Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
`-~ 21 ~21~l~
HOEC~ST AXTIENGESEL~SCE~ T ~OE 92/F 422 Dr~VF/PP
Description
2,4-Substituted 5-(N-substituted sulfamoyl)benzoylgua-
nidines, process for their preparation, their use as
drugs or diagnostic agents, and drugs in which they are
pre~ent
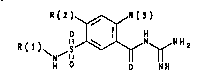
The invention relates to benzoylguanidinea of formula Io
R(2) ~ R(~)
)~N~S ~N~N H a
H o NH
in which
R(1) is R(4)R(5)N-C(X)-, where
X i~ oxygen, S or N-R(6) and
R(4) and R(5), which are identical or different,
are H, (Cl-Ca)-alkyl, (C3-C6)-alkenyl or -Cn~2n-
R(7), where n is equal to zero, 1, 2, 3 or 4 and
Rl7) i~ ~Cs-C~)-cycloalkyl or phenyl which is
unsubstituted or ubstituted by 1 - 3 substi-
tuents ~elected ~rom the group comprising ~ Cl,
CF3, methoxy and (C1-C4)-alkyl, or
R(4) and R(5) together arç 4 ox 5 methylene
groups of which one CH2 group can be replaced by
oxygen, S, NH, N-CH3 or N-benzyl~ and
R(6) i~ a~ defined for R(4) or i6 amidine,
R(2) is H, F, Cl, Br, I, (C1-Ca)-alkyl, alken-1-yl or
alkyn-1-yl, (C3-C8)-cycloalkyl, (C3-C6)-cyclo
alkyl-(C1-C4)-alkyl, phenyl, C6Hs-(C1-C4)-alkyl,
naphthyl, biphenylyl t 1,1-diphenyl-(C1-C4)-alkyl,
cyclopentadienyl, pyridyl, thiopyridyl, pyrrolyl,
furanyl, thienyl, thiazolyl, oxazolyl, indenyl,
quinolyl, indolyl, benzofuranyl, benzothienyl,
2~2 ~
benzothiazolyl, benzoxazolyl or -W-R~8), where
W i~ oxygen, S or ~R(9),
R(8) is ~, (C1-C6)-alkyl, ( C5_C7 ) cycloalkyl,
cyclohexylmethyl, cyclopentylmethyl, -(CH2)mCpF2~1
or ~Cq~2~~R(10), where m = zero or 1, p = 1, 2 or
3, q = zero, 1, 2, 3 or 4 and R(10) i~ phenyl
which i8 unsubstituted or substituted by 1 - 3
substituent~ selected from the group compri~ing
F, Cl, CF3, methyl, methoxy and NR(ll)R(12),
where R(11) and R(12) are ~ or (C1-C4)-~lkyl, and
R(9) i~ H or (C1-C3)-alkyl,
it also being po~sible for R(8) and R(9) together
to be 4 or 5 methylene group~ o which one C~2
group can be replaced by oxygen, S, ~H, N-CH3 or
~-benzyl, and
R(3) is H, F, Cl, ~r t I, (C1-C6)-alkyl or -W~R(8) a~
defined for R(2~, .
and to their pharmaceutically Acceptable ~alt
Preferred compounds I are tho~e in which
R(1) i~ R(4)R(5)N-C(X)- where
X i~ oxygen, 5 or N R(6),
R(4) and R(5), which are identical or different,
are H, (C1-C~)-alkyl or ~Cn~2~-R ( 7) where n i~
equal to zero, 1 or 2 and R(7) i~ (C5-C7)-cyclo~
~lkyl or phenyl whi~h i~ unsubstituted or
sub~tituted by 1 - 3 sub~tituent~ ~elected from
the group compri ing F, Cl, CF3, methoxy and (Cl-
C4 ) -a1kY1, or
R(4) and R(5) together are 4 or 5 methylene
yroup~ of whioh one CH2 group can be replaced by
oxygen, S, NH, N-CH3 or N-b nzyl, and
R(6) i~ as de~ined for R(4) or i~ amidine,
R ( 2 ) 15 H, F, C1 ~ ( C1_CB ) -alkyl~ ( C3_CB ) -cycloalkyl~
(C3-C8)-cycloalkyl-(C,-C2)-alkyl, phenyl,
naphthyl, biphenylyl, pyridyl, thiopyridyl,
pyrrolyl or -W-R(8) where
W i8 oxygen, S or NR(9),
R(8) i~ ~, (C1-C6)-alkyl~ -(CH2)~CpF2~, or CqH2g
R(10) where m = zero or 1, p = 1 t 2 or 3, q =
zero, 1, 2, 3 or 4 and R(10) = phenyl which i~
unsubstituted or sub~tituted by 1 - 3 sub-
stituents selected from the group compri~ing F,
Cl, CF3, methyl, methoxy and NR(ll)R(12~ where
R(11) and R(12) are H or (C1-C4)-alkyl, and
R(9) is N or (Cl-C3)-alkyl,
it also being possible for R(8) and R(9) together
to be 4 or 5 methylene group~ of which one CH2
group can be replaced by oxygen, S, NH, N-CH3 or
N-benzyl, and
R(3) is H, F, Cl, (Cl-C3)-alkyl or -W-R(8) as defined for
R~2).
