Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 93/12070 2 1 2 li 1 9 8 PCI/~IIU92/0005~3
NE'W TETRAHYDRONAPHTHALENE DE~IVATIVES
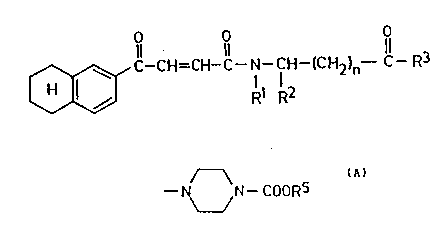
The invention relates to new tetrahydro-
naphthalene derivatives of general formula (I)
O O
Il 11 11
o ~C--CH=CH-C--N--CH--ICH2)n--C--R3
wherein
Rl represents a hydrogen atom,
R2 represents a hydrogen atom, a C1_4
alkoxycarbonylalkyl or benzyl group, or
R1 and R2 together represent a group of formula
- (CH2)3-~
R3 represents a hydroxy, C1_4 alkoxy or benzyl-
oxy group or a group of general formula (A),
~
--1~ N--COORS
/ (A
wherein
RS represents a Cl_4 alkyl group, and
n represents 0, 1, 2, 3 or 4,
and their pharmaceutically acceptable salts, further-
more to a process for preparing the same.
Compounds of general formula (I) may be in
SUBSTITUTE S~tEET
W093/l2070 212 6 19~ 2 - PCT/HU92/0~58
(E) configuration.
In the C1_4 alkyl or C1_4 alkoxy groups
the alkyl groups can be linear or branched saturated
hydrocarbon groups, i.e. methyl, ethyl, n- or isopro-
pyl, furthermore n-, sec- or t-butyl groups.
The new compounds of general formula ~I)
proved to be biologically active, they exerted
significant antiulcer, i.e. cytoprotective effect.
The invention relates furthermore to pharma-
ceutical compositions containing new tetrahydro-
naphthalene derivatives of general formula (I) -
wherein Rl, R2, R3 and n have the same meaning as
above - and their pharmaceutically acceptable salts,
furthermore to a process for preparing the same.
lS The therapeutic effect of the compounds of
general formula ~I) was studied in the following
tests.
Cytoprotective effect in the acidic ~lcohol i~uced
g~tric ulcer mo~el
(A. Robert: Gastroenterology, 77, 761-767, 1~79)
The study was performed in female rats
weighing 120 - lS0 g and starved for 24 hours. The
test compound was administered in a suspension
prepared with Tween 80, by gavage. After 30 minutes
acidic alcohol, in doses of 0.5 ml/lO0 g body weight,
was added similarly, by gavage. After one hour tbe
animals were Xilled, their stomach was removed and cut
along the curve. The reddish-brown strips ~hemorrhagic
lesions) were measured and their mean length pro
stomach was calculated. The protective effect of the
test compound was determined compared to the control
group. The effect of the compound described in Example
1: ED50- o.l mg/kg p.o.
SUBSTlTUrE SttEET
WO93/12070 2 1~ 8 PCT/HU92/00058
Cytoprotoctive effact in the acetic acid induced
chrouic gastric ulcer ~odQl
(Tagaki et al: Japanese Journal of Pharmacology 19,
418-426, 1969)
The study was performed in female rats
starved for 24 hours. The abdominal wall was opened
under ether a nesthesis, and 25 ~l of 20 % acetic
acid solution was injected in the subserous layer of
the glandular part of the stomach, near the pylorus.
Then the abdominal wall was closed and the animals
received food and water ad libitum; Treatment was
started on the 5th postoperative day and continued
with single daily doses for 10 days. The animals were
killed on the 15th postoperative day and their stomach
was removed. Evaluation was performed by measuring the
diameter of the necrotic areas and calculating their
surface. The therapeutic effect of the test compound
was calculated according to the following formula
0 ulcer surface ~control~ - ulcer surface (test compound)
ulcer surface (control)
The therapeutic effect of the compound of Example 1
amounted to 39 % at doses of 10 mg/kg p.o.
The pharmacological studies confirmed that the
compounds of the present invention as well as their
pharmaceutically acceptable salts are potent antiulcer
agents.
