Note: Descriptions are shown in the official language in which they were submitted.
WO 2023/091464
PCT/US2022/050069
USP9X INHIBITORS
Inventors: Richard Martinelli and Julian F. Bond
RELATED APPLICATION
This application claims the benefit of U.S. Provisional Application No.
63/279,783,
filed on November 16, 2021. The entire teachings of the above application are
incorporated
herein by reference.
BACKGROUND
Ubiquitination is a covalent post-translational modification of cellular
proteins
involving a complex enzymatic cascade. Emerging evidence suggests that many
enzymes of
the ubiquitination cascade are differentially expressed or activated in
several diseases and
may therefore be appropriate therapeutic targets.
Protein ubiquitination is a dynamic two-way process that can be reversed or
regulated
by deubiquitinating (deubiquitinase, DUB) enzymes. The human genome codes for
nearly
100 proteins with putative DUB activity which can be broadly divided into two
main sub-
groups: ubiquitin C-terminal hydrolase (UCH) and the ubiquitin-specific
proteases (USP).
USPs comprise the largest subclass of DUBs in humans, while only 4 known UCH
DUBs
have been described. DUBs primarily serve to counterbalance ubiquitin-protein
conjugation
and also facilitate the cleavage of ubiquitin from its precursors and
unanchored polyubiquitin
chains. Thus, DUBs regulate and maintain the homeostasis of free ubiquitin
pools in the cell.
Several DUBs have been reported to regulate deubiquitination of histones, DNA
damage
repair, cellular proliferation (USP2) and cytokine signaling (DUB-A). DUBs
such as USP14,
Uch37 and RPN11 have been shown to associate with the regulatory sub-unit of
the
proteasome (19S) and edit polyubiquitin chains on proteasome substrates.
SUMMARY
The invention relates to the discovery of compounds that inhibit DUBs,
particularly
USP9X. Inhibition of DUBs, particularly USP9X, induces the targeted
degradation of the
substrates of the deubiquitylase. These substrates include Mcl-1, erbB2, beta-
catenin and
aldehyde dehydrogenase 1A3. "lbe invention also includes methods of inhibiting
DUBs,
including USP9X. For example, the invention includes methods of treating,
inhibiting or
suppressing cancer, such as myeloma, lung cancer (e.g., non-small cell lung
cancer), colon
cancer, central nervous system (CNS) cancer, melanoma, ovarian cancer, renal
cancer,
- 1 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
prostate cancer, breast cancer (such as a triple negative breast cancer),
pancreatic cancer, a
virus-induced cancer, a Kaposi's sarcoma, a nasopharyngeal carcinoma (EBV),
leukemia, a
chronic myelogenous leukemia (CML), lymphoma, acute lymphocytic leukemia, a
chronic
lymphocytic leukemia, an acute myelogenous leukemia, a B-cell lymphoma, a
mantle cell
lymphoma, a multiple myeloma, a plasma cell dyscrasia, a myeloproliferative
disorder, or a
glioblastoma. In a preferable embodiment, provided is a method of treating,
inhibiting or
suppressing breast cancer or pancreatic cancer, more preferably, pancreatic
cancer.
The invention also includes methods of inducing degradation of a USP9X
substrate in
a cell, such as Mc-1 or erbB-2. For example, the invention includes methods of
treating a
condition in a subject wherein the condition is associated with a pathologic
cell that expresses
Mc1-1, comprising administrating to the subject an effective amount of the
compound.
Preferably, the condition is a cancer. In some embodiments, the degradation
level of the
USP9X substrate, such as Mc1-1, may be controlled, slowed down, or terminated
by co-
treating the cell with a proteasome inhibitor, such as Velcade.
Mc1-1 may serve as an anti -apoptotic factor that confers resistance, e.g.,
for a Bc1-2
family protein, to chemotherapy. The invention also includes methods of
enhancing potency
of a Bc1-2 family inhibitor in a cell expressing Mc-1 and at least one other
anti-apoptotic
Bc1-2 family protein (such Bc1-2, Bc1-xL, or both), comprising co-treating the
cell with the
compound and the Bc1-2 family inhibitor. Preferably, the Bc1-2 family
inhibitor is a BH3
mimetic such as Navitoclax or Venetoclax. The invention also includes methods
of treating a
condition in a subject wherein the condition is associated with a pathologic
cell that expresses
Mc-1 and at least one other anti-apoptotic Bc1-2 family protein, comprising co-
administrating to the subject an effective amount of a pharmaceutical
composition of the
compound and an effective amount of a pharmaceutical composition of a Bc1-2
family
inhibitor. In some embodiments, the condition is a cancer, more preferably,
acute myeloid
leukemia (AML) or breast cancer.
In some embodiments, the compound can be incorporated into a PROTAC construct
to target USP9X, leading to its ubiquitylation by an E3 ubiquitin ligase and
consequently
inducing the degradation of USP9X.
The invention also includes methods of treating inflammation, infection, such
as a
pathogenic infection, or a neurodegenerative disorder or symptoms of a
neurodegenerative
disorder, or a genetic disorder mediated to the DUB. Additionally, provided
are methods of
treating a condition arising from a pathogen infection comprising contacting
the pathogen or
a cell infected by the pathogen with the compound or composition as disclosed
herein. The
- 2 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
condition can be gastroenteritis, encephalitis, a respiratory tract infection,
SARS, virus-
induced cancer, rabies, a hemorrhagic fever, Rift valley fever, listeriosis,
or toxoplasmosis. In
some cases, the condition is meningitis, myocarditis. hepatitis, bacteremia,
or a skin infection.
The pathogen can be a virus, bacterium, fungus, or parasite. The virus can be
a calicivirus, a
norovirus, a sapovirus, a picornavirus, a Togavirus, a Bunyavirus, a
Rhabdovirus, a herpes
virus, an adenovirus, an arterivirus, a coronavirus, a flavivirus, a
paramyxovirus, a
papillomavirus, a virus encoding for an ovarian tumor (OTU)-like protease, a
baculovirus, or
a nairovirus. The virus can be a polyoma virus, a retrovirus or coronavirus.
In various cases,
the virus is selected from the group consisting of encephalomyocarditis virus
(EMCV),
Sindbis virus (SiNV), La Crosse virus (LaCV), Norwalk virus, Epstein-Barr
(EBV),
herpesvirus, Dengue virus, papillomavirus or coronavirus. The virus can be
cytomegalovirus,
BK virus, hepatitis C virus, or HIV. The bacterium can be Chlamydia,
Escherichia,
Salmonella, Yersiniaõ Burkholderia, Haemophilus, Listeria, or Mycobacterium.
In some
cases, the bacterium is Staphylococcus aureus. In various cases, the bacterium
is methicillin-
resistant Staph aureus (MRSA). The parasite or fungus can be Plasmodium
falciparum,
Toxoplasma gondii, Entamoeba histolytica, Giardia lamblia, Trypanosoma brucei,
Trypanosoma cruzi, Cestoda, Clonorchis, Opisthorchis, Strongylocides, Candida,
Aspergillus, or Cryptococcus.
The invention also includes methods of treating developmental disorders, such
as
intellectual disability, epilepsy, autism and developmental delay, and
neurodegeneration,
including Parkinson's and Alzheimer's disease, as well as autoimmune diseases
and
inflammation.
- 3 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Thus, provided herein area compound represented by Formula (I).
0
Rio Ri
\N
R 0
5 2 R2
".1\
X2
R3
C31.\
N-- R7 R5
RVCH 4
I A-711----1 Zi
R6 0
or optionally by a formula with a linear backbone structure obtained by
breaking a carbon-
carbon or carbon-nitrogen single bond on the cyclic backbone of Formula (1) at
any site, and
pharmaceutically acceptable salts thereof,
wherein Ri and Rio are each independently selected from H, 2H, and substituted
or
unsubstituted alkyl;
R2, R3, R4, R6, and R7 are each independently selected from H, 2H, halogen,
substituted or unsubstituted alkyl, and substituted or unsubstituted alkoxy;
and optionally, R3
and R4 together form a substituted or unsubstituted 5- or 6-membered
heterocyclic ring;
R5 and R9 are each independently selected from H, 2H, substituted or
unsubstituted
alkyl, substituted or unsubstituted alkoxy, and one or more aryl substituents,
such as halogen;
R8 is absent, or substituted or unsubstituted 1- to 4-carbon alkylene;
Ril is a hydrophobic alkyl selected from substituted or unsubstituted alkyl,
and
substituted or unsubstituted cycloalkyl;
Zi, Z2, and Z3 are each independently selected from CH and N;
Xi is selected from -0-, -NH-, -CH2-;
X2 is absent, or selected from -L-NRA-, -L-NRAC(0)-, -L-C(0)-, -L-0C(0)-, -L-
C(0)0-, -L-CH(COORA)-, -L-C(0)NRA-L-, -L-C(0)NRAC(0)-, wherein RA is selected
from
H, 2H, substituted or unsubstituted alkyl; and optionally, RA, Its, and X2
together with the aryl
- 4 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
to which Rs is attached to form a substituted or unsubstituted 5- or 6-
membered heterocyclic
ring;
L is a linker group ¨(CH2),ICRBRc(CH2)11-, wherein RB and Rc are each
independently
selected from H, 2H, substituted or unsubstituted alkyl; or RB and Rc together
with the carbon
to which they are attached form a substituted or unsubstituted C3-C6
cycloalkyl, or a
substituted or unsubstituted 3- to 6-membered heterocyclic ring; and wherein
each n is
independently 0, 1, or 2;
and m = 0, 1, 2, 3, or 4.
In preferable embodiments, Ri and R6 are H; R2, R3, R4, and Rs are each
independently selected from H, F, Cl, -CH3, and -OCH3; R7 is H or methyl (more
preferably
methyl); Rii is selected from -CH2C(CH3)3, -CH2CH(CH3)2, -CH2-cyclo-C3H5, and -
CH2-
cyclo-C4H2; and X is -0- or -CH2-.
In preferable embodiments, L is ¨CH2CH2CH2- or ¨CH2C(CH3)2CH2-, and more
preferably, ¨CH2C(CH3)2CH2-.
In preferable embodiments, X2 is ¨CH2C(CH3)2CH2C(0)N(CH3)-.
In some embodiments, Rs is a) a substituted 2-carbon alkene, wherein the
substituent
is selected from halogen such as F or Cl, oxo, -COOH, -CONH2, -CONH-C2-C12-
alkyl, -
CONH-C2-Cs-alkenyl, -CONH-C2-Cs-alkynyl, -CONH-C3-C12-cy cloalky 1, -CONH-
aryl, -
CONH-heteroaryl, -CONH-heterocycloalkyl, and -CONH-C2-C8-alkoxy; or b) -CH2CH2-
.
Preferably, Rs is selected from -CH(COOH)CH2-, -CHIC(0)NHCH2CH2OCH31CH2-, and -
CH2CH2-; and more preferably, is -CH2CH2-.
In some embodiments, each of Zi, Z2, and Z3 is CH; one of Z1, Z2, and Z3 can
be N; or
two of Zi, Z2, and Z3 can be N. Preferably, Zi is N, Zi and Z2 are CH.
In preferable embodiments, m =1, 2, or 3.
In preferable embodiments, provided herein are a compound represented by
Formula
(I) and pharmaceutically acceptable salts thereof
- 5 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
In a preferred embodiment, the compound can be of Formula (II) or (III):
x,
r/ \
NR,
717K-'=z3
R2HC
0>
NR3 R7N
\c¨sx
Z2
(:)NR4
X2 RoN
hR 0 \
na
0 (II) or
x,
R2HC rC'
0
NHR, R7N
Xi
\C/
NHRi o X2 R6N
___________________________________________________ \
5m (m)
and pharmaceutically acceptable salts thereof,
wherein Ri, R2, R3, R4, Rs, R6 , R7, and R8 are independently hydrogen or a
substituted
or unsubstituted alkyl,
R9, and Rio are independently hydrogen, substituted or unsubstituted alkyl, or
substituted or unsubstituted acyl,
L is a linker group, such as a substituted or unsubstituted, saturated or
unsaturated 1 to
10 carbon alkylene,
Z1, Z2, and Z3 are independently selected from N or CH,
- 6 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Xi, X2, X3 are independently H or one or more aryl substituents, such as a
halogen,
e.g., fluorine,
n and m are independently 0 or an integer, such as 1, 2, 3, 4, or 5.
Preferably, Ri is hydrogen, R2 is hydrogen, R3 is hydrogen or methyl (more
preferably
methyl), R4 is hydrogen or methyl (more preferably methyl), R5 is hydrogen, R6
is hydrogen
or methyl (more preferably hydrogen), and R7 is hydrogen or methyl (more
preferably
hydrogen). R8 is preferably an alkyl group, and more preferably a branched
alkyl, such as a
4, 5, or 6 carbon alkyl. -CH2C(CH3)3 is preferred. Where one or more of R2,
R5, and R8 are a
substituted or unsubstituted alkyl, a chiral center is formed. The invention
contemplates
racemates and purified or isolated stereoisomers, enantiomers and
diastereomers. Preferably,
the linker L is ¨(CH2)2C(CH3)2(CH2)2-. Alternatively, the linker can include
one or more
heteroatoms, such as an oxy, amine, amido or ester. For example, the linker
can be ¨
(CH2)20(CH2)2-, ¨(CH2)2NH(CH2)2-, ¨(CH2)2CONH (CH2)2-, or ¨(CH2)2CO2(CH2)2-.
The
linker can also be unsaturated or aromatic, such as a ¨(CH2)2CH=CH (CH2)2- or
a 1,4-
phenylene.
Preferably, each of Z1, Z2, and Z3 are CH. Preferably, one of Z1, Z2, and Z3
can be N.
Preferably, Zi and Z2 are CH and Z3 is CH or N.
In some embodiments, provided is a compound selected from those in Table 1.
Also
disclosed are pharmaceutical compositions comprising a compound as described
herein and a
pharmaceutically acceptable excipient. The pharmaceutical composition can be
formulated
for oral, topical, intravenous, subcutaneous, intramuscular, intrathecal,
ophthalmic, or
inhalational route of administration.
BRIEF DESCRIPTION OF THE DRAWINGS
The foregoing and other objects, features and advantages of the invention will
be
apparent from the following more particular description of preferred
embodiments of the
invention, as illustrated in the accompanying drawings in which like reference
characters
refer to the same parts throughout the different views. The drawings are not
necessarily to
scale, emphasis instead being placed upon illustrating the principles of the
invention.
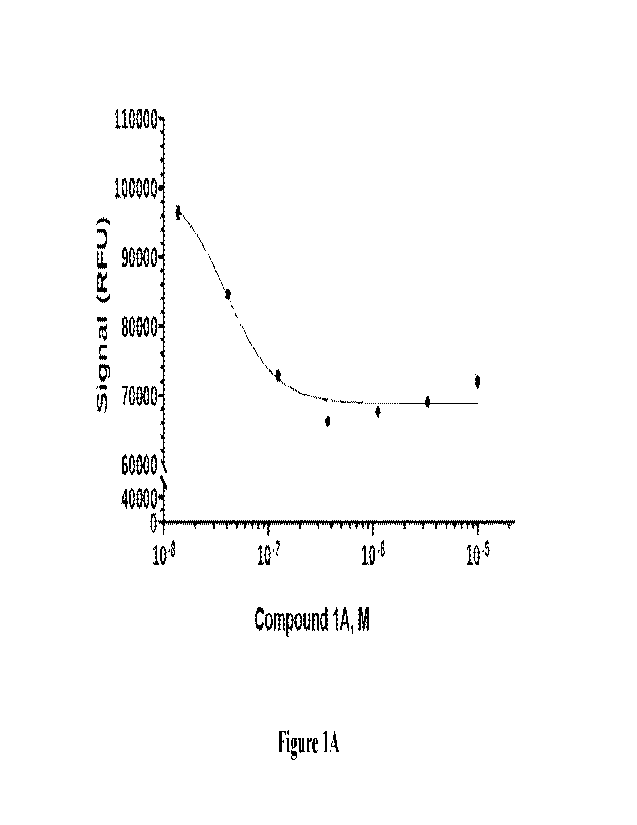
Figure 1A is a graph showing a curve of fluorescence (RFU) vs. the
concentration of
Compound 1A. The effect of Compound lA was evaluated in an assay measuring the
ability
of USP9X to catalyze the hydrolysis of the amide bond between the C terminal
carboxylate
of ubiquitin and rhodamine110 of the fluorogenic substrate ubiquitin-rhodamine
110.
- 7 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Figure 1B is a graph depicting relative response vs. time curves for Compound
2A
binding to USP9X as determined by the BIACORETM assay at Compound 2A
concentration
ranging from 0.3x 10 6 M01/1 to 20 x10 6 mo1/1 described in Example 2.
Figure IC is a graph depicting affinity determination of Compound 2A binding
to
USP9X as determined by the BIACORETM assay described in Example 2.
Figure 113 is a graph depicting the effects of compounds Compound 2A,
Comparator
1, Compound 3A, Comparator 2, and Compound 4A on the enzyme activity of USP9X
on the
fluorogenic substate ubiquitin-rhodamine110 (BPS-Biosciences). The fluorescent
signals
generated by the USP9X mediated generation of the fluorescent product were
monitored over
time at the presence of these compounds at varying concentrations and at the
presence of
ubiquitin aldehyde as a positive control, respectively.
Figure IE illustrates the set of reactions that comprise the inhibition of
enzyme
activity. The values of all the parameters may be deteriniiied by a series of
steady state
kinetic measurements. The values determined for the parameters a and j3
determine the
speei fie mode of inhibition
Figure IF is a graph comparing the experimental data of Compound 2A (solid
dot)
with the partial inhibition dose-response curve based on the kinetic model of
hyperbolic
mixed inhibition.
Figure 2A is a graph depicting a cell viability dose-response curve of
Compound 9A
inhibiting Mia-Paca-2 cell proliferation after 72-hour treatment at 37 C as
assessed by the
WST assay.
Figure 2B is a graph depicting a cell viability dose-response curve of
Compound 9A
inhibiting MDA-MB-231 cell proliferation after 72-hour treatment at 37 C as
assessed by the
WST assay.
Figure 2C is a graph depicting dose-response curves of RKO colon cell
viability for
Compound 7A, Compound 6A, Compound 8A in an RKO colon cell line.
Figure 2D is a graph depicting dose-response curves of BT-549 Breast cancer
cell
viability for Compound 7A, Compound 6A, Compound 8A in a BT-549 Breast cancer
cell
line.
Figure 2E is a graph depicting dose-response curves of MIA Paca-2 Pancreatic
cell
viability for Compound 7A, Compound 6A, Compound 8A in a MIA Paca-2 Pancreatic
cell
line.
Figure 3A is a graph showing normalized Mc1-1 levels at varying concentrations
of
Compound 7A after 24-hour treatment from western blot analysis. Displayed at
the upper
- 8 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
right corner is a representative blot of Mcl-1 at Compound 7A = 0 and 10 uM
with the
housekeeping protein tubulin as a reference.
Figure 3B is a graph depicting a cell viability dose-response curve of
Compound 7A
inhibiting U937 cell proliferation after 72-hour treatment, indicating IC50 =
0.27 uM as
assessed by the WST assay.
Figure 3C is a graph showing the erbB2 level changes over time during 72 hours
of
Compound 7A treatment from western blot analysis, normalized by the
housekeeping protein
actin as a reference, compared with that during 72 hours of vehicle treatment
only. Displayed
at the upper right corner is a representative blot of erbB2 treated with 10
?AM Compound 7A
after 24, 48, and 72 hours compared with erbB2 treated with vehicle only after
24, 48, and 72
hours.
Figure 3D is a graph depicting a cell viability dose-response curve of
Compound 7A
inhibiting SK-BR3 breast cancer cell proliferation after 72-hour treatment,
indicating IC50 =
2.7 1.1.1\4 as assessed by the WST assay.
Figure 4A is a western blot image showing Mcl-1 bands in U937 cell line at
different
stages of treatments. U937cells were treated with either 10 1.1g/mL
cycloheximide, 10 uM
MG132, the combination of cycloheximide and MG132, or vehicle (DMSO) for one
hour
prior to the treatment with Compound 5A for 24 hours. Bands of tubulin
undergoing the same
treatments are shown as a reference.
Figure 4B left is a graph of capture ELISA results showing the percent
decreases of
Mcl-1 in MIA-PaCa-2 cells treated with Compound 7A at 25 !AM, 12.5 uM, 6.25
uM, and
3.125 uM, respectively, for 24 hours. Right is graph of capture ELISA results
showing the
percent increases of ubiquitylated Mc1-1 in MIA-PaCa-2 cells treated with
Compound 7A at
uM, 12.5 uM, 6.25 uM, and 3.125 !AM, respectively, for 24 hours.
25 Figure 5A is a graph depicting cell viability dose-response curves, as
assessed by the
WST assay, of ABT737 inhibiting U937 cells with and without the co-treatment
of 0.5 tiM
Compound 7A, respectively. With the co-treatment of Compound 7A, the IC50
value of
ABT737 decreases to 0.061AM from 3.4 uM when treated with ABT737 alone. Co-
treatment
of 0.5 uM Compound 7A results in 57-fold enhancement in ABT737's potency for
inhibiting
U937 cells.
Figure 5B is a graph depicting cell viability dose-response curves of ABT737
inhibiting MDA-MB-231 cells with and without the co-treatment of 10 uM
Compound 5A,
respectively. With the co-treatment of Compound 5A, the IC5() value of ABT737
decreases to
- 9 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
1.8 !AM from 8.7 M when treated with ABT737 alone. Co-treatment of 10 !AM
Compound
5A results in 5-fold enhancement in ABT737's potency for inhibiting MDA-MB-231
cells.