Particularly preferred compounds of formula I are tho~e :~-
in which
,:
R(l) i~ R(4)R(5)N-C(X~- where
X is oxygen, S or NR(6),
R(4) and R(5), which are identical or different,
are H, (C1-C4)-alkyl or -CnH2n-R(7) where n is
equal to zero, 1 or 2 and R(7) is (C5-C,)-cyclo~
1 25 alkyl, or : ~ -
R(4) and R(5) together are 4 or 5 methylene
groups of which one C~2 group can be replaced by
oxygen, S, NH, N-C~3 or N benzyl, and
R(6) i~ as defined ~or R(4) or i~ amidine,
30 R(2) is hydrogen, F, Cl, (C1-C4)-alkyl, thiopyridyl or - ~ :~
W-R(8) where
W i~ oxygen, S or ~R(9)
R(8) i~ (C~C6)-alkyl or ~CgH29-R(10) where
q = zero, 1, 2, 3 or 4 and R(10) iB phenyl
which i~ unsubstituted or sub~tituted by 1 - 3
. :, . .- . . . , , - . ............ .
~,,¢ , ,;, ", ~
2 .1 ~`3 ~
substituents selected from the group comprising
F, Cl, CF3, methyl, methoxy and NR[ll)R(12) where
R(11) and R(12) are H or (C1-C4)~alkyl, and
R~9) is ~ or (C1-C3)-alkyl, or
R(8) and R(9) together are 4 or 5 methylene
groups of which one CH2 group ~an be replaced by
oxygen, S, NH, N-CH3 or N-benzyl, and
R(3) iB H, F, Cl or (C1-C3)-alkyl,
and their pharmaceutically compatihle salts.
If one of the three ~ubstituents R(1), R(2) and R(3)
contain a center of asymmetry, the invention includes
compounds of both the S and R configurations. The co~- -
pounds can exist as optical i~omer~, as diastereoi~omers,
as racemates or as mixture~ thereof.
Unless expre~sly stated otherwise, alkyl, alkenyl or
alkynyl are alway~ groups with a linear or branched
chain.
~;, :''
The compounds I are ~ubstituted acylguanidine~
The most prominent representative of the acylguanidine~
is the pyrazine derivative amiloride, which is used in
therapy as a potassium-sparing diuretic. Numerous other
compounds of the ~miloride type are de~cribed in the
literature, examples being dimethylamiloride or ethyliso-
propylamiloride.
R'
~"~N ~ ~ 2
c IJ~I~NH2
O ~ a
25 amilorides R', R'' = H
dimethylamiloride: R', R'' - CH3
ethylisopropylamiloride: R' ~ C2H5, R'' ~ C~(CH3)2
:,',~: ::::.''' ' ' '-. . : '''. - ': ': ' .:,
2 ~
-- 5 --
Furthermore, studies have been disclo~ed which indicate
antiarrhythmic properties of amiloride [Circulation 79,
1257-63 (1989)]. However, the obstacle to a broad
application as an antiarrhythmic is that this effect i5
only weak and i5 accompanied by a hypotensive and
saluretic action, these side-effect~ being undesirable in
the treatment of cardiac dysrhythmia,
Indications of antiarrhythmic properties of amiloride
have also been obtained from experiment~ on i olated
animal heart~ [~ur~ ~eart J. 9 (Suppl.): 167 (1988) tbook
of abstracts)]. Thus it hae been found on rat hearts,
for example, that an artificially produced ventricular
fibrillation can be fully ~uppreæ~ed by amiloride. The
abovementioned amiloride derivative ethylisopropyl-
amiloride wa~ even more potent than amiloride in this
model.
