Note: Descriptions are shown in the official language in which they were submitted.
CA 02738706 2011-06-13
SYNTHESIS OF BORONIC ESTER AND ACID COMPOUNDS
[000] This is a division of Canadian Patent Application No. 2.560.886
filed March
24, 2005.
BACKGROUND OF THE INVENTION
Field of the Invention
[001] This invention relates to the synthesis of boronic ester and acid
compounds.
More particularly, the invention relates to large-scale synthetic processes
for the preparation
of boronic ester and acid compounds by Lewis add promoted rearrangement of
boron "ate"
1 0
complexes.
Background of the Invention
[002] Boronic acid and ester compounds display a variety of
pharmaceutically
useful biological activities. Shenvi et al., U.S. Pat. No. 4,499,082 (1985),
discloses that peptide
boronic acids are inhibitors of certain proteolytic enzymes. Kettner and
Shenvi, U.S. Pat. No.
5,187,157 (1993), U.S. Pat. No. 5,242,904 (1993), and U.S. Pat. No. 5,250,720
(1993), describe a
class of peptide boronic acids that inhibit trypsin-like proteases. Kleeman et
al., U.S. Pat. No.
5,169,841 (1992), discloses N-terminally modified peptide boronic adds that
inhibit the
action of renin. Kinder et al., U.S. Pat. No. 5,106,948 (1992), discloses that
certain tripeptide
20 boronic acid compounds inhibit the growth of cancer cells.
[003] More recently, boronic acid and ester compounds have displayed
particular
promise as inhibitors of the proteasome, a multicatalytic protease responsible
for the
majority of intracellular protein turnover. Ciechanover, Cell, 79: 13-21
(1994), discloses that
the proteasome is the proteolytic component of the ubiquitin-proteasome
pathway, in which
proteins are targeted for degradation by conjugation to multiple molecules of
ubiquitin.
Ciechanover also discloses that the ubiquitin-proteasome pathway plays a key
role in a
variety of important physiological processes.
[004] Adams et al., U.S. Patent No. 5,780,454 (1998), U.S. Patent No.
6,066,730 (2000),
U.S. Patent No. 6,083,903 (2000), U.S. Patent No. 6,297,217 (2001), U.S.
Patent No. 6,548,668,
1
CA 02738706 2011-04-21
and U.S. Patent No. 6,617,317 (2003) describe peptide boronic ester and acid
compounds useful as proteasome inhibitors. The references also describe the
use of
boronic ester and acid compounds to reduce the rate of muscle protein
degradation,
to reduce the activity of NF-KB in a cell, to reduce the rate of degradation
of p53
protein in a cell, to inhibit cyclin degradation in a cell, to inhibit the
growth of a cancer
cell, to inhibit antigen presentation in a cell, to inhibit NF-KB dependent
cell adhesion,
and to inhibit HIV replication.
[005] Albanell and Adams, Drugs of the Future 27: 1079-1092 (2002),
discloses that
one such peptide boronic acid proteasome inhibitor, bortezomib (N-2-
pyrazinecarbonyl-L-
phenylalanine-L-leucineboronic acid), shows significant antitumor activity in
human tumor
xenograft models and is undergoing clinical evaluation. Richardson et al., New
Engl. J. Med.,
348:2609 (2003), reports the results of a Phase 2 study of bortezomib, showing
its
effectiveness in treating relapsed and refractory multiple myeloma.
[006] Studies of boronic acid protease inhibitors have been greatly
advanced by the
development of chemistry for the preparation of functionalized boronic acid
compounds,
particularly alpha-halo- and alpha-aminoboronic acids. Matteson and Majumdar,
J. Am.
Chem. Soc., 102:7590 (1980), discloses a method for preparing alpha-
chloroboronic esters by
homologation of boronic esters, and Matteson and Ray, J. Am. Chem. Soc.,
102:7591 (1980),
reports that chiral control of the homologation reaction can be achieved by
the use of
pinartediol boronic esters. The preparation of alpha-aminoboronic acid and
ester
compounds from the corresponding alpha-chloroboronic esters has also been
reported
(Matteson et al., J. Am. Chem. Soc., 103:5241 (1981); Shenvi, U.S. Patent No.
4,537,773 (1985)).
[007] Matteson and Sadhu, U.S. Patent No. 4,525,309 (1985), describes an
improved
procedure for the homologation of boronic esters by rearrangement of the
intermediate
boron "ate" complex in the presence of a Lewis acid catalyst. The Lewis acid
is reported to
promote the rearrangement reaction and to minimize epimerization at the alpha-
carbon
atom. Rigorous exclusion of water and careful control of Lewis acid
stoichiometry are
required for optimum results, however. These features render the reaction
difficult to
2
CA 02738706 2013-03-18
,
,
perform successfully on a production scale, and limit the availability of
pharmaceutically important boronic ester and acid compounds, such as
bortezomib.
Thus, there remains a need in the art for improved methods for the large-scale
production of boronic ester and acid compounds.
DESCRIPTION OF THE INVENTION
[008] The present invention provides improved synthetic processes for the
large-
scale production of boronic ester and acid compounds. These processes offer
increased yield and purity, increased throughput, and greater ease of handling
as
compared to prior art methods. Notably, the processes described herein are
suitable
for batch production on a large, multi-kilogram scale that is limited only by
the size
of the available manufacturing capabilities. The processes of the invention
are
particularly advantageous for the synthesis of chiral boronic ester and acid
compounds, including alpha-aminoboronic ester and acid compounds. Regardless
of scale, the desired products are produced with very high chemical and
stereochemical purity.
[009] The patent and scientific literature referred to herein establishes
knowledge
that is available to those with skill in the art. Unless otherwise defined,
all technical
and scientific terms used herein have the same meaning as commonly understood
by one of ordinary skill in the art to which this invention relates. In the
case of
inconsistencies, the present disclosure, including definitions, will control.
In addition,
the materials, methods, and examples are illustrative only and are not
intended to
be limiting.
[009-a]
An embodiment of the invention relates to a large-scale process for
forming a compound of formula (X/V):
3
CA 02738706 2013-03-18
0 OH
H
N B,
OH
0 y
(XIV)
or a boronic acid anhydride thereof comprising the steps:
(aa) coupling a compound of formula (XV///):
H2N B
(xvill)
or an acid addition salt thereof, with a compound of formula (X/X):
Fri-N
0 (XD)
wherein:
P1 is a cleavable amino group protecting moiety; and
X is OH or a leaving group;
to form a compound of formula (XX):
0
H
N
0
0 y
POO
wherein P1 is as defined above, said coupling step (aa) comprising the steps:
3a
CA 02738706 2013-03-18
(i) coupling the compound of formula (XV///) with a compound of
formula (X/X) wherein X is OH or a leaving group, in the presence of 2-(1H-
benzotriazol-1-y1)-1,1,3,3-tetramethyluronium tetrafluoroborate (TBTU) and a
tertiary amine in dichloromethane;
(ii) performing a solvent exchange to replace dichloromethane with
ethyl acetate; and
(iii) performing an aqueous wash of the ethyl acetate solution;
(bb) removing the protecting group P1 to form a compound of formula (XX/):
H
NõF3.0
H2N
o y
(XXI)
or an acid addition salt thereof, said protecting group removing step (bb)
comprising
the steps:
(i) treating the compound of formula (XX) with HCI in ethyl acetate;
(ii) adding heptane to the reaction mixture; and
(iii) isolating by crystallization the compound of formula (X)(/) as its
HCI addition salt;
(cc) coupling the compound of formula (XX/) with a reagent of formula
(XX//)
C )r x
0 (XX//)
wherein X is a OH or a leaving group, to form a compound of formula (XX///):
3b
CA 02738706 2014-05-21
0 0
H
B.
N N 0
N*. 0 y
said coupling step (cc) comprising the steps:
(i) coupling the compound of formula (X)(/) with the reagent ()0(//)
(e.g. 2-pyrazine-carboxylic acid) in the presence of TBTU and a tertiary amine
in
dichloromethane;
(ii) performing a solvent exchange to replace dichloromethane with
ethyl acetate; and
(iii) performing an aqueous wash of the ethyl acetate solution; and
(dd) deprotecting the boronic acid moiety to form the compound of formula
(X/V) or a boronic acid anhydride thereof, said deprotecting step (dd)
comprising the
steps:
(i) providing a biphasic mixture comprising the compound of
formula p0(I//), an organic boronic acid acceptor, a lower alkanol, a
C5_8 hydrocarbon solvent, and aqueous mineral acid;
(ii) stirring the biphasic mixture to afford the compound of formula
(XIV);
(iii) separating the solvent layers; and
(iv) extracting the compound of formula (X/V), or a boronic acid
anhydride thereof, into an organic solvent.
[009-b] Another embodiment of the invention relates to a process as defined
hereinabove, wherein step (dd)(iii) comprises the steps:
(1) separating the solvent layers;
3c
, CA 02738706 2013-03-18
(2) adjusting the aqueous layer to basic pH;
(3) washing the aqueous layer with an organic solvent; and
(4) adjusting the aqueous layer to a pH less than 8.
[009-c] Another embodiment of the invention relates to a process as
defined
hereinabove, wherein in step (dd)(iv), the compound of formula (X/V):
411
0 OH
H i
N N B.
N
:
0 -y
r N
(X/V)
or a boronic acid anhydride thereof, is extracted into dichloromethane, the
solvent is
exchanged to ethyl acetate, and the compound of formula (X/V), or a boronic
acid
anhydride thereof, is crystallized by addition of hexane or heptane.
[009-d] Another embodiment of the invention relates to a process as
defined
hereinabove, wherein in step (dd)(iv) the boronic acid anhydride of the
compound of
formula (X/V):
0 *OH
H 1
( ))(N OH
:
INI 0 y
(Xi V)
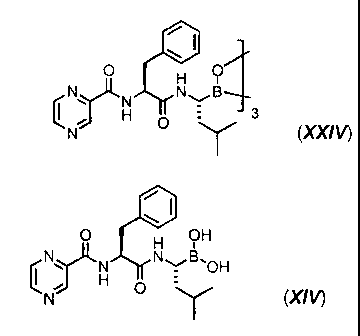
is a cyclic trimeric boronic acid anhydride of formula (XX/V):
3d
CA 02738706 2013-03-18
0 0
H
ii )
)ri N B - 3
lµr 0 y
(XXIV) ,
and wherein the cyclic trimeric boronic acid anhydride of formula (XX/V) is
extracted
into into dichloromethane, the solvent is exchanged to ethyl acetate, and then
the
cyclic trimeric boronic acid anhydride of formula (X)(/V) is crystallized by
addition of
hexane or heptane.
[010] The term "about" is used herein to mean approximately, in the region of,
roughly, or around. When the term "about" is used in conjunction with a
numerical
range, it modifies that range by extending the boundaries above and below the
numerical values set forth. In general, the term "about" is used herein to
modify a
numerical value above and below the stated value by a variance of 10%.
[011] The term "comprises" is used herein to mean "includes, but is not
limited to."
[012] The term "aliphatic", as used herein, means a straight-chain, branched
or
cyclic C1-12 hydrocarbon which is completely saturated or which contains one
or
more units of unsaturation, but which is not aromatic. For example, suitable
aliphatic
groups include substituted or unsubstituted linear, branched or cyclic alkyl,
alkenyl,
alkynyl groups and hybrids thereof, such as (cylcoalkyl)alkyl, (cycloalkenyl)
alkyl or
(cycloalkyl)alkenyl. In various embodiments, the aliphatic group has 1-12, 1-
8, 1-6,
or 1-4 carbons.
3e
CA 02738706 2011-04-21
[013] The terms "alkyl", "alkenyl", and "alkynyl", used alone or as part of
a larger
moiety, refer to a straight and branched chain aliphatic group having from 1
to 12 carbon
atoms, which is optionally substituted with one, two or three substituents.
For purposes of
the present invention, the term "alkyl" will be used when the carbon atom
attaching the
aliphatic group to the rest of the molecule is a saturated carbon atom.
However, an alkyl
group may include unsaturation at other carbon atoms. Thus, alkyl groups
include, without
limitation, methyl, ethyl, propyl, allyl, propargyl, butyl, pentyl, and hexyl.
[014] For purposes of the present invention, the term "alkenyl" will be
used when
the carbon atom attaching the aliphatic group to the rest of the molecule
forms part of a
carbon-carbon double bond. Alkenyl groups include, without limitation, vinyl,
1-propenyl,
1-butenyl, 1-pentenyl, and 1-hexenyl. For purposes of the present invention,
the term
"alkynyl" will be used when the carbon atom attaching the aliphatic group to
the rest of the
molecule forms part of a carbon-carbon triple bond. Alkynyl groups include,
without
limitation, ethynyl, 1-propynyl, 1-butynyl, 1-pentynyl, and 1-hexynyl.
[015] The terms "cycloalkyl", "carbocycle", "carbocyclyl", "carbocyclo", or
"carbocyclic", used alone or as part of a larger moiety, means a saturated or
partially
unsaturated cyclic aliphatic ring system having from 3 to about 14 members,
wherein the
aliphatic ring system is optionally substituted. Cycloalkyl groups include,
without
limitation, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl,
cyclohexenyl,
cycloheptyl, cycloheptenyl, cyclooctyl, cyclooctenyl, and cycloocladienyl. In
some
embodiments, the cycloalkyl has 3-6 carbons. The terms "cycloalkyl",
"carbocycle",
"carbocyclyl", "carbocyclo", or "carbocyclic" also include aliphatic rings
that are fused to one
or more aromatic or nonaromatic rings, such as decahydronaphthyl or
tetrahydronaphthyl,
where the radical or point of attachment is on the aliphatic ring.
[016] The terms "haloalkyl", "haloalkenyl" and "haloalkoxy" refer to an
alkyl,
alkenyl or alkoxy group, as the case may be, substituted with one or more
halogen atoms.
As used herein, the term "halogen" or "halo" means F, C, Br, or I. Unless
otherwise
indicated, the terms "alkyl", "alkenyl", and "alkoxy" include haloalkyl,
haloalkenyl and
haloalkoxy groups, including, in particular, those with 1-5 fluorine atoms.
[017] The terms "aryl" and "ar-'', used alone or as part of a larger
moiety, e.g.,
"aralkyl", "aralkoxy", or "aryloxyalkyl", refer to a C aromatic moiety
comprising one to
three aromatic rings, which are optionally substituted. Preferably, the aryl
group is a
aryl group. Aryl groups include, without limitation, phenyl, naphthyl, and
anthracenyl.
The term "aryl", as used herein, also includes groups in which an aromatic
ring is fused to
-4-
CA 02738706 2011-04-21
one or more non-aromatic rings, such as indanyl, phenanthridinyl, or
tetrahydronaphthyl,
where the radical or point of attachment is on the aromatic ring. The term
"aryl" may be
used interchangeably with the term "aryl ring".
