solution.
[00289] ANTIBODY-DRUG CONJUGATE TREATMENTS
[00290] It is contemplated that the antibody-drug conjugates (ADC) of
the present
invention may be used to treat various diseases or disorders, e.g.
characterized by the
overexpression of a tumor antigen. Exemplary conditions or hyperproliferative
disorders
include benign or malignant tumors; leukemia and lymphoid malignancies. Others
include
neuronal, glial, astrocytal, hypothalamic, glandular, macrophagal, epithelial,
stromal,
blastocoelic, inflammatory, angiogenic and immunologic, including autoimmune,
disorders.
[00291] Generally, the disease or disorder to be treated is a
hyperproliferative disease
such as cancer. Examples of cancer to be treated herein include, but are not
limited to,
carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies.
More
particular examples of such cancers include squamous cell cancer (e.g.
epithelial squamous
cell cancer), lung cancer including small-cell lung cancer, non-small cell
lung cancer,
adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the
peritoneum,
hepatocellular cancer, gastric or stomach cancer including gastrointestinal
cancer, pancreatic
cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder
cancer, hcpatoma,
breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial or
uterine
carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer,
vulval cancer,
thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, as well
as head and
neck cancer.
[00292] Autoimmune diseases for which the ADC compounds may be used in
treatment include rheumatologic disorders (such as, for example, rheumatoid
arthritis,
Sjogren's syndrome, scleroderma, lupus such as SLE and lupus nephritis,
polymyositis/dermatomyositis, cryoglobulinemia, anti-phospholipid antibody
syndrome, and
psoriatic arthritis), osteoarthritis, autoimmune gastrointestinal and liver
disorders (such as, for
example, inflammatory bowel diseases (e.g., ulcerative colitis and Crohn's
disease),
autoimmune gastritis and pernicious anemia, autoimmune hepatitis, primary
biliary cirrhosis,
primary sclerosing cholangitis, and celiac disease), vasculitis (such as, for
example, ANCA-
associated vasculitis, including Churg-Strauss vasculitis, Wegener's
granulomatosis, and
polyartcriitis), autoimmune neurological disorders (such as, for example,
multiple sclerosis,
opsoclonus myoclonus syndrome, myasthenia gravis, neuromyelitis optica,
Parkinson's
disease, Alzheimer's disease, and autoimmune polyneuropathies), renal
disorders (such as,
for example, glomerulonephritis, Goodpasture's syndrome, and Berger's
disease),
77
Date Recue/Date Received 2023-11-15
autoimmune dermatologic disorders (such as, for example, psoriasis, urticaria,
hives,
pcmphigus vulgaris, bullous pemphigoid, and cutaneous lupus erythematosus),
hematologic
disorders (such as, for example, thrombocytopenic purpura, thrombotic
thrombocytopenic
purpura, post-transfusion purpura, and autoimmune hemolytic anemia),
atherosclerosis,
uveitis, autoimmune hearing diseases (such as, for example, inner ear disease
and hearing
loss), Behcet's disease, Raynaud's syndrome, organ transplant, and autoimmune
endocrine
disorders (such as, for example, diabetic-related autoimmune diseases such as
insulin-
dependent diabetes mellitus (IDDM), Addison's disease, and autoimmune thyroid
disease
(e.g., Graves' disease and thyroiditis)). More preferred such diseases
include, for example,
rheumatoid arthritis, ulcerative colitis, ANCA-associated vasculitis, lupus,
multiple sclerosis,
Sjogren's syndrome, Graves' disease, IDDM, pernicious anemia, thyroiditis, and
glomerulonephritis.
1002931 For the prevention or treatment of disease, the appropriate
dosage of an ADC
will depend on the type of disease to be treated, as defined above, the
severity and course of
the disease, whether the molecule is administered for preventive or
therapeutic purposes,
previous therapy, the patient's clinical history and response to the antibody,
and the discretion
of the attending physician. The molecule is suitably administered to the
patient at one time or
over a series of treatments. Depending on the type and severity of the
disease, about 1 ig/kg
to 15 mg/kg (e.g. 0.1-20 mg/kg) of molecule is an initial candidate dosage for
administration
to the patient, whether, for example, by one or more separate administrations,
or by
continuous infusion. A typical daily dosage might range from about 1 pg/kg to
100 mg/kg or
more, depending on the factors mentioned above. An exemplary dosage of ADC to
be
administered to a patient is in the range of about 0.1 to about 10 mg/kg of
patient weight.