The invention also relates to a process for
preparing new tetràhydronaphthalene derivatives of
general formula (I) - wherein R1, R2, R3 and n have
the same meaning as above - w h i c h c o m p r i s -
e s - reacting an activated derivative of 4-oxo-4-
SUBSrlTUTE SHEET
W093~12070 PCT/HU92/00058
~61S8 - 4 -
(5,6,7,8-tetrahydro-2-napthyl)-2(E)- butenoic acid of
qeneral formula (II),
o
~C- CH=CH--COOH
with an amino acid derivative of general formula
(III),
NH--CH--ICH2)n--C--R4
Rl R2 ( ~
wherein R1 and R2 have the same meaning as above, and
R4 represents a Cl_4 alkoxy or benzyloxy group or a
group of formula A - whercin R5 represents a Cl_
alkyl group - , and
if desired, submitting to acidolysis the
compounds of general formula (I) - wherein Rl and R2
have the same meaning as above and R3 sta~ds for a t-
butyloxy group - andJor
if desired, reacting an activated derivative
of a compound of general formula (I) - wherein R1 and
R~ ha~e the same meaning as above and R3 stands for a
hydroxy group - with a piperazine derivative contain-
SV~E 5~EF~
WO93/12070 ~ 12 6 1 s ~ PCTtHU92/00058
ing a group of formula A - wherein R5 has the same
meaning as above - or are converted with an inorganic
or organic base to a salt.
From the starting materials applied in the
process the compounds of general formula (III) are
commercial products.
The 4-oxo-4-(5,6,7,8-tetrahydro-2-naphthyl-
2(E)-butenoic acid of general formula (II) can be
prepared from l,2,3,4-tetrahydronaphthalene and maleic
anhydride in a Friedel-Krafts reaction according to D.
Papa et al: J. Am. Chem. Soc.: 70, 3356, 1948.
According to a preferred embodiment of the
present invention the amide formation between the
compounds of general formulas (II) and (III) is
performed by activating in situ first 4-oxo-4-
~5,6,7,8-tetrahydro-2-naphthyl)-2(E)-butenoic a~id of
formula (II), and reacting it in an inert organic
solvent with the amine component of the formula (III).
The carboxy group of the compound of formula (II) can
be activated by not less than stoichio~etric amounts
of a carbodiimide type reagent, i. e. dicyclo~exyl-
carbodiimide, in an inert organic solvent, preferably
in a halogenated aliphatic hydrocarbon, advantageously
in dichloromethane, at a preferred temperature range
of 0C to 20C. An other preferred type of activation
is the mixed anhydride method, wherein a chloroformic
acid ester, preferably ethyl chloroformate is applied.
The reaction is performed in an inert organic solvent,
i. e. in a cyclic ether, preferably in tetrahydrofu-
ran, in the presence of stoichiometric amounts of a t-
amine base, preferably in triethylamine, at a
preferred temperature ran~e of -20QC to 0C. In the
case when a compound of general formula tI)- wherein
Rl and R2 have the same meaning as above and R3 is a
hydroxy group - is reacted with a piperazine
SUBSTITUTE SHEET
WO93/12070 PCTtHU92/00058
2 1 2 61-qg - 6 -
deriv ~ ~v~ acid is activated by either of theabove methods, i. e. either by the mixed anhydride or
the carbodiimide method.
If desired, the compounds of general formula
S (I) - wherein Rl and R2 have the same meaning as above
and R3 stands for a hydroxy group - can be converted
by known methods with an organic or inorganic base
into an alkali metal, alkali earth metal or zinc salt.
The compounds of general formula (I) - wherein
Rl, R2 and R3 have the same meaning as above - prepar-
ed by the process of the invention, can be isolated by
known methods, i. e. filtration, and purified by re-
crystallization.
The compounds of the invention can be
converted into pharmaceutical formulations suitable for
parenteral or enteral administration by mixing them
with non-toxic, inert solid or liquid carriers and/or
additives. Water, gelatin, lactose, starch, pectin,
magnesium stearate, stearic acid, talc, and plant
oils, such as peanut oil, olive oil, etc., can be used
as carriers. The active ingredient can be formulated
as is usual in pharmaceutical compositions, in solid
forms, i.e. round or rectangular tablets, coated
tablets, capsules, i. e. gelatine capsules, pills, or
suppositories.
The compositions can contain, if required, the
usual auxiliary materials, such a~ preservatives,
stabilizers, wetting agents, emulsifying agents, etc.