[01S] An "aralkyl" or "arylalkyl" group comprises an aryl group covalently
attached
to an alkyl group, either of which independently is optionally substituted.
Preferably, the
aralkyl group is C aryl(C1)alkyl, including, without limitation, benzyl,
phenethyl, and
naphthylmethyl.
[019] The terms "heteroaryl" and "heteroar-", used alone or as part of a
larger
moiety, e.g., heteroaralkyl, or "heteroaralkoxy", refer to groups having 5 to
14 ring atoms,
preferably 5, 6, 9, or 10 ring atoms; having 6, 10, or 14 it electrons shared
in a cyclic array;
and having, in addition to carbon atoms, from one to four heteroatoms selected
from the
group consisting of N, 0, and S. Heteroaryl groups include, without
limitation, thienyl,
tiffany', pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl,
isoxazolyl,
oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl,
pyrimidinyl,
pyrazinyl, indolyl, isoindolyl, benzothienyl, benzofuranyl, dibenzofuranyl,
indazolyl,
benzimidazolyl, benzthiazolyl, purinyl, quinolyl, isoquinolyl, cinnolinyl,
quinoxalinyl, naphthyridinyl, pteridinyl, carbazolyl, acridinyl, and
phenazinyl. The tern-is
"heteroaryl" and "heteroar-", as used herein, also include groups M which a
heteroaromatic
ring is fused to one or more nonaromatic rings, where the radical or point of
attachment is
on the heteroaromatic ring. Nonlimiting examples include tetrahydroquinolinyl,
tetrahydroisoquinolinyl, and pyrido[3,4-d]pyrimidinyl. The term "heteroaryl"
may be used
interchangeably with the term "heteroaryl ring" or the term "heteroaromatic",
any of which
terms include rings that are optionally substituted. The term "heteroaralkyl"
refers to an
alkyl group substituted by a heteroaryl, wherein the alkyl and heteroaryl
portions
independently are optionally substituted.
[020] As used herein, the terms "heterocycle", "heterocyclyl", or
"heterocyclic
radical" refer to a stable 5- to 7-membered monocyclic or 7- to 10-membered
bicyclic
heterocyclic moiety that is either saturated or partially unsaturated, and
having, in addition
to carbon atoms, one or more, preferably one to four, heteroatoms selected
from the group
consisting of N, 0, and S, wherein the nitrogen and sulfur heteroatoms are
optionally
oxidized and the nitrogen atoms are optionally quaternized. The heterocyclic
ring can be
attached to its pendant group at any heteroatom or carbon atom that results in
a stable
structure, and any of the ring atoms can be optionally substituted. Examples
of such
saturated or partially unsaturated heterocyclic radicals include, without
limitation,
- 5 -
CA 02738706 2011-04-21
tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, pyrrolidonyl, piperidinyI,
pyrrolinyl,
tetrahydroquinolinyl, tetrahydroisoquinolinyl, decahyciroquinolinyl,
oxazolidinyl,
piperazinyl, dioxanyl, dioxolanyl, diazepinyl, oxazepinyl, thiazepinyl, and
morpholinyl.
The terms "heterocycle", "heterocyclyl", and "heterocyclic radical", as used
herein, also
include groups in which a non-aromatic heteroatom-containing ring is fused to
one or more
aromatic or non-aromatic rings, such as indolinyl, chromanyl, phenanthridinyl,
or
tetrahydroquinolinyl, where the radical or point of attachment is on the non-
aromatic
heteroatom-containing ring. The term "heterocyclylalkyl" refers to an alkyl
group
substituted by a heterocyclyl, wherein the alkyl and heterocyclyl portions
independently are
optionally substituted.
[021] As used herein, the term "partially unsaturated" refers to a ring
moiety that
includes at least one double or triple bond between ring atoms. The term
"partially
unsaturated" is intended to encompass rings having one or multiple sites of
unsaturation,
but is not intended to include aryl or heteroaryl moieties, as herein defined.
[0221 The term "substituted", as used herein, means that one or more
hydrogen =
atoms of the designated moiety are replaced, provided that the substitution
results in a
stable or chemically feasible compound. A stable compound or chemically
feasible
compound is one in which the chemical structure is not substantially altered
when kept at a
temperature of 40 C or less, in the absence of moisture or other chemically
reactive
conditions, for at least a week, or a compound which maintains its integrity
long enough to
be useful for the synthetic processes of the invention. The phrase "one or
more substituents",
as used herein, refers to a number of substituents that equals from one to the
maximum
number of substituents possible based on the number of available bonding
sites, provided
that the above conditions of stability and chemical feasibility are met.
[023] An aryl (including the aryl moiety in aralkyl, aralkoxy,
aryloxyalkyl and the
like) or heteroaryl (including the heteroaryl moiety in heteroaralkyl and he
teroarylalkoxy
and the like) group may contain one or more substituents. Examples of suitable
substituents
on the unsaturated carbon atom of an aryl or heteroaryl group include -halo, -
NOõ -CN, -R*,
-OR*, -SR , -N(R+)õ -NR4C(0)R*, -N12.+C(0)N(R")õ -NR*CO,R , -0-CO2R*, -0-
C(0)1Z*, -CO.,R*,
-C(0)R*, -C(0)N(Rt)2, -0C(0)N(12')2, -S(0)2R , -SO,N(R.)õ -S(0)R , and -
NR4SO2N(RI. Each
R. independently is selected from the group consisting of R*, -C(0)R*, -CO,R*,
and -SO,R*,
or two R.* on the same nitrogen atom, taken together with the nitrogen atom,
form a 5-8
membered aromatic or non-aromatic ring having, in addition to the nitrogen, 0-
2 ring
heteroatoms selected from N, 0, and S. Each R* independently is hydrogen or an
optionally
- 6 -
CA 02738706 2011-04-21
substituted aliphatic, aryl, heteroaryl, or heterocyclyl group. Each X'
independently is an
optionally substituted aliphatic or aryl.group.
[024] An aliphatic group also may be substituted with one or more
substituents.
Examples of suitable substituents on the saturated carbon of an aliphatic
group or of a non-
aromatic heterocyclic ring include, without limitation, those listed above for
the unsaturated
carbon of an aryl or heteroaryl group.
[025] The present inventors have discovered that the requirement for
scrupulously
dry equipment, solvents, and reagents that characterized previously described
procedures
for the Lewis acid promoted rearrangement of boron "ate" complexes can be
obviated by use
of an ether solvent that has low miscibility with water. Remarkably, use of
such a solvent
permits the reaction to be run on a multi-kilogram scale without deterioration
in yield or
purity. In essence, the scale of the reaction is limited only by the size of
the available
manufacturing capabilities.
[0261 In one aspect, therefore, the invention provides a large-scale
process for
preparing a boronic ester compound of formula (I):
, R1
R3) B..OR4
OR5
wherein:
R is an optionally substituted aliphatic, aromatic, or heteroaromatic group;
R2 is hydrogen, a nucleofugic group, or an optionally substituted aliphatic,
aromatic,
or heteroaromatic group;
R5 is a nucleofugie group or an optionally substituted aliphatic, aromatic, or
heteroarornatic group; and
each of R4 and R5, independently, is an optionally substituted aliphatic,
aromatic, or
heteroaromatic group, or Wand R5, taken together with the intervening oxygen
and boron atoms, form an optionally substituted 5- to 10-membered ring having
0-2 additional ring heteroatoms selected from N, 0, or S.
[027] The process comprises the steps: =
-(a) - providing a boron "ate" complex of formula (II): -
- 7 -
CA 02738706 2011-04-21
R3
R2>01084
Y B -
M+
OR5 (Ti)
where
Y is a nucleofugic group;
1V1+ is a cation; and
each of R1 to R5 is as defined above; and
(b) contacting
the boron "ate" complex of formula (11) with a Lewis acid under
conditions that afford the boronic ester compound of formula (/), said
contacting step being
conducted in a reaction mixture comprising:
(i) a coordinating ether solvent that has low miscibility with water; or
(ii) an ether solvent that has low miscibility with water and a
coordinating co-solvent.
[028] The previously reported processes for Lewis acid promoted
rearrangement of
boron "ate" complexes employ tetTahydrofuran, an ether solvent that is fully
miscible with
water. Failure to employ rigorously dried equipment, solvents, and reagents in
these
processes results in a dramatic reduction in the diastereomeric ratio. The
hygroscopic Lewis
adds, in particular, typically must be flame-dried immediately prior to use in
the reaction.
Although techniques for running moisture-sensitive reactions are familiar to
those of skill in
the art and are routinely practiced on a laboratory scale, such reactions are
costly and
difficult to scale up.
[029] Moreover, attempted scale-up of the prior art process frequently
results in a
further deterioration in diastereomeric ratio during workup and isolation of
the product
boronic ester compound. Matteson and Erdiik, Organonzetallics, 2:1083 (1983),
reports that
exposure of alpha-haloboronic ester products to free halide ion results in
epimerization at
the alpha-carbon center. Without wishing to be bound by theory, the present
inventors
believe that epimerization is particularly problematic during reaction work-up
and/or
subsequent steps. For example, epimerization is believed to occur during
concentration of
the reaction mixture to remove the tetrallydrofuran solvent and exchange it
for a water-
immiscible solvent. Failure to completely remove the tetrahydroftu:an also
negatively
impacts diastereomeric ratio during the subsequent aqueous washes. Exposure of
the
- s -
CA 02738706 2011-04-21
=
=
product to halide ion during these steps is difficult to avoid, particularly
when the reaction
is performed on a large scale.
[030] The present inventors have discovered that the
rearrangement of boron "ate"
complexes is advantageously performed in an ether solvent that has low
miscibility with
water. Use of such solvents obviates the need for solvent exchange prior to
the aqueous
washes, and the organic-soluble product is effectively shielded from aqueous
halide ion
during the washes, even if performed on a large scale. Preferably, the
solubility of water in
the ether solvent is less than about 5% w/w, more preferably less than about
2% w/w. In
various embodiments, ether solvent that has low miscibility with water
constitutes at least
about 70%, at least about 80%, at least about 85%, at least about 90%, or at
least about 95%
v/v of the reaction mixture.
[031.] The ether solvent preferably is one that is suitable
for routine use in large-
scale production. As used herein, the term "large-scale" refers to a reaction
that utilizes at
least about five moles of at least one starting material. Preferably, a large-
scale process
utilizes at least about 10, 20, 50, or 100 moles of at least one starting
material.
[032] For purposes of the invention, the term "ether" refers to any of a
class of
chemical compounds characterized in having an oxygen atom attached to two
carbon atoms
. An "ether solvent" is an ether compound that exists in liquid form at the
desired reaction
temperature and is capable of dissolving the starting material(s) and/or
product(s) of the
reaction. Non-limiting examples of ether solvents suitable for use in the
process of the
invention include tert-butyl methyl ether, tert-butyl ethyl ether, tert-amyl
methyl ether, and
isopropyl ether.
[033] In one embodiment, the reaction mixture further comprises a
coordinating co-
solvent. In another embodiment, the ether solvent that has low miscibility
with water is
sufficiently coordinating that a coordinating co-solvent is not necessary. For
purposes of the
invention, the terms "coordinating co-solvent" and "coordinating solvent"
refer to a solvent
that is capable Of coordinating the Lewis add and solvating the ionic
components of the
reaction. Hindered ether solvents, such as tert-butyl methyl ether, are poorly
coordinating
and preferably are used with a coordinating co-solvent. Nonlimiting examples
of
coordinating co-solvents suitable for use in the practice of the invention
include
tetrahydrofuran, dioxane, water, and mixtures thereof.
= - [0341 - - In some embodiments, the reaction
mixture comprises at least about 5% or at - "-
least about 10% v/v of a coordinating co-solvent. Preferably, the amount of a
water-n-tiscible
-9-
CA 02738706 2011-04-21
coordinating co-solvent present in the reaction mixture is not so great as to
interfere with
phase separation during the reaction or workup. In various embodiments, the
coordinating
co-solvent constitutes no more than about 20%, about 15%, or about 10% v/v of
the reaction
mixture.
[035] As used herein, the term "nudeofugic" refers to any group that is
capable of
undergoing nucleophilic displacement under the rearrangement conditions of the
present
process. Such nucleofugic groups are known in the art. Preferably, the
nucleofugic group is
a halogen, more preferably chloro or bromo. In the course of the rearrangement
reaction
converting the boron "ate" complex of formula (II) into the boronic ester
compound of
formula (I), the nudeofugic group Y is released as Y. By way of example, when
Y is chloro,
chloride ion is released in step (b).
[036] The variable 1\4 is any cationic counterion for the negatively
charged
tetravalent boron atom in the boron "ate" complex of formula (/1). In some
preferred
embodiments, 1\4+ is selected from the group consisting of Li, Na, and K. One
of skill in
the art will recognize that the salt MY is formed as a byproduct in the
rearrangement
reaction of step (b).
[037] The variable R1 preferably is a group with good migratory aptitude.
In some
embodiments, R' is C,4 aliphatic, cio aryl, or (C6.10 ary1)(C1, aliphatic),
any of which groups is
optionally substituted. In certain embodiments, R1 is C, aliphatic,
particularly isobutyl.
[038] The variable le preferably is hydrogen, a nucleofugic group, or an
optionally
substituted C aliphatic, C610 aryl, or (C6.10 ary.1)(C1.6 aliphatic) group.
The variable R3 =
preferably is a nucleofugic group or an optionally substituted ca aliphatic, C
aryl, or
(c10 ary1)(c, aliphatic) group. One of skill in the art will recognize that
functional
substihients may be present on any of RI, le, or R3, provided that the
functional substituent
does not interfere with the formation of the boron "ate" complex of formula
(II).
[039] One embodiment of the invention relates to a process for preparing a
boronic
ester compound of formula (/), wherein R3 is a nucleofugic group. Such
compounds are
useful as intermediates for the synthesis of alpha-substituted boronic ester
and acid
compounds, including alpha-aminoboronic ester and acid compounds, as described
below.
In certain preferred embodiments, R3 is a nucleofugic group and Ie is
hydrogen.
[040] The variables R4 and R5 can be the same or different. In some
embodiments,
R4 and R5 are directly linked, so that R4 and R5, taken together with the
intervening oxygen
- 10-
CA 02738706 2011-04-21
and boron atoms, form an optionally substituted 5- to 10-membered ring, which
can have 0-
2 additional ring heteroatoms selected from N, 0, or S. In some embodiments,
the ring is a
5- or 6-membered ring, preferably a 5-membered ring.
[041] The present invention is particularly advantageous for the Lewis acid
promoted rearrangement of boron "ate" complexes of formula (II), wherein le
and re are
directly linked and together are a chiral moiety. One embodiment of the
invention relates to
the rearrangement of such chiral boron "ate" complexes to provide a boronic
ester
compound of formula (/) wherein the carbon atom bearing R', R, and re is a
chiral center.