US patent 5 091 394 (~OE 89/F 288) de~cribe~ benzoyl-
guanidines which carry a hydrogen in the posikion
corresponding to the radical R(l). German patent
application P 42 04 575.4 (~OE 92/034) proposes 3.5
substituted benzoylguanidines in which, however, the
substituents R(1) and R(2) are not defined as claimed in
the present invention. --
US patent 3 780 027 claims acylguanidines which are
~tructurally similar to the compounds of formula I. The
decisive difference from the compounds I i~ that said
acylguanidine~ are benzoylguanidines which in their
substitution pattern are derived from commercially
available diuretics ~uch as bumetanide and furosemide,
and carry an amino group in the 2- or 3- position
relative to the carbonylguanidine group, said amino group
being important for the salidiuretic action sought in
~aid patent. Accordingly, theqe compounds are reporked
to have a ~trong ealidiurekic activity.
It was therefore aurpri~ing that the compound~ according
6 2~2~9l~
to the invention do not have any undesirable and
disadvantageous salidiuretic properties, but do have very
good antiarrhythmic properties such as, for example,
those which occur in the case of anoxic manifestations.
On account of their pharmacological properties as anti-
arrhythmic drugs with a cardioprotective component, the
compounds are outntandingly suitable for the prophylaxis
and treatment of infarctus and for the treatment of
angina pectori~, and they also inhibit or greatly reduce,
in a preventive capacity, the pathophysiological
processes which occur during the development of
ischemically induced damage, especially during the
production of i~chemically induced cardiac arrhythmia.
Because of their protective effects against pathological
hypoxic and ischemic situations, the compounds of formula
I according to the invention, as a result of inhibiting
the cellular Na+/H~ exchange mechanism, can be u~ed as
drugs for the treatment of all acute or chronic damage
produced by ischemia or diseases induced thereby in a
primary or secondary process. This concern~ their use as
drugs for operative procedures, e.a. in or~an
transplants, it being posnible for the compounds to be
used not only for protecting the organs in the donor
before and during removal and for protecting removed
organs, for example in the case of treatment with or
without their storage in isotonic solutions, but also
during transfer into the recipient organism. The
compounds are also valuable drugs with a protective
action d~ring angioplastic operative procedure~, for
example on the heart and on peripheral vessels. By
virtue of their protective action against ischemically
induced damage, the compoundn are al80 auitable a~ drug~
for the treatment of ischemia of the nervous aystem,
especially the CNS, being uitable e.q. for the treatment
of stroke or cerebral edema. Furthermore, the compounds
of formula I according to the invention are also ~uitable
for the treatment of various foxms of shock, for example
allergic, cardiogenic, hypovolemic and bacterial shock.
'f;~ : ~ :: :: - : :
2~ ~ 2~9 ~
-- 7 --
In addition, the compounds of formula I according to the
invention are distinguished by a strong inhibitory action
on cell proliferation, for example fibroblast
proliferation and proliferation of the smooth vascular
muscle cells. The compounds of formula I are therefore
suitable as valuable therapeutic agents for diseases in
which cell proliferation is a primary or secondary cause,
and hence can be used a~ antiatherosclerotics and drugs
for diabetic late complication3, carcinosis, Eibrotic
diseases such as pulmonary fibrosis, hepatic fibrosis or
renal fibrosis, and organ hypertrophy and hyperplasia,
especially in cases of hyperpla~ia or hypertrophy of the
prostate.
The compounds according to the invention are effective
inhibitors of the cellular sodium/proton exchanger (~a~
exchanger), which is increased in numerous diseases
tessential hypertonia, atherosclerosis, diabetes etc.),
even in cells on which mea urements can easily be made,
for example in erythrocytes, thrombocytes or leukocytes.
The compounds according to the invention are therefore
suitable as outstanding and simple scientific tool6, for
example in their use as diagnostic agents for the deter-
mination and differentiation of particular f OrmB of
hypertonia, and al o of atherosclerosis, diabetes,
proliferative diseases etc. Furthermore, the compound~
of formula I are suitable for preventive ~herapy in
preventing the genesis of hypertension, for example
e sential hypertonia.
~he invention further relates to a process for the
preparation of the compounds I, which compri9es xeacting
compounds of formula II:
R(2) ~ R(3)
t 1 )~N~S~L I I
H O
.,~, " ~
2 ~ ~ 4
-- 8 --
with guanidine, R(l) to R(3~ being as defined and L bei~g
a leaving group readily 6usceptible to nucleophilic
substitution.
