Note: Descriptions are shown in the official language in which they were submitted.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
1
2,3-DIHYDROPORPHINE DERIVATIVES, PROCESSES FOR THEIR PREPARATION, AND PHARMA-
CEUTICAL COMPOSITIONS CONTAINING THEM
Technical field
The present invention relates to compounds of formula 3 or salts thereof,
processes
for the preparation of compounds of formula 3 or salts thereof, pharmaceutical
compositions comprising compounds of formula 3 or salts thereof, the use of
compounds of formula 3 or salts thereof as phototherapeutic or photodiagnostic
agents, and methods of treatment using compounds of formula 3 or salts
thereof.
Background art
Photodynamic therapy (PDT) is a known treatment that uses light to destroy,
for
example, cancer tissue. Cytoluminescent therapy (CLI) is a form of
photodynamic
therapy. In both photodynamic therapy and cytoluminescent therapy, a
photosensitizer is administered to a patient, generally orally or
intravenously. The
photosensitizer collects selectively in cancer tissue and, when exposed to
light,
becomes activated, releasing a highly energized, free radical form of oxygen
known
as singlet oxygen. Singlet oxygen destroys cancer cells from the inside out,
while
leaving normal tissues largely unaffected. The administered photosensitizer
can be
exposed to light and activated internally using fibre-optic catheters or
endoscopes
inserted into the body to bring the light directly to the seat of the tumour
or
externally using light of higher wavelengths, which allows a greater depth of
penetration into the body.
Most known photosensitizers have mayor drawbacks, for example, they may be
difficult to prepare and purify, or they may only accurnulate slowly in
tumours. For
example, Russian patent RU-2183956 discloses photosensitizers based on a
mixture
of alkali metal salts, chlorine-e6, purpurine-5 and purpurine-1 8, which is
obtained
by extracting Spirulina biomass. However, the photosensitizers disclosed in RU-
2183956 have a low selectivity for tumour tissues, a high toxicity to normal
organs
and tissues, and a low therapeutic photoactivity in tumour cells. Moreover,
they are
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
2
chemically and photochemically unstable, but are only slowly metabolised and
cleared from normal tissues.
It is therefore an object of the present invention to provide photosensitizers
with
certain desired physical, chemical, photophysical and biological properties,
such as
high selectivity for tumour tissue, optimum speed of accumulation in tumour
tissue,
rapid clearance from normal tissue, slow clearance from tumour tissue, high
photodynamic activity, low tendency to induce photosensitivity, low
cytotoxicity
towards normal tissue, homogeneity and chemical stability of inedicinal forms
during storage, and ease of preparation and purification of industrial
quantities.
The inventors of the present invention have investigated the compound of
formula
1, 18-carboxy-20-(carboxymethyl)-8-ethenyl-13-ethyl-2,3-dihydro-3,7,12,17-
tetramethyl-21 H,23H-porphine-2-propanoic acid, which is also known as
95 phytochlorin or chlorine-e6, and derivatives and metal complexes thereof.
~ ~ t \
NH
N NH
---u~õ \ \
HOOC COOH
COOH
The inventors of the present invention have further developed a process for
the
preparation of derivatives and metal complexes of chlorine-e6, which is simple
and
effective, and provides the derivatives and metal complexes without residual
toxic
reagents.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
3
Summary of the invention
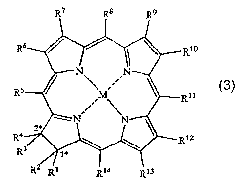
A first aspect of the present invention is a compound of formula 3
R7 R8 R9
R6 Rio
O N N
' ,
R5 M R"
:
N ' N 3
*
R4 2 ~ R12
R3
R2 Ri R14 R13
or a salt thereof,
wherein
M is a metal atom in the M(II) oxidation state, a metal halide or a metal
oxide, where the metal is Ca, Ti, V, Nb, Cr, Mo, Mn, Tc, Ru, Co, Rh, Ni, Pd,
Pt, Ag,
Au, Zn, Cd, Hg, Al, Ga, In, Ge, Pb or a lanthanide, or M is SiR2 where R is a
Ct-Ca
saturated or unsaturated alkyl group,
each R', R2, R3, R4, Rs, R6, R7, Rs, R9, Rlo, R", R12, R13 and R'4 is
independently hydrogen, (CH)õ-CHO, (CH)õ-CO2R15 or a Cl-C6 saturated or
unsaturated alkyl group optionally substituted with one or more of -OH and -
NH2,
n is 0, 1, 2 or 3, and
each R75 is independently hydrogen, lithium, sodium, potassium, magnesium,
calcium, a C1-C6 saturated or unsaturated alkyl group optionally substituted
with one
or more of -OH and -NHZ, or a naturally occurring amino acid.
The metal halide may be a metal fluoride, chloride, bromide, iodide or a
mixture
thereof. Preferably M is Zn, Cd, Ca, Mn, Au or Co. More preferably M is Zn.
Preferably the compound is immobilized on a protein, a polypeptide, a polymer
or
activated charcoal.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
4
A second aspect of the present invention is a compound of formula 3
R7 R8 R9
R6 ~ =~ t \ RiD
N \
/ N ~
11
R5 M R1i
;
: \
-N N 3
R4 2* gi2
R3 1*
R2 Rt Ri4 R13
or a salt thereof,
wherein
M is a metal atom in the M(II) oxidation state, a metal halide, a metal oxide
or a silicon with two axial substituents,
each R', R2, R3 > R 4, R5, R6, R7 > Raa R9, R10> R"> R12, R73 and R 14 is
independently hydrogen, (CH).-CHO, (CH2)õ-COZR75 or a C1-C6 saturated or
unsaturated alkyl group optionally substituted with one or more of -OH and -
NHZ,
n is 0, 1, 2 or 3,
each R15 is independently hydrogen, lithium, sodium, potassium, magnesium,
calcium, a Ct-C6 saturated or unsaturated alkyl group optionally substituted
with one
or more of -OH and -NH2, or a naturally occurring amino acid, and
wherein the compound is immobilized on a protein, a polypeptide, a polymer
or activated charcoal.
