Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
FLOW BATTERY AND REGENERATION SYSTEM WITH IMPROVED SAFETY
CROSS REFERENCE TO RELATED APPLICATIONS
[0001] This application is a PCT application of patent application number
14/184,702
titled "Flow Battery And Regeneration System With Improved Safety", filed in
the
United States Patent and Trademark Office on 19 February 2014, which is a
continuation-
in-part application of non-provisional patent application number 13/969,597
titled "Flow
Battery And Regeneration System", filed in the United States Patent and
Trademark
Office on 18 August 2013, which claims priority to and the benefit of
provisional patent
application number 61/684,805 titled "Fluid Battery With Water-compatible
Oxidants",
filed in the United States Patent and Trademark Office on 19 August 2012. The
specifications of the above referenced patent applications are incorporated
herein by
reference in their entirety.
BACKGROUND
[0002] The first widely commercialized automobiles at the dawn of the last
century
were electric and powered by lead acid batteries. Lead acid batteries are
currently used in
cars for starting, lighting, and ignition purposes. Lead acid batteries cost,
for example,
about 170 dollars/kilowatt hour (kWh) and are cheaper than many other
rechargeable
batteries known. However, the energy content of lead acid batteries is rather
low. The
specific energy of lead acid batteries is, for example, about 35 watt hour
(Wh)/kilogram
(Kg) or about 20% of their theoretical value. This is notably reflected in the
short driving
range provided by the lead acid batteries, for example, of about 30 km in
fully electric
vehicles. A long recharge time, for example, of about 2 hours required for
lead acid
batteries necessitates in many applications, a cumbersome mechanical swap of a
discharged battery by a charged battery.
[0003] By the year 1910, improvements in the performance of an internal
combustion
engine, the development of mechanical transmission, combined with a wide
availability
1
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
of liquid hydrocarbon fossils, resulted in the displacement of electric
vehicles by gasoline
vehicles in the terrestrial transportation market. Gasoline power systems
provide high
energy content, for example, about 4,000Wh/kg at wheels, that is, about 500
kilometres
driving range, and a quick mechanical refill. This provided gasoline power
systems an
advantage over batteries with solid electroactive materials (SEAM). Gasoline
cars were
widely used even through the oil crises of the 1970s. The oil crisis provoked
a concern
about the availability of hydrocarbon resources and promoted a short lasting
interest in
electric battery and hydrogen vehicles.
[0004] The current interest in electric cars started in 1990 with the passage
of the zero-
emissions vehicle mandate by the California Air Resources Board. Nickel-metal
hydride
batteries, commercialized around this time, were considered briefly for
automotive
applications. Although nickel-metal hydride batteries provided better
performance than
the lead acid batteries, for example, a driving range of about 60 km, a
specific energy of
about 60 Wh/Kg to about 90 Wh/Kg, an energy density of about 200 Wh/L -300
Wh/L, a
specific power of about 200 W/kg, and an electric recharge of about 3 hours,
albeit at a
higher cost of about $1,000/kWh, the nickel-metal hydride batteries were not
an
acceptable replacement for gasoline from the customer's perspective.
[0005] By the year 1990, hydrogen fuelled polymer electrolyte membrane fuel
cells
(PEMFCs), which were originally developed within American and Soviet space
exploration programs, became the leading contender among power sources for
electric
vehicles. The interest in PEMFCs was due to the following factors: the
perceived
availability of hydrogen fuel, a high specific energy, for example, of about
33.39
kWh/Kg for the low heating value of hydrogen (H2), a high specific power of
PEMFCs,
for example, about 0.7W/cm2 at about 60% efficiency and about 0.35 kW/Kg and
about
0.35 kW/L at the stack level, a competitive system energy density, for
example, about
1,000 Wh/L for a 700 bar gas, and about 1200 Wh/L for 1 atmospheric pressure
(atm)
liquid H2 allowing for a 600 km driving range, as well as a good energy
efficiency, for
example, about 60% for PEMFCs versus about 13% for an internal combustion
engine.
2
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0006] In the following 20 years, the idea of hydrogen economy and automotive
fuel
cells received a significant political and economic impetus which was
justified by the
concerns with the rising atmospheric carbon dioxide (CO2) levels and an
unstable supply
of liquid hydrocarbons. This was reflected in the statement by President G.W.
Bush in his
2003 State of the Union address: "a child born today will be driving a car, as
his or her
first car, which will be powered by hydrogen and pollution free."." In 2004
General
Motors was spending more than a quarter of its research budget on fuel cell
vehicles and
Larry Burns, GM's Vice President for R&D and Planning, said in February 2004
that the
company will have a commercially viable fuel cell vehicle by 2010. In 2004,
the State of
California said it would build a hydrogen highway, with hydrogen fueling
stations every
20 miles along major highways in the next few years. Despite the dedicated
work of
many scientists and engineers worldwide, the hydrogen fuelled polymer
electrolyte
membrane fuel cell (PEMFC) technology did not result in a market success of
electric
vehicles. The reasons are as follows: to achieve practically useful power
density on the
positive electrode, high platinum (Pt) loading is required which increases the
cost of the
PEMFCs; the dissolution of a Pt catalyst at positive potentials makes the
positive
electrode less durable; the lack of an inexpensive, sustainable, and a clean
hydrogen
source; and the lack of a hydrogen manufacturing and distribution
infrastructure. Hence,
there is a need for a technology that avoids the macro scale infrastructure
required for
hydrogen production and distribution and also reduces the amount of Pt
required for on-
board electricity generation.
[0007] Several revolutionary developments also occurred in the field of
batteries with
solid electroactive materials (SEAM). The advantages of a lithium (Li) metal
anode, for
example, a low equivalent weight, very negative redox potential, and a small
cation size,
allowing for an easy intercalation into cathode materials, were realized in
the early 1970s.
However, the first lithium batteries had a poor cycle life since the
electronically
insulating surface film formed on metallic lithium leads to dendritic Li
plating during
recharge. In 1981, researchers from Sony Corporation demonstrated a
rechargeable
lithium ion battery (LIB) with a graphite intercalation cathode. This lead to
the
commercialization of lithium batteries with a carbon anode in portable
applications,
3
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
within one decade. Since LIBs have a high energy density when compared to
other
commercialized room temperature batteries, LIB s have been used in commercial
electric
vehicles since the year 2010 despite a somewhat high cost, for example, of
about
$400/kW.
[0008] However, fully electric vehicles, unlike plug-in hybrids, based on
lithium ion
batteries (LIBs) did not achieve a widespread commercial success, primarily
due to a low
energy content, that is, a low driving range, and a high total cost of
ownership of the
batteries. For example, Nissan Leaf of Nissan Jidosha Kabushiki Kaisha DBA
Nissan
Motor Co. Ltd., has a battery that weighs about 20% of the total car weight
with about
200Wh/Kg, that is, about 53% of the theoretical value, and about 230 Wh/L, and
provides
about 60 Km to about 100 Km driving range depending on whether the air
conditioner is
on or off. A larger sport utility vehicle (SUV), for example, Toyota RAV4 EV
of Toyota
Jidosha Kabushiki Kaisha TA Toyota Motor Corporation, also shows a similar
performance. The often quoted statistics that 60% of daily car trips in the
United States
are less than 60 Km is apparently not helping the sales of lithium-ion battery
powered
cars as most drivers need the capability to make longer trips. Apart from the
low driving
range, the LIBs also have a low electric recharge rate, for example, the
Nissan Leaf
takes about 30 minutes for a charge of about 80% of full capacity, and the
construction of
a large scale battery swapping infrastructure is not justified due to the lack
of a sizable
LIB electric vehicle market, as illustrated by a recent bankruptcy of Better
Place. Also,
the capital cost of the LIBs needs to be reduced in the long term, for
example, from about
$500/kWh to $125/kWh and from about $30/kW to $8/kW at 250 Wh/kg, 400Wh/L, and
2 kW/kg.
[0009] The scientists at General Motors (GM) arrived at the same conclusion,
that is,
the battery electric vehicles based on current and targeted Li ion battery
technology will
be limited to small vehicle, low mileage-per-day applications due to
relatively low
specific energy and long recharge time constraints, and it is possible that
fundamental
physical limitations may prevent pure Li ion based battery electric vehicles
(BEVs) from
delivering the freedom of providing long trips, with intermittent quick
refills, that
4
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
consumers currently receive from their cars. According to Toyota spokesman
John
Hanson "We don't think that lithium-ion batteries are going to help us get to
a point
where we can dramatically increase volume and really call it a mass market.
We're going
to have a more significant breakthrough and probably go into some other area
of battery
chemistry." MIT's Yet-Ming Chiang concurs: "It is clear that long-term vehicle
electrification ¨ especially affordable 200 mile all-electric range ¨ will
require batteries
with approximately three times greater energy densities at about one third the
cost per
kWh than that of LIB s." Kevin See, analyst for Boston-based Lux Research,
said "It is
not realistic or feasible for automakers to significantly cut the price of
lithium-ion
batteries. There is going to be incremental improvement, but we don't believe
it will be
enough to spur the huge adjustment everyone was hoping for." Tesla Motors has
conceded that new technologies will eventually be required. According to Steve
Visco,
the founder of Polyplus: "What has happened over the past couple of years is
the growing
realization that lithium-ion chemistry will not take EVs to a mass adoption
vehicle. It is
just too expensive and they're too heavy."
[0010] Numerous attempts to commercialize lithium ion batteries (LIB s) for
use in fully
electric vehicles in the last 5 years failed as eloquently illustrated by the
mismatch of
large production capacities and negligible sales by all 9 award recipients of
the August
2009 $1.5 billion Department of Energy's (DOE) "Electric Vehicle Battery and
Component Manufacturing Initiative" who had a primary focus on electric
vehicle (EV)
batteries including Dow Kokam, Johnson Controls, A123 Systems, Compact Power,
EnerDel, General Motors, SAFT America, and LG Chem. The public's lack of
appetite
for battery-powered cars persuaded the Obama's administration in January of
2013 to
back away from its aggressive goal to put 1 million electric cars on U.S.
roads by 2015.
According to Takeshi Uchiyamada, Toyota's Vice Chairman, "the current
capabilities of
electric vehicles", based on fuel cells or lithium ion batteries, "do not meet
society's
needs, whether it may be the distance the cars can run, or the costs, or the
long time to
charge. Because of its shortcomings, that is, driving range, cost, and
recharging time, the
battery or fuel cell electric vehicle is not a viable replacement for most
conventional cars.
We need something entirely new". Thus, there is a need for a solution that
departs from
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
the currently available technologies and differs from others under
investigation in the
electric vehicle battery field. More specifically, there is a need for a power
source for
electric vehicles that provides a longer driving range, lower total cost of
ownership, and
allows for a quick recharge or refill than lithium-ion batteries.
[0011] The history of technology teaches that if the show stopping part in any
device is
identified and replaced with another part, then this may change the device
from a non-
functional device to a functional device, though the performance in one or
more
parameters may have to be sacrificed. In the case of lithium batteries, the
aforementioned
abandonment of the metallic lithium electrode in favor of lithium intercalated
into
graphite resulted in about 30% decrease in the theoretical energy density but
created a
marketable battery with a long cycle life. Flow systems such as fuel cells
(FCs) and redox
flow batteries (RFBs) allow an independent scaling on energy and power, and
are thus
better suited for transportation than batteries with solid electroactive
materials (SEAMs).
Other advantages of flow systems, when compared to SEAM batteries, are a
higher
system energy density, if the reactants are not too dilute, a quick refill
time, an intrinsic
fluid heat management, and a simple cell balancing. The advantages of redox
flow
batteries over fuel cells are: electric regeneration that does not require a
construction of a
new fuel distribution infrastructure, for example, a hydrogen distribution
infrastructure,
higher efficiency, and in general, a lower cost. Conventional redox flow
batteries such as
vanadium redox flow batteries have a low energy density that translates into a
short
driving range, because the components have low solubilities and a large amount
of an
otherwise useless solvent which has to be carried on-board to keep the
components in the
fluid state. For this reason, flow batteries have been considered mostly for
stationary
storage applications rather than for electric vehicles.
[0012] A Massachusetts based start-up, 24M, proposed a method that retains the
advantages of flow batteries while overcoming drawbacks of traditional
solution
chemistry, by developing a slurry flow battery based on the C6 ¨LiFePO4
chemistry used
by A123 Systems for batteries with solid electroactive materials (SEAM) or
SEAM
batteries. However, such a battery in an electric vehicle such as the Nissan
Leaf or the
6
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
Toyota RAV4 would provide only from about 90 Km to about 150 Km driving
range,
even if the battery reaches, for example, about 80% of its theoretical energy
density.
Improvements in packing factor, that is the ratio of practical to theoretical
energy density,
by using, for example, binder free SEAM batteries with a soluble mediator or a
soluble
redox couple or metal containing ionic liquid flow batteries or protected Li
metal anode,
run into the fundamental limitation that the intrinsic energy densities of
known battery
chemistries are not sufficiently high for fully electric vehicle applications.
Also, the cost
of such batteries is likely to stay above the mid-term target of about
$100/kWh and about
$30/kW, or about $2,250/car with about 100 horsepower. Hence, there is an
unmet need
for flow batteries with higher energy content and a lower cost in order to
gain market
acceptance of fully electric vehicles.
[0013] Polymer electrolyte membrane (PEM) fuel cells have high power and
energy
density at low operating temperatures as well as a flow design which makes the
PEM fuel
cells well suited for automotive applications. Furthermore, fuel cells provide
for a very
high system energy density since the oxidant, that is, 02 is not carried on-
board.
However, the fundamental problems related to the slow kinetics of the oxygen
electrode
result in high cost and poor durability of PEM fuel cells due to the necessity
of high Pt
loading in the case of near ambient temperature fuel cells. Another problem
with fuel
cells, in general, is the source of the fuel, for example, hydrogen. Hence,
there is a need
for a discharge flow battery that ensures a high energy density, high energy
efficiency,
generates a high electric power by replacing the free oxygen from air with a
high energy
density and kinetically fast on-board fluid oxidant, and allows for the
regeneration of a
fuel and an oxidant from the exhaust products.
[0014] Flow batteries use electrochemical power cells similar to fuel cells.
Flow batteries
also use fluid reactants, for example, liquid, gaseous, or suspended reactants
to store energy
and to generate electric power. However, instead of oxygen or air, a different
oxidant or a
solution of an oxidant can be employed. Due to the carrying of an on-board
oxidant, the
flow battery typically entails a lower system energy density than a fuel cell.
The reasons for
using the on-board oxidant method comprise, for example, increasing the
efficiency of
7
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
energy conversion, reduction in the amount of precious metal catalysts,
potential to change
the operating temperature of the electrochemical power cell, improved heat
management,
the possibilities of electric recharge and of mechanical refill, etc. When
compared to
batteries with solid electroactive materials (SEAM) or SEAM batteries, for
example,
lithium ion batteries, flow batteries offer an independent scaling of energy
and power, a
higher ratio of practical to theoretical energy density that is, packing
factor for systems with
a sufficiently long discharge time, a possibility of quick mechanical
recharge, intrinsic
liquid cooling, etc. Commercialized redox flow batteries, such as Vanadium
Redox Flow
Batteries have low energy densities because of the use of redox couples with
low
solubilities and with a low number of redox-active electrons per electroactive
atom. Paul
Zigouras, Director of New Business Development at EPC Corporation, eloquently
summarizes the status quo as: "Flow batteries are a great idea, but
unfortunately, no fluid
currently exists that will hold a decent amount of energy. Even the best
experimental
fluids have about 115th the energy density of the required value. I am
hopeful, but also
doubtful that a fluid will ever be developed that can effectively do this".
[0015] Hydrogen-halogen flow batteries employ fluid reagents and products, and
thus,
may avoid the aforementioned energy density dilution by a solvent. In the
series from
fluorine (F2) to iodine (12), the theoretical energy density decreases while
the efficiency,
cathode power, and exchange current increases. As a result, F2 has poor cycle
efficiency,
in addition to material compatibility issues, whereas 12 has a low energy
density in addition
to solubility problems. Hence, only bromine (Br2) and chlorine (C12) may be of
interest
for transportation applications. However, the chorine cells use an expensive
ruthenium
(Ru) - containing catalyst and provide poor energy efficiency. The theoretical
energy
density of hydrogen-bromine cells is only marginally better than that of
lithium-ion
batteries. The energy density becomes even lower if bromine is used as an
aqueous
solution with hydrogen bromide (HBr) to reduce the oxidant's cross over
through
membrane via the formation of Br3- anions and to lower the pressure of the Br2
vapour.
Hydrogen-bromine cells are therefore considered at present mostly for grid
storage
rather than for electric vehicles.
8
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0016] There is a need for resolving the aforementioned TRIZ contradiction
between
energy density and energy efficiency of halogens, for example, by introducing
a new
dimension to the choice of oxidants, for example, by adding a second dimension
of
oxocompounds such as oxides and oxoacids to the one dimensional space of
elements such
as halogens. Although hydrogen-oxoacid flow batteries such as H2-HNO3 have
been
considered in the past, these flow batteries have poor discharge efficiency
and lack the
ability of electrical recharge or regeneration of the reagents. The direct
electroreduction
of halogen oxoacids is highly irreversible under the polymer electrolyte
membrane fuel
cell (PEMFC) conditions. There is a need to overcome this problem, for
example, by
performing a slow reduction of an oxocompound in a solution, that is, in three
dimensions rather than on an electrode, that is, two dimensional.
[0017] Transition metal ion catalyzed electroreduction of oxoanions has been
known
for over 100 years. However, such reactions did not find applications in
energy storage
and conversion, mostly due to their poor reversibility. A more useful way to
facilitate the
electroreduction of halogen oxoanions is to employ a preceding homogeneous
reaction
such as comproportionation with a halide product as exemplary demonstrated for
a
halate by the equations below:
(1) X03- + 6e- + 6H+ = X- + 3H20 on the electrode, slow.
(2) X03- + 5X- + 6H+ = 3X2 + 3H20 in solution, fast.
(3) X2 2e- = 2X- on the electrode, fast.
where X= Cl, Br, I.
[0018] In practice, reaction (3) may precede reaction (1) during the initial
stage of the
cycle. Furthermore, at high concentrations of halogen oxoanion and of an acid
and for a
thick diffusion layer, the steady-state limiting current, determined by the
balance of the
rate of halogen, that is, X2 intermediate formation via comproportionation (2)
and by the
rate of halogen loss into the solution bulk, can reach enormous values over 1
A/cm2.
[0019] The reverse process of oxidation of halides is generally believed to
follow the
9
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
same pathway. For example, the oxidation of the halides such as iodide,
bromide, and
chloride at alkaline pH shows that the reverse of the chemical reaction
indicated by
equation (2) occurs through the formation of an intermediate hypohalate via a
homogeneous disproportionation: Here, R is a base:
(4) 2X2+2H20 + 2R- = 2HX0 + 2X- +2RH
followed by another homogeneous disproportionation:
(5) 5HOX (hypohalous acid) = 4X- + X03- + 1-1 + 2H20
or (4) and (5) combined
(6) 3X2 + 3H20 + 6R- = X03- + 5X- + 6RH
[0020] Thus, disproportionation, for example, reaction (6), can be used to
regenerate a
halogen oxoanion from a halide present in the discharge fluid via an
intermediate
halogen produced by one or several routes of oxidation of halide.
[0021] The occurrence of homogeneous disproportionation reactions (4), (5),
(6), and a
comproportionation reaction (2) facilitates discharge and regeneration
processes
respectively in the energy cycle. The occurrence of these reactions allows for
a high
power, high efficiency operation based on a fast electrode reaction (X2 + 2e-
= 2X-)
while performing slower steps such as reduction of the oxoanion with the
electro-
generated halide in the three dimensional bulk of the solution which can
accommodate a
higher reaction rate than the two dimensional electrode surface. Although the
use of a
mediator leads in theory to reduced energy efficiency compared to a direct
electrode
reaction, this thermodynamic loss of energy efficiency is often smaller than
the kinetic
loss associated with electrode over-voltage at the same power using oxidants
such as
oxygen or using direct electroreduction of the oxoanions.
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0022] The chemical methods of producing halogen oxoacids are used on an
industrial
scale. In the case of bromic acid, this chemical method consists of solution-
phase
disproportionation of bromine in Ba(OH)2, followed by Ba2+ precipitation with
sulfuric
acid and by evaporation of the excess water. However, this process
irreversibly
consumes Ba(OH)2, H2SO4 and generates BaSO4 waste. Also, this process does not
co-
produce a stoichiometric amount of hydrogen, which is required for the
complete energy
cycle of discharge and regeneration. Thus, this precipitation route does not
meet the
application requirements. An alternative method for preparing up to 40%-50%
bromic
acid via the electrooxidation of aqueous bromine solutions uses a lead dioxide
anode at
the current density of 10-20 mA/cm2 and a potential of +2.1 to +2.2 V versus a
normal
hydrogen electrode. Although this method is chemical and waste free, this
method has
poor energy efficiency and a low throughput.
[0023] Sunlight is a clean and carbon dioxide (CO2) free energy source and the
sun's
energy can be harvested thermally, photoelectrically, photochemically, or
photoelectrochemically. While about 120,000 terawatts (TW) of sunlight, year
averaged
power, reaches the earth, the current total energy consumption of human
civilization is
only about 13 TW. Currently, with a wide scale utilization of solar
technologies, there is
a TRIZ contradiction between cost and efficiency intrinsic to all
commercialized means
of sunlight energy conversion. For example, semiconductor based photovoltaic
solar
panels, for example, polycrystalline silicon photovoltaic solar panels,
multilayer
photovoltaic solar panels, InxGa (1-x) 5e2, etc., are either inefficient or
too expensive.
Photoelectrochemical water splitting into hydrogen (H2) and oxygen (02) using
anatase
TiO2 nanoparticles also suffers from a low efficiency due to the high over
voltage of the
oxygen production centers. Hence, there is a need for a method for converting
sunlight
energy into chemical energy or electric energy at low cost and without
producing any
chemical waste.
[0024] Hence, there is a long felt but unresolved need for an electrochemical
flow
battery that provides for a high energy density, that is, a long driving
range, a high
energy efficiency and power at a low operational and manufacturing cost, and
requires a
11
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
short refill time. Moreover, there is a need for a method and a system that
regenerates an
oxidant and a fuel simultaneously from a discharge fluid, in stoichiometric
amounts,
without consumption of extra chemicals and without generating chemical waste
and by
using electric or solar energy as the primary energy source. Furthermore,
there is a need
for an electrochemical flow battery that provides better safety and stability
by storing on-
board and off-board a stable form of the oxidant.
SUMMARY OF THE INVENTION
[0025] This summary is provided to introduce a selection of concepts in a
simplified
form that are further disclosed in the detailed description of the invention.
This summary
is not intended to identify key or essential inventive concepts of the claimed
subject
matter, nor is it intended for determining the scope of the claimed subject
matter.
[0026] The method and the discharge system disclosed herein address the above
stated
needs for a mechanically refillable, electrochemical flow battery that
provides a high
energy density, a high energy efficiency, and a high electric power at a low
cost, requires
a short refill time, reduces or completely eliminates usage of platinum and
other precious
materials in the electrodes, and reduces the size or completely eliminates the
humidification system. The method and the discharge system disclosed herein
produce
electric power from two fluids, namely, a reducer fluid also referred to as a
"fuel", and
an oxidant fluid comprising an aqueous multi-electron oxidant (AMO), and
release one
or more discharge fluids. The oxidant is an element or a compound in a
reduction-
oxidation reaction that receives one or more electrons from another species or
from an
electrode. The aqueous multi-electron oxidant (AMO) is an oxidant that, in at
least one
of its forms such as an acid form, has a high solubility in water, for
example, over 0.5 M,
and that transfers in a solution-phase redox reaction or in an electrochemical
reaction
more than 1 mole of electrons per 1 mole of the AMO. The AMO can be present in
one
or more of a salt form, an acid form, and other forms, and unless specified
otherwise, the
term "AMO" used herein refers to all these forms. The reducer is an element or
a
compound in a reduction-oxidation reaction that donates one or more electrons
to
12
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
another species or to an electrode. The methods and the systems disclosed
herein use an
aqueous multi-electron oxidant selected from oxides and oxoacids of non-metals
such as
halogens, for example, chlorine, bromine, and iodine in the form of gases,
liquids, melts,
low melting point solids, liquid solutions or suspensions.
[0027] Moreover, the method and the regeneration system disclosed herein
regenerate
an aqueous multi-electron oxidant in a salt form or other form and a reducer
simultaneously from a discharge fluid in a salt form or other form
simultaneously, in
stoichiometric amounts, without consumption of extra chemicals and without
generating
chemical waste. As used herein, the term "discharge fluid" refers to an
exhaust fluid
obtained as a result of an electrochemical discharge process, that is,
electric power
generation, in a flow battery or in a discharge system. In an embodiment, the
regeneration process consumes, for example, electric energy, solar energy,
thermal
energy, radiolytic energy, or any combination thereof. In another embodiment,
the
regeneration process comprises one or more of an electrochemical process,
photoelectrolysis, photolysis, thermolysis, radiolysis, etc. In another
embodiment, the
regeneration process is performed via chemical processes. In an embodiment,
the
method and the regeneration system disclosed herein regenerate a reducer and
an
aqueous multi-electron oxidant in one or more forms simultaneously and in
stoichiometric amounts from a discharge fluid by means of, for example,
electrolysis,
photoelectrolysis, homogeneous solution phase reaction, disproportionation, pH
change,
ion exchange, heterogeneous ion exchange such as using resins, homogeneous ion-
exchange such as via an orthogonal ion migration across laminar flow (OIMALF)
process, and if desired, concentration performed, for example, by evaporation
or reverse
osmosis. As used herein, the term "laminar flow" refers to a type of fluid
flow in which
directions and magnitudes of fluid velocity vectors in different points within
a fluid do
not change randomly in time and in space. Also, as used herein, the term,
"migration"
refers to a movement of an electrically charged object, such as an ion, due to
the action
of an external electric field. Disproportionation is a redox reaction in which
an element,
free or in a compound, is reduced and oxidized in the same reaction to form
different
products. For example, an element with an oxidation state A, not necessarily
A=0, on
13
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
disproportionation, is distributed between several species with different
oxidation states
B, C, etc., which differ from the element's initial oxidation state A, so that
B>A>C. As
used herein, the term "orthogonal" in the phrase "OIMALF", implies that the
vectors of
the laminar flow velocity and of the electric field are not parallel and not
anti-parallel. In
an embodiment, the methods and the systems disclosed herein facilitate halogen
oxoanion/halide conversion in both directions by means of electrochemical
reactions or
other reactions and pH-dependent homogeneous reactions. Disclosed herein is
also a
complete energy cycle comprising a method for generating electric power and a
discharge fluid from one or more forms of an aqueous multi-electron oxidant
and a
reducer using the discharge system, and a regeneration of the aqueous multi-
electron
oxidant and the reducer from the discharge fluid using the regeneration system
and
electric or other energy input. In the methods and systems disclosed herein,
multi-
electron redox couples with high solubilities of reagents and products are
used to
overcome low energy densities of known flow batteries.
[0028] Disclosed herein is a discharge system comprising an oxidant fluid
stored in an
oxidant fluid tank, a reducer fluid stored in a reducer fluid tank, and a
discharge unit.
The discharge unit is also referred to as a "flow battery". The oxidant fluid
is a chemical
or a mixture of chemicals that accepts electrons during a discharge process in
a
discharge mode of operation of the discharge unit. As used herein, the term
"the
discharge mode of operation" refers to a process of releasing the chemical
energy stored
in the discharge system in the form of sustainable electric current and
voltage. The acid
form of the oxidant fluid comprises one or more forms of an aqueous multi-
electron
oxidant (AMO), water, other solvents, an extra acid, and a buffer in their
acid forms. The
other solvent is, for example, a liquid other than water. The AMO is one or a
combination of an oxide of an element such as a halogen, an oxoanion of an
element
such as a halogen, etc. The buffer in the acid form is, for example, one or
more of
phosphoric acid, a dihydrogen phosphate of lithium, a dihydrogen phosphate of
another
cation, a substituted phosphonic acid, buffering agents such as Good's
buffers, and any
combination thereof, capable of maintaining pH of the oxidant fluid at a
value, for
example, below 4. In an embodiment, the buffer is in acid form during
discharge with a
14
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
pH < 7. The extra acid is a strong acid such as sulfuric acid, triflic acid,
another sulfonic
acid, halogen oxoacid, halic acid, etc. In an embodiment, the acid form of the
AMO
serves as the extra acid. The AMO can be pre-mixed with the buffer in the
oxidant fluid.
In an embodiment, the AMO is an oxide or an oxoacid of an element, for
example,
nitrogen, xenon, sulfur, etc. In another embodiment, the AMO is selected from
a group
consisting of a halogen compound such as a halogen oxide, a halogen oxoacid,
etc., an
interhalogen compound, a nitrogen compound, an oxide of nitrogen, a nitrogen
oxoacid,
an oxide of xenon, an oxoacid of xenon, an oxide of a chalcogen such as an
oxide of
sulfur, an oxide of nitrogen or another pnictogen, an oxoacid of nitrogen or
another
pnictogen, a volatile oxide of an element, a fluid oxide of an element, a
soluble oxide of
an element, a volatile oxoacid of an element, a fluid oxoacid of an element, a
soluble
oxoacid of an element, etc., any combination thereof.
[0029] The methods and the systems disclosed herein expand the choice of
oxidants
from one dimensional series of elements into a multidimensional matrix of
compounds,
and more specifically, into oxides of and oxoacids of a halogen, nitrogen and
other
pnictogens, sulfur and other chalcogens, and xenon. That is, the methods and
the systems
disclosed herein expand the one dimensional series of elements such as
halogens into a
multidimensional matrix of oxocompounds such as oxides and oxoacids. The oxide
is a
compound containing oxygen and another element. The halogen oxoacid is a
compound
having a formula HpXq0,-, where X is one of multiple halogens in particular
Cl, Br, and
I, 0 is oxygen, and 1 < p, q, r < 6. In one embodiment, the acid form of the
aqueous
multi-electron oxidant (AMO) is halogen oxoacid, for example, HBr03. The
reagents,
products, and intermediates of the halogen oxoacid reduction are either gases
or liquids
or are soluble in water. If the reagents and products are anions, their cross
over through a
cation exchange membrane is minimal. In an embodiment, the oxoacid is a
compound
having a formula HpX0r, where X is a halogen (C1, Br, I), 1 < p < 6, and 1 < r
< 6. In an
embodiment, the oxoacid is a compound having a formula HX0r, where X is a
halogen,
for example, Cl, Br, I, and 1 < r < 4.
[0030] In an embodiment, the aqueous multi-electron oxidant (AMO) is a
nitrogen
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
oxide having a formula Nx0õ, where x = 1 or 2 and 1 < n < 5. In another
embodiment,
the AMO is a nitrogen oxoacid having a formula HkNi0m, where 1 < k, 1, m < 3.
In
another embodiment, the nitrogen oxoacid is a compound having a formula HNOõ,
where 1 < n < 3. In an embodiment, the oxoacid is a compound having a formula
HpXgOr, where X is one of multiple halogens, nitrogen, other pnictogens,
chalcogens,
xenon, or other element, and where 1 < p, q, r < 6. In an embodiment, the acid
form of the
AMO is chloric acid which forms an aqueous room temperature solution, for
example, up
to about 40% w/w. In an embodiment, the acid form of the AMO is bromic acid
which
forms an aqueous room temperature solution, for example, up to about 55% w/w.
In
another embodiment, the acid form of the AMO is iodic acid which forms an
aqueous
room temperature solution, for example, up to about 74% w/w. In another
embodiment,
the acid form of the AMO is perchloric acid which forms an atmospheric aqueous
azeotrope, for example, about 72.5% w/w. In another embodiment, the AMO is
nitric acid
which forms an atmospheric aqueous azeotrope with, for example, about 68.4%
w/w.
Halogen oxoacids allow for energy-efficient and waste-free routes to their
regeneration
from the discharge fluid.
[0031] The reducer fluid, also referred herein as a "fuel", is a chemical that
donates
electrons during the discharge process. The reducer is, for example, hydrogen.
In an
embodiment, the reducer is selected from a group consisting of ammonia,
hydrazine,
hydroxylamine, phosphine, methane, a hydrocarbon, an alcohol such as methanol,
ethanol, etc., an aldehyde, a carbohydrate, a hydride, an oxide, a sulfide,
another organic
and inorganic compound, or any combination thereof, with each other, with
water, or
with another solvent. A hydrogen reducer is used herein because the hydrogen
reducer
can be regenerated from the discharge fluid along with the aqueous multi-
electron
oxidant (AMO) with a high efficiency and without irreversible consumption of
other
chemical and without generating chemical waste.
[0032] The discharge unit of the discharge system comprises a stack of
multiple
electrolytic cells also referred to as an "electrolytic cell stack". Each
electrolytic cell
comprises a 5-layer electrolyte-electrode assembly and half of a bipolar
plate/1 endplate.
16
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
The 5-layer electrolyte-electrode assembly is flanked on each side by a
bipolar plate or
an endplate. The 5-layer electrolyte-electrode assembly comprises a 3-layer
electrolyte-
electrode assembly flanked by a negative diffusion layer on the negative
electrode side and
a positive diffusion layer on the positive electrode side. The 3-layer
electrolyte-electrode
assembly comprises an electrolyte layer interposed between or flanked by a
positive
electrode layer and a negative electrode layer. The 3-layer electrolyte-
electrode assembly
and/or the 5-layer electrolyte-electrode assembly are herein referred to as an
"electrolyte-electrode assembly".
[0033] In an embodiment, the electrolytic cell stack is configured as a planar
cell stack
comprising planar electrolytic cells. The planar electrolytic cells in the
planar cell stack
are connected electrically in series so that the voltage of the electrolytic
cell stack is the
sum of the voltages of the electrolytic cells. Each planar electrolytic cell
shares one
bipolar plate with an adjacent planar electrolytic cell. One side of a bipolar
plate contacts
a positive side of one planar electrolytic cell and another side of the
bipolar plate contacts
a negative side of the adjacent planar electrolytic cell. The bipolar plates
and the
endplates are equipped with channels for delivering reagents, that is, the
oxidant fluid and
the reducer fluid to the electrolyte-electrode assemblies in the electrolytic
cell stack and
for removing the products, that is, one or more discharge fluids. The planar
cell stack is
further flanked by a pair of endplates. The endplates are further equipped
with ports for
the oxidant fluid, the reducer fluid, and the discharge fluid, and electric
connections.
[0034] In an embodiment, the electrolyte layer of the electrolyte-electrode
assembly is
composed of a material capable of ionic conduction, for example, protonic
conduction but
not electronic conduction. In another embodiment, the electrolyte layer of the
electrolyte-
electrode assembly is composed of an ionomer, a solid ion conductor, a solid
proton
conductor, or a liquid under laminar flow. The electrolyte is compatible with
water, the
aqueous multi-electron oxidant (AMO), the reducer, and the products. In
another
embodiment, the electrolyte layer of the electrolyte-electrode assembly is
composed of a
material comprising a chemical moiety selected from a group consisting of one
or more
proton donor moiety or proton acceptor moiety. In an embodiment, the
electrolyte
17
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
material is a cation-conductive polymer, for example, a polyperfluorosulfonic
acid such
as Nafion of E. I. du Pont de Nemours and Company Corporation, Hyflon Ion of
Ausimont S.R.L. Corporation, Aciplex of Asaki Kasei, Flemion of AsahiGlass,
Aquivion of Solvay-Solexis, etc. In another embodiment, the electrolyte layer
in the
electrolyte-electrode assembly is made of a composite material such as
GoreSe1ect of
W.L.Gore and Associates, Inc., or of an ionically conducting liquid retained
in the pores
of a solid matrix. In another embodiment, the electrolyte layer of the
electrolyte-electrode
assembly comprises a material with a cationic conduction exceeding an anionic
conduction of the material. Such cation-selective conductivity of the
electrolyte is
beneficial for both discharge and regeneration systems since electrolyte
reduces the
crossover of the AMO and of its reduction products and intermediates to the
negative
electrode.
[0035] In the discharge unit, during the discharge mode of operation, the
positive
electrodes of the electrolyte-electrode assemblies are supplied with the
oxidant fluid
containing one or more forms of the aqueous multi-electron oxidant and the
negative
electrodes of the electrolyte-electrode assemblies are supplied with the
reducer fluid
containing the reducer during the discharge mode of operation. The bipolar
plate provides
an electron pathway from one electrolytic cell in the electrolytic cell stack
to the next
electrolytic cell. The bipolar plates also supply reactants to the 5-layer
electrolyte-
electrode assemblies and remove the products. The endplates flank the
electrolytic cell
stack. The inner sides of the endplates operate in a manner similar to the
bipolar plates.
The endplates comprise inlet ports for adding reagents, outlet ports for
removing
products, and electric connections to an external electric circuit. The
endplates provide
electric connections and flow connections from the electrolytic cell stack to
the other
components of the discharge system.
[0036] During the discharge mode of operation, the reagents, that is, the
oxidant fluid
and the reducer fluid in the discharge system are converted into products to
produce
electric current through the electrolytic cell stack and through the external
electric circuit.
More specifically, the reagents in the oxidant fluid and in the reducer fluid
are converted
18
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
into products to produce an electric current through the external circuit and
through the
bipolar plates and an ionic current through the electrolyte layers. The
oxidant fluid and
the reducer fluid are supplied from their respective tanks which are
periodically filled
from an external source, for example, the regeneration system. The discharge
system
disclosed herein operates with an external electric circuit operably connected
to the
electrolytic cell stack of the discharge unit. During the discharge mode of
operation, the
external electric circuit comprising, for example, an electric engine
connected to the
discharge unit consumes the electric power generated by the discharge unit. In
the
discharge unit, the reducer is configured to donate the electrons to the
negative
electrodes, and the aqueous multi-electron oxidant (AMO) is configured to
accept the
electrons at the positive electrodes for producing an electric current in the
external
electric circuit that connects the positive endplate and the negative
endplate, and for
simultaneously producing an ionic current through the electrolyte layer of an
electrolytic
cell or the electrolyte layers of the electrolytic cells of the electrolytic
cell stack of the
discharge unit. In an embodiment, a solution-phase reaction facilitates one or
more
discharge reactions on the positive electrode of the electrolyte-electrode
assembly. In an
embodiment, the solution-phase reaction disclosed herein is, for example, a pH-
dependent solution-phase comproportionation, a solution-phase redox mediated
catalysis,
etc. As used herein, the term "comproportionation" is a redox reaction in
which an
element, free or in compounds, with oxidation states A and C, is converted
into another
substance or substances in which the element's oxidation states are B, such
that A>B>C.
In an embodiment, the rate of the solution-phase comproportionation depends on
the pH
of the solution.
[0037] The power generation in the discharge unit may benefit from a catalyst,
a redox
mediator, etc., for facilitating a charge transfer between the electrodes of
the electrolyte-
electrode assembly and the aqueous multi-electron oxidant (AMO) and the
reducer. In an
embodiment, a halide mediator, for example, a bromide mediator or a chloride
mediator
facilitates one or more discharge reactions on the positive electrode of the
electrolyte-
electrode assembly. For example, a redox mediator such as a halogen/halide
couple
facilitates a charge transfer between the positive electrode of the
electrolyte-electrode
19
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
assembly and the AMO. In another embodiment, multiple immobilized
heterogeneous
mediators, immobilized heterogeneous catalysts, homogeneous mediators, or
homogeneous catalysts facilitate a charge transfer between the positive
electrode of the
electrolyte-electrode assembly and the AMO. In another embodiment, a catalyst
selected
from a group consisting of a homogeneous catalyst, a heterogeneous catalyst, a
redox
mediator catalyst, or any combination thereof, facilitates one or more
discharge reactions
on the positive electrode of the electrolyte-electrode assembly. In another
embodiment,
one or more forms of a redox mediator, a product of an electrode reaction, an
acid, or any
combination thereof accelerates a rate of discharge of the AMO during one or
more
discharge reactions via a solution-phase reaction. In an embodiment, a product
of the
discharge reaction facilitates the discharge reaction via comproportionation.
In another
embodiment, a catalyst, for example, ruthenium dioxide (Ru02), lead dioxide
(Pb02), or
a platinoid electrocatalyst facilitates one or more electrochemical reactions
on the
positive electrode of the electrolyte-electrode assembly. In another
embodiment, a
platinoid electrocatalyst facilitates one or more electrochemical reactions on
the negative
electrode of the electrolyte-electrode assembly. The discharge system stores
the energy in
reducer and oxidant fluid tanks or containers and produces electric power on
demand
using the discharge unit, for stationary, mobile, and portable devices that
require
electrical power.
[0038] In an embodiment, the discharge unit disclosed herein operates in a
regenerative
mode or electric recharge mode or as a secondary flow battery. In the
regenerative mode
of operation, one or more of reagents and intermediates are regenerated within
the
discharge unit, by applying a voltage of the polarity opposite to the polarity
observed
during the discharge mode of operation to the terminals of the external
electric circuit.
For example, an intermediate such as bromine can be regenerated from bromide
present
in the discharge fluid using the discharge unit, if the discharge unit is
operated under
reverse polarity.
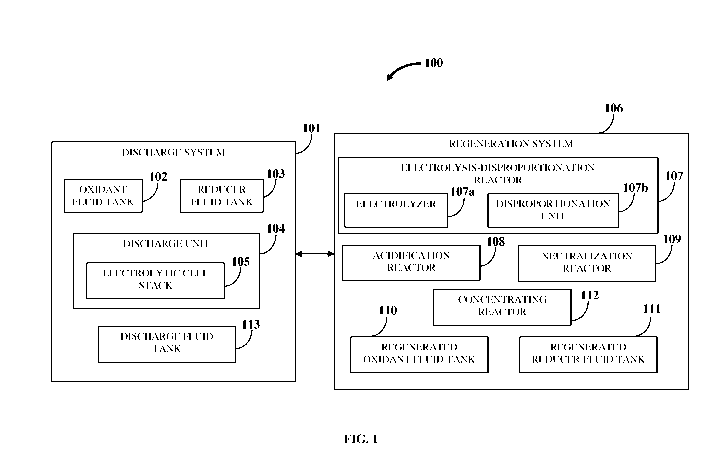
[0039] Also, disclosed herein is a regeneration system configured to
regenerate one or
more forms of the oxidant fluid and the reducer in stoichiometric amounts from
the
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
discharge fluid produced by the discharge unit using electric power. The
regeneration
system comprises, for example, an electrolysis-disproportionation (ED)
reactor, storage
tanks such as a regenerated oxidant fluid tank and a regenerated reducer fluid
tank for
storing the regenerated oxidant fluid and the regenerated reducer fluid
respectively,
optionally a neutralization reactor, for example, an ion exchange reactor such
as an
orthogonal ion migration across laminar flow (OIMALF) reactor, one or more
separation
reactors, and a concentrating reactor. In an embodiment, the neutralization
reactor
comprises a mixing reactor. In an embodiment, the regeneration system is
configured for
a batch mode of operation. In another embodiment, the regeneration system is
configured for a flow mode of operation. In another embodiment, the
regeneration
system is configured for a cyclic flow mode of operation. In another
embodiment, the
ED reactor is configured for a cascade flow mode of operation and comprises a
stack of
regeneration flow cells. The ED reactor performs either electrolysis or
electrolysis and a
solution phase reaction, for example, disproportionation, in one or more sub-
reactors.
The sub-reactors are also referred herein as individual cells of a stack or
regeneration
flow cells or cells. The separation reactors of the regeneration system are
gas-liquid
separators and are used to separate gases from the liquids during a
regeneration process.
[0040] In an embodiment, the electrolysis-disproportionation (ED) reactor
comprises,
for example, an electrolysis unit or an electrolyzer and a disproportionation
unit. In
another embodiment, both electrolysis and disproportionation are performed
within a
single ED flow cell which does not comprise separable electrolysis and
disproportionation units. Various configurations of the ED reactor can be
operated in a
batch mode, a cascade flow mode, a cyclic flow mode, and any combination
thereof. In
an embodiment, the configuration of the ED reactor is similar to that of a
polymer
membrane fuel cell stack with a modification of a graded catalytic layer on
the negative
electrode which prevents the electroreduction of relevant forms of the aqueous
multi-
electron oxidant (AMO) while allowing for the hydrogen evolution reaction and
alkalization to proceed and to the electrolytic cell or of the electrolytic
cell stack of the
discharge unit. The ED reactor comprises a number of flow cells connected, for
example, electrically in series and flow-wise in parallel. Such stack-type ED
reactor can
21
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
be operated in a cyclic flow mode, a cascade flow mode, a batch mode, and any
combination thereof. The ED reactor can be further configured for an AMO-on-
negative
electrode mode of operation, also referred to as an "AMO-on-negative mode of
operation", wherein the negative electrode comprises a multilayer or a graded
catalytic
layer configured to prevent the electroreduction of relevant forms of the AMO
while
allowing for the hydrogen evolution reaction and alkalization to proceed, or
for a no-
AMO-on-negative electrode mode of operation, also referred to as a "no-AMO-on-
negative mode of operation", wherein the base produced on the negative
electrode is
mixed with one or more forms of oxidant fluid or discharge fluid without
bringing the
AMO in contact with the negative electrode.
