Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02552099 2006-06-28
WO 2005/063693 PCT/IN2003/000401
1
IMPROVED PROCESS FOR THE PREPARATION OF ENTACAPONE .
The present invention relates to an improved process for the preparation of
ENTACAPONE. ENTACAPONE having the formula 1 is a potent and specific
peripheral catechol-O-methyltransferase (COMT) inhibitor. It is used in
combination
with levodopa / carbidopa to treat Parkinson's disease, sometime referred to
as
shaking palsy. Entacapone enhances the effect of levedopa / carbidopa by
improving
muscle control.
The invention also relates to a novel intermediate of the formula 4 where R
=ethyl
and a process for its preparation
The preparation of Entacapone of the formula (1) has been reported in GB
2200109
and US Patent No 4963590 (1987, 1990,Orion-Yhtymaeoy (Fl)) from two critical
intermediates viz; 3,4-dihydroxy-5-nitrobenzadehyde of the formula (5) and N,N-
diethylaminocyanoacetamide of the formula (3). The compound of the formula 5
is
condensed with the compound of the formula(3) in the presence of piperidine
acetate
and dry ethanoll as a solvent gave Entacapone of the formula (1). The 3,4-
dihydroxy-
5-nitrobenzaldehyde of the formula (5) in turn was prepared from 3-methoxy-
4,hydroxy-5-nitrobenzaldehyde( ) using acetic acid and hydrobromic acid as
described in scheme-1.
02N CHO 02N CHO
ACETICACID/HYDROBROMIC ACID
HO HO
We OH
6
O ~
02N CHO CN 0 N~
+ PIPERIDINE ACETATE 2N CN
HO O Ethanol
OH HO
OH
5 3
Scheme 1
CA 02552099 2006-06-28
WO 2005/063693 PCT/IN2003/000401
2
The above patent has described the preparation of the entacapone of the
formula (1)
without describing the stereochemistry or the polymorphism.
Subsequently it was described in the US Patent No 5135950(1992, Orion Orion-
Yhtymaeoy (FI)) about preparing E-isomer and polymorphism-A from the mixture
abtained from the reaction reported in the GB patent No 2200109.
The main disadvantage of this method , according tour findings , is that the
reaction
times are very long ranging about 84-100 hours and the reaction never goes to
completion.
Further the preparation of intermediate of the formula 5 from 3-hydroxy-4-
methoxy-
5-nitrobenzadehyde of the formula 6 as described in the scheme -1 has to be
purified
repeatedly and yield of the intermediate is only about 55%. Consequently the
yield of
the final product is also very low,.
The critical raw material 3,4-dihydroxy-5-nitrobenzaldehyde of the formula 5 a
catechol derivative, changes its colour from light yellow to dark colour on
storage at
room temperature in short period of time due to which the final product
entacapone
yield and quality is varies between the batches and there is no consistency in
yield and
quality. Hence storing of this compound required special conditions such as
below
15 C under dark room.
In addition it was known in the literature that the catechol derivatives are
known to
under goes the aerial oxidation and gives quinone derivatives which
contributes the
colour changes over the storage.
Hence it is of paramount important to find out the stable penultimate stage
intermediate which can be stable enough at room temperature for longer period
and it
can be stored and used as and when it is required and it was our target to
improve the
overall yield of the final product Entacapone which in the present route is
about 58%.
CA 02552099 2006-06-28
WO 2005/063693 PCT/IN2003/000401
3
Therefore the main objective of the present invention is to provide an
improved
process for the preparation of ENTACAPONE of the formula (1).
Another objective of the present invention is to provide an improved process
for the
preparation of ENTACAPONE of the formula (1) using 3-alkoxy-4-hydroxy-5-
nitrobenzadehyde of the formula (2) which is a guaiacol derivative and the
product of
the formula (4) wherer R = methyl or ethyl , which is also a guaiacol
derivative is
stable at room temperature. It is not found to change the color for two months
storage.
Yet another objective of the present invention is to provide an improved
process for
the preparation ENTACAPONE of the formula (1) by reducing the reaction time
and
making the reaction going to completion thereby making the process economical.