DETAILED DESCRIPTION
The ubiquitin-specific protease 9X (USP9X/FAM) is a substrate-specific DUB.
USP9X plays a significant role in cancer, both as an oncogene or tumor
suppressor,
developmental disorders, such as intellectual disability, epilepsy, autism and
developmental
delay, neurodegenerati on, including Parkinson's and Alzheimer's disease, and
inflammation
such as autoimmune diseases. Murtaza et al., Cell. Mol. Life Sci. (2015)
72:2075-2089, US
Patent Publication 2017/0204055 (Brandeis University) and US Patent
Publication
2016/0237082 (University of Michigan), which are incorporated herein by
reference.
The Compound of the Invention
Disclosed herein are compounds and methods of inhibiting a DUB, methods of
inhibiting DUB catalytic activity, methods of inhibiting Usp9x, methods of
inhibiting
survival or proliferation of a cell, such as a cancer cell, methods of
treating a
neurodegenerative disorder, methods of treating one or more symptoms of a
neurodegenerative disorder, methods of treating one or more symptoms of a
genetic disorder,
and methods of inhibiting or preventing a pathogenic infection. In some
embodiments, -the
USP9X inhibitor- and "the compound- as used herein are interchangeable. In
some
embodiments, -cancer cell- and -tumor cell- as used herein are
interchangeable. The
compounds of the invention include a compound of Formula (I), or optionally a
formula with
a linear backbone structure obtained by breaking a carbon-carbon or carbon-
nitrogen single
bond on the cyclic backbone of Formula (I) at any site, and salts thereof:
- 10 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
0
Rio Ri
Rs
R8 2 R11
0 R2
X2
R3
0.\\
N--- R7 R5
r CAM
R4
\.CH
-Xi
R6 0
(1),
The variables in Formula (I) (including Rt, R2, R3, R4, Rs, R6, R7, Rs, R9,
R10, R11, Z1,
Z2, Z3, Xi, X2, m, and L) are as defined above.
Prefen-ed alkyls include lower alkyls, such as methyl, ethyl, n-propyl, i-
propyl, n-
butyl, sec-butyl, i-butyl, tert-butyl, n-pentyl, s-pentyl, i-pentyl, or t-
amyl.
The invention contemplates racemates and purified or isolated stereoisomers,
enantiomers and diastereomers.
In some embodiments, the compounds of the invention include a compound of
Formula (II) or (III), and salts thereof:
x,
(NR R8
R2.HC\
0> <r
NR3 R7N
2/51
0 NR4 X2 ReN
I I4c
\
11 5m o
(11)
- II -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
x,
______________________________________ ¨).
/1
NR R8
(- >R2 HC
or
NHR, R7Nr/
NHRI 0 x2
R6 N
r`"I
0
- HR, zi
or (III)
and pharmaceutically acceptable salts thereof
The variables in Formula (II) and (III) (including Ri, R2, R3, R4, Rs, R6, R7,
R8, R9, R10, Z1,
Z2, Z3, Xi, X2, X3, L, m, and n) are as defined in Summary. Preferred alkyls
include lower
alkyls, such as methyl, ethyl, n-propyl, i-propyl, n-butyl, sec-butyl, i-
butyl, tert-butyl, n-
pentyl, s-pentyl, i-pentyl, or t-amyl. Preferred acyls include alkylcarbonyls,
such as acetyl or
t-boc. The invention contemplates racemates and purified or isolated
stereoisomers,
enantiomers and diastereomers.
The following Table 1 provides additional representative examples of the
invention.
Table 1. Exemplary Compounds
Compound # Structure
NH e
N¨ 0 HN¨
C)
IA
NH
/ 0 0
0 issiNs_i
¨ 1 2 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
0 NH X
HO NH2 1
2A
a 4. N
NH H 0
\ / _____________________________________________________ 0
0 = NH X,
(s)
- (s) 0
/¨NH -.NH 0 HN
0_/ 0
3A /
0 NH
HN 0 = 0
0 41. NH
(s)
0
HO -.NH 0 HN
4A
o /
NH NH
0 410
0 \---/ .
0
NH .
>(s)( 0
N¨ 0 HN---
0
5A
0
N/ NH
\ /0 . /
0
- 13 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
NH \K
0 H N
--N
6A
0 ----- N H
0
9
Cr 41.- F
7A
"
0
sszz .
0 )=======NH 5,}1.1
SA F
\).
410. NH
0
N¨ 0 HN¨
C)
9A
,NH
\ /0
=
0
NH
p
N-
0 H N 0
0
10A
,N H2
N 0¨(
/
N 0
¨ 1 4 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
NH
)(sj< 0
N- 0 HN-
C
11A
,NH
N 0
/ 0
N F\K
N-0) (4N 0
0
12A
0 N11\
N 0
/
0
HN= <
13A
\
'NH
/ / 0
Deubiquitinases (DUBs)
Deubiquitinating enzymes (i.e., deubiquitinases or DUBs) are typically a
cysteine
protease and may be classified into subgroups as ubiquitin-specific proteases
(USP) and
ubiquitin C-terminal hydrolases (UCH). Examples of DUBs include, for instance,
USP5,
USP6, USP4, USP5, USP13, USP2, USP11, USP14, USP7, USP9X, USP10, USP1, USP12,
USP16, USP15, USP17, USP19, USP20, USP3, USP9Y, USP18, USP21, USP22, USP33,
USP29, USP25, USP36, USP32, USP26, USP24, USP42, USP46, USP37, USP28, USP47,
USP38, USP44, USP50, USP35, USP30, Memame-AA088peptidase, Memame-AA091
peptidase, USP45, USP51, USP34, USP48, USP40, USP31, Mername-AA129peptidase,
- 15 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
USP49, USP17-like peptidase, USP54, USP53, USP39, UCH-L1, UCH-L3, UCH-BAP1,
UCH37, Cezanne deubiquitinating peptidase, Cezanne2, tumor necrosis factor
alpha-induced
protein 3. TRABID protein, VCP(p97)/p47-interacting protein, otubainl,
otubain2, CylD
protein, SENP1 peptidase, SENP3 peptidase, SENP6 peptidase, SENP2 peptidase,
SENP5peptidase, SENP7peptidase, SENP8peptidase, SENP4peptidase, Pohl
peptidase,
Jabl/MPN domain metalloenzyme, Memame-AA 165 peptidase, Memame-AA 166
peptidase, Memame-AA 167 peptidase, Mername-AA168 protein, COPS signalosome
subunit6, 26S proteasome non-ATPase regulatory subunit7, eukaryotic
translation initiation
factor3 subunit5, IFP38 peptidase homologue, autophagin (ATG), ovarian tumor
(OTU)
domain proteins, Josephin-domain (JD) or Machado-Joseph disease (MJD)
proteins,
ubiquitin-like protein-specific protease (ULP), and JAMM (Jabl/MPN domain-
associated
metalloisopeptidase) domain proteins. The compounds of the invention
preferably selectively
inhibit Usp9x.
The term "alkyl" as used herein, refers to saturated, unsaturated, straight-
or
branched-chain, or cyclic hydrocarbon radicals. "C i-C4 "Ci-C6 "CI-Cs
alkyl,"
"Ci-C 12 alkyl," "C2-C4 alkyl,- or "C3-C6 alkyl,- refer to alkyl groups
containing from one to
four, one to six, one to eight, one to twelve, 2 to 4 and 3 to 6 carbon atoms
respectively.
Examples of Ci-C8 alkyl radicals include, but are not limited to, methyl,
ethyl, propyl,
isopropyl, n-butyl, tert-butyl, neopentyl, n-hexyl, heptyl and octyl radicals.
The term "alkenyl- as used herein, denotes a monovalent group derived from a
hydrocarbon moiety having at least one carbon-carbon double bond by the
removal of a
single hydrogen atom. -C2-C8 alkenyl," -C2-C12 alkenyl," -C2-C4 alkenyl," -C3-
C4 alkenyl,"
or "C3-C6 alkenyl," refer to alkenyl groups containing from two to eight, two
to twelve, two
to four, three to four or three to six carbon atoms respectively. Alkenyl
groups include, but
are not limited to, for example, ethenyl, propenyl, butenyl, 2-methyl-2-buten-
2-v1, heptenyl,
octenyl, and the like.
The term "alkynyl" as used herein, denotes a monovalent group derived from a
hydrocarbon moiety having at least one carbon-carbon triple bond by the
removal of a single
hydrogen atom. "C2-Cs alkynyl," "C2-C12 alkyn"1," "C2-C4 alkynyl," "C3-C4
alkynyl," or
-C-C6 alkynyl," refer to alkynyl groups containing from two to eight, two to
twelve, two to
four, three to four or three to six carbon atoms respectively. Representative
alkynyl groups
include, but are not limited to, for example, ethynyl, 2-propynyl, 2-butynyl,
heptynyl,
octynyl, and the like. Alkenyls and alkynyls are "unsaturated alkyls."
- 16 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
The term "cycloalkyl", as used herein, refers to a monocyclic or polycyclic
saturated
(or unsaturated) carbocyclic ring or a hi- or tri-cyclic group fused, bridged
or Spiro system,
and the carbon atoms may be optionally oxo-substituted or optionally
substituted with
exocyclic olefinic double bond. Preferred cycloalkyl groups include C3-C12
cycloalkyl, C3-C6
cycloalkyl, C3-C8 cycloalkyl and C4-C7 cycloalkyl. Examples of C3-C12
cycloalkyl include,
but not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl,
cyclopentyl, cyclooctyl,
4-methylene-cyclohexyl, bicyclo[2.2.11heptyl, bicyclo[3.1.01hexyl,
spiro[2.51octyl, 3-
methy1enebicyclo[3.2.1loctyl, spiro[4.41nonanyl, and the like.
The term "cycloalkenyl", as used herein, refers to monocyclic or polycyclic
carbocyclic ring or a hi- or tri-cyclic group fused, bridged or spiro system
having at least one
carbon-carbon double bond and the carbon atoms may be optionally oxo-
substituted or
optionally substituted with exocyclic olefinic double bond. Preferred
cycloalkenyl groups
include C3-C12 cycloalkenyl, C3-C8 cycloalkenyl or C5-C7 cycloalkenyl groups.
Examples of
C3-C12 cycloalkenyl include, but not limited to, cyclopropenyl, cyclobutenyl,
cyclopentenyl,
cyclohexenyl, cycloheptenyl, cyclooctenyl, bicyclo[2.2.11hept-2-enyl,
bicyclo[3.1.01hex-2-
enyl, spiro[2.51oct-4-enyl, spiro[4.41non-2-enyl, bicyclo[4.2.11non-3-en-12-
yl, and the like.
The term "aryl," as used herein, refers to a mono- or polycyclic carbocyclic
ring
system having one or two aromatic rings including, but not limited to, phenyl,
naphthyl,
tetrahydronaphthyl, indanyl, idenyl and the like.
The term "heteroaryl," as used herein, refers to a mono- or polycyclic (e.g.
bi-, or tri-
cyclic or more) aromatic radical or ring having from five to ten ring atoms of
which one or
more ring atom is selected from, for example, S, 0 and N; zero, one or two
ring atoms are
additional heteroatoms independently selected from, for example, S, 0 and N;
and the
remaining ring atoms are carbon, wherein any N or S contained within the ring
may be
optionally oxidized. Heteroaryl includes, but is not limited to, pyridinyl,
pyrazinyl,
pyrimidinyl, pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl,
isooxazolyl, thiadiazolyl,
oxadiazolyl, thiophenyl, furanyl, quinolinyl, isoquinolinyl, benzimidazolyl,
benzooxazolyl,
quinoxalinyl, and the like.
As used herein, the term "arylalkyl" means a functional group wherein an
alkylene
chain is attached to an aryl group, e.g., -CH2CH2-phenyl. The term
"substituted arylalkyl"
means an arylalkyl functional group in which the aryl group is substituted.
Similarly, the term
"heteroarylalkyl- means a functional group wherein an alkylene chain is
attached to a
heteroaryl group. The term "substituted heteroarylalkyl" means a
heteroarylalkyl functional
group in which the heteroaryl group is substituted.
- 17 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
As used herein, the term "alkoxy" employed alone or in combination with other
terms
means, unless otherwise stated, an alkyl group having the designated number of
carbon atoms
connected to the rest of the molecule via an oxygen atom, such as, for
example, methoxy,
ethoxy, 2-propoxy, 2-propoxy (isopropoxy) and the higher homologs and isomers.
Preferred
alkoxy are (C2-C3) alkoxy.
It is understood that any alkyl, alkenyl, alkynyl, cycloalkyl, heterocyclic
and
cycloalkenyl moiety described herein can also be an aliphatic group or an
alicyclic group.
An -aliphatic- group is a non-aromatic moiety comprised of any combination of
carbon atoms, hydrogen atoms, halogen atoms, oxygen, nitrogen or other atoms,
and
optionally contains one or more units of unsaturation, e.g., double and/or
triple bonds.
Examples of aliphatic groups are functional groups, such as alkyl, alkenyl,
alkynyl, 0, OH,
NH, NH2, C(0), S(0)2, C(0)0, C(0)NH, OC(0)0, OC(0)NH, OC(0)NH2, S(0)2NH,
S(0)2NH2, NHC(0)NH2, NHC(0)C(0)NH, NHS(0)2NH, NHS(0)2NH2, C(0)NHS(0)2,
C(0)NHS(0)2NH or C(0)NHS(0)2NH2, and the like, groups comprising one or more
functional groups, non-aromatic hydrocarbons (optionally substituted), and
groups wherein
one or more carbons of a non-aromatic hydrocarbon (optionally substituted) is
replaced by a
functional group. Carbon atoms of an aliphatic group can be optionally oxo-
substituted. An
aliphatic group may be straight chained, branched, cyclic, or a combination
thereof and
preferably contains between about 1 and about 24 carbon atoms, more typically
between
about 1 and about 12 carbon atoms. In addition to aliphatic hydrocarbon
groups, as used
herein, aliphatic groups expressly include, for example, alkoxyalkyls,
polyalkoxyalkyls, such
as polyalkylene glycols, polyamines, and polyimines, for example. Aliphatic
groups may be
optionally substituted.
The terms "heterocyclic" or "heterocycloalkyl" can be used interchangeably and
referred to a non-aromatic ring or a bi- or tri-cyclic group fused, bridged or
Spiro system,
where (i) each ring system contains at least one heteroatom independently
selected from
oxygen, sulfur and nitrogen, (ii) each ring system can be saturated or
unsaturated (iii) the
nitrogen and sulfur heteroatoms may optionally be oxidized, (iv) the nitrogen
heteroatom
may optionally be quatemized, (v) any of the above rings may be fused to an
aromatic ring,
and (vi) the remaining ring atoms are carbon atoms which may be optionally oxo-
substituted
or optionally substituted with exocyclic olefinic double bond. Representative
heterocycloalkyl groups include, but are not limited to, 1,3-dioxolane,
pyrrolidinyl,
pyrazolinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, piperidinyl,
piperazinyl,
oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl,
quinoxalinyl,
- 18 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
pyridazinonyl, 2-azabicyclo[2.2.1]-heptyl, 8-azabicyclo[3.2. lioctyl, 5-
azaspiro[2.51octyl, 2-
oxa-7-azaspiro[4.41nonanyl, 7-oxooxepan-4-yl, and tetrahydrofuryl. Such
heterocyclic groups
may be further substituted. Heteroaryl or heterocyclic groups can be C-
attached or N-attached
(where possible).
It is understood that any alkyl, alkenyl, alkynyl, alicyclic, cycloalkyl,
cycloalkenyl,
aryl, heteroaryl, heterocyclic, aliphatic moiety or the like, described herein
can also be a
divalent or multivalent group when used as a linkage to connect two or more
groups or
substituents, which can be at the same or different atom(s). One of skill in
the art can readily
determine the valence of any such group from the context in which it occurs.
The term "substituted" refers to substitution by independent replacement of
one, two,
or three or more of the hydrogen atoms on an alkyl group (substituted alkyl)
or aryl group
(substituted aryl) with substituents including, but not limited to, -F, -Cl, -
Br, -I, -OH, Ci_C12-
alkyl; C2-C12-alkenyl, C2-C12-alkynyl, -C3-C12-cycloalkyl, protected hydroxy, -
NO2, -N3, -
CN, -NH2, protected amino, oxo, thioxo, -NH-C2-C12-alkyl, -NH-C2-C8-alkenyl, -
NH-C2-Cs-
alkynyl, -NH-C3-C12-cycloalkyl, -NH-aryl, -NH-heteroaryl, -NH-
heterocycloalkyl, -
dialkylamino, -diarylamino, -diheteroarylamino, -0-C2-C12-alkyl, -0-C2-Cs-
alkenyl, -0-C2-
C8-alkynyl, -0-C3-C12-cycloalkyl, -0-aryl, -0-heteroaryl, -0-heterocycloalkyl,
-C(0)-C2-C12-
alkyl, -C(0)-C2-C8-alkenyl, -C(0)-C2-C8-alkynyl, -C(0)-C3-C12-cycloalkyl, -
C(0)-aryl, -
C(0)-heteroaryl, -C(0)-heterocycloalkyl, -CONH2, -CONH-C2-C12-alkyl, -CONH-C2-
C8-
alkenyl, -CONH-C2-Cs-alkynyl, -CONH-C3-C12-cycloalkyl, -CONH-aryl, -CONH-
heteroaryl, -CONH-heterocycloalkyl, -0CO2-C2-C12-alkyl, -0CO2-C2-Cs-alkenyl, -
0CO2-C2-
C8-alkynyl, -0CO2-C3-C12-cycloalkyl, -0CO2-aryl, -0CO2-heteroaryl, -0CO2-
heterocycloalkyl, -0O2-C2-C12 alkyl, -0O2-C2-Cs alkenyl, -0O2-C2-C8 alkynyl,
CO2-C3-C12-
cycloalkyl, -0O2- aryl, CO2-heteroaryl, CO2-heterocyloalkyl, -000NH2, -000NH-
C2-C12-
alkyl, -000NH-C2-Cs-alkenyl, -OCONH-C2-C8-alkynyl, -000NH-C3-C12-cycloalk-yl, -
OCONH-aryl, -OCONH-heteroaryl, -OCONH- heterocyclo-alkyl, -NHC(0)H, -NHC(0)-C2-
C12-alkyl, -NHC(0)-C2-Cs-alkenyl, -NHC(0)-C2-C8-alkynyl, -NHC(0)-C3-C12-
cycloalkyl, -
NHC(0)-aryl, -NHC(0)-heteroaryl, -NHC(0)-heterocyclo-alkyl, -NHCO2-C2-C12-
alkyl, -
NHCO2-C2-Cs-alkenyl, -NHCO2- C2-Cs-alkynyl, -NHCO2-C3-C12-cycloalk-yl, -NHCO2-
aryl, -
NHCO2-heteroaryl, -NHCO2- heterocycloalkyl, -NHC(0)NH2, -NHC(0)NH-C2-C12-
alkyl, -
NHC(0)NH-C2-C8-alkenyl, -NHC(0)NH-C2-Cs-alkynyl, -NHC(0)NH-C3-C12-cycloalkyl, -
NHC(0)NH-aryl, -NHC(0)NH-heteroaryl, -NHC(0)NH-heterocycloalkyl, NHC(S)NH2, -
NHC(S)NH-C2_C12-alkyl, -NHC(S)NH-C2-C8-alkenyl, -NHC(S)NH-C2-C8-alkynyl, -
NHC(S)NH-C3-C12-cycloalkyl, -NHC(S)NH-aryl, -NHC(S)NH-heteroaryl, -NI-IC(S)NH-
-19-
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
heterocycloalkyl, -NHC(NH)NH2, -NHC(NH)NH-C2-C12-alky1, -NHC(NH)NH-C2-Cs-
alkenyl, -NHC(NH)NH-C2-C8-alkynyl, -NHC(NH)NH-C3-C12-cycloalkyl, -NHC(NH)NH-
aryl, -NHC(NH)NH-heteroaryl, -NHC(NH)NH-heterocycloalkyl, -NHC(NH)-C2-C12-
alkyl. -
NHC(NH)-C2-Cs-alkenyl, -NHC(NH)-C2-Cs-alkynyl, -NHC(NH)-C3-C12-cycloalkyl, -
NHC(NH)-aryl, -NHC(NH)-heteroaryl, -NHC(NH)-heterocycloalkyl, -C(NH)NH-C2-C12-
alkyl, -C (NI DNI I-C2-C g-alkenyl, -C (NI I)NI I-C2-C g-alkynyl, -C(NI I)NI I-
C 3-C 12-cy cloalkyl, -
C(NH)NH-aryl, -C(NH)NH-heteroaryl, -C (NH)NH-hetero cy cl o alkyl, -S (0)-C 2-
C 12- al ky 1, -
S(0)-C 2-C s-alkenyl, - S(0)-C2-C8-alkynyl, -S(0)-C3-C12-cycloalkyl, -S(0)-
aryl, -S(0)-
heteroaryl, -S(0)-heterocycloalkyl, -SO2NH2, -SO2NH-C2-C12-alkyl, -SO2NH-C2-Cs-
alkenyl,
-SO2NH- C2-Cs-alkynyl, -SO2NH-C3-C12-cycloalkyl, -SO2NH-aryl, -SO2NH-
heteroaryl, -
SO2NH- heterocycloalkyl, -NHS02-C2-C12-alkyl, -NHS02-C2-C8-alkenyl, - NHS02-C2-
C8-
alkynyl, -NHS02-C3-C12-cycloalkyl, -NHS02-aryl, -NHS02-heteroaryl, -NHS02-
heterocycloalkyl, -CH2NH2, -CH2S02CH3, -aryl, -arylalkyl, -heteroaryl, -
heteroatylalkyl, -
heterocycloalkyl, -C3-C12-CYcloalkyl, polyalkoxyalkyl, polyalkoxy, -
methoxymethoxy, -
methoxyethoxy, -SH, -S-C2-C12-alkyl, -S-C2-C8-alkenyl, -S-C2-C8-alkynyl, -S-C3-
C12-
cycloalkyl, -S-aryl, -S-heteroaryl, -S-heterocycloalkyl, or methylthio-methyl.
