Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 95/24389 PCTIUS9L1/02573
- 21~41 14
INDOLE DERIVATIVES AS CCK RECEPTOR ANTAGONISTS
~AAAAAA~AAAAAA~AAAAAA~AAAAAAAAAAAAAAAAAAAAAAAA
BACKGROUND OF THE INVENTION
Agents acting at central cholecystokinin (CCK)
receptors may induce satiety (Schick, Yaksh, and Go,
Regulatory Peptides 14:277-291, 1986). They are also
expected to act as analgesics (Hill, Hughes, and
Pittaway, Neuropharmacology 26:289-300, 1987), and as
anticonvulsants (MacVicar, Kerrin, and Davison, Brain
Research 406:130-135, 1987).
Reduced levels of CCK-peptides have been found in
the brains of schizophrenic patients compared with
controls (Roberts, Ferrier, Lee, Crow, Johnstone,
Owens, Bacarese-Hamilton, McGregor, O'Shaughnessey,
Polak, and Bloom, Brain Research 288:199-211, 1983).
It has been proposed that changes in the activity of
CCK neurones projecting to the nucleus accumbens may
play a role in schizophrenic processes by influencing
dopaminergic function (Totterdell and Smith,
Neuroscience 19:181-192, 1986). This is consistent
with numerous reports that CCK peptides modulate
dopaminergic function in the basal ganglia and
particularly the nucleus accumbens (Weiss, Tanzer, and
Ettenberg, Pharmacology, Biochemistry and Behaviour
30:309-317, 1988; Schneider, Allpert, and Iversen,
Peptides 4:749-753, 1983). It may therefore be
expected that agents modifying CCK receptor activity
may have therapeutic value in conditions associated
with disturbed function of central dopaminergic
function such as schizophrenia and Parkinson's disease.
CCK and gastrin peptides share a common carboxy
terminal pentapeptide sequence and CCK peptides can
bind to the gastrin receptor of the stomach mucosa and
elicit acid secretion in many species including human
W O 95/24389 2 1 8 4 1 1 ~ - PC~rnUS94/02573
(Konturek, Gastrointestinal Hormones, Ch. 23,
pp 529-564, 1980, ed. G. B. J. Glass, Raven Press, NY).
Antagonists of the CCK-B receptor would also be
expected to be antagonists at the stomach gastrin
receptor and this would also be of value for conditions
involving excessive acid secretion.
CCK and gastrin peptides have trophic effects on
the pancreas and various tissues of the gastro-
intestinal tract (Johnson, ibid., pp 507-527), actions
which are associated with increased DNA and RNA
synthesis. Moreover, gastrin secreting cells are
associated with certain gastrointestinal tumors as in
the Zollinger-Ellison syndrome (Stadil, ibid.,
pp 729-739), and some colorectal tumors may also be
gastrin/CCK dependent (Singh, Walker, Townsend, and
Thompson, Cancer Research 46:1612, 1986; Smith, J. P.,
Gastroenterology 95:1541, 1988). Antagonists of
CCK/gastrin receptors could therefore be of therapeutic
value as antitumor agents.
The CCK peptides are widely distributed in various
organs of the body including the gastrointestinal
tract, endocrine glands, and the nerves of the
peripheral and central nervous systems. Various
biologically active forms have been identified
including a 33-amino acid hormone and various carboxyl-
terminus fragments of this peptide (e.g., the
octapeptide CCK26-33 and the tetrapeptide CCK30-33).
(G. J. Dockray, Br. Med. Bull. 38(3):253-258, 1982).
The various CCK peptides are thought to be
involved in the control of smooth muscle contractility,
exocrine and endocrine gland secretion, sensory nerve
transmission, and numerous brain functions.
