Note: Descriptions are shown in the official language in which they were submitted.
WO 2023/053015
PCT/IB2022/059214
BICYCLIC AMINE DERIVATIVES
AS GABAA a5 RECEPTOR MODULATORS
THE FIELD OF THE INVENTION
The present invention provides compounds of formula (I) having affinity and
selectivity
for the gamma-aminobutyric acid A receptor subunit alpha 5 (GABAA a5) and act
as GABAA
a5 positive allosteric modulators (GABAA a5 PAMs), thereby useful in the
treatment or
prevention of diseases related to the GABAA a5 receptor, process for the
preparation and
intermediates of the preparation process thereof, pharmaceutical compositions
comprising
them and their use as medicaments.
THE BACKGROUND OF THE INVENTION
Gamma-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the
central nervous system. Receptors sensitive for GABA are divided into two main
families, the
ligand gated GABAA receptors and the G-protein coupled GABAB receptors.
The ligand gated GABAA receptor mediates the majority of inhibitory
neurotransmission in the adult mammalian brain. The receptor is composed by
the pentameric
assembly of multiple subunits (a1-6, 131-3, y1-3, 6, E, ii, A, p1-3) (Olsen
and Sieghart,
Pharmacol Rev 2008, 60:243-260) forming a ligand-gated chloride-channel.
Subunit
distribution varies developmentally and regionally in the brain. This high
variability leads to
broad variation in inhibitory and in certain conditions excitatory neural
mechanisms and
provides the possibility for specific therapeutic interventions (Fritschy and
Mohler, J Comp
Neurol 1995, 359:154-194; Jacob, Front Mol Neurosci 2019, 12: Art 179).
Physiological roles
and pharmacological profiles of GABAA receptors are strongly dependent on the
subunit
constitution. Studies on genetically modified mice have demonstrated that
receptor subunit
composition, especially regarding the a subtypes, considerably determines
pharmacology of
compounds acting on the benzodiazepine-sensitive allosteric modulatory site
(BDZ-site)
(Rudolph and Knoflach, Nat Rev Drug Discov 2011, 10:685-697). The widely
distributed al-
containing receptors mediate the sedative and amnesic effects, whereas the a2-
and a3-
containing receptors account for the anxiolytic, anticonvulsant and
myorelaxant effects
(Sieghart and Sperk, Curr Top Med Chem 2002, 2:795-816; Whiting et al, Drug
Discov Today
2003, 8:445-450). a5 subunit containing receptors (a5GABAARs) are
preferentially expressed
in the hippocampus, prefrontal cortex, amygdala and nucleus accumbens (Olsen
and
Sieghart, Neuropharmacology 2009, 56:141-148; Sur et al., Brain Res 1999,
822:265-270;
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
2
Martin et al., Biochem Soc Trans 2009, 37:1334-1337) and thought to be
involved in a variety
of CNS disorders.
a5-containing receptors are predominantly extrasynaptic and mediate tonic
inhibition
(Caraiscos et al., Proc Nat! Aced Sci USA 2004, 101:3662-3667). In contrast to
their inhibitory
role in the mature nervous system, a5GABAARs can provoke excitation in early
hippocampal
circuit development (Marchionni et al., J PhysioL 2007, 581:515-528). Their
modulatory effect
on the excitability of hippocampal and cortical principal neurons can explain
the significant
effect of a5GABAARs in neuronal development, cognition, learning and memory
and their
potential therapeutic usefulness in various disorders including stroke, mild
cognitive
impairment, schizophrenia, depression, dementia-related conditions or diseases
related to
impaired social cognition or neurodevelopmental disorders such as Down
syndrome or autism
spectrum disorder (ASD) (Jacob, Front Mol Neurosci2019, 12: Art 179; Mohamad
and Tarmizi
Che Has, J Mol Neurosci 2019, 67:343-351; Soh and Lynch, Curr Drug Targets
2015, 16:735-
746).
Genetic and pharmacological reduction in a5-mediated tonic inhibition may
improve
learning and memory (Mailer and Rudolph, F1000Res 2017 Feb 3;6. pii: F1000
Faculty Rev-
101) through enhanced neuronal plasticity (Martin et al., J Neurosci 2010,
30:5269-5282) and
network oscillatory activity (Towers et al, J Physiol 2004, 559:721-728;
Glykis and Mody,
Neurophysiol 2008, 95:2796-2807). However, hippocampal and cortical
hyperactivity arising
from reduced a5GABAAR function might also result hyperlocomotion and impaired
sensorimotor gating (Hauser at al., Mol Psychiatry 2005, 10:201-207), impaired
social
behaviour (Zurek et al., Ann Clin Trans! Neurol 2016, 3:392-398) and cognitive
deficit in
rodents (Engin et al., J Neurosci 2015, 35:13698-13712; Martin et al., J
(Veurosci 2010,
30:5269-5282; Prut et al., Genes Brain Behav 2010, 9:478-488), those
behavioural changes
characteristic in a variety of CNS disorders. In such a pathological condition
facilitation rather
than blockade of a5GABAAR function may be a promising treatment for positive,
negative and
cognitive symptoms associated with such diseases. In support of this idea,
virally--induced
overexpression of the ct5 subunit of the GABAA receptor in the ventral
hippocampus
normalized physiological and behavioural deficits in a rat model of
schizophrenia (Donegan et
al., Nature Communications 2019, 10:2819),
The University of Wisconsin-Milwaukee described certain 4H-benzo[f]imidazo[1,5-
a][1,4]diazepine derivatives (WO 2017/161370 Al) as a5-preferring PAM
compounds, such
as SH-053-2'F-R-CH3, MP-III-022 or GL-II-73 (Stamenio et al. Eur J Pharmacol
2016,
791:433-433; Savic et al., Neuropsychopharmacology 2008, 33:332-339; Prevot et
al., ACS
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
3
Chem. Neurosci. 2019, 10:2088-2090) that showed procognitive, anxiolytic and
antidepressant effects in mouse stress models and in aged mice (Prevot et al.,
Mol
Neuropsychiatry 2019, 5:84-97). MP-111-022 and the 6,7-dihydro-2-benzothiophen-
4(51-0-one
a5 PAM Compound 44 (Chambers et al., J Med Chem 2003, 46:2227-2240) improved
cognitive performance of young and aged rats, respectively (Poe, Michael M.,
Theses and
Dissertations. 1301(2016) https://dc.uwm.edu/etd/1301i Koh et al.
Neuropharmacology2013,
64:145:152. Acute treatment with GL-II-73 rescued chemogenetically induced
behavioural
deficits in a mouse model of depression (Fee et al., Int J
Neuropsychopharmacol 2021,
24:505-518), while chronic treatment with GL-II-73 reversed age-related
neuronal atrophy as
well as impairment in working memory in adult mice (Sibille et al., Biol
Psychiatry 2020,
87:Supp11, page S85). In addition, SH-053-2'F-R-CH3 and MP-III-022 attenuated
pathological
changes of locomotor activity of rats in developmental models of schizophrenia
(Gill et al.,
Neuropsychopharmacology 2011, 36:1903-1911; Batinic et al. Int J Dev
Neurosci 2017, 61:31-39).
AgeneBio Inc. described imidazo[1,5-a][1,2,4]-triazolo[1,5-
d][1,4]benzodiazepine
derivatives (WO 2015/095783 Al) as GABAA a5 PAMs and found in preclinical
proof of biology
studies of age-related cognitive impairment that such compounds occupy GABAA
a5 receptors
in the hippocampus under conditions of hippocampal overactivity (Press
release, AgeneBio,
11 Sep 2019; https://www.agenebio.com/agenebio-announces-additional-funding-to-
advance-novel-gaba-a-therapeutic-program-to-address-alzhei mers-and-other-cns-
conditions!), as their lead series has potent and selective compounds with
good in vivo efficacy
in age-impaired rats (https://grantome.com/grant/N1H/R44-AG063607-01).
The most preferred indication in accordance with the present invention is
autism
spectrum disorder (ASD). ASD is a complex, heterogeneous neurodevelopmental
disorder
characterized by a deterioration of social relationships, a decrease in
communication, typical
repetitive behaviours, and impairment in executive functions (Anagnostou et
al., CMAJ 2014,
186:509-519; Diagnostic and statistical manual of mental disorders. 5th ed.
Arlington, VA:
American Psychiatric Association; 2013 - Diagnostic Criteria for 299.00 Autism
Spectrum
Disorder). There are no medications approved for the treatment of core
symptoms of ASD.
Current pharmacological treatment is limited to atypical antipsychotics
risperidone and
aripiprazole which are approved for the treatment of ASD-associated aggression
and irritability
(Anagnostou et al., Curr Opin Neurol 2018, 31:119-125). Antidepressants are
used off-label
for alleviating obsessive/compulsive symptoms in ASD; the efficacy and the
tolerability of
these treatments are modest (Carrasco et al., Pediatrics 2012, 129:e1301-
e1310), so there is
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
4
an unmet need for more selective, pathophysiology-based treatment of the
aforementioned
conditions.
ASD can be associated with genomic alterations coupled with GABAAR subunits.
Chromosomal abnormalities, namely duplication of copy number variations in the
q11.2-13
region on chromosome 15 were reported in ASD patients. In humans, this region
contains
genes that encode the a5, 133 and 73 subunits of the GABAA receptor (Coghlan
et al., Neurosci
Biobehav Rev 2012, 36:2044-2055). An autism patient exome study identified
missense
mutations in Gabra5-/- and RDX, the genes for the a5GABAAR and its anchoring
protein
radixin, further supporting a a5GABAAR deficiency in ASD (Zurek et al., Ann
Clin Transl Neurol
2016, 3:392-398). There is increasing evidence for excitatory/inhibitory (E/I)
imbalance arising
from deteriorated GABAergic function in ASD. Reduced expression of the GABA
synthesizing
enzymes GAD65 and GAD67 and the reduction of GABAA receptor density have been
reported in post-mortem ASD brain (Fatemi et al., Biol Psychiatry 2002 52:805-
810; Oblak et
al, Autism Res 2009, 2:205-219). In imaging studies using positron emission
tomography
(PET) and magnetic resonance spectroscopy (MRS) reductions in GABA
concentration and
GABAA receptor availability have been reported in patients with ASD (Mori et
al., Brain Dev
2011, 34:648-654; Puts et al., Autism Res 2016, 10:608-619; Robertson et al.,
Curr Bio/ 2016,
26:80-85). A pilot PET study showed reduced binding of an a5GABAAR selective
tracer
[l 1
C]Ro154513 across multiple brain regions suggesting reduced level of a5GABAAR
in ASD
(Mendez at al., Neuropharmacology 2013, 68:195-201). Another study showed
changes in a
GABA-sensitive perceptual task in ASD patients (Horder et al., Se/ Trans/ Med
2018, pii:
eaam8434). In line with these observations, postmortem analyses revealed
reduced
expression of a5GABAAR (Blatt et al., J Autism Dev Disord 2001, 31:537-54;
Fatemi et al. J
Autism Dev Disord, 2010, 40:743-750). Impaired GABAergic function in ASD
patients can be
considered, thus facilitating cortical inhibition and restoring E/I balance by
a5 PAMs can be a
feasible therapeutic strategy in the treatment of the disease.
Increased neuronal excitability in the cortex may lead to autism-like
behavioural
deficits in rodents (Yizhar et al., Nature 2011, 477:171-178). Supporting the
clinical findings
genetic reduction of a5GABAAR exhibited a reduced tonic currents and increased
excitability
of principal hippocampal neurons in Gabra5-/-mice (Bonin et al., J
Neurophysiol 2007,
98:2244-2254). Besides the impairment in the executive function, robust autism-
like
behaviours and pathologies were observed in Gabra5-/- mice (Zurek et al., Ann
Ciin Transl
Neurol 2016, 3:392-398; Mesbah-Oskui et al., Neurotoxicol Teratol 2017, 61:115-
122).
Similarly, Fragile X syndrome model (Frnr1-") mice showed downreaulation of
a5GABAAR and
a deficit in tonic inhibition (Curia et al., Cereb Cortex 2009, 19:1515-1520)
which accompanied
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
with behavioural hallmarks of ASD (Bakker and Oostra, Cytogenet Genorne Res
2003,
100:111-123),
The prenatal valproate model has excellent construct and face validity,
therefore it is
a widely accepted disease model of ASD (Christensen et al,, JAMA 2013,
309:1696-1703;
5 Roullet et al., Neurotox leratoL 2013, 36:45-56). In this method, time-
mated female Wistar
rats are administered a single dose of valproic acid on gestational day 12.5.
After
investigational drug treatment, offspring are examined behaviorally in the
social preference
assay at postnatal day 59, The social preference test is a highly accepted
assay to assess
autistic behavior in rodents (Nadler et al., Genes Brain Behav 2007, 3:303-
314; Bambini-
Junior et eiL, Brain Res 2011, 1403:8-16). Briefly, in this assay a test
animal is allowed to
investigate a conspecific separated by a dividing perforated wall or a similar
area however,
without a target conspecific. An autistic animal (such as a prenatally
valproate-exposed rat)
spends little time with social investigation during a test session. It is
believed that the reduced
social behaviour of VPA-treated animals can be reversed to the normal level by
the restoration
of a5GABAA receptor mediated inhibitory synaptic transmission (Wang et al.,
Front Neurol
2018, 9:Article 1052). Thus, examples of the present invention may be of great
behavioral
benefit in this preolinical disease model that recapitulates the core symptoms
of ASD.
Therefore, it can be presented that the compounds of the invention,
specifically GABAA 05
PAMs, may have therapeutic potential for the core symptoms of autism spectrum
disorder in
humans.
GABA-A receptor positive modulators, such as the non-selective clonazepam in
low
dose, have also proven to ameliorate symptoms in preclinical models of ASD
(Han et al.,
Nature 2012, 489:385-390; Okamoto et alõ J Neuroimmunol 2018, 321:92-96)
increasing the
expectations that clinically used benzodiazepines could be used in extremely
low doses for
the treatment of the disease. Besides this strategy subunit selective
compounds, such as a2/3
modulators (AZD7325; https://www.clinicaltrials.gov/ct2/show/NCT03678129) or
a5 positive
allosteric modulators may offer an alternative approach for the treatment of
ASD possibly with
an improved therapeutic window. Accordingly, the a5 selective PAM compound
RG7816
(R07017773) is in Phase ll clinical development for the treatment of ASD
(https://www.clinicaltrials.govict2/show/NCT04299464).
Therefore, compounds having high affinity and selectivity for the a5GABAARs,
GABAA
a5 PAMs respectively, can be used, alone or in combination with one or more
other active
ingredients, for the treatment or prevention of disorders of the central
nervous system where
one of the symptoms and/or syndromes of the disease may be related to the
GABAA a5
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
6
receptor. These include, but not limited to neurodevelopmental disorders such
as autism
spectrum disorder (ASD) (Mendez et al., Neuropharmacology 2013, 68:195-201),
Fragile X
disorder (Curia et al, Cereb. Cortex 2009, 19:1515-1520), Prader-Willi
syndrome (Bittel et al.,
J Med Genet 2003, 40:568-574), or Down syndrome (Braudeau et al., J
Psychopharmacology
2011, 25:1030-1042; Martinez-Cue et al., J Neurosci 2013, 33: 953-966),
neurocognitive
disorders (Collinson et al., J Neurosci 2002, 22:5572-5580) such as
Alzheimer's disease (AD)
(Kwakowsky et al., J Neurochem 2018, 145:374-392; Solas et al., Curr Pharm Des
2015;
21:4960-4971; Wu et al., Nat Commun 2014, 4159), prodromal AD and mild
cognitive
impairment (Maubach, Curr Drug Targets CNS Neurol Disord 2003, 2:233-239),
vascular
cognitive impairment and vascular dementia (Gacsalyi et al., Eur J Pharmacol
2018, 834:118-
125), frontotemporal lobar degeneration including frontotemporal dementia,
progressive
supranuclear palsy and corticobasal syndrome (Murley and Rowe, Brain 2018,
5:1263-1285),
Lewy body dementia (Khundakar et al., Acta Neuropathol Commun 2016, 4:66), age-
associated memory impairment and cognitive decline (Koh et al.,
Neuropharmacology 2013,
64:142-152), cognitive impairment associated with brain cancers including, but
not limited to
medulloblastomas (Sengupta et al., CNS Oncol 2014, 3:245-247), post-operative
dementia
(Cheng et al., J Neurosci 2006, 26:3713-3720), inflammation-induced dementia
(Wang et al.,
Cell Rep 2012, 2: 488-496), HIV-Associated neurocognitive disorder (Green and
Thayer,
Neuropharmacology 2019, 149:161-168), cognitive impairments associated with
the diseases
including, but not limited to migraine and tension headache (Russo et al., Am
J Hum Genet
2005, 76:327-333), multiple sclerosis (Kammel et al., Neuroscience 2018,
395:89-100),
Parkinson's disease (Blaszczyk, Front Neurosci 2016, 10:269-277), epilepsy
(McGinnity et al.,
Brain Commun 2021, 3(1):fcaa190; Schipper et al., Mol Neurobiol 2016, 53:5252-
5265),
attention deficit hyperactivity disorder and adult attention deficiency
(Bollmann et al., Trans!
Psychiatry 2015, 8:e589; Edden et al., Arch Gen Psychiatry 2014, 69:750-753)
or other CNS
diseases including, but not limited to post-traumatic stress disorder (Lu et
al., Neuronal Plast
2017, 2017:5715816), schizophrenia (Guidotti et al., Psychopharmacology 2005,
180:191-
205), positive, negative and/or cognitive symptoms associated with
schizophrenia (Asai et al.,
Schizophrenia Res 2008, 99:333-340; Donegan et al., Nature Communications
2019, 10:
Article number 2819; Gill et al., Neuropsychopharmacology 2011, 36:1903-1911;
Hauser et
al., Mol Psychiatry 2005, 10:201-207; Marques et al., Mol Psychiatry 2021,
26:2616-2625;
Redrobe et al., Psychopharmacology 2012, 221: 451-468), bipolar disorders
(Otani et al.,
Neurosci Lett 2005, 381:108-113), Huntington's disease (Du et al., Front Mol
Neurosci. 2017,
10:198), neurofibromatosis type I (Ribeiro et al., Cortex 2015, 64:194-208),
sleep disorders
(Mesbah-Oskui et al., Neurotoxicol Teratol 2017, 61:115-122), substance-
related and
addictive disorders including, but not limited to alcohol use disorder or
gambling disorder (Mick
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
7
et al., Addict Biol 2017, 22:1601-1609; Stephens et al., Eur J Pharmacol 2005,
526:240-250),
fetal alcohol spectrum disorder (Toso et al., Am J Obstet Gynecol 2006,
195:522-527), mood
disorders (Bugay et al., Neuropsychopharmacology 2020, 45:2289-2298; Carreno
et al., Int J
Neuropsychopharmacology2017, 20:504-509; Choudary et al., Proc Nati Acad Sci
USA 2005,
102:15653-15658; Fische!l et al., Neuropsychopharmacology 2015; 40:2499-2509),
psychotic
disorders (Wearne et al., Neuropharmacology 2016, 111:107-118), substance-
induced
psychotic disorder (Neugebauer et al., Behav Brain Res 2018, 342:11-18),
anxiety disorders
(Behlke et al., Neuropsychopharmacology2016, 41:2492-2501; Botta et al., Nat
Neuroscience
2015, 18:1493-1500), fear related disorders (Botta et al., Nat Neuroscience
2015, 18:1493-
1500; Crestani et al., Proc Nall Acad Sci USA 2002, 99:8980-8985), stress
disorder (Fische!l
et al., Neuropsychopharmacology 2015; 40:2499-2509), Alzheimer's disease
related
neuropsychiatric symptoms (Xu et al., Psychopharmacology 2018, 235:1151-1161),
stroke
(Clarkson et al., Nature 2010, 468:305-309; Lake et al., J Cereb Blood Flow
Metab 2015,
35:1601-1609), traumatic brain injury (Khodaei et al., Grit Care Med 2020,
48:533-544),
neuropathic pain (Hernandez-Reyes et al., Pain 2019, 160:1448-1458) and
inflammatory pain
(Bravo-Hernandez et al., Eur J Pharmacol. 2014, 734:91-97; Munro et al.,
Neuropharmacology 2011, 61:121-132). Modulating a5GABAARs may also be
beneficial in
treating diseases and conditions including, but not limited to
bronchoconstrictive diseases
such as, but not limited to asthma, chronic obstructive pulmonary disease, and
bronchopulmonary dysplasia (Gallos et al., Am J Physiol Lung Cell Mol Physiol
2015,
308:L931-942; Mizuta et al., Am J Physiol Lung Cell Mol Physiol 2008,
294:L1206-1216) and
obesity (Xia et al., Mol Psychiatry 2021, doi: 10.1038/s41380-021-01053-w).
Compounds
capable of modulating a5GABAARs are in particular expected to be useful
candidates for the
treatment of neurodevelopmental disorders, neurocognitive disorders, mood
disorders and
schizophrenia.
Many structurally different compounds active on the a5 subunit of the GABAA
receptor
are known in the art (Guerrini et al., Expert Opin Ther Patents 2013,
23(7):843-866), including
isoxazole (e.g., WO 2009/071477 Al, WO 2018/104419 Al, WO 2019/238633 Al) and
triazole derivatives (e.g., WO 2012/062687 Al, WO 2014/001278 Al, WO
2014/001279 Al,
WO 2014/001282 Al, WO 2020/016443 Al).