In certain
embodiments, the substituents are independently selected from halo, preferably
Cl and F; C1-
C4-alkyl, preferably methyl and ethyl; halo-C1-C4-alkyl, such as fluoromethyl,
difluoromethyl, and trifluoromethyl; C2-C4-alkenyl; halo-C2-C4-alkenyl; C3-CG-
cycloalkyl,
such as cyclopropyl; C1_C4-alkoxy, such as methoxy and ethoxy; halo-C1-C4-
alkoxy, such as
fluoromethoxy, difluoromethoxy, and trifluoromethoxy, -CN; -OH; NH2; C1_C4-
allcylamino;
di(C1-C4-alkyl)amino; and NO2. It is understood that the aryls, heteroaryls,
alkyls, and the
like can be further substituted. In some cases, each substituent in a
substituted moiety is
additionally optionally substituted with one or more groups, each group being
independently
selected from CI-GI-alkyl; -CF3, -OCH3, -0CF3, -F, -Cl, -Br, -I, -OH, -NO2, -
CN, and -NH2.
Preferably, a substituted alkyl group is substituted with one or more halogen
atoms, more
preferably one or more fluorine or chlorine atoms.
The term "halo- or "halogen- alone or as part of another substituent, as used
herein,
refers to a fluorine, chlorine, bromine, or iodine atom.
The term -optionally substituted-, as used herein, means that the referenced
group
may be substituted or unsubstituted. In one embodiment, the referenced group
is optionally
substituted with zero substituents, i.e., the referenced group is
unsubstituted. In another
embodiment, the referenced group is optionally substituted with one or more
additional
group(s) individually and independently selected from groups described herein.
- 20 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
The term "hydrogen," Or H, includes hydrogen and deuterium (2H or D). In
addition,
the recitation of an atom includes other isotopes of that atom so long as the
resulting
compound is pharmaceutically acceptable.
The salts, e.g., pharmaceutically acceptable salts, of the disclosed
therapeutics or
compounds may be prepared by reacting the appropriate base or acid with a
stoichiometric
equivalent of the therapeutic or compound. Acids commonly employed to form
pharmaceutically acceptable salts include inorganic acids such as hydrogen
bisulfide,
hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid and
phosphoric acid, as
well as organic acids such as para-toluenesulfonic acid, salicylic acid,
tartaric acid, bitartaric
acid, ascorbic acid, maleic acid, besylic acid, fumaric acid, gluconic acid,
glucuronic acid,
formic acid, glutamic acid, methanesulfonic acid, ethanesulfonic acid,
benzenesulfonic acid,
lactic acid, oxalic acid, para-bromophenylsulfonic acid, carbonic acid,
succinic acid, citric
acid, benzoic acid and acetic acid, as well as related inorganic and organic
acids. Such
pharmaceutically acceptable salts thus include sulfate, pyrosulfate,
bisulfate, sulfite, bisulfate,
phosphate, monohydrogenphosphate, dihvdrogenphosphate, metaphosphate,
pyrophosphate,
chloride, bromide, iodide, acetate, propionate, decanoate, caprylate,
acrylate, formate,
isobutyrate, caprate, heptanoate, propiolate, oxalate, malonate, succinate,
suberate, sebacate,
fumarate, maleate, butyne-1,4-dioate, hexyne-1,6-dioate, benzoate,
chlorobenzoate,
methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, phthalate,
terephthalate, sulfonate, xylene sulfonate, phenylacetate, phenylpropionate,
phenylbutyrate,
citrate, lactate, 0-hydroxybutyrate, glycolate, maleate, tartrate,
methanesulfonate,
propanesulfonate, naphthalene-1-sulfonate, naphthalene-2-sulfonate, mandelate
and other
salts. In one embodiment, pharmaceutically acceptable acid addition salts
include those
formed with mineral acids such as hydrochloric acid and hydrobromic acid, and
especially
those formed with organic acids such as maleic acid.
The terms "inhibit" as used herein with respect to the activity of the USP9X
inhibitor,
refer to the ability to substantially antagonize, prohibit, prevent, restrain,
slow, disrupt, alter,
eliminate, stop, or reverse the progression or severity of, for example, the
catalytic activity of
a DUB such as USP9X, the survival or proliferation of a cancer cell, or a
disease or condition
associated with USP9X. The USP9X inhibitor of the invention preferably
inhibits the
catalytic activity of USP9X by at least about 20%, about 30%, about 40%, about
50%, about
60%, about 70%, about 80%, about 90%, about 95%, about 100%, or a range
defined by any
two of the foregoing values. In some embodiments, the compounds as described
can inhibit
USP9X by binding to an active site of USP9X. The compound binding to USP9X
according
-21 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
to the present disclosure will, in at least some embodiments, have a KD value
of 1.0x 10'
mo1/1 or lower at 25 C, in one embodiment a K D value of 1.0x10-6 mo1/1 or
lower at 25 C, in
another embodiment a KD value of 0.5x10 6 mo1/1 or lower at 25 C. The binding
activity is
determined with a standard binding assay, such as a Biacore surface plasmon
resonance
instrument. A method for determining the KD value of binding is described in
the Examples
Section. The binding affinity KD is determined using a Biacore T200 instrument
(GE
Healthcare, Biacore). Ubiquitin aldehyde can be used as a reference in
analyzing the
inhibition results determined via Biacore. Ubiquitin aldehyde is a potent
inhibitor of all
ubiquitin deconjugating enzymes, including UCHs (ubiquitin C-terminal
hydrolases), USPs
(ubiquitin-specific proteases) and DUBs (deubiquitinylating enzymes), and
binds covalently
to the thiol group of the active site Cys of USP9X. In some embodiments, the
compounds as
disclosed can partially inhibit USP9X. In some embodiments, the compounds as
disclosed
can partially inhibit USP9X, as compared with the complete inhibition of
ubiquitin aldehyde
(for example, as shown in Figure 1E; ubiquitin-aldehyde is an irreversible
inhibitor of
USP9X, forming a covalent bond with the thiol group of the active site Cys). A
partial
USP9X inhibition mechanism of the compounds as disclosed is proposed in the
present
application, which in some embodiments can be evaluated and confirmed by
comparing the
experimental data of the compounds with the partial inhibition dose-response
curve based on
the kinetic model of hyperbolic mixed inhibition (for example, as shown in
Figure 1F). The
partial inhibition dose-response curve can be generated using the rate
equation as shown
below:
is]
V = Vmax
1+¨Es]+M+[s][I]/alc5x,
Ks Ki
wherein a, 0, K1, Ks values can be determined from the experimental data for
each
compound (as described herein in Example 2).
As used herein, the term "IC50" or "the half maximal inhibitory concentration"
is used
as a measure of the potency of an inhibitor in inhibiting a specific
biological or biochemical
function. ICso is a quantitative measure that indicates how much of a
particular inhibitor is
needed to inhibit, in vitro, a given biological process or biological
component by 50%. In
some embodiments, the biological component is a DUB, especially a USP9X. ICso
values are
typically expressed as molar concentration. In some embodiments, the term -
IC50" and
"potency- can be used interchangeably.
- 22 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
In some embodiments, the compounds as disclosed have a IC5o value from 0.001
to 10
M. In some embodiments, the compounds as disclosed have a IC5o value from
0.005 to 10
M. In some embodiments, the compounds as disclosed have a IC5o value from 0.01
to 10
M. In some embodiments, the compounds as disclosed have a IC5o value from 1 to
10 M.
In some embodiments, the compounds as disclosed have a IC5o value from 0.001
to 1 M. In
some embodiments, the compounds as disclosed have a IC5o value from 0.001 to
0.1 M. In
some embodiments, the compounds as disclosed have a IC5o value from 0.001 to
0.01 M. In
some embodiments, the compounds as disclosed have a IC5o value from 0.01 to
0.1 M. In
some embodiments, the compounds as disclosed have a IC5o value from 0.1 to 1
M.
In some embodiments, the compounds as disclosed have lower IC5o values than
other
USP9X inhibitor in multiple cell lines, and such other USP9X inhibitor may be
FT709,
E0A13402143, or WP1130 (also called Degrasyn). In some cases, the compounds as
disclosed may be about 1- to about 10-fold more potent than other USP9X
inhibitor in
multiple cell lines. In some cases, the compounds as disclosed may be about 2-
fold, 3-fold,
about 4-fold, about 5-fold, about 6-fold. about 7-fold, about 8-fold, or about
9-fold more
potent than other USP9X inhibitor in multiple cell lines. In some cases, the
compounds as
disclosed may be about 4-fold more potent than WP1130 in multiple cell lines.
The compounds as disclosed also exhibit a high binding specificity for USP9X.
As
used herein, "specificity" and "selectivity" are interchangeable and describe
the selective
inhibition of the compounds towards a particular enzyme and minimal or no
detectable
influence on other enzymes' enzymatic activities when in contact over a
sufficient period of
time. In some embodiments, rather than USP9X, the compounds can inhibit one or
more
homologous enzymes' enzymatic activities by less than 10%. In some
embodiments, rather
than USP9X, the compounds can inhibit one or more homologous enzymes'
enzymatic
activities by less than 6%. In some embodiments, rather than USP9X, the
compounds can
inhibit one or more homologous enzymes' enzymatic activities by less than
about 5%, less
than about 4%, less than about 3%, less than about 2%, or less than about 1%.
In some
embodiments, rather than USP9X, the compounds have no detectable effect on one
or more
homologous enzymes' enzymatic activities, i.e., the compounds inhibit one or
more
homologous enzymes' enzymatic activities by 0%. Homologous enzymes include
other
DUBs rather than USP9X. For example, the compounds (such as Compound 5A as
shown in
Example 2) have no detectable effect on the enzymatic activities on DUBs
including, but not
limited to, CYLD, USP2, USP5, USP7, USP13, USP25, USP4, and UCH-L3. For
example,
the compounds (such as Compound 5A as shown in Example 2) have a minimal
detectable
- 23 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
effect on the enzymatic activities of other DUBs such as, but not limited to,
inhibiting
USP3O's enzymatic activity by less than about 5%, inhibiting USP1O's enzymatic
activity by
less than about 1%, inhibiting USP15's enzymatic activity by less than about
2%, inhibiting
VCPIP's enzymatic activity by less than about 3%, and inhibiting UCH-Li 's
enzymatic
activity by less than about 2%. The selectivity of the compound is determined
with a standard
screening assay, such as screening the compound on a DUB selectivity panel
using ubiquitin-
AMC.
In some embodiments, the compounds exhibit higher selectivity against USP9X
than
other USP9X inhibitors, such as E0A13402143 and WP1130. Besides USP9X,
E0A13402143 also inhibits USP9X, USP24, and USP5; while WP1130 also inhibits
USP5,
USP14, UCH-1L, and UCH37.
Methods disclosed herein include methods of treating a disorder, such as a
disorder
associated with DUB activity or a disorder affected by modulation of DUB
activity, or use of
a compound disclosed herein in the preparation of a medicament to treat a
disorder associated
with DUB activity and/or affected by modulation of DUB activity. Further
contemplated are
methods of treatment wherein DUB catalytic activity is inhibited. In some
cases, provided
herein are methods that further include identifying a subject having a
disorder affected by
modulation of activity of a DUB and administering to the subject a compound as
disclosed
herein.
In some cases, provided herein are methods of inhibiting proliferation of a
cell
comprising contacting the cell with an effective amount of a compound as
disclosed herein to
inhibit proliferation. In some cases, the cell is a cancer cell. Cancer cells
contemplated are
described elsewhere herein. In various cases, the compound inhibits a DUB
endogenous to
the cell and inhibits proliferation. In some cases, provided herein are
methods of inhibiting
Usp9x.
In some cases, provided herein are methods of treating, inhibiting, or
suppressing
cancer in a subject, comprising administrating to the subject an effective
amount of a
compound as disclosed herein to inhibit cancer cell proliferation, thereby
treating, inhibiting,
or suppressing cancer. Cancer contemplated are described elsewhere herein.
In various cases, the methods provided herein are prophylactic methods, and a
compound or composition as disclosed herein is administered prior to onset of
a disorder. In
certain cases, the method further comprises identifying a subject at risk of
contracting a
disorder associated with DUB activity and/or affected by DUB modulation (e.g.,
a virus,
- 24 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
bacterium; and/or parasite as disclosed herein), and administering an
effective amount of a
compound as disclosed herein.
In some cases, provided herein are methods of treating neuropathic or
inflammatory
pain comprising contacting a cell with a compound disclosed herein in an
amount sufficient
to reduce or alleviate the pain, or to inhibit Usp5 in the cell. In some
cases, the contacting
comprises administering the compound to a subject suffering from neuropathic
or
inflammatory pain.
In some cases, the methods disclosed herein further comprises administering a
second
therapeutic agent. The second therapeutic agent can be administered at the
same time as the
compound as disclosed herein, or at a different time (e.g., separated by a
time period of about
1 hour to about 12 hours). In cases where the agents are administered at the
same time, the
agents can be co-formulated, or formulated in separate formulations but given
at the same
time or within about 30 minutes of each other. Contemplated second agents
include, e.g., an
antiviral, antiparasitic, antibacterial, anticancer agent, agent that treats
one or more symptoms
of a genetic disorder, and/or an agent that treats a neurodegenerative
disorder.
Cancer
Cancer is a disease of the genome characterized by a diverse mutational
landscape
and genomic alterations that give rise to mutations that lead to abnormal cell
transduction
cascades. Signal transduction cascades relay growth signals from the cell
membrane into the
nucleus to initiate transcriptional responses or post-translational protein
modifications.
Dysregulation of signal transduction cascades in cancer ultimately results in
increased cell
survival and abnormal cell proliferation. Signal transduction cascades can be
regulated by
phosphorylation that controls protein function, and ubiquitination that
regulates protein
turnover and degradation.
Phosphorylation or kinase signaling cascades and the proteasome, a protein
complex
involved in ubiquitin mediated protein degradation, are major targets in
cancer therapy. The
anticancer activity of kinase and proteasome inhibitors arise from the
disruption of multiple
signaling pathways that support the growth, proliferation, and survival of
malignant cells.
In addition to chemotherapy and autologous stem-cell transplantation, current
therapy
for hematologic (B cell) cancers such as multiple myeloma (MM), mantle cell
lymphoma
(MCL) and chronic myeloid leukemia include the use of proteasome inhibitors
(bortezomib,
carfilzomib), immunomodulatory drugs (thalidomide, lenalidomide, pomalidomide)
and
inhibitors of kinase signal transduction cascades involved in B cell signaling
(Btk, mTOR
- 25 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
inhibitors). Although current treatment strategies for MM and MCL have
improved
management and overall survival of patients, the diseases remain incurable
with a significant
number of patients that eventually relapse and succumb to these diseases and
emphasizing the
need for more effective therapies.
Ubiquitin/proteasome-mediated protein degradation is one of the major
mechanisms
used by cells for protein turnover or degradation. It involves two successive
steps: 1) the
attachment of ubiquitin 76 amino acid polypeptide, to a protein substrate
mediated by the
ubiquitin activating, conjugating and ligating enzymes El, E2, and E3, and 2)
the degradation
of the tagged or poly-ubiquitinylated protein by the 26s proteasome complex or
lysosome.
(Oncogene (2012) 31, 2373-2388 and Acta Pharmacol Sin 2007 September; 28 (9):
1325-
1330).
Deubiquitylation is a reversible process where ubiquitin can be removed from
ubiquitinylated proteins by an enzymatic reaction catalyzed by deubiquitinases
(DUB).
Deubiquitinating enzymes are known to have important roles in the regulation
of protein
stability, proofreading of protein ubiquitination, recycling of ubiquitin and,
maintaining free
ubiquitin concentrations. DUBs can enhance protein stability by preventing
protein
degradation.
Consistent with the role of ubiquitination and DUBs in protein turnover and
stability,
dysregulation in the activity and expression of these enzymes have been linked
to cancer
development and progression. Due to their role in stabilizing the expression
of oncogenic or
tumor suppressor proteins, DUBs have been a focus of attention as drug targets
or as
diagnostic and prognostic biomarkers in oncology research. Several mutated
DUBs have been
found to act as oncogenes or tumor suppressors, and changes in the expression
levels of
DUBs were found in several hematologic and malignant solid tumors (lung,
pancreas,
prostate, colon, thyroid and breast). (Annu Rev Biochem. 2009; 78:363-97).
The DUB USP9X has recently received considerable attention as potential
therapeutic
target in several B cell malignancies (MM, MCL, chronic myeloid leukemia)
based on the
ability of USP9X to associate and stabilize the expression of the oncogenic
protein Myeloid
cell leukemia-1 (Mcl-1). (Nature. 2010 Jan. 7; 463(7277):103-7) The Mc1-1
protein is known
to promote tumor growth and survival by inhibiting apoptotic or cell death
pathways. Md-1
is overexpressed in MM, MCL and chronic myeloid leukemia. The Mc-1 gene was
found to
be amplified in 10.9% of cancers across multiple tissue types including
breast, lung, skin
(melanoma), neural tissue and sarcoma. (Nature. 2010 Feb. 18; 463(7283):899-
905).
- 26 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
In MM, protein expression levels of Mc-1 correlate with resistance to
chemotherapy, disease
relapse and poor survival. Similarly, high expression levels of USP9X were
also found in
MCL and MM which may be an underlying mechanism of increased Mcl-1 stability
in these
diseases. The removal of ubiquitin from Mcl-1 by USP9X rescues it from
proteasomal
degradation and helps promote high levels of Mc-1 inside of cells. Higher
levels of
intracellular Mc-1 helps confer resistance to apoptosis, which is a known
hallmark of cancer
cells. In theory, any inducement of lower levels of intracellular Mc-1 renders
may render
cell more susceptible to apoptosis, especially in response to standard
chemotherapeutic
agents. In support of this, knocking down USP9X expression in MM and MCL cells
reduced
Mc-1 levels, reduced MM cell survival and blocked cell proliferation.
(Leukemia. 2005 July;
19(7): 1248-52).
Also, degradation of Mc-1 via inhibition of USP9X can also be used in
preparation
for stem cell transplant for cancer patients. In current practice, prior to
the transplant, patients
are treated with a debilitating dose of chemotherapy and/or radiation to
ablate hematopoietic
cells. Since this stem cell population seems to uniquely dependent upon Mcl-1
for viability,
USP9X inhibition may provide a milder and more specific protocol for achieving
the same
result. One of the concerns about any therapeutic strategy that antagonizes Mc-
1 as a cancer
treatment is the attendant toxicity for normal cells. In order to curtail
unwanted toxicity, the
compounds of the invention may be conjugated to an antibody directed against a
surface
antigen (e.g., CD34) specific for these stem cells.
Mutations in USP9X gene and high USP9X protein expression were also found in
colorectal, breast, lung ovarian and non-small cell lung carcinoma. (Acta
Pharmacol. Sin
2007 28(9): 1325-1330). Inhibiting expression of USP9X in MM and colorectal
cancer
increased cell death, blocked cell proliferation and sensitized cells to
chemotherapy
suggesting an important role of USP9X in cancer pathology. (Nature, 2010,
463(7283):899-
905 and Cancer Biol. Ther. 2012 November; 13(13):1319-24).
USP9X was also found to be overexpressed in melanoma cells and in melanoma
patients. The use of compounds in melanoma cell lines resulted in the
increased expression of
the tumor suppressor p53, reduction in Mcl-1 protein, increased cell death,
suppression of
tumor cell invasiveness, and inhibition of cell proliferation. The compound
also enhanced and
further increased the apoptotic and anti-cell proliferation effect of the
kinase inhibitor
vemurafenib that was recently used in about. 60% of melanoma patients that
harbor a
mutation in BRAF, a component of kinase signaling cascade involved in cell
proliferation
- 27 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
and survival. In melanoma xenografts, use of monotherapy reduced tumor growth
and did not
have any notable side effects in animal weight, behavior and mobility.
Meanwhile, USP9X is implicated in regulating endocytosis of the breast cancer
oncogene ERBB2. The human protein erbB2 encoded by ERBB2 is also called HER2
(human epidermal growth factor receptor 2) or CD340 (cluster of
differentiation 340). In 5K-
BR3 breast cancer cells overexpressing erbB2 treated with the proteasome
inhibitor
bortezomib, erbB2 co-immunoprecipitates with a complex containing c-Cbl and
USP9X.
Reduction in USP9X levels increases bortezomib-induced downregulation of
erbB2,
suggesting that USP9X is associated with the internalisation and
ubiquitylation status of
erbB2. (Marx C et al., Cancer Res., 2010; 70:3709-3717.)
Overexpression of erbB2 is observed in breast and ovarian cancers (Slamon et
al.,
Science, 1987, 235:177-182; Slamon et al., Science, 1989, 244:707-712; and
U.S. Pat. No.