~m; ni stration of the native peptides cause gall
bladder contraction, amylase secretion, excitation of
central neurons, inhibition of feeding, anticonvulsive
actions and other behavioral effects.
w095/24389 ~ t &4 ~ I4 PCT~Sg4/02573
,
(Cholecystokinin: Isolation, Structure and Functions,
G. B. J. Glass, Ed., Raven Press, New York, 1980,
pp 169-221; J. E. Morley, Life Sciences 27:355-368,
1980; Cholecystokinin in the Nervous System,
J. de Belleroche and G. J. Dockray, Ed., Ellis Horwood,
Chichester, England, 1984, pp 110-127.)
The high concentrations of CCK peptides in many
brain areas also indicate major brain functions for
these peptides (G. J. Dockray, Br. Med. Bull.
38(3):253-258, 1982). The most abundant form of brain
CCK found is CCK26-33, although small quantities of
CCK30-33 exist (Rehfeld and Gotterman, J. Neurochem.
32:1339-1341, 1979). The role of central nervous
system CCK is not known with certainty, but it has been
implicated in the control of feeding ~Della-Fera and
Baile, Science 206:471-473, 1979).
Currently available appetite suppressant drugs
either act peripherally, by increasing energy
expenditure (such as thyroxine), or in some other
manner (such as the biguanides), or act by exerting a
central effect on appetite or satiety.
Centrally acting appetite suppressants either
potentiate central catecholamine pathways and tend to
be stimulants (for example, amphetamine), or influence
serotonergic pathways (for example, fenfluramine).
Other forms of drug therapy include bulking agents
which act by filling the stomach, thereby inducing a
"feeling" of satiety.
CCK is known to be present in some cortical inter-
neurones which also contain gamma-aminobutyric acid
(GABA) (H. Demeulemeester et al, J. Neuroscience
8:988-1000, 1988). Agents that modify GABA action may
have utility as anxiolytic or hypnotic agents
(S. C. Harvey, The Pharmacological Basis of
Therapeutics (7th ed.) 1985, pp 339-371, MacMillan).
Thus, agents which modify CCK action may have parallel
WO 95/24389 PCI/US94tO2573
218411 ~ -
--4--
anxiolytic or hypnotic activities. The role of CCK in
anxiety is disclosed in TIPS 11:271-273, 1990, and is
fully detailed in Woodruff, G. N. and Hughes, J., 1991,
Ann. Rev. Pharmacol. and Toxicol. 31, 469-501.
WO 92/04045 published March 19, 1992, concerns
dipeptoids useful, for example, as anxiolytics.
S~MMARY OF THE INVENTION
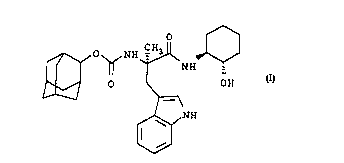
The invention relates to a novel compound of the
formula
O ~
~ O ~ NH ~ NH
OH
~l ~
~ NH
W
and the pharmaceutically acceptable salts thereof.
The present invention relates to a pharmaceutical
composition containing an effective amount of a
compound according to Formula I in combination with a
pharmaceutically acceptable carrier in unit dosage form
effective for appetite suppression.
The compounds are also useful as anxiolytics,
antipsychotics, especially for treating schizophrenic
behavior, as agents in treating disorders of the
extrapyramidal motor system, as agents for blocking the
trophic and growth stimulating actions of CCK and
gastrin, and as agents for treating gastrointestinal
motility.
WO 95124389 PCT/US94/02573
- 21841 14
Compounds of the invention are also useful as
analgesics, and they potentiate the effect of morphine.
They can be used as an adjunct to morphine and other
opioids in the treatment of severe pain such as cancer
pain, and reduce the dosage of morphine required in the
treatment of pain where morphine is contraindicated.
An additional use for compounds of the invention
is that a suitable radiolabeled isotope gives an agent
suitable for treatment of gastrin dependent tumors such
as those found in colonic cancers. I-125 radiolabeled
compounds of the invention can also be used as
diagnostic agents by localization of gastrin and CCK-B
receptors in both peripheral and central tissue.
The invention further relates to a method of
appetite suppression in mammals which comprises
administering an amount effective to suppress appetite
of the composition described above to a mammal in need
of such treatment.