[00294] For repeated administrations over several days or longer,
depending on the
condition, the treatment is sustained until a desired suppression of disease
symptoms occurs.
An exemplary dosing regimen comprises administering an initial loading dose of
about 4
mg/kg, followed by a weekly maintenance dose of about 2 mg/kg of an anti-ErbB2
antibody.
Other dosage regimens may be useful. The progress of this therapy is easily
monitored by
conventional techniques and assays.
[00295] LABELLED ANTIBODY IMAGING METHODS
1002961 In another embodiment of the invention, cysteine engineered
antibodies may
be labelled through the cysteine thiol with radionuclides, fluorescent dyes,
bioluminescence-
triggering substrate moieties, chemiluminescence-triggering substrate
moieties, enzymes, and
78
Date Recue/Date Received 2023-11-15
other detection labels for imaging experiments with diagnostic,
pharmacodynamic, and
therapeutic applications. Generally, the labelled cysteine engineered
antibody, i.e.
"biomarker" or "probe", is administered by injection, perfusion, or oral
ingestion to a living
organism, e.g. human, rodent, or other small animal, a perfused organ, or
tissue sample. The
distribution of the probe is detected over a time course and represented by an
image.
[00297] ARTICLES OF MANUFACTURE
[00298] In another embodiment of the invention, an article of
manufacture, or "kit",
containing materials useful for the treatment of the disorders described above
is provided.
The article of manufacture comprises a container and a label or package insert
on or
associated with the container. Suitable containers include, for example,
bottles, vials,
syringes, blister pack, etc. The containers may be formed from a variety of
materials such as
glass or plastic. The container holds an antibody-drug conjugate (ADC)
composition which
is effective for treating the condition and may have a sterile access port
(for example the
container may be an intravenous solution bag or a vial having a stopper
pierceable by a
hypodermic injection needle). At least one active agent in the composition is
an ADC. The
label or package insert indicates that the composition is used for treating
the condition of
choice, such as cancer. Alternatively, or additionally, the article of
manufacture may further
comprise a second (or third) container comprising a pharmaceutically-
acceptable buffer, such
as bacteriostatic water for injection (BWFI), phosphate-buffered saline,
Ringer's solution and
dextrose solution. It may further include other materials desirable from a
commercial and
user standpoint, including other buffers, diluents, filters, needles, and
syringes.
[00299] EXAMPLES
[00300] Example 1 - Preparation of Biotinylated ThioFab Phage
[00301] ThioFab-phage (5 x 1012 phage particles) were reacted with 150
fold excess of
biotin-PEO-maleimide ((+)-biotiny1-3-maleimidopropionamidy1-3,6-
dioxaoctainediamine,
Oda et al (2001) Nature Biotechnology 19:379-382, Pierce Biotechnology, Inc.)
for 3 hours at
room temperature. Excess biotin-PEO-maleimide was removed from biotin-
conjugated
phage by repeated PEG precipitations (3-4 times). Other commercially available
biotinylation reagents with electrophilic groups which are reactive with
cysteine thiol groups
may be used, including Biotin-BMCC, PEO-Iodoacetyl Biotin, Iodoacetyl-LC-
Biotin, and
Biotin-HPDP (Pierce Biotechnology, Inc.), and N 1-(3-
maleimidylpropionyl)biocytin (MPB,
Molecular Probes, Eugene, OR). Other commercial sources for biotinylation,
bifunctional
and multifunctional linker reagents include Molecular Probes, Eugene, OR, and
Sigma, St.
Louis, MO.
79
Date Recue/Date Received 2023-11-15
0
HIV NH
0 0 H
\
....._4z,
H
0 .
o
Biotin-PEO-maleimide
[00302] Example 2 - PIIESELECTOR Assay
[00303] Bovine serum albumin (BSA), erbB2 extracellular domain (HER2)
and
streptavi din (100 ul of 2 ug/m1) were separately coated on Maxisorp 96 well
plates. After
blocking with 0.5% Tween-207n PBS), biotinylated and non-biotinylated
hu4D5Fabv8-
ThioFab-Phage (2x101 phage particles) were incubated for 1 hour at room
temperature
followed by incubation with horseradish peroxidase (HRP) labeled secondary
antibody (anti-
M13 phage coat protein, pVIII protein antibody). Figure 8 illustrates the
PHESELECTOR
Assay by a schematic representation depicting the binding of Fab or ThioFab to
HER2 (top)
and biotinylated ThioFab to streptavidin (bottom).