The metal halide may be a metal fluoride, chloride, bromide, iodide or a
mixture
thereof. Preferably M is Mg, Ca, Ti, V, Nb, Cr, Mo, Mn, Tc, Fe, Ru, Co, Rh,
Ni,
Pd, Pt, Cu, Ag, Au, Zn, Cd, Hg, Al, Ga, In, Ge, Sn, Pb, a lanthanide or SiR2
where
R is a C,-Cg saturated or unsaturated alkyl group. More preferably M is Mg,
Ca, Ti,
V, Nb, Cr, Mo, Mn, Tc, Fe, Ru, Co, Rh, Ni, Pd, Pt, Cu, Ag, Au, Zn, Cd, Hg, Al,
Ga,
In, Ge, Sn, Pb or a lanthanide. Even more preferably M is Zn, Cu, Cd, Ca, Mn,
Au
or Co. Even more preferably M is Zn.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
The compound of the first aspect of the present invention is preferably
immobilized
on a protein, a polypeptide, a polymer or activated charcoal. The compound of
the
second aspect of the present invention is immobilized on a protein, a
polypeptide, a
polymer or activated charcoal. Either way, preferably the compound is
immobilized
5 in monomer form. Preferably the protein is serum humane albumin (SHA) or
bovine serum albumin (BSA), more preferably serum humane albumin (SHA).
Preferably the polypeptide is a low molecular weight polypeptide, more
preferably
polylysine or polyasparagine. Preferably the polymer is polyvinylpyrrolidone
(PVP).
For the purposes of this invention, a"saltaa of a compound of the present
invention
is formed between a carboxylic acid functionality of a compound of the present
invention and a suitable cation. Suitable cations include, but are not limited
to
lithium, sodium, potassium, magnesium, calcium and ammonium. Preferably the
salt is a pharmaceutically acceptable salt. The salt may be a mono-, di- or
tri-salt.
Preferably the salt is a mono- or di-lithium, sodium, potassium, magnesium,
calcium
or ammonium salt. More preferably the salt is a mono- or di-sodium salt.
Preferably each Rla R2f R3 > R > RSa R6 , R'a R8, R9, R10a R" f R'Z a R'3 and
R'4 is
independently hydrogen, methyl, ethyl, propyl, allyl, CO2H, CHZCO2H or
(CHDZCOZH. Preferably R' and R3 are hydrogen. Preferably R5, Rg and R" are
hydrogen.
Preferably Rt5 is hydrogen, sodium, a C,-C6 saturated or unsaturated alkyl
group or a
naturally occurring amino acid, such as aspartic acid or lysine.
The compound of formula 3 has two chiral centres, 1* and 2*, and can therefore
exist in the form of four stereoisomers. The present invention embraces all of
these
stereoisomers and mixtures thereof. Mixtures of the steteoisomers can be
resolved
by conventional methods, for example, chiral chromatography, fractional
recrystallisation, derivatisation to form diastereomers and subsequent
resolution,
and resolution using enzymes. Alternatively, the compound of formula 3 can be
prepared directly in substantially enantiomerically pure form by
enantioselective or
stereoselective synthesis.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
6
The compound of formula 3 preferably comprises at least 95% of one enantiomer,
preferably at least 98% of one enantiomer, and more preferably at least 99% of
one
enantiomer. Preferably the compound of formula 3 is substantially
enantiomerically
pure, which is defined for the purposes of the present invention as meaning
that the
compound of formula 3 comprises at least 99% of one enantiomer.
Preferably R' and R3 are hydrogen, and R' is in the down-configuration and R3
is in
the up-configuration in formula 3 as shown. More preferably R' and R3 are
hydrogen, R2 is (CHZ)ZCO2H, R' is COZH, and chiral centres 1* and 2* are in
the
(S)-configuration.
In the most preferred embodiment, the compound of the present invention is of
formula 2
/ N N
M
. ~' \
~-N N 2
-1t-~~~'
HOOC COOH
COOH
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
7
A third aspect of the present invention is a process for the preparation of a
compound of formula 3 or a salt thereof, comprising the step of mixing a
compound of formula 4
R7 R$ R?
Rs Rlo
NH N
R5 R11
---N HN 4
Ra \ Rl2
R3
R2 Rl Rl4 Rl3
or a salt thereof with a metal compound in an aqueous solution having a pH > 9
to
yield the compound of formula 3 or the salt thereof.
An irnmobilizer can be added to the compound of formula 3 upon formation.
Alternatively an immobilizer can be added to the compound of formula 4 prior
to
the mixing with the metal compound. Preferably the immobilizer is added to an
aqueous solution having a pH > 9.
The third aspect of the present invention further provides a process for the
preparation of a compound of formula 3 or a salt thereof, comprising the steps
of
(i) mixing a compound of formula 4
R7 Ra R9
R6 ~ ~ ~ '
/ NH N \
Ra ' R10
R5 Rl1
~~ N HN Rl2 4
3 \ ~
R
R2 Rl R 14 R13
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
8
or a salt thereof with an immobilizer in an aqueous solution having a pH > 9
to
yield an immobilized compound 4, and (ii) adding a metal compound to the
immobilized compound 4 to yield an immobilized compound of formula 3 or the
salt thereof.
Preferably the immobilizer is a protein, a polypeptide, a polymer or activated
charcoal. Preferably the protein is serum humane albumin (SHA) or bovine serum
albumin (BSA), more preferably serum humane albumin (SHA). Preferably the
polypeptide is a low molecular weight polypeptide, more preferably polylysine
or
polyasparagine. Preferably the polymer is polyvinylpyrrolidone (PVP).