[0041] In an electrolysis-disproportionation (ED) reactor configured for the
aqueous
multi-electron oxidant (AMO)-on-negative mode of operation in the cascade flow
mode
of operation, one or a mixture of a regenerated solution and the discharge
fluid passes
through a cascade or a series or stack of electrolysis-disproportionation (ED)
reactors,
that is, through the negative electrode of the first cell with a graded
catalytic layer to
allow for the hydrogen evolution reaction and the buffer alkalization to
proceed while
suppressing the electroreduction of all forms of the AMO, to the separator
that removes
H2 from the regenerated fluid, to the positive electrode of the first cell,
wherein the
process of electrolysis-disproportionation leading to one or more forms of the
AMO
takes place, then to the negative electrode of the second cell, then to the
positive
electrode of the second cell, and so on. The reducer and the base generated at
the
negative electrode of each of the ED reactors are separated in the separation
reactor,
where the base is returned into the mixing reactor preceding this ED reactor
and the
reducer is collected in a reducer container.
[0042] In the cyclic flow mode, as few as one regeneration flow cell can be
used with
an alternating flow between the negative and positive electrodes through the
valves
while releasing the produced H2 or other fuel through the separator. In
another
embodiment, referred herein as an aqueous multi-electron oxidant (AMO)-on-
negative
mode of operation, the problem of aqueous multi-electron oxidant (AMO)
reduction on
22
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
the negative electrode or electrodes in the electrolysis-disproportionation
(ED) reactor is
avoided by generating a base solution by passing a fluid such as pure water
free of the
AMO through the negative electrode. In this case, the base formed at the
negative
electrode via H20 + e- + M+ = 1/2 H2 MOH, where M is a cation, for example,
Li, is
mixed in one or more mixing reactors with the fluid produced at the positive
electrode
allowing the disproportionation to occur. This process can be performed in a
batch mode
of operation, a cascade flow mode of operation, and a cyclic flow mode of
operation.
This process avoids the possibility of AMO reduction on the negative electrode
but
requires removal of the excess water from the regenerated AMO. The water is
dragged
though the membrane along with M+ from the positive electrode to the negative
electrode and causes the dilution of the stock AMO solution such as LiBrO3
solution.
The water removal process can be performed by evaporation, reverse osmosis,
and other
methods. In an embodiment, the water removal process is performed in a
concentrating
reactor.
[0043] In the cyclic flow mode, a regenerated solution or the discharge fluid
is cycled
between the mixing reactor and the electrolysis-disproportionation (ED)
reactor until the
desired degree of conversion is achieved. An ED reactor configured for the
cyclic flow
mode has a lower capital cost but requires a longer regeneration time. The ED
reactor(s)
configured for the cascade flow mode has a higher capital cost but is capable
of a faster
regeneration. Multiple combinations of cyclic and cascade flow modes are
implemented
for a hardware combination that involves more than one series of
neutralization reactors,
ED reactors, and separation reactors of one series connected to the
neutralization reactor
of the same or the next series. The concentrating reactor concentrates a
solution of the
aqueous multi-electron oxidant (AMO) in a salt form or other forms to remove
water or
other solvents from a dilute fluid that enters the concentrating reactor and
releases a
concentrated fluid and water or another solvent.
[0044] Also, disclosed herein is a method for producing electric power from
the
reducer and the oxidant fluid comprising the aqueous multi-electron oxidant
(AMO) and
for simultaneously generating the discharge fluid. The method disclosed herein
provides
23
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
the discharge system comprising the oxidant fluid, the reducer fluid, and the
discharge
unit. The method for producing electric power facilitates electrochemical
reactions in the
discharge unit. Discharge occurs by transferring electrons, either directly or
via a
mediator, from the positive electrode of the 5-layer electrolyte-electrode
assembly to the
AMO and from the reducer to the negative electrode of the 5-layer electrolyte-
electrode
assembly to produce electric power, that is, a sustainable electric current
and a
sustainable electric voltage in the external electric circuit connected to the
terminals of
the discharge unit accompanied by electric current of ions through the
electrolyte layer
by electrochemical reactions on the electrodes. The discharge is facilitated
on the
positive electrode of the 5-layer electrolyte-electrode assembly, for example,
by one or
more of an electron transfer, electrolysis, electrocatalysis, a solution-phase
chemical
reaction, a solution-phase comproportionation, a solution-phase redox
catalysis, an acid-
base catalysis, lowering the solution pH, and any combination thereof.
[0045] The discharge unit consumes the aqueous multi-electron oxidant (AMO)
and
the reducer to produce electric power and to generate the discharge fluid. The
discharge
fluid comprises, for example, one or more of water, one or more forms of the
buffer, a
halogen, one or more halogen oxoanions, hydrogen ions, halide ions, a halogen
oxoacid,
a salt of halogen oxoacid, an extra acid, a counter cation, or any combination
thereof.
Since the discharge fluid coming out of the discharge unit is not water or not
only water,
the discharge fluid is not disposed into the surrounding environment but
collected in a
discharge container to be regenerated later into the reducer fluid and an
oxidant fluid
comprising the AMO. In an embodiment, a certain amount of intermediate oxidant
is
regenerated on the positive electrode in the discharge unit, for example, Br- -
le = 1/2 Br2
from the discharge fluid by reversing a polarity of an electric current
flowing through the
discharge unit during discharge of the discharge unit. This process is useful
for
regenerative breaking.
[0046] Also, disclosed herein is a method for regenerating the aqueous multi-
electron
oxidant (AMO) and the reducer in stoichiometric amounts from the discharge
fluid using
an external energy source. The method disclosed herein reuses all the required
chemicals
24
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
in the complete discharge-regeneration cycle, does not consume stoichiometric
amounts
of external chemicals, and does not generate stoichiometric amounts of
chemical waste.
The regeneration system is capable of performing the required electrochemical
and
chemical reactions for the conversion of the discharge fluid from the
discharge unit back
into the oxidant fluid and the reducer fluid. The regeneration system
neutralizes, if
necessary, the discharge fluid with an excess of a base form of a buffer in
the
neutralization reactor to produce a solution of a salt form of the discharge
fluid. The
regeneration system performs decomposition of one or more forms of the
discharge fluid
comprising, for example, water and bromide anion, into a reducer such as H2
and an
intermediate oxidant such as Br2. The decomposition can be performed by means
of one
or more of the following: electrolysis, photoelectrolysis, photolysis,
thermolysis,
radiolysis, etc. In an embodiment, the regeneration system electrolyzes one or
more
forms of the discharge fluid comprising, for example, bromide, yielding an
intermediate
oxidant such as bromine at a positive electrode in the electrolysis-
disproportionation
(ED) reactor. The decomposition process also releases the reducer such as H2
and a base
such as hydroxide MOH of the buffer's cation M or the basic form of the
buffer, for
example, M2HPO4. In the case where the decomposition is by electrolysis, the
reducer
and the base are released at the negative electrode of the ED reactor. The
reducer and the
base are separated in the separation reactor. In the no-AMO-on-negative mode
of
operation, the base is sent to the first mixing reactor or the neutralization
reactor to
neutralize the incoming discharge fluid to produce an alkaline discharge
fluid. At the
positive electrode of the ED reactor, the electrolysis process releases an
intermediate
oxidant, such as Br2, which reacts with the excess of the base to produce the
salt form of
the AMO such as MBr03. The conversion of the intermediate oxidant, for
example,
bromine into the original aqueous multi-electron oxidant (AMO) in the salt
form such as
bromate at the positive electrode of the ED reactor can be facilitated not
only by
disproportionation but also by a mediated oxidation using a solution phase
mediator such
as a chlorine/chloride couple, or electrocatalysts such as those comprising
one or more
of the following: lead dioxide, ruthenium dioxide, dimensionally stable anode
materials,
perovskites, graphite, glassy carbon, conductive diamond, other carbonaceous
materials,
etc. All these methods of facilitation can be used together.
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0047] The aqueous multi-electron oxidant (AMO) is regenerated via an electron
transfer at the positive electrode followed by disproportionation of the
intermediate
oxidant and the reducer is regenerated via an electron transfer at the
negative electrode
of the electrolysis-disproportionation (ED) reactor. The buffer maintains the
pH of the
discharge fluid in the optimal range, for example, between pH 7 and 9 for
disproportionation. The base component of the buffer is selected, for example,
from a
group comprising hydroxide, hydrogen phosphate, one or more forms of one or
more of
Good's buffers, an amine, a tertiary amine, a nitrogen heterocycle, a
substituted
phosphonate, and any combination thereof. The cation component of the buffer,
if
necessary, is selected, for example, from a group comprising lithium, sodium,
other
alkali metal cations, alkali earth metal cations, other inorganic cations,
organic cations,
etc.
[0048] In an embodiment, the oxidant fluid produced in the regeneration
system, for
example, comprising LiBr03, is further concentrated via the removal of water
within the
regeneration system to produce oxidant fluid for future use in the discharge
system. The
removal of water from the ionic components of the oxidant fluid, also referred
herein as
concentrating, is performed by one or a combination of the following:
evaporation,
pervaporation, reverse osmosis, dialysis, and other methods known in the art.
[0049] In an embodiment, the regeneration of the aqueous multi-electron
oxidant
(AMO) and/or the reducer is facilitated, for example, by an electrocatalyst, a
solution-
phase redox mediator, a pH-dependent solution-phase disproportionation, etc.,
or any
combination thereof. The conversion of the salt form of the AMO into the acid
form of
the AMO in the acidification reactor, also referred herein as the "ion
exchange reactor"
is facilitated by an acid, a buffer, hydrogen electrooxidation, other proton-
releasing
electrooxidation, electrochemical hydrogen evolution, ion-exchange on solids,
ion
exchange in solution, orthogonal ion migration across laminar flow, or any
combination
thereof.
26
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0050] In an embodiment, a mediator such as chlorine facilitates regeneration
of the
aqueous multi-electron oxidant (AMO) from one or more forms of discharge fluid
in the
electrolysis-disproportionation (ED) reactor. The ED reactor or the ED
reactors are
configured to operate in one of multiple modes comprising, for example, a
batch mode, a
single pass mode, a cyclic flow mode, and a combination thereof. If an
orthogonal ion
migration across laminar flow (OIMALF) reactor is used as ion exchange reactor
or as
an acidification-neutralization reactor, the regeneration system is configured
to support
the operation of the OIMALF reactor in a flow through mode, for example, using
additional storage tanks. An OIMALF reactor can work simultaneously as one or
more
OIMALF reactors are operated in a single pass flow-through mode or a cyclic
flow-
through mode but not in the batch mode, although an OIMALF reactor working in
one
of the flow modes can be used in combination with an ED reactor working in a
batch
mode.
[0051] In other embodiments, one or more forms of the aqueous multi-electron
oxidant
(AMO) and/or the reducer are regenerated, for example, using electrolysis, an
ion
exchange on solids, an ion exchange in a liquid, ion exchange in the discharge
fluid or in
an intermediate regenerated solution, pH-dependent solution-phase
disproportionation, or
any combination thereof. Ion exchange in a liquid such as water with a
dissolved salt
form of the AMO and the dissolved salt form of the buffer is performed, for
example, by
an electric field driven orthogonal ion migration across laminar flow (OIMALF)
process
which is substantially similar to eluent suppression in anion chromatography.
The ion
exchange process occurs before and/or after and outside of any series of the
neutralization-electrolysis-disproportionation loops. The regeneration of the
AMO from
the discharge fluid or from the intermediate regenerated solution comprises
neutralizing
an acid of the discharge fluid or the intermediate regenerated solution with a
base to
obtain an alkaline discharge fluid. The required base is produced, for
example, at the
negative electrode(s) of one or many electrolysis-disproportionation (ED)
reactors. The
regeneration system then converts the alkaline discharge fluid to the neutral
oxidant fluid,
that is, a liquid comprising water, the AMO, and one or more forms of the
buffer, for
27
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
example, via electrolysis, pH dependent solution phase disproportionation and
orthogonal
ion migration across laminar flow processes.
[0052] The reducer, for example, hydrogen, is co-produced in a stoichiometric
amount
with one or more forms of the aqueous multi-electron oxidant (AMO) in the
electrolysis-
disproportionation (ED) reactor. The conversion of the salt form of the AMO
into the
acid form of the AMO is performed using an acidification reactor such as the
ion
exchange reactor. If the ion exchange reactor is, for example, an orthogonal
ion migration
across laminar flow (OIMALF) reactor, the conversion comprises consuming
electric
power and recycling the hydrogen released on one or more negative electrodes
of the
OIMALF reactor and electro-oxidized on one or more positive electrodes of the
OIMALF
reactor. In an embodiment, the hydrogen produced in an ED reactor is flown
through the
flow field of the positive electrode of one or many OIMALF reactors and
combined with
the hydrogen produced at a negative electrode of one or many OIMALF reactors
either
before or after one or many OIMALF reactors. The regeneration of the reducer
and the
oxidant fluid by the ED-OIMALF method occurs by using an external electric
energy
input and without consumption or generation of external chemicals. Also,
disclosed
herein is the use of the pH-dependence of the spontaneous homogeneous
disproportionation of a halogen and comproportionation of a halide and a
halogen
oxoanion in order to facilitate the electrode reactions on the positive
electrodes during
regeneration and discharge. The method disclosed herein facilitates the
forward and
reverse halogen oxoanion/halide conversion and other redox processes involving
oxoanions via pH-dependent homogeneous reactions.
[0053] Also, disclosed herein is an embodiment of the discharge system
comprising one
or more forms of an oxidant fluid comprising one or more forms of an aqueous
multi-
electron oxidant (AMO), for example, an aqueous solution comprising LiBr03,
stored in
an oxidant fluid tank, one or more forms of a reducer fluid comprising one or
more forms
of a reducer such as hydrogen stored in a reducer fluid tank, an acidification
reactor,
optionally a neutralization reactor, a discharge unit, and a discharge fluid
tank to collect
the discharge fluid for future regeneration or disposal. In an embodiment, the
28
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
acidification reactor and the neutralization reactor are functionally combined
as an
orthogonal ion migration across laminar flow (OIMALF) reactor. In another
embodiment,
the neutralization reactor is integrated with the acidification reactor into
the OIMALF
reactor. In another embodiment, the neutralization reactor is an OIMALF
reactor. In this
embodiment, the acidification process, for example, an ion exchange process is
performed on-board in the discharge system rather than off-board, in order to
improve the
stability and safety of the systems disclosed herein. The discharge system
disclosed
herein is configured to operate in an electric partial recharge mode for
facilitating
regenerative breaking when the discharge system powers an electric vehicle.
During the
partial recharge mode, the reducer is produced on the negative electrode of
the electrolyte-
electrode assembly and an intermediate oxidant is produced on the positive
electrode of
the electrolyte-electrode assembly.
[0054] In the embodiment of the discharge system with improved safety, a
neutral
oxidant fluid comprising, for example, LiBrO3 is stored in the oxidant fluid
tank. The
discharge system initially converts the aqueous multi-electron oxidant (AMO)
in the salt
form such as LiBrO3 in the neutral oxidant fluid into the AMO in the acid form
such as
HBr03, found in the acidic oxidant fluid, using the acidification reactor. In
an
embodiment, the conversion of the salt form of the AMO into the acid form of
the AMO
is performed via an ion exchange process. The ion exchange process can be
performed
via a multiphase flow process, for example, based on ion-exchange resins or
via a single-
phase flow process such as an electric field driven orthogonal ion migration
across
laminar flow (OIMALF) process in the OIMALF reactor. In the case where the
acidification reactor is an OIMALF reactor, the acidification of the oxidant
fluid is
accompanied by a simultaneous neutralization of the acidic discharge fluid
while
recycling the reducer such as H2 produced at one or more negative electrodes
of the
OIMALF reactor and consumed at one or more positive electrodes of the OIMALF
reactor. The OIMALF process is substantially similar to eluent suppression of
ion
chromatography. The OIMALF reactor converts the neutral oxidant fluid into an
acidic
oxidant and an acidic discharge fluid into a neutral discharge fluid
simultaneously.
29
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0055] The orthogonal ion migration across laminar flow (OIMALF) reactor
comprises
an OIMALF cell stack which is configured similar to a polymer electrolyte fuel
cell
(PEFC) stack but with a liquid electrolyte flowing between two ionically
conducting
membranes. Only the outer sides, which are not in contact with the flowing
liquid, of the
membranes are coated with catalytic layers. The OIMALF reactor comprises flow
cell
assemblies, endplates, and bipolar plates. Each flow cell assembly of the
OIMALF
reactor comprises a couple of ion exchange membranes, an intermembrane flow
field, a
positive electrode layer and a negative electrode layer, and two porous
diffusion layers.
The ion exchange membranes are coated with a catalytic layer only on their
outer sides
which are not in contact with fluids comprising the aqueous multi-electron
oxidant
(AMO). The intermembrane flow field is interposed between the ion exchange
membranes and comprises multiple flow channels. The positive electrode layer
and the
negative electrode layer flank outer surfaces of the ion exchange membranes.
The two
porous diffusion layers flank the outer surfaces of the ion exchange membranes
and are in
an electric contact with the adjacent bipolar plates or endplates. The ion
exchange
membranes comprise a positive side ion exchange membrane and a negative side
ion
exchange membrane positioned parallel to each other. The positive electrode
layer is
configured for hydrogen oxidation reaction and the negative electrode layer is
configured
for hydrogen evolution reaction. Further variations of the electrode layers,
for example,
additional macro-porous and micro-porous layers are possible and known in the
art of
hydrogen polymer electrolyte fuel cell anodes and hydrogen polymer electrolyte
water
electrolyzer cathodes.
[0056] The acidic oxidant fluid comprises one or more of water, one or more
forms of
the aqueous multi-electron oxidant (AMO), for example, an acid or a salt form
or as a
combination thereof, an extra acid, and one or more of multiple counter
cations. The
AMO comprises one or a combination of halogens, halogen oxides, halogen
oxoanions,
and salts and acids of the halogen oxoanions. The halogen oxoanions comprise,
for
example, one or more of hypochlorite, chlorite, chlorate, perchlorate,
hypobromite,
bromite, perbromate, hypoiodite, iodite, iodate, and periodate. In an
embodiment, the
halogen oxoanion is bromate. The counter cations comprise, for example, alkali
metal
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
cations, alkali earth metal cations, and organic cations. In an embodiment,
one of the
counter cations is lithium. In another embodiment, one of the counter cations
is sodium.
A buffer may be present in the oxidant fluid if the buffer is carried over
from the
regeneration process. In an embodiment, the buffer functions as the extra
acid. The buffer
is in one of its forms, for example, one or more of monohydrogen phosphate, a
3-(N-
morpholino)propanesulfonate, a 3-(N-morpholino)ethanesulfonate, a substituted
phosphonate, an amine, a tertiary amine, a nitrogen heterocycle, other base
with an acid
dissociation constant pKa between, for example, 6 and 9. The extra acid is,
for example,
one or more of a phosphoric acid, a 3-(N-morpholino)propanesulfonic acid, a 3-
(N-
morpholino)ethanesulfonic acid, a methanesulfonic acid, a triflic acid, a
substituted
sulfonic acid, a substituted phosphonic acid, a perchloric acid, a sulfuric
acid, a molecule
comprising sulfonic moieties and phosphonic moieties, and an acid with a pKa <
2. The
AMO in one or several forms can be pre-mixed with one or several components of
the
buffer in the oxidant fluid in the storage tank, in the acidification reactor
or in both. In an
embodiment, the AMO is selected from a group consisting of a halogen compound
such
as a halogen oxide, a halogen oxoacid, a water-soluble salt of halogen
oxoacid, and any
combination thereof. The AMO can be stored on-board and off-board in the acid
or in
one or more salt forms on in a combination thereof. The salt forms of the AMO
are
considered over the acid form due their better stabilities, provided that they
have high
solubilities.
[0057] In an embodiment, the discharge system also performs complete or
partial
conversion of a stable form of the aqueous multi-electron oxidant (AMO), such
as
LiBr03, into an active form of the AMO, such as HBr03, using one or more
disclosed
acidification processes, for example, one or any combination of the following:
addition of
a stored acid, ion exchange on resins, and the orthogonal ion migration across
laminar
flow (OIMALF). The acidification process is performed either in a dedicated
acidification reactor, which can be an OIMALF reactor, or in a suitably
modified other
reactor, such as the discharge unit itself or in both. In an embodiment, the
discharge
system also performs complete or partial conversion of the discharged fluid,
such as one
containing HBr or another acid, into a less corrosive form, such as LiBr,
using one or
31
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
more disclosed neutralization processes, such addition of a stored base or/and
an
OIMALF process. The neutralization process is performed in a dedicated reactor
such as
a neutralization reactor, which can be an OIMALF reactor, or in a suitably
modified other
reactor, such as discharge unit. In an embodiment, the neutralization reactor
comprises a
mixing reactor.
[0058] Also, disclosed herein is an embodiment of the method for producing
electric
power from an aqueous multi-electron oxidant (AMO) and a reducer and for
simultaneously generating a discharge fluid. The method disclosed herein
provides the
discharge system comprising one or more forms of a reducer fluid, one or more
forms of
an oxidant fluid, a discharge unit, and an acidification reactor. The method
disclosed
herein facilitates discharge of the discharge unit for producing electric
power from a
neutral oxidant fluid comprising one or more forms of the aqueous multi-
electron
oxidant, and from the reducer fluid comprising one or more forms of the
reducer. The
facilitation of the discharge comprises: lowering pH of the neutral oxidant
fluid in the
acidification reactor for generating an acidic oxidant fluid; transferring
electrons from the
positive electrode of the electrolyte-electrode assembly to the aqueous multi-
electron
oxidant in the acidic oxidant fluid; and transferring electrons from the
reducer fluid to the
negative electrode of the electrolyte-electrode assembly to produce electric
power in the
external electric circuit operably connected to the terminals of the discharge
unit and to
generate an acidic discharge fluid on consumption of the acidic oxidant fluid
and the
reducer fluid. The transfer of the electrons from the positive electrode of
the electrolyte-
electrode assembly to the aqueous multi-electron oxidant in the acidic oxidant
fluid is
performed at a high current density and at low flow rates in an ignition mode
of operation
of the discharge system. A limiting current of the transfer of the electrons
from the
positive electrode of the electrolyte-electrode assembly to the aqueous multi-
electron
oxidant in the acidic oxidant fluid in an ignition regime is limited, for
example, by a
mass-transport of the aqueous multi-electron oxidant, a mass-transport of
acidic protons,
and a rate of comproportionation. The acidic discharge fluid comprises, for
example, one
or more of water, a halide, a hydroxonium cation, an extra acid, and one or
more counter
cations. In an embodiment, the stability of the acidic oxidant fluid is
maintained by
32
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
performing an ignition regime in the discharge system at low acid
concentrations in the
acidic oxidant fluid. In an embodiment, the method disclosed herein further
comprises
optionally neutralizing the acidic discharge fluid in the neutralization
reactor of the
discharge system to produce a neutral discharge fluid. The concentration of
one or more
forms of the aqueous multi-electron oxidant in the neutral oxidant fluid or
the acidic
oxidant fluid supplied to the discharge unit is, for example, above 1M, 2M,
5M, or 10M.
The concentration of acidic protons in the acidic oxidant fluid supplied to
the discharge
unit is, for example, below 0.1M, 0.5M, 1M, 2M, 5M, or 10M. The concentration
of
acidic protons in the acidic oxidant fluid stored in the discharge system is,
for example,
below 0.1M, 0.5M, 1M, 2M, or 5M. In an embodiment, the method disclosed herein
further comprises regenerating a certain amount of an intermediate oxidant and
the
reducer in the discharge unit from the acidic discharge fluid by applying an
electric
current of a polarity opposite to a polarity of electric current through the
discharge unit
during discharge.
[0059] In an embodiment, the generation of the acidic oxidant fluid from the
neutral
oxidant fluid is performed in the acidification reactor via an electric field
driven
orthogonal ion migration across laminar flow (OIMALF) process. In another
embodiment, the generation of the acidic oxidant fluid from the neutral
oxidant fluid is
performed, for example, by one or more of an ion exchange on solids, an ion
exchange in
liquids, electrolysis, and adding an extra acid to the neutral oxidant fluid
during discharge
of the discharge unit. In an embodiment, the discharge is facilitated on the
positive
electrode of the electrolyte-electrode assembly, for example, by one or more
of
electrocatalysis, a solution-phase chemical reaction, a solution-phase
comproportionation,
a solution-phase redox catalysis, a solution-phase redox mediator, an acid-
base catalysis,
and any combination thereof. In another embodiment, the discharge process is
facilitated
via a solution-phase comproportionation of the aqueous multi-electron oxidant
with a
final product of a reduction of the aqueous multi-electron oxidant. In an
embodiment, the
solution-phase comproportionation is pH-dependent and the discharge is
facilitated in the
presence of an acid.
33
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0060] Also, disclosed herein is an embodiment of the regeneration system
comprising
a splitting-disproportionation (SD) reactor, a concentrating reactor, multiple
separation
reactors, and storage tanks such as a regenerated oxidant fluid tank, a
regenerated reducer
fluid tank, a discharge fluid tank, and a water tank. In an embodiment, the SD
reactor is
configured as an electrolysis-disproportionation (ED) reactor comprising sub-
reactors, for
example, an electrolysis unit or an electrolyzer and a disproportionation
unit. In an
embodiment, the SD reactor is configured for an aqueous multi-electron oxidant
(AMO)-
on-negative mode of operation using a multilayer structure on a negative
electrode side of
the SD reactor. The multilayer structure on the negative electrode side of the
SD reactor
minimizes reduction of a regenerated AMO in a regenerated oxidant fluid on the
negative
electrode side while facilitating hydrogen evolution and an increase in pH of
the
regenerated oxidant fluid. In another embodiment, the SD reactor is configured
for the
no-AMO-on-negative mode of operation by transferring a base produced on one or
more
negative electrodes of the SD reactor to a regenerated oxidant fluid produced
at one or
more positive electrodes of the SD reactor and comprising one or more forms of
the
AMO and the intermediate oxidant. The SD reactor is configured to operate in
multiple
modes, for example, a batch mode, a cycle flow mode, a cascade flow mode, and
any
combination thereof.
[0061] The splitting-disproportionation (SD) reactor splits the alkaline
discharge fluid
into a reducer and an intermediate oxidant. The SD reactor converts the
intermediate
oxidant produced in the SD reactor into one or more forms of the aqueous multi-
electron
oxidant via disproportionation of the intermediate oxidant with the base. The
splitting
process releases a stoichiometric amount of the reducer and the base in the SD
reactor.
The SD reactor optimizes and stabilizes the pH of the alkaline discharge fluid
using a
buffer present in one or more forms of the discharge fluid to facilitate
disproportionation
of the intermediate oxidant into one or more forms of the aqueous multi-
electron
oxidant. The SD reactor continues the splitting and disproportionation
processes in a
batch mode of operation, a cyclic flow mode of operation, a cascade flow mode
of
operation, or a combination thereof, until a desired degree of conversion of a
discharge
product of the aqueous multi-electron oxidant into one or more forms of the
aqueous
34
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
multi-electron oxidant is achieved. The SD reactor splits one or more forms of
the
alkaline discharge fluid into the reducer and the intermediate oxidant, for
example, via
electrolysis, photolysis, photoelectrolysis, radiolysis, thermolysis, or any
combination
thereof. The process of photolysis and photoelectrolysis of the alkaline
discharge fluid is
performed in the presence or absence of a light adsorbing facilitator, a
semiconductor, a
catalyst, and any combination thereof.
[0062] In an embodiment, the splitting-disproportionation reactor is
configured as an
electrolysis-disproportionation (ED) reactor. The ED reactor converts a
neutral discharge
fluid into an alkaline discharge fluid by using an externally supplied base
and/or a base
produced at one or more negative electrodes of the ED reactor in an aqueous
multi-
electron oxidant-on-negative mode of operation, a no-aqueous multi-electron
oxidant-on-
negative mode of operation, or a combination thereof. The ED reactor splits
the alkaline
discharge fluid into a reducer and an intermediate oxidant via electrolysis.
The process of
electrolysis releases a stoichiometric amount of the reducer and the base at
one or more
negative electrodes of the ED reactor. The ED reactor converts the
intermediate oxidant
produced at one or more positive electrodes of the ED reactor into one or more
forms of
the aqueous multi-electron oxidant via disproportionation of the intermediate
oxidant
produced at one or more positive electrodes with the base. The ED reactor
continues the
electrolysis and disproportionation process in a batch mode of operation, a
cyclic flow
mode of operation, a cascade flow mode of operation, or any combination
thereof, until a
desired degree of conversion of a discharge product of the aqueous multi-
electron oxidant
(AMO) into one or more forms of the AMO is achieved.
[0063] Also, disclosed herein is an embodiment of the method for regenerating
an
aqueous multi-electron oxidant (AMO) and a reducer in stoichiometric amounts
from
one or more forms of a neutral discharge fluid using external power. The
discharge fluid
comprises, for example, one or more of water, a halide, a hydroxonium cation,
a buffer,
and one or more counter cations. The method disclosed herein comprises
converting the
neutral discharge fluid into an alkaline discharge fluid by using an
externally supplied
base and/or a base produced in the splitting-disproportionation (SD) reactor
in an
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
aqueous multi-electron oxidant-on-negative mode of operation, a no-aqueous
multi-
electron oxidant-on-negative mode of operation, or a combination thereof. The
pH of the
alkaline discharge fluid is, for example, between 6 and 9 or between 4 and 9.
The buffer
is configured to maintain the pH of the alkaline discharge fluid, for example,
between 6
and 9 or between 4 and 9. In an embodiment, the base component of the buffer
is
selected from a group comprising, for example, a hydroxide ion, hydrogen
phosphate, a
phosphate ester, a substituted phosphonate, alkylphosphonate, arylphosphonate,
a
deprotonated form of one or more of Good's buffers, an amine, a nitrogen
heterocycle,
and any combination thereof. In an embodiment, the cationic component of the
buffer
comprises a cation of lithium. In another embodiment, the cationic component
of the
buffer comprises a cation of sodium. In another embodiment, the anionic
component of
the buffer comprises one or more of w-(N-morpholino)alkanesulfonate, 2-(N-
morpholino)ethanesulfonate, 3-(N-morpholino)propanesulfonate, and 4-(N-
morpholino)butanesulfonate. In another embodiment, the anionic component of
the
buffer is one or more of w -(N-morpholino)alkanesulfonate, 2-(N-
morpholino)ethanesulfonate, 3-(N-morpholino)propanesulfonate, and 4-(N-
morpholino)butanesulfonate and the cationic component of the buffer is
lithium. In
another embodiment, the anionic component of the buffer comprises one or more
of an
alkylphosphonate or an arylphosphonate. In another embodiment, the anionic
component
of the buffer comprises one or more of an alkylphosphonate, an
arylphosphonate, and a
cationic component of the buffer is lithium. In an embodiment, the base
component of
the buffer is monohydrogen phosphate and a cationic component of the buffer is
sodium.
[0064] Also, disclosed herein is a method for producing electric power and
regenerating
an aqueous multi-electron oxidant (AMO) and a reducer in an energy storage
cycle. The
method disclosed herein provides the discharge system comprising one or more
forms of
a reducer fluid, one or more forms of an oxidant fluid, the discharge unit,
the acidification
reactor, optionally the neutralization reactor, and one or several storage
tanks. The
oxidant fluid comprising the AMO is converted into an acidic oxidant fluid. In
an
embodiment, the acidification of the oxidant fluid is performed by adding an
acid, for
example, sulfuric acid, triflic acid, phosphoric acid etc., to the oxidant
fluid stored in the
36
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
oxidant fluid tank before the oxidant fluid enters the discharge unit. In
another
embodiment, the acidification is performed using an orthogonal ion migration
across
laminar flow (OIMALF) reactor positioned between the AMO storage tank or the
oxidant
storage tank and the discharge unit. The method disclosed herein facilitates
discharge of
the discharge unit for producing electric power from the reducer and the
oxidant fluid
comprising the AMO, and generates the discharge fluid.
[0065] In an embodiment, one or more forms of the aqueous multi-electron
oxidant
(AMO) undergoes discharge in the ignition mode, that is, under the condition
when the
time required for the product such as a halide to comproportionate with the
AMO such as
a halate is shorter than the time required for the product to diffuse away
from the
electrode. The ignition mode assures a high power density of the discharge
unit. For a
sufficiently high concentration of the AMO such as provided by a highly
soluble LiBr03,
the ignition mode can be observed even when the ratio of the total
concentration of acid
protons to the total concentration of the AMO is below the stoichiometric
number
required by the chemical equation of the redox half-reaction. Herein, the
total
concentration of acid protons is the concentration of acid determined by
titration with a
strong aqueous base, such as NaOH, below the endpoint at pH 7Ø The AMO
reduction
can practically proceed in the ignition mode even when the ratio of the total
concentration
of acid protons to the total concentration of the AMO is below one and can be
as low as
0.05 when a high concentration of the AMO, a strong acid, and a thick
diffusion
boundary layer are employed at the same time.
[0066] The use of substoichiometric acid concentration for the
electroreduction of the
aqueous multi-electron oxidant (AMO) reduces energy and chemical expenses
associated
with the acidification of the oxidant fluid particularly when performed on-
board, reduces
system size, and improves safety. Furthermore, experimental data shows that at
least in
the case of the AMO being LiBr03, the ignition regime can be observed at low
acid
concentrations and acidic oxidant fluid remains stable as evidenced by very
low Br2
formation for over two weeks. This finding allows the elimination of the on-
board
acidification process and of the on-board acidification reactor.
37
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0067] The method disclosed herein further comprises optimizing and
stabilizing pH of
the acidic oxidant fluid in the splitting-disproportionation reactor using an
extra acid
present in the acidic oxidant fluid to facilitate comproportionation of the
aqueous multi-
electron oxidant with a final product of a reduction of the aqueous multi-
electron oxidant
into an intermediate oxidant. The pH of the acidic discharge fluid is, for
example, below
0, 1, 2, or 3. The concentration of acidic protons in the acidic discharge
fluid is, for
example, below 0.1M, 0.5M, 1M, 2M, 5M, or 10 M. The extra acid is one or a
combination of a phosphoric acid, a 3-(N-morpholino)propanesulfonic acid, a 3-
(N-
morpholino)ethanesulfonic acid, another w-(N-morpholino)propanesulfonic acid,
a
methanesulfonic acid, a triflic acid, a substituted sulfonic acid, a
substituted phosphonic
acid, a perchloric acid, a sulfuric acid, a molecule comprising sulfonic
moieties and
phosphonic acid moieties, and an acid with a pKa < 2.
[0068] If the acidic oxidant fluid is stored in the discharge system or
produced by the
addition of an extra acid, for example, H2SO4, F3CSO3H, etc., the discharge
fluid leaving
the discharge unit is in an acid including a partially acid form. In an
embodiment, the
acidic discharge fluid is neutralized with a base form of a buffer in the
neutralization
reactor of the discharge system to produce a solution of a neutral form of the
discharge
fluid. In this scenario, the discharge fluid leaving the discharge system is
in a neutralized
form including partially-neutralized form. The acidic discharge fluid
comprises one or
more of hydrogen bromide, hydrogen chloride, hydrogen iodide, and any
combination
thereof. In an embodiment, the acidic discharge fluid comprises one or more of
water, a
halide, a hydroxonium cation, and a counter cation. In the orthogonal ion
migration
across laminar flow (OIMALF) acidification embodiment, the discharge fluid
comprises
one or more of water, an extra acid, an acid form of the buffer, a discharge
acid, a
halogen, one or more forms of the aqueous multi-electron oxidant (AMO) such as
neutral, acidic or alkaline, and any combination thereof. The OIMALF reactor
replaces
acidic protons in the outgoing acidic discharge fluid for another cation such
as Li + present
in the incoming neutral oxidant fluid, while simultaneously converting an
incoming
38
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
neutral oxidant fluid into an outgoing acidic oxidant fluid and recycling H2
produced on
one or more negative electrodes and consumed on one or more positive
electrodes.
[0069] The aqueous multi-electron oxidant (AMO) and the reducer are
regenerated in
stoichiometric amounts from the discharge fluid in the regeneration system.
The method
and the system disclosed herein reduces the amount of electric energy utilized
by the
acidification reactor, for example, an orthogonal ion migration across laminar
flow
(OIMALF) reactor, for converting the salt form of the AMO into the acid form
of the
AMO by adding an extra acid, for example, one or more of triflic acid,
sulfuric acid,
perchloric acid, nitric acid, and any combination thereof to the oxidant fluid
before or
during the discharge process. The extra acid facilitates a faster
comproportionation, and
thus a higher power during discharge, for example, higher than H3PO4 alone can
cause,
and reduces the charge required for on board OIMALF. In an embodiment, the
acid form
of the buffer comprising, for example, a sulfonic acid group, is used as the
extra acid. In
an embodiment, the acid form of the AMO is used as the extra acid. The
regenerated
reducer fluid comprising the reducer and the regenerated one or more forms of
the
oxidant fluid comprising one or more forms of the AMO are supplied to the
discharge
system for facilitation of the discharge of the discharge unit. In an
embodiment, the heat
released during the discharge process is used to preheat one or more forms of
the oxidant
fluid prior to discharge.
[0070] In an embodiment, the regeneration system disclosed herein performs
regeneration of the oxidant and the fuel from the discharged solution via
photolysis,
photoelectrolysis, or any combination thereof. The reagents are regenerated
photoelectrochemically using sunlight and with semiconductor particles or
electrodes. In
this embodiment, the splitting-disproportionation reactor is configured as a
photoelectrolysis-disproportionation reactor. The photolysis and/or the
photoelectrolysis
of the alkaline discharge fluid is performed in the presence or absence of a
light
adsorbing facilitator, a catalyst, and any combination thereof, in the
photoelectrolysis-
disproportionation reactor. The method disclosed herein induces a splitting of
a discharge
product, for example, HBr in the photoelectrolysis-disproportionation reactor
by
39
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
irradiating the discharged solution with light. The regeneration system
comprising the
photoelectrolysis-disproportionation reactor regenerates one or more of the
oxidant, for
example, the aqueous multi-electron oxidant (AMO) and the fuel from the
discharged
solution.
[0071] Also, disclosed herein is a method for producing electric power and
regenerating
hydrogen and a neutral oxidant fluid comprising lithium bromate in an energy
storage
cycle. The method disclosed herein provides the discharge system comprising
the
discharge unit, the acidification reactor, and optionally the neutralization
reactor. The
discharge system comprises a neutral oxidant fluid comprising lithium bromate,
and
hydrogen. In an embodiment, the discharge system comprises one or more forms
of a
buffer. In another embodiment, the discharge system further comprises one or
more
forms of a base. In an embodiment, the cationic component of the buffer is
lithium and
the anionic component of the based form of the buffer is one or more of o)-(N-
morpholino)alkanesulfonate, 3-(N-morpholino)methanesulfonate, 3-(N-
morpholino)ethanesulfonate, 3-(N-morpholino)propanesulfonate, 3-(N-
morpholino)butanesulfonate, methylphosphonate, an alkylphosphonate, an
arylphosphonate, and a molecule comprising one or more of phosphonate moieties
and
sulfonate moieties. In another embodiment, the cationic component of the
buffer is
sodium, and the anionic component of the base form of the buffer is one or
more of o)-(N-
morpholino)alkanesulfonate, methylphosphonate, 3-(N-
morpholino)ethanesulfonate, 3-
(N-morpholino)propanesulfonate, an alkylphosphonate, an arylphosphonate, and a
molecule comprising phosphonate moieties and sulfonate moieties. In an
embodiment,
the discharge system further comprises a deprotionated form of an extra acid
comprising,
for example, one or more of an aqueous multi-electron oxidant (AMO) in the
acid form,
bromic acid, sulfuric acid, perchloric acid, triflic acid, a sulfonic acid,
molecules
comprising phosphonate moieties and sulfonate moieties, and an acid with a pKa
< 2. The
buffer is in an acid form during discharge with a pH < 4, and the acid form of
the buffer
comprises one or more of a phosphoric acid derivative, a phosphoric acid
ester, one or
more substituted phosphonic acids, one or more o)-(N-morpholino)
alkanesulfonic acids,
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
molecules comprising both phosphonate and sulfonate moieties, and buffers
capable of
maintaining pH between 4 and 9.
[0072] The concentration of lithium bromate dissolved in the neutral oxidant
fluid is,
for example, above 1M, 2M, 5M, or 10M. The acidification reactor converts the
neutral
oxidant fluid into an acidic oxidant fluid. The concentration of acidic
protons in the
acidic oxidant fluid is, for example, below 0.1M, 0.5M, 1M, 2M, 5M, or 10M.
The
method disclosed herein facilitates discharge of the discharge unit for
producing electric
power from the acidic oxidant fluid and from hydrogen and generates an acidic
discharge
fluid on consumption of the acidic oxidant fluid and hydrogen. The discharge
is
facilitated via a pH¨dependent solution-phase comproportionation of bromate
with
bromide formed via electroreduction of intermediate bromine. In an embodiment,
the
neutralization reactor optionally neutralizes the acidic discharge fluid to
produce one or
more forms of a neutral discharge fluid.
[0073] The regeneration system regenerates hydrogen and one or more forms of
the
oxidant fluid comprising lithium bromate in stoichiometric amounts from one or
more
forms of the neutral discharge using external power. The regeneration is
performed by
splitting one or more forms of the neutral discharge fluid into stoichiometric
amounts of
bromine, hydrogen, and a base form of the buffer using external power in the
splitting-
disproportionation reactor, and producing lithium bromate via
disproportionation of
bromine with the base form of the buffer. The splitting process is performed,
for
example, via electrolysis, photolysis, photoelectrolysis, radiolysis,
thermolysis, and other
methods know to those skilled in the art. The disproportionation reaction is
facilitated by
a buffer capable of maintaining a solution pH, for example, between 4 and 9.
The
splitting-disproportionation reactor continues splitting and
disproportionation in a no-
aqueous multi-electron oxidant-on-negative mode of operation and an aqueous
multi-
electron oxidant-on-negative electrode mode of operation until a desired
degree of
conversion of bromide into bromate is achieved. The splitting-
disproportionation reactor
is configured for a batch mode, a cyclic flow mode, a cascade flow mode, and
any
combination thereof. The regeneration system supplies the regenerated one or
more forms
41
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
of the oxidant fluid comprising bromate and the regenerated hydrogen to the
discharge
system for subsequent generation of electric power on demand.
BRIEF DESCRIPTION OF THE DRAWINGS
[0074] The foregoing summary, as well as the following detailed description of
the
invention, is better understood when read in conjunction with the appended
drawings. For
the purpose of illustrating the invention, exemplary constructions of the
invention are
shown in the drawings. However, the invention is not limited to the specific
methods and
components disclosed herein. The description of a structure or a method step
referenced
by a numeral in a drawing carries over to the description of that structure or
method step
shown by that same numeral in any subsequent drawing herein.
[0075] FIG. 1 illustrates a system for generating an electric power and a
discharge fluid
from an oxidant fluid and a reducer fluid using a discharge system and for
regenerating
an oxidant and/or a reducer from the discharge fluid using a regeneration
system.
[0076] FIG. 2 exemplarily illustrates a perspective view of a dissembled
single
electrolytic cell of an electrolytic cell stack of a discharge unit of the
discharge system
and of an electrolyzer of an electrolysis-disproportionation reactor of the
regeneration
system.
[0077] FIG. 3 exemplarily illustrates a perspective view of a planar cell
stack of the
discharge unit, showing three multi-layered electrolyte-electrode assemblies,
two bipolar
plates, and two endplates.
[0078] FIG. 4 exemplarily illustrates a discharge and regeneration cycle as
flows of
energy, materials, and processes, showing the discharge unit with hydrogen as
an
example of a reducer, an aqueous HX0r, as an example of an aqueous multi-
electron
oxidant, and a regeneration system using MZ as an example of a buffer in a
base form.
42
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0079] FIGS. 5A-5B exemplarily illustrate a table showing different reactions
used or
considered for electrochemical energy storage and energy conversion.
[0080] FIG. 6 exemplarily illustrates mass flows in a single electrolytic cell
of an
electrolytic cell stack of the discharge unit during discharge with H2 as the
fuel and
HX03 as the oxidant.
[0081] FIG. 7 illustrates a method for producing electric power from an
aqueous multi-
electron oxidant and a reducer and for simultaneously generating a discharge
fluid.
[0082] FIG. 8 illustrates a method for regenerating an aqueous multi-electron
oxidant
and a reducer in stoichiometric amounts from a discharge fluid using electric
power.
[0083] FIG. 9 exemplarily illustrates a negative-ion electrospray ionization-
mass
spectrometry spectrum of a 0.5M sodium phosphate pH 7.0 buffer solution after
addition
of 50mM of Br2.
[0084] FIGS. 10A-10B exemplary illustrate an electrolysis-disproportionation
orthogonal ion migration across laminar flow method for regenerating a reducer
(H2) and
an oxidant fluid comprising an aqueous multi-electron oxidant (HX03) from a
discharge
fluid (HX + H20) with MOH as a base.
[0085] FIGS. 11A-11B exemplary illustrate a cyclic operation of a flow-through
electrolysis-disproportionation reactor with bromate as an aqueous multi-
electron
oxidant, hydrogen phosphate as a base form of a buffer, and sodium as a
counter cation.
[0086] FIG. 12 exemplarily illustrates calculated and experimentally measured
limiting
currents on a rotating disk electrode in aqueous solutions of bromic acid of
various
concentrations.
[0087] FIG. 13 exemplary illustrates a graphical representation of a
power¨voltage
43
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
curve calculated for a H2-50% w/w HBrO3 discharge flow battery and measured
with a
glassy carbon rotating disk electrode, and with a platinum gauze electrode in
a flow cell,
and a corresponding curve for a commercial proton exchange membrane fuel cell
running
on hydrogen and air.
[0088] FIGS. 14A-14G exemplarily illustrate graphical representations showing
comparative performances of three on-board power sources at a nominal power of
130
kW: a gasoline-internal combustion engine, a lithium ion battery, and an H2-
aqueous
multi-electron oxidant discharge unit as well as the targets of the Advanced
Research
Projects Agency-Energy.
