Note: Descriptions are shown in the official language in which they were submitted.
~6~Z~
Summary of the Invention
.
This application relates to 7-[2-(5-amino-1,2,4-thiadiazol-3-
yl)-2-(substituted)iminoacetamido~-3-[3-(quaternaryammonio)-1-
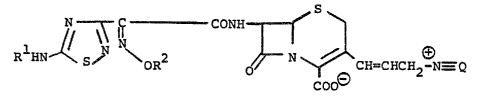
propen-l-yl]-3-cephem-4-carboxylates of the formula
C CON~ ~ ~
RlIIN~ oR2 'O ~CH=CHCH2-N_Q
O
in which Rl and R2 are as defined herein and -~ --Q is a quaternary
ammonio group as defined herein, and to salts and esters thereof.
This invention also relates to processes for the preparation of
the compounds of Formula I, to pharmaceutical compositions
containing at least one compound of ~ormula I, and to intermediates
in their preparation.
Back~round and Prior Art
A) U.S. Patent 4,390,534, issued June 28, 1983 to Tsutomu
Teraji _ al., discloses cephem and cepham compounds of the
formula
Rl ~ ~ C - CON~- ~
wheLein Rl is amino or protected amino; R~ is hydrogen, acyl,
optionally substituted aryl, substituted alkyl, alkenyl, alkynyl,
~, . .
~2'~6~3Z~
optionally substituted cycloalkyl, cycloalkenyl or an O- or
S-containing 5-membered heterocyclic ring substituted with oxo
group(s); R3 is hydrogen or alkyl; R4 is hydrogen, acyloxyalkyl,
acylthioalkyl, optionally substituted pyridinioalkyl, optionally
substituted heterocyclylthioalkyl, alkyl, halogen, hydroxy or
optionally substituted thiazolioalkyl; and R5 is carboxy or
protected carboxy; provided that RS is COO when R4 is optionally
substituted pyridinioalkyl or optionally substituted thiazolioalkyl;
and the dashed line indicates either a single or double bond.
European Patent ~pplication No. 13,762, published August 6,
1980 is concordant thereto and has a similar disclosure.
U.S. Patents 4,381,299 (issued April 26, 1983), 4,331,665
(issued May 25, 1982) and 4,332,798 (issued June 1, 1982) each
issued on parent applications of U.S. 4,390,534, and have similar
disclosures.
B) European Patent Application No. 62,321, published October
13, 1982, discloses cephem compounds of the formula
R~ CONH_~
~R2 ~
wherein Rl is amino or pro~ected amino; R2 is an optionally
subs~ituted lower aliphatic hydrocarbon group, or cycloalkenyl;
and the group of the formula
--N ~3
~,
:~76~9
is an optionally substituted heterocyclic cation group containing
more than one nitrogen atom; and pharmaceutically acceptable
salts thereof. Also disclosed are intermediates of the formula
R ~ ~ Il- CONH
CH N ~
wherein Rl and R2 are as defined above, R4 is a protected carboxyl
group and X is an acid residue.
C) European Patent Application No. 74,653, published March 23,
1983, discloses cephem compounds of the formula
Rl ~ I-CONH F ~ ~ 3
wherein Rl is amino or protècted amino;
R is an optionally substituted lower aliphatic hydrocarbon
group, cyclo(lower)alkyl or cyclo~lower)alkenyl;
R3 is (lower)alkylamino, N-protected(lower)alkylamino,
di(lower)alkylamino, suIfo(lower)alkylamino, hydroxy(lower~-
alkylamino, N-protected hydroxy(lower)alkylamino, acyloxy(lower)-
alkyl, (lower~alkoxy(lower)alkoxy(lower)alkyl, di(lower~alkylamino-
(lower)alkyl, (lower)alkylthio(lower)alkyl, (lower)alkylthio,
~;~ 76~29
(lower)alkoxy, tlower)alkoxy(lower)alkoxy, hydroxy(lower)alkoxy,
acyl(lower)alkyl, hydroxy(lower)alkylthio, di(lower)alkylamino-
(lower)alkylthio, N-containing unsaturated S-membered
heterocyclic group, N-containing unsaturated S-membered
heterocyclicthio, or N-containing unsaturated 5 or 6-membered
heterocyclicthio(lower)alkyl which may be substituted with
suitable substituent(s); and
R4 is hydrogen or (lower)alkyl; or a salt thereof.
D) U.S. Patent 4,332,800, iss~ed June 1, 1982 to Tsutomu Teraji
et al., discloses inter alia compounds of the formula
oR2 ~ ?--c~2~
wherein Rl is amino or protected amino; R2 is (lower)alkyl and X
is hydrogen or carbamoyl.
.
E) European Patent Application No. 47,977, published March 24,
1982, discloses cephem compounds of the formula
(~)m
Am- T ~11 - CONH
O-R ~ ~
2 O . ¦ CH2Rl
COO~
., .
~Z'716~Z~ .
wherein m is 0 or 1; Am is optionally substituted amino; T is a
thiadiazolyl moiety (attached to the other groups by two of its
carbon atoms); R2 is hydrogen, optionally substituted alkyl,
cycloalkyl or optionally substituted carbamoyl; and Rl is optionally
substituted thiazolio, optionally substituted pyrazolio, tri(lower)-
alkylammonio or a pyridinio group of the formula
R
~ ~ b
R
in which Ra is substituted (lower)alkyl [the substituent being
cycloalkyl, phenyl, hydroxy, alkoxy, halogen, cyano, carbamoyl,
carboxyl or sulfo], (lower)alkenyl or carboxy-substituted (lower)-
alkenyl, (lower)alkylthio or carboxy-substituted (lower)alkylthio,
ami.no or monosubsti~uted amino [the substituent being (lower)alkyl,
(lower)alkanoyl or aminobenzenesulfonyl], di(lower)alkylamino,
substituted carbamoyl [the substituent being (lower)alkyl,
hydroxy(lower)alkyl, (lower)alkoxy, hydroxy or cyano], di(lower)-
alkylcarbamoyl, thiocarbamoyl, cycloalkyl, phenyl, hydroxy,
(lower)alkoxy, halogen, (lower)alkoxycarbonyl, (lower)alkanoyloxy,
(lower)alkanoyl, carboxyl, sulfo, cyano, nitro or hydroxysulfo-
(lower)alkyl; Rb is hydrogen or carbamoyl, or has the same
meaning as Ra; and Rc is hydrogen or has the same meaning as Ra;
and salts thereof.
Although not formally related, European Patent Application
No..25,017, published March 11, 1981, has a similar disclosure.
F) European Patent Application No. 30~630, published June 24,
i981, discloses 3-vinyl cephem compounds of the formula
~7~;~2g~
. S
Rl _ A ~ CONH ~ ~
"~N~J~CH=CH2
12
wherein Rl is an optionally protected amino-substituted heterocyclic
group which may also have halogen, or a group of the formula
R SO2~N ~ ~
in which R3 is (lower)alkyl; R2 is carboxy or protected carboxy;
and A is lower alkylene which may have a substituent selected
from amino, protected amino, hydroxy, oxo and a group of the
formula =N~oR4, wherein R4 is hydrogen, cyclo(lower)alkenyl,
~lower)alkynyl, (lower)alkenyl [optionally substituted by carboxy
or protected carboxy], (lower)alkyl [optionally substituted by
one or more of carboxy, protected carboxy, amino, protected
amino, cyano, phosphono, protected phosphono and a heterocyclic
~roup which itself may be substituted]; and salts thereof.
This application specifically discloses compounds of the
formula
Cl CONI~S
H2N S/ \OR ~J\CH=CH;~
COOH
~2769Z9
in which oR4 is methoxy, carboxymethoxy, tert-butoxycarhonyl-
methoxy or l-tert-butoxycarbonylethoxy.
G) U.K. Patent Specification No. 1,399,0R6 published June 25,
1975, contains a generic disclosure encompassing a vast number of
cephalosporins of the formula
R-C-CO-N -
COOH
wherein R is hydrogen or an organic group, Ra is an etherifying
monovalent organic group linked to the oxygen through a carbon
atom, B is ~ S or ~ S-~O, and P is an organic group. In one
embodiment, P may be inter alia a vinyl group of the formula
R3
-CH=C
in which R3 and R4 independently may be hydrogen, nitrile,
(lower)alkoxycarbonyl, or substituted or unsubstitu~ed aliphatic,
cycloaliphatic, araliphatic or aromatic. However, the 5-amino-
1,2,4-thiadiazol-3-yl group is not identified as a possible R
substituent and there is no disclosure or suggestion that P may
be a ~uaternary ammonio-substituted propenyl group. ~.S. Patent
3,971,778 and its divisionals Nos. 4,024,133l 4,024,137, 4,064,346,
4,033,950, 4,079,178, 4l091,209, 4,092,477 and 4,093,303 have
similar disclosures.
~76~9
H) European Patent Application No. 88,385, published September
14, 1983, discloses compounds of the formula
R ~--CO~
in which Rl is (unsubstituted) ~hiadiazolyl; R2 is carboxy-
(lower)alkyl or protected carboxy(lower)alkyl; R3 is hydrogen,
halogen or (lower)alkenyl; and R4 is carboxy or protected carboxy.
Although l-propenyl is listed as one of the possible meanings of
R3, the applicatian only exemplifies compounds where R3 is
hydrogen, chloro or vinyl.
I) ~.S. Patent No. 4,307,233 issued to Daniel Farge et al. on
December 22, 1981, discloses inter alia, 3-vinylcephalosporin
derivatives of the formula
S
N~_ CONH ~ ~ R3
H2 S \oR5 o// ~ CH=CH N \R4
COOH
in which R5 inter alia may be alkyl, vinyl, cyanomethyl or a
protective group such as 2-methoxyprop-2-yl, and R3 and R4 are
alkyl groups (optionally substituted by hydroxy, alkoxy, amino,
alkylamino or dialkylamino) or phenyl groups, or R3 and R4, taken
together with the nitrogen to which they are attached, may form a
saturated heterocyclic ring of 5 or 6 members, optionally containing
another hetero-atom selected from N, O and S, and optionally
substituted by an alkyl group. The co~pounds are useful as
intermediates in the preparation of 3-thiovinyl cephalosporin
derivatives. There is no disclosure or suggestion of a 5-amino-
76~9
1,2,4-thiadiazol-3-yl moiety in place of the 2-aminothiazol-4-yl
substituent or of a quaternary ammonio-substituted propenyl
moiety for the 3-substituent. Published United Xingdom Patent
Application No. 2~051,062 is concordant thereto and has a similar
disclosure.