4,968,603), and in other carcinomas including carcinomas of the stomach,
endometrium,
salivary gland, lung, kidney, colon, thyroid, pancreas and bladder. (King et
al., Science, 1985,
229: 974; Yokota et al., Lancet, 1986, 1:765- 767; Fukushige et al., Mol Cell
Biol., 1986,
6:955- 958; Guerin et al., Oncogene Res., 1988, 3:21 -31; Cohen et al.,
Oncogene, 1989,
4:81 88; Yonemura et al., Cancer Res., 1991, 51:1 034:1 034; Borst et al.,
Gynecol. Oncol.,
1990, 38: 364; Weiner et al., Cancer Res., 1990, 50:421- 425; Kern et al.,
Cancer Res.,
1990, 50:5 184; Park et al., Cancer Res., 1989, 49:6 605; Zhau et al., Mol.
Carcinog., 1990,
3:254-257; Aasland et al. Br. J. Cancer, 1988, 57:358-363; Williams et al.
Pathobiology,
1991, 59:46-52; and McCann et al., Cancer, 1990, 65:88-92), Also, erbB2 may be
overexpressed in prostate cancer (Gu et al. Cancer Lett., 1996, 99:185-189;
Ross et al. Hum.
Pathol., 1997, 28:827-33; Ross et al. Cancer, 1997, 79:2162-70; and Sadasivan
et al. J. Urol.,
1993, 150:126-31). Specific cancers contemplated include, but are not limited
to, chronic
myelogenous leukemia (CML), melanoma, acute lymphocytic leukemia, chronic
lymphocytic
leukemia, acute myelogenous leukemia, B-cell lymphoma, mantle cell lymphoma,
multiple
myeloma, plasma cell dyscrasia, myeloproliferative disorders, glioblastoma,
Kapsi's sarcoma,
nasopharyngeal carcinoma (EBV), lung cancer, colon cancer, pancreatic cancer,
breast
cancer, prostate cancer, melanoma, and solid tumors.
The sensitivities of multiple cancer cells can be determined by screening the
compounds on a cancer cell line panel, such as an ONCOLINESIm Profiler
(Netherlands
Translational Research Center) assayed with various tumor cell lines. For more
information
on Oncolineslm methods, see https://www.oncolines.com/oncolinesioncolines-
explained-
facts/. In some embodiments wherein the ONCOLINES TM Profiler is used, the
compounds
- 28 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
inhibit the proliferation of a majority of the tumor cell lines being tested,
such as at least
about 80% of the tumor cell lines being tested or at least 50 of 66 tumor cell
lines being
tested. For example, Compound 7A inhibited the proliferation of 57 of the 66
cell lines being
tested. For a tumor cell line sensitive to the compounds, the compounds can
have a 1050 value
of no greater than 1 H.M. "A tumor cell line sensitive to the compounds- is
referred to that the
proliferation of a tumor cell line, when contacted with the compounds, is
inhibited by at least
50%, at least 60%, at least 70%, at least 80%, at least 85%, at least 90%, at
least 91%, at least
92%, at least 93%, at least 94%, at least 95%, at least 96%, at least 97%, at
least 98%, at least
99%, or 100%.
Any cancer cell associated with high expression of one or more USP9X
substrates,
such as Mcl-1 and/or erbB2, can be used as a therapeutic target for the
compounds as
disclosed. As a nonlimiting example, cancer cell line completed may include
the colon
carcinoma cell line RKO (that expresses high levels of Bc1-2 and very high
levels of Mc-1
but is resistant to the Bc1-2 and Bc1-xl inhibitor ABT737), the pancreatic
cancer cell line
MiaPaca-2 (that has mutations in both copies of Ras gene and over expresses
erbB2 and Mc-
1), the human breast cancer cell line SK-BR3 (that expresses erbB2 and Mc-1
and is
especially used in erbB2 targeting), the human breast ductal carcinoma cell
line BT-549, the
triple-negative breast cancer (TNBC) cell line MDA-MB-231, and the histiocytic
lymphoma
cell line U937.
In some embodiments, the induced degradations of USP9X substrates (such as Mc1-
1,
erbB2, beta-catenin and aldehyde dehydrogenase 1A3) in multiple cancer cell
lines by the
compounds, can be investigated using a standard analysis method, such as
western blot, flow
cytometry, or ELISA. After treated with the compounds for a certain period of
time (e.g.,
from about 1 hour to about 72 hours, such as 12, 24, 48, and 72 hours and
preferably 72
hours), the normalized signals from USP9X substrates, especially Mc-1 and/or
erbB2,
decrease by at least 40%, at least 50%, at least 55%, at least 60%, at least
65%, at least 70%,
at least 75%, at least 80%, at least 85%, at least 90%, at least 91%, at least
92%, at least 93%,
at least 94%, at least 95%, at least 96%, at least 97%, at least 98%, at least
99%, or 100%,
indicating that the compounds induce the substrate degradation by inhibiting
USP9X. The
induced degradation leads to the inhibition of cancer cell proliferation,
resulting in the
decrease in cell viability. In particular embodiments, even partial
degradation of the
substrates induced by the compounds can be sufficient to inhibit the
proliferation of cancer
cells completely, such as disclosed in Example 3 wherein the 50% decrease in
Mc-1 due to
- 29 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
the 24-hour treatment of the compound led to the inhibition the U937 cell
proliferation by
100% (i.e., cell viability decreases to 0%).
The mechanism of action for induced degradation of USP9X substrate can be
further
examined by comparing the combined effect of the compound and a proteasome
inhibitor
with the effect of the compound alone, on cells that express the USP9X
substrate. As
illustrated in Example 4, when U937 cells treated with both MG132 and Compound
5A,
decline in Mcl-1 was reduced compared to U937 cells treated with Compound 5A
only.
These results confirmed intracellular inhibition of USP9X by the compounds to
enhance
proteasomal degradation of Mc1-1. By varying the concentration ratios of the
compound and
proteasome inhibitor for treating cells, it is possible to control the
degradation of a USP9X
substrate so the USP9X substrate can decline to a desirable level rather than
degrade entirely.
The combined use of the compound and proteasome inhibitor provides a novel
approach for
inducing controlled degradation of a USP9X substrate wherein the USP9X
substrate declines
by about 10% or more, about 20% or more, about 30% or more, about 40% or more,
about
50% or more, about 60% or more, about 70% or more, about g0% or more, about
90% or
more.
Immune Oncology
Programmed death-ligand 1 (PD-L1), an immune-oncology target, has been
recently
identified as a substrate for USP9X. PD-Li as a transmembrane protein is
expressed on
tumor cells and capable of blunting the immune response to tumors by
interacting with PD1
on the surface of T cells that otherwise would attack tumor antigens on the
surface of these
cells and eradicate them. The PDL1-PD1 interaction can be blunted by
therapeutic antibodies
and has shown clinical benefit in cancer patients. The compounds as described
herein may be
capable of inducing PD-Li degradation by inhibiting USP9X and thus enhancing
the immune
response of T cells against tumor cells. People skilled in the art will
recognize that other
USP9X substrates, have been described and are continually being discovered.
All such
USP9X substrates can be therapeutic, prognostic, or research targets of the
compounds as
described herein and encompassed by the present invention.
Pathogenic Infections
The methods and compounds disclosed herein are useful in treating pathogenic
infections, e.g., preventing, inhibiting and/or ameliorating a pathogenic
infection or symptom
- 30 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
of a pathogenic infection. In some cases, the methods and compounds disclosed
herein are
useful in treating a condition due to a pathogenic infection.
Intentional contamination of the food and water supplies represents a major
threat to
the health and health-related services in the US population as a whole and to
our armed forces
serving throughout the world. Many of the category B water- and food-borne
pathogens have
specific properties, e.g. low infectious dose, high stability, that make them
attractive
candidates for this type of bioterrorism. To thwart this potential threat,
methods or agents that
provide protection or prophylaxis against these defined pathogens are urgently
needed.
Ideally, agents that provide protection against a wide spectrum of threats
would be desirable.
The compounds disclosed herein have broad activity against multiple pathogens.
For
example, a potent inhibitor of diverse category A and B pathogens, and related
family
members, e.g., murine norovirus, Tulane virus, Listeria monocytogenes,
Toxoplasma gondii
infection. Norwalk virus, Sindbis virus, La Crosse virus and coronavirus, such
as SARS,
MERS, COVID-19. COVID-19 replication requires deubiquitylase activity.
In certain cells the compounds disclosed herein inhibit a deubiquitinase and
this
action results in accumulation of ubiquitinated proteins in the cytoplasmic
and aggresomal
compartment of the cell. This can establish an inhospitable environment for
pathogen
infection or replication within the target cell. Thus, these compounds are
used as an
antimicrobial inhibitor that can effectively suppress multiple pathogens. The
compounds
disclosed herein block the infectivity of category A and/or B pathogens,
and/or related family
members.
Contemplated are pathogens that use a DUB in their infection mechanism. In
some
cases, the pathogen uses a DUB endogenous to the infected cell. In various
cases, the
pathogen uses a DUB endogenous to the pathogen.
Contemplated diseases or disorders due to a pathogenic infection include
gastroenteritis, encephalitis, respiratory tract infections (e.g., SARS),
virus-induced cancers,
rabies, hemorrhagic fevers (e.g., Crimean-Congo, Dengue), Rift valley fever,
listeriosis, or
toxoplasmosis. Also contemplated diseases or disorders due to a pathogenic
infection include
meningitis, myocarditis, hepatitis, bacteremi a, and skin infections.
Contemplated pathogens include viral, bacterial, fungal, and parasitic
pathogens.
Contemplated pathogenic viruses include a calicivirus (e.g., norovirus,
sapovirus), a
picomavirus, a Togavirus, a Bunyavirus, a Rhabdovirus, a herpes virus, an
adenovirus, an
arterivirus, a coronavirus, a flavivirus, a paramyxovirus, a papillomavirus, a
virus encoding
-31 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
for an ovarian tumor (OTU)-like protease, a baculovirus, or a nairovirus.
Other contemplated
pathogenic viruses include polyoma viruses and retroviruses.
Specific viruses contemplated include encephalomyocarditis virus (EMCV).
Sindbis
virus (SiNV), La Crosse virus (LaCV), Norwalk virus, Tulane virus, rotavirus,
Epstein-Barr
(EBV), herpesvirus, Dengue virus, and papillomavirus. Further specific viruses
contemplated
include cytomegalovirus, BK virus, hepatitis C virus, and IIIV.
Contemplated bacteria include Chlamydia, Escherichia, Salmonella, Yersinia,
Burkholderia, Haemophilus, Listeria, and Mycobacterium. Other bacteria
contemplated
include Staphylococcus aureus, or more specifically methicillin-resistant
Staph aureus
(MRSA).
Contemplated parasites or fungi include Plasmodium falciparum, Toxoplasma
gondii,
Entamoeba histolytica, Giardia lamblia, Trypanosoma brucei, Trypanosoma cruzi,
Cestoda,
Clonorchis, Opisthorchis, Strongylocides, Candida, Aspergillus, and
Cryptococcus.
Dosing and Pharmaceutical Formulations
The terms "therapeutically effective amount" and "prophylactically effective
amount,"
as used herein, refer to an amount of a compound sufficient to treat,
ameliorate, or prevent
the identified disease or condition, or to exhibit a detectable therapeutic,
prophylactic, or
inhibitory effect. The effect can be detected by, for example, an improvement
in clinical
condition, reduction in symptoms, or by any of the assays or clinical
diagnostic tests
described herein. The precise effective amount for a subject will depend upon
the subject's
body weight, size, and health; the nature and extent of the condition; and the
therapeutic or
combination of therapeutics selected for administration. Therapeutically and
prophylactically
effective amounts for a given situation can be determined by routine
experimentation that is
within the skill and judgment of the clinician.
Dosages of the therapeutic can alternately be administered as a dose measured
in
mg/kg. Contemplated mg/kg doses of the disclosed therapeutics include about
0.001 mg/kg to
about 1000 mg/kg. Specific ranges of doses in mg/kg include about 0.1 mg/kg to
about 500
mg/kg, about 0.5 mg/kg to about 200 mg/kg, about 1 mg/kg to about 100 mg/kg,
about 2
mg/kg to about 50 mg/kg, and about 5 mg/kg to about 30 mg/kg.
As herein, the compounds described herein may be formulated in pharmaceutical
compositions with a pharmaceutically acceptable excipient, carrier, or
diluent. The compound
or composition comprising the compound is administered by any route that
permits treatment
- 32 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
of the disease or condition. One route of administration is oral
administration. Additionally,
the compound or composition comprising the compound may be delivered to a
patient using
any standard route of administration, including parenterally, such as
intravenously,
intraperitoneally, intrapulmonary, subcutaneously or intramuscularly,
intrathecally, topically,
transdermally, rectally, orally, nasally or by inhalation. Slow release
formulations may also
be prepared from the agents described herein in order to achieve a controlled
release of the
active agent in contact with the body fluids in the gastro intestinal tract,
and to provide a
substantial constant and effective level of the active agent in the blood
plasma. The crystal
form may be embedded for this purpose in a polymer matrix of a biological
degradable
polymer, a water-soluble polymer or a mixture of both, and optionally suitable
surfactants.
Embedding can mean in this context the incorporation of micro-particles in a
matrix of
polymers. Controlled release formulations are also obtained through
encapsulation of
dispersed micro-particles or emulsified micro-droplets via known dispersion or
emulsion
coating technologies.
Administration may take the form of single dose administration, or a compound
as
disclosed herein can be administered over a period of time, either in divided
doses or in a
continuous-release formulation or administration method (e.g., a pump).
However, the
compounds of the embodiments are administered to the subject, the amounts of
compound
administered and the route of administration chosen should be selected to
permit efficacious
treatment of the disease condition.
In an embodiment, the pharmaceutical compositions are formulated with one or
more
pharmaceutically acceptable excipient, such as carriers, solvents,
stabilizers, adjuvants,
diluents, etc., depending upon the particular mode of administration and
dosage form. The
pharmaceutical compositions should generally be formulated to achieve a
physiologically
compatible pH, and may range from a pH of about 3 to a pH of about 11,
preferably about pH
3 to about pH 7, depending on the formulation and route of administration. In
alternative
embodiments, the pH is adjusted to a range from about pH 5.0 to about pH 8.
More
particularly, the pharmaceutical compositions may comprise a therapeutically
or
prophylactically effective amount of at least one compound as described
herein, together with
one or more pharmaceutically acceptable excipients. Optionally, the
pharmaceutical
compositions may comprise a combination of the compounds described herein, or
may
include a second active ingredient useful in the treatment or prevention of
bacterial infection
(e.g., anti-bacterial or anti-microbial agents).
- 33 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Formulations, e.g., for parenteral Or oral administration, are most typically
solids,
liquid solutions; emulsions or suspensions, while inhalable formulations for
pulmonary
administration are generally liquids or powders. A pharmaceutical composition
can also be
formulated as a lyophilized solid that is reconstituted with a physiologically
compatible
solvent prior to administration. Alternative pharmaceutical compositions may
be formulated
as syrups, creams, ointments, tablets, and the like.
The term "pharmaceutically acceptable excipient" refers to an excipient for
administration of a pharmaceutical agent, such as the compounds described
herein. The term
refers to any pharmaceutical excipient that may be administered without undue
toxicity.
Pharmaceutically acceptable excipients are determined in part by the
particular
composition being administered, as well as by the particular method used to
administer the
composition. Accordingly, there exists a wide variety of suitable formulations
of
pharmaceutical compositions (see, e.g., Remington's Pharmaceutical Sciences).
Suitable excipients may be carrier molecules that include large, slowly
metabolized
macromolecules such as proteins, polysaccharides, polylactic acids,
polyglycolic acids,
polymeric amino acids, amino acid copolymers, and inactive virus particles.
Other exemplary
excipients include antioxidants (e.g., ascorbic acid), chelating agents (e.g.,
EDTA),
carbohydrates (e.g., dextrin, hydroxyalkylcellulose, and/or
hydroxyalkylmethylcellulose),
stearic acid, liquids (e.g., oils, water, saline, glycerol and/or ethanol)
wetting or emulsifying
agents, pH buffering substances, and the like. Liposomes are also included
within the
definition of pharmaceutically acceptable excipients
The pharmaceutical compositions described herein are formulated in any form
suitable for an intended method of administration. When intended for oral use
for example,
tablets, troches, lozenges, aqueous or oil suspensions, non-aqueous solutions,
dispersible
powders or granules (including micronized particles or nanoparti cies),
emulsions, hard or soft
capsules, syrups or elixirs may be prepared. Compositions intended for oral
use may be
prepared according to any method known to the art for the manufacture of
pharmaceutical
compositions, and such compositions may contain one or more agents including
sweetening
agents, flavoring agents, coloring agents and preserving agents, in order to
provide a
palatable preparation.
Pharmaceutically acceptable excipients particularly suitable for use in
conjunction
with tablets include, for example, inert diluents, such as celluloses, calcium
or sodium
carbonate, lactose, calcium or sodium phosphate; disintegrating agents, such
as cross-linked
- 34 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
povidone, maize starch, or alginic acid; binding agents, such as povidone,
starch, gelatin or
acacia; and lubricating agents, such as magnesium stearate, stearic acid or
talc.
Tablets may be uncoated or may be coated by known techniques including
microencapsulation to delay disintegration and adsorption in the
gastrointestinal tract and
thereby provide a sustained action over a longer period. For example, a time
delay material
such as glyceryl monostearate or glyceryl distearate alone or with a wax may
be employed.
Formulations for oral use may be also presented as hard gelatin capsules
wherein the
active ingredient is mixed with an inert solid diluent, for example
celluloses, lactose, calcium
phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient
is mixed with
non-aqueous or oil medium, such as glycerin, propylene glycol, polyethylene
glycol, peanut
oil, liquid paraffin or olive oil.
In another embodiment, pharmaceutical compositions may be formulated as
suspensions comprising a compound of the embodiments in admixture with at
least one
pharmaceutically acceptable excipient suitable for the manufacture of a
suspension.
In yet another embodiment, pharmaceutical compositions may be formulated as
dispersible powders and granules suitable for preparation of a suspension by
the addition of
suitable excipients.
Excipients suitable for use in connection with suspensions include suspending
agents
(e.g., sodium carboxymethylcellulose, methylcellulose, hydroxypropyl
methylcellulose,
sodium alginate, polyvinylpyrrolidone, gum tragacanth, gum acacia); dispersing
or wetting
agents (e.g., a naturally occurring phosphatide (e.g., lecithin), a
condensation product of an
alkylene oxide with a fatty acid (e.g., polyoxy ethylene stearate), a
condensation product of
ethylene oxide with a long chain aliphatic alcohol (e.g.,
heptadecaethyleneoxycethanol), a
condensation product of ethylene oxide with a partial ester derived from a
fatty acid and a
hexitol anhydride (e.g., polyoxyethylene sorbitan monooleate)); and thickening
agents (e.g.,
carbomer, beeswax, hard paraffin or cetyl alcohol). The suspensions may also
contain one or
more preservatives (e.g., acetic acid, methyl or n-propyl p-hydroxy-benzoate);
one or more
coloring agents; one or more flavoring agents; and one or more sweetening
agents such as
sucrose or saccharin.
The pharmaceutical compositions may also be in the form of oil-in water
emulsions.
The oily phase may be a vegetable oil, such as olive oil or arachis oil, a
mineral oil, such as
liquid paraffin, or a mixture of these. Suitable emulsifying agents include
naturally-occurring
gums, such as gum acacia and gum tragacanth; naturally occurring phosphatides,
such as
soybean lecithin, esters or partial esters derived from fatty acids; hexitol
anhydrides, such as
- 35 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
sorbitan monooleate, and condensation products of these partial esters with
ethylene oxide,
such as polyoxyethylene sorbitan monooleate. The emulsion may also contain
sweetening
and flavoring agents. Syrups and elixirs may be formulated with sweetening
agents, such as
glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent,
a preservative,
a flavoring or a coloring agent.
Additionally, the pharmaceutical compositions may be in the form of a sterile
injectable preparation, such as a sterile injectable aqueous emulsion or
oleaginous
suspension. This emulsion or suspension may be formulated by a person of
ordinary skill in
the art using those suitable dispersing or wetting agents and suspending
agents, including
those mentioned above. The sterile injectable preparation may also be a
sterile injectable
solution or suspension in a non-toxic parenterally acceptable diluent or
solvent, such as a
solution in 1,2-propane-diol.
The sterile injectable preparation may also be prepared as a lyophilized
powder.
Among the acceptable vehicles and solvents that may be employed are water,
Ringer's
solution, and isotonic sodium chloride solution. In addition, sterile fixed
oils may be
employed as a solvent or suspending medium. For this purpose, any bland fixed
oil may be
employed including synthetic mono- or diglycerides. In addition, fatty acids
(e.g., oleic acid)
may likewise be used in the preparation of injectables.
To obtain a stable water-soluble dose form of a pharmaceutical composition, a
pharmaceutically acceptable salt of a compound described herein may be
dissolved in an
aqueous solution of an organic or inorganic acid, such as 0.3 M solution of
succinic acid, or
more preferably, citric acid. If a soluble salt form is not available, the
compound may be
dissolved in a suitable co-solvent or combination of co-solvents. Examples of
suitable co-
solvents include alcohol, propylene glycol, polyethylene glycol 300,
polysorbate 80, glycerin
and the like in concentrations ranging from about 0 to about 60% of the total
volume. In one
embodiment, the active compound is dissolved in DMSO and diluted with water.
The pharmaceutical composition may also be in the form of a solution of a salt
form
of the active ingredient in an appropriate aqueous vehicle, such as water or
isotonic saline or
dextrose solution. Also contemplated are compounds which have been modified by
substitutions or additions of chemical or biochemical moieties which make them
more
suitable for delivery (e.g., increase solubility, bioactivity, palatability,
decrease adverse
reactions, etc.), for example by esterification, glycosylation, PEGylation,
etc.
- 36 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
In some embodiments, the compounds described herein may be formulated for oral
administration in a lipid-based formulation suitable for low solubility
compounds. Lipid-
based formulations can generally enhance the oral bioavailability of such
compounds.