Despite the numerous studies and modulators of the GABAA a5 receptor, unmet
need
still persists to provide compounds that can be useful in the treatment or
prevention of
diseases related to the GABAA a5 receptor.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
8
SUMMARY OF THE INVENTION
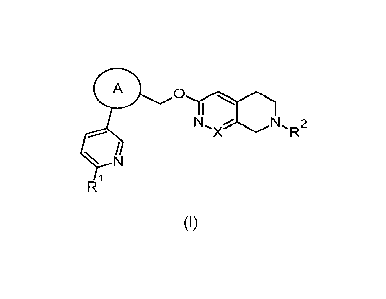
The present invention relates to compounds of formula (I)
A 0
NI, N, 2
-X R
N
R1
(I)
wherein
A is represented by
NPX,e,
.1;1 ss-
group, or "r"' group;
R1 is an alkyl, an alkoxy, or a haloalkyl group;
R2 is hydrogen; an alkyl group optionally substituted with -S(0)2-alkyl, a
cycloalkyl or a
heterocycle; a cycloalkyl group; a heterocycle group optionally substituted
with an alkyl; or a
heteroaryl group;
X is CH or N;
and/or salts thereof and/or stereoisomers thereof and/or enantiomers thereof
and/or
racemates thereof or diastereomers thereof and/or biologically active
metabolites thereof or
prodrugs thereof or solvates thereof or hydrates thereof and/or polymorphs
thereof.
The present invention provides a compound of formula (I), as defined above for
use as
medicament.
The present invention provides a compound of formula (I), as defined above for
use in
the treatment or prevention of diseases related to the GABAA a5 receptor.
The present invention provides the use of a compound of formula (I), as
defined above,
for the manufacture of a medicament for the treatment or prevention of
diseases related to the
GABAA a5 receptor.
The present invention provides a method of treating or preventing a disease
related to
the GABAA a5 receptor comprising administering to a subject, including humans,
in need of
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
9
such treatment or prevention an effective amount of at least one compound of
formula (I), as
defined above.
The present invention provides the combinational use of compounds of formula
(I) as
defined above, with one or more other active ingredients for the treatment or
prevention of
diseases related to the GABAA a5 receptor.
The present invention provides pharmaceutical compositions containing the
compound
of formula (I), as defined above as active ingredients.
The present invention provides medicaments (combinational pharmaceutical
compositions) comprising a combination of the compound of formula (I), as
defined above with
one or more other active ingredients.
The present invention provides pharmaceutical compositions containing the
compound
of formula (I), as defined above as active ingredients alone or in combination
with one or more
other active ingredients for use in the treatment or prevention of diseases
related to the GABAA
a5 receptor.
The present invention provides a process for the manufacture of the compounds
of
formula (I), as defined above and intemediates of the preparation process as
well.
The present invention also provides preparation of pharmaceutical compositions
containing the compounds of formula (I), as defined above alone, or in
combination with one
or more other active ingredients.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides compounds of formula (I) having affinity and
selectivity
for the alpha 5 subunit-containing gamma-aminobutyric acid A receptor (GABAA
a5 receptor)
and act as GABAA a5 receptor positive allosteric modulators, thereby useful in
the treatment
or prevention of diseases related to the GABAA a5 receptor, process for the
preparation
thereof, pharmaceutical compositions comprising them alone or in combination
with one or
more other active ingredients and their use as medicaments.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
The present invention relates to compounds of formula (I)
A OnciN 2
'X 'IR
N N
1
(I)
wherein
5 A is represented by
N
\
N "jr
µ1\1
WINN
group, or , group;
R1 is an alkyl, an alkoxy, or a haloalkyl group;
R2 is hydrogen; an alkyl group optionally substituted with -S(0)2-alkyl,
cycloalkyl or
heterocycle; a cycloalkyl group; a heterocycle group optionally substituted
with an alkyl; or a
10 heteroaryl group;
X is CH or N;
and/or salts thereof and/or stereoisomers thereof and/or enantiomers thereof
and/or
racemates thereof or diastereomers thereof and/or biologically active
metabolites thereof or
prodrugs thereof or solvates thereof or hydrates thereof and/or polymorphs
thereof.
Unless otherwise defined, all technical and scientific terms used herein have
the same
meaning as commonly understood by one of ordinary skill in the art to which
this invention
belongs. Although methods and materials similar or equivalent to those
described herein can
be used in the practice or testing of the invention, suitable methods and
materials are
described below.
The nomenclature used is based on IUPAC systematic nomenclature, unless
indicated
otherwise.
Any open valency appearing on a carbon, oxygen, sulfur or nitrogen atom in the
structures herein indicates the presence of a hydrogen, unless indicated
otherwise.
Definition of the general terms used herein, whether or not the terms in
question are
presented individually or in combination with other groups are described
below.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
11
"Optional" or "optionally" means that the subsequently described event or
circumstance may but need not occur, and that the description includes
instances where the
event or circumstance occurs and instances in which it does not.
The term "substituent" denotes an atom or a group of atoms replacing a
hydrogen atom
on the parent molecule.
The term "substituted" denotes that a specified group bears one or more
substituents.
Where any group may carry multiple substituents and a variety of possible
substituents
is provided, the substituents are independently selected and need not to be
the same.
The term "unsubstituted" means that the specified group bears no substituents.
The term "optionally substituted" means that any atom of the specified group
is
unsubstituted or substituted by one or more substituents, independently chosen
from the
group of possible substituents. When indicating the number of substituents,
the term "one or
more" means from one substituent to the highest possible number of
substitutions, i.e.,
replacement of one hydrogen up to replacement of all hydrogens by
substituents. The possible
substituents include, but are not limited to Ci_aalkyl, oxo and the like.
The term "alkyl" refers alone or in combination with other groups to a
straight or
branched, single or multiple branched, hydrocarbon radical and consists of 1
to 6 carbon
atoms. Preferably, an alkyl group consists of 1 to 4 carbon atoms. Examples
include, but are
not limited to methyl, ethyl, propyl, i-propyl (isopropyl), n-butyl, 2-butyl
(sec-butyl) or t-butyl
(tert-butyl) group. C1_2alkyl groups are more preferred. Methyl group is most
preferred.
The term "alkoxy" refers alone or in combination with other groups to -0-alkyl
group,
wherein the alkyl is as defined above. Preferably, an alkoxy group is a -0-
alkyl group wherein
the alkyl group consists of 1 to 4 carbon atoms. Examples include, but are not
limited to
methoxy, ethoxy, i-propoxy, n-propoxy or t-butoxy. C1_2alkoxy groups are more
preferred.
Methoxy group is most preferred.
The term "halogen", "halo" or "halide" refers alone or in combination with
other groups
to fluoro (fluorine), chloro (chlorine), bromo (bromine) or iodo (iodine).
Preferably, the halogen
is fluorine.
The term "haloalkyl" refers alone or in combination with other groups to an
alkyl as defined above substituted with one or more identical or different
halogens on any
carbon atoms of said alkyl, including vicinal and/or germinal halo-
substitutions as well, such
as perhaloalkyl groups. The term "perhaloalkyl" refers to an alkyl where all
hydrogen atoms
have been replaced by the same or different halogen atoms. Examples include,
but are not
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
12
limited to trihalo, dihalo-, or monohaloalkyl groups, for example 3,3,3-
trifluoropropyl, 2-
fluoroethyl, 2,2,2-trifluoroethyl, fluoromethyl, difluoromethyl or
trifluoromethyl. Preferably, the
haloalkyl group is a halo-C1_2alkyl group, more preferably difluoromethyl or
trifluoromethyl,
most preferably trifluoromethyl.
The term "cycloalkyl" refers to monovalent monocyclic saturated carbocyclic
groups
comprising 3 to 7 carbon ring atoms. Examples include cyclopropane,
cyclobutane,
cyclopentane, cyclohexane, cycloheptane. Preferably, the cycloalkyl group
comprises 4 to 6
carbon ring atoms. Most preferably the cycloalkyl is cyclobutane or
cyclopentane.
The term "heterocycle" refers alone or in combination with other groups to a
monovalent saturated or partly unsaturated monocyclic, bicyclic, fused,
bridged or spiro ring
system of 3 to 10 ring atoms comprising 1, 2, 3 or 4 ring heteroatoms
independently selected
from N, 0 and S, the remaining ring atoms being carbon. Examples for
monocyclic
heterocycles are aziridine, 2H-azirine, oxirane, thiirane, azetidine, oxetane,
thietane,
azetidine-2-one, pyrrolidine, pyrrolidinone, pyrroline, pyrazolidine,
imidazoline, pyrazoline,
tetrahydrofuran, dihydrofuran, dioxolane, tetrahydrothiophene, oxazolidine,
dihydro-oxazole,
isoxazolidine, oxathiolane, sulfolane, thiazolidine, thiazolidinedione,
succinimid, oxazolidone,
hydantoin, piperidine, piperidinone, piperazine, tetrahydropyran,
tetrahydrothiopyrane,
dihydropyrane, tetrahydropyridine, dioxane, thiane, dithiane, 1,1-dioxo-
thiane, morpholine,
thiomorpholine, 1,1-dioxo-thiomorpholin, azepane, diazepane, homopiperazine,
oxazepnayl
and the like. Preferably, the heterocycle refers alone or in combination with
other groups to a
monovalent saturated monocyclic ring of 3 to 7 ring atoms comprising 1, or 2
ring heteroatoms
independently selected from N, 0 and S, the remaining ring atoms being carbon.
More
preferably, the heterocycle refers alone or in combination with other groups
to a monovalent
saturated monocyclic ring of 3 to 7 ring atoms comprising one ring heteroatom
selected from
0 and S, the remaining ring atoms being carbon. Most preferably, the
heterocycle refers alone
or in combination with other groups to a monovalent saturated monocyclic ring
of 3 to 6 ring
atoms comprising one ring heteroatom selected from 0 and S, the remaining ring
atoms being
carbon such as oxetane, tetrahydrofuran, tetrahydrothiophene, tetrahydropyran.
The term "heteroaryl" refers alone or in combination with other groups to a
monovalent,
heterocyclic aromatic, mono- or bicyclic ring system of 5 to 10 ring atoms,
comprising 1, 2 or
3 heteroatoms independently selected from N, 0 and S, the remaining ring atoms
being
carbon. Examples for heteroaryl are pyrrole, furan, thiophene, imidazole,
oxazole, isoxazole,
thiazole, isothiazole, triazole, tetrazole, oxadiazole, thiadiazole,
tetrazole, pyridine, pyrazine,
pyrazole, pyridazine, pyrimidine, triazine, azepine, diazepine, benzofuran,
benzothiophene,
indole, isoindole, isobenzofuran, benzinnidazole, benzoxazole, benzoisoxazole,
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
13
benzothiazole, benzoisothiazole, benzooxadiazole, benzothiadiazole,
benzotriazole, purine,
quinoline, isoquinoline, quinazoline, quinoxaline, carbazole, or acridine.
Preferably, the
heteroaryl refers alone or in combination with other groups to a monovalent,
heterocyclic
aromatic, monocyclic ring system of 5 to 6 ring atoms, comprising 1, or 2
heteroatoms
independently selected from N, 0 and S, the remaining ring atoms being carbon.
More
preferably, the heteroaryl refers alone or in combination with other groups to
a monovalent,
heterocyclic aromatic, monocyclic ring system of 6 ring atoms, comprising 1,
or 2 heteroatoms
independently selected from N, 0 and S, the remaining ring atoms being carbon.
Most
preferably, the heteroaryl refers alone or in combination with other groups to
a monovalent,
heterocyclic aromatic, monocyclic ring system of 6 ring atoms, comprising 1,
or 2 heteroatoms
being N, the remaining ring atoms being carbon, such as pyridine, pyridazine,
pyrimidine,
pyrazine.
The terms "compound(s) of this invention", "compound(s) of the present
invention",
"compounds of formula (I), as defined above" refers to compounds of formula
(I) and/or salts
thereof and/or stereoisomers thereof and/or enantiomers thereof and/or
racemates thereof or
diastereomers thereof and/or biologically active metabolites thereof or
prodrugs thereof or
solvates thereof or hydrates thereof and/or polymorphs thereof.
The term "salt" refers to pharmaceutically acceptable or to pharmaceutically
non-
acceptable salts.
The term "pharmaceutically acceptable salt" refers to a conventional acid
addition or
base addition salt which preserves the biological efficacy and properties of
the compounds of
formula (I) and which can be formed with suitable non-toxic organic or
inorganic acids or
organic or inorganic bases. Examples of acid addition salts include salts
derived from
inorganic acids, such as, but not limited to hydrochloric acid, hydrobromic
acid, hydroiodic
acid, sulfuric acid, sulphamic acid, phosphoric acid, nitric acid and
perchloric acid and derived
from various organic acids, such as, but not limited to acetic acid, propionic
acid, benzoic acid,
glycolic acid, phenylacetic acid, salicylic acid, malonic acid, maleic acid,
oleic acid, pamoic
acid, palmitic acid, benzenesulfonic acid, toluenesulfonic acid,
methanesulfonic acid, oxalic
acid, tartaric acid, naphthalenedisulfonic acid, succinic acid, citric acid,
malic acid, lactic acid,
glutamic acid, fumaric acid and the like. Examples of base addition salts are
salts derived from
ammonium-, potassium-, sodium- and quaternary ammonium hydroxides such as
tetramethylammonium hydroxide.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
14
The "pharmaceutically non-acceptable salts" may be preferred for the
purification or
isolation of the compounds of formula (I) and are therefore also within the
scope of the
invention.
The term "prodrug" refers to derivatives of compounds of formula (I) according
to the
invention which themselves have no therapeutic effect but containing such
groups which, after
in vivo chemical or metabolic degradation (biotransformation) become
"biologically active
metabolite" which is responsible for the therapeutic effect.
Optical isomers can be prepared by resolving the racemic mixtures by known
methods,
for example, by using an optically active acid or base to form
diastereoisomeric salts or by
forming covalent diastereomers. Suitable acids include, for example, tartaric
acid,
diacetyltartaric acid, dibenzoyltartaric acid, ditoluoyltartaric acid and
camphorsulfonic acid.
Diastereoisomeric mixtures can be separated into individual diastereomers
based on their
physical and/or chemical differences, by methods known to those skilled in the
art, such as
chromatography or fractional crystallization. Subsequently, the optically
active bases or acids
are liberated from the separated diastereoisomeric salts. Various methods of
separating
optical isomers include chiral chromatography (e.g., chiral HPLC columns)
optionally used by
derivatization with the aim to maximize the separation of enantiomers.
Appropriate chiral
HPLC columns can be routinely chosen as desired. Where applicable, enzymatic
separations
carried out by derivatization may also be used. The optically active compounds
of general
formula (I) can also be prepared using optically active starting materials
using chiral synthesis
without racemization reaction conditions.
The absolute configuration of the chiral compounds can be determined e.g., by
optical
rotation, VCD (vibrational circular dichroism spectroscopy) and/or single
crystal X-ray
diffraction analysis, or 1H NMR spectroscopic assays of the diastereomeric
pair of compounds
synthesized from chiral compounds.
The compounds of formula (I) may exist in various polymorphic forms. As is
known in
the art, polymorphism is the ability of a compound to crystallize in more than
one crystalline
form, i.e., in polymorphic form. Polymorphic forms of a particular compound
can be defined
by identical chemical formula or composition and differ in their chemical
structure as crystalline
structures of two different chemical compounds.
The compounds of formula (I) and salts thereof may also be present as solvates
or
hydrates, which are also within the scope of the invention. The term "solvate"
refers to non-
covalent stoichiometric or nonstoichiometric combinations of solvent and
solute. The term
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
"hydrate" refers to non-covalent stoichiometric or nonstoichiometric
combinations of water and
solute.
The present invention provides pharmaceutical compositions comprising at least
one
compound of formula (I), as defined above as active ingredient.
5
The present invention provides pharmaceutical compositions comprising a
combination of the compound of formula (I), as defined above with one or more
other active
ingredients. The pharmaceutical composition may comprise at least one compound
of the
invention together with one or more other active ingredients in a single
dosage form or
separately. The combinational composition may be administered simultaneously,
separately
10 or sequentially.
The term "pharmaceutical composition" (or "composition") refers to a mixture
or
solution comprising a therapeutically effective amount of an active ingredient
together with
pharmaceutically acceptable excipients to be administered to a subject, e.g.,
a human in need
thereof.
15
The present invention also relates to the preparation of pharmaceutical
compositions.
The pharmaceutical compositions of the present invention may be formulated in
various pharmaceutical formulations, such as, but not limited to, solid oral
dosage forms such
as tablets (e.g., buccal, sublingual, effervescent, chewable, orally
dispersible), capsules, pills,
orally dispersible films, granules, powders; liquid formulations such as
solutions, emulsions,
suspensions, syrups, elixirs, drops; parenteral dosage forms such as
intravenous injections,
intramuscular injections, subcutaneous injections; other forms of medicine
such as eye drops,
semi-solid ophthalmic preparations, semi-solid dermal preparations (such as
ointments,
creams, pastes), transdermal therapeutic systems, suppositories, rectal
capsules, rectal
solutions, emulsions and suspensions, etc..
The pharmaceutical compositions of the present invention may be administered
in
various ways, such as, but not limited to oral, rectal, mucous, transdermal or
intestinal
administration, parenteral administration including intramuscular,
subcutaneous, intravenous,
intramedullary injections as well as intraarticular, intrathecal, direct
intraventricular,
intraperitoneal, intranasal or intraocular injections and eye drops.
Alternatively, the compounds may be administered locally and not systemically,
for
example by direct injection of the compound to the kidney or the heart, often
in a modified
release formulation. In addition, the drug may be administered in a targeted
carrier system,
for example in a tissue-specific antibody encapsulated liposome.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
16
The pharmaceutical composition may be administered in various ways and in
various
pharmaceutical forms. The compound of the invention may be administered alone
or in
combination with pharmaceutically acceptable excipients, in single or multiple
doses.
For simple administration, it is preferred that the pharmaceutical
compositions consist
of dosage units that contain the amount of active ingredient(s) to be
administered once, or a
small number of multiple, or half, one third, a quarter. Such dosage units
are, for example,
tablets that can be provided with a half or quarter groove to facilitate half
or quarter-splitting
of the tablet in order to weigh the required amount of active ingredient(s).
Pharmaceutical compositions containing the active ingredient(s) according to
the
invention generally contain from 0.001 to 500 mg of active ingredient(s) per
dosage unit. It is
of course also possible that the amount of active ingredient(s) in each
formulation exceeds
the above limit either up or down.
The present invention relates also to pharmaceutical compositions for use in
pediatric
use such as, but not limited to, solutions, syrups, elixirs, suspensions,
powders for the
preparation of suspensions, dispersible or effervescent tablets, chewable
tablets, orally
disintegrating tablets or granules, tablets or coated tablets, sparkling
powders or granules,
capsules.
The pharmaceutical compositions of the present invention may be prepared by
methods known per se such as conventional mixing, dissolution, emulsification,
suspending,
microencapsulation, freeze drying, extrusion and spheronization, lamination,
film coating,
granulation, encapsulation, pelletization or pressing.
The pharmaceutical compositions of the present invention may be formulated in
the
usual way using one or more physiologically or pharmaceutically acceptable
excipients which
promote the incorporation of the active ingredient into pharmaceutically
acceptable
pharmaceutical forms. The term "physiologically or pharmaceutically acceptable
excipient"
denotes any ingredient used in formulating pharmaceutical products which have
no
therapeutic activity and non-toxic. The proper formulation depends on the mode
of
administration chosen. Any of the techniques and excipients well known in the
art can be used.
The excipients applicable in the preparation may be selected from the
following
categories, such as, but not limited to fillers of tablets and capsules,
binders of tablets and
capsules, drug release modifying agents, disintegrants, glidants, lubricants,
sweeteners,
taste-masking agents, flavorants, coating materials, surfactants, stabilizers,
preservatives or
antioxidants, buffering agents, complexing agents, wetting or emulsifying
agents, salts for
adjusting the osmotic pressure, lyophilization excipients, microencapsulating
agents, ointment
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
17
materials, penetration enhancers, solubilizers, solvents, suppository
materials, suspending
agents.
The excipients described above and the various methods of preparation are only
representative examples. Other materials and process techniques known in the
art may also
be used.
