Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02621104 2008-03-03
- 1 -
DESCRIPTION
2'-DEOXYGUANOSINE HYBRID COMPOUND CONTAINING PYRROLE
AMIDE TETRAMER, METHOD OF PRODUCING THE SAME AND
PRODUCTION INTERMEDIATE THEREOF
Technical Field
The present invention relates to a novel hybrid
compound in which a pyrrole amide tetramer is introduced
via a linker to N2-position of 2'-deoxyguanosine and
which is useful as a medicinal agent for gene therapies,
production method and production intermediates therefor.
Background Art
Developments of recent molecular biology and genetic
technique are remarkable. Particularly, with the
development of genome science, human genome has been
analyzed, and further the genetic analyses of malignant
neoplasms and pathogenic viruses are in progress as post-
genome research. A number of causative genes responsible
for diseases whose etiology has been hitherto unknown
have been identified one after another, and the onset
mechanism thereof have become elucidated at molecule
level. There has been an idea of such gene therapies
since 1950's when existence of gene diseases came to be
known, but it has gradually become realistic as feasible
treatment means in consequence of the subsequent
CA 02621104 2008-03-03
- 2 -
development of recombinant DNA technique and gene
introduction technology.
As gene therapy methods, the three following methods
are known.
(1) Method of directly operating a gene itself;
This is a method of adding a normal gene to a cell
while leaving the abnormal gene untouched, and this
method was realized at early time and the practical use
thereof is now rapidly worked forward. The gene therapy
for "ADA (adenosine deaminase) deficiency" performed in
the past falls under the category of this method. In
addition to that, means to add respective normal gene to
a cell is under examination as gene therapies for
diseases such as familial hypercholesterolemia, cystic
fibrosis and Fanconi anemia.
It may be said that this method is a comparatively
simple method among gene therapies but there are
restrictive conditions for application that the abnormal
gene does not produce a harmfulness protein, that the
disease is caused by the mechanism that the gene does not
perform necessary functions, and so on.
(2) Method of stopping the abnormal gene from working;
This is a so-called "antisense DNA" therapy
comprising binding an artificial gene sequence to a
specific target gene sequence, thereby inhibiting the
transcription of the abnormal gene and inactivating the
function of the abnormal gene.
CA 02621104 2008-03-03
- 3 -
(3) Method of cutting off the abnormal part of the gene;
This is a method of cutting off the abnormal gene
using a "restriction enzyme". In the current technology,
however, restriction enzymes cannot cut off a target part
freely as desired and there is a limitation at the
cutting off position. Therefore, the application to
practical treatment is difficult at the current level.
The method of the above (2) is called antisense
method or antigene method, and expected as a gene therapy
method for various cancer diseases and viral diseases.
From this, the development of a compound which can
specifically recognize a particular etiologic target gene
sequence and inhibit the expression thereof is desired.
The characteristics demanded for such a compound are
as follows.
1. Ability of discerning a base sequence of a
sufficient length to specify a target gene in each cell.
2. High affinity only to the target base sequence.
3. Readiness of chemical synthesis.
4. Stability in the presence of various enzymes in the
living body.
5. Low cytotoxicity.
6. Ability of readily permeating the cell membrane and
nuclear membrane.
7. Ability of repeatedly acting (turnover) on a lot of
cells without being deactivated at one action.
CA 02621104 2008-03-03
- 4 -
On the other hand, genes having a base sequence to
be targeted include:
A. Double stranded DNA (reproduction and start of
transcription is inhibited: antigene method),
B. Single stranded DNA (transcription is inhibited:
antisense method), and
C. mRNA (translation is inhibited: antisense method,
RNAi method).
The above antisense method and antigene method are
methods which exhibit inhibitory effect on the
proliferation of cells and bacteria as well as viruses
having a target mRNA by binding a target mRNA, single
stranded or double stranded DNA with a DNA which has been
subjected to some kind of modification, thereby
inhibiting reproduction, transcription or translation or
promoting degradation by RNase H. Among compounds used
for the antisense method, phosphorothioate type
oligonucleotides in which one oxygen atom in the
phosphate group is replaced with its congener, a sulfur
atom are particularly extensively studied. These are
excellent in (1) decomposition activity by RNase H, (2)
resistance to nuclease, (3) cell membrane permeability
and (4) water solubility and are highly convenient in
that they can be used in combination with modification at
the other sugar moiety and the base region thereof.
The first antisense drug approved by FDA (American
Food and Drug Administration) in the application for
CA 02621104 2008-03-03
~ =
- 5 -
retinitis by cytomegalovirus is a phosphorothioate type
oligonucleotide ((21-mer) 21-meric compound) (Vitravene
(registered trademark)) and the effect on muscular
dystrophy has been also reported recently. The
phosphorothioate type oligonucleotides as therapeutic
drugs, however, have problems such as low thermodynamic
stability of double strands as compared with natural DNAs,
insufficient recognition ability of base sequences, and
besides, they may exhibit bioactivity not based on the
activity as an antisense due to their strong interaction
with proteins in the living body. In addition to these,
BNA (Bridged ring Nucleic Acid) in which puckering of the
furanose ring in the sugar moiety is fixed to 3'-endo
type (N type) to enhance affinity with mRNAs, those in
which a substituent group to enhance stacking effect is
introduced to the base region and PNA in which sugar is
replaced with an amide bond polymer in the main chain are
proposed as candidates of therapeutic drugs, but they are
still in a research stage at present.
Among therapies using DNA, a gene therapy using a
decoy type DNA which does not target a gene but a
sequence at a recognizing site in a protein referred to
as transcription modulating factor and inhibits the
function thereof draws attention.
Incidentally, technology using genes which has
greatly attracted attention in late years is RNA
interference (RNAi) method. This method uses a series of
CA 02621104 2008-03-03
- 6 -
flow starting from a cleavage of a long double stranded
RNA (dsRNA) into a short double stranded RNA (small
interfering RNAs; siRNAs) of 21 to 23 base pairs by an
enzyme called Dicer in the living body, formation of a
complex called RISC (RNA-induced silencing complex),
conversion of a taken-in double stranded siRNA to a
single stranded siRNA by RNA helicase, sequence selective
binding of RISC to a target mRNA using a single stranded
siRNA as a guide molecule and suppression of the gene
expression in consequence of cleavage of mRNA to which
RISC binds by RNase H. Practical applications can be
easily performed by directly introducing a dsRNA having
an arbitrary sequence into the cell or expressing it in
vivo using a vector but there somewhat remains a problem
of RNAs' stability.
As stated above, nucleic acids or the derivatives
thereof can be developed into applications as medicinal
drugs by making use of their intrinsic affinity to base
sequence selecting genes.
In the meantime, some compounds which base-sequence-
selectively act on the genes although they are not
derivatives of nucleic acids as mentioned above.
Distamycin A, an antibiotic isolated from a bacterium and
represented by the following formula, improves thermal
stability of double stranded DNAs having three or more
consecutive bases of adenine (A) or thymine (T).
CA 02621104 2008-03-03
- 7 -
H
~__ 0
HN
N~
HN 0
I\
NH2 H N
i VN ~'N CI- *NH2 0 The bow shaped molecular structure of distamycin A
matches the curve of minor groove of a B type DNA, and
the compound stabilizes the binding between the molecules
by forming hydrogen bonds between the inward-looking NH
groups slightly acidic with N3-position of adenine or
with the carbonyl oxygen of C2-position of thymine. When
the sequence contains a guanine (G) - cytidine (C) base
pair, there is caused a three-dimensional obstacle
between the amino group of the C2-position of guanine
protruding into the minor groove and the hydrogen of the
C3-position of 1-methylpyrrole (Py), and affinity to the
minor groove decreases thereby exhibiting base sequence
selectivity.
Subsequent studies have revealed that antiparallelly
disposed two molecules of distamycin A coordinate to a
single match site, and further that all of the four base
pair combinations (A-T, T-A, G-C and C-G) can be
recognized by replacing 1-methylpyrrole of the
antiparallelly disposed distamycin A derivative with 1-
CA 02621104 2008-03-03
- 8 -
methylimidazole (Im) or 3-hydroxy-l-methylpyrrole (Hp)
(for example, Non-Patent Document 1). In addition,
Dervan succeeded in binding antiparallelly disposed Py-Im
polyamide compounds via a linker and improving the
sequence selectivity and affinity to a double stranded
DNA (for example, Non-Patent Document 2).
Furthermore, now that automatic production process
thereof by solid phase method has been recently
established, the polyamide compounds which recognize
arbitrary base sequences is enabled to be easily
synthesized and they are attracting increasing attention
as novel base sequence recognizing molecules due to their
practical advantage that a compound which will be
adaptable to a target base sequence can be readily
synthesized and as well as their excellent stability
against nucleases which is one of the problems associated
with nucleic acid derivatives and their excellent
permeating properties through cell membrane and nuclear
membrane.
As for the studies using these polyamide compounds,
a number of attempts have been made to have a functional
molecule affecting DNA bind a sequence selective minor
groove binders (MGBs) polyamide and express the function
selectively on base sequence. The representative
examples used bleomycin, nitrogen mustard, cyclopropyl
indole, DNA intercalator and duocarmycin as functional
molecules. Of these, Sugiyama et al. have succeeded in
CA 02621104 2008-03-03
. =
- 9 -
having a hybrid compound of duocarmycin and the
derivatives thereof with an MGB polyamide selectively act
on an arbitrary target base sequence (Non-Patent Document
3). It has been thus demonstrated that the MGB polyamide
can add unique base sequence recognition ability while
maintaining a pharmacological activity of a functional
molecule in this way, and what can be to is clarified.