Still another objective of the present invention is to provide an improved
process for
the preparation of ENTACAPONE of the formula (1) by avoiding the use of 3,4-
dihydroxy-5-nitrobenzaldehyde of the formula (5
Yet another objective of the present- invention is to provide an improved
process for
the preparation of ENTACAPONE of the formula (1) employing intermediates of
the
formula 4 where R + methyl or ethyl
Another objective of the present invention is to provide a novel intermediate
of the
formula 4 where R = ethyl
Still another objective of the present invention is to provide a process for
the
preparation of novel intermediate of the formula 4 where R = methyl or ethyl
The compound of the formula 4 wherein R= methyl is known as metabolite in
Entacapone viz ;Drug Metabolism and Disposition (1993),21 , 81-92 but has
hitherto
been prepared by any chemical method . Further the compounds of the formula 4
where R =methyl or ethyl have not been hitherto known as an intermediate for
the
preparation of Entacapone.
CA 02552099 2006-06-28
WO 2005/063693 PCT/IN2003/000401
4
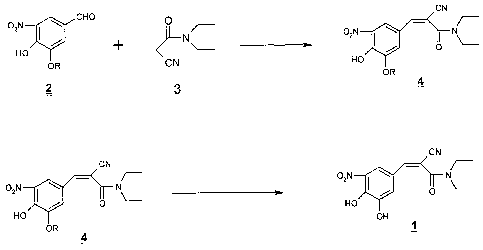
To achieve the above objectives , we devised an entirely different strategy
for the
preparation of the.Entacapone of the formula (1).
Accordingly the process envisaged and developed by us involves the reaction
shown
in the scheme 2
CN
CHO ~
OzN N 02N \ _I N
/ + I / 0
HO
~- <1
OR CN HO
OR
2 3 4
CN
CN
02N N
o2N I \ I N o
/ O HO
OH
H)P/
OR 1
4
R= Methyl or ethyl.
Scheme -2
Accordingly , the present invention provides an improved process for the
preparation
of Entacapone of the formula (1) which comprises.
(i)reacting 3-alkoxy- 4-hydroxy-5-nitrobenzadehyde of the formula (2 ) with
N,N-
diethylaminocyanoactamide of the formula (3) in the presence of mild acid
catalyst
and a solvent at a temperature in the range of 50-1150 C,to get the
intermediates of the
formula (4) wherein R is methyl, ethyl
(ii)Treating the 3- 0- alkylated ( methyl Or ethyl) Entacapone of the formula
(4) with
acid catalysts in the presence of organic base and solvents at a temperature
in the
range of 20-600 to get crude Entacapone of the formula (1)
CA 02552099 2010-12-17
(iii) purifying the crude entacapone obtained using solvent or a mixture
thereof.
In a preferred embodiment of the invention the Step (i) may be carried out
using solvents
such as alcohols with carbon (C l to C5) and toluene in presence of pyridine
and
piperidine salts such as piperidinium acetate, piperidinium propionate;
piperidinium para
toluene sulfonate, pyridinium para toluene sulfonate; pyridinium propionate
and
pyridinium acetate etc. The reaction temperature used in this step can be
preferably
between 70-115 C and more preferably between 110 to 115 C. The solvent which
can
be employed is selected from isopropyl alcohol, ethanol, n-butanol, and
toluene etc. The
duration of the reaction of this step can be between 15-25 hours.
We have observed hat the intermediate of the formula 4 where R =methyl or
ethyl exist in
two isomeric forms. Though we could separate the isomer in respect of the
compound
where R =methyl, we could not, in spite of our repeated and best efforts
separate the
isomers of the compound where R =ethyl.
However it is to be noted that the non separation of the isomers does not
afefcet the
conversion of the compound of the formual 4 where R =is methylor ethyl for its
conversion to Entacapone of the formula (1).
Another significant aspect of this invention is that intermediate of the
formula (4) where
R =methyl or ethyl having a cyano group is very susceptible for hydrolysis
even at mild
acidic and basic condition it can convert to corresponding amide or acid and
subsequently
we may get the starting material of the formula (2) but by careful designing
and execution
of the dealkylation to get ENTACAPONE in good yield by doing the reaction at
room
temperature between 25-50 C.
Another aspect of this invention is that the reaction time for condensation of
the formula
(2) and formula (3) is only 17 hours and the reaction goes to completion
thereby
increasing the yield to 86%. The overall yield from compound of the formula
(2) over
two stages is 80%.
Yet another importance of this invention is that the intermediate of the
formula (4) where
R =methyl or ethyl is stable and there is no colour change occurred during the
normal
storage condition.