Preferably
the immobilizer immobilizes the compound of formula 3 in monomer form.
Preferably the compound of formula 4 and the metal compound are mixed in a
ratio
of about 1:1. Preferably the compound of formula 4, the metal compound and the
immobilizer are mixed in a ratio of about 1:1:1.
Preferably the metal compound is an organometallic compound. Preferably the
metal compound is a carboxylic acid metal salt. Preferably the metal compound
is a
Zn compound, such as zinc acetate. Alternatively the metal compound may be a
Cd
or Cu compound, such as cadmium acetate or copper acetate.
Preferably the aqueous solution is provided with a pH ? 9 by the addition of
ammonia. Preferably the aqueous solution is provided with a pH of from 9 to
10.
Preferably the step of mixing the immobilized or non-immobilized compound of
formula 4 with the metal compound is carried out at a temperature of from IO C
to
100 C. Preferably the step of mixing the compound of formula 3 or 4 with the
immobilizer is carried out at a temperature of from IO C to 100 C. More
preferably
the steps are carried out at a temperature of from 15 C to 40 C, even more
preferably at a temperature of from 18 C to 20 C.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
9
A fourth aspect of the present invention is a pharmaceutical composition
comprising a compound of formula 3 or a salt thereof and a pharmaceutically
acceptable carrier or diluent.
Preferably the pharmaceutical composition is in a form suitable for oral,
parental
(including intravenous, subcutaneous, intramuscular, intradermal,
intratracheal,
intraperitoneal, intraarticular, intraabdominal, intracranial and epidural),
transdermal, airway (aerosol), rectal, vaginal or topical (including buccal,
mucosal
and sublingual) administration, most preferably in a form suitable for oral or
parental administration.
For oral administration, the pharmaceutical composition is preferably provided
in
the form of a tablet, capsule, hard or soft gelatine capsule, caplet, troche
or lozenge,
as a powder or granules, or as an aqueous solution, suspension or dispersion.
Moreover, the pharmaceutical composition is preferably in a form suitable for
providing 0.01 to 10 mg/kg/day of a compound of formula 3 or a salt thereof,
more
preferably 0.1 to 5 mg/kg/day, even more preferably about 2 mg/kg/day.
Alternatively, the pharmaceutical composition is in a form suitable for
parental, in
particular intravenous, administration, in which case the pharmaceutical
composition is preferably an aqueous solution or suspension having a pH of
from 6
to 8.5.
Preferably the pharmaceutical composition is suitable for use in the
photodynamic
therapy or cytoluminescent therapy of a human or animal disease. Preferably
the
human or animal disease is characterised by begin or malignant cellular
hyperproliferation or by areas of neovascularisation. More preferably the
human or
animal disease is a benign or malignant tumour.
Preferably the pharmaceutical composition is suitable for the treatment of
atherosclerosis, multiple sclerosis, diabetes, a benign or malignant tumour,
arthritis,
rheumatoid arthritis, a fungal, viral, chlamydial, bacterial, nanobacterial or
parasitic
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
infectious disease, HIV, hepatitis, herpes simplex, herpes zoster, psoriasis,
a
cardiovascular disease, or a dermatological condition.
A fifth aspect of the present invention is the use of a compound of formula 3
or a
5 salt thereof for the manufacture of a phototherapeutic agent for the use in
photodynamic therapy or cytoluminescent therapy. Preferably the
phototherapeutic
agent is used for the treatment of a disease characterised by begin or
malignant
cellular hyperproliferation or by areas of neovascularisation. More preferably
the
phototherapeutic agent is used for the treatment of a benign or malignant
tumour.
A sixth aspect of the present invention is the use of a compound of formula 3
or a
salt thereof for the manufacture of a medicament for the treatment of
atherosclerosis, multiple sclerosis, diabetes, a benign or malignant tumour,
arthritis,
rheumatoid arthritis, a fungal, viral, chlamydial, bacterial, nanobacterial or
parasitic
infectious disease, HIV, hepatitis, herpes simplex, herpes zoster, psoriasis,
a
cardiovascular disease, or a dermatological condition.
A seventh aspect of the present invention is the use of a compound of formula
3 or
a salt thereof for the manufacture of a photodiagnostic agent for the
identification
of an area that is affected by begin or malignant cellular hyperproliferation
or by
neovascularisation. Preferably the area is a begin or malignant tumour.
An eighth aspect of the present invention is a method of photodynamic therapy
or
cytoluminescent therapy of a human or animal disease, comprising administering
a
therapeutically effective amount of a compound of formula 3 or a salt thereof
to a
human or animal in need thereoà and subjecting the human or animal to
irradiation
or sound. Preferably the human or animal disease is characterised by begin or
malignant cellular hyperproliferation or by areas of neovascularisation. More
preferably the human or animal disease is a benign or malignant tumour. The
precise wavelength of the irradiation or sound used depends on the compound
administered to the human or animal. However, generally the irradiation is
electromagnetic radiation with a wavelength in the range of from 500nm to
1000nm,
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
11
preferably from 600nm to 900nm, more preferably from 620nm to 820nm, even
more preferably from 630nm to 710nm.
A ninth aspect of the present invention is a method of treating
atherosclerosis,
multiple sclerosis, diabetes, a benign or malignant tumour, arthritis,
rheumatoid
arthritis, a fungal, viral, chlamydial, bacterial, nanobacterial or parasitic
infectious
disease, HIV, hepatitis, herpes simplex, herpes zoster, psoriasis, a
cardiovascular
disease, or a dermatological condition, comprising administering a
therapeutically
effective amount of a compound of formula 3 or a salt thereof to a human or
animal in need thereof. Optionally the human or animal is further subjected to
irradiation or sound. The precise wavelength of the irradiation or sound used
depends on the compound administered to the human.or animal. However,
generally the irradiation is electromagnetic radiation with a wavelength in
the range
of from 500nm to 1000nm, preferably from 600nm to 900nm, more preferably from
620nm to 820nm, even more preferably from 630nm to 710nm.