The activated acid derivative~ of formula II, in which L
is an alkoxy group, preferably a methoxy group or ethoxy
group, a phenoxy group, a phenylthio, methylthio or 2-
pyridylthio group or a nitrogen heterocycle, preferably
imidazol-l-yl, are advantageou~ly obtained, in a manner
known per se, from the carboxylic acid chlorides on which
they are based ~formula II, L = Cl), which can in turn be
prepared, again in a manner known per se, from the
carboxylic acids on which they are ba~ed tformula II,
= OH), for example with thionyl chlorideO
Apart from the carboxylic acid chlorides o formula II (L
= Cl), other activated acid derivatives of formula II can
al~o be prepared, in a manner known pex_se, dixectly ~rom
the benzoic acid derivatives on which they are based
(formula II, L = OH~, exampleæ being the methyl ester~ or
ethyl esters of formula II, where L = OCH3 or OEt, by
treatment with gaseous HCl in methanol or ethanol, the
imidazolide~ of formula II by treatment with carbonyl-
diimidazole [L = imidazol-1-yl; Staab, AngewO Chem. Int.
Ed. Engl. 1, 351 - 367 (1962)~, the mixed anhydride~ II
by treatment with Cl-COOC2~5 or tosyl chloride in the
pre~ence of triethylamine in an inert ~olvent, and al~o
the benzoic acid derivatives activated with dicyclohexyl-
carbodiimide (DCC) or with O-[(cyano(~thoxycarbonyl)-
methylene)amino~-1,1,3,3-tetramethyluronium tetra-
fluoroborate ("TOTU") [Wei~s and Krommer, Chemiker
Zeitung 98, 817 (1974)]. A number of suitable methods
for the preparation of activated carboxylic acid
derivative~ of formula II are indicated in J. March,
Advanced Organic Chemistry, Third Edition (John Wiley &
Sons, 1985), p. 350, in which literature sources are
cited.
The reaction of an activated carboxylic acid derivative
9 2 1 1 ~, ~ .3 ~ -
of formula II with guanidine is carried out, in a manner
known per se, in a protic or aprotic polar but inert
organic solvent. Methanol or THF at between 20C and the
boiling point of the~e solvents have proved satisfactory
for the re~ction of the methyl benzoates (II, L = OMe)
with guanidine. Most of the reactions of compounds II
with salt-free guanidine have advantageously been carried
out in aprotic inert solvents such as THF, dimethoxy-
ethane or dioxane, although water can also be used as the
solvent.
If L = Clt the reaction is advantageously carried out
with the addition of an acid acceptor, e.q. in the form
of excess guanidins, in order to bind the hydrohalic
acid.
Some of the benzoic acid derivatives of formula II on
which the activated acid derivatives of formula II are
based are known and are described in the literature. ~he
unknown compounds of formula II can be prepared by
methods known in the literature, for example by con-
verting 2,4-dihalogeno-5-chlorosulfonylbenzoic acids to
5-aminosulfonyl-2,4-dihalogenobenzoic acids (IIIa, L =
OH) with ammonia and reacting the resultinq benzoic
acid , according to one of the process variants described
above, to give compounds I according to the invention.
~ 111
IIIa) R = NR(4)R(5)
IIIb) R = R(4)R(S)NC(X)
However, such carboxylic acids or their e~ters of formula
III, where L = -OH or, for example, ~O-methyl or -O-
ethyl, can also be used a~ ~tarting compounds for other
carboxylic acids, it being possible ~or the halogen in
2 ~ L 2 ~
- 10 - , ,
the R(2)- and/or R(3)-position to be exchanged very
conveniently, in known manner, with numerous nucleophilic
reagents such as mercaptans R(8)- SH, alcohols R(8)-OH or
amines R(8)R(9)NH.
The alkyl or aryl substituents are introduced by methodsl
known in the literature, involving the palladium-mediated
cross-coupling of aryl halides with, for example, organo-
zinc or organocopper compounds, organostannanes, organo-
boron acids or organoborane~.
The benzoic acid derivatives of formula III
(IIIb, X = O, S) are prepared by reacting the sodium
salts of IIIa (R4, Rs = H) with isocyanates or
carbamoyl chlorides (X = O) or isothiocyanates (X = S).
S-Alkylation of IIIb (X = S~ followed by exchange with an
amine [R(6)NH2] gives the benzoic acid derivative~ of
formula IIIb [X = NR(6)~.
