Note: Descriptions are shown in the official language in which they were submitted.
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
SYSTEMS AND METHODS FOR ARTIFICIAL INTELLIGENCE-GUIDED
BIOMOLECULE DESIGN AND ASSESSMENT
CROSS-REFERENCE TO RELATED APPLICATIONS
[0001] This application claims priority to U.S. Provisional Patent
Application No.
63/353,481, filed June 17, 2022 and entitled "Systems and Methods for
Artificial Intelligence-
Based Prediction of Amino Acid Sequences at a Binding Interface"; U.S. Patent
Application No.
17/384,104, filed July 23, 2021 and entitled "Systems and Methods for
Artificial Intelligence-
Guided Biomolecule Design and Assessment", and U.S. Provisional Patent
Application No.
63/224,801, filed July 22, 2021 and entitled "Systems and Methods for
Artificial Intelligence-
Guided Biomolecule Design and Assessment," the content of each of which is
incorporated
herein by reference in its entirety.
BACKGROUND
[0002] An increasing number of important drugs and vaccines are complex
biomolecules
referred to as biologics. For example, seven of the top ten best selling drugs
as of early 2020
were biologics, including the monoclonal antibody adalimumab (Humirag).
Biologics have
much more complex structure than traditional small molecule drugs. The process
of drug
discovery, drug development, and clinical trials require an enormous amount of
capital and time.
Typically, new drug candidates undergo in vitro testing, in vivo testing, then
clinical trials prior
to approval.
[0003] Software tools for in silico design and testing of new drug
candidates can cut the cost
and time of the preclinical pipeline. However, biologics often have hard-to-
predict properties
and molecular behavior. To date, software and computational tools (including
artificial
1
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
intelligence (AI) and machine learning) have been applied primarily to small
molecules, but,
despite extensive algorithmic advances, have achieved little success in
producing accurate
predictions for biologics due to their complexity.
SUMMARY
[0004] Described herein are systems and methods for designing and testing
custom biologic
molecules in sit/co which are useful, for example, for the treatment,
prevention, and diagnosis of
disease. In particular, in certain embodiments, the biomolecule engineering
technologies
described herein employ artificial intelligence (AI) software modules to
accurately predict
performance of candidate biomolecules and/or portions thereof (e.g., amino
acid backbones, sub-
regions of interest, etc.) with respect to particular design criteria. In
certain embodiments, the
AI-powered modules described herein determine performance scores with respect
to design
criteria such as binding to a particular target, which may be an individual
molecule, such as
protein or peptide monomer, or a complex, for example formed by multiple
protein and/or
peptide sub-units. The AI-computed performance scores may, for example, be
used as objective
functions for computer implemented optimization routines that efficiently
search a landscape of
potential protein backbone orientations and binding interface amino-acid
sequences. By virtue
of their modular design, as described herein, AI-powered scoring modules can
be used
separately, or in combination with each other, such as in a pipeline approach
where different
structural features of a custom biologic are optimized in succession.
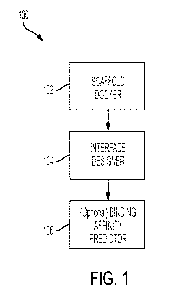
[0005] For example, presented herein is an AI-powered pipeline for
engineering a custom
biologic structure, said pipeline including (i) a scaffold docker, (ii) an
interface designer, and (iii)
a binding affinity predictor. The scaffold docker module determines favorable
three-dimensional
2
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
orientations ¨ also referred to herein as "poses" ¨ of candidate peptide
backbones susceptible to
interact with the target. In certain embodiments, candidate peptide backbones
correspond to
protein or peptide molecules with detailed structure of amino-acid side chains
stripped away, and
serve as molecular scaffolds that can be populated with amino acids to create
a custom biologic
structure. As such, these candidate peptide backbones together with the
favorable poses
determined via the scaffold docker module may be used as a starting point for
the interface
designer module, which is used to design an amino acid sequence a region of a
candidate peptide
backbone that is in proximity, and, accordingly, will influence binding to,
the target. Positions
along a particular candidate peptide backbone that, when occupied by amino
acids, will be in
proximity to the target are determined, for example, based on the geometry of
a particular
candidate peptide backbone and particular pose. The interface designer module
populates these
locations with amino acids, varying and evaluating different combinations of
amino acid types
and rotamers to generate candidate binding interfaces of the prospective
biomolecule. In certain
embodiments, a binding affinity predictor module is used to predict the
binding affinity of each
of a set of designed candidate interface regions to the target. The predicted
binding affinities
may be used to select a subset of the candidate interface regions, as well
make additional
refinements [e.g., by varying amino acids to modulate binding affinities
(e.g., in an interactive
fashion)], for use in creating a custom biologic structure for binding to the
target.
[0006] The scaffold docker module uses a candidate scaffold model, which is
a
representation of a candidate peptide backbone (e.g., a backbone of a protein
or peptide
molecule). As described in further detail herein, scaffold models used to
represent candidate
peptide backbones may also include representations of one or more side chain
atoms (e.g., atoms
that are common to a plurality of types of amino acid side chains, such as a
beta-carbon atoms).
3
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
These retained side chain atoms may act as, for example, placeholders,
identifying sites along a
candidate peptide backbone that may be occupied by amino acids. Accordingly,
in certain
embodiments, candidate scaffold models may be generated from structural models
of pre-
existing proteins or peptides (e.g., having a previously determined
crystallographic structure)],
for example by stripping away portions of amino acid side chains [e.g.,
retaining only one or
more (e.g., a single) side chain atoms, such as a beta carbon], or may be
newly generated, for
example via computational approaches. For a particular candidate scaffold
model, the scaffold
docker module generates a plurality of candidate poses with respect to the
target ("docked
poses") by rotating and/or translating the candidate scaffold model in three-
dimensional space.
[0007] In certain embodiments, candidate poses are filtered based on an
initial prediction of
whether they are likely to create a sufficient level of interaction between
the candidate scaffold
model and target (e.g., between atoms of the candidate scaffold model and
those of the target)
and/or cause clashes (e.g., excessive spatial overlap). In certain
embodiments, as described in
further detail herein, a Fast Fourier Transform (FFT) and shape map
representation approach can
be used to efficiently evaluate candidate poses in this manner.
[0008] In certain embodiments, the scaffold docker module uses a machine
learning
algorithm to evaluate and score the candidate poses and identify favorable
orientations. In certain
embodiments, a particular candidate pose is used to generate a corresponding
prospective
scaffold-target complex model that represents at least a portion of a complex
comprising the
candidate peptide backbone and target, with the candidate peptide backbone
oriented according
to the particular pose with respect to the target. The machine learning
algorithm receives the
prospective scaffold-target complex model as input, and determines a scaffold-
pose score that
measures a likelihood that the scaffold-target complex model could represent
to a viable,
4
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
physically occurring complex. The scaffold-pose score is determined by the
machine learning
algorithm based on a training procedure whereby the machine learning algorithm
is provided (i)
representations of existing, physically viable complexes as well as (ii)
artificially generated (e.g.,
computer generated), spurious, complexes that are not viable, and trained to
differentiate
between the two. In this manner, the scaffold docker module identifies
favorable poses ¨ i.e.,
three dimensional docked orientations of a candidate peptide backbone in
complex with a target¨
by assessing how 'plausible' they appear, based on the model's training. Among
other things, to
Applicant's knowledge, a machine learning approach has not been previously
applied in this
manner ¨ that is, in order to evaluate poses of and dock a scaffold model
representing a candidate
backbone, without a known amino acid sequence.
[0009] In certain embodiments, training data and the scaffold-target
complex models
received as input by the machine learning algorithm are three-dimensional
volumetric data, such
as electron density map (EDM) representations. In certain embodiments, the
machine learning
algorithm may utilize particular convolutional neural network (CNN)
architectures. In
particular, the present disclosure provides a spinal cord model (SCM)
architecture that offers
improved performance in capturing short-, middle- and long-range structural
features of an
interface (e.g., a protein-protein interface; e.g., a protein-peptide
interface). These specialized
features and approaches allow the machine learning algorithm of the scaffold
docker module to
evaluate candidate poses with a high degree of accuracy.
[0010] Accordingly, the scaffold docker module described herein utilizes
(a) efficient
sampling of the landscape of potential three-dimensional orientations of
candidate peptide
backbones for binding to a particular target along with (b) a specialized
machine learning model
trained to identify candidate peptide backbones and poses that are likely to
be viable. In this
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
manner, the scaffold docker module determines favorable orientations of
candidate scaffolds for
binding to a target.
[0011] In certain embodiments, once one or more favorable orientations ¨
also referred to as
docked poses ¨ are determined (e.g., via the scaffold docker module), an
interface designer
module is used to design an amino acid sequence of a prospective ligand (e.g.,
protein and/or
peptide) at an interface region that is in proximity to (e.g., and,
accordingly, influences binding
with) the target. In some embodiments, the interface designer module utilizes
a machine
learning algorithm that has been trained on a curated data set to accurately
predict which
interface sequences will be successful to bind the target. This training data
set may include both
existing, physically viable interfaces, for which structures have been
experimentally determined,
and also artificially generated (e.g., computer generated) mutant interfaces.
In certain
embodiments, mutant interfaces are generated by sampling both amino acid types
as well as
viable rotamers. Each interface is assigned a label that tallies the number of
mutations and
provides a measure of distance to an existing, physically viable interface.
Interfaces - both
existing and generated mutants ¨ can be binned according to a number of
mutations and the bins
sampled uniformly to generate a large uniform dataset that serves as a
foundation for training the
machine learning algorithm to make accurate predictions.
[0012] Once trained, the machine learning algorithm of the interface
designer module can be
used to score candidate interfaces. In certain embodiments, the machine
learning algorithm
determines an interface score that represents a measure of a difference
between the amino acid
sequence of a prospective interface and an interface of an existing,
physically viable complex
[e.g., a physical complex with a known interface structure (e.g., amino-acid
sequence)]. In
certain embodiments, the interface score is a predicted number of mutations
between the
6
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
prospective interface and an existing, e.g., native interface. Prospective
interfaces are thus
assessed based on their similarity or dissimilarity to native, wild-type
interfaces. Interfaces
scored as having a higher degree of similarity to native complexes (e.g., a
lower number of
predicted mutations) are identified as more likely to be successful at binding
the target and
selected, for example to be evaluated further or synthesized.
[0013] In certain embodiments, as soon as one or more candidate interfaces
are designed
(e.g., via the interface designer module), a binding affinity predictor module
is used to predict
the binding affinity of each candidate interface to the target. The predicted
binding affinities can
then be used to rank candidate interfaces, e.g., to select those with highest
predicted binding
affinities for synthesis or further evaluation. Predicting binding affinities
for large protein-
protein complexes is challenging in comparison with small-molecule binding
predictions.
Proteins are larger and more complex than small molecules and protein binding
data is also less
extensive, resulting in a smaller data set. Moreover, protein binding affinity
data can be highly
unbalanced since it relies on experimentally determined affinity values (e.g.,
Ka, K, or
logarithms thereof, such as pKa), which presents a challenge to providing
suitable training data
for machine learning techniques.
[0014] In certain embodiments, to address this challenge and allow for AI-
based prediction
of binding affinities, training approaches described herein include methods
for balancing the
dataset across a range of experimental pKa values via clustering and
differential augmentation
techniques.
[0015] Approaches described herein also may include a pre-training
technique (also referred
to as transfer learning) whereby the architecture of the machine learning
model utilized by the
binding affinity predictor module matches an architecture of a model
implemented in another,
7
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
different, module. Among other things, this allows model weights obtained when
training the
other machine learning model to be used as a starting point (e.g., pre-
training) for binding
affinity prediction. For example, in certain embodiments, both the scaffold
docker module and
the binding affinity predictor module utilize machine models that implement
the SCM
architecture described herein. Once the scaffold docker module's SCM is
trained, its weights
can be transferred to the binding affinity predictor's SCM. These initial
weights are then
adjusted by training the binding affinity predictor's SCM on experimentally
determined binding
affinity data. Among other things, this pre-training approach addresses
challenges associated
with the limited size of binding affinity data sets by leveraging training
performed to accomplish
a different (e.g., and, in certain embodiments, easier), but related, task.
[0016] Accordingly, the approaches described herein provide accurate
predictions of
biomolecule performance in a flexible modular framework that can be used to
engineer and
design custom biologics in-silico. In this manner, these tools disclosed
herein can facilitate drug
development, cutting the cost and time of the preclinical pipeline and
improving the speed and
efficiency with which new therapies are created and brought to market.
[0017] Also presented herein are systems and methods for prediction of
protein interfaces for
binding to target molecules. In certain embodiments, technologies described
herein utilize
graph-based neural networks to predict portions of protein/peptide structures
that are located at
an interface of custom biologic (e.g., a protein and/or peptide) that is being
designed for binding
to a target molecule, such as another protein or peptide. In certain
embodiments, graph-based
neural network models described herein may receive, as input, a representation
(e.g., a graph
representation) of a complex comprising a target and a partially-defined
custom biologic.
Portions of the partially-defined custom biologic may be known, while other
portions, such an
8
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
amino acid sequence and/or particular amino acid types at certain locations of
an interface, are
unknown and/or to be customized for binding to a particular target. A graph-
based neural
network model as described herein may then, based on the received input,
generate predictions of
likely acid sequences and/or types of particular amino acids at the unknown
portions. These
predictions can then be used to determine (e.g., fill in) amino acid sequences
and/or structures to
complete the custom biologic.
[0018] In one aspect, the invention is directed to a method for designing a
custom biologic
structure for binding to a target (e.g., a target molecule and/or complex) via
an artificial
intelligence (AI)-powered scaffold docker module, the method comprising: (a)
receiving and/or
generating, by a processor of a computing device, a candidate scaffold model,
wherein the
candidate scaffold model is a representation (e.g., a 3D representation) of a
candidate peptide
backbone; (b) generating, by the processor, for the candidate scaffold model,
one or more (e.g., a
plurality of) prospective scaffold-target complex models, each representing at
least a portion of a
complex comprising the candidate peptide backbone [e.g., or a variation
thereof (e.g., variations
accounting for backbone flexibility)] at a particular pose (e.g., three-
dimensional orientation)
with respect to the target; (c) for each of the one or more (e.g., plurality
of) prospective scaffold-
target complex models, determining, by the processor, a scaffold pose score
using a machine
learning model that receives, as input, a volumetric representation of at
least a portion of (e.g., an
extracted interface of) a particular prospective scaffold-target complex model
and outputs, for
the particular scaffold-target complex model, as the scaffold pose score, a
value representing a
measure of plausibility (e.g., quantifying a prediction, by the machine
learning model) [e.g., a
likelihood value representing a predicted probability or indicative thereof
(e.g., not necessarily
bounded between 0 and 1)] that the particular prospective scaffold-target
complex model
9
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
represents a native complex [e.g., such that the scaffold pose score value
represents a measure of
plausibility (e.g., a degree to which the scaffold-target complex model is
'native-like') of the
candidate peptide backbone and pose represented by the scaffold-target complex
model, as
determined by the machine learning model; e.g., wherein the scaffold pose
score is a measure of
similarity between the scaffold target complex and representations of native
complexes], thereby
determining one or more (e.g., a plurality of) scaffold pose scores; (d)
selecting, by the
processor, a subset of the one or more (e.g., plurality of) prospective
scaffold-target complex
models using the determined one or more (e.g., plurality of) scaffold pose
scores; and (e)
providing (e.g., by the processor) the selected subset of prospective scaffold-
target complex
models for use in designing the custom biologic structure and/or using the
selected subset of
prospective scaffold-target complex models to design the custom biologic
structure.
[0019] In certain embodiments, the method further comprises performing
steps (a) to (d) for
each of a plurality of candidate scaffold models [e.g., selected from a
library of scaffold models;
e.g., based on a library of protein structure models (e.g., experimentally
determined structures)],
wherein step (e) comprises designing the custom biologic structure for binding
to the target
molecule using the determined scaffold scores (e.g., testing multiple
candidate scaffolds each in
multiple orientations with respect to the target molecule).
[0020] In certain embodiments, step (b) comprises adjusting one or more
regions of the
candidate scaffold to represent variations in (e.g., portions of) the
candidate peptide backbone
accounting for backbone flexibility.
[0021] In certain embodiments, the candidate peptide backbone has a length
of less than
about 100 peptide bonds [e.g., less than about 50 peptide bonds (e.g., less
than about 20 peptide
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
bonds)] (e.g., the candidate peptide backbone has a relatively short length,
commensurate with a
peptide).
[0022] In certain embodiments, the candidate peptide backbone has a length
of greater than
about 20 peptide bonds [e.g., greater than about 50 peptide bonds (e.g.,
greater than about 100
peptide bonds)] (e.g., the candidate peptide backbone is relatively long,
commensurate with a
protein).
[0023] In certain embodiments, the candidate peptide backbone is a backbone
of a pre-
existing protein molecule (e.g., for which a crystallographic structure has
been previously
determined).
[0024] In certain embodiments, the candidate scaffold model corresponds to
a model of a
backbone of a template biologic (e.g., a protein or peptide) [e.g., from a
database (e.g., Protein
Data Bank (PDB))] {e.g., the candidate scaffold model having been generated,
and/or wherein
step (a) comprises generating the candidate scaffold model, by: receiving
and/or accessing a
structural model of a template biologic (e.g., a protein or peptide) [e.g.,
from a database (e.g.,
Protein Data Bank (PDB))]; and extracting, from the structural model, a model
of a backbone of
the template biologic [e.g., by stripping at least a portion of amino acid
side chain atoms (e.g.,
retaining a first side chain atom, such as a beta-carbon)] to generate the
candidate scaffold
model .
[0025] In certain embodiments, the template biologic comprises at least one
of a native
peptide, a native protein, an engineered protein, and an engineered peptide.
[0026] In certain embodiments, the candidate scaffold model is a
computationally generated
model (e.g., representing a candidate peptide backbone not necessarily
occurring in nature).
11
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0027] In certain embodiments, step (b) comprises applying one or more
(e.g., a plurality of)
three-dimensional rotational transforms to the candidate scaffold model,
wherein the one or more
(e.g., plurality of) rotational transformations are sampled uniformly from a
rotational space (e.g.,
using Hopf Fibration).
[0028] In certain embodiments, step (b) comprises generating a shape map
representation for
the scaffold and target molecule, wherein atoms are labeled based on their
solvent-accessible
surface area (e.g., labeled as surface or core according to their solvent-
accessible surface area
(SASA) value) (e.g., and performing a cross-correlation via a FFT to
distinguish poses that do
not cause contact, poses that do cause contact, and clashes).
[0029] In certain embodiments, the method comprises, for each particular
prospective
scaffold-target complex model of the one or more (e.g., plurality of)
prospective scaffold-target
complex models: receiving and/or generating, by the processor, a simulated
three-dimensional
electron density map (3D EDM) corresponding to (e.g., generated from) at least
a portion of the
particular prospective scaffold-target complex model; and using the simulated
3D EDM as the
volumetric representation of the particular prospective scaffold-target
complex model input to
the machine learning model.
[0030] In certain embodiments, the method comprises identifying, by the
processor, an
interface sub-region of the particular prospective scaffold-target complex
model, the interface
sub-region comprising representations of atoms of the candidate peptide
backbone and/or target
located in proximity to an interface between the candidate peptide backbone
and/or target.
[0031] In certain embodiments, identifying the interface sub-region
comprises: identifying,
as hotspots of the candidate peptide backbone, atoms (e.g., beta-carbons) of
the candidate
peptide backbone located within a threshold distance from an atom (e.g., a
beta-carbon) of the
12
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
target; identifying, as hotspots of the target molecule, atoms (e.g., beta-
carbons) of the target
located within a threshold distance from an atom (e.g., a beta-carbon) of the
candidate peptide
backbone; and determining, as the interface sub-region, a portion of the
scaffold-target complex
model representing [e.g., comprising (e.g., only) representations of and/or
bonds between] the
hotspots of the candidate peptide backbone and the hotspots of the target.
[0032] In certain embodiments, the method further comprises: identifying,
as context atoms
and/or residues of the candidate peptide backbone and/or target, atoms and/or
residues of the
candidate peptide backbone and/or target adjacent (e.g., bound) to a hotspot;
and expanding the
interface sub-region to incorporate the context atoms of the candidate peptide
backbone and/or
target.
[0033] In certain embodiments, the volumetric representation received by
the machine
learning model as input is a simulated 3D EDM.
[0034] In certain embodiments, the machine learning model comprises a
neural network
[e.g., a convolutional neural network (CNN)].
[0035] In certain embodiments, the machine learning model is a trained
model (e.g., a binary
classifier model), having been trained (e.g., using training data) to
determine a value representing
a measure of plausibility of a particular volumetric representation (e.g., 3D
EDM) of a scaffold-
target complex model received as input (e.g., wherein the value is a measure
of whether the
particular volumetric representation represents a plausible (e.g., a native
(e.g., wild-type)
complex) (e.g., a likelihood value, representing a predicted probability).
[0036] In certain embodiments, the machine learning model has been trained
(e.g., parameter
values of the machine learning model established) using training data
comprising: (A) a plurality
of native complex models, each native complex models representing at least a
portion of a native
13
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
complex based on [e.g., and determined from (e.g., allowing for
perturbations)] an
experimentally determined structural model of the native complex; and (B) a
plurality of
artificially generated variant complex models, each variant complex model
based on (e.g.,
generated from) structural models of one or more native ligands (e.g.,
proteins and/or peptides)
and/or complexes thereof {e.g., each variant complex generated by one or more
of (i), (ii), and
(iii) (including combinations thereof) as follows: (i) wherein each of at
least a portion (e.g., up to
all) of the variant complex models are generated from a structural model of a
native complex by
identifying a ligand portion and a target portion of the native complex and
applying one or more
3D rotation/translation operations to a representation of the ligand portion
to generate a variant
complex model that represents a variant of the native complex in which the
ligand portion is at a
different (e.g., new, artificial) 3D orientation with respect to the target
portion; (ii) wherein each
of at least a portion (e.g., up to all) of the variant complex models are
generated by combining
(e.g., two or more) monomeric structural models to generate variant complex
models that
represent combinations of monomers oriented [e.g., and applying one or more 3D
rotation/translations] at various poses with respect to each other; and (iii)
wherein each of at least
a portion (e.g., up to all) of the variant complexes are generated from a
structural model of a
native complex by altering a representation of a backbone of one or more of
its constituent
molecules}.
[0037] In certain embodiments, the method comprises using, by the
processor, a (e.g.,
computer implemented) optimization routine (e.g., simulated annealing) to
select the subset of
scaffold-target complex models (e.g., using the determined scaffold pose
scores) [e.g., wherein
steps (c) and/or (d) comprise using the determined scaffold pose scores as an
objective function
in a computer implemented optimization routine].
14
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0038] In certain embodiments, the target (e.g., molecule and/or complex)
comprises a
peptide and/or complex thereof
[0039] In certain embodiments, the target (e.g., molecule and/or complex)
comprises a
protein and/or complex thereof (e.g., a dimer, trimer, etc.).
[0040] In another aspect, the invention is directed to a method for
designing a custom
biologic structure for binding to a target (e.g., a target molecule and/or
complex) via an artificial
intelligence (AI)-powered interface designer module, the method comprising:
(a) receiving
and/or generating, by a processor of a computing device, a candidate scaffold-
target complex
model representing a candidate peptide backbone at a particular pose (e.g.,
three dimensional
orientation) with respect to the target; (b) receiving and/or generating, by
the processor, one or
more (e.g., a plurality of) prospective ligand-target complex models each
representing a
prospective ligand (e.g., protein and/or peptide) corresponding to the
candidate peptide backbone
[e.g., the prospective ligand having a peptide backbone corresponding to the
selected candidate
peptide backbone (e.g., wherein the peptide backbone of the prospective ligand
is the selected
candidate backbone or a version thereof (e.g., that accounts for backbone
flexibility, e.g.,
variation/movement in one or more flexible regions))] (i) comprising at least
an interface region
located in proximity to the target populated with amino acids and (ii)
positioned with respect to
the target based on the particular pose {e.g., wherein a pose of the
prospective ligand with
respect to the target is a modified version of the particular pose, accounting
for backbone
flexibility and/or allowing for rigid body perturbations (e.g., random
perturbations) [e.g., minor
translations and/or rotations [e.g., translations within about 10 angstroms or
less (e.g., about 5
angstroms or less, about 1 angstrom or less, about 0.5 angstroms or less)
along one or more
directions (e.g., an x-, y, or z-, direction) and/or rotations of
approximately 15 degrees or less
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
(e.g., approximately 5 degrees or less, e.g., approximately one or two degrees
or less about one
or more axes (e.g., x- and/or y- and/or z- axis))]]}, each prospective ligand
having a particular
(e.g., distinct) amino acid population at its interface region [e.g., a
particular (e.g., distinct)
combination (e.g., sequence) of amino acids and/or rotamers thereof at the
interface region of the
prospective ligand]; (c) for each of the one or more prospective ligand-target
complex models,
determining, by the processor, an interface score using a machine learning
model that receives,
as input, a volumetric representation of a particular prospective ligand-
target complex model and
outputs, for the particular prospective interface-target complex model, as the
interface score, a
measure of similarity and/or dissimilarity between an interface of the
particular prospective
ligand-target complex model and representations of native interfaces [e.g.,
such that the interface
score represents a measure of plausibility (e.g., a degree to which the ligand-
target complex
model is 'native-like', e.g., and/or is likely to form a viable complex when
tested experimentally)
of the amino acid interface represented by the ligand-target complex model, as
determined by the
machine learning model] [e.g., such that the interface score represents a
measure of distance
(e.g., a predicted number of amino acid mutations) between the interface of
the particular
prospective ligand-target complex and representations of native interfaces],
thereby determining
one or more (e.g., a plurality of) interface scores; (d) selecting, by the
processor, a subset of the
prospective ligand-target complex models based on the one or more (e.g.,
plurality of) interface
scores; and (e) providing (e.g., by the processor) the selected subset of
prospective ligand-target
complex models for use in designing the custom biologic structure for binding
to the target
and/or designing the custom biologic structure for binding to the target using
the selected subset
of prospective ligand-target complex models.
16
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0041] In certain embodiments, the method further comprises performing
steps (a) to (d) for
each of a plurality of candidate scaffold-target complex models.
[0042] In certain embodiments, each of at least a portion of the one or
more (e.g., plurality
of) candidate scaffold-target complex models represent a same candidate
peptide backbone at a
different particular pose with respect to the target molecule.
[0043] In certain embodiments, each of at least a portion of the one or
more (e.g., plurality
of) candidate scaffold-target complex models represent a different candidate
peptide backbone in
complex with the target molecule.
[0044] In certain embodiments, the a candidate scaffold-target complex
model is a member
of a subset of prospective scaffold-target complex models determined using an
artificial
intelligence (Al) powered scaffold docker module (e.g., that performs the
method of any one of
various aspects and embodiments described herein).
[0045] In certain embodiments, step (b) comprises assigning an initial
amino acid sequence
to an interface region of the candidate peptide backbone (e.g., a randomly
generate amino acid
sequence; e.g., based on a native protein or peptide from which the candidate
peptide backbone
was derived) and mutating amino acids to generate, for each prospective ligand-
target complex
models, the particular amino acid population at the interface region of the
prospective ligand.
[0046] In certain embodiments, the method comprises, for each particular
prospective ligand-
target complex model of the one or more (e.g., plurality of) prospective
ligand-target complex
models: receiving and/or generating, by the processor, a simulated three-
dimensional electron
density map (3D EDM) corresponding to (e.g., generated from) at least a
portion of the particular
prospective ligand-target complex model; and using the simulated 3D EDM as the
volumetric
17
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
representation of the particular prospective ligand-target complex model input
to the machine
learning model.
[0047] In certain embodiments, the method comprises identifying, by the
processor, an
interface sub-region of the particular prospective ligand-target complex
model, the interface sub-
region comprising representations of atoms of the prospective ligand and/or
target located in
proximity to an interface between the prospective ligand and target.
[0048] In certain embodiments, identifying the interface sub-region
comprises: identifying,
as hotspots of the prospective ligand, residues of the prospective ligand
located within a
threshold distance from a residue of the target; identifying, as hotspots of
the target molecule,
residues of the target located within a threshold distance from a residue of
the prospective ligand;
and determining, as the interface sub-region, a portion of the ligand-target
complex model
representing [e.g., comprising (e.g., only) representations of and/or bonds
between] the hotspots
of the prospective ligand and the hotspots of the target.
[0049] In certain embodiments, the method further comprises: identifying,
as context atoms
and/or residues of the prospective ligand and/or target, atoms and/or residues
of the prospective
ligand and/or target adjacent (e.g., bound) to a hotspot; and expanding the
interface sub-region to
incorporate the context atoms and/or residues of the prospective ligand and/or
target.
[0050] In certain embodiments, the volumetric representation received by
the machine
learning model as input comprises a simulated 3D EDM.
[0051] In certain embodiments, the machine learning model comprises a
neural network
[e.g., a convolutional neural network (CNN)].
[0052] In certain embodiments, the machine learning model is a trained
model (e.g., a
regression model), having been trained (e.g., using training data) to
determine (e.g., as the
18
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
measure of similarity and/or dissimilarity) a predicted number of mutations
between (i) an
interface that a particular volumetric representation (e.g., 3D EDM) of at
least a portion of a
ligand-target complex model received as input represents and (ii)
representations of native
interfaces.
[0053] In certain embodiments, the machine learning model has been trained
(e.g., parameter
values of the machine learning model established) using training data
comprising: (A) a plurality
of native interface models, each native interface models representing at least
a portion of a native
interface based on (e.g., and determined/derived from) an experimentally
determined structural
model of the native interface; (B) a plurality of artificially generated
mutant interface models,
each mutant interface model based on a mutated version of a native interface
[e.g., generated
from a structural model of a native interface by mutating amino acids of the
native interface
(e.g., changing an amino acid type and/or rotamer)].
[0054] In certain embodiments, the method comprises using, by the
processor, a (e.g.,
computer implemented) optimization routine (e.g., simulated annealing) to
select the subset of
the prospective ligand-target complex models [e.g., wherein steps (c) and/or
(d) comprise using
the determined interface scores as an objective function in a computer
implemented optimization
routine].
[0055] In another aspect, the invention is directed to a method for
engineering a custom
biologic structure for binding to a target (e.g., target molecule and/or
complex), via an artificial
intelligence (Al) powered binding affinity predictor module, the method
comprising: (a)
receiving and/or generating, by a processor of a computing device, one or more
prospective
ligand-target complex models, each representing at least a portion of a
complex comprising a
prospective (e.g., custom) biologic and the target, with the prospective
(e.g., custom) biologic
19
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
positioned at a particular pose (e.g., three-dimensional orientation) with
respect to the target; and
(b) for each of the one or more prospective ligand-target complex models,
determining, by the
processor, a binding affinity score using a machine learning model that
receives, as input, a
volumetric representation of a particular ligand-target complex model (e.g.,
determined using the
method of various aspects and/or embodiments described herein, for example
with respect to
various artificial intelligence (AI)-powered scaffold docker modules and/or
artificial intelligence
(AI)-powered interface designer modules for designing a custom biologic
structure for binding to
a target (e.g., a target molecule and/or complex), described herein) and
outputs, as the binding
affinity score, a value representing a predicted binding affinity between the
prospective (e.g.,
custom) biologic and the target molecule of the particular ligand-target
complex model.
[0056] In certain embodiments, the method comprises performing steps (a)
and (b) for a
plurality of ligand-target complex models, thereby determining a plurality of
binding affinity
scores and: (c) selecting, by the processor, a subset of the prospective
ligand-target complex
models based on the plurality of binding affinity scores; and (d) designing a
custom biologic
structure using the selected subset of prospective biologic-target complex
modules.
[0057] In certain embodiments, step (c) comprises selecting, as the subset,
those prospective
ligand-target complexes having binding affinity scores greater than a
particular binding affinity
threshold value, thereby selecting a high binding affinity subset.
[0058] In certain embodiments, step (c) comprises ranking the prospective
ligand-target
complexes according to the plurality of binding affinity scores and selecting
the subset based on
the ranking [e.g., selecting, as the subset, a portion of the ligand-target
complexes having a
higher binding affinity score than others (e.g., a top 1, top 2, top 5, etc.;
e.g., a top 10%, a top
quartile, etc.)].
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0059] In certain embodiments, the method comprises: performing steps (a)
and (b) for an
initial set of one or more ligand-target complex models, to determine an
initial set of binding
affinity scores; and (e.g., iteratively) updating ligand-target complex models
of the initial set to,
for each ligand-target complex model, mutate amino acids (e.g., type and/or
rotamer) of the
custom biologic and/or the target molecule, to generate a set of mutated
ligand-target complex
models and performing step (b) for the set of the mutated ligand-target
complex models to
determine an updated set of binding affinity scores [e.g., and comparing the
updated set of
binding affinity scores with the initial set of binding affinity scores (e.g.,
to predict stabilizing
and/or destabilizing mutations; e.g., to predict changes in binding affinity
after mutation; e.g., to
tune/modulate (e.g., increase or decrease) binding affinity))].
[0060] In certain embodiments, the binding affinity score is a predicted
dissociation constant
(e.g., pKd) value.
[0061] In another aspect, the invention is directed to a method for
engineering a custom
biologic structure for binding to a target molecule in silico, the method
comprising: (a) receiving
and/or generating, by a processor of a computing device, one or more candidate
scaffolds
models, wherein each of the one or more candidate scaffold models is a
representation of a
candidate peptide backbone; (b) determining, by the processor, based on the
one or more
candidate scaffold models, a set of one or more prospective scaffold-target
complex models
using a scaffold docker module (e.g., a machine learning software module,
e.g., software that
performs a method of any aspects and/or embodiments described herein, for
example, various
methods for designing a custom biologic structure for binding to a target
(e.g., a target molecule
and/or complex) via an artificial intelligence (AI)-powered scaffold docker
module, described
herein); (c) determining, by the processor, based on at least one member of
the set of prospective
21
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
scaffold-target complex models, a set of prospective ligand-target complex
models using an
interface designer module (e.g., a machine learning software module, e.g.,
software that performs
a method of any aspects and/or embodiments described herein, for example,
various methods for
designing a custom biologic structure for binding to a target (e.g., a target
molecule and/or
complex) via an artificial intelligence (AI)-powered interface designer
module, described
herein); and (d) providing (e.g., by the processor) the set of prospective
scaffold-target complex
models and/or the set of prospective ligand target complex models for use in
designing the
custom biologic structure for binding to the target molecule and/or designing
the custom biologic
structure using the determined set of prospective ligand-target complex models
and/or the set of
prospective scaffold-target complex models.
[0062] In certain embodiments, step (d) comprises: determining, by the
processor, for each
of at least a portion of the set of prospective ligand-target complex models
and/or the set of
prospective scaffold-target complex models, predicted binding affinity score
using a binding
affinity predictor module (e.g., a machine learning software module, e.g.,
software that performs
a method of any aspects or embodiments described herein, for example, various
methods for
designing a custom biologic structure for binding to a target (e.g., target
molecule and/or
complex), via an artificial intelligence (AI) powered binding affinity
predictor module, described
herein); and using the set of predicted binding affinity scores to design the
custom biologic
structure (e.g., to identify high-binding affinity biologic structures).
[0063] In certain embodiments, each of the scaffold docker module and the
interface
designer module comprises a machine learning software module.
[0064] In another aspect, the invention is directed to a method for
determining and/or
evaluating a predicted structure of a biologic complex, the method comprising:
(a) receiving
22
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
and/or generating, by the processor, one or more biologic complex models each
representing a
complex comprising a first biologic positioned at a (e.g., distinct)
particular pose (e.g., 3D
orientation) with respect to a second biologic; (b) for each of the one or
more biologic complex
models, determining, by the processor, a pose score using a machine learning
model that
receives, as input, a volumetric representation of a particular biologic
complex model and
outputs, for the particular biologic complex model, as the pose score, a value
representing a
measure of plausibility [e.g., likelihood value representing a predicted
probability or indicative
thereof (e.g., not necessarily bounded between 0 and I)] that the particular
biologic complex
model represents a native complex [e.g., such that the scaffold pose score
value represents a
measure of plausibility (e.g., a degree to which the scaffold-target complex
model is 'native-
like') of the pose represented by the biologic complex model, as determined by
the machine
learning model; e.g., wherein the scaffold pose score is a measure of
similarity between the
scaffold target complex and representations of native complexes], thereby
determining one or
more pose scores; (c) selecting, by the processor, a subset of the one or more
(e.g., plurality of)
biologic complex models using the determined one or more (e.g., plurality of)
pose scores; and
(d) storing and/or providing, by the processor, the selected subset for
display and/or further
processing.
[0065] In certain embodiments, each of the one or more biologic complex
models comprises:
a first biologic model representing at least a portion of the first biologic;
and a second biologic
model representing at least a portion of the second biologic.
[0066] In certain embodiments, the first biologic model is a scaffold model
representing a
backbone of the first biologic (e.g., and omitting at least a portion of amino
acid side-chain
23
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
atoms); and/or the second biologic model is a scaffold model representing a
backbone of the
second biologic (e.g., and omitting at least a portion of amino acid side-
chain atoms).
[0067] In certain embodiments, both the first and second biologic models
are scaffold
models.