Brief description of the drawings
Figure 1 shows the absorption spectra of (1) chlorine-e6 (Xm,X = 656nm), (2)
Zn-
chlorine-e6 complex = 632nm), and (3) Zn-chlorine-e6 complex immobilized
on SHA (km,X = 636nm), all in water.
Figure 2 shows the absorption spectrum of chlorine-e6 (~maX = 402, 502 and
656nm)
in water.
Figure 3 shows the absorption spectra of (1) chlorine-e6 (Xm. = 656nm), (2)
chlorine-e6 immobilized on SHA (km,X = 662nm), and (3) Zn-chlorine-e6 complex
immobilized on SHA (kmax = 636nm), all in water.
Figure 4 shows the absorption spectra of (1) chlorine-e6 (km,x = 656nm), (2)
Zn-
chlorine-e6 complex (~m,x = 632nm), and (3) Zn-chlorine-e6 complex immobilized
on PVP (~m,x = 638nm), all in water.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
12
Figure 5 shows the absorption spectra of (1) chlorine-e6 656nm), (2)
chlorine-e6 immobilized on PVP (k,õ,X = 662nm), and (3) Zn-chlorine-e6 complex
immobiiized on PVP (Xm,x = 638nm), all in water.
Figures 6 to 8 show the absorption spectra of Zn-chlorine-e6 complex 414
and 634nm), Zn-chlorine-e6 complex immobilized on SHA (~m,x = 418 and 636nm),
and Zn-chlorine-e6 complex immobilized on PVP Q%,,,,X = 416 and 638nm), all in
water, respectively.
Figures 9 and 10 show the fluorescence spectrum (km,X = 643nm) and the
fluorescence stimulation spectrum (Xm,x = 412 and 607nm) of Zn-chlorine-e6
complex in water respectively.
Figures 11 and 12 show the fluorescence spectrum 645nm) and the
fluorescence stimulation spectrum (Xm,x = 446 and 673nm) of Zn-chlorine-e6
complex immobilized on SHA in water respectively.
Figures 13 and 14 show the fluorescence spectrum (km,x = 645nm) and the
fluorescence stimulation spectrum (Xm,x = 429 and 727nm) of Zn-chlorine-e6
complex immobilized on PVP in water respectively.
Figures 15 and 16 show the fluorescence spectrum (,%m,X = 645nm) and the
fluorescence stimulation spectrum (Xmõ = 418 and 641nm) of a biological sample
taken from the liquid above the sediment of an ascite tumour taken from an
experimental animal (mouse), which had previously been injected
intraabdominally
with a preparation comprising Zn-chlorine-e6 complex immobilized on SHA.
Figure 17 shows the absorption spectra of (1) chlorine-e6 (km,X = 656nm), (2)
chlorine-e6 immobilized on PVP (%m,x = 662nm), and (3) Cd-chlorine-e6 complex
immobilized on PVP (~m,x = 646nm), all in water.
Figure 18 shows the absorption spectrum of Cd-chlorine-e6 complex immobilized
on PVP (~m,X = 424 and 646nm) in water.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
13
Figure 19 shows the absorption spectra of (1) chlorine-e6 (km,X = 656nm), (2)
chlorine-e6 immobilized on PVP (a.maX = 662nm), and (3) Cu-chlorine-e6 complex
immobilized on PVP 636nm), all in water.
S
Figure 20 shows the absorption spectrum of Cu-chlorine-e6 complex immobilized
on PVP (Xmax = 410, 505 and 636nm) in water.
Figure 21 shows the results of pharmacokinetic distribution studies. The
pharmacokinetic distribution of Zn-chlorine-e6 complex immobilized on SHA over
30 hours in organs, tissues, biological liquids and tumours (embryocarcinoma)
was
studied.
Detailed description of the invention
rs'
The present invention provides two routes to compounds of formula 3.
The first route (see Examples 1 and 2 below) comprises the step of mixing a
compound of formula 4, also called chlorine-e6, which is commercially
available,
with a metal compound in an aqueous solution having a pH _ 9 to yield the
compound of formula 3. The compound of formula 3 may be immobilized in
monomer form on an immobilizer, such as a protein, a polypeptide, a polymer or
activated charcoal, by adding the immobilizer to the compound of formula 3
upon
formation.
More specifically, chlorine-e6 is dissolved in an aqueous solution with a pH _
9. A
pH _ 9 can be achieved, for example, by adding ammonia to an aqueous solution.
Then an about equimolar quantity of a metal compound, for example zinc
acetate, is
added to the reaction mixture. When mixing the solution at about room
temperature, chlorine-e6 and the metal ion form a complex. The progress and
completion of the complex-formation reaction can be monitored with a
spectrophotometer.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
14
On completion of the complex-formation reaction, an about equimolar quantity
of
an immobilizer such as a protein, a polypeptide, a polymer or activated
charcoal, for
example serum humane albumin (SHA) or polyvinylpyrrolidone (PVP), is added to
the reaction mixture. The solution is mixed at about room temperature until
the
compound of formula 3 is immobilized on the immobilizer. The progress and
completion of the immobilization reaction can be monitored with the help of a
spectrophotometer.
The second route (see Examples 3 to 6 below) comprises the steps of (i) mixing
a
compound of formula 4 with an immobilizer in an aqueous solution having a pH
9 to yield an immobilized compound 4, and (ii) adding a metal compound to the
immobilized compound 4 to yield an immobilized compound of formula 3.
Preferably the compound of formula 4 is immobilized in monomer form on a
protein, a polypeptide, a polymer or activated charcoal. The progress and
completion of the immobilization and the complex-formation reaction can be
monitored with a spectrophotometer.