J) European Patent Application No. 53,537, published June 9,
1982, discloses, inter alia, 3-vinylcephalosporin derivatives of
the formula
S
CONH
~32~ O~COOR5 ~ \f\CE~=C~-N~
in which Ra5 and Rb are the same or different and are hydrogen or
alkyl, or taken together, form an alkylene group containing 2 or
3 carbon atoms, RC5 is an acid protecting group, R2 is an acid
protecting group such as an ester, R3 and R4 are the same or
different and are hydrogen, alkyl (optionally substituted by
hydroxy, alkoxy, amino, alkylamino or dialkylamino) or phenyl
groups, or R3 and R4, taken together with the nitrogen to which
they are attached, may form a saturated heterocyclic ring of 5 or
6 members, optionally containing another hetero-atom selected
from N, O and S, and optionally substituted by an alkyl group.
The compounds are useful as intermediates in the preparation of
3-thiovinyl cephalosporin derivatives. There is no disclosure or
suggestion of a 5-amino-1,2,4-thiadiazol-3-yl moiety in place of
the 2-aminothiazol-4-yl substituent or of a quaternary ammonio-
substituted propenyl group for the 3-substituent.
U.S. 4,423,214 is concordant thereto and has a similar
disclosure.
~Z'7~
11
~) European Patent Application No. 53,074, published ~une 2,
1982, generically discloses a vast number of 3-vinylcephalosporin
derivatives of the formula
(~)n
R la ~ S ~
O CH=C-R
COOR2a
wherein Rla (in one of several embodiments) may be
N~ ICl _CO_
H2 OR5
in which R5 inter alia may be hydrogen, alkyl, vinyl, cyanomethyl,
an oxime-protecting group such as trityl, etc., or a group of the
f ormula
/ C\OR 5
R S S
in which Ra5 and Rb5 are the same or different, and may be
hydrogen, alkyl or, taken together, an alkylene radical of 2 or 3
carbon atoms, and RC5 is hydrogen or an acid-protecting radical;
R2a is hydrogen or an acid-protecting radical such as methoxymethyl;
R (in one of several embodiments) may be a methyl group substitu~ed
by a 5- or 6-membered aromatic heterocyclic ring containing a
single hereto atom, such as 2- or 3-pyridyl, 2- or 3-thienyl or
2- or 3-furyl; and
t76~Z~ !
12
R3 is a group of the for~ula
R4S020-
in which R4 may be alkyl, trihalomethyl or optionally substituted
phenyl.
These compounds are stated to be intermediates in the
preparation of compounds in which the 3-substituent is a group of
the formula
R
-C~=C-SR
which are stated to have antibacterial activity.
Although this patent includes the possibility of R being a
methyl group substituted by an N-containing heterocyclic ring, in
both the intermediates and final products (thus giving a heterocyclic-
substituted propenyl moietyJ, it teaches only that the heterocyclic
ring is attached via one of its carbon atoms. Thus, there is no
suggestion of a quaternary ammonio-substituted propenyl group.
The reference exemplifies R~ in the intermediates and final
products only as methyl. Further, in both the intermediates and
final product, the propenyl group must contain a second substituent
(-03SR or -SR, respectively). Also there is no disclosure or
suggestion of a 5-amino-1,2,4-thiadiazol-3-yl moiety in place of
the 2-aminothiazol-4-yl substituent.
L) European Patent Application No. 53,538, published June 9,
1982! discloses, inter alia, 3-vinylcephalosporin intermediates
of the formula
.
(~)n
/~\~ /'/ ~H=CH-R
2~ O
t::OOH
in which n is O or 1, R5 is hydrogen, alkyl, vinyl, cyanomethyl
or an oxime-protecting group, and R3 is halogen. There is no
disclosure or suggestion of a 5-amino-1,2,4-thiadiazol-3-yl
moiety in place of the 2-aminothiazol-4-yl substituent, and no
disclosure or ~uggestion of a 3-halo-1-propen-1-yl substituent in
the 3-position.
6~g
14
Com~lete Dlsclosure
This applica~ion relates to novel cephalosporin deriva-
tives which are potent antibacterial agents. More particularly,
it relates to compounds of the formula
N ~ ~ CONH ~ ~ I
Rl~N S~ oR2 ~ N ~ C~=CH-CH2-N- Q
CO~
wherein ~1 is hydrsgen or a conventional amino-protecting group,
R2 is hydrogen, a straight or branched chain alkyl group contain-
ing from 1 to 4 carbon atoms, a cycloalkyl or cycloalkenyl ring
containing from 3 to 6 carbon atoms, or a group of the formula
R4 R4 COOH
-~-CH=C~-R , -~-C- C-R ' X or
R4
-C COOH
R5
in which R3 is hydrogen, (lower)alkyl or carboxyl, X is halogen,
hydroxy or (lower)alkoxy, and R4 and R5 are each independently
hydrogen~ methyl or ethyl, or R4 and R5, taken together with the
carbon atom to which they are attached, may be a cycloalkylidene
ring containing from 3 to 5 carbon atoms, and
0
-N - Q
~ILZ'7~!32~
is a quaternary ammonio group, and nontoxic pharmaceutically
acceptable salts and physiologically hydrolyzable esters thereof.
Also included within the scope of the invention are the solvates
(including hydrates) of the compounds of ~ormula I, as well as
the tautomeric forms of the compounds of Formula I~ e.g. the
2-iminothiazolin-4-yl form of the 2-aminothiazol-4-yl moiety.
In another aspect, this application relates to a
process for the preparation of the compounds of Formula I and to
cer~ain intermediates in their ~reparation.
As shown in the structural formula, the compounds of
Formula I have the ~syn" or HZ~ configuration with respect to the
alkoxyimino group. Because the compounds are geometric isomers,
some of the "antir isomer may also be present. This invention
comprises compounds of ~ormula I containing at least 90% of the
~syn~ isomer. Pre~erably the compounds of Formula I are rsyn~
isomers which are essentially free of the corresponding ~anti~
isomers.
In addition to geometric isomers possible with respect
to the alkoxyimino group, the compounds of Formula I (and the
intermediates of Formulae VIII and IX) also form geometric (cis
and trans) isomers about the double bond of the propenyl group.
Both the Ci5 ( rz~ ) and trans (rEr) isomers of these compounds are
specifically included within the scope of this invention.
The nontoxic pharmaceutically acceptable salts of the
compounds of Formula I include salts with mineral acids such as
hydrochloric, hydrobromic, phosphoric and sulfuric, or with
organic carboxylic acids or sulfonic acids such as acetic,
trifluoroacetic, citric, formic, maleic, oxalic, succinic,
benzoic, tartaric, fumaric, mandelic, ascorbic, malic, methane-
sulfonic, benzenesulfonic, p-toluenesulfonic and other acids
known and used in the penicillin and cephalosporin arts. Prepa-
ration of these acid addition salts is carried out by convention
al techniques.
.
16
Examples of physiologically hydrolyzable esters of the
compounds of Formula I include indanyl, phthalidyl, methoxy-
methyl, acetoxymethyl, pivaloyloxymethyl, glycyloxymethyl,
phenylglycyloxymethyl, S-methyl-2~oxo-1,3-dioxolen-4-ylmethyl and
other physiologically hydrolyzable esters known and used in the
penicillin and cephalosporin arts. Such esters are prepared by
conventional techniques known in the art.
The compounds of Formula I in which Rl is hydrogen
exhibit high antibacterial acti~ity against various Gram-positive
and Gram-negative bacteria, and are useful in the treatment of
bacterial infections in animals, including man. The compounds of
Formula I may be formulated for parenteral use in a conventional
manner utiliziny known pharmaceutical carriers and excipients,
and may be presented in unit dosage form or in multi-dosage
containers. The compositions may be in the form of solutions,
suspensions or emulsions in oily or aqueous vehicles, and may
contain conventional dispersing, suspending or stabilizing
agents. The compositions may also be in the form of a dry powder
for reconstitution before use, e.g. witk s~erile, pyrogen-free
water. The compounds of Formula I may also be formulated as
suppositories utilizing conventional suppository bases such as
cocoa butter or other glycerides. The compounds of this in-
vention may, if desired, be administered in combination with
other antibiotics such as penicillins or other cephalosporins.
When provided in unit dosage forms the compositions
will preferably contain from about 5n to about 1500 mg of the
active ingredient of ~ormula I. The dosage of the compounds of
Formula I is dependent on such factors as the weight and age of
the patient as well as the particular nature and severity of the
disease, and is within the discretion of the physician. However,
the dosage for adult human treatment will usually be in the range
of from about 500 to about 5000 mg per day, depending on the
frequency and route of administration. When administered intra-
muscularly or intravenously to an adult human, a total dosage of
from about 750 to about 3000 mg per day, in divided doses,
normally will be sufficient, although higher daily doses of some
~L~'7~i~Z~ ~
17
of ~he compounds may be desirable in the case of Pseudomon.as
infections.
The quaternary ammonio group of the formula
-N - Q
may be acyclic, cyclic, or a combination of ~he two, and may
contain one or more additional hetero atoms selected from nitro-
genr sulfur and oxygen.
An example of an acyclic quaternary ammonio group is a
group of the formula
R6
~91 7
-I-R
R~
6 7 8
in which R , R and R may be the same or different and may, for
example, be (lower)alkyl or substituted (lower)alkyl in which the
substituents are, for example, halogen, amino with the provision
that the amino group may not be on an a-carbon, hydroxy with the
provision that the hydrsxy group may not be on an ~-carbon,
(lower~alkoxy with the provision tha~ the alkoxy group may not be
Qn an -carbon, (lower)alkylthio, (lower)alkylamino, di(lower)-
alkylamino, carbamoyl, (lower)alkenyl, phenyl(lower)alkyl, phenyl
or substituted phenyl (in which the substituents may be, for
example, halogen, hydroxy, amino, (lower)alkylamino, di(lower)-
alkylamino, acylamino, (lower)alkyl, (lower)alkylthio, (lower)-
alkoxy or the like).
Examples of cyclic quaternary ammonio groups are fully
unsaturated monocyclic heterocyclic ring systems, and bicyclic
heterocyclic ring systems in which at least one N-containing ring
is fully unsaturated. Suitable cyclic guaternary ammonio ring
~.~7~
18
systems include, for example, those of the formulae
-N~10 ~R10
R9 ,j~R9 , ~ ~R9
SR109 RlD S R10
~R~ ~10
oRltl 0 ~5 S
RlO~R9 R10
RlO~R9 -N~
N~VN
~O~R9
Rl
and the like, in which R9 and R10 are the same or different and
~1~27~
19
may be, for example, hydrogen, halogen, amino, (lower)alkyl,
(lower)alkenyl, (lower)alkylthio, carboxy, hydroxy, (lower)-
alkoxy, ~lower)alkoxy(lower)alkyl, halo(lower)alkyl, hydroxy-
(lower)alkyl, amino(lower)alkyl, (lower)alkylamino(lower)alkyl,
di(lower)alkylamino(lower)alkyl, (lower)alkylamino, di(lower)-
alkylamino, carboxy~lower)alkyl, carboxy(lower)alkylamino,
carboxy(lower)alkylthio, carbamoyl, N-~lower)alkylcarbamoyl,
formylamino, acylamino, acyloxy, phenyl, pyridyl, amidino,
guanidino and the like. Where the structure of the heterocyclic
ring permits, R9 and R10, taken~together, may be an alkylene
group containing from 3 to 5 carbon atoms, e.g. propylene.