[0089] FIG. 15 illustrates an embodiment of the system for generating electric
power
and a discharge fluid from an oxidant fluid and a reducer fluid using a
discharge system
comprising an orthogonal ion migration across laminar flow reactor and for
regenerating
an oxidant and/or a reducer from the discharge fluid using a regeneration
system.
[0090] FIG. 16 exemplarily illustrates a process flow diagram showing mass and
electricity flows in an energy cycle between the discharge unit, an
acidification reactor,
and a neutralization reactor of the discharge system.
[0091] FIGS. 17A-17B exemplarily illustrate mass flows in a single cell of an
electrolysis-disproportionation reactor configured for regeneration in an
aqueous multi-
electron oxidant-on-negative electrode mode of operation.
[0092] FIG. 18 exemplarily illustrates mass flows in a single cell of an
electrolysis-
disproportionation reactor configured for regeneration in a no-aqueous multi-
electron
oxidant-on-negative electrode mode of operation and a batch mode.
[0093] FIG. 19 exemplary illustrates a mass and electricity flow diagram of a
discharge
system comprising a single cell discharge unit and an orthogonal ion migration
across
laminar flow reactor.
44
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0094] FIG. 20A illustrates a method for producing electric power from an
aqueous
multi-electron oxidant and a reducer and for simultaneously generating a
discharge fluid.
[0095] FIG. 20B illustrates a method for regenerating an aqueous multi-
electron
oxidant and a reducer in stoichiometric amounts from one or more forms of a
neutral
discharge fluid using external power.
[0096] FIG. 20C illustrates a method for producing electric power and
regenerating an
aqueous multi-electron oxidant and a reducer in an energy storage cycle.
[0097] FIG. 20D illustrates a method for producing electric power and
regenerating
hydrogen and an oxidant fluid comprising lithium bromate in an energy storage
cycle.
[0098] FIG. 21A exemplary illustrates polarization curves of a glassy carbon
rotating
disk electrode in solutions comprising 5M LiBr03+50%w H3PO4+1 mM LiBr at
different
rotation rates and 20 C.
[0099] FIG. 21B exemplary illustrates polarization curves of a glassy carbon
rotating
disk electrode in a solution comprising 30% H2SO4 + 166mM LiBrO3 + 5mM NaBr.
[0100] FIG. 22 exemplarily illustrates Pourbaix diagrams for bromine in
aqueous media
at pH 0 and pH 14.
[0101] FIG. 23A exemplarily illustrates a solar radiation spectrum at sea
level and the
positions of the silicon (Si) band-gap, bromine and/or bromide, and bromate
and/or
bromide standard electrode potentials.
[0102] FIG. 23B exemplarily illustrates a batch mode of a photoelectrolysis-
disproportionation method for regenerating a halate from a halide.
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0103] FIG. 24 exemplarily illustrates a graphical representation showing
background-
subtracted limiting currents of bromide electrooxidation-disproportionation on
a glassy
carbon rotating disk electrode in a 0.5M sodium phosphate buffer at various
rotation
rates.
[0104] FIG. 25 exemplarily illustrates a staircase cyclic voltammetry on a
glassy carbon
rotating disk electrode in a 2 hour aged solution containing 2.0 M H2SO4 and
approximately 5M LiBrO3 at various rotation rates.
[0105] FIG. 26 exemplarily illustrates an electrospray ionization-mass
spectrometry
(MS) spectrum, showing experimental data demonstrating the feasibility of a
regeneration process.
DETAILED DESCRIPTION OF THE INVENTION
[0106] FIG. 1 illustrates a system 100 for generating an electric power and a
discharge
fluid from an oxidant fluid and a reducer fluid using a discharge system 101
and for
regenerating an oxidant and/or a reducer from the discharge fluid using a
regeneration
system 106. The oxidant fluid is a chemical or a mixture of chemicals that
accepts
electrons during a discharge process in a discharge mode of operation of a
discharge unit
104 of the discharge system 101. As used herein, the term "the discharge mode
of
operation" refers to a process of releasing chemical energy stored in the
discharge unit
104 in the form of sustainable electric current and voltage, for example,
direct current
(DC). The discharge unit 104 disclosed herein is also referred to as a "flow
battery". The
oxidant fluid comprises one or more forms of an aqueous multi-electron oxidant
(AMO),
water, other solvents, acids, bases, catalysts, and one or more forms of a
buffer or buffers.
The AMO may be present at various stages in the methods disclosed herein in
one or
several forms, for example, acid forms, salt forms such an Li form, etc.,
differing in
composition, concentration, etc. The phrase "aqueous multi-electron oxidant"
or "AMO"
refers collectively to all such forms and any combination thereof. The other
solvent is, for
example, a liquid other than water. The reducer fluid, also referred herein as
a "fuel", is a
46
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
chemical that donates electrons during the discharge process. The reducer
fluid is, for
example, hydrogen gas. The discharge fluid is an exhaust fluid obtained as a
result of an
electrochemical discharge process. The discharge fluid comprises, for example,
water,
other solvents, hydrogen cations, lithium cations, other cations, halide
anions, one of
more forms of the buffer, and the unreacted AMO.
[0107] The system 100 disclosed herein comprises the discharge system 101 and
the
regeneration system 106. The discharge system 101 disclosed herein comprises
an
oxidant fluid tank 102 comprising oxidant fluid comprising aqueous multi-
electron
oxidant (AMO), a reducer fluid tank 103 comprising a reducer, a discharge
fluid tank 113
for collecting discharge fluid, and a discharge unit 104. The AMO is a
chemical that
accepts electrons from an electrode during the electrochemical discharge
process and acts
as an oxidizing agent. The reducer is a chemical that donates electrons to an
electrode
during the electrochemical discharge process and acts as a reducing agent. The
discharge
system 101 disclosed herein can be technically classified as a type of a redox
flow
battery. Unlike conventional redox flow battery systems, the discharge system
101
disclosed herein carries a minimal amount of a solvent and thus provides a
higher system
energy density. Also, unlike conventional redox flow battery, the discharge
unit 104 is
not intended for complete regeneration of oxidant fluid and reducer fluid by
reversing the
flow of electric current and of reagents through the discharge unit 104,
although partial
regeneration, for example, by producing intermediate oxidant such as Br2 is
possible and
recommended, for example for regenerative breaking when used in an electric
vehicle
such as an electric car. Also, unlike conventional fuel cell systems that
carry a reducer
but not oxidant, the discharge system 101 disclosed herein carries both the
reducer and
the AMO in reducer fluid tanks 103 and oxidant fluid tanks 102 respectively.
In an
embodiment, the AMO and the reducer are stored in reagent containers or
supplied via
multiple oxidant fluid tanks 102 and reducer fluid tanks 103 respectively.
[0108] The aqueous multi-electron oxidant (AMO) is an oxidant that, in at
least one of
its forms such as an acid form or a salt form, for example, a Li salt has a
high solubility in
water, for example, over 1M, and that transfers in a solution-phase redox
reaction or in an
47
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
electrochemical reaction more than 1 mole of electrons per 1 mole of the AMO.
The
AMO comprises one or more of halogens, halogen oxoacids, halogen oxoanions,
and
other oxoanions. The AMO is one or more of an oxide of an element such as a
halogen,
an oxoacid of an element such as a halogen oxoacid. The halogen is, for
example, one or
more of chlorine, bromine, and iodine. An oxoanion is an anion comprising one
or more
oxygen atoms and one or more atoms of another element. An oxoacid is a
compound
comprising an oxoanion and one or more forms of hydrogen cation. In the energy
cycle
disclosed herein, the AMO is present in the charged oxidant fluid along with
water and
one or more forms of a buffer. The buffer in the base form is used during
regeneration to
maintain pH of the AMO at an appropriate value, for example, greater than 7,
while
providing sufficient solubility, for example, > 1M for the salt form of the
AMO. The
buffer is chemically compatible with the AMO, the intermediate oxidant, the
discharge
fluid, the electrolysis process, etc. The buffer in the base form comprises,
for example,
anions such as OH-, a monohydrogen phosphate, a substituted phosphonate, an
amine, a
tertiary amine, one or more of a buffering agent described as Good's buffers,
etc. Good's
buffers comprise about twenty buffering agents for biochemical and biological
research
selected and described by Norman Good and others. In addition to a group
defining its
buffering property, the buffer comprises a strong acidic group such as a
sulfonate which
is beneficial for the buffer as the strong acidic group reduces its crossover
throughout the
cation exchange membrane during discharge and electrolysis-regeneration.
[0109] The cation component of the buffer is one or more of lithium (Li),
other alkali
metals, alkali earth elements, other elements, protonated nitrogen bases,
quaternary
nitrogen cations, quaternary phosphorous cations, etc. Li + provides a
substantially high
solubility for bromate and bromide. Li + does create problems with poor
solubility of
lithium phosphate which forms upon decomposition and/or precipitation of its
base form
Li2HPO4 (= '1/2 Li3PO4+ '1/2 LiH2PO4), if hydrogen phosphate is used as the
buffer, but
since this may happen only during off-board regeneration and only in no-
aqueous multi-
electron oxidant (AMO)-on-negative electrode mode of operation also referred
to as a
"no-AMO-on-negative mode of operation", use of Li + will not create a safety
problem.
The AMO in one or more forms can be pre-mixed with the buffer. In an
embodiment, the
48
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
AMO is an oxide or an oxoacid of an element, for example, nitrogen, xenon,
sulfur, etc.
In another embodiment, the AMO is selected from a group consisting of, for
example, a
halogen compound such as a halogen oxide, a halogen oxoacid, etc., an
interhalogen
compound, an oxide of nitrogen, a nitrogen oxoacid, an oxide of xenon, an
oxoacid of
xenon, an oxide of sulfur, an oxoacid of sulfur, an oxide of a chalcogen, an
oxoacid of a
chalcogen, an oxide of a pnictogen, an oxoacid of a pnictogen, a volatile
oxide of an
element, a fluid oxide of an element, a soluble oxide of an element, a
volatile oxoacid of
an element, a fluid oxoacid of an element, a soluble oxoacid of an element,
and any
combination thereof.
[0110] The oxide is a compound having a formula XmOn, where X is one or more
chemical elements, and where 0 is oxygen, and m and n are integers. In an
embodiment,
1<m<2 and I< n < 7. For example, the aqueous multi-electron oxidant (AMO) is a
halogen oxide having a formula XmOn, where X is one of multiple halogens, 0 is
oxygen, and 1<m<2, and 1 < n < 7. The oxoacid is a compound having a formula
HpXgOr, where X is one of multiple halogens, nitrogen, chalcogens, xenon, or
other
element, and 1 < p, q, r < 6. In an example, the halogen oxoacid is a compound
having a
formula HpXq0r, where X is one of multiple halogens, 0 is oxygen, and 1 < p,
q, r < 6
such as HBrO3 or bromic acid. The reagents, products, and intermediaries of
the
reduction of halogen oxoacids are either gases, liquids or are soluble in
water. If the
reagents, intermediates, and products are anions, their cross over through a
cation
exchange membrane is minimal. In an embodiment, the oxoacid is a compound
having a
formula HpX0r, where X is a halogen, H is hydrogen, 0 is oxygen, 1 < p < 6,
and 1 < r <
6. In an embodiment, the AMO is a nitrogen oxide having a formula N,(0õ, where
x = 1
or 2 and 1 < n < 5. In another embodiment, the AMO is a nitrogen oxoacid
having a
formula HkNi0m, where H is hydrogen, N is nitrogen, 0 is oxygen, and 1 < k, 1,
m < 3.
In another embodiment, the AMO is a nitrogen oxoacid having a formula HNOõ,
where
H is hydrogen, N is nitrogen, 0 is oxygen, and 1 < n < 3. In another
embodiment, the
AMO in acid form is chloric acid which forms a stable aqueous room temperature
solution, for example, up to about 40% w/w. Chloric acid can be used, for
example, for
military and aerospace applications where high energy density is needed. In
another
49
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
embodiment, the AMO in acid form is bromic acid (HBr03) which forms a stable
aqueous
room temperature solution, for example, up to about 55% w/w. Bromic acid
and/or its salt
is convenient for the regeneration part of the energy cycle and thus used, for
example, in
automotive applications. In another embodiment, the AMO in acid form is iodic
acid
which forms a stable aqueous room temperature solution, for example, up to
about 74%
w/w. In another embodiment, the AMO is nitric acid which forms an atmospheric
aqueous
azeotrope with, for example, about 68.4% w/w. The AMO can be used as an
aqueous or
non-aqueous solution. Other examples of the AMO in acid form are hypochlorous
acid,
hypobromous acid, perbromic acid, perchloric acid, periodic acid, etc. A
subgroup of the
AMO comprising oxoacids (and salts of oxoanions) of halogens (C1, Br, I) is of
special
interest in energy storage applications since the latter AMOs can be
regenerated from
discharge fluid with full recycling of all chemicals.
[0111] In an embodiment, high energy oxidants rather than oxygen or air are
used with
the discharge system 101 which is otherwise similar to a polymer electrolyte
membrane
fuel cell (PEMFC) system, except for a difference in the structures of one or
more
electrodes. The high energy density aqueous multi-electron oxidant (AMO) and a
mediator are components of the oxidant fluid which is stored in the oxidant
container or
the oxidant fluid tank 102. The reducer is, for example, hydrogen. The use of
hydrogen
as the reducer imparts a benefit of an efficient regeneration via electric
energy, solar
energy, etc., in a regeneration system 106 or in the discharge system 101 or
in both. In
an embodiment, the reducer is selected from a group consisting of, for
example,
ammonia, hydrazine, hydroxylamine, phosphine, methane, a hydrocarbon, an
alcohol
such as methanol, ethanol, etc., an aldehyde, a carbohydrate, a hydride, an
oxide, a
chalcogenide, another organic and inorganic compound and any combination
thereof.
The oxide is, for example, carbon monoxide (CO), nitrous oxide (N20), nitric
oxide
(NO), sulfur dioxide (802), etc.
[0112] The discharge unit 104 of the discharge system 101 comprises an
electrolytic
cell stack 105. The electrolytic cell stack 105 comprises multiple
electrolytic cells 200.
Each electrolytic cell 200 comprises a 5-layer electrolyte-electrode assembly
206
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
exemplarily illustrated in FIG. 2. The 5-layer electrolyte-electrode assembly
206
comprises a 3-layer electrolyte-electrode assembly 205 flanked by two
diffusion layers
201a and 201b exemplarily illustrated in FIG. 2. The 3-layer electrolyte-
electrode
assembly 205 comprises a positive electrode 205a, a negative electrode 205b,
and an
electrolyte layer 205c interposed between the positive electrode 205a and the
negative
electrode 205b. The positive electrode 205a and the negative electrode 205b
are herein
collectively referred to as "electrodes". The term "electrode" refers to an
electronic
conductor or a mixed electronic-ionic conductor, the surface of which is in
contact with an
ionically conducting medium. The 3-layer electrolyte-electrode assembly 205 is
flanked
by a positive diffusion layer 201a on the positive side and a negative
diffusion layer
201b on the negative side forming the 5-layer electrolyte-electrode assembly
206. The S-
layer electrolyte-electrode assembly 206 is flanked on each side by a bipolar
plate 202 or
an endplate 301, exemplarily illustrated in FIG. 3. The electrolytic cell
stack 105 with the
oxidant fluid tank 102, the reducer fluid tank 103, a discharge fluid tank
113, and
connecting lines form the discharge system 101. In an embodiment, the
discharge unit 104
comprises the electrolytic cell stack 105, an enclosure, electric leads, gas
hoses and/or
liquid hoses. In an embodiment, the electrolytic cell stack 105 is configured
as a planar
cell stack 300 exemplarily illustrated in FIG. 3, comprising electrolytic
cells 200
exemplarily illustrated in FIG. 2.
[0113] The theoretical standard equilibrium single cell voltages and tanks'
energy
densities of the discharge system 101 using various combinations of reducers
and
aqueous multi-electron oxidants as well as of other more commonly used battery
materials are exemplarily illustrated in FIGS. 5A-5B. The halogens, the
halogen
oxoacids, and discharge products, for example, hydrogen halides and water are
present
as liquids, gases, or liquid solutions, thereby simplifying mass transport
processes in the
discharge system 101 and the regeneration system 106.
[0114] The chemistry of the oxides and oxoacids of halogens, of chalcogens,
and of
pnictogens may pose problems such as disproportionation of lower oxides and
oxoacids,
and precipitation of solid phases. Disproportionation is a redox reaction in
which an
51
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
element, free or in a compound, is reduced and oxidized in the same reaction
to form
different products. For example, an element with an oxidation state A, not
necessarily
A=0, on disproportionation is distributed between several species with
different
oxidation states B, C, etc., which differ from the element's initial oxidation
state A, so
that B>A>C. For example, the formation of 12 may result in phase-segregation
such as
pore blocking and manifold blocking, when the temperature (T) is low, for
example, for
iodine below its melting point of about 114 C. To keep all the compounds, for
example,
12, in a fluid state, T>120 C may be desired. The high temperature also
benefits ionic
conductivity, reaction kinetics, and the rate of heat rejection. However,
other factors, for
example, startup time, materials corrosion, and pressure limits of the seals
may favor a
lower temperature for operation, for example, about 60 C. Since the discharge
system
101 disclosed herein comprising the oxidant fluid tank 102 and the reducer
fluid tank
103, and a discharge unit 104 with the electrolytic cell stack 105 can be
enclosed, the
operation of the discharge system 101 at such elevated temperatures and/or
pressures is
relatively easier than in the case of regular fuel cells that use 02 from air.
[0115] The fast kinetics on the positive electrode 205a such as bromine-
bromide
reactions, assures high power density and efficiency of the discharge unit 104
as well as
the possibility of partial electric recharge which conventional fuel cells
lack. Aqueous
multi-electron oxidants (AMOs) with high energy content, for example, above
400 watt-
hour (Wh)/kilogram (kg) and above 200 Wh/litre (L) are used to ensure a
driving range
of about 200-300 kilometres or more. Although the required energy densities
can be
achieved with many highly soluble or fluid in the pure state and multi-
electron redox
couples, for example, nitric acid, the requirements for fast reversible
kinetics and high
faradaic efficiency of both electroreduction on the positive electrode 205a of
the
discharge unit 104 and electro-oxidation on the positive electrode of the
electrolyzer 107a
of the regeneration system 106 rules out most of such oxidants. Suitable AMOs
must
assure that the reagents, products, and intermediates of the reduction of the
AMOs are
gases, liquids, or are highly soluble and compatible with the entire group
consisting of
water, electrolyte layer materials, electrode materials, hose materials, and
all other
materials that come in contact with the oxidant fluid, the discharge fluid,
and the reducer.
52
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
Also, the reagents and the products of the process of reduction or
electroreduction of the
AMOs can be anions which provide an additional benefit of a reduced crossover
if a
cation exchange membrane is used as the electrolyte layer 205c.
[0116] In an embodiment, the discharge unit 104 disclosed herein operates in
the
discharge mode. In the discharge mode of operation, the discharge unit 104
produces the
electric power in an external electric circuit 203, exemplarily illustrated in
FIG. 4, when
supplied with the reducer 401 and the aqueous multi-electron oxidant (AMO) 402
from
external reducer fluid tanks 103 and oxidant fluid tanks 102 respectively,
that can be
periodically refilled by pumping the reducer and the AMO from a refueling
station or
multiple reagent sources into their respective reagent containers or tanks 103
and 102.
[0117] In an embodiment, the discharge unit 104 operates in a regenerative
mode, also
referred herein as an "electric recharge mode". In the electric recharge mode
of operation,
the discharge unit 104 produces a reducer or an intermediate reducer and an
intermediate
oxidant which may or may not be the same as the reducer and the aqueous multi-
electron
oxidant (AMO) used during the discharge. The discharge unit 104 operating in
the electric
recharge mode produces an oxidant or an intermediate oxidant, for example, a
halogen or a
halogen compound, and the reducer, for example, hydrogen by consuming a
sustainable
electric current from an external power source or external electric circuit
203, exemplarily
illustrated in FIG. 2, and by splitting the discharge products in the
discharge fluid, for
example, hydrogen halides. The method of regeneration uses, in combination
with other
steps or by itself, electrolysis, that is, with consumption of electric
energy. In the electric
recharge mode or the electric recuperation mode of operation of the discharge
unit 104, the
reducer or the intermediate reducer is produced on the negative electrode
205b, and the
AMO or the intermediate oxidant is generated on the positive electrode 205a,
when the
electric current is forced through the electrodes 205a and 205b of the
discharge unit 104
and/or the 5-layer electrolyte-electrode assembly 206, also referred herein as
the discharge
cell, in a direction opposite to the direction of the electric current during
the discharge
mode of operation, provided that proper chemicals, for example, the discharge
products
are supplied to the respective electrodes 205a and 205b. The electric recharge
mode or the
53
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
electric recuperation mode is useful for regenerative breaking when discharge
system 101
is used to power a vehicle.
[0118] In an embodiment, a solution-phase reaction facilitates one or more
discharge
reactions on the positive electrode 205a of the electrolyte-electrode assembly
205. In an
embodiment, the solution-phase reaction disclosed herein is, for example, a pH-
dependent solution-phase comproportionation, a solution-phase redox catalysis,
etc.
Comproportionation is a redox reaction in which an element, free or in
compounds, with
oxidation states A and C is converted into another substance or substances in
which the
element's oxidation states are B, such that A>B>C. In an embodiment, the rate
of the
solution-phase comproportionation depends on the pH of the solution. In an
embodiment, an electrocatalyst, for example, lead oxide, ruthenium oxide
(Ru02) or a
platinoid facilitates one or more discharge reactions on the positive
electrode 205a of the
electrolyte-electrode assembly 205. Such facilitation may occur via a direct
electroreduction of an aqueous multi-electron oxidant (AMO) such as bromate,
or via
electroreduction of an intermediate oxidant such as bromine on the positive
electrode
205a. In another embodiment, a platinoid electrocatalyst facilitates one or
more
discharge reactions on the negative electrode 205b of the electrolyte-
electrode assembly
205. In another embodiment, a redox mediator facilitates a charge transfer
between the
positive electrodes 205a of the electrolyte-electrode assemblies 205 and the
AMO. The
redox mediator is a halogen/halide couple, for example, C12/CF. In another
embodiment,
a chloride mediator facilitates one or more discharge or regeneration
reactions on the
positive electrode 205a of the electrolyte-electrode assembly 205, for example
via a
reaction: Br03- + 5C1- + 6H+ = BrC1+ 2C12+ 3H20.
[0119] In another embodiment, one or more of multiple immobilized
heterogeneous
mediators, immobilized heterogeneous catalysts, electrocatalysts, homogeneous
mediators, or homogeneous catalysts facilitate a charge transfer between the
positive
electrodes 205a of the electrolyte-electrode assemblies 205 and the oxidant
fluid. In
another embodiment, a catalyst selected from a group consisting of, for
example, a
homogeneous catalyst, a heterogeneous catalyst, a redox mediator catalyst, or
a
54
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
combination thereof, facilitates one or more discharge or charge reactions on
the positive
electrodes 205a of the electrolyte-electrode assemblies 205. In another
embodiment, a
reduced form of a homogeneous solution-phase mediator, a product of an
electrode
reaction or any combination thereof, accelerates a rate of discharge during
one or more
discharge reactions via a solution-phase comproportionation, which may or may
not be
pH-dependent. For example, pH-dependent solution-phase comproportionation of
the
aqueous multi-electron oxidant (AMO) such as bromate with a final product of a
reduction of the AMO such as bromide accelerates the rate of discharge of the
discharge
unit 104.
[0120] The regeneration system 106 of the system 100 disclosed herein is
configured
to regenerate the aqueous multi-electron oxidant (AMO) and the reducer from
the
discharge fluid produced by the discharge unit 104. The regeneration system
106
comprises, for example, an electrolysis-disproportionation (ED) reactor 107,
an
acidification reactor, also referred herein as an "ion exchange reactor" and
referenced by
the numeral 108, such as an orthogonal ion migration across laminar flow
(OIMALF)
reactor, a neutralization reactor 109, a concentrating reactor 112, multiple
separation
reactors 1006, 1007, and 1010 exemplarily illustrated in FIG. 10B, storage
tanks such as
a regenerated oxidant fluid tank 110 and a regenerated reducer fluid tank 111.
The ED
reactor 107 comprises sub-reactors, for example, an electrolysis unit or an
electrolyzer
107a and a disproportionation unit 107b which can be configured in one ED
reactor 107.
The configuration of the electrolyzer 107a of the ED reactor 107 is similar to
that of an
electrolytic cell 200 of the electrolytic cell stack 105 of the discharge unit
104
exemplarily illustrated in FIG. 2. In an embodiment, the electrolyzer 107a and
the
disproportionation unit 107b as well as the neutralization reactor 109 are
physically
combined in the same hardware.
[0121] The neutralization reactor 109 is configured to neutralize the
discharge fluid,
for example, hydrogen halide produced by the discharge unit 104 with a base
form of a
buffer to produce a solution of a neutral or base form of the discharge fluid.
In an
embodiment, the neutralization reactor 109 comprises a mixing reactor. The
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
neutralization reactor 109 is configured to maintain an optimal pH during the
conversion
of the discharge fluid into the oxidant fluid. For example, in the case of a
halate as the
aqueous multi-electron oxidant (AMO), the value of the optimal pH is limited
at the low
end by the reverse reaction of comproportionation between halate and halide,
and the
upper end by the stability of the intermediate hypohalate toward further
disproportionation. In the case of bromate, the optimal pH range is, for
example,
between 7 and 9. The electrolysis-disproportionation (ED) reactor 107 is
configured to
electrolyze the solution of the salt form of the discharge fluid into an
intermediate
oxidant such as a halogen at a positive electrode of the ED reactor 107
accompanied by a
release of the reducer such as hydrogen and a base form of the buffer at a
negative
electrode of the ED reactor 107, while producing a salt form of the aqueous
multi-
electron oxidant (AMO) at the positive electrode via disproportionation of the
intermediate oxidant produced at the positive electrode with an excess of the
base form
of the buffer, and simultaneously releasing a stoichiometric amount of the
reducer and
the base form of the buffer for neutralization. The ED reactor 107 can be
configured to
operate, for example, in a batch mode, as exemplarily illustrated in FIG. 10A
a single
pass flow-through cascade mode, and in a multi-pass cyclic flow mode, as
exemplarily
illustrated in FIG. 10B.
[0122] The ED reactor 107 is used in series with the ion exchange reactor 108.
The ion
exchange reactor 108 is configured to convert the aqueous multi-electron
oxidant
(AMO) in a salt form such as halate into an acid form of the AMO such as a
halic acid.
The storage tanks, for example, the regenerated oxidant fluid tank 110, the
regenerated
reducer fluid tank 111, and a buffer tank (not shown) are used to store the
regenerated
oxidant, the regenerated reducer, and the buffer respectively. The separation
reactors
1006, 1007, and 1010, exemplarily illustrated in FIG. 10B are gas-liquid
separators and
are used to separate gases from the liquids during the regeneration process.
[0123] The electrolysis-disproportionation (ED) reactor 107 or reactors can be
operated in a cyclic flow mode or in a cascade flow mode. In the cyclic flow
mode, the
regenerated solution or the discharge fluid is cycled between a mixing reactor
or the
56
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
neutralization reactor 109, a three-way valve 1004, and another three-way
valve 1005
exemplarily illustrated in FIG. 10B, through the ED reactor 107. In the
cascade flow
mode, the regenerated solution flows through a cascade (not shown) of
functionally
identical mixing reactors of the neutralization reactor 109 and ED reactors
107, and
three-way valves 1004 and 1005. An ED reactor 107 configured for the cyclic
flow
mode has a lower upfront cost but requires a longer regeneration time. The ED
reactor
107 configured for the cascade flow mode has a higher upfront cost but is
capable of a
faster regeneration or higher throughput.
[0124] An exemplary operation of the electrolysis-disproportionation (ED)
reactor 107
in the cyclic flow mode is disclosed in the detailed description of FIGS. 11A-
11B. A
loop within the ED step including the ED reactor 107, the ion exchange reactor
108 such
as the orthogonal ion migration across laminar flow (OIMALF) reactor, and the
mixing
reactor or the neutralization reactor 109 is disclosed in the detailed
description of FIG.
10B. As used herein, the term "laminar flow" refers to a type of fluid flow,
for example,
a liquid flow or a gas flow, in which directions and magnitudes of fluid
velocity vectors
in different points within a fluid do not change randomly in time and in
space. Also, as
used herein, the term "migration" refers to a movement of an electrically
charged object
such as an ion due to the action of an external electric field. In the OIMALF
process, the
vectors of the laminar flow velocity and the electric field are not parallel
and not anti-
parallel. The concentrating reactor 112 concentrates the acid form of the
aqueous multi-
electron oxidant (AMO) to remove the excess water produced on the positive
electrode
205a during the discharge and to remove water introduced with the buffer
during
electrolysis-disproportionation. The concentrating reactor 112 removes water
or other
solvents from a dilute fluid that enters the concentrating reactor 112 and
releases a
concentrated fluid and water or another solvent. The concentrating reactor 112
performs
concentration, for example, by evaporation or reverse osmosis.
[0125] The discharge system 101 and the regeneration system 106 can be used
together
in a complete energy cycle that recycles all the chemicals, does not consume
external
chemicals, and does not generate chemical waste. The complete energy cycle
employs the
57
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
regeneration system 106 in addition to the discharge system 101. The discharge
products
such as LiBr and H20 produced in the discharge unit 104 of the discharge
system 101 are
converted back to intermediates such as Br2, and/or stable reactants such as
LiBrO3 and
H2 of the reactants in the ED reactor 107, and to the active form such as
HBrO3 in the
ion exchange reactor 108, for example, the orthogonal ion migration across
laminar flow
(OIMALF) reactor.
[0126] The reverse transformation of the cathodic discharge product, for
example,
LiBr, into the aqueous multi-electron oxidant (AMO), for example, LiBrO3 in
the
regeneration system 106 is accompanied by the release of the reducer, for
example,
hydrogen in a stoichiometric amount, as exemplified by equations (18)-(21) for
a
particular lithium bromate-phosphate chemistry. As a result, the regeneration
system 106
can produce simultaneously both the AMO and hydrogen, in stoichiometric
amounts,
which can be used again as reactants during the direct mode of operation of
the
discharge unit 104 of the discharge system 101. In an embodiment, the
regeneration of
the AMO from the spent discharge fluid or from the intermediate oxidant is
catalyzed by
a homogeneous catalyst such as chlorine, polyvalent metal ions, etc., or by a
heterogeneous electrocatalyst such as ruthenium dioxide, lead dioxide, and
their
derivatives. The energy cycle based on the discharge unit 104 and the process
of on-site
regeneration disclosed herein eliminates the need for a macro scale
infrastructure for the
production, transportation and storage of the reducer, for example, hydrogen
in contrast
to applications based on fuel cells.
[0127] The discharge unit 104 and the electrolysis-disproportionation (ED)
reactor 107
disclosed herein are implemented with aqueous multi-electron oxidants (AM0s)
compatible with water and with cation¨exchange membranes such as commercially
available polyperfluorosulfonic acids. The aqueous multi-electron oxidants
are, for
example, halogens, halogen oxides, halogen oxoanions, and halogen oxoacids. In
an
embodiment, the aqueous multi-electron oxidants are, for example, oxides,
oxoanions, and
oxoacids of chalcogens, of pnictogens, of xenon, etc. The listed compounds can
assure a
higher theoretical energy density than the elemental halogens and batteries
with solid
58
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
electroactive materials such as lithium ion batteries, but at the expense of
lower energy
efficiency and lower power density and a higher cost as an expensive catalyst
may be
required. In this embodiment, homogeneous reactions near the positive
electrode 205a
are utilized in order to achieve a higher power from the positive electrode
205a. The
discharge system 101 disclosed herein circumvents the drawback of lower energy
efficiency and power density and of higher cost by using a solution¨phase
redox
mediator. The solution-phase redox mediator is an Oxmed/Redmed couple which is
subject
to a rapid and reversible transformation at an electrode and is capable of a
quick
homogeneous redox reaction with the aqueous multi-electron oxidant. A solution-
phase
redox mediator is a redox couple dissolved in a solution, for example, in the
oxidant
fluid, that is capable of relatively fast electron transfer reactions both at
the electrode and
with a primary aqueous multi-electron oxidant (AMO), for example,
bromine/bromide
couple. At the same time, the reduced form of the Oxmed/Redmed couple
participates in a
rapid redox reaction with the high energy but electrochemically inactive AMO:
AMO + Redmed Red + 0Xmed
0Xmed limed e = Redmed
[0128] The solution-phase redox mediators help to realize the electrochemical
process
at a low over-voltage on the electrodes 205a with or without a low amount of
platinum
(Pt) and other expensive catalyst. The solution-phase redox mediator is stable
with
respect to side reactions and hence allows the discharge unit 104 to be used
for many
days or cycles. The solution-phase redox mediator can be present only within
the
positive electrode space of the discharge unit 104 with minimal cross-over to
the
negative electrode space. The solution¨phase redox-mediator helps to realize a
high rate
of electron transfer from the principal aqueous multi-electron oxidant (AMO)
to the
positive electrode 205a on discharge. The reduced form of the solution¨phase
redox
mediator (Redmed) causes a rapid solution-phase chemical reaction during
discharge and
can be regenerated from the oxidized form of the solution¨phase redox mediator
(Oxmed). In an embodiment, to mediate AMO reduction in the discharge unit 104,
a
solution-phase mediator, for example, polyoxometallates is used to facilitate
the
59
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
electrode reaction on the positive electrode 205a. In this embodiment, the
regenerating
couple is suspended or immobilized polyoxometallates which do not cross the
membrane
and do not discharge at the negative electrode 205b due to their large size,
negative
charge or a combination thereof. In an embodiment, the regeneration process is
based on
the redox-mediated catalysis by the redox couple:
AMO + Redmed -> Discharge Product + Oxmed Red.,' <-1 Oxmed + nmed e
[0129] In an embodiment, the reduced form of the mediator is the final product
of the
reduction of the aqueous multi-electron oxidant (AMO) and the homogeneous
reaction
facilitating a discharge of the AMO is a comproportionation reaction.
[0130] In a reduction of aqueous multi-electron oxidants (AMOs), a large
number of
protons are consumed. The discharge unit 104 disclosed herein produces protons
at the
negative electrode 205b by electro-oxidation of hydrogen or a hydride and
transfers the
protons to the positive electrode 205a across the electrolyte layer 205c. The
hydrogen
reducer is automatically co-regenerated with the aqueous multi-electron
oxidant (AMO),
or an intermediate, Oxmed, during the regeneration process. Thus, the
regeneration
system 106 restores back both the components of the oxidant fluid, that is,
the AMO,
Ox, or the oxidized intermediate, Oxmed; and the fuel or the reducer such as
H2. The
discharge unit 104 disclosed herein uses AMOs. A homogeneous redox mediator is
added to or generated within the discharge unit 104 to perform the reduction
of the AMO
during the discharge process in the bulk of the solution rather than on the
surface of the
electrode 205a where the number of active sites is lower. The homogeneous
redox
mediators allow for the use of AMOs in electrochemical power sources and
resolve the
issue of the slow and irreversible direct electrode reactions of the AMO.
[0131] The discharge system 101 disclosed herein therefore provides a long
driving
range, a high energy density, a high power, and a high energy efficiency at a
lower cost
than proton exchange membrane fuel cells (PEMFCs). The discharge system 101
requires a short refill time and can be operationally combined with the
regeneration
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
system 106 to enable an electric energy cycle based on the H2- aqueous multi-
electron
oxidant (AMO) chemical matter cycle. Other combinations of discharge system
101 with
various regeneration systems 106 can use other types of energy, such as solar
energy as
the input in the chemical cycle. The discharge unit 104 disclosed herein
avoids the need
for a large amount of platinum or other expensive metals required for the
electroreduction of oxygen. Since the discharge unit 104 does not consume
oxygen, the
discharge system 101 can be used in enclosed environments such as submarines,
space
ships, etc.
[0132] In an embodiment, the discharge unit 104 employs the ultimate reduction
product as the reduced form of the intermediate, for example by taking
advantage of the
homogeneous comproportionation between an oxoanion and a free halide, leading
to an
electrochemically active halogen on discharge in an ignition type cycle. In
the case of
bromate (Br03- ) as an aqueous multi-electron oxidant (AMO):
On the negative electrode 3 H2 - 6 e- = 6 1-1 , fast
On the positive electrode 3 Br2 + 6 e- = 6 Br- , fast
In the catholyte 5Br- + Br03- + 6H+ = 3H20 + 3Br2 @ pH<4.
[0133] The discharge unit 104 allows for a fast reversible reaction on the 2D
surface of
an inexpensive electrode such as a carbon-based electrode while performing the
slower
comproportionation step utilizing the actual energy storing species, for
example, the
aqueous multi-electron oxidant (AMO) such as bromate or other halogen
oxoanion, in
the three-dimensional (3D) bulk of the solution where a higher reaction rate
can be
sustained. The reagent and the product of the discharge are anions which
result in their
low crossover from the positive electrode 205a through a cation-exchange
membrane
205c to the negative electrode 205b. Among the bromine oxoacids HBrOn, 1< n<4,
bromic acid (HBr03) presents a useful compromise between the energy density
and the
energy efficiency. The theoretical energy efficiency of a H2-HBrO3 discharge
unit 104
on discharge can be estimated as the ratio of the standard equilibrium
potential of the
bromine/bromide couple, for example, about 1.07V and the standard equilibrium
61
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
potential of the bromate/bromide couple, for example, at about 1.42V, measured
with
respect to the standard hydrogen electrode and is equal to about 75%, which is
acceptable for transportation applications. The bromate/bromide direct
electroreduction
is slower than the iodate/iodide direct electroreduction. At pH 10, the
difference between
the onset potentials of bromate reduction to bromide and bromide oxidation to
bromate
on Pt amounts to 0.4 V. In an acidic solution, the reduction of iodate follows
the same
pathway as the reduction of bromate, that is, via a homogeneous
comproportionation to
bromine.
[0134] The method and the system 100 disclosed herein use halic acids or
halate
anions as the aqueous multi-electron oxidant (AMO) among halogen oxoacids due
to a
number of reasons and/or factors. One of the factors is, for example:
perhalates are inert
kinetically, both in direct reduction on an electrode 205a and in homogeneous
comproportionation, whereas halites and hypohalites have lower energy
densities. Other
factors are considered too. For example: during the discharge, both the
efficiency of the
halogen electrode kinetics, that is, the halogen/halide exchange current and
the ratio of
the standard electrode potentials of halogen/halide to oxohalate/halide are
important in
the overall cycle energy efficiency. Due to the first factor, bromine oxoacids
are used
instead of chlorine oxoacids and due to the second factor, bromine oxoacids
are used
instead of iodine oxoacids. The discharge system 101 can be used on-board, for
example, a vehicle. The regeneration system 106 can be used on-board or off-
board. The
structures of the discharge unit 104 or the electrolytic cell stack 105 are
based on the
corresponding structures in proton exchange membrane fuel cells (PEMFCs).
[0135] In an embodiment, the reagent containers, for example, the reducer
fluid tanks
103, and the oxidant fluid tanks 102, exemplarily illustrated in FIG. 1, are
refilled by
pumping the reducer and the oxidant fluid comprising the aqueous multi-
electron oxidant
(AMO) from their respective stationary storage facilities such as an off-road
fueling
station. In an embodiment, the reagent containers, for example, 102 and 103
are located
outside the discharge unit 104 and are connected to the electrolytic cell
stack 105 via the
ports 302 and 303. In another embodiment, the reagent containers, for example,
102 and
62
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
103 are refilled by regenerating or partially regenerating the intermediate
oxidant and the
reducer, for example, by electrolysis, by applying an electric current of a
polarity opposite
to the polarity of the electric current that the discharge unit 104 generates
during the
discharge mode of operation, etc. This partially regenerated AMO is useful for
regenerative braking while driving an electric vehicle, load leveling, etc.
[0136] FIG. 2 exemplarily illustrates a perspective view of a dissembled
single
electrolytic cell 200 of an electrolytic cell stack 105 of the discharge unit
104 of the
discharge system 101 and of the electrolyzer 107a of the electrolysis-
disproportionation
(ED) reactor 107 of the regeneration system 106 exemplarily illustrated in
FIG. 1. Each
electrolytic cell 200 comprises the 3-layer electrolyte-electrode assembly
205. The 3-
layer electrolyte-electrode assembly 205 of the electrolytic cell stack 105 is
flanked by
pair of diffusion layers 201a and 201b, where the pair of diffusion layers
201a and 201b
is flanked by a pair of bipolar plates 202. The diffusion layers 201a and 201b
are
electronically conducting and porous. The diffusion layers 201a and 201b are
sheets
capable of gas transport or liquid transport through pores of the diffusion
layers 201a
and 201b or though the bulk of the diffusion layers 201a and 201b. Moreover,
the
diffusion layers 201a and 201b are capable of electronic conductivity through
their bulk.
The diffusion layers 201a and 201b are positioned on either side of the 3-
layer
electrolyte-electrode assembly 205 in order to facilitate a uniform
distribution of the
reactants and removal of the discharge products over the areas of the
electrodes 205a
and 205b. The 3-layer electrolyte-electrode assembly 205 flanked by a negative
diffusion
layer 201b on the negative electrode side and a positive diffusion layer 201a
on the
positive electrode side forms a 5-layer electrolyte-electrode assembly 206.
The 5-layer
electrolyte-electrode assembly 206 flanked by two bipolar plates 202 or a
bipolar plate 202
and an endplate 301, exemplarily illustrated in FIG. 3, forms a single
electrolytic cell 200.
Multiple electrolytic cells 200 connected electrically in series and flanked
by endplates
301 form the electrolytic cell stack 105 so that any two adjacent electrolytic
cells 200
share a common bipolar plate 202.
[0137] The diffusion layers 201a and 201b are made of, for example, porous
carbon,
63
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
composites containing carbon particles and fibers, and carbon cloths such as
those used
for hydrogen-air proton exchange membrane fuel cells (PEMFCs) and for redox
flow
batteries. The bipolar plates 202 comprise flow channels 202a exemplarily
illustrated in
FIG. 3, for supplying the reducer and the aqueous multi-electron oxidant (AMO)
from the
storage tanks 103 and 102 respectively, into the electrolytic cell stack 105
and for
removing the discharge products from the electrolytic cell stack 105. The
bipolar plates
202 are made of, for example, graphite, other carbonaceous materials, carbon-
polymer
composites, metals, alloys, or electrically conductive ceramic. The 3-layer
electrolyte-
electrode assembly 205 and/or the 5-layer electrolyte-electrode assembly 206
are
hereafter referred to as "electrolyte-electrode assembly".
[0138] The 3-layer electrolyte-electrode assembly 205 comprises the
electrolyte layer
205c flanked by the positive electrode layer 205a and the negative electrode
layer 205b as
disclosed in the detailed description of FIG. 2. The positive electrode 205a
is supplied
with the oxidant fluid comprising the aqueous multi-electron oxidant (AMO) and
the
negative electrode 205b is supplied with the reducer fluid during the
discharge mode of
operation of the discharge unit 104. The positive electrode 205a produces the
intermediate
oxidant such as Br2 and the negative electrode 205b produces the reducer such
as H2 on
partial recharge, that is, when electric current is forced through the
discharge unit 104 in a
direction opposite to the direction of the electric current during discharge.
A certain amount
of the intermediate oxidant in the discharge unit 104 is regenerated from the
discharge fluid
by reversing a polarity of the electric current flowing through the discharge
unit 104 during
discharge. In an embodiment, the electrodes 205a and 205b are multiphase
systems
comprising an electron conducting phase, an ion conducting phase, an
electrocatalyst
phase that can be functionally combined with an electron conductor, and a
reactant/product- transporting porous phase that can be functionally combined
with an
ion conductor. The discharge unit 104 is a device that converts chemical
energy of the
reducer and the AMO into electrical energy by means of electrochemical
reactions on the
two electrodes 205a and 205b and an ion transport through the electrolyte
layer 205c.
[0139] The electrolyte layer 205c of the electrolyte-electrode assembly 205 in
the
64
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
discharge unit 104 acts as an ion conductor, as well as an electronically non-
conducting
mechanical barrier separating the negative electrode 205b and the positive
electrode 205a
of the electrolytic cells 200, thereby precluding an internal electrical and
chemical short
circuit from being established between the positive electrode 205a and the
negative
electrode 205b as well as between the aqueous multi-electron oxidant (AMO) and
the
reducer. In an embodiment, the electrolyte layer 205c of the electrolyte-
electrode
assembly 205 is composed of a material, for example, a solid, a gel, a liquid,
a polymer,
an ionomer, a solid ion conductor, or a solid proton conductor or a
combination thereof,
that is capable of protonic conduction or, more generally, of ionic conduction
but not
electronic conduction. The electrolyte layer 205c conducts ions but not
electrons. The
electrolyte layer 205c with a higher permeability and/or conductivity to
cations than to
anions has an additional advantage of reducing the chemical short-circuiting
during
discharge via the reduction of the AMO on the negative electrode 205b. The
electrolyte
layer 205c is compatible with water, with the AMO, with the reducer, with the
buffer,
and with the discharge products. Furthermore, since durable fluorinated
polymer cation
selective fuel cell membranes are available commercially, the discharge unit
104 disclosed
herein uses such cation-conductive fluorinated polymer electrolytes. In
another
embodiment, the electrolyte layer 205c of the electrolyte-electrode assembly
205 is
composed of a material with a cationic conduction exceeding an anionic
conduction of the
material. In an embodiment, the electrolyte layer 205c is composed of a
material that
contains one or more proton donor groups or proton acceptor groups, for
example, sulfonic,
phosphonic, boronic or nitrogen-base groups. In an embodiment, the electrolyte
layer 205c
is a solid in which hydrogen ions are mobile. In another embodiment, the
electrolyte layer
205c is a liquid or a gel in which hydrogen ions are mobile.