Thus a water-soluble immobilizer, for example serum humane albumin (SHA) or
polyvinylpyrrolidone (PVP), is added to the reaction mixture in an about
equimolar
quantity relative to chlorine-e6, either before (route 2) or after (route 1)
carrying out
the complex-formation reaction.
The fact that compounds of formula 3 can be immobilized in monomolecular form
on the immobilizer is surprising, since monomeric compounds of formula 3 axe
not
particularly stable in aqueous solution. The quantity of the immobilizer
required is
defined by the number of sites on the molecule to be immobilized, which is one
for
compounds of formula 3.
Without wishing to be bound by theory, it is believed that it is the monomer
form
of the compounds of formula 3, which is the photoactive form, which may be
useful
as a phototherapeutic or photodiagnostic agent. However, compounds of formula
3, which have not been immobilized, have a tendency to form aggregates
(dimers,
trimers and oligomers of unknown structure) with unpredictable physical,
chemical,
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
photophysical and biological properties, in particular when the compounds of
formula 3 are subjected to pHs lower than 9. For example, aggregates of Zn-
chlorine-e6 are chemically very stable and attempts to disaggregate the Zn-
chlorine-
e6 aggregates, for example, by increasing pH, heating, using polar solvents,
etc. have
5 failed. Thus the aggregation process is difficult, if not impossible, to
reverse. The
present invention solves this problem by immobilizing the compounds of formula
3
in monomeric form prior to any aggregation occurring.
The compounds of formula 3 are photosensitizers and therefore useful in
10 pharmaceutical compositions and medicaments for the use in photodynamic
therapy. Moreover the photosensitizers of formula 3 can be used as
photodiagnostic agents for the identification of areas that are affected by
begin or
malignant cellular hyperproliferation or by neovascularisation.
15 The pharmaceutical composition or medicament employed in the present
invention
can be administered by oral, parental (including intravenous, subcutaneous,
intramuscular, intradermal, intratracheal, intraperitoneal, intraarticular,
intraabdominal, intracranial and epidural), transdermal, airway (aerosol),
rectal,
vaginal or topical (including buccal, mucosal and sublingual) administration.
For oral administration, the compounds of the invention will generally be
provided
in the form of tablets, capsules, hard or soft gelatine capsules, caplets,
troches or
lozenges, as a powder or granules, or as an aqueous solution, suspension or
dispersion.
Tablets for oral use may include the active ingredient mixed with
pharmaceutically
acceptable excipients such as inert diluents, disintegrating agents, binding
agents,
lubricating agents, sweetening agents, flavouring agents, colouring agents and
preservatives. Suitable inert diluents include sodium and calcium carbonate,
sodium
and calcium phosphate, and lactose. Corn starch and alginic acid are suitable
disintegrating agents. Binding agents may include starch and gelatine. The
lubricating agent, if present, may be magnesium stearate, stearic acid ox
talc. If
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
16
desired, the tablets may be coated with a material, such as glyceryl
monostearate or
glyceryl distearate, to delay absorption in the gastrointestinal tract.
Capsules for oral use include hard gelatine capsules in which the active
ingredient is
mixed with a solid diluent, and soft gelatine capsules wherein the active
ingredient is
mixed with water or an oil such as peanut oil, liquid paraffin or olive oil.
Formulations for rectal administration may be presented as a suppository with
a
suitable base comprising, for example, cocoa butter or a salicylate.
Formulations suitable for vaginal administration may be presented as
pessaries,
'tampons, creams, gels, pastes, foams or spray formulations containing in
addition to
the active ingredient such carriers as are known in the art to be appropriate.
For parenteral use, the compounds of the present invention will generally be
provided in a sterile aqueous solution or suspension, buffered to an
appropriate pH
and isotonicity. Suitable aqueous vehicles include Ringer's solution and
isotonic
sodium chloride or glucose. Aqueous suspensions according to the invention may
include suspending agents such as cellulose derivatives, sodium alginate,
polyvinylpyrrolidone and gum tragacanth, and a wetting agent such as lecithin.
Suitable preservatives for aqueous suspensions include ethyl and n-propyl p-
hydroxybenzoate. The compounds of the invention may also be presented as
liposome formulations.
For topical and transdermal administration, the compounds of the invention
will
generally be provided in the form of ointments, cataplasms (poultices),
pastes,
powders, dressings, creams, plasters or patches.
Suitable suspensions and solutions can be used in inhalers for airway
(aerosol)
administration.
In general, a suitable dose will be in the range of 0.01 to 10 mg per kilogram
body
weight of the recipient per day, preferably in the range of 0.1 to 5 mg per
kilogram
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
17
body weight per day, more preferably about 2 mg per kilogram body weight per
day.
The desired dose is preferably presented once a day, but may be dosed as two,
three,
four, five, six or more sub-doses administered at appropriate intervals
thxoughout
the day. These sub-doses may be administered in unit dosage forms, for
example,
containing 1 to 1500 mg, preferably 10 to 1000 mg, and most preferably 20 to
500
mg of active ingredient per unit dosage form.
The invention will now be described with reference to the following examples.
It
will be appreciated that what follows is by way of example only and that
modifications to detail may be made whilst still falling within the scope of
the
invention.
Synthetic experimental details
Example 1
Ammonia was added to water until the pH of the solution was not less than 9.
Then chlorine-e6 (1.0g) was dissolved in the aqueous solution. An equimolar
quantity of zinc acetate (0.22g) was added and the reaction mixture was
stirred for
15 minutes at about 20 C to achieve the complex-formation reaction. The
progress
and completion of the reaction was monitored with the help of a
spectrophotometer. On completion of the complex-formation reaction, serum
humane albumin (SHA) (71g) was added to the reaction mixture as an
immobilizer.
On completion of the immobilization reaction, which was monitored with a
spectrophotometer, the product of the reaction, Zn-chlorine-e6 complex
immobilized on SHA, was purified by dialysis.