As such, pharmaceutical compositions comprise a therapeutically or
prophylactically
effective amount of a compound described herein, together with at least one
pharmaceutically
acceptable excipient selected from the group consisting of medium chain fatty
acids and
propylene glycol esters thereof (e.g., propylene glycol esters of edible fatty
acids, such as
caprylic and capric fatty acids) and pharmaceutically acceptable surfactants,
such as polyoxyl
40 hydrogenated castor oil.
In some embodiments, cyclodextrins may be added as aqueous solubility
enhancers.
Exemplary cyclodextrins include hydroxypropyl, hydroxyethyl, glucosyl,
maltosyl and
maltotriosyl derivatives of alpha-, beta-, and gamma-cyclodextrin. A specific
cyclodextrin
solubility enhancer is hydroxypropyl-o-cyclodextrin (BPBC), which may be added
to any of
the above-described compositions to further improve the aqueous solubility
characteristics of
the compounds of the embodiments. In one embodiment, the composition comprises
about
0.1% to about 20% hydroxypropyl-o-cyclodextrin, more preferably about 1% to
about 15%
hydroxypropyl-o-cyclodextrin, and even more preferably from about 2.5% to
about 10%
hydroxypropyl-o-cyclodextrin. The amount of solubility enhancer employed will
depend on
the amount of the compound of the invention in the composition.
Combination Therapy
The methods of the embodiments also include the use of a compound or compounds
as described herein together with one or more additional therapeutic agents
for the treatment
of disease conditions. Thus, for example, the combination of active
ingredients may be: (1)
conjugated, (2) co-formulated and administered or delivered simultaneously in
a combined
formulation; (3) delivered by alternation or in parallel as separate
formulations; or (4) by any
other combination therapy regimen known in the art. When delivered in
alternation therapy,
the methods described herein may comprise administering or delivering the
active ingredients
sequentially, e.g., in separate solution, emulsion, suspension, tablets, pills
or capsules, or by
different injections in separate syringes. In general, during alternation
therapy, an effective
dosage of each active ingredient is administered sequentially, i.e., serially,
whereas in
simultaneous therapy, effective dosages of two or more active ingredients are
administered
together. Various sequences of intermittent combination therapy may also be
used.
- 37 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
In some cases, a compowid disclosed herein is administered and/or formulated
with a
second therapeutic--e.g., a chemotherapeutic.
Chemotherapeutic agents contemplated for use include, without limitation,
alkylating
agents including: nitrogen mustards, such as mechlor-ethamine,
cyclophosphamide,
ifosfamide, melphalan and chlorambucil; nitrosoureas, such as carmustine
(BCNU),
lomustine (CCNU), and semustine (methyl-CCNU); ethylenimines/methylmelamine
such as
thriethylenemelamine (TEM), triethylene, thiophosphoramide (thiotepa),
hexamethylmelamine (HMM, altretamine); alkyl sulfonates such as busulfan;
triazines such
as dacarbazine (DTIC); antimetabolites including folic acid analogs such as
methotrexate and
trimetrexate, pyrimidine analogs such as 5-fluorouracil, fluorodeoxyuridine,
gemcitabine,
cytosine arabinoside (AraC, cytarabine), 5-azacytidine, 2,2'-
difluorodeoxycytidine, purine
analogs such as 6-mercaptopurine, 6-thioguanine, azathioprine, 21-
deoxycoformycin
(pentostatin), erythrohydroxynonyladenine (EHNA), fludarabine phosphate, and 2-
chlorodeoxyadenosine (dadri bine, 2-CdA); natural products including
antimitotic drugs such
as paclitaxel, vinca alkaloids including vinblastine (VLB), vincristine, and
vinorelbine,
taxotere, estramustine, and estramustine phosphate; epipodophylotoxins such as
etoposide
and teniposide; antibiotics such as actinomycin D, daunomycin (rubidomycin),
doxorubicin,
mitoxantrone, idarubicin, bleomycins, plicamycin (mithramycin), mitomycin C,
and
actinomycin; enzymes such as L-asparaginase; biological response modifiers
such as
interferon-alpha, IL-2, G-CSF and GM-CSF; miscellaneous agents including
platinum
coordination complexes such as cisplatin and carboplatin, anthracenediones
such as
mitoxantrone, substituted urea such as hydroxyurea, methylhydrazine
derivatives including
N-methylhydrazine (MIH) and procarbazine, adrenocortical suppressants such as
mitotane
(o,p'-DDD) and aminoglutethimide; hormones and antagonists including
adrenocorticosteroid
antagonists such as prednisone and equivalents, dexamethasone and
aminoglutethimide;
progestin such as hydroxyprogesterone caproate, medroxyprogesterone acetate
and megestrol
acetate; estrogen such as diethylstilbestrol and ethinyl estradiol
equivalents; antiestrogen such
as tamoxifen; androgens including testosterone propionate and
fluoxymesterone/equivalents;
antiandrogens such as flutami de, gonadotropin-releasing hormone analogs and
leuproli de;
non-steroidal antiandrogens such as flutamide; kinase inhibitors, histone
deacetylase
inhibitors, methylation inhibitors, proteasome inhibitors, monoclonal
antibodies, oxidants,
anti-oxidants, telomerase inhibitors, BH3 mimetics, ubiquitin ligase
inhibitors, Stat inhibitors,
and nanoparti cl es.
- 38 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Protein and antibody chemotherapeutics are well suited for combination
therapies
and/or chemical conjugation to the compounds of the invention. For example,
the
combination therapy can include aldesleukin (e.g., PROLEUKIN), alemtuzumab
(e.g.,
CAMPATH), asparaginase Erwinia chrysanthemi (e.g., ERWINAZE), bevacizumab
(e.g.,
AVASTIN), blinatumomab (e.g., BLINCYTO), brentuximab vedotin (e.g., ADCETRIS),
cetuximab (e.g., ERBITUX), denosumab (e.g., PROLIA, XGEVA), Dinutuximab (e.g.,
UNITUXIN), ibritumomab tiuxctan (e.g., ZEVAL1N), ipilimumab (e.g., YERVOY),
leuprolide acetate (e.g., LUPRON DEPOT, LUPRON DEPOT-3 MONTH, LUPRON
DEPOT-4 MONTH, LUPRON DEPOT-PED, LUPRON, VIADUR), niv-olumab (e.g.,
OPDIVO), obinutuzumab (e.g., GAZYVA), OEPA, ofatumumab (e.g., ARZERRA),
panitumumab (e.g., VECTIBIX), pegaspargase (e.g., ONCASPAR), peginterferon
alfa-2b
(e.g., PEG-INTRON), peginterferon alia-2b (e.g., SYLATRON), pembrolizumab
(e.g.,
K_EYTRUDA), ramucirumab (e.g., CYRAMZA), R-CHOP, recombinant HPV bivalent
vaccine (e.g., CERVARIX), recombinant human papillomavirus (e.g., HPV)
nonavalent
vaccine (e.g., GARDASIL 9), recombinant human papillomavirus (e.g., HPV)
quadrivalent
vaccine (e.g., GARDASIL), recombinant interferon alfa-2b (e.g., INTRON A),
rituximab
RITUXAN), siltuximab (e.g., SYLVANT), tositumomab and iodine 1 131 tositumomab
(e.g., BEXXAR), and trastuzumab (e.g., HERCEPTIN).
Conjugation can be achieved by covalently reacting a moiety on the conjugate
or
protein and a moiety substituted on the compound of formula (I). For example,
the
compound of formula (I) can be substituted by an attachment group, such as an
electrophilic
group or group that can react with a crosslinking agent. For example,
conjugation moieties
include thiols, hydroxy groups and amines. For example, maleimide groups,
activated
disulfides, active esters such as NHS esters and HOBt esters, haloformates,
acid halides, alkyl
and benzyl halides such as haloacetamides can be used. Self-stabilizing
maleimides and
bridging disulfides can also be used in accordance with the disclosure.
One example of a "self-stabilizing" maleimide group that hydrolyzes
spontaneously
under antibody conjugation conditions to give an ADC species with improved
stability is
depicted in the schematic below. See U.S. Published Application No.
2013/0309256,
International Application Publication No. WO 2013/173337, Tumey et al., 2014,
Bioconjugate Chem. 25: 1871-1880, and Lyon et al., 2014, Nat. Biotechnol. 32:
1059-1062.
Thus, the maleimide attachment group is reacted with a sulfhydryl of an
antibody to give an
intermediate succinimide ring. The hydrolyzed form of the attachment group is
resistant to
- 39 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
deconjugation in the presence of plasma proteins. Other crosslinking moieties
such as
epichlorohydrin can also be used.
Also using conjugation technology, targeting agents, such as, peptides,
proteins, small
molecules, antibodies or ligands specific to cell receptors, can be linked to
a compound of the
invention. For example, the compounds as described can be incorporated into
PROTA.0
constructs to target USP9X, leading to its ubiquitylati on by an 3 ubiquitin
ligase and
consequently inducing the degradation of 1JSP9X. For example, cell receptors
on cancer
cells can be targeted. For example, antibodies or ligands that bind one of the
following
antigens can be used: Aminopeptidase N (CD13), Annexin Al, B7-H3 (CD276,
various
cancers), CA125, CA15-3 (carcinomas), CA19-9 (carcinomas), L6 (carcinomas),
Lewis Y
(carcinomas), Lewis X (carcinomas), alpha fetoprotein (carcinomas), CA242,
placental
alkaline phosphatase (carcinomas), prostate specific antigen (prostate),
prostatic acid
phosphatase (prostate), epidermal growth factor (carcinomas), CD2 (Hodgkin's
disease, NHL
lymphoma, multiple myeloma), CD3 epsilon (T cell lymphoma, lung, breast,
gastric, ovarian
cancers, autoimmune diseases, malignant ascites), CD19 (B cell malignancies),
CD20 (non-
Hodgkin's lymphoma), CD22 (leukemia, lymphoma, multiple myeloma, SLE), CD30,
CD33,
CD38 (multiple myeloma), CD40 (lymphoma, multiple myeloma, leukemia), CD51
(Metastatic melanoma, sarcoma), CD52, CD56 (small cell lung cancers, ovarian
cancer,
Merkel cell carcinoma, and the liquid tumor, multiple myeloma), CD66e
(cancers), CD70
(metastatic renal cell carcinoma and non-Hodgkin lymphoma), CD74 (multiple
myeloma),
CD80 (lymphoma), CD98 (cancers), mucin (carcinomas), CD221 (solid tumors),
CD227
(breast, ovarian cancers), CD262 (NSCLC and other cancers), CD309 (ovarian
cancers),
CD326 (solid tumors), CEACAM3 (colorectal, gastric cancers), CEACAM5
(carcinoembryonic antigen; CEA, CD66e) (breast, colorectal and lung cancers),
DLL4
(.DELTA.-like-4), EGFR (Epidermal Growth Factor Receptor, various cancers),
CTLA4
(melanoma), CXCR4 (CD184, Heme-oncology, solid tumors), Endoglin (CD105, solid
tumors), EPCAM (epithelial cell adhesion molecule, bladder, head, neck, colon,
NHL
prostate, and ovarian cancers), ERBB2 (Epidermal Growth Factor Receptor 2;
lung, breast,
prostate cancers), FCGR1 (autoimmune diseases), FOLR (folate receptor, ovarian
cancers),
GD2 ganglioside (cancers), G-28 (a cell surface antigen glycolipid, melanoma),
GD3 idiotype
(cancers), Heat shock proteins (cancers), HER1 (lung, stomach cancers), HER2
(breast, lung
and ovarian cancers), HLA-DR10 (NHL), HLA-DRB (NHL, B cell leukemia), human
chorionic gonadotropin (carcinoma), IGF1R (insulin-like growth factor 1
receptor, solid
tumors, blood cancers), 1L-2 receptor (interleukin 2 receptor, T-cell leukemia
and
- 40 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
lymphomas), IL-6R (interleukin 6 receptor, multiple myeloma, RA, Castleman's
disease, IL6
dependent tumors), Integrins (.alpha.v.beta.3, .alpha.5.beta.1,
.alpha.'.beta.4, .alpha.11.beta.3,
.alpha.5.beta.5, .alpha.v.beta.5, for various cancers), MAGE-1 (carcinomas),
MAGE-2
(carcinomas), MAGE-3 (carcinomas), MAGE 4 (carcinomas), anti-transferrin
receptor
(carcinomas), p97 (melanoma), MS4A1 (membrane-spanning 4-domains subfamily A
member 1, Non-IIodgkin's D cell lymphoma, leukemia), MUC1 or MUC1-KLII
(breast,
ovarian, cervix, bronchus and gastrointestinal cancer), MUC16 (CA125) (Ovarian
cancers),
CEA (colorectal), gp100 (melanoma), MARTI (melanoma), MPG (melanoma), MS4A1
(membrane-spanning 4-domains subfamily A, small cell lung cancers, NHL),
Nucleolin, Neu
oncogene product (carcinomas), P21 (carcinomas), Paratope of anti-(N-
glycolylneuraminic
acid, Breast, Melanoma cancers), PLAP-like testicular alkaline phosphatase
(ovarian,
testicular cancers), PSMA (prostate tumors), PSA (prostate), ROB04, TAG 72
(tumor
associated glycoprotein 72, AML, gastric, colorectal, ovarian cancers), T cell
transmembrane
protein (cancers), Tie (CD202b), TNERSFIOB (tumor necrosis factor receptor
superfamily
member 10B, cancers), TNFRSF13B (tumor necrosis factor receptor superfamily
member
13B, multiple myeloma, NHL, other cancers, RA and SLE), TPBG (trophoblast
glycoprotein,
Renal cell carcinoma), TRAIL-R1 (Tumor necrosis apoptosis Inducing ligand
Receptor 1,
lymphoma, NHL, colorectal, lung cancers), VCAM-1 (CD106, Melanoma), VEGF, VEGF-
A,
VEGF-2 (CD309) (various cancers). Some other tumor associated antigens
recognized by
antibodies have been reviewed (Gerber, et al, mAbs 1:3, 247-253 (2009);
Novellino et al,
Cancer Immunol Immunother. 54(3), 187-207 (2005). Franke, et al, Cancer
Biother
Radiopharm. 2000, 15, 459-76). Many other antigens are: many other Cluster of
Differentiations (CD4, CD5, CD6, CD7, CD8, CD9, CD10, CD11a, CD11b, CD11c,
CD12w,
CD14, CD15, CD16, CDw17, CD18, CD21, CD23, CD24, CD25, CD26, CD27, CD28,
CD29, CD31, CD32, CD34, CD35, CD36, CD37, CD41, CD42, CD43, CD44, CD45, CD46,
CD47, CD48, CD49b, CD49c, CD53, CD54, CD55, CD58, CD59, CD61, CD62E, CD62L,
CD62P, CD63, CD68, CD69, CD71, CD72, CD79, CD79a, CD79b, CD81, CD82, CD83,
CD86, CD87, CD88, CD89, CD90, CD91, CD95, CD96, CD100, CD103, CD105, CD106,
CD109, CD117, CD120, CD127, CD133, CD134, CD135, CD138, CD141, CD142, CD143,
CD144, CD147, CD151, CD152, CD154, CD156, CD158, CD163, CD166, CD168, CD184,
CDw186, CD195, CD202 (a, b), CD209, CD235a, CD271, CD303, CD304), Apo2,
ASLG659, BMPR1B (bone morphogenetic protein receptor), CRIPTO, Annexin Al,
Nucleolin, Endoglin (CD105), ROB04, Amino-peptidase N, .DELTA.-like-4 (DLL4),
VEGFR-2 (CD309), CXCR4 9CD184), Tic2, B7-H3, WT1, MUC1, LMP2, HPV E6 E7,
-41 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
EGFRvIII, HER-2/neu, Idiotype, MAGE A3, p53 nonmutant, NY-ES0-1, GD2, CEA,
MelanA/MART1; Napi3b (NAPI-3B, NPTIIb, SLC34A2, solute carrier family 34,
member 2,
type II sodium-dependent phosphate transporter 3b), Ras mutant, gp100, p53
mutant,
Proteinase3 (PR1), bcr-abl, Tetratocarcinoma-derived growth factors), EphA
receptors, EphB
receptors, EGFr, EGFRvIII, ETBR (Endothelin), HER2/neu, HER3, HLA-DOB (MHC
class
II molecule Ia antigen), integrins, IRTA2, MPF (MPF, MSLN, SMR, megakaryocyte
potentiating factor, mesothelin), cripto, Sema 5b (FLJ10372, KIAA1445,
Mm42015,
SEMA5B, 5EMAG, semaphoring 5 bHlog, sdema domain, seven thrombospondin
repeats,
cytoplasmic domain), PSCA, STEAP1 (six transmembrane epithelial antigen of
prostate), and
STEAP2 (HGNC 8639, IPCA-1, PCANP1, STAMP1, STEAP2, STMP, prostate)Tyrosinase,
Survivin, hTERL Sarcoma translocation breakpoints, EphA2, PAP, ML-IAP, AFP,
EpCAM,
ERG (TMPRSS2 ETS fusion gene), NA17, PAX3, ALK, Androgen receptor, Cyclin Bl,
Polysialic acid, MYCN, RhoC, TRP-2, GD3, Fucosyl GM1, Mesothelin, PSCA; MAGE
Al,
sLe(a), CYP1B1, PLAC1, GM3, BORIS, Tn, GloboH, ETV6-AML, NY-BR-1, RGS5,
SART3, STn, Carbonic anhydrase IX, PAX5, 0Y-TES1, Sperm protein 17, LCK,
HMWMAA, AKAP-4, SSX2, XAGE 1, B7H3, Legumain, Tie 2, Page4, VEGFR2, MAD-
CT-1, FAP, PDGFR-.beta, MAD-CT-2, Fos-related antigen 1.
Targeting agents that can target cells implicated in autoimmune diseases
include:
anti-elastin antibody; Abys against epithelial cells antibody; Anti-Basement
Membrane
Collagen Type IV Protein antibody; Anti-Nuclear Antibody; Anti ds DNA; Anti ss
DNA,
Anti Cardiolipin Antibody IgM, IgG; anti-celiac antibody; Anti Phospholipid
Antibody IgK,
IgG; Anti SM Antibody; Anti Mitochondrial Antibody; Thyroid Antibody;
Microsomal
Antibody, T-cells antibody; Thyroglobulin Antibody, Anti SCL-70; Anti-Jo; Anti-
U<sub>1RNP</sub>; Anti-La/SSB; Anti SSA; Anti SSB; Anti Perital Cells Antibody; Anti
Histones;
Anti RNP; C-ANCA; P-ANCA; Anti centromere; Anti-Fibrillarin, and Anti GBM
Antibody,
Anti-ganglioside antibody; Anti-Desmogein 3 antibody; Anti-p62 antibody; Anti-
sp100
antibody; Anti-Mitochondrial (M2) antibody; Rheumatoid factor antibody; Anti-
MCV
antibody; Anti-topoisomerase antibody; Anti-neutrophil cytoplasmic (cANCA)
antibody.
Targeting agents for infectious diseases include antibodies that bind
pathogens, such
as Poxyiridae, Herpesviridae, Adenoviridae, Papovaviridae, Enteroviridae,
Picornaviridae,
Parvoviridae, Reoviridae, Retroviridae, influenza viruses, parainfluenza
viruses, mumps,
measles, respiratory syncytial virus, rubella, Arboviridae, Rhabdoviridae,
Arenaviridae, Non-
A/Non-B Hepatitis virus, Rhinoviridae, Coronaviridae (including COVID-19 such
as
targeting-he so-called spike protein), Rotoviridac, Oncovirus [such as, HBV
(FlepatoccIlular
- 42 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
carcinoma), HPV (Cervical cancer, Anal cancer), Kaposi's sarcoma-associated
herpesvirus
(Kaposi's sarcoma), Epstein-Barr virus (Nasopharyngeal carcinoma, Burkitt's
lymphoma,
Primary central nervous system lymphoma), MCPyV (Merkel cell cancer), SV40
(Simian
virus 40), HCV (Hepatocellular carcinoma), HTLV-I (Adult T-cell
leukemia/lymphoma)],
Immune disorders caused virus: [such as Human Immunodeficiency Virus (AIDS)];
Central
nervous system virus: [such as, JCV (Progressive multifocal
leukoencephalopathy), MeV
(Subacute sclerosing panencephalitis), LCV (Lymphocytic choriomeningitis),
Arbovirus
encephalitis, Orthomvxoviridae (probable) (Encephalitis lethargica), RV
(Rabies),
Chandipura virus, Herpesviral meningitis, Ramsay Hunt syndrome type II;
Poliovirus
(Poliomyelitis, Post-polio syndrome), HTLV-I (Tropical spastic paraparesis)];
Cytomegalovirus (Cytomegalovirus retinitis, HSV (Herpetic keratitis));
Cardiovascular virus
[such as CBV (Pericarditis, Myocarditis)]; Respiratory system/acute viral
nasopharyngitis/viral pneumonia: [Epstein-Barr virus (EBV infection/Infectious
mononucleosis), Cytomegalovirus; SARS coronavirus (Severe acute respiratory
syndrome)
Orthomyxoviridae: Influenzavirus A/B/C (Influenza/Avian influenza),
Paramyxovirus:
Human parainfluenza viruses (Parainfluenza), RSV (Human respiratory syncytial
virus),
hMPV]; Digestive system virus [MuV (Mumps), Cytomegalovirus (Cytomegalovirus
esophagitis); Adenovirus (Adenovirus infection); Rotavirus, Norovirus,
Astrovirus,
Coronavirus; HBV (Hepatitis B virus), CBV, HAY (Hepatitis A virus), HCV
(Hepatitis C
virus), HDV (Hepatitis D virus), HEV (Hepatitis E virus), HGV (Hepatitis G
virus)];
Urogenital virus [such as, BK virus, MuV (Mumps)].