The present inventors also synthesized and reported
hybrid compounds in which a pyrrole polyamide trimer was
introduced to N2-position of 2'-deoxyguanosine via a
linker having an amide bond and hybrid compounds in which
a pyrrole amide trimer was introduced via a linker by N-
alkylation as shown below aiming at creating a genetic
information control molecule based on genome chemistry
under the above background (Non-Patent Document 4, Non-
Patent Document 5).
H
~=0
HN
N",
0 HN
0
~N : NH
HO 0 N N~NH H N
O~N~ / ~ 0
OH " N" (1)
0
CA 02621104 2008-03-03
- 10 -
H
>=0
HN
N~
0 HN
0
N
</ ~ ~ \
HO N N NH H N I N
~ V N' 0
OH N"
0 (20)
When these hybrid compounds of a nucleoside and an
MGB polyamide are taken into a DNA by a biosynthetic
pathway like normal nucleic acids or modified nucleosides
used as anticancer agents, it is supposed that they
exhibit stabilization of the double stranded DNA by
forming an extremely tight and stable binding to the
minor groove thereby inhibiting replication and
transcription of the DNA in the case that a site which
the MGB polyamide specifically recognizes is present in
the neighboring sequence.
On the other hand, in the case that a site which the
MGB polyamide does not specifically recognize is present
in the neighboring region, it can be expected that the
compound does not exhibit stabilization of the double
stranded DNA, thereby inhibiting no reproduction or
transcription. In addition, when an MGB polyamide-
nucleoside hybrid compound acts on a double stranded DNA
by itself, it is supposed that the nucleoside in the
hybrid forms a Hoogsteen type hydrogen bond and has an
CA 02621104 2008-03-03
- 11 -
antigene-like effect of exhibiting affinity and base
selectivity by the MGB polyamide.
As described above, hybrid compounds of the MGB
polyamide have all of the characteristics such as high
base sequence recognition ability, high stability of MGB
polyamide against enzymes in the living body and
excellent membrane permeability and can be expected as
candidates as genetic information control molecules.
[Non-Patent Document 1] White, S.; Baird, E. E.; Dervan,
P. B. Chem. Biol., 1997, 4, 569.
[Non-Patent Document 2] Trauger, J. W.; Baird, E. E.;
Dervan, P. B. J. Am. Chem. Soc., 1996,118, 6160.
[Non-Patent Document 3] Tao, Z.; Fujiwara, T.; Sugiyama,
H. J. Am. Chem.Soc., 1999,121,4961.
[Non-Patent Document 4] Y.; Ohba, Y.; Terui, K.; Kamaike,
T.; Oshima, E.; Kawashima, Nucleic Acids Symposium Series
No. 48, 2004, p.55-56.
[Non-Patent Document 4] Y.; Ohba, Y.; Terui, K.; Kamaike,
T.; Oshima, E.; Kawashima, Nucleic Acids Symposium Series
No. 48, 2004,p.55-56.
[Non-Patent Document 5] "Abstracts of 14th antisense
symposium" Antisense DNA/RNA Research Group, December 2,
2004, p. 55.
Disclosure of the Invention
The MGB polyamide can add unique base sequence
recognition ability while maintaining the pharmacological
CA 02621104 2008-03-03
- 12 -
activity of various functional molecules by forming
hybrids with such molecules. Furthermore, since the MGB
polyamide has high stability against enzymes in the
living body and excellent membrane permeability in
addition to the high base sequence recognition ability, a
hybrid compound with the MGB polyamide can be expected to
have all of these characteristics.
These hybrid compounds have an effect of selectively
stabilizing dsDNA containing a target base sequence
(antigene type approach) . Alternatively, the hybrid
compound itself is incorporated in a DNA via the
biosynthetic pathway and have a selectively stabilizing
effect by primarily coordinating only to the minor groove
having a site which the MGB polyamide in this DNA
specifically recognizes, and therefore, they can be
expected to inhibit reproduction and transcription of the
target gene.
Incidentally, when the present inventors evaluated
the stability against the ammonia treatment necessary in
the oligomer synthesis of the above hybrid compound (20),
2'-deoxyguanosine, which was detected by the ammonia
treatment of the hybrid compound (1) via a linker by
amide bonding, was not detected in the examination by TLC
and 'H-NMR. It has become clear from this that the
stability at the N2-position of the hybrid compound
against the ammonia treatment can be enhanced by changing
binding form to the linker by N-alkylation. However, a
CA 02621104 2008-03-03
- 13 -
spot having an Rf value lower than that of (20) was just
slightly caused on TLC. From 'H-NMR data, this was
supposed to be a compound having lost a formyl group of
the N-terminal end. That is, although 2'-deoxyguanosine
was not detected in the ammonia treatment, dropout of a
formyl group of the N-terminal end was observed, which
means that the above compound (20) does not necessarily
have satisfactory stability.
Therefore, an object of the present invention is to
provide an MGB polyamide hybrid compound excellent in
stability which enables to synthesize the oligomer
effectively while maintaining the performance of the MGB
polyamide hybrid compound. Another object of the present
invention is to provide a production method of the MGB
polyamide hybrid compound and the intermediates useful
for that purpose.
The present inventors have conducted intensive
studies for finding an MGB polyamide hybrid compound
which has high base sequence recognition ability, high
stability against enzymes in the living body and
excellent membrane permeability and enables to synthesize
the oligomer effectively, and consequently have found
that it is enabled to synthesize the oligomer effectively
while maintaining excellent base sequence recognition
ability, excellent resistance to enzymes in the living
body and excellent membrane permeability by introducing a
1-methylpyrrole-2-carbonyl group in place of a formyl
CA 02621104 2008-03-03
- 14 -
group at the N-terminal end to form a pyrrole amide
tetramer, and thus completed the present invention.
That is, the present invention is directed to the
following.
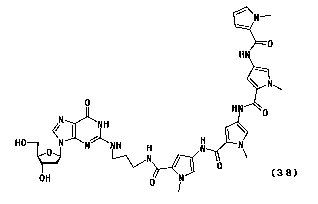
1. A 21-deoxyguanosine hybrid compound represented by
following general formula (38) characterized in that the
compound contains a pyrrole amide tetramer. 0
HN
\ N~
0 HN
0
~/ " J H \
HO N N NH H N I \
0
OH 0 N (38)
2. A 2'-deoxyguanosine hybrid compound having protected
hydroxyl groups and represented by following general
formula (37) characterized in that the compound contains
a pyrrole amide tetramer,
CA 02621104 2008-03-03
- 15 - 0
HN
\ N~
0 HN
0
N
, ~ ~
RO O N N~!NH H N I\
N 0 (37)
R20 0
wherein R' and R2 each mean the same or different
hydroxyl group protecting groups.
3. A protected 2'-deoxyguanosine hybrid compound
represented by following general formula (36)
characterized in that the compound contains a pyrrole
amide tetramer, 0
HN
\ N~
OR3 HN
/N \N 0
Rl0 0\N N5:~NH H N I \ 11
/ 0
R2 0 0 N (36)
wherein R', R 2 and R3 each mean the same or different
hydroxyl group protecting groups.
CA 02621104 2008-03-03
- 16 -
4. A production method of a protected 2'-deoxyguanosine
hybrid compound represented by above general formula (36)
characterized by reacting a diamine compound represented
by following general formula (35) with a compound
represented by following general formula (23),
0-
HN
\ N~
HN 0
H
~N H N I\N
Fmoc ~N 0 ~ (35)
~
0
OR3
/
N
Rl0 N N~Hal
(23)
~
R2 0
wherein Fmoc means a 9-fluorenylmethoxycarbonyl group;
and Rl, R 2 and R3 are the same as above.
5. A diamine compound represented by following general
formula (35) characterized in that the compound contains
a pyrrole amide tetramer,
CA 02621104 2008-03-03
- 17 - 0
HN
\ N~
HN 0
H H N N ~ \
Fmoc -N
0 (35)
0
wherein Fmoc is the same as above.
6. A production method of a diamine compound
represented by above general formula (35) characterized
by reacting a carbamate compound represented by following
formula (26) with a carboxylic acid compound containing a
pyrrole amide tetramer represented by following formula
(34). 0
HN
N~
HN 0
H
N
HO I 0
0 N (34)
CA 02621104 2008-03-03
- 18 -
\ I I /
H
0 'Y N~,_/ N*H3 -CI
0 (26)
7. A carbamate compound represented by following
formula (26) for the production of a diamine compound
(35) represented by above general formula (35).
H
0 'Y N,_,-,,/ N=H3 -CI
0 (26)
The pyrrole amide tetramer-2'-deoxyguanosine hybrid
compound of the present invention has a 1-methylpyrrole-
2-carbonyl group at the N-terminal end as one of the
characteristics thereof and the compound has excellent
base sequence recognition ability as compared with
distamycin A, excellent resistance to enzymes in the
living body and excellent membrane permeability and
enables to synthesize the oligomer effectively.
Therefore, the MGB polyamide-nucleoside hybrid
compound of the present invention has high base sequence
selecting ability, and utility as a novel genetic
information control molecule is greatly expected.