The invention also relates to a pharmaceutical
composition for reducing gastric acid secretion
containing an effective amount of a compound of
Formula I in combination with a pharmaceutically
acceptable carrier in unit dosage form effective for
reducing gastric acid secretion.
The invention also relates to a method for
reducing gastric acid secretion in mammals which
comprises administering an amount effective for gastric
acid secretion reduction of the composition described
above to a mammal in need of such treatment.
The invention also relates to a pharmaceutical
composition containing an effective amount of a
compound of Formula I in combination with a
pharmaceutically acceptable carrier in unit dosage form
effective for reducing anxiety.
3S The invention also relates to a method for
reducing anxiety in mammals which compri~es
W O 95/24389 PC~rnUS94/02573
2184~ 1~
administering an amount effective for anxiety reduction
of the composition described above to a mam.mal in need
of such treatment.
The invention also relates to a pharmaceutical
composition containing an effective amount of a
compound of Formula I in combination with a
pharmaceutically acceptable carrier in unit dosage form
effective for treating gastrointestinal ulcers.
The invention further relates to a method for
treating gastrointestinal ulcers in mammals which
comprises administering an amount effective for
gastrointestinal ulcer treatment of the composition as
described above to a m~mm~l in need of such treatment.
The invention also relates to a pharmaceutical
composition containing an effective amount of a
compound of Formula I in combination with a pharma-
ceutically acceptable carrier in unit dosage form
effective for treating psychosis, i.e., schizophrenia.
The invention further relates to a method for
treating psychosis in m~mm~l S which comprises
administering an amount effective for treating
psychoses of a composition as described above to a
m~mm~l in need of such treatment.
The invention also relates to pharmaceutical
compositions effective for stimulating or blocking CCK
or gastrin receptors, for altering the activity of
brain neurons, for schizophrenia, for treating
disorders of the extrapyramidal motor system, for
blocking the trophic and growth stimulating actions of
CCK and gastrin, and for treating gastrointestinal
motility.
The invention also relates to a pharmaceutical
composition for preventing the withdrawal response
produced by chronic treatment for abuse of drugs or
alcohol.
woss/2438s PCT~Sg4/02573
- 21~411~
The invention further relates to a method for
treating the withdrawal response produced by withdrawal
from chronic treatment or withdrawal from abuse of
drugs or alcohol. Such drugs include benzodiazepines,
- 5 especially diazepam, cocaine, alcohol, and nicotine.
Withdrawal symptoms are treated by administration of an
effective withdrawal treating amount of a compound of
the invention.
The invention also relates to a pharmaceutical
composition containing an effective amount of a
compound of Formula I in combination with a
pharmaceutically acceptable carrier in unit dosage form
effective for treating and/or preventing panic.
The invention also relates to a method for
treating and/or preventing panic in mammals which
comprises administering an amount effective for panic
treatment and/or prevention of the composition
described above to a m~mm~l in need of such treatment.
The invention further relates to the use of the
compounds of Formula I to prepare pharmaceutical and
diagnostic compositions for the treatment and diagnosis
of the conditions described above.
The invention further provides processes for the
preparation of compounds of Formula I.
The compound of Formula I and its pharmaceutically
acceptable salts have an absolute oral bioavailability
in rats of 30%. This bioavailability is substantially
higher than other CCK-B antagonists of the dipeptoid
type.
.
WO 95124389 PCI`/US94/02573
218~1.1.4
_~_
DETAILED DESCRIPTION
The compounds of the present invention are
represented by the formula
s
O ~
O ~ NH$1~ NH~J
O ~ OH IA
_ ~
O, NH
and its pharmaceutically acceptable salts. Any
solvates or hydrates of the compounds are also
included. The compounds of the present invention have
multiple chiral centers. In particular, the compounds
of the present invention exist as diastereomers,
mixtures of diastereomers, or as the mixed or the
individual optical enantiomers. The present invention
contemplates all such forms of the compounds. The
mixtures of diastereomers are typically obtained as a
result of the reactions described more fully below.