The rearrangement reaction preferably proceeds with a high degree of
stereodirection by the
R4-1: chiral moiety to provide the boronic ester compound of formula (/)
having a
diastereomeric ratio at the carbon atom bearing R, R2, and R2 of at least
about 96:4 relative to
a chiral center in the R4-R5 chiral moiety. Preferably, the diastereomeric
ratio is at least about
97:3.
[042] The terms "stereoisomer", "enantiomer", "diastereomer", "epimer", and
"chiral
center", are used herein in accordance with the meaning each is given in
ordinary usage by
those of ordinary skill in the art. Thus, stercoisomers are compounds that
have the same
atomic connectivity, but differ in the spatial arrangement of the atoms.
Enantiomers are
stereoisomers that have a mirror image relationship, that is, the
stereochemical configuration
at all corresponding chiral centers is opposite. Diastereomers are
stereoisomers having more
than one chiral center, which differ from one another in that the
stereochemical
configuration of at least one, but not all, of the corresponding chiral
centers is opposite.
Epimers are diastereomers that differ in stereochemical configuration at only
one chiral
center.
[043] As used herein, the term "diastereomeric ratio" refers to the ratio
between
diastereomers which differ in the stereochemical configuration at one chiral
center, relative
to a second chiral center in the same molecule. By way of example, a chemical
structure
with two chiral centers provides four possible stereoisomers: R*R, R*S, S*R,
and SS,
wherein the asterisk denotes the corresponding chiral center in each
stereoisomer. The
diastereomeric ratio for such a mixture of stereoisomers is the ratio of one
diastereomer and
its enantiomer to the other diastereomer and its enantiomer = (R*R S*S) : (R*S
S*R).
[044) One of ordinary skill in the art will recognize that additional
stereoisomers
are possible when the molecule has more than two chiral centers.. For.
purposes of the _
_ .
present invention, the term "diastereomeric ratio" has identical meaning in
reference to
-11-
CA 02738706 2011-04-21
compounds witn muinpie crural centers as if does in reference to compounds
having two
chiral centers. Thus, the term "diastereomeric ratio" refers to the ratio of
all compounds
having R*R or SS configuration at the specified chiral centers to all
compounds having R*S
or S*R configuration at the specified chiral centers. For convenience, this
ratio is referred to
herein as the diastereomeric ratio at the asterisked carbon, relative to the
second specified
chiral center.
[045] The diastereomeric ratio can be measured by any analytical method
suitable
for distinguishing between diastereomeric compounds having different relative
stereochemical configurations at the specified chiral centers. Such methods
include, without
limitation, nuclear magnetic resonance (NMR), gas chromatography (GC), and
high
performance liquid chromatography (HPLC) methods.
[046] As discussed above, one embodiment of the invention is directed to
processes
that provide a boronic ester compound of formula (1) having a diastereomeric
ratio at the
carbon atom bearing R', R2, and le of at least about 96:4 relative to a chiral
center in the RI-R5
chiral moiety. One of skill in the art will recognize that the R4-R5 chiral
moiety may itself
contain more than one chiral center. When le-le does have more than one chiral
center, it
preferably has high diastereomeric purity, and the diastereomeric ratio at the
carbon atom
bearing and R3 can be measured relative to any one of the chiral
centers in R4-Rs.
[047] In the processes of the invention, the R4-le chiral moiety preferably
has a high
level of enantiomeric purity. For purposes of the invention, the term
"enantiomeric purity"
is used to mean "enantiomeric excess", which is the amount by which the major
enantiomer
is in excess of the minor enantiomer, expressed as a percentage of the total.
Preferably, the
R1--R5 chiral moiety has an enantiomeric purity of at least about 98%, more
preferably at least
about 99%, still more preferably at least about 99.5%, and most preferably at
least about
99.9%.
[048] When the le-R5 chiral moiety has very high enantiomeric purity, the
diastereomeric ratio at the carbon atom bearing R1, R2, R3 approximates the
epimeric ratio at
that center, i.e., diastereomeric ratio :4 (R*R) : (S*R) or (R*S) : (S*S)a,
(R*) (S*). As used
herein, the term "epimeric ratio" refers to the ratio of product having one
absolute
stereochemical configuration at a given chiral center to product having the
opposite absolute
stereochemical configuration at the corresponding chiral center. Preferably,
the products
_ . have identical stereochemical.config-uration at
allbther_corresponding chiral centers. In one
embodiment, therefore, the invention relates to the rearrangement of a chiral
boron "ate"
-12-
CA 02738706 2011-04-21
complex of formula (II) to provide a boronic ester compound of formula (I)
wherein the
epimeric ratio at the carbon atom bearing R', le, and le is at least about
96:4, more preferably
at least about 97:3.
[049] Lewis acids suitable for use in the practice of the invention are
those capable
of complexing with the nucleofugic group to facilitate its displacement upon
migration of le.
Preferably, the Lewis acid is additionally capable of coordinating with art
oxygen atom
attached to boron. Nonlimiting examples of suitable Lewis acids include zinc
bromide, zinc
chloride, ferric bromide, and ferric chloride. In certain preferred
embodiments, the Lewis
acid is zinc chloride.
[050] The contacting step preferably is performed at low temperature, but
may be
performed at ambient or elevated temperature. The selection of an appropriate
reaction
temperature will depend largely on the Lewis acid employed, as well as the
migratory
aptitude of the R' moiety. One skilled in the art will be able to select a
suitable temperature
in view of the reaction conditions being used.
[051] In some embodiments, the contacting step is performed at a reaction
temperature of at least about -100 "C, -78 "C, or -60 C. In some embodiments,
the contacting
step is performed at a reaction temperature that is no greater than about SO
C, 40 C, or 30
C. Any range encompassing these high and low temperatures are included within
the
scope of the invention. Preferably, the contacting step is performed at a
reaction
temperature in the range of about -100 C to about 80 C, about -70 C to about
40 C, about
-60 C to about 30 C, or about -50 C to about 30 C. In certain preferred
embodiments, the
contacting step is begun at low temperature, preferably in the range of about -
70 C to about
-30 C, and then the reaction mixture is allowed to warm, preferably to
ambient temperature.
[052] Surprisingly, the process of the present invention requires no
special
precautions to avoid the presence of water during the rearrangement reaction
itself. In some
embodiments, moist Lewis acid is employed, with minimal deterioration in
diastereomeric
ratio. When used in reference to the Lewis acid, the term "moist" means that
the water
content of the Lewis acid is greater than about 100, 200, 500, or 1,000 ppm.
Remarkably, the
Lewis acid even can be added to the reaction mixture in the form of an aqueous
solution
without deleterious impact on diastereomeric ratio.
[053] In some embodiments, therefore, the process of the invention
comprises the
steps: -
(a) providing a solution comprising a boron "ate" complex of formula
(//) and
-13-
CA 02738706 2011-04-21
(1) a coordinating ether solvent that has low miscibility with
water; or
(ii) an ether solvent that has low miscibility with water and a
coordinating co-solvent; and
(b) adding to the solution of step (a) a Lewis add solution comprising
water and
a Lewis acid.
[054] In some other embodiments, the Lewis acid solution comprises
tetrahydrofuran and a Lewis acid.
[055] Thus, unlike the prior art process, the process of the invention is
readily
amenable to large-scale production. In various embodiments, at least about 5,
10, 20, 50, 100,
500, or 1000 moles of boron "ate complex of formula (II) is contacted with a
Lewis acid
under conditions that afford the boronic ester compound of formula (1). The
invention
further provides a composition comprising a boronic ester compound of formula
(.0, as
described herein, and an ether solvent that has low miscibility with water.
The composition
preferably comprises at least about 5, 10, 20, 50, 100, 500, or 1000 moles of
the boronic ester
compound of formula (I). In certain embodiments, R4 and R5 together are a
chiral moiety,
and the compound of formula (1) present in the composition has a
diastereomeric ratio of at
least about 96:4 at the carbon atom bearing R1, R2, and R3, relative to a
chiral center in the
R4-R5 chiral moiety.
[056] Workup of the reaction preferably comprises washing the reaction
mixture
with an aqueous solution and concentrating the washed reaction mixture by
removal of
solvents to afford a residue comprising the boronic ester compound of formula
(I).
Preferably, the residue comprises at least about 5, 10, 20, 50, 100, 500, or
1000 moles of the
boronic ester compound of formula (1). In those embodiments wherein 1<4-1Z' is
a chiral
moiety, the boronic ester compound of formula (I) present in the residue
preferably has a
diastereomeric ratio of at least about 96:4 at the carbon atom bearing R, R2,
and R3, relative
to a chiral center in the R4-R5 chiral moiety. More preferably, the
diastereomeric ratio is at
least about 97:3.
[0571 The boron "ate" complex of formula (II) can be prepared by any
known
method, but preferably is prepared by reaction of a boronic ester of formula
(M):
R1..õ0R4
'?
OR5 (M)
with a reagent of formula (IV):
-14-
CA 02738706 2011-04-21
R3
M+ (IV)
wherein each of Mt, Y, and le to R5 are as defined above for the boron "ate"
complex of
formula (II). In some embodiments, the reaction is performed at a reaction
temperature of at
least about -100 C, -78 C, or -60 C. In some embodiments, the reaction is
performed at a
reaction temperature no greater than about 0 C, -20 C, or -40 C. Any range
encompassing
these high and low temperatures are included within the scope of the
invention. The
reaction preferably is performed at a reaction temperature in the range of
about -100 C to
about 0 C, about -78 C to about -20 C, or about -60 C to about -40 C. In
some
embodiments, the boron "ate" complex of formula (II) is prepared in a solution
comprising
an ether solvent having low miscibility with water, and the reaction mixture
is directly
treated with a Lewis acid to effect rearrangement to the boron ester compound
of formula
(1)-
[0581 In some embodiments, the reagent of formula (IV) is formed in
situ. Such
embodiments include the steps:
(i) providing a solution comprising a boronic ester of formula (III), as
defined
above, and a compound of formula (V):
192.õ,..193
(V)
wherein Ie and R5 are as defined above for the reagent of formula (IV); and
(ii) treating the solution with a strong, sterically hindered base to form
the boron
"ate" complex of formula (II).
[059) In some embodiments, the sterically hindered base is an alkali
metal
dialkylamide bases of formula M2N(le)2, where M2 is Li, Na, or IC, and each
R4,
independently is a branched or cyclic Cõ aliphatic. In situ formation of the
reagent of
formula (IV) is especially advantageous in those embodiments wherein? is a
nucleofugic
group, due to the instability of the reagent of formula (IV).
[0601 The boronic ester of formula (III) can be prepared by any
known method, but
typically is prepared by esterification of the corresponding boronic acid
compound, e.g., by
, methods described in Brown et al.,.Organometallics, 2:1311-1316 (1983)._
Cyclic boronic esters - =
_
of formula (III) preferably are prepared by:
- 15-
CA 02738706 2011-04-21
(a) = providing a solution comprising:
(1) a boronic acid compound of formula R1-B(OH).2;
(ii) a compound of formula HO-R4-R5-OH, wherein R4 and 11..5, taken
together, are an optionally substituted linking chain comprising 2-5 carbon
atoms
and 0-2 heteroatorns selected from the group consisting of 0, N, and S; and
(iii) an organic solvent that forms an azeotrope with water; and
(b) heating the solution at reflux with azeotropic removal of water.
[061] As used in reference to Wand R5, the term 'slinking chain" refers to
the
shortest linear chain of atoms connecting the oxygen atoms to which R4 and le
are attached.
The linking chain optionally is substituted at any chain atom, and one or more
chain atoms
also may form part of a ring system that is spiro to, fused to, or bridging
the linear linking
chain. By way of example, but not limitation, in some embodiments, the
compound of
formula HO-R4-R5-OH is pirianediol, having the structure:
HO
In such embodiments, the linking chain R4-R5 comprises two carbon atoms, which
together
form one side of the bicyclo[3.1.11heptarte ring system, and one of which
additionally is
substituted with a methyl group.
[062] In some embodiments, the compound of formula HO-R4-R5-0H is a chiral
diol, preferably one having high diastereomeric and enantiomeric purity. One
of skill in the
art will appreciate that in such embodiment, the compound of formula H0-R1-R5-
0H is
employed as a chiral auxiliary to direct the stereochemical configuration at
the carbon
bearing R2, R2, and Ie. Chiral diols useful as chiral auxiliaries in organic
synthesis are well-
known in the art. Nonlimiting examples include 2,3-butanediol, preferably
(2R,3R)-(-)-2,3-
butanediol or (2S,3S)-(+)-2,3-butanediol; pinanediol, preferably (1R,2R,3R,5S)-
(-)-pinanediol
or (1S,2S,3S,5R)-(+)-pinanediol; 1,2-cyclopentanediol, preferably (1S,2S)-(+)-
trans-1,2-
cyclopentanediol or (1R,2R)-(-)-trans-1,2-cyclopentanediol; 2,5-hexanediol,
preferably
(2S,5S)-2,5-hexanediol or (2R,5R)-2,5-hexanediol; 1,2-dicyclohexy1-1,2-
ethanediol, preferably
(1R,21)-1,2-dicyclohexy1-1,2-ethanediol or (15,25)-1,2-dicydohexy1-1,2-
ethanediol;
hydrobenzoin,preferably.(S,S)-(-)-hydrobenzoin or.(R,R)-(+)-hydrobe.nzoin;
2,4=pentanecliol,._
preferably (R,R)-(+2,4-pentanediol or (S,S,)-( )-2,4-pentanediol; erythronic y-
lactone,
-16-
CA 02738706 2011-04-21
preferably D-erythronic y-lactone. Carbohydrates, e.g. a 1,2,5,6-symmetrically
protected
rnannitol, also may be used as chiral diols.
[063] Nonlimiting examples of organic solvents suitable for use in
the esterification
reaction include ace tonitrile, toluene, hexane, heptane, and mixtures
thereof. In some
embodiments, the organic solvent is an ether solvent, preferably an ether
solvent that has
low miscibility with water. In certain preferred embodiments, the
esterification reaction is
performed in an ether solvent that has low miscibility with water, and the
product solution
comprising the boronic ester of formula (M) is used directly in the next step,
without
isolation of the boronic ester.
[064] As noted above, the process of the present invention for the
first time permits
Nvorkup of large-scale reactions without significant deterioration in
diastereomeric ratio. In
another aspect, therefore, the invention provides a composition comprising at
least about 5,
10, 20, 50, 100 , 500, or 1000 moles of a boronic ester compound of formula
(/):
R2*
R3 B-OR4
OR5 (.1)
wherein:
R1 is an optionally substituted aliphatic, aromatic, or heteroarornatic group;
R2 is hydrogen, a nucleofugic group, or an optionally substituted aliphatic,
aromatic,
or heteroaromatic group;
R is a nucleofugic group or an optionally substituted aliphatic, aromatic, or
laeteroaromatic group; and
R4 and Rs, taken together with the intervening oxygen and boron atoms, form an
optionally substituted 5- to 10-membered chiral ring having 0-2 additional
ring
he teroatams selected from N, 0, or S;
wherein the carbon atom to which RI, R2, and R.' are attached is a chiral
center, having
a diastereomeric ratio of at least about 96:4, preferably at least about 97:3,
relative
to a chiral center in the R4-R5 chiral moiety.