CA 02621104 2008-03-03
- 19 -
Best Mode for Carrying Out the Invention
In the previous hybrid compound invention by the
present inventors, the pyrrole polyamide trimer of the
MGB polyamide moiety can be readily synthesized by
purification method utilizing partition by acid-base
distribution based on the report of Boger et al.
Subsequently, the present inventors have considered
stability when a hybrid compound is incorporated into a
DNA and performed designing and synthesis of a hybrid
compound which is stable under the deprotection condition
at the time of the DNA synthesis by changing the binding
form at N2-position from amide bond to a binding by N-
alkylation. In order to selectively protect the amino
group of one side of diaminopropane used as a linker,
Fmoc phenylcarbonate was synthesized as a chemical
reagent for forming a mono-Fmoc compound, and just one
equivalent was allowed to act on the diaminoalkane. As a
result, conversion to a mono-Fmoc compound was
successfully achieved with good yield by converting one
amino group to a hydrochloride salt. This synthetic
process can be widely used as a method for introducing a
protecting group which is stable against acid in
synthesis researches of polyamine compounds and the like,
and therefore it is considered to highly useful.
Subsequently, an MGB polyamide was condensed with the
mono-Fmoc-diaminopropane and after the removal of the
Fmoc group, it was reacted with a 2-fluoroinosine
CA 02621104 2008-03-03
- 20 -
derivative to achieve the synthesis of the hybrid
compound (N-terminal-l-methylpyrrole-2-carbonyl
compound/pyrrole amide tetramer) linked by N-alkylation.
Ammonia treatment of a hybrid compound (pyrrole amide
tetramer) linked by N-alkylation, and as a result of the
examination, it was revealed that the compound was stable
and that oligomer synthesis was possible.
The "hydroxyl group protecting group" in R' and R2,
which may be the same or different, is acyl, carbamoyl or
aralkyl. The said acyl group is preferably formyl, C1-lo
alkylcarbonyl, C1-6 alkoxycarbonyl, C6_14 arylcarbonyl, C7-
13 aralkylcarbonyl, aromatic heterocycle-carbonyl, 5- to
7-membered non-aromatic heterocycle-carbonyl, C3-lo
cycloalkylcarbonyl (e.g., cyclopentane carbonyl,
cyclohexane carbonyl) ; C8-13 arylalkenylcarbonyl (e.g.,
styrylcarbonyl); C8-13 arylalkynylcarbonyl (e.g.,
phenylethynylcarbonyl); C6-14 arylsulfonyl (e.g.,
phenylsulfonyl); mono- or di-C1-6 alkylcarbamoyl (e.g.,
methylcarbamoyl, tert-butyl carbamoyl); C3-10
cycloalkylcarbamoyl (e.g., cyclopropylcarbamoyl,
cyclopentylcarbamoyl, cyclohexylcarbamoyl); C6-14
arylcarbamoyl ( e. g., phenylcarbamoyl); C7-14
aralkylcarbamoyl (e.g., benzylcarbamoyl,
phenethylcarbamoyl, diphenylethylcarbamoyl); C4-13
cycloalkylalkylcarbamoyl (e.g., cyclohexylmethyl
carbamoyl), aromatic heterocycle-carbamoyl (e.g.,
isoxazolyl carbamoyl, benzothiazolylcarbamoyl); non-
CA 02621104 2008-03-03
- 21 -
aromatic heterocycle-carbamoyl (e.g., pyrrolidinyl
carbamoyl ) , C7_14 aralkyloxycarbamoyl (e . g . ,
benzyloxycarbamoyl); etc. Examples of the "acyl group
having 1 to 15 carbon atoms" include C1_6 alkoxy-carbamoyl
(e.g., methoxycarbamoyl), C1_6 alkoxycarbonylcarbamoyl
(e.g., methoxycarbonylcarbamoyl, ethoxycarbonylcarbamoyl).
Preferably the "hydroxyl group protecting groups" are the
same, and particularly preferably it is an acetyl group.
The "hydroxyl group protecting group" may be TIPDS in
which R' and R 2 bind together.
The production method of the compound of the present
invention is as follows. Refer to Examples for details.
In the previously synthesized hybrid compounds (1)
and (20), the amide bond between the pyrrole rings in
itself was stable against ammonia treatment. This is
supposed to be attributable to stabilization of n-bonds
in the amide by resonance with the pyrrole ring.
Therefore, an attempt was made to achieve stabilization
against ammonia treatment by binding a 1-methylpyrrole-2-
carbonyl group via an amide bond as the N-terminal end
group in place of a formyl group so that stability of the
hybrid compound against ammonia treatment performed in
the subsequent oligomer synthesis might be improved.
At first, compound 3 was converted to carboxylic
acid 32 with aqueous sodium hydroxide and the latter was
condensed with a compound in which a Boc group was
removed from pyrrole amide trimer 8 to obtain compound 33.
CA 02621104 2008-03-03
- 22 -
Then, after ester 33 was converted to compound 34 by
hydrolyzation, the latter was condensed with mono-Fmoc
diaminopropane 26 to obtain compound 35.
CI3C /\ 2 M NaOH aq. HO /\
N MeOH,60 C
O O
3 HN Boc 32 89% N
HN O HN
N
\ 1) AcCI, MeOH, AcOEt, 0 C N- 2 M NaOH aq. N-
HN
O 2) 32, EDCI, DMAP HN MeOH, 60 C HN
O
DMF, R to 80 C 0
N N H ~~ H ~~
Me0
Me0 N
O 0 O N I O N HO O N
/ O N
8 Fmoc-NH 33 73% N\ 34 quant.
26, DCC, HOBT, DIEA ~ HN 0
NH H ~
DMF, rt O /N ~ NH N I
0 N\
O N
35 86%
Here, pyrrole amide trimer 8 protected with a Boc
group was synthesized by condensation of 4-(tert-
butoxycarbonyl)amino-l-methylpyrrole-2-carboxylic acid 6
with the amino compound removed the Boc group of pyrrole
amide dimer 7 under acidic condition. The pyrrole amide
dimer 7 was prepared by condensation of the carboxylic
acid 6 with the amino compound derived by the catalytic
reduction of the nitro group of compound 5.
CA 02621104 2008-03-03
- 23 -
NOz 1) H2, 10% Pd/C, AcOEt, rt MeO N-Boc
Me0 ~~ 2) 6, EDCI, DMAP, DMF, rt N
O I ' N O 1
7 99% HN-Boc
OMe
1) AcCI, MeOH, AcOEt, rt O NH N I N
2) 6, EDCI, DMAP, DMF, rt O
O N
~ 8 98%
The above synthetic process uses l-ethyl-3-(3-
dimethylaminopropyl)carbodiimide hydrochloride (EDCI) as
a condensation agent, and therefore purification of the
reaction product can be performed only by liquid
separation operation by acid-base, and therefore, target
pyrrole polyamide trimer 8 can be synthesized at a high
yield without purification by column chromatography.
Subsequently, compound 36 was obtained by removing a
Fmoc group of compound 35 with triethylamine and
performing substitution reaction with a fluorine atom of
2'-deoxyinosine derivative 23. Finally, 2'-
deoxyguanosine hybrid compounds 38 containing a pyrrole
amide tetramer can be obtained by removing 2-(4-
nitrophenyl)ethyl group at 06-position, and acetyl groups
at 3'-position and 5'-position of compound 36.
CA 02621104 2008-03-03
- 24 -
O
p HN
HN
N-
p HN O
HN triethylamine
\ N DMF, rt or 60 C \ N
H \ HN
O
H H O H2N,N N
Fmoc'N,N N p i
0 NO2
O
HN p
23, triethylamine N ~ DBU
DMF, rt to 60 C N N~NH \ N
~ pyridine, rt
AcO HN
NH N O
~ N
Aco p N p
36 79% / -
~ N-
O
HN O
NH
N N"~NH \ N
AcO ~-NH HN N 0
AcO p N/ N ~N-
i p 0
37 80% N NH HN O
NaOMe ~/ ~ ~
MeOH, pyridine, rt N N NH N
HO p L-L HN \
NH N 0
~
HO p N/ p
38 97%
Hereinbelow, the present invention is specifically
described by way of Examples.
CA 02621104 2008-03-03
- 25 -
The analytical instruments and so on used in this
experiment are as follows.(1) 1H nuclear magnetic
resonance spectrum (1H-NMR),
* Bruker DPX 400 NMR spectrometer
* Measurement solvent; heavy chloroform, heavy
dimethylsulfoxide, heavy methanol.
(2) 13C nuclear magnetic resonance spectrum (13C-NMR),
* Bruker DPX 400 NMR spectrometer
* Measurement solvent; heavy chloroform, heavy
dimethylsulfoxide, heavy methanol.
(3) 31P nuclear magnetic resonance spectrum (31P-NMR),
* Bruker DPX 400 NMR spectrometer
* Measurement solvent; heavy chloroform, heavy
dimethylsulfoxide.
(4) Mass spectrum (MS),
* VG Auto SpecE (Micro Mass)
* TSQ-700 (Thermoquest)
(5) Elemental analysis,
* Elemental Vavio EL
(6) Thin-layer chromatography (TLC),
* Merck Kieselgel 60 F254 (developed by ascending method)
* Detection method: UV absorption (5% sulfuric acid -
methanol solution was atomized and heated at 100 to 150
C. After immersed in a mixture of anisic aldehyde,
ethanol, sulfuric acid, water and acetic acid the mixture
was heated at 100 to 150 C) .