[0068] In certain embodiments, the first biologic model includes
representations of at least a
portion of amino-acid side chains of the first biologic (e.g., within an sub-
region in proximity to
the second biologic; e.g., over the entire first biologic) (e.g., is a ligand
model, having at least a
portion populated with amino acids); and/or the second biologic model includes
representations
of at least a portion of amino-acid side chains of the second biologic (e.g.,
within an sub-region
in proximity to the first biologic; e.g., over the entire second biologic)
(e.g., is a ligand model,
having at least a portion populated with amino acids).
[0069] In certain embodiments, both the first and second biologic models
include
representations of at least a portion of amino acid side chains of the
respective biologic that they
represent.
[0070] In certain embodiments, the method further comprises using the
selected subset of
biologic complex models as an initial starting point for one or more physics-
based (e.g., force-
field; e.g., energy functional) docking routines.
[0071] In certain embodiments, step (a) comprises receiving the one or more
biologic
complex models, and the received one or more biologic complex models having
been determined
using one or more physics-based docking routines.
[0072] In another aspect, the invention is directed to a method for
determining a predicted
structure of a biologic complex, the method comprising: (a) receiving and/or
generating, by the
processor, one or more biologic complex models, each representing a complex
comprising a first
24
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
biologic having a first amino acid sequence and positioned at a particular
pose (e.g., 3D
orientation) with respect to a second biologic having a second amino acid
sequence; (b) for each
of the one or more biologic complex models, determining, by the processor, an
sequence score
using a machine learning model that receives, as input, a volumetric
representation of a particular
biologic complex model and outputs, for the particular biologic complex model,
as the sequence
score, a measure of similarity and/or dissimilarity between an interface
between the first and
second biologic [e.g., the interface formed first and second amino acid
sequences of the
particular biologic complex in combination, e.g., including spatial
relationships] and
representations of native interfaces [e.g., such that the sequence score
represents a measure of
plausibility (e.g., a degree to which the biologic complex model is 'native-
like') of the amino
acid sequences represented by the biologic complex model, as determined by the
machine
learning model] [e.g., such that the sequence score represents a measure of
distance (e.g., a
predicted number of amino acid mutations) between the interface of the
particular prospective
ligand-target complex and representations of native interfaces]; (c)
selecting, by the processor, a
subset of the one or more (e.g., a plurality of) biologic complex models using
the determined one
or more (e.g., a plurality of) sequence scores; and (d) storing and/or
providing, by the processor,
the selected subset for display and/or further processing.
[0073] In certain embodiments, step (a) comprises receiving and/or
generating a plurality of
biologic complex models wherein, for each particular biologic complex model,
the first biologic
is a distinct first biologic (e.g., having a distinct first amino acid
sequence).
[0074] In certain embodiments, for each particular biologic complex model,
the second
biologic is a same second biologic (e.g., having a same second amino acid
sequence).
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0075] In certain embodiments, for each particular biologic complex model,
the second
biologic is a distinct second biologic (e.g., having a distinct second amino
acid sequence).
[0076] In certain embodiments, step (a) comprises varying the first and/or
second amino acid
sequence to generate a plurality of distinct biologic complex models.
[0077] In certain embodiments, variations in the first amino acid sequence
are restricted to a
portion of the first amino acid sequence that corresponds to an interface sub-
region of the first
biologic in proximity to the second biologic; and/or variations in the second
amino acid sequence
are restricted to a portion of the second amino acid sequence that corresponds
to an interface sub-
region of the second biologic in proximity to the first biologic.
[0078] In certain embodiments, variations in the first and/or second amino
acid sequences
are not limited to an interface sub-region of the first and/or second
biologic, such that the method
provides for designing an entire biologic (e.g., not just an interface; e.g.,
a complete protein or
peptide).
[0079] In another aspect, the invention is directed to a pipeline (e.g., a
computer architecture
pipeline) for designing custom biologic structures in silico, said pipeline
comprising a plurality
of AI-powered modules, wherein each module in the pipeline optimizes candidate
custom
biologic structural features with respect to a particular criteria [e.g.,
using a machine learning
model that receives, as input, a representation of the candidate custom
biologic structural
features and generates, as output, a score representing a measure of
performance with respect to
the particular criteria].
[0080] In another aspect, the invention is directed to a system comprising:
a processor of a
computing device; and a memory having instructions stored thereon, wherein the
instructions,
26
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
when executed by the processor, cause the processor to perform the method of
any one of the
aspects and/or embodiments described herein.
[0081] In another aspect, the invention is directed to a method for
designing a custom
biologic structure for binding to a target (e.g., a target molecule and/or
complex) in-silico via a
pipeline of artificial intelligence (AI)-powered modules, the method
comprising: (a) receiving
and/or generating, by a processor of a computing device, one or more (e.g., a
plurality of)
prospective scaffold-target complex models (e.g., 3D representations), each
representing at least
a portion of a complex comprising a candidate peptide backbone [e.g., at least
a portion of the
scaffold-target complex models representing a same candidate peptide backbone
and/or
variations thereof (e.g., variations accounting for backbone flexibility) at
various different poses;
e.g., at least a portion of the scaffold-target complex models representing
distinct candidate
peptide backbones, e.g., so as to evaluate a library of different candidate
peptide backbones] at a
particular pose (e.g., three-dimensional orientation) with respect to the
target; (b) for each of the
one or more (e.g., plurality of) prospective scaffold-target complex models,
determining, by the
processor, a scaffold pose score using a first machine learning model, thereby
determining one or
more (e.g., a plurality of) scaffold pose scores; (c) selecting, by the
processor, a scaffold-target
complex model of the one or more (e.g., plurality of) prospective scaffold-
target complex models
using the determined one or more (e.g., plurality of) scaffold pose scores,
thereby identifying a
selected candidate peptide backbone and a selected pose represented by the
selected scaffold-
target complex model as a backbone and pose on which to build a custom
interface portion of a
ligand for binding to the target molecule; (d) generating, by the processor,
based on the selected
scaffold-target complex model, one or more (e.g., a plurality of) prospective
ligand-target
complex models (e.g., 3D representations), each representing a prospective
ligand (e.g., protein
27
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
and/or peptide) corresponding to the selected candidate peptide backbone
[e.g., the prospective
ligand having a peptide backbone corresponding to the selected candidate
peptide backbone
(e.g., wherein the peptide backbone of the prospective ligand is the selected
candidate backbone
or a version thereof (e.g., that accounts for backbone flexibility, e.g.,
variation/movement in one
or more flexible regions))] (i) comprising at least an interface region
located in proximity to the
target populated with amino acids and (ii) positioned with respect to the
target based on the
selected pose {e.g., wherein a pose of the prospective ligand with respect to
the target is a
modified version of the selected pose, accounting for backbone flexibility
and/or allowing for
rigid body perturbations (e.g., random perturbations) [e.g., minor
translations and/or rotations
[e.g., translations within 10 angstroms or less (e.g., 5 angstroms or less, 1
angstrom or less, 0.5
angstroms or less) along one or more directions (e.g., an x-, y, or z-,
direction) and/or rotations of
15 degrees or less (e.g., 5 degrees or less, e.g., about one or two degrees or
less about one or
more axes (e.g., x- and/or y- and/or z- axis))]]}, each prospective ligand
comprising a particular
(e.g., distinct) amino acid population at its interface region [e.g., a
particular (e.g., distinct)
combination (e.g., sequence) of amino acids and/or rotamers thereof at the
interface region of the
prospective ligand]; (e) for each of the one or more (e.g., plurality)
prospective ligand-target
complex models, determining, by the processor, an interface score using a
second machine
learning model, thereby determining one or more (e.g., a plurality) of
interface scores; (f)
selecting, by the processor, a subset of the prospective ligand-target complex
models based on at
least a portion of the one or more (e.g., plurality of) interface scores; and
(g) providing (e.g., by
the processor) the selected subset of prospective ligand-target complex models
for use in
designing the custom biologic structure for binding to the target and/or
designing the custom
28
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
biologic structure for binding to the target using the selected subset of
prospective ligand-target
complex models.
[0082] In certain embodiments, the candidate peptide backbone comprises a
length of less
than about 100 peptide bonds [e.g., less than about 50 peptide bonds (e.g.,
less than about 20
peptide bonds)] (e.g., the candidate peptide backbone has a relatively short
length, commensurate
with a peptide).
[0083] In certain embodiments, the candidate peptide backbone comprises a
length of greater
than about 20 peptide bonds [e.g., greater than about 50 peptide bonds (e.g.,
greater than about
100 peptide bonds)] (e.g., the candidate peptide backbone is relatively long,
commensurate with
a protein).
[0084] In certain embodiments, the candidate peptide backbone is a backbone
of a pre-
existing protein molecule (e.g., for which a crystallographic structure has
been previously
determined).
[0085] In certain embodiments, step (a) comprises receiving and/or
generating a candidate
scaffold model representing at least a portion of the candidate peptide
backbone, wherein the
candidate scaffold model corresponds to a model of a backbone of a template
biologic (e.g., a
protein or peptide) [e.g., from a database (e.g., Protein Data Bank (PDB))]
{e.g., the candidate
scaffold having been generated, and/or wherein step (a) comprises generating
the candidate
scaffold model, by: receiving and/or accessing a structural model of a
template biologic (e.g., a
protein or peptide) [e.g., from a database (e.g., Protein Data Bank (PDB))];
and extracting, from
the structural model, a model of a backbone of the template biologic [e.g., by
stripping at least a
portion of amino acid side chain atoms (e.g., retaining a first side chain
atom, such as a beta-
carbon)] to generate the candidate scaffold model}.
29
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0086] In certain embodiments, the template biologic comprises at least one
of a wild-type
peptide, a wild-type protein, an engineered protein, and an engineered
peptide.
[0087] In certain embodiments, step (a) comprises receiving a
computationally generated
candidate scaffold model and/or generating a candidate scaffold model
representing the
candidate peptide backbone via a computational approach (e.g., thereby
representing a candidate
peptide backbone not necessarily occurring in nature).
[0088] In certain embodiments, step (a) comprises applying a one or more
(e.g., plurality of)
three-dimensional rotational transforms to a candidate scaffold model
representing the candidate
peptide backbone, wherein the one or more (e.g., plurality of) three-
dimensional rotational
transformations are sampled uniformly from a rotational space (e.g., using
Hopf Fibration).
[0089] In certain embodiments, step (a) comprises generating a shape map
representation for
each of the candidate peptide backbone and the target molecule, the shape map
representation
comprising representations of atoms that are labeled based on their solvent-
accessible surface
area (e.g., labeled as surface or core according to their solvent-accessible
surface area (SASA)
value) (e.g., and performing a cross-correlation via FFT to distinguish poses
that do not cause
contact, poses that do cause contact, and clashes).
[0090] In certain embodiments, the first machine learning model receives,
as input, for each
particular prospective scaffold-target complex model, a volumetric
representation of at least a
portion of (e.g., an extracted interface of) the particular prospective
scaffold-target complex
model and outputs, for the particular scaffold-target complex model, as the
scaffold pose score, a
value representing a measure of plausibility (e.g., quantifying a prediction,
by the first machine
learning model) [e.g., a likelihood value representing a predicted probability
or indicative thereof
(e.g., not necessarily bounded between 0 and 1)] that the particular
prospective scaffold-target
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
complex model represents a native complex [e.g., such that the scaffold pose
score represents a
measure of plausibility (e.g., a degree to which the scaffold-target complex
model is 'native-
like') of the candidate peptide backbone and pose represented by the scaffold-
target complex
model, as determined by the machine learning model; e.g., wherein the scaffold
pose score is a
measure of similarity between the scaffold target complex and representations
of native
complexes], thereby determining the one or more (e.g., plurality of) scaffold
pose scores.
[0091] In certain embodiments, the method comprises, for each particular
prospective
scaffold-target complex model of the one or more (e.g., plurality of)
prospective scaffold-target
complex models: receiving and/or generating, by the processor, a simulated
three-dimensional
electron density map (3D EDM) corresponding to (e.g., generated from) at least
a portion of the
particular prospective scaffold-target complex model; and using the simulated
3D EDM as the
volumetric representation of the particular prospective scaffold-target
complex model input to
the fist machine learning model.
[0092] In certain embodiments, the method comprises identifying, by the
processor, an
interface sub-region of the particular prospective scaffold-target complex
model, the interface
sub-region comprising representations of atoms of the candidate peptide
backbone and/or target
located in proximity to an interface between the candidate peptide backbone
and/or target.
[0093] In certain embodiments, the first machine learning model is a
trained model (e.g., a
binary classifier model), having been trained (e.g., using training data) to
determine a value
representing a measure of plausibility of a particular volumetric
representation (e.g., 3D EDM)
of a scaffold-target complex model received as input (e.g., wherein the value
is a measure of
whether the particular volumetric representation represents a plausible (e.g.,
a native (e.g., wild-
type) complex) (e.g., a likelihood value, representing a predicted
probability).
31
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0094] In certain embodiments, the first machine learning model has been
trained (e.g.,
parameter values of the machine learning model established) using training
data comprising: (A)
a plurality of native complex models, each native complex models representing
at least a portion
of a native complex based on [e.g., and determined from (e.g., allowing for
perturbations)] an
experimentally determined structural model of the native complex; and (B) a
plurality of
artificially generated variant complex models, each variant complex model
based on (e.g.,
generated from) structural models of one or more native ligands and/or
complexes thereof {e.g.,
each variant complex generated by one or more of (i), (ii), and (iii)
(including combinations
thereof) as follows: (i) wherein each of at least a portion (e.g., up to all)
of the variant complex
models are generated from a structural model of a native complex by
identifying a ligand portion
and a target portion of the native complex and applying one or more 3D
rotation/translation
operations to a representation of the ligand portion to generate a variant
complex model that
represents a variant of the native complex in which the ligand portion is at a
different (e.g., new,
artificial) 3D orientation with respect to the target portion; (ii) wherein
each of at least a portion
(e.g., up to all) of the variant complex models are generated by combining
(e.g., two or more)
monomeric structural models to generate variant complex models that represent
combinations of
monomers oriented [e.g., and applying one or more 3D rotation/translations] at
various poses
with respect to each other; and (iii) wherein each of at least a portion
(e.g., up to all) of the
variant complexes are generated from a structural model of a native complex by
altering a
representation of a backbone of one or more of its constituent molecules} .
[0095] In certain embodiments, step (d) comprises assigning an initial
amino acid sequence
to an interface region of the candidate peptide backbone (e.g., a randomly
generated amino acid
sequence; e.g., based on a native protein or peptide from which the candidate
peptide backbone
32
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
was derived) and mutating amino acids to generate, for each prospective ligand-
target complex
model, the particular amino acid population at the interface region of the
prospective ligand.
[0096] In certain embodiments, the second machine learning model receives,
as input, for
each particular prospective ligand-target complex model, a volumetric
representation of at least a
portion of the particular prospective ligand-target complex model and outputs,
for the particular
prospective ligand-target complex model, as the interface score, a measure of
similarity and/or
dissimilarity between an interface of the particular prospective ligand-target
complex model and
representations of native interfaces [e.g., such that the interface score
represents a measure of
plausibility (e.g., a degree to which the ligand-target complex model is
'native-like', e.g., and/or
is likely to form a viable complex when tested experimentally) of the amino
acid interface
represented by the ligand-target complex model, as determined by the machine
learning
model][e.g., such that the interface score represents a measure of distance
(e.g., a predicted
number of amino acid mutations) between the particular prospective ligand-
target complex and a
native complex].
[0097] In certain embodiments, the second machine learning model is a
trained model (e.g., a
regression model), having been trained (e.g., using training data) to
determine (e.g., as the
measure of similarity and/or dissimilarity) a predicted number of mutations
between (i) an
interface that a particular volumetric representation (e.g., 3D EDM) of at
least a portion of a
ligand-target complex model received as input represents and (ii)
representations of native
interfaces.
[0098] In certain embodiments, the second machine learning model has been
trained (e.g.,
parameter values of the machine learning model established) using training
data comprising: (A)
a plurality of native interface models, each native interface models
representing at least a portion
33
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
of a native interface based on (e.g., determined / derived from) an
experimentally determined
structural model of the native interface; and (B) a plurality of artificially
generated mutant
interface models, each mutant interface model based on a mutated version of a
native interface
[e.g., generated from a structural model of a native interface by mutating
amino acids of the
native interface (e.g., changing an amino acid type and/or rotamer)].
[0099] In certain embodiments, at least one of the first machine learning
model and the
second machine learning model comprises a neural network [e.g., a
convolutional neural network
(CNN)].
[0100] In certain embodiments, the method comprises: using, by the
processor, a (e.g.,
computer implemented) optimization routine (e.g., simulated annealing) to
select the scaffold-
target complex model of the prospective scaffold-target complex models using
the determined
scaffold pose scores [e.g., wherein steps (b) and/or (c) comprise using the
determined scaffold
pose scores as an objective function in a computer implemented optimization
routine]; and/or
using, by the processor, a (e.g., computer implemented) optimization routine
(e.g., simulated
annealing) to select the subset of the prospective ligand-target complex
models [e.g., wherein
steps (e) and/or (f) comprise using the determined interface scores as an
objective function in a
computer implemented optimization routine].
[0101] In certain embodiments, the target (e.g., molecule and/or complex)
comprises is a
peptide and/or complex thereof
[0102] In certain embodiments, the target (e.g., molecule and/or complex)
comprises a
protein and/or a protein complex (e.g., a dimer, trimer, etc.).
[0103] In certain embodiments, the method further comprises: for each of at
least a portion
of the subset of the prospective ligand-target complex models determined at
step (f),
34
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
determining, by the processor, a binding affinity score using a third machine
learning model that
receives, as input, a volumetric representation of at least a portion of a
particular ligand-target
complex model and outputs, as the binding affinity score, a value representing
a predicted
binding affinity between the prospective custom biologic structure and the
target molecule of the
particular ligand-target complex model; and at step (g), using the one or more
binding affinity
scores to design the custom biologic structure.
[0104] In certain embodiments, the method comprises: selecting one or more
high binding
affinity ligand-target complex models based on the one or more binding
affinity scores; and
providing the one or more high binding affinity ligand-target complex models
for use in
designing the custom biologic structure and/or designing the custom biologic
structure using the
one or more high binding affinity ligand-target complex models.
[0105] In certain embodiments, the method comprises comparing the one or
more binding
affinity scores to a threshold value.
[0106] In certain embodiments, the method comprises ranking the prospective
ligand target-
complex models of the subset determined at step (f) according to the one or
more determined
binding affinity scores.
[0107] In certain embodiments, the binding affinity score is a predicted
dissociation constant
(e.g., a pKd value).
[0108] In certain embodiments, the third machine learning model comprises a
neural network
(e.g., a CNN).
[0109] In another aspect, the invention is directed to a system for
designing a custom
biologic structure for binding to a target (e.g., a target molecule and/or
complex) in-silico via a
pipeline of artificial intelligence (AI)-powered modules, the system
comprising: a processor of a
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
computing device; and a memory having instructions stored thereon, wherein the
instructions,
when executed by the processor, cause the processor to: (a) receive and/or
generate one or more
(e.g., a plurality of) prospective scaffold-target complex models, each
representing at least a
portion of a complex comprising a candidate peptide backbone at a particular
pose (e.g., three-
dimensional orientation) with respect to the target [e.g., at least a portion
of the scaffold-target
complex models representing a same candidate peptide backbone and/or
variations thereof (e.g.,
variations accounting for backbone flexibility) at various different poses;
e.g., at least a portion
of the scaffold-target complex models representing distinct candidate peptide
backbones, e.g., so
as to evaluate a library of different candidate peptide backbones]; (b) for
each of the one or more
(e.g., plurality of) prospective scaffold-target complex models, determine a
scaffold pose score
using a first machine learning model, thereby determining one or more (e.g., a
plurality of)
scaffold pose scores; (c) select a scaffold-target complex model of the one or
more (e.g., plurality
of) prospective scaffold-target complex models using the determined one or
more (e.g., plurality
of) scaffold pose scores, thereby identifying a selected candidate peptide
backbone and a selected
pose represented by the selected scaffold-target complex model as a backbone
and pose on which
to build a custom interface portion of a ligand for binding to the target
molecule; (d) generate,
based on the selected scaffold-target complex model, one or more (e.g., a
plurality of)
prospective ligand-target complex models, each representing a prospective
ligand (e.g., protein
and/or peptide) corresponding to the selected candidate peptide backbone
[e.g., the prospective
ligand having a peptide backbone corresponding to the selected candidate
peptide backbone
(e.g., wherein the peptide backbone of the prospective ligand is the selected
candidate backbone
or a version thereof (e.g., that accounts for backbone flexibility, e.g.,
variation/movement in one
or more flexible regions))] (i) comprising at least an interface region
located in proximity to the
36
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
target molecule populated with amino acids and (ii) positioned with respect to
the target based on
the selected pose {e.g., wherein a pose of the prospective ligand with respect
to the target is a
modified version of the selected pose, accounting for backbone flexibility
and/or allowing for
rigid body perturbations (e.g., random perturbations) [e.g., minor
translations and/or rotations
[e.g., translations within 10 angstroms or less (e.g., 5 angstroms or less, 1
angstrom or less, 0.5
angstroms or less) along one or more directions (e.g., an x-, y, or z-,
direction) and/or rotations of
15 degrees or less (e.g., 5 degrees or less, e.g., about one or two degrees or
less about one or
more axes (e.g., x- and/or y- and/or z- axis))]]}, each prospective ligand
comprising a particular
(e.g., distinct) amino acid population at its interface region [e.g., a
particular (e.g., distinct)
combination (e.g., sequence) of amino acids and/or rotamers thereof at the
interface region of the
prospective ligand]; (e) for each of the one or more (e.g., plurality of)
prospective ligand-target
complex models, determine an interface score using a second machine learning
model, thereby
determining one or more (e.g., a plurality of) interface scores; (f) select a
subset of the
prospective ligand-target complex models based on the one or more (e.g.,
plurality of) interface
scores; and (g) provide the selected subset of prospective ligand-target
complex models for use
in designing the custom biologic structure for binding to the target and/or
design the custom
biologic structure for binding to the target using the selected subset of
prospective ligand-target
complex models.
[0110] In another aspect, the invention is directed to a method for
designing a custom
biologic structure for binding to a target (e.g., a target molecule and/or
complex) via an artificial
intelligence (AI)-powered scaffold docker module, the method comprising: (a)
receiving and/or
generating, by a processor of a computing device, a candidate scaffold model,
wherein the
candidate scaffold model is a representation (e.g., a 3D representation) of a
candidate peptide
37
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
backbone; (b) generating, by the processor, for the candidate scaffold model,
one or more (e.g., a
plurality of) prospective scaffold-target complex models, each representing at
least a portion of a
complex comprising the candidate peptide backbone [e.g., or a variation
thereof (e.g., variations
accounting for backbone flexibility); e.g., wherein step (b) comprises
adjusting one or more
regions of the candidate scaffold to represent variations in (e.g., portions
of) the candidate
peptide backbone accounting for backbone flexibility] at a particular pose
(e.g., three-
dimensional orientation) with respect to the target; (c) for each of the one
or more (e.g., plurality
of) prospective scaffold-target complex models, determining, by the processor,
a scaffold pose
score using a machine learning model that receives, as input, a volumetric
representation of at
least a portion of (e.g., an extracted interface of) a particular prospective
scaffold-target complex
model and outputs, for the particular scaffold-target complex model, as the
scaffold pose score, a
value representing a measure of plausibility (e.g., quantifying a prediction,
by the machine
learning model) [e.g., a likelihood value representing a predicted probability
or indicative thereof
(e.g., not necessarily bounded between 0 and 1)] that the particular
prospective scaffold-target
complex model represents a native complex [e.g., such that the scaffold pose
score value
represents a measure of plausibility (e.g., a degree to which the scaffold-
target complex model is
'native-like') of the candidate peptide backbone and pose represented by the
scaffold-target
complex model, as determined by the machine learning model; e.g., wherein the
scaffold pose
score is a measure of similarity between the scaffold target complex and
representations of native
complexes], thereby determining one or more (e.g., a plurality of) scaffold
pose scores; (d)
selecting, by the processor, a subset of the one or more (e.g., plurality of)
prospective scaffold-
target complex models using the determined one or more (e.g., plurality of)
scaffold pose scores;
and (e) providing (e.g., by the processor) the selected subset of prospective
scaffold-target
38
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
complex models for use in designing the custom biologic structure and/or using
the selected
subset of prospective scaffold-target complex models to design the custom
biologic structure.
[0111] In another aspect, the invention is directed to a method for
designing a custom
biologic structure for effectively binding to a target in-silico via a
pipeline of artificial
intelligence (AI)-powered modules, the method comprising: (a) receiving and/or
generating, by
a processor of a computing device, one or more prospective scaffold-target
complex models,
each representing at least a portion of a complex comprising a candidate
peptide backbone at a
particular pose with respect to the target; (b) selecting, by the processor, a
scaffold-target
complex model of the one or more prospective scaffold-target complex models
using a first
machine learning model, thereby identifying a selected candidate peptide
backbone and a
selected pose represented by the selected scaffold-target complex model as a
backbone and pose
on which to build a custom interface portion of a ligand for binding to the
target molecule; (c)
generating, by the processor, based on the selected scaffold-target complex
model, one or more
prospective ligand-target complex models, each representing a prospective
ligand corresponding
to the selected candidate peptide backbone (i) comprising at least an
interface region located in
proximity to the target populated with amino acids and (ii) positioned with
respect to the target
based on the selected pose, each prospective ligand comprising a particular
amino acid
population at its interface region; (d) selecting, by the processor, a subset
of the prospective
ligand-target complex models using a second machine learning model; and (e)
providing the
selected subset of prospective ligand-target complex models for use in
designing the custom
biologic structure for binding to the target.
[0112] In certain embodiments, the first machine learning model has been
trained to evaluate
plausibility of a particular scaffold-target complex model using training data
comprising: (A) a
39
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
plurality of native complex models, each native complex model representing at
least a portion of
a native complex based on an experimentally determined structural model of the
native complex;
and (B) a plurality of artificially generated variant complex models, each
variant complex model
based on structural models of one or more native ligands and/or complexes
thereof.
[0113] In certain embodiments, the second machine learning model is a
trained model,
having been trained to determine a predicted number of mutations between (i)
an interface that a
particular volumetric representation of at least a portion of a ligand-target
complex model
received as input represents and (ii) representations of native interfaces. In
certain embodiments,
the second machine learning model has been trained using training data
comprising: (A) a
plurality of native interface models, each native interface model representing
at least a portion of
a native interface based on an experimentally determined structural model of
the native interface;
and (B) a plurality of artificially generated mutant interface models, each
mutant interface model
based on a mutated version of a native interface.
[0114] In another aspect, the invention is directed to a system for
designing a custom
biologic structure for binding to a target in-silico via a pipeline of
artificial intelligence (AI)-
powered modules, the system comprising: a processor of a computing device; and
a memory
having instructions stored thereon, wherein the instructions, when executed by
the processor,
cause the processor to: (a) receive and/or generate one or more prospective
scaffold-target
complex models, each representing at least a portion of a complex comprising a
candidate
peptide backbone at a particular pose with respect to the target; (b) select a
scaffold-target
complex model of the one or more prospective scaffold-target complex models
using a first
machine learning model, thereby identifying a selected candidate peptide
backbone and a
selected pose represented by the selected scaffold-target complex model as a
backbone and pose
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
on which to build a custom interface portion of a ligand for binding to the
target molecule; (c)
generate, based on the selected scaffold-target complex model, one or more
prospective ligand-
target complex models, each representing a prospective ligand corresponding to
the selected
candidate peptide backbone (i) comprising at least an interface region located
in proximity to the
target molecule populated with amino acids and (ii) positioned with respect to
the target based on
the selected pose, each prospective ligand comprising a particular amino acid
population at its
interface region; (d) select a subset of the prospective ligand-target complex
models using a
second machine learning model; and (e) provide the selected subset of
prospective ligand-target
complex models for use in designing the custom biologic structure for binding
to the target.
[0115] In another aspect, the invention is directed to a method for
designing a custom
biologic structure for binding to a target via an artificial intelligence (AI)-
powered scaffold
docker module, the method comprising: (a) receiving and/or generating, by a
processor of a
computing device, a candidate scaffold model, wherein the candidate scaffold
model is a
representation of a candidate peptide backbone; (b) generating, by the
processor, for the
candidate scaffold model, one or more prospective scaffold-target complex
models, each
representing at least a portion of a complex comprising the candidate peptide
backbone at a
particular pose with respect to the target; (c) selecting, by the processor, a
subset of the one or
more prospective scaffold-target complex models using a machine learning model
that evaluates
plausibility that each prospective scaffold-target complex model represents a
native complex;
and (d) providing the selected subset of prospective scaffold-target complex
models for use in
designing the custom biologic structure for binding to the target.
[0116] In another aspect, the invention is directed to a method for
designing a custom
biologic structure for binding to a target in-silico via a pipeline of
artificial intelligence (AI)-
41
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
powered modules, the method comprising: (a) receiving and/or generating, by a
processor of a
computing device, one or more prospective scaffold-target complex models, each
representing at
least a portion of a complex comprising a candidate peptide backbone at a
particular pose with
respect to the target, wherein the candidate peptide backbone is a prospective
backbone of the
custom biologic structure being designed and is represented using a scaffold
model that identifies
types and locations of peptide backbone atoms while omitting amino-acid side
chain atoms; (b)
for each of the one or more prospective scaffold-target complex models,
determining, by the
processor, a scaffold pose score, wherein determining the scaffold pose score
for each particular
one of the one or more prospective scaffold-target complex models comprises:
generating, based
on the particular scaffold-target complex model, a corresponding
representation (e.g., a graph
representation or a 3D volumetric representation); and using the corresponding
representation as
input to a first machine learning model that determines, as output, the
scaffold pose score for the
particular scaffold-target complex model; (c) selecting, by the processor, a
scaffold-target
complex model of the one or more prospective scaffold-target complex models
using the
determined one or more scaffold pose scores, thereby identifying a selected
candidate peptide
backbone, oriented at a selected pose, on which to build a custom interface
portion of a ligand
for binding to the target; (d) generating, by the processor, based on the
selected scaffold-target
complex model, one or more prospective ligand-target complex models, each
representing a
prospective ligand corresponding to the selected candidate peptide backbone
(i) comprising at
least an interface region located in proximity to the target populated with
amino acids and (ii)
positioned with respect to the target based on the selected pose, each
prospective ligand
comprising a particular amino acid population at its interface region; (e) for
each of the one or
more prospective ligand-target complex models, determining, by the processor,
an interface
42
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
score using a second machine learning model, thereby determining one or more
interface scores;
(f) selecting, by the processor, a subset of the prospective ligand-target
complex models based on
at least a portion of the one or more interface scores; and (g) providing the
selected subset of
prospective ligand-target complex models for use in designing the custom
biologic structure for
binding to the target.
[0117] In certain embodiments, the candidate peptide backbone comprises a
length of less
than about 100 peptide bonds.
[0118] In certain embodiments, the candidate peptide backbone comprises a
length of greater
than about 20 peptide bonds.
[0119] In certain embodiments, the candidate peptide backbone is a backbone
of a pre-
existing protein molecule.
[0120] In certain embodiments, step (a) comprises receiving and/or
generating a candidate
scaffold model representing at least a portion of the candidate peptide
backbone, wherein the
candidate scaffold model corresponds to a model of a backbone of a template
biologic.
[0121] In certain embodiments, the template biologic comprises at least one
of a wild-type
peptide, a wild-type protein, an engineered protein, and an engineered
peptide.
[0122] In certain embodiments, step (a) comprises receiving a
computationally generated
candidate scaffold model and/or generating a candidate scaffold model
representing the
candidate peptide backbone via a computational approach.
[0123] In certain embodiments, step (a) comprises applying a one or more
three-dimensional
rotational transforms to a candidate scaffold model representing the candidate
peptide backbone,
wherein the one or more three-dimensional rotational transformations are
sampled uniformly
from a rotational space.
43
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0124] In certain embodiments, step (a) comprises generating a shape map
representation for
each of the candidate peptide backbone and the target molecule, the shape map
representation
comprising representations of atoms that are labeled based on their solvent-
accessible surface
area.
[0125] In certain embodiments, the first machine learning model-outputs,
for the particular
scaffold-target complex model, as the scaffold pose score, a value
representing a measure of
plausibility that the particular prospective scaffold-target complex model
represents a native
complex, thereby determining the one or more scaffold pose scores.
[0126] In certain embodiments, step (b) comprises generating, as the
representation based on
the particular scaffold-target complex model, a simulated three-dimensional
electron density map
(3D EDM) corresponding to at least a portion of the particular prospective
scaffold-target
complex model; and using the simulated 3D EDM as the representation of the
particular
prospective scaffold-target complex model input to the first machine learning
model.
[0127] In certain embodiments, the method comprises identifying, by the
processor, an
interface sub-region of the particular prospective scaffold-target complex
model, the interface
sub-region comprising representations of atoms of the candidate peptide
backbone and/or target
located in proximity to an interface between the candidate peptide backbone
and/or target.
[0128] In certain embodiments, the first machine learning model is a
trained model, having
been trained to determine a value representing a measure of plausibility of a
particular
representation of a scaffold-target complex model received as input.
[0129] In certain embodiments, the first machine learning model has been
trained using
training data comprising: (A) a plurality of native complex models, each
native complex model
representing at least a portion of a native complex based on an experimentally
determined
44
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
structural model of the native complex; and (B) a plurality of artificially
generated variant
complex models, each variant complex model based on structural models of one
or more native
ligands and/or complexes thereof.
[0130] In certain embodiments, step (d) comprises assigning an initial
amino acid sequence
to an interface region of the candidate peptide backbone and mutating amino
acids to generate,
for each prospective ligand-target complex model, the particular amino acid
population at the
interface region of the prospective ligand.
[0131] In certain embodiments, the second machine learning model receives,
as input, for
each particular prospective ligand-target complex model, a representation of
at least a portion of
the particular prospective ligand-target complex model and outputs, for the
particular prospective
ligand-target complex model, as the interface score, a measure of similarity
and/or dissimilarity
between an interface of the particular prospective ligand-target complex model
and
representations of native interfaces.
[0132] In certain embodiments, the second machine learning model is a
trained model,
having been trained to determine a predicted number of mutations between (i)
an interface that a
particular representation of at least a portion of a ligand-target complex
model received as input
represents and (ii) representations of native interfaces.
[0133] In certain embodiments, the second machine learning model has been
trained using
training data comprising: (A) a plurality of native interface models, each
native interface model
representing at least a portion of a native interface based on an
experimentally determined
structural model of the native interface; and (B) a plurality of artificially
generated mutant
interface models, each mutant interface model based on a mutated version of a
native interface.
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0134] In certain embodiments, at least one of the first machine learning
model and the
second machine learning model comprises a neural network.
[0135] In certain embodiments, the method comprises: using, by the
processor, an
optimization routine to select the scaffold-target complex model of the
prospective scaffold-
target complex models using the determined scaffold pose scores; and/or using,
by the processor,
an optimization routine to select the subset of the prospective ligand-target
complex models.
[0136] In certain embodiments, the target comprises a peptide and/or a
complex thereof.
[0137] In certain embodiments, the method further comprises: for each of at
least a portion
of the subset of the prospective ligand-target complex models determined at
step (f),
determining, by the processor, a binding affinity score using a third machine
learning model that
receives, as input, a representation of at least a portion of a particular
ligand-target complex
model and outputs, as the binding affinity score, a value representing a
predicted binding affinity
between the prospective custom biologic structure and the target molecule of
the particular
ligand-target complex model; and, at step (g), using the one or more binding
affinity scores to
design the custom biologic structure. In certain embodiments, the method
comprises: selecting
one or more high binding affinity ligand-target complex models based on the
one or more
binding affinity scores; and providing the one or more high binding affinity
ligand-target
complex models for use in designing the custom biologic structure. In certain
embodiments, the
method comprises comparing the one or more binding affinity scores to a
threshold value. In
certain embodiments, the method comprises ranking the prospective ligand
target-complex
models of the subset determined at step (f) according to the one or more
determined binding
affinity scores.
46
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0138] In another aspect, the invention is directed to a system for
designing a custom
biologic structure for binding to a target in-silico via a pipeline of
artificial intelligence (AI)-
powered modules, the system comprising: a processor of a computing device; and
a memory
having instructions stored thereon, wherein the instructions, when executed by
the processor,
cause the processor to: (a) receive and/or generate one or more prospective
scaffold-target
complex models, each representing at least a portion of a complex comprising a
candidate
peptide backbone at a particular pose with respect to the target, wherein the
candidate peptide
backbone is a prospective backbone of the custom biologic structure being
designed and is
represented using a scaffold model that identifies types and locations of
peptide backbone items
while omitting amino-acid side chain atoms; (b) for each of the one or more
prospective
scaffold-target complex models, determine a scaffold pose score, wherein
determining the
scaffold pose score for each particular one of the one or more prospective
scaffold-target
complex models comprises: generating, based on the particular scaffold-target
complex model, a
corresponding representation; and using the corresponding representation as
input to a first
machine learning model that determines, as output, the scaffold pose score for
the particular
scaffold-target complex model; (c) select a scaffold-target complex model of
the one or more
prospective scaffold-target complex models using the determined one or more
scaffold pose
scores, thereby identifying a selected candidate peptide backbone, oriented at
a selected pose, on
which to build a custom interface portion of a ligand for binding to the
target; (d) generate, based
on the selected scaffold-target complex model, one or more prospective ligand-
target complex
models, each representing a prospective ligand corresponding to the selected
candidate peptide
backbone (i) comprising at least an interface region located in proximity to
the target molecule
populated with amino acids and (ii) positioned with respect to the target
based on the selected
47
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
pose, each prospective ligand comprising a particular amino acid population at
its interface
region; (e) for each of the one or more prospective ligand-target complex
models, determine an
interface score using a second machine learning model, thereby determining one
or more
interface scores; (f) select a subset of the prospective ligand-target complex
models based on the
one or more interface scores; and (g) provide the selected subset of
prospective ligand-target
complex models for use in designing the custom biologic structure for binding
to the target.