They can be prepared by usual methods, so in the case
of solid formulations, by sieving, mixing, granulating
and pressing. The formulations can be submitted to the
usual pharmaceutical technological procedures, i. e.
sterilization.
The following examples are illustrating but
3~ not limiting the scope of the invention.
~UBSTITUTE SHEET
W093/l~070 PCT/HU92/00058
_ 7 _ 212~1~8
Exam~le 1
4-Oxo-4-(5,6,7,8-tetrahydro-2-naphthyl)-2(E)-butenoyl-
L-proline ethyl ester
4.6 g (0.02 mole) of 4-oxo-4-(5,6,7,8-tetra-
hydro-2-naphthyl-2(E)-butenoic acid are dissolved in
100 ml of anhydrous dichloromethane, then 5.6 ml (0.04
mole) of triethylamine are added. The solution is
cooled to -15~C, thereafter 2.01 ml (0.022 mole) of
ethyl chloroformate, dissolved in 10 ml of anhydrous
dichloromethane, then at -15C 4.0 g (0.022 mole) of
L-proline ethyl ester hydrochloride, dissolved in 10
ml of anhydrous dichloromethane are added. The
reaction mixture is stirred first at -15C for 30
minutes, then at room temperature for 2 hours, and
the mixture is extracted with the following solutions
in the order listed: 1 N hydrochloric acid, water, 5 %
sodium carbonate and saturated sodium chloride solu- -~
tion. The organic layer is dried over sodium sulfate
and evaporated. The evaporation residue is
crystallized from a mixture of cyclohexane-petroleum
ether (1:1).
Yield: 5.03 g ~70 %)
M.p.: 63-66C
[~]D25: -78.8C (c=0.5, chloroform)
Exam~le 2
N-~4-Oxo-4-(5,6,7,8-tetrahydro-2-naphthyl)-2(E)-
-butenoyl]-L-proline
7.66 g t0.02 mole) of 4-oxo-4-~5,6,7,8-
tetrahydro-2-naphthyl)-2(E)-butenoyl-L-proline t-
butyl ester are stirred in 200 ml of 2.4 N hydro-
chloric acid solution in dioxane for 24 hours, then
the mixture is evaporated. The evaporation residue
is dissolved in 200 ml of dichloromethane and the
SUBSTrTUTE SHEET
WO93/1~070 2 1 ~ 61 3 ~ - 8 - PCr/HU92/00058
solution is extracted with water and saturated sodium
chloride solution. The organic layer is dried over
anhydrous magnesium sulfate and the solvent is evapo-
rate~, to give an oil.
Yield: 6.50 g (g9 %).
ExamPle 3
N-t4-Oxo-4-~5,6,7,8-tetrahydro-2-naphthyl~
2(E)-butenoyl]-L-proline zinc salt tetrahydrate
6.5 g of N-[4-oxo-4-(5,6,7,8-tetrahydro-2-
naphthyl)-2(E)-butenoyl]-L-proline, prepared according
to the process of Example 2, are dissolved in a mix-
ture of 10 ml of water and 2.78 ml (0.02 mole~ of tri-
ethylamine, then 1.36 g of zinc chloride in 10 ml of
water are added. The precipitated compound is filter-
ed, washed with ice-water, ethanol and diethyl ether.
The product has light yellow colour.
Yield: 4.9 g (62 %)
M.p.: 122-124C
I~D25 -82.4 (c-1, methanol).
Example 4
N-t4-Oxo-4-(5,6,7,8-tetrahydro-2-naphthyl)-2(E)-
butenoyl]-D-aspartic acid diethyl ester
4.6 g (0.02 mole) of 4-oxo-4-(5,6,7,8-
-~etrahydro-2-naphthyl-2(E)-butenoic acid are dis-
solved in 50 ml of anhydrous tetrahydrofuran, 5~6
ml (0.04 mo~e) of triethylamine are added and the
solution is cooled to -10C. To this solution are
added slowly 2.01 ml (22 mmole) of ethyl chloroformate
in 20 ml of anhydrous tetrahydrofuran. Thereafter,
maintaining the temperature of the reaction mixture at
-10C, 4.5 g (0.02 mole) of D-aspartic acid diethyl
esther are added in 20 ml of anhydrous dichloromethane
solution. The reaction mixture is stirred for 30
SlJBSTlTUTE SHEET
WO93/12070 2 ~ ?. ~ PCT/HU92~00058
_ g _
minutes at 10C, sub~equently for one hour at room
temperature, then it is poured on 200 ml of water. The
mixture is extracted with dichloromethane. The organic
layer is repeatedly extracted with 5 % sodium carbona-
5 te and saturated sodium chloride solutions. The organic
layer is dried over anhydrous magnesium sulfate and the
solvent is evaporated. Light yellow crystals are formed
from the evaporation residue after adding petroleum
ether.