(7) Column chromatography,
CA 02621104 2008-03-03
- 26 -
* Wako Pure Chemical Industries Wakogel C-300
* Kanto Chemical silica gel 60N
(8) Melting point measurement,
* Micro-melting-point apparatus of Yanagimoto Mfg. Co.,
Ltd. (uncorrected values are described)
Example 1
[Pre-step 1]
Synthesis of 2-trichloroacetyl-l-methylpyrrole (3);
O
11
CI3CCI CIgC YQ
I Et20, rt
O
2 3
Trichloroacetyl chloride (130 mL, 1.16 mol) was
dissolved in ether (250 mL) under argon atmosphere and
cooled to 0 C. 1-methylpyrrole (100 mL, 1.12 mol),
which was dissolved in ether (250 mL) beforehand, was
added dropwise using a dripping funnel, while taking care
of heat generation. After the dropwise addition was
completed, the mixture was stirred for one hour while
gradually elevating the temperature to room temperature.
The reaction was terminated by adding 2 M potassium
carbonate aqueous solutions (300 mL) and the organic
layer was removed by liquid separation operation. After
extracted from the aqueous layer with ether (300 mLx2),
all the organic layers were combined and washed with a
CA 02621104 2008-03-03
- 27 -
saturated sodium chloride aqueous solution (300 mL). The
organic layer was dried over anhydrous magnesium sulfate
and the anhydrous magnesium sulfate was removed by
filtration, and 244 g(96%) of compound 3 was obtained by
concentrating the filtrate under reduced pressure.
1H-NMR (DMSO-d6) :8 7.44 (s, 1H, Py-H) , 7.43 (d, 1H, J
=1 . 18Hz, Py-H) , 6. 30 (dd, 1H, J1',2'=2 . 57Hz, J2',2"=1 . 63 Hz, Py-
H),3.91(s,3H,Py-CH3)
13C-NMR(DMSO-d6):5171.84,135.30, 123.68, 120.68, 109.10,
96.04, 37.93
FAB-MS:m/z 225 (M+H)
Molecular weight (theoretical value) C7H6N1O1C13 : C,
37.13, H, 2.67, N, 6.18.
Molecular weight (observed value): C, 37.14: H, 2.82: N,
6.15.
[Pre-step 2]
Synthesis of 2-trichloroacetyl-l-methyl-4-nitropyrrole
(4) ;
N Oz
C13C ~ HN03 C13C
O I Ac2O, 40 C
3 4
The above compound 3 (2.14 g, 9.46 mmol) was
dissolved in acetic anhydride (12 mL) and cooled to -40
C. Fuming nitric acid (0.83 mL) was added dropwise to
this solution while keeping the temperature at -40 C.
The reaction solution was gradually warmed to room
CA 02621104 2008-03-03
- 28 -
temperature and stirred for another hour. The reaction
solution was cooled to -20 C again and after added with
isopropanol (12 mL), stirred for 15 minutes. Then the
solution was allowed to stand still for 15 minutes while
keeping the temperature at -20 C and the resulted
crystals in the solution were separated by suction
filtration. After the crystals were washed with
isopropanol and dried under reduced pressure to obtain
white crystals. The filtrate was further concentrated
under reduced pressure, added with isopropanol again,
stirred for 10 minutes at -20 C and the resulted
crystals were separated by suction filtration. After the
crystals were washed with isopropanol and dried under
reduced pressure to obtain 2.01 g of compound 4 (78%)
together with the crystals obtained above in total.
1H-NMR (DMSO-d6) : 88.55 (d, 1H, J= 1.44 Hz, Py-H), 7.80
(d, 1H, J= 1.91 Hz, Py-H), 4.00 (s, 3H, Py-CH3)
13C-NMR (DMSO-d6) : 8172.79, 134.25, 132.57, 120.60,
116.30, 94.54
m.p.: 134-136 C
[Pre-step 31
Synthesis of methyl 1-methyl-4-nitropyrrole-2-carboxylate
(5) ;
NOZ N02
NaH
CI3C Me0
1
N MeOH, rt
N
4 5
CA 02621104 2008-03-03
- 29 -
The above compound 4 (15.0 g, 55.3 mmol) was
dissolved in methanol (20 mL) under argon atmosphere, and
sodium hydride (60%) (0.2 g, 5.5 mmol)dissolved in
methanol (5.0 mL) beforehand was added dropwise thereto.
After the reaction solution was stirred at room
temperature for one hour, strong sulfuric acid (0.3 mL,
5.5 mmol) was added to terminate the reaction. After
neutralization with a saturated sodium hydrogen carbonate
aqueous solution (10 mL), the residue was diluted with
ethyl acetate (200 mL) and the reaction solution was
washed with a saturated sodium hydrogen carbonate aqueous
solution (100 mLx2) and a saturated sodium chloride
aqueous solution (100 mL) The organic layer was dried
over anhydrous magnesium sulfate and concentrated under
reduced pressure after the anhydrous magnesium sulfate
was removed by filtration. The residue was crystallized
form ethyl acetate and 6.02 g (61%) of compound 5 was
obtained by collecting crystals by suction filtration.
The filtrate was further concentrated under reduced
pressure, the residue was purified by silica gel column
chromatography (hexane:ethyl acetate =5:1) to obtain 9.78
g(96%) of compound 5 in total.
1H-NMR (DMSO-d6) : 58.27 (d, 1H, J= 1.97 Hz, Py-H), 7.30
(d, 1H, J= 2.07 Hz, Py-H), 3.92 (s, 3H, Py-CH3), 3.80 (s,
3H, -OCH3)
13C-NMR (DMSO-d6) : 6159.83, 134.16 129.42, 122.59, 111.52,
51.77, 37.44
CA 02621104 2008-03-03
- 30 -
[Pre-step 4]
Synthesis of 4-(tert-butoxycarbonyl)amino-l-
methylpyrrole-2-carboxylic acid (6);
H
N0z N
Me0 1) H2, 10% Pd/C, AcOEt, rt' HO ' Boc
TN 2)(Boc)20, Et20,rt N
1 3) 2 M NaOH aq., MeOH, 60 C O O
6
The above compound 5 (6.00 g, 32.6 mmol) was
dissolved in ethyl acetate (200 mL) and added with 10%
palladium carbon (2.00 g) . The suspension was vigorously
agitated under hydrogen atmosphere at room temperature
for 12 hours and then palladium carbon was removed by
filtration with celite. Immediately after concentrating
the filtrate under reduced pressure, the residue was
dissolved in ether (30 mL) under argon atmosphere. Di-
tert-butyldicarbonate (7.46 g, 34.2 mmol) which was
dissolved in ether (20 mL) beforehand was added dropwise
and the mixture was stirred at room temperature for two
hours. After concentrated under reduced pressure, the
reaction solution was dissolved in methanol (100 mL),
added with 2 M aqueous sodium hydroxide (100 mL) and
stirred at 60 C for two hours. After methanol was
removed by concentration under reduced pressure,
remaining aqueous layer was washed with ether (50 mLx2).
After the aqueous layer was adjusted to pH 3 with 10%
hydrochloric acid, extraction with ethyl acetate (200
mLx3) was performed. The organic layers were collected
CA 02621104 2008-03-03
- 31 -
all together, washed with a saturated sodium chloride
aqueous solution (200 mL) and dried over anhydrous
magnesium sulfate. After the anhydrous magnesium sulfate
was removed by filtration, the organic layer was
concentrated under reduced pressure and the residue was
crystallized from ethyl acetate-hexane to obtain 6.97 g
of compound 6 (89%).
1H-NMR (DMSO-d6) : 812 . 08 (s, 1H, -COOH) , 9.03 (s, 1H,
Boc-NH), 7.04 (s, 1H, Py-H), 6.58 (d, 1H, J = 1.04 Hz,
Py-H), 3.77 (s, 3H, Py-CH3) , 1.44 (s, 9H, CH3- of Boc
group)
13C-NMR (DMSO-d6) : 5161.86, 152.70, 122.80, 119.64,
118.72, 107.45, 78.39, 36.01, 28.14
[Pre-step 5]
Synthesis of methyl 4-{[4-(tert-butyloxycarbonyl)amino-l-
methylpyrrol-2-yl]carbonyl}amino-l-methylpyrrole-2-
carboxylate (7);
NOz 1) H2110% Pd/C, AcOEt, rt Me0 NH
-goc
N
Me0 Z) 6, EDCI, DMAP, DMF, rt
O YI/~
0 I N / 0
7
The above compound 5 (3.00 g, 16.3 mmol) was
dissolved in ethyl acetate (100 mL) and added with 10%
palladium carbon (1.00 g) The suspension was agitated
vigorously under hydrogen atmosphere at room temperature
for 12 hours and then palladium carbon was removed by
filtration with celite. Immediately after concentrating
CA 02621104 2008-03-03
- 32 -
the filtrate under reduced pressure, carboxylic acid 6
(3.92 g, 16.3 mmol), EDCI (6.25 g, 32.6 mmol) and DMAP
(3.98 g, 32.6 mmol) were added thereto and the mixture
was dissolved in DMF (100 mL) under argon atmosphere and
stirred at room temperature for three hours. The
reaction solution was concentrated under reduced pressure
to about one-third and diluted with a mixed solvent of
ethyl acetate (200 mL) and methanol (20 mL). The organic
layer was washed with 10% hydrochloric acid (100 mLx3), a
saturated sodium hydrogen carbonate aqueous solution (100
mLx3), a saturated sodium chloride aqueous solution (100
mL) and dried over anhydrous magnesium sulfate. The
anhydrous magnesium sulfate was removed by filtration and
the organic layer was concentrated under reduced pressure
to obtain 6.07 g of compound 7(99%).