[0139] In another aspect, the invention is directed to a method for
designing a custom
biologic structure for binding to a target via an artificial intelligence (AI)-
powered scaffold
docker module, the method comprising: (a) receiving and/or generating, by a
processor of a
computing device, a candidate scaffold model, wherein the candidate scaffold
model is a
representation of at least a portion of a candidate peptide backbone, wherein
the candidate
peptide backbone is a prospective backbone of the custom biologic structure
being designed and
wherein the candidate scaffold model represents the candidate peptide backbone
by identifying
types and locations of peptide backbone atoms while omitting amino-acid side
chain atoms; (b)
generating, by the processor, for the candidate scaffold model, one or more
prospective scaffold-
target complex models, each representing at least a portion of a complex
comprising the
candidate peptide backbone at a particular pose with respect to the target;
(c) for each of the one
or more prospective scaffold-target complex models, determining, by the
processor, a scaffold
pose score, wherein determining the scaffold pose score for each particular
one of the one or
more prospective scaffold-target complex models comprises: generating, based
on the particular
scaffold-target complex model, a corresponding representation (e.g., a graph
model or a 3D
volumetric representation); and using the corresponding representation as
input to a machine
learning model that determines, as output, the scaffold pose score, wherein
the scaffold pose
48
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
score is a value representing a measure of plausibility that the particular
prospective scaffold-
target complex model represents a native complex, thereby determining one or
more scaffold
pose scores; (d) selecting, by the processor, a subset of the one or more
prospective scaffold-
target complex models using the determined one or more scaffold pose scores;
and (e) providing
the selected subset of prospective scaffold-target complex models for use in
designing the
custom biologic structure for binding to the target.
[0140] In certain embodiments, step (e) comprises populating at least an
interface region of
one or more of the selected subset of prospective scaffold-target complex
models with amino
acid side chains to generate one or more ligand models for use in designing
the custom biologic
structure.
[0141] In another aspect, the invention is directed to a system for
designing a custom
biologic structure for binding to a target via an artificial intelligence (AI)-
powered scaffold
docker module, the system comprising: a processor of a computing device; and a
memory
having instructions stored thereon, wherein the instructions, when executed by
the processor,
cause the processor to: (a) receive and/or generate a candidate scaffold
model, wherein the
candidate scaffold model is a representation of at least a portion of a
candidate peptide backbone,
wherein the candidate peptide backbone is a prospective backbone of the custom
biologic
structure being designed and wherein the candidate scaffold model represents
the candidate
peptide backbone by identifying types and locations of peptide backbone atoms
while omitting
amino-acid side chain atoms; (b) generate, for the candidate scaffold model,
one or more
prospective scaffold-target complex models, each representing at least a
portion of a complex
comprising the candidate peptide backbone at a particular pose with respect to
the target; (c) for
each of the one or more prospective scaffold-target complex models, determine
a scaffold pose
49
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
score, wherein determining the scaffold pose score for each particular one of
the one or more
prospective scaffold-target complex models comprises: generating, based on the
particular
scaffold-target complex model, a corresponding representation (e.g., a graph
model or a 3D
volumetric representation); and using the corresponding representation as
input to a machine
learning model that determines, as output, the scaffold pose score, wherein
the scaffold pose
score is a value representing a measure of plausibility that the particular
prospective scaffold-
target complex model represents a native complex, thereby determining one or
more scaffold
pose scores; (d) select a subset of the one or more prospective scaffold-
target complex models
using the determined one or more scaffold pose scores; and (e) provide the
selected subset of
prospective scaffold-target complex models for use in designing the custom
biologic structure for
binding to the target.
[0142] In certain embodiments, the instructions, when executed by the
processor, cause the
processor to, in step (e), populate at least an interface region of one or
more of the selected
subset of prospective scaffold-target complex models with amino acid side
chains to generate
one or more ligand models for use in designing the custom biologic structure.
[0143] In another aspect, the invention is directed to a method for
generating an amino acid
interface of a custom biologic for binding to a target molecule in silico, the
method comprising:
(a) receiving (e.g., and/or accessing), by a processor of a computing device,
a preliminary graph
representation of a complex comprising (i) at least a portion of a target
molecule and (ii) at least
a portion of the custom biologic; (b) using, by the processor, the preliminary
graph
representation as input to a machine learning model (e.g., a graph neural
network model) that
generates, as output, a structural prediction for at least a portion of the
complex (e.g., a graph
representation comprising a probability distribution at each node) comprising
(e.g., but not
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
limited to) a prediction of an amino acid type and/or structure for each of
one or more amino acid
positions within an interface region of the custom biologic; and (c) using, by
the processor, the
interface prediction to determine the amino acid interface for the custom
biologic.
[0144] In another aspect, the invention is directed to a system for
generating an amino acid
interface of a custom biologic, the system comprising a processor of a
computing device and
memory having instructions stored thereon, wherein the instructions, when
executed by the
processor, cause the processor to perform the method described above.
[0145] In one aspect, the invention is directed to a method for the in-
silico design of an
amino acid interface of a biologic for binding to a target (e.g., wherein the
biologic is an in-
progress custom biologic being designed for binding to an identified target),
the method
comprising: (a) receiving (e.g., and/or accessing), by a processor of a
computing device, an
initial scaffold-target complex graph comprising a graph representation of at
least a portion of a
biologic complex comprising the target and a peptide backbone of the in-
progress custom
biologic, the initial scaffold-target complex graph comprising: a target graph
representing at least
a portion of the target; and a scaffold graph representing at least a portion
of the peptide
backbone of the in-progress custom biologic, the scaffold graph comprising a
plurality of
scaffold nodes, a subset of which are unknown interface nodes, wherein each of
said unknown
interface nodes: (i) represents a particular (amino acid) interface site,
along the peptide
backbone of the in-progress custom biologic, that is [e.g., is a-priori known
to be, or has been
determined (e.g., by the processor) to be] located in proximity to one or more
amino acids of the
target, and (ii) has a corresponding node feature vector comprising a side
chain type component
vector (e.g., and/or side chain structure component vector) populated with one
or more masking
values, thereby representing an unknown, to-be determined, amino acid side
chain [e.g., wherein
51
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
the node feature vector further comprises (i) a constituent vector
representing a local backbone
geometry (e.g., representing three torsional angles of backbone atoms, e.g.,
using two elements
for ¨ a sine and a cosine of¨ each angle) and/or (ii) a constituent vector
representing a side chain
geometry (e.g., one or more chi angles)]; (b) generating, by the processor,
using a machine
learning model, one or more likelihood graphs based on the initial scaffold-
target complex graph,
each of the one or more likelihood graphs comprising a plurality of nodes, a
subset of which are
classified interface nodes, each of which: (i) corresponds to a particular
unknown interface node
of the scaffold graph and represents a same particular interface site along
the peptide backbone
of the in-progress custom biologic as the corresponding particular interface
node, and (ii) has a
corresponding node feature vector comprising a side chain component vector
populated with one
or more likelihood values (e.g., representing a likelihood that a side chain
at the particular amino
acid site is of a particular type); (c) using, by the processor, the one or
more likelihood graphs to
determine a predicted interface comprising, for each interface site, an
identification of a
particular amino acid side chain type; and, optionally, (d) providing (e.g.,
by the processor) the
predicted interface for use in designing the amino acid interface of the in-
progress custom
biologic and/or using the predicted interface to design the amino acid
interface of the in-progress
custom biologic.
[0146] In certain embodiments, the target graph comprises a plurality of
target nodes, each
representing a particular (amino acid) site of the target and having a
corresponding node feature
vector comprising one or more constituent vectors (e.g., a plurality of
concatenated constituent
vectors), each constituent vector representing a particular (e.g., physical;
e.g., structural) feature
of the particular (amino acid) site. In certain embodiments, for each node
feature vector of a
target node, the one or more constituent vectors comprise one or more members
selected from
52
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
the group consisting of: a side chain type, representing a particular type of
side chain (e.g., via a
one-hot encoding scheme); a local backbone geometry [e.g., representing three
torsional angles
of backbone atoms (e.g., using two elements for ¨ a sine and a cosine of¨ each
angle)]; and a
side chain geometry (e.g., one or more chi angles).
[0147] In certain embodiments, the target graph comprises a plurality of
target edges, each
associated with two particular target nodes and having a corresponding edge
feature vector
comprising one or more constituent vectors representing a relative position
and/or orientation of
two (amino acid) sites represented by the two particular target nodes.
[0148] In certain embodiments, the node feature vectors and/or edge feature
vectors of the
target graph are invariant with respect to three-dimensional translation
and/or rotation of the
target.
[0149] In certain embodiments, for each node feature vector of a target
node, at least a subset
of the one or more constituent vectors comprise absolute coordinate values
(e.g., on a particular
coordinate frame) of one or more atoms (e.g., backbone atoms; e.g., a beta
carbon atom) of the
particular amino acid site represented by the target node.
[0150] In certain embodiments, each of the plurality of scaffold nodes of
the scaffold graph
represents a particular (amino acid) site along the peptide backbone of the in-
progress custom
biologic and has a corresponding node feature vector comprising one or more
constituent
vectors, each constituent vector representing a particular (e.g., physical;
e.g., structural) feature
of the particular (amino acid) site. In certain embodiments, for each node
feature vector of a
scaffold node, the one or more constituent vectors comprise one or more
members selected from
the group consisting of: a side chain type, representing a particular type of
side chain (e.g., via a
one-hot encoding scheme); a local backbone geometry [e.g., representing three
torsional angles
53
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
of backbone atoms (e.g., using two elements for ¨ a sine and a cosine of¨ each
angle)]; and a
side chain geometry (e.g., one or more chi angles).
[0151] In certain embodiments, the scaffold graph comprises a plurality of
scaffold edges,
each associated with two particular scaffold nodes and having a corresponding
edge feature
vector comprising one or more constituent vectors representing a relative
position and/or
orientation of two (amino acid) sites represented by the two particular
scaffold nodes. In certain
embodiments, the initial scaffold-target complex graph comprises a plurality
of scaffold-target
edges, each corresponding to (e.g., connecting) a particular scaffold node and
a particular target
node and having a corresponding edge feature vector comprising one or more
constituent vectors
representing a relative position and/or orientation of two (amino acid) sites
represented by the
particular scaffold node and the particular target node.
[0152] In certain embodiments, the node feature vectors and/or edge feature
vectors of the
scaffold graph are invariant with respect to three-dimensional translation
and/or rotation of the
peptide backbone of the in-progress custom biologic.
[0153] In certain embodiments, for each node feature vector of a target
node, at least a subset
of the one or more constituent vectors comprise absolute coordinate values
(e.g., on a particular
coordinate frame) of one or more atoms (e.g., backbone atoms; e.g., a beta
carbon atom) of the
particular amino acid site represented by the target node.
[0154] In certain embodiments, a subset of the scaffold nodes are known
scaffold nodes,
each having a node feature vector comprising a known side chain component
representing a
(e.g., a-priori known and/or previously determined) side chain type.
[0155] In certain embodiments, the machine learning model is or comprises a
graph neural
network.
54
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0156] In certain embodiments, step (b) comprises generating a plurality of
likelihood graphs
in an iterative fashion: in a first iteration, using the initial scaffold-
target complex graph as an
initial input to generate an initial likelihood graph; in a second, subsequent
iteration, using the
initial likelihood graph and/or an initial interface prediction based thereon,
as input to the
machine learning model, to generate a refined likelihood graph and/or a
refined interface
prediction based thereon; and repeatedly using the refined likelihood graph
and/or refined
interface prediction generated by the machine learning model at one iteration
as input to the
machine learning model for a subsequent iteration, thereby repeatedly refining
the likelihood
graph and or an interface prediction based thereon.
[0157] In another aspect, the invention is directed to a system for the in-
silico design of an
amino acid interface of a biologic for binding to a target (e.g., wherein the
biologic is an in-
progress custom biologic being designed for binding to an identified target),
the system
comprising: a processor of a computing device; and a memory having
instructions stored
thereon, wherein the instructions, when executed by the processor, cause the
processor to: (a)
receive (e.g., and/or access) an initial scaffold-target complex graph
comprising a graph
representation of at least a portion of a biologic complex comprising the
target and a peptide
backbone of the in-progress custom biologic, the initial scaffold-target
complex graph
comprising: a target graph representing at least a portion of the target; and
a scaffold graph
representing at least a portion of the peptide backbone of the in-progress
custom biologic, the
scaffold graph comprising a plurality of scaffold nodes, a subset of which are
unknown interface
nodes, wherein each of said unknown interface nodes: (i) represents a
particular (amino acid)
interface site, along the peptide backbone of the in-progress custom biologic,
that is [e.g., is a-
priori known to be, or has been determined (e.g., by the processor) to be]
located in proximity to
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
one or more amino acids of the target, and (ii) has a corresponding node
feature vector
comprising a side chain type component vector (e.g., and/or side chain
structure component
vector) populated with one or more masking values, thereby representing an
unknown, to-be
determined, amino acid side chain [e.g., wherein the node feature vector
further comprises (i) a
constituent vector representing a local backbone geometry (e.g., representing
three torsional
angles of backbone atoms, e.g., using two elements for ¨ a sine and a cosine
of¨ each angle)
and/or (ii) a constituent vector representing a side chain geometry (e.g., one
or more chi angles)];
(b) generate, using a machine learning model, one or more likelihood graphs
based on the initial
scaffold-target complex graph, each of the one or more likelihood graphs
comprising a plurality
of nodes, a subset of which are classified interface nodes, each of which: (i)
corresponds to a
particular unknown interface node of the scaffold graph and represents a same
particular
interface site along the peptide backbone of the in-progress custom biologic
as the corresponding
particular interface node, and (ii) has a corresponding node feature vector
comprising a side
chain component vector populated with one or more likelihood values (e.g.,
representing a
likelihood that a side chain at the particular amino acid site is of a
particular type); (c) use the
one or more likelihood graphs to determine a predicted interface comprising,
for each interface
site, an identification of a particular amino acid side chain type; and,
optionally, (d) provide the
predicted interface for use in designing the amino acid interface of the in-
progress custom
biologic and/or using the predicted interface to design the amino acid
interface of the in-progress
custom biologic.
[0158] In certain embodiments, the target graph comprises a plurality of
target nodes, each
representing a particular (amino acid) site of the target and having a
corresponding node feature
vector comprising one or more constituent vectors (e.g., a plurality of
concatenated constituent
56
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
vectors), each constituent vector representing a particular (e.g., physical;
e.g., structural) feature
of the particular (amino acid) site. In certain embodiments, for each node
feature vector of a
target node, the one or more constituent vectors comprise one or more members
selected from
the group consisting of: a side chain type, representing a particular type of
side chain (e.g., via a
one-hot encoding scheme); a local backbone geometry [e.g., representing three
torsional angles
of backbone atoms (e.g., using two elements for ¨ a sine and a cosine of¨ each
angle)]; and a
side chain geometry (e.g., one or more chi angles).
[0159] In certain embodiments, the target graph comprises a plurality of
target edges, each
associated with two particular target nodes and having a corresponding edge
feature vector
comprising one or more constituent vectors representing a relative position
and/or orientation of
two (amino acid) sites represented by the two particular target nodes.
[0160] In certain embodiments, the node feature vectors and/or edge feature
vectors of the
target graph are invariant with respect to three-dimensional translation
and/or rotation of the
target.
[0161] In certain embodiments, for each node feature vector of a target
node, at least a subset
of the one or more constituent vectors comprise an absolute (e.g., on a
particular coordinate
frame) of one or more atoms (e.g., backbone atoms; e.g., a beta carbon atom)
of the particular
amino acid site represented by the target node.
[0162] In certain embodiments, each of the plurality of scaffold nodes of
the scaffold graph
represents a particular (amino acid) site along the peptide backbone of the in-
progress custom
biologic and has a corresponding node feature vector comprising one or more
constituent
vectors, each constituent vector representing a particular (e.g., physical;
e.g., structural) feature
of the particular (amino acid) site. In certain embodiments, for each node
feature vector of a
57
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
scaffold node, the one or more constituent vectors comprise one or more
members selected from
the group consisting of: a side chain type, representing a particular type of
side chain (e.g., via a
one-hot encoding scheme); a local backbone geometry [e.g., representing three
torsional angles
of backbone atoms (e.g., using two elements for ¨ a sine and a cosine of¨ each
angle)]; and a
side chain geometry (e.g., one or more chi angles).
[0163] In certain embodiments, the scaffold graph comprises a plurality of
scaffold edges,
each associated with two particular scaffold nodes and having a corresponding
edge feature
vector comprising one or more constituent vectors representing a relative
position and/or
orientation of two (amino acid) sites represented by the two particular
scaffold nodes. In certain
embodiments, the initial scaffold-target complex graph comprises a plurality
of scaffold-target
edges, each corresponding to (e.g., connecting) a particular scaffold node and
a particular target
node and having a corresponding edge feature vector comprising one or more
constituent vectors
representing a relative position and/or orientation of two (amino acid) sites
represented by the
particular scaffold node and the particular target node.
[0164] In certain embodiments, the node feature vectors and/or edge feature
vectors of the
scaffold graph are invariant with respect to three-dimensional translation
and/or rotation of the
peptide backbone of the in-progress custom biologic.
[0165] In certain embodiments, for each node feature vector of a target
node, at least a subset
of the one or more constituent vectors comprise absolute coordinate values
(e.g., on a particular
coordinate frame) of one or more atoms (e.g., backbone atoms; e.g., a beta
carbon atom) of the
particular amino acid site represented by the target node.
58
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0166] In certain embodiments, a subset of the scaffold nodes are known
scaffold nodes,
each having a node feature vector comprising a known side chain component
representing a
(e.g., a-priori known and/or previously determined) side chain type.
[0167] In certain embodiments, the machine learning model is or comprises a
graph neural
network.
[0168] In certain embodiments, the instructions, when executed by the
processor, cause the
processor to, in step (b), generate a plurality of likelihood graphs in an
iterative fashion: in a first
iteration, use the initial scaffold-target complex graph as an initial input
to generate an initial
likelihood graph; in a second, subsequent iteration, use the initial
likelihood graph and/or an
initial interface prediction based thereon, as input to the machine learning
model, to generate a
refined likelihood graph and/or a refined interface prediction based thereon;
and repeatedly use
the refined likelihood graph and/or refined interface prediction generated by
the machine
learning model at one iteration as input to the machine learning model for a
subsequent iteration,
thereby repeatedly refining the likelihood graph and or an interface
prediction based thereon.
[0169] In another aspect, the invention is directed to a method for the in-
silico design of an
amino acid interface of a biologic for binding to a target (e.g., wherein the
biologic is an in-
progress custom biologic being designed for binding to an identified target),
the method
comprising: (a) receiving (e.g., and/or accessing), by a processor of a
computing device, an
initial scaffold-target complex graph comprising a graph representation (e.g.,
comprising nodes
and edges) of at least a portion of a biologic complex comprising the target
and a peptide
backbone of the in-progress custom biologic; (b) generating, by the processor,
using a machine
learning model, a predicted interface comprising, for each of a plurality of
interface sites, an
identification of a particular amino acid side chain type; and (c) providing
(e.g., by the processor)
59
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
the predicted interface for use in designing the amino acid interface of the
in-progress custom
biologic and/or using the predicted interface to design the amino acid
interface of the in-progress
custom biologic.
[0170] In another aspect, the invention is directed to a system for the in-
silico design of an
amino acid interface of a biologic for binding to a target (e.g., wherein the
biologic is an in-
progress custom biologic being designed for binding to an identified target),
the system
comprising: a processor of a computing device; and a memory having
instructions stored
thereon, wherein the instructions, when executed by the processor, cause the
processor to: (a)
receive (e.g., and/or access) an initial scaffold-target complex graph
comprising a graph
representation (e.g., comprising nodes and edges) of at least a portion of a
biologic complex
comprising the target and a peptide backbone of the in-progress custom
biologic; (b) generate,
using a machine learning model, a predicted interface comprising, for each of
a plurality of
interface sites, an identification of a particular amino acid side chain type;
and (c) provide the
predicted interface for use in designing the amino acid interface of the in-
progress custom
biologic and/or use the predicted interface to design the amino acid interface
of the in-progress
custom biologic.
[0171] In another aspect, the invention is directed to a method for in
silico design of a
custom biologic structure for binding to a target, the method comprising: (a)
receiving and/or
generating, by a processor of a computing device, a scaffold-target complex
model
corresponding to a selected candidate peptide backbone, oriented at a selected
pose, on which to
build a custom interface portion of a ligand for binding to the target; (b)
generating, by the
processor, based on the scaffold-target complex model, one or more prospective
ligand-target
complex models, each representing a prospective ligand corresponding to the
selected candidate
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
peptide backbone and each (i) comprising at least an interface region located
in proximity to the
target populated with amino acids and (ii) positioned with respect to the
target based on the
selected pose, each prospective ligand comprising a particular amino acid
population at its
interface region; (c) for each of the one or more prospective ligand-target
complex models,
determining, by the processor, an interface score using a machine learning
model, thereby
determining one or more interface scores; (d) selecting, by the processor, a
subset of the
prospective ligand-target complex models based on at least a portion of the
one or more interface
scores; and, optionally, (e) providing the selected subset of prospective
ligand-target complex
models for use in designing the custom biologic structure for binding to the
target.
[0172] In certain embodiments, the candidate peptide backbone comprises a
length of less
than about 100 peptide bonds and/or greater than about 20 peptide bonds.
[0173] In certain embodiments, the candidate peptide backbone is a backbone
of a pre-
existing protein molecule.
[0174] In certain embodiments, step (a) comprises receiving and/or
generating a simulated
three-dimensional electron density map (3D EDM) corresponding to at least a
portion of the
scaffold-target complex model.
[0175] In certain embodiments, the method comprises identifying, by the
processor, an
interface sub-region of the scaffold-target complex model, the interface sub-
region comprising
representations of atoms of the candidate peptide backbone and/or the target
located in proximity
to an interface between the candidate peptide backbone and/or the target.
[0176] In certain embodiments, step (b) comprises assigning an initial
amino acid sequence
to an interface region of the candidate peptide backbone and mutating amino
acids to generate,
61
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
for each prospective ligand-target complex model, the particular amino acid
population at the
interface region of the prospective ligand.
[0177] In certain embodiments, the machine learning model receives, as
input, for each
particular prospective ligand-target complex model, a representation of at
least a portion of the
particular prospective ligand-target complex model and outputs, for the
particular prospective
ligand-target complex model, as the interface score, a measure of similarity
and/or dissimilarity
between an interface of the particular prospective ligand-target complex model
and
representations of native interfaces.
[0178] In certain embodiments, the machine learning model is a trained
model, having been
trained to determine a predicted number of mutations between (i) an interface
that a particular
representation of at least a portion of a ligand-target complex model received
as input represents
and (ii) representations of native interfaces.
[0179] In certain embodiments, the machine learning model has been trained
using training
data comprising: (A) a plurality of native interface models, each native
interface model
representing at least a portion of a native interface based on an
experimentally determined
structural model of the native interface; and (B) a plurality of artificially
generated mutant
interface models, each mutant interface model based on a mutated version of a
native interface.
[0180] In certain embodiments, the machine learning model comprises a
neural network.
[0181] In certain embodiments, the method comprises using, by the
processor, an
optimization routine to select the subset of the prospective ligand-target
complex models.
[0182] In certain embodiments, the target comprises a peptide and/or a
complex thereof.
[0183] In certain embodiments, the method further comprises, for each of at
least a portion of
the prospective ligand-target complex models, determining, by the processor, a
binding affinity
62
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
score using a machine learning model that receives, as input, a representation
of at least a portion
of a particular ligand-target complex model and outputs, as the binding
affinity score, a value
representing a predicted binding affinity between the prospective custom
biologic structure and
the target molecule of the particular ligand-target complex model; and using
the one or more
binding affinity scores to design the custom biologic structure.
[0184] In certain embodiments, the method comprises: selecting one or more
high binding
affinity ligand-target complex models based on the one or more binding
affinity scores; and
providing the one or more high binding affinity ligand-target complex models
for use in
designing the custom biologic structure.
[0185] In certain embodiments, the method comprises comparing the one or
more binding
affinity scores to a threshold value and/or ranking the prospective ligand
target-complex models
according to the one or more determined binding affinity scores.
[0186] In another aspect, the invention is directed to a system for in
sit/co design of a custom
biologic structure for binding to a target, the system comprising: a processor
of a computing
device; and a memory having instructions stored thereon, wherein the
instructions, when
executed by the processor, cause the processor to: (a) receive and/or generate
a scaffold-target
complex model corresponding to a selected candidate peptide backbone, oriented
at a selected
pose, on which to build a custom interface portion of a ligand for binding to
the target; (b)
generate, based on the scaffold-target complex model, one or more prospective
ligand-target
complex models, each representing a prospective ligand corresponding to the
selected candidate
peptide backbone and each (i) comprising at least an interface region located
in proximity to the
target populated with amino acids and (ii) positioned with respect to the
target based on the
selected pose, each prospective ligand comprising a particular amino acid
population at its
63
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
interface region; (c) for each of the one or more prospective ligand-target
complex models,
determine an interface score using a machine learning model, thereby
determining one or more
interface scores; (d) select a subset of the prospective ligand-target complex
models based on at
least a portion of the one or more interface scores; and, optionally, (e)
provide the selected subset
of prospective ligand-target complex models for use in designing the custom
biologic structure
for binding to the target.
[0187] In certain embodiments, the candidate peptide backbone comprises a
length of less
than about 100 peptide bonds and/or greater than about 20 peptide bonds.
[0188] In certain embodiments, the candidate peptide backbone is a backbone
of a pre-
existing protein molecule.
[0189] In certain embodiments, the instructions cause the processor to, at
step (a), receive
and/or generate a simulated three-dimensional electron density map (3D EDM)
corresponding to
at least a portion of the scaffold-target complex model.
[0190] In certain embodiments, the instructions cause the processor to
identify an interface
sub-region of the scaffold-target complex model, the interface sub-region
comprising
representations of atoms of the candidate peptide backbone and/or the target
located in proximity
to an interface between the candidate peptide backbone and/or the target.
[0191] In certain embodiments, the instructions cause the processor to, at
step (b), assign an
initial amino acid sequence to an interface region of the candidate peptide
backbone and mutate
amino acids to generate, for each prospective ligand-target complex model, the
particular amino
acid population at the interface region of the prospective ligand.
[0192] In certain embodiments, the machine learning model receives, as
input, for each
particular prospective ligand-target complex model, a representation of at
least a portion of the
64
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
particular prospective ligand-target complex model and outputs, for the
particular prospective
ligand-target complex model, as the interface score, a measure of similarity
and/or dissimilarity
between an interface of the particular prospective ligand-target complex model
and
representations of native interfaces.
[0193] In certain embodiments, the machine learning model is a trained
model, having been
trained to determine a predicted number of mutations between (i) an interface
that a particular
representation of at least a portion of a ligand-target complex model received
as input represents
and (ii) representations of native interfaces.
[0194] In certain embodiments, the machine learning model has been trained
using training
data comprising: (A) a plurality of native interface models, each native
interface model
representing at least a portion of a native interface based on an
experimentally determined
structural model of the native interface; and (B) a plurality of artificially
generated mutant
interface models, each mutant interface model based on a mutated version of a
native interface.
[0195] In certain embodiments, the machine learning model comprises a
neural network.
[0196] In certain embodiments, the instructions cause the processor to use
an optimization
routine to select the subset of the prospective ligand-target complex models.
[0197] In certain embodiments, the target comprises a peptide and/or a
complex thereof.
[0198] In certain embodiments, the instructions cause the processor to, for
each of at least a
portion of the prospective ligand-target complex models, determine a binding
affinity score using
a machine learning model that receives, as input, a representation of at least
a portion of a
particular ligand-target complex model and outputs, as the binding affinity
score, a value
representing a predicted binding affinity between the prospective custom
biologic structure and
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
the target molecule of the particular ligand-target complex model; and use the
one or more
binding affinity scores to design the custom biologic structure.
[0199] In certain embodiments, the instructions cause the processor to:
select one or more
high binding affinity ligand-target complex models based on the one or more
binding affinity
scores; and provide the one or more high binding affinity ligand-target
complex models for use
in designing the custom biologic structure.
[0200] In certain embodiments, the instructions cause the processor to
compare the one or
more binding affinity scores to a threshold value and/or rank the prospective
ligand target-
complex models according to the one or more determined binding affinity
scores.
[0201] Features of embodiments described with respect to one aspect of the
invention may
be applied with respect to another aspect of the invention.
[0202] Throughout the description, where devices, systems, procedures,
and/or methods are
described as having, including, or comprising specific components, or where
methods are
described as having, including, or comprising specific steps, it is
contemplated that, additionally,
there are devices, systems, procedures, and/or methods of the present
disclosure that consist
essentially of, or consist of, the recited components, and that there are
methods according to the
present disclosure that consist essentially of, or consist of, the recited
processing steps.
[0203] It should be understood that the order of steps or order for
performing certain actions
is immaterial as long as the method remains operable. Moreover, two or more
steps or actions
may be conducted simultaneously.
[0204] The following description is for illustration and exemplification of
the disclosure
only, and is not intended to limit the disclosure to the specific embodiments
described.
66
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0205] The mention herein of any publication, for example, in the
Background section, is not
an admission that the publication serves as prior art with respect to any of
the present claims.
The Background section is presented for purposes of clarity and is not meant
as a description of
prior art with respect to any claim.
BRIEF DESCRIPTION OF THE DRAWING
[0206] The patent or application file contains at least one drawing
executed in color. Copies
of this patent or patent application publication with color drawing(s) will be
provided by the
Office upon request and payment of the necessary fee.
[0207] The foregoing and other objects, aspects, features, and advantages
of the present
disclosure will become more apparent and better understood by referring to the
following
description taken in conjunction with the accompanying drawing, in which:
[0208] FIG. 1 is a block flow diagram of an example process for designing
custom biologic
structures for binding to a target, according to an illustrative embodiment.
[0209] FIG. 2 is a block flow diagram of an example process for training
and testing a
machine learning model, according to an illustrative embodiment.
[0210] FIG. 3 is a schematic illustration of various example data
preparation steps used in
various embodiments described herein.
[0211] FIG. 4 is a diagram of an example procedure for splitting a dataset
comprising
biological data into training and testing datasets, according to an
illustrative embodiment.
[0212] FIG. 5 is a schematic of an example approach for identifying and/or
extracting an
interface sub-region of a model of a biological complex, according to an
illustrative embodiment.
67
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0213] FIG. 6 is a schematic of an example process for creating a
volumetric representation
of an interface sub-region of a biological complex model, according to an
illustrative
embodiment.
[0214] FIG. 7 is a schematic of a machine learning model that determines a
scoring function
used for evaluating performance of a particular biological complex model,
according to an
illustrative embodiment.
[0215] FIG. 8 is a schematic illustration of a scaffold docking approach,
according to an
illustrative embodiment.
[0216] FIG. 9 is a block flow diagram of an example process for identifying
favorable
peptide backbones and poses thereof (e.g., for use in connection with a
scaffold docking module
as described herein), according to an illustrative embodiment.
[0217] FIG. 10 is a schematic of a representation target (e.g., a
particular receptor) in a
complex with a scaffold model, according to an illustrative embodiment.
[0218] FIG. 11 is a schematic of a representation target molecule (e.g., a
particular receptor)
in a complex with a scaffold model, according to an illustrative embodiment.
[0219] FIG. 12 is a diagram of comparing certain features of various pose
quality metrics
described herein, according to an illustrative embodiment.
[0220] FIG. 13 is a schematic illustrating calculation of a root mean
squared distance
(RMSD) value, according to an illustrative embodiment.
[0221] FIG. 14 is a pair of illustrative graphs showing variation in two
pose quality metrics
for different types of complexes, according to an illustrative embodiment.
[0222] FIG. 15 is a block flow diagram of an example process for creation
of a training
dataset for use in training a machine learning model, according to an
illustrative embodiment.
68
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0223] FIG. 16A is a block flow diagram of an example pose generation
process, according
to an illustrative embodiment.
[0224] FIG. 16B is a schematic illustrating an approach to pose generation,
according to an
illustrative embodiment.
[0225] FIG. 17 is a schematic illustrating an example sampling process,
according to an
illustrative embodiment.
[0226] FIG. 18 is a schematic illustrating an example approach to creation
of various datasets
comprising labeled examples of two classes, based on pose quality metrics,
according to an
illustrative embodiment.
[0227] FIG. 19A is a schematic illustrating a spinal cord model (SCM)
machine learning
architecture, according to an illustrative embodiment.
[0228] FIG. 19B is portion of a network diagram of an example SCM
architecture, according
to an illustrative embodiment.
[0229] FIG. 19C a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19C continues from FIG. 19B.
[0230] FIG. 19D a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19D continues from FIG. 19C.
[0231] FIG. 19E a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19E continues from FIG. 19D.
[0232] FIG. 19F a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19F continues from FIG. 19E.
[0233] FIG. 19G a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19G continues from FIG. 19F.
69
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0234] FIG. 19H a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19H continues from FIG. 19G.
[0235] FIG. 191 a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19I continues from FIG. 19H.
[0236] FIG. 19J a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19J continues from FIG. 191.
[0237] FIG. 19K a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19K continues from FIG. 19J.
[0238] FIG. 19L a portion of the network diagram of the example SCM
architecture,
according to an illustrative embodiment. FIG19L continues from FIG. 19K.
[0239] FIG. 20 is a schematic illustrating an example transfer learning
approach for training
a machine learning model, according to an illustrative embodiment.
[0240] FIG. 21 shows three tables presenting results demonstrating
performance of three
trained machine learning models created in accordance with certain embodiments
described
herein.
[0241] FIG. 22A is a graph showing receiver operating characteristic (ROC)
curves for one
of the three machine learning models of FIG. 21.
[0242] FIG. 22B is a graph showing receiver operating characteristic (ROC)
curves for one
of the three machine learning models of FIG. 21.
[0243] FIG. 22C is a graph showing receiver operating characteristic (ROC)
curves for one
of the three machine learning models of FIG. 21.
[0244] FIG. 23 is a schematic showing an example process for evaluating
candidate scaffold
model poses, according to an illustrative embodiment.
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0245] FIG. 24 is a schematic illustrating an example interface design
approach, according to
an illustrative embodiment.
[0246] FIG. 25 is a block flow diagram of an example process for creating
and evaluating
candidate interface designs, according to an illustrative embodiment.
[0247] FIG. 26 is a schematic illustrating an approach for obtaining and
curating an initial
dataset for training a machine learning model to evaluate prospective
interface designs,
according to an illustrative embodiment.
[0248] FIG. 27 is a schematic illustrating an approach for creation of a
training dataset for
training a machine learning model to evaluate prospective interface designs,
according to an
illustrative embodiment.
[0249] FIG. 28 is a schematic illustrating an approach for creation of a
training dataset for
training a machine learning model to evaluate prospective interface designs,
according to an
illustrative embodiment.
[0250] FIG. 29 is a schematic diagram illustrating approaches for mutating
amino acid side
chains to create candidate interfaces, according to an illustrative
embodiment.
[0251] FIG. 30 is a schematic illustrating certain features of a dataset
comprising examples
of interfaces created in accordance with various embodiments described herein.
[0252] FIG. 31A is a schematic of an example architecture of a machine
learning model for
evaluating candidate interface designs, according to an illustrative
embodiment.
[0253] FIG. 31B is a network diagram of an example regression model machine
learning
architecture, according to an illustrative embodiment.
71
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0254] FIG. 32A is a candlestick chart demonstrating performance of a
machine learning
model used for computing interface scores in accordance with various
embodiments described
herein.
[0255] FIG. 32B is a graph showing a receiver operating characteristic
(ROC) curve
demonstrating performance of interface scores computed by a machine learning
model as
described herein for differentiation between native and non-native interfaces,
in accordance with
various embodiments described herein.
[0256] FIG. 33 is a diagram of an example process for designing candidate
interfaces using
an optimization algorithm, according to an illustrative embodiment.
[0257] FIG. 34 is a schematic showing an example approach for evaluating
performance of a
binder candidate, according to an illustrative embodiment.
[0258] FIG. 35 is a block flow diagram of an example process for using
predicted binding
affinity scores to select a subset and/or refine candidate interface designs,
according to an
illustrative embodiment.
[0259] FIG. 36 is a schematic of an example approach for creating of a
dataset for training a
machine learning model for determining binding affinity predictions, according
to an illustrative
embodiment.
[0260] FIG. 37 is a schematic of an example approach, including balancing
and data
augmentation steps, for creating a dataset for training a machine learning
model for determining
binding affinity predictions, according to an illustrative embodiment.
[0261] FIG. 38 is a schematic of a two-step transfer learning approach for
training a machine
learning model, according to an illustrative embodiment.
72
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0262] FIG. 39 is a graph demonstrating performance of a machine learning
model used for
determining binding affinity predictions, according to an illustrative
embodiment.
[0263] FIG. 40 is a schematic showing an example modular approach for
designing custom
biologics, according to an illustrative embodiment.
[0264] FIG. 41 is a block diagram of an exemplary cloud computing
environment, used in
certain embodiments.