Individual diastereomers may be separated from mixtures
of the diastereomers by conventional techniques such as
column chromatography, HPLC, or repetitive
recrystallizations. Individual enantiomers may be
separated by convention method well known in the art
such as conversion to a salt with an optically active
compound, followed by separation by chromatography or
recrystallization and reconversion to the nonsalt form.
Alternatively, they were synthesized by utilizing
enantiomerically pure trans-(S,S)-2-aminocyclohexanol
and trans-(R,R)-2-aminocyclohexanol prepared as shown
in Scheme I (see Overman L.E., et. al., J. Org. Chem.
wossl24389 PCT~Sg4/02573
2I~1~ 14
50:4154, 1985; also Aube J., et. al., Syn. Comm.
22~20):3003, 1992). Coupling reaction of the trans-
~S,S)-2-aminocyclohexanol with R-2-Adoc-~-methyl-Trp
using coupling reagent such as DCC, HOBt afforded
compound of Example 6.
W095/24389 2 1 ~ 4 1 1 ~ PCTnUS94/02573
--1 0--
SCHEME I
~2 ~ Me3Al/CH2C12 ~ ~
H3C Ph 78%C NH NH
~"1 J~
H3C ~ Ph H3C ~ Ph
A 1:1 B
Pd/C ~ ~OH DCC/HOBt/EtOAc
A ~ Example 6
NH4 COO- ~ NH2 (R)-2-AdoC-a-Me-Trp
-
w095/24389 2 1 8 ~ PCT~Sg4/02573
The individual substltuted alpha amino acid
starting materials are generally known or, if not
- known, may be synthesized and, if desired, resolved by
methods within the skill of the art. (Synthesis of
racemic [DL]-a-methyl tryptophan methyl ester - see
Brana, M. F., et al, J. Heterocyclic Chem., 1980,
17:829.)
A key intermediate in the preparation of compounds
of Formula I is a compound of formula
H
~ II
ROCONH COOH
Me
wherein R is selected from 9-fluorenylmethyl, Bz, and
other suitable N-blocking groups. These are useful as
intermediates in the preparation of compounds of
Formula I. The compounds wherein R is 1-adamantyl,
2-adamantyl, 4-protoadamantyl, exo-bornyl, endo-bornyl,
exo-norbornyl, endo-norbornyl, 2-methylcyclohexyl,
2-chlorocyclohexyl, or camphoryl are novel and are
preferred.
The disclosure of U.S. 4,757,151 is hereby
incorporated by reference. It describes the
9-fluorenylmethyl blocking group.
Compounds of Formula II are prepared by reacting
ROH III
WO 95/24389 ` PCT/US94/02573
2184~ 14
-12-
wherein R is as defined above, with phosgene or a
phosgene substitute to produce a corresponding compound
of formula
ROCOC1 IV
and then reacting a compound of Formula IV with
a-methyltryptophan to produce the desired compound of
Formula II above.
Alternatively, a compound of Formula IV can be
reacted with an a-methyltryptophan methyl ester to
produce
~ V
ROCONH COOMe
Me
which can be converted to a compound of Formula II by
known means such as hydrolysis with aqueous lithium
hydroxide.
The compound of Formula I exhibits an absolute
bioavailability of 30~. It is, therefore, unique in
the area of CCK selective dipeptoids.
Single 20-mg/kg PO gavage or 20-mg/kg IV doses of
the compound were given to fasted male Wistar rats
(four per route). For both routes, drug was
administered as a solution of the compound in 0.lN HCl
in ethanol:50~ w/v hydroxypropyl-~-cyclodextrin in
normal saline (30:70). Blood samples were drawn from
jugular vein cannulae into syringes containing heparin
before dosing and at different times up to 24 hours
postdose. Plasma was harvested from blood by
centrifugation and stored frozen until analysis.