In general, benzoylguanidines I are weak bases and can
bind acid to fonm salts. Suitable acid addition ~alts
are salts of all pharmacologically compatible acids, for
example halides, especially hydrochloride~, lactates,
sulfates, citrates, tartrates, acetates, phosphates,
methylsulfonates and p-toluenesulfonates.
Drugs containing a compound I can be administered orally,
parenterally, intravenously, rectally or by inhalation,
the preferred form of administration depending on the
particular clinical picture of the disease. The
compounds I can be used by themselves or together with
galenic adjunct~, in both veterinary and human medicine.
On the basis of their expert knowledge, those skilled in
the art are familiar with which adjuncts are suitable for
the desired drug formulation. In addition to solvents,
gel-forming agents, ~upposikory bases, tableting aids and
other active ingredient excipients, it i8 po~sible for
example to use antioxidants, dispersants, emulsifier~,
L~
antifoams, taste correctors, preservatives, solubilizing
agents or colorants~
For an oral form of administration, the active compounds
are mixed with the appropriate additives such a~
excipients, 6tabilizers or inert diluents, and converted
by the conventional methods to the appropriate form~ of
administration, such as tablets, coated tablets, hard
capsules or aqueous, alcoholic or oily ~olution~.
Example6 of inert excipients which can be u~ed are gum
arabic, magnesium hydroxide, magnesium carbonate,
potassium pho~phate, lacto~e, glucose or ~tarch,
especially corn ætarch. The preparation can take the
form of either dry or moi~t granules. Examples of
suitable oily excipients or solvents are vegetable or
animal oils such as sunflower oil or cod liver oil.
For subcutaneous or intravenous administration, the
active compounds are dis~olved, suspended or emulsified,
if de ired together with the su~stance~ conventionally
u~ed for this purpose, ~uch a~ ~olubilizing agents,
emul~ifiers or other adjuncts. Example~ of ~uitable
solvent~ are water, i~otonic ~olution or alcohols, e.q.
ethanol, propanol or glycerol, a~ well as eugar solution~
such as glucose or mannitol solutions ~ or else a mixture
of the variou~ 601vents mentioned.
Examples of suitable pharmaceutical formulations for
admini~tration in the form of aero~ol3 or sprays are
~olution~, ~uspensions or emul~ions of the active
ingredient of formula I in a pharmaceutically acceptable
solvent such a~, in particular, ethanol or water, or a
mixture of such ~olvents. A~ reguired, the formulation
can al50 contain other pharmaceutical adjuncts such as
~urfactants, emulsifiers and ~tabilizers, as well a~ a
propellant. Such a preparation conventionally contain~
the active ingredient in a concentration of about 0 1 to
10, especially of about 0.3 to 3% by weight.
- 2 ~ f-~
~ 12 -
The dosage of the active ingredient of formula I to be
administered, and the frequency of admini~tration, depend
on the potency and duration of action of the compound~
used; they further depend on the nature and ~everity of
the disease to be treated and on the sex, age, weight and
individual responsiveness of the mammal to be treated
On average, the daily do~e of a compound of formula I in
the ca~e of a patient weighing ~bout 75 kg i~ at l~ast
0.001 mg, pre~erably 0.01 mg to 10 mg and particularly
preferably 1 mg. For acute onsets of the di~ease, ~or
instance immediately after a cardiac infarctus has been
suffered, it may be nece~sary to admini ter even higher
and, in particular, more frequent doses, e.q. up to 4
~ingle doses per day~ ~pecially in the ca~e of i.v.
administration, for instance for an infarctus patient in
intensive care, it may be necessary to administer up to
100 mg per day.
Experimental section:
General instructions for the preparatisn of benzoyl-
guanidines (I) from methyl ~or ~thyl) benzoates (II, L =OMe or OEt)
0.01 mol of the methyl (or ethyl) benzoate derivative of
general formula II i~ dis~olved or ~uspended in 60 ml of
anhydrous tetrahydrofuran (THF), and 0.05 mol of
guanidine is then added. After refluxing for a few
hour6, the T~F is distilled off, water is added, the pH
i~ adjueted to 6 - 7 with 2 N HCl and the corresponding
benzoylguanidine (I) i~ filtered off. The acylyuanidi~es
obtained in this way can be converted to the
¦30 corresponding salts by treatment with aqueou~ or metha-
¦nolic hydrochloric acid or other pharmacologically
compatible acids.