[0140] Examples of the electrolytes 205c used in the electrolyte-electrode
assembly 205
disclosed herein comprise polymers such as Nafion of E. I. du Pont de Nemours
and
Company Corporation, Flemion series of polymers of Asahi Glass Company,
Aciplex of
Asahi Kasei Chemicals Corporation, short-chained trifluorovyniloxy polymers
from Dow
Chemicals, Hyflon -Ion of Solvay Specialty Polymers, Aquivion of Solvay SA
Corporation, a polymer with ¨0¨(CF2)4-503H pendant groups developed by 3M
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
Company, BAM membrane from Ballard Advances Materials Corp., sulfonamide based
polymers developed by DesMarteau, reinforced membranes from W. L. Gore &
Associates, Inc., polybenzimidazole, and other polymers with acidic groups,
basic groups
or a combination thereof. The acidic groups comprise, for example, sulfonic,
phosphonic,
boronic, and carboxylic groups. In an embodiment, the electrolyte 205c is a
polymer
capable of anionic conduction, for example, polymers with quaternary nitrogen
and
phosphorus groups such as polymers employed in alkaline membrane fuel cells.
Another
example of an electrolyte 205c employed in the electrolyte-electrode assembly
205
disclosed herein is an ionically conducting liquid retained in the pores of a
solid matrix.
Examples of such ionically conducting liquid electrolytes comprise phosphoric
acid in a
silicon carbide (SiC) matrix, hydroxide melts, and electrolyte solutions
comprising, for
example, solid oxide matrices, polymer matrices, and a combination thereof.
Another
example of an electrolyte 205c employed in the electrolyte-electrode assembly
205
disclosed herein is a solid proton conductor such as CsH2PO4, CsHSO4, and
related
materials, alkaline-earth cerate- and zirconate- based perovskite materials
such as doped
SrCe03, BaCe03, and BaZr03, as well as rare-earth niobates, tantalates, and
tungstates.
Polymer electrolytes are considered due to their mechanical properties. Cation-
conductive electrolytes are considered due to their ability to reduce
crossover such as
self-discharge.
[0141] In an embodiment, the electrolyte layer 205c is a porous solid matrix
imbibed
with a liquid or gel or solid ion conducting material. That is, the
electrolyte layer 205c is
a composite material comprising an ion conducting liquid or gel or solid
within pores of
the porous solid matrix. The liquid in the electrolyte layer 205c is, for
example,
phosphoric acid or an aqueous solution of phosphoric acid, a hydroxide or an
aqueous
solution of a hydroxide, molten carbonates, molten hydroxides, a molten salt,
etc. The
conducting ion in the electrolyte layer 205c is, for example, fl+, 0H-, F, Cl-
, Br-, E, or a
combination thereof. The porous solid matrix is, for example,
polytetrafluoroethylene
(PTFE), polyvinylidene fluoride (PVDF), a dielectric such as silicon carbide,
silicon
dioxide, a silicate, other ceramic materials, other polymer materials, etc.
The ion
conducting liquid is, for example, water, an acid, a base, a salt, a molten
electrolyte, an
66
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
organic solvent, or a combination thereof.
[0142] In another embodiment, the electrolyte layer 205c of the electrolyte-
electrode
assembly 205 is composed of a material, for example, a solid membrane, capable
of
protonic conduction. The solid membrane is, for example, Nafion of E. I. du
Pont de
Nemours and Company Corporation and related sulfonic acid polymers, a
sulfonamide
polymer, acid doped polybenzimidazole, alkali hydrogen sulfates, phosphates
such as
cesium hydrogen sulfate (CsHSO4), other solid proton conductors, etc. In an
example, the
material used as the electrolyte layer 205c is CsH2PO4, other solid proton
conductors,
etc., when the reducer used in the discharge unit 104 is hydrogen or a
hydride. Although
selective ionic conduction of the electrolyte layer 205c is not required, an
fr conducting
membrane confers the benefit of a more complete reduction of the aqueous multi-
electron
oxidant, and higher solubility of the discharge products, that is, of a larger
energy density
of the discharge unit 104 and the regeneration system 106. In another
embodiment, the
electrolyte layer 205c is a liquid under laminar flow.
[0143] In an embodiment, the electrolyte-electrode assembly 205 of the
discharge unit
104 further comprises electrodes or electrode layers 205a and 205b disposed on
each of
the electrolyte layers 205c. The electrode layers 205a and 205b comprise, for
example,
catalysts, carbon particles or fibers, a binder, a pore-forming agent, etc. In
an
embodiment, the catalyst in the electrode layer disposed on the negative
hydrogen
electrode 205b is platinum (Pt) and Pt nanoparticles on carbon microparticles
or on
carbon microfibers. In an embodiment, the catalyst in the electrode layer
disposed on the
positive electrode 205a is one or more carbonaceous materials with or without
metals,
metal oxides, such as Ru02 and dimensionally stable anodes (DSAs), other
metallic and
non-metallic materials, etc.
[0144] The discharge unit 104 disclosed herein produces, in the discharge
mode, an
electric power, that is, sustainable electrical current and electric voltage,
via an
electrochemical reaction using two reactants: the reducer and the aqueous
multi-electron
oxidant (AMO) on spatially separated electrodes 205a and 205b. During the
discharge
67
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
mode of operation of the discharge unit 104, the negative electrodes 205b are
supplied
with the reducer, such as H2 and the positive electrodes 205a are supplied
with the AMO,
such as bromate resulting in a sustainable voltage difference and sustainable
electric
current between the electrodes 205a and 205b. The discharge mode of operation
of the
discharge unit 104 is also known as a power generating mode of operation or a
direct
mode of operation. The discharge unit 104 produces electric potential
difference between
the electrodes 205a and 205b, which in turn produces an electric potential
difference
between the diffusion layers 201a and 201b and between the bipolar plates 202,
when the
reactants are supplied to their respective electrodes 205a and 205b. The
reducer donates
electrons to the negative electrode 205b and produces ions. The external
electric circuit
203 conducts or transfers electrons from the negative electrode 205b to the
positive
electrode 205a. The aqueous multi-electron oxidant, either directly or via an
intermediate,
accepts the electrons from the positive electrode 205a for producing the
electric current
in the external electric circuit 203. The electrolyte layer 205c provides for
a movement of
the ions between the negative electrode 205b and the positive electrode 205a,
thereby
maintaining electroneutrality of the electrolyte layer 205c and conservation
of charge in
the discharge unit 104, and producing a sustainable current and sustainable
voltage
between the electrodes 205a and 205b and between the bipolar plates 202.
[0145] When a load 204, for example, a light bulb, is attached between the
terminals of
the electrolytic cell 200 or between the endplate terminals of the discharge
unit 104, the
electric current flows for as long as the reactants are supplied to the
electrodes 205a and
205b and the discharge products are removed from the electrodes 205a and 205b.
In the
discharge mode of operation, the discharge unit 104 consumes the reducer and
the
aqueous multi-electron oxidant that are produced from the discharge fluid
outside the
discharge unit 104 or fully or partially regenerated inside the discharge unit
104. As used
herein, the term "partially regenerated" refers to the number of electrons
donated by the
discharge fluid being less the number of electrons lost by the parent oxidant
fluid
regardless of how these electrons are distributed between various chemical
species.
[0146] FIG. 3 exemplarily illustrates a perspective view of a planar cell
stack 300 of the
68
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
discharge unit 104 exemplarily illustrated in FIG. 1, showing three 5-layer
electrolyte-
electrode assemblies 206, two bipolar plates 202, and two endplates 301. The
planar cell
stack 300 comprises multiple electrolytic cells 200 combined electrically in a
series
combination for delivering a larger electric power than a single electrolytic
cell 200
exemplarily illustrated in FIG. 2. When individual electrolytic cells 200 are
connected
electrically in series, the planar cell stack 300 produces more electric power
via a large
voltage with about the same current, than the electric power produced by a
single
electrolytic cell 200. In an embodiment, each 5-layer electrolyte-electrode
assembly 206
of the planar cell stack 300 comprises a 3-layer electrolyte-electrode
assembly 205
flanked by the diffusion layers 201a and 201b. Each stack in the planar cell
stack 300
comprises the 5-layer electrolyte-electrode assembly 206 positioned between
two bipolar
plates 202 or between a bipolar plate 202 and an endplate 301 that conduct
electrons.
[0147] The bipolar plates 202 in the planar cell stack 300 comprise flow
channels 202a.
The flow channels 202a are grooves which allow the reactants to be delivered
to the
electrodes 205b and 205a and for the discharge products of the electrochemical
reaction
from the electrodes 205b and 205a through the diffusion layers 201b and 201a
to be
removed. The flow channels 202a of the bipolar plates 202 allow transport of
the reagents
and products to and from the electrodes 205b and 205a and to and from the
endplates 301.
The planar cell stack 300 terminates with the endplates 301. The endplates 301
are similar
in structure to the bipolar plates 202 but do not comprise the flow channels
202a on the
outer surfaces 301b of the endplates 301. The endplates 301 comprise the flow
channels
202a on the inner surfaces 301a of the endplates 301. Moreover, the endplates
301
comprise connecting ports 302 and 303, for example, inlet ports and outlet
ports on the
outer surfaces 301b of the endplates 301 for facilitating movement of the
reducer fluid and
the oxidant fluid into the planar cell stack 300 and for the discharge fluid
to be moved out
of the planar cell stack 300. Furthermore, the endplates 301 comprise electric
contacts on
the outer surfaces 301b of the endplates 301.
[0148] Each electrolytic cell 200 shares one or two bipolar plates 202 with an
adjacent
electrolytic cell or cells 200. One side of each bipolar plate 202 contacts a
positive side of
69
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
one electrolytic cell 200 and another side of each bipolar plate 202 contacts
a negative
side of the adjacent electrolytic cell 200. The electrolytic cells 200 in the
planar cell stack
300 are stacked electrically in series such that each bipolar plate 202 faces
a diffusion
layer 201a of the positive electrode 205a of one electrolytic cell 200 on one
side and the
diffusion layer 201b of the negative electrode 205b of another electrolytic
cell 200 on the
other side. The individual electrolytic cells 200 are stacked together such
that each bipolar
plate 202 contacts the negative side of the electrolytic cell 200 at the left
of the bipolar
plate 202 and contacts the positive side of the electrolytic cell 200 at the
right of the
bipolar plate 202. The electrolytic cells 200 in the planar cell stack 300 are
stacked
electrically in series such that each bipolar plate 202 serves as the positive
side of one
electrolytic cell 200 and as the negative side of the next electrolytic cell
200. Moreover,
the bipolar plates 202 are equipped with through channels (not shown) that
provide for
transport of the reducer, the aqueous multi-electron oxidant (AMO) and the
discharge
products from the electrolytic cell 200 to the next electrolytic cell 200 in
the planar cell
stack 300 or to the connecting ports 302 and 303. The number of repeat units
or
electrolytic cells 200 in the planar cell stack 300 can be adjusted according
to the desired
power or voltage. The endplates 301 and the bipolar plates 202 are made of
chemically
inert electronically conducting materials, for example, carbon or carbon
composite, and
are equipped with flow channels 202a for supplying the reactants and removing
the
products.
[0149] The oxidant fluid and the reducer fluid are stored in reagent
containers, for
example, the oxidant fluid tanks 102 and the reducer fluid tanks 103
exemplarily
illustrated in FIG. 1. The reagent containers or tanks 102 and 103 are
connected to the
endplates 301 of the planar cell stack 300 via pipes 302 and 303. In a small
planar cell
stack 300, the reagent containers or tanks 102 and 103 can be placed above the
planar cell
stack 300 for gravity feeding the reactants to the electrolyte-electrode
assembly 205. In
an embodiment, in order to overcome the friction in the flowing fluids,
pressurized
reagent containers are used or pumps are inserted into the connecting lines.
In a large
planar cell stack 300, the reagent containers or tanks 102 and 103 are placed
at some
distance from the planar cell stack 300 and may include heat transfer loops
(not shown)
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
for cooling or heating the reactants and the discharge products. For purposes
of
illustration, the detailed description refers to a planar electrolytic cell
200 and planar cell
stacks 300; however, the scope of the method and the system 100 disclosed
herein is not
limited to the planar electrolytic cell 200 or planar cell stacks 300 but may
be extended to
other configurations of flow batteries and fuel cells known in the art, for
example, a
tubular stack.
[0150] FIG. 4 exemplarily illustrates a discharge and regeneration cycle as
flows of
energy, materials, and processes, showing the discharge unit 104 with hydrogen
as an
example of the reducer 401, an aqueous HX0r, as an example of an aqueous multi-
electron oxidant (AMO), and the regeneration system 106 using MZ as an example
of a
buffer in a base form. In FIG. 4, HX0r, refers to the AMO in the acid form,
MX0r, refers
to the AMO in the salt form, HZ refers to the buffer in the acid form, and MZ
refers to
the buffer in the base form. The flow of materials is represented using solid
arrows and
the flow of electric energy is represented using dotted arrows. Electric power
is used
during the process of concentrating 412 by reverse osmosis although other
sources, for
example, heat can also be used, for example, for evaporation.
[0151] The discharge unit 104 is similar to the polymer electrolyte membrane
fuel cells
(PEMFCs), with a low cost Pt- free porous carbon positive electrode 205a,
hydrophilic
positive diffusion layer 201a, and with the air feed line replaced by an
aqueous multi-
electron oxidant (AMO) line, for example, a HBrO3 line. This combination may
provide
about 1, 200 Ah/kg x 1.42 V=1,704 Wh/kg theoretical energy density, and about
426
Wh/kg system-level energy density for about 5% w/w compressed H2, and about
50%
w/w aqueous HBr03. The pH-driven disproportionation reactions allow solution-
phase
transformation from a high energy bromate to high power bromine during
discharge, for
example, at a pH<2. The discharge unit 104 also allows for a partial recharge
via
electrooxidation of bromide into bromine in the discharge fluid which is
useful, for
example, for regenerative breaking.
[0152] During the discharge process, the discharge unit 104 is supplied with
the reducer
71
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
401, for example, H2, and the acidic oxidant fluid comprising the aqueous
multi-electron
oxidant (AMO) in acid form HX0r, 402, for example, HBr03,. In an embodiment,
the
AMO, for example, HX0r, 402 is mixed with a buffer in acid form HZ such as
H3PO4,
carried over from the regeneration step. The reducer 401 donates electrons to
the negative
electrode 205b, also referred to as an "anode", and splits into ions. The
reaction at the
negative electrode 205b is, for example, 3H2 ¨ 6e- = 6H+. The external
electric circuit 203
conducts and transfers electrons from the negative electrode 205b to the
positive
electrode 205a. The reaction at the positive electrode 205a, also referred to
as a
"cathode", is, for example, 3Br2 + 6 e- = 6 Br- , or when combined with the
comproportionation reaction the catholyte, for example, Br03- + 6e- +6H+ = Br-
+ 3H20.
The aqueous multi-electron oxidant accepts the electrons at the positive
electrode 205a
for producing the electric current in the external electric circuit 1 203. The
discharge unit
104 releases 403 HX, for example, HBr and the buffer HZ in the acidic form, if
the buffer
HZ is added initially, and generates electric current in the external electric
circuit 1 203.
The electrolyte layer 205c provides for a movement of the ions between the
negative
electrode 205b and the positive electrode 205a. At a steady state, the
electric current
transferred through the discharge unit 104 is equal to the electric current
through the
external electric circuit 1 203.
[0153] The thermodynamics of the discharge process is illustrated herein using
the
example of H2-HBrO3 reaction. Bromate is a good aqueous multi-electron oxidant
(AMO) since it provides a good thermodynamic efficiency (Ebromate/Ebromine)
and the
corresponding bromine/bromide couple has a fast electrode kinetics even on
inexpensive
carbonaceous electrodes. Since bromine reacts on the electrode 205a and
bromate is the
energy storing species in the oxidant fluid tank 102 exemplarily illustrated
in FIG. 1, the
fraction of the hydrogen-bromate system energy that can actually be converted
into
electrical energy is less than 100%. To estimate the fraction of energy, that
is, the
projected energy efficiency, the standard potentials of the couples of
interest are used:
5Br2 + 10e- = 10 Br- E A = +1.0873 V (7)
5Br2 + 5H2 = 10 HBr AG A = 1OF E A (8)
72
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
and
2Br03- + 12H+ + 10e- = Br2 (1) + 6H20 E B =+1.48 V (9)
2HBrO3 + 5H2 = Br2 (1) + 6H20 AG B = 1OF E B (10)
[0154] The energy stored on-board is given by:
3H2 + HBrO3 = HBr +3H20 AG c = AG A /5+ AG B (11)
[0155] The electric power produced by the discharge unit 104 is given by
equation (8).
The ratio of the electric power produced in the discharge unit 104 to the
chemical energy
of the reagents in the tanks 102 and 103 exemplarily illustrated in FIG. 1,
gives the
projected discharge efficiency:
MDE = AG A /AG c = AG A / (AG A /5+ AG B) = 1OF E A / (2F PA +10F E B) = E A/
(E B+ E A/5) = 1.0873 /(1.0873 +1.48/5) = 78%
[0156] For the homogeneous disproportionation and/or comproportionation:
HBrO3 + 5HBr = 3Br2 + 6H20
AG D = (AG B - AG A)/2 = 5F (E B - PA ) = 5F*0.3927V = 379 kJ/mol
Kc = [Br2]3 / [H+ 16 [Br031[Br15 = exp (-AG D /RT) = exp (-153) = 1 0-66 4
The equilibrium constant Kc comprises [H+] and can be used at any pH.
[0157] For RT = 2.479 kJ/mol, the critical pH at which [Br2]3/[H +
]6[Br031[Br15= 1, is
11. Thus, for the comproportionation reaction to occur, the solution pH can be
brought
below 11; however due to the formation of an intermediate hypobromite, which
is
kinetically stable above the acid dissociation constant pKa (HBrO) = 8.6, and
due to a
slow rate of comproportionation at neutral pHs, a lower pH value such as below
3, is
used. In acidic solutions in the discharge unit 104, the comproportionation
reaction is
strongly favored.
73
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0158] Several embodiments of the method of regeneration of the H2-aqueous
multi-
electron oxidant (AMO) chemistry are disclosed herein. For purposes of
illustration, the
detailed description refers to a method of regeneration using HBrO3 as the AMO
in the
acid form, however the scope of the method and the system 100 disclosed herein
is not
limited to HBrO3 but can be extended to include other AMOs such as HC103,
HC104,
HBr04, HI03, HI04, etc. The regeneration process starts with neutralization
404 of the
acid in the discharge fluid with a base, for example, HBr with LiOH or another
base such
as Li-3-(N-morpholino) propanesulfonic acid (MOPS) in the neutralization
reactor 109 of
the regeneration system 106 exemplarily illustrated in FIG. 1. Neutralization
404 is a
chemical reaction in which a base and an acid react to form a salt. The
neutralization 404
of the discharge fluid, HX, with the base, MOH, is performed in the
neutralization reactor
109. In an embodiment, some process steps of the energy cycle, for example,
neutralization 404, and electrolysis and disproportionation 406 can be
combined in a
single reactor. The base is regenerated at the negative electrode of the
electrolyzer 107 of
the regeneration system 106 during the electrolysis process.
[0159] The neutralization 404 of the discharge fluid acid with a base, for
example, HBr
with LiOH or Li-3-(N-morpholino) propanesulfonic acid (MOPS) produces 405 a
solution of a salt MX such as LiBr. The solution of a salt such as LiBr and
H20
undergoes electrooxidation into the intermediate oxidant such as Br2 at the
positive
electrode while H2 and LiOH or H2 and Li-MOPS are produced at the negative
electrode.
The process of electrolysis 406 is accompanied by the release of the reducer
401, for
example, hydrogen in stoichiometric amounts which is used as the reducer 401
in the
discharge unit 104. In the case of Br2, if the pH at the positive electrode is
maintained
near 8, a disproportionation 406 to bromate occurs, for example, with a LiOH
base:
3Br2 + 6LiOH = 5LiBr + LiBrO3 + 3H20 (12)
[0160] Electrolysis 406 of the LiBr + H20 solution and the disproportionation
406
reactions proceed in a batch mode, a cascade flow mode, or a cyclic flow mode
till most
of the LiBr is converted into LiBr03. The residual LiBr may or may not be
removed. In
74
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
the latter case, the product LiBrO3 will have some LiBr present. In an
embodiment, a
provision to remove the residual LiBr is provided. In an embodiment, a buffer
is used
during the cyclic process in order to maintain the pH at a near constant value
which is
optimal for the disproportionation 406, for example, 6<pH<10 or near 8. In
another
embodiment, the buffer comprises hydrogen phosphate and dihydrogen phosphate.
In
another embodiment, the buffer comprises one or more of Good's buffers, other
amines,
other tertiary amines, and nitrogen heterocycles. In another embodiment, the
buffer
comprises a phosphonic acid derivative. In another embodiment, the buffer
comprises a
lithium counter-cation. H2P042- has a proper pH for disproportionation and is
chemically
compatible with the rest of the chemistry throughout the whole energy cycle
if, for
example, sodium is used as the counter cation.
[0161] In the regeneration process, the electrooxidation step or electrolysis
406 is
followed by the disproportionation 406 of the intermediate oxidant such as
bromine. The
disproportionation 406 is the reverse of the comproportionation of the aqueous
multi-
electron oxidant (AMO) discharge and is favored at a higher pH than the
comproportionation of discharge. In the beginning of electrolysis 406 of
hydrobromic
acid, Br2 and H2 are formed in the equal molar amounts:
HBr = y2H2+ y2Br2 (13)
[0162] If there is no buffer present, the anolyte turns acidic due to
hydrolysis:
Br2 + H20 = HBr + HBrO (14)
[0163] In a reactor with a cation¨selective membrane, the anolyte compartment
turns
into a solution of HBrO through the equations (13) to (14) route. A further
oxidation of
HBrO does not proceed on a carbon electrode at low over-voltages; however, a
further
disproportionation 406 of HBrO can occur in the aqueous phase yielding
bromate:
3H0Br = 2HBr + HBrO3 (15)
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
or combined: 3Br2 + 60H- = 5Br- + Br03- + 3H20 (16)
[0164] The disproportionation 406 of Br2 to Br03- and Br- is strongly favored
thermodynamically at pH above 11, which is equivalent to 1 mM of OH-, although
this
reaction has the maximal rate at pH near 8 due to the formation of an
intermediate
hypobromite which is stable toward further disproportionation 406 at pH >
pKa(HBrO) =
8.8. However, even if the HBr produced in equation (15) is consumed in
equation (13),
one proton per Br03- will not get electro-reduced due to the lack of an anodic
counter-
process unless both faradaic and voltage efficiency are sacrificed by running
oxygen
evolution reactions (OER) or other parasitic process to make 02 and OH-. The
resulting
pH drop due to the formation of a strong acid HBrO3 will cause equations (15)
and (16)
to proceed to the left thus ceasing the regeneration when bromine's average
oxidation
state is around +1. Thus, a disproportionation 406 of Br2 to Br (+5) requires
an
introduction of an external base. In the case of an anionic base with a
counter cation, this
will result in formation of a bromate salt rather than of bromic acid. The
hydroxide
generated during hydrogen evolution reaction (HER) on the negative counter
electrode
can be used as the needed base or to make the needed base. Li + can be used as
a counter-
cation to achieve high solubilities of the salts involved such as bromide and
bromate. A
pH buffer comprising, for example, a dissolved phosphonate and/or one or more
of
Good's buffers is used to prevent spatial and temporal deviations of pH from
the value of
near 8 within the electrolysis-disproportionation (ED) reactor 107. The
resulting product,
for example, LiBr03, 407 needs to be converted or partially converted to the
electrochemically active aqueous multi-electron oxidant (AMO), for example,
HBrO3.
This can be accomplished via a solution-phase cation exchange process in the
ion
exchange reactor 108, for example, the orthogonal ion migration across laminar
flow
(OIMALF) reactor with a simultaneous conversion of the input discharge fluid
into a salt,
for example, HBr into LiBr. LiBrO3 is converted into HBrO3 using the
orthogonal ion
migration across laminar flow (OIMALF) or the ion exchange process 408. The
buffer is
converted from the acid form into a base form simultaneously.
[0165] The continuous electrolysis-disproportionation (ED) 406-orthogonal ion
76
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
migration across laminar flow (OIMALF) process 408 disclosed herein for the
regeneration of HBrO3 from HBr ends with an ion exchange of the base form of
the
oxidant fluid comprising, for example, LiBrO3 into the acid form of the
oxidant fluid
comprising, for example, HBrO3 in the ion exchange reactor 108 while realizing
hydrogen at the negative electrode and consuming hydrogen at the positive
electrode as
disclosed in the detailed description of FIG. 10B. The principle of OIMALF 408
is
identical to ion suppression in anion chromatography. In an embodiment, The
OIMALF
process 408 generates and consumes H2 within the OIMALF reactor or the ion
exchange
reactor 108. The OIMALF process 408 of converting MX0r, into HX0õ, for
example,
LiBrO3 into HBrO3 avoids cumbersome chemical separation and ion exchanger
regeneration steps. The net reaction of the ion exchange or the OIMALF process
408 is,
for example, LiBrO3 +HA = HBrO3 + LiA, where HA is a source of protons, for
example, water, phosphoric acid, dihydrogen phosphate, one or more of Good's
buffers,
etc. The regeneration system 106 is connected to an external electric circuit
2 409 which
provides electric power for the OIMALF process 408. The base, for example, MOH
or
LiA 410 generated as a result of the OIMALF process 408 is used during the
process of
neutralization 404 of the discharge fluid, for example, HBr. In an embodiment,
LiBrO3 is
converted into HBrO3 using ion exchange on resins. This is followed by
electrolysis (E)
406 of LiBr into bromine and, in the same ED reactor 107 or another reactor,
by
disproportionation (D) 406 of the halogen into halate and halide in a suitable
buffer, for
example, a lithium hydrogen phosphate buffer, one or more of Good's buffers,
or any
combination thereof, near pH 8. The electrolysis-disproportionation 406 cycle
continues
in the same flow or batch ED reactor 107 till [bromide]/[bromatel ratio
decreases, for
example, below 0.05. The resulting solution can be concentrated 412, for
example, using
reverse osmosis or evaporation. The concentrated solution, for example,
approximately
10M LiBrO3 solution, the concentration of which is limited by the solubility
of LiBrO3 at
the operating temperature, for example, of about 20 C, then goes back into the
ion
exchange reactor 108 such as the OIMALF reactor, where Li + in LiBrO3 is
exchanged for
1-1+ from the incoming HBr, thus producing, for example, a solution comprising
0.5M
HBrO3 and 9.5M LiBrO3. The hardware components of the hydrogen-bromate energy
cycle disclosed herein comprise analytical chemical detectors (not shown) used
for
77
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
process monitoring and control.
[0166] The resulting concentrated HBrO3 solution is used as the aqueous multi-
electron
oxidant (AMO) for the discharge unit 104. The net result of regeneration for
an
exemplary combination of the AMO and the buffer LiA is:
HBr + 3H20 = (electricity in two places, LiA recycled) = 3H2 + HBrO3 (17)
[0167] The electrolysis-disproportionation (ED) 406-orthogonal ion migration
across
laminar flow (OIMALF) 408 process has a reasonably high projected energy
efficiency
of about 70%. The oxidant fluid comprising one or more forms of the aqueous
multi-
electron oxidant (AMO) may be further concentrated. The commercial process of
concentrating 412 HBrO3 uses evaporation, with an estimated energy loss of
approximately 10-15% if heat exchangers are used. The evaporation is likely to
lead to
the loss of volatile bromine species and evaporation may be less energy
efficient than
reverse osmosis (RO). The reverse osmosis process requires overcoming of the
osmotic
pressure, for example, of 536 bars, which is possible in a cascade flow mode
with
commercial supported ion exchange membranes. The minimal energy expense at an
infinitely slow filtration rate is 6.6% of the energy content of the product
50% w/w
HBrO3 and 3H2. Due to a finite flow rate, the regeneration process disclosed
herein uses
optimization of the unit size, power, and operating pressure in terms of the
energy
efficiency and capital cost.
[0168] Since the kinetics of all the processes involved in the chemical cycle
of the
discharge unit 104 and the economic figures for polymer electrolyte membrane
fuel cells
(PEMFCs) are well known, quantitative predictions on the performance of the
discharge
unit 104 disclosed herein can be derived. The data for the discharge unit 104
disclosed
herein, also referred to as a flow battery or a discharge flow cell, is
calculated for a one-
dimensional model with a flow-by smooth carbon cathode for a constant solution
composition outside of the diffusion boundary layer as well as from the
experimental data
disclosed in the detailed description of FIG. 13 and using other relevant
performance
78
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
figures from the PEMFCs literature are compared with the Advanced Research
Projects
Agency-Energy (ARPA-E) targets. The projected performance of the discharge
unit 104
and the ARPA-E targets are shown in Table 1 below.
Table 1:
ARPA-E Projected
Parameter Units
Target Value
Manufacturing cost $/kWh <100-125 140
Effective specific energy Wh/kg >150 570
Effective energy density Wh/L >230 900
Effective specific power on W/kg >300 690
discharge, 80% DOD/30 s
Cycle life at 80% depth of cycles > 1000 1000
discharge (DOD)
Calendar life years > 10 6
Operating temperature C >-30 -40
[0169] The discharge unit 104 meets the requirements as the primary power
source for
electric vehicles (EVs). The one-way discharge efficiency of about 85% at the
target
power of about 0.05 W/cm2 is found using a precious metal free smooth glassy
carbon
rotating disk electrode (RDE) as disclosed in the detailed description of FIG.
12, FIG. 13,
FIG. 21, and FIG. 25. The discharge unit 104 disclosed herein has a short
refueling time
in EV applications when combined with off-board regeneration, which is based
on the
disproportionation 406 of Br2 electrochemically regenerated from the
discharged LiBr,
HBr, etc.
[0170] In Table 1, the projected temperature refers to a cold-start up and is
limited by
the aqueous multi-electron oxidant (AMO)'s freezing point. The cost figures
are
calculated based on the design of modern polymer electrolyte membrane fuel
cells
(PEMFCs) minus the cost of the Pt catalyst on the positive electrode 205a. The
cost
figures do not account for the economy-of-scale discount. The parameters refer
to the
system 100 exemplarily illustrated in FIG. 1, with H2 storage as a 5% w/w
metal hydride
79
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
and 50% w/w/ HBrO3 and 78% discharge efficiency at 0.5 W/cm2 power. The power
is
calculated for a smooth flow-by carbon cathode on the basis of kinetic
parameters
reported in the literature and by assuming membrane resistance of 0.1 ohm/cm2
as
exemplarily illustrated in FIG. 13. The durability number is the operational
life and not
the calendar life. The projected durability of the discharge unit 104 is
limited by the
degradation of Pt on the hydrogen anode accounting for the aqueous multi-
electron
oxidant cross-over at open circuit potential (OCP) on the basis of relevant
data for the
PEMFCs. Purging both the electrodes 205b and 205a with on-board water on shut-
downs
can increase the projected durability.
[0171] The results of system level modeling in the Advanced Research Projects
Agency-Energy (APRA-E) metrics show that the most conservative estimate for
the
energy density of the 5% H2-50% HBrO3 on-board system is 426 Wh/kg, which is
2.8
times larger than the ARPA-E target of 150 Wh/kg and 6.5 times larger than the
corresponding number for lithium iron phosphate (LFP) batteries in Tesla
Roadster of
Tesla Motors, Inc. The estimate of the specific energy of the discharge system
101
disclosed herein depends on the type of H2 storage and varies from 208 Wh/L
for 350 bar
gas, 339 Wh/L for 5% w/w metal hydride and 400 Wh/L for liquid H2. For a 150
kWh
sport utility vehicle (SUV), the system volume is 750, 970, and 2,000 L for
liquid, metal
hydride and compressed H2, respectively, of which only 300L is the aqueous
multi-
electron oxidant (AMO) tank. These values fall in between the volumes of the
combination of a gasoline tank with an internal combustion engine (ICE) and
the
combination of a lithium ion battery (LIB) with an electric engine. Regardless
of the H2
storage method, the system-level energy density of the discharge system 101
meets the
ARPA-E target of 230 Wh/L.
[0172] The energy and material cycle exemplarily illustrated in FIG. 4
incorporates an
affordable method to regenerate both the reducer such as hydrogen (H2) reducer
401 and
the aqueous multi-electron oxidant (AMO), for example, bromate (Br03-) from
the
discharge fluid, for example, aqueous solution comprising bromide (Br-),
without reliance
on fossil fuels, thus resolving the need for a hydrogen source which is
injurious to the
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
development of hydrogen economy. Although the gravimetric specific energy of
hydrogen is high, the volumetric energy density of hydrogen is low even at the
highest
practically achievable pressures and hydrogen storage. Both high pressure
carbon
composite cylinder and metal hydrides tank may satisfy the mass and the volume
requirements. In addition, 20% of the required H2 can be regenerated on board
from the
discharge fluid using metals, which in turn can be regenerated by electrolysis
406 of
MBr2 off-board:
M (M=Zn, Sn, Fe, etc.) +2HBr MBr2+H2
[0173] The safety of the H2-aqueous multi-electron oxidant (AMO) discharge
system
101 is also considered. Since the two reagents, that is, the AMO and hydrogen
do not
come in contact under normal operating conditions and only small amounts of H2
and
AMO may contact each other without reaction in an accident within the
discharge unit
104, the safety of H2 and of the AMO such as 5-20 M aqueous LiBr03, is
individually
considered. On-board hydrogen is safer than gasoline systems due to faster
escape in an
open space. Bromates are moderately toxic, comparable to nitrites, and
although
suspected carcinogens, are widely used as additives in bread flour in the
United States. In
an outdoor environment bromates eventually turn into benign bromides.
Moreover,
bromates are listed as oxidants and are corrosive but not explosive. The
intermediate
bromic acid, present in the ion exchange reactor 108 and discharge unit 104,
is classified
as an oxidizer, but not as an explosive. Bromic acid can be safely
concentrated 412 by
vacuum-distillation at 80 C up to 50% w/w. From a practical handling
viewpoint, HBrO3
is similar to HNO3 although the former does not stain skin. HBr is a well
known
corrosive agent having a long history of safe use in various applications. The
system
energy density of the H2-AMO discharge system 101 disclosed herein is about 6
times
larger when compared to the Li-ion battery pack of the Tesla Roaster and
hence allows
for the incorporation of additional safety features such as a collision and/or
spill-proof
enclosure without jeopardizing the driving range and power of the electric
vehicle. The
risk of using such a corrosive oxidant, that is, HBrO3 if it is present on-
board only in a
small amount in the discharge unit 104 can be mitigated.
81
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0174] The H2-aqueous multi-electron oxidant (AMO) discharge system 101
disclosed
herein has the following advantages: high system energy content, for example,
about 426
Wh/kg and about 200-400 Wh/L, which is 6 times greater than that of a lithium
ion
battery (LIB) pack; high power density, for example, of about 690 W/kg, which
exceeds
the Advanced Research Projects Agency-Energy (ARPA-E) target more than twice;
mechanic refill: can be refilled at a pump in less than 5 min; long range, for
example, of
about 300 miles per refill with about 120L storage; aqueous chemistry which is
intrinsically safer than Li-ion batteries; low materials and manufacturing
cost, for
example, of about $120/kWh and about $115/kW; low total cost of ownership
(TOC), for
example, of about $0.15/mile for a 6 year lifetime of the discharge system 101
and the
regeneration system 106; simultaneous stoichiometric regeneration of H2 and
AMO using
electricity as the only input and without irreversibly consuming other
chemicals and
without generating chemical waste.
[0175] FIGS. 5A-5B exemplarily illustrate a table showing different reactions
used or
considered for electrochemical energy storage and energy conversion. The
characteristics
of the redox reactions comprise, for example, theoretical charge density in
(ampere*
hour)/kilogram (A*h/kg), standard equilibrium cell potential (Eeq) in volts
(V), the
reactants' theoretical energy density in watt-hour per kilogram (Wh/kg), the
oxidant's
solubility (weight percentage %), maximum practical energy density (ED) in
Wh/kg,
exchange current in milliampere (mA)/ square centimeter (cm2), energy
efficiency in %,
and practical energy efficiency x energy efficiency in Wh/kg. As exemplarily
illustrated
in FIGS. 5A-5B, some H2- aqueous multi-electron oxidant (AMO) chemistries can
afford
four times higher theoretical energy densities than batteries with solid
electroactive
materials, for example, lithium ion batteries. The practical ratio may be as
much as 10
due to a higher packing ratio in a flow battery or in the discharge system 101
exemplarily
illustrated in FIG. 1, but not in batteries with solid electroactive materials
(SEAM) such
as lithium ion batteries. The practical energy density includes water in the
concentrated
aqueous multi-electron oxidant (AMO). The maximum energy density includes
oxidant
solubility but not H2 storage and energy efficiency in %. For oxohalic acids,
the projected
82
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
energy efficiency is defined as the ratio of the standard equilibrium
potentials of
halogen/halide and oxohalate/halide.
[0176] FIG. 6 exemplarily illustrates mass flows in a single electrolytic cell
200
exemplarily illustrated in FIG. 2, of an electrolytic cell stack 105 of the
discharge unit
104 exemplarily illustrated in FIG. 1, during discharge with H2 as the fuel
and HX03 as
the oxidant. In this example, the reducer is H2. The aqueous multi-electron
oxidant (AMO)
is HBr03. The standard equilibrium potential for this combination is 1.42 V
and the
theoretical energy density is, for example, about 1,705 Wh/kg, which is 4.4
times higher
than that of lithium iron phosphate/graphite chemistry. HBrO3 can be used, for
example,
pumped as an aqueous solution which is stable up to, for example, about 55%
w/w
concentration (938Wh/kg). In another embodiment, HI03 can be used as the AMO.
The
equilibrium voltage is 1.19V and the energy density is 1,052 Wh/kg and the
room
temperature solubility is 74% at 20 C. HI03 has faster kinetics, that is,
electrolytic cell
power, but the intermediate 12 is solid and reduces at a lower potential thus
lowering the
efficiency of the energy cycle. The discharge unit 104 is configured similar
to a polymer
electrolyte fuel cell but with a hydrophilic liquid diffusion layer on the
positive electrode
205a. A cation exchange membrane, for example, Nafion of E. I. du Pont de
Nemours and
Company Corporation is used as the electrolyte 205c. The cation exchange
membrane
minimizes the crossover or rejects anionic species and assures nearly single
or exclusive
hydrogen ion conductivity and high power density. The operating temperature of
the
discharge unit 104 is maintained above the ambient temperature to facilitate
heat rejection
and electrode kinetics. The negative electrode 205b, for example, the hydrogen
side of the
electrolyte-electrode assembly 205 has a standard design and prepared by
standard
methods known to those skilled in the art of polymer electrolyte fuel cells
(PEFCs).
[0177] The design of the positive electrode 205a is also similar to polymer
electrolyte
fuel cell (PEFC) electrodes but the positive electrode layer 205a is paired
with a liquid
diffusion layer on the back since the reagents and products on the positive
electrodes 205a
are in the liquid phase in contrast to an air-supplied proton exchange
membrane fuel cell
(PEMFC). In an embodiment, a parallel flow field is used, although numerous
other
83
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
designs, for example, single, multiple, serpentine, meander, inter-digitated,
etc., known to
those skilled in the art are employed. In an embodiment, the walls of the flow
field of the
positive electrode 205a are made of a porous carbon and used without a liquid
diffusion
layer, or the whole flow field can be made of a solid material and a liquid
diffusion layer,
for example, made of a porous carbon sheet and can be placed between the flow
field and
the electrolyte-electrode assembly 205. The positive side of the membrane and
the
positive wall of the bipolar plate 202 exemplarily illustrated in FIGS. 2-3,
can be coated
with catalytic layers. The intermediate oxidant, for example, Br2 can be
regenerated by a
direct electrochemical process on the positive electrode 205a. Suitable
positive electrodes
205a comprise, for example, one or any combination of carbon, platinum, Pb02,
Ru02,
dimensionally stable anode (DSA), and other oxides, metals and non-metals,
including
conductive diamond.
[0178] FIG. 7 illustrates a method for producing electric power from an
aqueous multi-
electron oxidant (AMO) and a reducer and for simultaneously generating a
discharge
fluid. The method disclosed herein provides 701 the discharge system 101
comprising the
oxidant fluid tank 102 comprising the AMO, the reducer fluid tank 103
containing the
reducer, and the discharge unit 104 as exemplarily illustrated and disclosed
in the detailed
description of FIG. 1. The method for producing electric power facilitates 702
discharge
of the discharge unit 104. Discharge occurs by transferring 702a electrons
from the
positive electrode 205a of the 5-layer electrolyte-electrode assembly 206
exemplarily
illustrated in FIG. 2, to the AMO and transferring 702b electrons from the
reducer to the
negative electrode 205b of the 5-layer electrolyte-electrode assembly 206
exemplarily
illustrated in FIG. 2, to produce 702c an electric power or (I*U#0) or a
sustainable
electric current, that is, a direct current (DC) in an external electric
circuit 203 connected
to the terminals of the discharge unit 104 and transferring ions between the
positive
electrodes 205a and the negative electrode 205b of the 5-layer electrolyte-
electrode
assembly 206, thus conserving the charge. The discharge is facilitated on the
positive
electrode 205a of the 5-layer electrolyte-electrode assembly 206, for example,
by one or
more of electrolysis, electrocatalysis, a solution-phase chemical reaction, a
solution-phase
comproportionation, a solution-phase redox catalysis, an acid-base catalysis,
and any
84
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
combination thereof.
[0179] The discharge unit 104 consumes the aqueous multi-electron oxidant
(AMO)
and the reducer supplied from their respective storage tanks 102 and 103 to
generate the
discharge fluid stored in a discharge fluid storage tank (not shown) and
electric power in
the external electric circuit 203. The discharge fluid comprises, for example,
one or more
of water, an acid form of the buffer, a base form of the buffer, a halogen, a
hydrogen
halide, a halogen oxoacid, and any combination thereof. Since the discharge
fluid coming
out of the discharge unit 104 is not water or not only water, the discharge
fluid is not
disposed into surroundings but collected in a discharge fluid storage tank or
container
(not shown) to be regenerated later into the reducer and the AMO. The buffer
is in the
acid form during the discharge with a pH < 7. The acid form of the buffer is,
for example,
one or more of phosphoric acid, a dihydrogen phosphate of lithium, Good's
buffers, and
any combination thereof.
[0180] Consider an example where the aqueous multi-electron oxidant (AMO) is
bromic acid and the reducer is hydrogen. The processes in the discharge unit
104 are:
oxidation of hydrogen on the negative electrode 205b, transport of a hydrogen
ion with
water from the negative electrode 205b to the positive electrode 205a through
a cation
exchange membrane, comproportionation of bromate with bromide in the fluid
near the
positive electrode 205a, and reduction of bromine on the positive electrode
205a,.
Protons or other positive ions are transferred through the cation exchange
membrane
from the negative electrode 205b to the positive electrode 205a, for example,
due to a
concentration gradient. Electrons are transferred from the negative electrode
205b to the
positive electrode 205a, thus producing electric power, that is, current and
voltage in the
external electrically conducting connecting circuit, that is, the external
electric circuit
203.
[0181] FIG. 8 illustrates a method for regenerating the aqueous multi-electron
oxidant
(AMO) and the reducer in stoichiometric amounts from a discharge fluid using
electric
power. The method disclosed herein provides the regeneration system 106
comprising the
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
neutralization reactor 109, the electrolysis-disproportionation (ED) reactor
107, the ion
exchange reactor 108, and the concentrating reactor 112 exemplarily
illustrated in FIG. 1.
The neutralization reactor 109 neutralizes 801 the discharge fluid, produced
by the
discharge unit 104 exemplarily illustrated in FIG. 1. The electrolysis-
disproportionation
proceeds in the ED reactor 107 configured to electrolyze 802 the solution of
the salt form
of the discharge fluid into an intermediate oxidant, for example, Br2, at a
positive
electrode in the ED reactor 107. The regeneration system 106 performs
electrolytic
decomposition of the discharge fluid, for example, HBr into the reducer, for
example, H2
and the intermediate oxidant, for example, Br2. The electrolysis process
releases the
reducer and the base form of the buffer at a negative electrode of the ED
reactor 107
while producing a salt form of the AMO at the positive electrode via a series
of chemical
and electrochemical reactions. The ED reactor 107 is further configured to
disproportionate 802 the intermediate oxidant produced at the positive
electrode with an
excess of the base form of the buffer to produce the salt form of the AMO,
while
simultaneously releasing a stoichiometric amount of the base form of the
buffer for
neutralization. The base form of the buffer is, for example, phosphonate,
hydrogen
phosphate, an amine, a tertiary amine, a morpholine derivative, etc. The
cation of the
buffer is, for example, lithium, other alkali metal, substituted ammonium,
imidazolium,
organic cation, etc. Other examples of the buffer components are hydroxide, a
lithium
cation, a magnesium cation, etc. In an embodiment, the buffer is one or more
of the
Good's buffers. The regeneration system 106 continues 803 the cycle of
electrolysis-
disproportionation in a single ED reactor 107 of a cascade of ED reactors till
the desired
degree of conversion of, for example, bromide into bromate is achieved.