[0265] FIG. 42 is a block diagram of an example computing device and an
example mobile
computing device, used in certain embodiments.
[0266] FIG. 43 is a block flow diagram of an example process for generating
a predicted
interface for use in design of a custom biologic, according to an illustrative
embodiment;
[0267] FIG. 44A is a ribbon diagram of a biologic complex, according to an
illustrative
embodiment;
[0268] FIG. 44B is a diagram of a graph representation of a biologic
complex, according to
an illustrative embodiment;
[0269] FIG. 45A is a diagram illustrating representation of amino acid
sites of a biologic
complex via nodes in a graph representation, according to an illustrative
embodiment;
[0270] FIG. 45B is a diagram illustrating an example approach for encoding
structural
information of amino acid sites of a biologic via a node feature vector of a
graph representation,
according to an illustrative embodiment;
[0271] FIG. 45C is a diagram illustrating an example approach for encoding
relational
information (e.g., interactions and/or relative positioning between) two amino
acid sites of a
biologic via an edge feature vector of a graph representation, according to an
illustrative
embodiment;
73
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0272] FIG. 46A is a diagram illustrating an initial complex graph
comprising a target graph
and a scaffold graph comprising unknown interface nodes and known scaffold
nodes, according
to an illustrative embodiment;
[0273] FIG. 46B is a diagram illustrating a masked component vector,
according to an
illustrative embodiment;
[0274] FIG. 46C is a block flow diagram of an example process for
generating a predicted
interface for use in design of a custom biologic, according to an illustrative
embodiment;
[0275] FIG. 46D is a block flow diagram of an example process for
generating a predicted
interface for use in design of a custom biologic, according to an illustrative
embodiment;
[0276] FIG. 46E is a schematic of a multi-headed neural network
architecture, according to
an illustrative embodiment;
[0277] FIG. 46F is a schematic of a multi-headed neural network
architecture with a graph
featurizer module, according to an illustrative embodiment;
[0278] FIG. 47A is a block flow diagram showing an example training
procedure for training
a machine learning model to generate predicted interfaces for use in design of
a custom biologic,
according to an illustrative embodiment;
[0279] FIG. 47B is a diagram showing an example training procedure for
training a machine
learning model to generate predicted interfaces for use in design of a custom
biologic, according
to an illustrative embodiment;
[0280] FIG. 47C is a schematic of a (e.g., stackable) block for use in a
graph network
approach, according to an illustrative embodiment;
[0281] FIG. 48A is bar graph showing accuracy of predictions for 20 amino
acid side chain
types evaluated using a full molecule test dataset;
74
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0282] FIG. 48B is bar graph showing Fl-scores for predictions for 20 amino
acid side chain
types evaluated using a full molecule test dataset;
[0283] FIG. 48C is bar graph showing Area Under the Curve (AUC) values for
predictions
for 20 amino acid side chain types evaluated using a full molecule test
dataset;
[0284] FIG. 49A is bar graph showing accuracy of predictions for 20 amino
acid side chain
types evaluated using an interface specific test dataset;
[0285] FIG. 49B is bar graph showing Fl-scores for predictions for 20 amino
acid side chain
types evaluated using an interface specific test dataset;
[0286] FIG. 49C is bar graph showing Area Under the Curve (AUC) values for
predictions
for 20 amino acid side chain types evaluated using an interface specific test
dataset;
[0287] Features and advantages of the present disclosure will become more
apparent from
the detailed description of certain embodiments that is set forth below,
particularly when taken in
conjunction with the figures, in which like reference characters identify
corresponding elements
throughout. In the figures, like reference numbers generally indicate
identical, functionally
similar, and/or structurally similar elements.
CERTAIN DEFINITIONS
[0288] In order for the present disclosure to be more readily understood,
certain terms are
first defined below. Additional definitions for the following terms and other
terms are set forth
throughout the specification.
[0289] A device, composition, system, or method described herein as
"comprising" one or
more named elements or steps is open-ended, meaning that the named elements or
steps are
essential, but other elements or steps may be added within the scope of the
composition or
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
method. To avoid prolixity, it is also understood that any device,
composition, or method
described as "comprising" (or which "comprises") one or more named elements or
steps also
describes the corresponding, more limited composition or method "consisting
essentially of' (or
which "consists essentially of') the same named elements or steps, meaning
that the composition
or method includes the named essential elements or steps and may also include
additional
elements or steps that do not materially affect the basic and novel
characteristic(s) of the
composition or method. It is also understood that any device, composition, or
method described
herein as "comprising" or "consisting essentially of' one or more named
elements or steps also
describes the corresponding, more limited, and closed-ended composition or
method "consisting
of' (or "consists of') the named elements or steps to the exclusion of any
other unnamed element
or step. In any composition or method disclosed herein, known or disclosed
equivalents of any
named essential element or step may be substituted for that element or step.
[0290] As used herein, "a" or "an" with reference to a claim feature means
"one or more," or
"at least one."
[0291] Administration: As used herein, the term "administration" typically
refers to the
administration of a composition to a subject or system. Those of ordinary
skill in the art will be
aware of a variety of routes that may, in appropriate circumstances, be
utilized for administration
to a subject, for example a human. For example, in some embodiments,
administration may be
ocular, oral, parenteral, topical, etc. In some particular embodiments,
administration may be
bronchial (e.g., by bronchial instillation), buccal, dermal (which may be or
comprise, for
example, one or more of topical to the dermis, intradermal, interdermal,
transdermal, etc.),
enteral, intra-arterial, intradermal, intragastric, intramedullary,
intramuscular, intranasal,
intraperitoneal, intrathecal, intravenous, intraventricular, within a specific
organ (e.g.,
76
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
intrahepatic), mucosal, nasal, oral, rectal, subcutaneous, sublingual,
topical, tracheal (e.g., by
intratracheal instillation), vaginal, vitreal, etc. In some embodiments,
administration may
involve dosing that is intermittent (e.g., a plurality of doses separated in
time) and/or periodic
(e.g., individual doses separated by a common period of time) dosing. In some
embodiments,
administration may involve continuous dosing (e.g., perfusion) for at least a
selected period of
time.
[0292] Affinity: As is known in the art, "affinity" is a measure of the
tightness with which
two or more binding partners associate with one another. Those skilled in the
art are aware of a
variety of assays that can be used to assess affinity, and will furthermore be
aware of appropriate
controls for such assays. In some embodiments, affinity is assessed in a
quantitative assay. In
some embodiments, affinity is assessed over a plurality of concentrations
(e.g., of one binding
partner at a time). In some embodiments, affinity is assessed in the presence
of one or more
potential competitor entities (e.g., that might be present in a relevant ¨
e.g., physiological ¨
setting). In some embodiments, affinity is assessed relative to a reference
(e.g., that has a known
affinity above a particular threshold [a "positive control" reference] or that
has a known affinity
below a particular threshold [ a "negative control" reference"]. In some
embodiments, affinity
may be assessed relative to a contemporaneous reference; in some embodiments,
affinity may be
assessed relative to a historical reference. Typically, when affinity is
assessed relative to a
reference, it is assessed under comparable conditions.
[0293] Amino acid: in its broadest sense, as used herein, refers to any
compound and/or
substance that can be incorporated into a polypeptide chain, e.g., through
formation of one or
more peptide bonds. In some embodiments, an amino acid has the general
structure H2N¨
C(H)(R)¨COOH. In some embodiments, an amino acid is a naturally-occurring
amino acid. In
77
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
some embodiments, an amino acid is a non-natural amino acid; in some
embodiments, an amino
acid is a D-amino acid; in some embodiments, an amino acid is an L-amino acid.
"Standard
amino acid" refers to any of the twenty standard L-amino acids commonly found
in naturally
occurring peptides. "Nonstandard amino acid" refers to any amino acid, other
than the standard
amino acids, regardless of whether it is prepared synthetically or obtained
from a natural source.
In some embodiments, an amino acid, including a carboxy- and/or amino-terminal
amino acid in
a polypeptide, can contain a structural modification as compared with the
general structure
above. For example, in some embodiments, an amino acid may be modified by
methylation,
amidation, acetylation, pegylation, glycosylation, phosphorylation, and/or
substitution (e.g., of
the amino group, the carboxylic acid group, one or more protons, and/or the
hydroxyl group) as
compared with the general structure. In some embodiments, such modification
may, for
example, alter the circulating half-life of a polypeptide containing the
modified amino acid as
compared with one containing an otherwise identical unmodified amino acid. In
some
embodiments, such modification does not significantly alter a relevant
activity of a polypeptide
containing the modified amino acid, as compared with one containing an
otherwise identical
unmodified amino acid. As will be clear from context, in some embodiments, the
term "amino
acid" may be used to refer to a free amino acid; in some embodiments it may be
used to refer to
an amino acid residue of a polypeptide.
[0294] Antibody, Antibody polypeptide: As used herein, the terms "antibody
polypeptide"
or "antibody", or "antigen-binding fragment thereof', which may be used
interchangeably, refer
to polypeptide(s) capable of binding to an epitope. In some embodiments, an
antibody
polypeptide is a full-length antibody, and in some embodiments, is less than
full length but
includes at least one binding site (comprising at least one, and preferably at
least two sequences
78
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
with structure of antibody "variable regions"). In some embodiments, the term
"antibody
polypeptide" encompasses any protein having a binding domain which is
homologous or largely
homologous to an immunoglobulin-binding domain. In particular embodiments,
"antibody
polypeptides" encompasses polypeptides having a binding domain that shows at
least 99%
identity with an immunoglobulin binding domain. In some embodiments, "antibody
polypeptide" is any protein having a binding domain that shows at least 70%,
80%, 85%, 90%,
or 95% identity with an immuglobulin binding domain, for example a reference
immunoglobulin
binding domain. An included "antibody polypeptide" may have an amino acid
sequence
identical to that of an antibody that is found in a natural source. Antibody
polypeptides in
accordance with the present invention may be prepared by any available means
including, for
example, isolation from a natural source or antibody library, recombinant
production in or with a
host system, chemical synthesis, etc., or combinations thereof. An antibody
polypeptide may be
monoclonal or polyclonal. An antibody polypeptide may be a member of any
immunoglobulin
class, including any of the human classes: IgG, IgM, IgA, IgD, and IgE. In
certain embodiments,
an antibody may be a member of the IgG immunoglobulin class. As used herein,
the terms
"antibody polypeptide" or "characteristic portion of an antibody" are used
interchangeably and
refer to any derivative of an antibody that possesses the ability to bind to
an epitope of interest.
In certain embodiments, the "antibody polypeptide" is an antibody fragment
that retains at least a
significant portion of the full-length antibody's specific binding ability.
Examples of antibody
fragments include, but are not limited to, Fab, Fab', F(ab')2, scFv, Fv, dsFy
diabody, and Fd
fragments. Alternatively or additionally, an antibody fragment may comprise
multiple chains
that are linked together, for example, by disulfide linkages. In some
embodiments, an antibody
polypeptide may be a human antibody. In some embodiments, the antibody
polypeptides may be
79
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
a humanized. Humanized antibody polypeptides include may be chimeric
immunoglobulins,
immunoglobulin chains or antibody polypeptides (such as Fv, Fab, Fab', F(ab')2
or other antigen-
binding subsequences of antibodies) that contain minimal sequence derived from
non-human
immunoglobulin. In general, humanized antibodies are human immunoglobulins
(recipient
antibody) in which residues from a complementary-determining region (CDR) of
the recipient
are replaced by residues from a CDR of a non-human species (donor antibody)
such as mouse,
rat or rabbit having the desired specificity, affinity, and capacity.
[0295] Approximately: As used herein, the term "approximately" or "about,"
as applied to
one or more values of interest, refers to a value that is similar to a stated
reference value. In
certain embodiments, the term "approximately" or "about" refers to a range of
values that fall
within 25%, 20%, 19%, 18%, 17%, 16%, 15%, 14%, 13%, 12%, 11%, 10%, 9%, 8%, 7%,
6%,
5%, 4%, 3%, 2%, 1%, or less in either direction (greater than or less than) of
the stated reference
value unless otherwise stated or otherwise evident from the context (except
where such number
would exceed 100% of a possible value).
[0296] Backbone, peptide backbone: As used herein, the term "backbone," for
example, as
in a backbone or a peptide or polypeptide, refers to the portion of the
peptide or polypeptide
chain that comprises the links between amino acid of the chain but excludes
side chains. In other
words, a backbone refers to the part of a peptide or polypeptide that would
remain if side chains
were removed. In certain embodiments, the backbone is a chain comprising a
carboxyl group of
one amino acid bound via a peptide bond to an amino group of a next amino
acid, and so on.
Backbone may also be referred to as "peptide backbone". It should be
understood that, where
the term "peptide backbone" is used, it is used for clarity, and is not
intended to limit a length of
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
a particular backbone. That is, the term "peptide backbone" may be used to
describe a peptide
backbone of a peptide and/or a protein.
[02971 Biologic: As used herein, the term "biologic" rell-,rs to a
composition that is or may
be produced by recombinant DNA technologies, peptide synthesis, or purified
from natural
sources and that has a desired biological activity. A biologic can be, for
example, a protein,
peptide, glycoprotein, polysaccharide, a mixture of proteins or peptides, a
mixture of
glycoproteins, a mixture of polysaccharides, a mixture of one or more of a
protein, peptide,
glycoprotein or polysaccharide, or a derivatized form of any of the foregoing
entities. Molecular
weight of biologics can vary widely, from about 1000 Da for small peptides
such as peptide
hormones to one thousand kDa or more for complex polysaccharides, mucins, and
other heavily
glycosylated proteins. in certain embodiments, a biologic is a drug used for
treatment of
diseases and/or medical conditions. Examples of biologic drugs include,
without limitation,
native or engineered antibodies or antigen binding fragments thereof, and
antibody-drug
conjugates, which comprise an antibody or antigen binding fragments thereof
conjugated directly
or indirectly (e.g., via a linker) to a drug of interest, such as a cytotoxic
drug or toxin. in certain
embodiments, a biologic is a diagnostic, used to diagnose diseases and/or
medical conditions.
For example, allergen patch tests utilize biologics (e.g., biologics
manufactured from natural
substances) that are known to cause contact dermatitis. Diagnostic biologics
may also include
medical imaging agents, such as proteins that are labelled with agents that
provide a detectable
signal that facilitates imaging such as fluorescent markers, dyes,
radionuclides, and the like.
[0298] In vitro: The term "in vitro" as used herein refers to events that
occur in an artificial
environment, e.g., in a test tube or reaction vessel, in cell culture, etc.,
rather than within a multi-
cellular organism.
81
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0299] In vivo: As used herein, the term "in vivo" refers to events that
occur within a multi-
cellular organism, such as a human and a non-human animal. In the context of
cell-based
systems, the term may be used to refer to events that occur within a living
cell (as opposed to, for
example, in vitro systems).
[0300] Native, wild-type (WT): As used herein, the terms "native" and "wild-
type" are used
interchangeably to refer to biological structures and/or computer
representations thereof that
have been identified and demonstrated to exist in the physical, real world
(e.g., as opposed to in
computer abstractions). The terms, native and wild-type may refer to
structures including
naturally occurring biological structures, but do not necessarily require that
a particular structure
be naturally occurring. For example, the terms native and wild-type may also
refer to structures
including engineered structures that are man-made, and do not occur in nature,
but have
nonetheless been created and (e.g., experimentally) demonstrated to exist. In
certain
embodiments, the terms native and wild-type refer to structures that have been
characterized
experimentally, and for which an experimental determination of molecular
structure (e.g., via x-
ray crystallography) has been made.
[0301] Patient: As used herein, the term "patient" refers to any organism
to which a
provided composition is or may be administered, e.g., for experimental,
diagnostic, prophylactic,
cosmetic, and/or therapeutic purposes. Typical patients include animals (e.g.,
mammals such as
mice, rats, rabbits, non-human primates, and/or humans). In some embodiments,
a patient is a
human. In some embodiments, a patient is suffering from or susceptible to one
or more disorders
or conditions. In some embodiments, a patient displays one or more symptoms of
a disorder or
condition. In some embodiments, a patient has been diagnosed with one or more
disorders or
conditions. In some embodiments, the disorder or condition is or includes
cancer, or presence of
82
CA 03226172 2024-01-03
WO 2023/004116
PCT/US2022/038014
one or more tumors. In some embodiments, the patient is receiving or has
received certain
therapy to diagnose and/or to treat a disease, disorder, or condition.
[0302]
Peptide: The term "peptide" as used herein refers to a polypeptide that is
typically
relatively short, for example having a length of less than about 100 amino
acids, less than about
50 amino acids, less than about 40 amino acids less than about 30 amino acids,
less than about 25
amino acids, less than about 20 amino acids, less than about 15 amino acids,
or less than 10
amino acids.
[0303]
Polypeptide: As used herein refers to a polymeric chain of amino acids. In
some
embodiments, a polypeptide has an amino acid sequence that occurs in nature.
In some
embodiments, a polypeptide has an amino acid sequence that does not occur in
nature. In some
embodiments, a polypeptide has an amino acid sequence that is engineered in
that it is designed
and/or produced through action of the hand of man. In some embodiments, a
polypeptide may
comprise or consist of natural amino acids, non-natural amino acids, or both.
In some
embodiments, a polypeptide may comprise or consist of only natural amino acids
or only non-
natural amino acids. In some embodiments, a polypeptide may comprise D-amino
acids, L-
amino acids, or both. In some embodiments, a polypeptide may comprise only D-
amino acids.
In some embodiments, a polypeptide may comprise only L-amino acids. In some
embodiments,
a polypeptide may include one or more pendant groups or other modifications,
e.g., modifying or
attached to one or more amino acid side chains, at the polypeptide's N-
terminus, at the
polypeptide's C-terminus, or any combination thereof. In some embodiments,
such pendant
groups or modifications may be selected from the group consisting of
acetylation, amidation,
lipidation, methylation, pegylation, etc., including combinations thereof. In
some embodiments,
a polypeptide may be cyclic, and/or may comprise a cyclic portion. In some
embodiments, a
83
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
polypeptide is not cyclic and/or does not comprise any cyclic portion. In some
embodiments, a
polypeptide is linear. In some embodiments, a polypeptide may be or comprise a
stapled
polypeptide. In some embodiments, the term "polypeptide" may be appended to a
name of a
reference polypeptide, activity, or structure; in such instances it is used
herein to refer to
polypeptides that share the relevant activity or structure and thus can be
considered to be
members of the same class or family of polypeptides. For each such class, the
present
specification provides and/or those skilled in the art will be aware of
exemplary polypeptides
within the class whose amino acid sequences and/or functions are known; in
some embodiments,
such exemplary polypeptides are reference polypeptides for the polypeptide
class or family. In
some embodiments, a member of a polypeptide class or family shows significant
sequence
homology or identity with, shares a common sequence motif (e.g., a
characteristic sequence
element) with, and/or shares a common activity (in some embodiments at a
comparable level or
within a designated range) with a reference polypeptide of the class; in some
embodiments with
all polypeptides within the class). For example, in some embodiments, a member
polypeptide
shows an overall degree of sequence homology or identity with a reference
polypeptide that is at
least about 30-40%, and is often greater than about 50%, 60%, 70%, 80%, 90%,
91%, 92%,
93%, 94%, 95%, 96%, 97%, 98%, 99% or more and/or includes at least one region
(e.g., a
conserved region that may in some embodiments be or comprise a characteristic
sequence
element) that shows very high sequence identity, often greater than 90% or
even 95%, 96%,
97%, 98%, or 99%. Such a conserved region usually encompasses at least 3-4 and
often up to 20
or more amino acids; in some embodiments, a conserved region encompasses at
least one stretch
of at least 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 or more contiguous
amino acids. In some
embodiments, a relevant polypeptide may comprise or consist of a fragment of a
parent
84
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
polypeptide. In some embodiments, a useful polypeptide as may comprise or
consist of a
plurality of fragments, each of which is found in the same parent polypeptide
in a different
spatial arrangement relative to one another than is found in the polypeptide
of interest (e.g.,
fragments that are directly linked in the parent may be spatially separated in
the polypeptide of
interest or vice versa, and/or fragments may be present in a different order
in the polypeptide of
interest than in the parent), so that the polypeptide of interest is a
derivative of its parent
polypeptide.
[0304] Protein: As used herein, the term "protein" refers to a polypeptide
(i.e., a string of at
least two amino acids linked to one another by peptide bonds). Proteins may
include moieties
other than amino acids (e.g., may be glycoproteins, proteoglycans, etc.)
and/or may be otherwise
processed or modified. Those of ordinary skill in the art will appreciate that
a "protein" can be a
complete polypeptide chain as produced by a cell (with or without a signal
sequence), or can be a
characteristic portion thereof. Those of ordinary skill will appreciate that a
protein can
sometimes include more than one polypeptide chain, for example linked by one
or more disulfide
bonds or associated by other means. Polypeptides may contain L-amino acids, D-
amino acids, or
both and may contain any of a variety of amino acid modifications or analogs
known in the art.
Useful modifications include, e.g., terminal acetylation, amidation,
methylation, etc. In some
embodiments, proteins may comprise natural amino acids, non-natural amino
acids, synthetic
amino acids, and combinations thereof The term "peptide" is generally used to
refer to a
polypeptide having a length of less than about 100 amino acids, less than
about 50 amino acids,
less than 20 amino acids, or less than 10 amino acids. In some embodiments,
proteins are
antibodies, antibody fragments, biologically active portions thereof, and/or
characteristic
portions thereof.
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0305] Target: As used herein, the terms "target," and "receptor" are used
interchangeably
and refer to one or more molecules or portions thereof to which a binding
agent ¨ e.g., a custom
biologic, such as a protein or peptide, to be designed ¨ binds. In certain
embodiments, the target
is or comprises a protein and/or peptide. In certain embodiments, the target
is a molecule, such
as an individual protein or peptide (e.g., a protein or peptide monomer), or
portion thereof. In
certain embodiments, the target is a complex, such as a complex of two or more
proteins or
peptides, for example, a macromolecular complex formed by two or more protein
or peptide
monomers. For example, a target may be a protein or peptide dimer, trimer,
tetramer, etc. or
other oligomeric complex. In certain embodiments, the target is a drug target,
e.g., a molecule in
the body, usually a protein, that is intrinsically associated with a
particular disease process and
that could be addressed by a drug to produce a desired therapeutic effect. In
certain
embodiments, a custom biologic is engineered to bind to a particular target.
While the structure
of the target remains fixed, structural features of the custom biologic may be
varied to allow it to
bind (e.g., at high specificity) to the target.
[0306] Treat: As used herein, the term "treat" (also "treatment" or
"treating") refers to any
administration of a therapeutic agent (also "therapy") that partially or
completely alleviates,
ameliorates, eliminates, reverses, relieves, inhibits, delays onset of,
reduces severity of, and/or
reduces incidence of one or more symptoms, features, and/or causes of a
particular disease,
disorder, and/or condition. In some embodiments, such treatment may be of a
patient who does
not exhibit signs of the relevant disease, disorder and/or condition and/or of
a patient who
exhibits only early signs of the disease, disorder, and/or condition.
Alternatively, or additionally,
such treatment may be of a patient who exhibits one or more established signs
of the relevant
disease, disorder and/or condition. In some embodiments, treatment may be of a
patient who has
86
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
been diagnosed as suffering from the relevant disease, disorder, and/or
condition. In some
embodiments, treatment may be of a patient known to have one or more
susceptibility factors
that are statistically correlated with increased risk of development of a
given disease, disorder,
and/or condition. In some embodiments the patient may be a human.
[0307] Machine learning module, machine learning model: As used herein, the
terms
"machine learning module" and "machine learning model" are used
interchangeably and refer to
a computer implemented process (e.g., a software function) that implements one
or more
particular machine learning algorithms, such as an artificial neural networks
(ANN),
convolutional neural networks (CNNs), random forest, decision trees, support
vector machines,
and the like, in order to determine, for a given input, one or more output
values. In some
embodiments, machine learning modules implementing machine learning techniques
are trained,
for example using curated and/or manually annotated datasets. Such training
may be used to
determine various parameters of machine learning algorithms implemented by a
machine
learning module, such as weights associated with layers in neural networks. In
some
embodiments, once a machine learning module is trained, e.g., to accomplish a
specific task such
as determining scoring metrics as described herein, values of determined
parameters are fixed
and the (e.g., unchanging, static) machine learning module is used to process
new data (e.g.,
different from the training data) and accomplish its trained task without
further updates to its
parameters (e.g., the machine learning module does not receive feedback and/or
updates). In
some embodiments, machine learning modules may receive feedback, e.g., based
on user review
of accuracy, and such feedback may be used as additional training data, for
example to
dynamically update the machine learning module. In some embodiments, a trained
machine
learning module is a classification algorithm with adjustable and/or fixed
(e.g., locked)
87
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
parameters, e.g., a random forest classifier. In some embodiments, two or more
machine
learning modules may be combined and implemented as a single module and/or a
single software
application. In some embodiments, two or more machine learning modules may
also be
implemented separately, e.g., as separate software applications. A machine
learning module may
be software and/or hardware. For example, a machine learning module may be
implemented
entirely as software, or certain functions of a ANN module may be carried out
via specialized
hardware (e.g., via an application specific integrated circuit (ASIC), field
programmable gate
arrays (FPGAs), and the like).
[0308] Substantially: As used herein, the term "substantially" refers to
the qualitative
condition of exhibiting total or near-total extent or degree of a
characteristic or property of
interest.
[0309] Scaffold Model: As used herein, the term "scaffold model" refers to
a computer
representation of at least a portion of a peptide backbone of a particular
protein and/or peptide.
In certain embodiments, a scaffold model represents a peptide backbone of a
protein and/or
peptide and omits detailed information about amino acid side chains. Such
scaffold models,
may, nevertheless, include various mechanisms for representing sites (e.g.,
locations along a
peptide backbone) that may be occupied by prospective amino acid side chains.
In certain
embodiments, a particular scaffold models may represent such sites in a manner
that allows
determining regions in space that may be occupied by prospective amino acid
side chains and/or
approximate proximity to representations of other amino acids, sites, portions
of the peptide
backbone, and other molecules that may interact with (e.g., bind, so as to
form a complex with) a
biologic having the peptide backbone represented by the particular scaffold
model. For example,
in certain embodiments, a scaffold model may include a representation of a
first side chain atom,
88
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
such as a representation of a beta-carbon, which can be used to identify sites
and/ approximate
locations of amino acid side chains. For example, a scaffold model can be
populated with amino
acid side chains (e.g., to create a ligand model that represents at least a
portion of protein and/or
peptide) by creating full representations of various amino acids about beta-
carbon atoms of the
scaffold model (e.g., the beta-carbon atoms acting as 'anchors' or
`placeholders' for amino acid
side chains). In certain embodiments, locations of sites and/or approximate
regions (e.g.,
volumes) that may be occupied by amino acid side chains may be identified
and/or determined
via other manners of representation for example based on locations of an alpha-
carbons,
hydrogen atoms, etc. In certain embodiments, scaffold models may be created
from structural
representations of existing proteins and/or peptides, for example by stripping
amino acid side
chains. In certain embodiments, scaffold models created in this manner may
retain a first atom
of stripped side chains, such as a beta-carbon atom, which is common to all
side chains apart
from Glycine. As described herein, retained beta-carbon atoms may be used,
e.g., as a
placeholder for identification of sites that can be occupied by amino acid
side chains. In certain
embodiments, where an initially existing side chain was Glycine, the first
atom of glycine, which
is hydrogen, can be used in place of a beta-carbon and/or, in certain
embodiments, a beta carbon
(e.g., though not naturally occurring in the full protein used to create a
scaffold model) may be
added to the representation (e.g., artificially). In certain embodiments, for
example where
hydrogen atoms are not included in a scaffold model, a site initially occupied
by a Glycine may
be identified based on an alpha-carbon. In certain embodiments, scaffold
models may be
computer generated (e.g., and not based on an existing protein and/or
peptide). In certain
embodiments, computer generate scaffold models may also include first side
chain atoms, e.g.,
beta carbons, e.g., as placeholders of potential side chains to be added.
89
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
DESCRIPTION
[0310] Reference will now be made in detail to the present disclosed
embodiments, one or
more examples of which are illustrated in the accompanying drawing. The
detailed description
uses numerical and/or letter designations to refer to features in the drawing.
Like or similar
designations in the drawing and description have been used to refer to like or
similar parts of the
present embodiments.
[0311] It is contemplated that systems, architectures, devices, methods,
and processes of the
claimed invention encompass variations and adaptations developed using
information from the
embodiments described herein. Adaptation and/or modification of the systems,
architectures,
devices, methods, and processes described herein may be performed, as
contemplated by this
description.
[0312] Throughout the description, where articles, devices, systems, and
architectures are
described as having, including, or comprising specific components, or where
processes and
methods are described as having, including, or comprising specific steps, it
is contemplated that,
additionally, there are articles, devices, systems, and architectures of the
present invention that
consist essentially of, or consist of, the recited components, and that there
are processes and
methods according to the present invention that consist essentially of, or
consist of, the recited
processing steps.
[0313] It should be understood that the order of steps or order for
performing certain action
is immaterial so long as the invention remains operable. Moreover, two or more
steps or actions
may be conducted simultaneously.
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0314] The mention herein of any publication, for example, in the
Background section, is not
an admission that the publication serves as prior art with respect to any of
the claims presented
herein. The Background section is presented for purposes of clarity and is not
meant as a
description of prior art with respect to any claim.
[0315] Headers are provided for the convenience of the reader ¨ the
presence and/or
placement of a header is not intended to limit the scope of the subject matter
described herein.
[0316] Computer-aided design of candidate molecules for use as new drugs
can facilitate the
drug discovery process, increasing the speed at which new drugs are
identified, tested, and
brought to market and reducing costs associated with, e.g., experimental trial-
and-error. Such in-
silico molecule design approaches are, however, challenging and limited in
their accuracy,
especially when applied to design of large molecules, such as proteins and/or
peptides. These
molecules are typically on the order of several kilo-Daltons (kDa) in terms of
molecular weight,
and have complex and hierarchical three-dimensional structures that influence
their behavior,
making functionality difficult to predict computationally. Accordingly,
success rates of existing
computational approaches to design of large molecules, such as proteins and
peptides, is limited,
and extensive experimental verification is often required.
[0317] In certain embodiments, technologies described herein provide, among
other things,
systems, methods, and architectures that address challenges associated with
generating accurate
predictions of structural features, properties, and functions of large
molecules, thereby providing
an improved toolkit for in-silico design of biologics, for example proteins
and peptides. In
particular, in certain embodiments, systems and methods described herein
include artificial
intelligence (AI) ¨ based software modules that can accurately predict
performance of candidate
biomolecules and/or portions thereof (e.g., amino acid backbones, sub-regions
of interest, etc.)
91
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
with respect to particular design criteria. In certain embodiments, design
criteria and
performance metrics that are evaluated by AI-powered modules described herein
are tailored
based on structural biology considerations relevant to large molecule design,
for example,
reflecting hierarchical organization of protein and peptide structures. In
this manner,
technologies described herein provide an improved toolkit for in-silico
biomolecule design,
thereby increase a likelihood of generating viable options for use in real
world applications such
as in disease treatment, prevention, and diagnosis. Accordingly, approaches
described herein can
reduce experimentation costs and cycle time associated with verifying
biomolecule properties.
A. In-Silico Design and Engineering of Custom Biomolecules
[0318] In certain embodiments, designing a particular biologic structure
(e.g., protein and/or
peptide) with various desired structural features and, e.g., ultimately,
properties in-silico involves
using computer-generated predictions to examine how changes to structural
features of the
biologic impact desired functionality and properties and, for example, making
adjustments
according to achieve desired performance.
[0319] A variety of structural features may be varied and examined. These
include, for
example, without limitation, amino acid sequences in various regions of the
biologic, rotamer
variations for one or more amino acids, post-translational modifications
(PTMs) and
conformations of a protein and/or peptide molecule's peptide backbone.
Structural features may
also include properties that relate to a way the biologic interacts with other
molecules. For
example, as described in further detail herein, approaches that aim to design
biologics for
binding to particular targets (e.g., molecules and/or complexes formed
thereof), a three-
dimensional orientation of the biologic with respect to a particular target
(referred to herein as a
92
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
"pose") molecule may be varied so as to allow different poses of the biologic
in relation to the
target to be evaluated. Accordingly, especially for large, biologic
structures, a wide range of
structural features, both of the biologic itself as well as in relation to how
it orients and forms a
complex with respect to another, e.g., target, molecule exist and can be
adjusted to influence
performance. The landscape of variations in and/or combinations of these
structural features
creates an extensive search space to be explored in order to identify and
design features of a
prospective custom biologic structure that will result in desired properties
and functionality.
Doing so in an efficient manner presents a significant challenge.
[0320] Moreover, in certain embodiments, navigating this search space to
identify those
favorable structural features that create performance improvements in sit/co
relies on use of
computational tools to generate predictions, for example, of how changing one
or more particular
structural features influences a desired property, such as binding affinity to
a particular target,
thermal stability, aggregation, etc. For example, in designing a biomolecule
for binding to a
particular target, a computer generated prediction of binding affinity may be
used as a
performance metric to compare different biomolecule structure designs.
[0321] Accordingly, the ability to (i) efficiently explore a vast search
space of structural
features of large molecules and (ii) generate accurate predictions of how
changes in structural
features of a biologic design impact properties and performance with respect
to desired design
criteria are key capabilities that allow for successful in silico design of
biologic molecules.
[0322] In particular, managing the size of the potential search space is
non-trivial. First, in
certain embodiments, intelligent sampling techniques are typically used in
order to optimize an
objective function that measures performance with respect to a desired design
criteria. Brute
force, e.g., random, sampling approaches may not viable in certain
embodiments. For example,
93
CA 03226172 2024-01-03
WO 2023/004116
PCT/US2022/038014
brute force approaches exhaustively explore a search space. When a search
space is large, its
exploration via a brute force approach can become intractable. Accordingly, in
certain
embodiments, approaches such as simulated annealing may be used in connection
with tools
described herein. Second, additionally or alternatively, in certain
embodiments, approaches
described herein may leverage insight based on structural biology
considerations to reduce sizes
and/or dimensionality of potential search spaces. For example, as described
herein, in certain
embodiments, tools described herein utilize and/or provide for a step-wise,
modular approach
whereby, particular structural features ¨ such as backbone orientation and
amino acid sequences
- are optimized separately, one after the other. In certain embodiments, this
modular approach
reflects a hierarchical organization of protein and/or peptide structures.
[0323]
Additionally or alternatively, accurately predicting performance of structures
in a
manner that allows different structural designs scored in a quantifiable (or
objective) fashion and,
accordingly, compared, is also challenging. In certain embodiments, tools
described herein
address limitations of physics-based, empirical, and knowledge-based (for
example, machine
learning optimized around one or more handpicked features) approaches by
leveraging a deep
learning approach that utilizes AI-computed scoring functions.
[0324] In
particular, in certain embodiments, for example, tools described herein
leverage
insight that computed scores need not necessarily correspond to experimentally
measurable
performance metrics. For example, in certain embodiments, levels of similarity
between key
features prospective custom biologic structures and those of pre-existing,
successful biological
structures and assemblies can be useful and accurate predictors of success.
Moreover, in certain
embodiments, machine learning models can be used to accurately identify these
key features, and
determine levels of similarity in a quantitative fashion. Accordingly, in
certain embodiments,
94
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
approaches such as AI-based classification and/or regression can be used to
create scoring
functions that accurately measure a likelihood that particular designs will be
successful.
[0325] Accordingly, approaches described herein may utilize computational
tools such as
artificial intelligence (AI), neural networks, artificial neural networks
(ANN), convolutional
neural networks (CNN), generative adversarial networks (GAN), deep learning
models, and
others to explore the search space and generate predictions for large molecule
and other
biomolecule function, structure, and/or properties. As machine learning
techniques typically rely
on training procedures in order to establish model parameters (e.g., weights)
and allow models to
make accurate predictions, embodiments described herein, may utilize a variety
of data sources
for training, such as, without limitation, public databases such as the
protein databank (PDB),
publicly available binding affinity databases, data from other biological
databases, proprietary
databases, as well examples generated from other sources of data including
laboratory data,
academic research, and open literature. As described in further detail herein,
approaches
described herein may also include data augmentation approaches and use of
computer generated
training examples to supplement data on pre-existing structures and to tailor
training data sets to
a particular types of structural features (e.g., backbone conformation, amino
acid sequence, etc.)
and/or performance being evaluated by a particular model.
B. Example Pipeline for Designing Custom Binders
[0326] FIG. 1 illustrates an example process 100 for designing a custom
biologic structure
for binding to a target. Example process 100 shown in FIG. 1 utilizes a
scaffold docker module
102, an interface designer module 104, and, optionally, a binding affinity
predictor module 106.
In example process 100, scaffold docker module 102, interface designer module
104, and
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
binding affinity predictor module 106 are arranged sequentially, in a
pipeline, with results
obtained from scaffold docker module 102 used as input for interface designer
module 104.
Other arrangements of these and/or other modules are also possible and are
contemplated by the
present disclosure.
[0327] As explained in further detail herein, each of the three modules
(scaffold docker
module 102, interface designer module 104, and binding affinity predictor
module 106) utilizes a
particular machine learning model to evaluate and score certain structural
features of a
prospective custom biologic with respect to particular performance metrics.