13 2~2~n1~ ~
Example 1:
4-Chloro-3-(N-propylcarbamoylsulfamoyl)benzoylguanidine
hydrochloride: colorless crystals, m.p. 205 - 207~C
~ 1
tl H
N~l~N~5~Nq~
o 2 0 NH2
Synthesi~ route:
a)Methyl4-chloro-3-tN-propylcarbamoylsulfamoyl)benzoate
Addition of methyl 4-chloro-3-sulfamoylbenzoate to
propyl isocyanate with 2 equivalents of potassium
carbonate in acetone (reflux, 1 hour), acidification
with HCl and filtration of the precipitate, colorless
crystals, m.p. 185C. :~
b) 4-Chloro-3-(N-propylcarbamoylsulfamoyl)benzoylguani- : :
dine hydrochloride
from a) according to general instructions.
Example 2:
15 4-Chloro-3-(N-ethylthiocarbamoylsulfamoyl)benzoylguani- -:
dine hydrochloride: colorless crystals, m.p. 175C
~ I
Eth ~ N~ ~ ~ N~2
Synthesi~ route:
a) Methyl 4 chloro-3-(N-ethylthiocarbamoylsul~amoyl)-
benzoate
'~,, ~ ~, , " . . ' - ' '
- 14 - 2~2~
Addition of methyl 4-chloro-3-~ulfamoylbenzoate to
ethyl isothiocyanate with 2 equivalents of potassium
carbonate in acetone (reflux, 2.5 hours), aci~ifica-
tion with ~C1 and filtration of the precipitate, m.p.
175C.
b) 4-Chloro-3-(N-ethylthiocarbamoylsulfamoyl)benzoyl-
guanidine hydrochloride
from a) according to general in~tructionsO
Example 3:
3-(N-Cyclohexylcarbamoyl6ulfamoyl)-4-phenoxybenzoylgua-
nidine hydrochloride: colorless crystals, m~p. 231C
P h 0~
o, N~ ; =Nq~ N H 2
Synthesi~ route~
a) Methyl 4-phenoxy-3-sulamoylbenzoate
Phenol and pota6sium hydroxide are boiled until khe
mixture i~ homogeneoue, after which methyl 4-fluoro-
3- ulfamoylb~nzoate i added. After 45 min at 130~C,
the reaction ~olution i~ poured into water/ ice and
the preaipitate i~ filtered off. m.p. 145C.
b) Methyl 3-(N-cyclohexylcarbamoyl~ulfamoyl)-4-phenoxy-
benzoate
:
~rom a) with cyclohexyl i~ocyanate analogously to
¦ Example la), colorles~ cry~tals, m.p. 1~2C.
. c) 4-Ph2noxy-3-(N-cyclohexylcarbamoylsul~amoyl)benzoyl-
guanidine hydrochloride
'i :
~,~f,,~f, ,. i ~ ~,, , ", . - ,,
- 15
from b) according to general inst~uctions.
Example 4:
4-Isopropyl-3-tN cyclohexylcarbamoylsulfamoyl)benzoyl-
guanidine hydrochloride: colorless crystals, m.p. 232 -
236C
H H ~
~ N ~ N`S ~ N ~ NH2
Synthesis route:
a) 4~Isopropyl-3-sulfamoylbenzoic acid
from 3-chlorosul~onyl-4-isopropylbenzoic acid with
ammonia (20 hours, room temperature), acidification
with HCl, extraction with ethyl acetate and di~til~
lation of the solvent, colorle~s crystals, m.p. 231~C.
b) Ethyl 4-isopropyl-3-sulfamoylbenzoate
from a) with ~Cl/ethanol solution, distillation of the
~olvent and crystallization of the re~idue from
diethyl ether/petroleum ether, colorless crystals,
m.p. 110 - 113C.
c) Ethyl 3-(N-cyclohexylcarbamoyl~ulfamoyl)-4-i60pro
pylbenzoate
from b) with cyclohexyl i~ocyanate analogously ~o la),
colorless oil.
d) 3-(N-Cyclohexylcarbamoylsulfamoyl)-4-isopropylben-
zoylguanidine hydrochloride
according to general instructions.
'.I
:,
~, ~ .~ . : . .