WO 95/24389 21 8 ~1 I 1 4 - PCT/US94/02573
Plasma samples were analyzed for the compound using a
validated liquid chromatographic method. The method
involved liquid-liquid extraction of the compound and
internal standard carbamic acid, (R)-[2-[(hexahydro-
lH-azepin-1-yl)amino]-1-(lH-indol-3-ylmethyl)-l-methyl-
2-oxoethyl]-, tricyclo[3.3 . 1 . 13'7] dec-2-yl ester,
liquid chromatographic separation of the analytes on a
C-18 column, and quantitation by fluorescence
detection.
Mean (+SD) pharmacokinetic parameters are
presented in Table 1. Following IV dosing, the
compound plasma concentrations declined rapidly in a
multiexponential manner with a harmonic mean terminal
elimination half-life of 0.87 (+0.02) hours. Following
PO administration, absorption was generally rapid as
demonstrated by a mean (iSD) tmax value of 1.8 (+1.6)
hours. Corresponding mean (iSD) Cmax value was
756 (i264) ng/mL. Harmonic mean terminal elimination
half-life after PO dosing was 0.99 (iO.28) hours,
comparable to the IV half-life. Mean (+SD) absolute PO
bioavailability, based on ratio of mean PO and IV
AUC(0-~) values, was 30 (i8)%.
Absorption of the compound in rats following oral
solution administration was generally rapid. Absolute
oral bioavailability was 30%.
W 095/24389 PCTnUS94/02S73
2i8411q
-14-
TABLE I. Summary of Mean (~SD) Compound 6
Pharmacokinetic Parameters in
Fasted Male Wistar Rats Following a
Single 20-mg/kg PO or IV Dose
Route of Admini~tration
ParameterIV PO
N 4 4
Dose 20 20
Cmax -- 756 (+264)
tmax -- 1.8 ~+1.6)
0 t~ 0.87 ~iO.02) 0.99 ~+0.28)
AUC(0-tldc) 7220 (+1630) 2130 (~257)
AUC(0-~)7230 (+1630) 2160 (+268)
%F -- 30 (+8)
Doce = mg/kg.
Cmax = ~xi observed drug plasma
concentration (ng/mL).
tmax = Time to reach Cmax (hr).
t~ = Terminal elimination half-life, harmonic
mean (hr).
AUC(0-tldc) = Area under the plasma concentration-time
curve from zero to time of last
detectable concentration ~ng-hr/mL).
AUC(0-~) = Area under the plasma concentration-time
curve from zero to infinite time
(ng hr/mL).
%F = Absolute bioavailability based on ratio
of mean PO and IV AUC~0-~) values.
wossl24389 PCT~Sg4/02573
2184~ 1~
For preparing pharmaceutical compositions from the
compounds of this invention, inert, pharmaceutically
- acceptable carriers can be either solid or liquid.
Solid form preparations include powders, tablets,
- 5 dispersible granules, capsules, cachets, and
suppositories.
A solid carrier can be one or more substances
which may also act as diluents, flavoring agents,
solubilizers, lubricants, suspending agents, binders,
or tablet disintegrating agents; it can also be an
encapsulating material.
In powders, the carrier is a finely divided solid
which is in a mixture with the finely divided active
component. In tablets, the active component is mixed
with the carrier having the necessary binding
properties in suitable proportions and compacted in the
shape and size desired.
For preparing suppository preparations, a
low-melting wax such as a mixture of fatty acid
glycerides and cocoa butter is first melted and the
active ingredient is dispersed therein by, for example,
stirring. The molten homogeneous mixture is then
poured into convenient sized molds and allowed to cool
and solidify.
The powders and tablets preferably contain 5% to
about 70% of the active component. Suitable carriers
are magnesium carbonate, magnesium stearate, talc,
lactose, sugar, pectin, dextrin, starch, tragacanth,
methyl cellulose, sodium carboxymethyl cellulose, a
low-melting wax, cocoa butter, and the like.
Preferred pharmaceutically acceptable salts are
the N-methyl glucamine salt and sodium.