Examples of combined acyclic/cyclic quaternary ammonio
groups include, for example, those of the formulae
12 ~ ~ 12 0 ~ 12
-N ~ R , -N ~ ~ R , -N
" Rll Rll ,Rll
~ 12 ~ R12 ~12
-N ~ O ~ -N S
Rl~ ~,11/ li/ \--/
~ 2
Rll ~ ~ Rll ~ ~ 1 ~ R
0 ~ 12 N N
-N N~ wer)alkyl, -N ~ t R , ~ ~ S ~ ~12
3LZ~
2~
and the like, in which Rll may be, for example, (lower)alkyl,
(lower)alkoxy(lower)alkyl, hydroxy(lower)alkyl with the provision
that the hydroxy may not be on an a-carbon, carboxy(lower)alkyl,
amino(lower)alkyl with ~he provision that the amino may not be on
an ~-carbon, (lower)alkenyl, halo(lower)alkyl, allyl and the
like, and R12 may be, for example, hydrogen, hydroxy, halogen,
(lower)alkyl, hydroxy(lower)alkyl, (lower)alkoxy(lower)alkyl,
halo(lower)alkyl, amino(lower)alkyl/ (lower)alkoxy, (lower)-
alkylthio, (lower)alkenyl, amino~ (lower)alkylamino, di(lower)-
alkylamino, acylamino, acyloxy,-carbamoyl, amidino(lower)alkyl,
phenyl, pyridyl, amidino, guanidino and the like.
.
Preferred quaternary-ammonio groups are those of the
formulae
R13 - ~ ~ 16 (3 ~ R16
17 R17
~ ~ 18 - (CN2)n - ~ R~
wherein R13, R14 and R15 are the same or different and are
(lower)alkyl, (lower)alkenyl, amino(lower)alkyl with the pro-
vision that the a~ino may not be on an a-carbon, or hydroxy-
(lower)alkyl with the provision that the hydroxy group may not be
on an ~-carbon;
R15 is hydrogen, ~lower)alkyl, (lower)alkoxy, (lower~-
alkylthio, amino, (lower)alkylamino, di(lower)alkylamino, formyl-
amino, (lower)alkanoylamino, carboxy, hydroxy, carboxy(lower)-
alkyl, carboxy(lower)alkylthio, hydroxy(lower)alkyl, halo(lower)-
;aZ'76~
`` 21
alkyl, amino(lower)alkyl, (lower)alkoxy(lower)alkyl, carbamoyl orN-~lower)alkylcarbamoyl, or R16 may represent a divalent alkylene
group having 3 to 5 carbon atoms;
R17 is (lower)alkyl, (lower)alkoxy(lower)alkyl, halo-
(lower)alkyl, allyl, hydroxy~lower)alkyl with the provision that
the hydroxy group is not on the a-carbon, amino(lower)alkyl with
the provision that the amino group is not on the ~-carbon, or
phenyl(lower)alkyl;
R18 is hydrogen, (lower)alkyl, (lower)alkoxyl (lower)-
alkoxy(lower)alkyl, (lower)alkyithio, amino, (lower)alkylamino,
di(lower)alkylamino, carboxy, hydroxy, carboxy(lower)alkyll
kydroxy(lower)alkyl, amino(lower)alkyl, formylamino, (lower)-
alkanoylamino, carbamoyl or N-(lower)alkylcarbamoyl;
n is an integer of from 1 to 3, inclusive;
Z is CH2 or, when n is 2, Z also may be S, O or N-~l9,
in which Rl is hydrogen or (lower)alkyl; and
R20 and R~l are the same or different and are hydrogen,
(lower)alkyl, (lower)alkoxy, (lower)alkylthio, amino, (lower)-
alkylamino, di(lower)alkylamino, carboxy, hydroxy, hydroxy-
(lower)alkyl, amino(lower)alkyl, (lower)alkoxy(lower)alkyl,
carboxy(lower)alkyl, carboxy(lower)alkylamino, (lower)alkanoyl-
amino, carboxy(lower)alkanoylamino, carbamoyl or N-(lower)alkyl-
carbamoyl.
Particularly preferred quaternary ammonio groups are
N-(lower)alkylpyrrolidinio (and especially N-methylpyrrolidinio),
tri(lower)alkylammonio (and especially trimethylammonio), pyri-
dinio, aminopyridinio, formylaminopyridinio, carbamoylpyridinio,
amino(lower)alkylpyridinio, carboxypyridinio, hydroxy(lower)-
alkylpyridinio, N-(lower)alkylcarbamoylpyridinio, (lower)-
alkylenepyridinio, 2-methylthiazolio and 2-amino-5-thiazolo-
[4,5-c]pyridinio.
76~
In the compounds of Formula I, particularly preferred
values of R2 are (lower)alkyl (and especially methyl), cycloalkyl
containing from 3 ~o 5 carbon atoms, l-carboxycycloalk-l-yl
containing from 3 to 5 carbon atoms, allyl, propargyl and
carboxy(lower)alkyl (and especially 2-carboxyprop-2-yl)~ The
most preferred compounds of the invention are
1) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl~-2-methoxyiminoace~amido]-
3-[3-(trime~hylammonio)-1-propen-1-yl]-3-cephem-4-carboxylate,
2) 7-[2-(5-amino-1,2,4-thiadiaz~1-3-yl)-2-methoxyiminoacetamido]-
3-[3-(1-methylpyrrolidinio)-1-propen-1-yl]-3-cephem-4-car~oxy-
late,
3) 7-[2-(5-amino 1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido~-
3-[3-pyridinio-1-propen-1-yl]-3-cephem-4-carboxylate,
4) 7-[2-(5 amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(3-aminopyridinio)-1-propen-1-yl]-3-cephem-4-carboxylate,
5) 7-[2-(S-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(3-formylaminopyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
6) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl~-2-methoxyiminoacetamido]-
3-[3-(3-aminomethylpyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
7) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(3-carbamoylpyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
8) 7-[2-(5-amino-1,2,4-thiadiaæol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(4-carbamoylpyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
9) 7-[2-t5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(2-methylthiazolio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
10) 7-~2-(5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(2-amino-5-thiazolo[4,5-cjpyridinio)-1-propen-1-yl]-3-
cephem 4-carboxylate,
11) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(4-hydroxymethylpyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
12) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
"`` 1~6~
23
3-[3-(3-hydroxymethylpyridinio)~l-propen-1-yl]-3-cephem-4
carboxylate,
13) 7-[2-~5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(4-{N-methylcarbamoyl}pyridinio)-l-propen-l-yl]-3-
cephem-4-carboxylate,
14) 7-[2-~5-amino-1,2,4-~hiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(2,3-propylenepyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
15) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-ethoxyiminoace~amido]-
3-[3-(4-carbamoylpyridinio)-~-propen-1-yl]-3-cephem-4-
carboxylate,
16) 7-[2-(5-amino-1,2,4- thiadiazol-3-yl)-2-cyclopentyloxyimino-
acetamido]-3-[3-(4-carbamoylpyridinio)-1-propen-1-yll-3-
cephem-4-carboxylate,
17) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-allyloxyimino-
acetamido]-3-[3-(4-carbamoylpyridinio)-1-propen-1-yl~-3-
cephem-4-carboxylate,
18) 7-[2-(S-amino-1,2,4-thiadlazol-3-yl)-2-propargyloxyimino-
acetamido]-3-[3-(4-carbamoylpyridinio)-1-propen-1-yl]-3-
cephem-4-carboxylate,
19) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(4-carboxypyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate,
20) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-ethoxyiminoacetamido]-
3-[3-(4-carboxypyridinio)-2-propen-1-yl]-3-cephem-4-
carboxylate,
21~ 7-[2-(5-amino-1,2,4~-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(3-carboxymethylpyridinio)-1-pEopen-l-yl]-3-cephem-4-
carboxylate and
22) 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-
3-[3-(4-carboxym~thylthiopyridinio)-1-propen-1-yl]-3-cephem-4-
carboxylate.
The numbering system utilized herein for the various
reactants, intermediates and final products is as follows:
. .
~lLz76~!291
[Roman Numeral] - rArabic Numeral~ r Letter
~ f appropriate~ ~ f appropriate)J .
The Roman Numeral designates whether the compound is a final
product [I] or an intermediate or other reactant [all other Roman
Numerals]. The Arabic Numerals and Letters are not used in those
instances where the overall class ~genus) of compounds is meant.
.
The Arabic Numeral designates the particular meaning of
substituent R2. If the particular R2 group contains a carboxyl
group which is protected by a conventional carboxyl-protecting
group, a ~prime~ (') is used after the Arabic Numeral to indicate
this fact. No ~prime~ is used if the carboxyl group is unpro-
tected. A ~primer also is used with the generic R2 substituent
~i.e. R ) when generically referring to an R2 group containing a
protected carboxyl group.
The Le~ter at the end of the compound number refers to
the particular meaning of the quaternary ammonio group
-N -- Q -
For convenience, the Arabic Numerals and Letters
assigned to some of the preferred R2 groups and quaternary
ammonio groups are set forth below.
Arabic Numeral R2
1 = methyl
2 = ethyl
3 = allyl
4 = propargyl
S = cyclopentyl
. .
Letter ~N - Q
_ _ _
A 8 l-methylpyrrolidinio
B = pyridinio
C = 2-amino-5-thiazolo[4,5-c]pyridinio
D = trimethylammonio
E = 3-aminopyridinio
F = 3-formylaminopyridinio
G = 3-carbamoylpyridinio
- H = 4-carbamoylpyridinio
I = 3-amin~methylpyridinio
J = 2-methylthiazolio
K = 3--hydroxymethylpyridinio
L = 4-hydroxymethylpyridinio
M = 4-(N-methylcarbamoyl)pyridinio
N = 4-carboxypyridinio
O = 2,3-propylenepyridinio
P - 3-carboxymethylpyridinio
Q = 4-carboxymethylthiopyridinio
In the primary evaluation of the compounds of this
invention, the Minimum Inhibitory Concentrations (MIC's) of the
compounds were determined by the two-fold serial agar dilution
method in Mueller-Hinton agar agains~ 32 strains of test orga-
nisms in six groups. The geometric means of the MIC's determined
in these tests are shown in Table 1.