- 16 - ~ ~?~ D~
Example 5:
3-(N-Cycloh2xylcarbamoyl~ulfamoyl)-4-(2'-mercaptopyri-
dinyl)benzoylguanidine hydrochloride:
H ~ S
~ N ~ N`S ~ NH~
colorle~s crystals, m.p. 152 - 155C
Synthesis route:
a) Ethyl 3-sulfamoyl-4-(2~-mercaptopyridinyl)benzoate
from ethyl 4-fluoro-3-~ulfamoylbenzoate with 1 equi-
valent of 2-mercaptopyridine and 3 equivalent~ of
potassium carbonate in DMF (reflux, 6 hours), treat~
ment with water, acidification with ~Cl, extraction
with ethyl acetate, distillation of the solvent and
column chromatography on silica gel with ethyl ace-
tate/methanol (9:1), brownish cry~tals, m.p. 94 -
95C.
b) Ethyl 3-(N-cyclohexylcarbamoylsulfamoyl)-4-~2' mer-
captopyridinyl)benzoate
from a) with cyclohexyl i~ocyanate analogously to la),
colorle~s cry tals, m.p. 145 - 150C,
c) 3-(N-Cyclohexylcarbamoyl6ulfamoyl)-4-(2'-mercapto~
pyridinyl)benzoylguanidine hydrochloride
from b) according to general in~tructionR.
i *:::?
2~12~ A~
-- 17 --
Example 6:
2,4-Dichloro-5-(N-phenylcarbamoylsul~amoyl)benzoylgua-
nidine hydrochloride: colorle~s cry~tals, m.p. 215 -
218JC
H H ~C I
P h ~ O~Nq~N H 2
o 2 0 NH2
Synthesis route:
a) Methyl 2,4-dichloro-5-(N-phenylcarbamoyl~ulfamoyl)~
benzoate
from methyl 2,4-dichloro-5-sulfamoylbenzoate and ::
phenyl isocyanate analogously tu la~, colorle~s :
crystals, m.p. 187 - 189C.
b) 2,4-Dichloro-5-(N-phenylcarbamoylsul~amoyl)benzoyl :~
guanidine hydrochloride
from a) according to general instruction I (~ee ;~
above).
Example 7:
5-(N~Cyclohexylcarbamoylsulfamoyl)-2,4-diohlorobenzoyl
guanidine hydrochloride: colorle~ cry~tal~, m.p. 211
215C
C I ~C l
O~N~N`S~,Nq~H NH2
2~ ~2 ~
- 18 -
Synthesis route:
a) Methyl 5-(N-cyclohexylcarbamoyl~ulfamoyl)-2,4-dichlo-
robenzoate
from methyl 2,4-dichloro-5-sulfamoylbenzoate with
cyclohexyl i~ocyanate analogously to la), colorle6s
crystals, m.p. 211 - 214C.
b) 5-(N-Cyclohexylcarbamoylsulfamoyl)~2,A-dichloroben-
zoylguanidine hydrochloride ~ -
from a) according to general in~tructions.
Example 8~
4-Chloro-3-[~-morpholinocarbo~yl) 6ul famoyl]benzoylgua- -
nidine hydrochloride. colorless crystals, m.p. 155C :~.
H ~Nq~ N H 2
2 o NH2
Synthesis route:
a) Methyl 4-chloro-3-[~N-morpholinocarbonyl)sulfamoyl]-
benzoate
from mekhyl 4-chloro-3-~ulfamoylbenzoate with 1.5 ~ -
equivalents of morpholine-N-carboxylic acid chloride
and 2 equivalents o~ potassium carbonate (reflux, 2
hour~), treatment with water, acidification with HCl,
extraction with ethyl acetate and distillation
colorless cry~tal~, m.p. 162C.
b) 4-Chloro-3-[(N-morpholinocarbonyl)eulfamoyl]benzoyl-
guanidine hydrochloride
r . " ,, , ~
,~",,,,, ., . , , , : " - : , ,~
,~ ., . :
, . . , ~,, , . ,. ,. ": ~ , ~ .
~ ~ ~ ?~ }
- 19 -
from a) according to g0neral instructions.
Example 9:
3-[N-(N'-Butyl-N''-ethylguanidino)~ulfonyl]benzoylguani-
dine hydrochloride- amorphou~
H H ~ - -
E t N~N~S~N~,N ~ 2
N 2 0 NH2
n Bu
Synthe~is route:
a) ~ethyl 3-[(N-ethylthiocarbamoyl) ulfamoyl]benzoate :
from methyl 3-sulfamoylbenzoate and ethyl isothio~
cyanate analogously to 2a), colorless cry~tals, m.p
126 - 128 C.