[0182] The regeneration system 106 also converts the intermediate oxidant, for
example, bromine produced at the positive electrode or electrodes of the
electrolysis-
disproportionation (ED) reactor 107 into the aqueous multi-electron oxidant
(AMO) in
the salt form, such as bromate, using a chemical process, for example, a
homogeneous
chemical reaction such as a disproportionation reaction driven by a pH change,
or a
homogeneous oxidation by a mediator. The electrolysis-disproportionation (ED)
reactor
107 of the regeneration system 106 simultaneously releases on its negative
electrode or
86
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
electrodes a stoichiometric amount of hydrogen and of the base form of the
buffer for the
disproportionation. In an embodiment, the salt form of the AMO is converted
into the
acid form of the AMO in an orthogonal ion migration across laminar flow
(OIMALF)
reactor by recycling the reducer, for example, H2 produced at the negative
electrode and
consumed at the positive electrode of the OIMALF reactor. Other embodiments
for
recycling or partially recycling H2 during the regeneration are possible as
disclosed in the
detailed description of FIGS. 10A-10B. The ion exchange process proceeds in
the ion
exchange reactor 108 configured to convert 804 the AMO in the salt form, for
example,
LiBrO3 into the AMO in the acid form, for example, HBr03. All forms of the AMO
is
referred herein as the AMO. The conversion of the salt form of the AMO
produced at the
positive electrode into the acid form of the AMO is performed by an ion
exchange
process, for example, an electric field driven orthogonal ion migration across
laminar
flow (OIMALF) method known to those skilled in ion chromatography, in the ion
exchange reactor 108. In an embodiment, the conversion of the salt form of the
AMO
produced at the positive electrode into the acid form of the AMO is
accompanied by a
conversion of the base form of the buffer into the acid form of the buffer.
The AMO and
the reducer are stored in the regeneration system 106 until they are
transferred to the
discharge system 101 exemplarily illustrated in FIG. 1. The acid or salt form
of the AMO
is concentrated 805 in the concentrating reactor 112 to remove water produced
on the
positive electrode during the discharge and to remove water introduced with
the buffer
during electrolysis-disproportionation. The AMO is regenerated via an electron
transfer
to the positive electrode with or without a combination with a solution-phase
process
such as disproportionation; and the reducer is regenerated at the negative
electrode of the
ED reactor 107. The buffer maintains or stabilizes the pH of the discharge
fluid at an
optimal level or a constant value, for example, between 7 and 11 or at pH 8
for
disproportionation in the ED reactor 107. The buffer in the base form is
selected from a
group comprising, for example, an alkali metal hydroxide, an alkali metal
hydrogen
phosphate, an alkali metal salt of one of Good's buffers, substituted
phosphonic acid, and
any combination thereof. The alkali metal is, for example, lithium or sodium.
The base
form of the buffer, if its structure permits, is converted into an acid form
or a neutral form
during ion exchange.
87
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0183] In an embodiment, the regeneration of the aqueous multi-electron
oxidant
(AMO) and/or the reducer is facilitated, for example, by an electrocatalyst, a
solution-
phase redox mediator such as chlorine/chloride, a pH-dependent solution-phase
disproportionation, etc., or any combination thereof. In an embodiment, the
conversion of
the intermediate oxidant, for example, bromine, into one or more forms of the
AMO is
facilitated by a buffer in the disproportionation unit 107b. In another
embodiment, a
chloride mediator facilitates regeneration of the AMO from the discharge
fluid. In
another embodiment, the regeneration of the AMO and/or the reducer from the
discharge
fluid is facilitated by adding a base to the discharge fluid. The electrolysis-
disproportionation (ED) reactor 107 is configured to operate in one of
multiple modes
comprising, for example, a batch mode, a cascade flow mode, and a cyclic flow
mode.
The regeneration system 106 is configured for batch, cyclic or cascade flow
modes of
operation, or any combination thereof.
[0184] The electrolysis-disproportionation (ED) reactor 107 converts a
discharged
product such as bromide, into a salt form of the aqueous multi-electron
oxidant, for
example, bromate. The ion exchange reactor 108 converts the aqueous multi-
electron
oxidant such as bromate from the salt form into the acid form. The ion
exchange reactor
108 also converts the discharge fluid from the acid form into a neutral form.
The ED
reactor 107 adds a base, for example, HP042- to the discharge fluid
comprising, for
example, bromide as exemplary illustrated for one specific chemistry in the no-
aqueous
multi-electron oxidant (AMO)-on-negative mode of operation in equation (18)
below:
Br- + H2P042- + OH-= Br- +HP042 (18)
and electrolyzes the resulting alkaline discharge fluid to produce hydrogen
(H2) and the
intermediate oxidant such as Br2.
anode: Br- -e- = '1/2 Br2; (19)
cathode: H20 + e- = OH- + 1/2 H2 (20)
88
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0185] The counter cation used in the process shown in equations (18)-(19) is
one or a
combination of an alkali metal, for example, Na-F, an alkali earth metal,
quaternary
nitrogen or phosphorus cations, derivatives of nitrogen heterocycles, and
other organic
and inorganic cations.
[0186] The base is regenerated in the catholyte in the course of the hydrogen
evolution
reaction. The intermediate oxidant such as bromine further disproportionates
via a
reaction with the base, for example, as follows:
3Br2 + 6HP042- + 3H20 = Br03- + 5Br- + 6H2P042 (21)
[0187] The process of electrolysis-disproportionation as shown in equations
(18)-(21)
above continues in a cyclic flow mode or a cascade flow mode until all or
almost all the
bromide is converted into bromate. In the next stage, which can be performed
either on-
board within the discharge system 101 or off-board within the regeneration
system 106 or
in both systems, the bromate is converted into bromic acid in the ion exchange
reactor
108, for example, an orthogonal ion migration across laminar flow (OIMALF)
reactor.
The salt left over from the disproportionation buffer such as lithium
dihydrogen
phosphate is, for example, also converted into an acid such as phosphoric acid
and for
example, and is used in the oxidant fluid with the aqueous multi-electron
oxidant without
separation.
[0188] In an embodiment, the intermediate oxidant, for example, a halogen, is
regenerated via an electron transfer at the positive electrode, and the
reducer such as
hydrogen is regenerated at the negative electrode of the electrolyzer 107a of
the
electrolysis-disproportionation (ED) reactor 107. In another embodiment, the
intermediate
oxidant disproportionates during the process of regeneration by consuming a
base and
provides the final aqueous multi-electron oxidant, for example, a halate in
the form of a
salt. The base required for the disproportionation of the intermediate oxidant
can be
supplied externally or can be produced in the course of the cathodic counter
reaction, such
89
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
as hydrogen evolution, in the electrolyzer 107a of the ED reactor 107. A
buffer is added to
either the catholyte chamber or the anolyte chamber or in both the catholyte
chamber and
the anolyte chamber of electrolyzer 107a in order to stabilize the pH at the
level optimal for
the disproportionation. A suitable pH of the buffer is between, for example, 7
and 11
depending on the target aqueous multi-electron oxidant (AMO). A suitable
buffer is, for
example, a solution of Na2HPO4 and NaH2PO4 in various ratios and
concentrations.
Another suitable buffer is one or more of the Good's buffers, other secondary
amine, other
amine, substituted phosphonate, and a nitrogen heterocycle. During the
disproportionation
reaction, in the presence of a buffer or a base containing a cation other than
hydrogen, a salt
form of the aqueous multi-electron oxidant, for example, NaBrO3 is produced.
[0189] The intermediate product, that is, the salt of the aqueous multi-
electron oxidant
(AMO) is converted into the acid form in the ion exchange reactor 108, for
example, the
orthogonal ion migration across laminar flow (OIMALF) reactor using one or a
combination of electrolysis, ion exchange on solids, ion exchange in a fluid,
and an
electric-field driven OIMALF process. The ion exchange occurs after the
electrolysis-
disproportionation (ED) loop or cascade as exemplarily illustrated in FIGS.
10A-10B.
The ED loop is a cyclic process involving oxidation of the salt form or other
forms of the
discharged oxidant, for example, bromide, on the positive electrode of the
electrolyzer
107a of the ED reactor 107 into the intermediate oxidant, for example,
bromine; a
disproportionation reaction that converts the intermediate oxidant such as
bromine into
the salt form of the AMO such as bromate, and into the salt form of the
discharged
oxidant such as bromide; oxidation of the salt form of the discharged oxidant
on the
positive electrode of the electrolyzer 107a into the intermediate oxidant,
etc.
[0190] In an embodiment, the regeneration of the aqueous multi-electron
oxidant from
the discharge fluid occurs by reverse transformation of a cathodic discharge
product in
the discharge fluid and without oxygen consumption or evolution. In another
embodiment, the regeneration of the aqueous multi-electron oxidant from the
discharge
fluid comprises neutralizing an acid of the discharge fluid, for example, via
an ion
exchange such as orthogonal ion migration across laminar flow (OIMALF). The
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
regeneration system 106 then converts the neutralized discharge solution into
an
intermediate oxidant and the reducer by means of electrolysis. The
intermediate oxidant
is further converted into the salt form of the aqueous multi-electron oxidant
(AMO) via
pH dependent solution phase disproportionation and the salt form of the AMO is
converted into to the acid form of the AMO via ion exchange such as orthogonal
ion
migration across laminar flow process. The regeneration process on the
positive electrode
of the electrolyzer 107a of the electrolysis-disproportionation (ED) reactor
107 is
facilitated by using one or a combination of an electrocatalyst, a solution-
phase catalyst,
an ion exchange on solids, an ion exchange in a liquid, a pH¨dependent
disproportionation, and an orthogonal ion migration across laminar flow in one
ED
reactor 107 or separate reactors in series and/or in parallel. For the H2-
HBrO3 regeneration,
different embodiments of the methods or routes of electrochemical regeneration
of
hydrogen and bromic acid from aqueous hydrogen bromide are disclosed herein.
Direct
electrolysis such as with Pb02 and Ru02- based anodes and mediated
electrolysis such as
with C12-mediator are also implemented.
[0191] FIG. 9 exemplarily illustrates a negative-ion electrospray ionization-
mass
spectrometry spectrum of a 0.5M sodium phosphate pH 7.0 buffer solution after
addition
of 50mM of Br2. Bromide and bromate are the only detectable negative Br
species with
2Da 1:1 doublets. These data affirm that bromine disproportionates only into
bromide and
bromate in a pH 7 buffer. The labeled signals of bromide and bromate prove the
occurrence of the regeneration reaction (6) in this buffer. The kinetics of
the bromine
disproportionation has been studied mostly in near neutral media 4 < pH < 8,
where the
rates of various steps fall in the range convenient for experimental
measurements. The
disproportionation of Br2 in water may go all the way to bromate and even to
perbromate.
The first step occurs at near neutral 4 < pH < 8 via the following pathway:
Br2 +H20 = HBrO + H++Br- (22)
Br2 +OH- = HBrO + Br - (23)
[0192] Herein, bromine disproportionates into bromide and hypobromite in two
parallel
91
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
reactions with water and with another base such as hydroxide, that is, via a
general base
mechanism. The equilibrium constant at 25 C for equation (22) at 0.5M ionic
strength is
6.1x10-9 M2. The first order rate constant for the forward reaction for
equation (22) is 97
s-1, while for the reverse comproportionation reaction with H+ it is 1.6x101
M-2s-1. The
bromine disproportionation has not been studied computationally, but molecular
dynamics show that the homologous chlorine reaction in water clusters proceeds
as a
bimolecular a+ transfer between C12 and H20. The chlorine disproportionation
in acidic
solutions also follows a general acid-base catalysis route, first order in C12
and in the
general base, while the reverse comproportionation reaction is first order in
HOC1, CF
and in the general acid.
[0193] The hypobromous acid formed in reactions (22) and (23) above undergoes
a
further disproportionation which is strongly pH dependent. At a low pH bromine
and
bromate are formed:
5H0Br 4¨* 2Br2 + Br03- +2H20 + H+ pH<4 (24)
[0194] The bimolecular rate constant with respect to the total Br(I) is
approximately
2.2x10-3 M-ls-1 when extrapolated to pH 0 and increases at higher pH due to
the
participation of a deprotonated hypobromite in the rate limiting step. At a
higher pH,
bromide and bromate are formed:
3H0Br 4¨* 2Br- + Br03- +3H+ pH>4 (25)
and the rate of the reaction decreases with pH above the pKa of hypobromous
acid of 8.8,
although the kinetic equation retains the second order in total Br(I) and the
general base
catalysis is operative. Both reactions (24) and (25) occur in parallel at the
intermediate
4<pH<8 where the formal second-order rate constant is the highest. Thus, the
optimal pH
for the regeneration process
3Br2 + 60H- = Br03- + 5Br- + 3H20 (26)
92
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
is 4-9. For example, the optimal pH for the regeneration process is between 6-
8. The
reaction is slower at higher pH since an intermediate hypobromite is more
stable, and at a
lower pH, the equilibrium shifts towards Br2. These considerations based on a
literature
analysis are confirmed in ESI-MS data, as exemplarily illustrated in FIG. 9
and FIG. 26,
which prove the feasibility of the regeneration process as per equation (26).
[0195] FIGS. 10A-10B exemplary illustrate an electrolysis-disproportionation
(ED)-
orthogonal ion migration across laminar flow (OIMALF) method for regenerating
a
reducer (H2) and an oxidant fluid comprising an aqueous multi-electron oxidant
(HX03)
from a discharge fluid comprising HX and H20 with MOH as the base. FIG. 10A
illustrates electrolysis-disproportionation in the batch mode with no-aqueous
multi-
electron oxidant (AMO)-on-negative mode of operation. FIG. 10B illustrates
electrolysis-
disproportionation in the cyclic flow mode with no-AMO-on-negative mode of
operation.
FIG. 10A exemplarily illustrates a method for regenerating halic acid and
hydrogen from
discharged hydrogen halide with a batch ED reactor 107. FIG. 10A exemplary
illustrates
a regeneration system 106 comprising an electrolysis-disproportionation
reactor 107 and
an ion exchange reactor 108 in a batch mode of operation for regenerating
reducer (H2)
and acidic oxidant fluid comprising an aqueous multi-electron oxidant (HX03)
from the
discharge fluid (HX + H20) with MOH as a base configured for the no-AMO-on-
negative mode of operation. FIG. 10B exemplarily illustrates a method for
regenerating
halic acid and hydrogen from discharged hydrogen halide with a flow-through ED
reactor
107 in a cyclic flow mode. FIG. 10B exemplary illustrates a regeneration
system 106
comprising a flow-type electrolysis-disproportionation reactor 107 configured
for the no-
AMO-on-negative mode of operation and an ion exchange reactor 108 for
regenerating
reducer (H2) and the oxidant fluid comprising the aqueous multi-electron
oxidant (HX03)
from the discharge fluid (HX + H20) with MOH as a base. For purposes of
illustration
and not of limitation, the concentrating reactor 112 is exemplarily
illustrated after rather
than before the ion exchange reactor 108. The regeneration system 106 is
equipped with
the ion exchange reactor 108, in addition to the electrolysis-
disproportionation (ED)
reactor 107, that converts salts into acids, for example, an aqueous solution
comprising
93
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
lithium bromate and a 3-(N-morpholino) propanesulfonic acid (MOPS) anion into
an
aqueous solution comprising bromic acid and protonated MOPS using a flow-
through
OIMALF reactor, which is similar to an eluent suppression reactor employed in
ion
chromatography The OIMALF process of converting salts into acids avoids
chemical
separation and ion exchanger regeneration steps. The OIMALF reactor can employ
hydrogen produced in the ED reactor 107 as exemplarily illustrated in FIGS.
10A and
10B, or hydrogen stored on board as exemplarily illustrated in FIG. 19, or
recycle the
hydrogen produced on the negative electrode and consumed on the positive
electrode of
the ED reactor 107.
[0196] The regeneration system 106 converts the discharge fluid back into the
aqueous
multi-electron oxidant (AMO) and the reducer using the ED reactor 107 and
depending
on the preferred options, the ion exchange reactor 108, a mixing reactor or
the
neutralization reactor 109, and separation reactors, for example, 1006, 1007,
and 1010 as
exemplarily illustrated in FIG. 10B, if needed, are not counted as parts of
the other
devices. In an embodiment, the ED reactor 107 is configured and operated as a
batch
reactor or a flow-through reactor. In the batch ED reactor, also referred to
as a stirred
reactor, the liquid in the positive electrode compartment is stirred to
achieve a uniform
composition. The batch ED reactor operates in the start-stop batch regime till
the desired
degree of conversion of bromide into bromate is achieved.
[0197] A series of a single neutralization reactor 109, a single flow-through
ED reactor
107, and a single H2/base separation reactor 1006 can operate in the cyclic
regime till the
desired degree of conversion of bromide into bromate is achieved. The
feedbacks 1009
and 1003 return the base from the H2 separation reactor 1006 and partially
regenerated
oxidant fluid from the positive loop valve 1004, respectively. The H2 is
accumulated in
the H2 container during this cycle.
[0198] A series of a single neutralization reactor 109, a single flow-through
the ED
reactor 107, and a single H2/base separation reactor 1006 can operate in the
cascade
regime, wherein the discharge fluid HX is first neutralized with an excess of
a base
94
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
generated earlier in the ion exchange reactor 108, for example, the orthogonal
ion
migration across laminar flow (OIMALF) reactor. The first flow-through ED
reactor 107
then converts some Br- into Br03- on the positive electrode while releasing H2
and base
on the negative electrode. The H2 goes into an H2 storage container (not
shown), and the
base from the separation reactors 1006, 1007, and 1010 is returned to the
mixing reactor
of the neutralization reactor 109 preceding this H2 separation reactor 1006 in
the series.
The partially regenerated oxidant fluid, for example, comprising LiBr and
LiBrO3 in
some ratio, instead of going into one preceding mixing reactor of the
neutralization
reactor 109 as in the cyclic flow mode, goes into the second mixing reactor in
the
cascade, where LiBr+ LiBrO3 is neutralized by the base produced in the second
mixing
reactor and so on. The number of repeated mixing reactor-ED-separation reactor
series in
the cascade is, for example, between 5 and 8, and is determined by desired
throughput,
power, cost, degree of conversion, etc.
[0199] The regeneration process comprises the steps of neutralization in the
neutralization reactor 109, electrolysis-disproportionation in the ED reactor
107,
separation of the reducer, that is, H2 gas from the aqueous multi-electron
oxidant (AMO)
species in water in the separation reactor 1006, and ion exchange via an
orthogonal ion
migration across laminar flow (OIMALF) in the ion exchange reactor 108 as
disclosed in
the detailed description of FIG. 8. The regeneration system 106 takes HX +
3H20 from
the discharge fluid and produces 3H2 + HX03, while recycling within itself,
water, and
the buffer. The separation reactor 1006 separates the liquid solution with the
base from
the hydrogen gas reducer. The base component of the buffer is represented as
OH-.
[0200] The ED reactor 107 has an electrolysis unit or an electrolyzer 107a
with
multilayer electrolyte-electrode assemblies (not shown), a number of bipolar
plates, and
two endplates. The discharge fluid from the discharge fluid storage tank (not
shown) is
mixed in the neutralization reactor 109, with the solution of the buffer in
the base form
coming out of the gas-liquid separation reactors 1006, 1007, and 1010 through
the return
lines 1009, 1002, and 1003, and then sent to the positive compartment of the
ED reactor
107. The neutralized discharge fluid MX is electro-oxidized into the
intermediate oxidant
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
X2 at the positive electrode of the ED reactor 107. The intermediate oxidant
X2 reacts
with the base form of the buffer to produce a salt form of the aqueous multi-
electron
oxidant (AMO), for example, LiBr03. On the negative electrode of the ED
reactor 107,
hydrogen gas is produced upon electrolysis, and a base, for example, an amine,
a
phosphonate, or a hydroxide is formed in the negative electrode. The base from
the
negative compartment is mixed with the discharge fluid in the mixing reactor
or the
neutralization reactor 109 prior to or directly in the positive compartment of
the ED
reactor 107. On the positive electrode, an intermediate oxidant such as
bromine is
produced during electrolysis and reacts with the base introduced from the
negative
compartment yielding, for example, a bromate and a bromide via a
disproportionation
reaction. The remaining halide is oxidized into halogen on the positive
electrode of the
ED reactor 107 and disproportionates in a reaction with the base in the next
ED cycle.
[0201] The electrolysis-disproportionation (ED) process can proceed as a
single pass
process with a three-way valve 1004 for a sufficiently long ED reactor 107 and
sufficiently high amount of the buffer in the base form added in the
neutralization reactor
109. In an embodiment, the ED process can run in a cyclic flow mode with two
three-way
valves 1004 and 1005 in the loop, which is useful for a shorter ED reactor
107, which
increases the regeneration time but saves on capital expenses. The three-way
valves 1004
and 1005 are exemplarily illustrated in FIG. 10B in positions for the single
pass mode of
operation of the ED reactor 107. The three-way valves 1004 and 1005 send the
aqueous
multi-electron oxidant (AMO) in the salt form, that is, MX03, for ion exchange
via an
orthogonal ion migration across laminar flow (OIMALF) in the ion exchange
reactor 108
to produce the acid form of the AMO, that is, HX03.
[0202] The electrolysis-disproportionation (ED) reactor 107 can be configured
and
operated in a batch mode or in a flow- through mode. The flow- through mode
can be a
cycle with one or more units or a cascade with 2 or more units. When a
sufficient degree
of conversion, that is, ratio of bromate concentration to the total
concentration of Br in all
forms is achieved in the ED reactor 107, after a certain charge, that is, time
or number of
cycles passed, the electrolysis is completed. The liquid from the positive
electrode
96
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
chamber of the ED reactor 107 goes into the ion exchange reactor 108 where, in
the
middle chamber in the exemplary case of the Li-3-(N-morpholino)
propanesulfonic acid
(MOPS) base form of the buffer, the bromate is converted into bromic acid, Li-
MOPS
buffer is converted into a protonated MOPS zwitter-ion, and hydrogen is
consumed in the
positive chamber, and hydrogen is produced in the negative chamber. The base,
for
example, Li-MOPS or its equivalent is generated in the negative chamber along
with
hydrogen. The base solution is used to neutralize the acidic discharge fluid,
for example,
comprising HBr, incoming from the discharge unit 104 exemplarily illustrated
in FIG. 1.
The separation reactors 1006, 1007, and 1010 are used to separate the gases
from the
liquids.
[0203] Halates are produced by disproportionation of a halogen in the presence
of a
base. The process of disproportionation of halogens is described by the
following
equation using hydroxide as an example of a base:
3X2 + 6 MOH = MX03 + 5MX + 3H20. (27)
[0204] In the electrolysis-disproportionation (ED) reactor 107, exemplarily
illustrated
in FIGS. 10A-10B, if the liquid in the positive electrode chamber and the
liquid in the
negative electrode chamber are allowed to mix, the halogen produced on the
positive
electrode can react with the base produced on the negative, yielding, for
example, a
halate and a halide. The halide is oxidized on the anode, thus initiating the
new cycle of
the loop:
MX +H20 +electrolysis = (0.5H2 + MOH) negative electrode chamber +0.5X2
positive electrode chamber, (28)
3X2 + 6MOH = 5MX+ MX03 + H20 after mixing in the positive electrode
chamber, (29)
where the disproportionation described in equation (29) can be performed
either in a
97
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
flow-through process or a batch process with or without assistance of a buffer
such as a
phosphate buffer. The net equation of the regeneration process, that is, the
ED process is:
6MX + 3H20 = MX03+ 3H2 (30)
with the electrolysis-disproportionation loop 109 to 1007 as exemplarily
illustrated in
FIGS. 10A-10B. The reduction of X03- on the negative electrode in the
electrolyzer 107a
is prevented, for example, by using a negative electrode with a multilayer
structure with a
cation-selective membrane facing the solution comprising the aqueous multi-
electron
oxidant (AMO). In an embodiment, a membraneless reactor can also be used if
the
negative electrode comprises, for example, Ni capable of selective reduction
of water into
hydrogen without reducing X03-, or if the electrolyte layer 205c is not a
solid membrane
but a laminar flow electrolyte. The optional concentrating reactor 112 removes
water
introduced with the buffer during electrolysis-disproportionation. A portion
of the water
is transferred to the neutralization reactor 109 via the water return line
1008 exemplarily
illustrated in FIG. 10B.
[0205] The choice of the counter-cation in the regeneration schemes of FIG. 8
and
FIGS. 10A-10B depends on the solubility of the counter-cation' s halide,
halate, and
buffer salts such as a phosphonate, a Good's buffer, etc., since circulating a
small volume
of a liquid and minimizing the energy and capital expenses of water removal in
making a
concentrated aqueous multi-electron oxidant (AMO) solution is beneficial.
These
considerations are relaxed in the case of an off-board regeneration system 106
as
compared to an on-board regeneration system 106 exemplarily illustrated in
FIG. 1.
Lithium bromide (18.4m) and bromate (13.3m) have substantially high
solubilities in
water at 20 C and so does lithium hydroxide (5.3m). Li salts with a suspension
of
hydroxide or phosphate or with addition of a complexing agent such as 15-crown-
5
(15C5), benzo-15-crown-5 (B15C5), dicyclohexano-18-crown-6 (DC18C6),18-crown-6
(18C6), 12-crown-4 (12C4), dibenzo-18-crown-6 (DB18C6), and their more water-
soluble derivatives are also considered. For I( , bromate solubility is low,
for example,
about 0.41m at about 20 C. Na + salts have intermediate solubilities in
water, for
98
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
example, of about 2.4m for bromate, and about 8.8m for bromide at about 20 C.
The "m"
does not have to be a monovalent cation. For example, magnesium bromate has a
good
solubility in water (2.5m at 0 C, 5.36m at 60 C). Calcium bromate has also a
good
solubility (1.86m at 40 C) that shows only a weak dependence on temperature.
Also,
Good's buffers have high solubility often above 2m. The symbol "m" refers to
the molal
concentration, that is, the moles of solute per kg of solvent.
[0206] FIGS. 11A-11B exemplary illustrate a cyclic operation of a flow-through
electrolysis-disproportionation (ED) reactor 107 with bromate as an aqueous
multi-
electron oxidant (AMO), hydrogen phosphate as a base form of a buffer, and
sodium as
the counter cation configured for no-AMO-on-negative mode of operation. FIGS.
11A-
11B exemplarily illustrate a method for regenerating sodium bromate and
hydrogen from
sodium bromide and water. The charge of one electron per bromide is shown in
each
cycle for the sake of illustration not of limitation. Theoretically estimated
water transfer
numbers are shown for the sake of illustration and not of limitation. The
balance of water
dragged with ions is not shown. A buffer can be used instead of the hydroxide
or in
addition to the hydroxide. This method allows for minimizing the spatial and
temporal
variations of pH outside of the range between 7 and 9. For example, a solution
comprising Na + cation and any of the forms of deprotonated phosphoric acid
can be used
as a component of the buffer. FIGS. 11A-11B exemplarily illustrate a method to
execute
the electrolysis-disproportionation (ED) regeneration step based on a cyclic
operation of
the ED reactor 107 with a cation exchange membrane or in a row of, for
example, 6 cells
connected in series. For purposes of illustration, the detailed description
refers to the
bromate chemistry, the Na + cation, and a phosphate buffer; however the scope
of the
method and the system 100 disclosed herein is not limited to the bromate
chemistry, the
Na + cation, and the phosphate buffer but can be extended to include other
aqueous multi-
electron oxidants (AM0s), cations including Li, and buffers including Good's
buffers.
[0207] FIG. 12 exemplarily illustrates calculated and experimentally measured
limiting
currents on a rotating disk electrode in aqueous solutions of bromic acid of
various
concentrations. FIG. 12 exemplarily illustrates the calculated kinetic
limiting current of
99
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
bromate comproportionation in a 0.1 M HBrO3 + 1mM Br2 and experimentally
measured
limiting currents on a glassy carbon rotating disk electrode for the 1st wave
in 50% w
HBrO3 attributed to the electroreduction of bromine generated via equation (2)
and for
the 2nd wave in 50% w HBrO3 attributed an unidentified intermediate of
equation (2)
comprising Br in the oxidation state 0<OS<5. The rational for using a diluted
acidic
aqueous multi-electron oxidant (AMO) solution for the 2nd wave measurements
was the
current range limitation of the potentiostat. The calculated current and
experimental
current of the first wave is due to the reduction of bromine and its value is
determined by
the rate of the comproportionation of bromate with electrogenerated bromide
near the
electrode surface. The current of the second wave is tentatively attributed to
an
intermediate in the comproportionation reaction, such as BrO,,- with x = 1 or
2. A direct
electrochemical reduction of bromate at room temperature occurs with a
significant
overvoltage on all electrode materials, and a bromate reduction on an
electrode can be
facilitated via a homogeneous comproportionation with bromide into highly
electrochemically active bromine:
In the catholyte: 5HBr + HBrO3 = 3H20 + 3Br2 (31)
On the cathode: Br2 + 6 e- = 6 Br- (32)
[0208] The cathode refers to the electrode where the electroreduction takes
place, that
is, the positive electrode in this case.
[0209] The comproportionation reaction (31) is known to be first order in
[Br031 and
[Br] and second order in [H+] at pH below 2. An additional term with a second
order in
bromide is apparent at high bromide concentrations. The actual mechanism
involves
several serial and parallel steps that show general acid catalysis effects,
and the
mechanism is similar to the homologous chlorine and iodine processes. Chloride
accelerates reaction (31). The effect of the addition of chloride species on
both the
discharge and regeneration processes is also disclosed herein since the
intermediate
chlorine increases the energy and power densities of the system 100 with Br2
as the
intermediate oxidant due to a complex interplay between the aqueous
chemistries of the
100
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
two halogens. The electroreduction of Br2 as per equation (32) is a first
order process
with a high exchange current even on carbon electrodes which are used in Zn-
Br2 and
NaSõ-Br2 batteries.
[0210] The calculated dependence of the limiting current on the rotation rate
in a 0.1M
aqueous multi-electron oxidant (AMO) solution is represented in FIG. 12 as a
solid line,
the experimental data on a glassy carbon rotating disk electrode for the main
wave in
approximately 20% AMO solution is represented as a dotted line, and the
prewave in
approximately 50% AMO solution is represented as a dashed line. Currents over
0.5A/cm2 are obtained on a smooth carbon electrode. The limiting current shows
a
decrease with the rotation rate due to the loss of the intermediate bromine
into the
solution, when the thickness of the diffusion boundary layer is smaller than
the thickness
of the kinetic boundary layer. The dependence of the limiting current on a log
of the
rotation rate in HBrO3 solutions is also exemplarily illustrated in FIG. 12.
[0211] While the theoretical energy density of the H2/HBrO3 system 100 is, for
example, about 1,951 Wh/kg, the limited stability of bromic acid solution with
the
concentration above 55% w/w, makes 938 Wh/kg, that is, 3.25 times higher than
the
theoretical value for the lithium iron phosphate (LFP) chemistry, a more
realistic
estimate. Taking into account the 5% w/w H2 content for high-pressure storage
and the
flow design, about 426 Wh/kg is obtained as a realistic target value at the
system level,
which is 6 times larger than the corresponding number for the LFP battery
pack, for
example, in Tesla Roadster of Tesla Motors, Inc.
[0212] FIG. 13 exemplary illustrates a graphical representation of a
power¨voltage
curve calculated for a H2-50% w/w HBrO3 discharge unit 104 and measured with a
glassy
carbon rotating disk electrode, and with a platinum gauze electrode in a flow
cell and a
corresponding curve for a commercial proton exchange membrane fuel cell
running on
hydrogen and air. The +1.4 V onset of HBrO3 electroreduction on Pt implies a
direct
process rather than comproportionation-mediated electrode process. The
reduction of
bromate on the positive electrode is modeled for one dimensional (1D)
diffusion normal
101
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
to the electrode and for constant thicknesses of the kinetic and hydrodynamic
boundary
layers. As used herein, the term "diffusion" refers to mass transport due to a
concentration gradient.
[0213] In FIG. 13, which exemplary illustrates the calculated power versus
voltage
plots for a hydrogen-bromate discharge unit 104, the term "Zo" refers to the
ratio of the
hydrodynamic boundary layer thickness to the kinetic boundary layer thickness
and under
the conditions of the experiment, the latter is equal to approximately 1.5 pm.
The term
"Co" exemplarily illustrated in FIG. 13 refers to the bulk concentration of
free
intermediate oxidant such as bromine. A typical curve for a H2-air polymer
electrolyte
membrane fuel cell (PEMFC) is also shown in FIG. 13 for comparison. The
membrane
resistance for the solid line is equal to 0.1 Ohm/cm2 and the membrane
resistance for the
dotted line is equal to 0.25 Ohm/cm2. The lines with circles represent
experimental data
in 50% HBrO3 aqueous multi-electron oxidant (AMO) solution. The solid lines
and the
dashed lines represent experimental data with the glassy carbon rotating disk
electrode
(GCRDE) at different rotation rates, and the dashed-double dotted line
represents
experimental data in a proton exchange membrane (PEM) type flow cell, for
example, a
Fuel Cell StoreTM #1071025 with Pt gauze electrodes on both sides, powered by
H2 and
50% HBrO3.
[0214] The experimental data of FIG. 13 with glassy carbon (GC) electrodes in
an
aqueous solution of HBrO3 shows three regions in the power-voltage curve - a
cathodic
pre-wave at +1.15 V versus a standard hydrogen electrode (SHE) with a 42
mV/decade
Tafel slope, a main cathodic wave at +0.7 V versus SHE with a 208 mV/decade
Tafel
slope, and an anodic wave. Both cathodic waves show a decrease in the limiting
current
at higher rotation rates as approximately i¨l/oil and at lower aqueous multi-
electron
oxidant (AMO) concentrations as i¨CAm03. The more positive wave, that is, the
1st wave
on GC is attributed to the predicted reduction of the intermediate Br2 since
the positive
wave on GC occurs at the appropriate potential and has a low Tafel slope,
close to 60
mV/dec that is usually observed, whereas the more negative wave, that is, the
2nd wave is
likely due to an intermediate with a lower exchange current such as BAT or
Br02-,
102
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
formed during the comproportionation before Br2. The small but unambiguous
anodic
wave positive to E (Br2/2Br-) with a very high formal Tafel slope of 332
mV/dec is
likely due to the oxidation of bromide which is slowly formed via the
reversible
disproportionation of bromine present in equilibrium with HBr03. A discharge
power of
mW/cm2 at 70% efficiency with respect to E of Br037Br- is achieved with a
smooth
carbon electrode and dilute 20% AMO.
[0215] The 1W/cm2 target can be achieved by using a high area porous
electrode,
increasing the concentration of the aqueous multi-electron oxidant (AMO) and
by adding
additional proton donors such as an extra acid, to the AMO stock. Unlike the
case of the
glassy carbon rotating disk electrode (GC-RDE), the onset of bromate reduction
on Pt,
exemplarily illustrated in FIG. 13, occurs at 1.42 V versus reversible
hydrogen electrode
(RHE) at pH-0, which is more positive than the E (Br2/Br- ) =1.066 V. This is
attributed
to the direct electroreduction of bromate on Pt in acid. Despite the
possibility of having a
higher efficiency discharge unit 104 exemplarily illustrated in FIG. 1, the
preliminary
economic analysis suggests against the use of Pt at the 0.2 mg/cm2 loading in
the cathode
of the discharge unit 104 in automotive applications due to an increased
upfront cost
which will not amortize over 3 years, which is the projected Pt cathode
durability, by the
lower operational cost and energy efficiency. A Pt or another catalyst can be
used on the
positive electrode 205a exemplarily illustrated in FIG. 2 in other high-end
applications
such as in military applications and aerospace applications. Oxide catalysts
such as Ru02
and its derivates are suitable for the use on the positive electrode 205a of
the discharge
unit 104.
[0216] FIGS. 14A-14G that exemplarily illustrate graphical representations
showing
comparative performances of three on-board power sources at a nominal power of
130
kW: a gasoline-internal combustion engine, a lithium ion battery, and an H2-
aqueous
multi-electron oxidant discharge unit as well as the targets of the Advanced
Research
Projects Agency-Energy are disclosed along with the examples enumerated later
in this
description.
103
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0217] FIG. 15 illustrates an embodiment of the system 100 for generating
electric
power and a discharge fluid from an oxidant fluid and a reducer fluid using a
discharge
system 101 comprising an orthogonal ion migration across laminar flow (OIMALF)
reactor 1501, and for regenerating an oxidant and/or a reducer from the
discharge fluid
using a regeneration system 106. The system 100 disclosed herein cyclically
discharges
and recharges or refills the discharge system 101 in an energy storage cycle.
In this
embodiment, the acidification process such as the ion exchange process is
performed
within the discharge system 101 rather than within the regeneration system 106
in order
to improve the stability and safety of the systems disclosed herein. The
discharge system
101 comprises the discharge unit 104, an acidification reactor 1501a,
optionally a
neutralization reactor 1501b, the oxidant fluid tank 102, the reducer fluid
tank 103, and a
discharge fluid tank 113. The acidification reactor 1501a comprises one or
more of an
acid storage tank (not shown) storing H2SO4, TfOH, etc., and an acid mixing
tank (not
shown) in the OIMALF reactor 1501. The acidification reactor 1501a converts
the
neutral oxidant fluid stored in oxidant fluid tank 102 into an acidic oxidant
fluid by
lowering the pH of the acidic oxidant fluid for facilitating further
electroreduction of
acidic oxidant fluid at one or more positive electrodes 205a of the discharge
unit 104 via
a pH-dependent comproportionation.
[0218] The acidic oxidant fluid comprises, for example, one or more of water,
one or
more forms of the aqueous multi-electron oxidant (AMO), for example, an acid
or a salt
form or as a combination thereof, an extra acid, and one or more of multiple
counter
cations. The AMO comprises one or a combination of halogens, halogen oxides,
halogen
oxoanions, and salts and acids of the halogen oxoanions. The extra acid is,
for example,
one or more of a phosphoric acid, a 3-(N-morpholino)propanesulfonic acid, a 3-
(N-
morpholino)ethanesulfonic acid, a methanesulfonic acid, a triflic acid, a
substituted
sulfonic acid, a substituted phosphonic acid, a perchloric acid, a sulfuric
acid, a molecule
comprising sulfonic moieties and phosphonic moieties, and an acid with a pKa <
2. The
halogen oxoanions comprise, for example, one or more of hypochlorite,
chlorite, chlorate,
perchlorate, hypobromite, bromite, perbromate, hypoiodite, iodite, iodate, and
periodate.
In an embodiment, the halogen oxoanion is bromate. The counter cations
comprise alkali
104
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
metal cations, alkali earth metal cations, and organic cations. In an
embodiment, one of
the counter cations is lithium. In another embodiment, one of the counter
cations is
sodium. The acidic oxidant fluid has a sufficient chemical reactivity to cause
an ignition
regime of electroreduction on the positive electrodes 205a of the discharge
unit 104. The
neutralization reactor 1501b neutralizes the discharge fluid, for example,
hydrogen halide
produced by the discharge unit 104 with a base form of a buffer to produce a
solution of a
salt form of the discharge fluid also referred to herein as a "neutral
discharge fluid". In an
embodiment, the neutralization reactor 1501b comprises a mixing reactor. The
discharge
fluid tank 113 is used to collect the discharge fluid for future regeneration
or disposal.
[0219] In an embodiment, the acidification reactor 1501a and the
neutralization reactor
1501b are functionally combined as an orthogonal ion migration across laminar
flow
(OIMALF) reactor 1501. In another embodiment, the neutralization reactor 1501b
is
integrated with the acidification reactor 1501a into the OIMALF reactor 1501
as
exemplarily illustrated in FIG. 15 and FIG. 19. The OIMALF reactor 1501
comprises an
OIMALF cell stack (not shown) which is configured similar to a polymer
electrolyte fuel
cell (PEFC) stack but with a liquid electrolyte flowing between two ionically
conducting
membranes. The OIMALF reactor 1501 comprises flow cell assemblies, endplates,
and
bipolar plates. Each flow cell assembly of the OIMALF reactor 1501 comprises a
couple
of ion exchange membranes comprising a positive side ion exchange membrane and
a
negative side ion exchange positioned parallel to each other, an intermembrane
flow field
interposed between the ion exchange membranes and comprising multiple flow
channels,
a positive electrode layer and a negative electrode layer flanking outer
surfaces of the ion
exchange membranes, and porous diffusion layers flanking the outer surfaces of
the
positive and negative electrode layers. The porous diffusion layers are in
electric contact
with the adjacent bipolar plates or endplates. The positive electrode layer is
configured
for hydrogen oxidation reaction and the negative electrode layer is configured
for
hydrogen evolution reaction. Although, the on-board OIMALF reactor 1501 adds
to the
weight of the discharge system 101, this addition can be tolerated due to the
high power
density and low energy consumption of the OIMALF reactor 1501. Moreover, only
10%
or less of the electric power generated by the discharge unit 104 is required
to support the
105
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
OIMALF reactor 1501. Also, the estimated weight of the OIMALF reactor 1501 for
a
130 kW discharge system 101 is about 54.2 kg which is only about 50% of the
weight of
the discharge unit 104 and 30% of the weight of the oxidant and the reducer,
and thus
adds only approximately 14% to the weight of the discharge system 101.
[0220] The discharge system 101 disclosed herein is configured to operate in
an electric
partial recharge mode for facilitating regenerative breaking when the
discharge system 101
powers an electric vehicle. During the partial recharge mode, the reactions on
the positive
and negative electrode reverse their directions, that is, the reducer is
produced on the
negative electrode 205b of the electrolyte-electrode assembly 205 and an
intermediate
oxidant is produced on the positive electrode 205a of the electrolyte-
electrode assembly
205. For example, H2 is produced on the negative electrode 205b and Br2 is
produced on
the positive electrode 205a. Since the pH of the oxidant fluid is acidic
during the
discharge, the disproportionation does not occur and the aqueous multi-
electron oxidant
(AMO), that is, bromate is not formed. The regeneration stops at the bromine
which is the
intermediate oxidant and can be easily consumed to provide power when the
current
direction goes back to the discharge mode.
[0221] The discharge unit 104 disclosed herein reduces the crossover of the
anionic
oxidants and products from the positive cathode to the negative hydrogen anode
by
employing a cation-exchange membrane between the electrodes. In contrast to a
polymer
electrolyte fuel cell, the discharge system 101 reduces or completely
eliminates platinum
from the positive electrode 205a, uses a thicker hydrophilic porous electrode
(HPE)
instead of a thin catalytic layer and a hydrophobic gas diffusion layer on the
positive
electrode 205a which assures a higher power per cross-sectional area, reduces
the size or
completely eliminates the humidification system due to back diffusion of water
from the
aqueous multi-electron oxidant (AMO) solution to the hydrogen electrode within
each
electrolytic cell 200, and allows for energy recuperation by oxidation on the
positive
electrode 205a of bromide in the discharge fluid into bromine with
simultaneous
hydrogen evolution on the negative electrode 205b.
106
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0222] The regeneration system 106 of the system 100 disclosed herein is
configured to
regenerate the aqueous multi-electron oxidant (AMO) and the reducer from the
discharge
fluid produced by the discharge unit 104. The regeneration system 106
comprises, for
example, a splitting-disproportionation (SD) reactor 1502, a concentrating
reactor 112,
multiple separation reactors 1010, and storage tanks such as a regenerated
oxidant fluid
tank 110, a regenerated reducer fluid tank 111, a discharge fluid tank 1503,
and a water
tank 1504. An electrolysis-disproportionation reactor 107 is an example of the
splitting-
disproportionation reactor 1502. In an embodiment, the SD reactor 1502 is
configured as
the electrolysis-disproportionation (ED) reactor 107, exemplarily illustrated
in FIG. 1,
comprising sub-reactors, for example, an electrolysis unit or an electrolyzer
107a and a
disproportionation unit 107b, exemplarily illustrated in FIG. 1. In an
embodiment, the
electrolyzer 107a and the disproportionation unit 107b are physically combined
in the
same hardware.
[0223] In an embodiment, the splitting-disproportionation (SD) reactor 1502
uses
electrolytic splitting and is configured for flow modes of operation. The SD
reactor 1502
comprises a stack of SD flow cells configured similar to a conventional
polymer
electrolyte fuel cell (PEFC) bipolar stack so that one side of every inner
bipolar plate
serves the current collector of a negative electrode and the other side serves
as the current
collector of a positive electrode. Several SD flow cells can be stacked and
operated in a
cascade flow mode. Each SD flow cell has a structure similar to a polymer
electrolyte
membrane fuel cell (PEMFC) with a 5-layer membrane-electrode assembly, where
the
gas diffusion layer on the positive side is replaced with a hydrophilic porous
layer.
Furthermore, the stack and the negative electrodes of the 5-layer membrane-
electrode
assembly are configured for either the aqueous multi-electron oxidant (AMO)-on-
negative electrode mode of operation also referred to as the "AMO-on-negative
mode of
operation", or the no-AMO-on-negative electrode mode of operation also
referred to as
the "no-AMO-on-negative mode of operation". The individual SD flow cells in
the
bipolar stack are connected electrically in series so that the bipolar stack
voltage is the
sum of the individual SD flow cell voltages. The individual SD flow cells in
the bipolar
stack are connected flow-wise in parallel or series, with a parallel
connection affording
107
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
more uniform voltages in different SD flow cells in the bipolar stack. In an
embodiment,
the SD reactor 1502 is configured for the AMO-on-negative mode of operation
using a
multilayer structure on a negative electrode side of the SD reactor 1502. The
multilayer
structure on the negative electrode side minimizes reduction of a regenerated
aqueous
multi-electron oxidant in a regenerated oxidant fluid on the negative
electrode side while
facilitating hydrogen evolution and increase in pH of the regenerated oxidant
fluid. In
another embodiment, the SD reactor 1502 is configured for the no-AMO-on-
negative
mode of operation by transferring a base produced on one or more negative
electrodes of
the SD reactor 1502 to a regenerated oxidant fluid produced at one or more
positive
electrodes of the SD reactor 1502 and comprising one or more forms of the
aqueous
multi-electron oxidant and the intermediate oxidant.