Figure 1 shows the long-wave region of the visible absorption spectra of (1)
the
starting material chlorine-e6 656nm), (2) Zn-chlorine-e6 complex (km,x =
632nm), and (3) Zn-chlorine-e6 complex immobilized on SHA (7~max = 636nm), all
in
water.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
18
As can be seen in Figure 1, the formation of the Zn-chlorine-e6 complex is
accompanied by a 24nm short-wave shift of the long-wave absorption peak, and
the
immobilization of Zn-chlorine-e6 on protein causes a 4nm long-wave shift. Such
shifts of the long-wave peak are typical for both complex-formation with metal
and
immobilization on protein and prove the completeness and purity of the
reactions.
Moreover, the characteristic absorption peak of chlorine-e6 of medium
intensity at
%m,x = 502nm practically disappears for Zn-chlorine-e6, and instead a weak
peak at
km,x = 514nm appears, which also demonstrates the completeness and purity of
the
reaction.
For comparison, Figure 2 shows the visible absorption spectrum of the starting
material chlorine-e6 in water down to 350nm. The maxima of the main absorption
peaks are at kmaX = 402, 502 and 656nm.
Example 2
The synthesis of immobilized Zn-chlorine-e6 was carried out as described in
Example 1, except that as immobilizer polyvinylpyrrolidone (PVP) (62g) was
used
instead of SHA.
As can be seen in Figure 4, the spectral picture of the visible absorption
spectra of
(1) the starting material chlorine-e6 (,%m,X = 656nm), (2) Zn-chlorine-e6
complex
(,Xm,X = 632nm), and (3) Zn-chlorine-e6 complex immobilized on PVP (X,,,a. =
638nm) are practically identical to the ones depicted in Figure 1. One
observes a
significant 24nm short-wave shift of the long-wave peak upon metal complex
formation and a small 6nm long-wave shift upon immobilization on polymer PVP.
The medium intensity peak of chlorine-e6 at km,X = 502nm practically
disappears,
when forming the Zn-chlorine-e6 complex. All of these changes prove the
completeness of the reactions and the purity and homogeneity of the products
obtained.
Example 3
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
19
Ammonia was added to water until the pH of the solution was not less than 9.
Then chlorine-e6 (1.0g) was dissolved in the aqueous solution. An equimolar
quantity of SHA (71g) was added and the reaction mixture was stirred for 17
minutes at about 20 C to immobilize chlorine-e6 on SHA. Then an equimolar
quantity of zinc acetate (0.22g) was added and the reaction mixture was
stirred at
room temperature to complex Zn into the chlorine-e6, which was monitored with
a
spectrophotometer. The product of the reaction, Zn-chlorine-e6 complex
immobilized on SHA, was purified by dialysis.
Figure 3 shows the long-wave region of the visible absorption spectra of (1)
the
starting material chlorine-e6 (~m,x = 656nm), (2) chlorine-e6 immobilized on
SHA
(X.õx = 662nm), and (3) Zn-chlorine-e6 complex immobilized on SHA (~m,x =
636nm). Unlike the first method of synthesis (see Example 1), when forming
chlorine-e6 immobilized on protein, frst a 6nm long-wave shift of the
absorption
peak occurs, and then a 26nm short-wave shift, when forming Zn-chlorine-e6
immobilized on SHA. Such shifts of the absorption peak agree with the
properties
of the synthesized products and prove the completeness of the reactions and
the
purity of the products obtained. Moreover, the medium intensity peak of
chlorine-
e6 (~m,X = 502nm) is observed in the spectra of chlorine-e6 as well as of
chlorine-e6
immobilized on protein, but then it disappears in the spectrum of Zn-chlorine-
e6
complex immobilized on protein and gets transformed into a peak at Xm,X =
514nm.
Example 4
The synthesis of immobilized Zn-chlorine-e6 was carried out as described in
Example 3, except that as immobilizer polyvinylpyrrolidone (PVP) (62g) was
used
instead of SHA.
Figure 5 shows the long-wave region of the visible absorption spectra of (1)
the
starting material chlorine-e6 = 656nm), (2) chlorine-e6 immobilized on PVP
(~m,x = 662nm), and (3) Zn-chlorine-e6 complex immobilized on PVP (~max =
638nm). As in Example 3, when immobilising chlorine-e6 on PVP, a 6nm long-
wave shift of the absorption peak takes place, and then after introduction of
Zn
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
ions into chlorine-e6 and formation of the Zn-chlorine-e6 complex immobilized
on
PVP, a 24nm short-wave shift of the absorption peak occurs. These results
demonstrate the completeness of the reactions and the purity of the products
obtained. They are also evidenced by the behaviour of the medium intensity
peak
5 of chlorine-e6 at Xm,X = 502nm, which is present in the spectra of chlorine-
e6 as weII
as of chlorine-e6 immobilized on PVP, but disappears in the spectrum of Zn-
chlorine-e6 complex immobilized on PVP.
The fact that the spectra of the products, synthesised by the two different
routes
10 discussed above (route 1: Examples 1 and 2, route 2: Examples 3 and 4), are
identical proves that the conclusions drawn in the final paragraphs of
Examples 1 to
4 are correct.
Discussion of further spectra
Figures 6 to 8, with a spectral range of 350-700nm, show visible absorption
spectra
of Zn-chlorine-e6 complex, Zn-chlorine-e6 complex immobilized on SHA and Zn-
chlorine-e6 complex immobilized on PVP, all in water, respectively. The
absorption
spectra have main absorption peaks at ).m,X = 414 and 634nm for Zn-chlorine-e6
complex, Xmõ = 418 and 636nm for Zn-chlorine-e6 complex immobilized on SHA,
and = 416 and 638nm for Zn-chlorine-e6 complex immobilized on PVP. The
conclusions, drawn from these absorption spectra regarding the purity and
stability
of the monomeric products, were confirmed at every stage of the synthesis with
the
help of the highly sensitive analytical method of fluorescence spectroscopy
(see
Figures 9 to 14, discussed below).