10 b) Methyl [(N-ethyl-S-methylthioguanidino)sulfonyl]ben- : ;
zoate
from a) with 4 equivalents of methyl iodide in
methanol (reflux, 24 hour~) ~ di~tillation of the
solvent, taking-up of the reeidue with ethyl acetate,
washing with ~odium carbonate, di~tillation and
crystallization of the re~idue from diisopropyl ether,
colorless crystals, m.p. 178 - 181C.
c) Methyl 3-[N-(N'-butyl-N''-ethylgua~idino)~ulfonyl]-
benzoate
from b) with n-butylamine (0C, 2.5 hour~), di til-
lation of the n-butylamine and crystallization of the
re~idue from diisopropyl e~her/ethyl aceka~e, m.p. 8G
- 85C .
.$,", J "
,,7j~r . i: i, ' , ~ :, ,:, , , ' , ' . . . .
1 9 ~
- 2~ -
d) 3-[N-(N'-Butyl-N''-ethylguanidino)~ulfonyl]benzoyl-
guanidine hydrochloride
from c) according to general instructions.
: ::
Ex~mple 10: :
5 3-[N-(N'-Butyl-N''-ethylguanidino)6ul~onyl]-4-chloroben- ~ :
zoylguanidine hydrochloride: colorless cry8tal8 ~ m.p.
212C
C I ~,:
H H
E ~ N~N~S,~ Nq~N H
N 2 0 NH2
\n B u
, ~
Synthesis route: ;
a) Methyl 4-chloro-3-[(N-ethyl-S methylthioguanidino-
sulfonyl]benzoate : :;~
from 2a) with methyl iodide analogou~ly to 9b),
colorlese cry~tal~, mOp. 175C.
b) Methyl 3- r N-(N'-butyl-N''-ethylguanidino)sulfonyl~-4-
chlorobenzoate
15from a) with n-butylamine analogou~ly to 9c), color-
le~s crystal~, m.p. 82C.
1 c) 3-[N-(N~-~utyl-N~ ethylguanidino)~ulfonyl]-4-chloro-
benzoylguanidine hydrochloride
¦ from b) according to general in~truction~.
.- ~, ~ " . , ,, , , ., , , : ~
- 21 ~ 2 ~ l2
Example 11:
4-Chloro-3-[N-(N'-ethyl-N''-amidino)sulfonylguanidino]-
benzoylguanidine hydrochloride
Cl
H H ~Nq~N H 2
N~ N H o N H2
N H
colorless crystal~, m.pt 233 - 235C, from lOa) with 10
equivalents of guanidine according to general instruc-
tions.
Pharmacological data:
Inhibitors of the Na+t~ exchanger of rabbit erythrocyte~
New Zealand white rabbits (Ivanovas) received a standard
diet with 2~ of chole~terol for ~ix week~ to activate the
Na+/H+ exchange, thereby making it pos~ible to determinet
by flame photometry, the Na~ influx into the erythrocyte
vla Na+/H+ exchange. The blood wa~ t~ken from the auri-
cular arteries and rendered incoagulable with 25 IU/ml of
heparin potas~ium. Part of each sample was uRed for
double determination of the hematocrit by centrifugation.
100 ~l measurement of the initial Na~ content of the
erythrocyte~. For determination of the amiloride-
~ensitive sodium influx, 100 ~l of each blood ~ample were
incubated in 5 ml of a hypero~molar salt/sucrose medium
(mmol/l: 140 NaCl, 3 KCll 150 sucro~e, 0.1 ouabain, 20
tris(hydroxymethyl)aminomethane) at pH 7.4 and 37C. The
erythrocytes were then wa~hed thxee times with ic~-o~ld
MgCl2/ouabain solution (mmol/l: 112 MgCl2, 0.1 ouabain)
- 22 - '~ L ~
and hemolyzed in 2.0 ml of distilled water. The
intracellular sodium ~ontent was determined by flame
photometry.
The net Na' influx was calculated from the difference
between initial sodium value6 and the sodium content of
the erythrocytes after incubation. The sodium influx
which can be inhibited by amiloride was obtained ~rom the
dif~erence in the sodium content of the erythrocytes
after incubation with and without 3 x 10-4 mol/l of
amiloride. This procedure was also adopted for ~he
compounds according to the invention.
Results
Inhibition of the Na+/H~ exchanger: -
i
Example IC50 (mol/l)
2 1 x 10-5
_
6 2 x 10-5
7 2xlO-s
r,.~?. ~. . . , .: ~ . :
~,,~',:: ...... ,-: ' , :