`` ~276~
26
Table i
Config, Geometric Mean of MIC ~mcg/ml)
~mpd. double (G~)-Ia (G+)-Ib (G-)-Ia (G-~-Ib (G-)-II (G-)-III
No. bond (5) (5) ~5) (5) (5) (7)
_ _ _ ~ _ _ _
I-lA E/Z=l/l 0.26 0.70 0.05 0.15 0.23 2.4
I-lA E/Z=7/1 0.13 0.35 0.029 0.05 0.17 1.4
I-lB E 0.20 0.40 0.016 0.044 0.11 1.6
I-IB E/Z=1/4 0.35 0.80 0.05 0.11 0.35 3.5
I-lC E 0.10 0.20 0.0071 0.033 0.087 3.8
I-lD E/Z=l/l 0.61 1.4 0.10 0.26 0.46 2.4
I-lD E/Z=10/1 0.30 0.53 0.05 0.076 0.26 1.3
I-lE E 0.20 0.40 0.0094 0.029 0.10 1.4
I-lF E 0.15 0.40 0.0094 0.033 0.099 1.2
I-lG E 0.20 0.35 0.0094 0.033 0. io 1 . 4
I-lH E 0.20 0.40 0.013 0.043 0.10 0.97
I-lI E 0.80 1.6 0.10 0.20 0.69 3.1
I-lJ E 0.17 0.35 0.025 0.076 0.15 1.6
I-lK E 0.35 0.80 0.029 0.044 0.20 3.5
I-lL E 0.26 0O61 0.029 0.088 0.15 2O6
I-lM E 0.35 0.70 0.029 0.10 0.17 2.3
I-lN E/2=7/1 1.2 1.6 0.013 0.066 0.30 5.7
I-lO E 0.17 0.35 0.029 0.033 0.11 14
I-2H E 0.20 0.40 0.014 0.057 0.15 1.4
I-2N E 1.2 2.1 0.016 0.11 0.35 4.7
I-2N . Z 1.4 3.1 0.044 0.15 0.69 10
I-3H E 0.23 0.40 0.057 0.10 0.52 1.9
I-4H E 0.26 0.46 0.06Ç 0.11 0.60 2.6
I-5H E 0.13 0.40 0.20 0.46 2.1 4.2
I-lP E 0.8 1.6 0.013 0.087 0.34 14
I-lQ 0.7 0.92 0.0095 0.044 0.23 14
27
~G~)-Ia : Penicillin-sensitive S. aureus (5 strains)
(G+)-Ib : Penicillin resistant S. aureus (5 strains)G-)-Ia : Cephalothin-sensitive E. coli (2 strains), Rl.
pneumoniae (1 str~in) and Pr. mirabilis (2 strains)
(G-)-Ib : Cephalothin-resistant E. coli (3 strains) and Kl.
~neumoniae (2 strains)G-)-II : M. ~ (1 strain), Ent. cloacae (2 strains) and
Ser. marcescens (2 strains)
(G-)-III : Ps. aeru~inosa (7 strains~
Table 2, below, gives the Protective Dose50 (PD50) in
mice for a number of the compounds of Formula I against selected
microorganisms. Table 3 gives blood levels of various compounds
of Formula I upon intramuscular administration of the test
compounds to mice at a dosage of 20 mg/kg.
Table 2
PD50 (mg/kg)
5. aureus E._coli P. aeruginosa
5~ Smith Juhl A9843A
I-lB 0.44 0.028 7.7
I-lB 0.65 0.072 NT
. I-lC 0O22 0.013 NT
I-lG 0.96 0.021 5.92
I-lH 0.39 0.015 3.9
I-lJ 0.35 0O029 NT
I-lK 0.53 NT NT
I-lM 0.96 NT NT
I-lN 2.0 NT NT
I-lO 0.26 0.17 NT
I-2N 5.0 NT NT
NT = Not Tested
6~
2R
Table 3
max Tl/2 AUC
~y~ (mcg/ml) (min) mcg hr/ml)
I-lB 17 21 11
I-lC 21 32 18
I-lD 20 19 11
I-lH 23 16 14
I lJ 19 16 9.7
I-lR 24 ~ 14 14
I-lM 20 23 14
I-lN 24 19 18
I-lO 28 32 17
I-2N 22 20 12
I-3H 19 47 25
I-4H 27 22 16
I-5H 22 32 18
In another aspect, this invention relates to processes
for the preparation of the compounds of Formula I. The preferred
procedures are shown below in Reaction Schemes la, lb and lc,
while an alternative procedure is shown in Reaction Scheme 2.
. The abbreviation "Ph" represents the phenyl group. Thus, the
-CH(Ph)2 moiety is the benzhydryl group, which is a preferred
carboxyl-protecting group. When R contains a carboxyl group, it
is desirable to protect the carboxyl group with a conventional
carboxyl-protecting group such as the t-butvl moiety. Y
represents chloro, bromo or iodo.
Reaction Scheme la
~N ~ ~ II
o~ N~CH;i!
CoocH(ph)2
¦ H2N ~ ~ N \ 2 III
.. . ...
,7~9
29
~ICI--CON~
COOC~I ( Ph ) 2
~1, NaX or RI
N~;~C----CONEI~ ~ \
~2N S ~P ~bR2 ~ N~c~2 I
. v ~oc~c~(Ph)2 P(Ph)3
P (Ph~ 3
N ~C--C:ONH ~ S 1~ VI
~2N S ~ o:R2~ ~2P (Ph) 3
COOCH (Ph) 2
~base
N~ CONHF 5 ~ VII
H2 S ~ \oR2N~CH=P(Ph) 3
OOC H 5 Ph ) 2
~ ClCH2CHO
~ t7~
N fi __--CONH T~ ~ VIII
~ S~N \oR2 ~N~C~I=CHCH2C
2 . COOCE~ (Ph ) 2
NaI or KI
~ ~ '
N.~C----CCN~ 1
H2 ~ C~ IC~2 I ~
IX CC)C)CH (Ph) 2 \
~R . Q ~N
J,~secondary am~n ) XI
~tert-
N~ CON}~ ~ iary
H22~ 5~ \oR2 ~ N~H CHCEI2 N\ amine)
COOC~ h) 2 R'
'X
~ R . . .
N ~C;----CONH Tl ~ ~
X2N S oR2 o~ ~3=CHCE2 N~Q
XII COOCB ~Ph ) 2
31
oeblock
R ~ ~ ~ ~=C~c~2~
Reaction Scheme la shows two alternate means of going
from Compound IX to Compound XII. The direct route, utilizing a
tertiary amine (XI), is applicable for the preparation of all
compounds of Formula Io The indirect route, via Compound X,
utilizes a secondary amine as reactant, and is quaternized in the
following step. The secondary amine RR'NH may be acyclic (e.g.
dimethylamine) or cyclic (e.g. pyrrolidine), and this indirect
procedure therefore is suitable for the preparation of compounds
of Formula I in which the quaternary ammonio group is acyclic or
"mixed~ acyclic/cyclic. This indirect route is not suitable for
the preparation of compounds of Formula I wherein the quaternary
nitrogen is in a fully unsaturated heterocyclic ring (e.g.
pyridinio, thiazolio, 2-amino-5-thiazolo[4,5-c]pyridinio, and the
like).
Reac~ion Scheme lb
o
7~9
32
~3CH~N,~ S ~ XIII
N ~ CH2Cl
COOCH (Ph ) 2
Na I or XI
'
~3CE~=N~ ~
XIV ~H2 I P t Ph ) 3
.,
P (Ph ) 3
~ /
~3 CH--NF,~S ~
N~ H2P(Ph)3
COOC:II ( Ph ) 2
¦ ba~e
<~CH-~S ~ XVI
N ~CH-P (Ph ) 3
COOCH (Ph~ 2
33
I ClCH2CHO
~ /
. ~ CH= ~ ~ XVII
~CH=C~ H2Cl
oOCH(Ph)2
¦ ~irard Reagent T
or
/ HCl
H2N ~ ~ XVIII
0 ~ ~ CH=CHCH2Cl
COOCH(Ph)2
~ IIT
_ __~ ----------- I
VIII as in Schemela
Reaction Scheme lb is a variation of Reaction Scheme la
in that the 7-amino group of the starting material (II) is
protected as a Schiff base during most of the reaction steps, and
the desired 7-side chain acid is added later in the synthesis.
9therwise, the yeneral procedure is similar.
2~
Rea ct i on 5ch eme lc
~-CH=N~/ ~
N~ CH=CH-CH2Cl XVI I
COOCH ( Ph ) 2
NaI or KI
~CH=N F,'~,
oCH=CH-CH2 I XIX
COOCH (Ph) 2
R
HN~R,
(secondary amine)
~3--C~ CH=CHCH~ (terti.ry
XX O R ~amine )
COOCH (Ph) 2
~"Y
~H=N~.S ~
XXI O \~CR=CHCH2 ~--Q
~ COOCH (Ph) 2
~ 76~
S~
H2N~
~ ~3
O I CH=CHCH2-N_ Q
XXII
COO ~'
N-acylation
\ with III
N ~ CONH
H2N ~ \oR2 ~ N ~
O ¦ CH=CHCH2-N - Q
. COO ~)
Reaction Scheme lc is a further variation of Reaction Scheme
lbo In Reaction Schemes la and lb~ quaternization of the 3-side
Ghain is the last step, but in Reaction Scheme lc tbe last step
is acylation of the 7-amino group. The relationship between
Reaction Schemes la, lb and lc is shown in the following
flowchart.
~'7i6~
36
H2NT~S~
0~ CH2cl
la / II COOCH~Ph)2 \ lb
\lc
N ~ fi;CONH ~ ~ ~ H-N
IV COOCH(Ph) XIIX H2Cl
2 COOCH(Ph)2
la ~ ~ lb
~lc
/ ,~
C - CONH ~ ~ S
N~ 1 7-N-Acylation H~N~~r--f~ ~
H2N ~ S ~ \oR2 g ----N ~ CH2Cl lb ~ ~ CH=CKC~ Cl
COOCH(Ph)2 2
YIII XVIII co Wl(Ph)2
.
Quaternization Quaternization
lb Deblocking lC Deblocking
7-N-~cylation
Comp ound I H ~5 ~CII
~r ~Q
XXII
. , .
~76~2~
~7
In Reaction Schemes la, lb and lc, the benzhydryl group
was shown as the preferred carboxyl-protecting group. It will be
appreciated by those skilled in the art that other carboxyl
protecting groups, well-known in the art, may be used. The
acylating acid III may be used in the form of a derivative such
as its acid halide, activated ester, mixed acid anhydride, etc.,
all of which are well-known in the art. We prefer to utilize it
in the form of its acid chloride. Acylating acid III also may
have its amino group protected ~y any of the common amino-
protecting groups, e.g. N-trityl, N-formyl, or the like. The
base used to conveet the phosphonium iodide (VI or XV) to give
the phosphorylide (VII or XVI) may be NaOH, Na2CO3l IRA-410 (OH )
resin, IRA(CO3 ) resin, or the like, or a mixture thereof. The
chloroacetaldehyde used to convert the phosphorylide VII to the
3-chloropropenyl-3-cephem compound VIII (or Compound XVI to Com-
pound XVII) may be the commercially available 40-50% aqueous
solution, a distilled solution (e.g. 70%) or the anhydrous
aldehyde.