Yield: 5.6 g (70 %)
M.p.: 95-97C
t~]D25: -0.33O (~=0.5, chloroform).
Example 5
1-tN-/4-Oxo-4-(5,6~7,8-tetrahydro-2-naphthyl)-2(E)-
butenoyl/-L-prolyl]-4-ethoxycarbonyl-piperazine
4.24 g (18.4 mmole) of 4-oxo-4-(5,6,7,8-
tetrahydro-2-naphthyl)-2(E)-butenoic acid are
dissolved in 80 ml of anhydrous tetrahydrofuran, 2.57
ml (18.4 mmole) of triethylamine are added, then 1.76 --
ml (18.4 mmole) of ethyl chloroformate in 10 ml of
anhydrous tetrahydrofuran. To this mixture are added
4.7 g of 1-L-prolyl-4-ethoxycarbonylpiperazine -
prepared according to paragraph b) of the production
process of starting materials - i~ 30 ml of anhydrous
tetrahydrofuran. The reaction mixture is stirred at -
15~C, then at -10C for 30 minutes and at room
temperature for one hour. Subsequently the mixture is
poured on 400 ml of water and extra ted with dichloro-
methane. The organic layer is extracted first with
5 % sodium carbonate solution, then with water, finally
with saturated sodium chloride solution. The organic
layer is dried over anhydrous magnesium sulfate and
the solvent is evaporated. The residual oil is
~5 recrystallized from a mixture of diethyl ether and
SUBSTITUTE SHEET
W093/12070 PCT~HU92/00058
~1261.38 - lo-
isopropanol.
Yield: 4.87 g (57 %)
M . P .: 110-112 C
t~]D25: +9.4O (c=1, chloroform).
ExamPle 6
N-~4-Oxo-4-(5,6,7,8-tetrahydro-2-naphthyl)-2(E)-
butenoyl]-~-proline benzyl ester
14.5 g (0.06 mole) of L-proline benzyl ester
hydrochloride and 8.3 g (0.06 mole) of potassium
carbonate are dissolved in a mixture of 100 ml of
water and 100 ml of dichloromethane. The organic layer
is dried and the solvent is evaporated. The residual
oil is directly processed in the next step.
13.8 g (0.06 mole) of 4-oxo-4 (5,6,7,8-
tetrahydro-2-naphthyl)-2(E)butenoic acid are
dissolved in 150 ml of anhydrous tetrahydrofuran and
the solution is cooled to -15C. Then 8.4 ml (0.06
mole) of triethylamine and subsequently 6.1 ml (66
mmole~ of ethyl chloroformate in 20 ml of anhydrous
tetrahydrofuran are added. Thereafter the solution of
L-proline benzyl ester base in 20 ml of anhydrous
tetrahydrofuran, obtained in the previous step, is
added. The reaction mixture is stirred for 30 minutes
at -15C, for a further hour at room temperature, then
it is pou~ed on 400 ml of water and extracted with
~ichloromethane. The organic layer is washed first
with 5 % sodium carbonate solution, then with water
and finally with a saturated sodium chloride solution.
The organic layer is dried over anhydrous magnesium
sulfate and the solvent is evaporated. The residue is
crystallized from petroleum ether and recrystallized
from a mixture of acetone-n-hexan.
Yield: 12.0 g (4~ %).
M.p.: 95-96C.
SUBSrlTUTE SHEET
WO93/12070 2 12 ~ 1 3 8 PCT/HU92/00058
~]D25: -411 (c-1, chloroform) -
~xam~le 7
N-~4-Oxo-4-(5,6,7,8-tetrahydro-2-naphthyl)-2(E)-
butenoyl]-B-alanine ethyl ester
2.3 g (0.01 mole) of 4-oxo-4-(5,6,7,8-
tetrahydro-2-naphthyl)-2(E)-butenoic acid and 1.53 g
(0.01 mole) of B-alanine ethyl ester hydrochloride are
suspended in 50 ml of anhydrous dichloromethane and
cooled to 0OC. Then 1.01 g t0.01 mole) of N-methyl-
morpholine in 20 ml of anhydrous dichloromethane and
finally 2.06 g (0.01 mole3 of dicyclohexylcarbodiimide
are added to the suspension. The reaction mixture is
stirred for one hour at 0C and for one hour at room
temperature.