1H-NMR (DMSO-d6) : 59.85 (s, 1H, -CONH-), 9.09 (s, 1H,
Boc-NH), 7.45 (d, 1H, J = 1.86 Hz, Py-H), 6.90 (d, 1H, J
= 1.90 Hz, Py-H), 6.89 (s, 1H, Py-H), 6.84 (s, 1H, Py-H),
3.83 (s, 3H, Py-CH3), 3.81 (s, 3H, Py-CH3), 3.73 (s, 3H,
-OCH3) , 1.45 (s, 9H, CH3- of Boc group)
13C-NMR (DMSO-d6) : 6160.80, 158.42, 152.85, 122.98,
122.60, 122.42, 120.72, 118.49, 117.15, 108.37,103.82,
78.30, 50.90, 36.13, 36.13, 36.01, 28.19
[Pre-step 61
Synthesis of methyl 4-[(4-{[4-(tert-
butyloxycarbonyl)amino-l-methylpyrrol-2-
CA 02621104 2008-03-03
- 33 -
yl]carbonyl}amino-l-methylpyrrol-2-yl]carbonyl]amino-l-
methylpyrrole-2-carboxylate (8);
H N- Boc
MeO N-goc OMe
H
N 1) AcCI, MeOH, ACOEt, rt 0 N I N
N O i 2) 6, EDCI, DMAP, DMF, rt
NH O ~
0 N
~
7 g
Compound 7 (3.00 g, 7.97 mmol) was dissolved in
ethyl acetate (20 mL) under argon atmosphere and cooled
to 0 C. Methanol (4.1 mL, 100 mmol) was added and
subsequently acetyl chloride (5.7 mL, 80 mmol) was added
dropwise while taking care of heat generation. After
stirred at 0 C for 30 minutes, the solvent and the
reaction reagents were removed by concentration under
reduced pressure. Carboxylic acid 6(2.11 g, 8.77 mmol),
EDCI (3.06 g, 15.94 mmol) and DMAP (2.92 g, 23.91 mmol)
were added to the residue and dissolved in DMF (50 mL)
under argon atmosphere and stirred at room temperature
for three hours. The reaction solution was concentrated
under reduced pressure to about a one-third and diluted
with a mixed solvent of ethyl acetate (200 mL) and
methanol (20 mL) . The organic layer was washed with 10%
hydrochloric acid (100 mLx3), a saturated sodium hydrogen
carbonate aqueous solution(100 mLx3) and a saturated
sodium chloride aqueous solution (100 mL) and dried over
anhydrous magnesium sulfate. The anhydrous magnesium
sulfate was removed by filtration and the organic layer
CA 02621104 2008-03-03
- 34 -
was concentrated under reduced pressure to obtain 3.90 g
of compound 8 (98%).
1H-NMR (DMSO-d6) :89. 92 (s, 1H, -CONH-) , 9. 87 (s, 1H, -CONH-
),9.08(s,1H,Boc-NH), 7.47 (d,1H,J =1.85 Hz,Py-
H),7.22(d,1H,J=1.61 Hz,Py-H),7.08(d,1H,J =1.59 Hz,Py-H),
6.92(d,1H,J=1.93 Hz,Py-H),6.90(s,1H,Py-H),6.85(s,1H,Py-
H),3.85(s,3H,Py-CH3),3.84(s,3H,Py-CH3),3.82(s,3H,Py-
CH3) , 3. 74 (s, 3H, -OCH3) , 1.46 (s, 9H, CH3-of Boc group)
13C-NMR(DMSO-d6):8160.81, 158.52, 158.44, 152.88, 123.01,
122.83, 122.52, 122.34, 20.74, 118.52, 117.08, 108.38,
104.83, 103.85, 78.29, 50.91, 36.15, 36.04, 28.20
[Pre-step 7]
Synthesis of 3',5'-di-O-acetyl-2-fluoro-06-[2-(4-
nitrophenyl)ethyl]-2'-dioxyguanosine (23);
[Pre-step 7-1]
Synthesis of 31,5'-di-O-acetyl-2'-dioxyguanosine (21);
0 0
N N:]
N \ N I i\
N NH2 Ac20, DMAP N NHZ
HO 0 DMF, rt Ac0 0
HO AcO 21
2'-Deoxyguanosine
2'-Deoxyguanosine (8.02 g, 30 mmol) was
azeotropically dried with pyridine three times and
dissolved in DMF (24 mL) under argon atmosphere.
Subsequently, 4-dimethylaminopyridine (0.37 g, 3.0 mmol),
pyridine (24 mL) and acetic anhydride (22.1 mL, 240 mmol)
CA 02621104 2008-03-03
- 35 -
were added and the mixture was stirred at room
temperature for three hours. Then, water (10 mL) was
added to the reaction solution to terminate the reaction
and the mixture was stirred with ethanol (200 mL) for 30
minutes and allowed to stand still at room temperature.
The resulted crystals were suction filtered and the
crystals ware washed with ethanol (10 mLx3), and after
further washed with 2-propanol (100 mL), dried under
reduced pressure to obtain 8.92 g of compound 21 (85%).
1H-NMR (DMSO-d6) :81 . 87 and 2. 03 (2s, 6H, CH3COx2) ,
2.45 (ddd, 1H, J1',Z"= 6.0 z, JZ',Z"= 2.3 Hz, J2",3,=4 . 0 Hz,H-2"),
2.91(m,1H,H-2'), 4.17-4.29(m,3H,H-4',5',5"), 5.29(m,1H,H-
3' ), 6.13 (dd, 1H, J1',2'8.6 Hz, J1',2"=6.0 Hz,H-1' ),
6.52 (brs, 2H,N2-H2) 7. 90 (s, 1H,H-8), 10.74 (brs, 1H,N1-H) .
[Pre-step 7-2]
Synthesis of 3' , 5' -di-O-acetyl-O5- [2- (4-
nitrophenyl)ethyl]-2'-dioxyguanosine (22);
NO2
O
N O
~ NH
I 2-(4-nitrophenyl)ethanol N N
N N H2 Ph3P, DEAD I N I ~
O
Ac0 1,4-dioxane, rt ' N NHZ
Ac0 O
AcO 21
AcO 22
Compound 21 (1.75 g, 5.0 mmol) was azeotropically
dried by 1,4-dioxan three times, added with
CA 02621104 2008-03-03
- 36 -
triphenylphosphine (2.62 g, 10.0 mmol) and 2-(4-
nitrophenyl)ethanol (1.67 g, 10.0 mmol) and the mixture
was dissolved in 1,4-dioxan under argon atmosphere (50
mL) and stirred at room temperature for 15 minutes.
Subsequently, the reaction solution was added dropwise in
azodicarboxylic acid diphenyl ester 40% toluene solution
(4.5 mL, 10.0 mmol) and stirred at room temperature for
30 minutes. After concentrated under reduce pressure and
added with ethyl acetate (50 mL), the reaction solution
was washed with a saturated solution of sodium hydrogen
carbonate (50 mL) twice, water (50 mL) and a saturated
brine (50 mL) once respectively and after the organic
layer was dried over anhydrous magnesium sulfate, the
anhydrous magnesium sulfate was removed by filtration and
the filtrate was concentrated under reduced pressure.
The residue was purified by silica gel column
chromatography (chloroform: ethyl acetate =1:3) to obtain
2.30 g of compound 22 (92%).
1H-NMR (CDC13) :52.08 and 2.13 (2s, 6H, CH3COx2) , 2.52 (ddd,
1H, J1',2 = 6.1 Hz, J2',2" = 14.1 Hz, J2",3, = 2.5 Hz, H-2") ,
2.98(ddd, 1H, J1',2' = 7.8 Hz, Jz,,Zõ = 14.1 Hz, J2,,3,=4.9 Hz,
H-2' 1) 3.27 (t, 2H, J = 6. 8 Hz, CH2CH2PhNO2) , 4. 31-4.47 (m,
3H, H-4',51,5"), 4.73(t, 2H, J= 6.8 Hz, CHZCHzPhNOz),
4. 89 (brs, 2H, N2-H2) , 5.42 (m, 1H, H-3' ), 6.27 (dd, iH,
J1,,2, = 7.8 Hz, Jl',Z = 6. 1 Hz, H-1' ), 7.48 (d, 2H, J 8. 6
Hz, Ph-H of 2-(4-nitrophenyl)ethyl group), 7.74(s, 1H, H-
CA 02621104 2008-03-03
- 37 -
8), 8.16 (d, 2H, J = 8.6 Hz, Ph-H of 2-(4-
nitrophenyl)ethyl group).