The term "other active ingredient" refers to therapeutic agents including, but
not limited
to 5-HT1A antagonists or agonists (such as lecozotan, NLX 101, sarizotan); 5-
HT1B and 5-H-r1p
agonists (such as rizatriptan, zolmitriptan, naratriptan and sumatriptan); 5-
HT2 antagonists; 5-
HT4 agonists (such as PRX-03140); 5-HT6 antagonists (such as GSK 742467, SGS-
518, FK-
962, SL-65.0155, SRA-333 and xaliproden); A2a adenosine receptor antagonists;
acetylcholinesterase inhibitors (such as galantamine, rivastigmine, donepezil,
tacrine,
phenserine, ladostigil and ABT-089); ADAM-10 ligands; alpha adrenoceptor
agonists; AMPA
agonists or modulators (such as CX-717, LY 451395, LY404187 and S-18986);
androgen
receptor modulators (such as SFX 01); anti-amyloid antibodies including anti-
amyloid
humanized monoclonal antibodies (such as bapineuzumab, ACC001, CAD 106,
AZD3102,
H12A11V1); anticholinergics (such as biperiden); anticonvulsants (such as
acetazolamide,
carbamazepine, eslicarbazepine acetate, ethosuximide, lacosamide, nitrazepam,
oxcarbazepine, perampanel, phenobarbital, phenytoin, prim idone, rufinamide,
stiripentol,
topiramate, valproate); anti-inflammatory compounds (such as (R)-flurbiprofen,
nitroflurbiprofen, ND-1251, VP-025, HT-0712, and EHT-202); ApoE4 conformation
modulators; atypical antipsychotics (such as aripiprazole, asenapine,
brexpiprazole,
brilaroxazine, cariprazine, iloperidone, loxapine, lumateperone tosylate,
lurasidone
hydrochloride, molindone, olanzapine, paliperidone, quetiapine, risperidone,
sulpiride and
ziprasidone); barbiturates; beta- (such as verubecestat, and AZD3293) and
gamma-secretase
inhibitors (such as LY450139 and TAK 070) or modulators; blockers of Ap
oligomer formation;
bradykinin B1 receptor antagonists (such as SSR240612, NVPSAA164 or any of
those
compounds described in WO 2007/072092 A2, WO 2008/068540 Al, WO 2008/050167
Al,
WO 2008/050168 Al); butyrophenone (such as haloperidol); calcium channel
blockers (such
as ziconotide and NMED160); CB-1 receptor antagonists or inverse agonists
(such as
drinabant, cannabidiol); CB-2 agonists (such as GW-842166X and SAB378) or CB
modulators
(cannabidivarin, TI/C20, tetrahydrocannabinol conjugate, ZYN-002); cholinergic
agonist;
phenothiazines (such as chlorpromazine, fluphenazine, mesoridazine,
perphenazine,
thioridazine, trifluoperazine); thioxanthenes (such as chlorprothixene and
thiothixene); COMT
inhibitors (such as entacapone); cyclopyrrolones; diphenylbutylpiperidine
(such as pimozide)
and indolone (such as molindolone) classes of neuroleptic agents; DNA-directed
DNA
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
18
polymerase inhibitors (such as suramin sodium); dopamine agonists and partial
agonists
(such as pramipexole, ropinirole); dopamine precursors (such as carbidopa,
levodopa);
dopamine transport inhibitors; enzyme modulators or replacements (such as CM-
AT, CM-
4612 and CM-182); fatty acid amide hydrolase inhibitors (such as JNJ
42165279); fatty acid
or triglyceride replacements (such as triheptanoin); fenamate compounds (such
as ASD-002);
GABAA blockers (such as S44819, NGD 97-1, a5IA, a51A-II, MRK-016, basmisanil
or any
those compounds described in PCT/IB2019/058208); GABAA receptor agonists (such
as
acamprosate); GABAA signaling enhancers (such as AZD-7325, PF-06372865, L-
838,417,
TPA-023, brexanolone, zuranolone, alphaxalone, ganaxolone, gaboxadol,
tiagabine,
vigabatrine, bunnetanide); GABAB receptor agonists (such as arbaclofen or any
of those
compounds described in WO 2018/167629 Al or WO 2018/167630 Al); gabapentinoids
(such
as pregabalin, gabapentin); glutamate modulators (such as AMO 04); glycine
transport
inhibitors; glycogen synthase kinase 3 beta inhibitors (such as tideglusib,
AZD1080,
SAR502250 and CEP16805); growth hormone secretagogues (such as ibutamoren,
ibutamoren mesylate, and capromorelin); HDAC inhibitors; heterocyclic
dibenzazepines (such
as clozapine); histamine H3 receptor antagonists and inverse agonists (such as
S38093, ABT-
834, ABT 829, GSK 189254, CEP16795 or any of those compounds described in WO
2014/136075 Al); HMG-CoA reductase inhibitors; imidazopyridines (such as
zolpidem);
immunomodulators (such as IMM-124E); KCNQ antagonists; lithium; LRRK2
inhibitors; LXR
p agonists; lysine specific demethylase 1 inhibitors (such as vafidemstat); M1
or M4 mAChR
agonists or PAMs; MARK ligands; melatonergic agents; melatonin agonists and
antagonists;
methyl-CpG binding protein 2 (MECP2) gene replacement therapy (such as AVXS
201);
mGluR2 antagonists or modulators; mGluR4 positive allosteric modulators (such
as ADX-
88178, foliglurax); mGluR5 antagonists (such as HTL-14242, AZD9272,
mavoglurant);
microbiome modulators (such as AB-2004, CP-101, SB-121); minor tranquilizers;
MMP
inhibitors; a7 nAChR agonists or positive allosteric modulators (such as ABT-
126, AZD0328,
EVP-6124, AVL-3288, PNU-120596 or any of those compounds described in WO
2020/012422 Al, WO 2020/012423 Al or WO 2020/012424 Al) or antagonist (such as
mecamylamine hydrochloride); neuropeptide receptor modulators (such as
trofinetide,
davunetide, NNZ-2591); neutrophil inhibitory factor; NK1/NK3 receptor
antagonists; NMDA
receptor agonists or antagonists (such as memantine, neramexane, EVT101,
AZD4282, BHV
5000); noradrenaline transport inhibitors; norepinephrine modulators; NOS
inhibitors (such as
SD6010 and 274150); NQ01 modulators (such as vatiquinone); NR2B antagonists
(such as
radiprodil); NSAIDs (such as ibuprofen); opioid analgesics (such as codeine,
fentanyl,
hydromorphone, levorphanol, meperidine, methadone, morphine, oxycodone,
oxymorphone,
pentazocine, propoxyphene); orexin antagonists and agonists; oxytocin;
p25/CDK5 inhibitors;
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
19
PDE10 inhibitors; PDE4 inhibitors (such as HT0712); PDE9 inhibitors (such as
BI40936);
PI3KB inhibitors (such as BBP-472); potassium channel openers; PPAR gamma
agonists
(such as pioglitazone and rosiglitazone); prokineticin agonists and
antagonists;
pyrazolopyrimidines; pyrrolidone compounds modulating cholinergic/metabotropic
glutamate
receptors (such as fasoracetam, levetiracetam, brivaracetam, piracetam); sigma-
1 receptor
agonists (such as blarcamesine); sodium channel blockers and antagonists (such
as
lamotrigine, VX409 and SPI860); sphingosine 1 phosphate receptor modulators
(such as
fingolimod, ozanimod, siponimod, ponesimod); SSRIs or SNRIs (such as
fluoxetine,
citalopram, escitalopram, fluvoxamine, paroxetine, sertraline; or
desvenlafaxine, duloxetine,
venlafaxine); sulfonamides (such as zonisannide); tau phosphorylation
inhibitors; thronnbolytic
agents; triazolopyridines; benzodiazepines; tricyclic antidepressant drugs; T-
type calcium
channel antagonists; tyrosine hydroxylase inhibitors (such as L1-79);
vasopressin; Via
receptor antagonists (such as balovaptan, BTRX-323511 or any of those
compounds
described in WO 2019/116324 Al or WO 2019/116325 Al); vitamin E; VR-1
antagonists (such
as AMG517, 705498, 782443, PAC20030, VI 14380 and A425619) or other drugs that
affect
receptors or enzymes that either increase the efficacy, safety, convenience,
or reduce
unwanted side effects or toxicity of the compounds of the present invention.
In one embodiment, the other active ingredient refers to 5-HT1A antagonists or
agonists
(such as lecozotan, NLX 101, sarizotan); atypical antipsychotics (such as
aripiprazole,
asenapine, brexpiprazole, brilaroxazine, cariprazine, iloperidone, loxapine,
lumateperone
tosylate, lurasidone hydrochloride, molindone, olanzapine, paliperidone,
quetiapine,
risperidone, sulpiride and ziprasidone); CB-1 receptor antagonists or inverse
agonists (such
as drinabant, cannabidiol); CB-2 agonists (such as GW-842166X and SAB378) or
CB
modulators (cannabidivarin, T1 /C20, tetrahydrocannabinol conjugate, ZYN-002);
DNA-
directed DNA polymerase inhibitors (such as Suramin sodium); fatty acid amide
hydrolase
inhibitors (such as JNJ 42165279); fatty acid or triglyceride replacements
(such as
triheptanoin); GABAA receptor agonists (such as acamprosate); GABAA signaling
enhancers
(such as AZD-7325, PF-06372865, L-838,417, TPA-023, brexanolone, zuranolone,
alphaxalone, ganaxolone, gaboxadol, tiagabine, vigabatrine, bumetanide); GABAB
receptor
agonists (such as arbaclofen or any of those compounds described in WO
2018/167629 Al
or WO 2018/167630 Al); glutamate modulators (such as AMO 04); glycogen
synthase kinase
3 beta inhibitors (such as tideglusib, AZD1080, SAR502250 and 0EP16805);
lysine specific
demethylase 1 inhibitors (such as vafidemstat); methyl-CpG binding protein 2
(MECP2) gene
replacement therapy (such as AVXS 201); microbiome modulators (such as AB-
2004, CP-
101, SB-121); neuropeptide receptor modulators (such as trofinetide,
davunetide, NNZ-2591);
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
NMDA receptor agonists or antagonists (such as nnemantine, neramexane, EVT101,
AZD4282, BHV 5000); NQ01 modulators (such as vatiquinone); oxytocin;
pyrrolidone
compounds modulating cholinergic/metabotropic glutamate receptors (such as
fasoracetam,
levetiracetam, brivaracetam, piracetam); sigma-1 receptor agonists (such as
blarcamesine);
5 sphingosine 1 phosphate receptor modulators (such as fingolimod,
ozanimod, siponimod,
ponesimod); SSRIs or SNRIs (such as fluoxetine, citalopram, escitalopram,
fluvoxamine,
paroxetine, sertraline; or desvenlafaxine, duloxetine, venlafaxine); tyrosine
hydroxylase
inhibitors (such as L1-79) vasopressin; or Via receptor antagonists (such as
balovaptan,
BTRX-323511 or any of those compounds described in WO 2019/116324 Al or WO
10 2019/116325A1).
The term "modulators" refers to molecules interacting with the target
receptor, wherein
the interaction can be e.g., agonistic, antagonistic or inverse agonistic.
The term "inhibitors" referes to molecules competing with, reducing or
preventing the
binding of a particular ligand to a particular receptor or reducing or
preventing the inhibition of
15 the function of a particular protein.
The term "agonists" refers to compounds having affinity to a receptor binding
site and
enhancing the activity of the receptor-mediated response. "Full-agonists"
effect a full
response, "partial agonists" effects less than full activation even when
occupying the total
receptor population.
20 The term "inverse agonists" refers to compounds producing an effect
opposite to that
of an agonist by binding to the same agonist binding site, or reducing the
effect of an agonist
by binding at a different allosteric binding site.
The term "antagonists" refers to compounds diminishing or preventing the
action of
another compound or receptor site, or attenuating the effect of an agonist.
"Competitive
antagonists" bind to the same site as the agonist but does not activate it,
thus blocks the
agonists' action. "Non-competitive antagonists" binds to an allosteric site on
the receptor to
prevent activation of the receptor. Binding of "reversible antagonists" to a
receptor is non-
covalent (can be washed out), while binding of "irreversible antagonists" is
covalent (cannot
be washed out).
The term "allosteric modulators" refers to compounds binding to a receptor at
a site
distinct from the agonist binding site, i.e., to the allosteric site, wherein
by inducing
conformational change in the receptor, alter the affinity and/or activity of
the receptor for the
endogenous ligand or agonist. "Positive allosteric modulators" or "PAMs"
increase the affinity
and/or activity, whilst "negative allosteric modulators" or "NAMs" decrease
the affinity and/or
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
21
activity of a receptor. The compounds of formula (I), as defined above are
positive allosteric
modulators.
The term "inhibition constant" (K) refers to the absolute binding affinity of
a particular
inhibitor to a receptor. It is measured using competition binding assays and
is calculated from
the concentration where the particular inhibitor would occupy half of the
receptors (IC5o) if no
competing ligand was present using the Cheng Prusoff relationship: K =
IC50/[1+([11/KD)],
where EL] is the radioligand concentration and KD the affinity of the labeled
ligand for the
receptor binding site. K values can be converted logarithmically to pki values
(-logKi) in which
higher values indicate exponentially greater potency.
The term "submaximal effective concentration" refers to the concentration of a
particular compound required for obtaining 10% of the maximum of a particular
effect.
The terms "condition", "defect", "deficit", "disability", "disorder, "disease"
or "disease
state" are used interchangeably to denote any disease, condition, symptom,
syndrome,
disorder or indication.
The term "disease related to the GABAA a5 receptor refers to a disease,
condition or
disorder of the central nervous system where one of the symptoms and/or
syndromes of the
disease may be related to the GABAA a5 receptor. Such a disease includes, but
not limited to
a neurodevelopmental disorder, a neurodegenerative disorder, a neurocognitive
disorder,
schizophrenia, a mood disorder, a pain disorder, a substance-related and
addictive disorder
or other diseases.
The diseases related to the GABAA a5 receptor may show comorbidity with each
other.
Comorbidity indicates a medical condition existing simultaneously but
independently with
another condition in a patient, or a medical condition in a patient that
causes, is caused by, or
is otherwise related to another condition in the same patient. However, in
psychiatric,
psychologic, or mental health diseases comorbidity does not necessarily imply
the presence
of multiple diseases, but instead can reflect our current inability to supply
a single diagnosis
that accounts for all symptoms.
The term "neurodevelopmental disorder" includes, but not limited to autism
spectrum
disorder (ASD), Angelman syndrome, Fragile X disorder, Prader-Willi syndrome,
Rett
syndrome or Down syndrome.
The term "neurodegenerative disorder" includes, but not limited to Alzheimer's
disease
(AD), Huntington's disease (HD), Parkinson's disease (PD), or amyotrophic
lateral sclerosis
(ALS).
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
22
The term "neurocognitive disorder" includes, but not limited to cognition
deficiency
disorders, memory deficits, age-associated memory impairment or cognitive
decline,
dementia (or different forms thereof such as dementia in Alzheimer's disease,
Niemann Pick-
disease, Parkinson's disease, or Huntington's disease, dementia with Lewy
bodies (DLB),
frontotemporal dementia, vascular dementia (VaD), subcortical dementia, mixed
vascular and
subcortical dementia, multi-infarct dementia, post-operative dementia, or
inflammation-
induced dementia), Alzheimer's disease related neuropsychiatric symptoms, mild
cognitive
impairment (MCI), vascular cognitive impairment (VC!), CNS conditions
occurring after stroke,
cognitive impairment associated with brain cancers (including, but not limited
to
nnedulloblastonnas), cognitive decline in Down Syndrome (DS), cognitive
dysfunction in major
depressive disorder (MDD) or HIV-Associated neurocognitive disorder. The term
"schizophrenia" includes, but not limited to, different forms of
schizophrenia, positive, negative
and/or cognitive symptoms associated with schizophrenia, schizotypal and
delusional
disorders.
The term "pain disorder" includes, but not limited to nociceptive, neuropathic
or
inflammatory pain.
The term "mood disorder" includes, but not limited to depression-related
disorders
(such as major depressive disorder (MDD), dysthymia, cyclothymic disorder,
seasonal
affective disorder/seasonal depression, depression after traumatic brain
injury (TBI),
postpartum depression, premenstrual dysphoric disorder, depressive symptoms
associated
with menopause, depression following substance abuse/withdrawal, bipolar
disorders (bipolar
disorder in remission, or depressive episodes of bipolar disorder), substance
(alcohol or drug)
induced, or not otherwise specified mood disorders (MD-NOS).
The term "other disease" includes, but not limited to attention deficit
hyperactivity
disorder and adult attention deficiency, other stress related conditions,
stroke,
neurofibromatosis type I, multiple sclerosis, acute meningitis, alcohol use
disorder, fetal
alcohol spectrum disorder, bronchoconstrictive diseases (such as asthma,
chronic obstructive
pulmonary disease, and bronchopulmonary dysplasia) or obesity.
In one embodiment, the disease related to the GABAA a5 receptor refers to
autism
spectrum disorder (ASD); Angelman syndrome, Fragile X disorder, Prader-Willi
syndrome,
Rett syndrome, Down syndrome, Alzheimer's disease (AD), Huntington's disease
(HD),
Parkinson's disease, amyotrophic lateral sclerosis (ALS), cognition deficiency
disorders,
memory deficits, age-associated memory impairment or cognitive decline,
dementia or
different forms thereof such as dementia in Alzheimer's disease, Niemann Pick-
disease,
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
23
Parkinson's disease, or Huntington's disease, dementia with Lewy bodies (DLB),
frontotemporal dementia, vascular dementia (VaD), subcortical dementia, mixed
vascular and
subcortical dementia, multi-infarct dementia, post-operative dementia, or
inflammation-
induced dementia), Alzheimer's disease related neuropsychiatric symptoms, mild
cognitive
impairment (MCI), vascular cognitive impairment (VCI), CNS conditions
occurring after stroke,
cognitive impairment associated with brain cancers (including but not limited
to
medulloblastomas), cognitive decline in Down Syndrome (DS), cognitive
dysfunction in major
depressive disorder (M DD), HIV-Associated neurocognitive disorder; different
forms of
schizophrenia, positive, negative and/or cognitive symptoms associated with
schizophrenia,
schizotypal and delusional disorders; nociceptive, neuropathic or inflammatory
pain;
depression-related disorders (such as major depressive disorder (MDD),
dysthymia,
cyclothymic disorder, seasonal affective disorder/seasonal depression,
depression after
traumatic brain injury (TB!), postpartum depression, premenstrual dysphoric
disorder,
depressive symptoms associated with menopause, depression following substance
abuse/withdrawal, bipolar disorders (bipolar disorder in remission, or
depressive episodes of
bipolar disorder), substance (alcohol or drug) induced, not otherwise
specified mood disorders
(MD-NOS); attention deficit hyperactivity disorder and adult attention
deficiency, other stress
related conditions, stroke, neurofibromatosis type I, multiple sclerosis,
acute meningitis,
alcohol use disorder, fetal alcohol spectrum disorder, bronchoconstrictive
diseases (such as
asthma, chronic obstructive pulmonary disease, and bronchopulmonary dysplasia)
or obesity.
In a preferred embodiment, the disease related to the GABAA a5 receptor refers
to
autism spectrum disorder (ASD), Angelman syndrome, Fragile X disorder, Prader-
Willi
syndrome, Rett syndrome, Alzheimer's disease (AD), cognition deficiency
disorders, memory
deficits, age-associated memory impairment or cognitive decline, dementia,
mild cognitive
impairment (MCI), bipolar disorders, negative and/or cognitive symptoms
associated with
schizophrenia, epilepsy, post-traumatic stress disorder, amyotrophic lateral
sclerosis.
The present invention provides a method of treating or preventing a disease
related to
the GABAA a5 receptor comprising administering to a subject, preferably a
mammal, more
preferably a human being, in need of such treatment or prevention,
therapeutically effective
amount of a compound of formula (I), as defined above alone or with at least
one
pharmaceutically acceptable excipient in the form of a pharmaceutical
formulation.
The present invention provides a method of treating or preventing a disease
related to
the GABAA a5 receptor comprising administering to a subject, preferably a
mammal, more
preferably a human being, in need of such treatment or prevention,
therapeutically effective
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
24
amount of a compound of formula (I), as defined above in combination with one
or more other
active ingredients.
The present invention provides a method of treating or preventing of a
neurodevelopmental disorder, neurodegenerative disorder, neurocognitive
disorder,
schizophrenia, a mood disorder, a pain disorder, a substance-related and
addictive disorder
or other disease, or at least one of the symptoms and/or syndromes thereof,
where one of the
symptoms and/or syndromes of the disease may be related to the GABAA a5
receptor, in a
subject, preferably a mammal, more preferably a human being, suffering
therefrom. This
method of treatment comprises administering to a subject, preferably a mammal,
more
preferably a human being, in need of such treatment or prevention,
therapeutically effective
amount of the compound of formula (I), as defined above. The method of
treatment may
include administering to a subject preferably a mammal, more preferably a
human being, in
need of such treatment therapeutically effective amount of a pharmaceutical
composition
comprising the compound of formula (I), as defined above.
The present invention provides a method of treating or preventing autism
spectrum
disorder (ASD), Angelman syndrome, Fragile X disorder, Prader-Willi syndrome,
Rett
syndrome, Alzheimer's disease (AD), cognition deficiency disorders, memory
deficits, age-
associated memory impairment or cognitive decline, dementia, mild cognitive
impairment
(MCI), bipolar disorders, negative and/or cognitive symptoms associated with
schizophrenia,
epilepsy, post-traumatic stress disorder, amyotrophic lateral sclerosis, or at
least one of the
symptoms and/or syndromes thereof, in a subject, preferably a mammal, more
preferably a
human being, suffering therefrom comprising administering a therapeutically
effective amount
of the compound of formula (I), as defined above.
The present invention provides the compound of formula (I), as defined above
for use
in the treatment or prevention of diseases related to the GABAA a5 receptor.
The present invention provides the compound of formula (I), as defined above
in
combination with one or more other active ingredients for use in the treatment
or prevention
of diseases related to the GABAA a5 receptor.
The present invention provides the compound of formula (I), as defined above
for use
in the treatment or prevention of a neurodevelopmental disorder, a
neurodegenerative
disorder, a neurocognitive disorder, schizophrenia, a mood disorder, a pain
disorder, a
substance-related and addictive disorder or other disease, or at least one of
the symptoms
and/or syndromes thereof.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
The present invention provides the compound of formula (I), as defined above
for use
in the treatment or prevention of autism spectrum disorder (ASD), Angelman
syndrome,
Fragile X disorder, Prader-Willi syndrome, Rett syndrome, Alzheimer's disease
(AD), cognition
deficiency disorders, memory deficits, age-associated memory impairment or
cognitive
5 decline, dementia, mild cognitive impairment (MCI), bipolar disorders,
negative and/or
cognitive symptoms associated with schizophrenia, epilepsy, post-traumatic
stress disorder,
amyotrophic lateral sclerosis, or at least one of the symptoms and/or
syndromes thereof.
The present invention provides the use of the compound of formula (I), as
defined
above for the manufacture of a medicament for the treatment or prevention of
diseases related
10 to the GABAA a5 receptor.
The present invention provides the use of the compound of formula (I), as
defined
above in combination with one or more other active ingredients, for the
manufacture of a
medicament for the treatment or prevention of diseases related to the GABAA a5
receptor.
The present invention provides the use of the compound of formula (I), as
defined
15 above for the manufacture of a medicament for the treatment or
prevention of a
neurodevelopmental disorder, a neurodegenerative disorder, a neurocognitive
disorder,
schizophrenia, a mood disorder, a pain disorder, a substance-related and
addictive disorders
or other disease, or at least one of the symptoms and/or syndromes thereof.