Figures 9 and 10 show the fluorescence spectrum and the fluorescence
stimulation
spectrum of Zn-chlorine-e6 complex in water respectively. The monomeric Zn-
chlorine-e6 complex has a characteristic fluorescence spectrum with X,õ,x =
643nm,
and a fluorescence stimulation spectrum with main peaks at Xm,X = 412 and
607nm,
i.e. analogous to the peaks observed in the absorption spectrum. This shows
that
the fluorescence belongs to the monomeric Zn-chlorine-e6 complex and the
fluorescence data prove the high purity and homogeneity of the studied
product.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
21
Figures 11 and 12 show the fluorescence spectrum and the fluorescence
stimulation
spectrum of Zn-chlorine-e6 complex immobilized on SHA in water-respectively.
The fluorescence spectrum is similar to the fluorescence spectrum of Zn-
chlorine-
e6 complex in water, though slightly shifted into the red region (a.m,x =
645nm) and
with peaks of a smaller half-width, which demonstrates the great structural
similarity
between the centres of Zn-chlorine-e6 complex and Zn-chlorine-e6 complex
immobilized on SHA observed in these spectra. The fluorescence stimulation
spectrum of Zn-chlorine-e6 complex immobilized on SHA, shown in Figure 12, is
very similar to its absorption spectrum shown in Figure 7 and shows two main
peaks at Xm,x = 446 and 673nm with a smaller half-width and a more regular
shape
compared to the peaks in the absorption spectrum. This proves that the
fluorescence belongs to monomeric Zn-chlorine-e6 complex immobilized on SHA
and that the studied product has a high homogeneity and purity.
Figures 13 and 14 show the fluorescence spectrum and the fluorescence
stimulation
spectrum of Zn-chlorine-e6 complex immobilized on PVP in water respectively.
The shape of the fluorescence spectrum is very similar to the fluorescence
spectra
discussed above and has a peak at ~max = 645nm as in the spectrum of Zn-
chlorine-
e6 complex imrnobilized on SHA. The fluorescence stimulation spectrum has main
peaks at 7,m,' = 429 and 727nm, which agrees with its absorption spectrum and
shows that the fluorescence belongs to Zn-chlorine-e6 complex immobilized on
PVP and that the product is highly pure.
Figures 15 and 16 show the fluorescence spectrum and the fluorescence
stimulation
spectrum of a biological sample taken from the liquid above the sediment of an
ascite tumour taken from an experimental animal (mouse), which had previously
been injected intraabdominally with a preparation comprising Zn-chlorine-e6
complex immobilized on SHA. As can be seen by comparing the spectra of the
biological sample shown in Figures 15 and 16 with the corresponding spectra of
the
models shown in Figures 9 to 14, the peaks in the spectra of the biological
sample
occur at similar XRõx (fluorescence spectrum in Figure 15: km,x = 645nm;
fluorescence stimulation spectrum in Figure 16: ~max = 418 and 641nm) and have
a
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
22
similar peak shape and peak intensity ratio as the peaks in the spectra of the
models.
This means that the preparation injected into the experimental animal did not
undergo substantial structural changes and comprises Zn-chlorine-e6 with a
high
structural homogeneity of the absorbing and fluorescent centre as was observed
for
Zn-chlorine-e6 complex immobilized on SHA.
Example 5
Cd-chlorine-e6 complex immobilized on PVP was synthesized in a similar way to
Zn-chlorine-e6 complex immobilized on PVP (see Example 4). Figure 17 shows the
long-wave part of the visible absorption spectra of (1) the starting material
chlorine-
e6 (Xmõ, = 656nm), (2) chlorine-e6 immobilized on PVP (Xm,x = 662nm), and (3)
Cd-
chlorine-e6 complex immobilized on PVP (Xnõx = 646nm). Figure 18 shows the
absorption spectrum in the range of 350-750nm of the monomer form of Cd-
chlorine-e6 complex immobilized on PVP in water. As can be seen in Figure 18,
the spectrum of Cd-chlorine-e6 complex immobilized on PVP in monomer form
has two main peaks at 1,m,X = 424 and 646nm respectively.
Example 6
Cd-chlorine-e6 complex immobilized on PVP was synthesized in a similar way to
Zn-chlorine-e6 complex immobilized on PVP (see Example 4). Figure 19 shows the
long-wave part of the visible absorption spectra of (1) the starting material
chlorine-
e6 (~maX = 656nm), (2) chlorine-e6 immobilized on PVP 662nm), and (3) Cu-
chlorine-e6 complex immobilized on PVP (km,X = 636nm). Figure 20 shows the
absorption spectrum in the range of 350-750nm of the monomer form of Cu-
chlorine-e6 complex immobilized on PVP in water. As can be seen in Figure 20,
the absorption spectrum of Cu-chlorine-e6 complex immobilized on PVP in
monomer form differs from the monomer spectra of Zn-chlorine-e6 immobilized
complex and Cd-chlorine-e6 immobilized complex. The absorption spectrum of
Cu-chlorine-e6 complex immobilized on PVP in monomer form has three main
peaks at kmax = 410, 505 and 636nm respectively.
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
23
Preclinical pharmacolcinetic studies
A remarkable and important feature of the immobilized monomer Zn-chlorine-e6
complex is the possibility of preparing a stable form of the monomeric Zn-
chlorine-
e6 complex at a pH of from 6 to 8.5, which is required for injection usage. Zn-
chlorine-e6 complex preparations suitable for injection may be prepared by
acidifying the reaction medium after completion of the synthesis.