[0328] In particular, in certain embodiments, scaffold docker 102 may be
used to first
identify particular designs of peptide backbones and ways in which they can be
oriented, with
respect to the target, that are favorable for binding. Once identified, such
favorable backbones
can be populated with amino acids to create a custom biologic structures
(e.g., in silico, via use
of various computer representations and approaches described herein) via
downstream modules,
e.g., reflecting the hierarchical manner of protein structures.
[0329] In particular, in certain embodiments, scaffold docker module 102
evaluates
candidate scaffolds models and particular three-dimensional orientations ¨
referred to herein as
poses - thereof for predicted suitability (e.g., a likelihood of success) in
binding to a particular
target. Candidate scaffold models are representations of candidate peptide
backbones, which can
be populated with amino acids to create a custom biologic structures. Scaffold
docker module
102 generates a plurality of prospective scaffold-target complex models, each
representing a
particular candidate peptide backbone positioned at a particular pose with
respect to the target.
Scaffold docker module 102 utilizes a machine learning algorithm to compute
scaffold-pose
scores for the prospective scaffold-target complex models. As explained in
further detail herein,
96
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
a scaffold-pose score for a prospective scaffold-target complex model is a
value that provides a
measure of suitability of the particular candidate peptide backbone and pose
represented by the
prospective scaffold target-complex model. Accordingly, scaffold docker module
102 uses
computed scaffold-pose scores to select a subset of prospective scaffold-
target complex models,
each representing a particular candidate peptide backbone at a particular pose
determined (e.g.,
based on the scaffold-pose scores) to be suitable for binding to the target.
[0330] In certain embodiments, prospective scaffold-target complex models
determined via
scaffold docker module 102 can be used as a starting point for interface
designer module 104,
which populates candidate peptide backbones with amino acids to generate
candidate interfaces
comprising various combinations of amino acid types and rotamers at sites
located in proximity
to the target (e.g., the target molecule and/or e.g., in the case of a
complex, one or more
constituent molecules thereof)(e.g., hotspot locations and/or context
locations). In this manner,
interface designer module 104 generates a plurality of prospective ligand-
target complex models,
each representing a particular candidate peptide backbone positioned at a
particular pose with
respect to the target (e.g., as determined by scaffold docker module 102) and
having a particular
amino acid interface. Interface designer module 104 generates and evaluates
prospective ligand-
target complex models to determine interface designs likely to be successful
for binding to the
target. In particular, interface designer module 104 utilizes a machine
learning algorithm to
compute an interface score for each prospective ligand-target complex model.
Interface scores
are described in further detail herein, and provide a measure of suitability
of a particular interface
design for binding to the target. Interface designer module 104 selects a
subset of the
prospective ligand-target complex models based on the computed interface
scores, thereby
identifying ligand structures predicted to be successful for binding to the
target. These identified
97
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
ligand structures, or portions thereof (e.g., sub-regions in proximity to the
target) can be used to
create custom biologics.
[0331] In certain embodiments, a binding affinity predictor module 106 may
also be used to
predict binding affinities between designer biologic structures and the
target. In certain
embodiments, binding affinity predictor module 106 may be used to evaluate
and/or refine
prospective ligand-target complex models determined via interface designer
module 104. For
example, in certain embodiments, binding affinity module 106 may receive a set
of candidate
ligand-target complex models from interface designer module 104 and generate
binding affinity
predictions based on the set of candidate ligand-target complex models. As
described in further
detail herein, these predicted binding affinities can be used, for example, to
sort and/or identify a
subset of candidate designs, as well as to refine and/or modulate structural
designs further.
C. Dataset Creation and Data Representation Techniques
I. Dataset Creation
[0332] In certain embodiments, scaffold-docker module 102, interface
designer module 104,
and binding affinity predictor module 106 each utilize a machine learning
model as a scoring
function that predicts performance of various structural modifications and
designs en route to
creating a custom biologic structure.
[0333] Turning to FIG. 2, in certain embodiments, each machine learning
model is trained,
for example, using structural data (e.g., representing experimentally
determined crystallographic
structures) for existing biological complexes obtained from public databases
or elsewhere. As
described in further detail herein, depending on a particular type of
structural feature and/or
scoring function to be evaluated, examples of existing biological complexes
may be
98
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
supplemented with computer generated representations of artificial biological
complexes that
have not been demonstrated to exist physically.
[0334] FIG. 2 shows an example training and validation workflow 200 used in
certain
embodiments. Method 200 may be used to develop a machine learning model that
can be used
as a scoring function, for example to predict performance of particular design
elements of a
custom biologic being created, for use in various modules described herein
(e.g., scaffold docker
module, interface designer module, binding affinity predictor module, etc.).
In certain
embodiments, training various machine learning models described herein may
generally include
one or more data preparation steps 210, including, for example, collection of
an initial dataset
212 (e.g., from various databases) and data augmentation steps 214. These data
preparation steps
210 may be used to create a robust and/or non-biased) training dataset that
includes, for example,
a sufficient number and/or variety of examples to allow a machine learning
model to be trained
to make accurate assessments of structural designs and their predicted
performance. As
illustrated in FIG. 2, in certain embodiments, a portion of examples may be
set aside or split off
216 to create a testing dataset 218b, distinct from training dataset 218a.
Training dataset 218b
may be used for training (e.g., to establish weights) 220 and create a trained
machine learning
model 222. Testing dataset 218b can be used to validate a trained machine
learning model 222,
for example to identify and/or avoid overfitting.
[0335] FIG. 3 illustrates various data preparation steps in further detail.
For example, a step
of collecting an initial dataset 320, may include collecting data, for
example, from one or more
public databases such as the protein databank (PDB) and/or other biological
databases) as well as
curating an initial dataset based on a specific task or goal. In certain
embodiments, data curation
may include filtering the data based on various criteria, such as a minimum
resolution (e.g., such
99
CA 03226172 2024-01-03
WO 2023/004116
PCT/US2022/038014
that structures for which a resolution is above a particular threshold value
are not included). For
example, various datasets in example implementations of embodiments described
herein were
created using resolutions better than 2.7A. Individual data elements may be
pre-labeled and/or
assigned labels, for example to identify each data element as belonging to a
particular class. In
certain embodiments, various steps may be performed to produce a balanced
dataset 340 from an
initial dataset. For example, sequence clustering may be performed to identify
clusters of similar
examples, and a subset (e.g., smaller, limited number) of representative
examples from each
cluster be selected for inclusion in a balanced data set. Additionally or
alternatively, in certain
embodiments, balancing techniques including up-sampling and down-sampling, may
be used.
[0336] In
certain embodiments, balancing may include a data augmentation step 214. For
example, in certain embodiments, neural networks require a large amount of
training data. In
certain cases, datasets that are available (e.g., initial datasets) are small,
e.g., and may not be of
sufficient size for training. Accordingly, in certain embodiments, data
augmentation techniques
can be used to artificially generate more data from an existing (e.g.,
initial) dataset. Additionally
or alternatively, in certain embodiments, available datasets may be
imbalanced. For example, in
certain embodiments, binding affinity datasets may contain many examples of
complexes with
mid-range affinities whereas high and low affinity complexes may be under-
represented. In
certain embodiments, data augmentation may also be used to balance a dataset.
Classes can be
differentially augmented to try to limit class imbalance. Additionally or
alternatively, in certain
embodiments, data augmentation utilized herein may apply rotations to
structural data used. For
example, in certain embodiments, various machine learning models utilized
herein comprise
convolutional neural networks (CNNs). CNN's may 'perceive' rotated versions of
otherwise
identical structures differently. Accordingly, generating multiple training
examples from one
100
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
structure by rotating it in different ways can be used to avoid inadvertently
training a CNN to
learn to differentiate otherwise identical structures on the basis of
rotational variations. In
certain embodiments, to generate rotational examples for data augmentation,
sampling is
performed via a Hopf Fibration, e.g., using a HEALPix grid, since sampling
along a
conventional three-axis grid may produce non-uniform sampling of rotational
vectors. Hopf
Fibration is described in Gorski et al. arXiv:astro-ph/0409513. 2005 and
Yershova et al. Int J
Rob Res. 2010 Jun 1; 29(7): 801-812.
[0337] In certain embodiments, a dataset (e.g., a balanced dataset 340) may
be divided 216
into training sets 218a and testing sets 218b (e.g., collectively, 360) for
training and validating a
machine learning model. For example, where individual data elements represent
biologic
structures, a dataset may be split into training and testing datasets based on
sequence similarities.
For example, in certain embodiments a data set may be split such that there is
from about 20% to
about 80% sequence similarity between the training and testing sets. In some
embodiments, the
sequencing similarity may be from about 10% to about 90%, or from about 25% to
about 70%,
or from about 30% to about 60%, or from about 35% to about 50%, or from about
35% to about
45%.
[0338] An example process 500 for splitting a dataset comprising examples
of protein and/or
peptide complexes (e.g., interfaces) based on sequence clustering is shown in
FIG. 4. An
approach such as example process 500, and/or variations thereof may be used in
various
embodiments of training procedures described herein. Other approaches for
splitting a dataset
may also be utilized in accordance with embodiments described herein.
ii. Data Representations
101
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0339] In certain embodiments, various modules (e.g., a scaffold docker
module 102, an
interface designer module 104, a binding affinity predictor module 106) and/or
machine learning
models (e.g., utilized by various modules) described herein operate on and
analyze
representations of biologic structures and compute values of scoring functions
based thereon. In
certain embodiments, representations include structural models of a biologic,
or portion thereof
(e.g., a scaffold model, representing a peptide backbone of a protein and/or
peptide). In certain
embodiments, representations may also include models of a biologic or portion
thereof together
with one or more other molecules, such as a target, in a complex.
[0340] For example, in certain embodiments various technologies and tools
described herein
utilize, manipulate, evaluate, etc., structural models of proteins and/or
peptides. In certain
embodiments, such structural models include models of proteins and/or peptides
in complex with
other molecules. In certain embodiments, these include models of a ligand and
a receptor, and
are referred to as a ligand-receptor complex model, which comprises a ligand
model ¨ a
computer representation of at least a portion of the ligand - and a receptor
model ¨ a computer
representation of at least a portion of the receptor.
[0341] In certain embodiments, various modules and approaches described
herein may
utilize a scaffold model representation that represents a peptide backbone of
a particular protein
and/or peptide. In certain embodiments, scaffold models may be created from
structural
representations of existing proteins and/or peptides, for example by stripping
amino acid side
chains. In certain embodiments, while scaffold models omit detailed amino acid
side chain
structure, they may nevertheless retain a first atom of a side chain, such as
a beta-carbon atom,
which is common to all side chains apart from Glycine, and may be used, e.g.,
as a placeholder
for identification of sites that can be occupied by amino acid side chains. In
certain
102
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
embodiments, where an initially existing side chain was Glycine, the first
atom of glycine, which
is hydrogen, can be used in place of a beta-carbon. In certain embodiments,
scaffold models
may be computer generated (e.g., and not based on an existing protein and/or
peptide). In certain
embodiments, computer generate scaffold models may also include first side
chain atoms, e.g.,
beta carbons, e.g., as placeholders of potential side chains to be added.
[0342] Accordingly, in certain embodiments, rather than represent an entire
ligand of a
particular biological complex, a scaffold model can be used in combination
with a model of a
receptor, creating a scaffold-receptor complex model.
[0343] Various structural models described herein may be implemented in a
variety of
manners, via a variety of data representations. In certain embodiments, a
structural model may
be represented as a listing of types and coordinates of various atoms in
space, such as, for
example, PDB files. In certain embodiments, structural models may include
additional
information, such as an indication of which atoms belong to which particular
amino acid residue
or portion of peptide backbone, an indication of secondary structure motifs,
etc.
[0344] Turning to FIG. 5, in certain embodiments, while an overall complex
comprising, for
example, a ligand and a receptor molecule may be large, behavior such as
binding may be
influenced primarily by a smaller sub-region 640 of the complex, about an
interface where atoms
and/or amino acid side chains of the ligand and receptor are located in
proximity to each other.
Accordingly, in certain embodiments, approaches described herein include
and/or utilize various
interface extraction steps, used to identify interface sub-regions comprising
portions of a ligand
and/or receptor of a complex. Representations (e.g., complex models) 660 of
identified interface
sub-regions may be utilized, e.g., as opposed to models of a larger portion
610 (e.g., though not
necessarily entire) of a complex, for (e.g., to facilitate) further
processing, such as identifying
103
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
particular amino acid sites to limit sequence design to, and/or to provide
more manageable input
to a machine learning model.
[0345] For example, in certain embodiments, interface extraction may be
based on and/or
include steps of identifying particular amino acid sites of a ligand and/or
receptor determined to
be relevant to influencing binding. For example, in certain embodiments, sites
referred to as
"hotspots" may be identified on a ligand and/or receptor. For a ligand,
hotspots refer to sites
which, when occupied by an amino acid side chain, place at least a portion of
the amino acid side
chain in proximity to one or more side chains and/or atoms of the receptor.
Likewise, for a
receptor, hotspots are sites which, when occupied via an amino acid side
chain, place at least a
portion of the amino acid side chain in proximity to one or more side chains
and/or atoms of the
ligand.
[0346] In certain embodiments, for example since size, geometry, and
orientation of various
acid side chains may vary, hotspots may be identified based on distances
between beta carbon
(CP) atoms of a ligand and receptor of a complex. For example, a ligand
hotspot may be
identified as a particular site on the ligand that, when occupied by an amino
acid side chain, will
place a Cfl atom of the side chain located at the site within a threshold
distance of a Cfl atom of
the receptor. Receptor hotspots may be identified analogously. Since Cfl atoms
are common to
every amino acid side chain apart from Glycine, this approach provides a
uniform criteria for
identifying hotspots, independent of a particular amino acid that occupies a
particular site. In
certain embodiments, in the singular case where a Glycine residue occupies a
particular site,
Glycine's hydrogen atom may be used in place of a Cfl, but hotspots identified
in an otherwise
identical fashion. Additionally or alternatively, in certain embodiments,
distances between
alpha-carbons (Ca) associated with amino-acid sites may be determined, e.g.,
in a similar manner
104
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
to which distances between CP atoms are determined. In this manner, Ca
distances may be
compared with various threshold values to identify hotspots.
[0347] Various threshold distances may be used for identification of
hotspots. For example,
in certain embodiments, a hotspot threshold distance of 8A (i.e., 8 Angstroms)
is used. In some
embodiments, other thresholds may be used for defining a hotspot (such as less
than 3A, less
than 4A, less than 5A, less than 6A, less than 7A, less than 9A, less than
10A, less than 12A, less
than 15A, less than 20A, as well as other suitable thresholds).
[0348] In certain embodiments, hotspots may be identified based on
comparison of values
computed by various functions ¨ e.g., of one or both of a Ca and CP distance ¨
with one or more
threshold values. Such functions may take into account features such as bond
angles, surface
area, etc.
[0349] Additionally, or alternatively, approaches described herein may also
identify sites
referred to as context sites, which are not hotspots themselves, but are
located near (e.g., and on a
same peptide or polypeptide chain) hotspots. In certain embodiments, for a
particular hotspot,
one or more context sites about the hotspot are identified as those sites for
which a CP atom of a
residue located at the site (or H atom, where the residue is Glycine) is
within a threshold distance
(e.g., a context threshold distance) of a CP atom (or H atom) of a residue
occupying the
particular hotspot. In this manner, for a particular hotspot, one or more
associated context sites
may be identified.
[0350] In certain embodiments, as with hotspot threshold distances, various
context threshold
distances may be used to identify context sites. For example, as shown in FIG.
5, in certain
embodiments, a context threshold distance of 5A (i.e., 5 Angstroms) is used.
In some
embodiments, other thresholds may be used for defining a hotspot (such as less
than 3A, less
105
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
than 4A, less than 5A, less than 6A, less than 7A, less than 9A, less than
10A, less than 12A, less
than 15A, less than 20A, as well as other suitable thresholds). In certain
embodiments, a context
threshold distance is less than a hotspot threshold distance.
[0351] In certain embodiments, hotspot and context site identification
(and, accordingly,
interface extraction) may be performed for scaffold models as well as ligand
models. For
example, as described herein, scaffold models may retain first side chain
atoms ¨ beta carbons
and/or hydrogens ¨ and, accordingly, hotspot and context site identification
as described herein
may be performed for ligand and scaffold models alike.
[0352] In certain embodiments, interface extraction may be used to identify
certain portions
of a computer representation of a biological complex comprising at least a
portion of ligand and
a receptor. For example, in certain embodiments, portions of a biological
complex model (e.g.,
representations of amino acid side chains, voxels of a three dimensional grid
or matrix, etc.)
corresponding to hotspot and/or context sites may be identified. For example,
in certain
embodiments, an interface portion of a biological complex model may include
representations of
amino acid side chains located at hotspot and/or context sites, and exclude
other portions of the
complex model. In certain embodiments, an interface portion may include
representations of
portions of a peptide backbone of a ligand and/or receptor that are associated
with hotspot and/or
context sites. For example, an interface portion may include representations
of side chains
located at hotspot and/or context sites as well as adjacent atoms of a peptide
backbone (e.g.,
alpha carbon, hydrogen, and peptide bond). In certain embodiments, an
interface portion may
include portions of a complex model corresponding to locations within a
continuous volume
comprising identified hotspot and/or context sites, such as a smallest
rectangular volume
comprising identified hotspot and/or context sites.
106
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0353] Turning to FIG. 6, in certain embodiments, biological molecules
and/or complexes
thereof may be represented via structural models that, among other things,
identify types and
locations of atoms in physical space, for example via coordinate files such as
those used for PDB
entries. In certain embodiments, approaches described herein may also utilize
volumetric
representations, whereby a three-dimensional data representation (e.g.,
matrix) is used to
represent a physical three-dimensional space. In certain embodiments,
approaches described
herein create, as a volumetric representation of a particular biological
molecule and/or complex,
a three dimensional electron density map (EDM) 710. In certain embodiments, a
3D EDM may
be created from a structural model, for example, by simulating x-ray
diffraction and scattering.
For example, in certain embodiments, approaches described herein generate 3D
EDMs from
structural models (e.g., atomic coordinates) based on a five-term Gaussian
approximation and
atomic scattering factors as found in International Tables for X-ray Cryst.
Vol.IV. A similar
approach is implemented in CCP4 (see, e.g., ccp4.ac.uk). In certain
embodiments, other
Gaussian approximations, such as a two-term Gaussian approximation, may be
used. In certain
embodiments, such EDM representations, as described and utilized herein, may,
have a size of
64x64x64 A3 (i.e., cubic Angstroms) with a lA (one Angstrom) grid spacing,
though various
embodiments and implementations may utilize other input sizes and resolution.
[0354] Turning to FIG. 7, in certain embodiments, volumetric
representations, such as EDMs
810, are used as input to machine learning models 820 used to evaluate and
score various
structural designs for creating custom biologics as described herein. In this
manner, in certain
embodiments, a machine learning model 820 receives a 3D EDM representing at
least a portion
of a biological complex (e.g., a sub-region about an interface) and
determines, as output a score
830. In certain embodiments, the score quantifies a measure of similarity
between the biological
107
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
complex and native and/or otherwise successful complexes, as determined by the
machine
learning model. In certain embodiments, the score is a predicted physical
property, such as a
predicted binding affinity. In certain embodiments, machine learning models as
described herein
are trained using thousands of curated example representations of biological
complexes, allowing
them to make accurate inferences and predictions.
[0355] Without wishing to be bound to any particular theory, it is believed
that use of 3D
EDMs as input to machine learning models as described herein may be
advantageous in that it
allows for use of CNNs and facilitates incorporation of three-dimensional
spatial relationships
into AI-based learning procedures. Additionally or alternatively, electron
density maps provide
an accurate way of representing three dimensional structure, as well as
physical and chemical
properties, of biological complexes, such as receptors-ligand complex and/or,
more particularly,
complexes formed by prospective custom biologic designs intended for binding
to target
molecules and/or target complexes as described herein.
[0356] In certain embodiments, among other things, use of 3D EDMs as
volumetric input to
a machine learning model is distinct from other approaches, which convert
atomic coordinates to
abstract representations of each atom and interpolate their positions into 3D
grids.
[0357] In certain embodiments, for example as shown in FIG. 6, interface
extraction may be
used to identify an interface portion of a biological complex model, and the
identified interface
portion 660 (e.g., rather than the entire biological complex model) used to
generate a 3D EDM
710 for use as input to a machine learning model.
108
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
D. Scaffold Docker Module
[0358] FIG. 8 is a schematic illustration of a scaffold docking approach,
which, in certain
embodiments, may be performed by a scaffold docker module 102 as described
herein. As
shown in FIG. 8, a large molecule, such as a biologic may present to a
particular target at a wide
variety of different three-dimensional orientations ¨ i.e., poses. Different
poses place different
portions of the biologic in proximity to the target, and, among other things,
certain orientations
may be favorable for binding and forming a complex with the target, while
others are not.
Without wishing to be bound to any particular theory, in certain embodiments,
depending on a
particular biologic's peptide backbone, certain poses may orient particular
sub-regions, e.g.,
having particular local geometries, in a favorable manner with respect to a
target or binding
pocket thereof, so as to, for example, place a sufficient and/or maximal
number of amino acid in
proximity to atoms of the target. Moreover, due to, for example, particular
amino acid sequences
at various portions of the target, various physiochemical properties and/or
features may be
present and, accordingly, may influence interaction with backbone structures
and potential amino
acid interfaces created thereon in a complex fashion.
[0359] Accordingly, in certain embodiments, designing a custom biologic
suitable for
binding to a particular target begins with identifying one or more candidate
peptide backbones
and, for each, determining which, if any, poses provide favorable orientations
for binding to the
target. Candidate peptide backbones and poses thereof that are identified as
favorable can then
be used as a starting point ¨ for example, a molecular scaffold ¨ for
downstream design steps that
tailor amino acid side chain sequences to optimize molecular interactions with
the target and
design a binding interface of the custom biologic.
109
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0360] Accordingly, in certain embodiments, custom biologic design tools
described herein
include and/or provide for a scaffold docker module that can be used to
identify favorable
candidate peptide backbones and poses thereof for binding to a desired target.
Turning to FIG. 9,
in certain embodiments, a scaffold docker module receives as input, accesses,
or otherwise
obtains structural models that represent candidate ligands and/or their
peptide backbones 1010.
In certain embodiments, structural models utilized by a scaffold docker module
represent (e.g.,
solely) a peptide backbone of a protein or peptide molecule, omitting amino
acid side chains, and
are referred to herein as scaffold models.
[0361] In certain embodiments, scaffold docker module generates and
evaluates multiple
poses for a particular candidate peptide backbone by creating and/or accessing
a plurality of
scaffold-target complex models 1020. Each scaffold-target complex model
comprises a
corresponding candidate scaffold model and structural model of the target and
represents the
candidate peptide backbone at a particular pose with respect to the target. In
certain
embodiments, scaffold-target complex models to be evaluated are generated by
applying three-
dimensional rotation and/or translation operations to scaffold model to
represent various poses.
Rotation and/or translation operations to be applied to a scaffold model may
be determined, for
example, via a random sampling approach, or, additionally or alternatively, in
certain
embodiments, via certain pose generation processes described herein. In
certain embodiments,
rotation is homogeneously sampled, for example via sampling along a (e.g.,
fixed) interval (e.g.,
degrees, 10 degrees, 15 degrees, 20 degrees, etc.). In certain embodiments, a
particular
rotational space, such as using a Hopf Fibration, as described herein, is
used. In certain
embodiments, use of a Hopf Fibration does not rely on degrees, but rather on
the number of
points that will homogeneously sample a rotation sphere.
110
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0362] In certain embodiments, scaffold docker module may evaluate
generated scaffold-
target complex models and determine 1030 scaffold-pose scores ¨ e.g.,
numerical values that
provide a quantitative measure of suitability or favorability of particular
complex models and the
poses that they represent. Based on the determined scaffold-pose scores, a
scaffold docker
module may then select a subset of scaffold-target complex models, e.g., as
representing
favorable candidate peptide backbones and poses thereof 1040. A selected
subset may then be
provided to and/or used a starting point for other modules, such as an
interface designer module
as described herein.
[0363] For example, as shown in FIG. 8, both position and orientation of a
particular
candidate scaffold model 902 with respect to the target 904 may be varied, to
generate multiple
candidate poses and thereby sample a search space of three dimensional
orientations and
positions of the candidate scaffold model with respect to the target. In
certain embodiments, one
or more regions of interest of the target are identified and candidate poses
are generated and
evaluated so as to orient and assess viability / potential performance, as
described in further
detail herein, of the candidate scaffold model with respect to the one or more
regions of interest
of the target. These target regions of interest may be, for example, putative
binding sites and
may be, in certain embodiments, pre-selected by a user and/or automatically
identified, e.g.,
based on known binding sites, structural features, output of other modules,
etc.
[0364] The scaffold docker module computes scores based on the sampled
poses, in order to
identify those most favorable for binding. For example, as illustrated in FIG.
9, a low score is
computed for pose 920 (e.g., in pose 920, neither the location nor orientation
of scaffold model
902 are favorable), an intermediate score is computed for pose 940 (e.g., in
pose 940, a position
111
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
of scaffold model 902 is favorable, but its orientation is not) and a high
score computed for pose
960, e.g., due to a favorable orientation and position of the candidate
scaffold.
[0365] In certain embodiments, a scaffold docker module utilizes an AI-
based scoring
approached whereby a machine learning model is used to evaluate prospective
scaffold-target
complex models and determine scaffold-pose scores. In this manner, approaches
described
herein leverage extensive structural data on existing native protein-protein
and/or protein-peptide
complexes along with tailored training procedures to create a scaffold
predictor model that
implements a trained machine learning algorithm to assess which candidate
peptide backbones
and poses thereof (as represented via scaffold-target complex models) are
favorable for binding
to a particular target.
i. Training Data Set Construction
Native and Artificial Scaffold-Receptor Complex Models
[0366] In certain embodiments, a scaffold predictor model is a machine
learning model that
receives, as input, a representation of at least a portion of a particular
scaffold-target complex
model and determines, as output, a scaffold pose score. In certain
embodiments, a scaffold pose
score is a numerical value, for example a probability value ranging from zero
to one. In certain
embodiments (e.g., where a binary classifier is used), scaffold pose score may
further
transformed into a Boolean value, e.g., based on a comparison of with a
threshold value.
[0367] For example, in certain embodiments, as described herein, a scaffold
predictor model
may be trained to differentiate between and/or determine a measure of
similarity between
representations of native complexes (e.g., which have been demonstrated to be
physically viable)
and artificially generated complexes that have varying features, which may not
be suitable for
112
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
binding. In particular, in certain embodiments, in order to train a machine
learning model in this
manner, examples of both native and artificial are used as training data.
Native complex
examples may be obtained and curated from datasets of existing biological
complexes.
Representations of native complexes are, by definition, examples of physically
viable complexes,
and represent candidate peptide backbones and poses that are suitable for
binding. In certain
embodiments, for example to provide examples of complexes that have structural
features of
varying degrees of suitability for binding, artificial complex models are
generated. As described
herein, artificial complex models may be generated by perturbing native
complex models. In
certain embodiments, measures of an extent of the perturbation are determined.
These measures,
referred to as pose quality metrics, allow various artificial complex models
to be labeled,
selected, sorted, etc., based on their similarity to existing native complex
models. Accordingly,
together with examples of native complexes, creating and labeling artificial
complex models in
this manner provides for creation of a labeled dataset that can be used to
train a machine learning
model to differentiate between and/or quantify similarities between successful
native complexes,
native-like artificial complexes that may reflect successful features, and
artificial complexes that
are do not have features suitable for binding. Such a machine learning model,
when presented
with new data, such as a candidate scaffold-target complex model, may then be
used to generate
a scaffold-pose score that reflects the machine learning model's assessment of
how 'native-like'
the structure appears. In this manner, scaffold-pose scores can be utilized to
identify and design
candidate peptide backbones and poses that will be suitable for binding to a
particular target.
Pose Quality Metrics
[0368] In certain embodiments, one or more pose quality metrics are
computed for scaffold-
target complex models used as training data. In certain embodiments, pose
quality metrics are
113
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
computed for native scaffold-target complex models as well as artificial
scaffold-target complex
models. As explained in further detail herein, pose quality metrics can be
used to ensure training
examples to be used to train the scaffold docker's machine learning model are
sufficiently
varied.
[0369] Turning to FIGs. 10 and 11, in certain embodiments, pose quality
metrics are
determined based on a number of identified contact locations between a
scaffold and target in a
complex. In certain embodiments, contact locations may be identified as
locations wherein a
first side chain atom (e.g., beta carbon and/or hydrogen) of a scaffold is
within a particular
contact threshold distance of a first side chain atom of a target (e.g.,
contact locations do not
necessarily refer to points of physical contact, but rather locations of sites
on a scaffold and
target that are in sufficient proximity to each other to be likely to
influence binding). In certain
embodiments, the contact threshold value is 8A (i.e., 8 Angstroms) is used. In
some
embodiments, other thresholds may be used for defining a contact (such as less
than 3A, less
than 4A, less than 5A, less than 6A, less than 7A, less than 9A, less than
10A, less than 12A, less
than 15A, less than 20A, as well as other suitable thresholds). In certain
embodiments, contact
locations may be identified in a manner analogous to that described herein
with respect to
identification of hotspots.
[0370] In certain embodiments, a native contact number (NCN) is determined
to quantify the
number of native contacts in a particular scaffold-target complex model. As
used herein, a
native contact refers to a contact present in a native complex. In certain
embodiments, a total
contact number (TCN) is determined to quantify a total number of contacts in a
particular
scaffold-target complex model. FIG. 10 shows an example of a native scaffold-
target complex
model 1100. As shown in FIG. 10, five contacts (red circles) are identified
between scaffold
114
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
1104 and target molecule 1102. Since FIG. 10 shows a native scaffold-target
complex model
1100, each contact is a native contact, and both NCN and TCN for scaffold-
target complex
model 1100 equal five.
[0371] FIG. 11 shows an example of an artificial scaffold-target complex
model 1120.
Scaffold-target complex model 1120 is derived from native scaffold-target
complex model 1100.
In particular, it utilizes a same scaffold model 1104 and target 1102, but a
pose of scaffold model
1104 is varied (e.g., by applying a random three-dimensional rotation and/or
translation
operation to scaffold 1104) to create a new, non-native pose. As shown in FIG.
10, with the new
pose, three of the original, native contacts are maintained (red circles) and
two native contacts
are removed (open circles). The new pose also results in two new, non-native
contacts (purple
circles). Accordingly, NCN and TCN values for generated artificial scaffold-
target complex
model 1120 are three and five, respectively.
[0372] According, in certain embodiments, a training data set may be
constructed by (i)
obtaining native scaffold-target complex models based on experimentally
derived structural data
and (ii) generating artificial scaffold-target complex models, for example by
rotating and/or
translating scaffold models of native-scaffold-target complex models to
generate new poses. In
certain embodiments, values of pose quality metrics such as NCN and TCN can be
computed for
each (native and artificial) scaffold-target complex model.
[0373] For example, FIG. 12 summarizes and compares certain features of NCN
and TCN
values computed for native and artificial scaffold-target complex models. In
certain
embodiments, as shown in FIG. 12, pose quality metrics can be combined to
yield additional
metrics. For example, NCN and TCN may be combined into a single pose quality
metric
computed as the ratio, NCN/TCN. In certain embodiments, pose quality metrics
such as NCN
115
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
and TCN can be used to evaluate and select particular native and artificial
scaffold-target
complex models for inclusion and/or exclusion from a training data set. For
example, as shown
in FIG. 12, in one example, only scaffold-target complex models with TCN
values above a
threshold value (e.g., five) were selected for inclusion in the training data
set. In this manner,
potentially spurious native complex examples with a small number of contacts
were filtered out.
Other filtering approaches, threshold values, etc. based on NCN, TCN, NCN/TCN
values as well
as other pose quality metrics may be used additionally or alternatively.
[0374] In certain embodiments, a pose quality metric may provide a measure
of similarity
between an artificial scaffold-target complex model and a native scaffold-
target complex model
from which it is derived. For example, FIG. 13 illustrates calculation of a
root mean squared
distance (RMSD) between atoms of two structures. In certain embodiments, an
RMSD between
atoms of (i) a particular native scaffold-target complex model and (ii) a
particular artificial
scaffold-target complex model derived from the particular native scaffold-
target complex model
may be used as a pose quality metric. In particular, as explained herein, in
certain embodiments
an artificial scaffold-target complex model may be derived from a native
scaffold-target complex
model by applying three-dimensional rotations and/or translations to a
scaffold model of the
native-scaffold complex model in order to place it in a new, artificial, pose
relative to the target.
Accordingly, in certain embodiments an RMSD(Native, Pose) value can be
computed as follows:
RMSD(Native, Pose) = \A Eni_illNative,¨ Poseill2
= jn
1 ¨n 1 (Nativei, ¨ Pose,,)2 + (Native,, , ¨ Pose07, )2 + (Native, , ¨ P, )2
1=1 z ose,z
[0375] Accordingly, in certain embodiments, RMSD(Native, Pose) computes the
average of
the distances between locations of atoms of the initial, native complex model
and their new,
116
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
shifted locations, as they are in the new pose that the artificial complex
model represents. In this
manner, RMSD(Native, Pose) may provide a measure of similarity between an
artificial scaffold-
target complex model and a native scaffold-target complex model from which it
was derived.
[0376] Accordingly, as illustrated in FIG. 14, in certain embodiments, pose
quality metrics
such as those described herein reflect quality of a particular pose
represented by a scaffold-target
complex model. In certain embodiments, a pose quality metric provides a
numerical measure of
similarity between a pose represented by a particular scaffold-target complex
model and a native
pose (e.g., "native-ness"). Pose quality metrics may vary with, or inversely
to a level of
similarity to a native pose. For example, schematic 1420 illustrates how pose
quality or
similarity to a native complex varies with NCN/TCN value. Pose quality metric
NCN/TCN has
values ranging from zero to one. A value of NCN/TCN increases, approaching
one, with
increasing similarity between a pose represented by a particular complex model
and a native
pose. As explained herein, since NCN = TCN for a native complex model, a
NCN/TCN value of
one indicates a native complex model. For example, schematic 1440 illustrates
how pose quality
or similarity to a native complex varies with RMSD value. As explained herein,
RMSD reflects
a relative geometric distance of atoms of a particular complex model to a
native complex model.
An RMSD of zero indicates a particular complex model is a native complex
model. Increasing
RMSD reflects increasing distance between molecular structures represented in
an artificial
complex model and their native positions and orientations. As explained in
further detail herein,
pose quality metrics such as RMSD and NCN/TCN can be used to label potential
training
examples. By virtue of this labeling approach, examples can be selected to
sample a variety of
RMSD and NCN/TCN values (e.g., a uniform sample across a particular range of
values),
117
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
thereby providing training data that exposes a machine learning model to a
sufficient degree of
variation in pose qualities.
[0377] In certain embodiments, values such as TCN, NCN, RMSD can be used in
a
preliminary filtering step, e.g., to filter out irrelevant poses. For example,
in certain
embodiments, structures (e.g., obtained from databases) having computed RMSD
values above a
particular threshold are excluded from further evaluation. Such filtering
approaches may be used
at various steps in processes described herein.
[0378] In certain embodiments, artificial scaffold-target complex models
may be generated
via approaches other than that described with respect to FIG. 11 and may be
used additionally or
alternatively to the approach described herein with regard to FIG. 11. For
example, in certain
embodiments, artificial scaffold-target complex models may be created by
combining structural
representations of two or more monomers, e.g., structural models representing
native peptide
and/or protein monomers, and, e.g., arranging each monomer at various poses
with respect to
each other. In certain embodiments, artificial scaffold-target complex models
may be derived
from native scaffold-target complex models by altering a scaffold model
portion of the native
scaffold-target complex model, to produce an artificial scaffold-target
complex model wherein
the scaffold portion represents a non-native (e.g., artificial), perturbed,
backbone.
Training Dataset Generation
[0379] For example, FIG. 15 shows an example process 1500 for generating
datasets for
training and testing (e.g., validating) a machine learning model to determine
scaffold pose
scores, used in certain embodiments. Specific numbers and description (e.g.,
in boxes) in FIG.
118
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
15 describe values and steps carried out in a particular exemplary
implementation of process
1500, but values and particular approaches may vary from implementation to
implementation.
[0380] In example process 1500, an initial dataset (e.g., of native complex
models) is
obtained from one or more databases 1510, and the initial dataset may be split
into initial training
and testing datasets 1520, for example according to process 400 described
herein. Complex
models of the initial training and testing datasets may be used to create new,
artificial complex
models in a pose generation step 1530, for example by applying three
dimensional rotation
and/or translations to scaffold models of native complex models. In certain
embodiments, pose
generation step 1530 encompasses an initial filtering approach used to exclude
poses that
generate highly improbable and/or non-viable complexes, such as translations
that would
generate significant overlap between a scaffold model and target, or place
them too far apart to
interact/bind. One such approach is described in further detail below, with
regard to FIGs. 16A
and 16B (e.g., process 1600). In certain embodiments, one or more pose quality
metrics are
calculated for complex models generated via pose generation step 1530 and
computed pose
quality metrics are used to label the generated complex models 1540. Complex
models may then
be selected for inclusion in final training and/or testing data sets according
to computed pose
quality metrics via a sampling step 1550. For example, in certain embodiments,
complex models
are selected so as to uniformly sample a range of one or more particular pose
quality metrics,
such as NCN/TCN and/or RMSD.