[065] Preferred values for R' to le are as described above.
Preferably, solvents
- constitute less than-about 30% w/w, 20% w/w, 10% w/w, or 5% w/w of the
composition =
according to this aspect of the invention. In some embodiments, the boronic
ester
- 17 -
CA 02738706 2011-04-21
compound of formula (/) constitutes at least about 70% w/w, 80% w/w, 90% w/w,
or 95%
w/w of the composition.
[066] One embodiment relates to the composition described above, wherein
at least
one of the following features is present:
(a) R3 is chloro;
(b) the boronic ester compound (1) is
Dl
R1
112 .*
R3 Er4< R3 B4.
,
or
(c) R2 is hydrogen; and
(d) R' is C,, aliphatic.
[0671 All of the boronic ester compound of formula (1) present in the
composition
may be produced in a single batch run. For purposes of the invention, the term
"batch run"
refers to execution of a synthetic process, wherein each step of the process
is performed only
once. Preferably, the boronic ester compound of formula (1) present in the
composition is
prepared in a single batch run of the process according to the first aspect of
the invention.
One of ordinary skill in the art will appreciate that preparation of a given
quantity of
product by a single batch run of a large-scale process is more efficient and
provides a more
homogeneous product than preparation of the same quantity of product by
repeated
execution of a small-scale process.
[0681 The boronic ester compounds of formula (I) wherein le is a
nueleofugic
group are useful as intermediates for the synthesis of alpha-aminoboronic
ester compounds.
In another aspect, therefore, the invention provides a large-scale process for
preparing an
alpha-aminoboronic ester, preferably by a process comprising the steps:
(a) providing a boron "ate" complex of formula (II):
R3
R2.).,..F.110R4
Y B -
1 m+
OR' (11)
where
Y is a nucleofugic group;
- 18-
CA 02738706 2011-04-21
=
I'Vr is a cation;
RI is an optionally substituted aliphatic, aromatic, or heteroaromatic group;
R2 is hydrogen;
is a nucleofugic group; and
each of IZ4 and R5, independently, is an optionally substituted aliphatic,
aromatic, or
heieroaromatic group, or RI and R5, taken together with the intervening oxygen
and boron atoms, form an optionally substituted 5- to 10-membered ring having
0-2 additional ring heteroatoms selected from N, 0, or S;
(b) contacting the boron "ate" complex of formula (II)
with a Lewis acid under
conditions that afford the boronic ester compound of formula (I):
R1
R3 B-C)R4
OR5
(1)
where each of R to R5 is as defined above, said contacting step being
conducted in a reaction
mixture comprising:
(i) a coordinating ether solvent that has low miscibility with water; or
(ii) an ether solvent that has low miscibility with water and a
coordinating co-solvent; and
(c) treating the boronic ester compound of formula (I)
with a reagent of formula
M-N(G)2, where lv11 is an alkali metal and each G individually or together is
an amino group
protecting group to form a byproduct of formula M'-R3 and a compound of
formula (VIII):
R1
(G)2NOR4
OR5
(Viii)
wherein each G and R' to R5 are as defined above; and
(d) removing the G groups to form a compound of formula
(VD):
F11
ORS (
or an acid addition salt thereof.
- 19 -
CA 02738706 2011-04-21
[069] In some embodiments, in step (c), the boronic ester compound of
formula (1)
is treated with a reagent of formula M'-N(Si(R6)3)2 where is an alkali
metal and each R6
independently is selected from the group consisting of alkyl, aralkyl, and
aryl, where the
aryl or aryl portion of the aralkyl is optionally substituted.
[070] Reaction of the boronic ester compound of formula (1) with the
reagent of
formula M'-N(G)2 preferably is conducted at a reaction temperature in the
range of about
-100 C to about 50 C, preferably about -50 C to about 25 C, and more
preferably about -30
C to about 0 C. In some embodiments, R3 is halo, preferably cl-iloro, and M1
is Li. To
facilitate isolation of the product of formula (VIII), the reaction mixture
preferably
comprises an organic solvent in which the byproduct M'-R3 has low solubility.
Nonlimiting
examples of suitable organic solvents include methylcyclohexane, cyclohexane,
heptane,
hexane, and toluene. In some embodiments, step (c) further comprises filtering
the reaction
mixture to remove NT-Wand provide a filtrate comprising the compound of
formula (VIII).
Preferably, the filtrate is used directly in step (d).
[071] In those embodiments wherein the reaction mixture comprises an
organic
solvent in which the byproduct M'-le has low solubility, the reaction mixture
may
additionally comprise a solvent in which the byproduct IVII-R3 has high
solubility. In such
cases, the solvent in which the byproduct M'-R3 has high solubility preferably
is removed
prior to filtration of the reaction mixture. By way of example, in some
embodiments, a
reagent of formula IvV-N(Si(Ie),), is added to the reaction mixture as a
solution comprising
tetrahydrofiu:an. In such embodiments, step (c) preferably further comprises
removing the
tetrahydrofuran before filtering the reaction mixture.
[072] Those of skill in the art are aware of various methods that can be
used to
remove the protecting groups G M. the compound of formula (VIII), including,
e.g., aqueous
hydrolysis or treatment with acid. The product alpha-aminoboronic ester of
formula (V11)
has low stability and preferably is immediately derivatized (Matteson et al.,
J. Am. Chem.
Soc., 103:5241 (1981)) or is isolated as an acid addition salt. In some
embodiments, step (d)
comprises treating the compound of formula (VIII) with an acid and isolating
the compound
of formula (VII) as the acid addition salt. In certain preferred embodiments,
the acid is
trifluoroacetic acid, and the compound of formula (VII) is isolated as the
trifluoroacetic acid
addition salt.
[073].. As.discussed above, the processes of theinven.tion are particularly
well- -
suited for preparing alpha-aminoboronic ester compounds of formula (VII),
wherein the
-20-
CA 02738706 2011-04-21
WO 2005/097809 PCPUS2005/009774
alpha carbon is a chiral center. Thus, one embodiment of the invention relates
to a large-
scale process for preparing an alpha-aminoborortic ester compound of formula
(Vila) or
(Mb):
R1 Rl
H2N R4H2 R4
N1-C)
OR5 (Vila) OR5 (VIlb)
or an acid addition salt thereof, wherein:
R1 is an optionally substituted aliphatic, aromatic, or heteroaromatic group;
and
R and R5, taken together with the intervening oxygen and boron atoms, form an
optionally substituted chiral cyclic boronic ester;
said process comprising:
(a) providing a boron "ate" complex of formula (Ha) or (11b):
R3 R3
R- R.OR4 R2,1 4
= , 1,OR
Y B Y B
M+ m
6R5 (Ha) OR (ID)
where
Y is a nucleofugic group;
is a cation;
R2 is hydrogen;
R3 is a nucleofugic group; and
R4 and le are as defined above;
(b) contacting the boron "ate" complex of formula (MI) or (Ilb) with a
Lewis acid
under conditions that afford a boronic ester compound of formula (la) or (lb):
Ri 191
R2* R2 -
R3 B..0R4
R3f3'"OR4
OR5 (Ia) OR5 (lb)
where each of R to R5 is as defined above, said contacting step being
conducted in a reaction
mixture comprising:
. _
= " = (i) d-cobrdinating ether solvent that has low
miscibility with water; Or
-21-
CA 02738706 2011-04-21
WO 2005/097809 PCTIUS2005/009774
(ii) an ether solvent that has low miscibility with water and a
coordinating co-solvent; and
(c) treating the boronic ester compound of formula (In) or (lb) with a
reagent of
formula M1-N(G)2, where M' is an alkali metal and G is an amino group
protecting moiety, to
form a compound of formula (Villa) or (VIIlb):
R1
(G)2N,J.,..0R4
0R4
OR5 (Villa) OR (VIllb)
wherein each G and R to Ware as defined above; and
(d) removing the G groups to form a compound of formula (Vila) or (VIM):
R1 R1
HN_oR4
H2NB'OR4
OR5 (Vila) OR5 (VIIb)
or an acid addition salt thereof.
[074] Preferred values for Y, M, R1 to R5, and G are as described above.
The
compound of formula (Vila) or (VIM) preferably has a diastereorneric ratio at
the alpha-
carbon of at least about 96:4, more preferably at least about 97:3, relative
to a chiral center in
the W-R5 chiral moiety.
[075] The alpha-aminoboronic ester compounds of formula (V1.0 are useful
synthetic intermediates for the preparation of peptidyl boronic ester
compounds. In some
embodiments, therefore, the process according to this aspect of the invention
further
comprises coupling the compound of formula (VII) with a compound of formula
(IX):
R7
X
P -N
0 (IX)
wherein:
P' is an amino group blocking moiety;
R7 is selected from the group consisting of hydrogen, C,.,aaliphatic,
optionally
substituted Coaryl, or C,,aliphatic-R'; and
- 22 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/009774
Ra is selected from the group consisting of alkoxy, alkylthio, optionally
substituted
aryl, heteroaryl, and heterocyclyl groups, and optionally protected amino,
hydroxy, and guanidino groups; and
X is OH or a leaving group;
to form a compound of formula (X):
R7 H OR4
6,
P -N N OR5
0 R1 (X)
wherein each of P', R', 12.4, R5, and R7 is as defined above.
[076] The leaving group X is any group capable of nucleophilic displacement
by the
alpha-amino group of the compound of formula (V//). In some embodiments, the
moiety ¨
C(0)-X is an activated ester, such as an 0-(N-hydroxysucccinimide) ester. In
some
embodiments, an activated ester is generated in sit-u by contacting a compound
of formula
Ca'), wherein X is OH, with a peptide coupling reagent. Examples of suitable
peptide
coupling reagents include, without limitation, carbodiimide reagents, e.g.,
dicyclohexylcarbodiimide (DCC) or 1-(3-dimethylaminopropy1)-3-
ethylcarbodiimide (EDC);
phosphonium reagents, e.g., benzotriazol-1-yloxytals(dimethylamino)phosphonium
hexafluorophosphate (BOP reagent); and uronium reagents, e.g., 0-(1H-
benzotriazol-1-y1)-
N,N,N',N'-tetramethylurortiurn tetrafluoroborate (TBTU).
[077] Those of skill in the art also are aware of procedures that permit
the direct
coupling of sily-1 protected amines, without a prior deprotection step. In
such procedures,
the silyl groups are removed in situ under the coupling reaction conditions.
In some
embodiments of the present invention, therefore, a compound of formula (VIII)
is contacted
with a compound of formula (a) under conditions that remove the (126)3Si
groups in situ and
form a compound of formula (X).
[078] For purposes of the invention, the term "amino-group blocking moiety"
refers
to any group used to derivatize an amino group, especially an N-terminal amino
group of a
peptide or amino acid. The term "amino-group blocking moiety includes, but is
not limited
to, protecting groups that are commonly employed in organic synthesis,
especially peptide
synthesis. See, for example, Gross and Mienhoffer, eds., The Peptides, Vol. 3,
Academic Press,
. .
New York, 1981, pp. 3-88; Green and Wuts, Protective Groups in Organic
Synthesis, rd edition,
John Wiley and Sons, Inc., New York, 1999. Unless otherwise specified,
however, it is not
= -23-
CA 02738706 2011-04-21
WO 2005/097809
PCT/US20051009774
necessary for an amino-group blocking moiety to be readily cleavable. Amino-
group
blocking moieties include, e.g., alkyl, acyl, alkoxycarbonyl, aminocarbonyl,
and sulfonyl
moieties. In some embodiments, the amino-group blocking moiety is an acyl
moiety derived
from an amino acid or peptide, or a derivative or analog thereof.
[079] As used herein, the term "amino acid" includes both naturally
occurring and
unnatural amino adds. For purposes of the invention, a "derivative" of an
amino acid or
peptide is one in which a functional group, e.g., a hydroxy, amino, carboxy,
or guanidino
group at the N-terminus or on a side chain, is modified with a blocking group.
As used
herein, an "analog" of an amino acid or peptide is one which includes a
modified backbone
or side chain. The term "peptide analog" is intended to include peptides
wherein one or
more stereocenters are inverted and one or more peptide bonds are replaced
with a peptide
isostere.
[080] In some embodiments, P is a cleavable protecting group. Examples of
cleavable protecting groups include, without limitation, acyl protecting
groups, e.g., formyl,
acetyl (Ac), succinyl (Suc), or methoxysuccinyl (Me0Suc), and urethane
protecting groups,
e.g., tert-butoxycarbonyl (Boc), benzyloxycarbonyl (Cbz), or
fluorenylmethoxycarbonyl
(Fmoc).
[081] In some such embodiments, the process according to this aspect of the
invention further comprises the steps:
(f) removing the protecting group P1 to form a compound of formula (Xi):
R7 1.1 OR4
H2N y
0 R1 (A-1)
or an acid addition salt thereof, wherein each of re, re, and R7 is as defined
above; and
(g) coupling the compound of formula (XI) with a reagent of formula 13'-X,
wherein P.' is any amino group blocking moiety, as described above, and X is a
leaving
group, to form a compound of formula (XII):
R7 vi OR4
P2-N))..iN yi-o R5
o R' CA21)
wherein each of P?, R1, R4, R51 and R7 are as defined above; One-of skill in
the art will - -
recognize that in those embodiments wherein P' is an acyl group, including,
e.g., an acyl
- 24 -
CA 02738706 2011-04-21
WO 2(1(15/1)97809 ITT/t1S2005/009774
moiety derived from an amino acid or peptide, or an analog or derivative
thereof, the
leaving group X may be generated in situ, as discussed above for the compound
of formula
(IX).
[082] In each of the compounds (X) and (.121), the boronic acid moiety
is protected
as a boronic ester. If desired, the boronic acid moiety can be deprotected by
any method
known in the art. Preferably, the boronic acid moiety is deprotected by
transesterification in
a biphasic mixture. More preferably, the boronic acid deprotecting step
comprises the steps:
(i) providing a biphasic mixture comprising the boronic ester compound of
formula (X) or (Ku), an organic boronic acid acceptor, a lower alkartol, a C,õ
hydrocarbon
solvent, and aqueous mineral acid;
(ii) stirring the biphasic mixture to afford the corresponding deprotected
boronic
acid compound of formula (Xa) or (XIII):
R7 1_, OH R7
H 91-1
N 13,
P -N y OH P2-Nr N ya'OH
H
0 R1 (Xa)
0 R1 (XIII);
(iii) separating the solvent layers; and
(iv) extracting the compound of formula (Xa), (XIII), or a boronic acid
anhydride
thereof, into an organic solvent.