We have found that Compound VIII prepared from Compound
VII (Scheme la) typically had a Z:E ratio of about 2:1 at the
propenyl double bond. Compound VIII prepared from Compound XVIII
(Scheme lb), on the other hand, typically was almost exclusively
the Z isomer. The difference may not be in the route used, but
in the conditions utilized in the Wittig reaction (VII to VIII or
XVI to XVII). We have also found that the use of an appropriate
silyl reagent such as N,O bis(trimethylsilyl)acetamide in the
Wittig reaction (VII to VIII in Scheme la and XVI to XVII in
Scheme lb) caused improYement of the yields and purity of VIII
and XVII. The reaction is preferably carried out with 2-5
equivalents of the silyl reagent. When the chloropropenyl cephem
(VIII) was reacted with NaI in acetone to give the iodopropenyl
cep~.em (IX), the double bond in the propenyl group WAS isomerized
from Z to E during the iodination. A short reaction period
retained the configuration of the parent Compound VIII to a large
extent, while a long reaction period gave primarily the ~ isomer
of Compound IX. However, an excessive reaction time at high
~2'7~9
3~
temperature gives a lower purity compound IX. We find that abo~t
10 minutes at 25C and 2 hours at 5C gives pure IX in good
yield. When utilizing Reaction Scheme Ic, we have found that,
when iodinating compound XIV with Nal, a purer compound is
obtained if the acetone solution is diluted with CC14 when
iodination is essentially completed, and the isomerization
portion of the reaction is conducted in the acetone-CC14 mixture.
When iodination of the chloropropenyl cephem (XVII) to the
iodopropenyl cephem (XIX) was performed with KI in DMF, the
isomerization of the double bon* from Z to E proceeded as fast as
the iodination did. The whole reaction completed within 45
minutes at room temperature to give pure XIX without dilution
with CC14 in the course of the reaction.
Compound XII normally was deblocked without purifica-
tion, and the final product (I) was purified by reverse phase
column chromatography utilizing a glass column containing the
packing removed from a Waters' ~ssociates PrepPAK-500/C18*car-
tridge.
* Trade Mark
~.2~
39
Reection Scheme 2
N ~ I ~ CONH~
Rl-HNJ~ S ~ \oR2 ~ N~71`CH=CH-CH2I XXIII
COOCH (Ph) 2
R ~1 5 ~OR oF_~C~I=CH_
COOCH ~Ph) ;~
~CONH~ ~1 ~3 XXV
R --HN S ~ oR2 ~ H=CH-CH2N----Q
COOCH (Ph) 2
Reduc:tion
Rl-H~l~C--CONH~ CH=CH-CH~N---Q XXVI
COOCH (Ph) 2
~7~ 2~ i
4~
deblock
1 fi ~ CONH ~ ~ ~ I
H2N ~S N ~OR2 ~ ~ N ~--CH=CH2N--Q
CO~
Reaction Scheme 2, shown in brief outline form above,
is similar to Reaction Scheme la except that Compound XXIII
(equivalent to Compound IX of Reaction Scheme la) is converted ~o
its S-oxide prior to quaterniæation. Compound XXV is subsequently
reduced, and the remainder of Reaction Scheme 2 is as Reaction
Scheme la. In Reaction Scheme 2, it is preferred to protect the
amino group of the 7-side chain with a known amino-protecting
group such as the trityl group.
.
The acylating acids of Formula I~I herein are either known
compounds or are readily prepared by published procedures.
European Patent Specification 7,470 published October 12, 1983
(application published February 6, 1980) exemplifies the preparation
of compounds of Formula III wherein R2 is methyl, ethyl, propyl
and isopropyl. U.S. Patent 4,390,534, referred to in the Prior
Art section, above, exemplifies the preparation of a wide variety
of compounds of Formula III wherein R is, for example, cyclopentyl,
2-cyclopenten-1-yl, allyl, 2-propynyl, l-tert. butyloxycarbonyl-l-
methylethyl, l-tert. butyloxycarbonyl-l-cyclopentyl, l-ethoxy-
carbonyl-l-methylethyl, tert. butyloxycarbonylmethyl, l-tert.
butyloxycarbonyl-2-methylpropyl, trityl, and the like.
Compound II herein (7-amino-3-chloromethyl-3 cephem-4-
~2~7~
41
carboxylate), used as a starting material in Reaction Schemes la,
lb and lc, is a known compound.
~ he tertiary amines of Formula XI (and the secondary amines
RR'NH) utilized in preparing the ~uaternary ammonio compounds of
this invention are either known compounds or are readily prepared
by those of ordinary skill in the art. Many of the amines are
commercially available.
The present invention also provides a process for the
preparation of compounds of the formula
_~ S ~
N~~CONBTl I I
~ A--N ~ ~E3
RlHN~S ~~oR2 C~ ~ ~CH--C~--C~I2-N~Q
~:od3
,
wherein Rl is hydrogen or a conventional amino-protecting group,
R2 is hydrogen, a straight or branched chain alkyl group contain-
ing from 1 to 4 carbon atoms, a cycloalkyl or cycloalkenyl ring
containing from 3 ~o 6 carbon atoms, or a group of the formula
R4 R4 COOH
-C-C~-CH R3 , -C-C _C-R , ~ or
R4 X
-1-COOH
in which R3 is hydrogen, (lower)alkyl or carboxyl, X is halogen,
hydroxy or (lower)alkoxy, and R4 and R5 are each independently
hydrogen, methyl or ethyl, or R4 and R5, taken together with the
-
~2~;~6~:~9
42
carbon atom to which ~hey are attached, may be a cycloalkylidene
ring containing from 3 to 5 carbon atoms, and
-N _ Q
is a quaternary ammonio group, and nontoxic pharmaceutically
acceptable salts and physiologically hydrolyzable esters thereof,
which process comprises reacting a compound of the formula
~)m
2 ~ / ~O~R~
O CH=CHCH2 Z
COOBl
wherein R~ is the same as.R2 or is a group of the formula
X COOBl 14
< > or -C--COOB
Y
X R5
in which X, R4 and R5 are as defined above, Bl is a conventional
carboxyl-protecting group, B2 is hydrogen or a conventional
amino-protecting group, Z is chloro, bromo or iodo, and m is zero
or onet with a tertiary amine Q -~N (or sequentially with a
secondary amine R2'NH and a compound of the formula R~Z), an~d, if
m is 1, reducing the sulfoxide by conventional means, and
subsequently removing all blocking groups by conventional means.
The.present invention also provides a process for the
preparation of co~pounds of the formula
....
~;Z7~
43
~ ~ ~ ~ C~
Rl~N l S OR~ ~ N ~ HCCR-CH2-~--Q
cod~
wherein Rl is hydrogen or a conventional amino-protecting group,
R2 is hydrogen, a straight or branched chain alkyl group contain-
ing from 1 to 4 carbon atoms, a cycloalkyl or cycloalkenyl ring
containing from 3 to 6 carbon atoms, or a group o~ the formula
R4 R4 COOH
-C-CH-CH-R3 , -C-C -~C-R3 , ~ ~
R4 X
-C-COOH
R~
i-n which R3 is hydrogen~ (lower)alkyl or carboxyl, X is halogen,
hydroxy or (lower)alkoxy, and R4 and RS are each independently
hydrogen, methyl or ethyl, or R4 and R5, taken together with the
carbon atom to which they are attached, may be a cycloalkylidene
ring containing from 3 to 5 carbon atoms, and
-N -.Q
is a quaternary ammonio group, and nontoxic pharmaceutically
acceptable salts and physiologically hydrolyzable esters thereof,
which process comprises acylating a compound of the formula
,
1~716~aZ9
s
2N~ ~
O ~ CH=CHC~2-N- Q
. COo ~ XXII
with an acid of the formula
B HN ~ 5 ~ ICl - COOH III
or with an acylating derivative of said acid, wherein R is the
same as R or is a group of the formula
.
COOB R4
X
~ ~ or -C---COOBl
r
X RS
in which X, R4 and R5 are as defined above, Bl is a conventional
carboxyl-protecting group and B2 is hydrogen or a conventional
amino-protecting group~
The reactions are carried out in a non-aqueous organic
solvent such as dimethyl sulfoxide, hexamethylphosphoramide,
methylene chloride, chloroform, ethyl ether, hexane, ethyl
a~etate, tetrahydrofuran, acetonitrile and the like, or mixtures
of such solvents. The reactions are conveniently carried out a~
a temperature of from about -10C to about +50C; we normally
prefer to conduct the reactions at room temperature. During the
quaternization step, at least one mole of the tertiary amine
should be used per mole of Compound IX, XIX, XXIII or XXIV; we
.. . .
~2~2g
normally prefer to utilize from about 25% to 100~ excess of the
tertiary amine.
Carboxyl~protecting groups suitable for use as Bl in the
above reactions are well-known to those skilled in the art and
include aralkyl groups such as benzyl, p-methoxybenzyl,
p-nitrobenzyl and diphenylmethyl (benzhydryl); alkyl groups such
as t-butyl; haloalkyl groups such as 2,2,2-trichloroethyl, and
other carboxyl protecting groups described in the literature,
e.g. in V.K. Patent 1,399,086. We prefer to utili3e
carboxyl-protecting groups which are readily removed by treatment
with acid. Particularly preferred carboxyl-protecting groups are
the benzhydryl and t-butyl moieties.
Amino-protecting groups suitable for use as B2 are also
well-known in the art, and include the trityl group and acyl
groups such as chloroacetyl, formyl and trichloroethoxycarbonyl.
Amino-protecting groups which are readily removed by treatment
with acid, e.g. the trityl group, are preferred.
.
When the cephalosporin nucleus is utilized in the form of
the 1-oxide (m = 1), the l-oxide is prepared by known procedures
such as oxidation with m-chloroperbenzoic acid, peracetic acid,
sodium tungstate, etc. The l-oxide subsequently may be reduced
by known procedures, e.g. reduction of the corresponding
alkoxysulfonium salt with iodide ion in an aqueous medium. The
alkoxysulfonium salt itself is readily prepared by treatment of
the l-oxide with, for example, acetyl chloride.
In another aspect, this invention relates to novel
intermediates of the formula
76~29
46
H N 1 5 ~ \oR2 ~ ~ CH=C~CH2Z
COOH
. XXVIII
wherein Z is chloro, bromo, or iodo, R? is hydrogen, a straight
or branched chain alkyl group containing from 1 to 4 carbon
atoms, a cycloalkyl or cycloalkenyl ring containing from 3 to 6
carbon atoms, or a group of the formula
R4 R4 COOH
-C-C~=C~-R , -C-C -- C-R3 , ~ ~
R4 X
-C-COOH ,
R5
in which R3 is hydrogen, (lower)alkyl or carboxyl, X is halogen,
hydroxy-or (lower)alkoxy, and R4 and R5 are each independently
hyd.rogen, methyl or ethyl, or R4 and R5r taken together with the
carbon atom to which they are attached~ may be a cycloalkylidene
ring containing from 3 to 5 carbon atoms, and salts and esters
thereof. Also included are compounds of Formula XXVIII in which
the amino and/or carboxyl groups are protec~ed by conventional
amino-protecting or carboxyl-protecting groups.