CA 02552099 2010-12-17
6
The another aspect of this invention is that the step (ii) is that the
dealkylation which
normally required higher temperature is presently carried out at of about 25-
50 . The
solvents which can be employed is selected from chloroform, methylene
dichloride and
ethylene dichloride and tetahydrofuran etc:
The solvents which can be employed purification of the crude entacapone may be
selected from like toluene, isopropylalchlol, methanol and acetone or their
mixtures
thereof.
In accordance with an aspect of the present invention, there is provide a
process for the
preparation of entacapone of the formula (1)
CN
OZN N
1 O
HO
OH f
(1)
which comprises:
(i) reacting 3-alkoxy-4-hydroxy-5-nitrobenzadehyde of the formula (2) with N,
N-
diethylaminocyanoactamide of the formula O in the presence of an acid catalyst
and a solvent at a temperature in the range of 50 - 115 C to get the
intermediates of
the formula (4);
CN
02N CHO 0 02N N -
HO CN HO
OR OR
~a (3) (4)
R = methyl or ethyl
(ii) treating the 3-0-alkylated entacapone of the formula (1) with an acid
catalyst in the
presence of an organic base and a solvent at a temperature in the range of 20 -
60 C to
get crude entacapone of the formula (1); and
CA 02552099 2010-12-17
6a
CN CN
O2N N O2N N
0
HO HO
OR OH
(4) (1)
(iii) optionally purifying the crude entacapone of formula (1) by using at
least one
solvent.
In accordance with another aspect of the present invention, there is provide a
process
for the preparation of an intermediate of the formula (4)
CN
O2N N
HO
OR
(4)
R = methyl or ethyl
which comprises: reacting a compound of the formula ()
CHO
O2N I
HO
OR
4
R = methyl or ethyl
with N, N-diethylaminocyanoactamide of the formula (3)
O tNr
CN
(3)
in the presence of an acid catalyst and a solvent at a temperature in the
range of 50 -
115 C, to get the intermediate of the formula (4).
CA 02552099 2010-12-17
6b
The details of the process of this invention are described in the Examples
given below
which are provided only by way of illustration and therefore should not be
construed to
limit the scope of the invention.
EXAMPLE-1
STEP-1: PREPARATION OF N, N-DIETHYL-2-CYANO-3- (-3-ETHOXY- 4-
HYDROXY-5 NITROPHENYL) ACRYLAMIDEOF THE FORMULA 4 WHERE R
= ETHYL:
0
N(C2H5)2
H5C20 CN
HO
N O2
Charge 3-ethoxy-4-hydroxy-5-nitrobenzaldehyde 20g (0. 0947mo1e) of the formula
2
where R= ethyl and N, N-diethylamino cyanoacetamide of the formula (3) 14. 6g,
(0.1042), acetic acid 3.13g and piperidine 4.45g along with toluene 200m1 and
heated
to reflux temperature of about 100-115 C with continuous removal of water
azeotrophically 15 hours. After the reaction is over the reaction mixture is
concentrated to a volume of 20-30m1 quenched in to dilute hydrochloric acid
and
chilled water 200m1 and stirred for 60 minutes. The precipitated solid was
filtered and
dried to get 30g (95%), the HPLC purity is 94% (including isomer) of N, N-
DIETHYL
- 2-CYANO-3- (-3-ETHOXY-4-HYDROXY-5 NITROPHENYL)
CA 02552099 2006-06-28
WO 2005/063693 PCT/IN2003/000401
7
ACRYLAMIDE. The product is used as such directly to the-next stage. MR = 110.1-
117.2 C.
STEP-2: PREPARATION OF N,N-DIETHYL-2-CYANO-3-(3,4-DIHYDROXY-5-
NITROPHENYL)ACRYLAMIDE( ENTACAPONE)
O
N (C2H5)2
HO CN
HO
N02
N,N-diethyl-2-cyano-3-(3-ethoxy-4-hydroxy-nitrophenyl) acrylamide 5g (0.0150
mole) of the formula 4 where-, R= ethyl obtained by the process described in
step(i)
above is charged with pyridine 12.5m1 along with dichloromethane 50 ml and
stirred
and cooled to 0-5 degrees and slowly charged the aluminium chloride
10g(0.751m)
keeping the temperature below 5 and stirred at 0-50 for 30 minutes . After
maintaining
for 30 minutes the reaction mixture was slowly raised to 40-45 C and stirred
at that
temperature for 50 hours. After reaction completion the solvent is removed to
get a
residual volume of 10 ml and quenched in to dilute hydrochloric acid and
chilled
water (50m1) and stirred for 30 minutes. The formed product was filtered and
dried.