[00304] Standard HRP reaction was carried out and the absorbance was
measured at
450 rim, 'Iltiol reactivity was measured by calculating the ratio between
0D450 for
streptavidin/OD450 for HER2. A thiol reactivity value of 1 indicates complete
biotinylation of
the cysteine thiol. In the case of Fab protein binding measurements,
hu4D5Fabv8 (2-20 ng)
was used followed by incubation with HRP labeled goat polyclonal anti-Fab
antibodies.
[00305] Example 3a - Expression and Purification of ThioFabs
[00306] ThioFabs were expressed upon induction in 34B8, a non-
suppressor E. coli
strain (Baca et al (1997) Journal Biological Chemistry 272(16):10678-84). The
harvested
cell pellet was resuspended in PBS (phosphate buffered saline), total cell
lysis was performed
by passing through a microfluidizer and the ThioFabs were purified by affinity
chromatography with protein G SEPHAROSETM (Amersham).
[00307] ThioFabs L-V15C, L-VI 10C, H-A88C, and H-A121C were expressed
and
purified by Protein-G SEPHAROSETM column chromatography. Oligomeric-Fab was
present in fractions 26 to 30, and most of the monomeric form was in fractions
31-34.
Fractions consisting of the monomeric form were pooled and analyzed by SDS-
PAGE along
with wild type hu4D5Fabv8and analyzed on SDS-PAGE gel in reducing (with DTT or
BME)
and non-reducing (without DTT or BME) conditions. Gel filtration fractions of
A121C-
ThioFab were analyzed on non-reducing SDS-PAGE.
Date Recue/Date Received 2023-11-15
[00308] ThioFabs were conjugated with biotin-PEO-maleimide as described
above and
the biotinylated-ThioFabs were further purified by Superdex-200Th (Amersham)
gel filtration
chromatography, which eliminated the free biotin-PEO-maleimide and the
oligomeric
fraction of ThioFabs. Wild type hu4D5Fabv8 and hu4D5Fabv8 A121C-ThioFab (0.5
mg in
quantity) were each and separately incubated with 100 fold molar excess of
biotin-PEO-
maleimide for 3 hours at room temperature and loaded onto a Superdex-200 gel
filtration
column to separate free biotin as well as oligomeric Fabs from the monomeric
form.
[00309] Example 3b - Analysis of ThioFabs
1003101 Enzymatic digest fragments of biotinylated hu4D5Fabv8 (A121C)
ThioFab
and wild type hu4D5Fabv8 were analyzed by liquid chromatography electrospray
ionization
mass spectroscopy (LS-ES1-MS) The difference between the 48294.5 primary mass
of
biotinylated hu4D5Fabv8 (A121C) and the 47737.0 primary mass of wild type
hu4D5Fabv8
was 557.5 mass units. This fragment indicates the presence of a single biotin-
PEO-
maleimide moiety (C211-116N507S2). Table 8 shows assignment of the
fragmentation values
which confirms the sequence.
81
Date Recue/Date Received 2023-11-15
Table 8. LC-ESI-Mass spec analysis of biotinylated hu4D5Fabv8 ThioFab
A121C after
tryptic digestion
Amino acid b Fragment y Fragment
A (Alanine) 72
M (Methionine) 203 2505
D (Aspartic acid) 318 2374
Y (Tyrosine) 481 2259
W (Tryptophan) 667 2096
G (Glycine) 724 1910
Q (glutamine) 852 1853
G (Glycine) 909 1725
T (Threonine) 1010 1668
L (Leucine) 1123 1567
V (Valine) 1222 1454
T (Threonine) 1323 1355
V (Valine) 1422 1254
S (Serine) 1509 1155
S(Scrim) 1596 1068
C (Cysteine) + biotin 2242 981
S (Serine) 2329 335
T (Threonine) 2430 248
K (Lysine) 175
[00311] Before and after Superdex-200 gel filtration, SDS-PAGE gel
analyses, with
and without reduction by DTT or BME, of biotinylated ABP- hu4D5Fabv8-A121C,
biotinylated ABP- hu4D5Fabv8-V110C, biotinylated double Cys ABP-hu4D5Fabv8-
(V110C-A88C), and biotinylated double Cys ABP-hu4D5Fabv8-(V110C-A121C) were
conducted.