In still another aspect, this invention relates to novel
intermediates of the formula
~Z76~
R23
R24~ /--CH=N ~S~
R25 \~ CH=CHC}1
COOR
XXIX
wherein R22 is hydrogen or a conventional carboxyl-protecting
group, and R23, R24 and ~25 are the same or different and are
hydrogen, hydroxy, (lower)alkyl or (lower)alkoxy; or a salt,
solvate, hydrate or ester thereofO
In still another aspect, this invention relates to novel
intermediates of the formula
~\~\ CH=CH--CH2-N_C;!
~0 ~
XXII
wherein ~ is a quaternary ammonio group; or a salt, ester
solvate or hydrate thereof.
As used herein, the terms acylamino and acyloxy refer to an
acylated amino or acylated hydroxy group in ~hich the acyl moiety
is (lower)alkanoyl (e.g. formyl, acetyl, propionyl, butyryl,
isobutyryl, isovaleryl, etc.), aroyl ~e.g~ benzoyl, etc.),
(lower)alkanesulfonyl (e.g. mesyl, ethanesulfonyl, etc.) or
arylsulfonyl (e.g. benzenesulfonyl, tosyl, etc.).
.
7~9~ ,
48
As used herein, the terms ~(lower)alkyl~, ~(lower)alkoxy~,
~(lower)alkylthio~ (or the like) mean straight or branohed chain
alkyl, alkoxy, alkylthio (or the like) groups containing from 1
to 6 carbon atoms, inclusive. Similarly, the terms
(lower)alkenyl and (lower)alkynyl mean alkenyl or alkynyl groups
containing from 2 to 6 carbon atoms.
... .
6~9
~9
ExamPle 1
N ~ ~ ~ CONH
J~ N~ ~ N,~ C~--CH~,N~
C}13
I-lA *Z/l =l/l
7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxYiminoacetamido]-3-
[3-(1-methylPyrrolid _io)-l- ~open-l-yl]-3-cephem-4-car_ xYlate
(I-lA)
To a solution of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadiazol-3-yl)-2-methoxyiminoacetamido]-3~(3-iodo-1-propen-1-
yl)-3-cephem-4-carboxylate (IX-l~ (Z/E=2/1, 150 mg, 0.21 mmole)
in ethyl acetate (2 ml) was added a solution of
l-methylpyrrolidine (36 mg, 0.42 mmole) in ethyl acetate (1 ml)
in one portion with stirring. The mixture was stirred for 15
minutes and diluted with isopropyl ether (10 ml) to form a
precipitate, which was collected b~ filtration. A mixture of the
solid (130 mg), formic acid (1 ml) and concentrated HCl (0.1 ml)
was stirred at room temperature. Af~er 1 hour, the reaction
mixture wa~ concentrated under reduced pressure, diluted with
water (20 ml) and filtered. The aqueous solution was passed
through a reverse phase column (the packing of PrepPAK-500/C18
cartridge, 100 ml), elu~ing with water and 10% CH30H. The
desired fractions were collected, and concentrated in vacuo to a
small volume and ~reeze-dried to give 13 mg (12~) of the title
compound (I-lA) (Z/E=lfl), melting at ~280C (dec.).
IR : ~K~r cm 1 3400, 1760, 1660, 1610.
max
UV ~Phosphate buffer (pH 7~ nm (El~ ) 236 (372), 288 (322)-
max 1 cm
NMR :~D2O 2.31 (4~1, m, ~ )~ 3.12 (3H, s, N-CH3), 3.6 (5H,
ppm ,~
m, 2-H & N ~ ~, 3.79 (lH, s, 2-H), 4.1 (2H, d,
J=8, CH2N)~ 4.2 ~3H, s, OCH3), 5.36 (lH, d, J=4.5,
6-H), 5.95 (3H, m, 7-H ~ 3-CH=CH), 6.66 (1/2H, d,
J=10, 3-CH cis), 7.0 (1/2H, d, J=16, 3-C~ trans).
Example 2
~ ~ C ~ CONH ~ S
H2 S ~ \ OC~3 ~ N ~ CH=CH-CH2-N
I-lB *E
7-[2-(5-Amino-1,2,4~thiadiazol-~yl)-2-methoxyiminoac_tamido]-3-
[3-pyridinio-1-propen-1-yl]-3-cephem-4-carboxylate (I-lB)
A mixture of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-propen-1-
yl)~3-cephem-4-carboxylate ~IX-l) (E, 716 mg, 1 mmole~, pyridine
1158 mg, 2 mmoles) in dimethylsulfoxide (DMSO) ~1 ml) was stirred
for 1 hour at room temperature. To the mixture was added ethyl
acetate (20 ml) to precipitate a solid (620 mg), which was added
to formic acid (6 ml) containing sodium bisul~ite (60 mg). The
mixture was stirred for 30 minutes at 40JC and concentrated to
dryness. The residue was dissolved in H2O (40 ml) and some
insolubles were removed. The aqueous solution was charged on a
column of reverse phase (PrepPAK-500/C18, 100 ml~ eluting with
., , ~ - . . .
~L~7~i~9 1
51
H2O (300 ml) and 5% aqueous CH30~ ~800 ml), and the eluate was
monitored by uv (254 nm) and HPLC. The fractions (5% aqueous
CH30H) containing the desired product were combined, concentrated
to a small volume and lyophilized to yield 40 mg (8%) of the
title compound ~I-lB), melting at >200C (dec.).
IR : v~Br cm 1 3350, 1760, 1660, 1600.
max
UV ~Phosphate buffer (pH 7~ nm (El% ) 240 (352)~ 258 (366)~
max 1 cm 267 (279), ~90 (469)-
NMR : ~ 2 D 6 3.74 (2H, br-s, 2-H), 4.20 (3~, s, OCH3),
ppm 5.92 (lH, d, J-4.5~ 7-H), 6.15 (lH, m,
3-CH=CH), 7.04 (lH, d, J=16, 3-CH ~rans),
8.2 (2~, m, Py-H3 5), 8.62 (lH, m, Py-H4),
8.97 (2H, m, Py-H2 6)
Example 3
N ~ C ~ CONH
H2N 5 OC~3 o ~ ~ C~-CH-o~3
I-lB *Z/E=4/1
7-lk~ 1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-
[3-Pyridinio-l-propen-1-~1]-3-cephem-4-carboxylate (I-lB)
The chloropropenyl compound, diphenylmethyl 7-[2-(5-
amino-1,2,4-thiadiazol-3-yl)-2~methoxyiminoacetamido]-3-(3-
chloxo-l-propen-l-yl)-3-cephem-4-carboxylate (VIII-l) (Z, 937 mg,
1.5 mmoles) was added to a s~irred sslution oP pyridine (237 mg,
;76~
52
3 mmoles) in DMSO (3 ml) containins NaI (11 mg, 0.075 mmole).
The mixture was allowed to stand overnight at room temperature in
the dark. The mixture was diluted with ethyl acetate (30 ml) to
separate the precipitate which was then collected by filtration,
washed with ethyl acetate (10 ml) and dried to give 350 mg of the
blocked product. The precipitate was ~reated with formic acid
~3.4 ml) containing sodium bisulfite (34 mg) for 30 minutes at
40C. After removal of the formic acid, the residue was purified
by reverse phase column chromatography (packiny of
PrepPAK-500/C~8 cartridge, 100 ml) by eluting with 5% aqueous
CH30H. The fractions containing the desired product were
combined on the basis of HPLC analysis, evaporated under reduced
pressure and lyophilized to give 41 mg (5.5~) of the title
compound (I-lB) (Z/E=4/1). Mp. >200C (dec.).
IR : vKBr cm 1 3300, 1760, 1660, 1600-
max
UV ~phosphate buffer (pH 7) nm (El% ) 237 (386), 250 (377),
max 1 cm 258 (369), 265 (347)~
280 (311).
N~R : ~D2O 3.45 & 3.76 (each lH, d, J=16, 2-H), 4.18 (3H, s,
ppm OCH3), 5.34 (3H, m, CH=CH-CH2 & 6-H), 5.92 (lH, d,
J=4.5, 7-H), 6.5R (4/5H, d, J=ll, 3-CH cis), 7.03
(l/SH, d, J=16, 3-CH trans), 8.12 (2H, m, Py-H3 5),
8.56 (lH, m, Py-H4), 8.82 (2H, m, Py-H2 6)
7~i~Z~
53
S
~ CON~ ~ ~
H2N S OCH3 N ~ CH=CHCII~- ~ S ~ NH2
I-lC *~
7-~2-(5-Amino-1,2,4-thiadiazol-3-~1~-2-methoxyiminoacetamido]-3-
[3-(2-amino-5-thiazolo~4,5-c]pyridinio)-1-propen-1-yl]-3-cephem-
4-carboxylate (I-lC)
A stirred solutlon of diphenylmethyl 7-[2-(5-amino~
1,2,4-thiadiazol 3-yl) 2-methoxyiminoacetamido]-3 (3-iodo-1-
pr~pen-l-yl)-3-cephem-4-carboxylate (IX-l) (E isomer, 714 mg, 1
mmole), 2-aminothiazolo[4,5-c]pyridine [prepared according to the
procedure of T. Takahashi et al., Pharm. Bull (Japan), 2, 34
(1954)] and dry DMSO ~1 ml) was kept for 1 hour at room tempera-
ture. To the reaction mixture was added ethyl acetate (20 ml) to
give a yellow powder (710 mg). Formic acid (7 ml) and sodium
bisulfite (70 mg) were added to the powder (700 mg), and the
mixture was stirred for 30 minutes at 40-45C. After evapo-
ration, the residue was triturated with ~2 (40 ml). Insolubles
were filtered off, and the filtrate was chromatographed over a
reverse phase column (PrepPA~-500/C18, 100 ml), with H2O and 10%
CH30H as eluant. The fractions containing the desired product
were combined, and the solvent was removed under reduced pres-
sure. Lyophilization gave the desired product (I-lC) as a
colorless amorphous powder of the E isomer. Yield 110 mg (19~.
Mp. ~200C (dec.).
, .
76;~329
54
I~ : vK r cm 1 3300, 1760, 1660, 1630, 1600.
max
UV ~Phosphate buffer (pH 7) nm (El% ) 245 (499)~ 285 (286)-
max 1 cm
NMR ~ SO d6 2 3.86 (3H, s, OC~3), 4~98 (lH, d, J-4.5,
ppm 6-H), 5.2 (2H, m, CH=CH-CH2), 5.57 (lH, m,
3-CH=CH), 5.96 (lH, m~ 7-H), 7.16 (lH, d,
J=16, 3-C~ trans), 8.36 & 8.45 (each lH, d,
J=7, Py-~), 8.~2 (lH, s, Py-H).