Pharmaceutically
acceptable additives, which do not interfere with the structural stability of
the Zn-
chlorine-e6 complex and the homogeneity of the preparation, may be added to
such
preparations suitable for injection.
The pharmacokinetic distribution of Zn-chlorine-e6 complex immobilized on SHA
over 30 hours in organs, tissues, biological liquids and tumours
(embryocarcinoma)
was studied. Female mice of the line Balb/c weighing 20-21g were used as
experimental animals. The pharmacokinetic studies were carried out using a
Perkin-
Elmer spectrofluorimeter on homogetates of organs and tumours, taken after the
intraabdominal injection of Zn-chlorine-e6 complex immobilized on SHA at a
dose
of 25 mg/kg weight. The results of these pharmacokinetic distribution studies
are
depicted in Figure 21 and summarised in Table 1 below.
Organ Amount of Zn-chlorine-e6 complex immobilized on SHA
accumulated in organ X hours after injection in relative
units
X=1 X=5 X=15 X=24 X=30
1. BIood 4 9 6 1 1
2. Urine 7 0 0 0 0
3. Sma11 intestine 98 175 136 90 46
4. Liver 86 147 66 49 33
5. Spleen 11 17 17 12 13
6. Kidney 38 64 25 11 8
7. Lungs 18 15 26 11 8
8. Tumour 8 40 99 67 79
9. Skin 9 8 19 7 15
10. Muscle 25 7 33 6 18
Table 1
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
24
Results:
A. Intraabdominal injection of Zn-chlorine-e6 complex immobilized on SHA at
a dose of 25 mg/kg weight was well endured by the animals without any signs of
toxicity and did not affect their behavioural reaction, both immediately and
30
hours after the injection.
B. The immobilized complex was rapidly absorbed from the abdominal cavity
into the blood and was deposited in the liver during the first hours after
injection.
Its content in the liver tissues was 10-14 times higher than its level in the
blood.
C. A significant quantity of the immobilized complex was also accumulated in
the kidneys in the first 12 hours after injection (only 2-2.5 times less than
in the
liver), however, the immobilized complex was practically absent from the
urine.
During the next 18 hours, the immobilized complex was washed out intensely
from
the kidney tissue into the blood. The kidneys' secretion function was not
affected
during the whole observation period.
D. The maximum concentrations of the immobilized complex in the liver were
found during the first 8 hours after injection. During the next 24 hours, the
surplus
of the immobiiized complex was discharged intensely into the small intestines.
The
dynamics of the distribution curves of the liver and small intestines
correlate
precisely with one another. It may be sufficient to inject 5-10 times smaller
doses
of the immobilized complex in order to achieve maximum concentrations in the
tumour.
E. 5-8 times Iess of the immobilized complex accumulated in the spleen and the
lungs compared to the liver or tumour, and 24 hours after injection the spleen
and
lungs had phone readings.
F. Skin and muscle tissue both had practically the same accumulation dynamics,
the only difference being that the immobilized complex content in the muscle
was
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
1.5 times higher in the first 15 hours than the immobilized complex content in
the
skin.
G. The accumulation of the immobilized complex in tumour increased
5 progressively from the moment of injection and reached its maximum 15 hours
after
injection. The maximum concentration plateau (12-20 hours) was found to be
much
longer than after chlorine-e6 injection, and after an insignificant fall by
the end of
the first 24 hours, a second increase of immobilized complex concentration up
to
the maximum readings of the concentration plateau was observed between 24 and
10 30 hours. A "scissors" effect (the immobilized complex concentration in the
tumour is increasing, while the immobilized complex concentration in the liver
is
decreasing) was observed twice for the liver and tumour, once 12 hours after
injection and once, even more pronounced, 24 hours after injection.
15 To summarise, after the absorption of the immobilized complex in the
abdominal
cavity, redistribution from the blood into the organs and washing out of the
irnmobilized complex surplus by the liver during the first 24 hours, the
immobilized
complex accumulated in the tumour tissue in a concentration of 2.5 times
greater
than in the liver and 6 times greater than in the skin, muscle and other
parenchymal
20 organs. In comparison with the pharmacokinetics of the dimer form, the
monomer
form demonstrated much greater tumour selectivity and stability of the
chemical
structure in tissues.
The spectroscopic data (see Figures 1 to 20) and pharmacokinetic data (see
Figure
25 21) discussed above show that the ixnmobilized preparations preserve the
monomeric structure, purity and chemical stability of the porphyrin nucleus of
the
Zn-chlorine-e6 complex.
Definition of acute toxicity parameters:
To define parameters LDlO and LDso three preparation doses were chosen (100,
125
and 150 mg/kg weight) for single intraabdominal injection. Chlorine-e6
readings
CA 02582437 2007-04-05
WO 2005/033111 PCT/IB2004/051998
26
were taken as a prototype, where LDIo is 119 mg/kg weight and LD50 is 160
mg/kg
weight.
After injection of Zn-chlorine-e6 complex immobilized on SHA in the above
stated
doses, a first reaction to the injection was observed only with the third
animal group
(150 mg/kg weight), because the preparation was injected in 3ml physiological-
solution, which caused temporal animal stillness due to abdominal swelling.
After
the absorption of the surplus liquid, however, these animals did not differ
from the
animals in the other two groups in their behavioural reactions (moving
activity,
defence reflex, food reflex, coat condition).
During the following 72 hours, signs of acute toxicity (slow reaction, hollow
sides,
diarrhoea, defence and food reflex absence) did not appear. The animals were
kept
under observation for a further fortnight.
Further tests were carried out similarly with intraabdominal injections of
175, 200
and 225 mg/kg weight, as well as 300, 350 and 450 mg/kg weight. None of these
concentrations proved toxic.
It wilI be understood that the present invention has been described above by
way of
example only. The examples are not intended to limit the scope of the
invention.
Various modifications and embodiments can be made without departing from the
scope of the invention, which is defined by the following claims.