Pharmaceutically acceptable salts are acetate,
benzenesulfonate, benzoate, bicarbonate, bitartrate,
bromide, calcium acetate, camsylate, carbonate,
chloride, citrate, dihydrochloride, edetate, edisylate,
w09s/24389 PCT~Sg4/02573
?,~8~
-16-
estolate, esylate, fumarate, glucaptate, gluconate,
glutamate, glycollylarsanilate, hexylresorcinate,
hydrabamine, hydrobromide, hydrochloride,
hydrochloride, hydroxynaphthoate, iodide, isethionate,
lactate, lactobionate, malate, maleate, mandelate
mesylate, methylbromide, methylnitrate, methylsulfate,
mucate, napsylate, nitrate, pamoate (embonate),
pantothenate, phosphate/diphosphate, polygalacturonate,
salicylate, stearate, subacetate, succinate, sulfate,
tannata, tartrate, teoclate, triethiodide, benzathine,
chloroprocaine, choline, diethanolamine,
ethylenediamine, meglumine, procaine, aluminum,
calcium, lithium, magnesium, potassium, sodium, and
zinc.
The term "preparation" is intended to include the
formulation of the active component with encapsulating
material as a carrier providing a capsule in which the
active component (with or without other carriers) is
surrounded by a carrier which is thus in association
with it. Similarly, cachets are included.
Tablets, powders, cachets, and capsules can be
used as solid dosage forms suitable for oral
administration.
Liquid form preparations include solutions,
suspensions, and emulsions. Sterile water or water-
propylene glycol solutions of the active compounds may
be mentioned as an example of liquid preparations
suitable for parenteral administration. Liquid
preparations can also be formulated in solution in
aqueous polyethylene glycol solution.
Aqueous solutions for oral administration can be
prepared by dissolving the active component in water
and adding suitable colorants, flavoring agents,
stabilizers, and thickening agents as desired. Aqueous
suspensions for oral use can be made by dispersing the
finely divided active component in water together with
w09sl24389 2 1 8 4 1 1 4 PCT~Sg4/02573
a viscous material such as natural synthetic gums,
resins, methyl cellulose, sodium carboxymethyl
~ cellulose, and other suspending agents known to the
pharmaceutical formulation art.
~ 5 Preferably the pharmaceutical preparation is in
unit dosage form. In such form, the preparation is
divided into unit doses containing appropriate
quantities of the active component. The unit dosage
form can be a packaged preparation, the package
containing discrete quantities of the preparation, for
example, packeted tablets, capsules, and powders in
vials or ampoules. The unit dosage form can also be a
capsule, cachet, or tablet itself, or it can be the
appropriate number of any of these packaged forms.
EXAMPLES
EXAMPLE 1
Carbamic acid, N-[2-[(2-hydroxycyclohexyl)amino]-1-(lH-
indol-3-ylmethyl)-1-methyl-2-oxoethyl]-,tricyclo-
[3.3.1.13~7]dec-2-ylester,[l~(R),2~]
The title compound was synthesized by coupling
trans-2-aminocyclohexanol with
(R)-2-adamantyloxycarbonyl-2-methyltryptophan using DCC
and HOBt.
Analysis for C29H39N3O4-0.5 H2O:
Calc.: C, 69.29; H, 8.02; N, 8.35.
Found: C, 69.38; H, 8.46; N, 8.00.
EXAMPLES 2 AND 3
Carbamic acid, [2-[(2-hydroxycyclohexyl)amino]-1-(lH-
indol-3-ylmethyl)-1-methyl-2-oxoethyl]-,tricyclo-
[3.3.1.13~7]dec-2-ylester Isomer 1
TRP Center is R; Ring center unknown
WO 95/24389 PCTIUS94/02573
2184t~
-18-
Carbamic acid, [2-[(2-hydroxycyclohexyl)amino]-1-(lH-
indol-3-ylmethyl)-1-methyl-2-oxoethyl]-,tricyclo-
[3.3.1. 13' 7 ] dec-2-ylester Isomer 2
TRP Center is R; Other center unknown
Individual diastereomers of Example 1 were
separated by Waters Model 46K HPLC using 6 ~m silica
column (Prep Nova Pak~). The mobile phase was ethyl
acetate-hexane (60:40) and flow rate was 15 mL/minute.