The present invention provides the use of the compound of formula (I), as
defined
20 above for the manufacture of a medicament for the treatment or
prevention of autism spectrum
disorder (ASD), Angelman syndrome, Fragile X disorder, Prader-Willi syndrome,
Rett
syndrome, Alzheimer's disease (AD), cognition deficiency disorders, memory
deficits, age-
associated memory impairment or cognitive decline, dementia, mild cognitive
impairment
(MCI), bipolar disorders, negative and/or cognitive symptoms associated with
schizophrenia,
25 epilepsy, post-traumatic stress disorder, amyotrophic lateral sclerosis,
or at least one of the
symptoms and/or syndromes thereof.
The present invention also relates to pharmaceutical composition comprising
the
compound of formula (I), as defined above for use in the treatment or
prevention of diseases
related to the GABAA a5 receptor.
The present invention also relates to pharmaceutical composition comprising
the
compound of formula (I), as defined above with one or more other active
ingredients for use
in the treatment or prevention of diseases related to the GABAA a5 receptor.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
26
The term "treatment" refers to the alleviation of a specific pathological
condition, the
elimination or reduction of one or more of the symptoms of the condition, the
slowing or
elimination of the progression of the disease state, and the prevention or
delay of recurrency
of the pathological condition of a patient or subject already suffering from
or diagnosed with
the disease. The "prevention" (or prophylaxis or delay of action of the
disease) is typically
performed by administering the drug in the same or similar way as if it were
given to a patient
with a disease or condition already developed.
The term "therapeutically effective amount" refers to the amount of active
ingredient - in
comparison with the corresponding subject who did not receive such amount -
which results
in the treatment, cure, prevention or improvement of the disease or disease
state or side
effect, and reduces the progression of the disease or pathological condition.
The term also
includes effective amounts to enhance normal physiological function. For use
in therapy the
compound of formula (I), as defined above as well as any salts thereof and/or
salts thereof
and/or stereoisomers thereof and/or enantiomers thereof and/or racemates
thereof or
diastereomers thereof and/or biologically active metabolites thereof or
prodrugs thereof or
solvates thereof or hydrates thereof and/or polymorphs thereof may be
administered in a
therapeutically effective amount as a raw chemical. In addition, the active
ingredient is
available as a pharmaceutical formulation.
The term "subject" refers to a vertebrate. In certain embodiments, the
vertebrate is a
mammal. Mammals include humans, non-human primates such as chimpanzees and
other
apes and monkey species, farm animals such as cattle, horses, sheep, goats,
and swine,
domestic animals such as rabbits, dogs, and cats, laboratory animals including
rodents, such
as rats, mice, and guinea pigs. In certain embodiments, a mammal is a human.
The term
subject does not denote a particular age or sex.
In one embodiment, the present invention relates to compounds of formula (I')
A a2
b2 0N ====(0 2
'')(
X N
R1
(r)
wherein
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
27
A is represented by
N%al --5"
N
'N21
bl Art` bl Awn
group, or group;
wherein site "al" of any ring A is attached to site "a2" and wherein site "bl"
of any ring A is
attached to site "b2"; R1, R2 and X are as defined above for the compounds of
formula (I)
and/or salts thereof and/or stereoisomers thereof and/or enantiomers thereof
and/or
racemates thereof or diastereomers thereof and/or biologically active
metabolites thereof or
prodrugs thereof or solvates thereof or hydrates thereof and/or polymorphs
thereof.
In one embodiment, the present invention relates to compounds of formula (I-a)
0
\
0
2
R
/
N
R
N
(I-a)
wherein R1, R2 and X are as defined above for the compounds of formula (I)
and/or salts
thereof and/or stereoisomers thereof and/or enantiomers thereof and/or
racemates thereof or
diastereomers thereof and/or biologically active metabolites thereof or
prodrugs thereof or
solvates thereof or hydrates thereof and/or polymorphs thereof.
In one embodiment, the present invention relates to compounds of formula (I-b)
N/
iN
N
01--1
(I-b)
wherein R1, R2 and X are as defined above for the compounds of formula (I)
and/or salts
thereof and/or stereoisomers thereof and/or enantiomers thereof and/or
racemates thereof or
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
28
diastereomers thereof and/or biologically active metabolites thereof or
prodrugs thereof or
solvates thereof or hydrates thereof and/or polymorphs thereof.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R1 is a C1_6alkyl, a C1_6alkoxy, or a halo-C1_6alkyl group.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R2 is hydrogen; a C1_6alkyl group optionally substituted with -S(0)2-
C1_6alkyl, C3_7cycloalkyl or
a monovalent saturated or partly unsaturated monocyclic, bicyclic, fused,
bridged or spiro ring
system of 3 to 10 ring atoms comprising 1, 2, 3 or 4 ring heteroatoms
independently selected
from N, 0 and S, the remaining ring atoms being carbon; a C3_7cycloalkyl
group; a monovalent
saturated or partly unsaturated monocyclic, bicyclic, fused, bridged or spiro
ring system of 3
to 10 ring atoms comprising 1, 2, 3 or 4 ring heteroatoms independently
selected from N, 0
and S, the remaining ring atoms being carbon optionally substituted with a
C1_6alkyl; or a
monovalent, heterocyclic aromatic, mono- or bicyclic ring system of 5 to 10
ring atoms,
comprising 1, 2 or 3 heteroatoms independently selected from N, 0 and S, the
remaining ring
atoms being carbon.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R1 is a Ci_6alkyl, a Ci_6alkoxy, or a halo-Ci_6alkyl group;
R2 is hydrogen; a Ci_ealkyl group optionally substituted with -S(0)2-
Ci_ealkyl, C3_7cycloalkyl or
a monovalent saturated or partly unsaturated monocyclic, bicyclic, fused,
bridged or spiro ring
system of 3 to 10 ring atoms comprising 1, 2, 3 or 4 ring heteroatoms
independently selected
from N, 0 and S, the remaining ring atoms being carbon; a C3_7cycloalkyl
group; a monovalent
saturated or partly unsaturated monocyclic, bicyclic, fused, bridged or spiro
ring system of 3
to 10 ring atoms comprising 1, 2, 3 or 4 ring heteroatoms independently
selected from N, 0
and S, the remaining ring atoms being carbon optionally substituted with a
C1_6alkyl; or a
monovalent, heterocyclic aromatic, mono- or bicyclic ring system of 5 to 10
ring atoms,
comprising 1, 2 or 3 heteroatoms independently selected from N, 0 and S, the
remaining ring
atoms being carbon.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R1 is a C1_4alkyl, a Ci_aalkoxy, or a halo-C1_4alkyl group.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
29
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R2 is hydrogen; a Ci_aalkyl group optionally substituted with -S(0)2-
Ci_4alkyl, a Ca_scycloalkyl
or a monovalent saturated monocyclic ring of 3 to 7 ring atoms comprising 1,
or 2 ring
heteroatoms independently selected from N, 0 and S, the remaining ring atoms
being carbon;
a C4_6cycloalkyl group; a monovalent saturated monocyclic ring of 3 to 7 ring
atoms comprising
1, or 2 ring heteroatoms independently selected from N, 0 and S, the remaining
ring atoms
being carbon optionally substituted with a Ci_aalkyl; or a monovalent,
heterocyclic aromatic,
monocyclic ring system of 5 to 6 ring atoms, comprising 1, or 2 heteroatoms
independently
selected from N, 0 and S, the remaining ring atoms being carbon.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R1 is a C1_4alkyl, a Cl_aalkoxy, or a halo-C1_4alkyl group;
R2 is hydrogen; a Ci_aalkyl group optionally substituted with -S(0)2-
C1_4alkyl, a Ca_Bcycloalkyl
or a monovalent saturated monocyclic ring of 3 to 7 ring atoms comprising 1,
or 2 ring
heteroatoms independently selected from N, 0 and S, the remaining ring atoms
being carbon;
a C4_6cycloalkyl group; a monovalent saturated monocyclic ring of 3 to 7 ring
atoms comprising
1, or 2 ring heteroatoms independently selected from N, 0 and S, the remaining
ring atoms
being carbon optionally substituted with a Cl_aalkyl; or a monovalent,
heterocyclic aromatic,
monocyclic ring system of 5 to 6 ring atoms, comprising 1, or 2 heteroatoms
independently
selected from N, 0 and S, the remaining ring atoms being carbon.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R1 is a Ci_2alkyl, a Ci_2alkoxy, or a halo-C1_2alkyl group.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R2 is hydrogen; a Ci_aalkyl group optionally substituted with -S(0)2-
Ci_2alkyl, C4_6cycloalkyl or
a a monovalent saturated monocyclic ring of 3 to 7 ring atoms comprising one
ring heteroatom
selected from 0 and S, the remaining ring atoms being carbon; a C4_6cycloalkyl
group; a
monovalent saturated monocyclic ring of 3 to 7 ring atoms comprising one ring
heteroatom
selected from 0 and S, the remaining ring atoms being carbon optionally
substituted with a
Ci_aalkyl; or a monovalent, heterocyclic aromatic, monocyclic ring system of 6
ring atoms,
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
comprising 1, or 2 heteroatonns independently selected from N, 0 and S, the
remaining ring
atoms being carbon.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
5 R1 is a C1_2alkyl, a Ci_zalkoxy, or a halo-C1_2alkyl group;
R2 is hydrogen; a Ci_aalkyl group optionally substituted with -S(0)2-
Ci_2alkyl, C4_6cycloalkyl or
a a monovalent saturated monocyclic ring of 3 to 7 ring atoms comprising one
ring heteroatom
selected from 0 and S, the remaining ring atoms being carbon; a C4_6cycloalkyl
group; a
monovalent saturated monocyclic ring of 3 to 7 ring atoms comprising one ring
heteroatom
10
selected from 0 and S, the remaining ring atoms being carbon optionally
substituted with a
Ci_aalkyl; or a monovalent, heterocyclic aromatic, monocyclic ring system of 6
ring atoms,
comprising 1, or 2 heteroatoms independently selected from N, 0 and S, the
remaining ring
atoms being carbon.
15
In one embodiment, the present invention relates to compounds of formula (I)
wherein
X is CH.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
Xis N.
20
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R2 is hydrogen.
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R1 is an alkyl, an alkoxy, or a haloalkyl group; R2 is hydrogen; and X is CH
or N.
25
In one embodiment, the present invention relates to compounds of formula (I)
wherein
R1 is a C1_4alkyl, a Ci_aalkoxy, or a halo-C1_4alkyl group; R2 is hydrogen;
and X is CH or N.
In one embodiment, the present invention relates to compounds of formula (I-a)
wherein R1 is a C1_2alkyl, or a halo-C1_2alkyl group; R2 is hydrogen; and X is
CH or N.
30
In one embodiment, the present invention relates to compounds of formula (I),
as
defined above selected from the group consisting of:
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
31
6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-1,2,3,4-
tetrahydro-2,7-
naphthyridine,
6-({5-methy1-3-[6-(trifluoromethyl)pyridin-3-y1]-1,2-oxazol-4-yl}methoxy)-
1,2,3,4-tetrahydro-
2,7-naphthyridine,
2-methy1-6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-
1,2,3,4-tetrahydro-
2,7-naphthyridine,
2-cyclobuty1-64[5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
2-(cyclobutylmethyl)-64[5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-
yl]methoxy}-1,2,3,4-
tetrahydro-2,7-naphthyridine,
2-cyclopenty1-6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-({5-methy1-346-(trifluoromethyppyridin-3-y1]-1,2-oxazol-4-yl}methoxy)-2-
(oxan-4-y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-({5-methy1-346-(trifluoromethyppyridin-3-y1]-1,2-oxazol-4-yllmethoxy)-2-
(oxolan-3-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-2-(oxolan-3-y1)-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-2-(oxetan-3-y1)-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-2-(oxan-4-y1)-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
2-(1-methanesulfonylpropan-2-y1)-6-({5-methy1-346-(trifluoromethyppyridin-3-
y1]-1,2-oxazol-
4-yl}methoxy)-1,2,3,4-tetrahydro-2,7-naphthyridine,
6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-2-(pyridin-2-
y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine,
2-methy1-545-methy1-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-yloxy}methyl)-1,2-
oxazol-3-
yl]pyridine,
545-methy1-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-yloxy}methyl)-1,2-oxazol-3-
y1]-2-
(trifluoromethyl)pyridine,
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
32
2-methy1-5-{5-methy1-4-[({7-methyl-5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-
yl}oxy)methyl]-1,2-
oxazol-3-yl}pyridine,
545-methy1-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-yloxy}methyl)-1,2-oxazol-3-
y1]-2-
(trifluoromethyppyridine,
5[5-methy1-4-({[7-(oxolan-3-y1)-5H ,6H ,7H ,8H-pyrido[3,4-c]pyridazi n-3-
yl]oxy}methyl)-1,2-
oxazol-3-y1]-2-(trifluoromethyl)pyridine,
3-([3-({5-methyl-346-(trifluoromethyppyridin-3-y1]-1,2-oxazol-4-yl}methoxy)-
5H,6H,7H,8H-
pyrido[3,4-c]pyridazin-7-yl]methy1}-11ambda6-thiolane-1,1-dione,
64[4-methyl-I -(6-methylpyridi n-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-1,2,3,4-
tetrahydro-2 ,7-
naphthyridine,
2-methy1-64[4-methyl-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
64[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-(propan-
2-y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-({146-(difluoromethyppyridin-3-y1]-4-methy1-1H-1,2,3-triazol-5-yllmethoxy)-2-
methyl-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-({4-methy1-1-[6-(trifluoromethyppyridin-3-y1]-1H-1,2,3-triazol-5-yl}methoxy)-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-({1-[6-(difluoromethyppyridin-3-y1]-4-methy1-1H-1,2,3-triazol-5-y1}methoxy)-
1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-({4-methy1-1-[6-(trifluoromethyppyridin-3-y1]-1H-1,2,3-triazol-5-yl}methoxy)-
2-(propan-2-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
2-methy1-6-({4-methy1-146-(trifluoromethyl)pyridin-3-y1]-1H-1,2,3-triazol-5-
y1}methoxy)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-({1-[6-(difluoromethyppyridin-3-y1]-4-methy1-1H-1,2,3-triazol-5-y1}methoxy)-
2-(propan-2-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-(oxolan-
3-y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-({146-(difluoromethyl)pyridin-3-y1]-4-methy1-1H-1,2,3-triazol-5-yllmethoxy)-
2-(oxolan-3-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
33
6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-(oxetan-
3-y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-({4-methy1-1-[6-(trifluoromethyl)pyridin-3-y1]-1H-1,2,3-triazol-5-
yl}methoxy)-2-(oxolan-3-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-U4-methyl-I -(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-(oxan-4-
y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-([1-(6-methoxypyridin-3-y1)-4-methyl-1H-1,2,3-triazol-5-yl]methoxy}-1,2,3,4-
tetrahydro-2,7-
naphthyridine,
6-({4-methy1-146-(trifluoromethyppyridin-3-y1]-1H-1,2,3-triazol-5-yllmethoxy)-
2-(oxan-4-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
34[6-({4-methyl-146-(trifluoromethyl)pyridin-3-y1]-1H-1,2,3-triazol-5-
yl}methoxy)-1,2,3,4-
tetrahydro-2,7-naphthyridin-2-Amethylplambda6-thiolane-1,1-dione,
6-({4-methy1-146-(trifluoromethyppyridin-3-y1]-1H-1,2,3-triazol-5-yl}methoxy)-
2-(pyridin-3-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
64[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-[(3S)-
oxolan-3-y1]-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-[(3R)-
oxolan-3-y1]-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-{[1-(6-methoxypyridin-3-y1)-4-methy1-1H-1,2,3-triazol-5-yl]methoxy}-2-(oxan-
4-y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine,
6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-(2-
methylpropy1)-
1,2,3,4-tetrahydro-2,7-naphthyridine,
6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-243-
(propan-2-yl)oxetan-
3-y1]-1,2,3,4-tetrahydro-2,7-naphthyridine,
2-(3-ethyloxetan-3-y1)-6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-
5-yl]methoxy}-
1,2,3,4-tetrahydro-2,7-naphthyridine,
2-methy1-544-methy1-5-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-yloxy}methyl)-1H-
1,2,3-
triazol-1-yl]pyridine,
545-({[7-(cyclobutylmethyl)-5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-
yl]oxylmethyl)-4-methyl-
1H-1,2,3-triazol-1-y1]-2-methylpyridine, and
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
34
5-{54({7-cyclobuty1-5H,61-1,7H,8H-pyrido[3,4-c]pyridazin-3-yl}oxy)methyl]-4-
methyl-1H-1,2,3-
triazol-1-y1}-2-methylpyridine
and/or salts thereof and/or stereoisomers thereof and/or enantiomers thereof
and/or
racemates thereof or diastereomers thereof and/or biologically active
metabolites thereof or
prodrugs thereof or solvates thereof or hydrates thereof and/or polymorphs
thereof.
In describing the general synthesis of the compounds of formula (I), the
biological assays,
Intermediates and Examples, the following abbreviations have been used:
Cs2003= cesium carbonate Na2SO4. = sodium sulfate
DCM = dichloromethane Pd(OAc)2 = palladium(II)
acetate
DIBAL-H = diisobutylaluminium hydride POCI3= phosphorus oxychloride
DMSO = dimethyl sulfoxide TBHP = tert-butyl hydroperoxide
Et0Ac = ethyl acetate TFA = trifluoroacetic acid
K2CO3= potassium carbonate THF = tetrahydrofuran
Me0H = methanol TLC = thin layer chromatography
MgSO4 = magnesium sulfate brine = high-concentration
solution of salt
Na2003 = sodium carbonate (usually sodium chloride)
NaHCO3 = sodium bicarbonate rt = room temperature, 25 C
Process for the preparation of the compounds of formula (I)
The compounds of formula (I) of the present invention can be synthesized
according
to the reaction sequence depicted in Scheme 1, 2, 3, 4 and 5.
The compounds of formula (1-a) wherein X=CH, and R1 and R2 are as defined in
any
of the embodiments described above can be prepared according to Scheme 1 and
2.
N
OH N
CI
N 15 R (II) N N (III)
R
Scheme 1
According to Scheme 1, reacting a compound of formula (II) with a chlorinating
agent,
such as POCI3 provides intermediates of formula (111). Hydroxy derivatives of
formula (II) are
known in the art (WO 2018/104419 Al) or can be synthesized by conventional
methods.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
HO
N, I N
X 130C 0
(IV) 14 1
\ CI
i (III)
Ni N
0 0
N N N N
R1
RI
(V) (I-a)
Scheme 2
According to Scheme 2, etherification between alcohols of formula (IV) and
intermediates of formula (III) can be accomplished in the presence of a
suitable base, such as
5 K2CO3 in a suitable solvent, such as acetonitrile to form a compound of
formula (V).
Compounds of the general formula (I-a), wherein R2=H were obtained after
removal of the
protective group of formula (V) using acid, such as ethyl acetate saturated
with hydrogen
chloride or TFA in dichloromethane. Compounds of the general formula (I-a),
wherein R2=
alkyl, optionally substituted with -S(0)2-alkyl, cycloalkyl or heterocycle;
cycloalkyl; heterocycle
10 were obtained from those compounds of the general formula (I-a), wherein
R2=H by alkylation.
Compounds of the general formula (I-a), wherein R2= heteroaryl were obtained
from those
compounds of the general formula (I-a), wherein R2=H by arylation. Compounds
of the general
formula (I-a), wherein R2= heterocycle, optionally substituted with alkyl were
obtained from
those compounds of the general formula (I-a), wherein R2=H by condensation
with
15 benzotriazole and a carbonyl compound, followed by a nucleophilic
reaction using Grignard
reagents. Alcohol of formula (IV) can be purchased or can be prepared by
conventional
methods.
The compounds of formula (I-a) wherein X=N, R1 and R2 are as defined in any of
the
embodiments described above can be prepared according to Scheme 3.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
36
CI
N
0
(VI) OH
/ (II)
\N: N
0 0
0 \ 0
N
N 2
'130C
`IR
N N N
R
(VII) Ri (I-a)
Scheme 3
According to Scheme 3, etherification between chloro derivatives of formula
(VI) and
hydroxy derivatives of formula (II) can be carried out by a palladium-mediated
process in the
presence of a suitable base, such as Cs2CO3 to provide a compound of formula
(VII).
Compounds of the general formula (I-a), wherein R2=H were obtained after
removal of the
protective group of formula (VII) using acid, such as ethyl acetate saturated
with hydrogen
chloride or TFA in dichloromethane. Compounds of the general formula (I-a),
wherein R2=
alkyl, optionally substituted with -S(0)2-alkyl, cycloalkyl or heterocycle;
cycloalkyl; heterocycle
were obtained from those compounds of the general formula (I-a), wherein R2=H
by alkylation.
Compounds of the general formula (I-a), wherein R2= heteroaryl were obtained
from those
compounds of the general formula (I-a), wherein R2=H by arylation. Compounds
of the general
formula (I-a), wherein R2= heterocycle, optionally substituted with alkyl were
obtained from
those compounds of the general formula (I-a), wherein R2=H by condensation
with
benzotriazole and a carbonyl compound, followed by a nucleophilic reaction
using Grignard
reagents. Chloro derivative of formula (VI) can be purchased or can be
prepared by
conventional methods.
The compounds of formula (I-b) wherein X, R1 and R2 are as defined in any of
the
embodiments described above can be prepared according to Scheme 4 and 5.