Controlling Degradation of USP9X Substrates
Provided herein is also a method of controlling induced degradation of a U
SP9X
substrate in a cell by co-treating the cell with an effective amount of a
proteasome inhibitor
and an effective amount of the compound as disclosed. In some embodiments, the
ratio of the
proteasome inhibitor and the compound may be selected from any values between
0.01 and
100, preferably, between 0.1 and 10. The proteasome inhibitor used herein may
be, as a
nonlimiting example, MG132. In some embodiments, the USP9X substrate is Mcl-1.
In some
embodiments, the induced degradation can be controlled to reach a desirable
level, at which,
for example, the USP9X substrate declines by about 10% or more, about 20% or
more, about
30% or more, about 40% or more, about 50% or more, about 60% or more, about
70% or
more, about 80% or more, or about 90% or more.
- 43 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
The proteasome inhibitor MG132 has been frequently used as a laboratory
research
tool to examine the consequences of blocking the ubiquitin¨proteasome pathway.
For clinical
use, MG132 has been modified (by introducing a boronate group to the peptide
backbone) to
yield the proteasome inhibitor bortezomib which is marketed as Velcade. A
proteasome
inhibitor drug such as Velcade may take antagonistic action against a drug of
the compounds
as described. In some embodiments, provided may also be methods of treating a
subject with
a condition by co-administering the compounds as described and subsequently a
proteasome
inhibitor drug (such as Velcade) to the subject, wherein a USP9X substrate
(preferably Mcl-
1) in a cell of the subject is induced to degrade by the drug of the compounds
as described
and the induced degradation is controlled, slowed down, or terminated by the
proteasome
inhibitor drug.
Combination Therapy with Other Inhibitors
The USP9X substrate Mel-1 may serve as an anti-apoptotic factor conferring
resistance to chemotherapy, e.g., providing protection for other pathologic
proteins against
their inhibitors. These pathologic proteins may be an overexpressed anti-
apoptotic protein
such as, but are not limited to, Bc1-2 and Bc1-xL.
ABT737 is a prototype of new class of anti-cancer drugs known as BH3 mimetics,
such as Navitoclax or Venetoclax. This class of drugs functions by
antagonizing the action of
anti-apoptotic proteins such as Bc12, Bc1-xL, and Mel-1 by blocking their
ability to prevent
the activation of pro-apoptotic proteins such as Bak and Bax. ABT737 is a
small molecule
that binds to Bc12 and Bc1-xL but not Mel-i. The binding site for ABT737 on
Bc12 and Bel-
xL is the same surface groove employed by Bc12 and Bc1-xL to bind the BH3
domains of
pro-apoptotic proteins culminating in the activation of Bak and Bax, the
permeabilization of
the outer membrane of the mitochondrion and the activation of caspases to
dismantle the cell.
Treatment of cancer cells with ABT737 alone, may induce apoptosis but only if
the
intracellular level of Mc1-1 is low enough. This suggests that treatment of
ABT737-resistant
cells with USP9X inhibitors may augment the cell killing potential of ABT737
and similar
compounds.
Provided herein is a method of inhibiting a cancer cell expressing Mel-1 and
at least
one other anti-apoptotic Bc1-2 family protein, by co-treating the cell with an
effective amount
of the compound as disclosed and an effective amount of the Bc1-2 family
inhibitor. The at
least one other anti-apoptotic Bc1-2 family protein may be Bc1-2, Bc1-xL, or
both Bc1-2 and
- 44 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Bc1-xL. The cancer cell may be selected from, but not limited to, acute
myeloid leukemia
(AML) cell and breast cancer cell. In some embodiments, the Bc1-2 family
inhibitor is a Bcl-
2/Bc1-xL inhibitor, such as ABT737, Navitoclax or Venetoclax. Provided herein
is also a
method of sensitizing a cell expressing Mc-1 and at least one other anti-
apoptotic Bc1-2
family protein for a Bc1-2 family inhibitor, by co-treating the cell with an
effective amount of
the compound as disclosed and an effective amount of the Bc1-2 family
inhibitor. The at least
one other anti-apoptotic Bc1-2 family protein may be Bc1-2, Bc1-xL, or both
Bc1-2 and Bc1-
xL. Preferably, the cell is a cancer cell, such as acute myeloid leukemia
(AML) cell or breast
cancer cell. In some embodiments, the Bc1-2 family inhibitor is a Bc1-2/Bc1-xL
inhibitor, such
as ABT737, Navitoclax or Venetoclax.
Provided herein is also a method of enhancing potency of a Bc1-2 family
inhibitor in a
cell expressing Mc-1 and at least one other anti-apoptotic Bc1-2 family
protein, by co-
treating the cell with an effective amount of the compound as disclosed and an
effective
amount of the Bc1-2 family inhibitor. In some embodiments, the potency of the
Bc1-2 family
inhibitor may be enhanced by about 2 to about 100 folds. in some embodiments,
the potency
of the Bc1-2 family inhibitor may be enhanced by about 2, about 5, about 10,
about 20, about
30, about 40, about 50, about 60, about 70, about 80 folds, or about 90 folds.
In some
embodiments, the potency of the Bc1-2 family inhibitor may be enhanced by
about 50 folds.
Preferably, the Bc1-2 family inhibitor is a Bc1-2/Bc1-xL inhibitor, such as
ABT737,
Navitoclax or Venetoclax.
Provided herein is also a method of treating a condition in a subject wherein
the
condition is associated with a pathologic cell that expresses Mc1-1 and at
least one other anti-
apoptotic Bc1-2 family protein, by co-administrating to the subject an
effective amount of a
pharmaceutical composition of the compound as disclosed and an effective
amount of a
pharmaceutical composition of a Bc1-2 family inhibitor. In some embodiments,
the condition
is a cancer, such as acute myeloid leukemia (AML) or breast cancer. In some
embodiments,
the at least one other anti-apoptotic Bc1-2 family protein may be Bc1-2, Bc1-
xL, or both Bc1-2
and Bc1-xL. In some embodiments, the Bc1-2 family inhibitor is a Bc1-2/Bc1-xL
inhibitor,
such as a clinically acceptable derivative of ABT737 (e.g., Venetoclax and
Navitoclax).
The invention will be more fully understood by reference to the following
examples
which detail exemplary embodiments of the invention. They should not, however,
be
construed as limiting the scope of the invention. All citations throughout the
disclosure are
hereby expressly incorporated by reference.
- 45 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
EXAMPLES
Example 1: Synthesis of Compounds
Materials and Methods
Unless otherwise noted, reagents and solvents were used as purchased from
commercial suppliers. Solvent removal was accomplished usually using a rotary
evaporator at
¨15 mm Hg pressure unless otherwise specified. Thin-layer chromatography (TLC)
was
performed using silica-gel 60 plates (F254. Merck) and visualized by UV light
(254 nm).
Column chromatography and flash column chromatography were carried out using
silica gel
(60-203 mesh and 40-60 mesh, respectively) unless otherwise specified.
Synthetic Route for Compound 7A
Preferred compounds of the invention include:
==
0
====
- ..............
so, <,
er.
:NH
07-4
Building block synthesis -
NO2 NHFmoc
NO2
1) H2, Pd/C
00 2) FmocCI,
DI PEA N K2CO3
OTZ0 0
0 OH 0 OH
Step 1: N-methy1-2-(4-nitrophenyl)ethan-1-amine hydrochloride (500 mg, 2.3
mmol) was
dissolved in DCM (23 mL) before DlEA (2.0 mL, 11.5 mmol) and 4,4-
dimethyldihydro-2H-
pyran-2,6(3H)-dione (361 mg, 2.5 mmol) were added. The mixture was stirred at
room
temperature and monitored by LCMS. After 15 mm, the reaction complete. The
reaction was
washed with 1.2N HC1 solution and brine, dried over MgSO4, filtered and
concentrated to
- 46 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
give 3,3-dimethy1-5-(methyl(4-nitrophenethyl)amino)-5-oxopentanoic acid (616
mg, 83 %
yield), which was used as is.
Step 2: 3,3-dimethy1-5-(methyl(4-nitrophenethyl)amino)-5-oxopentanoic acid
(0.616 g, 1.9
mmol) and 10% Pd/C (0.2 g, 0.19 mmol) were stirred in Et0Ac (19 ml) under a
hydrogen
atmosphere (1 atm). After the reaction was complete as determined by LCMS, the
mixture
was filtered and concentrated to give 5-((4-aminophenethyl)(methyl)amino)-3,3-
dimethy1-5-
oxopentanoic acid (0.373 g, 66.8 % yield) as a yellow oil, which was used as
is.
Step 3: 5-((4-aminophenethyl)(methyl)amino)-3,3-dimethy1-5-oxopentanoic acid
(0.373 g,
1.3 mmol) and potassium carbonate (0.212 g, 1.5 mmol) were dissolved in Water
(6 ml). The
mixture was cooled to 0 C before a solution of (9H-fluoren-9-yl)methyl
carbonochloridate
(0.330 g, 1.3 mmol) in acetonitrile (6 ml) was added dropwise. The reaction
was warmed to
room temperature slowly and monitored by LCMS. After 30 min, the reaction was
acidified
with 1M HC1 and extracted with ether. The combined ether extracts were dried
over MgSO4,
filtered and concentrated. Purification by silica gel chromatography gave 5-
((4-((((9H-
fluoren-9-yfimethoxy)carbonyl)amino)phenethyl)(rnethyfiamino)-3,3-dimethyl-5-
oxopentanoic acid (0.174 g, 26.5 % yield) as a white solid.
OH F OH F
OH F
Fmoc0Su
o H2, Pd/C
-71 0 io K2c.3 0
Fmoc,N
CN H2N
Step 1: 5-Cyano-2-fluorobenzoic acid (3.0 g, 18.3 mmol) and 10% palladium on
carbon (0.3
g, 2.7 11111101) were shaken in methanol (200mL) under a hydrogen atmosphere
(20 PSI).
After 3h, the reaction was complete by LCMS. The mixture is filtered and
concentrated to
give 2.1 g of the desired product (68%), which was used as is.
Step 2: 5-(aminomethyl)-2-fluorobenzoic acid (2 g, 11.8 mmol) and (9H-fluoren-
9-yOmethyl
(2,5-dioxopyrrolidin-1-y1) carbonate (3.99 g, 11.8 mmol) were dissolved in THF
(250mL). A
saturated K2CO3 solution was added until the pH was non-acidic. The mixture
was stirred at
room temperature and monitored by LCMS. After 2h, the mixture was concentrated
and the
residue was washed with water and DCM (2x) and dried overnight to give the
desired product
in >75% yield.
1) DOH
2) TFA
OMe OMe 3) Fmoc0Su, OH
DIAD, PPhi Na2003
41101 0
Boo...N., 01:- Boo¨NZ o
Fmoc,N, 0 11110 0
HO
- 47 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Step 1. Methyl 4-hydroxybenzoate (0.250 g, 1.6 mmol) was dissolved in THF (16
ml) before
tert-butyl (2-hydroxyethyl)(methyl)carbamate (0.333 ml, 2.0 mmol) and Ply313
(0.560 g, 2.1
mmol) were added. The reaction was cooled to 0 C and DIAD (0.383 ml, 2.0 mmol)
was
added dropwise and the reaction was monitored by LCMS. After 40 mm, the
reaction was
concentrated the crude residue was purified by silica gel chromatography to
give 0.440 g
(87%) of desired product.
Step 2: 4- Methyl 4-(2-((tert-butoxycarbonyl)(methyl)amino)ethoxy)benzoate
(0.440 g, 1.4
mmol) and 1M aqueous LiOH (7.11 ml, 14.2 mmol) were stirred in THF (14 ml) at
60 C and
the reaction was monitored by LCMS. After stirring overnight, the reaction was
complete.
The mixture was acidified (pH 2-3) with 1M aqueous HCl and extracted with DCM
(3x). The
combined organic extracts were dried over Na2SO4, filtered and concentrated to
give crude 4-
(2-((tert-butoxycarbonyl)(methypamino)ethoxy)benzoic acid, which was
subsequently
dissolved in DCM (14 ml) before TFA (2.191 ml, 28.4 mmol) was added. The
reaction was
stirred at room temperature and monitored by LCMS. After 45 minutes, complete
conversion
was observed. The mixture was concentrated and then azeotroped with toluene to
remove
excess TFA. The product, 4-(2-(methylamino)ethoxy)benzoic acid, was dissolved
in
acetonitrile (11 ml) and water (3.5 ml) before sodium carbonate (0.603 g, 5.7
mmol) was
added. Fmoc-OS u (0.504 g, 1.5 mmol) was then added and the reaction stirred
at room
temperature. After the reaction was complete (1h), the mixture was acidified
(pH 2-3) with
1M aqueous HC1 and extracted with DCM (x3). The combined organic extracts were
dried
over Na2SO4, filtered and concentrated. The crude product was purified by
silica gel
chromatography to give 515 mg (87% over 3 steps) of the desired product.
H
40 NHFmoc op NiNH F H
r
F
solid-phase 0 40
HATU, 1-10A1 Nyrq'NH
0
synthesis N DIPEA, DMF 0 lip
_______________________________________________________ Yob- ===-.N
NH
=HN
0 OH 0H 0 0
0 N 0
NH
Typical synthesis of analogs ¨
Loading: 2-Chloro-trityl chloride resin (1.2 equiv) was swelled in DCM for 10
mm and then
filtered and washed with DCM. The appropriate Fmoc protected amino acid (1.0
equiv) and
- 48 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
N-ethyl-N-isopropylpropati-2-amine (6.0 equiv) were dissolved in DCM. The
resulting
solution was added to the swelled resin and agitated for 2 hours. The resin
was then washed
with 85:10:5 DCM:MeOH:DIPEA (10 mL x 3); DCM (10 mL x 3), DMF (10 mL x 3), DCM
(20 mL x 3). After flushing with argon and dried under vacuum, compound linked
resin was
obtained.
Solid-phase Fmoc-deprotection-Resin (1.0 equiv) was suspended in DMF (4 mL x 5
min) and
mixed with a stream of N2 every 30 seconds. The Fmoc group was removed from
the resin-
supported building block by mixing the resin twice with a solution of 2% DBU,
2%
piperidine in DMF (4 mL x 5 min) while agitating with a stream of N2 every 30
seconds.
The resin was washed six times with DMF (4 mL x 30 sec) and used in the next
step as is.
Solid-phase amide formation ¨ The appropriate carboxylic acid (0.1 M solution
in DMF, 2
equiv), followed by HATU (0.2M solution in DMF, 2.1 equiv) and N-methyl
morpholine (1.0
M in DMF, 6.7 equiv) were added to the resin (1.0 equiv). The reaction mixture
was agitated
by a stream of nitrogen for 2 hours. The reagents were drained from the
reaction vessel, and
the resin was washed with six times DMF (4 mL x 30 sec) and used in the next
step as is.
Cleavage: Resin (1.0 equiv) was treated with 5% TFA in CH2C12 (8 mL x 5 min)
then washed
with DCM (8 mL). This was repeated two more times. Solvent was removed by
evaporation
using a Genevac EZ2.2 evaporator. The crude reaction mixture was carried on to
the next
reaction.
Cyclization: Dissolved the linear amino acid (1.0 equiv) in DMF (0.05 M)
before N-ethyl-N-
isopropylpropan-2-amine (5.0 equiv) was added. The resulting solution was
added to a pre-
mixed solution of HATU (1.2 equiv) and HOAt (1.2 equiv) in DMF (0.011 M) at ¨2
mL/min.
The reaction was monitored by LCMS and additional HATU was added until
complete
conversion to the desired product was observed. The mixture was concentrated
and purified
by either preparative HPLC (small-scale) or reverse-phase silica gel
chromatography to give
the desired product.
Synthetic Route for Compound lA
Synthesis of intermediate A
Preparation of 4,4-Dimethylheptanedioic acid
- 49 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
0 0
N N KOH, water HO OH
80 C, 20 h
1 2
4,4-Dimethylheptanedinitrile (1, 500 mg, 3.3 mmol, 1.0 equiv) was suspended in
water (10
mL) and KOH (930 mg, 16.6 mmol, 5.0 equiv) was added. The reaction mixture was
stirred
at 80 C for 20 h. The progress of the reaction was monitored by TLC
(CHC13:Me01-l=20: 1 +
anisaldehyde) and LCMS analyses. After completion, the mixture was diluted
with water and
its pH was adjusted to 3-4 by the addition of 2 M aq. HC1 solution, then
extracted with
Et0Ac twice. The organic phase was dried over MgSO4, filtered and concentrated
in vacuo to
obtain 4,4-Dimethylheptanedioic acid.
Preparation of 4,4-Dimethy1-7-(methyl(4-nitrophenethyl)amino)-7-oxoheptanoic
acid
0 \ HCI = NO2
HN
3 OH
0 OH C)
HATU, DIPEA
OH DMF, rt, 20 h
2 4
4,4-Dimethylheptanedioic acid (1.1 g, 5.8 mmol, 1.0 equiv) was dissolved in
anhydrous DMF
(23 mL) then DIPEA (1.9 g, 14.5 mmol, 2.5 equiv) and HATU (1.1 g, 2.9 mmol,
0.5 equiv)
were added. The resulting mixture was stirred at rt for 30 min then reagent
(3, 630 mg, 2.9
mmol, 0.5 equiv) was added and stirring was continued at rt for 4 h. As no
full conversion
was detected by TLC and LCMS analyses, another portion of HATU (660 mg, 1.7
mmol, 0.3
equiv) and - after 10 mm stirring - reagent (3, 380 mg, 1.7 mmol, 0.3 equiv)
were added. The
reaction mixture was stirred at rt for 20 h. The progress of the reaction was
monitored by
TLC (CHC13:Me0H=5:1 + bromocresol green) and LCMS analyses. After completion,
the
reaction mixture was concentrated in vacuo, the residue was dissolved in Et0Ac
and washed
with 10 w/vv% aq. Na2CO3 solution. The pH of the aqueous phase was adjusted to
3-4 by the
addition of 1 M aq. HC1 solution and extracted with Et0Ac twice. The combined
organic
phase was dried over MgSO4, filtered and concentrated in vacuo. The crude
product was
purified by column chromatography (eluent: 0 to 10 % Me0H in CHC13) to obtain
Compound 4.
- 50 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Preparation of Ethyl 4,4-dimethy1-7-(methyl(4-nitrophenethyl)amino)-7-
oxoheptanoate
\N = NO2 \N 11 NO2
OH ______________________________________ 0
SOCl2, Et0H
rt, 20 h 0
4 5
Starting acid compound 4 (1.17 g, 3.34 mmol, 1.0 equiv) was dissolved in Et0H
(10 mL)
then S0C12 (1.26 mL, 16.69 mmol, 5.0 equiv) was added. The resultant mixture
was stirred at
rt for 2 h. The progress of the reaction was monitored by TLC (DCM:Me0H=20:1)
and
LCMS analyses. After completion, the solvent was evaporated and the residue
was taken up
in Et0Ac and washed with sat. aq. NaHCO3 solution. The organic phase was dried
over
MgSO4, filtered and concentrated under reduced pressure to obtain the crude
product 5,
which was used as is.
Preparation of Ethyl 7-((4-aminophenethyl)(methypamino)-4,4-dimethyl-7-
oxohcptanoatc
ID NO2 = NH2
0 _______________________________________ _ 0
Pd/C, H2
0 Me0H, rt, 3 h 0
5 6
Starting compound 5 (829 mg, 2.19 mmol, 1.0 equiv) was dissolved in ElOH (22
mL) then
Pd/C (10% Pd on C, 82.9 mg, 0.078 mmol, 0.036 equiv) was added. The vessel was
closed,
purged with argon then pressurized with 3 bar H2 and stirred at rt for 5 h.
The progress of the
reaction was monitored by TLC (CHC13:Me0H=20:1) and LCMS analyses. After
completion, the mixture was filtered through a short pad of celite and the
filtrate was
evaporated. The crude product was purified by flash chromatography (eluent: 0
to 70%
Et0Ac in n-heptane) to obtain compound 6, which was used as is.
-51 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Preparation of Ethyl (S)-7-((4-(2-((tert-butoxycarbonyl)amino)-4,4-
dimethylpentanamido)phenethyl)(methyDamino)-4,4-dimethy1-7-oxoheptanoate
(poi
0 _____________________________________________________________________
_______________________________________________________________________ (
1, NH2 )\-NI-1.."
4. NH NH
0¨/ 7
0
/¨µ
HODt, EDCI, DIPEA CD
DCM, rt, 20 h 0
0
6 8
Acid reagent (7, 314 mg, 1.3 mmol, 1.1 equiv) was dissolved in anhydrous DCM
(3.5 mL)
then D1PEA (301 mg, 2.33 mmol, 2.0 equiv), HOBt (190 mg, 1.4 mmol, 1.2 equiv)
and EDC1
(268 mg, 1.4 mmol, 1.2 equiv) were added. The resulting mixture was stirred at
rt for 30 min
then starting compound 6 (406 mg, 1.16 mmol, 1.0 equiv) was added and stirring
was
continued at rt for 20 h. As no full conversion was detected by TLC and LCMS
analyses,
another portion of D1PEA (60 mg. 0.46 mmol, (14 equiv), H013t (63 mg. 0.46
mmol, 0.4
equiv) and EDCI (89 mg, 0.46 mmol, 0.4 equiv) were added and stirring was
continued for
h. The progress of the reaction was monitored by TLC (CHC13:Me0H=20:1) and
LCMS
analyses. After completion, the reaction mixture was diluted with DCM and
washed with
water and brine. The organic phase was dried over MgSO4, filtered and
concentrated in
vacuo. The crude product was purified by column chromatography (eluent: 0 to
80% Et0Ac
15 in n-heptane) to obtain Compound 8.