The dried weight is 4g (87.3%) ( with HPLC purity of about 93.25%. it was
purified
using methanol and toluene as a solvent to get the ENTACAPONE as a pure
product
having HPLC purity of 99.5% and the IR matching with the reported spectrum.
MR = 162-163 C
HPLC = 99.76%
PMR = 200MHz (DMSO-d6): 6 value; 1.26 (m, 6H), 3.49( q, 4H), 7.50(s, 1H,),
7.905 (d, 1H,J=0.01), 7.99( 1H, J=0.01).
MS = M/Z = 306.4 (M+1),
EXAMPLE-2
STEP-1: PREPARATION OF N,N-DIETHYL -2-CYANO -3-(-3- ETHOXY -4-
HYDROXY-5-NITROPHENYL) ACRYLAMIDEOF THE FORMULA 4 WHERE R
= METHYL :
CA 02552099 2006-06-28
WO 2005/063693 PCT/IN2003/000401
8
0
N(C2H5)2
MeO I cN
HO
NO2
Charged 3-methoxy4-hydroxy-5-nitrobenzaldehyde 25g (0.126 mole) of the formula
2, where R= methyl ) and N,N-diethylamino cyanoacetamide of the formula (3) )
22.2g,( 0.158 mole) acetic acid 4.18g and piperidine 5.94g along with toluene
250m1
and heated to reflux temperature of about 105-110 and remove the water
azeotrophically for 15 hours. The reaction mixture is concentrated and
quenched in to
dilute hydrochloric acid and chilled water 375m1 and stirred for 3 hours. The
precipitated solid was filtered and dried to get 36g (88.95%) with HPLC purity
is
94.2% (including isomer) and the N,N-DIETHYL -2-CYANO -3-(-3- METHOXY
-4-HYDROXY-5-NITROPHENYL) ACRYLAMIDE which is used as such for the
next stage.
Small sample was purified to get the pure single isomer (HPLC) by
crystallising form
methanol. The pure product is having the following properties.
HPLC PURITY : 99.63%
MR ; 130-132 C
IR: (Cm"'): 2204 ( -CN), 1637 ( -C=O),
PMR( 200MHz), 6 value; 1.28(m, ,6H), 3.49 (q, 4H), 4.02(s, 3H), 7.608(s, 1H,),
8.02(d, 1H, J=0.01), 7.98(s, 1H, J=0.01),
MS= M/Z= 320.2 (M+')
CA 02552099 2006-06-28
WO 2005/063693 PCT/IN2003/000401
9
STEP-2 PREPARATION OF N,N- DIETHYL-2-CYANO-3-(3,4-DIHYDROXY-5-
NITROPHENYL) ACRYLAMIDE (ENTACAPONE):
0
N(C2H5)2
HO CN
HO
NO2
N,N-diethyl-2-cyano-3-(3-methoxy-4-hydroxy-5-nitrophenyl) acrylamide (20g)
(0.062mole) of the formula 4 where , R=methyl obtained by the process
described in
step(i) of Example -2 is charged with pyridine 50m1 along with dichloromethane
120
ml and stirred and cooled to 0-5 degrees and slowly charged the aluminium
chloride
32g (0.239 mole) keeping the temperature below 5 and stirred at 0-5 C for 30
minutes . After maintaining for 30 minutes the reaction mixture was slowly
raised to
room temperature to 40-45 C and stirred for 2 hours. The reaction mixture
quenched
in dilute hydrochloric acid and ice water (50m1) after removing the methylene
chloride and stirred for 60 minutes. The formed product was filtered and
dried. The
dried weight is 18g (94.1 %) with HPLC purity of about 94.42%.
It was purified using methanol and toluene as a solvent to get the ENTACAPONE
as
a pure product having HPLC purity of 99.5%. it is matching with in all respect
with
the product obtained from the Example-1 (step-2).
Advantages of the invention
The process is simple and economical as the reaction time reduced and the
reaction
goes to completion.
The process avoids the use of 3,4-dihydroxy-5-nitrobenzaldehyde of the formula
(5~
The process results in a new intermediate of the formula 4 Wherein R= ethyl