The precipitated dicyclohexylurea is
filtered, and the filtrate is extracted with the
following solvents in the order listed: 1 ~
hydrochloric acid, water, saturated sodium carbonate
solution and finally sodium chloride solution. The
organic layer is dried over anhydrous magnesium
sulfate and the solvent is evaporated. The residue
crystallizes upon the addition of diethyl ether.
Yi~ld: 1.3 g (41 %).
M.p.: 112-114C.
Example 8
N-t4-Oxo-4-(5,6,7,~-tetrahydro-2-naphthyl)-2(E)-
butenoyl]-6-aminohexanoic acid ethyl ester
2.3 g (0.01 mole) of 4-oxo-4-(5,6,7,8-
- -tetrahydro-2-naphthyl)-2-(E)-butenoic acid and 1.95 g
(1.01 mole) of 6-aminohexanoic acid ethyl ester are
suspended in 60 ml of anhydrous dichloromethane and
cooled to 0C. To this mixture first a solution of
1.01 g (0.01 mole) of N-methylmorpholine in 20 ml ~f
SUBSTrrUTE SHEET
W093/12070 PCT/HU92/00058
2 1 ~ 8 - 12 -
anhydrous dichloromethane, then 2.06 g (0.01 mole) of
dicyclohexylcarbodiimide are added. The reaction
mixture is stirred for one hour at 0C and for one
hour at room temperature. The precipitated di-
cyclohexylurea is filtered and the filtrate is
extracted with the following solutions in the order
listed: 1 N hydrochloric acid, water, saturated sodium
carbonate solution and finally saturated sodium
chloride solution. The organic layer is dried over
anhydrous magnesium sulfate and the solvent is
evaporated. The residue is crystallized from a mixture
of diethyl ether and n-hexane.
Yield~ 8 g (40 %).
M.p.: 74-76OC.
Example 9
N-[4-Oxo-4-(5,6,7,8-tetrahydro-2-naphthyl)-2( F ) -
butenoyl3-L-phenylalanine methyl ester
4.3 g (0.02 mole) of L-phenylalanine methyl
ester hydrochloride and 4.6 g (0.02 mole) of 4-oxo-4~
(5,6,7,8-tetrahydro-2-naphthyl)-2(E)-butenoic acid are
suspended in 80 ml of anhydrous dichloromethane and
cooled to 0C. To this mixture a solution of 2.03 g
(0.02 mole) of N-methylmorpholine in 30 ml of an-
hydrous dichloromethane, then 4.12 g (0.02 mole) of
dicyclo-hexylcarbodiimide are added. The reaction
mixture is stirred for one hour at 0C and f or one
hour at room temperature. The precipitated dicyclo-
hexylurea is filtered and the filtrate is extracted
with the following solutions in the order listed: 1 N
hydrochloric acid, water, saturated sodium carbonate
solution and finally saturated sodium chloride
solution.
The organic layer is dried over anhydrous
magnesium sulfate and the solvent is evaporated. The
SU8STITVTE 5HEET
W093/l2070 - 13 - 2 12 6 1~ ~
evaporation residue is crystallized from diethyl
ether, and recrystallized from a mixture of ethyl
acetate and diethyl ether.
Yield: 3.7 g (47 %).
M.p.: 145-147C.
[~]D25: -22.5 (c=1, methanol).
Example 10
1-[N-/4-Oxo-4-~5,6,7,8-tetrahydro-2-naphthyl)-2(E)-
butenoyl/-L-prolyl~-4-ethoxycarbonylpiperazine
6.5 g of the oily N-[4-oxo-4-(5,6,7,8-
tetrahydro-2-naphthyl)-2(E)-butenoyl]-L-proline -
prepared according to the process described in Example
2 - are dissolved in 150 ml of anhydrous dichloro- -
methane, cooled to OoC, then 3.16 g (0.02 mole) of 1-
ethoxycarbonylpiperazine and catalytic amounts of 4-
dimethylaminopyridine are added. To this mixture a
solution of 4.54 q (22 mmole) of dicyclohexylcarbo-
diimide in 20 ml of anhydrous dichloromethane are
added under constant stirring. The reaction mixture is
stirred for 24 hours at room temperature. The precipi-
tated dicyclohexylurea is filtered and washed with
with dichloromethane. The organic layer is extracted
with the following solu~ions in the order listed: l N
hydrochloric acid, water, 10 ~ sodium carbonate
solution, water, and finally saturated ~odiu~ chloride
solution. The organic layer is dried over anhydrous
magnesium sulfate, filtered and the solvent is
evaporated. The resulting oil is recrystallized from a
3iO mixture of diethyl ether and isopropanol.