[0381] In certain embodiments, in sampling step 1550, complex models are
also assigned to
two or more classes and labelled accordingly. For example, for a machine
learning model that
performs a binary classification function, complex models may be assigned to
one of two classes
(e.g., each complex model identified as an example of one class or another),
and used to train the
119
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
machine learning model to differentiate between the two classes. In certain
embodiments, more
than two classes may be used, for example to train a machine learning model
that performs non-
binary classification. In certain embodiments, complex models are assigned to
two or more
classes based on threshold values for one or more pose quality metrics. In
certain embodiments,
complex models are not sorted into classes, but instead labeled with a
numerical value, for
example determined from (e.g., including equal to) a pose quality metric. Such
an approach may
be used, for example, to train a regression model type of machine learning
model.
[0382] In certain embodiments, an additional, data augmentation step 1560
is performed. In
certain embodiments, as described herein, data augmentation step 1560 creates
additional
versions of examples of complex models by rotating entire models in three
dimensions. As
described herein, this approach can be used to account for the propensity of
CNNs to perceive
rotated versions of otherwise identical structures differently.
[0383] In this manner, final training and testing datasets may be generated
1570 and used to
establish weights of a machine learning model for use in evaluating candidate
scaffold-target
complex models.
Example Training Dataset Construction Implementations
[0384] FIGs. 16A to 20 show exemplary implementations of various steps in
process 1500,
for building training datasets. The implementation shown in FIGs. 16A to 20 is
used to generate
training data in which complex models are assigned to two classes, and was
used to train a binary
classifier machine learning model to distinguish between complex models that
were likely to
represent native complex, and complex models that were likely to represent
artificial structures,
as perceived via the machine learning model. As described in further detail
herein, FIGs. 16A
and 16B show an example approach for generating poses, suitable for use at
step 1630, and
120
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
filtering to exclude those that would produce highly non-physical complexes in
a rapid and
efficient manner. FIG. 17 describes a particular implementation of sampling
step 1550, which
may be used in certain embodiments. FIG. 18 describes an approach for
assigning complex
models to two classes.
[0385] FIGs. 16A and 16B illustrate an example process 1600, whereby ligand
and receptor
models can be represented via matrices (e.g., 3D matrices or tensors) of
complex numbers, and
an efficient cross-correlation approach used to identify poses that are
predicted to place molecule
surfaces in sufficient proximity for binding, and filter out those that likely
create non-physical
clashes and/or place a receptor and ligand too far apart for binding to occur.
In certain
embodiments, process 1600 begins with receiving and/or accessing, as input
1610, a ligand
model 1612 and a receptor model 1614. In certain embodiments, amino acid side
chains are
removed 1620 from ligand model 1612 to create a scaffold model 1622.
[0386] In certain embodiments, a shape map representation 1632 is created
from receptor
model 1614 via shape map generation step 1630a. In certain embodiments, as
shown in FIG.
16B, a shape map representation may be created from a particular constituent
partner (e.g., a
ligand or receptor) of a biological complex by labeling each atom in the
particular partner as
surface or core according to their solvent-accessible surface area (SASA)
value. A shape map
representation is then created 1630 by representing the particular partner as
centered on a three-
dimensional grid (e.g., matrix), and for each labelled atom, assigning a
complex number to grid
points within a pre-defined radius (e.g., surrounding the atom). In certain
embodiments, the pre-
defined radius for a particular atom is or is based on a Van Der Waals radius
for the particular
atom. In certain embodiments, the particular complex number assigned to grid
points associated
with a particular is determined based on a label of the particular atom. For
example, in certain
121
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
embodiments, grid points associated with core and surface atoms of a scaffold
model
representing a backbone of a ligand are assigned values of 0 + 100i and 1 +
0i, respectively. In
certain embodiments, grid points associated with core and surface atoms of a
receptor model
representing a receptor of a complex are assigned values of 0 ¨ 100i and 1 +
0i, respectively.
[0387] Turning to FIG. 16A, in this manner, receptor shape map 1632 may be
created from
receptor model 1614. Scaffold model 1622 may then be rotated via a rotation
{r} 1640 to create
a rotated scaffold model 1642, from which a shape map representation ¨ rotated
scaffold shape
map 1662 ¨ is then created 1630b.
[0388] In certain embodiments, rotated scaffold shape map 1662 and receptor
shape map
1632 are then cross correlated 1672. In certain embodiments, cross-correlation
1672 is
performed via a Fast Fourier Transform (FFT). In certain embodiments, cross
correlation scans
rotated scaffold shape map 1662 and receptor shape map 1632 across each other,
calculating a
cross-correlation value at a particular translation {t} of rotated scaffold
shape map 1662 relative
to receptor shape map 1632. In this manner, for a particular rotation {r},
cross-correlation 1672
samples a range of possible translations, computing, for each rotation-
translation pair, {ri, tj}, a
corresponding cross-correlation value ccu. In particular, in certain
embodiments, cross-
correlation step 1672 outputs a grid where each point corresponds to a
different translation of a
same rotated pose and holds a cross-correlation (cc) value.
[0389] In certain embodiments, a cross-correlation value calculated for a
particular rotation-
translation pair can be used to infer whether a particular pose represented by
the particular
rotation-translation pair would result in one of three outcomes 1674 ¨ no
contact 1674a, contact
1674b, or a clash 1674c. As illustrated in FIG. 16B, a no contact 1674a
outcome indicates that a
complex model formed by orienting scaffold model 1622 according to the
particular rotation-
122
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
translation pair with respect to receptor model 1614 would place the two too
far apart for binding
to be feasible (e.g., sufficiently likely). In certain embodiments, a no
contact 1674a outcome can
be identified via a cross-correlation value having a real part equal to zero.
In certain
embodiments, clash outcomes 1674c have a large real negative contribution to
their
corresponding cross-correlation value, while contact outcomes 1674b have small
real positive
contribution. As illustrated in FIG. 16B, clash outcomes indicate placements
of a scaffold model
and a receptor model that cause excessive overlap, which would also not likely
result in a viable
complex. Contact outcomes are indicative of poses that place a scaffold model
in sufficient
proximity (e.g., not necessarily perfect physical contact) to a receptor model
to correspond to a
complex with potential for binding to occur. Accordingly, contact outcomes are
desirable, while
clashes and no contact outcomes are not.
[0390] Accordingly, in certain embodiments, {r,t} pairs that result in
clash and/or no contact
outcomes are filtered out, and only contact outcomes are retained 1680. In
certain embodiments,
other filters may also be utilized 1690, for example to retain poses with a
high likelihood of
being successful. For example, in certain embodiments a threshold value may be
determined
empirically, for example by evaluating cross-correlation values obtained from
shape map
representations of successful native complex models. For example, in one
embodiment, it was
found that an empirically determined threshold of 1100 captured 90% of WT
Poses.
Accordingly, by filtering poses ({r,t} pairs) having real parts of their cross-
correlation value
below 1100, only poses closely resembling native poses can be retained.
Accordingly, in this
manner, for a particular rotation, a set of filtered poses can be generated.
In certain
embodiments, as illustrated in FIG. 16B, this approach (e.g., steps 1640
through 1690) can be
iteratively applied to multiple rotations 1645 to generate, for each rotation,
a set of filtered poses.
123
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
Sets of filtered poses generated in this manner can then be combined to create
a final set of
filtered poses for multiple rotations and translations.
[0391] FIG. 17 illustrates an example sampling approach, used in certain
embodiments. In
example sampling process 1700, at a variable definition step, examples are
labeled wildtype
(WT) or non-wildtype (non-WT) based on a threshold RMSD variable, or other
pose quality
metrics as described herein. During a binning process poses may be grouped
into classes
according to one or more of the WT variable definition, a RMSD category, a TCN
category, and
a protein database (PDB) category, among other possible categories. During a
sampling process,
a single pose from each bin is sampled sequentially, one at a time,
alternating between bins, and
not returning to a given bin until each of the other bins have been sampled
from in the interim.
This sampling process continues until a given bin is empty, at which point it
may be refilled with
its original dataset. The alternating of bins during model training prevents
the model from
becoming overly constrained and/or "over-tuned" to a specific sub-set of the
overall dataset.
During pure-wild type injection, about 50% (e.g., or from about 40% to 60%, or
from about 30%
to about 70%, e.g., up to 100%) of the cross-correlated generated poses in the
WT class may be
replaced with WT poses (i.e., the original, native, poses). During interface
rotation, poses may
be grouped by PDB category and may be assigned a homogenously sampled
augmentation
rotation that may be applied to the entire pose.
[0392] Without wishing to be bound to any particular theory, it is believed
this type of
sampling procedure removes biases, promotes generalization and prevents
undesired correlation.
For example, this approach may decorrelate contacts seen by model (TCN) and
label (e.g.,
native/wild-type or non-native/non-wild-type), so that the model does not
learn to count contacts
and/or is not biased by a size of molecules. In particular, as described
herein, the label is a
124
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
metric that represents the quality of the pose ¨ e.g., how likely it is to be
a pose with native like
interface properties and therefore how likely it is for the receptor and the
ligand to bind. The
model needs to predict this by learning a set of features from the data (e.g.,
training data). The
TCN metric is essentially a number of contacts between a receptor and ligand
in a particular
pose. It is believed that the model should in theory learn this feature quite
easily as it will "see"
that there are many atoms close to each other (i.e. in contact) at the
interface. It is believed that a
CNN models will be quite good at identifying this type of feature.
[0393] Again, without wishing to be bound to any particular theory, a model
may, in certain
embodiments, learn to identify TCN and to use it (e.g., alone, excessively) to
predict the label.
However, predicting a label based, for example solely on a learned correlation
between a TCN
feature and label and/or excessively weighting/relying on the TCN feature may
be undesirable.
[0394] In particular, non-native poses can have both large and small
interface contact areas,
so it is not a predictive feature of how native the interface is. Moreover,
large molecules tend to
have larger contact area than small molecules and by correlating the TCN and
the label the
model will tend to predict higher label values for large molecules (e.g.,
without regard to whether
they are a native or non-native pose). Accordingly, more accurate predictions
and performance
are obtained by avoiding and/or limiting an extent to which a model learns
this correlation, as it
can lead to unintended biases.
[0395] Accordingly, in certain embodiments, approaches described herein
address this
challenge by creating datasets in which these two metrics are purposely
uncorrelated (at least to
the extent to which this is possible given the data at hand). Such training
data set provide
examples of poses with high TCN and low label, high TCN and high label, low
TCN and high
label, etc. By providing multiple combinations in examples where, e.g., high
TCN is not
125
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
necessarily associated with a high label value, it is believed that the model
does not learn to
correlate high TCN with label, and rather learns other more relevant features
to make an
appropriate prediction.
[0396] Additionally or alternatively, the approach aims to reduce PDB
category redundancy
to prevent memorization of specific PDB categories by the model, and uniformly
distributes
labels to prevent bias in the model predictions. For example, it is believed
that having a dataset
with a uniform label distribution prevents the model from learning biases
during training.
[0397] In certain embodiments, a labeled dataset constructed in this
fashion may be
combined with one or more additional labeled datasets, e.g., created via other
sampling
procedures. For example, an additional labeled dataset may be created by
random sampling
(e.g., of bins).
[0398] FIG. 18 illustrates an approach to generating various training
datasets, used in certain
embodiments. In particular, in certain embodiments, training examples may be
labeled as native-
like (e.g., also referred to as wild-type) and non-native (e.g., also referred
to as non-wild type)
based on one or more pose quality metrics, such as RMSD and NCN/TCN. For
example, in
certain embodiments, various complex models to be used training examples can
be labeled as
native-like or non-native by comparing their RMSD values to a particular RMSD
threshold
value. In certain embodiments, structures may be discarded from the training
dataset based on a
comparison with a (e.g., different, higher) exclusion threshold. For example,
in the RMSD-
based dataset shown in FIG. 18, structures having an RMSD value above a 74A.
threshold were
discarded. In certain embodiments, complex models to be used training examples
can be labeled
as native-like or non-native by comparing their NCN/TCN values to a particular
NCN/TCN
threshold value. As described in further detail herein, in one implementation
multiple training
126
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
datasets were generated in this manner and used to train and test multiple
machine learning
models for evaluating scaffold-pose scores. For example, as shown in FIG. 18,
in one
implementation 214,000 example poses (e.g., represented by scaffold-receptor
complex models)
were used to create training and testing datasets of 170,000 and 44,000
examples, respectively.
ii. Example Machine Learning Model Architecture
[0399] Turning to FIG. 19A, machine learning models of AI-powered modules
described
herein implement a variety of different architectures, including various
artificial neural networks
(ANN), convolutional neural networks (CNN), and others. In certain
embodiments, a machine
learning model utilized herein implements a spinal cord model architecture
(SCM, which may be
considered a type of CNN). In certain embodiments, a machine learning model
used within
scaffold docker module to compute scaffold-pose scores as described herein
implements a SCM
architecture.
[0400] FIG. 19A illustrate an example SCM architecture that receives a
three-dimensional
EDM as input (e.g., a 64 x 64 x 64 EDM) and includes three parallel
convolutional networks,
each of which uses a different kernel size. In particular, in SCM, a first
convolutional network
utilizes a 3A resolution kernel, a second convolutional network utilizes a 5A
kernel, and a third
convolutional network utilizes a 7A kernel. In certain embodiments, multiple
kernel sizes are
utilized in this manner to capture short-, middle- and long-range features of
an interface region.
SCM may also include a main central network that integrates the respective
outputs of the 3
parallel networks at each layer. Without wishing to be bound to any particular
theory, this
approach may be considered similar to a spinal cord integrating information
from peripheral
nerves. In certain embodiments, main central network also features 3 x 3 x 3
kernels. In certain
127
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
embodiments, parallel layer level operations performed by the three parallel
convolutional
network and integration performed by the main central network are treated as a
group ¨ e.g.,
corresponding to single a "vertebra."
[0401] In certain embodiments, this "vertebra" grouping is repeated,
resulting in multiple
integrations from parallel networks. For example, in certain embodiments, a
vertebra pattern is
repeated up to 6 times, resulting in up to five integrations from parallel
networks. The SCM may
include any suitable number of iterations including 1, 2, 3, 4, 5, 6, 7, 8, 9,
10, 20, 50, 100, 1000,
10,000, 100,000, 1,000,000, and more than 1,000,000 iterations, as well as
other numbers and
subranges of iterations therebetween. In certain embodiments, fully connected
layers further
reduce a shape of output from 1024 to 1 (i.e., a single numerical value). In
certain embodiments,
this last (output) value represents a probability of the input pose to feature
native-like properties.
A detailed network architecture diagram of an exemplary SCM in accordance with
embodiments
described herein is shown in FIGs. 19B-L.
iii. Example Transfer Learning Training Procedure
[0402] Turning to FIG. 20, in certain embodiments multiple (e.g., two or
more) machine
learning models are trained. For example, as shown in FIG. 20, multiple models
may be trained
utilizing different training datasets. In certain embodiments, a transfer
learning approach is used.
For example, FIG. 20 illustrates an example approach that utilizes a four-
stage transfer learning
approach wherein four models are trained in successive fashion, each using a
different training
dataset. In the example shown in FIG. 20, each machine learning model shares a
common
architecture ¨ a SCM ¨ and performs binary classification. In particular, each
machine learning
model is trained to evaluate an input representation ¨ such as 3D EDM - of a
scaffold-target
128
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
complex model and output a value that represents a probability that the input
represents a native
complex. In this manner, the machine learning model output can be used as a
scaffold pose
score that measures a degree of similarity between structural features of the
complex represented
by the input and those of native or native-like complexes that the machine
learning model has
been provided as training data.
[0403] Accordingly, each training data set used in the example shown in
FIG. 20 comprises a
plurality of complex models that are assigned to one of two classes ¨ a native-
like class and a
non-native class. In the four training datasets, values of pose quality
metrics computed for each
complex are compared to a threshold value in order to sort example complex
models into the two
classes. A first training data set, "NT1", utilizes the NCN/TCN value and
assigns complex
models to the native-like class if their NCN/TCN value is one. The second,
third, and fourth
datasets ¨ "RMO," "RM1," and "RM2," respectively - are created by assigning
complex models
to the native-like or non-native class according to their computed RMSD
values, based on a
comparison with a particular threshold value. In a particular RMSD dataset,
complex models
having a RMSD value below the threshold value are assigned to the native-like
class, and those
with RMSD values above the RMSD value are assigned to the non-native class.
Each dataset
uses a different threshold value ¨ RMO uses a threshold of OA, RM1 uses a
threshold of 1A, and
RM2 uses a threshold of 2A. In this manner, training different models using
different datasets
can be used to create models that impose varying degrees of stringency when
scoring a particular
input representation.
[0404] In certain embodiments, training multiple models in this fashion may
utilize a transfer
learning approach, whereby model parameters (e.g., weights) determined via
training one model
are used as initial starting points for training another. For example, as
shown in FIG. 20, the four
129
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
models are trained in succession, with more stringent models trained first,
and their parameters
utilized as starting points for increasingly relaxed models. Once trained, the
three RMSD-based
models were validated using testing datasets as described herein. Validation
results for each of
the three RMSD models are shown in FIGs. 21 and 22A-C. FIG. 21 provides tables
of
performance metrics and FIGs. 22A-C compares ROC curves for each of the three
models. As
shown in FIG. 21 and FIGs. 22A-C: for the RMO model, an Area under the Curve
(AUC) of 0.94
was obtained, along with a true positive rate (TPR) of 0.87, a false positive
rate (FPR) of 0.14, an
Fl score of 0.87 and an accuracy of 0.87; for the R1\41 model, an AUC of 0.95,
TPR of 0.87,
FPR of 0.14, Fl score of 0.88, and accuracy of 0.87 were obtained; and for the
R1\42 model, an
AUC of 0.88, TPR of 0.75, FPR of 0.15, Fl score of 0.80 and accuracy of 0.80
were obtained.
iv. Example Scaffold Docking Process
[0405] In certain embodiments, trained machine learning models as described
herein are
utilized in a scaffold docker module in order to evaluate candidate scaffold
models and poses
thereof for use in designing a custom biologic for binding to a target. As
described herein, a
scaffold docker module aims to identify favorable peptide backbones
(represented by scaffold
models) and orientations thereof that can be used as molecular scaffolds and
populated with
amino acids to design a binding interface.
[0406] FIG. 23 illustrates an example process 2300 for identifying
favorable candidate
scaffold models and poses thereof In certain embodiments, a candidate scaffold
model 2302
representing a particular candidate peptide backbone and a structural model of
a target (e.g., a
particular receptor; e.g., a target molecule or target complex) 2304 are
received as input 2310.
Pose transformations are generated 2320 (e.g., via process 1600) and applied
2330 to scaffold
130
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
model 2302 to orient and position it in different poses with respect to target
2304. In certain
embodiments, this approach creates a plurality of candidate scaffold-target
complex models,
each representing a complex comprising the particular candidate peptide
backbone oriented at a
particular pose with respect to the target.
[0407] In certain embodiments, for example in addition or alternatively to
orienting and/or
positioning a scaffold model in different poses with respect to a target,
generation of scaffold-
target complex models may also include adjustments the scaffold model and/or
portions thereof.
Such adjustments may, for example, be used to account for and/or model
backbone flexibility,
wherein certain sub-regions of peptide backbones may, naturally, move, flex,
etc. in space.
[0408] In certain embodiments, scaffold-target complex models generated in
this manner are
then evaluated and scored by a machine learning model ¨ scaffold predictor
model 2380. In
particular, in certain embodiments, for each scaffold-target complex model, an
interface
extraction step (e.g., as described herein, with respect to FIGs. 5 and 6) is
performed 2340 to
identify an interface sub-region of the scaffold-target complex model
comprising a portion of the
scaffold and target that are in proximity to each other. In certain
embodiments, a three-
dimensional EDM is generated 2360 based on the identified interface sub-region
and provided as
input to scaffold predictor model 2380. Based on the received EDM, scaffold
predictor model
2380 determines a scaffold-pose score 2382 for the particular scaffold-target
complex model. In
certain embodiments, as described herein, scaffold predictor model is trained
to perform a binary
classification and, accordingly, outputs, as the scaffold-pose score, a
likelihood value
representing a probability that the EDM representation of the scaffold-target
complex model
represents a native complex. In this manner, although the input scaffold-
target complex is
artificially generated, the scaffold-pose score determined by the scaffold
predictor model
131
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
provides a quantitative assessment of a degree to which the input scaffold-
target complex model
and, accordingly, the particular candidate peptide backbone and pose it
represents, exhibits
properties and/or has key structural features resembling those of native,
physically viable
structures. For example, a scaffold-pose score may range from zero to one
(e.g., representing a
probability) with values closer to one indicative of more favorable scaffold
models and poses
thereof.
[0409] Accordingly, the scaffold docker may use its machine learning model
to evaluate and
score a plurality of scaffold-target complex models and use the determined
scaffold-pose scores
to select a subset of scaffold-target complex models that represent favorable
poses of a particular
candidate peptide backbone.
[0410] Without wishing to be bound to any particular theory, while, in
certain embodiments,
a scaffold model of a scaffold-target complex model does not represent
detailed amino acid
structure of a candidate peptide backbone, the representation of the target
may be a full
representation of a protein and/or peptide and, accordingly, include
representations of amino acid
side chains. Accordingly, the machine learning based approach utilized herein
may account for,
not only a geometrical relation between a target and various backbones and
poses thereof, but
also complex physiochemical behavior due to a detailed amino acid structure
and atoms of the
target. Leveraging deep learning to train machine learning models of the
scaffold docker module
as described herein, scaffold-pose scores computed and used to evaluate
candidate backbones
and poses thereof may thus reflect and be based on detailed geometric and
physiochemical
features 'learned' (e.g., via the training process) by the machine learning
models.
[0411] In certain embodiments, poses and scaffold-target complex models are
generated in
batches, and then scored 2390a. In certain embodiments, poses and scaffold-
target complex
132
CA 03226172 2024-01-03
WO 2023/004116
PCT/US2022/038014
models are generated and scored in an interactive fashion 2390b, whereby a
pose is generated,
used to create a scaffold-target complex model that is then scored, and the
determined score used
as feedback for generation of a new pose which is evaluated in a subsequent
iteration. In certain
embodiments, this iterative approach utilizes optimization algorithms, such as
simulated
annealing, with the scaffold predictor model acting as an objective function
whose output is
maximized.
[0412]
Computationally, in certain embodiments, a scaffold docker module in
accordance
with the present disclosure can evaluate about 1 million poses in about 36
hours using a graphics
processing unit (GPU), in particular, as tested using a GPU xl: NVIDIA TITAN
RTX, 24 GB
and CPU core x10: Intel(R) Xeon(R) CPU E5-2609 v4 @ 1.70GHz. In certain
embodiments, a
particular sampling and/or optimization approach, such as a brute force
approach, simulated
annealing approach, etc., may be selected based on a number of poses to be
evaluated (e.g.,
determined poses, e.g., based on various approaches described herein). For
example, in certain
embodiments, a brute force approach may be used when a number of poses to be
evaluated is
below (e.g., or equal to) a particular pose threshold value and another, such
as a simulated
annealing, approach used when a number of poses to be evaluated is greater
than (e.g., or equal
to) the particular pose threshold value. Selection of a particular
sampling/optimization approach
may be performed automatically, e.g., based on a comparison with a particular
pose threshold
value. One or more pose threshold values may be used to select between various
(e.g., two or
more) sampling and/or optimization techniques. Pose threshold values used in
this manner may
be determined and/or set based on various criteria, such as computer hardware
properties, desired
execution times, etc. and/or via a user interaction (e.g., as a user-defined
parameter). For
example, in the example approach shown in FIG. 23, a brute force approach
2390a was used
133
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
when a number of poses to be evaluated is about 8 million or less, and when a
number of poses
to be evaluated exceeded 8 million, an optimization approach 2390b such as
simulated
annealing, was used, in order to decrease a number of cases. In some
embodiments, other
computational and/or hardware configurations that make use of application-
specific integrated
circuits (ASIC), multiple GPUs, one or more tensor processing units (TPU),
and/or other scheme
that employ parallelization may be used.
[0413] In certain embodiments, scaffold docking approaches described herein
utilize one or
more clustering methods to reduce a number of poses, for example (i) for
evaluation and scoring
by a machine learning model and/or thereafter ¨ pre-scoring clustering, and/or
(ii) for evaluation
and/or further processing in design of a custom biologic ¨ post-scoring
clustering. In certain
embodiments, this is achieved by clustering/grouping the poses based on an
RMSD distance
metric and selecting only centroids of various clusters as representative
poses (e.g., selecting, for
each cluster, a centroid of the cluster as representative of all poses in the
particular cluster). In
certain embodiments, by reducing an initial set of poses to a smaller subset
of representative
cluster centroid poses, the number of poses used in a scaffold docker module
pipeline as
described herein can be significantly reduced. This reduction offers benefits
in terms of
computational time for downstream processing steps, and, additionally or
alternatively,
facilitates analysis for users.
[0414] Pre-scoring clustering: In certain embodiments, pre-scoring
clustering is performed,
wherein poses are clustered after a pose generation step (e.g., such as cross-
correlation, and/or
other steps such as, but not limited to, steps of process 1600 described
herein), but before they
have been scored (e.g., by a machine learning model). This approach reduces a
number of poses
that are evaluated and scored by a machine learning model and may provide
significant benefit in
134
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
computational time as this step may be one of the slowest steps of the
process, e.g., especially a
number of poses to test is on the order of millions. Additionally or
alternatively, native-like, and
hence high scoring poses, are likely to be located in a same neighboring space
and, accordingly,
once one of these poses is identified in that space, others are not
necessarily required (e.g., for
example, for a binding site region on a receptor, most poses in that proximity
are likely to be
more native like).
[0415] Post-scoring clustering: In certain embodiments, post-scoring
clustering is
performed, wherein, poses are clustered after they have been scored, e.g., by
a machine learning
model as described herein. In certain embodiments, this approach reduces a
number of poses
provided, for example as output of a scaffold docker module as described
herein. Where such
poses are reviewed and/or otherwise evaluated/analysed by a human operator,
this provides a
more manageable number of poses for the human operator to analyse.
Additionally or
alternatively, wherein favourable poses are provided to downstream modules in
a pipeline, such
as an interface designer module and/or binding affinity predictor module as
described herein, this
approach provides a reduced set of poses for downstream processing. As
described herein, in
this approach takes advantage of a likelihood that native-like poses tend to
be concentrated in
certain spatial regions.
[0416] Various methods for clustering may be implemented to perform pre-
scoring
clustering and/or post-scoring clustering as described herein.
E. Interface Designer Module
[0417] FIG. 24 is a schematic illustration of an approach to interface
design, which, in
certain embodiments, may be performed by an interface designer module as
described herein. In
135
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
particular, in certain embodiments, interface design begins with a candidate
peptide backbone
oriented in a particular pose with respect to a target and aims to design a
binding interface for
interacting with the target. In particular, in certain embodiments, a
candidate peptide backbone
serves as a molecular scaffold which can be populated with amino acid side
chains to create a
binding interface. As illustrated in FIG. 24, a structural model representing
a candidate peptide
backbone 2402 in a favorable pose may be populated with varying types and
orientations (e.g.,
rotamers) of amino acid side chains (e.g., 2406a,b,c,d,e) along a region in
proximity to the target
2404. Amino acids may be varied, and resulting complex models scored to design
a favorable
interface.
[0418] FIG. 25 shows an example process 2500 for designing candidate
interfaces, described
in further detail herein.
i. Training Data Set Construction
[0419] In certain embodiments, an interface designer module utilizes a
machine learning
model to determine an interface score that quantifies a level of similarity
between a
representation of a prospective interface received as input and a native
interface. In certain
embodiments, an interface score is a numerical value that represents a
predicted number of
mutations between a prospective interface and a native interface, as
determined by the machine
learning model. In certain embodiments, in order to train a machine learning
model to generate
interface scores in this manner, systems and methods described herein utilize
a training dataset
construction approach that uses (i) examples of native interfaces obtained
from structural data of
native complexes and (ii) artificially generated mutant interfaces.
136
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0420] Turning to FIG. 26, for example, in certain embodiments, native
interface models
may be obtained and/or created by obtaining structural models of various
ligand-receptor
complexes (e.g., biological assemblies) from one or more databases. These may
include, without
limitation, public databases such as PDB, as well as other database sources,
such as proprietary
databases. For example, as shown in FIG. 26, the September 2018 PDB release
provides access
to structural models of over one hundred and forty thousand native biological
assemblies. In
certain embodiments, a subset of the available structural models are filtered
and selected based
on various selection criteria in a data curation step to produce a curated
dataset. For example, in
certain embodiments, a minimum resolution criteria is imposed. In certain
embodiments,
additionally or alternatively, clustering analysis may be used to select a
subset of structural
models based on sequence similarity.
[0421] For example, FIG. 26 shows an illustrative implementation whereby a
data curation
step imposed a minimum resolution requirement to select a subset of
crystallographic structures
of protein complexes with a resolution of < 2.7A (i.e., 2.7 Angstroms) and
performed clustering
analysis to group structures based on sequence similarity. Representative
structures of resulting
clusters were selected. This approach resulted in selection of a subset of
13,566 particular
structural models from the 148,827 available in the PDB database. In various
embodiments and
implementations, other resolution thresholds such as from about 0.1A to about
10A, or from
about 0.2A to about 8A, or from about 0.3A to about 7A, or from about 0.4A to
about 6A, or
from about 0.5A to about 5A, or from about 1.0A to about 4A, or from about 2A
to about 3.5A,
or from about 2.5A to about 3A may be used as minimum crystallographic
resolutions.
[0422] In certain embodiments, an interface extraction step is performed on
each structural
model of the curated dataset to obtain a plurality of interface models, each a
representing a
137
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
portion of a complex comprising a ligand and a receptor, said portion an
interface sub-region
about an interface between the ligand and receptor. In certain embodiments, a
particular
structural model may yield more than one extracted interface. For example, the
example
implementation shown in FIG. 26 resulted in 14,778 interfaces.
[0423] Approaches described above with respect to FIG. 26, e.g., for
interface extraction
and/or data curation may also be used in connection with steps and processes
for creating
training data for other approaches described herein, for example with respect
to creation of
training data for interface designer module and/or binding affinity predictor
module, described in
further detail herein.
[0424] Turning to FIG. 27, in certain embodiments, interface models may be
binned
according to a number of identified hotspots. For example, in certain
embodiments, each
interface model is analyzed to identify hotspots available on a particular
constituting chain. An
interface model is then assigned to one or more bins according to a number of
identified hotspots
on a particular constituting chain ¨ e.g., a ligand-side or receptor-side of
the interface model.
[0425] In certain embodiments, each interface model includes two
constituting chains, and
hotspots are identified on each constituting chain, such that two sets of
hotspots are identified
(e.g., treating one chain as the ligand and the other as the receptor, and
then switching). In
certain embodiments, a particular constituting chain to use for identifying
hotspots for purposes
of assignment to one or more bins is selected randomly. In one example
process, an interface
model was randomly assigned as a putative candidate to a specific class/bin
based on a number
of hotspots found on each chain. This assignment is made with the prerequisite
that for a
particular chain, a number of hotspots could not be smaller than the bin label
¨ for example, if
one chain of a particular interface model was identified as having 6 hotspots
it would not be
138
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
placed in a bin associated with class 9 (e.g., chains with 9 hotspots), but if
the other chain was
identified as having 9 or more hotspots, it would be. In the specific case
where an interface has
both chains assigned to the same bin, the generation procedure after picking
the interface once as
a candidate will prioritize the random selection of other interfaces and will
only select the second
chain if needed.
[0426] Turning to FIG. 28, in certain embodiments, artificial mutant
interfaces may be
generated from native interfaces by mutating (i.e., varying) amino acids in
one or more hotspot
locations on a particular chain. In certain embodiments, mutant interfaces are
generated to create
examples of mutant interfaces having a number of mutations spanning particular
(e.g.,
predefined) range. For example, as shown in FIG. 28, mutant interface 2820 is
generated from
native interface 2802 by mutating amino acid side chains in two hotspots
(shown in red along
mutant interface 2802) and retaining an amino acid side chain in a third
hotspot. In certain
embodiments, a uniform sampling and binning approach such as the approach
shown in FIG. 28
is used.
[0427] Turning to FIG. 29, in certain embodiments, mutating an amino acid
may comprise
varying a particular type and/or rotamer of an amino acid side chain in a
hotspot. In particular,
FIG. 29 illustrates an approach to the random mutation procedure illustrated
in FIG. 28. In
certain embodiments, random mutation procedure may generally include AA (amino
acid) type
sampling followed by rotamer sampling. AA type sampling may include randomly
selecting a
natural amino acid from a given hotspot. In certain embodiments, amino acid
types are sampled
according to their naturally occurring frequencies (e.g., as opposed to from a
uniform
distribution), for example via a frequency table as shown in FIG. 29. In
certain embodiments, a
frequency table such as that shown in FIG. 29 accounts for a particular type
of secondary
139
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
structure (e.g., providing for different frequencies based on a particular
secondary structure motif
associated with the interface). In certain embodiments, once a particular
amino acid type is
selected for a particular hotspot site, allowed rotamers for the particular
amino acid type and
hotspot site are computed based on a library of rotamer probabilities (e.g.,
torsion angles and
probabilities thereof for particular amino acid types), for example such as a
Dunbrack Rotamer
Library. In certain embodiments, rotamers that induce clashes with the rest of
the molecule are
excluded. In certain embodiments, for each combination of amino acid type and
phi/psi
backbone torsion angles, the Dunbrack library yields probabilities of rotamers
that have been
derived from (for example) existing PDB structures.
Example Training Dataset
[0428] FIG. 30 illustrates results of an example training dataset
constructed via approaches
described herein, for example with respect to FIGs. 26 through 29. In
particular, the example
training dataset described in FIG. 30 was constructed by identifying hotspots
on 14,778 curated
native interfaces and assigning each of the native interfaces to one or more
of 15 bins labeled 0
to 14 according to a number of identified hotspots. In particular, in
accordance with the hotspot
binning approaches described with respect to FIGs. 27 and 28, an interface
with n identified
hotspots on a constituting chain was assigned each of bins labeled zero to n.
As shown in FIG.
28, for each interface in a particular bin, mutant versions of the interface
were generated by
randomly mutating n hotspots of the interface. In this manner, interfaces in
bin 0 provided
examples of un-mutated, native interfaces (i.e., 0 mutations), interfaces in
bin 1 had a single
mutation, interfaces in bin 2 had two mutations, and so on, through bin 14.
Each interface in
each bin was used to generate 10 mutant interfaces to create a final dataset
which was split into
(i) a training set comprising 3.6 million examples and (ii) a testing dataset
comprising 382,000
140
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
examples to validate a machine learning model once trained. Graph 3020 of FIG.
30 plots a
distribution of training examples according to their label ¨ i.e., with points
representing examples
located according to a number of mutations and hotspots. The uniform
distribution of interfaces
across all 15 bins limits possible correlations between the number of hotspots
and the labels,
thereby preventing biasing of the model during training.
[0429] In order to train a machine learning model, each interface example
had interface
extraction performed and was used to generate a 3D EDM representation for
input to the
machine learning model.
ii. Example Machine Learning Model Architecture
[0430] In certain embodiments, interface designer modules as described
herein utilize an
interface scoring machine learning model to determine an interface score that
quantifies a
measure of similarity between a representation of a candidate interface and a
native-like
interface. In certain embodiments, an interface scoring machine learning model
implements a
regression model architecture. In certain embodiments the interface scoring
model determines,
as an interface score, a predicted number of mutations. In certain
embodiments, an interface
scoring machine learning model implements a classifier architecture, such as a
multi-class (e.g.,
non-binary, having greater than two classes). In certain embodiments, a
classifier architecture
computes one or more classifier probabilities (e.g., likelihoods of belonging
to a particular class)
which can, in turn, be used to generate a continuous score, e.g., by computing
an expected value
using the classifier probabilities and a class label value.
[0431] As an illustrative example, two classes representing two intervals
e.g. class 0,
representing a number of mutations in an interval [0, 4] and class 1,
representing a number of
141
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
mutation interval [4, 8] can be used to create a continuous score as follows
by associated each
class with a representative value, based on the interval it represents. For
example, a mean value
of the interval can be used, such that class 0 can be associated with to a
mean value for its
interval, i.e. 2, and class 1 can be likewise associated with a mean value of
6. Other values /
manners of converting an interval to a representative value, e.g., use of a
median, mode, etc.,
may be used. An expectation value for a model prediction can then be
determined based on the
probabilities predicted for each class and their representative values (e.g.,
as the sum of the
probability-weighted representative values). For example, if a machine
learning model predicts a
probability of 0.2 and 0.8 respectively for the two classes, one can then
compute a score
corresponding to an expected value as follows: score = 2 x 0.2 + 6 x 0.8 =
5.2. Other approaches
for generating a continuous score from a classifier may be used, additionally
or alternatively.
For example, one approach is to pre-calculate a regression between the
predicted and true labels
using the test dataset. The pre-calculated regression function can then be
used compute a
continuous score. In another approach, a distribution over the different
classes may be predicted
and then used to derive a mean value.
[0432] FIG. 31A shows an example regression model architecture that may be
implemented
via an interface scoring model to compute interface scores as described
herein. Example
regression model 3100 takes a three-dimensional EDM representation of an
interface region of
an unknown ligand-target complex model as input and outputs a value
representing a predicted
number of mutations (e.g., an integer value). The particular implementation
shown in FIG. 31
receives an input EDM 3102 having a size of 64x64x64 A3 (i.e., cubic
Angstroms) with a lA
(one Angstrom) grid spacing, though various embodiments and implementations
may utilize
other input sizes and resolution. Regression model 3100 includes multiple
convolution layers
142
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
that progressively collapse output size before passing through a series of
fully connected layers
to produce the final output value 3104. In certain embodiments, FIG. 31B shows
a detailed
network diagram of example architecture 3100.