CA 03231776 2024- 3- 13
WO 2023/053015 PCT/IB2022/059214
37
0
___________________________ :H(1(0--N,
)11.-
sN
0
-i'
NO (2)
(3) N
01
NH Ri R
i 0
H
IN
01
N
----
N (1) ri\i
(VIII)
Ri
R1
..........N 3
01 t
1
R (4)
Scheme 4
In a first step, a compound of formula (1) is reacted with ethyl acetoacetate
in a suitable
solvent, such as DMSO to give a compound of formula (2) which is coupled with
N-
tosylhydrazide in the presence of KI and TBHP to give a compound of formula
(3) (Huang et
al. Adv. Synth. Catal. 2018, 360:3117-3123). Treatment of a compound of
formula (3) with a
reducing agent such as DIBAL-H in a suitable solvent such as toluene gives a
compound of
formula (VIII). Alternatively, a compound of formula (1) is converted to a
diazonium salt, which
is further reacted with trimethylsilyl azide to give a compound of formula
(4). Compounds of
formula (4) reacted with 2-butyn-1-ol give a compound of formula (VIII).
N, I N
`x 'BOG
N
(VI) W -K., OH
N
01 (Vill) \
R1
N N
Nil -K(:)
_____________________________________________________________ 3.-
01 01
R1
1
(IX) R (I-b)
Scheme 5
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
38
According to Scheme 5, etherification between chloro derivatives of formula
(VI) and
hydroxy derivatives of formula (VIII) can be carried out by a palladium-
mediated process in
the presence of a suitable base, such as Cs2CO3 to provide a compound of
formula (IX).
Compounds of the general formula (I-b), wherein R2=H were obtained after
removal of the
protective group of formula (IX) using acid, such as ethyl acetate saturated
with hydrogen
chloride or TFA in dichloromethane. Compounds of the general formula (I-b),
wherein R2=
alkyl, optionally substituted with -S(0)2-alkyl, cycloalkyl or heterocycle;
cycloalkyl; heterocycle
were obtained from those compounds of the general formula (I-b), wherein R2=H
by alkylation.
Compounds of the general formula (I-b), wherein R2= heteroaryl were obtained
from those
compounds of the general formula (I-b), wherein R2=H by arylation. Compounds
of the general
formula (I-b), wherein R2= heterocycle, optionally substituted with alkyl were
obtained from
those compounds of the general formula (I-b), wherein R2=H by condensation
with
benzotriazole and a carbonyl compound, followed by a nucleophilic reaction
using Grignard
reagents. Chloro derivative of formula (VI) can be purchased or can be
prepared by
conventional methods.
The reagents and detailed process steps required for the above reactions are
set forth
in the Intermediates and Examples.
The present invention thus relates to a process for the preparation of
compounds of
formula (I) as defined above, comprising
step (i) a coupling reaction, selected from the group consisting of
(a-1) reacting a compound of formula (IV) with a compound of formula (III), to
give a
compound of formula (V), wherein X=CH and R1 and R2 are as defined above;
(a-2) reacting a compound of formula (VI) with a compound of formula (II), to
give a
compound of formula (VII), wherein X=N and R1 and R2 are as defined above; and
(b) reacting a compound of formula (VI) with a compound of
formula (VIII), to give
a compound of formula (IX), wherein X, R1 and R2 are as defined above
step (ii) deprotection of a compound of formula (V), (VII) or (IX) to obtain a
compound of
formula (I) wherein A, X, and R1 are as defined above and R2 is hydrogen, and
step (iii) optionally transforming a compound of formula (I) wherein R2 is
hydrogen to a
compound of formula (I) wherein A, X, and R1 are as defined as above and R2 is
an alkyl group
optionally substituted with -S(0)2-alkyl, a cycloalkyl or a heterocycle; a
cycloalkyl group; a
heterocycle group optionally substituted with an alkyl; or a heteroaryl group.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
39
In an aspect, the present invention provides novel intermediates of formula
(I")
synthesised in the process for preparing the compound of general formula (I)
wherein A, X,
and R1 are as defined above and R2 is an amino protecting group (Peter G. M.
Wuts: Greene's
Protective Groups in Organic Synthesis: Fifth Edition, Chapter 7. Protection
for the Amino
Group, pages 895-1193), such as a carbamate (methyl, 9-fluorenylmethyl, 2,2,2-
trichloroethyl,
tert-butyl, 2-(trimethylsilyl)ethyl, ally!, benzyl), trifluoroacetamide,
benzylamine, allylamine, or
tritylamine, preferably a carbamate, most preferably tert-butyloxycarbonyl
protecting group.
A 0
N, 2
'X R
N
1
(r)
In a further aspect, the present invention provides novel intermediates of
formula (V)
synthesised in the process for preparing the compound of general formula (I)
wherein X is CH,
R1 and R2 are as defined above with the proviso that the compound is not tert-
butyl 6-{[5-
methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]nethoxy}-1,2,3,4-tetrahydro-
2,7-
naphthyridine-2-carboxylate, or tert-butyl 6-(15-methyl-3[6-
(trifluoromethyppyridin-5 3-y11-1,2-
oxazol-4-yl}methoxy)-1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate.
In another further aspect, the present invention provides novel intermediates
of formula
(VII) synthesised in the process for preparing the compound of general formula
(I) wherein X
is N, R1 and R2 are as defined above.
In one embodiment, the present invention relates to the intermediates of
formula (VII)
selected from the group consisting of:
tert-butyl 2-methyl-5-[5-methyl-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-
yloxy}methyl)-1,2-
oxazol-3-yl]pyridine-2-carboxylate, and
tert-butyl 545-methyl-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-yloxy}methyl)-
1,2-oxazol-3-
y1]-2-(trifluoromethyppyridine-2-carboxylate.
In yet another aspect, the present invention provides novel intermediates of
formula
(IX) synthesised in the process for preparing the compound of general formula
(I) wherein X,
R1 and R2 are as defined above.
In one embodiment, the present invention relates to the intermediates of
formula (IX)
selected from the group consisting of:
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
tert-butyl
6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-
1,2,3,4-
tetrahydro-2,7-naphthyridine-2-carboxylate,
tert-butyl
6-({4-methy1-1-[6-(trifluoromethyl)pyridi n-3-yI]-1H-1,2, 3-triazol-5-
yl}methoxy)-
1,2, 3,4-tetrahyd ro-2 , 7-naphthyri di ne-2-carboxyl ate,
5 tert-butyl
6-({1-[6-(difluoromethyppyridin-3-y1]-4-methy1-1H-1,2 , 3-triazol-5-
yl}methoxy)-
1,2, 3,4-tetrahyd ro-2 , 7-naphthyri di ne-2-carboxyl ate,
tert-butyl
6-{[1-(6-methoxypyridin-3-y1)-4-methy1-1H-1,2,3-triazol-5-yl]methoxy}-
1,2,3,4-
tetrahydro-2,7-naphthyridine-2-carboxylate, and
tert-butyl 2-methyl-5-[4-methyl-5-({5H ,6H , 7H ,8 H-pyrido[3,4-c]pyridazi n-3-
yloxy}methyl)-1H-
10 1,2,3-triazol-1-yl]pyridine-2-carboxylate.
The activity data of each of the compounds of formula (1) of the present
invention are
determined in vitro by the methods described below.
15 Biological example 1: Binding assay
The GABAA 05133y2 protein used for the receptor binding assay was derived from
membranes produced from HEK cells (Millipore CYL3073) expressing the human
recombinant
GABAA a5133y2 receptor. Cells were stored and cultured in-house according to
the instructions
provided by the vendor (Millipore). Cell pellet was homogenized in 10 times
modified Krebs
20 Henseleit buffer (membrane preparation buffer): 20 mM Tris, 120 mM NaCI,
100 mM KCI, 25
mM CaCl2 and 25 mM MgCl2 pH=7.4 at 4 C using Ultra Turrax (Janke&Kunkel)
maximal
speed for 15 seconds. The homogenate was centrifuged at 40,000 g for 30
minutes at 4 C.
Supernatant was discarded and the resulting pellet was washed in membrane
preparation
buffer. Pellet was resuspended in membrane preparation buffer and aliquots of
1.4 mL
25 ampules were stored at -70 C until use.
Receptor binding assays were performed in 96-well format in deep-well plates.
For
each 96-well plate one ampule of membrane homogenate was thawed and diluted in
binding
buffer (50 mM Tris pH=7.4, 100 mM KCI) and 200 pL was dispensed into each
well.
Radioligand [3H]Ro151788 (Perkin Elmer: NET757250UC) was prepared in binding
buffer and
30 added to each well in 50 pL volume to give final concentration of 0.5
nM. Test compounds in
suitable concentration(s) were added in additional 50 pL. The final assay
volume was 300 pL.
Incubation was carried out for 60 minutes at 4 C. For non-specific binding 10
pM unlabeled
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
41
diazepam was used. After incubation samples were filtered over UniFilter
GF/BTM using
Filtermate Harvester (Perkin Elmer) and washed with 5x1 mL binding buffer. The
plate was
dried at 40 C for an hour and 40 pL Microscint (Perkin Elmer) scintillation
cocktail was added
to each well. The plate was read in Microbeta (Perkin Elmer).
The specific radioligand binding (SB) was defined as the difference between
total
binding (Tot) and the non-specific binding (NSB). Results are expressed as a
percent inhibition
of specific binding obtained in the presence of compound of interest.
For IC50 and K determination a minimum of six drug concentrations in
triplicate were
used. IC50 values (i.e., concentration of compound giving 50% inhibition of
specific binding)
were calculated from concentration-displacement curves by sigmoidal fitting
using Origin 7.5
software. K, values (i.e., inhibition constants) were calculated using the
Cheng-Prusoff
equation KJ = IC50/[1+(LJKD)], where [L] is the radioligand concentration and
KD the affinity of
the labelled ligand for receptor. KD was determined from the Saturation
analyses.
The compounds of the present invention were tested in the above described
assay,
and all were found to have high affinity for the GABAA a5 receptor (K< 150
nM).
Table 1 showing representative hGABAA a5 KJ test results, obtained by the
above
described binding assay:
Ex. hGABAA a5 KJ (nM) Ex. hGABAA a5 Ki (nM) Ex. hGABAA a5 Ki
(nM)
1 3.9 17 17.6 33
29.5
2 37.0 18 12.7 34
51.6
3 4.2 19 35.5 35
88.8
4 4.5 20 26.5 36
53.5
5 7.4 21 23.0 37
37.9
6 8.4 22 22.7 38
20.9
7 29.3 23 7.3 39
21.4
8 11.2 24 8.3 40
28.5
9 4.9 25 128 41
61.5
10 4.5 26 93.8 42
28.0
11 7.4 27 63.2 43
19.0
12 28.2 28 5.9 44
40.4
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
42
13 6.6 29 29.7 45
99.5
14 3.2 30 3.3 46
61.5
15 17.1 31 36.0
16 5.6 32 44.5
Biological example 2: Functional assay
Human HEK293 cell lines expressing GABAA a5(33y2 receptors were used in
functional
assays using the QPatch automated patch clamp system.
HEK293 cell lines stably expressing human recombinant GABAA a5133y2 receptor
subunits (Millipore, CYL3053) were cultured in DMEM supplemented with 10% FBS
(Gibco),
passed two times per week and plated on Petri dishes previously coated with
poly-d-lysine.
Automated whole-cell patch clamp recordings were made from cells 2-4 days
after
plating. Cells were detached using trypsin/EDTA (Sigma) treatment (2 minutes
in 0.25%
trypsin at 37 C), then, after centrifugation (125 g, 3 min, 2x), resuspended
in a serum-free
based media (Gibco, CHO-S-SFM-II) containing 12.5 mM HEPES, lx penicillin-
streptomycin-
amphotericin (SigmaMix) and soybean trypsin inhibitor (Sigma, 0.04 mg/ml).
Cell suspension, as well as the extracellular solution (130 mM NaCI, 5 mM KCI,
5.1
mM HEPES, 4.9 mM HEPES-Na, 10 mM CaCl2, 2 mM MgCl2, 10 mM glucose and 0.1%
DMSO, pH=7.35-7.4) and the intracellular solution (80 mM KCI, 50 mM KF, 36 mM
KOH, 10
mM EGTA, 10 mM HEPES, 1.75 mM MgCl2, 0.5 mM CaCl2, 4 mM Na2ATP. 14 mM
phosphocreatine, 50 Wm! creatine-phosphokinase, 0.3 mM GTP, pH=7.25-7.3) were
added
to the QPatch-HTX automated patch clamp system (Sophion) in single-cell mode
at room
temperature. Inward currents were evoked at a holding potential of -80 mV by 3-
s-long
applications of the control agonist GABA at 1 pM at 2-4-min intervals first in
concentration-
matched DMSO (0.1 or 0.3%) control solution for five times, then in the
presence of the test
compound for four times, finally in control solution again for three times
(wash-out). At the end
of the experiment 100 pM GABA was applied to saturate the GABA-response and to
assess
the efficacy of the control GABA application. Current signals were low-pass
filtered at 100 Hz
and recorded at a sampling rate of 1 kHz.
The percentage modulation was calculated from the comparison of GABA-evoked
peak current amplitudes in the presence and absence of the test compound.
The compounds of the present invention were tested at 1 pM in the above
described
assay, and all were found to possess GABAA a5 positive allosteric modulator
activity.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
43
Table 2 showing representative hGABAA a5 functional efficacy test results,
obtained by the
above described assay:
hGABAA a5 hGABAA a5 hGABAA a5
Ex. Ex. Ex.
efficacy (%) efficacy CYO efficacy
CYO
1 155 16 146 32 155
2 100 19 129 33 71
7 125 20 111 34 62
8 133 21 134 37 53
9 106 22 68 40 86
131 24 88 42 99
11 114 25 73 43 85
13 129 29 102 44 96
14 126 30 59 45 97
115 31 115 46 76
Examples
The present invention will be further illustrated by the following
Intermediates and
5 Examples without limiting the scope of the present invention to them.
From the above
description and from the Intermediates and Examples, the person skilled in the
art may
ascertain the essential features of the invention and without departing from
its essence and
scope, may make certain changes and modifications in order to adapt the
invention to various
applications and conditions. As a result, the invention is not limited to the
following illustrative
10 examples, but rather to the scope determined by the appended claims.
In general, the compounds of formula (I) can be prepared according to the
common
general knowledge of the person skilled in the art and/or the methods
described for the
working examples and/or intermediates. Solvents, temperatures, pressures and
other reaction
conditions can be easily selected by the person skilled in the art. Starting
materials are
15 commercially available and/or can be easily prepared by the person
skilled in the art according
to literature procedure. During the preparation of compounds combinatorial
techniques can
be used, for example, where intermediates are suitable for the use of these
methods.
Intermediate 1
5[4-(chloromethyl)-5-methyl-1 ,2-oxazol-3-y11-2-methyl pyri dine
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
44
0
1\i\ CI
N N
1.00 g (4.89 mmol) of [5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-
yl]nethanol (WO
2018/104419 Al, Hoffmann-La Roche) was dissolved in 30 mL of phosphorus
oxychloride.
The reaction mixture was stirred for 2 hours at 115 C, then evaporated to
dryness. Ethyl
acetate was added and washed with saturated sodium hydrogen carbonate solution
and with
water, dried over anhydrous sodium sulfate, and evaporated to obtain 0.95 g
(87%) of the title
compound. MS (ESI) m/z: 223.1 [M+H].
Intermediate 2
5-[4-(chloromethyl)-5-methyl-1,2-oxazol-3-y1]-2-(trifluoromethyl)pyridine
0
14\ \ CI
N N
In analogy of Intermediate 1, {5-methyl-346-(trifluoromethyppyridin-3-y1]-1,2-
oxazol-4-
yl}methanol (WO 2018/104419 Al, Hoffmann-La Roche) was converted into the
title
compound. MS (ESI) m/z: 277.1 [M-'-H].
Intermediate 3
14-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yllmethanol
NCOH
Method A
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
a: methyl (2E)-3-[(6-methylpyridin-3-yl)amino]but-2-enoate
To a mixture of 1.00 g (9.20 mmol) of commercially available 6-methylpyridine-
3-amine and
1.40 mL (1.11 mmol) of ethyl acetoacetate in 30 mL of ethanol, 1.67 g (13.9
mmol) of
anhydrous magnesium sulfate and 0.10 mL (1.85 mmol) of acetic acid was added.
The
5 reaction mixture was refluxed for 10 hours. After cooling, filtration of
inorganics and
concentration of the filtrate under reduced pressure afforded the residue
which was used in
the next step without further purification. MS (ES!) m/z: 207.1 [M+H]t
b: ethyl 4-methyl-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazole-5-carboxylate
To a mixture of 8.31 g (37.7 mmol) of methyl (2E)-3-[(6-methylpyridin-3-
yl)amino]but-2-
10 enoate, 8.43 g (45.3 mmol) of methylbenzenesulfonehydrazide, 6.26 g
(37.7 mmol) of
potassium iodide in 70 mL of DMSO, 7.31 mL (75.5 mmol) of TBHP (70% solution
in water)
was added slowly. Then the mixture was stirred at 70 C for 24 hours. After the
reaction was
completed (monitored by TLC), 140 g of sodium dithionite dissolved in 300 mL
of water was
added to the reaction mixture, and the resulting mixture was extracted with
ethyl acetate. The
15 combined organic layers were then dried over MgSO4., filtered, and then
concentrated in
vacuo. Purification of the residue by flash coloumn chromatography (silica
gel, eluent:
DCM:Me0H, 0-10% gradient) afforded the desired product. Yield: 6.35 g (68 %),
MS (ESI)
m/z: 247.1 [M+Hr.
c:[4-methy1-1-(6-methvIpvridin-3-v1)-1H-1,2,3-triazol-5-vI]methanol
20 6.35 g (25.8 mmol) of ethyl 4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-
triazole-5-carboxylate
was dissolved in 80 mL of anhydrous THF and cooled to 0 C. 103 mL of DIBAL-H
(1 M
solution in toluene) was added dropwise under argon and the reaction mixture
was stirred at
room temperature for 1 hour. After cooling it was quenched with 71 mL of water
and
acidificated with 135 mL of 1M HCI. The combined organic layers were washed
with brine,
25 dried over Na2SO4, filtered and evaporated in vacuo. The crude product
was crystallised from
isopropanol to obtain the title compound as a white solid. Yield: 3.42 g,
(65%), MS (ESI) m/z:
205.1 [M+H]+.
Method B
30 a: 5-azido-2-methvIpvridine
5.0 g (46 mmol) of commercially available 6-methylpyridine-3-amine was
diisolved in a mixture
of 14 mL of cc. HCI and 14 mL of water and cooled to 0 C. 3.19 g (46.2 mmol)
of NaNO2
dissolved in 12 mL of water was added dropwise. The reaction mixture was
stirred at 0 C for
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
46
20 min then 10.6 mL (80 mmol) of trimethylsylil azide was added dropwise
slowly and the
reaction mixture was stirred at room temperature for 1.5 hour. After
completion 70 mL of ethyl
acetate was added and washed three times with 30 mL of saturated sodium
carbonate solution
and with water, dried over anhydrous sodium sulfate, and evaporated. The crude
product was
used in the next step without further purification.
b: [4-methyl-1-(6-methvIpvridi n-3-v1)-1H-1,2,3-triazol-5-vIlmethanol
5.81 g (43.3 mmol) of 5-azido-2-methylpyridine was dissolved in 3.24 mL (43.3
mmol) of 2-
butyn-1-ol and the reaction mixture was stirred at 100 C for 10 h. The residue
was purified by
flash coloumn chromatography (silica gel, eluent: cyclohexane:Et0Ac 40-80 %
gradient).
Yield: 2.30 g (26 c/0), white solid. MS (ESI) m/z: 205.1 [M+H]+.
Intermediate 4
14-methyll F6-(trifluoromethyppyridin-3-y11-1H-1,2,3-triazol-5-yllmethanol
NN OH
N N
The compound was synthesized according to the procedure described for
intermediate 3 using
commercially available 6-(trifluoromethyl)pyridin-3-amine in step a. MS (ESI)
m/z: 259.1
[M+H]+.
Intermediate 5
{1-[6-(difluoromethyppyridin-3-y11-4-methyl-1H-1,2,3-triazol-5-yl}methanol
1\1,1\OH
N N
FF
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
47
The compound was synthesized according to the procedure described for
intermediate 3 using
commercially available 6-(difluoromethyl)pyridin-3-amine in step a. MS (ESI)
rn/z: 241.1
[M+H]+.
Intermediate 6
ri-(6-methoxvpyridin-3-v1)-4-methyl-1H-1,2,3-triazol-5-vIlmethanol
Nsi\IN-COH
N N
The compound was synthesized according to the procedure described for
intermediate 3 using
commercially available 6-methoxypyridin-3-amine in step a. MS (ESI) rn/z:
221.1 [M+H]+.
Example 1
6-{[5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yllmethoxy}-1,2,3,4-
tetrahydro-2,7-
naphthyridine trifluoroacetic acid salt
N
o
N NH F 0
F
N F
A: tert-butyl 64[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yllmethoxyl-
1,2,3,4-
tetrahydro-2,7-naphthyridine-2-carboxylate
1.96 g (8.80 mmol) of 5[4-(chloromethyl)-5-methyl-1,2-oxazol-3-y1]-2-
methylpyridine
(Intermediate 1), and 2.20 mg (8.80 mmol) of commercially available tert-butyl
6-hydroxy-3,4-
dihydro-2,7-naphthyridine-2(1H)-carboxylate were dissolved in 120 mL of
anhydrous
acetonitrile. Then, 3.65 mg (26.40 mmol) of anhydrous potassium-carbonate was
added to
the solution, and the suspension was stirred under reflux for 12 h. The
conversion was
followed by TLC (Et0Ac:cyclohexane=1:1 as el uent, silica plate). After the
reaction completed,
the mixture was filtered, and evaporated to give an oily crude product, which
was purified by
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
48
flash coloumn chromatography (silica gel, eluent: Et0Ac:cyclohexane=1:1).
Yield: 640 mg
(16.6 %) white solid. MS (ESI) m/z: 437.3 [M+H].