[0224] The splitting-disproportionation (SD) reactor 1502 or reactors can be
configured
and operated in a batch mode, in a cyclic flow mode or in a cascade flow mode.
An SD
reactor 1502 configured for the cyclic flow mode has a lower upfront cost but
requires a
longer regeneration time. Such an SD reactor 1502 may be utilized for at-home-
garage
regeneration. The SD reactor 1502 configured for the cascade flow mode has a
higher
upfront cost but is capable of a faster regeneration. This SD reactor 1502 may
be utilized
at multi-user charging stations. A combination of cyclic and cascade flow
modes in the
same regeneration unit allows for an optimization of the capital cost and
throughput and
it is recommended for most applications.
[0225] In an embodiment, the concentrating reactor 112 is placed between the
splitting-
disproportionation (SD) reactor 1502 and the orthogonal ion migration across
laminar
flow (OIMALF) reactor 1501 whereby the concentrating reactor 112 produces a
concentrated solution of neutral oxidant fluid comprising a salt from of the
aqueous
multi-electron oxidant (AMO). The concentrating reactor 112 increases the
concentrations of one or more forms of the AMO as well as the total AMO
concentration
in the oxidant fluid produced by the splitting-disproportionation (SD) reactor
1502 before
the AMO is stored in the regenerated oxidant fluid tank 110. The concentrating
reactor
112 removes water or other solvents from a dilute fluid that enters the
concentrating
108
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
reactor 112 and releases a concentrated fluid and water or another solvent.
The
concentrating reactor 112 performs concentration, for example, by evaporation,
pervaporation, reverse osmosis, and other known methods. The storage tanks,
for
example, the regenerated oxidant fluid tank 110, the regenerated reducer fluid
tank 111,
the water tank 1504, and a buffer tank (not shown) are used to store the
regenerated
neutral oxidant fluid, the regenerated reducer, water, and the buffer
respectively. The
separation reactors 1010 are gas-liquid separators or separation reactors 1010
and are
used to separate gases from the liquids during the regeneration process.
[0226] FIG. 16 exemplarily illustrates a process flow diagram showing mass and
electricity flows in an energy cycle between the discharge unit 104, the
acidification
reactor 1501a, and the neutralization reactor 1501b of the discharge system
101. The on
board system is enclosed in a dotted frame with the reducer fluid, oxidant
fluid, and
discharge fluid shared by both on-board and off-board systems. In an
embodiment, the
discharge system 101 comprises a single reactor such as an orthogonal ion
migration
across laminar flow (OIMALF) reactor 1501 which performs both acidification
1602 and
neutralization 1606. In FIG. 16, HX0r, refers to the aqueous multi-electron
oxidant
(AMO) in the acid form, MX0r, refers to the AMO in the salt form, HA refers to
the
buffer in the acid form, and MA refers to the buffer in the base form. The
flow of
materials is represented using solid arrows and the flow of electric power is
represented
using dotted arrows. The aqueous multi-electron oxidant (AMO) may be present
at
various stages in the discharge and regeneration energy cycle in one or
several forms, for
example, acid form, salt forms such Li form, etc., differing in composition,
concentration, etc. If not specified, the term "aqueous-multi-electron
oxidant" or "AMO"
refers collectively to all such forms and any combination thereof.
[0227] Certain salts of both the aqueous multi-electron oxidant (AMO) and the
discharge product of the AMO have high aqueous solubilities as well as high
rates of
homogeneous disproportionation and comproportionation. Such a combination can
be
obtained, for example, when the AMO salt is lithium bromate with a solubility
of, for
example, over 10 molal at 20 C and over 20 molal at 80 C and the discharge
salt is
109
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
lithium bromide with the solubility of, for example, over 15 molal at 20 C and
over 25
molal at 80 C. Although a salt form of the AMO can be used directly in a
discharge unit
104 to produce electric power, the slow kinetics of the direct
electroreduction of the salt
form of the AMO requires the use of expensive platinoid catalysts and, even
then, occurs
with poor energy efficiency. The electroreduction of the salt form of the AMO
can
process more efficiently, even on a bare carbon electrode, when it is mediated
by a
soluble mediator. In an embodiment, the electroreduction product, for example,
bromide,
is utilized as the reduced form of the mediator. In this case, the mediation
reaction is a
comproportionation reaction. The reduction of the AMO in general and the
comproportionation reaction in particular requires proton donors to proceed at
a useful
rate. Proton donors can be introduced into a stable stock solution of the salt
form of the
AMO in a process referred herein as acidification. Also, in the method and
systems or
energy cycle disclosed herein, a pH manipulation and/or change is used to
facilitate the
conversion between a stable but low-power salt form of the AMO and a high-
power but
poorly stable acid form of the AMO.
[0228] The neutral oxidant fluid 1601 has a high energy density due to the
high
solubility of the aqueous multi-electron oxidant (AMO) such as LiBrO3 and due
to the
multi-electron oxidant property of the AMO: 6 electrons are transferred during
the
reduction of one bromate ion into one bromide ion. Thus, the discharge unit
104 can
store a large amount of energy or charge per unit of weight or volume and this
storage is
safe due to a low reactivity of the AMO at neutral and alkaline pH. However,
to achieve
a high power, for example during an on-board discharge process, the AMO needs
to be
present in an acidic form that is at a low pH. This can be achieved by
converting the
neutral oxidant fluid 1601 into an acidic oxidant fluid 1603 in the
acidification reactor
1501a. The process of acidification 1602 can be performed via ion exchange on
solids,
ion exchange in solution or by any other known acidification method, and by
any
combination thereof. In an embodiment, the acidification is performed via the
orthogonal
ion migration across laminar flow (OIMALF) process. The use of the OIMALF
process
confers an additional benefit of being free of input and output chemicals, as
well as the
benefits of high power density and of high energy efficiency. In another
embodiment,
110
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
the acidification is performed by adding an extra acid HA such as phosphoric
acid
H3PO4, carried over from the regeneration step, sulfuric acid, triflic acid,
other strong
acid, etc., to the neutral oxidant fluid. Also, the on-board storage of a salt
form of the
AMO is used over an acid form of the AMO for safety reasons. The use of salts
forms
rather than of acid forms puts forward additional requirements for high
solubilities of the
AMO and its discharge product(s) in their salt forms. The complete
acidification with
1:1 stoichiometric ratio of acidic protons to the AMO, for example, bromate,
is not
necessary for the ignition regime of the AMO electroreduction to occur, and a
partial
acidification suffices. This finding confers the benefits of improved safety,
energy
efficiency, and reduced size of the discharge system 101, which facilitate
application of
the discharge system 101 in automotive applications.
[0229] In an embodiment, the stability of the acidic oxidant fluid is
maintained by
performing an ignition regime in the discharge system 101 at low acid
concentrations of
the acidic oxidant fluid. The concentration of one or more forms of the
aqueous multi-
electron oxidant in the neutral oxidant fluid or the acidic oxidant fluid
supplied to the
discharge unit 104 is, for example, above 1M, 2M, 5M, or 10M. The
concentration of
acidic protons in the acidic oxidant fluid supplied to the discharge unit 104
is, for
example, below 0.1M, 0.5M, 1M, 2M, 5M, or 10M. The concentration of acidic
protons
in the acidic oxidant fluid stored in the discharge system 101 is, for
example, below
0.1M, 0.5M, 1M, 2M, or 5M. In an embodiment, the acidification process is
performed
off-board in the discharge system 101, yielding a weakly acidic solution that
is capable
of ignition-like electro-reduction yet is sufficiently stable on the week time
scale for
automotive applications. In the discharge system 101 disclosed herein, the
concentration
of acid that is required to cause ignition with a practically suitable power
in a highly
concentrated aqueous multi-electron oxidant [AMO] > 10M is very low about 5mM.
The
AMO does not decompose for over a week. This allows the acidification process
such as
orthogonal ion migration across laminar flow (OIMALF) process to be performed
off-
board and also allows storage of the acidic oxidant fluid on-board in the
oxidant fluid
tank 102 of the discharge system 101 for almost a week. The stored AMO is a
stable
solution capable of ignition. The method and the discharge system 101
disclosed herein
111
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
allow the storage of a more stable form of the AMO on-board which is achieved
with an
acceptable sacrifice in the system energy density.
[0230] The discharge system 101 uses the acidification reactor 1501a to
convert the
neutral oxidant fluid 1601 into acidic oxidant fluid 1603 which has sufficient
chemical
reactivity to cause an ignition regime of electroreduction on the positive
electrodes 205a
of the discharge unit 104. During the acidification process 1602, a stable
aqueous multi-
electron oxidant (AMO) stock stored on board, such as neutral oxidant fluid
1601
comprising LiBrO3 is converted into a chemically reactive form of the AMO, for
example, acidic oxidant fluid 1603 comprising HBrO3. This can be accomplished
via a
solution-phase cation exchange process in the orthogonal ion migration across
laminar
flow (OIMALF) reactor 1501 with a simultaneous conversion of the outgoing
acidic
discharge fluid 1605 into a neutral form 1607, for example, HBr into LiBr.
LiBrO3 is
converted into HBrO3 using the OIMALF process or another ion exchange process
or
direct addition of an extra acid. In an embodiment, the OIMALF process
generates and
consumes H2 within the OIMALF reactor 1501. The OIMALF process of converting,
including partially converting, MX0r, into HX0õ, for example, LiBrO3 into
HBrO3
avoids cumbersome chemical separation and ion exchange regeneration steps. The
choice
of the acid form of the AMO can be expanded beyond HBrO3 to other AMOs
comprising,
for example, HC104, HC103, HC102, HC10, HBr04, HBr02, HBrO, etc. Phosphoric
acid
will be present in the oxidant fluid if a phosphate buffer is used during
regeneration. The
net reaction of the ion exchange or the OIMALF process is: LiBrO3 +HA = HBrO3
+
LiA, where HA is a source of protons comprising, for example, one or more of
the
following: water, phosphoric acid, dihydrogen phosphate, one or more of Good's
buffers,
one or more derivatives of sulfonic acid, sulfuric acid, triflic acid,
perchloric acid, etc.
For on-board operation, the OIMALF reactor 1501 is operably connected to an on-
board
power source such as discharge unit 104 or a battery (not shown) which
provides electric
power for the OIMALF process.
[0231] During the discharge process, the discharge unit 104 is supplied with
the reducer
1604, for example, H2, and the acidic oxidant fluid 1603 comprising the
aqueous multi-
112
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
electron oxidant (AMO) in the acid form, HX0õ, for example, HBr03. The reducer
1604
donates electrons to the negative electrode 205b, and splits into ions. The
reaction at the
negative electrode 205b is, for example, 3H2 ¨ 6e- = 6H+. The on-board
electric circuit
203 conducts and transfers electrons from the negative electrode 205b to the
positive
electrode 205a. The reaction at the positive electrode 205a, for example, 3Br2
+ 6 C = 6
Br- , or when combined with the comproportionation reaction the catholyte, for
example,
Br03- + 6C + 6H+ = Br- + 3H20. The aqueous multi-electron oxidant accepts the
electrons at the positive electrode 205a for producing the electric current in
the on-board
electric circuit 203. The discharge unit 104 releases the acidic discharge
fluid HX 1605
comprising, for example, HBr and the buffer HA in the acidic form and
generates electric
current in the on-board electric circuit 203. The cation-selective electrolyte
layer 205c
provides for a movement of cations, such as hydronium ions, between the
negative
electrode 205b and the positive electrode 205a.
[0232] The generation of electric power using the aqueous multi-electron
oxidant
(AMO), for example, bromate during the discharge is accompanied by the
following
chemical transformations.
Negative Electrode: 3H2 + 6e = 6H+ (33)
Positive Electrode: Br03- + 6H+ - 6e = Br- + 3H20 (34)
[0233] The latter electrode half-reaction may proceed not by a direct
electroreduction of
a bromate species on the electrode but via the formation of a Br2 intermediate
in a
homogeneous comproportionation reaction between bromate and bromide as shown
below:
Comproportionation: Br03- + 5Br- + 6H+ = 3Br2 + 3H20 (35)
Reduction: 3 Br2 + 6 C = 6 Br- (36)
[0234] An extra acid, for example a strong acid, HA, such as H2SO4, LiHSO4,
HC1,
HNO3, HC104, F3CSO3H, F3CCOOH, etc., can be added in a small concentrations
113
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
compared to the total aqueous multi-electron oxidant (AMO) concentration to
accelerate
the rate of reaction (20) on discharge. The use of such an extra acid may be
more
advantageous than an increase in the phosphoric acid (H3PO4) concentration,
which is a
weak acid and which is limited by the properties of Li2HPO4 decomposing in
water into a
very soluble LiH2PO4 and a poorly soluble Li3PO4. In an embodiment, bromic
acid itself
is used as the extra acid. The use of a higher acid concentration, afforded by
adding the
extra acid, facilitates the rate of the comproportionation because for a
general acid-
catalyzed reaction such as Br03- + 5Br- + 6H+ = 3Br2 + 3H20, the same rate can
be
obtained with a lower concentration of a strong extra acid such as HC104 than
with a
weaker acid such as H3PO4. A smaller concentration of the extra acid, compared
to the
concentration of phosphoric acid that shows comparable rate constant for the
comproportionation, requires a smaller charge in the orthogonal ion migration
across
laminar flow (OIMALF) process, thus reducing the energy expenses and the size
of the
OIMALF reactor 1501. For purposes of illustration, the detailed description is
described
with reference to an OIMALF process for conversion of the salt form of the AMO
into
the acid form of the AMO; however the scope of the method and system disclosed
herein
is not limited to the OIMALF process but can be extended to include other
processes
such as a ion exchange on resins, a direct addition of the extra acid, and can
be justified
in other applications.
[0235] In an embodiment, aqueous multi-electron oxidant (AMO) in a stable
form, for
example, LiBrO3 is converted, at least partially, into an active form, for
example, HBr03,
using, for example, ion exchange on resins or ion exchange in solution such as
an
orthogonal ion migration across laminar flow (OIMALF) within the discharge
system
101. The resulting acidic oxidant fluid 1603 comprising bromate as the aqueous
multi-
electron oxidant (AMO) is used in the discharge unit 104. This is followed by
discharge
of hydrogen on negative electrodes 205b of discharge cells and bromate on the
positive
electrodes 205a of discharge cells of the electrolytic cell stack 105, with a
release of
bromide and water on the positive electrodes 205a of discharge cells, provided
that the
discharge cells are equipped with cation-conductive membranes such as Nafion
or its
analogues. In an embodiment, the discharge on the positive electrodes 205a is
facilitated
114
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
by a homogeneous comproportionation of bromide product with bromate oxidant,
or in
general of a halide with a halogen oxoanion. The discharge process based on
the
sequence of orthogonal ion migration across laminar flow (OIMALF),
comproportionation, and electroreduction process has a reasonably high
projected energy
efficiency of about 70%. For on-board operation, the OIMALF reactor 1501 is
operably
connected to an on-board power source such as the discharge unit 104 or a
battery (not
shown) which provides electric power for the OIMALF process.
[0236] The regeneration process is preceded by raising the pH of one or more
forms of
the discharge fluid 1605 with a base, for example, Na2HPO4, LiOH or Li-3-(N-
morpholino) propanesulfonic acid (MOPS) in the neutralization reactor 1501b of
the
discharge system 101 exemplarily illustrated in FIG. 15. The acidic discharge
fluid
comprises one or more of water, a halide, a hydroxonium cation, an extra acid,
and a
counter cation. Neutralization 1606 is a chemical reaction in which a base and
an acid
react to form a salt. The neutralization reactor 1501b neutralizes 1606 the
acidic
discharge fluid 1605 into neutral discharge fluid 1607 which is safe to
handle, for
example, to transfer to an off-board regeneration system 106. The base
generated as a
result of the orthogonal ion migration across laminar flow (OIMALF) process is
used
during the process of neutralization 1606 of the acidic discharge fluid 1605,
for example,
comprising HBr. The neutralization 1606 can be performed using an OIMALF
reactor
1501. In an embodiment, some process steps of the energy cycle, for example,
neutralization 1606 and acidification 1602 can be combined in a single reactor
such as
1501. In another embodiment, the concentration can precede conversion to acid.
[0237] The aqueous multi-electron oxidant (AMO) and the reducer are
regenerated in
stoichiometric amounts from the discharge fluid in the regeneration system
106. The
splitting-disproportionation (SD) 1608 process disclosed herein for the
regeneration of
the oxidant fluid comprising the AMO, for example, bromate from the neutral
discharge
fluid 1607 comprising, for example, bromide starts with an optional pH
optimization of
the discharge fluid for the disproportionation step. The pH optimization can
be performed
within the discharge system 101 or within the regeneration system 106 or in
both by
115
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
adding acid or base to the discharge fluid in question via electrolysis, ion
exchange on
solids, ion exchange in solution such as orthogonal ion migration across
laminar flow
(OIMALF), etc. and any combination thereof. A buffer present in one or more
forms of
the oxidant fluid and/or the discharge fluid is used to facilitate the pH
optimization.
During the regeneration of the AMO and the reducer, the splitting-
disproportionation
(SD) reactor 1502 of the regeneration system 106 splits 1608 the neutral
discharge fluid
1607 comprising halide into an intermediate oxidant such as a halogen
accompanied by a
release of the reducer 1604 such as hydrogen and a base form of the buffer. In
the case of
splitting being electrolysis, the intermediate oxidant is produced at the
positive electrode
of the SD reactor 1502, and the reducer and the base are produced at a
negative electrode
of the SD reactor 1502. In an embodiment, the SD reactor 1502 is configured as
an
electrolysis-disproportionation reactor 107 and is powered by the off-board
electric
circuit 409. The neutral discharge fluid 1607 comprising, for example, LiBr
and H20
undergoes electrolysis, photolysis, photoelectrolysis, radiolysis, or
thermolysis to the
intermediate oxidant such as Br2 at the positive electrode and, for example,
H2 and LiOH
or H2 and Li-3-(N-morpholino) propanesulfonic acid (MOPS) at the negative
electrode.
The process of splitting 1608 is accompanied by the release of the reducer
1604, for
example, hydrogen in stoichiometric amounts which is used as the reducer 1604
in the
discharge unit 104. H2 is produced on the negative electrode, configured for
use with a
liquid electrolyte, leaving behind an aqueous base solution, for example LiOH:
6H20 + 6e- + 6Li+ (aq.) = 6LiOH + 3H2 (37)
[0238] The liquid containing the base, such as LiOH, and the hydrogen gas are
separated in separation reactors 1010. The regenerated hydrogen is collected
in fuel
storage tank or the regenerated reducer fluid tank 111, while the base-
containing liquid is
pumped into the positive electrode compartment. On the positive electrode,
halogen X2 is
produced:
6X- - 6e- = 3 X2 (aq.) (38)
116
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0239] In the presence of the base, provided that the pH of the liquid in the
positive
electrode compartment is maintained at a proper level, for example, between 4
and 9, or
between 6 and 8, using an appropriate buffer, such as monohydrogen phosphate,
a
substituted phosphonate, amine, one or more of Good's buffers, or a
combination thereof,
the halogen disproportionates producing the desired aqueous multi-electron
oxidant
(AMO) such as a halate. For example, with A- as the base:
3 X2 (aq.) + 6LiA + 3H20 = LiX03 +5LiX + 6HA (39)
[0240] In an embodiment, the base form of the buffer is obtained by a reaction
of the
neutral form of the buffer generated in the disproportionation reaction with
the base
produced at the negative electrode: 6LiOH + 6HA = 6LiA. The water necessary to
prevent drying and LiOH precipitation on the negative electrode in the no-
aqueous multi-
electron oxidant (AMO)-on-negative mode of operation is supplied from the
positive
electrode compartment via electro osmotic drag by Li + cations, by pressure-
driven flow
through the membrane, etc., or from a separate water tank 1504. This excess
water can be
removed from the regenerated fluid using the concentrating reactor 112 using
reverse
osmosis, evaporation, pervaporation, etc. and stored in water tank 1504.
[0241] In the case of Br2, if the pH of the anolyte is maintained between 6
and 8, or
between 4 and 9, a disproportionation 1608 to bromate occurs, for example,
with a LiOH
base: 3Br2 + 6LiOH = 5LiBr + LiBrO3 + 3H20. Splitting 1608 of the LiBr + H20
solution and the disproportionation 1608 reactions proceed in a cyclic fashion
or in a
cascade, in batches or continuous modes, till most of the LiBr is converted
into LiBr03.
The disproportionation of the intermediate oxidant such as halogen into
aqueous multi-
electron oxidant (AMO) can be implemented in AMO-on-negative mode of operation
and
in no-AMO-on-negative mode of operation which require different hardware
designs.
The base required for the disproportion of halogen produced on the positive
electrode
during regeneration is conveniently produced as a by-product of hydrogen
evolution on
the negative electrode. There are two possible methods for introducing the
base into the
regenerated solution, that is, the AMO-on-negative electrode mode of operation
and the
117
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
no-AMO-on-negative electrode mode of operation as well as multiple
combinations of the
two. These are illustrated in FIGS. 17A-17B and FIG. 18 using X03- or bromate
as AMO,
M+ or Li + as the counter-cation, and A- as the base form of the buffer. The
AMO-on-
negative mode of operation is exemplarily illustrated in FIGS. 17A-17B for a
cyclic flow
mode. The multilayer structure of the negative electrode configured for this
mode and the
operation of the SD reactor 1502 is disclosed in the detailed description of
FIGS. 17A-
17B. The no-AMO-on-negative mode of operation is exemplarily illustrated in
FIG. 18
for a batch mode. Various modes of regeneration namely batch, flow-cycle, flow-
cascade
can be combined with either the AMO-on-negative and no-AMO-on-negative modes
of
operation.
[0242] Li + can be used as a counter-cation to achieve high solubilities of
the salts
involved, such as bromide and bromate. A pH buffer comprising, for example, a
dissolved base, LiA, such as a lithium alkylphosphonate or arylphosphonate, an
amine or
amines such as one or more of Good's buffers is used to prevent spatial and
temporal
deviations of pH from the range between 4 and 9, for example, between 6 and 8,
within
the disproportionation reactor. The resulting product, for example, LiBrO3, in
the off-
board neutral oxidant fluid 1601, if needed or desired, can be concentrated
off-board in
the neutral oxidant fluid 1601 using the concentrating reactor 112 before the
neutral
oxidant fluid 1601 is placed on-board. The neutral oxidant fluid is stable,
non-corrosive
and safe to handle, thus allowing for it transfer between off-board and on-
board tanks and
on-board storage without undue risk and without extraordinary precautions.
Furthermore,
the on-board storage of the neutral oxidant fluid 1601 mitigates the risk of
spillage of the
neutral oxidant fluid 1601 in the case of an accident. The net balanced
chemical equation
of regeneration for an exemplary combination of the aqueous multi-electron
oxidant
(AMO) and the buffer is:
LiBr + 3H20 = (electricity in two places, LiA recycled) = 3H2 + LiBrO3 (40)
[0243] The splitting-disproportionation (SD) process converts, for example,
concentrated LiBr in the neutral discharge fluid 1607 into a concentrated
LiBrO3 in the
118
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
neutral oxidant fluid 1601. Nevertheless, upon numerous discharge-regeneration
cycles
the solutions get diluted due to accumulation of water. To keep the energy
density of the
neutral oxidant fluid 1601 high, a water removal process is performed
occasionally, for
example, as part of the off-board regeneration before placing the neutral
oxidant fluid
1601 on board. The commercial process of concentrating salts uses evaporation,
with an
estimated energy loss of approximately 10-15% if heat exchangers are used. The
reverse
osmosis process requires overcoming of the osmotic pressure, for example, of
about 536
bars, which is possible in a cascade flow mode with commercial supported ion
exchange
membranes. The minimal energy expense at an infinitely slow filtration rate
is, for
example, about 7% of the energy content of the product 10M LiBrO3 and 3H2. Due
to a
finite flow rate, the regeneration process disclosed herein uses optimization
of the unit
size, power, and operating pressure in terms of the energy efficiency and
capital cost.
[0244] The splitting-disproportionation 1608 cycle continues in the same flow
or batch
SD reactor 1502 till the [bromide]/[bromatel concentration ratio decreases to
the desired
value, for example, below 0.05. The resulting neutral oxidant fluid 1601, for
example,
approximately 5-10 M LiBrO3, can be further concentrated, for example, to
about 10-20
M, using reverse osmosis, evaporation or other methods known in the art. The
use of
evaporation for concentrating has an additional advantage of producing a hot
solution of
LiBrO3 which has almost twice the solubility of a cold solution of LiBrO3. The
concentrated solution, for example, approximately 10M LiBrO3 solution, the
concentration of which is limited by the solubility of LiBrO3 at the operating
temperature,
for example, about 20 C, then goes back into the orthogonal ion migration
across laminar
flow (OIMALF) reactor 1501, where Li + in LiBrO3 is exchanged for fr from the
incoming HBr, thus producing for example, a solution comprising 0.5M HBrO3 and
9.5M
LiBrO3. Further exchange for Li + for fr is unnecessary since the ignition
regime of
electroreduction is already observed at such composition and may be
detrimental due to
reduced stability of bromate, which decomposes with oxygen evolution in highly
acidic
solutions.
[0245] The hot solution of LiBrO3 can be pumped to an on-board oxidant storage
tank
119
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
102 where it may be allowed to cool with precipitation of solid LiBrO3, thus
increasing
the theoretical energy density of the on-board discharge system 101. The heat
released
during the cooling and precipitation of the hot concentrated solution of
LiBrO3 can be
used to preheat the neutral discharge fluid 1607 or the neutral oxidant fluid
1601 prior to
their use. The neutral oxidant fluid 1601 undergoes acidification 1602 in the
acidification
reactor 1501a. The precipitated LiBrO3 can be re-dissolved in water or in an
acidic
discharge fluid and delivered as the acidic oxidant fluid 1603 to the
discharge unit 104
for producing electric energy. The hardware components of the hydrogen-bromate
energy
cycle disclosed herein comprise analytical chemical detectors (not shown) used
for
process monitoring and control.
[0246] In an embodiment, in the first step in the scheme of regeneration, the
halogen and
a stoichiometric amount of hydrogen are regenerated by sunlight energy
harvesting, that is,
via photolysis or photoelectrolysis of the spent hydrogen halide. In this
embodiment, the
splitting-disproportionation reactor 1502 is configured as a photoelectrolysis-
disproportionation reactor (not shown). A decomposition into H2 and X2 is
induced in the
discharge fluid in the photoelectrolysis-disproportionation reactor by
irradiating the
discharge fluid with sunlight in the presence of a photocatalyst such as a
semiconductor.
The regeneration system 106 disclosed herein comprising the photoelectrolysis-
disproportionation reactor, regenerates the aqueous multi-electron oxidant
(AMO) and the
reducer during the induced reverse electrochemical process by consuming solar
energy
and the discharge products.
[0247] Since the regeneration system 106 replaces 02 with the aqueous multi-
electron
oxidant (AMO), the sunlight energy harvesting method acquires a different
perspective. A
halogen, for example, bromine, the first intermediate in the regeneration
process is
produced from the spent hydrogen halide photoelectrochemically with a higher
efficiency than water splitting achieves since there is no oxygen evolution
over-voltage,
and at a lower cost than photoelectrochemical water splitting as the Pt
catalyst is not
required for oxygen evolution. The photolysis process and the
photoelectrolysis process
involve irradiation of the hydrogen halide solution with light or without the
presence of a
120
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
light adsorbing facilitator, a catalyst, or a combination thereof. The light
adsorbing
facilitator is, for example, a semiconductor, a dye, a transition metal
complex or a
combination thereof. A semiconductor is, for example, TiO2 in an anatase or
rutile form
and preferably in the form of particles suspended in the solution to be
oxidized. The
particle surface is also coated by one or several catalysts to facilitate
evolution of hydrogen
and/or halogen.
[0248] The projected performance of the H2-aqueous multi-electron oxidant
(AMO)
discharge system 101 versus a 2012 Toyota RAV4EV lithium ion battery pack and
the
2013 ARPA-E targets are shown in Table 2 below.
Table 2:
Toyota
Parameter Units Target H2-AMO System
RAV4 EV
Manufacturing cost $/kWh <100-125 500 120
Effective specific energy, system Wh/kg >150 110 426
level
Effective energy density, system Wh/L >230 <200 200-400
level
Effective specific power on W/kg >300 303 690
discharge 80% DOD/30 s
Cycle life at 80% depth of Cycles > 1000 > 1000 1000
discharge (DOD)
Years > 10 <8 years >10 (6
Calendar life
operational)
Operating temperature C >-30 >-30 >-40
[0249] In Table 2, the projected low temperature limit refers to a cold-start
up and it is
limited by the freezing or precipitation point of the neutral oxidant fluid.
The cost figures
are calculated based on the design of modern proton exchange membrane fuel
cells
(PEMFCs) minus the cost of Pt catalyst on the cathode. The cost figures do not
account
121
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
for the economy-of-scale discount. The parameters refer to the system 100 with
5% w/w
H2 storage and 50% w/w/ aqueous multi-electron oxidant (AMO) at 78% discharge
efficiency corresponding to 0.5 W/cm2 power. The power is calculated for a
smooth
flow-by carbon cathode on the basis of kinetic parameters reported in the
literature and
assuming membrane resistance of 0.1 ohm/cm2. A five times higher power can be
reasonably expected from a flow-through porous electrode. The number shown is
the
operational not calendar life if the discharge flow battery is limited by the
degradation of
Pt on the hydrogen anode accounting for the oxidant cross-over at open circuit
potential
(OCP) on the basis of relevant data for PEFCs. Purging both electrodes with on-
board
water on shut-downs can increase the projected durability. System energy
density
increases for H2 storage methods in the order of: 350 bar gas < cryo-liquid
<5% metal
hydride. Although the gravimetric specific energy of hydrogen is high, the
volumetric
energy density of hydrogen is low even at the highest practically achievable
pressures
and hydrogen storage. Both high pressure carbon composite cylinder and metal
hydrides
tank may satisfy the mass and the volume requirements.
[0250] FIGS. 17A-17B exemplarily illustrate mass flows in a single cell 1700
of a
splitting-disproportionation reactor 1502, more specifically, an electrolysis-
disproportionation (ED) reactor 107 configured for regeneration in an aqueous
multi-
electron oxidant (AMO)-on-negative electrode mode of operation. FIG. 17A
exemplarily
illustrates an operation of a single regeneration flow cell 1700. LiA is the
buffer in the
base form, for example, Li-3-(N-morpholino) propanesulfonic acid (MOPS). In an
embodiment, the ED reactor 107 is configured for the AMO-on-negative mode of
operation using a modified membrane-electrode assembly (MEA) 1701. The
negative
electrode layer 1702 of the MEA 1701 of the ED cell 1700 when configured for
the
AMO-on-negative mode of operation has a graded and/or multilayer structure
and/or
composition in order to avoid and/or minimize on the negative electrode 1702,
the
reduction of the AMO regenerated on the positive electrode 1703 while allowing
for H2
evolution and for maintaining the pH in the optimal basic range. The side or
the negative
electrode layer 1702a closer to a cation-conductive polymer electrolyte
membrane 1704
which is the inner layer, is a catalytic layer comprising Pt/C embedded into a
cation-
122
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
conducting polymer electrolyte (CCPE) such as Nafion . A Pt catalyst is
supplied to
facilitate the reduction of 1-1+ into 1/2 H2. Cations, such as Li + and 1-1+
and neutral species,
such as H2 and H20 can permeate through the CCPE. Anions, such as halate and
halide,
permeate the CCPE to a much smaller extent. The outer layer of the MEA
catalytic layer
comprises CCPE and carbon but not Pt thus allowing for the transport of
electrons and
cations but preventing the reduction of the AMO species on the negative
electrode 1702
during the regeneration.
[0251] In the cyclic flow mode under the aqueous multi-electron oxidant (AMO)-
on-
negative mode of operation for a single cell electrolysis-disproportionation
(ED) reactor
107, the solution containing the AMO is cycled between the negative electrode
1702 of
the ED reactor 107 where neutralization and/or alkalization occurs and the
positive
electrode 1703 where electrooxidation and disproportionation occur. In the
cascade flow
mode under the AMO-on-negative mode of operation, the solution containing the
AMO
moves between the negative electrode 1702 of one ED flow cell 1700 where
neutralization occurs to the positive electrode 1703 of an adjacent ED flow
cell 1700
where electrooxidation and disproportionation occur. In the cascade flow mode,
the
regenerated AMO solution flows through a cascade of functionally identical ED
reactors
107 such as positive electrode compartments of individual cells 1700.
[0252] An operation of the aqueous multi-electron oxidant (AMO)-on-negative
mode of
operation is exemplarily illustrated in FIG. 17A with an electrolysis-
disproportionation
(ED) reactor 107 represented by a single cell 1700 operating in the cyclic
flow mode. A
neutral discharge fluid, for example, from a car's discharge tank, or a
neutral partially
regenerated oxidant fluid, for example, from a previous regeneration cycle,
passes
through a negative compartment and a negative electrode 1702 of the
regeneration flow
cell or the ED cell 1700 where hydrogen is produced and the pH of the
discharge fluid is
raised. The AMO-on-negative mode of operation is facilitated via the use of
the outer
negative electrode layer 1702b to prevent the access of AMO anions to the
surface of
electrocatalysts in the negative electrode layer 1702a. If the discharge fluid
is flushed in
the second and subsequent cycles through the negative electrode 1702 to lower
the pH, an
123
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
undesirable electroreduction of bromate on an electrocatalyst may occur:
LiBrO3 + 6e- + 6H+ = LiBr +3H20 (41)
[0253] In this aqueous multi-electron oxidant (AMO)-on-negative mode of
operation,
the reduction of the AMO anion species on the electrocatalysts such as Pt in
the negative
electrode 1702 can be minimized or prevented by blocking the surface of Pt by
a cation-
selective coating, such as Nafion polymer, by using, for example, a two layer
electrode
1702, with only the inner layer 1702a containing a catalyst, for example, Pt
capable of
hydrogen evolution reaction; and the outer layer 1702b exposed to the flowing
electrolyte
comprising, for example, a Pt-free porous carbon containing an electron-
conducting
component such as carbon particles and fibers, for providing electronic
current between
the inner layer 1702a and a current collector 1705, a cation-selective
component such as
Nafion polymer, which allows for cation transport between the flowing liquid
1009 in the
negative electrode compartment and flowing liquid in the positive electrode
compartment. The structure of the inner layer 1702b is similar to the modern
generation
of the catalytic layers of the membrane-electrode assemblies of polymer
electrolyte fuel
cells. The inner layer 1702b allows for a transport of electrons, protons, and
other cations
to the Pt electrocatalyst but of anions, thereby selectively allowing hydrogen
production
and suppressing AMO reduction.
[0254] A more detailed illustration of the chemistry aspects of the aqueous
multi-
electron oxidant (AMO)-on-negative mode of operation at the membrane-electrode
assembly 1701 level is exemplarily illustrated in FIG. 17B using the first
cycle with the
charge of one electron per bromide. FIG. 17B shows an operation of an ED
reactor 107 in
the flow modes and the AMO-on-negative mode of operation showing
neutralization
performed at the negative electrode 1702 with a multilayer structure. Water
flux through
membrane is not shown. 1 electron per cycle is shown as a means of
illustration not of
limitation. HA is, for example, Li-3-(N-morpholino) propanesulfonic acid
(MOPS). The
two layer negative electrode 1702 is exemplarily illustrated in FIG. 17B with
the inner
layer 1702a containing Pt on carbon fibers or particles embedded into a cation-
124
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
conductive membrane and the outer layer 1702b made of carbon fibers or
particles
without Pt and embedded into a cation-conductive membrane. The inner layer
1702a with
Pt allows for hydrogen evolution reaction to occur while preventing the
electroreduction
of bromate on Pt.
[0255] FIG. 18 exemplarily illustrates mass flows in a single cell 1700 of a
splitting-
disproportionation reactor 1502, more specifically, an electrolysis-
disproportionation
(ED) reactor 107 configured for regeneration in a no-aqueous multi-electron
oxidant
(AMO)-on-negative electrode mode of operation and a batch mode. Only the first
two e-
/X- cycles are shown. There are two modes of operation for proceeding with the
ED
regeneration cycle: with and without passing AMO through the negative
electrode 1702.
Furthermore, each of these two modes of operation can be implemented in a
batch mode
or in a flow mode. The flow mode can be implemented in a cyclic flow mode or
in a
cascade flow mode. Furthermore, these different modes of operation can be
combined
within one ED cell 1700, within a single ED reactor 107, and within one
regeneration
system 106. In an embodiment, the ED reactor 107 is configured for the no-AMO-
on-
negative mode of operation using an additional mixing reactor (not shown) to
add a base
produced on the negative electrode 1702 to the AMO containing fluid on the
positive
electrode 1703. The no-AMO-on-negative mode of operation avoids exposure of
the
AMO to the negative electrode(s) 1702 in the ED reactor 107 and instead relies
on the
transfer of a base produced on the negative electrode 1702 during the hydrogen
evolution
or generation reaction, for example,
H20 +e- +M = V2H2 + MOH (42)
to the disproportionation reactor which can be the positive electrode
compartment as
exemplarily illustrated in FIG. 18. Only shown are the first two electrolysis-
disproportionation (ED) cycles. Water fluxes are not shown.
[0256] In this no-aqueous multi-electron oxidant (AMO)-on-negative mode of
operation, the electroreduction of the AMO on the negative electrode 1702 of
the ED cell
125
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
1700 can be prevented by preventing the flow of the AMO-containing fluid
through the
negative electrode 1702. The base such as MOH shown in FIG. 18, produced on
the
negative electrode 1702 in reaction (42) and required for the
disproportionation can be
carried over from the negative electrode 1702 to the positive electrode 1703
with a solvent
such as water. During the regeneration ED cycles, this water can be supplied
to the
negative electrode 1702 from an external source or from the positive electrode
1703
through the cation-conductive polymer electrolyte membrane 1704 by one or a
combination of the following: electro osmotic drag with M , by applying
pressure to the
positive electrode 1703, by other methods known in the art. This excess water
may be
separated from ionic components in the oxidant fluid, yielding concentrated
AMO
solution suitable for an on-board use, produced on the positive electrode
1703, using one
or more of the following: distillation, reverse osmosis, evaporation,
nanofiltration,
pervaporation, ion exchange, freezing, other methods known in the art, and by
any
combination thereof.
[0257] The no-aqueous multi-electron oxidant (AMO)-on-negative mode of
operation
uses a less complicated structure of the positive electrodes 1703 of the ED
reactor 107,
and when a LiOH base with solubility over 5 molal is used, it can provide a
practical and
useful system power density which, nevertheless, can be limited by the maximal
sustainable pH gradient across the cation-conductive polymer electrolyte
membrane
1704. On the other hand, the AMO-on-negative mode of operation does not suffer
from
poor solubility of the base transferred and it overcomes a potential problem
of the
instability of aqueous Li2HPO4 toward decomposition into Li3PO4 (solid) and
LiH2PO4
(solute) by consuming hydrogen phosphate in the disproportionation before the
aqueous
Li2HPO4 decomposes.
[0258] FIG. 19 exemplary illustrates a mass and electricity flow diagram of a
discharge
system 101 comprising a single cell discharge unit 104 and an orthogonal ion
migration
across laminar flow (OIMALF) reactor 1501, exemplarily illustrated in FIG. 1
and FIG.
15. The aqueous multi-electron oxidant (AMO)-on-negative mode of operation is
represented using dash-dotted lines and the no-AMO-on-negative mode of
operation is
126
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
represented using dotted lines. LiBr03, H3PO4, LiZ chemistry is exemplarily
illustrated
for the sake of illustration and not as a limitation. The discharge system 101
comprises
the OIMALF reactor 1501 represented by a single orthogonal ion migration
across
laminar flow (OIMALF) cell 1900, the discharge unit 104 represented by a
single
discharge cell 104a, connecting electric lines, hoses, valves, and an electric
management
system (not shown). The OIMALF reactor 1501 comprises an OIMALF cell stack
(not
shown) which is configured similar to a polymer electrolyte fuel cell (PEFC)
stack but
with a liquid electrolyte flowing between two ionically conducting membranes.
The
OIMALF reactor 1501 comprises endplates 1902a and 1902b and bipolar plates
(not
shown), and the OIMALF flow cell assembly 1901 as disclosed in the detailed
description of FIG. 15. Each flow cell assembly 1901 of the OIMALF reactor
1501
comprises an intermembrane flow field (not shown) with multiple OIMALF flow
channels 1903, two layers of an ion exchange membrane comprising a positive
side ion
exchange membrane 1904a and a negative side ion exchange membrane 1904b
positioned parallel to each other on each side of the intermembrane flow
field, a positive
electrode layer 1905a and a negative electrode layer 1905b flanking outer
surfaces of the
ion exchange membranes, and porous diffusion layers 1905a and 1905b flanking
the
outer surfaces of the ion exchange membranes and in electric contact with the
adjacent
bipolar plates or endplates 1902a and 1902b. The positive electrode layer
1905a is
configured for a hydrogen oxidation reaction and the negative electrode layer
1905b is
configured for a hydrogen evolution reaction. Two modes of neutralizing the
discharge
fluid are exemplarily illustrated in FIG. 19: (i) directly at the negative
electrode(s) 1905b
in the OIMALF flow cell 1900 which requires graded/multilayer negative
electrode
layers 1905b in the OIMALF reactor 1501 to prevent the reduction of an AMO
anion on
the catalyst surface of the negative electrode(s) 1905b, and (ii) indirectly
in a
neutralization reactor 1501b, using the base such as LiOH produced at the
negative
electrode(s) 1905b of the OIMALF cell stack.
[0259] The orthogonal ion migration across laminar flow (OIMALF) reactor 1501
or
the OIMALF cell 1900 converts the salt forms of the aqueous multi-electron
oxidant
(AMO), for example, aqueous LiBrO3 and of the other components of the neutral
oxidant
127
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
fluid, for example, LiH2PO4, and of the extra acid, for example, LiZ into
acidic oxidant
fluid which comprises their acid forms, for example, HBr03, HA, etc. A
complete
conversion of the salt form of the AMO into the acidic form is not necessary
and a partial
conversion is suitable in many applications. An acid concentration, for
example, below
1M may cause an ignition regime provided that the total concentration of all
forms of the
aqueous multi-electron oxidant (AMO) is maintained high, for example, over lm
and the
thickness of the diffusion boundary layer is large, for example, over 1
micron. The buffer,
for example, one or more forms of phosphate is present in the acidic oxidant
fluid
because it is carried over from the splitting-disproportionation (SD) reactor
1502 of the
regeneration system 106 where the base form of the buffer is used in the
disproportionation reaction such as the one shown below:
3Br2 + 6LiA + 3H20 = 5 LiBr + LiBrO3 + 6HA (43)
[0260] An extra acid, for example, HA, such as H2SO4, LiHSO4, HC1, HNO3,
HC104,
CF3S03H, etc., can be added to accelerate the rate of comproportionation as
shown in
equation (44) below on discharge. The use of such an extra acid may be more
advantageous than an increase in the phosphoric acid (H3PO4) concentration
which is a
weak acid. The use of a higher acid concentration, afforded by adding the
extra acid,
facilitates the rate of the comproportionation because for a general acid-
catalyzed
reaction such as:
Br03- + 5Br- + 6H+ = 3Br2 + 3H20 (44)
[0261] The same rate can be obtained with a lower concentration of a strong
extra acid,
such as HC104 or F3CSO3H than with a weaker acid such as H3PO4. A smaller
concentration of the extra acid, compared to the concentration of phosphoric
acid that
shows comparable rate constant for the comproportionation, requires a smaller
charge in
the orthogonal ion migration across laminar flow (OIMALF) process, thus
reducing the
energy expenses and the size of the OIMALF reactor 1501. For purposes of
illustration,
the detailed description is described with reference to an OIMALF process for
conversion
128
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
of the salt form of the aqueous multi-electron oxidant (AMO) into the acid
form of the
AMO; however the scope of the method and system disclosed herein is not
limited to the
OIMALF process but can be extended to include other processes such as a ion
exchange
on resins and in other applications.
[0262] The acidic oxidant fluid travels from a central compartment 1903 of the
orthogonal ion migration across laminar flow (OIMALF) cell 1900 to the
positive
electrode compartment of the discharge cell 104a of the discharge unit 104
where the
acidic oxidant fluid undergoes electroreduction and comproportionation as
shown below.
3 Br2 + 6 e- = 6 Br- (45)
Br03- + 5Br- + 6H+ = 3Br2 + 3H20 (46)
[0263] The reducer, for example, H2, undergoes electrooxidation, represented
by: 3H2 -
6 e- = 6 6H+, at the negative electrode 205b of the discharge cell 104a. The
discharge
system 101 produces electric power for the consumer and, if needed, for
operating the
orthogonal ion migration across laminar flow (OIMALF) reactor 1501.