[0433] FIG. 32A shows a candlestick chart of the resulting performance of
an interface
scoring model implementing the example architecture shown in FIGs. 31A and B.
Candlestick
chart of FIG. 32A plots resulting label as a function of what was predicted.
The performance, in
this example, included an R value of 0.91, a MAE (mean absolute error, which
is a measure of
the number of mutations needed to get to a wild type interface) of 1.39, and
an RMSE (root mean
squared error) of 1.80. FIG. 32B demonstrates performance of an example use of
an interface
designer module in classifying interfaces as native or non-native (e.g., as a
binary classifier)
based on a predicted number of mutations predicted by an interface scoring
model as shown in
FIGs. 321A and B (a same model as used to generate the graph in FIG. 32A). The
interface
scoring model was tested with a testing dataset comprising 1,000 native/wild-
type interfaces and
1,000 non-native interfaces, with a number of non-native examples for each bin
(e.g., number of
mutations) divided equally between the bins. The interface scoring model
determined a
predicted number of mutations for each example, and examples determined (by
the interface
scoring model) to have mutations below a selected threshold value were
classified as native, and
others, with a number of mutations above the selected threshold value were
classified as non-
native. As shown in FIG. 32B, for a selected threshold value of 0.859, use of
the model
predictions as a binary classifier resulted in an AUC of 0.96, a TPR of 0.94,
accuracy of 0.91 and
a FPR Of 0.13.
[0434] As described herein, the regression model architectures shown in and
described with
respect to FIGs. 31A and 31B may be used to implement an interface scoring
model, for
143
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
example, for use in an interface designer module as described herein. It
should be understood,
however, that particular model features and parameters, such as input size and
resolution, kernel
sizes, number of layers, etc. are exemplary and may be varied and used in
accordance with
various embodiments described herein. Such variations are contemplated in
accordance with
various embodiments described herein. Additionally or alternatively, in
certain embodiments,
such regression model architectures are not limited in use to interface
scoring approaches and/or
use within an interface designer module, and may be used in connection with
other models, to
generate other predictions, for example relevant to other scoring approaches
(e.g., scaffold-pose
scoring, binding affinity prediction, etc.) described herein. Additionally or
alternatively, in
certain embodiments, other regression model architectures may also be used,
for example for
determining interface scores and/or other scores described herein (e.g.,
scaffold pose scores,
binding affinities, etc.).
iii. Example Interface Design Process
[0435] Turning to FIGs. 25 and FIG. 33, in certain embodiments a trained
interface scoring
model as described herein can be used in an interface designer module to
design one or more
ligand interfaces for binding to a particular target. As described herein, an
interface designer
module may utilize a scaffold-target complex model 2502 as a starting point.
The interface
designer module may then populate an interface region of the scaffold model of
the scaffold-
target complex model with amino acid side chains to create a ligand-target
complex model which
corresponds to the scaffold-target complex model, but with a ligand model
representing a portion
of the candidate peptide backbone with amino acid side chains populating sites
that are located in
proximity to the target. In certain embodiments, these populated sites
comprise hotspots. In
144
CA 03226172 2024-01-03
WO 2023/004116
PCT/US2022/038014
certain embodiments, populated sites also comprise context regions. The
interface designer
module may mutate amino acids in various sites (e.g., hotspots and/or context
regions) of the
ligand model to create a plurality of candidate ligand-target complex models,
each representing a
different candidate interface in a complex with the target 2510. In certain
embodiments,
generation of candidate ligand-target complex models may represent a ligand
having a peptide
backbone and pose thereof with respect to the target that is based on, but not
necessarily identical
to the candidate peptide backbone and pose of the scaffold-target complex
model used as a
starting point. For example, various ligand-target complex model may be
created to account for,
and represent variations of the candidate peptide backbone accounting for
backbone flexibility.
Additionally or alternatively, a pose of the initial scaffold-target complex
model may be varied,
for example via rigid body perturbations (e.g., random perturbations to one or
more (e.g., of six)
degrees of translational and/or rotations freedom. Such perturbations may
allow for minor
translations and/or rotations along one or more axis. In certain embodiments,
such translations
are within about 10 angstroms or less, (e.g., about 5 angstroms or less, about
1 angstrom or less,
about 0.5 angstroms or less) along one or more directions (e.g., an x-, y, or
z-, direction). In
certain embodiments, rotational perturbations may be approximately 15 degrees
or less (e.g.,
approximately 5 degrees or less, e.g., approximately one or two degrees or
less) about one or
more axes (e.g., x- and/or y- and/or z- axis).
[0436] In
certain embodiments, volumetric representations, such as 3D EDMs are created
from candidate ligand-target complex models and provided as input to a machine
learning model
such as an interface scoring model as described herein, thereby determining
interface scores
2520 which can be used to rank and/or select a subset of interface designs
that are likely to be
successful 2530.
145
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0437] In certain embodiments, for example in order to efficiently search a
landscape of
possible interface designs, an interface designer module may leverage an
optimization algorithm,
such as simulated annealing, using an interface scoring model as an objective
function whose
output the optimization algorithm seeks to optimize. FIG. 33 shows an example
process 3300
that utilizes a simulated annealing algorithm with an interface scoring model
as an objective
function. Process 3300 includes an input preparation step 3320 in which
hotspots on a ligand-
side of a ligand-target complex are identified, and certain pre-calculations,
such as calculation of
allowed rotamers at the identified hotspot locations, are performed. Following
input preparation
step 3320, a simulated annealing procedure is used to iteratively mutate amino
acids at the
identified hotspot locations to create a new candidate ligand-target complex
model that
represents the mutated interface and evaluate the new ligand target-complex
using an interface
scoring model to compute an interface score as described herein until a
termination criteria is
reached. For example, simulated annealing algorithm may terminate when one or
more
thresholds have been met; for example, a total of 6000 iterations have been
run, or an interface
score determined by the interface scoring model is less than 1 for twenty
consecutive iterations.
[0438] In this manner, in certain embodiments, a subset of one or more
ligand-target
complex models, each representing a candidate interface determined, e.g.,
based on computed
interface scores, as favorable. In certain embodiments, the subset of ligand-
target complex
models may then be used to design a custom biologic. In certain embodiments,
one or more
additional modules may be used to further refine designs of candidate
interfaces based on the
subset of ligand-target complex models.
146
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
F. Binding Affinity Predictor
[0439] Turning to FIG. 34, in certain embodiments, in sit/co biologic
design tools described
herein include a binding affinity predictor module that can be used to predict
a binding affinity
between a particular ligand 3402 and target 3404. In certain embodiments, a
binding affinity
predictor module as described herein evaluates one or more ligand-target
complex models and
determines, for each, a predicted binding affinity score 3406. In certain
embodiments, the
predicted binding affinity score is a numerical value representing a predicted
pKavalue. In
certain embodiments, the predicted binding affinity score is a classification
(e.g., as determined
via a machine learning model that acts as a classifier) and/or a value on a
scale that is related
(e.g., correlated with, proportional to, indicative of a range of) a binding
affinity, e.g., pKa, value.
[0440] In certain embodiments, a binding affinity predictor module utilizes
a machine
learning model to evaluate a particular ligand-target complex model and
determine, as output, a
binding affinity score. In certain embodiments, a binding affinity predictor's
machine learning
model receives, as input, a volumetric representation of at least a portion of
the particular ligand-
target complex model. For example, a 3D EDM may be generated from at least a
portion, such
as an extracted interface, of the particular ligand-target complex model, and
used as input to the
binding affinity predictor's machine learning model. In certain embodiments, a
binding affinity
score determined by the machine learning model corresponds directly to a
(e.g., is a predicted)
pKd value.
[0441] Turning to FIG. 35, in certain embodiments, which shows an example
process 3500
for determining and using predicted binding affinity scores, a binding
affinity module may
receive, as input, a plurality of candidate ligand-target complex models, each
representing a
prospective custom biologic design 3502. For example, in certain embodiments,
candidate
147
CA 03226172 2024-01-03
WO 2023/004116
PCT/US2022/038014
ligand-target complex models are produced via other modules described herein,
such as an
interface designer module, and received as input by the binding affinity
module. The binding
affinity module may then use its machine learning model to determine predicted
binding affinity
scores 3510 for each of the candidate ligand-target complex models. In certain
embodiments, a
binding affinity module may select a subset of the candidate ligand-target
complex models,
based on the predicted binding affinity scores 3520, e.g., to determine a
final set of designs for
use in creating one or more new, engineered, custom biologics to be
synthesized and
experimentally tested. For example, the binding affinity module may rank
candidate ligand-
target complex models according to their predicted binding affinities, and
select a portion, for
example those having highest predicted affinities (e.g., a top 1, a top 5,
those, lying in a
particular upper percentile, etc.). Additionally or alternatively, in certain
embodiments, a subset
of ligand-target complex models may be selected and/or filtered by comparing
predicted binding
affinities to one or more threshold values.
[0442]
Additionally or alternatively, in certain embodiments, a binding affinity
predictor
module may utilize predicted binding affinities to refine one or more received
ligand-target
complex models 3540. For example, in certain embodiments, one or more amino
acids of a
candidate ligand-target complex model may be mutated, and evaluated by a
machine learning
model to determine effects of various mutations on predicted binding
affinities. In certain
embodiments, mutations that improve binding affinity can be identified in this
manner, and
applied to an initial candidate ligand-target complex model to generate a find
candidate, with
improved binding affinity. In certain embodiments, optimization algorithms,
for example as
described herein, may be used to optimize binding affinity as a function of
amino acid mutations,
for example using received candidate ligand-target complex models as initial
input and using
148
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
predicted binding affinities generated via a machine learning model as an
objective function to
be optimized.
[0443] Accordingly, as described herein, binding affinity modules described
herein may be
utilized alone and/or incorporated in various custom biologic design pipelines
and workflows to
predict binding affinities for and/or further refine candidate biologic
designs.
i. Example Machine Learning Model for Predicting Binding Affinities
[0444] As described herein, developing a machine learning model to generate
accurate
predictions and perform scoring functions as described herein involves steps
and procedures
including construction of an appropriate (e.g., balanced, sufficiently varied,
etc.) training dataset,
selecting a particular machine learning architecture and applying a training
procedure, as well as
validating performance. FIG. 36 through 39 describe an example implementation
of a machine
learning model used for predicting binding affinities, in accordance with
certain embodiments
described herein. Example implementation shown in FIG. 36 through 39 includes
steps of
training dataset construction, training, and validation. Accordingly, this
example implementation
demonstrates accurate predictions of binding affinities via machine learning
approaches as
described herein.
Training Dataset Construction
[0445] Turning to FIG. 36, construction of a training dataset for binding
affinity prediction
may generally include data collection, data curation, data extraction and
binning. Data collection
may entail gathering binding affinity data from public databases such as PDB
Bind and MOAD.
. In certain embodiments, K (inhibition constant) and Ka (dissociation
constant) values are used
and converted into pKa(-logK) values to determine a measure of binding
affinity for each ligand
149
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
in the training set. In certain embodiments, only K and Ka values are used for
determining
binding affinity and IC50 values are not used/excluded. In certain
embodiments, receptor and
ligand chains are identified manually. Data extraction may be multi-chain
meaning that a single
interface may yield multiple side chains. The interfaces may be distributed
across 15 bins based
on pKa units.
[0446] Turning to FIG. 36, creation of a training dataset used in binding
affinity prediction
may include clustering protein-protein interfaces into bins that share 95%
sequence similarity. A
single interface may then be selected from each of the (for example, 25) bins
to act as a
representative for that bin to be used in a uniformly distributed test set
(i.e., for testing the
model). The remaining interfaces may be used for training. In order to achieve
a balanced set, a
differential augmentation on a per-bin basis (for example, based on pKa value)
may be used to
obtain an evenly distributed training set.
[0447] In an example implementation, a training set created in this manner
included about
7250 entries per bin while the testing set included about 1000 entries per bin
with labels ranging
uniformly from 0 to 15. In this example implementation, this approach resulted
in a total of
about 65,184 entries in the training data set and t a total of about 8,928
entries in the testing data
set. Interface extraction was performed and each extracted interface converted
to a 3D EDM.
Accordingly, in certain embodiments, the training set may include from about 5
to about 10
times (or from about 6 to about 9 times, or from about 7 time to about 8
times) more entries than
the testing set. Stated otherwise, from about 70% to about 95% (or from about
75% to about
90%, or from about 80% to about 90%) of the total number of entries may be
used for model
training, while the remaining entries (or data points) may be used for
testing.
150
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
Example Training Approach
[0448] FIG. 38 illustrates an approach to two-step training used in binding
affinity
prediction, used in certain embodiments. The approach to two-step training may
include a first
step in which weights from a best scaffold docker model trained on RMO (i.e.,
one of the RMSD
models) may be transferred to the binding affinity spinal cord model (SCM).
The approach to
two-step training may also include a second step in which the model is trained
and
hyperparameters are optimized for binding affinity prediction. Weights for
layers of an SCM
trained for a scaffold docker model were transferred, apart from a final,
fully-connected layer
that outputs a single value and was trained on a binding affinity dataset
created as described
herein. Without wishing to be bound to any particular theory, in certain
embodiments a transfer
learning approach such as the approach shown in FIG. 38 provides for accurate
training of a
machine learning model even when a limited dataset is used, by leveraging
training performed
on a related (though not identical) task for which a more extensive dataset is
available. In
certain embodiments, this approaches allows for creation of an accurate
binding affinity
predictor machine learning model, despite relatively limited experimental
binding affinity data.
Performance Example
[0449] FIG. 39 illustrates performance results for binding affinity
predictions, used in certain
embodiments. The performance, in this example, included an R value of 0.7, a
MAE (mean
absolute error, which is a measure of the number of mutations needed to get to
a wild type
interface) of 1.54, and an RMSE (root mean squared error) of 1.98.
151
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
[0450] Accordingly, embodiments of the binding affinity module described
herein include
various features that facilitate accurate prediction of binding affinity, and
generate models
capable of more accurate predictions than other (e.g., previous) approaches.
G. Additional Modules and Flexible, Modular, Pipeline Architecture
[0451] As described herein, embodiments of various modules described herein
¨ such as the
scaffold docker module, interface designer, and binding affinity predictor may
be utilized
separate and/or in combination to engineer structural features of custom
biologics with respect
particular criteria (e.g., each module evaluating and facilitating design with
respect to a particular
criteria). In certain embodiments, these modules, as well as various other
modules may be used
individually or combined with each other, in pipeline architectures as
described herein, e.g., with
respect to design of custom binders, as well as other architectures and
organizations.
[0452] For example, as shown in FIG. 40 a modular approach as described
herein allows, in
certain embodiments, for creation of various custom pipelines 4010, tailored
for a particular
design task, to create various custom biologic designs 4012 having particular
desired
functionality. Various pipelines can be created in a flexible manner, via
selection and
arrangement of various modules from a collection of modules 4020. In certain
embodiments,
module collection may include one or more of a scaffold docker module 4020a,
an interface
designer module, 4020e, and a binding affinity predictor module 4020f as
described herein. In
certain embodiments, module collection 4020 may include various other modules,
such, without
limitation, any of a rotamer relax module 4020h, a solubility prediction
module 4020b, a
thermodynamic stability prediction module 4020c, an immunogenicity prediction
module 4020d,
and a thermostability prediction module 4020g. In certain embodiments, as with
the scaffold
152
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
docker, interface designer, and binding affinity predictor modules, these
modules may be used,
along with the scaffold docker, interface designer and binding affinity
predictor modules, in
various combinations, sequentially or in parallel fashion, depending on a
particular application.
[0453] In certain embodiments, approaches described herein with regard to a
scaffold docker
module may be used to create a ligand docking module that identifies docking
configurations of
two peptide and/or polypeptide chains. In particular, instead of operating on
scaffold models,
various dataset creation, training, sampling and scoring procedures described
herein with regard
to a scaffold docker module may be performed using two or more ligand models,
each of which
represents a full ligand ¨ i.e., including amino acid side chains. In this
manner, favorable
orientations of full ligands for binding with respect to each other may be
identified ¨ providing a
tool for, among other things, handling classical protein and/or peptide
docking.
H. Computer System and Network Environment
[0454] Turning to FIG. 41, an implementation of a network environment 4100
for use in
providing systems, methods, and architectures as described herein is shown and
described. In
brief overview, referring now to FIG. 41, a block diagram of an exemplary
cloud computing
environment 4100 is shown and described. The cloud computing environment 4100
may include
one or more resource providers 4102a, 4102b, 4102c (collectively, 4102). Each
resource
provider 4102 may include computing resources. In some implementations,
computing
resources may include any hardware and/or software used to process data. For
example,
computing resources may include hardware and/or software capable of executing
algorithms,
computer programs, and/or computer applications. In some implementations,
exemplary
computing resources may include application servers and/or databases with
storage and retrieval
153
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
capabilities. Each resource provider 4102 may be connected to any other
resource provider 4102
in the cloud computing environment 4100. In some implementations, the resource
providers
4102 may be connected over a computer network 4108. Each resource provider
4102 may be
connected to one or more computing device 4104a, 4104b, 4104c (collectively,
4104), over the
computer network 4108.
[0455] The cloud computing environment 4100 may include a resource manager
4106. The
resource manager 4106 may be connected to the resource providers 4102 and the
computing
devices 4104 over the computer network 4108. In some implementations, the
resource manager
4106 may facilitate the provision of computing resources by one or more
resource providers
4102 to one or more computing devices 4104. The resource manager 4106 may
receive a request
for a computing resource from a particular computing device 4104. The resource
manager 4106
may identify one or more resource providers 4102 capable of providing the
computing resource
requested by the computing device 4104. The resource manager 4106 may select a
resource
provider 4102 to provide the computing resource. The resource manager 4106 may
facilitate a
connection between the resource provider 4102 and a particular computing
device 4104. In
some implementations, the resource manager 4106 may establish a connection
between a
particular resource provider 4102 and a particular computing device 4104. In
some
implementations, the resource manager 4106 may redirect a particular computing
device 4104 to
a particular resource provider 4102 with the requested computing resource.
[0456] FIG. 42 shows an example of a computing device 4200 and a mobile
computing
device 4250 that can be used to implement the techniques described in this
disclosure. The
computing device 4200 is intended to represent various forms of digital
computers, such as
laptops, desktops, workstations, personal digital assistants, servers, blade
servers, mainframes,
154
CA 03226172 2024-01-03
WO 2023/004116
PCT/US2022/038014
and other appropriate computers. The mobile computing device 4250 is intended
to represent
various forms of mobile devices, such as personal digital assistants, cellular
telephones, smart-
phones, and other similar computing devices. The components shown here, their
connections
and relationships, and their functions, are meant to be examples only, and are
not meant to be
limiting.
[0457] The
computing device 4200 includes a processor 4202, a memory 4204, a storage
device 4206, a high-speed interface 4208 connecting to the memory 4204 and
multiple high-
speed expansion ports 4210, and a low-speed interface 4212 connecting to a low-
speed
expansion port 4214 and the storage device 4206. Each of the processor 4202,
the memory
4204, the storage device 4206, the high-speed interface 4208, the high-speed
expansion ports
4210, and the low-speed interface 4212, are interconnected using various
busses, and may be
mounted on a common motherboard or in other manners as appropriate. The
processor 4202 can
process instructions for execution within the computing device 4200, including
instructions
stored in the memory 4204 or on the storage device 4206 to display graphical
information for a
GUI on an external input/output device, such as a display 4216 coupled to the
high-speed
interface 4208. In other implementations, multiple processors and/or multiple
buses may be
used, as appropriate, along with multiple memories and types of memory. Also,
multiple
computing devices may be connected, with each device providing portions of the
necessary
operations (e.g., as a server bank, a group of blade servers, or a multi-
processor system). Thus,
as the term is used herein, where a plurality of functions are described as
being performed by "a
processor", this encompasses embodiments wherein the plurality of functions
are performed by
any number of processors (one or more) of any number of computing devices (one
or more).
Furthermore, where a function is described as being performed by "a
processor", this
155
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
encompasses embodiments wherein the function is performed by any number of
processors (one
or more) of any number of computing devices (one or more) (e.g., in a
distributed computing
system).
[0458] The memory 4204 stores information within the computing device 4200.
In some
implementations, the memory 4204 is a volatile memory unit or units. In some
implementations,
the memory 4204 is a non-volatile memory unit or units. The memory 4204 may
also be another
form of computer-readable medium, such as a magnetic or optical disk.
[0459] The storage device 4206 is capable of providing mass storage for the
computing
device 4200. In some implementations, the storage device 4206 may be or
contain a computer-
readable medium, such as a floppy disk device, a hard disk device, an optical
disk device, or a
tape device, a flash memory or other similar solid state memory device, or an
array of devices,
including devices in a storage area network or other configurations.
Instructions can be stored in
an information carrier. The instructions, when executed by one or more
processing devices (for
example, processor 4202), perform one or more methods, such as those described
above. The
instructions can also be stored by one or more storage devices such as
computer- or machine-
readable mediums (for example, the memory 4204, the storage device 4206, or
memory on the
processor 4202).
[0460] The high-speed interface 4208 manages bandwidth-intensive operations
for the
computing device 4200, while the low-speed interface 4212 manages lower
bandwidth-intensive
operations. Such allocation of functions is an example only. In some
implementations, the high-
speed interface 4208 is coupled to the memory 4204, the display 4216 (e.g.,
through a graphics
processor or accelerator), and to the high-speed expansion ports 4210, which
may accept various
expansion cards (not shown). In the implementation, the low-speed interface
4212 is coupled to
156
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
the storage device 4206 and the low-speed expansion port 4214. The low-speed
expansion port
4214, which may include various communication ports (e.g., USB, Bluetoothg,
Ethernet,
wireless Ethernet) may be coupled to one or more input/output devices, such as
a keyboard, a
pointing device, a scanner, or a networking device such as a switch or router,
e.g., through a
network adapter.
[0461] The computing device 4200 may be implemented in a number of
different forms, as
shown in the figure. For example, it may be implemented as a standard server
4220, or multiple
times in a group of such servers. In addition, it may be implemented in a
personal computer such
as a laptop computer 4222. It may also be implemented as part of a rack server
system 4224.
Alternatively, components from the computing device 4200 may be combined with
other
components in a mobile device (not shown), such as a mobile computing device
4250. Each of
such devices may contain one or more of the computing device 4200 and the
mobile computing
device 4250, and an entire system may be made up of multiple computing devices
communicating with each other.
[0462] The mobile computing device 4250 includes a processor 4252, a memory
4264, an
input/output device such as a display 4254, a communication interface 4266,
and a transceiver
4268, among other components. The mobile computing device 4250 may also be
provided with
a storage device, such as a micro-drive or other device, to provide additional
storage. Each of
the processor 4252, the memory 4264, the display 4254, the communication
interface 4266, and
the transceiver 4268, are interconnected using various buses, and several of
the components may
be mounted on a common motherboard or in other manners as appropriate.
[0463] The processor 4252 can execute instructions within the mobile
computing device
4250, including instructions stored in the memory 4264. The processor 4252 may
be
157
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
implemented as a chipset of chips that include separate and multiple analog
and digital
processors. The processor 4252 may provide, for example, for coordination of
the other
components of the mobile computing device 4250, such as control of user
interfaces,
applications run by the mobile computing device 4250, and wireless
communication by the
mobile computing device 4250.
[0464] The processor 4252 may communicate with a user through a control
interface 4258
and a display interface 4256 coupled to the display 4254. The display 4254 may
be, for example,
a TFT (Thin-Film-Transistor Liquid Crystal Display) display or an OLED
(Organic Light
Emitting Diode) display, or other appropriate display technology. The display
interface 4256
may comprise appropriate circuitry for driving the display 4254 to present
graphical and other
information to a user. The control interface 4258 may receive commands from a
user and
convert them for submission to the processor 4252. In addition, an external
interface 4262 may
provide communication with the processor 4252, so as to enable near area
communication of the
mobile computing device 4250 with other devices. The external interface 4262
may provide, for
example, for wired communication in some implementations, or for wireless
communication in
other implementations, and multiple interfaces may also be used.
[0465] The memory 4264 stores information within the mobile computing
device 4250. The
memory 4264 can be implemented as one or more of a computer-readable medium or
media, a
volatile memory unit or units, or a non-volatile memory unit or units. An
expansion memory
4274 may also be provided and connected to the mobile computing device 4250
through an
expansion interface 4272, which may include, for example, a SIMM (Single In
Line Memory
Module) card interface. The expansion memory 4274 may provide extra storage
space for the
mobile computing device 4250, or may also store applications or other
information for the
158
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
mobile computing device 4250. Specifically, the expansion memory 4274 may
include
instructions to carry out or supplement the processes described above, and may
include secure
information also. Thus, for example, the expansion memory 4274 may be provide
as a security
module for the mobile computing device 4250, and may be programmed with
instructions that
permit secure use of the mobile computing device 4250. In addition, secure
applications may be
provided via the SIMM cards, along with additional information, such as
placing identifying
information on the SIMIVI card in a non-hackable manner.
[0466] The memory may include, for example, flash memory and/or NVRAM
memory (non-
volatile random access memory), as discussed below. In some implementations,
instructions are
stored in an information carrier. The instructions, when executed by one or
more processing
devices (for example, processor 4252), perform one or more methods, such as
those described
above. The instructions can also be stored by one or more storage devices,
such as one or more
computer- or machine-readable mediums (for example, the memory 4264, the
expansion
memory 4274, or memory on the processor 4252). In some implementations, the
instructions
can be received in a propagated signal, for example, over the transceiver 4268
or the external
interface 4262.
[0467] The mobile computing device 4250 may communicate wirelessly through
the
communication interface 4266, which may include digital signal processing
circuitry where
necessary. The communication interface 4266 may provide for communications
under various
modes or protocols, such as GSM voice calls (Global System for Mobile
communications), SMS
(Short Message Service), EMS (Enhanced Messaging Service), or MMS messaging
(Multimedia
Messaging Service), CDMA (code division multiple access), TDMA (time division
multiple
access), PDC (Personal Digital Cellular), WCDMA (Wideband Code Division
Multiple Access),
159
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
CDMA2000, or GPRS (General Packet Radio Service), among others. Such
communication
may occur, for example, through the transceiver 4268 using a radio-frequency.
In addition,
short-range communication may occur, such as using a Bluetoothg, Wi-FiTM, or
other such
transceiver (not shown). In addition, a GPS (Global Positioning System)
receiver module 4270
may provide additional navigation- and location-related wireless data to the
mobile computing
device 4250, which may be used as appropriate by applications running on the
mobile computing
device 4250.
[0468] The mobile computing device 4250 may also communicate audibly using
an audio
codec 4260, which may receive spoken information from a user and convert it to
usable digital
information. The audio codec 4260 may likewise generate audible sound for a
user, such as
through a speaker, e.g., in a handset of the mobile computing device 4250.
Such sound may
include sound from voice telephone calls, may include recorded sound (e.g.,
voice messages,
music files, etc.) and may also include sound generated by applications
operating on the mobile
computing device 4250.
[0469] The mobile computing device 4250 may be implemented in a number of
different
forms, as shown in the figure. For example, it may be implemented as a
cellular telephone 4280.
It may also be implemented as part of a smart-phone 4282, personal digital
assistant, or other
similar mobile device.
[0470] Various implementations of the systems and techniques described here
can be
realized in digital electronic circuitry, integrated circuitry, specially
designed ASICs (application
specific integrated circuits), computer hardware, firmware, software, and/or
combinations
thereof. These various implementations can include implementation in one or
more computer
programs that are executable and/or interpretable on a programmable system
including at least
160
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
one programmable processor, which may be special or general purpose, coupled
to receive data
and instructions from, and to transmit data and instructions to, a storage
system, at least one
input device, and at least one output device.
[0471] Actions associated with implementing the systems may be performed by
one or more
programmable processors executing one or more computer programs. All or part
of the systems
may be implemented as special purpose logic circuitry, for example, a field
programmable gate
array (FPGA) or an application-specific integrated circuit (ASIC), or both.
All or part of the
systems may also be implemented as special purpose logic circuitry, for
example, a specially
designed (or configured) central processing unit (CPU), conventional central
processing units
(CPU) a graphics processing unit (GPU), and/or a tensor processing unit (TPU).
[0472] These computer programs (also known as programs, software, software
applications
or code) include machine instructions for a programmable processor, and can be
implemented in
a high-level procedural and/or object-oriented programming language, and/or in
assembly/machine language. As used herein, the terms machine-readable medium
and
computer-readable medium refer to any computer program product, apparatus
and/or device
(e.g., magnetic discs, optical disks, memory, Programmable Logic Devices
(PLDs)) used to
provide machine instructions and/or data to a programmable processor,
including a machine-
readable medium that receives machine instructions as a machine-readable
signal. The term
machine-readable signal refers to any signal used to provide machine
instructions and/or data to
a programmable processor.
[0473] To provide for interaction with a user, the systems and techniques
described here can
be implemented on a computer having a display device (e.g., a CRT (cathode ray
tube) or LCD
(liquid crystal display) monitor) for displaying information to the user and a
keyboard and a
161
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
pointing device (e.g., a mouse or a trackball) by which the user can provide
input to the
computer. Other kinds of devices can be used to provide for interaction with a
user as well; for
example, feedback provided to the user can be any form of sensory feedback
(e.g., visual
feedback, auditory feedback, or tactile feedback); and input from the user can
be received in any
form, including acoustic, speech, or tactile input.
[0474] The systems and techniques described here can be implemented in a
computing
system that includes a back end component (e.g., as a data server), or that
includes a middleware
component (e.g., an application server), or that includes a front end
component (e.g., a client
computer having a graphical user interface or a Web browser through which a
user can interact
with an implementation of the systems and techniques described here), or any
combination of
such back end, middleware, or front end components. The components of the
system can be
interconnected by any form or medium of digital data communication (e.g., a
communication
network). Examples of communication networks include a local area network
(LAN), a wide
area network (WAN), and the Internet.
[0475] The computing system can include clients and servers. A client and
server are
generally remote from each other and typically interact through a
communication network. The
relationship of client and server arises by virtue of computer programs
running on the respective
computers and having a client-server relationship to each other.
[0476] In some implementations, modules described herein can be separated,
combined or
incorporated into single or combined modules. The modules depicted in the
figures are not
intended to limit the systems described herein to the software architectures
shown therein.
I. Interface Designer Module ¨ embodiments using graph representations
162
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
[0477] Described in this section are further methods, systems, and
architectures for designing
interfaces of custom biologic structures for binding to particular targets of
interest. In particular,
as described in further detail herein, artificial-intelligence (AI)-based
interface designer
technologies of the present disclosure begin with a structural model of a
particular target of
interest and a partial, or incomplete, structural model of a custom biologic
that is being / in the
progress of being designed, for the purpose of binding to the target. The
partial structural model
of the in-progress custom biologic may include certain, for example,
previously determined or
known information about the custom biologic, but does not include an
identification of a type
(e.g., and/or rotamer structure) of one or more amino acid side chains within
an interface region
that is expected to interact and influence binding with the target. That is,
while structural
features, such as a backbone geometry, of the in-progress custom biologic may
be determined
and/or known, an amino acid sequence within an interface region of the to-be
designed custom
biologic is as yet unknown, and to-be determined.
[0478] Interface designer technologies described in this section utilize
trained machine
learning models in combination with a graph representation to generate, based
on the structure of
the particular target together with the partial model of the in-progress
custom biologic, predicted
interfaces ¨ i.e., partial amino acid sequences within an interface region,
that are determined, by
the machine learning model, to bind (e.g., with high affinity) to a target.
[0479] FIG. 43 shows a schematic of an example overall approach 4300 for
generating
predicted interfaces in accordance with the AI-based techniques described
herein. For example,
an interface design approach 4300 in accordance with the present disclosure
may use, as a
starting point, a structural model of a complex 4302 comprising at least a
portion of a particular
target and a portion of an in-progress custom biologic. In certain
embodiments, a peptide
- 163 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
backbone structure of the in-progress custom biologic is known and/or has been
previously
determined. As described herein, as well as, for example in U.S. Patent
Application No.
17/384,104, entitled "Systems and Methods for Artificial Intelligence-Guided
Biomolecule
Design and Assessment," filed July 23, 2021, incorporated herein by reference
in its entirety,
peptide backbone structures may be represented via scaffold models, which
identify locations of
backbone atoms, but leave amino acid side chains open / undefined. For
example, at each of one
or more amino acid sites, instead of including a representation of a
particular side chain, a
scaffold model may use a placeholder, such as a beta-carbon (Ca) atom. In
certain embodiments,
candidate peptide backbones for use in designing a custom biologic may be
generated via
machine learning techniques, such as a scaffold docker approach, described in
further detain in
U.S. Patent Application No. 17/384,104, entitled "Systems and Methods for
Artificial
Intelligence-Guided Biomolecule Design and Assessment," filed July 23, 2021. A
candidate
peptide backbone may, accordingly, be used as a starting point or foundation,
that can
subsequently be populated with amino acids in an interface region to create a
final custom
biologic structure.
[0480] Accordingly, in certain embodiments, as shown in FIG. 43, an initial
scaffold-target
complex model 4302, which includes a representation of the particular target
along with a
scaffold model representation of a candidate peptide backbone is received
(e.g., from another
computer module, such as a scaffold docker module) and/or accessed. As
explained in further
detail, interface designer technologies in certain embodiments described
herein represent protein
complexes as graphs, encoding structural features in vectors associated with
nodes and edges.
Accordingly, the initial scaffold-target complex model 4302 may itself be, or
used to generate, a
- 164 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
scaffold-target graph 4304, which is then used as input to a machine-learning
step 4306 that
generates a predicted interface 4310 (e.g., graph).
[0481] As described in further detail herein, machine learning step 4306
utilizes a machine
learning model 4308 to perform a node classification operation that is used to
generate the
predicted interface 4310. Predicted interface 4310 may be a direct output of
machine learning
model 4308, or, in certain embodiments, additional processing (e.g., post
processing steps) is
used to create a final predicted interface 4310 from the output of machine
learning model 4308.
Additionally or alternatively, multiple iterations and feedback loops may be
used within machine
learning step 4306.
[0482] By utilizing a graph representation in conjunction with a machine
learning model that
performs a node classification operation, interface designer technologies
described herein are
able to generate direct predictions of amino acid interface sequences that are
likely to be
successful in binding to a particular target. This approach, accordingly, does
not use the machine
learning model as a scoring function, to evaluate candidate interface designs,
but instead directly
predicts a single interface. Directly predicting interfaces in this manner
simplifies the AI-based
biologic design process, reduces computational load, and facilitates training
of the machine
learning model itself.
[0483] Without wishing to be bound to any particular theory, it is believed
that this approach
of directly predicting interfaces as described herein provides several
benefits over searching and
scoring approaches. First, rather than generate numerous "guesses" of possible
structures, and
evaluating them via a machine learning model-based scoring function, direct
prediction
approaches as described herein generate one (or a few, if used in an iterative
procedure)
predictions of amino acid sequences at an interface. There is no need to
generate guesses or
- 165 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
search a landscape, thereby avoiding any need to employ complex searching
routines such as
simulated annealing to ensure a global, rather than local, optimum is
obtained. Second, in a
related benefit, direct prediction approaches can reduce the number of runs of
a machine learning
algorithm, since no searching is required. Third, since the direct prediction
approaches described
herein do not score an overall structure, so as to distinguish between
structures that are or are not
physically viable, there is no need to create any artificial training data
(e.g., representing
structures that are not-physically viable). Instead, structures from
databases, such as the protein
data bank (PDB) are sufficient. Training data can be created by masking a
portion of a known
structure, and having the machine learning algorithm attempt to recreate the
ground truth.
Accordingly, by allowing for direct prediction of amino acid interfaces,
approaches described
herein facilitate design of custom biologic structures.
a. Graph-Based Representation of Protein/Peptide Structure
[0484] In certain embodiments, structures of proteins and/or peptides, or
portions thereof,
may be represented using graph representations. Biological complexes, for
example comprising
multiple proteins and/or peptides, as well as, in certain embodiments small
molecules, may also
be represented using graph representations. An entire complex may be
represented via a graph
representation, or, in certain embodiments, a graph representation may be used
to represent
structure of a particular portion, such as in a vicinity of an interface
between two or more
molecules (e.g., constituent proteins and/or peptides of the complex).
[0485] For example, FIGs. 44A and 44B illustrate an approach for
representing a portion of
complex comprising a particular biologic (a protein or peptide) interacting
with a target, which
may be another protein or peptide, such as a particular receptor. FIG. 44A
shows a ribbon
- 166 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
diagram of the portion of the biological complex 4400, comprising a portion of
a particular
biologic 4404 (shown in green) together with a portion of the target 4402
(shown in blue).
[0486] FIG. 44B shows a schematic of a graph representation 4420 of the
biological complex
shown in FIG. 44A. As shown in FIG. 44B, in certain embodiments, the target
and particular
biologic may each be represented as a graph ¨ a target graph 4422 and a custom
biologic graph
4424. Each of graphs 4422 and 4424 comprise a plurality of nodes and, in
certain embodiments,
edges. In FIG. 44B, each node is illustrated as a circle and each edge is
shown as a line
connecting two nodes. The target graph is shown in blue, with nodes 4422a,
4422b, 4422c and
the biologic graph 4424 is shown in green, with nodes 4424a, 4424b, 4424c.