[Pre-step 7-3]
Synthesis of 3',5'-di-O-acetyl-2-fluoro-OS-[2-(4-
nitrophenyl)ethyl]-2'-dioxyguanosine (23);
2
NOz NO
\ I \ I
0 0
N ~ N 45% HF-pyridine N
t-butyl nitrite _
N N NH2 -45 C to rt N N!\F
Ac0 O Ac0 O
Ac0 22 AcO 23
Compound 22 (0.50 g, 1.00 mmol) was azeotropically
dried with pyridine three times and added with 45%
hydrogen fluoride/pyridine solution (4.8 mL) at -30 C
and added with t-butyl nitrite (0.36 mL, 3.00 mmol) under
argon atmosphere and stirred for 30 minutes after resumed
to room temperature. Then the mixture was neutralized
with a saturated sodium hydrogen carbonate aqueous
solution (15 mL), extracted with chloroform (30 mL) three
times and washed with water (50 mL) twice. The organic
layer was dried over anhydrous magnesium sulfate and the
anhydrous magnesium sulfate was removed by filtration,
and the filtrate was concentrated under reduced pressure.
The residue was purified by silica gel column
chromatography (chloroform: ethyl acetate =1:3) to obtain
0.44 g of compound 23 (87%).
CA 02621104 2008-03-03
- 38 -
1H-NMR (CDC13) :52.08 and 2.11 (2s, 6H, CH3COx2) , 2.60 (ddd,
1H, J1',2" = 5.9 Hz, J2,,2" = 14.1 Hz, J2,,,3' = 2.5 Hz, H-2") ,
2.87 (ddd, 1H, J1',2' = 7.9 Hz, J2',2" = 14.2 Hz, J2',3' = 6.4
Hz, H-2'), 3.30 (t, 2H, J= 6.7 Hz, CH2CH2PhNO2) , 4.30-4.39
(m, 3H, H-4' , 5' , 5") , 4. 82 (t, 2H, J= 6. 7 Hz, CH2CH2PhNO2)
5.39 (m, 1H, H-3' ), 6.35 (dd, 1H, J1,,2' = 7. 9 Hz, J1',2" =
5.9 Hz, H-1'), 7.48 (d, 2H, J = 8.7 Hz, Ph-H of 2-(4-
nitrophenyl)ethyl group), 8.06 (s, 1H, H-8), 8.15 (d, 2H,
J = 8.7 Hz, Ph-H of 2-(4-nitrophenyl)ethyl group).
[Pre-step 8]
Synthesis of 9-fluorenylmethyl 3-aminopropyl-i-carbamate
hydrochloride (26);
/1,3-diaminopropane(1.0 eq.) ONH
u O then HCI-pyridine, rt
II
O ~ N'H3 CI
25 26
Compound 25 (3.80 g, 12.0 mmol) was suspended in
methanol (50 mL) Immediately after that, 1,3-
diaminopropane (1.00 mL, 12.0 mmol) was added and the
mixture was stirred at room temperature for four hours.
After pyridine hydrochloride (3.00 g, 26.0 mmol) was
added to the reaction solution which became slightly
transparent, the mixture was stirred for 10 minutes and
then concentrated under reduced pressure. The residue
was suspended in eluent (chloroform:methanol =4:1) for
silica gel column chromatography and purified by column
CA 02621104 2008-03-03
- 39 -
chromatography shortly loaded with silica gel
(chloroform:methanol =4:1) to obtain 3.25 g of compound
26 (820). Although it was usually difficult to protect
one of the diamine with a protecting group, this purpose
was able to be achieved with the above compound.
1H-NMR (DMSO-d6) :57.79 (d, 2H, J = 7.52 Hz, Fmoc-H) , 7.63
(d, 2H, J = 7.39 Hz, Fmoc-H), 7.39 (t, 2H, J = 7.38 Hz
Ph-H), 7.31 (t, 2H, J = 7.45 Hz Ph-H), 4.40 (d, 2H, J
6.61 Hz, Fmoc-H), 4.20 (t, 1H, J = 6.44 Hz, Fmoc-H), 3.20
(d, 2H, J = 6.44 Hz, CH2NH), 2.91 (t, 2H, J = 7.34 Hz,
NHCH2) , 1.82. (tt, 2H, J = 7.11 Hz, J = 6.83 Hz, -CH2CH2-)
13C-NMR (DMSO-d6) :6159.49, 145.41, 142.80, 128.94, 128.27,
126.21, 121.10, 67.83, 48.64, 38.42, 29.32
HRESIMS m/z : 297. 1615 (Calcd for C1aH2102N2:297.1603)
Anal. Calcd for C18H2ON20zCl + H20 : C, 61 . 62 ; H, 6. 61 ; N,
7.98
Found. C, 61.29; H, 6.83; N, 7.27
m.p.: 129-130 C
[Step 11
Synthesis of 1-methylpyrrole-2-carboxylic acid (32);
CI3C 2 M NaOH aq. HO
N
N MeOH, 60 C O
O
3 32
Compound 3 (5.00 g, 22.0 mmol) synthesized in Pre-
step 1 was dissolved in methanol (50 mL) and added with
2.0 M aqueous sodium hydroxide (50 mL) . The reaction
CA 02621104 2008-03-03
- 40 -
solution was warmed to 60 C and stirred for one hour.
Methanol was removed under reduced pressure and the
remaining solution was diluted with water (50 mL) and
washed with ether (50 mL). The aqueous layer was
adjusted to approximately pH 3 with 10% hydrochloric acid
and extracted with ethyl acetate (100 mLx3) . After the
organic layers were collected all together, washed with a
saturated sodium chloride aqueous solution (150 mL) and
dried over anhydrous magnesium sulfate, the anhydrous
magnesium sulfate was removed by filtration. The
filtrate was concentrated under reduced pressure and the
residue was dried under reduced pressure to obtain 2.45 g
of compound 32 (89%).
1 H-NMR (DMSO-d6) :512 .23 (br. , 1H, -COOH) , 6. 99 (d, 1H, J =
1.94 Hz, Py-H), 6.80 (d, 1H, J = 1.80 Hz, Py-H),
6. 04 (d, 1H, J = 2.58 Hz, Py-H), 3.84 (s, 3H, Py-CH3),
13C-NMR (1JMSO-d6):6162.14, 129.82, 122.67, 117.37, 107.42,
36.34
FAB-MS m/z:125.1
Anal. Calcd for C6H7N102: C, 57 . 59; H, 5. 64 ; N, 11.19
Found. C.57.59; H,5.64; N,11.06
m.p.: 127-130 C
[Step 21
Synthesis of methyl 1-methyl-4-{[4-({4-[(1-methylpyrrol-
2-yl)carbonyl)amino-l-methylpyrrol-2-yl}carbonyl)amino-l-
methylpyrrol-2-yl]carbonyl}aminopyrrole-2-carboxylate
(33) ;
CA 02621104 2008-03-03
- 41 -
N-
Boc
HN
HN O
N~
HN 1) AcCI, MeOH, AcOEt, 0 C N--
O 2) 32, EDCI, DMAP HN
H DMF, rt to 80 C 0
~ \
MeO N 0 N MeO N N
p N" \~ J O \
O
/ N
8 33
Compound 8 (100 mg, 0.20 mmol) synthesized in Pre-
step 6 was dissolved in methanol (5.0 mL) under argon
atmosphere and cooled to 0 C. Subsequently, acetyl
chloride (1.7 mL, 23.9 mmol) was added dropwise while
taking care of heat generation and after stirred at 0 C
for 30 minutes, the solvents and the reaction reagents
were removed by performing concentration under reduced
pressure. The above compound 32 (27.7 mg, 0.22 mmol),
EDCI(84.5 mg, 0.44 mmol) and DMAP (53.4 mg, 0.44 mmol)
were added to the residue, dissolved in DMF (2.0 mL)
under argon atmosphere and stirred at 80 C for one hour.
The reaction solution was concentrated under reduced
pressure and the residue was purified by silica gel
column chromatography (chloroform:methanol =19:1) to
obtain 71.4 mg of compound 33 (73%).