[0264] The final step performed by the discharge system 101 is neutralization
of the
acidic discharge fluid. For the aqueous multi-electron oxidant (AMO)-on-
negative mode
of operation to neutralization, the acidic discharge fluid comprises, for
example, one or
more forms of water, HBr, H3PO4, and HA such as H2SO4, F3CSO3H, etc., in
concentrations between, for example, about 1mM and 20 M. In an embodiment, the
acidic discharge fluid comprises, for example, one or more of water, a halide,
a
hydroxonium cation, and a counter cation. In an embodiment, the acidic
discharge fluid
produced at the positive electrode compartment of the discharge cell 104 flows
through
or by the negative electrode 1905b of an orthogonal ion migration across
laminar flow
(OIMALF) cell 1900 where a hydrogen evolution or production reaction and pH
increase
occur as shown by the equations below:
129
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
HA+ e- + Li+ = LiA + 1/2 H2 (47)
[0265] The negative electrodes 1905b of the orthogonal ion migration across
laminar
flow (OIMALF) reactor 1501 can take advantage of the Pt-free electron and
cation
conductive inner layer similar to the negative electrodes 1702 of the
regeneration cells or
the SD cells 1700 configured for the aqueous multi-electron oxidant (AMO)-on-
negative
mode of operation, to prevent the electroreduction of residual AMO in the
discharge
fluid. Such a layer, however, is not necessary if the discharge of the AMO in
the
discharge unit 104 proceeds to near completion so that little AMO is present
in the acidic
discharge fluid.
[0266] In other words, as exemplarily illustrated in FIG. 19, the acidic
discharge fluid is
passed over the negative electrode 1905b of the orthogonal ion migration
across laminar
flow (OIMALF) reactor 1501 where the acidity of the discharge fluid is lowered
via a
hydrogen evolution or production reaction with a simultaneous replacement of
H+ with a
cation from the salt of the aqueous multi-electron oxidant (AMO), for example,
a Li+
cation. This produces a neutralized discharge fluid and avoids the formation
and handling
of corrosive and moderately soluble LiOH and is utilized in on-board
applications.
[0267] Alternatively, if the no-aqueous multi-electron oxidant (AMO)-on-
negative
mode of operation is implemented in the SD reactor 1502, a base such as LiOH,
produced
in the negative electrode compartment of the SD reactor 1502 is mixed with the
acidic
discharge fluid allowing for the following chemical processes to occur:
HBr +xHA+ (1+x) LiOH = LiBr + (1+x) H20+ xLiA (48)
[0268] The neutralized discharge fluid passes first through a negative
electrode 1702 of
a regeneration flow cell or SD cell 1700, where the neutralized discharge
fluid is
converted into alkaline regenerated fluid and H2 as shown below. The alkaline
regenerated fluid and H2 are separated in the separation reactor 1010,
exemplarily
illustrated in FIG. 10B and FIG. 17A.
130
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
LiBr+ xLiH2PO4 +zLiZ + xe- + xLi+ = LiBr+ xLi2HPO4 +zLiZ + x/2 H2 (49)
[0269] During the regeneration process the neutral discharge fluid passes
through the
positive electrodes 1703 of the SD reactor 1502 or the regeneration system
106. At the
positive electrode 1703 of the SD cells 1700 also referred herein as the
"regeneration
flow cell", bromide is oxidized into bromine and bromine disproportionates
into bromide
and bromate by reacting with water in the presence of an alkaline form of the
buffer, for
example, A-
LiBr + - xe- - xLi+ = (1-x) LiBr+ x/2 Br2 (50)
(1-x)LiBr + x/2 Br2 + xLiA + x/2 H20 = (1-x/6) LiBr + x/6 LiBrO3 + xHA (51)
[0270] A complete regeneration of the aqueous multi-electron oxidant (AMO) may
not
be necessary and a partially regenerated, that is with LiBrO and LiBr present,
neutral or
near-neutral oxidant fluid can be loaded on-board. The cycle of
electrooxidation-
disproportionation (ED) can be continued in a batch mode, cyclic flow mode,
cascade
flow mode or in any combination thereof using one or more regeneration systems
106
configured for such a mode. The cycle or cascade of regeneration is continued
till the
desired ratio of [Br-V[Br031 is obtained. The cascade flow mode provides a
higher
throughput and the cyclic flow mode provides a lower capital cost. The cascade
flow
mode of regeneration is utilized for multi-user facilities and the cyclic flow
mode is
utilized for at-home regeneration. Based on the equations (23)-(24), 6 cycles
are needed
to convert all bromide into bromate. However, a smaller or larger number can
be used in
practice since a 100% conversion of bromide to bromate is not necessary either
in a
single SD cycle or in a complete regeneration process for the working of the
disclosed
energy cycle.
[0271] FIG. 20A illustrates a method for producing electric power from an
aqueous
multi-electron oxidant and a reducer and for simultaneously generating a
discharge fluid.
The method disclosed herein provides 2001 the discharge system 101 comprising
one or
131
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
more forms of a reducer fluid, one or more forms of an oxidant fluid, the
discharge unit
104, and the acidification reactor 1501a exemplarily illustrated in FIG. 15.
The method
disclosed herein facilitates 2002 discharge of the discharge unit 104 for
producing
electric power from a neutral oxidant fluid comprising one or more forms of
the aqueous
multi-electron oxidant, and from the reducer fluid comprising one or more
forms of the
reducer, for example, hydrogen. In an embodiment, the reducer is selected from
a group
consisting of ammonia, hydrazine, hydroxylamine, phosphine, methane, a
hydrocarbon,
an alcohol such as methanol, ethanol, etc., an aldehyde, a carbohydrate, a
hydride, an
oxide, a sulfide, an organic compound, an inorganic compound, and any
combination
thereof, with each other, with water, or with another solvent. The
facilitation of discharge
comprises lowering 2002a pH of the neutral oxidant fluid in the acidification
reactor
1501a for generating an acidic oxidant fluid, transferring 2002b electrons
from the
positive electrode 205a of the electrolyte-electrode assembly 205 to the
aqueous multi-
electron oxidant in the acidic oxidant fluid, and transferring electrons from
the reducer
fluid to the negative electrode 205b of the electrolyte-electrode assembly 205
to produce
electric power in the external electric circuit operably connected to the
terminals of the
discharge unit 104 and to generate an acidic discharge fluid on consumption of
the acidic
oxidant fluid and the reducer fluid. A limiting current of the transfer of the
electrons from
the positive electrode 205a of the electrolyte-electrode assembly 205 to the
aqueous
multi-electron oxidant in the acidic oxidant fluid in an ignition regime is
limited, for
example, by a mass-transport of the aqueous multi-electron oxidant, a mass-
transport of
acidic protons, and a rate of comproportionation. The transfer of electrons
from the
positive electrode 205a of the electrolyte-electrode assembly 205 to the
aqueous multi-
electron oxidant in the acidic oxidant fluid is performed at a high current
density and at
low flow rates in an ignition mode of operation of the discharge system 101.
The acidic
discharge fluid comprises, for example, one or more of water, a halide, a
hydroxonium
cation, an extra acid, and one or more counter cations. In an embodiment, the
method
disclosed herein further comprises optionally neutralizing the acidic
discharge fluid in the
neutralization reactor 1501b to produce a neutral discharge fluid. In an
embodiment, the
method disclosed herein further comprises regenerating a certain amount of an
intermediate oxidant and the reducer in the discharge unit 104 from the acidic
discharge
132
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
fluid by applying an electric current of a polarity opposite to the polarity
of electric
current through the discharge unit 104 during discharge.
[0272] In an embodiment, the generation of the acidic oxidant fluid from the
neutral
oxidant fluid is performed in the acidification reactor 1501a via an electric
field driven
orthogonal ion migration across laminar flow process. In another embodiment,
the
generation of the acidic oxidant fluid from the neutral oxidant fluid is
performed, for
example, by one or more of an ion exchange on solids, an ion exchange in
liquids,
electrolysis, and adding an extra acid to the neutral oxidant fluid during
discharge of the
discharge unit 104. In an embodiment, the discharge is facilitated on the
positive
electrode 205a of the electrolyte-electrode assembly 205, for example, by one
or more of
electrocatalysis, a solution-phase chemical reaction, a solution-phase
comproportionation,
a solution-phase redox catalysis, a solution-phase redox mediator, an acid-
base catalysis,
and any combination thereof. In another embodiment, the discharge process is
facilitated
via a solution-phase comproportionation of the aqueous multi-electron oxidant
with a
final product of a reduction of the aqueous multi-electron oxidant. In an
embodiment, the
solution-phase comproportionation is pH-dependent and the discharge is
facilitated in the
presence of an acid.
[0273] FIG. 20B illustrates a method for regenerating an aqueous multi-
electron
oxidant and a reducer in stoichiometric amounts from one or more forms of a
neutral
discharge fluid using external power. The discharge fluid comprises, for
example, one or
more of water, a halide, a hydroxonium cation, a buffer, and one or more
counter
cations. In the method disclosed herein, one or more forms of a buffer are
present in the
oxidant fluid and in the discharge fluid, but the buffer is not essential for
the discharge.
The method disclosed herein comprises converting 2003 the neutral discharge
fluid into
an alkaline discharge fluid by using an externally supplied base and/or a base
produced
in the splitting-disproportionation reactor 1502 exemplarily illustrated in
FIG. 15,
configured for an aqueous multi-electron oxidant-on-negative mode of
operation, a no-
aqueous multi-electron oxidant-on-negative mode of operation, or a combination
thereof.
133
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0274] The pH of the alkaline discharge fluid is optimized and stabilized in
the
splitting-disproportionation reactor 1502 using a buffer present in one or
more forms of
the discharge fluid to facilitate disproportionation of the intermediate
oxidant into one or
more forms of the aqueous multi-electron oxidant. The pH of the alkaline
discharge fluid
is maintained between 6 and 10, for example, between 4 and 9. The buffer is
configured
to maintain the pH of the alkaline discharge fluid between 6 and 10, for
example,
between 4 and 9. In an embodiment, the base component of the buffer is
selected from a
group comprising a hydroxide ion, hydrogen phosphate, a phosphate ester, a
substituted
phosphonate, an alkylphosphonate, an arylphosphonate, a deprotonated form of
one or
more of Good's buffers, an amine, a nitrogen heterocycle, and any combination
thereof.
In an embodiment, the cationic component of the buffer comprises a cation of
lithium. In
another embodiment, the cationic component of the buffer comprises a cation of
sodium.
In another embodiment, the anionic component of the buffer comprises one or
more of
w-(N-morpholino)alkanesulfonate, 2-(N-morpholino)ethanesulfonate, 3-(N-
morpholino)propanesulfonate, and 4-(N-morpholino)butanesulfonate. In another
embodiment, the anionic component of the buffer is one or more of w -(N-
morpholino)alkanesulfonate, 2-(N-morpholino)ethanesulfonate, 3-(N-
morpholino)propanesulfonate, and 4-(N-morpholino)butanesulfonate and the
cationic
component of the buffer is lithium. In another embodiment, the anionic
component of
the buffer comprises one or more of an alkylphosphonate and an
arylphosphonate. In
another embodiment, the anionic component of the buffer comprises one or more
of an
alkylphosphonate, an arylphosphonate, and a cationic component of the buffer
is lithium.
In an embodiment, the base component of the buffer is monohydrogen phosphate
and a
cationic component of the buffer is sodium.
[0275] The splitting-disproportionation reactor 1502 splits 2004 the alkaline
discharge
fluid into a reducer and an intermediate oxidant. The splitting-
disproportionation reactor
1502 converts the intermediate oxidant into one or more forms of the aqueous
multi-
electron oxidant via disproportionation of the intermediate oxidant with the
base. The
splitting process releases a stoichiometric amount of the reducer and the base
in the
splitting-disproportionation reactor 1502. The splitting-disproportionation
reactor 1502
134
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
continues 2005 the splitting process and disproportionation in a batch mode of
operation,
or a cyclic flow mode of operation, or a cascade flow mode of operation, or a
combination thereof, until a desired degree of conversion of a discharge
product of the
aqueous multi-electron oxidant into one or more forms of the aqueous multi-
electron
oxidant is achieved. The splitting-disproportionation reactor 1502 splits the
alkaline
discharge fluid into the reducer and the intermediate oxidant, for example,
via electrolysis,
photolysis, photoelectrolysis, radiolysis, thermolysis, or any combination
thereof. The
process of photolysis and photoelectrolysis of the alkaline discharge fluid is
performed in
the presence or absence of a light adsorbing facilitator, a semiconductor, a
catalyst, and
any combination thereof.
[0276] In an embodiment, the splitting-disproportionation reactor 1502 is
configured
as an electrolysis-disproportionation reactor 107. The electrolysis-
disproportionation
reactor 107 converts a neutral discharge fluid into an alkaline discharge
fluid by using an
externally supplied base and/or a base produced at one or more negative
electrodes of
the electrolysis-disproportionation reactor 107 in an aqueous multi-electron
oxidant-on-
negative mode of operation, a no-aqueous multi-electron oxidant-on-negative
mode of
operation, or a combination thereof. The electrolysis-disproportionation
reactor 107
splits or electrolyzes the alkaline discharge fluid into a reducer and an
intermediate
oxidant via electrolysis. The process of electrolysis releases a
stoichiometric amount of
the reducer and the base at one or more negative electrodes of the
electrolysis-
disproportionation reactor 107. The electrolysis-disproportionation reactor
107 converts
the intermediate oxidant produced at one or more positive electrodes of the
electrolysis-
disproportionation reactor 107 into one or more forms of the aqueous multi-
electron
oxidant via disproportionation of the intermediate oxidant produced at one or
more
positive electrodes with the base. The electrolysis-disproportionation reactor
107
continues the electrolysis and disproportionation processes in a batch mode of
operation,
or a cyclic flow mode of operation, or a cascade flow mode of operation, or a
combination thereof, until a desired degree of conversion of a discharge
product of the
aqueous multi-electron oxidant into one or more forms of the aqueous multi-
electron
oxidant is achieved.
135
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0277] FIG. 20C illustrates a method for producing electric power and
regenerating an
aqueous multi-electron oxidant and a reducer in an energy storage cycle. The
method
disclosed herein provides 2001 the discharge system 101 comprising one or more
forms
of a reducer fluid, one or more forms of an oxidant fluid, the discharge unit
104, the
acidification reactor 1501a, and optionally a neutralization reactor 1501b
exemplarily
illustrated in FIG. 15. The method disclosed herein facilitates 2002 discharge
in the
discharge unit 104 for producing electric power from a neutral oxidant fluid
comprising
one or more forms of the aqueous multi-electron oxidant (AMO) and from the
reducer
fluid comprising one or more forms of the reducer. The pH of the oxidant fluid
is lowered
2002a in the acidification reactor 1501a such as the orthogonal ion migration
across
laminar flow (OIMALF) reactor 1501. The oxidant fluid is converted into an
acidic
oxidant fluid via an electric field driven OIMALF process. The discharge
system 101
converts or partially converts the AMO in the salt form such as LiBrO3 into
the AMO in
the acid form such as HBrO3 in the acidification reactor 1501a. When OIMALF
process
is employed in the acidification reactor 1501a and the neutralization
reactor(s) 1501b, the
conversion of the AMO from the salt form to the acid form is accompanied by a
simultaneous release of stoichiometric amount of the base form of the buffer
for
neutralization of the discharge fluid. The conversion of the salt form of the
AMO
produced at the positive electrode into the acid form of the AMO is performed
via an
addition of an acid, ion exchange on resins, ion exchange in solution, for
example, an
electric field driven orthogonal ion migration across laminar flow (OIMALF)
process in
the acidification reactor 1501a. The conversion of the salt form of the AMO
into the acid
form of the AMO in the acidification reactor 1501a is facilitated by an acid,
a buffer,
electrolysis, ion exchange in solution, ion exchange on surfaces, or any
combination
thereof. In an embodiment, the choice of the acid form of the AMO can be
expanded
beyond HBrO3 to other AMOs comprising, for example, HC104, HC103, HC102, HC10,
HBr04, HBr02, HBrO, etc. Phosphoric acid will be present in the oxidant fluid
if
phosphate buffer is used during the regeneration.
136
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0278] In an embodiment, the conversion of the salt form of the aqueous multi-
electron
oxidant (AMO) into the acid form of the AMO occurs within the acidification
reactor
1501a which is used to produce electric power in combination with the
discharge unit
104 and located, for example, on-board of a vehicle. If the acidification
reactor 1501a is
an orthogonal ion migration across laminar flow (OIMALF) reactor, the
acidification
process occurs by consuming electric power and by recycling the hydrogen
released on
the negative electrode of the acidification reactor 1501a and electro-oxidized
on the
positive electrode of the acidification reactor 1501a. In an embodiment, the
hydrogen
produced at the negative electrode or electrodes 1702 in the SD reactor 1502
is combined
with the hydrogen produced at the negative electrode 1905b of one or many
OIMALF
reactors 1501 either before or after one or many OIMALF reactors 1501 or at
the
negative electrode or electrodes 1905b of the OIMALF reactor 1501, and the
hydrogen is
flown through the flow field of the positive electrode 1905a of one or many
OIMALF
reactors 1501. The method disclosed herein reduces the amount of electric
charge utilized
by the acidification reactor 1501a for converting the salt form of the AMO
into the acid
form of the AMO by adding another acid to the AMO during the discharge
process. In
order to reduce the electric charge required by the acidification reactor
1501a and the
degree of conversion required in the OIMALF process to convert the salt form
of the
AMO into the acid form of the AMO, another acid or its anion, for example,
H2SO4,
HC104, F3CSO3H, another strong acid, etc., can be co-present with the AMO
during the
discharge in all stages of the energy cycle.
[0279] In another embodiment, the process of on-board acidification does not
comprise
orthogonal ion migration across laminar flow (OIMALF) but rather an addition
on an
acid stored within the discharge unit 104. Furthermore, the requirement for
storing a
stoichiometric amount of H2 in the discharge system 101 can be reduced by up
to 20% if
an extra H2 is produced from the acidic discharge fluid using metals stored in
the
discharge system 101 as shown in the reaction below.
M (M=Zn, Sn, Fe, etc.) +2HBr MBr2+H2
137
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0280] Such a metal can be used in a complete energy cycle, with regeneration
performed by splitting of MBr2 off-board:
MBr2 M + Br2
[0281] The discharge unit 104 facilitates discharge by simultaneously
transferring
2002b electrons from a positive electrode 205a of the 5-layer electrolyte-
electrode
assembly 206 exemplarily illustrated in FIG. 2, to the aqueous multi-electron
oxidant
(AMO) in the acidic oxidant fluid, and transferring electrons from the reducer
fluid to a
negative electrode 205b of the 5-layer electrolyte-electrode assembly 206 to
produce
electric power in an external electric circuit operably connected to the
terminals of the
discharge unit 104 and to generate an acidic discharge fluid on consumption of
the acidic
oxidant fluid and the reducer fluid. The pH of the acidic discharge fluid in
the
acidification reactor 1501a is optionally raised 2002c for generating a
neutral discharge
fluid. The generation of electric power using the AMO, for example, bromine
during the
discharge is accompanied by the following half-cell electrochemical reactions:
Negative Electrode: 3H2 + 6e- = 6H+ (52)
Positive Electrode: Br03- + 6H+ - 6e- = Br- + 3H20 (53)
[0282] The positive electrode half-reaction (53) may proceed not only by a
direct
electroreduction of the aqueous multi-electron oxidant (AMO), for example,
bromate
species on the electrode but rather facilitated via the formation of an
intermediate, for
example, bromine in a homogeneous comproportionation reaction, for example,
between
bromate and bromide (54) as shown below:
Comproportionation: Br03- + 5Br- + 6H+ = 3Br2 + 3H20 (54)
Electroreduction: 3 Br2 + 6 e- = 6 Br- (55)
[0283] In an embodiment, the neutralization reactor 1501b neutralizes the
acidic
discharge fluid, for example, by using the orthogonal ion migration across
laminar flow
138
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
(OIMALF) reactor to raise the pH of the discharge fluid and produce neutral
discharge
fluid. The discharge fluid is then collected in the discharge fluid tank 113
for subsequent
regeneration. The aqueous multi-electron oxidant (AMO) and the reducer are
regenerated
2006 in stoichiometric amounts from the discharge fluid in the regeneration
system 106.
In an embodiment, the pH of the discharge fluid is optimized by adding or
generating an
acid or a base to the discharge fluid. During the regeneration of the AMO and
the
reducer, the neutral discharge fluid is converted 2003 into an alkaline
discharge fluid by
using an externally supplied base and/or a base produced in the splitting-
disproportionation (SD) reactor 1502 of the regeneration system 106. The SD
reactor
1502 splits 2004 the alkaline discharge fluid at the selected pH into a
reducer and an
intermediate oxidant in the SD reactor 1502. The intermediate oxidant is
converted into
one or more forms of the AMO via disproportionation of the intermediate
oxidant with
the base. The splitting process releases a stoichiometric amount of the
reducer and the
base in the SD reactor 1502. The intermediate oxidant disproportionates when
contacted
with a base such as the base form of the buffer produced at the negative
electrode 1702.
The disproportion reaction produces the desired AMO in a stable salt form, for
example,
LiBrO3 as well as discharged oxidant, for example, LiBr which undergoes
further cycles
of splitting-disproportionation until the desired degree of conversion, for
example, [Br03-
]/([Br031 + [Br0-1 + 2[Br2] + [Br-1) > 0.95 is achieved. The cycle of
splitting-
disproportionation is continued 2005 till the desired degree of conversion of
the discharge
product of the aqueous multi-electron oxidant into one or more forms of the
aqueous
multi-electron oxidant is achieved. The regenerated one or more forms of the
oxidant
fluid comprising the AMO and the regenerated reducer fluid comprising the
reducer are
then supplied 2007 to the discharge system 101 for facilitating discharge of
the discharge
unit 104.
[0284] The pH of the acidic oxidant fluid in the splitting-disproportionation
(SD)
reactor 1502 of the regeneration system 106 exemplarily illustrated in FIG.
15, is
optimized and stabilized using an extra acid present in the acidic oxidant
fluid to facilitate
comproportionation of the aqueous multi-electron oxidant with a final product
of a
reduction of the aqueous multi-electron oxidant into the intermediate oxidant.
The extra
139
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
acid is, for example, one or more of a phosphoric acid, a 3-(N-
morpholino)propanesulfonic acid, a 3-(N-morpholino)ethanesulfonic acid,
another a-(N-
morpholino)propanesulfonic acid, a methanesulfonic acid, triflic acid, a
substituted
sulfonic acid, a substituted phosphonic acid, a perchloric acid, a sulfuric
acid, a molecule
comprising sulfonic moieties and phosphonic acid moieties, and an acid with a
pKa < 2.
The pH of the acidic discharge fluid is, for example, below 0, 1, 2, or 3. The
concentration of acidic protons in the acidic discharge fluid is, for example,
below one of
0.1M, 0.5M, 1M, 2M, 5M, or 10M.
[0285] FIG. 20D illustrates a method for producing electric power and
regenerating
hydrogen and an oxidant fluid comprising lithium bromate in an energy storage
cycle.
The method disclosed herein provides 2001a the discharge system 101 comprising
the
discharge unit 104, the acidification reactor 1501a, and optionally the
neutralization
reactor 1501b as exemplarily illustrated in FIG. 15. The discharge system 101
comprises
a neutral oxidant fluid comprising lithium bromate, and hydrogen. In an
embodiment, the
discharge system 101 comprises one or more forms of a buffer. In another
embodiment,
the discharge system 101 further comprises one or more forms of a base. In an
embodiment, the cationic component of the buffer is lithium and the anionic
component
of the buffer is, for example, one or more of w-(N-morpholino)alkanesulfonate,
3-(N-
morpholino)methanesulfonate, 3-(N-morpholino)ethanesulfonate, 3-(N-
morpholino)propanesulfonate, 3-(N-morpholino)butanesulfonate, other amines,
monohydrogen phosphate, methylphosphonate, an alkylphosphonate, an
arylphosphonate,
and a molecule comprising phosphonate moieties and sulfonate moieties. In
another
embodiment, the cationic component of the buffer is sodium, and the anionic
component
of the buffer is, for example, one or more of a-(N-morpholino)alkanesulfonate,
methylphosphonate, 3-(N-morpholino)ethanesulfonate, 3-(N-
morpholino)propanesulfonate, an alkylphosphonate, an arylphosphonate, and a
molecule
comprising phosphonate moieties and sulfonate moieties. In the discharge
process only
the extra acid is relevant not the buffer. Some molecules can function as both
the buffer
and the extra acid. Those comprising both phosphonic and sulfonic moieties are
utilized
here. The discharge system 101 further comprises a deprotionated form of an
extra acid.
140
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
The extra acid comprises, for example, one or more of bromic acid, sulfuric
acid,
perchloric acid, triflic acid, a sulfonic acid, molecules comprising
phosphonate moieties
and sulfonate moieties, and an acid with a pKa < 2. The buffer is in an acid
form during
discharge with a pH < 4 and the acid form of the buffer comprises one or more
of a
phosphoric acid derivative, substituted phosphonic acids, 2-(N-morpholino)
alkanesulfonic acid(s), molecules comprising both phosphonate and sulfonate
moieties,
amines and buffers capable of maintaining pH between 4 and 9. The base form of
the
buffer is, for example, one or more of w-(N-morpholino)alkanesulfonate, 2-(N-
morpholino)ethanesulfonate, 3-(N-morpholino)propanesulfonate, 4-(N-
morpholino)butanesulfonate, a phosphoric acid derivative, an alkylphosphonate,
an
arylphosphonate, a molecule comprising phosphonate moieties and sulfonate
moieties, an
amine, a nitrogen heterocycle, and a base with a pKa between 4 and 9.
[0286] The concentration of lithium bromate dissolved in the neutral oxidant
fluid is,
for example, above 1M, 2M, 5M, or 10M. The acidification reactor 1501a
converts 2008
the neutral oxidant fluid into an acidic oxidant fluid. The concentration of
acidic protons
in the acidic oxidant fluid is, for example, below 0.1M, 0.5M, 1M, 2M, 5M, or
10M. The
method disclosed herein facilitates 2009 discharge of the discharge unit 104
for
producing electric power from the acidic oxidant fluid and from hydrogen and
generates
an acidic discharge fluid on consumption of the acidic oxidant fluid
comprising lithium
bromate and hydrogen. The discharge is facilitated via a pH¨dependent solution-
phase
comproportionation of bromate with bromide formed via electroreduction of
intermediate
bromine. In an embodiment, the neutralization reactor 1501b neutralizes 2010
the acidic
discharge fluid to produce one or more forms of a neutral discharge fluid.
[0287] The method disclosed herein further comprises optimizing and
stabilizing the
pH of the acidic oxidant fluid in the splitting-disproportionation reactor
1502 using an
extra acid present in the acidic oxidant fluid to facilitate
comproportionation of the
aqueous multi-electron oxidant with a final product of a reduction of the
aqueous multi-
electron oxidant into an intermediate oxidant. The pH of the acidic discharge
fluid is
below 3, 2, 1 or O. The extra acid is one or a combination of bromic acid,
another
141
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
aqueous multi-electron oxidant (AMO) acid, phosphoric acid, 3-(N-
morpholino)propanesulfonic acid, 3-(N-morpholino)ethanesulfonic acid, other o)-
(N-
morpholino)propanesulfonic acid, methanesulfonic acid, triflic acid,
substituted sulfonic
acid, substituted phosphonic acid, perchloric acid, sulfuric acid, a molecule
comprising
sulfonic moieties and phosphonic acid moieties, and an acid with a pKa < 2.
[0288] The regeneration system 106 regenerates 2011 hydrogen and one or more
forms
of the oxidant fluid comprising lithium bromate in stoichiometric amounts from
one or
more forms of the neutral discharge using external power. The regeneration is
performed
by splitting 2011a one or more forms of the neutral discharge fluid into
stoichiometric
amounts of bromine, hydrogen, and a base form of a buffer using external power
in the
splitting-disproportionation reactor 1502, and producing lithium bromate via
disproportionation of bromine with the base form of the buffer. The splitting
process is
performed via electrolysis, photolysis, photoelectrolysis, radiolysis, or
thermolysis. In the
case of splitting being electrolysis, bromine is produced on a positive
electrode of the
electrolysis-disproportionation reactor 107 and hydrogen and a base are
produced at a
negative electrode of the electrolysis-disproportionation reactor 107. The
disproportionation reaction is facilitated by a buffer capable of maintaining
a solution pH
between 4 and 9 or between 6 and 8. The splitting-disproportionation reactor
1502
continues 2011b splitting and disproportionation in a cyclic manner in the no-
aqueous
multi-electron oxidant-on-negative mode of operation or the aqueous multi-
electron
oxidant-on-negative mode of operation in one of multiple modes until a desired
degree of
conversion of bromide into bromate is achieved. The modes comprise, for
example, a
batch mode, a cyclic flow mode, a cascade flow mode, and any combination
thereof. The
regeneration system 106 supplies 2012 the regenerated one or more forms of the
oxidant
fluid comprising bromate and the regenerated hydrogen to the discharge system
101 for
subsequent generation of electric power on demand.
[0289] FIG. 21A exemplary illustrates polarization curves of a glassy carbon
rotating
disk electrode in solutions comprising 5M LiBr03+50%w H3PO4+1 mM LiBr at
different
rotation rates in rpm and 20 C. The decrease of the limiting current at higher
rotation
142
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
rates is noticeable as exemplarily illustrated in FIG. 12. Such a regime is
observed when
the concentration of acid is sufficiently high so that the limiting current is
controlled by
the kinetics of the comproportionation rather than by the mass-transport of
protons. As
the concentration and the dissociation constant of the acids such as H3PO4,
H2SO4,
F3CSO3H, etc., in the oxidant fluid is increased, the limiting current on
discharge also
increases. However, the lifetime of the aqueous multi-electron oxidant (AMO)
stock
solution decreases. For example, a limiting current of about 50 mA/cm2 for a
50% w
H3PO4 in combination with 5M LiBrO3 on a smooth carbon electrode is produced
while
the lifetime of this AMO stock or acidic oxidant fluid is about 10 days at 20
C. A shorter
lifetime is obtained with 30% w H2SO4. Therefore, when the orthogonal ion
migration
across laminar flow (OIMALF) process is performed on-board rather than off-
board, a
TRIZ contradiction between the power and stability of the acidic oxidant fluid
can be
resolved with an additional benefit of improved safety of the on-board
discharge system
101. This way only safe and stable AMO salt solution, for example, LiBrO3 with
a high
energy density and room temperature solubility of about 13.27 molal and charge
density
of ca. 660 Ah/kg is stored on-board and off-board, and the reactive acid form
of the AMO
(H-AMO) is generated on-board just before it is consumed by the discharge unit
104.
One feature that enables a practical use of an on-board acidification system
is that the
ignition regime of bromate electroreduction can be observed under low ratios
of proton to
bromate concentrations, about [H ]/[Br031< 0.1, if the total concentration of
bromate is
high, about over 10 molal which is possible with LiBrO3. Also, the base, for
example,
Li0H, Li-3-(N-morpholino) propanesulfonic acid (MOPS), etc., produced at the
negative
electrode during the on-board OIMALF process is used to neutralize the acid,
for
example, HBr produced in the discharge unit 104, so that the amount of
dangerous
materials, for example, HBr03, HBr, Li0H, etc., present on board at any time
is
minimized.
[0290] FIG. 21B exemplary illustrates polarization curves of a glassy carbon
rotating
disk electrode in a solution comprising 30% H2504 + 166mM LiBrO3 + 5mM NaBr.
Limiting current similar to that found in phosphoric acid at a much higher
bromate
concentration exemplarily illustrated in FIG. 21A, is observed which is
interpreted as
143
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
evidence of a higher disproportionation rate constant when a stronger sulfuric
acid is used
rather than when a weaker phosphoric acid is used. In both cases, the limiting
current
decreases with the rotation rate suggesting that the limiting current is
controlled by the
rate of the disproportionation rather than by mass transport or, for example,
acidic
protons.
[0291] FIG. 22 exemplarily illustrates Pourbaix diagrams for bromine in
aqueous media
at pH 0 and pH 14. FIG. 22 exemplarily illustrates the concept of the
possibility of
shifting from disproportionation to comproportionation by changing the pH of
the
oxidant fluid. The numbers near the lines denote the standard potentials of
the
corresponding electrochemical reactions. When the potential to the left is
lower than the
potential to the right, the species is stable, for example, Br2 in acid. When
the potential to
the left is higher than the potential to the right, the species
disproportionates, for example,
HBrO in acid or Br2 in alkali.
[0292] FIG. 23A exemplarily illustrates a solar radiation spectrum at sea
level and the
positions of a silicon (Si) band-gap, bromine and/or bromide, and bromate
and/or
bromide standard electrode potentials. The solar photo electrochemical
regeneration of
H2 and Br2 from HBr followed by a conversion of Br2 into HBrO3 enables the use
of
sunlight at a cost similar to the cost involved in traditional methods such as
natural gas
combustion and uranium fission. An open-circuit potential photoelectrolysis of
aqueous
HBr on semiconductor particles can be performed with a lower cost and higher
efficiency
than the photoelectrolysis of water advocated by the proponents of hydrogen
economy.
FIG. 23A exemplarily illustrates that while the Si band-gap cannot provide
enough
energy to convert HBr + 3H20 into 3H2+ HBrO3 directly, the reaction 2HBr =
H2+Br2
can be driven by the Si band-gap energy. The further uphill conversion of
bromine into
bromate is performed via disproportionation driven by a pH change, for
example, 3Br2+
30H- = 5Br- + Br03- with hydroxide as a base. The uphill pH change, in turn,
is driven
electrochemically by the hydrogen evolution or production reaction of the
negative
electrode(s) in an electrolyzer (not shown) of the photoelectrolysis-
disproportionation
(ED) reactor.
144
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0293] FIG. 23B exemplarily illustrates a batch mode of a photoelectrolysis-
disproportionation method for regenerating a halate from a halide.
[0294] Example 1: FIGS. 14A-14G exemplarily illustrate graphical
representations
showing comparative performances of three on-board power sources at a nominal
power
of 130 kW: a gasoline-internal combustion engine, a lithium ion battery (LIB),
and an H2-
aqueous multi-electron oxidant (AMO) discharge unit 104 or flow battery
exemplarily
illustrated in FIG. 1, as well as the targets of the Advanced Research
Projects Agency-
Energy (ARPA-E). Table 2 exemplarily compares the projected performance of an
H2-
AMO discharge system 101 at a nominal power of 130 kW with the performance of
2012
Toyota RAV4EV and with 2013 ARPA-E goals for a battery for a Fully Electric
Vehicle.
The AMO is 50% w/w aqueous HBr03. The Toyota RAV4 EV of Toyota Jidosha
Kabushiki Kaisha TA Toyota Motor Corporation is chosen as an example of a
sport utility
vehicle, which is or was available in gasoline and in lithium-ion battery
(LIB) versions, to
illustrate the capabilities of the discharge unit 104. A sport utility vehicle
(SUV) is selected
because it is a large vehicle that presents a greater challenge for
electrification than a small
urban vehicle. The data are available for Toyota RAV4 in both gasoline and
electric
vehicle lithium ion battery (LIB) versions. All calculations are based on the
rated power of
about 130 kW=174 hp. The size of the storage unit in the vehicle using the
discharge unit
104 of the discharge system 101 exemplarily illustrated in FIG. 1, is adjusted
to give the
same driving range as the gasoline power system, rather than the electric
version. H2 is
stored in the vehicle using the discharge system 101 as a metal hydride to
minimize the H2
tank volume.
[0295] In the vehicle using the discharge system 101 disclosed herein, both
the reagent, for
example, bromate and the product such as bromide of the discharge are anions,
which
minimizes their cross-over through a cation -exchange membrane such as Nafion
and
other poly perfluorosulfonic acid (pPFSA) membranes and prevents a parasitic
self-
discharge. Also, the electrode reaction of bromine/bromide does not require an
expensive
catalyst and the electrode reaction occurs with an acceptable rate even on
carbon
145
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
electrodes. It is also estimated that the capital cost of the discharge unit
104 can be as low
as 120 $/kW which is less than a half of the lithium-ion battery cost in the
Nissan Leaf of
Nissan Jidosha Kabushiki Kaisha DBA Nissan Motor Co. Ltd., and the Toyota RAV4
.
The lithium ion battery (LIB) takes up about 20% of the vehicle weight while
the discharge
system 101, takes about 10% of the vehicle weight, similar to, for example,
the internal
combustion engine (ICE)-gas system as exemplarily illustrated in FIG. 14A. In
the
Advanced Research Projects Agency-Energy (ARPA-E) metrics, the energy density
of the
on-board discharge unit 101 is, for example, about 426 Wh/kg, which is about
2.8 times
larger than the ARPA-E target of 150 Wh/kg.
[0296] The volume of the discharge system 101 is, for example, twice the
volume of the
gas tank including the internal combustion engine (ICE) and half of the
lithium-ion battery
(LIB) and the electric engine as exemplarily illustrated in FIG. 14B. The
energy density of
the discharge unit 104 depends on the method of hydrogen storage and it is,
for example,
200-400 Wh/L, which exceeds the Advanced Research Projects Agency-Energy (ARPA-
E)
target of 230 Wh/L. Both the gasoline power system and the discharge unit 104
can provide
a driving range of about 300 miles as exemplarily illustrated in FIG. 14C,
while Toyota
RAV4 EV has a range of about 92 miles, according to the Environment Protection
Agency
(EPA) criteria, which comes from its low battery stack energy of about 41.8
kWh. The
manufacturing cost of the discharge unit 104 is about $15,000 based on the
current prices
proton exchange membrane fuel cells (PEMFCs) produced in low volumes
accounting for
the absence of Pt on the positive electrode 205a in the H2-aqueous multi-
electron oxidant
(AMO) system, or about 120$/kWh and 115 $/kW as exemplarily illustrated in
FIG. 14D,
and is more than the manufacturing cost for the ICE, which is about $5,000,
but is close to
the Advanced Research Projects Agency-Energy (ARPA-E) target of <140/kWh, and
is
three times lower than the cost per mile drive of the LIB system. The
projected tank- to-
wheel efficiency of the discharge unit 104 under realistic operating
conditions is slightly
lower than that of lithium ion batteries (LIBs) but much higher than that of
internal
combustion engines (ICEs) as exemplarily illustrated in FIG. 14E. Both the
gasoline power
system and the discharge unit 104 can be refilled mechanically within minutes,
while
Toyota RAV4 EV needs about 5 hours for electric recharge as exemplarily
illustrated in
146
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
FIG. 14F. The standard discharge efficiency of the discharge unit 104 is about
78% and
such efficiency can be a practical target at about 0.5 W/cm2. For the power of
about 1
W/cm2, the finite rate of the comproportionation and the non-negligible
membrane
resistance make 65% a more realistic target.
[0297] Platinum on the H2 electrode is used at the same loading as the PEMFC
but the
loading is between 1/10 and 1/20 of what is used on the air electrode in the
PEMFCs, and
the loading has been shown to be sustainable economically, and is not a large
contributor to
the cost. FIG. 14G exemplarily illustrates the projected competitive positions
of the H2-
aqueous multi-electron oxidant (AMO) discharge unit 104 on the Advanced
Research
Projects Agency-Energy (ARPA-E) price-range plot for different vehicle power
sources.
The discharge system 101 disclosed herein can meet the range, cost, cost and
safety
targets for fully electric vehicles (FEVs) defined by the ARPA-E's Robust
Affordable
Next Generation Electric Vehicle (RANGE) program as exemplarily illustrated in
FIG.
14G.
[0298] Example 2: The comparison of a gasoline engine, a lithium ion battery,
and two
hydrogen-bromate batteries with different methods of hydrogen storage, that
is, 700 bar
compressed and 9% w/w metal hydride is provided in the table below.
50% aqueous multi-electron
H2 storage oxidant (AMO)
350 liqui 5%
bar d MH 5.74M
theoretical limit g/L 25 70 125 1.48
100kg,300 kW real
systems g/L 10 26 20
Ah/ 1,87
theoretical limit L 670 5 3,350 923
charge per mass of Ah/k 26,8 26,7 2680
pure H2 g 00 86 0 623.7
147
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
real system w% 5 5 5
Ah/k 1,34 1,34
real system g 0 0 1,340 623.7
Ah/
real system L 268 697 536 923
vol.% for storage 77.4
system H2 5 57.0 63.3
wt. % for storage
system H2 2.28 2.28 2.27
mL/
volume/charge Ah 3.73 1.44 1.87 1.08
0.03 0.03 0.037
mass/charge g/Ah 73 73 3 1.60
4h drive
RAV4=520 kWh kg 388 388 388 834
4h drive 1,94
RAV4=520 kWh L 0 746 970 563
system energy Wh/
density L 208 397 339
specific energy Wh/
5%w/w H2 kg 426 426 426
[0299] The parameters used for lithium ion batteries (LIB s) are 230 Wh/L, 128
Wh/kg,
and $0.47/Wh. The parameters used for H2 storage are 50 g/L compressed 125 g/L
MH.
The LiBrO3 solution density is assumed as 1.49 g/cm3, the same as for 48% w/w
aqueous
HBr. The cost of 50% HBr = $2000/ton =$2/kg.
[0300] Example 3: Reactions at a positive electrode during discharge of
bromate using
a vanadium redox mediator are provided below:
HBrO3 + 5V0+2 + 5H+ = '1/2Br2 + 5V02+ + 3H20 in solution
148
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
5V02+ + 5e- + 10H+ = 5\70+2 + 5H20 on the
positive electrode
'1/2Br2 + 1e- = Br- on the
positive electrode
[0301] Example 4: A bromine/bromide couple is used as a mediator for a bromate
reduction "r" on discharge as shown below:
HBrO3 + 5Br- + 5H+ = 3Br2 + 3H20; in solution;
3Br2 + 6 e- = 6Br-; on the positive electrode.
[0302] Example 5: A chlorine/chloride couple is used as the mediator for
bromide/bromate on charge as shown below:
cr- i e = v2 C12; on the positive electrode; (56)
V2Br2 + 2.5C12 + 3H20 = HBrO3 + 5HC1 in solution. (57)
[0303] Example 6: The conversion of hydrobromic acid to bromic acid using a
resin-
type ion exchange reactor is shown below, where M refers to a cation such as
an alkali
,an alkali earth metal ,or organic cation, and "solid" refers to an ion
exchanging material
such as a resin:
M+(solid) + HBr(spent) = H+(solid) +MBr resin regeneration
MBr + 6MOH - 6 e- = MBrO3 + 3H20 + 6M+ positive electrode
3H20 + 6e- +6M+ = 3H2 + 6MOH negative electrode
MBrO3 + H+(solid) = HBrO3 + M+(solid) ion exchange on the resin
[0304] The above method for regenerating the aqueous multi-electron oxidant
from the
spent discharge fluid may result in the incomplete exchange of M+ for H+ under
stoichiometric conditions, which results in an overuse of the acid regenerant
and of the
energy needed to produce the acid regenerant. However, a complete exchange of
M+ for
H+ is not required for the ignition to occur.
149
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0305] Example 7: H2-aqueous multi-electron oxidant (AMO) discharge redox flow
battery: In an embodiment, in H2-LiBrO3 discharge flow batteries, modified
single and
multiple stack type proton exchange membrane fuel cells (PEMFCs) are employed.
The
electrolyte-electrode assemblies are fabricated using a polyperfluorosulfonic
acid
(pPFSA) membrane, with a conventional negative electrode layer 205b
exemplarily
illustrated in FIG. 2, comprising Pt, C, and pPFSA, and a conventional gas
diffusion layer
(GDL) used for H2 oxidation on the negative anode side. The positive cathode
design,
however, is different from the proton exchange membrane (PEM) air cathode,
since
neither bromate nor bromide are soluble in the pPFSA, which completely
surrounds the
Pt/C electrocatalyst in modern thin-film PEMFC catalytic layers. A porous flow-
through
or flow-by media, for example, porous hydrophilic carbon or carbon cloth, is
used for the
positive electrode 205a in a H2-HBrO3discharge flow battery.
[0306] Under operating conditions at a high acid concentration, a slower yet
above
stoichiometric flow rate of the aqueous multi-electron oxidant (AMO)-
containing acidic
oxidant fluid leads to a higher cell power in contrast with fuel cells and
conventional
redox flow batteries. This is due to a larger fraction of the intermediate
such as bromine
escaping the kinetic boundary layer into the solution bulk as the diffusion
boundary layer
gets thinner. This finding suggests that the cell operation at high power does
not require
significant energy expenses on pumping and that, in contrast to fuel cells, a
near
stoichiometric supply of the aqueous multi-electron oxidant may provide an
optimal
performance in terms of the power, energy efficiency, and system size. Also, a
quick
depletion of bromate in the ignition regime and the higher viscosity of the
aqueous multi-
electron oxidant (AMO) compared to air implies a preference for short
channels, which,
in combination with a parallel-channel flow field and slow flow rates, also
leads to a
lower pressure drop. Also, the absence of the gas phase in the cathode stream,
the large
heat capacity, and the high water content of the AMO supply simplify the
design,
manufacture, and operation of the cathode side as well as of the discharge
unit 104 and of
the whole discharge system 101.
[0307] Example 8: Power and efficiency of the hydrogen-bromate discharge unit
104:
150
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
In order to estimate the power and voltage of the hydrogen-bromate discharge
unit 104
during discharge, the following model is used: The discharge unit 104
comprises a single
electrolyte-electrode assembly 205 exemplarily illustrated in FIG. 2. Pure
humidified
hydrogen is supplied to the anode or the negative electrode 205b. The anode
polarization
losses and reagent cross-over are ignored. The cell or membrane ohmic
resistance is, for
example, set to 0.1 Ohm/cm2. The cathode or the positive electrode 205a is
smooth and is
supplied with 50 % w/w/ HBrO3 containing a few mM of Br2, Co=[Br2]0, to
initiate the
electroreduction cycle. Electrochemical polarization of the cathode is
ignored, that is,
bromine/bromide exchange current is large compared to the applied currents.