[0487] In certain embodiments, each node in a graph representation , such
as target graph
4422 and/or biologic graph 4424, represents a particular amino acid site in
the target or custom
biologic and has a node feature vector 4440 that is used to represent certain
information about
the particular amino acid site. For example, a node feature vector may
represent information
such as an amino acid side chain type, a local backbone geometry, a side chain
rotamer structure,
as well as other features such as a number of neighbors, an extent to which
the particular amino
acid site is buried or accessible, a local geometry, etc. Node feature vectors
are described in
further detail, for example, in section A.i below.
[0488] Edges in a graph representation may be used to represent
interactions and/or relative
positions between amino acids. Edges may be used to represent interactions
and/or relative
positioning between amino acids that are located within a same protein or
peptide, as well as
interactions between amino acids of different molecules, for example between
the custom
biologic and the target. As with nodes, each edge may have an edge feature
vector 4460. An
edge feature vector may be used to represent certain information about an
interaction and/or
- 167 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
relative positioning between two amino acid sites, such as a distance, their
relative orientation,
etc. Edge feature vectors are described in further detail in section A.ii
below.
[0489] In FIG. 44B, nodes representing amino acid sites of a (e.g., known)
target molecule
are shown in blue, and nodes representing amino acid sites of a custom
biologic being designed
are shown in green. Edges representing interactions within (i.e., between
amino acids of) the
target and the biologic are shown in blue and green, respectively. Edges
representing an
interaction and/or relative positioning between an amino acid of the target
and one of the
biologic ¨ inter-chain edges ¨ are shown in red.
a.i Node Features
[0490] Turning to FIG. 45A, as described herein, nodes represent amino acid
sites on a
biologic and/or target, such as a protein or peptide. In certain embodiments,
each amino acid site
includes peptide backbone atoms (e.g., N, Ca, C, 0, as shown in FIG. 45A)
together with a side
chain, which may be known, or as yet unknown, to-be-determined. For example,
as shown in
FIG. 45A, nodes 4524a and 4524b represent amino acid sites 4504a and 4504b of
a particular
custom biologic, each of which includes peptide backbone atoms along with a
side chain, 4508a
and 4508b, respectively. Side chains 4508a and 4508b may be unknown and/or to-
be-
determined, but can, for example, be approximately located by virtue of the
beta-Carbon (q3)
atoms as shown in FIG. 45A. Similarly, node 4522a may be used to represent
amino acid site
4502a (which includes side chain 4506a) of a target.
[0491] A node feature vector may be used to represent information about a
particular amino
acid site, such as side chain type (if known), local backbone geometry (e.g.,
torsional angles
describing orientations of backbone atoms), rotamer information, as well as
other features such
- 168 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
as a number of neighbors, an extent to which the particular amino acid is
buried or accessible, a
local geometry, and the like. Various approaches for encoding such information
may be used in
accordance with technologies described herein.
[0492] For example, in certain embodiments, a node feature vector comprises
one or more
component vectors, each component vector representing a particular structural
feature at a
particular amino acid location, as illustrated in FIG. 45B. That is, a node
feature vector may be
thought of as several component vectors 'stitched', or concatenated, together.
Each component
vector may include one or more elements, whose values encode a particular type
of structural
information. For example, as shown in FIG. 45B, one component vector 4552 may
be used to
represent a type of side chain 4508a, another component vector 4554 used to
encode local
backbone geometry, another component vector 4556 to encode rotamer structure
of side chain
308a, and so on.
[0493] In certain embodiments, side chain type may be represented via a one-
hot encoding
technique, whereby each node feature vector comprises a twenty element side
chain component
vector 4552 comprising 19 "0's" and a single "1," with the position of the "1"
representing the
particular side chain type (e.g., glycine, arginine, histidine, lysine,
serine, glutamine, etc.) at a
particular node/amino acid site. In certain embodiments, local backbone
geometry may be
represented using three torsion angles (e.g., the phi (cp), psi (w), and omega
(w) representation).
In certain embodiments, a node feature vector may include a component vector
representing a
rotamer, for example a vector of chi angles. In certain embodiments, each
angle may be
represented by two numbers ¨ e.g., a sine of the angle and a cosine of the
angle.
- 169 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
a.ii Edges and Features
[0494] In certain embodiments, as described herein, edges may be used to
represent
interactions between and/or a relative positioning between two amino acid
sites. A graph
representation accounting for interactions between every amino acid could
include, for each
particular node representing a particular amino acid site, an edge between
that node and every
other node (e.g., creating a fully connected graph). In certain embodiments, a
number of edges
for each node may be limited (e.g., selected) using certain criteria such that
each node need not
be connected to every other node and/or only certain, significant,
interactions are represented.
For example, in certain embodiments, a k-nearest neighbor approach may be
used, wherein
interactions between a particular amino acid and its k nearest neighbors (k
being an integer, e.g.,
1, 2, 4, 8, 16, 32, etc.) are accounted for in a graph representation, such
that each node is
connected to k other nodes via k edges. In certain embodiments, a graph
representation may only
include edges for interactions between amino acids that are separated by a
distance that is below
a particular (e.g., predefined) threshold distance (e.g., 2 angstroms, 5
angstroms, 10 angstroms,
etc.).
[0495] Turning to FIG. 45C, in certain embodiments, an edge feature vector
includes a
representation of a relative distance and orientation between two amino acid
sites. For example,
an edge feature vector may include a value representing a distance 4572
between beta-Carbon
atoms of the two amino acid sites, along with values representing the three
dihedral angles and
two planar angles that represent their relative orientations. In certain
embodiments, an edge
feature vector may also include a value indicating whether the two nodes it
connects represent
amino acid sites on a same or different molecule.
- 170 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
a.iii Relative and Absolute Spatial Encoding Features
[0496] In certain embodiments, a graph representation may include only
features that are
invariant with respect to rotation and translation in three dimensional space.
For example, as
described above and illustrated in FIGs. 45A-C, local backbone torsion angles
do not change
when an entire biological complex is rotated and/or translated in 3D space.
Likewise, edge
feature vectors that represent relative distances between two amino acids, and
their relative
orientations with respect to each other also do not change when an entire
biological complex is
rotated and/or translated in 3D space. In certain embodiments, use of relative
features, which are
invariant under 3D translation/rotation is advantageous in that it obviates a
need to train a
machine learning model to avoid interpreting versions of a single structure
that are rotated and/or
translated as different structures.
[0497] Additionally or alternatively, in certain embodiments, absolute
coordinate values,
such as Cartesian x,y,z coordinates may be used in node feature vectors. In
certain embodiments,
this approach simplifies structural representations, for example allowing a
graph to represent a
3D protein and/or peptide structure with only nodes. In certain embodiments,
when absolute (as
opposed to relative) coordinates are used, node features may no longer be
invariant with respect
to 3D rotation and/or translation and, accordingly, a training approach that
ensures a machine
learning model is equivariant to rotations and translations in 3D space is
used.
b. Interface Prediction Using Graph Networks
[0498] Turning to FIGs. 46A-46C, graph representations of complexes
comprising a partially
unknown custom biologic may be used as input to a machine learning model
(e.g., a graph neural
network) in order to generate a predicted amino acid interface for binding to
a particular target.
171
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
In certain embodiments, a graph neural network is used to predict an amino
acid interface by
performing node classification and/or edge classification.
b.i Input Graph Representations and Component Masking
[04991 Turning to FIG. 46A, a graph neural network may receive a graph
representation that
comprises one or more unknown or partially unknown nodes and/or edges and be
used to
generate a prediction for the unknown nodes and/or edges. In certain
embodiments, a portion of
a biologic complex comprising a particular target together with an in-progress
custom biologic
that is being designed for binding to the target is represented via an initial
complex graph 400.
[05001 In certain embodiments, the in-progress custom biologic is at a
stage where its
peptide backbone structure within and/or about its prospective binding
interface has been
designed and/or is known, but particular amino acid side chain types at
interface sites, located in
proximity to (e.g., one or more amino acids of) the target, are unknown, and
to-be determined.
For example, a scaffold model representing a prospective peptide backbone for
the in-progress
custom biologic may have been generated via an upstream process or software
module, or
accessed from a library of previously generated scaffold models. In certain
embodiments, a
scaffold docker module as described in U.S. Patent Application No. 17/384,104,
filed July 23,
2021, the content of which is hereby incorporated by reference in its
entirety, may be used or
may have been used to generate a scaffold model representing a prospective
peptide backbone
for the in-progress custom biologic.
[05011 Accordingly, initial complex graph 4600 may include a target graph,
representing at
least a portion of the target, and a scaffold graph, representing at least a
portion of the peptide
backbone of the in-progress custom biologic. A scaffold graph may include a
plurality of nodes,
- 172 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket
No.: 2013969-0027
at least a portion of which are unknown interface nodes. Each unknown
interface node (e.g.,
4604) represents a particular interface site along the peptide backbone of the
in-progress custom
biologic. Interface sites are amino acid sites that are either a-priori known
or are/have been
determined to be located in proximity to, and, accordingly, are expected to
influence binding
with, the target.
[0502] As
illustrated in FIG. 46B, unknown interface nodes have node feature vectors
with a
side chain component vector that is masked so as to represent an unknown, to-
be-determined
amino acid side chain. Rather than being populated with a particular value or
set of values that
represents a particular type of amino acid side chain, a masked side chain
component vector is
populated with one or more masking values, that provide an indication that a
particular side
chain type is unknown or subject to change (e.g., by the machine learning
model). A masked
side chain component vector may be populated with one or more masking values.
A variety of
schemes with various combinations of masking values may be used to mask a side
chain
component vector. For example, in the context of the one-hot encoding scheme,
describe herein
with respect to FIG. 45B, as illustrated in FIG. 46B, a masked side chain
component vector may
be a zero vector. That is, while a particular side chain type can be
represented by setting one
element of a 20-length vector to "1", and the rest of the elements to "0", a
masked side chain
component can be represented via a 20-length zero vector. Additionally or
alternatively, other
values may be used, such as another integer (e.g., other than 1), or a null,
or 1/20 (e.g., indicating
a uniform probability of each side chain type). In certain embodiments, a 21-
element side chain
component could be used, with the first 20 elements representing particular
physical side chain
types and the 21st corresponding to an unknown side chain type.
- 173 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
[0503] In certain embodiments, node feature vectors of unknown interface
nodes may also
include components that represent information that is known, such as a local
backbone geometry
as described, e.g., in section A, herein. In certain embodiments, a scaffold
graph may also
include known scaffold nodes (e.g., 4606) representing a portion of the in-
progress custom
biologic for which amino acid side chain types are known and/or desired to be
fixed. A target
graph may include a plurality of nodes (e.g., 4602) each of which represents
an amino acid site
of the target and encodes structural information as described herein (e.g., in
section A, above).
[0504] In certain embodiments, a scaffold graph may include edges. In
certain
embodiments, edges of a scaffold graph may all be known and/or fixed, or
certain edges may be
unknown and/or allowed to change. Such edges may have feature vectors that are
completely or
partially masked, using masking values in an analogous fashion to that
described herein with
respect to masked side chain components.
b.ii Machine Learning Model Output and Processing
[0505] FIG. 46C shows an example process 4620 by which a machine learning
model may
be used to generate a predicted interface for an in-progress custom biologic
using a graph
representation approach as described herein. Machine learning model 4624 may
receive, as
input, initial complex graph 4622, comprising a target graph and scaffold
graph.
[0506] Machine learning model 4624 may include a plurality of layers and/or
implement
various architectures, examples of which are described in further detail
herein. In certain
embodiments, the machine learning model includes layers such as transformer
layers, graph
convolution layers, linear layers, and the like. In certain embodiments, the
machine learning
model is or includes a graph neural network that performs node and/or edge
classification. In
- 174 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
certain embodiments, a graph neural network may, for example, output a
probability distribution
for values of one or more unknown features of nodes and/or edges, which can
then be evaluated
to select a particular value for each unknown feature of interest.
[0507] For example, machine learning model 4624 may receive initial complex
graph 4622
as input and generate, as output, a likelihood graph 4630. Illustrative
likelihood graph 4630
comprises, for each unknown interface node of input scaffold graph portion of
initial complex
graph 4622, a corresponding classified interface node 4632 (shown with stripe
fill). For a
particular unknown interface node of the input scaffold graph, the
corresponding classified
interface node 4632 has a node feature vector comprising a side chain
component 4634 that is
populated with likelihood values 4636. Likelihood values of classified
interface node 4634's
node feature vector provide a measure of a predicted likelihood (e.g., of
suitability for binding)
for each particular side chain type, as determined by machine learning model
4624. As
illustrated in FIG. 46C, such likelihood values may, for example, be floating
point number
between zero and 1, thereby indicating a probability distribution for
potential side chain types of
classified interface node 4634.
[0508] In certain embodiments, likelihood graph 4630 may then be used to
select 4640, for
each classified interface node, a determined side chain type, to create a
predicted interface 4650.
For example, predicted interface 4650 may be a graph, for which each node of
the custom
biologic is known ¨ i.e., has a known side chain type. For example, values
4656 of a side chain
component vector 4654 that represent a particular side chain type may be
determined from
likelihood values 4636 by setting an element having a maximum likelihood to
"1" and the rest to
"0", thereby creating a known interface node 4652 from a classified interface
node 4632.
Likelihood values may be determined and used to create classified and known
nodes in
- 175 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
accordance with a variety of approaches and are not limited to the 0 to 1
probability distribution
approach illustrated in FIG. 46C. For example, values ranging from 0 to 100,
or on other scales
may be used. Scales may be linear or non-linear. In certain embodiments,
likelihood values may
be output in a binary (e.g., 0 or 1) fashion, such that, for example, side
chain components of
classified nodes 4632 are directly output in a one-hot encoding scheme and no
selection step is
needed to determine a final side chain component 4656 from a classified node's
likelihood
values.
[0509] In certain embodiments, other information represented in components
of node and/or
edge feature vectors may be predicted in a likelihood graph by machine
learning model 4624.
For example, likelihood values for rotamer structures of side chains, as well
as orientations
and/or distances encoded in edge feature vectors, may also be generated.
[0510] In certain embodiments, machine learning model 4624 may generate
predictions for
node and/or edge features for an entire graph representation, e.g., including
nodes / edges that are
a priori known. That is, likelihood graph 4630 may include classified
interface nodes, as well as
classified nodes that correspond to nodes of the input scaffold graph and/or
target graph for
which a side chain type was not masked, and previously known. In certain
embodiments, to
determine a final custom biologic interface, predictions for unknown /
partially known nodes
and/or edges are used to determine final feature values, while predictions for
nodes and/or edges
that are already known may be discarded, and a priori known values used. For
example,
selection step 4640 may also reset side chain components of known scaffold
nodes to their
previously known values.
[0511] In certain embodiments, a neural network may be restricted to
generate predictions
for only a portion of a graph representation, for example, only for nodes
(e.g., performing solely
- 176 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
node classification), only for edges (e.g., performing solely edge
classification), only for
unknown features, or the like.
b.iii Single Run and Iteratively Refined Predictions
[0512] Turning to FIG. 46D, in certain embodiments, as explained herein, a
neural network
may generate, as output, a structural prediction for an input graph
representation. In certain
embodiments, the structural prediction comprises, for each of one or more
nodes and/or edges of
the input graph representation, a prediction of one or more component features
of an associated
feature vector. For example, as explained herein, in the context of a
prediction of a type of an
amino acid, a neural network may generate a probability distribution
comprising, for each
possible type of amino acid, a likelihood that an amino acid represented by a
particular node is of
a particular type (e.g., glycine, arginine, histidine, lysine, serine,
glutaniine, etc.). In certain
embodiments, such structural predictions may then be used to determine a final
value of each
component feature, for example, by identifying an amino acid type, for each
node, predicted as
having a maximum likelihood.
[0513] In certain embodiments, as shown in FIG. 46D, in an illustrative
process 4670,
multiple iterations are performed, whereby a structural prediction generated
from one iteration is
fed back into the neural network as input for a subsequent iteration. Such
structural predictions
may be a likelihood graph 4630, or intermediate predicted interfaces derived
from a likelihood
graph, via a selection and/or set/reset step 4640 as described above.
[0514] That is, in certain embodiments, in an initial iteration, the
machine learning model
4624 receives, as input, initial complex graph 4622 and generates as output
initial likelihood
graph 4630. Then, initial likelihood graph itself is fed back into machine
learning model 4624,
- 177 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
as input, to generate a refined likelihood graph. This process may be repeated
in an iterative
fashion, to successively refine likelihood graphs, with each iteration using a
likelihood graph
generated via a previous iteration as input. After the final iteration,
predicted interface 4650 is
determined from a final likelihood graph.
[0515] In certain embodiments, at each iteration, rather than use a
likelihood graph from a
previous iteration as input, an intermediate predicted interface is generated
and used as input.
For example, in certain embodiments, in an initial iteration, machine learning
model 4624
receives, as input, initial complex graph 4622 and generates as output initial
likelihood graph
4630. Initial likelihood graph 4630 may then be used to generate an
intermediate predicted
interface, for example, by using classified nodes from likelihood graph to
determine particular
side chain types as described above with respect to FIG. 46C. The intermediate
predicted
interface may then be fed back into machine learning model 4624, as input, to
generate a refined
likelihood graph, which, in turn, may be used to generate a refined predicted
interface. This
process may be repeated in an iterative fashion, to successively refine
likelihood graphs and
interface predictions, with each iteration using an interface prediction
generated via a previous
iteration as input. A final predicted interface 4650 is produced on the final
iteration.
[0516] Various numbers of iterations may be used. For example, two, five,
ten twenty, fifty,
100, 250, 500, 1,000 or more iterations may be used. In certain embodiments,
one or more
thresholds are set to determine whether further iteration is necessary.
b. iv Neural Network Architectures
[0517] As shown in FIG. 46C and 46D, a particular neural network model may
comprise one
or more (e.g., a plurality of) layers, including, for example, various
transformer layers, graph
- 178 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
convolutional layers, linear layers, etc. Each layer need not be of a same
type, and various types
of layers (e.g., transformer, graph convolutional, linear) may be combined in
a particular neural
network model.
[0518] Turning to FIG. 46E, in certain embodiments, a neural network model
may be a
multi-headed model that utilizes multiple 'input heads' ¨ parallel sets of
neurons within each of
one or more particular layers ¨ to separately process different classes of
interactions between
amino acids. As opposed to 'attention heads' which are sets of neurons
(learnable parameters)
that receive the same input and generate a corresponding output, these 'input
heads' operate on
different inputs with each head specialized for its own particular kind of
input. For example, in
certain embodiments, a three-headed network model may be used in which each of
one or more
layers of a neural network model comprises three parallel sets of neurons,
each associated with a
different type of interaction. In certain embodiments, other approaches
comprising more or less
than three 'input heads' may be used. For example, each input head may be
specialized for a
certain edge type (e.g., where each input head has neurons/weights that are
specialized on a
specific edge type), and they can be concatenated or otherwise combined.
[0519] In this way, multiple input heads are allocated to receive different
'versions' of the
same graph. For example, each version could include a certain subset of the
edges in the graph,
for example, and omit other edges. For example, in certain embodiments, a
first set of neurons
may, for example, evaluate, for each node, ki edges and corresponding neighbor
nodes that
represent the ki nearest neighbor amino acids. A second set of neurons may
then be associated
with, and process, for each node, k2 edges and corresponding neighbor nodes
that represent the
interactions between k2 nearest neighboring amino acids. Finally, a third set
of neurons may then
be associated with, and process, for each node, k3 edges and corresponding
neighbor nodes that
179
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
represent the interactions between k3 nearest neighboring amino acids. ki, k2,
and k3 may be
integers, with ki <k2 <k3, (e.g., ki = 8, k2 = 16, and k3= 32) such that the
first set of neurons
tends to be associated with short range interactions, the second set of
neurons tends to be
associated with intermediate range interactions, and the third set of neurons
tends to be
associated with long range interactions.
[0520] Additionally or alternatively, in certain embodiments various sets
of neurons in a
multi-headed network may be associated with different types of interactions
between amino
acids based on other criteria. For example, three different sets of neurons
may be associated with
(i) peptide bond interactions, (ii) intra-chain interactions (e.g.,
interactions between amino acids
within a same molecule) and (iii) inter-chain interactions (e.g., interactions
between amino acids
on different molecules), respectively. Thus, for example, where three input
heads are used, one
input head might only consider edges that represent peptide bonds, another
input head only
considers edges that represent intra-chain interactions, and another input
head only considers
edges that represent inter-chain interactions.
[0521] In certain examples, other ways of organizing/defining input heads
are implemented
according to what a particular input head is dedicated to. For example, there
could be one or
more input heads, each of which only considers edges that represent
interactions between amino
acid sites that are within a particular threshold distance of each other
(e.g., a first input head for 5
angstroms or less, a second input head for 10 angstroms or less, and a third
input head for 15
angstroms or less). In another example, there could be one or more input
heads, each of which
considers a first k (where k is an integer) edges that are the k nearest
neighbors (e.g., a first input
head that considers the 5 nearest neighbors, a second input head that
considers the 15 nearest
neighbors, and a third input head that considers the 30 nearest neighbors).
180
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
[0522] Furthermore, in an alternative embodiment, both inter and intra-
chain interactions can
be combined in one input head (receives both inter and intra chain edges), for
example, with an
additional value on the end of each edge feature vector that serves as a
"chain label" ¨ e.g., "1" if
the edge is an inter-chain edge and "0" if the edge is an intra chain edge.
Moreover, in certain
embodiments, redundant information could be eliminated, thereby simplifying
the task for the
neural network. For example, backbone torsion angles have some redundancy
according to the
edge definitions - certain edges may be simplified by removing degrees of
freedom, and certain
angles may be computed using information about the orientation of neighboring
amino acids.
[0523] The sets of edges considered by different input heads may be
overlapping or non-
overlapping sets. For example, a set of intra-chain edges and a set of inter-
chain edges are
generally non-overlapping, while a set of edges representing sites within 5
angstroms or less and
a set of edges representing sites within 10 angstroms or less are overlapping
(the second set
includes the first). In certain embodiments, various input heads may be used
in different
combinations in a single machine learning model.
[0524] In certain embodiments, an ensemble machine learning model is
created as a
collection of multiple subsidiary machine learning models, where each
subsidiary machine
learning model receives input and creates output, then the outputs are
combined (e.g., a voting
model). For example, in certain embodiments, a voting ensemble machine
learning model may
be used wherein a likelihood value is an integer, such as a sum of votes of
multiple machine
learning models. For example, as applied in the method illustrated in FIG.
46C, the values 4636
of predicted likelihood (e.g., of suitability for binding) for each particular
side chain type, as
determined by machine learning model 4624, may be integers representing sums
of votes of
multiple machine learning models in a voting ensemble machine learning model,
thereby
- 181 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
indicating a probability distribution for potential side chain types of the
classified interface node
4634 in the example. Certain embodiments use different ways of combining
subsidiary machine
learning model output in a voting model. For example, a simple average may be
taken, a
weighted average may be taken (e.g., where some models are weighted more
heavily than
others), votes may be counted (e.g., integers), and the like. Where subsidiary
machine learning
models are weighted, a weighting function may be used according not only to
the model, but also
according to the particular side chain. For instance, for a first model,
predictions made of
hydrophobic side chains may be weighted heavily, whereas, for a second model,
predictions of
hydrophilic side chains are weighted heavily.
[0525] In the schematic of FIG. 46E, three 'input heads' are depicted
4682a, 4682b, and
4682c, where each input head receives and processes a portion of the edges of
the scaffold-target
graph 4680 and generates output vectors 4684a, 4684b, and 4684c, allowing the
processing of
different ranges or scales of information. For example, input head 4682a may
process inter-
chain edges, input head 4682b may process intra-chain edges, and input head
4682c may process
edges that represent peptide bonds, i.e., connecting neighboring amino acid
sites. At step 4686,
the output may be concatenated, averaged, added, weighted, and/or otherwise
processed to
produce combined output vector 4688.
[0526] The schematic of FIG. 46F depicts how graph versions for input may
be created from
an initial graph (e.g., by selecting and retaining various sets of edges) or
may be created directly
from a structural model of the biologic complex 4690. For example, as shown in
FIG. 46F, a
graph featurizer module 4692 may operate on a biologic complex model 4690
(e.g., a protein
data bank (PDB) file) and generate multiple graph representations 4694a,
4694b, and 4694c,
each used as input to a corresponding input head 4682a, 4682b, and 4682c. In
certain
- 182 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
embodiments, two or more of the generated graph representations may use the
same edge feature
vector scheme. In certain embodiments, two or more generated graphs may use a
different
approach for representing edge feature vectors, e.g., to encode different
types of information.
For example, graph 4694a and 4694b might both include edges that have feature
vectors
conveying the structural information as shown in FIG. 45C (except that one
graph might include
k = 5 nearest neighbors and the other graph might include the k = 10 nearest
neighbors, for
instance), while graph 4694c may use a different scheme for encoding structure
information in
an edge feature vector (for instance, where edges are limited to peptide
bonds, with each edge
having a feature vector that has two elements indicating which amino acid is
upstream from the
other). At step 4686, the output may be concatenated, averaged, added,
weighted, and/or
otherwise processed to produce combined output vector 4688.
c. Example Training and Performance of a Side Chain Classification Network
[0527] This example shows a training procedure, and performance results for
an example
graph network approach for predicting side chain types in accordance with the
embodiments
described herein.
c.i Example Training Procedure
[0528] FIGs. 47A-47C illustrate an approach for training a graph-based
neural network used
in certain embodiments described herein. The particular network utilized in
this example
comprises multiple blocks, which may be combined together in a pipeline. FIG.
47A shows an
outline of an illustrative process 4700 for training a graph-based neural
network comprising
multiple blocks. In certain embodiments, a graph based neural network may be
trained using
- 183 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
data from structural models of proteins and/or peptides, for example obtained
from the protein
data bank (PDB). In certain embodiments, input graph representations are
created from
structural models, according to the approaches for representing amino acids
and their interactions
via nodes and edges as described herein. In certain embodiments, training data
is created by
masking various nodes and/or edges of a known structural model, leaving the
masked portions
for the neural network to predict during the training process.
[0529] For example, as shown in FIG. 47A, training data set 4710 was
created using graph
representations created from structural models obtained from the PDB. Various
training
representations were created by randomly selecting portions of a molecule
and/or complex to
mask (e.g., such that a predefined amount, e.g., 1/3 of the molecule or
complex, is masked). The
particular amino acid sites that were masked were not restricted to a
particular region, such as an
interface, of a molecule or complex, but were allowed to vary randomly
throughout the full
molecule or complex. This "full molecule" training dataset 4710 was then used
for a first round
of training, that used multiple steps to successively add blocks to and train
a multi-block machine
learning model. As shown in FIG. 47A, in a first step, full molecule training
dataset 4710 was
used to train a single block model 4720a, generating a first set of layer
weights for the single
block. These layer weights were then used to initialize 4730a a two block
model 4720b, which
was then trained, again using training dataset 4710, to determine a second set
of layer weights.
These second set of layer weights were used to initialize a three block model.
This process was
repeated, adding an additional block at each step, and initializing layer
weights using weights
from a preceding step, was repeated, out to n (a desired number of) iterations
and size (i.e.,
number of blocks) in an nth model 4720n. At each step, training was performed
using a cross
entropy loss function. A variety of size models and iterations, for example,
two, five, ten twenty,
- 184 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
fifty, 100, 250, 500, 1,000 or more may be used. In certain embodiments, one
or more thresholds
are set to determine whether further iteration is necessary.
[0530] A final, second round of training was performed to further refine
nth model 4720n for
the ultimate purpose of predicting side chain types at an interface, rather
than arbitrary positions
within one or more molecules. Accordingly, a second, interface specific
training dataset 4740
was created, this time using graph representations of complexes where masked
side chain
components were restricted to interface nodes. Training dataset 4740 was used
to train nth model
4720n, to create a final model 4750.
[0531] FIG. 47B illustrates, schematically, how each block may receive, as
input, various
types of feature vectors, including known and unknown features, and be used to
predict new
distributions, similar to the approach described above with respect to FIGs.
46C and 46D. In
FIG. 47B, each node representation includes amino acid (AA) encoding
indicating type of amino
acid (in green) and structural descriptors (in blue). The illustrative process
for predicting new
amino acid distributions is iterative. The distributions of amino acids are
initialized from the
empirical distribution in the molecule (i=0), where the graph is composed of a
set of known and
unknown nodes labels. During each pass through the machine learning
architecture detailed in
FIG. 47C (the "AH bloc"), (i to i+1), the distributions are updated, with the
distributions of
known nodes reset as in the input. The process may be repeated as much as
desired.
[0532] FIG. 47C shows a schematic of an illustrative architecture used to
create a block
("AH bloc") used in the present example. Other architectures, using different
types of layers,
organizations, and the like, are also contemplated. FIG. 47C shows the overall
process flow
4760 and details of the "AH bloc" architecture 4770, 4780 depicted in FIGs.
47B and 47C and
used in the examples whose results are shown below.
- 185 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
c.ii Results
[0533] Table 1 below shows overall performance of the approach for
classifying amino acid
side chain types over a full molecule test set, created analogously to full
molecule training
dataset 4710 (i.e., not necessarily restricted to an interface specific test
set), described above with
respect to FIGs. 47A-47C. Overall perfounance may be quantified using, among
other things, an
identity score and a similarity score. Identity score measures the fraction of
predicted side chain
types that were identical to the ground truth, while similarity score accounts
for similarities
between certain types of amino acids (as determined according to the BLOSUM 62
matrix) (e.g.,
while a predicted side chain type might not be identical to the ground truth,
it may be a type that
would work just as well in the structure).
Table 1: Overall Performance Evaluated on a Full Molecule Test Dataset
Overall performances
Identity: 0.47438593137723406
Similarity: 0.6653624677434771
Total number of predictions: 157408
Total number of molecules: 835
[0534] Table 2 displays performance metrics evaluated on a full molecule
test dataset,
broken down by side chain type.
- 186 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
PCT/US 2022/038 014- 18.05.2023
CA 03226172 2024-01-03
Replacement Pages Docket No.: 2013969-0027
Table 2: Individual Side Chain Performance Evaluated on a Full Molecule Test
Dataset
precision recall 1'1 -score support pied AUC accuracy
GLN 0.199264 01419111 :11165767.: 64900 4622.a]
]'0.808566 0141911
MET 0.135765 0.221763 0.168421 3391.0 5539.0 0.802935 0.221763
LEU 0.632664 0.584545 0.607653 16435.M 400;0406520 0.584545
VAL 0.563306 0.557207 0.555250 11773.0 11866.0 0.941588 0.557207
SER 0.495857 0.348429 0.409271 10820.0 7603.0 0.874407 0.348429
GLY 0.992451 0.998553 0.995493 11060.0 11128.0 0.999976 0.998553
THR 0.456399 0,501720 0.477987 9013.0 9908.0 0.905942 0,501720
MU 0,323074 0.320018 0.321539 11415.0 11307.0 0.855357 0.320018
INS 0.269124 0,252146 0.260359 10018.0 9386.0 0.848505 0,252140:
PRO 0.950005 0.984458 0.968925 7528,0 7801.0 0.999760 0.984458
ILE 0.471653 0,601909 0.528879 9744.0 12435.0 0.945871 0.6019:
CYS 0.329253 0.562566 0.415391 2821,0 4820,0 0.903031 0.562566
TYR 0277.* 0216527 0,322026 6208.0 59960 0899680 A016527:
PHE 0.436844 0.388787 0.411417 7063.0 6286.0 0.916806 0.388787
TRP 043154 0.23371184182640 22610 22710 0.87500 02331181
AEG 0.263850 0.238223 0.250382 8597.0 7782.0 0.837169 0.P389.03
ASP 0.454535 0,421467 0.437377 9773.0 9082,0 0.904510 0.421467.:
MA 0.587853 0.536639 0.561189 12134.0 11081,0 0.920588 0.536839
ASN 0.344873 0.251297 0.348055 7515.0 7655.0 0.877052 0.351227:
HIS 0.163275 0.264017 0.201770 3799.0 6143.0 0,826591 0,264017
;;:tivg I total 0.481851 0.475308 0.475714 167847.0 167847.0 0.919044 0.475308
[0535] FIGs. 648A-48C show accuracy, Fl-score, and AUC metrics for
predictions of each
particular side chain type (20 canonical amino acids) obtained using the graph
neural network
approach of the present example.
[0536] Performance was also evaluated using an interface specific
test data set, created
analogously to interface specific training dataset 4740. The interface
specific test dataset
- 187 -
AMENDED SHEET
Date Recue/Date Received 2024-01-03
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
performance for predicting amino acid side chain types for unknown interface
nodes to be
evaluated.
[0537] Tables 3 and 4 below shows overall performance of the approach for
classifying
amino acid side chain types over the interface specific test set, and broken
down by particular
side chain type, respectively, conveying the same information as in Tables 1
and 2 above, but for
the interface specific test dataset).
Table 3: Overall Performance Evaluated on an Interface Specific Test Dataset
Overall performances
Identity: 0.4412931105215655
Simi1arity: 0.6527090227825945
Total number of predictions: 36563
Total number of interfaces: 835
188
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
Table 4: Individual Side Chain Performance Evaluated on an Interface Specific
Test Dataset
precision recall fl -score support preci AUG
accuracy
GLN 0.181713 0.110098 0.137118 1426.0 864.0 0.791252 0.110098
MET 0.140365 0.204624 0.166510 865.0 1261.0 0.807995
0.204624
LEO 0,515658 0.563605 0.538566 3506.0 3832.0 0.897962 0.563605
VAL 0,520308 0.427365 0.469279 2368,0 1945.0 0.921030 0.427365
SEA 0.474289 0.306370 0.372270 2559,0 1653.0 0.851905 0.306370
GLY 0.991312 0.996782 0.994039 2175,0 2187.0 0,999880 0.996782
THR 0.489415 0.383228 0.429861 2051,0 1606.0 0.877177 0.383228
GLU 0.278485 0.347461 0.309173 2265.0 2826.0 0.841155 0.347461
LYS 0.209687 0.386424 0.271856 1871.0 3448.0 0.847177 0.386424
PRO 0.953555 0.967986 0.960716 1718.0 1744.0 0.998978 0.967986
ILE 0.447398 0.566888 0.500105 2108.0 2671.0 0.936182 0.566888
CYS 0.399746 0.519802 0.451937 606.0 788,0 0.891913 0.519802
TYR 0.287647 0.314874 0.300646 1553.0 1700,0 0.881837 0.314874
PHE 0.479428 0.321729 0,385057 1666.0 1118,0 0.907990 0.321729
TRP 0.177072 0.289963 0,219873 538.0 881.0 0.878418 0.289963
ARG 0.300469 0.181818 0.226549 2112.0 1278.0 0.834137 0.181818
ASP 0.463065 0.396975 0,427481 2116.0 1814.0 0.891215 0,396975
ALA 0.507514 0.527433 0,517282 2497.0 2595.0 0.900312 0.527433
ASN 0.316699 0.298552 0.307358 1658.0 1563.0 0.845206 0.298552
HIS 0.221800 0.193370 0.206612 905.0 789.0 0.822973 0.193370
avg I total 0.453710 0.441293 0.440905 36563.0 36563.0 0.903696 0.441293
[0538] FIGs. 49A-C are analogous to FIGs. 48A-C, but show results obtained
for predictions
over the interface specific dataset.
[0539] These results, in particular the area under the curve (AUC) metrics
shown in FIGs.
48C and 49C demonstrate accurate performance of the approaches described
herein.
[0540] Elements of different implementations described herein may be
combined to form
other implementations not specifically set forth above. Elements may be left
out of the
processes, computer programs, databases, etc. described herein without
adversely affecting their
operation. In addition, the logic flows depicted in the figures do not require
the particular order
189
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
shown, or sequential order, to achieve desirable results. Various separate
elements may be
combined into one or more individual elements to perform the functions
described herein.
[0541] Throughout the description, where apparatus and systems are
described as having,
including, or comprising specific components, or where processes and methods
are described as
having, including, or comprising specific steps, it is contemplated that,
additionally, there are
apparatus, and systems of the present invention that consist essentially of,
or consist of, the
recited components, and that there are processes and methods according to the
present invention
that consist essentially of, or consist of, the recited processing steps.
[0542] It should be understood that the order of steps or order for
performing certain action
is immaterial so long as the invention remains operable. Moreover, two or more
steps or actions
may be conducted simultaneously.
[0543] While the invention has been particularly shown and described with
reference to
specific preferred embodiments, it should be understood by those skilled in
the art that various
changes in form and detail may be made therein without departing from the
spirit and scope of
the invention as defined by the appended claims.
EQUIVALENTS
[0544] It is to be understood that while the disclosure has been described
in conjunction with
the detailed description thereof, the foregoing description is intended to
illustrate and not limit
the scope of the claims. Other aspects, advantages, and modifications are
within the scope of the
claims.
[0545] This written description uses examples to disclose the invention,
including the best
mode, and also to enable any person skilled in the art to practice the present
embodiments,
190
CA 03226172 2024-01-03
WO 2023/004116 PCT/US2022/038014
including making and using any devices or systems and performing any
incorporated methods.
The patentable scope of the present embodiments is defined by the claims, and
may include other
examples that occur to those skilled in the art. Such other examples are
intended to be within the
scope of the claims if they include structural elements that do not differ
from the literal language
of the claims, or if they include equivalent structural elements with
insubstantial differences from
the literal languages of the claims.
191