[00312] Mass spectroscopy analysis (MS/MS) of of hu4D5Fabv8-(V110C)-
BMPEO-
DM1 (after Superdex-200 gel filtration purification): Fab+1 51607.5, Fab
50515.5. This data
shows 91.2% conjugation. MS/MS analysis of hu4D5Fabv8-(V110C)-BMPEO-DM1
(reduced): LC 23447.2, LC+1 24537.3, HC (Fab) 27072.5. This data shows that
all DM1
conjugation is on the light chain of the Fab.
82
Date Recue/Date Received 2023-11-15
[00313] Example 4 - Preparation of ABP-hu4D5Fabv8-(V110C)-MC-MMAE by
conjugation of ABP-hu4D5Fabv8-(V110C) and MC-MMAE
1003141 The drug linker reagent, maleimidocaproyl-monomethyl auristatin
E
(MMAE), i.e. MC-MMAE, dissolved in DMSO, is diluted in acetonitrile and water
at known
concentration, and added to chilled ABP-hu4D5Fabv8-(V110C) ThioFab in
phosphate
buffered saline (PBS) according to US 7521541, US 7659241, and US 7498298.
After about
one hour, an excess of maleimide is added to quench the reaction and cap any
unreacted
antibody thiol groups. The reaction mixture is concentrated by centrifugal
ultrafiltration and
ABP-hu4D5Fabv8-(V110C)-MC-MMAE is purified and desalted by elution through G25
resin in PBS, filtered througjh 0.2 jam filters under sterile conditions, and
frozen for storage.
[00315] Example 5 - Preparation of ABP-hu4D5Fabv8-(LC V1I0C)-MC-MMAF by
conjugation of ABP-hu4D5Fabv8-(LC V110C) and MC-MMAF
[00316] ABP-hu4D5Fabv8-(LC V110C)-MC-MMAF is prepared by conjugation of
ABP-hu4D5Fabv8-(LC V110C) ThioFab and MC-MMAF following the procedure of
Example 4.
[00317] Example 6 - Preparation of ABP-HC A121C-ThioFab -MC- val-cit-
PAB-
MMAE by conjugation of ABP-HC A121C-ThioFab and MC-val-cit-PAB-MMAE
[00318] ABP-hu4D5Fabv8-(HC A121C)-MC-val-cit-PAB-MMAE is prepared by
conjugation of ABP-hu4D5Fabv8-(HC A121C) and MC-val-cit-PAB-MMAE following the
procedure of Example 4.
1003191 Example 7 - Preparation of ABP-HC A121C-ThioFab -MC- val-cit-
PAB-
MMAF by conjugation of ABP-HC A121C-ThioFab and MC-val-cit-PAB-MMAF
[00320] ABP-hu4D5Fabv8-(HC A121C)-MC-val-cit-PAB-MMAF is prepared by
conjugation of ABP-hu4D5Fabv8-(HC A121C) and MC-val-cit-PAB-MMAF following the
procedure of Example 4.
H 0
o y NH 0.
I 0 0
0 H 0
0 H 0 H OCH3 OCH3
(NH
0\
NH2
MC-val-cit-PAB-MMAF
[00321] Example 8 - Preparation of hu4D5Fabv8-(LC V110C) ThioFab-BMPEO-
DM 1
83
Date Recue/Date Received 2023-11-15
1003221 The free cysteine on hu4D5Fabv8-(VI10C) ThioFab was modified by
the bis-
maleimido reagent BM(PEO)3 (Pierce Chemical), leaving an unreacted maleimido
group on
the surface of the antibody. This was accomplished by dissolving BM(PEO)4 in a
50%
ethanol/water mixture to a concentration of 10 mM and adding a tenfold molar
excess of
BM(PEO)3 to a solution containing hu4D5Faby8-(V110C) ThioFab in phosphate
buffered
saline at a concentration of approximately 1.6 mg/ml (10 micromolar) and
allowing it to react
for 1 hour. Excess BM(PEO)3 was removed by gel filtration (HiTrap column,
Pharmacia) in
30 mM citrate, pH 6 with 150 mM NaCl buffer. An approximate 10 fold molar
excess DM I
dissolved in dimethyl acetamide (DMA) was added to the hu4D5Fabv8-(LC V11 0C)
ThioFab-BMPEO intermediate. Dimethylformamide (DMF) may also be employed to
dissolve the drug moiety reagent. The reaction mixture was allowed to react
overnight before
gel filtration or dialysis into PBS to remove unreacted drug. Gel filtration
on S200 columns
in PBS was used to remove high molecular weight aggregates and furnish
purified
hu4D5Fabv8-(LC V11 0C) ThioFab-BMPEO-DM1.