[083] The organic boronic acid acceptor in step (i) preferably is an
aliphatic, aryl, or
ar(aliphatic)boronic acid. In some embodiments, the boronic acid acceptor is
selected from
the group consisting of phenylboronic acid, benzylboronic acid, butylboronic
acid,
pentylboronic acid, hexylboronic acid, and cyclohexylboronic acid. In certain
embodiments,
the boronic acid acceptor is isobutylboronic acid. In some embodiments, the
boronic acid
acceptor is selected so that the boronic ester compound of formula (11I) is
formed as a
byproduct of the deprotection reaction. The boronic ester compound of formula
(III) can
then be used in another batch run of the process described above. In such
embodiments, the
moiety 1.24-R5 is effectively recycled, which may be particularly advantageous
if 124-R5 is an
expensive chiral moiety.
[084] To enhance the purity of the product, the aqueous layer
containing the
compound of formula (Xa) or (XIII) preferably is washed to remove neutral
organic
- impurities prior to the extracting step (iv).- In Stich ethbodiments, step
(iii) preferably -
comprises the steps:
- 25 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/009774
(1) separating the solvent layers;
(2) adjusting the aqueous layer to basic pH;
(3) washing the aqueous layer with an organic solvent; and
(4) adjusting the aqueous layer to a pH less than about 6.
[085] In some embodiments, the invention relates to an improved process
for
manufacturing the proteasome inhibitor bortezomib. Thus, in one embodiment,
the
invention provides a large-scale process for forming a compound of formula
(.X7V):
0 OH
H
N B,
- OH
1)/1
(XIV)
or a boronic acid anhydride thereof. The process comprises the steps:
(a) providing a boron "ate" complex of formula (XV):
R3T,C):31õ
H¨)¨B
Y b
M+ (XV)
wherein:
R3 is a nucleofugic group;
Y is a nucleofugic group; and
IV.r is an alkali metal;
(b) contacting the boron "ate" complex of formula (XV) with a Lewis acid
under
conditions that afford a boronic ester compound of formula (KW):
(xvi)
said contacting step being conducted in -a -reaction mixture comprising:
a coordinating ether solvent that has low miscibility with water; or
-26 -
CA 02738706 2011-04-21
peruS21105/00977-1
WO 2005/097809
(ii) an ether solvent that has low miscibility with water and a
coordinating co-solvent;
(c) treating the boronic ester compound of formula (XVI) with a reagent of
formula NI1-N(G)2, where IVI1 is an alkali metal and each G individually or
together is an
amino group protecting group, to form a compound of formula (XVII):
1-11
(G)2N B
(.1.7771)
(d) removing the G groups to form a compound of formula (XVIII):
,CLO s
H2N, x
(xv/n)
or an acid addition salt thereof;
(e) coupling the compound of formula (.1.1/211) with a compound of formula
(A2X);
X
P1-N
0 (.,CLX)
wherein:
P is a cleavable amino group protecting moiety; and
X is OH or a leaving group;
to form a compound of formula (MY):
H 9
N B,
0
0 y=
(Aa)
wherein 1)' is as defined above;
- 27 -
CA 02738706 2011-04-21
WO 2005/097809
ITT/US2005/009774
=
(f) removing the protecting group P1 to form a compound
of formula (2M):
4,
0
H
112N
0 y
(1M)
or an acid addition salt thereof;
(g) coupling the compound of formula (xx-r) with a
reagent of formula (.=/)
=
0
wherein X is a OH or a leaving group, to form a compound of formula
1100
0 0
H
N
0
0 y
(=/) ; and
(h) deprotecting the boronic acid moiety to form the
compound of formula (XIV)
or a boronic acid anhydride thereof.
[086] In some embodiments, the process is characterized by
at least one of the
following features (1)-(5). In certain preferred embodiments, the process is
characterized by
all five features (1)-(5) below.
(1) In the boron "ate" complex of formula (XV), 1-Z.3 and Y both are
ch_loro.
(2) The coupling step (c) comprises the steps:
(i) coupling the compound of formula (XVIII) with a
compound of
formula (XIX) wherein X is OH in the presence of 2-(1H-benzotriazol-1-y1)-
1,1,3,3-
tetran-letlayluronitun tetrafluoroborate (TBTU) and a tertiary amine in
dichloromethane;
_ _ (ii) performing a solvent-exchange to replace
dichloromethane with ethyl - -
acetate; and
-2e -
CA 02738706 2011-04-21
PCPUS2005/009774
WO 2005/097809
(iii) performing an aqueous wash of the ethyl acetate solution.
(3) The protecting group removing step (f) comprises the steps:
(1) treating the compound of formula (LX) with HC1 in ethyl
acetate;
(ii) adding heptane to the reaction mixture; and
(iii) isolating by crystallization the compound of formula (XXI) as its HC1
addition salt.
(4) The coupling step (g) comprises the steps:
(1) coupling the compound of formula (XXI) with 2-
pyrazinecarboxylic
acid in the presence of TBTU and a tertiary amine in dichloromethane;
= (ii) performing a solvent exchange to replace
dichloromethane with ethyl
acetate; and
(iii) performing an aqueous wash of the ethyl acetate solution.
(5) The boronic acid deprotecting step (h) comprises the steps:
(1) providing a biphasic mixture comprising the compound of
formula
(XXIII), an organic boronic acid acceptor, a lower alkanol, a C., hydrocarbon
solvent,
and aqueous mineral acid;
(ii) stirring the biphasic mixture to afford the compound of formula
(XIV);
(iii) separating the solvent layers; and
(iv) extracting the compound of formula (XIV), or a boronic acid
anhydride thereof, into an organic solvent.
[0871 Preferably, step (h)(iii) comprises the steps:
(1) separating the solvent layers;
(2) adjusting the aqueous layer to basic pH;
(3) washing the aqueous layer with an organic solvent; and
(4) adjusting the aqueous layer to a pH less than about 6;
- 29 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/009774
[088] In another embodiment, the invention relates to a large-scale
process for
forming a compound of formula (XIV)
41:1
0 OH
H
Nj-L N`;'B'OH
N
(xav)
or a boronic acid anhydride thereof, comprising the steps:
(aa) coupling a compound of formula (XVIII):
H2N
Gx-vin)
or an acid addition salt thereof, with a compound of formula (X/X):
410
X
P1-N
0 (A7X)
wherein:
P' is a cleavable amino group protecting moiety; and
X is OH or a leaving group;
to form a compound of formula (a'):
H 9
Pl.N N B.
0
0 y =
( X X )
wherein? is as defined above, said coupling step (aa) comprising the steps:
(i) coupling the compound of formula (XVIII) with a compound
of
= formula (,-yrx) wherein X is OH in the Presence of 2-(1H-benzotriazol-1-
y1)-1,1,3,3-
- 30 -
CA 02738706 2011-04-21
WO 2005/097809 PCl/US20051009774
tetramethyluroniurn tetrafluoroborate (TBTU) and a tertiary amine in
dichloromethane;
(ii) performing a solvent exchange to replace dichloromethane with ethyl
acetate; and
(iii) performing an aqueous wash of the ethyl acetate solution;
(bb) removing the protecting group I') to form a compound of formula (XXI):
41:1
H 9
H2N N `..?" 0
0 y
(MIT
or an acid addition salt thereof, said protecting group removing step (bb)
comprising the
steps:
(i) treating the compound of formula (XX) with HC1 in ethyl acetate;
(ii) adding heptane to the reaction mixture; and
(iii) isolating by crystallization the compound of formula (XXI) as its HC1
addition salt;
(cc) coupling the compound of formula (Xal) with a reagent of formula
(XA.7.1)
(N.,.
o
X
(XXII)
wherein X is a OH or a leaving group, to form a compound of formula ca/10:
õ
NiLN
0 H 9
N B..
-%-"' 0
0
(XXII/),
said coupling step (cc) comprising the steps:
(i) coupling the compound of formula (M.11) with
27pyrazinecarboxylic
acid in the presence of TBTU and a tertiary amine in dichloromethane;
- 31 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/00977-1
(ii) performing a solvent exchange to replace dichloromethane with ethyl
acetate; and
(iii) performing an aqueous wash of the ethyl acetate solution; and
(dd) deprotecting the boronic acid moiety to form the compound of formula
(17V)
or a boronic acid anhydride thereof, said deprotecting step (dd) comprising
the steps:
(i) providing a biphasic mixture comprising the compound of formula
(70011), an organic boronic acid acceptor, a lower alkanol, a cs hydrocarbon
solvent,
and aqueous mineral acid;
(ii) stirring the biphasic mixture to afford the compound of formula
(iii) separating the solvent layers; and
(iv) extracting the compound of formula (XIV), or a boronic acid
anhydride thereof, into an organic solvent.
[089] Preferably, step (dd)(iii) comprises the steps:
(1) separating the solvent layers;
(2) adjusting the aqueous layer to basic pH;
(3) washing the aqueous layer with an organic solvent; and
(4) adjusting the aqueous layer to a pH less than about 6;
[090] The efficiency of the processes described above is further enhanced
by
telescoping steps, for example, by carrying a reaction mixture or worked-up
product
solution from one reaction directly into the following reaction, without
isolation of the
intermediate product. For example, in some embodiments, step (c)(iii) or
(aa)(iii) affords an
ethyl acetate solution comprising a compound of formula MO, and the ethyl
acetate
solution is directly subjected in step (f) or (bb) to conditions effective to
remove the
protecting group P. In some such embodiments, the protecting group 13 is an
acid-labile
protecting group, for example, tert-butoxycarbonyl (Boc), and the ethyl
acetate solution from
step (e)(iii) or (aa)(iii) is treated with acid. In certain preferred
embodiments, the ethyl
acetate solution from step (e)(iii) or (aa)(iii) is dried azeotropically and
then treated with
gaseous HC1.
- 32-
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/009774
[091] When the deprotecting step (f) or (bb) is performed under anhydrous
conditions, as described above, the product of formula (XXI) can be isolated
by
crystallization from the reaction mixture as its HC1 addition salt.
Crystallization of the
product salt is promoted by addition of a hydrocarbon solvent such as n-
heplane. In some
embodiments, the reaction mixture is partially concentrated prior to addition
of the
hydrocarbon solvent. The present inventors have discovered that
crystallization of the
compound of formula (M7) in this manner efficiently removes any tripeptide
impurity that
may have formed during the coupling step (e) or (aa). Such impurities are
difficult to
remove at later stages in the synthesis.
[092] Further telescoping of the process is possible by carrying the
product mixture
from the coupling step (g) or (cc) directly into the boronic acid moiety
deprotecting step (h)
or (dd). Preferably, the organic solvent from the coupling reaction is first
replaced with
ethyl acetate in order to facilitate aqueous washes. A second solvent exchange
into a
hydrocarbon solvent then permits the product solution from step (g) or (cc) to
be used
directly in the biphasic boronic acid deprotecting step (h) or (dd), without
isolation of the
compound of formula (=II).
[093] Alternatively, a more convergent approach may be adopted for the
synthesis
of the compound of formula (XIV). Thus, in yet another embodiment, the
invention
provides a large-scale process for forming a compound of formula (UV)
401
0
H OH
N B.
OH
0
(-UV)
or a boronic acid anhydride thereof. The process comprises the steps:
(a) providing a boron "ate" complex of formula (KV):
R3T03:31..õ
Y 0
(XV)
wherein:
R3 is a nucleofugic group;
- 33 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/LIS20051009774
Y is a nucleofugic group; and
M+ is an alkali metal;
(b) contacting the boron "ate" complex of formula (XV) with a Lewis
acid under
conditions that afford a boronic ester compound of formula (X'V/):
QS
(xrv-r)
said contacting step being conducted in a reaction mixture comprising:
(i) a coordinating ether solvent that has low miscibility with water; or
(ii) an ether solvent that has low miscibility with water and a
coordinating co-solvent;
(c) treating the boronic ester compound of formula (.X'VI) with a
reagent of
formula 1\41-N(Si(126)3)2, where NI' is an alkali metal and each R6
independently is selected
from the group consisting of alkyl, aralkyl, and aryl, where the aryl or aryl
portion of the
aralkyl is optionally substituted, to form a compound of formula (-XVII):
-
((R6)3S1)2N
0
WWI)
(d) removing the (R6)3Si groups to form a compound of formula (XVH/):
1-12N
0
(XVIII)
or an acid addition salt thereof;
(e') coupling the compound of formula (XV/II) with a compound of formula
(X1Xa):
- 34 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/009774
0
N X
ii:))111\-11
0
(.1.7X.a)
wherein X is 0I-1 or a leaving group, to form a compound of formula (=IT):
0 0
H
N 13.
0
0 y
(=I) ; and
(f) deprotecting the boronic acid moiety to form the compound of
formula (X7V)
or a boronic acid anhydride thereof.
[094] In some embodiments, the process is characterized by at least one
of the
following features (1)-(3). In certain preferred embodiments, the process is
characterized by
all three features (1)-(3) below.
(1) In the boron "ate" complex of formula (XV), Ie and Y both are
chloro.
(2) The coupling step (e') comprises the steps:
(i) coupling the compound of formula (XVIII) with a compound of
formula (XIX-a) wherein X is OH in the presence of 2-(1H-benzotriazol-1-y1)-
1,1,3,3-
tetramethyluronium tetrafluoroborate (IBM) and a tertiary amine in
dichloromethane;
(ii) performing a solvent exchange to replace dichloromethane with ethyl
acetate; and
(iii) performing an aqueous wash of the ethyl acetate solution.
(3) The boronic acid deprotecting step (f') comprises the steps:
(i) providing a biphasic mixture comprising the compound of formula
(=II), an organic boronic acid acceptor, a lower aLkanol, a C5, hydrocarbon
solvent,
and aqueous mineral add;
(ii) stirring the biphasic mixture to afford the compound of formula
(X-717);
- 35 -
CA 02738706 2011-04-21
WO 2005;097809
PCPUS201)5/009774
(iii) separating the solvent layers; and
(iv) extracting the compound of formula (1.1V), or a boronic acid
anhydride thereof, into an organic solvent.
[0951 Preferably, step (f)(iii) comprises the steps:
(1) separating the solvent layers;
(2) adjusting the aqueous layer to basic pH;
(3) washing the aqueous layer with an organic solvent; and
(4) adjusting the aqueous layer to a pH less than about 6;
[0961 In step (h)(iv), (dd)(iv), or (1')(iv) of the processes
described above, the
= compound of formula (2ffV), or a boronic acid anhydride thereof,
preferably is extracted into
ethyl acetate and crystallized by addition of hexane or heptane. In some
embodiments, the
process further comprises isolation of a boronic acid anhydride of the
compound of formula
(KW), preferably a trimeric boronic acid anhydride of formula (XXIV):
0 0
NAN
0 y
(XXIV)
[097] The processes of the invention permit the large-scale
manufacture of
bortezomib of very high chemical and stereochemical purity. Prior art
processes were
limited in scale and afforded product of lower overall purity. In yet another
aspect,
therefore, the invention provides a composition comprising at least one
kilogram of a
compound of formula
,
0
N?H
N B---_
HI - 3
0 y
(XXIV)
- 36 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/009774
The compound of formula (MV) preferably is prepared according to the process
described
above, and preferably constitutes at least 99% w/w of the composition
according to this
aspect of the invention.