B: 6-{[5-methyl-3-(6-m ethylpyridin-3-yI)- 1,2-oxazol-4-
yl]methoxy)- 1,2,3,4-tetrahydro-2, 7-
naphthyridine trifluoroacetic acid salt
97.97 mg (0.22 mmol) of tert-butyl 6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-
oxazol-4-
yl]methoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate was dissolved
in 10 mL of
DCM. Then, 1489 mg (13.06 mmol) of trifluoroacetic acid was added to the
solution, and the
suspension was stirred at rt for 6 h. After the reaction completed, the
mixture was evaporated
to give the title compound. Yield: 90 mg (91%) yellow solid. MS (ESI) m/z:
337.1 [M--H]. 1H
NMR (DMSO-d6, 400 MHz) 6 (ppm): 8.96-9.07 (br m, 2H), 8.81 (br d, J=2.0 Hz,
1H), 8.12 (dd,
J=8.1, 2.3 Hz, 1H), 8.06 (s, 1H), 7.49 (d, J=8.1 Hz, 1H), 6.73 (s, 1H), 5.27
(s, 2H), 4.25 (br t,
J=4.5 Hz, 2H), 3.31-3.39 (m, 2H), 2.95 (t, J=6.3 Hz, 2H), 2.57 (s, 3H), 2.56
(s, 3H).
Example 2
6-1{5-methyl-3l6-(trifl uoromethyppyridi n-3-y11-1,2-oxazol-4-yl}methoxy)-1,
2,3,4-
tetrahydro-2,7-naphthyridine trifluoroacetic acid salt
P
N \ 1 0
F
F i<ruj
FF
N N F
The title compound prepared according to the procedure described for Example 1
using 544-
(chloromethyl)-5-methy1-1,2-oxazol-3-y1]-2-(trifluoromethyppyridine
(Intermediate 2) in step a.
MS (ESI) m/z: 391.2 [M-'-H]t 1H NMR (DMSO-d6, 400 MHz) 6 (ppm): 9.11 (d, J=1.9
Hz, 1H),
8.82-9.03 (br m, 2H), 8.48 (dd, J=8.1, 1.7 Hz, 1H), 8.11 (d, J=8.1, 1H), 8.04
(s, 1H), 6.73 (s,
1H), 5.33(s, 2H), 4.24 (br t, 2H), 3.31-3.38 (br m, 2H), 2.94 (t, J=6.3 Hz,
2H), 2.61 (s, 3H).
Example 3
2-methyl-64[5-methyl-3-(6-methyl pyrid I n-3-y1)--1,2-oxazol-4-yllmethoxy}-
1,2, 3,4-
tetrahydro-2,7-naphthyridi ne
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
49
0
\
X1N
N
450 mg (1.0 mmol) of 6-{[5-methy1-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-
yl]methoxy}-1,2,3,4-
tetrahydro-2,7-naphthyridine trifluoroacetic acid salt (Example 1) was added
to a solution of
saturated NaHCO3 and extracted with Et0Ac. The organic layer was separated,
dried over
MgSO4, filtered and evaporated in vacuo. The obtained base was dissolved in 2
mL of water
and 240 mg (4.0 mmol) of acetic acid, 122 mg (1.5 mmol) of formaldehyde
solution (37% in
water) and 131 mg (2.0 mmol) of zinc powder was added. The reaction mixture
was stirred at
30 C for 48 hours. After the reaction was completed (monitored by TLC), the
reaction mixture
was neutralized with ammonia solution, and the resulting mixture was extracted
with DCM.
The combined organic layers were then dried over MgSO4., filtered, and then
concentrated in
vacuo. Purification of the residue by flash coloumn chromatography (silica
gel, eluent:
DCM:Me0H=10:1) afforded the desired product. Yield: 59.3 mg (16.9 %), MS (ESI)
rn/z: 351.2
[M+1-1]+.
Example 4
2-cyclobutv1-6-{f5-methyl-3-(6-methylpyridin-3-v1)-1,2-oxazol-4-vIlmethoxv}-
1.2,3,4-
tetrahydro-2,7-naphthyridine
0
0 N
N
To a solution of 200 mg (0.44 mmol) of 64[5-methy1-3-(6-methylpyridin-3-y1)-
1,2-oxazol-4-
yl]nethoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine trifluoroacetic acid salt
(Example 1) in 4 mL
of 2,2,2-triluoroethanol 112 mg (1.33 mmol) of NaHCO3 was added and stirred
for 30 min,
then 32 mg (0.44 mmol) of cyclobutanone was added in one portion and the
reaction mixture
was warmed up to 45 C. The so obtained solution was stirred for 5 min, then
16.8 mg (0.44
mmol) of sodium borohydride was added. The reaction mixture was stirred at 45
C for 3 hours.
After completion the solvent was evaporated, the residue was dissolved in DCM
and washed
with brine. The organic layer was separated, dried over MgSO4, filtered and
evaporated in
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
vacua. Purification of the residue by flash coloumn chromatography (silica
gel, eluent:
Et0Ac:Me0H=10:1) afforded the desired product. Yield: 28.1 mg (16.1 %), MS
(ESI) rn/z:
393.2 [M+H].
5 Example 5
2-(cyclobutylmethvI)-6-{f5-methvl-3-(6-methylpyridin-3-v1)-1,2-oxazol-4-
vIlmethoxv}-
1,2,3,4-tetrahydro-2,7-naphthyridine
P
N\ oCC
N NJ:3
--N
10 The title compound prepared according to the procedure described for
Example 4 using
commercially available cyclobutanecarbaldehyde. MS (ESI) mk: 405.2 [m+H].
Example 6
2-cyclopenty1-64f5-methyl-3-(6-methylpyridin-3-v1)-1,2-oxazol-4-yllmethoxyl-
1,2,3,4-
15 tetrahydro-2,7-naphthyridine
N\ 0
N N
--N
The title compound prepared according to the procedure described for Example 4
using
commercially available cyclopentanone. MS (ESI) m/z: 405.2 [M-F1-1]+.
20 Example 7
6-({5-methy1-3-1-6-(trifluoromethyl)pyridin-3-v11-1,2-oxazol-4-yl}methoxy)-2-
(oxan-4-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
51
0
14\ 0
)0 N N
/
CIC N
The title compound prepared according to the procedure described for Example 4
using 64{5-
methyl-346-(trifluoromethyppyridin-3-y1]-1,2-oxazol-4-yl}methoxy)-1,2,3,4-
tetrahydro-2,7-
naphthyridine trifluoroacetic acid salt (Example 2) and commercially available
tetrahydropyran-4-one. MS (ESI) m/z: 475.2 [M+H]t
Example 8
6-1{5-methy1-346-(trifluoromethyppyridin-3-y11-1,2-oxazol-4-v1}methoxv)-2-
(oxolan-3-
v11-1,2,3,4-tetrahydro-2,7-naphthwidine
N \)(
N N
N N
The title compound prepared according to the procedure described for Example 4
using 64{5-
methyl-346-(trifluoromethyppyridin-3-y1]-1,2-oxazol-4-yllmethoxy)-1,2,3,4-
tetrahydro-2,7-
naphthyridine trifluoroacetic acid salt (Example 2) and commercially available
3-
oxotetrahydrofuran. MS (ESI) m/z: 461.2 [M+H].
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
52
Example 9
6-{1.5-methy1-3-(6-methylpyridin-3-v1)-1,2-oxazol-4-yllmethoxyl-2-(oxolan-3-
y1)-1,2,3,4-
tetralwdro-2,7-naphthyridine
,0
N\ 0
N N
N N
The title compound prepared according to the procedure described for Example 4
using
commercially available 3-oxotetrahydrofuran. MS (ESI) m/z: 407.2 [M+H].
Example 10
6-{[5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yllmethoxy}-2-(oxetan-3-y1)-
1,2,3,4-
tetrahydro-2,7-naphthyridine
P ,
N\
N N
N
--N
The title compound prepared according to the procedure described for Example 4
using
commercially available 3-oxetanone. MS (ESI) m/z: 393.2 [M+H]t
Example 11
6-{[5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yllmethoxy}-2-(oxan-4-y1)-
1,2,3,4-
tetrahydro-2,7-naphthyridine
,0
N \ 0
o
/
N
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
53
The title compound prepared according to the procedure described for Example 4
using
commercially available 4-oxotetrahydropyran. MS (ESI) m/z: 421.2 [M+H].
Example 12
2-(1-methanesulforwlpropan-2-v1)-6-({5-methyl-346-(trifluoromethyl)pyridin-3-
v11-1.2-
oxazol-4-v1}methoxv)-1,2,3,4-tetrahvdro-2,7-naphthyridine
N
0
I N
0
`.0
183.6 mg (0.36 mmol) of 6-({5-methyl-346-(trifluoromethyppyridin-3-y1]-1,2-
oxazol-4-
yl}methoxy)-1,2,3,4-tetrahydro-2,7-naphthyridine trifluoroacetic acid salt
(Example 2) was
added to a solution of saturated Na2CO3 and extracted with DCM. The organic
layer was
separated, dried over MgSO4., filtered and evaporated in vacua. The obtained
base was added
to a stirred solution of 49 mg (0.36 mmol) of methanesulfonylacetone in 1 mL
of methanol and
1 mL of 2,2,2-triluoroethanol at room temperature. The mixture was stirred for
1 h. 84 mg (0.72
mmol) of triethylsilicon was added by syringe and followed by 57 mg (0.26
mmol) of indium(III)
chloride (Lee et al., J Org. Chem. 2008, 73, 22, 8829-8837) The reaction was
allowed to stir
at room temperature and was monitored by TLC. When the reaction was completed,
the
mixture was quenched by 1 mL of saturated K2CO3 solution. The mixture was
extracted with
Et0Ac. The combined organic layer was washed with brine and finally was dried
over Na2SO4.
The crude product was purified by flash column chromatography (silica gel,
eluent:
cyclohexane:EtA0c=1:1). Yield: 21 mg (11 %), MS (ESI) m/z: 511.1 [M+H].
Example 13
6-{[5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-yl]methoxy}-2-(pyridin-2-
y1)-1,2,3,4-
tetrahydro-2,7-naphthyridine
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
54
0
0
I
Ni;H\L'
N N
283 mg (0.63 mmol) of 6-{[5-methyl-3-(6-methylpyridin-3-y1)-1,2-oxazol-4-
yl]methoxy}-1,2,3,4-
tetrahydro-2,7-naphthyridine trifluoroacetic acid salt (Example 1) was
dissolved in 2 mL of 2-
fluoropyridine the reaction mixture was stirred at 120 C for 3 h. The residue
was purified by
flash coloumn chromatography (silica gel, eluent: DCM:Me0H=10:1). Yield: 50 mg
(19.2 %).
MS (ESI) m/z: 414.2 [M+H]t
Example 14
2-methv1-545-methyl-4-({5H,6H,7H,8H-pyridor3,4-clpyridazin-3-vloxv}methvI)-1,2-
oxazol-3-vIlpyridine
0
14\ 0
N;1.N I NH
N N
A: tert-butyl 2-methyl-545-methyl-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-
yloxy}methyl)-
1,2-oxazol-3-yl]pyridine-2-carboxylate
Under argon atmosphere a flask was charged with 660 mg (2.45 mmol) of
commercially
available tert-butyl 3-chloro-5,8-dihydropyrido[3,4-c]pyridazine-7(6H)-
carboxylate, 500 mg
(2.45 mmol) of {5-methyl-3[6-(trifluoromethyppyridin-3-y1]-1,2-oxazol-4-
yl}methanol (WO
2018/104419 Al, Hoffmann-La Roche), 1595 mg (4.89 mmol) of Cs2CO3, 98 mg (0.25
mmol)
of rac-2-(di-tert-butylphosphino)-1,11-binaphthyl, 55 mg (0.24 mmol) of
Pd(OAc)2 and 20 mL
of anhydrous toluene. The mixture was stirred at 100 C for 12 h. The
conversion was checked
by TLC (cyclohexane:Et0Ac=1:1 as eluent, silica plate). The reaction mixture
was filtered
through a celite pad, washed with acetone, dried over anhydrous sodium
sulfate, and
evaporated. The residue was purified by flash coloumn chromatography (silica
gel, eluent:
cyclohexane:EtA0c=1:1). Yield: 342 mg (32 %), white, amorphous solid. MS (ESI)
m/z: 438.2
[M+H].
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
B: 2-methyl-545-methyl-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-yloxy}methyl)-
1,2-oxazol-
3-yl]pyridine
342 mg (0.78 mmol) of tert-butyl 2-methyl-545-methyl-4-({5H,6H,7H,8H-
pyrido[3,4-
c]pyridazin-3-yloxy}methyl)-1,2-oxazol-3-yl]pyridine-2-carboxylate was
dissolved in 50 mL of
5 DCM. Then, 1782 mg (15.63 mmol) of trifluoroacetic acid was added to the
solution, and the
suspension was stirred at rt for 24 h. After completion the mixture was
evaporated, the residue
was dissolved in DCM and washed with saturated Na2CO3 solution and water. The
organic
layer was separated, dried over MgSO4, filtered and evaporated in vacuo.
Purification of the
residue by flash coloumn chromatography (silica gel, eluent: Et0Ac:Me0H=10:1)
afforded the
10 desired product. Yield: 132 mg (50 %), MS (ESI) m/z: 338.2 [M+H]t
Example 15
545-methy1-4-({5H,6H,7H,8H-pyridor3,4-clpyridazin-3-vloxylmethyl)-1,2-oxazol-3-
v11-2-
(trifluoromethyppyridine
P,
N I
I NH
N N
A: tert-butvl 5[5-methvI-4-({5H,6H,7H,8H-pyrido[3,4-clpyridazi n-3-
vloxylmethyl)-1,2-oxazol-
3-v11-2-(trifluoromethvl)pyridine-2-carboxylate
Under argon atmosphere a flask was charged with 668 mg (2.48 mmol) of
commercially
available tert-butyl 3-chloro-5,8-dihydropyrido[3,4-c]pyridazine-7(6H)-
carboxylate, 639 mg
(2.48 mmol) of 5-methyl-3[6-(trifluoromethyl)pyridin-3-y1]-1,2-oxazol-4-
yllmethanol (WO
2018/104419 Al, Hoffmann-La Roche), 1614 mg (4.95 mmol) of Cs2CO3, 99 mg (0.25
mmol)
of rac-2-(di-tert-butylphosphino)-1,11-binaphthyl, 56 mg (0.25 mmol) of
Pd(OAc)2 and 20 mL
of anhydrous toluene. The mixture was stirred at 100 C for 12 h. The
conversion was checked
by TLC (cyclohexane:Et0Ac=1:1 as eluent, silica plate). The reaction mixture
was filtered
through a celite pad, washed with acetone, dried over anhydrous sodium
sulfate, and
evaporated. The residue was purified by flash coloumn chromatography (silica
gel, eluent:
cyclohexane:EtA0c=1:1). Yield: 395 mg (32.5 %). MS (ESI) m/z: 492.2 [M+H].
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
56
B: 545-methy1-4-({5H,6H,7H,8H-pyrido[3,4-c]pyridazin-3-
yloxy}methyl)-1,2-oxazol-3-y1]-2-
(trifluoromethyppyridine
395 mg (0.80 mmol) of tert-butyl 5-[5-methy1-4-({5H,6H,7H,8H-pyrido[3,4-
c]pyridazin-3-
yloxy}methyl)-1,2-oxazol-3-y1]-2-(trifluoromethyppyridine-2-carboxylate was
dissolved in 20
mL of DCM. Then, 916 mg (8.03 mmol) of trifluoroacetic acid was added to the
solution, and
the suspension was stirred at rt for 24 h. After completion the mixture was
evaporated, the
residue was dissolved in DCM and washed with saturated Na2CO3 solution and
water. The
organic layer was separated, dried over MgSO4, filtered and evaporated in
vacuo. Purification
of the residue by flash coloumn chromatography (silica gel, eluent:
Et0Ac:Me0H=10:1)
afforded the desired product. Yield: 175 mg (56 %), MS (ESI) m/z: 392.1
[M+H]+.
Example 16
2-methy1-545-methyl-4-11(7-methyl-5H,6H,7H,8H-pyridor3,4-elpyridazin-3-
vIloxv)methyll-1,2-oxazol-3-vIlpyridine
0
14\ 0
N
N N
To a solution of 74 mg (0.22 mmol) of 2-methy1-545-methy1-4-({5H,6H,7H,8H-
pyrido[3,4-
c]pyridazin-3-yloxy}methyl)-1,2-oxazol-3-yl]pyridine (Example 14) in 5 mL of
methanol 27 mg
(0.33 mmol) of formaldehyde solution (37% in water) was added and the reaction
mixture was
warmed up to 50 C, then 93 mg (0.44 mmol) of sodium triacetoxyborohydride was
added in
one portion. The reaction mixture was stirred at 50 C for 5 hours. After
completion the solvent
was evaporated, the residue was dissolved in Et0Ac and washed with saturated
NaHCO3
solution. The organic layer was separated, dried over MgSO4, filtered and
evaporated in
vacuo. Purification of the residue by flash coloumn chromatography (silica
gel, eluent:
Et0Ac:Me0H=10:1) afforded the desired product. Yield: 40 mg (52 %), MS (ESI)
m/z: 352.2
[m+H]t
Example 17
5-15-methv1-4-({5H,6H,7H,8H-pyrido[3.4-clpyridazin-3-vloxylmethyl)-1,2-oxazol-
3-v11-2-
(trifluoromethyl)pyridine
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
57
0
14\ 0
X N
The title compound prepared according to the procedure described for Example
16 using 5-
[5-methyl-4-({5H ,6H , 7H , 8H-pyrido[3,4-c]pyridazin-3-yloxy}methyl)-1,2-
oxazol-3-y1]-2-
(trifluoromethyl)pyridine (Example 15). MS (ESI) m/z: 406.1 [M+H].
Example 18
545-methY1-4-({17-(oxolan-3-y1)-5H,6H,7H,8H-pyridor3,4-clpyridazin-3-
ylloxy}methyl)-
1,2-oxazol-3-y11-2-(trifluoromethyl)pyridine hem i napadisylate salt
0 OH
N'\ 0 0=S=0
N. I 0.5 1011101
'CO
N
0=S=0
OH
A: Synthesis of the free base
To a solution of 130 mg (0.33 mmol) of 545-methyl-4-({5H,6H,7H,8H-pyrido[3,4-
c]pyridazin-
3-yloxy}methyl)-1,2-oxazol-3-y1]-2-(trifluoromethyppyridine (Example 15) in 5
mL of 2,2,2-
trifluoroethanol 29 mg (0.34 mmol) of 3-oxotetrahydrofuran and 13 mg (0.34
mmol) of sodium
borohydride was added. The reaction mixture was stirred at 45 C for 12 hours.
After
completion the solvent was evaporated, the residue was dissolved in DCM and
washed with
water. The organic layer was separated, dried over MgSO4, filtered and
evaporated in vacuo.
Purification of the residue by flash coloumn chromatography (silica gel,
eluent:
DCM:Me0H=10:1) afforded the free base as an oil. Yield: 23 mg (15 c/o), MS
(ESI) m/z: 462.2
[M+H]
B: Synthesis of the hem inapadisylate salt
23 mg (0.05 mmol) of 545-methyl-4-({[7-(oxolan-3-y1)-5H,6H,7H,8H-pyrido[3,4-
c]pyridazin-3-
yl]oxy}methyl)-1,2-oxazol-3-y1]-2-(trifluoromethyppyridine was dissolved in 2
mL of ethanol
and 18 mg (0.05 mmol) of 1,5-naphthalenedisulfonic acid tetrahydrate was added
and stirred
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
58
at 60 C for 10 minutes, then allowed to cool to rt. The precipitated product
was collected by
filtration, washed with cold ethanol and dried in vacuum to obtain the title
compound as a white
solid. Yield: 17 mg (56 %), MS (ESI) m/z: 462.2 [M+H]. 1H NMR (DMSO-d6, 400
MHz) 5
(ppm): 10.05-10.50 (br m, 1H), 9.12 (d, J=1.7 Hz, 1H), 8.49 (dd, J=8.1, 1.7
Hz, 1H), 8.10 (br
d, J=8.2 Hz, 1H), 7.21 (br s, 1H), 4.40-4.85 (br m, 2H), 4.07-4.34 (br m, 2H),
3.91-4.06 (br m,
1H), 3.76-3.89 (m, 1H), 3.30-3.74 (br m, 5H), 3.00-3.18 (br m, 2H), 2.64 (s,
3H), 2.12-2.43 (br
m, 2H); napadisylate (acid/base molar ratio 1:2) signals: 8.85 (dd, J=8.5, -1
Hz, 2H), 7.91 (dd,
J=7.0 Hz, 1.1 Hz, 2H), 7.38 (dd, J=8.5, 7.1 Hz, 2H).
Example 19
3413-({5-methyl-3-1-6-(trifluoromethyl)pyridin-3-y11-1,2-oxazol-4-y1}methoxy)-
5H,6H,7H,8H-pyridor3,4-clpyridazin-7-vIlmethvl}-11ambda6-thiolane-1,1-dione
tartarate
salt
0
N/\ 0
0 OH
CSe:1
'0 HO,ily1õTrOH
N N OH 0
A: Synthesis of the free base
In a microwave tube 100 mg (0.256 mmol) of 545-methyl-4-({5H,6H,7H,8H-
pyrido[3,4-
c]pyridazin-3-yloxy}methyl)-1,2-oxazol-3-y1]-2-(trifluoromethyppyridine
(Example 15) was
dissolved in 3 mL of acetonitrile, then 66 mg (0.51 mmol) of N,N-
diisopropylethylamine and
54.6 mg (0.256 mmol) of 3-bromomethyltetrahydrothiophene 1,1-dioxide was
added. The tube
was placed in a microwave reactor and heated at 100 C with stirring for 3
hours. After the
reaction completed, the mixture was evaporated and purified by flash coloumn
chromatography (silica gel, eluent: DCM:Me0H=10:1) to obtain 34 mg product as
an oil. Yield:
38 mg (28.4 %), MS (ESI) m/z: 524.1 [M+H].