1003231 By the same protocol, hu4D5Fabv8 (HC A121C) ThioFab-BMPEO-DM I
was
prepared.
1003241 Example 9 - In vitro cell proliferation assay
1003251 Efficacy of ADC were measured by a cell proliferation assay
employing the
following protocol (CellTiter Glo Luminiscent Cell Viability Assay, Promega
Corp.
Technical Bulletin TB288; Mendoza et al (2002) Cancer Res. 62:5485-5488):
1. An aliquot of 100 j.il of cell culture containing about 104 cells (SKBR-
3, BT474,
MCF7 or MDA-MB-468) in medium was deposited in each well of a 96-well, opaque-
walled plate.
2. Control wells were prepared containing medium and without cells.
3. ADC was added to the experimental wells and incubated for 3-5 days.
4. The plates were equilibrated to room temperature for approximately 30
minutes.
5. A volume of CellTiter-Glo Reagent equal to the volume of cell culture
medium
present in each well was added.
6. The contents were mixed for 2 minutes on an orbital shaker to induce
cell lysis.
7. The plate was incubated at room temperature for 10 minutes to stabilize
the
luminescence signal.
8. Luminescence was recorded and reported in graphs as RLU = relative
luminescence
units.
84
Date Recue/Date Received 2023-11-15
[00326] Certain cells are seeded at 1000-2000/well (PC3 lines) or 2000-
3000/well
(OVCAR-3) in a 96-well plate, 50 uL/well. After one (PC3) or two (OVCAR-3)
days, ADC
are added in 50 FtL volumes to final concentration of 9000, 3000, 1000, 333,
111, 37, 12.4,
4.1, or 1.4 ng/mL, with "no ADC" control wells receiving medium alone.
Conditions are in
duplicate or triplicate After 3 (PC3) or 4-5 (OVCAR-3) days, 100 4/well Cell
TiterGlo II is
added (luciferase-based assay; proliferation measured by ATP levels) and cell
counts are
determined using a luminometer. Data are plotted as the mean of luminescence
for each set of
replicates, with standard deviation error bars. The protocol is a modification
of the CellTiter
Glo Luminiscent Cell Viability Assay (Promega):
1. Plate 1000 cells/ well of PC3/Mucl6 , PC3/ neo (in 50 ItL/well) of media.
0vcar3
cells should be plated at 2000 cells/ well (in 50 4) of their media. (recipes
below) Allow
cells to attach overnight.
2. ADC is serially diluted 1:3 in media beginning at at working concentration
18 iu.g/m1
(this results in a final concentration of 9 ig/m1). 50 pi_ of diluted ADC is
added to the 50 jut
of cells and media already in the well.
3. Incubate 72-96 hrs (the standard is 72 hours, but watch the 0 ug/mL
concentration to
stop assay when the cells are 85-95% confluent).
4. Add 100 p.L/well of Promega Cell Titer Glo reagent, shake 3 min. and read
on
luminometer
Media: PC3/ neo and PC3/MUC16 grow in 50/50/10%FBS/glutamine/250 lig/mL G-
418 OVCAR-3 grow in RPMI/20%FBS/glutamine
[00327] Example 10 - Tumor growth inhibition, in vivo efficacy in high
expressing
HER2 transgenic explant mice
[00328] Animals suitable for transgenic experiments can be obtained
from standard
commercial sources such as Taconic (Germantown, N.Y.). Many strains are
suitable, but
FVB female mice are preferred because of their higher susceptibility to tumor
formation.
FVB males were used for mating and vasectomized CD.1 studs were used to
stimulate
pseudopregnancy. Vasectomized mice can be obtained from any commercial
supplier.
Founders were bred with either FVB mice or with 129/BL6 x FVB p53 heterozygous
mice.