Yield: 5.1 g (55 ~).
M.p.: 110-112C.
~]D25 ~9.2 (c-l, chloroform).
Pre~aration of startina materials
SUE~STIlUTE SHEET
W093/l2070 PCT/HU92/0~58
2 1 ? 6 1 9 8
a.) N-~4-Oxo-4-~S,6,7,8-tetrahydro-2-naphthyl)-2(E)-
-butenoyl]-L-proline t-butyl ester
5.23 g (0.02 mole) of L-proline t-butyl
ester oxalate are suspended in 50 ml of dichlorome-
thane, and the mixture is extracted with a solution of2.76 g (0.02 mole) of potassium carbonate in S0 ml of
water up to the point the solution gets clear. The
organic layer is separated, dried over anhydrous
sodium sulfate, and evaporated.
Yield: 3.7 g of L-proline t-butyl ester.
4.6 g (0.02 mole) of 4-oxo-4-(5,6,7,8-tetra-
hydro-2-naphthyl)-2(E)-butenoic acid are dissolved in
50 ml of anhydrous tetrahydrofuran and 2.8 ml (0.02
lS mole) of triethylamine are added. The reaction mixture
is cooled to -15C, then a solution of 2.01 ml (22
mmole) ethyl chloroformate in 20 ml of anhydrous
tetrahydrofuran are added, 2nd thereafter the above
prepared solution of 3.7 g (22 mmoie) of L-proline t- -
butyl ester in 20 ml of anhydrous tetrahydrofuran. The
reaction mixture is stirred for 30 minutes at -15C,
for 30 minutes at room temperature, then it is poured
on 200 ml of water and extracted with dichloromethane.
The organic layer is extracted with the followlng
solutions in the order listed: 5 % sodium carbonate
solution, saturated sodium chloride solution and
finally water. The organic layer is dried over
anhydrous magnesium suIfate and the solvent is
evaporated. The residue crystallizes upon the addition
of petroleum ether.
Yield: 3.89 g t51 %).
M.p.: 9s-98C.
[~D25: -81.2 (c=0.5, chloroform).
b) 1-(N-8enzyloxycarbonyl-L-prolyl)-4-ethoxycarbonyl-
SUBSrlTUTE SHEET
WO93/12070 - 15 - ~ 1 2 61 ~ 8
pipera z ine
12.45 g (0.05 mole) of N-benzyloxycarbonyl-
L-proline and 7.91 g (O.05 mole) of 1 ethoxycarbonyl-
piperazine are dissolved in 150 ml of anhydrous
dichloromethane, cooled to +5C, then a solution of
11.35 g (55 mmole) of dicyclohexylcarbodiimide in 50
ml of anhydrous dichloromethane are added. The mixture
is stirred for 24 hours at room temperature, the
precipitated dicyclohexylurea is filtered and the
filtrate is extracted with the following solutions in
the order listed: 1 N hydrochloric acid, water, lO %
sodium carbonate solution, and finally water. The
organic layer is dried over anhydrous magnesium
sulfate, and the solvent is evaporated. The product
starts to crystallize upon refrigerating. The white
crystals are filtered and washed with cool diethyl
ether.
Yield: 14.0 g (72 %).
M.p.: 90.5-92C.
t~]D30: -11.4 (c=1, methanol).
c) l-L-Prolyl-4-ethoxycarbonylpiperazine
7.4 g (19 mmole) of l-(N-benzyloxycarbonyl-
L-prolyl)-4-ethoxycarbonylpiperazine - prepared above
- are dissolved in 150 ml of anhydrous methanol and
the solution is submitted to hydrogenation for 2 hours
in the presence of a Pd-C catalyst. The solution is
evaporated after the removal of the catalyst.
Yield: 4.7 g (96.~ %).
SUBSTITUTE SltEET