Preparation of (S)-7-04-(2-((Tert-butoxycarbonyl)amino)-4,4-
dimethylpentanamido)phenethyl)(methypamino)-4,4-dimethyl-7-oxoheptanoic acid
(Intermediate A)
R\ ______________________________ R\ ____
- 7 - _____
N_/0¨¨NH NH N/(>
NH
CD 0--õ,õ. 0 TBAH, THE 0µ OH
to it, 2 h
0
20 8 Intermediate A
Starting compound 8 (518 mg, 0.9 mmol) was dissolved in THF (8.9 mL) and
cooled to 0 C
then TBAH (2042 mg, 40 w/w% in water, 3.15 mmol, 3.5 equiv) was added
dropwise. The
resulting mixture was let to warm up to rt and stirred for 2 h. The progress
of the reaction was
- 52 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
monitored by TLC (CHC13.Me0H-20.1) and LCMS analyses. After completion, the
volatile
solvent was evaporated in vacuo, the residue was diluted with water and its pH
was adjusted
to 3-4 by the addition of 10 w/w% aq. citric acid solution and extracted with
Et0Ac twice.
The organic phase was dried over MgSO4, filtered and concentrated under
reduced pressure
to obtain Intermediate A, which was used as is.
Synthesis of Intermediate B
Preparation of Methyl 4-(2-((tert-butoxycarbonyl)(methyl)amino)ethoxy)benzoate
HO 0
io
Boc Co''.
TPP, DIAD
0 THF, 0 C to it, 20 h
9 11
Starting compound 9 (2.0 g, 13.1 mmol, 1.0 equiv) was dissolved in anhydrous
THF (52 mL)
then hydroxy reagent (10, 3.45 g, 19.7 mmol, 1.5 equiv) and TPP (5.17 g, 19.7
mmol, 1.5
equiv) were added under Ar atmosphere. The resulting mixture was cooled to 0
C then a
solution of DIAD (3.98 g, 19.7 mmol, 1.5 equiv) in anhydrous THF (19 mL) was
added
dropwise. The reaction mixture was allowed to warm up to rt and stirred at
this temperature
for 20 h. The progress of the reaction was monitored by TLC (n-
heptane:Et0Ac=4:1) and
LCMS analyses. After completion, the reaction mixture was diluted with Et0Ac
and washed
with water and brine. The organic phase was dried over MgSO4, filtered and
evaporated. The
crude product was purified by column chromatography (eluent: 0 to 40 % Et0Ac
in n-
heptane) to obtain the compound 11.
Preparation of 4-(2-((Tert-butoxycarbonyl)(methyl)amino)ethoxy)benzoic acid
0 0
Boc 0 ___________ Boc Na0H, THF
50 C, 20 h OH
11 12
Starting compound 11 (3.25 g, 10.5 mmol, 1.0 equiv) was dissolved in THF (32.5
mL) then 1
M aq. NaOH solution (1.26 g, 31.5 mmol, 3 equiv) was added. The resulting
mixture was
stirred at 50 C for 20 h. The progress of the reaction was monitored by TLC
(n-
heptane:E10 Ac=2: 1) and LCMS analyses. After completion, the volatile solvent
was
- 53 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
removed in vacuo, the residue was diluted with water and extracted with Et0Ac.
The pH of
the aqueous phase was adjusted to 3-4 by addition the addition of 1 M aq. HC1
solution, then
extracted with Et0Ac. The organic phase was dried over MgSO4, filtered and
evaporated
under reduced pressure to obtain the compound 12, which was used as is.
Preparation of Methyl 5-(aminomethyl)-2-fluorobenzoate
0 0
0 0
N Pd/C, Me0H
rt, 3 h H2N
13 14
Starting compound 13 (3.0 g, 16.7 mmol) was dissolved in Me0H (168 mL), then
Pd/C (10%
Pd on C, 300 mg, 2.82 mmol, 0.17 equiv) was added. The vessel was closed and
stirred under
atmospheric pressure H2 (balloon) at rt for 3 h. The progress of the reaction
was monitored by
TLC (CHC13:Me0H=20:1) and LCMS analyses. After completion, the mixture was
filtered
through a short pad of celite and the filtrate was evaporated to obtain the
compound 14.
Preparation of Methyl 5-((4-(2-((tert-
butoxycarbonyl)(methyDamino)ethoxy)benzamido)methyl)-2-fluorobenzo ate
0
0 H2N 0 0
14
Boc Boc
SHi
HATU, DI A
DCM, rt, 20PE h
12 OH ___________________________ 15
Starting compound 12 (2.7 g, 9.4 mmol, 1.0 equiv) was dissolved in anhydrous
DCM (23
mL), then DIPEA (3.0 g, 23.5 mmol, 2.5 equiv) and HATU (3.9 g, 10.3 mmol, 1.1
equiv)
were added. After 10 min stirring, reagent (14, 2.7 g, 70 w/w%, 10.3 mmol, 1.1
equiv) was
added. The resulting mixture was stirred at rt for 20 h. The progress of the
reaction was
monitored by TLC (n-heptane:Et0Ac=2:1) and LCMS analyses. After completion,
the
mixture was diluted with DCM and washed with water and brine. The organic
phase was
dried over MgSO4, filtered and evaporated under reduced pressure. The crude
product was
- 54 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
purified by column chromatography (eluent: 0 to 60 % Et0Ac in n-heptane) to
obtain the
compound 15.
Preparation of Methyl 2-fluoro-5-((4-(2-
(methylamino)ethoxy)benzamido)methyl)benzoate (Intermediate B)
0 0 0 0
Boc
[1101 40
0 _______________________________________________________________ 1 11
HCl/1,4-dioxane
rt, 1 h
15 Intermediate B
Starting compound 15, (2.28 g, 4.9 mmol, 1.0 equiv) was dissolved in 1,4-
dioxane saturated
with HC1 gas (49 mL). The resulting mixture was stirred at rt for 1 h. The
progress of the
reaction was monitored by TLC (CHC13:Me0H=20:1) and LCMS analyses. After
completion, the reaction mixture was concentrated in vactto, the residue was
dissolved in
water and extracted with Et0Ac. The pH of the aqueous phase was adjusted to 9-
10 by
addition of 10 w/w% aq. Na2CO3 solution, then extracted with Et0Ac. The
combined organic
phase was dried over MgSO4, filtered and evaporated under reduced pressure.
The crude
product was purified by column chromatography (eluent: 0 to 5 % Me0H in CHC13)
to obtain
Intermediate B.
Synthesis of Compound 1A
Preparation of Ethyl (S)-5-44-(2-(7-04-(2-((tert-butoxycarbonyl)amino)-4,4-
dimethylpentanamido)phenethyl)(methyDamino)-N,4,4-trimethy1-7-
oxoheptanamido)ethoxy)benzamido)methyl)-2-fluorobenzoate
- 55 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
o (
¨
NH
'3
srs1¨
0, (
* NH NH ON 00
0
10 __
0
/., OH _µ0 0 "
o
HOBt, EDCI, DIPEA
110 DUM, rt, 20 h
0
Intermediate A Intermediate B
HN
0/
0
16
Intermediate A (500 mg, 0.82 mmol, 1.0 equiv) was dissolved in anhydrous DCM
(3.3 mL)
then DIPEA (265 mg, 2.05 mmol, 2.5 equiv), HOBt (133 mg, 0.99 mmol, 1.2 equiv)
and
EDCI (189 mg, 0.99 mmol, 1.2 equiv) were added. The resulting mixture was
stirred at rt for
30 min then amine reagent (Intermediate B, 355 mg, 0.99 mmol, 1.2 equiv) was
added and
stirring was continued at rt for 20 h. The progress of the reaction was
monitored by TLC
(CHC13:Me0H=10:1) and LCMS analyses. After completion, the reaction mixture
was
diluted with DCM and washed with water and brine. The organic phase was dried
over
MgSO4, filtered and concentrated in vacuo. The crude product was purified by
column
chromatography (eluent: 0 to 6% Me0H in chloroform) to obtain the compound 16
(460 mg.
63%).
Preparation of Ethyl (S)-544-(2-(7-((4-(2-amino-4,4-
dimethylpentanamido)phenethyl)(methyDamino)-N,4,4-trimethyl-7-
oxoheptanamido)ethoxy)benzamido)methyl)-2-fluorobenzoate
- 56 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Czx __________________________________________________ R\ __
¨ ________________________________________________________
_/-0¨NH NH _/-0¨NH NH2
0 0 ______________________________________________ _e0
) ) \N¨
S
0 0
HCl/1,4-dioxane
C, 2 h
) HN HN
0 0
0 0
16 17
Starting compound 16(460 mg, 0.52 mmol, 1.0 equiv) was dissolved in 1,4-
dioxane saturated
with HC1 gas (2 mL). The resulting mixture was stirred at 10 C for 2 h. The
progress of the
reaction was monitored by TLC (CHC13:Me0H=10:1) and LCMS analyses. After
5 completion, the reaction mixture was evaporated in vacuo, the
residue was dissolved in water
and extracted with DCM. The pH of the aqueous phase was adjusted to 10-11 by
addition the
addition of 10% aq. Na2CO3 solution, then extracted with DCM. The organic
phase was dried
over MgSO4, filtered and evaporated under reduced pressure to give the
compound 17 (408
mg, quant. yield) %), which was used as is.
- 57 -
CA 03237744 2024- 5- 8
WO 2023/091464
PCT/US2022/050069
Preparation of (S)-5-04-(2-(7-04-(2-amino-4,4-
dimethylpentanamido)phenethyl)(methyDamino)-N,4,4-trimethyl-7-
oxoheptanamido)ethoxy)benzamido)methyl)-2-fluorobenzoic acid (18)
o . __
o¨
) * NH NH2 NH:(
)
0
0
110 TBAH, THF
0 C to rt, 2 h
0
0 HN
\) HN
HO 400
110 0
0
17 18
Starting compound 17 (408 mg, 0.52 mmol, 1.0 equiv) was dissolved in THF (5.1
mL) and
cooled to 0 C. TBAH (838 mg, 40 w/w% in water, 1.29 mmol, 2.5 equiv) was added
dropwise. The resulting mixture was let to warm up to rt and stirred for 2 h.
The progress of
the reaction was monitored by TLC (CHC13:Me0H=10:1) and LCMS analyses. After
completion, the volatile solvent was evaporated in vacuo, the residue was
diluted with water
and its pH was adjusted to 3-4 by the addition of 10% aq. citric acid
solution. The precipitate
was collected by filtration and washed with DCM to obtain the compound 18,
which was
used as is.
- 58 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Preparation of (S)-54-fluoro-14,18,18,22-tetramethy1-8-neopentyl-25-oxa-
3,7,10,14,22-
pentaaza-1,11(1,4),5(1,3)-tribenzenacyclopentacosaphane-2,6,9,15,21-pentaone
(Compound 1A)
o (
>
NH2
OK0
N-
0
0
HN
0
N\
HOAt, EDCI DIPEA 53
DCM, rt, 20 h
0 0
HN NH
0 \I
HO 0
0
18 Compound 1A
DIPEA (100 mg, 0.77 mmol, 6 equiv) was combined with HOAt (52.6 mg, 0.39 mmol,
3.0
equiv) and EDCI (74.1 mg, 0.39 mmol, 3 equiv) in anhydrous DCM (20 mL), then a
solution
of starting amino acid 18(100 mg, 0.13 mmol, 1.0 equiv) dissolved in a mixture
of anhydrous
DCM (20 mL) and DIPEA (50 mg, 0.39 mmol, 3.0 equiv) was added at rt, via a
syringe
pump over 3 h. The resulting mixture was stirred at it overnight. The progress
of the reaction
was monitored by TLC (CHC13:Me0H=10:1) and LCMS analyses. After completion,
the
reaction mixture was washed with water and brine. The organic phase was dried
over MgSO4,
filtered and concentrated in vacua The crude product was purified by column
chromatography (eluent: 0 to 6% Me0H in chloroform) then by preparative HPLC
to obtain
the Compound 1A. ES+ MS m/z 758 [M+Hr.
Example 2: Assessing Compounds for Activities Against DUBs
Inhibiting the catalytic activity of USP9X
The inhibition property of compound Compound lA was evaluated in assays
measuring the ability of USP9X to catalyze the hydrolysis of the amide bond
between the C
- 59 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
terminal carboxylate of ubiquitin and diodamine110 of the fluorogenic
substrate ubiquitin-
rhodamine 110. To the wells of a 96 well reaction plate was added 12.5
USP9X solution
(1.3 nM) in assay buffer (100 mM NaC1, 1 mM DTT, 0.1% bovine serum albumin,
0.05%
Tween 20, 50 mM Tris, pH 7.5). 12.5 !AL of Compound 1A in assay buffer (4><
final
concentration) was added to the wells containing the USP9X solution, and the
solution was
gently mixed by pipetting up and down several times. The plate was sealed and
incubated at
room temperature for 20 minutes. Then 25 uL of the 1 tiM substrate (ubiquitin-
rhodamine110) solution was added in assay buffer. The final substrate
concentration was 0.5
iuM. The fluorescence from each of the wells was determined using 485 nm
excitation and
535 nm emission for a series of time points. The slope for the rate of change
of the
fluorescence was determined and plotted vs the concentration of Compound 1A.
The
fluorescence signal decreased and plateaued when increasing the concentration
of Compound
1A. The results indicated that Compound 1A partially inhibited the activity of
USP9X.
Binding to USP9X and Inhibiting USP9X Enzymatic Activities
Direct binding of the compounds (Compound 2A, Compound 3A, and Compound 4A)
to USP9X was evaluated using a Biacore '1200 instrument at 25 C according to
the
manufacturer's instructions. USP9X was immobilized on a chip for surface
plasmon
resonance measurements via Biacore T200. Solutions of compound Compound 2A at
various
concentrations were flowed over the immobilized USP9X and the change in
refractive index
was monitored. The displayed traces indicated the binding of Compound 2A to
USP9X and
its subsequent dissociation. A value (0.48 iuM) for the equilibrium
dissociation constant, KD,
were determined from the ratio of the dissociation rate constant to the
association rate
constant The values of the maximum response for each concentration of Compound
2A were
plotted and were consistent with a binding of the compound to a single site on
USP9X with a
dissociation equilibrium constant of 1.9 tiM in reasonable agreement with
value determined
from the kinetic association and dissociation rates. Compound 2A did not bind
to carbonic
anhydrase immobilized upon a chip, indicating that non-specific binding was
negligible.
USP9X enzyme assays were set up in a 96 well plate to assess the abilities of
Compound 3A and Compound 4A to inhibit the activity of USP9X, respectively.
Ubiquitin-
rhodamine110 was used as the substrate and ubiquitin aldehyde was included in
the assay as
a positive control. Ubiquitin aldehyde binds covalently to the thiol group of
the active site
Cys of USP9X. The activity of USP9X was completely abolished by incubation
with
ubiquitin aldehyde. The activity of USP9X was reduced approximately 60% with
the higher
- 60 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
concentrations of Compound 2A, Compound 3A, and Compound 4A (> 10 uM). The
partial
inhibition of USP9X was not due to the lack of solubility of these compounds
at high
concentrations but instead was shown to have a mechanistic basis. The ICso for
the inhibition
of USP9X activity by Compound 2A was calculated from a four parameter,
variable slope fit
of the data in GraphPad Prism to be 0.39 04. This value was similar to the
values of the
dissociation equilibrium constant determined for this compound from the
surface plasmon
resonance spectrometry. The ICso values of Compound 2A, Compound 5A, Compound
7A,
and Compound 11A against USP9X were determined to be 0.39 uM, 0.03 p..M, 0.011
uM,
and 0.048 p,M, respectively.
Mechanism of Inhibition of USP9X by the Compounds
In order to determine whether there was a mechanistic basis for the observed
partial
inhibition of USP9X activity by Compound 2A, steady-state enzyme inhibition
kinetic assays
were carried out in which the activity of USP9X was determined as a function
of the substrate
ubiquitin-rhodamine110 concentration at various concentrations of the
inhibitor Compound
2A. The data were fit to a general model for enzyme inhibition (Figure 1E) so
the values of
the parameters determined from the assays specified the mode of inhibition.
For example, in
the case of a = 1 and fi = 0, it corresponded to purely non-competitive
inhibition. For a =
infinity and f3 = 1, it corresponded to purely competitive inhibition. The
values of all the
parameters, Ks, KT, kcal, a, and f3 could be determined from the steady state
measurements.
The enzyme reactions were set up as described in the Inhibiting the catalytic
activity of
USP9X section. The concentration of the substrate ubqiutin-rhodamine110 was
varied. The
initial rates of the reactions were determined at several concentrations of
the inhibitor
Compound 2A. In order to determine the values of the parameters Ks and kcat,
the reaction
rates in the absence of the inhibitor Compound 2A, the initial reaction rate
data were fit to the
standard Michaelis-Menten equation v = Vmax [S]/ (Km + [S]). The values of a
and 13 could
then be determined by fitting the initial rate data of the enzyme reaction at
various
concentrations of Compound 2A to a modified version of the Michaelis-Menten
equation,
designating both Vmax and Ks apparent to distinguish the apparent values of
the parameters
determined from these plots to the true values for these parameters determined
from the
steady state kinetics in the absence of Compound 2A. The values of a and f3
could be
determined from the variation of the slopes and intercepts of the series of
double reciprocal
plots of 1/v versus 1/[S] for each concentration of Compound 2A. The plots of
1/A slope vs.
1/[ Compound 2A1 or 1/A intercept vs. 1/Compound 2A were linear and from these
plots
-61 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
values of it, 13, and Kt could be determined. The results of this analysis
were shown in Table
2. The two values determined for Ki were from the two different plots. The
value of a, 1.73,
was >1 and indicated that the affinity of the USP9X- Compound 2A complex for
the
substrate was weaker than that for USP9X itself, suggesting a competitive mode
of inhibition.
The value of f3, 0.71, was <1 and indicated that the rate of the conversion of
substrate to
product was slower for the USP9X- Compound 2A complex with the substrate than
for the
USP9X-substrate complex itself, suggesting a non-competitive inhibition.
Together these
values of the inhibition parameters served to classify the mode of inhibition
as hyperbolic
mixed inhibition (See Segel I. H. Enzyme kinetics: behavior and analysis of
rapid equilibrium
and steady-state enzyme systems. New York: Wiley-Interscience).
Inhibition Kinetic Parameters Predict Partial Inhibition of the Compounds
against
USP9X
To determine whether the values of the parameters determined from enzyme
inhibition kinetics in Table 2 predicts the observed partial inhibition of
USP9X by
compounds in the previous figures, a theoretical dose-response curve for the
activity of
USP9X vs. [Compound 2A1 was constructed using the Michaelis-Menten equation
for the
reaction. As shown in Figure 1F, the theoretical dose-response curve (solid
line) was
compared with the actual dose-response curve (filled circles) obtained for
Compound 2A as
described in the Binding to USP9X and Inhibiting USP9X Enzymatic Activities
section. The
results were in support of a mechanistic basis for the observed partial
inhibition of USP9X by
Compound 2A.
Compounds Have a Common Mechanism of Inhibition of USP9X
In addition to Compound 2A as described above, Compound 5A, Compound 7A, and
Compound 11A were evaluated in order to determine whether they exhibited a
common
mechanism of inhibition of USP9X. These results of inhibition kinetic
parameters were
summarized in Table 2.
- 62 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
Table 2. Inhibition Kinetic Parameters of Compound 2A, Compound 5A, Compound
7A, and Compound 11A
Compound Compound Compound Compound
Compound
2A 5A 7A HA
USP9X
390 30 11 48
ICso nM
Ki,nM 1201, 2902 6.71, 242 9.41, 10.32
7.01, 692
1.73 2.3 1.85 2
0.71 0.78 0.72 0.88
Ks, PM 0.95 1.2 1.87 0.91
VMAX
310 335 589 408
RFU/min
The two values for Ki were determined by two different methods for determining
this
parameter from the kinetic data: 1 represented a value determined from the
slope of the 1/A
intercept vs 141] plot; 2 represented a value determined from they intercept
of the 1/A slope
vs 1/[I] plot. These four compounds inhibited USP9X catalytic activity with
the same
mechanism ¨ hyperbolic mixed inhibition. Only the values of the parameters
varied for the
compounds. The potency of the inhibition of USP9X improved significantly for
this series of
compounds beginning with several hundred nM for Compound 2A and increasing by
nearly
two orders of magnitude for Compound 7A, whether comparing the 1050s from dose-
response curves or the values for Ki determined from steady-state enzyme
inhibition kinetics.
Binding Specificity
Compounds were screened for DUB inhibitory and apoptotic activity against
other
deubiquitylases in a DUB selectivity panel using ubiquitin-AMC (BPS Bioscience
Inc.). The
same assay for evaluating the inhibition property of compound Compound lA
described
above was carried out but only USP9X was replaced with other deubiquitylases.
The
compounds showed minimal or no detectable effect on DUBs tested. Specifically,
5 iuM
Compound 5A screened using 500 nM Ubiquitin-AMC exhibited no detectable effect
on
CYLD, USP2, USP5, USP7, USP13, USP25, USP4, and UCH-L3 (i.e., 0% inhibition);
and
5% inhibition for USP30, 1% inhibition for USP10, 2% inhibition for USP15, 3%
inhibition
for VCPIP, and 5% inhibition for UCH-Li. An attempt was made to determine an
IC50 value
- 63 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
for inhibition of Compound 5A against several DUBS and IC5o was determined to
be above
50 luM for USP7, above 50 pM for USP2, and 15 trM for USP28.