The mice with heterozygosity at p53 allele were used to potentially increase
tumor formation.
However, this has proven unnecessary. Therefore, some F1 tumors are of mixed
strain.
Founder tumors are FVB only. Six founders were obtained with some developing
tumors
without having litters.
Date Recue/Date Received 2023-11-15
[00329] Animals having tumors (allograft propagated from Fo5 mmtv
transgenic mice)
were treated with a single or multiple dose by IV injection of ADC. Tumor
volume was
assessed at various time points after injection.
[00330] Tumors arise readily in transgenic mice that express a
mutationally activated
form of neu, the rat homolog of HER2, but the HER2 that is overexpressed in
human breast
cancers is not mutated and tumor formation is much less robust in transgenic
mice that
overexpress nonmutated HER2 (Webster et al (1994) Semin. Cancer Biol. 5:69-
76).
[00331] To improve tumor formation with nonmutated HER2, transgenic
mice were
produced using a HER2 cDNA plasmid in which an upstream ATG was deleted in
order to
prevent initiation of translation at such upstream ATG codons, which would
otherwise reduce
the frequency of translation initiation from the downstream authentic
initiation codon of
HER2 (for example, see Child et al (1999) J. Biol. Chem. 274: 24335-24341).
Additionally, a
chimeric intron was added to the 5' end, which should also enhance the level
of expression as
reported earlier (Neuberger and Williams (1988) Nucleic Acids Res. 16:6713;
Buchman and
Berg (1988) Mol. Cell. Biol. 8:4395; Brinster et al (1988) Proc. Natl. Acad.
Sci. USA
85:836). The chimeric intron was derived from a Promega vector, Pci-neo
mammalian
expression vector (bp 890-1022). The cDNA 3'-end is flanked by human growth
hot mone
exons 4 and 5, and polyadenylation sequences. Moreover, FVB mice were used
because this
strain is more susceptible to tumor development. The promoter from MMTV-LTR
was used
to ensure tissue-specific HER2 expression in the mammary gland. Animals were
fed the AN
76A diet in order to increase susceptibility to tumor foiination (Rao et al
(1997) Breast
Cancer Res. and Treatment 45:149-158).
[00332] Example 11 - Reduction/Oxidation of ThioMabs for Conjugation
[00333] Full length, cysteine engineered monoclonal antibodies
(ThioMabs) expressed
in CHO cells were reduced with about a 50 fold excess of TCEP (tris(2-
carboxyethyl)phosphine hydrochloride; Getz et al (1999) Anal. Biochem. Vol
273:73-80;
Soltec Ventures, Beverly, MA) for 3 hrs at 37 C. The reduced ThioMab (Figure
11) was
diluted and loaded onto a HiTrap S column in 10 mM sodium acetate, pH 5, and
eluted with
PBS containing 0.3M sodium chloride. The eluted reduced ThioMab was treated
with 200
nM aqueous copper sulfate (CuSO4) at room temperature, overnight.
Dehydroascorbic acid
(DHAA) and ambient air oxidation arc also effective oxidants.
[00334] Example 12 - Conjugation of ThioMabs
[00335] The reoxidized ThioMabs from Example 11, including thio-
trastuzumab (HC
A121C), thio-2119 (A121C), and thio-3A5 (A121C), were combined with a 10 fold
excess of
86
Date Recue/Date Received 2023-11-15
drug-linker intermediate, BM(PEO)3-DM1, mixed, and let stand for about an hour
at room
temperature to effect conjugation and form the ThioMab antibody-drug
conjugates, including
thio-trastuzumab (HC A121C)-BMPEO-DM1, thio-2H9 (HC Al 21C)-BMPEO-DM1, and
thi0-3A5 (HC A121C)-BMPEO-DM1. The conjugation mixture was gel filtered, or
loaded
and clutcd through a HiTrap S column to remove excess drug-linker intermediate
and other
impurities.
[003361 The present invention is not to be limited in scope by the
specific
embodiments disclosed in the examples which are intended as illustrations of a
few aspects of
the invention and any embodiments that arc functionally equivalent are within
the scope of
this invention. Indeed, various modifications of the invention in addition to
those shown and
described herein will become apparent to those skilled in the art and are
intended to fall
within the scope of the appended claims.
87
Date Recue/Date Received 2023-11-15