Example S
N ~ ~ ~ CONH ~ ~
H2N S \ OCH3 ~ CH=C~-CH2-N(~H3)3
., COO~)
I-lD *Z~E ~ 1~1
7 [2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-
(3-trimethylammonio-1-propen-1-~1)-3-cephem-4-carboxyl te (I-lD)
To a solution of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadiaæol-3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-propen-1
yl)-3-cephem-4-carboxylate (IX-l) (Z/E=2/1, 490 mg, ~.68 mmole~
in ethyl acetate (14 ml) was added a 0.1 M trimethylamine so-
lution in ether (13.6 ml) in one portion. The mixture was
stirred for 10 minutes and evaporated to dryness, and the residue
was triturated with ether (20 ml). The resulting solid (490 mg)
was added to trifluoroacetic acid (0.2 ml) containing one drop of
anisole. After 1.5 hours' stirring, the mixture was evaporated
to dryness under reduced pressure and the residual oil was
triturated with ether (20 ml). The resulting precipitate was
collected by filtration and dissolved in ~2 (20 ml). Some
insolubles were removed, and the aqueous solution was eluted on a
~27~
C18 reverse phase column (the packing of PrepPAK-500/C18 car-
tridge, Waters' 30 ml) using wa~er as eluant. Fractions contain-
ing the desired c~mpound were combined and concentrated to a
small volume and lyophilized to afford 30 mg (9.2%) of the title
compound (I-lD) ~/E = 1/1) a~ a colorless amorphous powder,
melting at >150C ~dec.).
IR : vKBr cm 1 3300, 1770, 1670, 1605.
max
UV ~Phosphate buffer (pM 7) nm (El~ ) 236 (389), 287 (343)-
max 1 cm
NMR : ~D2O 3.45 & 3.7 (lH, d, J=16, 2-H), 3.81 (lH, s, 2-H),
ppm
4.1 (2H, d, J=8, -CH2N)~ 4.21 (3H, s, OCH3), 5.39
(lH, d, J=4.5, 6-H), 5.95 (2H, m, 3-CH=CH ~ 7-H),
6.61 (1/2H, d, J=ll, 3-C~ cis), 7.05 (1/2H, d,
J=16, 3-CH trans~.
Example 6
N _ ----CON~
3 ~ C~ CHC32-
I-lE *E
7-[2-(5-Amino-1,2,4-thaadiazol-3-yl)-2-methoxyiminoacetamido]-3-
[3-(3-amino~yridinio)-1-propen-1-yl]-3-cephem-4-carboxylate
(I-lE)
Diphenylmethyl 7-[2-(5-amino-1,2,4-thiadiazol-3-yl)-2-
methoxyiminoacetamido]-3-(3 iodo-1-propen-1-yl)~3-cephem-4-
carboxylate (IX-l) (E, 716 mg, 1 mmole) was added to a stirred
solution of 3-aminopyridine (188 mg, 2 mmoles) in DMSO (1 ml).
~6~32~3
56
The mixture was stirred for 1 hour and diluted with ethyl acetate
(20 ml). The resulting precipitate was collected by filtration,
washed with ethyl acetate and dried to give 520 mg of yellow
powder. A mixture of the powder (500 mg), formic acid (5 ml) and
sodium bisulfite (50 mg) was stirred for 30 minutes at 40C. The
ixture was concentrated in vacuo, dissolved in H O (40 ml) and
filtered to remove insolubles. The aqueous solution was
chromatographed on a column of reverse phase (packing of
PrepPAK-500/C18, 100 ml), with 7.5% aqueous CH30H elution. The
fractions containing the desired compound were evaporated and
lyophilized to give the title compound (I-lE) (7 mg, 1.4~),
melting at >185C (dec.).
IR : v cm 1 3400, 1765, 1675, 1620, 1600.
max
UV ~Pho~phate buffer (pH 7) nm (El% ) 246 (403), 290 (468)-
max lcm
NMR : ~D2O 3.72 (2H, m, 2-H), 4.14 (3H, s, OCH3), 5.35 (3H, m,
ppm 6-H & CH=CH-CH2), 5.9 (lH, d, J=4.5, 7-H), 6.1 (lH,
m, 3-CH=CH), 7.05 (lH, d, J=16, 3-CH, trans), 8.1
(lH, m, Py-H5), 8.54 (lH, br-s, Py-H6), 8.68 (lH, m,
Py-H4), 9.4 (lH, m~ PY H2)
Treatment of IX-l (716 mg, 1 mmole) with 3-t-butoxy-
carbonylaminopyridine (324 mg, 2 mmolesl by a procedure similar
to that described above gave 12 mg (2.3%) of I-lE.
~Z 76~Z9
5~'
ExamPle ?
N fi - CON
~ ~\ ~ 1 * ~, ,7~ NH2
H2 S ~ OCH3 ~ CH=CHCH2-N
I-lE *Z/E G 1/1
7-[2-(S-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-
[3-(3-amino-1-pyridinio)-1-propen-1-yl]-3-cephem-4-carboxylate
(I-lE)
~ mixture of diphenylmethyl 7-~2-(5-amino-1,2,4-
thiadiazol-3-yl)-2-methoxyiminoacetamido] 3-(3-iodo-1-propen-1-
yll-3-cephem-4-carboxylate (IX-l) (Z/E=2/1, 500 mg, 0.7 mmole)
and 3-aminopyridine (66 mg, 0.7 mmole) in dimethylsulfoxide (1
ml) was stirred for 20 minutes at room temperature. The mixture
was diluted with ethyl acetate (10 ml) and ether (10 ml), and the
resulting precipitate was collected by filtration, washed with
ether (10 ml) and dried. The quaternized salt was dissolved in
formic acid (3 ml) containing concentrated HCl (0.3 ml) and
stirred for 1.5 hours at room temperature. The mixture was
concentrated to dryness under reduced pressure. The residue was
dissolved in 2% HCl (10 mlj and filtered. The aqueous layer was
chromatographed on a reverse phase column (PrepPAK-500/C18, 100
ml). After washing with water (500 ml), the column was eluted
with 5~ aqueous CH30H. The fractions containing the title
compound were combined, concentrated ln vacuo and freeze-dried to
give 15 mg (4.2%) of the title compound (I-lE) (Z/E=l/l) as a
colorless amorphous powder. Mp. >160C (dec.).
~ 76~
5~
IR : ~KBr cm 1 3400~ 1765, lG75, 1620, 1600.
max
UV ~Phosphate bUffer (pH 7~ nm (El% ~ 244 (434), 287 ~333)-
max 1 cm
NMR ~DMS-d6+D2 3.73 (2H, m), 4.14 ~3~, s, OCH3),
ppm m, 6-H & CH-CH-CH2), 6.0 (2H, m, 7 H &
3-CH=CH), 6.6 (1/2H, d, J=ll, 3-CH cis),
7.05 (1/2H, d, J=16, 3-CH trans), 8.08 (lH,
m, Py-H5), 8.6 (2H, m, Py-H4 6)~ 9 4 (lH, m,
Py-~12 ) .
S
H2N ~ ~ \ OC~3 O ~ ~ C~-CHCH2-
I-lF *F
7-~2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-
[.3-(3-fo~ylaminopyridinio)-1-propen-1-yl]-3-cephem-4-carboxylate
(I-lF)
A mix~ure of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-propen-1-
yl)-3-cephem-4-carboxylate (IX-l) (E, 716 mg, 1 mmole) and 3-
formylaminopyridine [prepared according to the procedure of N.
Enomoto et al., Bull. Chem. Soc. Japan, 45, 2665 (1972)] (244 mg,
2 mmoles) in DMSO (2 ml) was stirred at room temperature for 1
hour, and poured into ethyl acetate (200 ml). The precipitate
was oollec~ed by filtration, washed well with ethyl acetate and
dried A mix~ure of the quaternized salt (500 mg) and sodium
bisulfite (50 mg) in HCOOH (5 ml) was stirred at 40-50~C for 80
minutes and evaporated to dryness in vacuo. The residue was
.~. ,, , ~ .
6~
59
dissolved in water (40 ml), neutralized with NaHCO3 and then
filtered to remove insoluble material~ The clear filtrate was
chromatographed on a reverse phase column, PrepPAK-500/C18 (100
ml), with water and 5~ CH30H, 10~ CH30H, 20% CH30H and 30% CH30H.
The frac~ions containing the desired compound were combined,
concentrated in vacuo and lyophilized to give 16 mg (2.9~) of the
title compound (I-lF) (E) as a tan powder. Mp. ~170C (dec.).
IR : ~K r cm 1 3340(br)l 1760, 1670, 1620(br), 1590.
max
UV ~Phosphate buffer (pH 7) nm (El% ) 218 (428), 248 (362),
max 1 cm 290 (474).
NMR : ~D2O+NaHCo3 3.68 (2H, br, 2-H), 4.15 (3H, s, OCH3), 5.~1
ppm ~lH, d, J=4.5, 7-H), 6.25 (lH~ m, CH=CH-CH2),
6.98 (lH, d, J=16, 3-CH trans), 8.8-7.9 (4H,
m, Py-H), 9.38 (lH, br, NHCHO).
.
' Example 9
N ~ CONH
H~N S ~ ~r~3 ~ ~ CH CHCH2-
I-lG *E
7-[2-(5~Amino-1,2,4-thiadiazol-3-yl)-2-methoxyimin acetamido]-3-
[3-(3-carbamoylpyridinio~-1-propen-1 yl~-3-cephem-4-carboxylate
~I-lG~
To a solution of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadiazol~3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-propen-1-
yl)-3-cephem-4-carboxylate (IX-l) (E, 716 mg, 1 mmole) in DMSO (2
ml) was added nicotinamide (244 mg, 2 mmoles), and the mixture
was stirred at ambient temperature for 1.5 hours and poured into
~o
ethyl acetate (200 ml) with stirring. The resulting precipitate
was collected by filtration. The quaternized salt (500 mg) was
dissolved in HCOOH (5 ml) in the presence of sodium bisulfite (50
mg), and the mixture was heated at 40-50C for 40 minutes, with
stirring, and evaporated to dryness. The residue was dissolved
in water (40 ml) and an insoluble solid was filtered off and
washed with a small amount of water. The filtrate and wash were
combined and chromatographed on a reverse phase column, PrepPAK-
500/ClB ~100 ml). After elution with water and 5%, 10% and 20%
aqueous CH30H, successively, the fractions containing the desired
material were combined, concentrated in vacuo and freeze-dried to
yield 21 mg (3.8%) of the title compound (I-lG) (E) as a yellow
powder. Mp. >175C (dec.3.
IR : vKBr cm 1 3340(br), 1760, 1670, 1600.
max
UY ~Phosphate buffer (pH 7~ nm (El~ ) ~35 (326), 274 (sh,
max 1 cm 405), 290 (446).