B: Synthesis of the tartarate salt
11.2 mg (0.021 mmol) of 3-{[3-({5-methyl-346-(trifluoromethyppyridin-3-y1]-1,2-
oxazol-4-
yl}methoxy)-5H,6H,7H,8H-pyrido[3,4-c]pyridazin-7-yl]methy11-11ambda6-thiolane-
1,1-dione
was dissolved in 1 mL of ethanol and 3.2 mg (0.021 mmol) of L-(+)-tartaric
acid was added
and stirred at 60 C for 10 minutes, then allowed to cool to rt. The
precipitated product was
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
59
collected by filtration, washed with cold ethanol and dried in vacuum to
obtain the title
compound as a white solid. Yield: 12.5 mg (86.7 %), MS (ESI) m/z: 524.1 [M+H].
1H NMR
(DMSO-d6, 400 MHz) 5 (ppm): 11.40-13.60 (br m, 1H), 9.12 (d, J=1.7 Hz, 1H),
8.46 (dd, J=8.0
Hz, 1.8 Hz, 1H), 8.09 (d, J=8.0 Hz, 1H), 7.00 (s, 1H), 5.48 (s, 2H), 3.75 (s,
2H), 3.14-3.26 (m,
2H), 3.00-3.09 (m, 1H), 2.82 (t, J=5.4 Hz, 2H), 2.74-2.81 (m, 2H), 2.54-2.73
(m, 4H), 2.63 (s,
3H), 2.20-2.29 (m, 1H), 1.73-1.83 (m, 1H); tartarate (acid/base ratio 1:1)
signal: 4.28 (s, 2H).
Example 20
641'4-methyl-I -(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yllmethoxy}-1,2,3,4-
tetrahydro-
2,7-naphthyridine
N -1.00N
I NH
A: tert-butyl 6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-
yl]methoxy}-1,2,3,4-
tetrahydro-2,7-naphthyridine-2-carboxylate
Under argon atmosphere a flask was charged with 504 mg (1.88 mmol) of
commercially
available tert-butyl 6-chloro-3,4-dihydro-2,7-naphthyridine-2(1H)-carboxylate,
383 mg (1.88
mmol) of [4-methyl-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methanol
(Intermediate 3),
1220 mg (3.75 mmol) of Cs2003, 74.7 mg (0.18 mmol) of rac-2-(di-tert-
butylphosphino)-1,11-
binaphthyl, 42 mg (0.18 mmol) of Pd(OAc)2 and 20 mL of anhydrous toluene. The
mixture was
stirred at 100 C for 12 h. The conversion was checked by TLC
(cyclohexane:Et0Ac=1:1 as
eluent, silica plate). The reaction mixture was filtered through a celite pad,
washed with
acetone, dried over anhydrous sodium sulfate, and evaporated. The residue was
purified by
flash coloumn chromatography (silica gel, eluent: cyclohexane:EtA0c 30-70%
gradient).
Yield: 287 mg (35 %). MS (ESI) m/z: 437.2 [M+H]4.
B: 6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-
1,2,3,4-tetrahydro-2,7-
naphthyridine
287 mg (0.65 mmol) of tert-butyl 64[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-
triazol-5-
yl]methoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate was dissolved
in 12 mL of
ethyl acetate. 12 mL of ethyl acetate saturated with hydrogen chloride was
added dropwise to
the solution. The reaction mixture was stirred for 30 minutes at room
temperature. The white
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
precipitate formed was filtered out, washed with small portion of ethyl
acetate. The
hydrochloride salt was added to a solution of saturated NaHCO3 and extracted
with Et0Ac.
The organic layer was separated, dried over MgSO4, filtered and evaporated in
vacuo.
Purification of the residue by flash coloumn chromatography (silica gel,
eluent:
5 DCM:Me0H=10:1) afforded the desired product. Yield: 78 mg (35 %), MS
(ESI) m/z: 337.2
[M+H]+.
Example 21
2-methyl-6-(14-methy1-1-(6-methyl pyrid
10 tetrahydro-2,7-naphthyridine
The title compound prepared according to the procedure described for Example
16 using 6-
{[4-methy1-1-(6-m ethyl pyrid i n-3-yI)-1H- 1,2, 3-triazol-5-yl]methoxy}-1,2 ,
3,4-tetrahyd ro-2, 7-
naphthyridine (Example 20). MS (ESI) m/z: 351.1 [M+H].
Example 22
641'4-methyl-I -(6-methylpyridin-3-v1)-1H-1,2,3-triazol-5-vIlmethoxv}-2-
(propan-2-v1)-
1,2,3,4-tetrahydro-2,7-naphthwidine
0
The title compound prepared according to the procedure described for Example
18, Step A
using 6-{[4-methyl-1-(6-m ethyl pyri d n-3-yI)-1H-1,2 , 3-triazol-5-
yl]methoxy}-1,2 ,3,4-tetrahyd ro-
2,7-naphthyridine (Example 20) and commercially available acetone. MS (ESI)
m/z: 379.2
[M+H]t
Example 23
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
61
6-1{1 -r6-(difl uoromethyl)pyridin-3-y11-4-methy1-1 H-1 ,2,3-triazol-5-
vIlmethoxy)-1 ,2,3,4-
tetrahydro-2,7-naphthyridi ne
N NH
NH
FF
N
A: tert-butyl 6-({146-(difl uoromethyl)pyridi n-3-y1]-4-methy1-1H-1,2,3-
triazol-5-yl}methoxy)-
1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate
Under argon atmosphere a flask was charged with 91.3 mg (0.34 mmol) of
commercially
available tert-butyl 6-chloro-3,4-dihydro-2,7-naphthyridine-2(1H)-carboxylate,
81.6 mg (0.34
mmol) of {1[6-(difluoromethyl)pyridin-3-y1]-4-methy1-1H-
1,2,3-triazol-5-yl}methanol
(Intermediate 5), 226 mg (0.69 mmol) of Cs2CO3, 13.8 mg (0.034 mmol) of rac-2-
(di-tert-
butylphosphino)-1,11-binaphthyl, 7.8 mg (0.034 mmol) of Pd(OAc)2 and 10 mL of
anhydrous
toluene. The mixture was stirred at 100 C for 12 h. The conversion was checked
by TLC
(cyclohexane:Et0Ac=1:2 as eluent, silica plate). The reaction mixture was
filtered through a
celite pad, washed with acetone, dried over anhydrous sodium sulfate, and
evaporated. The
residue was purified by flash coloumn chromatography (silica gel, eluent:
cyclohexane:EtA0c=1:2). Yield: 90 mg (56 %). MS (ESI) m/z: 473.2 [M+H].
B: 6-({146-(difluoromethyppyridin-341-4-methyl-1H-1,2,3-triazol-5-
yllmethoxy)-12,3,4-
tetrahydro-2,7-naphthyridine
90 mg (0.19 mmol) of tert-butyl 6-({1-[6-(difluoromethyl)pyridin-3-y1]-4-
methy1-1H-1,2,3-triazol-
5-yllmethoxy)-1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate was dissolved
in 10 mL of
DCM. Then, 652 mg (5.71 mmol) of trifluoroacetic acid was added to the
solution, and the
suspension was stirred at rt for 3 h. After completion the mixture was
evaporated, the residue
was dissolved in DCM and washed with saturated Na2CO3 solution and water. The
organic
layer was separated, dried over MgSO4, filtered and evaporated in vacuo.
Purification of the
residue by flash coloumn chromatography (silica gel, eluent: DCM:Me0H=10:1)
afforded the
desired product. Yield: 28.4 mg (40 %), MS (ESI) m/z: 373.2 [M+H]+.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
62
Example 24
6-1{146-(difluoromethyl)pyridin-3-v11-4-methyl-1H-1,2,3-triazol-5-vIlmethoxv)-
2-methyl-
1,2,3,4-tetrahydro-2,7-naphthyridine
--c)
FNF
N
The title compound prepared according to the procedure described for Example
16 using 6-
({146-(difluoromethyl)pyridi n-3-yI]-4-methyl-1H -1,2 ,3-triazol-5-yl}methoxy)-
1,2,3,4-
tetrahydro-2 ,7-naphthyridine (Example 23). MS (ESI) m/z: 387.2 [M+H]t
Example 25
6-({4-methyl-146-(trifluoromethyl)pyridin-3-y1]-1H-1,2,3-triazol-5-yl}methoxy)-
1,2,3,4-
tetrahydro-2,7-naphthyridine
I NH
N
A: tert-butyl 6-({4-methyl-146-(trifluoromethyppyridin-3-y1]-1H-1,2,3-triazol-
5-yl}methoxy)-
1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate
Under argon atmosphere a flask was charged with 521 mg (1.94 mmol) of
commercially
available tert-butyl 6-chloro-3,4-dihydro-2,7-naphthyridine-2(1H)-carboxylate,
500 mg (1.94
mmol) of 4-methyl-116-(trifluoromethyppyridin-3-y1]-1H-1,2,3-
triazol-5-yl}methanol
(intermediate 4), 1260 mg (3.87 mmol) of Cs2CO3, 77.2 mg (0.194 mmol) of rac-2-
(di-tert-
butylphosphino)-1,11-binaphthyl, 43.5 mg (0.194 mmol) of Pd(OAc)2 and 30 mL of
anhydrous
toluene. The mixture was stirred at 100 C for 12 h. The conversion was checked
by TLC
(DCM:Me0H=9:1 as eluent, silica plate). The reaction mixture was filtered
through a celite
pad, washed with acetone, dried over anhydrous sodium sulfate, and evaporated.
The residue
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
63
was purified by flash coloumn chromatography (silica gel, eluent:
DCM:Me0H=9:1). Yield: 710
mg (74.8 %), amorphous solid. MS (ESI) m/z: 491.2 [M+H].
B: 6-({4-methyl-146-(trifluoromethyppyridin-3-y1]-1H-1,2,3-
triazol-5-yl}methoxy)-1,2,3,4-
tetrahydro-2,7-naphthyridine
710 mg (1.45 mmol) of tert-butyl 6-({4-methyl-146-(trifluoromethyl)pyridin-3-
y1]-1H-1,2,3-
triazol-5-yl}methoxy)-1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate was
dissolved in 15
mL of DCM. Then, 3300 mg (29 mmol) of trifluoroacetic acid was added to the
solution, and
the suspension was stirred at it for 24 h. After completion the mixture was
evaporated, the
residue was dissolved in DCM and washed with saturated Na2CO3 solution and
water. The
organic layer was separated, dried over MgSO4, filtered, and evaporated in
vacuo. Purification
of the residue by flash coloumn chromatography (silica gel, eluent:
DCM:Me0H=9:1) afforded
the desired product. Yield: 320 mg (56.6 %), MS (ESI) m/z: 391.2 [M+H].
Example 26
6-1{4-methyl-146-(trifluoromethyppyridin-3-v11-1H-1,2,3-triazol-5-yl}methoxy)-
2-
(Propan-2-v1)-1,2,3,4-tetrahydro-2,7-naphthyridine
N" JILQ
.5\,) N=., I
N N
To a solution of 160 mg (0.41 mmol) of 6-({4-methyl-146-
(trifluoromethyl)pyridin-3-y1]-1H-
1,2,3-triazol-5-yllmethoxy)-1,2,3,4-tetrahydro-2,7-naphthyridine (Example 25)
in 5 mL of
2,2,2-trifluoroethanol 23.8 mg (0.41 mmol) of acetone and 15.5 mg (0.41 mmol)
of sodium
borohydride was added. The reaction mixture was stirred at 45 C for 12 hours.
After
completion the solvent was evaporated, the residue was dissolved in DCM and
washed with
water. The organic layer was separated, dried over MgSO4., filtered, and
evaporated in vacua.
Purification of the residue by flash coloumn chromatography (silica gel,
eluent:
DCM:Me0H=9:1) afforded the title compound. Yield: 61 mg (34 %), MS (ESI) m/z:
433.2
[M+H].
Example 27
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
64
2-methy1-6-({4-methyl-1 -f6-(trifl uoromethyl)pyridi n-3-y11-1 H-1 ,2,3-
triazol-5-ylImethoxy)-
1 ,23,4-tetrahydro-2,7-naphthyridine
N., I
N N
The title compound prepared according to the procedure described for Example
16 using 6-
({4-methyl-146-(trifluoromethyl)pyridin-3-y1]-1H-1,2,3-triazol-5-yl}methoxy)-
1,2,3,4-
tetrahydro-2,7-naphthyridine (Example 25, Step B). MS (ESI) m/z: 405.1 [M+H].
Example 28
6-1{1 F6-(difluoromethyl)pyridin-3-y11-4-methy1-1 H-1 ,2,3-triazol-5-
vIlmethoxy)-2-
(propan-2-v1)-1,2,3,4-tetrahyd ro-2,7-naphthyridi ne
N
oCC1
N NT.
N
F-1\)
The title compound prepared according to the procedure described for Example
18, Step A
using 6-({146-(difluoromethyppyridin-3-y1]-4-methy1-1H-1,2,3-
triazol-5-yl}methoxy)-1,2,3,4-
tetrahydro-2,7-naphthyridine (Example 23) and commercially available acetone.
MS (ESI)
m/z: 415.2 [M+H]t
Example 29
6-{14-methvI-1-(6-methylvvridin-3-v1)-1 H-1 .2.3-triazol-5-vIlmethoxv}-2-
(oxolan-3-v1)-
1,2,3,4-tetrahydro-2,7-naphthyridine
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
caco
The title compound prepared according to the procedure described for Example
18, Step A
using 6-1[4-methyl-1-(6-methylpyridin-3-0-1H-1,2,3-triazol-5-yl]methoxy}-
1,2,3,4-tetrahydro-
2,7-naphthyridine (Example 20) and commercially available 3-
oxotetrahydrofuran. MS (ESI)
5 m/z: 407.2 [M+H].
Example 30
6-1(146-(difluoromethyl)pyridin-3-y11-4-methy1-1H-1,2,3-triazol-5-vIlmethoxy)-
2-(oxolan-
3-v1)-1,2,3,4-tetrahydro-2,7-naphthyridine
-c)
CC.1".4
N
The title compound prepared according to the procedure described for Example
18, Step A
using 6-({146-(difluoromethyl)pyridin-3-y1]-4-methy1-1H-1,2,3-
triazol-5-yl}methoxy)-1,2,3,4-
tetrahydro-2,7-naphthyridine (Example 23) and commercially available 3-
oxotetrahydrofuran.
MS (ESI) m/z: 443.2 [M-'-H].
Example 31
6-f 14-methvl-1-(6-methvl Dvridi n-3-v1)-1H-1 .2.3-triazol-5-vIlmethoxv}-2-
(oxetan-3-v1)-
1,2,3,4-tetrahydro-2,7-naphthyridine
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
66
The title compound prepared according to the procedure described for Example
18, Step A
using 6-{[4-methyl-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-
1,2,3,4-tetrahydro-
2,7-naphthyridine (Example 20) and commercially available 3-oxetanone. MS
(ESI) rn/z: 393.2
[WEN+.
Example 32
64{4-methyl-1-1-6-(trifluoromethyl)pyridin-3-y1-1-1 H-1 ,2,3-triazol-5-
yl}methoxy)-2-
(oxolan-3-yI)-1,2,3,4-tetrahydro-2,7-naphthyridine
N1j0
X N
The title compound prepared according to the procedure described for Example
26 using
commercially available 3-oxotetrahydrofuran. MS (ESI) rn/z: 461.2 [m+H].
Example 33
64[4-methyl-1-(6-methylpyridin-3-y1)-1 H-1 ,2,3-triazol-5-yllmethoxy}-2-(oxan-
4-y1)-
1,2,3,4-tetrahydro-2,7-naphthyridine
--K0
ctCL
The title compound prepared according to the procedure described for Example
18, Step A
using 6-{[4-methyl-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]nethoxy}-
1,2,3,4-tetrahydro-
2,7-naphthyridine (Example 20) and commercially available 4-
oxotetrahydropyran. MS (ESI)
rn/z: 421.2 [M+H].
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
67
Example 34
641'1 -(6-methoxypyridi n-3-yI)-4-methyl-1 methoxy}-1,2,3,4-
tetrahydro-2,7-naphthyridi ne
Ni a
ECNH
A: tert-butvl 6-411-(6-methoxviovridin-3-v1)-4-methvI-1H-1,2,3-triazol-5-
vIlmethoxv}-1,2,3,4-
tetrahvdro-2,7-naphthvridine-2-carboxvlate
Under argon atmosphere a flask was charged with 300 mg (1.12 mmol) of
commercially
available tert-butyl 6-chloro-3,4-dihydro-2,7-naphthyridine-2(1H)-carboxylate,
246 mg (1.12
mmol) of [1-(6-methoxypyridin-3-y1)-4-methy1-1H-1,2,3-triazol-5-yl]methanol
(Intermediate 6),
727 mg (2.23 mmol) of Cs2CO3, 44.5 mg (0.11 mmol) of rac-2-(di-tert-
butylphosphino)-1,11-
binaphthyl, 25 mg (0.11 mmol) of Pd(OAc)2 and 20 mL of anhydrous toluene. The
mixture was
stirred at 100 C for 12 h. The conversion was checked by TLC
(cyclohexane:Et0Ac=1:1 as
eluent, silica plate). The reaction mixture was filtered through a celite pad,
washed with
acetone, dried over anhydrous sodium sulfate, and evaporated. The residue was
purified by
flash coloumn chromatography (silica gel, eluent: cyclohexane:EtA0c=1:1).
Yield: 200 mg
(39.5 %). MS (ES1) rrilz: 453.2 [M-'-H]t
B: 6-{[1-(6-methoxypyridin-3-y1)-4-methy1-1H-1,2,3-triazol-5-yl]methoxy}-
1,2,3,4-tetrahydro-
2,7-naphthyridine
200 mg (0.44 mmol) of tert-butyl 6-{[1-(6-rnethoxypyridin-3-y1)-4-methyl-1H-
1,2,3-triazol-5-
yl]methoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine-2-carboxylate was dissolved
in in 7 mL of
ethyl acetate. 7 mL of ethyl acetate saturated with hydrogen chloride was
added dropwise to
the solution. The reaction mixture was stirred for 30 minutes at room
temperature. The white
precipitate formed was filtered out, washed with small portion of ethyl
acetate. The
hydrochloride salt was added to a solution of saturated NaHCO3 and extracted
with Et0Ac.
The organic layer was separated, dried over MgSO4., filtered and evaporated in
vacuo.
Purification of the residue by flash coloumn chromatography (silica gel,
eluent:
DCM:Me0H=10:1) afforded the desired product. Yield: 115 mg (74 %), MS (ES1)
m/z: 353.2
[M+H]+.
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
68
Example 35
6-1{4-methy1-1F6-(trifluoromethyl)pyridin-3-v11-1 H-1 ,2,3-triazol-5-
yllmethoxy)-2-(oxan-
4-v1)-1,2,3,4-tetrahydro-2,7-naphthyridine
N
The title compound prepared according to the procedure described for Example
26 using
commercially available 4-oxotetrahydropyran. MS (ESI) m/z: 475.3 [m+H]t
Example 36
3-{16-({4-methv1-1-1-6-(trifluoromethvflpyridin-3-v11-1 H-1 ,2,3-triazol-5-
v1}methoxv)-
1,2,3,4-tetrahydro-2,7-naphthyridin-2-yllmethylM1ambda6-thiolane-1,1-dione
heminapadisylate salt
OH
0=S=0
fc
,0
I 0.5 00
X N 0=S=0
FLF
The free base of the title compound prepared according to the procedure
described for
Example 19, Step A using 6-({4-methyl-1-[6-(trifluoromethyppyridin-3-y1]-1H-
1,2,3-triazol-5-
yllmethoxy)-1,2,3,4-tetrahydro-2,7-naphthyridine (Example 25). The
heminapadisylate salt
prepared according to the procedure described for Example 18 in Step B. MS
(ESI) m/z: 523.2
[M+H]. 1H NMR (DMSO-d6, 400 MHz) 6 (ppm): 9.45-9.70 (br m, 1H), 9.09 (d, J=2.4
Hz, 1H),
8.47 (dd, J=8.3 Hz, 2.2 Hz, 1H), 8.23 (d, J=8.3 Hz, 1H), 7.93-7.99 (br m, 1H),
6.72 (br s, 1H),
5.51 (s, 2H), 4.50-4.68 (m, 1H), 4.14-4.29 (br m, 1H), 3.60-3.76 (br m, 1H),
3.21-3.54 (br m,
5H), 3.00-3.12 (m, 3H), 2.84-3.00 (br m, 2H), 2.43 (s, 3H), 2.29-2.40 (br m,
1H), 1.77-1.91 (br
m, 1H); napadisylate (acid/base molar ratio 1:2) signals: 8.85 (dd, J=8.5, -1
Hz, 2H), 7.91 (dd,
J=7.0 Hz, 1.1 Hz, 2H), 7.39 (dd, J=8.5, 7.1 Hz, 2H).
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
69
Example 37
6-U4-methyl-I 46-(trifl uoromethyl)pyridi n-3-vI1-1 H-1 ,2,3-triazol-5-
yllmethoxy)-2-
(Pvridin-3-vI)-1,2,3,4-tetrahvdro-2,7-naphthvridine napadvsilate salt
OH
0=S=0
NI/ NOS
N N
0=S=0
F-5\1
01 H
A: Synthesis of the free base
In a microwave tube, under argon atmosphere 239 mg (0.612 mmol) of 6-({4-
methyl-146-
(trifluoromethyppyridin-3-y1]-1H-1,2,3-triazol-5-yllmethoxy)-1,2,3,4-
tetrahydro-2,7-
naphthyridine (Example 25), 117 mg (0.741 mmol) of 3-bromopyridine, 141 mg
(1.26 mmol)
of potassium tert-butoxide, 38 mg (0.061 mmol) of 2,2-bis(Diphenylphosphino)-
1,1'-
binaphthalene, 13.7 mg (0.061 mmol) of Pd(OAc)2 and 5 mL of anhydrous toluene.