[0308] The homogeneous kinetics of the comproportionation is incorporated
through
the use of kinetic boundary layer thickness, Lo= (Dbromide/5 k0 Cbromate)1/2 =
1.5 pm,
where kcon is the appropriate rate constant for the homogeneous
comproportionation. At
currents above 1 A/cm2, further correction becomes important, i.e. L = Lo /(1-
(iz0
/5DbromateCbromate))112. The effect of convection is incorporated through the
use of the
diffusion boundary layer thickness, zo Zo = zo/L. Its value is selected on the
basis of
common values of the respective quantities for the rotating disk and channel
electrodes in
aqueous electrolytes, that is, 15 pm and 150 pm. Diffusion coefficients for
bromide and
bromine are set to 1.5 x10-5 cm2/s and 1.0 x10-5 cm2/s, respectively. Activity
coefficients
of all species are set to 1.
[0309] A more detailed analysis leads to the following formula for a
polarization curve
for bromate comproportionation-electroreduction on a smooth electrode:
Exp{2(E-E )F/RT} = [Br2]0 + (iL/FDbromme)(0.1Z0-0.6thZ0)1(FDbromidefiLthZ0)2
[0310] The corresponding plots for power are exemplarily illustrated in FIG.
13.
Although the experimental data that is reported in FIG. 13 are for much higher
rotation
rates (low Zo), the data in FIGS. 21A-21B for lower rotation rates support the
conclusion
that the comproportionation reaction can sustain large currents in the
discharge unit 104.
The hydrogen-bromate discharge unit 104 can achieve under very realistic
conditions,
151
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
even with a smooth carbon electrode, a power, for example, about 1 W/cm2 at
around 1.0
V, which corresponds to the energy efficiency, for example, of about 68% with
respect to
the standard electrode potential of bromate/bromide, that is, about 1.48 V.
Such
performance compares favorably with the performance of state-of-the-art
hydrogen¨air
fuel cell, yet it can be achieved with about a 10 times smaller Pt loading and
with electric
or solar regeneration of the reducer and the aqueous multi-electron oxidant.
Under
operating conditions at a high acid concentration, the concentration of free
bromine has
little effect on the cell performance, whereas a stronger convection decreases
the cell
power in contrast with conventional fuel cells. This is due to a faster escape
of
intermediate Br2, homogeneously produced in the vicinity of the electrode,
into the bulk
of the solution at smaller hydrodynamic boundary layer thicknesses. Such an
effect is not
observed in conventional fuel cells and flow batteries since the electroactive
reagent is
delivered from the bulk of the solution rather than formed near the electrode.
Also, the
kinetic layer thickness, which determines the minimal meaningful pore diameter
in the
porous electrode, is L= 1.5 i.tm in 50% w/w HBr03, and a thicker 6L =9 iim
hydrodynamic boundary layer is needed for the ignition to occur.
[0311] The 1D model disclosed herein assumes a constant solution composition
outside
the hydrodynamic boundary. The model disclosed herein shows that a low near-
stoichiometric flow rate is appropriate for the operation of the discharge
unit 104 with
reduced energy losses entailed. The parallel flow field with a channel length
longer than
the ignition length but shorter than the depletion length with a flow rate
slightly above the
stoichiometric can provide maximal power while simultaneously reducing pumping
losses.
[0312] Example 9: In an embodiment, the regeneration system 106 produces a
dilute
aqueous multi-electron oxidant (AMO) solution, for example 0.5M, which needs
to be
concentrated, for example, to about 3.88M. Water evaporation, vacuum
distillation,
pervaporation are suitable means of concentrating the AMO solution. Heat
exchangers
are used to transfer heat from the concentrated product to dilute input
solution if the
water removal is performed at an elevated temperature. The energy expenses of
152
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
concentrating dilute AMO produced in the orthogonal ion migration across
laminar flow
(OIMALF) step should be compared with the energy of a H2-AMO battery. In the
case of
bromic acid, the stored electric power is:
1,705 Wh/kg * 0.135 kg/mol = 230 Wh/mol = (1 Wh = 3.6kJ) = 829 kJ/mol
[0313] The evaporation of excess water is also possible, more efficiently with
heat
exchangers, but it leads to the loss of volatile bromine species. In the case
of reverse
osmosis (RO) process of concentrating the aqueous multi-electron oxidant (AMO)
solution, the osmotic pressure difference between the dilute and concentrated
solutions of
the AMO such as bromic acid can be estimated via the Morse equation. Molality
is
assumed the same as molarity and dissociation is complete:
II = iMRT = 2 * (3.88-0.5) mol/L*103 L/m3* 300K *8.3145 J K-1 mo1-1 = 16.8 MPa
=
168 bar
[0314] This pressure falls within the range of commercial cascade reverse
osmosis
units, thus, such a process is technically feasible. The minimal energy
expense for reverse
osmosis (RO) concentrating can be estimated as 1.742 kg of water per 1 mole of
HBrO3
needs to be removed. This corresponds to 1.742 10-31113 * 16.8 106 Pa = 2.93
104 J = 29.3
kJ/mol HBr03. This is only 3.3% of the battery energy per 1 mole of bromic
acid. This
number is the lower limit at the infinitely slow rate of water permeation and
the number
will be higher in practice. For example, sea water desalination requires
usually 5 times
more energy than the theoretical value. Using the factor of 5, about 16.5%
battery energy
is obtained which is acceptable in practice.
[0315] Example 10: The molal solubilities, that is, moles of solute per kg of
water of
some compounds of interest in the electrolysis-disproportionation (ED)-
orthogonal ion
migration across laminar flow (OIMALF) process at 20 C and 60 C are provided
in the
table below:
153
CA 02929694 2016-02-08
WO 2015/026393 PCT/US2014/019170
moles of solute per kg of solvent, m
bromide bromide bromate bromate Hydroxide Hydroxide
cation
20 C 60 co 20 C 60 C 20 C 60 C
Li + 18.4 25.7 13.3 19.9 5.3 5.8
Na + 8.8 11.5 2.4 4.1 27.3 43.5
K+ 5.5 7.2 0.4 1.4 20 27.4
NMe4+ 7.79E-03 n/a n/a n/a Mp 67C, 50% = 5H20
Ba2+ 3.7 4.1 1.65E-02 5.77E-02 2.27E-01
1.2
Sr2+ 4.1 6.1 0.9 01.05 1.46E-01 0.7
Ca2+ 7.2 13.9 7.8 n/a 2.33E-02 n/a
mg2+
5.5 6.1 20.7 n/a 1.71E-03 n/a
[0316] The data in the table above suggests that Li + cation provides a high
molal
solubility for bromide and bromate. The limited solubility of LiOH is
irrelevant since it
does not appear in the laminar flow of the orthogonal ion migration across
laminar flow
(OIMALF) reactor 1501 where solids can disrupt the process. Also, if a buffer
such as 3-
(N-morpholino) propanesulfonic acid (MOPS) is used, LiOH will react with the
buffer.
[0317] Example 11: In an embodiment, in the case of a redox couple with both
components being anions, for example, halate and halide, the cross-over of the
oxidant
couple to the negative electrode 205b exemplarily illustrated in FIG. 2, can
be prevented
with a cation exchange membrane. In the case of lithium bromate, the discharge
process
on the positive electrode 205a is as shown below:
Br2+2e- +2Li+ = 2LiBr on the electrode. (58)
LiBrO3 +5LiBr +6HA = 3Br2+3H20 +6LiA in solution (59)
where HA represents the acid present in the acidic oxidant fluid such as
bromic acid,
phosphoric acid, and/or the extra acid.
154
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0318] The concentration of the neutral intermediate Br2 is maintained
sufficiently low,
so that its cross-over to the negative electrode 205b makes a negligible
contribution
compared to the current of the electrolytic cell 200. The ratio of the
standard redox
potentials of bromate/bromide and bromine/bromide suggests, for example, only
about
25% loss of efficiency when performing comproportionation mediated rather than
direct
discharge of bromic acid at the equilibrium potential. The regeneration of
bromate and
hydrogen from bromide and water or, in general, oxidant fluid from discharge
fluid can
be performed off-board. Direct electrolytic regeneration can be performed with
an anode
such as Pb02 or a dimensionally stable anode (DSA).
[0319] In an embodiment, a solution-phase mediator, for example, a redox
couple is
used to expedite the rates of an otherwise slow electrode reaction and thus to
increase the
system power and efficiency. A redox couple that undergoes electron exchange
with
both an electrode and a reduced or an oxidized form of the aqueous multi-
electron
oxidant can be used to accelerate the rates of charge or discharge, thereby
improving
efficiency. Different redox mediators can be employed in the charge and
discharge
processes. In an embodiment, C12/2C1- can be used as a solution-phase mediator
in the
electrochemical regeneration process. Since oxidations, for example, electro-
oxidation of
a halide to a halate, are more facile in alkaline solutions, performing
regeneration at high
pH and then, for usage in the discharge unit 104, converting the salt into
acid, for
example, by means of the orthogonal ion migration across laminar flow (OIMALF)
process are considered.
[0320] In an embodiment, pH-dependent disproportionation and pH-dependent
comproportionation reactions involving halogens and their compounds are used
to
facilitate the discharge and regeneration of the aqueous multi-electron
oxidants. The
rate(s) and the equilibrium constant(s) of the disproportionation reaction(s)
in some
cases may show a dependence of the solution pH. The rate(s) and the
equilibrium
constant(s) of the comproportionation reaction(s) in some cases may show
dependence
of the solution pH.
155
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0321] In an embodiment, the aqueous multi-electron oxidant (AMO) can be
regenerated by reacting the halide with ozone or by photolytic oxidation on a
suitable
semiconductor such as Ti02. In another embodiment, a mediator is used for
oxidation at
the positive electrode during regeneration. The preferences for a suitable
mediator in the
halide oxidation are a standard redox potential of about 0.1V-0.4V more
positive than
the standard redox potential of the halate, the electrode reaction of the
mediator having a
high exchange current, the homogeneous reaction between the mediator and the
halide
being fast, the mediator couple not involving cationic species capable of
crossing the
membrane, etc. Chlorine is, for example, a mediator for iodate or iodide at
all pH levels
but chlorine evolution requires an electrocatalyst, for example, dimensionally
stable
anode (DSA) which can make this process more expensive than electro-oxidation-
disproportionation. Chlorine is a mediator for bromide oxidation into bromate
only in
neural and alkaline media.
[0322] Ozone is a suitable mediator for oxidation or a charge reaction, though
with less
than 50% energy efficiency for oxidizing halides into halates and perhalates
or
corresponding acids. This regeneration process can be performed in acidic
media by
electrolysis using a proton exchange membrane (PEM) electrolyzer or a similar
device.
The co-produced H2 can be used later as a reducer in the discharge unit 104
exemplarily
illustrated in FIG. 1, while the ozone reacts with the spent hydrogen halide
in a separate
vessel to yield the halic acid oxidant. In an embodiment, the ozone for
regeneration can
be produced by a gas discharge according to commercialized methods. Other
suitable
mediators comprise, for example, transition metal ion and their compounds such
as
negatively charged polyoxometallates to prevent their cross-over through the
cation
exchange membrane. In an embodiment, a direct electrolytic oxidation of
halides, for
example, bromide to bromate is performed, for example, with a Pb02, Ru02,
dimensionally stable anode (DSA) or a conductive diamond electrode.
[0323] Example 12: In an embodiment, the discharge unit 104 is a modified
version of
a polymer electrolyte fuel cell. A membrane electrode assembly (MEA) uncoated
on the
positive side is used in the discharge unit 104. The diffusion layer on the
positive side is
156
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
replaced with a hydrophilic porous carbon cloth. The flow field on the
positive side of a
carbon bipolar plate 202 exemplarily illustrated in FIG. 2, is of a double
serpentine type
but other types known in the arts of fuel cells and flow batteries are also
employed. In
another embodiment, the discharge unit 104 is equipped with an MEA coated on
the
positive side with a Pt-free and perfluorosulfonic acid (PFSA) free carbon
fiber layer
replacing a catalyst layer in the conventional polymer electrolyte membrane
fuel cell
(PEMFC), thereby reducing the ohmic resistance between the points where the
bromate
reduction occurs and the hydrogen electrode. In an embodiment, a grid with
interdigitated
millimeter deep channels in one direction and with thinner channels in the
perpendicular
direction can be used for the positive electrode flow field.
[0324] In another embodiment, for the positive electrode 205a exemplarily
illustrated in
FIG. 2, a hydrophilic porous electrode (HPE) replacing the hydrophobic gas
diffusion
layer in the conventional 5-layer proton exchange membrane (PEM)-membrane
electrode
assembly (MEA) design with or without a carbon-ionomer layer (CIL) coating on
the
positive side of the membrane is designed. Such an HPE can either be used as a
flow-
through with an inter-digitated or with a flow-by or with a parallel channel
flow field. A
pore diameter above 12L that is 18 i.tm is beneficial, and the layer thickness
or pore
length does not need to be much larger. A suitable channel width can be larger
than the
inter-channel spacing, and a parallel channel flow field with relatively short
channels is
longer than the ignition length, and shorter than the depletion length with a
low pressure
drop and a near stoichiometric flow rate. As used herein, the term "ignition
length" refers
to the distance from the opening of the channel where the current density on
the positive
electrode reaches 1/2 of its maximal value. In the case of bromate as the
aqueous multi-
electron oxidant (AMO), the current increase along the channel is due to
accumulation of
bromide and bromine and the resulting increase in the rate of the
disproportionation.
Also, as used herein, the term "depletion length" refers to the distance along
the channel
past the maximum current density point, where the current density decreases to
1/2 of its
maximal value. This decrease is due to the depletion of the AMO in the bulk of
the
solution as well as due to an increase in the diffusion boundary layer
thickness.
157
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0325] Suitable carbonaceous materials for the porous electrode are available
commercially. One suitable carbon cloth is, for example, pyrolysed PAN AvCarb
1071
HCB 80045-001 with about 350 i.tm thickness, about 7.5 i.tm fiber diameter,
about
19.3/cm warp, about 18.5/cm weft, and about 10-3 ohm=cm conductivity. A
thinner
unidirectional carbon fabric, for example, about 152 i.tm thickness is
available from Fibre
Glast Developments Corporation. Some suitable carbon cloth are, for example,
potential
hydrophilic carbon cloth with approximately 18 i.tm diameter for the
hydrophilic porous
electrode (HPE), commercial carbon cloth as thin as 700 i.tm, cloth made of
electrospun
carbon fibers as thin as 20 nm, Zoflex of Xilor, Inc., weaved carbon down to
400 um,
etc. Surface modification such as sulfonation of carbon can be used to improve
the
hydrophilicity.
[0326] A conventional bipolar stack polymer electrolyte membrane fuel cell
(PEMFC)
with a hydrophilic porous layer modification on the positive side of the
membrane
electrode assembly (MEA) and a Pt-free positive electrode layer is used. Since
the
aqueous multi-electron oxidant (AMO), in contrast to air, is ionically
conducting, shunt
currents in a bipolar stack have to be considered. Methods for minimizing
shunt currents
are known and include: increasing ionic resistance between the electrolytic
cells 200 in a
stack 300 exemplarily illustrated in FIG. 3, for example, by increasing the
length and
decreasing the width of the flow channels within the bipolar plates connecting
the
electrolytic cells 200, reducing the number of single electrolytic cells 200
in series,
decreasing the resistances of manifold and channel, increasing the power of
single
electrolytic cell 200, placing shunt resistors in the electrolyte paths, and
any combination
thereof. The operating temperature of the discharge unit 104 is between 0 C
and 100 C,
for example, between 10 C and 60 C.
[0327] Example 13: A steady-state one-dimensional model was developed for a
comproportionation-mediated discharge of bromate with a Nernstian hydrodynamic
boundary layer of a fixed thickness. Such a model is an adequate first-order
approximation for the discharge at the rotating disk and at channel flow
electrodes. For a
sufficiently high rate of the comproportionation reaction ensured by high
concentrations
158
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
of bromate and protons in bulk solution, there are three different regimes
determined by
the ratio diffusion to kinetic boundary layer thicknesses as exemplarily
illustrated in FIG.
12. The latter decreases as the disproportionation rate gets larger, for
example, at lower
pH and higher bulk aqueous multi-electron oxidant (AMO) concentration and it
is equal
to 1.5 i.tm in 50 % w/w HBr03.
Lo= (Dbromicle/5 kconCbromate)1/2 = 1.5 t.tm (60)
[0328] During electroreduction of the aqueous multi-electron oxidant (AMO)
such as
bromate mediated by homogeneous comproportionation when the diffusion boundary
layer is thin compared to the kinetic boundary layer, that is, at high flow or
stirring rates,
the intermediate bromide formed via electroreduction of the initial bromine
escapes the
hydrodynamic boundary layer before the intermediate bromide comproportionates
with
bromate to form more bromine near the electrode. In this non-ignition (normal)
regime
(not shown), the limiting current is the same as it would be in a solution
with only
bromine and no bromate present. When the diffusion boundary layer is thick
compared to
the kinetic boundary layer, that is, at low flow and/or rotation rates, the
intermediate
bromide has enough time to react with bromate near the electrode producing
more
bromine as exemplarily illustrated in FIG. 12, resulting in an ignition regime
with the
limiting current significantly exceeding the bromine limiting current found in
the non-
ignition regime. The limiting current in the ignition regime can be limited by
the rate of
comproportionation as exemplarily illustrated in FIGS. 12, 21A-21B or by the
mass-
transport of protons as exemplary illustrated in FIG. 25. The nature of the
limiting current
depends on the relative concentrations of acidic protons and bromate. The
behaviour
when the limiting current abnormally decreases with the rotation flow rate as
exemplarily
illustrated in FIGS. 12, 21A-21B contrasts that of other flow batteries and
fuel cells
which show a higher current and power upon increased flow rate, and such a
regime is
useful for practical applications since the regime allows for a high power at
low pumping
rates.
[0329] An additional confirmation of the comproportionation mechanism
disclosed
159
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
herein is obtained through a direct observation of a brown colored bromine in
a layer near
the rotating disk electrode (RDE). The brown cloud (not shown) attached to the
electrode
is the intermediate bromine formed during the comproportionation of bromate
with
electro-generated bromide as in equation (2). The current is negative that is
cathodic. The
visible thickness of the colored layer and the measured current at constant
potential
decreases with the electrode rotation rate (not shown).
[0330] In the intermediate regime, the limiting current decreases with flow
and/or
rotation rate as exemplarily illustrated in FIG. 12, due to the escape of the
intermediate
bromine. The ignition regime observed at low mass-transport rates is
particularly
interesting for practical applications as it affords a high generated peak
electric power
even on a smooth carbon electrode, that is, over 0.1 A/cm2 and 0.1 W/cm2, as
exemplarily
illustrated in FIG. 13, at low consumed pumping power in contrast to other
fuel cells and
redox flow batteries. The fast kinetics of the bromine/bromide electrode
reaction assures
that the energy efficiency of the discharge unit 104 at high power is over
60%.
[0331] Example 14: The power required for an on-board orthogonal ion migration
across laminar flow (OIMALF) is calculated. The balance of charge in the
OIMALF
reactor 1501 and the discharge unit 104 is exemplarily illustrated in FIG. 19.
The
matching ratio of currents in charges per unit time through the OIMALF reactor
1501 and
the discharge unit 104 are (1+x+z+y-w) / (6+x+z-y) =1. In the simplest case, x
= y = z =
w = 1, thus the charge ratio is 1: 6. Assuming the single cell voltage
produced in the
discharge unit 104 as 1.0 V, the current density in the OIMALF reactor 1501 as
0.5
A/cm2, and the areal cell resistance as 0.15 cm2, which is three times the
areal
resistance of 601.tm thick Nafion 112, we obtain 0.5x0.195/1.0 = 10%,
justifying a small
sacrifice in energy efficiency while making a significant improvement in the
safety on the
on-board system and the complete energy cycle.
[0332] Example 15: The energy and power density of the of the on-board
orthogonal
ion migration across laminar flow (OIMALF) discharge system 101: The Toyota
RAV4
EV of Toyota Jidosha Kabushiki Kaisha TA Toyota Motor Corporation is chosen as
an
160
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
example of a sport utility vehicle to illustrate the capabilities of the
discharge unit 104 with
the on-board OIMALF reactor 1501. In order to compare H2-Li aqueous multi-
electron
oxidant (AMO) on-board discharge unit 104 with a lithium-ion battery system,
the Toyota
RAV4 with rated power of 174 hp, that is about 130 kW and target driving
range of 311
miles or 500 Km is selected. Considering an experimental value of 40 mA/cm2 at
0.9V
for a smooth carbon electrode in about 5M LiBr03+50% w H3PO4+1 mM LiBr, and
multiplying it by a roughness factor of 25 for a porous carbon electrode and a
factor of 2
for a near-saturated LiBrO3 solution and without considering additional
acceleration due
to a high proton concentration in the on-board OIMALF reactor 1501, a current
of 2
A/cm2 for a smooth carbon electrode in case of 10M bromate and > 0.5M acid, a
cell
voltage with an IR drop of 0.8 V and 0.05 S2 cm2 areal resistance, a cell
power of 1.6
W/cm2, and discharge energy efficiency of 61% with respect to standard
electrode
potential bromate/bromide, that is, 1.48 V are obtained for the discharge unit
104 with the
on-board OIMALF reactor 1501. Using the same area-to-volume conversion factor
as the
fuel cell stack in Ballard's HD6 0.5 W/cm2 to 371 W/kg, a power density of 1.2
kW/kg
and a weight of 108 kg is obtained to ensure the needed 130 kW of the rated
power for
the on-board discharge unit 104.
[0333] Since automotive fuel cell stacks are usually designed for 130 V, the
required
number of cells in the discharge unit 104 is equal to 130 V/0.9V= 144 cells.
This
translates for the 130 kW/130V=1kA current into lkA/2 A/cm2 =500 cm2 total
area of all
electrodes in the fuel cell stack and to 500 cm2/144= 3.46 cm2 z 2x2 cm2
membrane
electrode assembly (MEA), which is reasonable considering the slower diffusion
and the
shorter depletion length expected for an aqueous multi-electron oxidant (AMO)
compared to 02 in gaseous air.
[0334] The weight of the on-board orthogonal ion migration across laminar flow
(OIMALF) reactor 1501 can be estimated as follows. The stoichiometry of the
OIMALF
process requires about 1/6 of the current produced in the discharge unit 104
which is
lkA/6= 167A. Assuming that the OIMALF reactor 1501 has one third of the
current-to-
weight ratio, for example, 1/3x1000A/108kg= 3.08A/kg as the discharge flow
battery, we
161
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
obtain 167A/3.08A/kg = 54.2 kg for the weight of the on-board OIMALF reactor
1501.
The weight of discharge system 101 obtained by combining the weights of the
discharge
unit 104 and the OIMALF reactor 1501 is 108+54.2=162.2 kg and the power ¨to-
weight
ratio is 130kW/162.2kg = 800 W/kg which compares favorably with Li ion battery
with
power density, for example, 100 W/kg at 1C rate and polymer electrolyte
membrane fuel
cell (PEMFC) stack with power density, for example, 100 W/kg at 50%
efficiency. The
weight of the power-generating discharge system 101 needs to be combined with
the
weight of the reagents that determine the on-board energy, for example, the
driving
range.
[0335] Using the data for Toyota RAV4 EV with 166 km driving range and 41.8
kWh
battery, a 500 km driving range would require 126 kWh of energy. For a single
cell
voltage of 0.9V, this translates into 140 kAh or 5.22 kmoles of electrons.
This in turn
requires 2.61 kmole = 5.22 kg of H2 or 104.4 kg of 5% w H2 storage system. The
equivalent amount of LiBrO3 required is 870 moles or 90.8 kg of solid or 181.6
kg or
50% w solution, that is 78% of saturated solution at 20 C. The combined
weight of the
oxidants and the discharge system 101 for 500 km is 181.6+104.4+162.2 = 448
kg, that is
0.896 km/kg which compares favorably at 2.05 times higher at the system level
with 380
kg of Toyota RAV4 EV's battery pack that provides only 166 km range, that is
0.437
km/kg at a significantly higher upfront cost.
[0336] The high solubility of LiBrO3 at 64% w at 20 C and the multi-electron
oxidizing nature lead to equivalent molal concentration of electrons of
13.27M*6z 80 N
which is more than 3 times higher than that of solid LiFePO4 used in a flow
suspension
battery under development by 24M, a Massachusetts based start-up. At the tank
level, the
combination of 5% w/w H2 with 64% LiBrO3 gives 487 Ah/kg, that is 521 Wh/kg
whereas the LiFePO4 + C6 battery gives 117 Ah/kg, that is 384 Wh/kg at the
reagent level
and 31 Ah/kg, that is 100 Wh/kg at the cell level. The discharge system 101
with the on-
board orthogonal ion migration across laminar flow (OIMALF) reactor 1501
reduces the
energy density of the discharge system 101 by approximately 10% and the
efficiency of
the discharge system 101 to 80% from 90%. However, in many automotive
applications,
162
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
this new performance metrics is acceptable and the improved safety fully
justifies a small
decrease in the system energy density. Furthermore, the possibility to use
higher acid
concentrations during discharge allows for the discharge flow battery to
produce a higher
power thus reducing the system power density dilution and lowering the system
cost.
[0337] Example 16: Lithium bromate chemistry with a 3-(N-morpholino)
propanesulfonic acid (MOPS) buffer: In this example, lithium bromate chemistry
that
follows a cyclic or cascade rather than a batch mode is illustrated. In an
embodiment, that
is, in the aqueous multi-electron oxidant (AMO)-on-negative mode of operation,
the
regenerated solution or the discharge fluid is cycled between a negative
compartment and
a negative electrode 1702 of the SD flow cell 1700 where hydrogen evolution
occurs with
a resulting increase in the pH of the regenerated solution.
Li-MOPS+ Br2 + H20 = 5/3LiBr + 1/3 LiBrO3 + H-MOPS (61)
[0338] Experimental data demonstrating the feasibility of reaction (61) is
exemplarily
illustrated in FIG. 26.
[0339] The negative electrode 1702 is configured to support the hydrogen
evolution
reaction by employing a hydrogen-evolution catalyst, for example Pt or other
platinoid,
using porous carbon flow-through or flow-by support or any combination
thereof, etc. At
the same time the negative electrode 1702 is configured to prevent the
electroreduction of
bromate, if the aqueous multi-electron oxidant (AMO)-on-negative mode of
operation is
used. The hydrogen gas produced in (27) is separated from the liquid oxidant
solution in
the separation reactor 1010 and collected for future use, for example, in a
discharge
system 101. The liquid comprising LiBr and LiA is further carried over to the
positive
electrode 1703 where electrooxidation of bromide followed by bromine
disproportionation occurs:
LiBr + e- = 1/2Br2+ Li + (62)
163
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
1/2Br2+ LiA + 1/2H20 = 5/6 LiBr +1/6 LiBrO3 + HA (63)
[0340] Upon the completion of the first SD cycle only up to 1/6 of the
original bromide
can be converted to bromate. Thus, further cycles or cascade of splitting-
disproportionation (SD) is used.
[0341] Example 17: Electric energy cycle with a LiBrO3 regeneration using an
anionic
buffer base and the aqueous multi-electron oxidant (AMO)-on-negative mode of
operation. Lithium bromate and bromide are well suited for the energy cycle
disclosed
herein due to their high aqueous solubilities. Phosphate buffer is utilized
due to the
appropriate pH and chemical compatibility with other ingredients. However, the
intermediate acid form of the phosphate buffer H2PO4- produced in the
disproportionation
is not the final acid form H3PO4 used in the discharge unit 104. The
conversion of the
intermediate acid form of phosphate into the final acid form requires extra
expenses of
chemical or energy which may not be the preferred mode under on-board
acidification
scenarios. Also, the possibility of the formation of a poorly soluble Li3PO4
in the case of
phosphate buffer, limits the flexibility of the design of the regeneration
system 106. For
these reasons other buffers are considered.
[0342] For purposes of illustration, this example refers to a Good's buffer
HA, for
example, Me2NCH2CH2S03H or 3-(N-morpholino) propanesulfonic acid (MOPS) with
pKa= 7.2 or 4-(N-morpholino) butanesulfonic acid (MOBS) with pKa = 7.6
available
from Sigma-Aldrich. One advantage of such buffers is that in their acidic form
HMe2N-
R-503H, they can perform the function of the strong extra acid in the ignition
mode of
discharge, eliminating the need for an additional chemical component. Another
advantage
is their anionic state which reduced their cross-over through a cation-
exchange
membrane. Two commercially available compounds are of particular interest. The
propyl
version, MOPS, is inexpensive at about 390 $/kg since MOPS is easily produced
by
reaction of morpholine and propane sultone, both being readily available, but
MOPS has
a pKa of 7.2 which is within the suitable range. The use of Li-MOPS for
bromine
disproportionation is exemplarily illustrated in FIG. 26. The butane version,
MOBS has a
164
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
higher pKa=7.6, requiring a shorter regeneration time, but has a significantly
higher cost
of about 16,000 $/kg due to the higher cost and/or difficult synthesis of the
butane sultone
precursor.
[0343] The energy and matter cycle starting with neutral discharge fluid
comprising
LiBr and the buffer acid HA is disclosed herein. In the regeneration system
106, the
neutral discharge fluid is first converted into alkaline discharge by passing
thru the
negative electrode compartment of the SD reactor 1502 configured for the
aqueous multi-
electron oxidant (AMO)-on-negative mode of operation, producing H2 and
alkaline
discharge fluid comprising LiBr and LiA.
LiBr + HA+le-+ Li+= LiBr + LiA + 1/2 H2 on the negative electrode (64)
[0344] In the separation reactor 1010 the H2 is separated from the alkaline
discharge
fluid and the latter is pumped into the positive electrode compartment wherein
bromide
electrooxidation (65) and disproportionation (66) take place:
LiBr + LiA -1e-+ = 1/2 Br2 + LiA + Li + (65)
1/2 Br2 + LiA + 1/2H20 = 5/6 LiBr + 1/6 LiBrO3 + HA (66)
while the counter cation such as Li + released at the positive electrode 1703
in (65) moves
through the cation-exchange membrane to the negative electrode 1702, wherein
electroreduction and neutralization shown in (64) take place.
[0345] The partially regenerated neutral oxidant fluid formed in (66) at the
positive
electrode 1703 is transferred again to the negative electrode compartment
where the
partially regenerated neutral oxidant fluid enters a new cycle of alkalization
(64), splitting
(65), and disproportionation (66). In this example, the negative electrode
1702 is
configured for the aqueous multi-electron oxidant (AMO)-on-negative mode of
operation
using a cation conductive layer and an electron conductive layer 1702b which
prevents
165
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
the electroreduction of the AMO such as bromate on the negative electrode
1702. The
cycle is continued until the desired ratio of bromate to all bromine species
in the neutral
oxidant fluid reaches a predetermined value, for example 0.95. This
regenerated neutral
oxidant fluid and hydrogen are stored in the regeneration system 106 until
they are
transferred into a discharge system 101 such as in an electric vehicle.
[0346] In the discharge system 101, the neutral oxidant fluid is converted
first into
acidic oxidant fluid using, for example, an orthogonal ion migration across
laminar flow
(OIMALF) reactor 1501. The chemical transformations in the OIMALF reactor 1501
can
be illustrated by the following examples:
On the positive electrode: 1/2 H2 ¨ e- = H+
(67)
In the central channel: LiBrO3 + HA +HBr = HBrO3 + HA +LiBr- (68)
On the negative electrode: HBrO3 + HA + 6H+ + 6e--= HBr + 3H20 + HA (69)
wherein reaction (68) represents the ion exchange process such as the
orthogonal ion
migration across laminar flow (OIMALF). In an embodiment, the H2 produced on
the
negative electrode 205b in (68) is consumed on the positive electrode 205a in
(67).
[0347] The acidic oxidant fluid produced in reaction (68) is supplied to the
positive
electrode 205a of the discharge cell 104a wherein the discharge proceeds via
the
electroreduction (70)¨comproportionation (71) cycle:
2.5 Br2 +e- = 5Br- (70)
5Br- + HBrO3 + HA +5H+- = 3Br2 +3H20+ HA (71)
while hydrogen electrooxidation on the negative electrode 205b supplies the
protons
consumed in (71):
2.5 H2 ¨ e- = 5H+ (72)
166
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
[0348] The reaction (71) produces more Br2 than reaction (70) consumes for the
same
amount of bromide. This feature leads to the possibility of the ignition
regime where the
electrode current increases as the convection rate decreases. A useful feature
of the ED-
cycle (70)-(71) is that the use of high acid concentration is not necessary
for the cycle to
proceed in the ignition mode. In the case of highly soluble LiBr03, the ratio
[H ]/[Br03-1
as low as 0.05 may suffice. The low acid concentration in the acidic discharge
fluid is
critical for the practical applications of the disclosed technology since it
assures a low
rate of the decomposition reaction (73) which competes with the desired
comproportionation reaction (71):
2HBrO3 = Br2 + 2.502 + H20 (73)
[0349] The gross equation for the chemical process in the discharge unit 104
is:
HBrO3 + 3H2+HA= HBr+ 3H20+HA (74)
and the gross equation for the chemical process in the discharge system 101
is:
LiBr03+HA + 3H2 = LiBr +HA+ 3H20 (75)
[0350] The neutral discharge fluid produced in (74) is used to start a new
energy cycle
with process (64) in the regeneration system 106.
[0351] Example 18: Solar regeneration of LiBrO3 from LiBr using an anionic
buffer
base and the aqueous multi-electron oxidant (AMO)-on-negative mode of
operation:
Unlike the hydrogen economy scenario, where the poor efficiency of solar water
splitting,
either photoelectrochemically, photothermally or some other way, prevents a
large-scale
use of sunlight as the primary energy source, the energy cycle disclosed
herein employs
splitting of a hydrogen halide, for example, HBr as the main input step for
external
energy. Energy and cost efficient routes to the reaction 2HBr = H2 Br2 using
solar
power, particularly, photoelectrolysis, have been reported or are known in the
art. For
167
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
example, a method for decomposing a solution of HBr using a platinum cathode
and
platinum-coated n-type amorphous silicon photo-anode and a red light and
approximately
0.5% conversion efficiency is known in the art. Also, a system with a higher
efficiency,
for example, approximately 8%, that utilizes a p-GaInP2(Pt)/GaAs
photoelectrochemical/photovoltaic device is also known in the art.
[0352] In the solar regeneration example disclosed herein, splitting of one or
more
forms of the discharge fluid into hydrogen and bromine is performed via
photoelectrolysis. In an embodiment, the neutral discharge fluid comprising
LiBr and the
buffer acid HA is first converted into alkaline discharge fluid by passing the
neutral
discharge fluid through the negative electrode compartment of a
photoelectrolysis-
disproportionation reactor (not shown) configured for the aqueous multi-
electron oxidant
(AMO) on-negative mode of operation, producing H2 and alkaline discharge fluid
comprising LiBr and LiA.
LiBr + HA +1e-+ Li + = LiBr + LiA + 1/2 H2 on the negative electrode (76)
[0353] In the separation reactor 1010 H2 is separated from the alkaline
discharge fluid
and the latter is pumped into the positive electrode compartment wherein
bromide
electrooxidation (77) and disproportionation (78) take place:
LiBr + LiA -1e-+ = 1/2 Br2 + LiA + Li + (77)
1/2 Br2 + LiA + 1/2H20 = 5/6 LiBr + 1/6 LiBrO3 + HA (78)
while the counter cation such as Li + released at the positive electrode 1703
in (77) moves
through the cation-exchange membrane 1704 to the negative electrode 1702,
wherein
electroreduction and neutralization shown in (76) take place.
[0354] The partially regenerated neutral oxidant fluid formed in (78) at the
positive
electrode 1703 goes again to the negative electrode compartment where it
enters a new
168
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
cycle of alkalization (76), splitting (77), and disproportionation (78). In
this example, the
negative electrode 1702 is configured for the aqueous multi-electron oxidant
(AMO)-on-
negative mode of operation using a cation- and electron-conductive layer which
prevents
the electroreduction of the AMO such as bromate on the negative electrode
1902. The
cycle is continued until the desired ratio of bromate to all bromine species
in the neutral
oxidant fluid reaches a predetermined value, for example 0.95. This
regenerated neutral
oxidant fluid and hydrogen are stored in the regeneration system 106 until
they are
transferred into a discharge system 101 such as in an electric vehicle.
[0355] Example 19: Decomposition of bromate in acid: The discharge process
disclosed
herein faces a TRIZ contradiction between the discharge cell power and the
stability of
the acidic aqueous multi-electron oxidant (AMO) solution, that is, upon
increasing the
acid concentration in the acidic oxidant fluid, the electroreduction of the
AMO is
facilitated while the stability of the AMO deteriorates. The existence of a
composition
meeting both requirements for a high discharge power and stability cannot be
predicted
theoretically. Experimental studies were conducted to find a composition of
acidic
discharge fluid which meets both requirements for stability and for discharge
power.
Solutions of sulfuric acids of various compositions were prepared by mixing
98% w/w
H2SO4 and water to 5 mL volumes. Noticeable heating was observed in all cases.
While
the solutions were still hot an excess of solid LiBrO3 was added to each of
the solutions.
The experimental observations of decomposition of bromate introduced as an
excess of
solid LiBrO3 in various acidic solutions are summarized in Table 3 below:
Table 3:
H20:
H2S 04 H2504 02 Br2 Final
H2504 H2504 M
discharge
w% density evolution evolution color
v/v
dark
5:5 65 1.55 10.3 noticeable vigorous
brown
light
6:4 55 1.45 8.1 starts first starts later
brown
169
CA 02929694 2016-02-08
WO 2015/026393 PCT/US2014/019170
6.25: dark
52 1.415 7.5 noticeable slow
3.75 yellow
medium
7:3 44 1.34 6.0 noticeable slower
yellow
light
8:2 31.5 1.22 4.0 slow limited
yellow
not very light
9:1 17 1.17 1.9
observed limited yellow
[0356] Two parallel decomposition pathways were observed: one leading to
oxygen
evolution or production and the other leading to bromine evolution or
production. The
oxygen evolution pathway dominates at the lower acidities which are of
interest to the
disclosed technology. The data disclosed in Table 3 suggest that acidic
bromate solutions
are sufficiently stable to be used in a discharge system 101 when the
concentration of a
strong acid is below 4M. Furthermore, as exemplarily illustrated in FIGS. 12-
13, FIGS.
21A-21B, and FIG. 25 a 2M concentration of acidic protons is sufficient to
provide a
practically useful discharge power when bromate is used as the aqueous multi-
electron
oxidant (AMO) as disclosed in Example 13. Hence, the acidification process can
be
performed off-board and a week's supply of the acidic oxidant fluid can be
stored on-
board. The concentrated HBrO3 stored on-board is a stable solution yet still
capable of
discharge with a high power.
[0357] Example 20: FIG. 24 exemplarily illustrates a graphical representation
showing
background-subtracted limiting currents in mA/cm2 of bromide electrooxidation-
disproportionation on a glassy carbon rotating disk electrode in a 0.5M sodium
phosphate
buffer at various rotation rates in rpm. The sodium phosphate buffer has a pH
of 8.0 and
comprises about 5mM NaBr. The dotted line in FIG. 24 represents the calculated
Levich
plot for the diffusion limited current of bromide.
[0358] Electrooxidation-disproportionation of bromide on a glassy carbon
rotating disk
electrode: An experiment to demonstrate the feasibility of the
electrooxidation-
170
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
disproportionation step in the regeneration process using a phosphate buffer
which has a
suitable pH and to determine the time scale of this process was conducted. In
this
experiment, a 3-compartment glass cell equipped with a glassy carbon rotating
disk
electrode of Pine Instruments with about 5.0 mm diameter, a Ag/AgC1 reference
electrode in 3.0 M NaC1 connected via a Luggin capillary, and a Pt counter
electrode
were used. The background electrolyte was 0.5M sodium phosphate buffer with a
Ph of
about 8.0 procured from Teknova to which about 5mM NaBr was added. The
background-subtracted limiting currents at +1.30 Vv Ag/AgC1 obtained in this
experiment are exemplarily illustrated in FIG. 24. At high rotation rates, the
limiting
current follows the Levich behavior that is, the limiting current increases
linearly with the
square root of the rotation rate, as expected for a diffusion-limited process.
At low
rotation rates a positive deviation from the Levich behavior is observed which
agrees
with the occurrence of the disproportionation (16). The time scale of the
disproportionation in this buffer can be estimated as the diffusion time
across the
diffusion boundary layer at 900 rpm which is a characteristic point of
deviation.
According to the Levich equation, the thickness of the diffusion boundary
layer at this
rotation rate in water is ca. 20 iim, which translates via Fick's 2nd Law into
the diffusion
time of 0.5s. Thus, 0.5s is the characteristic time of the disproportionation
of bromine in
0.5 M sodium phosphate buffer. This time-scale is well -suited for a
commercial
regeneration process.
[0359] Example 21: FIG. 25 exemplarily illustrates a staircase cyclic
voltammetry on a
glassy carbon rotating disk electrode of about 0.283 cm2 area in a 2 hour aged
solution
containing 2.0 M H2SO4 and approximately 5M LiBr03. The electrode rotation
rates and
scan directions are exemplarily illustrated near the curves. The reference
electrode is
Ag/AgC1 in 3 M NaCl.
[0360] Electroreduction of bromate in acid on a carbon rotating disk
electrode: An
experiment was conducted to determine practically achievable limits of power
per
electrode area upon discharge imposed by the aforementioned TRIZ contradiction
between the stability and the limiting current in the acidic oxidant fluid.
Although
171
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
numerous compositions were tested, only the data for a 2.0 M H2SO4 solution
are shown
in FIG. 25 since this acid concentration was found to be near-optimal. In
order to
minimize the decomposition of the aqueous multi-electron oxidant (AMO) before
the
measurements, a solid LiBrO3 was added to the acid solution in the
electrochemical cell.
As noticed in previous experiments, in this aged solution the more positive
wave
attributed to the electroreduction of bromine produced via the
comproportionation is
followed by a more negative wave attributed to the electroreduction of a
bromate
decomposition intermediate tentatively, hypobromite. Only the more positive
wave is
observed in fresh solutions. The limiting currents of both waves seem to be
controlled by
the concentration of acidic protons rather than that of bromate since the
latter is present in
a large stoichiometric excess. This also explains why the decrease in the
limiting current
with the rotation rate similar to the one shown in FIG. 25 is not observed.
The solutions
become yellow during such experiments in a batch cell due to the
comproportionation of
product bromide with bromate. As Example 11 shows, in the absence of bromide
the
stability of bromate in acids is better. The problem of the parasitic bromate
comproportionation with bromide is not present in the discharge flow cells
disclosed
herein elsewhere.
[0361] Example 22: Disproportionation of bromine in Li-3-(N-morpholino)
propanesulfonic acid (MOPS) buffer: 1.0 M Li-MOPS solution was prepared from H-
MOPS and Li0HxH20. The pH of the resulting solution is 7.2 and the density is
1.11
g/mL. 2 moles of this solution (2mL) was mixed with lmmole of Br2 which is
about 160
mg and about 52 ilL. One week later, the solution composition was analyzed
using
negative mode electrospray ionization (ESI) - mass spectrometry (MS). A sample
of
unreacted Li-MOPS was used as a control. The expected chemical reaction is
given by:
1/2 Br2 + Li-MOPS + 1/2H20 = 5/6 LiBr + 1/6 LiBrO3 + H-MOPS
[0362] FIG. 26 exemplarily illustrates an electrospray ionization (ESI)-mass
spectrometry (MS) spectrum, showing experimental data demonstrating the
feasibility of
a regeneration process. The ESI-MS spectrum exemplarily illustrated in FIG. 26
confirms
172
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
the formation of bromate and bromide.
Sample # Composition
1 '1/2 Br2 + Li-MOPS + 1/2H20 = 5/6 LiBr + 1/6 LiBrO3 + H-
MOPS
2 Li-MOPS only
3 Li-MOPS + NaBr
4 Li-MOPS+ Br2 in excess, red liquid
[0363] Similar experiments were carried out using a lithium-phosphate buffer.
0.2 mole
of Li0HxH20 (8.392g) was dissolved in 100 mL of water to which 0.1 mole of
H3PO4 of
about 6.22 mL of 80% w was added. A white precipitate was formed due to the
following reaction:
2Li0HxH20 + H3PO4 = 3H20 + Li2HPO4 (= 1/2Li3PO4 ,I, +1/2LiH2PO4)
[0364] 10 mL, that is, 0.010 moles of Li2HPO4 equivalent of the resulting
white slurry
was sampled into a separate vial and treated with 0.0050 of bromine of about
0.25 mL.
The following reaction:
Li3PO4 ,I, + LiH2PO4 + Br2 + H20 = 5/3 LiBr + 1/3 LiBrO3 + 2LiH2PO4
proceeds even at 60 C which is too slow for practical applications.
[0365] The foregoing examples have been provided merely for the purpose of
explanation and are in no way to be construed as limiting of the present
invention
disclosed herein. While the invention has been described with reference to
various
embodiments, it is understood that the words, which have been used herein, are
words of
description and illustration, rather than words of limitation. Further,
although the
invention has been described herein with reference to particular means,
materials, and
embodiments, the invention is not intended to be limited to the particulars
disclosed
173
CA 02929694 2016-02-08
WO 2015/026393
PCT/US2014/019170
herein; rather, the invention extends to all functionally equivalent
structures, methods and
uses, such as are within the scope of the appended claims. Those skilled in
the art, having
the benefit of the teachings of this specification, may affect numerous
modifications
thereto and changes may be made without departing from the scope and spirit of
the
invention in its aspects.
174