EXAMPLES
Abbreviations
BOC ter t-butoxycarbonyl
de-ionized
DIvIF N,N-dimethylformamide
GC gas chromatography
GC-MS gas chromatography-mass spectrometry
Hours
HDPE high density polyethylene
HPLC high performance liquid chromatography
LDA lithium diisopropylamide
LOD loss on drying
min Minutes
MTBE t-butyl methyl ether
RP-HPLC reverse phase high performance liquid chromatography
RPM revolutions per minute
T13TU 0-benzotriazol-1-yl-N,N,N',N'-tetramethyluronium
tetrafluoroborate
THF tetrah.ydrofuran
Example 1: (1R)-(S)-Pinanediol 1-anunonium trifluoroacetate-3-methylbutane-1-
boronate Manufacturing Process
(IS)-(S)-Pinanediol 1-chloro--3-methylbutane-1.-boronate
1. - (S)-Pinanedio1-2-Methylpropane-1-boronate (12.0 kg, 50.8 moles) was
charged to a
reaction vessel maintained under a nitrogen atmosphere.
- 37.
CA 02738706 2011-04-21
=
WO 20(15/097809
PCT/US2005/00977-1
2. tert-Butyl methyl ether (53 kg)' and dichloromethane (22.5 kg) were
charged and the
resultant mixture was cooled to ¨57 C with stirring.
3. Diisopropylamine, (6.7 kg) was charged to another reaction vessel
maintained under
a nitrogen atmosphere.
4. fert-Butyl methyl ether (27 kg) was charged to the diisopropylamine and
the
resultant mixture was cooled to ¨10 C with stirring. .
5. n-Hexyllithium in hexane (33.2 weight% solution) (17.6 kg) was added to
the
diisopropylamin.e mixture over a period of 57 minutes, while the reaction
temperature was maintained at ¨10 C to ¨7 C.
6. This mixture (LDA-mixture) was stirred for 33 minutes at ¨9 C to ¨7 C
before it was
used.
7. Zinc chloride, (12.1 kg) was charged to a third reaction vessel
maintained under a
nitrogen atmosphere.
S. tert-Butyl methyl ether (16 kg) was charged to the zinc
chloride and the resultant
mixture was warmed to 30 C with stirring.
9. Tetrahydrofuran (53 kg) was added to the zinc chloride suspension over a
period of
18 minutes, while the reaction temperature was maintained at 35 C to 40 C.
10. This mixture (ZnC12-mixture) was stirred for 4 hours and 28 minutes at
38 C to 39 C
until it was used.
11. The LDA-mixture (from #3 - 6) was added over a period of 60 minutes to
the
reaction vessel containing (S)-pinariedio1-2-methylpropane-1-boronate, while
the
reaction temperature was maintained at -60 C to ¨55 C.
12. A tert-butyl methyl ether rinse (10 kg) was used to complete the
addition.
13. The reaction mixture was stirred for an additional 20 minutes at -59 C
to ¨55 C.
14. The reaction mixture was warmed to -50 C over a period of 11 minutes.
15. The ZnC1,-mixture (from # 7 - 10) was added over a period of 48 minutes
to the
reaction vessel containing (S)--pinanedio1-2-methylpropane-1-boronate and the
LDA-
mixture, while the reaction temperature was maintained at -50 C to ¨45 C.
16. A tert-butyl methyl ether rinse (10 kg) was used to complete the
addition. -
_
- 38 -
=
CA 02738706 2011-04-21
WO 2005/097809
PCT/1JS2005/009774
17. The reaction mixture was stirred for an additional 30 minutes at -45 C
to ¨40 C and
then warmed to 10 C over a period of 81 minutes.
18. A 10% sulfuric acid solution (72 kg) was added over a period of 40
minutes to the
reaction vessel, while the reaction temperature was maintained at 10 C to 21
C.
19. The reaction mixture was stirred for 16 minutes at ambient temperature,
before the
aqueous phase was separated.
20. The organic phase was washed successively with deionized (D.I.) water
(32 kg), and
10% sodium chloride solution (26.7 kg), each wash involved vigorous stirring
for 15
to 17 minutes at ambient temperature.
21. The reaction mixture was concentrated under reduced pressure (p.õ = 81
mbar),
maintaining an external (jacket/bath) temperature of 50 C to 55 C, providing
a
residue which was dissolved in methylcyclohexane (56 kg).
22. The reaction mixture was refluxed (in a Dean-Stark type condenser for
water
separation) under reduced pressure (põ, = 67 mbar), maintaining an external
(jacket/bath) temperature of 50 C to 55 C for 2 hours and 7 minutes, until
no more
water was separated.
23. About 35 L of.the solvents were distilled off under reduced pressure
(põ, -= 81 mbar),
maintaining an external (jacket/bath) temperature of 50 C to 55 C.
24. The resultant dry methylcyclohexane mixture containing (1S)-(S)-
pinanedio11-
chloro-3-methylbutane-1-boronate was cooled to 14 C.
(1R)-(S)-Pinanediol 1-bis(trimethylsilyl)amino-3-methylbutane-1-boronate =
1. Lithium bis(trimethylsilyl)amicie in tetrahydrofuran (19.4 weight%
solution), (41.3
kg) was charged to a reaction vessel maintained under a nitrogen atmosphere
and
cooled to -19 C with stirring.
2. The methylcyclohexane mixture containing (1S)-(S)-pinanediol 1-chloro-3-
methylbutane-1-boronate was added over a period of 55 minutes, while the
reaction
temperature was maintained at ¨19 C to ¨13 C.
3. A methylcyclohexane rinse (5 kg) was used to complete the addition.
4. The reaction mixture was stirred for an additional 65 minutes at -13 C
to -12 C and
- =.
= = then Warnied to 25 C over 'a period of 25 minutes.
-39-
CA 02738706 2011-04-21
WO 2005/097809
ITT/US2005/009774
5. A suspension of Celite (2.5 kg) in methylcyclohexane (22 kg) was added
to the
reaction mixture.
6. The reaction mixture was concentrated under reduced pressure (p.õ,= 25
mbar),
maintaining an external (jacket/bath) temperature of 45 C to 50 C, providing
a
residue which was dissolved in methylcyclohexane (36 kg).
7. A sample was then removed for in-process testing for tetrahydrofuran
content by
GC.
8. The tetrahydrofuran assay was 0.58%.
9. The solids were removed by filtration, the filtrate was filtered through
a plug of
Silica Gel (2.0 kg).
10. Both filter units were washed with isopropyl ether (30 kg).
11. The resultant methylcyclohexane/isopropyl ether mixture containing (1R)-
(S)-
pinarteclio11-bis(trimethylsilyl)amino-3-methylbutane-1-boronate was stored in
a
container at ambient temperature until it was used in the next step.
(1R)-(S)-Pinanediol 1-ammonium trifluoroacetate-3-n-tethylbutane-1-boronate
1. Trifluoroace tic acid, (12 kg) was charged to another reaction vessel
maintained under
a nitrogen atmosphere.
2. Isopropyl ether (78 kg) was charged to the tifluoroacetie acid and the
resultant
mixture was cooled to ¨10 C with stirring.
3. The methylcyclohexane /isopropyl ether mixture containing (1R)-(S)-
pinanediol 1-
bis(timethylsilyl)ainino-3-methylbutane-1-boronate was added over a period of
53
minutes causing product precipitation, while the reaction temperature was
maintained at ¨10 C to ¨5 'C.
4. An isopropyl ether rinse (5 kg) was used to complete the addition.
5. The reaction mixture was stirred for an additional 8 hours and 20
minutes at -9 C to
-7 C.
6. The solid was collected by filtration, washed with isopropyl ether (70
kg) in two
portions, and dried under reduced pressure (pmin = 56 mbar) at 41 C to 42 C
for 2
hours and 15 minutes.
- 40 -
CA 02738706 2011-04-21
WO 2005/097809 PCT/US2005/009774
7. The solid was stirred with D.I. water (60 kg) for 24 minutes at ambient
temperature,
before the D.I. water was removed by filtration.
S. The solid was washed with D.I. water (12 kg).
9. The solid was then dried under vacuum (pmin = 4 mbar) at 40 C to 44 C
for 9 hours
and 22 minutes, after that time the loss on drying was 0.51%, which meets the
5_ 1%
requirement.
10. The intermediate, (1R)-(S)-pinanediol 1-ammonium trifluoroacetate-3-
methylbutane-
1-boronate, crude, was then packaged into single polyethylene bags in
polypropylene drums and labeled. The yield was 72%.
Recrystallization of (1R)-(S)-pinanediol 1-ammonium trifluoroacetate-3-
methylbutane-1-
boronate, crude
1. (1R)-(S)-Pinanediol i-ammoniuin trifluoroacetate-3-methylbutane-1-
boronate, crude,
(13 kg) was charged to a reaction vessel maintained under a nitrogen
atmosphere.
2. Trifluoroacetic acid (31 kg) was charged to the reaction vessel and the
resultant
mixture was cooled to 4 C with stirring.
3. Once all of the solid was dissolved leaving a slightly turbid mixture,
isopropyl ether
(29 kg) was added over a period of 57 minutes, while the reaction temperature
was
maintained at 2 C to 3 C.
4. After complete addition the mixture was filtered through a filter into a
receiving
vessel maintained under a nitrogen atmosphere.
5. Reactor and filter were rinsed with a mixture of trifluoroacetic acid
(3.8 kg) and
isopropyl ether (5 kg). The rinse was added to the filtrate.
6. Isopropyl ether (126 kg) was added over a period of 15 minutes causing
product
precipitation, while the reaction temperature was maintained at 16 C to 18
C.
7. The mixture was stirred at 16 C to 18 C for 15 min, then cooled to -5
C over a
period of 67 minutes, and stirred at -3 C to -5 C under a nitrogen
atmosphere for 89 =
minutes.
8. The solid was then isolated by filtration, washed with isopropyl ether
(48 kg) in two
portions, and dried under vacuum (pmin = 2 mbar) at 34 C to 40 C for 2 hours
and
55 minutes afterthat-time the loss on-drying-was 0.32%, which meets the 0.5%
requirement.
-41-
CA 02738706 2011-04-21
WO 2005/097809 1)CT/ VS2005/009774
9. The product, (1R)-(S)-pinanediol 1-ammonium trifluoroacetate-3-
methylbutane-l-
boronate, was then packaged into double polyethylene bags in fiber drums and
labeled. The yield was 36%.
Example 2: N-(2-Pyrazinecarbony1)-L-phenylalanine-L-leucine boronic anhydride
=
Manufacturing Process
(1S,25,3R,5S1-Pinanediol N-B0C-L-phenylalanine-L-leucine boronate
1. In a fume hood, a three-necked glass reaction flask equipped with a
Claisen head
temperature recorder and a mechanical stirrer was flushed with nitrogen.
2. (1R)-(S)-Pinanediol 1-ammonium trifluoroacetate-3-methylbutane-1-
boronate (2.0
kg), was charged to the flask.
3. BOC-L-phenylalanine (1.398 kg) was charged to the flask.
4. 2-(1H-13enzotriazol-1-y1)-1,1,3,3-tetramethyl uronium tetrafluoroborate,
TBTU (1.864
kg) was charged to the flask.
5. Dichlorornethane (15.3 L) was charged to the flask.
6. The stirring motor was adjusted to provide stirring at 260 RPM.
7. Using an ice/water cooling bath, the reaction mixture was cooled to 1.0
C,
maintaining a nitrogen atmosphere.
3. N,N-Diisopropylethylamine (2.773 L) was charged to a glass flask and
transferred to
the reaction mixture over a period of 117 minutes using a peristaltic pump
maintaining a reaction temperature range of 0.7 C ¨ 2.1 C. The overall
addition rate
was 23.7 rnL/rnin.
9. A dichloromethane (0.2 L) rinse of the flask into the reaction mixture
was used to
complete the addition.
10. The reaction mixture was stirred for an additional 33 minutes. The
temperature at the
start of the stir time was 1.8 C, and 2.5 C at the end.
11. A sample was then removed for in-process testing by reverse phase high
performance liquid chromatography (RP-HPLC). The percent conversion was
determined to be 99.3%.
12. The reaction mixture was transferred in approximately two equal
halves to two . =
. .
rotary evaporator flasks. The reaction mixture was concentrated under reduced
- 42 -
CA 02738706 2011-04-21
WO 2005/097809 PCT/US2005/009774
pressure using a rotary evaporator, maintaining an external bath temperature
of 29-
30 C.
13. Ethyl acetate (4.0 L) was divided into two approximately equal portions
and charged
to the two rotary evaporator flasks.
14. The mixtures in each flask were again concentrated under reduced
pressure using a
rotary evaporator, maintaining an external bath temperature of 29-30 'C.
= 15. The residues in each rotary evaporator flask were then
transferred back to the
reaction flask using ethyl acetate (13.34 L).
16. In a glass flask equipped with a stirrer, a 1% aqueous phosphoric acid
solution was
prepared by mixing D.I. water (13.18 L) and phosphoric acid (0.160 kg).
17. In a glass flask equipped with a stirrer, a 2% aqueous potassium
carbonate solution
(12.0 L) was prepared by mixing D.I. water (11.76 L) and potassium carbonate
(0.24
kg).
18. In a glass flask equipped with a stirrer, a 10% aqueous sodium chloride
solution
(13.34 L) was prepared by mixing D.I. water (13.34 L) and sodium chloride
(1.334 kg).
19. D.I. water (13.34 L) was charged to the reaction flask containing the
ethyl acetate
solution and the mixture stirred at 380 RPM for 7 minutes. The layers were
allowed
to separate and the aqueous phase (bottom layer) was transferred under vacuum
to a
suitable flask and discarded.
20. Again, DJ. water (13.34 L) was charged to the reaction flask containing
the ethyl
acetate solution and the mixture stirred at 385 RPM for 7 minutes. The layers
were
allowed to separate and the aqueous phase (bottom layer) was transferred under
vacuum to a suitable flask and discarded.
21. The 1% phosphoric acid solution prepared in Step 16 was charged to the
reaction
flask containing the ethyl acetate solution and the mixture stirred at 365 RPM
for 7
minutes. The layers were allowed to separate and the acidic aqueous phase
(bottom
layer) was transferred to a suitable flask and discarded.
22. The 2% potassium carbonate solution prepared in Step 17 was charged to
the
reaction flask containing the ethyl acetate solution and the mixture stirred
at 367
RPM for 7 minutes. The layers were allowed to separate and the basic aqueous
phase .