The tube
was placed in a microwave reactor and heated at 120 C with stirring for 1
hours. After the
reaction completed, the mixture was evaporated and purified by flash coloumn
chromatography (silica gel, eluent: DCM:Me0H=10:1) to obtain 19 mg product as
an oil. Yield:
19 mg (6.6 %), MS (ESI) m/z: 468.2 [M+H]t
B: Synthesis of the napadisylate salt
19 mg (0.041mmol) of 6-({4-methyl-1-[6-(trifluoromethyppyridin-3-y1]-1H-1,2,3-
triazol-5-
yllmethoxy)-2-(pyridin-3-y1)-1,2,3,4-tetrahydro-2,7-naphthyridine was
dissolved in 2 mL of
methanol and 14.7 mg (0.041 mmol) of 1,5-naphthalenedisulfonic acid
tetrahydrate was
added and stirred at 60 C for 10 minutes, then allowed to cool to rt. The
precipitated product
was collected by filtration, washed with cold methanol, and dried in vacuum to
obtain the title
compound as a yellow solid. Yield: 11 mg (36 %), MS (ESI) m/z: 468.2 [M-'-H].
1H NMR
(DMSO-d6, 400 MHz) 6 (ppm): 9.09 (d, J=2.4 Hz, 1H), 8.46 (dd, J=8.4 Hz, 2.2
Hz, 1H), 8.43
(d, J=2.8 Hz, 1H), 8.21(d, J=8.4 Hz, 1H), 8.17 (d, J=5.3 Hz, 1H), 8.06 (dd,
J=8.8 Hz, 2.7 Hz,
1H), 7.96 (s, 1H), 7.85 (dd, J=8.9 Hz, 5.4 Hz, 1H), 6.69 (s, 1H), 5.49 (s,
2H), 4.55 (s, 2H), 3.65
(t, J=6.0 Hz, 2H), 2.92 (t, J=6.0 Hz, 2H), 2.43 (s, 3H); napadisylate
(acid/base molar ratio 1:1)
signals: 8.86 (dd, J=8.5 Hz, -1 Hz, 2H), 7.92 (dd, J=7.0 Hz, 1.1 Hz, 2H), 7.40
(dd, J=8.5 Hz,
7.1 Hz, 2H).
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
Example 38
6-114-methyl-I -(6-methyl pyridi n-3-vI)-1 H-1 ,2,3-triazol-5-vIlmethoxyl-2-
113S)-oxolan-3-
v11-1 ,2,3,4-tetrahydro-2,7-naphthyridine or enantiomer, tartarate salt
0 OH
H0)-Lyiy0H
[W1 OH 0
5 Separation of the enantiomers of the racemic 6-{[4-methyl-1-(6-
methylpyridin-3-yI)-1H-1,2,3-
triazol-5-yl]methoxy}-2-(oxolan-3-y1)-1,2,3,4-tetrahydro-2,7-naphthyridine
(Example 29) by
chiral HPLC (column: Lux i-Amylose-1 5pm 150x21,2mm) afforded the enantiopure
title
compound. MS (ESI) rn/z: 407.2 [M-I-H]. The tartarate salt prepared according
to the
procedure described for Example 19 in Step B. MS (ESI) miz: 407.2 [M+H]t
Example 39
641'4-methyl-I -(6-methyl pyridi n-3-vI)-1 H-1 ,2,3-triazol-5-yllmethoxy}-2-
113R)-oxolan-3-
V11-1 ,2,3,4-tetrahydro-2,7-naphthyridine or enantiomer, tartarate salt
0 OH
HOJ-L,fAir,OH
.CO OH 0
Separation of the enantiomers of the racemic 6-{[4-methyl-1-(6-methylpyridin-3-
y1)-1H-1,2,3-
triazol-5-yl]nethoxy}-2-(oxolan-3-y1)-1,2,3,4-tetrahydro-2,7-naphthyridine
(Example 29) by
chiral HPLC (column: Lux i-Amylose-1 5pm 150x21,2mm) afforded the enantiopure
title
compound. MS (ESI) rn/z: 407.2 [M+H]. The tartarate salt prepared according to
the
procedure described for Example 19 in Step B. MS (ESI) m/z: 407.2 [M+H].
Example 40
64[1 -(6-methoxypyridi n-3-yI)-4-methyl-1
methoxy}-2-(oxan-4-yI)-
1,2,3,4-tetrahydro-2,7-naphthyridine heminapadisylate salt
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
71
OH
0=S=0
0.5 001
\ N
N N
0=3=0
0H
The free base of the title compound prepared according to the procedure
described for
Example 18, Step A using 6-{[1-(6-methoxypyridin-3-y1)-4-methy1-1H-1,2,3-
triazol-5-
yl]methoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine (Example 34) and commercially
available 4-
oxotetrahydropyran. MS (ESI) m/z: 437.2 [M+H]4. The heminapadisylate salt
prepared
according to the procedure described for Example 18 in Step B. MS (ESI) m/z:
437.2 [m+H].
1H NMR (DMSO-d6, 800 MHz) 5 (ppm): 9.70-9.77 (br m, 1H), 8.41 (d, J=2.8 Hz,
1H), 8.01 (s,
1H), 7.97 (dd, J=8.8, 2.7 Hz, 1H), 7.06 (d, J=8.8 Hz, 1H), 6.74 (s, 1H), 5.35-
5.41 (AB d, J=13.5
Hz, 2H), 4.52 (d, J=14.6, 1H), 4.31 (dd, J=15.0, 8.3 Hz, 1H), 3.98 (br d,
J=11.1 Hz, 2H), 3.94
(s, 3H), 3.70-3.74 (m, 1H), 3.49-3.55 (m, 1H), 3.26-3.35 (m, 3H), 3.01-3.10
(m, 2H), 2.39 (s,
3H), 2.04 (br d, J=12.0 Hz, 1H), 1.99 (br d, J=12.2 Hz, 1H), 1.63-1.73 (m,
2H); napadisylate
(acid/base molar ratio 1:2) signals: 8.85 (dd, J=8.4, 1.0 Hz, 2H), 7.91 (dd,
J=7.0, 1.0 Hz, 2H),
7.38 (dd, J=8.4, 7.0 Hz, 2H).
Example 41
6414-methyl-1-(6-methy1pyr1d1n-3-y1)-1H-1,2,3-triazol-5-yllmethoxy}-2-(2-
methylpropy1)-
1,2,3,4-tetrahydro-Z7-naphthyridine heminapadisylate salt
OH
0=3=0
cj- N 0.5 O.
/
OH
N
0=3=0
The free base of the title compound prepared according to the procedure
described for
Example 18, Step A using 6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-
triazol-5-
yl]methoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine (Example 20) and commercially
available
isobutyraldehyde. MS (ESI) m/z: 393.3 [M+H]. The heminapadisylate salt
prepared according
to the procedure described for Example 18 in Step B. MS (ESI) m/z: 393.3
[M+H]. 1H NMR
(DMSO-d6, 400 MHz) 5 (ppm): 9.22-9.40 (br m, 1H), 867(d, J=2.4 Hz, 1H), 799(s,
1H), 7.98
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
72
(dd, J=8.3, 2.6 Hz, 1H), 7.52 (d, J=8.3 Hz, 1H), 6.72 (s, 1H), 5.39 (s, 2H),
4.56 (bid, J=14.43
Hz, 1H), 4.19 (dd, J=15.1, 7.7 Hz, 1H), 3.62-3.71 (m, 1H), 3.22-3.36 (m, 1H),
2.97-3.15 (m,
4H), 2.58 (s, 3H), 2.39 (s, 3H), 2.15 (sep, J=6.7, 1H), 0.98 (t, J=6.1 Hz,
6H); napadisylate
(acid/base molar ratio 1:2) signals: 8.85 (dd, J=8.4, 1.2 Hz, 2H), 7.91 (dd,
J=7.0, 1.2 Hz, 2H),
7.38 (dd, J=8.4, 7.0 Hz, 2H).
Example 42
641'4-methyl-I -(6-methyl pyridi n-3-yI)-1 H-1 ,2,3-triazol-5-yllmethoxyl-243-
(propan-2-
vl)oxetan-3-y11-1 ,2,3,4-tetrahydro-2,7-naphthyridine napadisylate salt
OH
1
0=S=0
0 0=S=0
01 H
A: 2-[3-(1H- 1,2,3- benzotriazol- 1-y0oxetan-3-y1]-6-{[4-methyl- 1-(6-
methylpyridin-3-y1)- 1H-
1,2,3-triazol-5-yl]methoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine
To a solution of 1030 mg (3.06 mmol) of 64[4-methy1-1-(6-methylpyridin-3-y1)-
1H-1,2,3-triazol-
5-yl]methoxy}-1,2,3,4-tetrahydro-2,7-naphthyridine (Example 20) in 30 mL of
DCM 243 mg
(3.37 mmol) of 3-oxetanone and 383 mg (3.21 mmol) of 1H-benzotriazole was
added. The
reaction mixture was stirred at rt for 12 hours. After completion the solvent
was evaporated to
dryness to obtain the title compound as a white solid. Yield: 1540 mg (98.7
%), MS (ESI) m/z:
510.2 [M+H]t
B: 64[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-yl]methoxy}-2-[3-
(propan-2-
yl)oxetan-3-yI]-1,2,3,4-tetrahydro-2,7-naphthyridine
Under argon atmosphere a solution of 520 mg (1.02 mmol) of 243-(1H-1,2,3-
benzotriazol-1-
yl)oxetan-3-y1]-6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-
yl]nethoxy}-1,2,3,4-
tetrahydro-2,7-naphthyridine in 10 mL of THF was added to 593 mg (4.08 mmol)
of
isopropylmagnesium chloride lithium chloride complex solution in one portion.
The reaction
mixture was stirred at rt for 10 min. After the reaction completed, the
mixture was quenched
with water and extracted with Et0Ac. The organic layer was separated, dried
over MgSO4.,
filtered, and evaporated in vacuo. Purification of the residue by flash
coloumn chromatography
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
73
(silica gel, eluent: hexane:Et0Ac: 2c/oEt3N, 30-60% gradient) afforded the
title compound.
Yield: 177 mg (40 %), MS (ESI) m/z: 435.2 [M+H]4.
C: 6-{[4-methy1-1-(6-methylpyridin-3-y1)-1H-1,2,3-triazol-5-
yl]methoxy}-2-[3-(propan-2-
y0oxetan-3-y1]-1,2,3,4-tetrahydro-2,7-naphthyridine napadisylate salt
The heminapadisylate salt prepared according to the procedure described for
Example 36 in
Step B. MS (ESI) m/z: 435.2 [M+H]t 1H NMR (DMSO-d6, 500 MHz) 5 (ppm): 9.60-
10.50 (br
m, 1H), 8.71 (d, J=2.4 Hz, 1H), 8.04 (dd, J=8.3 Hz, 2.4 Hz, 1H), 7.99 (s, 1H),
7.57 (d, J=8.3
Hz, 1H), 6.75 (s, 1H), 5.41 (s, 2H), 4.69 (AB d, J=8.8 Hz, 2H), 4.66 (AB d,
J=8.8 Hz, 2H), 4.35-
4.61 (br m, 2H), 3.44-3.83 (br m, 2H), 3.04-3.17 (br m, 2H), 2.60 (s, 3H),
2.40 (s, 3H), 2.34-
2.44(m, 1H), 1.13(d, J=6.7 Hz, 6H); napadisylate (acid/base molar ratio 1:1)
signals: 8.85 (br
d, J=8.6 Hz, 2H), 7.91 (d, J=7.0 Hz, 2H), 7.40 (dd, J=8.4 Hz, 7.3 Hz, 2H).
Example 43
2-(3-ethvloxetan-3-v1)-6414-methvI-1-(6-methvl pvridi n-3-v1)-1H-1,2,3-triazol-
5-
vIlmethoxv}-1,2,3,4-tetrahvdro-2,7-naphthvridine napadisvlate salt
OH
1\1/ j 0=S=0
N
0 0=S=0
01H
The title compound prepared according to the procedure described for Example
42 using
ethylmagnesium bromide solution in Step B. MS (ESI) m/z: 421.2 [M+H]t 1H NMR
(DMS0-
d8, 500 MHz) 5 (ppm): 10.54-10.96 (br m, 1H), 8.70 (d, J=2.4 Hz, 1H), 8.02
(dd, J=8.3 Hz, 2.6
Hz, 1H), 7.95 (s, 1H), 7.56 (d, J=8.3 Hz, 1H), 6.75 (s, 1H), 5.41 (s, 2H),
4.80 (br d, 2H), 4.57
(d, J=8.1 Hz, 2H), 4.24-4.44 (br m, 2H), 3.26-3.52 (br m, 2H), 3.02-3.18 (br
m, 2H), 2.60 (s,
3H), 2.40 (s, 3H), 1.78-1.96 (br m, 2H), 1.23 (t, J=7.3 Hz, 3H); napadisylate
(acid/base molar
ratio 1:1) signals: 8.85 (br d, J=8.5 Hz, 2H), 7.91 (dd, J=7.0 Hz, 0.9 Hz,
2H), 7.39 (dd, J=8.5
Hz, 7.1 Hz, 2H).
Example 44
2-methy1-5-14-methvI-5-({5H,6H,7H,8H-pvridor3,4-clpvridazin-3-yloxy}methvI)-1H-
1,2,3-
triazol-1-vIlpvridine
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
74
fJN'nCINH
'N
A: tert-butyl 2-methyl-544-methyl-5-(4.5H ,6H ,7H ,8H-pyrido[3,4-
c1pyridazin-3-yloxylmethyl)-
1H-1,2, 3-triazol-1-yllpyridine-2-carboxylate
In a microwave tube, under argon atmosphere 135 mg (0.50 mmol) of commercially
available
tert-butyl 3-chloro-5,8-dihydropyrido[3,4-c]pyridazine-7(6H)-carboxylate, 102
mg (0.50 mmol)
of [4-methyl-1-(6-methylpyridin-3-yI)-1H-1,2,3-triazol-5-yl]methanol
(Intermediate 3), 112 mg
(1.00 mmol) of potassium tert-butoxide, 20 mg (0.05 mmol) of rac-2-(di-tert-
butylphosphino)-
1,11-binaphthyl, 11.2 mg (0.05 mmol) of Pd(OAc)2 and 10 mL of anhydrous
toluene was
added. The tube was placed in a microwave reactor and heated at 120 C with
stirring for 3
hours. After the reaction completed, the mixture was filtered through a celite
pad, washed with
acetone, dried over anhydrous sodium sulfate, and evaporated. The residue was
purified by
flash coloumn chromatography (silica gel, eluent: cyclohexane:EtA0c=1:1).
Yield: 57 mg (26
%). MS (ESI) m/z: 438.2 [M+H]t
B: 2-methyl-544-methyl-5-({5H ,6H ,7H ,8H-pyrido[3,4-c]pyridazi n-3-
yloxy}methyl)-1H-1,2, 3-
triazol-1-yl]pyridine
138 mg (0.31 mmol) of tert-butyl 2-methyl-544-methyl-5-({5H,6H,7H,8H-
pyrido[3,4-
c]pyridazin-3-yloxy}methyl)-1H-1,2,3-triazol-1-yl]pyridine-2-carboxylate was
dissolved in 10
mL of DCM. Then, 360 mg (3.16 mmol) of trifluoroacetic acid was added to the
solution, and
the suspension was stirred at rt for 48 h. After completion the mixture was
evaporated, the
residue was dissolved in DCM and washed with saturated Na2003 solution and
water. The
organic layer was separated, dried over MgSO4, filtered and evaporated in
vacuo. Purification
of the residue by flash coloumn chromatography (silica gel, eluent:
DCM:Me0H=10:1)
afforded the desired product. Yield: 77 mg (72 %), MS (ESI) m/z: 338.1 [M+H]+.
Example 45
5-15-({17-(cyclobutylmethyl)-5H,6H,7H,8H-pyrido[3,4-clpyridazin-3-
ylloxylmethyl)-4-
methyl-1H-1,2,3-triazol-1-y11-2-methylpyridine tartarate salt
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
0 OH
XNO HO)lyrOH
The free base of the title compound prepared according to the procedure
described for
Example 18, Step A using 2-methyl-544-methyl-5-({5H,6H,7H,8H-pyrido[3,4-
c]pyridazin-3-
yloxy}methyl)-1H-1,2,3-triazol-1-yl]pyridine (Example 44) and commercially
available
5 cyclobutanecarboxaldehyde. MS (ESI) m/z: 406.3 [M+H]. The tartarate salt
prepared
according to the procedure described for Example 19 in Step B. MS (ESI) m/z:
406.3 [M+H].
1H NMR (DMSO-d6, 500 MHz) 6 (ppm): 8.67 (d, J=2.5 Hz, 1H), 7.97 (dd, J=8.3 Hz,
2.6 Hz,
1H), 7.49 (d, J=8.3 Hz, 1H), 6.96 (s, 1H), 5.53 (s, 2H), 3.68 (s, 2H), 2.80
(t, J=5.8 Hz, 2H),
2.64 (t, J=5.9 Hz, 2H), 2.55-2.62 (m, 1H), 2.57 (d, J=7.0 Hz, 2H), 2.56 (s,
3H), 2.41 (s, 3H),
10 2.00-2.08 (m, 2H), 1.75-1.93 (m, 2H), 1.64-1.73 (m, 2H); tartarate
(acid/base ratio 1:1) signal:
4.28 (s, 2H).
Example 46
545-11(7-cyclobutyl-5H ,6H ,7H ,8H-pyridor3,4-clpyridazi n-3-ylloxy)methy11-4-
methy1-1H-
15 1,2,3-triazol-1-y11-2-methylpyridine tartarate salt
Ni1Y-K0
0 OH
1W1HOJ-yrOH
OHO
The free base of the title compound prepared according to the procedure
described for
Example 18, Step A using 2-methyl-544-methyl-5-({5H,6H,7H,8H-pyrido[3,4-
c]pyridazin-3-
yloxy}methyl)-1H-1,2,3-triazol-1-yl]pyridine (Example 44) and commercially
available
20 cyclobutanone. MS (ESI) m/z: 392.2 [M+H]t The tartarate salt prepared
according to the
procedure described for Example 19 in Step B. MS (ESI) m/z: 392.2 [M+H]. 1H
NMR (DMSO-
d6, 500 MHz) 6 (ppm): 8.67 (d, J=2.5 Hz, 1H), 7.98 (dd, J=8.3 Hz, 2.6 Hz, 1H),
7.50 (d, J=8.3
Hz, 1H), 6.97 (s, 1H), 553(s, 2H), 3.58 (s, 2H), 2.95 (qui, J=7.6 Hz, 1H),
2.81 (br t, J=5.8 Hz,
2H), 2.57 (s, 3H), 2.52 (br t, J=5.8 Hz, 2H), 2.41 (s, 3H), 2.03-2.11 (m, 2H),
1.82-1.91 (m, 2H),
25 1.63-1.71 (m, 2H); tartarate (acid/base ratio 1:1) signal: 4.28 (s, 2H).
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
76
Pharmaceutical preparation examples
The following formulation examples illustrate representative pharmaceutical
compositions of this invention. The present invention however is not limited
to the following
pharmaceutical compositions.
A) Solid oral dosage forms
I., Tablets
Active ingredient(s) 0.01 ¨ 90%
Filler 1 ¨ 99.9%
Binder 0 ¨ 20%
Disintegrant 0 ¨ 20%
Lubricant 0¨ 10%
Other specific excipient(s) 0 ¨ 50%
II., Orodispersible films
Active ingredient(s) 0.01 ¨ 90%
Film forming agent 1 ¨ 99.9%
Plasticizer 0 ¨ 40%
Other specific excipient(s) 0 ¨ 50%
B) Liquid oral dosage forms
III., Oral suspensions
Active ingredient(s) 0.01 ¨ 50%
Liquid vehicle 10 ¨ 99.9%
Wetting agent 0 ¨ 50%
Thickener 0 ¨ 50%
Buffering agent q.s.
Osmotic agent 0 ¨ 50%
Preservatives q.s.
IV., Syrups
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
77
Active ingredient(s) 0.01 ¨ 50%
Solvent 10 ¨ 99.9%
Sugar component 1 ¨ 20%
Flavouring agents 0 ¨ 10%
C) Parenteral dosage forms
V., Intravenous injections
Active ingredient(s) 0.01 ¨ 50%
Solvent 10 ¨ 99.9%
Co-solvent 0 ¨ 99.9%
Osmotic agent 0 ¨ 50%
Buffering agent q.s.
D) Other dosage forms
VI., Suppositories
Active ingredient(s) 0.01 ¨ 50%
Suppository base 1 ¨ 99.9%
Surface-active agents 0 ¨ 20%
Lubricants 0 ¨ 20%
Preservatives q.s.
VII., Eye drops
Active ingredient(s) 0.01 ¨ 50%
Water 0 ¨ 99.9%
Solvent 0 ¨ 99.9%
Osmotic agent 0 ¨ 20%
Viscosity enhancer 0 ¨ 20%
Buffering agent q.s.
Preservatives q.s.
VIII., Nasal drops or spray
CA 03231776 2024- 3- 13
WO 2023/053015
PCT/IB2022/059214
78
Active ingredient(s) 0.01 ¨ 50%
Water 0 ¨ 99.9%
Solvent 0 ¨ 99.9%
Osmotic agent 0 ¨ 20%
Viscosity enhancer 0 ¨ 20%
Co-solvent q.s.
Buffering agent q.s.
Preservatives q.s.
CA 03231776 2024- 3- 13