1H-NMR (DMSO-d6):59.96 (s, 1H, Py-NH) , 9.95 (s, 1H, Py-
NH), 9.85 (s, 1H, Py-NH), 7.48 (d, 1H, J = 1.89 Hz, Py-H),
7.25 (d, 1H, J = 1.74 Hz, Py-H), 7.24 (d, 1H, J = 1.73 Hz,
CA 02621104 2008-03-03
- 42 -
Py-H), 7.07 (d, 1H, J = 1.77 Hz, Py-H), 7.05 (d, 1H, J
1.76 Hz, Py-H), 6.95 (d, 1H, J = 1.89 Hz, Py-H), 6.93 (d,
1H, J = 1.66 Hz, Py-H), 6.91 (d, 1H, J = 1.66 Hz, Py-H),
6.06 (d, 1H, J = 2.58 Hz, Py-H), 3.88 (s, 3H, Py-CH3),
3.86 (s, 3H, Py-CH3), 3.85 (s, 3H, Py-CH3), 3.84 (s, 3H,
Py-CH3) , 3.74 (s, 3H, O-CH3)
13C-NMR (DMSO-d6):5160.82, 158.61, 158.50, 128.16, 125.45,
122.99, 122.73, 122.49, 122.32, 122.11, 120.76, 118.57,
118.48, 112.67, 108.34, 106.66, 104.77, 104.69, 50.97,
36.27, 36.20, 36.14
HRESIMS m/z :506.2156 (Calcd for C25H28N705 :506.2152)
Anal Calcd for C25H27N705: C, 59.40; H, 5.38; N, 19.40
Found. C, 59.22; H, 5.45; N,19.38
m.p.: 229-231 C
[Step 31
Synthesis of 1-methyl-4-{[4-({4-[(l-methylpyrrol-2-
yl)carbonyl)amino-l-methylpyrrol-2-yl}carbonyl)amino-l-
methylpyrrol-2-yl]carbonyl}aminopyrrole-2-carboxylic acid
(34) ;
\
HN 0 HN
2 M NaOH aq. N-
\
HN MeOH, 60 C HN 0
0
MeO N I N N N
0 N 0 HO 0
0 N
33 34
CA 02621104 2008-03-03
- 43 -
Compound 33 (127 mg, 0.25 mmol) was dissolved in
methanol (2.0 mL) and added with 2.0 M aqueous sodium
hydroxide (2 mL) . The reaction solution was warmed to 60
C and stirred for two hours. Methanol was removed under
reduced pressure and the remaining solution was diluted
with water (5 mL) and washed with ether (3 mL). The
aqueous layer was adjusted to approximately pH 3 with 10%
hydrochloric acid and extracted with ethyl acetate (10
mLx3) . After the organic layers were collected all
together, washed with a saturated sodium chloride aqueous
solution (15 mL) and dried over anhydrous magnesium
sulfate, the anhydrous magnesium sulfate was removed by
filtration. The filtrate was concentrated under reduced
pressure and the residue was dried under reduced pressure
to obtain 123 mg of compound 34 (quant.).
1H-NMR (DMSO-d6) :812 . 13 (brs, 1H, COOH) , 9. 93 (s, 1H, Py-
NH), 9.89 (s, 1H, Py-NH), 9.82 (s, 1H, Py-NH), 7.43 (d,
1H, J = 1.91 Hz, Py-H), 7.24 (d, 2H, J = 2.10 Hz, Py-H),
7.07 (d, 1H, J = 1.83 Hz, Py-H), 7.04 (d, 1H, J = 1.84 Hz,
Py-H), 6.95 (d, 1H, J = 1.81 Hz, Py-H), 6.92 (d, 1H, J =
1.73 Hz, Py-H), 6.85 (d, 1H, J = 1.94 Hz, Py-H), 6.06 (d,
1H, J = 2.55 Hz, Py-H), 6.06 (d, 1H, J = 2.58 Hz, Py-H),
3.88 (s, 3H, Py-CH3) , 3.86 (s, 3H, Py-CH3) , 3.85 (s, 3H,
Py-CH3) , 3 .83 (s, 3H, Py-CH3) ,
13C-NMR (DMSO-d6):5161.94, 158.59, 158.47, 158.43, 128.09,
125.45, 122.75, 122.68, 122.58, 122.26, 122.08, 120.24,
CA 02621104 2008-03-03
- 44 -
117.50, 118.45, 112.63, 108.38, 106.62, 104.69, 36.20,
36.08
HRESIMS m/z :492.1989 (Calcd for C24H26N705 : 492.1995)
Anal Calcd for C24H25N705 + MeOH : C, 57. 35; H, 5.58;
N,18.73
Found. C,56.95; H, 5.64; N, 18.56
m.p.: 180-182 C
[Step 41
Synthesis of 9-fluorenylmethyl 3-[(4-{[4-{4-[(1-
methylpyrrol-2-yl)carbonyl]amino-l-methylpyrrol-2-
yl}carbonyl)amino-l-methylpyrrol-2-yl]carbonyl}amino-l-
methylpyrrol2-yl)carbonyl]aminopropyl-l-carbamate (35);
N Fmoc-NH
~
HN HN
O O
HO 26, DCC, HOBT, DIEA NH H
N ~ N N
O N~ NH O \ DMF, rt N NH O
O N O N
34 35
Compound 34 (1.00 g, 2.04 mmol) and compound 26
(0.74 g, 2.24 mmol) synthesized in the above Pre-step 8
were dissolved in DMF (10 mL) along with
dicyclohexylcarbodiimide (0.63 g, 3.06 mmol) and 1-
hydroxybenztriazole (0.41 g, 3.06 mmol) under argon
atmosphere and after added with diisopropylethylamine
(0.39 ml, 2.24 mmol), stirred at room temperature for 12
hours. Subsequently, the reaction solution was diluted
with ethyl acetate (200 mL) and washed with 10%
CA 02621104 2008-03-03
- 45 -
hydrochloric acid (50 mLx3), a saturated sodium hydrogen
carbonate aqueous solution (50 mLx3) and a saturated
sodium chloride aqueous solution (50 mL) and dried over
anhydrous magnesium sulfate. After the anhydrous
magnesium sulfate was removed by filtration and the
filtrate was concentrated under reduced pressure, the
residue was purified by silica gel column chromatography
(chloroform:methanol =19:1) to obtain 1.35 g of compound
35 (86%) ) .
1H-NMR (DMSO-d6):89.93 (s, 1H, Py-NH) , 9.89 (s, 1H, Py-
NH), 9.82 (s, 1H, Py-NH), 8.31 (t, 1H, J = 5.55 Hz, Boc-
NH), 7.90 (d, 2H, J = 7.48 Hz, Fmoc-H), 7.70 (d, 2H, J =
7.31 Hz, Fmoc-H), 7.41 (t, 2H, J = 7.37 Hz, Fmoc-H), 7.33
(t, 2H, J = 7.42 Hz, Fmoc-H), 7.28 (t, 1H, J = 5.67 Hz,
Py-CONH), 7.25 (d, 2H, J = 1.65 Hz, Py-H), 7.19 (d, 1H, J
= 1.17 Hz, Py-H), 7.06 (d, 2H, Py-H), 6.95 (d, 1H, J =
1.79 Hz, Py-H), 6.93 (d, 1H, J = 1.64 Hz, Py-H), 6.88 (d,
1H, J = 1.32 Hz, Py-H), 6.06 (d, 1H, J = 2.57 Hz, Py-H)
4.32 (d, 2H, J = 6.84 Hz, Fmoc-CH2), 4.22 (t, 1H, J =
6.64 Hz, Fmoc-CH), 3.89 (s, 3H, Py-CH3), 3.86 (s, 3H, Py-
CH3) , 3.85 (s, 3H, Py-CH3) , 3.80 (s, 3H, Py-CH3) , 3.17 (dt,
2H, J = 8.15 Hz, J = 5.27 Hz, -NHCH2-), 3.02 (dt, 2H, J
6.44 Hz, J = 6.28 Hz, -CH2NH-), 1.62 (tt, 2H, J = 6.68
Hz, -CH2CH2CH2-)
13C-NMR (DMSO-d6):8161.27, 158.50, 158.46, 156.12, 143.93,
140.74, 128.11, 127.58, 128.04, 125.47, 125.11, 122.91,
122.79, 122.19, 122.16, 122.09, 120.10, 118.43, 117.80,
CA 02621104 2008-03-03
- 46 -
112.64, 106.64, 104, 71, 104.13, 65.21, 48.59, 46.78,
38.05, 36.21, 36.07.35.99, 29.66
HRESIMS m/z :770.3400 (Calcd for C42H44N9O6 :770.3415)
Anal Calcd for C42H43N9O6 + MeOH : C, 62 . 99; H, 6. 02 ; N, 15 . 37
Found. C,62.92; H, 5.79; N, 15.19
[Step 5]
Synthesis of 3' , 5' -di-O-acetyl-N2- {3- [ (4- { [4- ( {4- [ (1-
methylpyrrol-2-yl)carbonyl]amino-i-methylpyrrol-2-
yl}carbonyl)amino-l-methylpyrrol-2-yl]carbonyl}amino-l-
methylpyrrol - 2 -yl ) carbonyl ] aminopropyl } - O6- [2 - ( 4 -
nitrophenyl)ethyl-2'-deoxyguanosine (36);
~ N~
-
O N-
HN NOZ O
N_ I HN
\ -
O 1) triethylamine O N_
HN
DMF, rt or 60 C N ~ N HN
N 2) 23, triethylamine 0
HN DMF, rt to 60 N N NlH H I N
H H O Ac0 O N N \
~ 0
Fmoc NN N
O Ac0 O
35 36
Compound 35 (1.00 g, 1.30 mmol) and compound 23
(0.69 g, 1.36 mmol) synthesized in the above Pre-step 7-3
were dissolved in DMF (5.0 mL) under argon atmosphere.
Subsequently, the mixture was added with triethylamine
(1.0 mL) and stirred at 60 C for 12 hours. After the
reaction solution was concentrated under reduced pressure,
the residue was purified by silica gel column
CA 02621104 2008-03-03
~
- 47 -
chromatography (chloroform:methanol =29:1) to obtain 1.07
g of compound 36 (79%).