(bottorn layer) was 'transferred to a suitable flask and discarded.
- 43 -
CA 02738706 2011-04-21
WO 200.5/097809 ITT/US2005/1109774
23. The 10% sodium chloride solution prepared in Step 18 was charged to the
reaction
flask containing the ethyl acetate solution and the mixture stirred at 373 RPM
for 6
minutes. The layers were allowed to separate and the aqueous phase (bottom
layer)
was transferred to a suitable flask and discarded.
24. The ethyl acetate solution was transferred to a rotary evaporator flask
and
concentrated under reduced pressure using a rotary evaporator, maintaining a
bath
temperature of 29-30 C, to provide a residue.
25. The residue was then redissolved in ethyl acetate (4.68 L).
26. The solution was concentrated under vacuum using a rotary evaporator,
maintaining
a bath temperature of 29-30 C, to provide a residue once more.
27. Again, the residue was then redissolved in ethyl acetate (4.68 L) and
two samples
taken for determination of water content by Karl Fisher titration. The waler
content
of two samples was determined as 0.216 `)/0 and 0.207 %.
28. Using a further quantity of ethyl acetate (12.66 L), the mixture was
transferred from
the rotary evaporator flask to a dry reaction flask equipped with a
temperature
recorder, a mechanical stirrer, and a fritted gas dispersion tube, and purged
with
nitrogen.
(1S,2S,31Z,5S)-Pinanediol L-phenylalanine-L-leucine boronate, HC1 salt
1. The ethyl acetate solution containing (1S,2S,3R,5S)-pinanediol N-BOC-L-
phenylalanine-L-leucine boronate was cooled using an ice/water cooling bath to
- =
0.9 C.
2. Hydrogen chloride (1.115 kg) gas was bubbled into the reaction mixture
over a
period of 1.48 hours. The temperature at the start of the addition was -0.9
C, and
6.8 C at the end.
3. The reaction was then allowed to warm to 14.4 C over 50 minutes, while
maintaining a nitrogen atmosphere.
4. A sample was removed for in-process testing by RP-EIPLC. The percent
conversion
was 68.9 % (area %).
5. The reaction was stirred for 35 minutes. The temperature at the start
was 14 C, and
14.8 C at the end.
- 44 -
CA 02738706 2011-04-21
WO 2005/097809
PCT/US2005/009774
6. A sample was removed for in-process testing by RP-I-IPLC. The percent
conversion
was 94.7% (area /0).
7. The reaction was stirred for approximately a further 50 minutes,
maintaining a
temperature of 10 C 5 C.
8. A sample was removed for in-process testing by RP-I-I2LC. The percent
conversion
was 97.3%.
9. The reaction was stirred for approximately a further 50 minutes,
maintaining a
temperature of 10 C 5 C. The final temperature was 14.6 C.
10. A sample was removed for in-process testing by RP-I-EPLC. The total
reaction time
after addition of hydrogen chloride gas was four (4) hours.
11. The percent conversion was 99%.
12. A slurry was observed.
13. n-I-Ieptane (8.8 L) was charged to the reaction mixture.
14. The slurry was stirred for 2 hours. The temperature at the start of the
stir time was
12.7 C, and 15.3 C at the end.
15. The solid was isolated by filtration on a Buchner funnel lined with a
polypropylene
felt filter pad.
16. The solid was washed with n-heptane (4.68 L).
17. In a hood, the solid was transferred to three drying trays at not more
than 1" deep
and air-dried for 1 hour.
18. The solid was then dried at <35 C under a vacuum of 27" of Hg for 16
hours 28
minutes in a vacuum oven equipped with a vacuum gauge and a temperature
recorder.
19. The solid was sampled from each drying tray to determine the % Loss on
Drying.
The LOD was determined to be 0 %, 0.02 A), and 0.02 % on the three samples
taken.
20. (1S,2S,3R,5S)-Pinanediol L-phenylalanine-L-leucine boronate, NCI salt
was then
packaged into double poly bags in fiber drums and labeled, and sampled.
21. The isolated yield was 1.57 kg, 79.1%. The intermediate was stored at 2-
8 C until
used in further manufacturing. .
- 45 -
CA 02738706 2011-04-21
WO 2005/097809
S2005/009774
(2S,25,3R,55)-Pinanediol N-(2-pyrazinecarbony1)-L-phenylalanine-L-leucine
boronate
1. In a fume hood a three-necked glass reaction flask equipped with a
Claisen head,
temperature recorder and a mechanical stirrer was flushed with nitrogen.
2. (/S,2S,3R,5S)-Pinanediol L-phenylalanine-L-leucine boronate, HG! salt
(1.85 kg) was
charged to the flask.
3. 2-Pyrazinecarboxylic acid (0.564 kg) was charged to the flask.
4. 2-(H-Benzotriazol-1-y1)-1,1,3,3-tetramethyl tuonitun tetrafluoroborate,
.1.13TU (1.460
kg) was charged to the flask.
5. Dic_hloromethane (18.13 L) was charged to the flask.
6. The stirring motor was adjusted to provide stirring at 272 RPM.
7. Using a cooling bath, the reaction mixture was cooled to -1.2 C.
8. N,N-Diisopropylethylarnine (1.865 kg) was charged to a glass flask and
transferred
to the reaction over a period of 50 minutes using a peristaltic pump
maintaining a
reaction temperature range of -1.2 C to 2.8 C.
9. A dichloromethane rinse (0.37 L) of the flask into the reaction mixture
was used to
complete the addition.
10. The reaction mixture was allowed to warm and stirred for an additional
81 minutes.
11. The temperature at the start of the stir time was 15 C, and 24.9 C at
the end.
12. A sample was then removed for in-process testing by RP-I-IPLC. The
percent
conversion was determined to be 99.9%.
13. The reaction mixture was transferred in approximately two equal halves
to two
rotary evaporator flasks. The reaction mixture was concentrated under reduced
pressure using two rotary evaporators, maintaining an external bath
temperature of
33-34 C.
14. Ethyl acetate (12.95 L) was divided into two approximately equal
portions and
charged to the two rotary evaporator flasks.
15. The mixtures in each flask were then concentrated under reduced
pressure using a
rotary evaporator, maintaining an external bath temperature of 33-34 C.
16. - The residues in each rotary evaporator flask were then transferred back
to the
reaction flask using ethyl acetate (12.95 L).
- 46 -
CA 02738706 2011-04-21
WO 2005/0978(19
PCT/US2005/00977-1
17. In a glass flask equipped with a stirrer, a 1% aqueous phosphoric acid
solution (12.34
L) was prepared by mixing D.I. water (12.19 L) and phosphoric acid (0.148 kg).
18. In a glass flask equipped with a stirrer, a 2% aqueous potassium
carbonate solution
(12.34 L) was prepared by mixing D.I. water (12.09 L) and potassium carbonate
(0.247
kg).
19. In a glass flask equipped with a stirrer, a 10% aqueous sodium chloride
solution
(12.34 L) was prepared by mixing D.I. water (12.34 L) and sodium chloride
(1.234 kg).
20. D.I. water (12.34 L) was charged to the reaction flask containing the
ethyl acetate
solution and the mixture stirred at 382 RPM for 7 minutes. The layers were
allowed
to separate and the aqueous phase (bottom layer) was transferred to a suitable
flask
and discarded.
21. Again, D.I. water (12.34 L) was charged to the reaction flask
containing the ethyl
acetate solution and the mixture stirred at 398 RPM for 7 minutes. The layers
were
allowed to separate and the aqueous phase (bottom layer) was transferred to a
suitable flask and discarded.
22. The 1% phosphoric acid solution prepared in Step 17 was charged to the
reaction
flask containing the ethyl acetate solution and the mixture stirred at 364 RPM
for 8
minutes. The layers were allowed to separate and the acidic aqueous phase
(bottom
layer) was transferred to a suitable flask and discarded.
23. The 2% potassium carbonate solution prepared in Step 18 was charged to
the
reaction flask containing the ethyl acetate solution and the mixture stirred
at 367
RPM for 8 minutes. The layers were allowed to separate and the basic aqueous
phase
(bottom layer) was transferred to a suitable flask and discarded.
24. The 10% sodium chloride solution prepared in Step 19 was charged to the
reaction
flask containing the ethyl acetate solution and the mixture stirred at 374
RP1v1 for 8
minutes. The layers were allowed to separate and the aqueous phase (bottom
layer)
was transferred to a suitable flask and discarded.
25. The ethyl acetate solution was transferred under vacuum in
approximately two equal
halves to two rotary evaporator flasks and concentrated under reduced pressure
using a rotary evaporator, maintaining an external bath temperature of 34 C.
26. n-Heptane (143 L)-Nvas divided-intd two approximately equal portions
and charged
to the two rotary evaporator flasks. The mixtures in each flask were then
- 47 -
CA 02738706 2011-04-21
WO 201)5/097309
PCT/US2005/009774
=
concentrated under reduced pressure using a rotary evaporator, maintaining an
external bath temperature of 34 C.
N-(2-Pvrazinecarbony1)-L-Dhenyialanine-L-leticine boronic anhydride, crude
1. In a glass flask equipped with a stirrer, a 1N solution of hydrochloric
acid (22.2 L)
was prepared by mixing D.I. water (20.36 L) and hydrochloric acid (1.84 kg).
2. In a glass flask equipped with a stirrer, a 2N sodium hydroxide solution
(12.03 L)
was prepared by mixing DI water (12.03 L) and sodium hydroxide (0.962 kg).
3. The residues containing (2S,2S,3R,55)-pinanediol N-(2-pyrazinecarbony1)-
L-
phenylalanine-L-leudne boronate in each rotary evaporator flask were then
transferred to a three-necked glass reaction flask equipped with a temperature
recorder and a mechanical stirrer, using n-heptane (14.8 L) and methanol (14.8
L).
4. The stirring motor was adjusted to provide stirring at 284 RPM.
5. 2-1vIethylproparieboronic acid (0.672 kg) was charged to the flask.
6. 1N hydrochloric acid prepared in Step 1(11.2 L) was charged to the
flask.
7. The stirring motor was adjusted to provide stirring at 326 RPM.
S. The reaction mixture was stirred for 16.38 hours The start
batch temperature was
28.6 C, and the end batch temperature was 21.6 C.
9. A sample was then removed for in-process testing by RP-I-IPLC.
10. The percent conversion was determined to be 100%.
11. Stirring was stopped and the biphasic mixture allowed to separate.
12. The n-heptane layer (upper layer) was transferred to a suitable flask
and discarded.
13. n-Heptane (5.37 L) was charged to the reaction flask and the mixture
stirred at 351
RPM for 6 minutes. The layers were allowed to separate and the n-heptane phase
(upper layer) was transferred to a suitable flask and discarded.
14. Again, n-hep Lane (5.37 L) was charged to the reaction flask and the
mixture stirred at
340 RPM for 6 minutes. The layers were allowed to separate and the n-heptane
phase
(upper layer) was transferred to a suitable flask and discarded.
15. The aqueous methanol solution was transferred in approximately two
equal halyes .
. .
to two rotary evaporator flasks and concentrated under reduced pressure using
a
- 48 -
CA 02738706 2011-04-21
WO 2005/097809
PCITUS2005/009774
rotary evaporator, maintaining an external bath temperature of 33-34 C. 15 L
of
methanol were collected.
16. Dichloromethane (5.37 L) was used to transfer the residue from the
rotary evaporator
flasks back into the reaction flask.
17. 2N sodium hydroxide (11.2 L) prepared in Step 2 was charged to the
flask.
18. The dichloromethane layer (lower layer) was transferred to a suitable
flask and
discarded.
19. DicMoromethane (5.37 L) was charged to the flask and the mixture
stirred at 374
RPM for 6 minutes. The phases were allowed to separate and the dichloromethane
layer (lower layer) was transferred to a suitable flask and discarded.
20. Again, dichloromethane, (5.37 L) was charged to the flask and the
mixture stirred at
368 RP1v1 for 8 minutes. The phases were allowed to separate and the
dichloromethane layer (lower layer) was transferred to a suitable flask and
discarded.
21. Dichloromethane (5.37 L) was charged to the flask.
22. 1N hydrochloric acid (10.7 L) was charged to the flask with stirring.
The pH of the
aqueous phase was determined to be 6.
23. Stirring was discontinued and the phases allowed to separate.
24. The dichloromethane phase (lower layer) was transferred under vacuum to
a glass
receiving flask.
25. Dichloromethane (5.37 L) was charged to the flask and the mixture
stirred at 330
RPM for 6 minutes. The phases were allowed to separate and the
dichlorornethane
layer (lower layer) was transferred to the glass receiving flask.
26. Again, clichlorome thane, (5.37 L) was charged to the flask and the
mixture stirred at
335 RPM for 6 minutes. The phases were allowed to separate and the
dichloromethane layer (lower layer) was transferred to the glass receiving
flask.
27. The dichioromethane extracts were combined and transferred in
approximately two
equal halves to two rotary evaporator flasks and concentrated under reduced
pressure using a rotary evaporator, maintaining an external bath temperature
of 33-
34 C.
-49 -
CA 02738706 2011-04-21
periUS2005/009774
WO 2005/097809
28. Ethyl acetate (12.95 L) was divided into two approximately equal
portions and
charged to the two rotary evaporator flasks. The mixtures in each flask were
then
concentrated under reduced pressure using a rotary evaporator, maintaining an
external bath temperature of 45-46 C.
29. Again, ethyl acetate (12.95 L) was divided into two approximately equal
portions and
charged to the two rotary evaporator flasks. The mixtures in each flask were
then
concentrated under reduced pressure using a rotary evaporator, maintaining an
external bath temperature of 45-46 C, until approximately 10% of the original
volume remained.
30. n-Ilep lane (10.2 L) was divided into two approximately equal portions
and charged
to the two rotary evaporator flasks, and the slurry stirred under a nitrogen
atmosphere for 2.67 hours at 22-23 C.
31. The solid was isolated by filtration on a Buchner funnel, lined with a
polypropylene
felt, filter pad.
32. The solid was washed with n-heptane (2.96 L).
33. In a hood, the solid was transferred to four drying trays and air-dried
for 1.25 hours.
34. The solid was then dried at 36 - 50 C under a vacuum of 27" of lig for
18 hours 27
minutes in a vacuum oven equipped with a vacuum gauge and a temperature
recorder.
35. The solid was sampled from each tray to determine the % Loss on Drying
(LOD). The
LOD was determined to be 0.38%, 0.62%, 0.71%, and 0.63% on the four samples
taken.
36. N-(2-Pyrazinecarbony1)-L-phenylalartine-L-leucine boronic anhydride,
crude was
packaged into two 5L, HDPE, tamper-proof wide-mouth bottles and labeled.
37. The isolated yield was 1.314 kg, 83%.
Recrystallization of N-(2-Pyrazinecarbony1)-L-phenylalanine-L-leucine boronic
anhydride,
crude
1. In a hood a glass reaction flask equipped with a mechanical stirrer, a
reflux
condenser and a temperature recorder was flushed with nitrogen.