The results from the assessments for activities against DUBs indicated that
the
compounds partially inhibited USP9X and were selective for USP9X. In contrast,
another
partially selective USP9X inhibitor, WP1130, inhibited not only USP9x but also
USP5;
USP14, UCH-L1, and UCH37.
Screening for various cell lines
Compounds can be screened for DUB inhibitory and apoptotic activity in a panel
of CML,
myeloma and Mantle cell lymphoma cell lines. Selected compounds can also be
tested for
DUB inhibition in intact cells and in isolated DUB (USP9X-UCH domain) enzyme
preparations. General descriptions of the methods employed in these assays can
be found,
e.g., in Kapuria; et al., A novel small molecule deubiquitinase inhibitor
blocks Jak2 signaling
through Jak2 ubiquitination, Cell Signal, 2011, 23(12):2076-85; Kapuria, et
al.,
Deubiquitinase inhibition by small-molecule WP 1130 triggers aggresome
formation and
tumor cell apoptosis. Cancer Res, 2010. 70(22): p. 9265-76; Sun, et al., Bcr-
Abl
ubiquitination and Usp9x inhibition block kinase signaling and promote CML
cell apoptosis.
Blood, 2011. 117(11): p. 3151-62; Kapuria, et al., Protein cross-linking as a
novel mechanism
of action of a ubiquitin-activating enzyme inhibitor with anti-tumor activity.
Biochem
Pharmacol, 2011. 82(4): p. 341-9; and Bartholomeusz; et al., Activation of a
novel Bcr/Abl
destruction pathway by WP1130 induces apoptosis of chronic myelogenous
leukemia cells.
Blood, 2007. 109(8): p. 3470-8. Purified recombinant USPx9 (R&D Systems) in
50mM Ins
buffer pH 7.5, 1 mM DTT, 100 mM NaC1, 0.05% Tween 20 and 0/1 % BSA is used at
a
concentration of 0.325 nM with 0.5 nM ubiquitin-rhodamine (R&D Systems).
Chemical structures can be screened for inhibitory activity using this assay.
Fluorescent scans are used to assess inhibitory activity in this enzyme assay.
Example 3: Assessing Compounds for Inhibiting Cancer Cells
Inhibitory Activities against the Proliferation of Mia-Paca-2 and Other Cancer
Cells
Compound Compound 9A was tested for the ability to inhibit the proliferation
of Mia-
Paca-2 pancreatic cancer cells. Mia-Paca-2 and MDA-MB-231 cells were seeded
into the
wells of a 96 well tissue culture plate in complete medium (10% fetal calf
serum) and
incubated overnight at 37 C/5% CO2). The medium was exchanged for fresh
medium
containing varying concentrations of Compound 9A and the plate was incubated
for 72 hours
- 64 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
at 37 C/5% CO2. The viability of cells at the end of this incubation was
assessed with the
WST-1 reagent. To 10 ml of warm (37 C) complete medium, 1 ml WST-1 reagent
was
added. The medium from the wells of the plate with the Mia-Paca-2 cells was
removed and
replaced with 100 i.tL of the WST-1 reagent in complete medium. The plate was
incubated at
37 37 C/5% CO2. At timed intervals the absorbance at 450 nm was determined
for each well
in a UVNis plate reader. For each well the slope of the plot of the absorbance
450 nm vs
time was determined. The readings were normalized to that of the wells for the
untreated
control and a dose-response curve was plotted. A fit to the data was made from
a four
parameter, variable slope program (GraphPad Prism) and the ICso was
determined.
Compound 9A inhibited the proliferation of both types of cells, with an ICso
of 10 nM for Mi-
aPaCa-2 cells and 2 iuM for MDA-MB-231 cells.
The same procedures were also applied to the compounds Compound 7A, Compound
6A, and Compound 8A to test their inhibitory activities against RKO colon, Mia-
Paca-2
pancreatic and BT-549 cancer cells. The results showed the significant
decreases of cell
viability in all three cancer cells treated with Compound 7A, Compound 6A, and
Compound
8A each (Figure 2C-2E).
Sensitivities of Multiple Cancer Cell Lines to the Compounds
The compounds were profiled on an OncolinesTM Profiler (Netherlands
Translational
Research Center) assayed with 66 cancer cell lines to determine if they were
capable of
inhibiting the proliferation of these cancer cells. The results showed 57 of
66 cancer cell lines
were sensitive to Compound 7A, wherein Compound 7A had ICso values < 1 litM in
9 cancer
cell lines. Compound 7A had an ICso value over 32 tIM in the malignant
melanoma cell line
MeWo. Compound 7A had an ICso value of 2.4 jiM in the ovarian cancer cell line
OVCAR-3.
Compound 7A had an ICso value of 0.88 M in the colon cancer cell line RKO.
Compound
7A had an ICso value of 0.79 !AM in the pancreatic cancer cell line MIA PaCa-
2. Compound
7A had an ICso value of 0.2 uM in the triple negative breast cancer cell line
BT-549. Table 3
provided ICso values for cell lines wherein Compound 7A were active at ICso <
5 M.
Table 3. ICso of Compound 7A in Multiple Cell Lines
Cell Line ATCC ref Disease ICso
jtM
A-172 CRL-1620 Glioblastoma
1.067
A-204 HTB-82 Rhabdomyosarcoma
1.360
- 65 -
CA 03237744 2024- 5-8
WO 2023/091464 PCT/US2022/050069
A375 CRL-1619 Malignant melanoma
2.017
A388 CRL-7905 Epidermal carcinoma
2.394
AN3 CA HTB-111 Endometrium adenocarcinoma
0.660
BT-20 HTB-19 Breast carcinoma
1.394
BT-549 HTB-122 Breast ductal carcinoma
0.199
CCRF-CEM CCL-119 Acute lymphoblastic
leukemia, Tcell 0.247
Daoy HTB-186 Medulloblastoma, CNS
3.379
DoTc2 4510 CRL-7920 Cervix carcinoma 0.963
DU 145 HTB 81 Prostate carcinoma
2.014
HCT-116 CCL-247 Colon carcinoma
2.390
Hs 57BT HTB-126 Breast carcinoma
1.414
Jurkat E6.1 TIB-152 Leukemia, T cell 0.548
K-562 CCL-243 Chronic myelogenous leukemia (CML)
0.486
LoVo CCL-229 Colorectal adenocarcinoma
0.635
MIA PaCa-2 CRL-1420 Pancreatic ductal carcinoma
0.790
MOLT-4 CRL-1582 Acute lymphoblastic leukemia
0.594
NCI-H460 HTB-177 Large cell lung carcinoma
2.093
OVCAR-3 HTB-161 Ovary adenocarcinoma
2.400
PA-1 CRL-1572 Ovary teratocarcinoma
2.073
RKO CRL-2577 Colon carcinoma
0.683
RPM1-7951 HTB-66 Malignant melanoma 1.197
SK-N -AS CRL-2137 Neuroblastoma
1.720
SR CRL-2262 Large cell immunoblastic lymphoma
1.584
SUP-Ti CRL-1942 T-cell lymphoblastic lymphoma
4.289
VA-ES-BJ CRL-2138 Epithelioid carcinoma, bone
0.682
Compound 7A had an ICso value of 0.486 uM in the CML cell line K-562.
According
to BH3 profiling (a functional assay to determine which of the pro-survival
Bc1-2 proteins
that a cell depends upon for survival), Mc-1 was not essential for survival of
K562.
Inhibition of USP9X in K562 cells led to the K63 polyubiquitylation of Bcr-Abl
(the
oncogenic driver in K562) with consequent relocation to pen-nuclear aggresomes
and thus
loss of activity.
- 66 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
The profiling results from Oncolines Profiler indicated that various cancer
cells
were sensitive to the compounds and thus the compounds can be used as
therapeutics for
treating, inhibiting, or suppressing those cancers in Table 3.
Comparing The Compounds with Another USP9X Inhibitor
To further evaluate potency of the compounds, they were compared with the
USP9X
inhibitor WP1130 by screening both the compounds and WP1130 on a 66-cancer
cell line
panel ONCOLINESTm Profiler.
IC50 ratios of WP1130/ Compound 7A in 12 cancer cell lines were shown in Table
4.
Table 4. IC50 Ratios of WP1130/ Compound 7A in cancer cell lines
Compound 7A WP 1130
Cell Line Type 100 Ratio
IC5o 1-tM IC50 IAM
LoVo colon 0.64 5.5 8.6
RKO colon 0.88 4.3 4.9
A-172 glioblastoma 1.10 5.6 5.1
RPMI7951 melanoma 1.20 4.2 3.5
A549 lung 3.00 14 4.7
H82 small cell lung 1.10 6.6 6.0
BT-20 breast 1.40 5.1 3.6
BT-549 breast 0.20 5.4 27.0
Hs578T breast 1.40 6.3 4.5
D U145 prostate 2.00 9.6 4.8
CCRF-CEM ALL 0.25 4.4 17.6
MOLT-4 ALL 0.59 4.0 6.8
In 12 of 66 cell lines, Compound 7A was about 4-fold more potent than WP1130.
In 3
of 66 cell lines, Compound 7A appeared to be inactive and WP1130 was above 4-
fold more
potent than Compound 7A. The 3 cell lines expressed high levels of the anti-
apoptotic Bel-
xL, known to correlate with resistance to antagonism of Mcl-1. Further, as a
less selective
inhibitor of USP9X, WP1130 also inhibited the activity of other
deubiquitvlases, which might
account for the observed higher potencies of WP1130 in certain cell lines. The
results
indicated that the compound was generally more potent than WP1130.
Inhibition against U937 Cells and intracellular Mcl-1
The effect of Compound 7A on U937 cells was assessed using the same experiment
described in the Inhibitory Activities against the Proliferation of Mia-Paca-2
and Other
Cancer Cells section above. In response to treatment with Compound 7A,
proliferation of
U937 leukemia cells was inhibited and induced apoptosis was obsessed by trypan
blue
- 67 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
staining. The ICso for the inhibition of U937 by Compound 7A for 72-how-
treatment was 270
nM. Similar inhibiting effects of Compound 7A were observed in multiple cancer
cell lines
(e.g., cancer cell lines listed in Table 3).
After 24 hours treatment with Compound 7A, U937 cells were collected by
centrifugation and whole cell lysates were prepared. These lysates were
analyzed by Western
blot to determine the level of Mc1-1. Western blot analysis was carried out as
previously
described (Wang et al., 2014 Journal of the American Society of Nephrology
DOI:
10.1681/asn.2013060665). The band intensity for Mel-1 in these samples were
normalized to
that of tubulin to correct for any inconsistencies in total protein in loading
and processing the
whole cell lysates. The insert in Figure 3A showed Western blot band
intensities for Mc-1
and tubulin observed for the untreated U937 cells and those treated with
101.1.M Compound
7A and illustrated the decline in Mc-1 levels induced by the treatment.
In U937 cell line, after 24-hour treatment of Compound 7A at varying
concentrations,
the normalized level of Mc-1 decreased by 50% when increasing Compound 7A's
concentration from 0 to 33 ium, indicating that the compound successfully
induced
degradation of Mc-1 in U937 cells. And cell viability decreased to 0 when Mc-1
level
decreased by 50%, indicating that the 50% decrease in Mc-1 induced by the
compound
within 24 hours was sufficient to inhibit the proliferation of U937 cells by
100%.
The results indicated that the compound induced partial degradation of Mc-1 in
U937
cells and resulted in the complete inhibition of U937 cell proliferation.
Inhibition against SK-BR3 Cells and intracellular erbB2
Another substrate of USP9X is the receptor tyrosine kinase erbB2. The
involvement
of USP9X in the regulation of erbB2 suggested that its degradation may be
induced by
treatment with USP9X inhibitors. The breast cancer cell line SK-BR3 expresses
high levels
of erbB2. This cell line was treated with Compound 7A and the effects on cell
viability as
well as expression levels of erbB2 were examined.
Compound 7A inhibited the viability of SK-BR3. The levels of erbB2 were
determined by Western blot and found to decrease in parallel with the decline
in cell viability.
After 72-hour treatment of 10 m Compound 7A, the level of erbB2 declined to
about zero.
The loss of erbB2 from the plasma membrane was also confirmed by flow
cytometry.
The results from both western blot and flow cytometry indicated the compound
induced the degradation of erbB2 in SK-BR3 breast cancer cells. From cell
viability results
- 68 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
after 72-hour treatment with Compound 7A, the compound exhibited an ICso value
of 2.7 M
for inhibiting the proliferation of SK-BR3 breast cancer cells by 100%.
Capture ELISA
In addition to Western blot analysis, direct effects of the compounds on
cancer related
cellular substrates were determined using capture ELI SA techniques. Following
the treatment
of the compound, decreases were detected in erbB2 levels in SK-BR-3, MIA-PaCa-
2, BT-
549, and MDA-MB-231 cells; and in Mc-1 levels in SK-BR-3, MIA-PaCa-2, BT-549,
and
U937 cells. Increases were detected in the level of ubiquitylatedeErbB2 in SK-
BR-3 cells and
in the level of ubiquitylated Mc1-1 in SK-BR-3 cells, MIA-PaCa-2, and BT-549
cells. For
SK-BR-3 cells treated, it was also detected the loss of erbB2/erbB3
heterodimers and erbB2
homodimers.
Example 4: Investigating Mechanism of Action of Compounds Using Protein
Synthesis
Inhibitor and Proteasome Inhibitor
Western Blot Analysis
To investigate the mechanism of action, Western blot analysis was carried out
to test
the induced degradation of USP9X substrate Mc-1 by the compounds under the
effects of
protein synthesis inhibitor cycloheximide and/or proteasome inhibitor MG132.
U937 cells
were treated with 10 M cycloheximide for one hour, with 101..iM MG132 for one
hour, with
the combination of 10 M cycloheximide and 10 M MG132 for one hour, and with
vehicle
(DMSO) for one hour, respectively, prior to the treatment with Compound 5A for
24 hrs. The
U937 cells were harvested after treatment and whole cell lysates were
prepared. These lysates
were analyzed by Western blot to determine the abundance of Mc1-1 as well as
that of tubulin
as a normalization control. Lanes 1 and 7 of Figure 4A showed the intensity of
the Mc-1 and
tubulin bands for vehicle treated U937 cells. The band intensity of the Mc-1
in cells treated
with cycloheximide alone (Lane 2) was reduced relative to that for tubulin.
Intracellular Mc-
1 has a short half-life (approximately 30 minutes). Global blockade of protein
synthesis by
cycloheximide led to a significant decrease in the expression levels,
especially for short lived
Mc1-1. The retention of a reduced portion of Mc-1 indicated the immunity of
this portion to
proteasomal degradation. The treatment of U937 cells with Compound 5A (Lane 3)
resulted
in an about 50% reduction of Mc1-1. In contrast, the band intensity of Mc1-1
relative to that
of tubulin increased upon treatment of U937 cells with the proteasome
inhibitor MG132
(Lane 4). The ability of Compound 5A to induce the degradation of Mc1-1 was
blocked by
- 69 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
addition of MG132 (Lanes 5 and 6), suggesting that the mechanism for the
induced
degradation of Mcl-1 was via the proteosome. Together, the results of this
Example are
consistent with the compounds acting on the proposed mechanism that
intracellular inhibition
of the deubiquitylase activity of USP9X blocks its ability to rescue USP9X
substrates such as
Mc-1 from proteasomal degradation. In other aspects, by varying the
concentration ratios of
the compound and proteasome inhibitor for treating cells, it was possible to
achieve
controlled degradation of a USP9X substrate so the substrate could decline to
a desirable
level rather than degrade entirely.
ELISA Analysis
MIA-PaCa-2 cells were treated with Compound 7A at 25 !AM, 12.5 !AM, 6.25 1,1M,
and
3.125 1.tM, respectively, for 24 hours. Capture ELISA of Mc-1 was performed
using a
dilution series of MIA-PaCa-2 cell lysates equalized for total protein in a 96-
well plate coated
with sheep anti-human Mc-1 (R&D Systems, #AF8281). Detection of Mc-1 was
performed
using biotinylated sheep anti-human Mcl-1 followed by streptavidin-horseradish
peroxidase
and TMB development. For detection of ubiquitylated Mcl-1 the same capture
conditions
were used but employing a biotinylated mouse anti-human ubiquitin antibody
(R&D
Systems, #MAB701), again detecting with streptavidin-horseradish peroxidase
and TMB
development. Lysate samples were run in triplicate and percentages were
calculated based on
the signal generated from cell lysates with no compound added (vehicle control
samples).
After treading cells with 25 i.tM Compound 7A for 24 hours, Mc-1 declined by
about 23.0%
whereas ubiquitylated Mc-1 increased by about 27.5%. With 12.51.tM Compound
7A, Mcl-1
declined by about 25.0% whereas ubiquitylated Mc-1 increased by about 14.0%.
Treatment of cells with Compound 7A induced the degradation of USP9X substrate
Mcl-1. The level of ubiquitylation of Mc-1 increased even as the level of Mc1-
1 expression
declined. These results were consistent with the inhibition of USP9X within
cells, causing
the level of Mc-1 ubiquitylation to rise and its subsequent degradation in the
proteasome.
Example 5: Sensitizing Cells and Enhancing Potency for Other Protein Inhibitor
Mc-1 can serve as an anti-apoptotic factor conferring resistance to
chemotherapy.
Western blot analysis was carried out to investigate the effect of the
compounds on other
inhibitors of which the targeted proteins could otherwise be protected by Mc-1
and thus
resistant to these inhibitors.
- 70 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
In the AML cell lisle U937, western blot bands showed high intensity for Bc1-
xL,
medium intensity for Mc1-1, and low intensity for Bc1-2. U937 cells were
treated with the
Bc1-2/Bc1-xL inhibitor ABT737 72 hours at varying concentrations and underwent
western
blot analysis. The IC50 value of ABT737 was determined to be 3.4 !AM from the
dose-
response curve. In contrast, after adding 0.5 [i.M Compound 7A to the same
treatment, the
IC50 value of ABT737 was determined to be 0.06 j.tM from the dose-response
curve. Co-
treatment of 0.5 pM Compound 7A resulted in 57-fold enhancement in the
sensitivity of
U937 cells to ABT737.
The TNBC cell line MDA-MB-231 express high levels of Mc1-1 and USP9X in
addition to Bc1-2 and Bc1-xL. MDA-MB-231 cells were treated with ABT737 for 72
hours at
varying concentrations and underwent western blot analysis. The ICso value of
ABT737 was
determined to be 8.71,1M from the dose-response curve. In contrast, when co-
treated with 10
iuM Compound 5A and underwent the same experiments, the IC50 value of ABT737
was
determined to be 1.8 M from the dose-response curve. Co-treatment of 10 iuM
Compound
5A resulted in 5-fold enhancement in the sensitivity of MDA-MB-231 cells to
ABT737.
Example 6: Downregulating PD-Li
Programmed death-ligand 1 (PD-L1), an immune-oncology target, has been
identified
as a substrate for USP9X. PDL1 as a transmembrane protein is expressed on
tumor cells and
capable of blunting the immune response to tumors by interacting with PD1 on
the surface of
T cells that otherwise would attack tumor antigens on the surface of these
cells and eradicate
them. The compounds as described herein induce the targeted degradation of
PDL1 from
tumor cells and enhance the immune response of T cells against the tumor
cells.
MIA PaCa-2 cells expressing detectable levels of PD-Li are treated with
Compound
7A at various concentrations for 24 hours, and the ensuing level of PD-Li are
determined by
Western blot to decrease by about 50% after the treatment.
Example 7: In vivo animal model test
USP9X is highly expressed and activated in melanoma cells. The effect of
compounds
on USP9X activity in a representative melanoma cell line, A375, and an A375
variant cell
line that is resistant to the BRAF kinase inhibitor, vemurafenib, can be
examined. Treatment
results in inhibition of USP9X activity in either cell type. To determine anti-
tumor activity in
animals, mice are treated intravenously (IV) or by oral savage (PO). Compound
is
-71 -
CA 03237744 2024- 5-8
WO 2023/091464
PCT/US2022/050069
administered once to two mice per group at the indicated dosage level and
route (IV or PO)
and plasma is collected after administration. Compound concentration in the
plasma is
measured by high performance liquid chromatography coupled with mass
spectroscopy
detection (LC/MS).
Tumor cells are injected into the dorsal region of twenty female
NOD/SCID/gamma-
2 knockout mice (NSG) weighing about 20 grams each. After 3 weeks tumors
become visible
and measurable with calipers. Mice are separated into four groups of 5 mice
each and IP
injected with the compound of the invention dissolved in 55% dimethyl
sulfoxide, 25%
polyethylene glycol 300, 20% phosphate-buffered saline at dose levels of 0,
2.5, 5 and 10
mg/kg mouse body weight. Animals are injected once per day for 14 days and
tumor growth
(measured with calipers) and animal weight are monitored over the treatment
interval.
Results show that tumor growth in mice treated with the compound at dose
levels of 2.5, 5
and 10 mg/kg are inhibited at different levels, compared with that in the
group of untreated
mice; and the dose of 10 mg/kg of the compound lead to the maximum inhibition
of tumor
growth by about 80% among the three treated groups, increasing the life span
by about 200%
compared with untreated mice.
While this invention has been particularly shown and described with references
to
preferred embodiments thereof, it will be understood by those skilled in the
art that various
changes in form and details may be made therein without departing from the
scope of the
invention encompassed by the appended claims_
- 72 -
CA 03237744 2024- 5-8