1H-NMR (DMSO-d6):89.93(s, 1H, Py-NH) , 9.88 (s, 1H, Py-NH)
9.82 (s, 1H, Py-NH), 8.18 (d, 2H, J = 8.50 Hz, NPE-H),
8.03 (s, 1H, H-8), 8.02 (t, 1H, J = 5.42 Hz, -CH2NH-CO-)
7.60 (d, 2H, J = 8.05 Hz, NPE-H), 7.24 (d, 2H, J = 1.69
Hz, Py-H), 7.17 (d, 1H, J = 1.59 Hz, Py-H), 7.06 (s, 1H,
Py-H), 7.05 (s, 1H, Py-H), 7.03 (t, 1H, J = 5.9 Hz, NZ-H),
6.95-6.92 (m, 3H, Py-H), 6.25 (t, 1H, J = 6.78 Hz, H-1'),
6.07 (dd, 1H, J = 2.65 Hz, J = 3.86 Hz, Py-H), 5.43 (m,
1H, H-3'), 4.69 (t, 2H, J = 6.59 Hz, NPE-H), 4.31 (t, 1H,
J = 7.59 Hz, H-4'), 4.22 (m, 2H, H-5'), 3.89 (s, 3H, Py-
CH3) , 3.86 (s, 3H, Py-CH3) , 3.85 (s, 3H, Py-CH3) , 3. 80 (s,
3H, Py-CH3), 3.37 (m, 2H, -CH2NH-CO-), 3.27 (m, 4H, -COCH2,
NPE-CH2), 3.18 (m, 1H, H-2' ), 2.46 (m, 1H, H-2") , 2.08 (s,
3H, OAc), 1.99 (s, 3H,OAc),1.77 (tt, 2H, J = 6.75 Hz,-
CH2CH2CH2)
13C-NMR (DMSO-d6):5170.01, 161.33, 160.01, 158.72, 158.60,
158.50, 157.86, 146.72, 146.24, 130.22, 128.10, 125.47,
123.39, 122.96, 122.79, 122.20, 122.09, 118.43, 117.75,
112.64, 106.63, 104.71, 104.16, 81.37, 74.24, 65.42,
63.65, 36.21, 36.07, 35.91, 34.33, 20.78, 20.48
HRESIMS m/z :1031.4148 (Calcd for C49H55N14012 : 1031.4124)
[Step 6]
Synthesis of 3' , 5' -di-O-acetyl-NZ-{3- [ (4-{ [4-{4- [ (1-
methylpyrrol-2-yl)carbonyl]amino-l-methylpyrrol-2-
yl}carbonyl)amino-l-methylpyrrol-2-yl]carbonyl}amino-l-
CA 02621104 2008-03-03
- 48 -
methylpyrrol-2-yl)carbonyl]aminopropyl}-21-deoxyguanosine
(37) ;
O
N- H N
iN NO z -
~ HN 0
I DBU HN O
0 N\ pyridine, rt ~
N ~ N
~~ ~ N HN O N~NH H
N N NH H ~ N N N NH N I N O
Ac0 O ~ N N 0 AcO N
Ac0 0
36 AcO 37
Compound 36 (22.8 mg, 22 mol) was dissolved in 0.5 M
DBU/pyridine solution (1.0 mL) under argon atmosphere.
After stirring at room temperature for 12 hours, ammonium
chloride (50.0 mg, 0.94 mmol) was added thereto to
terminate the reaction. The reaction solution was
concentrated under reduced pressure and the residue was
purified by silica gel column chromatography
(chloroform:methanol =4:1) to obtain 15.6 mg of compound
37 (80%).
1 H-NMR (DMSO-d6) :610. 67 (br, 1H,H-1), 9.93 (s, 1H, Py-NH)
9.88 (s, 1H, Py-NH),9.82 (s, 1H, Py-NH), 8.05 (t, 1H, J
5.61 Hz, Py-CONH), 7.87 (s, 1H, H-8), 7.24 (d, 1H2H, J =
1.52 Hz, Py-H), 7.18 (d, 1H, J = 1.62 Hz, Py-H), 7.05 (dm,
1H2H, J = 1.58, Py-H), 6.95-6.90 (m, 3H, Py-H), 6.53 (brs,
1H,N2-H),6.17 (d, 1H, J = 7.00 Hz, H-11), 6.06 (dd, 1H, J
CA 02621104 2008-03-03
- 49 -
= 2.61 Hz, J = 3.88 Hz, H-1'), 5.41 (m, 1H, H-3'), 4.30
(m, 1H, H-4'), 4,18 (m, 2H, H-5', H-5"), 3.89 (s, 3H, Py-
CH3), 3.86 (s, 3H, Py-CH3), 3.85 (s, 3H, Py-CH3), 3.81 (s,
3H, Py-CH3) 3.36 (m, 2H, -CH2NH-) , 3.25 (m, 4H, -COCH2, ),
3.06 (m, 1H, H-2'), 2.47 (m, 1H, H-2"), 2.08 (s, 3H, OAc)11.99(s, 3H, OAc),
1.75 (s, 3H,OAc).
13C-NMR (DMSO-d6):8170.12, 170.01, 161.43, 158.61, 158.50,
158.46, 156.73, 152.58, 150.45, 136.25, 128.10, 125.46,
122.89, 122.78, 122.18,146.72, 146.24, 130.22, 128.10,
125.47, 123.39, 122.96, 122.79, 122.20, 122.09, 118.42,
117.28, 112.63, 106.64, 104.72, 104.22, 83.28, 81.33,
79.16, 74.12, 63.67, 38.18, 36.21, 36.07, 35.90, 35.05,
29.18, 20.76, 20.47
HRESIMS m/z :882 .3661 (Calcd for C41H48N13010 :881.89)
[Step 7]
Synthesis of N2-{3- [ (4-{ [4-{4- [ (1-methylpyrrol-2-
yl)carbonyl]amino-l-methylpyrrol-2-yl}carbonyl)amino-l-
methylpyrrol-2-yl]carbonyl}amino-l-methylpyrrol-2-
yl)carbonyl]aminopropyl}-2'-deoxyguanosine (38);
0
HN HN
5NO- N-
NaOMe
MeOH, pyridine, rt HN O
O ~ 0
\ N ~ N
~
I NH HN N I NH H
N N~NH H O N N~NH H I\ O
AcO ~-O N ~N HO O N N
O 0
AcO 37 HO 38
CA 02621104 2008-03-03
- 50 -
Compound 37 (60.0 mg, 76 mol) was dissolved in
pyridine (0.2 mL) under argon atmosphere. Subsequently,
the mixture was added with 0.0 M sodium
methoxide/methanol solution (0.8 mL) and stirred at room
temperature for two hours. The reaction solution was
adjusted to pH 6 with Dowex-50(H+ form) and the resin was
removed by filtration. After the filtrate was
concentrated under reduced pressure, the residue was
added with methanol to perform recrystallization. The
resulted crystals were collected by suction filtration to
obtain 51.6 mg of compound 38 (97%).
1H-NMR (DMSO-d6) :510.56 (brs, 1H, H-1) , 9.93 (s, 1H, Py-
NH), 9.89 (s, 1H, Py-NH), 9.84 (s, 1H, Py-NH), 8. 08 (s, 1H,
H-8), 7.90 (t, 1H, J = 5.49 Hz, -CH2NH-) , 7.24(d, 1H, J
1.66 Hz, Py-H), 7.20 (m, 2H, Py-H), 7.05(d, 1H, J = 1.65
Hz, Py-H), 6.95-6.89 (m, 3H, Py-H), 6.52 (brs, 1H, N2-H),
6.15 (t, 1H, J 6.67 Hz, H-i'), 6.06(d, 1H, J = 1.65 Hz,
Py-H) 5.27 (br, 1H, 5'-OH), 4.86(t, 1H, J 5.4 Hz, H-3'),
4.36 (br, iH, 3'-OH), 3.88 (s, 3H, Py-CH3), 3.86 (s, 3H,
Py-CH3') , 3 .85 (s, 3H, Py-CH3' ) , 3. 82 (s, 3H, Py-CH3' ) ,
3.81 (m, 1H, H-4'), 3.55 (m, 1H, H-5'), 3.51 (m, H1, H-
5"), 3.32 (dt, 2H, J = 5.47 Hz, J = 6.49 Hz, -CH2NHCO-),
3.25 (m, 2H, -CH2N2H), 2.60 (m, 1H, H-2'), 2.21 (m, iH,
H-2 " ) , 1.75 (m, 2H, -CH2CH2CH2-)
13C-NMR (DMSO-d6) :8161.44, 158.60, 158.46, 156.75, 152.54,
150.50, 135.75, 128.11, 125.45, 122.75, 122.08, 118.42,
CA 02621104 2008-03-03
- 51 -
117.84, 116.87, 112.63, 106.64, 104.71, 104.24, 87.57,
82.76, 70.86, 61.83, 38.18, 36.21, 36.07, 35.92, 29.18
HRESIMS m/z :798.3445 (Calcd for C37H44N1308 : 798.3436)
Anal. Calcd for C37H43N13O8 + 3H20 + MeOH : C, 51 . 64 ; H,
6.04; N, 20.60
Found. C, 51.85; H,5.88;N,20.48
As above, the present inventors have synthesized MGB
polyamide-nucleoside hybrid compounds for which utility
as novel genetic information control molecules were
expected.
Industrial Applicability
Pyrrole amide-2'-deoxyguanosine hybrid compound 38
of the present invention forms a stable double bond to
ssDNA having a site which a pyrrole polyamide
specifically recognizes when it is incorporated in ssDNA
by biosynthesis. In addition, the compound has an
extremely high base sequence selection ability. Thus,
the MGB polyamide-nucleoside hybrid compound of the
present invention is a novel genetic information control
molecule having high base sequence selection ability and
effective applications to antisense drugs can be expected.