2. Ethyl acetate.(21.L) was charged-to the flask.-- - - -
- 50 -
CA 02738706 2011-04-21
WO 2005/097809 ITT/
82005/009774
3. The ethyl acetate was heated to 66.8 C under a nitrogen atmosphere,
using a hot
water/steam bath.
4. N-(2-Pyrazinecarbony1)-L-phenylalanine-L-leucine boronic anhydride,
crude (1.311
kg) was slowly charged to the reaction flask. Charging occurred over a period
of 3
minutes.
5. The mixture was stirred for 1 minute until all the solid had dissolved.
The
temperature of the solution was 64 'C.
6. The heat source was removed and the mixture was slowly cooled to 60 C
using a
cold bath. =
7. The hot ethyl acetate solution was transferred into a receiving flask
via poly tubing
and a polypropylene in-line filter capsule using a peristaltic pump.
8. The mixture was allowed to cool to 27.2 C, and allowed to stand under a
nitrogen
atmosphere without stirring, for 17.75 hours. The final temperature was
recorded as
20.5 C.
9. The mixture was cooled using an ice/water bath with stirring for 2.33
hours. The
temperature at the start of the stir time was 3.8 C, and -2.8 C at the end.
10. The solid was isolated by filtration on a Buchner funnel lined with a
polypropylene
felt filter pad. The filtrate was collected in a collection flask.
11. The solid was washed with ethyl acetate (2.62 L), cooled to 4.7 C.
12. In a hood, the solid was transferred to two drying trays.
13. The solid was then dried at 51-65 C under a vacuum of 27" of Hg for 19
hours 10
minutes in a vacuum oven equipped with a vacuum gauge and a temperature
recorder.
14. The solid was sampled to determine the % Loss on Drying (LOD). The LOD
was
determined to be 0.65 To and 0.62 % on the two samples taken.
15. N-(2-Pyrazinecarbony1)-L-phenylalanine-L-leucine boronic anhydride was
packaged
into four 1L, Type 3, Amber Wide-Mouth Bottles with Teflon-Lined Caps and
labeled.
16. The isolated yield was 1.132 kg, 86.3%.
17. N-(2-Pyrazinecarbony1)-L-phenylalanine-L-leucine boronic anhydride was
stored at
¨ 25 to -15 'C.
-51 -
CA 02738706 2011-04-21
WO 20(15/097809
PCT/US21105/009774
Example 3: N-(2-PyrazinecarbonyI)-L-phenylalanine-L-leucine boronic anhydride
Convergent Synthesis
US 2S 3R 5S -Pinanediol vrazinecarlanine-L-leucine boronatg
[098] A solution of (1R)-(S)-Pinanediol 1-ammonium trifluoroacetate-3-
methylbutane-1-boronate (13.97 g) and N-hydroxysuccirtimide (6.23 g) of in 66
mL of DMF
was cooled to ¨5 C, followed by the addition of dicyclohexylcarbodiimide
(10.83 g). The
resulting suspension was stirred for one hour at a temperature of ¨5 to 0 C.
To a solution of
N-(2-pyrazinecarbony1)-L-phenylalanine (19.52 g; prepared by coupling the
preformed
succinimide ester of pyrazinecarboxylic acid with L-phenylalardne in dioxarte-
water) in 62
mL of D/vIF was added N-methylniorpholine (5.7 mL) at a temperature of 0 C,
and the
resulting solution was added to the suspension. The suspension was adjusted to
pH 7 by
the addition of another 5.7 mL of N-rnethylmorpholine and stirred overnight,
raising the
temperature slowly to 21 'C. After filtration, the filtercake was washed twice
with MTBE
and the combined filtrates were diluted with 950 mL of MTBE. The organic layer
was
washed with 20% aqueous citric acid (3 x 150 mL), 20% aqueous NaHCO, (3 x 150
mL), and
brine (2x). The organic layer was dried over Na,SOõ filtered, and
concentrated, yielding 25.5
g (95.5 /0) of the title compound as a foam. As indicated by tic this
material contained some
minor impurities, induding approximately 2% of cyclohexyl urea.
N-(2-Pyrazinecarbony1)-L-phenylalanine-L-leucine boronic anhydride
[099] A solution of (1S,2S,3R,5S)-Pinanediol N-(2-pyrazinecarbony1)-L-
phenylalanine-L-leueine boronate (25.2 g) in 207 mL of IVIe0.H and 190 rilL of
hexane was
cooled to 15 C, and 109.4 mL of 1N I-ICI were added in portions, keeping the
temperature
between 15 and 25 C. 2-IvIethylpropaneboronic acid (8.67 g) was then added
under
vigorous stirring, and the stirring of the biphasic mixture was continued over
night. After
separation of the two phases, the lower layer was extracted once with 75 mL of
hexane. The
lower layer was then concentrated in vacuo until it became cloudy, followed by
the addition
of 109.4 mL of 2N NaOH and 100 mL of Et,O. The two phases were separated the
lower
layer was extracted with Etp (4 x 100 mL each), and then brought to pH 6.0 by
the addition
of 109 n-11, of 1N HCI. After extraction with 100 mL of ethyl acetate, the
lower layer was
adjusted to 6.0 with 1N 1-ICI and extracted one more time with 75 mL of
ethyl acetate.
The combined ethyl acetate layers were washed with semi-saturated brine (2 x
25 mL) and
brine (2 x 25 mL), dried over Na,SO4, filtered, and concentrated to afford
15:3 g (81.8 To) of
crude N-(2-PyrazinecarbonyI)-L-phenylalanine-L-leucine boronic anhydride as a
foam. The
- 52 -
CA 02738706 2011-04-21
WO 2005/097809
ITT/URN5/009774
crude material was dissolved in 150 mL of ethyl acetate and concentrated in
vacuo to a
suspension, followed by the addition of 150 iriL of MTBE. The suspension was
stored
between 2 and 8 C over night, filtered, washed twice with MTI3E, and dried
under high
vacuum, yielding 10.69 g (57.2 A) of N-(2-pyrazinecarbony1)-L-phenylalanine-L-
leucine
boronic anhydride as a white solid.
Example 4: Measurement of Diastereomeric Ratio of (1R)-(1S,2S,3R,55)-
Pinanediol-1-
ammoniumtrifluoroacetate-3-methylbutane-1-boronate
[0100] The diastereomeric purity of (1R)-(1S,2S,3R,5S)-pinanedio1-1-
amrnoniumtrifluoroacetate-3-methylbutane-1-boronate (compound 1) was
determined by
non-chiral gas chromatography (GC).
Chemicals: Acetonitrile (p.a. Bruker or equivalent)
Tetradecane (internal standard) (Fluka puriss. or equivalent)
Trifluoroacetic anhydride (TFAA) (p.a. Merck or equivalent)
Instrument: Trace-GC 2000 system or equivalent =
Mobile phase:
Solvent A (with Approximately 300 mg of tetradecane were weighed with an
internal standard) accuracy of 0.1 mg into a 100-mL volumetric flask. 1.5
mL of
TFAA were added and the flask was brought to volume with
acetonitrile.
Sample Preparation: About 150 mg of the sample were exactly weighed (within
0.1
mg) into a 10-mL volumetric flask. The flask was brought to
volume with Solvent A. The solution was stored for 15 minutes
before injection.
GC Parameters:
Column: Rtx-200; 105m x 0.25 mm i.d. x 0.25 pm film
Mobile phase:
Temp. program: 130 C (0.5 min); 0.5 C/min to 200 C (0 min); 30 C/min
to 300
C (2 min)
Flow: 0.9 nAL/min (const. flow)
Injector temperature: 250 C
Detector temperature: 250 CC (FM)
Split: 1: 50
Injection volume: 1 u,L
- 53 -
CA 02738706 2011-04-21
WO 2005/097809
PCIAS21m5/0o9774
Substances
Compound 1 (1R)-(1S,25,3R,55)-pinanedio1-1-
arnmoniurntrifluoroacetate-3-
methylbutane-1-boronate
yyt13-0
NH2 F3c-co0H
Compound 2 (1S)-(1S,2S,3R,5S)-pinanedio1-1-ammoniwntrifluoroacetate-
3-
methylbutane-1-boronate
NH2 F3c-co0H
Stability of the solution
[0101] A stock solution of compound 1 was prepared by weighing 150.13 mg
of
compound 1 into a 10-inL volumetric flask and bringing it to volume with
Solvent A.
Stability of this solution was tested at ambient temperature over 48 hours.
The stock
solution was filled in 6 separate GC vials. Injections onto the GC system were
carried out
from these vials after 0, 12,24, 48, arid 72 hours (double injection out of
each vial. The area%
of compound 1 and compound 2 were determined. No changes in area% were
observed,
indicating that the solution is stable over 72 hours at ambient temperature.
Specificity
[0102] Approximately 150 mg of a sample comprising compound 1 and compound
2
were dissolved in Solvent A and injected to the GC chromatographic system. The
peak for
compound 1 was well separated from the peak for compound 2. Peak purity check
by GC-
MS showed no other components co-eluting with compound 1 or compound 2.
Limit of Detection
[0103] The limit of detection (LOD) was defined to be that concentration
where the
signal of compound 1 showed a signal to noise ratio of at least 3:1. A
previous blank
measurement was carried out to show that no other peaks interfered. The signal
to noise
ratio was calculated by the equation:
- 54 -
CA 02738706 2013-03-18
,
,
,
H (signal)
S/N - _________________________________________________
H (baseline)
S/N = signal to noise ratio
H(signal) = height of signal for compound 1 [mm]
H(baseline) = height of signal baseline [mm]
[0104]
A sample concentration of 0.05% of the standard test sample
concentration was injected and showed a signal to noise ratio of 4.3.
Therefore, the
limit of detection is 0.0075 mg/mL.
Limit of Quantitation
[0105]
The limit of quantitation (LOQ) was defined to be that concentration
where the signal of compound 1 showed a signal to noise ratio of at least
10:1.
Signal to noise ratio was calculated as described above. A sample
concentration of
0.1% of the standard sample concentration was injected and showed a signal to
noise ratio of 10.1. Therefore, the limit of quantitation is 0.015 mg/mL.
Example 5: Purity Assay for N-(2-Pyrazinecarbony1)-L-phenylalanine-L-
leucine boronic anhydride
[0106]
The purity of N-(2-pyrazinecarbony1)-L-phenylalanine-L-leucine boronic
anhydride (compound 3) was assayed by reverse phase HPLC.
Reagents: Water, HPLC grade
Acetonitrile, HPLC grade
Formic acid, ACS grade, > 98% pure
3% Hydrogen peroxide, ACS grade or equivalent
Instrument
High
performance Autosampler capable of delivering 20-uL injections and
liquid chromato graph maintaining a temperature of 5 C
Pump capable of gradient delivery at 1.0 mL/min
UV detector capable of monitoring effluent at 270 nm
Column Symmetry* C18 chromatographic column, 250 mm x 4.6
mm ID, 5-pm, Waters, cat# WAT054275.
*Trademark
CA 02738706 2011-04-21
=
Sample Preparation: Approximately 50 mg of compound 3 were accurately
weighed
into a 50-mL volumetric flask. Mobile Phase B (5 mL) was
added and the mixture was sonicated to dissolve compound 3
(approximately 30-60 seconds). The solution was allowed to
reach room temperature, diluted to volume with Mobile Phase
A, and mixed well. Each sample was prepared in duplicate and
was stable for 7 days when stored at 2-8 C protected from light.
HPLC Parameters:
Mobile phase A: Acetonitrile /water/formic acid, 30:70:0.1 (v/v/v),
degassed
Mobile phase B: Acetonitrile/water /formic acid, 80:20:0.1 (v/v/v),
degassed
Flow rate: 1.0 mL/min
Detector: UV at 270 nm
Injection Volume: 20 ).).1_,
Column Tenzp: Ambient
Sample Tray Temp: 5 C
Gradient Program: Time
0 100 0
15 100 0
30 0 100
45 0 100
47 100 0
55 100 0
Substances
Compound 3 N-(2-Pyrazinecarbony1)-L-phenylalanine-L-leucine boronic
anhydride
0 0
H
N
))LN - 3
0 y
Compound 4 N-(2-Pyrazinecarbony1)-D-phenylalanine-L-leucine
boronic anhydride
0 2 0
H 3
14
56
CA 02738706 2011-04-21
WO 2005/097809
ITT/US20057009774
Compound 5 N-(2-Pyrazinecarbony1)-L-phenylalanine-D4eucine
boronic
anhydride
411 _
0 0
H
N
[0107] The retention time of compound 3 was typically between 10
and 14 minutes
when using an HPLC system with a 1.3 minute dwell volume. Compounds 4 and 5 co-
eluted at longer retention time, with a resolution of 2Ø
[0103] The relative retention of compound 3 in a sample
chromatogram to that in the
standard chromatogram was calculated according to the following equation:
tS11171
Rt. =--
tstd
= Where:
relative retention
C111 retention time of compound 3 peak in the
sample
chromatogram, minutes
1:;td= retention time of the drug substance peak
in the
closest preceding standard chromatogram, minutes
[0109] Assay results were calculated for each sample according to
the following
equation:
Asam Wsul X P 1
%assay = ___________________________ x x100
A./ Warn 100
100
Where:
A= peak area response of compound 3 in the
sample
preparation
Aõ = mean peak area response of compound 3 in
the
working standard preparation
Wstd= weight of the standard, mg
assigned purity of the standard (decimal format)
_
= weight of the sample, mg
moisture content of the sample, %
-57 -
CA 02738706 2011-04-21
WO 2005/097809 PCTIUS20075/009774
100 = conversion to percent
[0110] Relative retention and impurity levels in each sample were
calculated
according to the following equations:
ti
Rr
tds
Where:
= relative retention
t;= retention time of the individual impurity
t,= retention time of the compound 3 peak
% ¨ Ai x147 x Px DF xRFi
x100
Aste1,17oXWRIM
Where:
individual impurity
.A1 peak area response of individual impurity in the
sample preparation
Amd,i% = average peak area response of compound 3 in the 1%
standard preparation
= weight of the standard, mg
SARI weight of sample, mg
assigned purity of the standard (decimal format)
DF = dilution factor, 1/100
RF, = relative response factor of individual impurity
100 = conversion to percentage factor
(0111] When assayed by this method, N-(2-pyrazinecarbony1)-L-
pheny1alanine-L-
leucineboronic anhydride from Example 2 showed total impurities of less than
1%.
[01121 While the foregoing invention has been described in some detail
for purposes
of clarity and understanding, these particular embodiments are to be
considered as
illustrative and not restrictive. It will be appreciated by one skilled in the
art from a reading
of this disclosure that various changes in form and detail can be made without
departing
from the true scope of the invention and appended claims.
. = -
- 58 -