The first diastereomer (Example 2) (mp 130-137C) had
retention time of 11.92 minutes. The second
diastereomer (Example 3) (mp 125-130C) had retention
time of 13.87 minutes.
EXAMPLE 4
(lS,2S)-trans-2-[(R)-(a-Methylbenzyl)amino]cyclohexanol
(A) and (lR,2R)-trans-2-[(R)-(a)-Methylbenzyl)amino]-
cyclohexanol (B)
To a well stirred solution of (R)-a-methyl-
benzylamine (6.06 g, .005 mmol) in methylene chloride
(60 mL) was added dropwise a solution of Me3Al
(27.5 mL, 2.0 M solution in toluene, 0.055 mol) at
0-5C. The resulting solution was stirred at 0C for
additional 45 minutes, and then a solution of
cyclohexene epoxide in methylene chloride (15 mL) was
added dropwise at 0C. The reaction mixture was
stirred overnight at room temperature. The aluminate
salt was decomposed at 0C by adding NaF (8.82 g,
0.21 mol) followed by 6.0 mL of H20. After stirring at
room temperature for 1 hour, the reaction mixture was
passed through celite, dried over MgSO4 and
concentrated. Flash column chromatography (ethyl
acetate/hexane/Et3N 1:1:0.1) gave Compound A (4.5 g,
39.2% Rf = 0.5) in first fraction and Compound B
(4.5 g, 39.2% Rf = 0.32) in the later fraction.
W O 95/24389 2 1 ~ 4 1 1 g PC~rrUS94/02573
--1 9--
EXAMPLE 5
(lS,2S)-trans-2-Aminocyclohexanol
- A mixture of (lS,2S)-trans-2-[(R)-(~-methyl-
benzyl)amino]cyclohexanol (2.2 g, 0.01 mol), 10~ Pd/C
S and ammonium formate (O.38 g, 0.06 mol) in methanol
(75 mL) was stirred overnight at room temperature. The
reaction mixture was passed through celite and
concentrated. The residue was then diluted with ethyl
acetate, washed with water, dried and concentrated to
give 0.92 g (80%) of the titled compound.
EXAMPLE 6
Tricyclo[3.3.1.1. 3~7] dec-2-yl ester
[lS-[l~(S*)2~][2-[(2-hydroxycyclohexyl)amino]-
1-(lH-indol-3-ylmethyl)-1-methyl-2-oxoethyl]carbamate
A solution of (R)-2-adamantyloxycarbonyl-
a-methyltryptophan (0.79 g, 0.002 mol) in ethyl acetate
(60 mL) was treated with dicyclohexylcarbodiimide
(0.495 g, 0.002 mol) and l-hydroxybenzotriazole hydrate
(0.35 g, 0.0022 mol). After stirring for 2 hours at
room temperature, the precipitated dicyclohexyl urea
was removed by filtration. To the clear filtrate
(lS,2S)-trans-2-aminocyclohexanol (0.23 g, 0.002 mol)
was added. The reaction mixture was stirred at room
temperature overnight. The ethyl acetate solution was
washed with 5% HCl, 5% NaHCO3, and brine. The organic
phase was dried over MgSO4 and concentrated to yield a
white foam. The crude product was chromatographed over
silica gel using 50% ethyl acetate/hexane to give
0.72 g of white solid which was further purified by
Waters HPLC (Model 46K) using 6 ~m silica column (Prep
Nova Pak). The mobile phase was ethyl acetate/hexane
(1:1), flow rate was 15 mL/minute, and retention time
of 9.54 minutes. Yield of the title compound 0.4 g
(39.5~), mp 137-142C.
t ~ A Pcrluss4lo2573
Wo 95/24389 2 1 ~ 4 1 1 ~
--2 0--
Analysis for C29H39N3O:
Calc.: C, 70 . 56; H, 7 . 96; N, 8 . 51 .
Found: C, 70.76; H, 8.31; N, 7.94.