MR :~D2O NaHC8 3.68 (2H, br, 2-H), 4.15 (3H, s, OCH3), 5.32
ppm (lH, d, J=4.5, 6-H), 5.45 (lH, d, J=7,
CH=CH CH2), 5.88 (lH, d, J-4.5, 7-H~, 6.15
(lH, d-t, J=16 & 7, 3-CH=CH), 7.00 (lH, d,
J=16, 3-CH trans), 8.23 (lH, m, Py-H5), 9.03
(2H, m, Py-H4&6), 9.34 (lH, s, Py-H2).
6~
61
ExamPle 10
- ~ ~ CONH ~ ~
\ ~ N ~ CH-CHCH2- ~ CONH2
I~lH *E
7-~2-(5-Amino-1,2,4-thiadiazo~ y~ methoxyiminoacetamido]-3-
[3-(4-carbamoylpyridinio?-1-propen-1-yl]-3-cephem 4-carboxylate
(I-lH)
To a stirred solution of diphenylmethyl 7-[2-(5-amino
1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-
pr~pen-l-yl)-3-cephem-4-carboxylate (IX-l) (E~ 716 mg, 1 mmole)
in dry DMSO (2 ml) was added isonicotinamide (244 mg, 2 mmoles).
The mixture was stirred at room temperature for 1 hour and then
poured into ethyl acetate (200 ml). The resulting precipitate
was collected by fil~ration, washed well with ethyl acetate and
dried. A stirred mixture of the quaternized material (400 mg)
and sodium bisulfite (40 mg) in HCOOH (4 ml) was heated at
40-$0C for 1 hour, and evaporated to dryness under reduced
pressure. The crude solid was dissolved in water (40 ml). After
filtration of an insoluble material, the filtrate was chromato-
graphed on a reverse phase column (packing of PrepPAK/Cl~, 100
ml) using water and 5~, 10~, 20% and 30% aqueous CH30H as eluant.
The fractions containing the desired compound were combined,
evaporated and lyophilized to ~ive 21 mg (3.8%) of the title
compound (I-lH) (E) as a pale yellow powder. Mp. ~180C (dec.).
62
IR : ~ cm 1 3340(br), 1760, 1670, 1500.
max
UV ~Phosphate buffer (pH 7) nm (El% ) 222 (362), 285 (452)-
max 1 cm
N~lR : ~D2O NaHCO3 3.68 (2H, br, 2-H), 4.15 (3H, s, OCH3), 5.33
ppm(1~, d, J=4.5, 6-H), 5.46 (2~, d, J=7~
CH=CH-CH2), 5.90 (1~, d, J=4.5~ 7~~)l 6.17
(1~, d-t, J=16 ~ 7r 3-CH-C~), 7.02 (lH~ d,
J=16, 3-CH trans), 8.43 & 9.09 (each 2H, d,
Ja7~ py H).
Example 11
N ~ N ~ CONH ~ ~ C~2N~2
~2~ ~ S ~ \ OCH3 ~ N ~ CH-CHCHz-N
COO~)
,. .
I-lI *E
7-[2-(S-Amino-1,2 ! 4-thiadiazol-3-yl)-2-methoxyimlnoacetamido]-3-
[3-(3-aminometh~lpYrldinio)-l-propen-l-yl]-3-cephem-4-carbo~ylate
(I-lI)
A mixture of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadiazol-3-yl3-2-methoxyiminoacetamido]-3-(3-iodo-1-propen-1-
yl)-3-cephem-4-carboxylate (IX-1) (E, 716 mg, 1 mmole) and 3-(t-
butyloxycarbonylaminomethyl)pyridine (516 mg, 2 mmoles) in DMSO
(2 ml) was stirred at ambient temperature for 30 minutes. The
mixture was poured into ethyl acetate ~200 ml), and the precipi-
tate was collected by filtration, washed well with ethyl acetate
and dried. A mixture of the quaternized salt (500 mg), sodium
bisulfite (50 mg) in HCOOH (5 ml) was ~tirred at 40-50C for 30
minutes and evaporated to dryness under reduced pressureO The
residual solid was dissolved in water (40 ml), and the mixture
'
63
was neutralized with NaHCO3. Insoluble material was filtered
off, and the filtrate was chromatographed on a reverse phase
column (packing of PrepP~K-500/C18 cartridge, 100 ml), eluting
with water, 5%, 10%, 20% and 30~ aqueous CH30H, successively.
The fractions containing the desired compound were combined,
evaporated and lyophilized to provide 10 mg (l.e%) of the title
compound (I-lI) (E) as a tan powder.
IR : vK3r cm 1 3380(br), 1760, 1650(sh), 1620(sh).
max
UV ~Phosphate buffer (pH 7) nm (El% ) 235 (sh, 260)~ 286
max 1 cm (370).
NMR :~ 2 3 3.68 (2H, br, 2-H), 4.16 (3~, s, OCH3), 6.98
PP (lH, d, J=16, 3-CH trans), 8.05 (lH, m,
Py-H5), 8.50 (lH, m, Py-H4), 8.80 (2H, m,
Py-H2 6)
Ex~ple 12
CON~
- N ~,~ * ~3 ~
N' ` S ~ OCH3 ~ ~ CH=CHCH2- ~ -CONH2
~ *E
7-[2-(5-Amino-1,2,4-thiadia~ol-3-yl)-2-methoxyim noacetamido]-3-
[3-(4-carbamoylpyridinio)-1-propen-1-yl]-3=cephem-4-carboxylate
~I-lH)
A mixt~re of diphenylmethyl 7-[2-(5-amino~ ,4-
thiadiazol-3-yl)-2-methoxyiminoacetamidoJ-3-(3-iodo-1-propen-1-
yl)-3-cephem-4-carboxylate (IX-l~ (E isomer, 4.1 g, 5.7 mmoles)
and ~sonicotinamide (1.4 g, 11 mmoles) in dry DMSO (6 ml) was
12~ 29 ?
64
stirred for 2 hours at room temperature while monitoring by TLC
(silica gel plate, CHC13:CH30H = 3:1). The reaction mixture was
diluted with ethyl acetate (100 ml) to separate a yellow gum,
which was treated with formic acid (40 ml) and sodium bisulfite
(390 mg) at 45~C for 30 minutes. The resulting solution was
concentrated to dryness. The residue was dissolved in H20 (100
ml) and insolubles were removed by filtration. The combined
filtrate and water wash was applied to the top of a column
containing reverse phase packing (PrepPAK-500/C18j 120 ml). The
column was eluted with H20. The eluate was collected in 300 ml
fractions and monitored by uv (254 nm) and HPLC (Lichrosorb
RP-18, 4 x 300 mm, 0.01 M ammonium phosphate buffer, pH 7.2
containing 20% CH30H). Fraction Nos. 4 and 5 were combined and
concentrated to a small volume. Lyophilization gave 250 mg
(8.1~) of the title compound I-lH, melting at >180~C (dec.).
The spectra indicated that tbe product was identical to
that obtained in Example 10.
Preparation of the hydrochloride - To a suspension of
Compound I-lH (98 mg, 0.18 mmole) in CH30H (1 ml~ was added 10%
HCl (0.1 ml), and the mixture was stirred for 5 minutes. To the
resulting yellow solution was added acetone (100 ml) to give a
precipitate, which was collected by filtration, washed with
acetone (2 x 10 ml) and dried in vacuo to give the hydrochloride
salt of I-lH as a colorless powder. Yield 88 mg (79%). Mp.
>l90~C (dec.).
~5
IR : v cm 3300, 1770f 1680, 1620.
max
UV ~Phosphate buffer (pH 7) nm (E 1% ) 227 (385), 286
max 1 cm (374).
N~IR : ~D20 2.32 (lH, s, acetone-H), 3.79 (2H, br-s, 2-H), 4.17
ppm 3H, s, OCH3), 5.34 (lH, d, J=4.5, 6-H), 5.49 (2H, d,
J=7, CH=CH-CH2~, 5.93 (lH, d, J=4.5, 7-H), 6.28 (lH,
d-t, J=16 & 7, 3-CH=CH), 7.15 (lH, d, J=16, 3-CH),
8.43 & 9.1 (each 2H, d, J=7, Py-H).
. S
I ~ ~ ~ CONH
H2N ~ S~' OCH3 ~ ~ CH-CHCH
I-lJ *E
7-12-(5-Amino--1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-
[3-(2-methylthiazolio)-1-propen-1-yl]-3-cephem-4-carboxylate
(I-lJ)
To a mixture of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-propen-1-
yl)-3-cephem-4-carboxylate ~IX-l) (E, 714 mg, 1 mmole) and 2-
met~ylthiazole EPrePared according to the procedure of R. P.
Kurkjy, E. V. Brown, J. Am. Chem. Soc., 74, 5778 (1952)] (198 mg,
2 mmoles) in dry CH2C12 (10 ml) was added AgBF4 (90~ pure, 217
mg, 1 mmole) at 20C. The mixture was stirred for 30 minutes at
room temperature and filtered. The precipitate was extracted
with 10% CH30H-CHC13 (3 x 20 ml). The combined extracts were
washed with brine (2 x 5 ml), dried over MgS04 and evaporated to
dryness to give a yellow residue, which was triturated with
isopropyl e~her and riltered to yield 350 mg of the quaternized
~L27~ 9
66
product. A mixture of this solid, sodium bisulfite (35 mg) and
formic acid (3.5 ml) was stirred at 40C for 30 minutes. The
mixture was concentrated to remove the formic acid, and the
residue was diluted with H2O (40 ml). Some insolubles were
removed by filtration. The filtrate was placed on a reverse
phase column (PrepPAK-500/C18, 100 ml). The column was eluted
with H2O (200 ml), 5% aqueous CH30H (400 ml) and 10% aqueous
CH30EI (300 ml), successively. The fractions containing the
desired product were pooled on the basis of HPLC analysis
(Lichrosorb RP-18, 4 x 300 mm, 0.01 M ammonium pbosphate buffer
pH 7.2, containing 20% CH30H). The combined solution was concen-
trated to a small volume and lyophilized to give 40 mg (7.7%) of
the title compound (I-lJ) (E). Mp. >195C (dec.).
IR : v cm 3300, 1760, 1660, 1600.
max
UV ~Phosphate buffer (pH 7) nm (El% ) 238 (442), 292 (421)-
max 1 cm
NMR : ~ 2 6 3.06 (3H, s, thiazole-CH3), 3.74 (2H, br-s,
ppm 2-H), 4.19 (3H, s, OCH3), 5.92 (lH, d,
J=4.5, 7-H), 6.1 (lH, m, 3-CH=CH), 6.8
(lH, d, J=16, 3-CH trans), 8004 & 8.23
(each lH, d, Ja4~ thiazole-H).
~276~;~9
67
.
CON~
,~ ~CH3 ~ CH=C~2--
I-lL *E
7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2~methoxyiminoacetamido]-3-
ate ~I-lL)
A mixture of diphenyl 7-[2-(5-amino-1,2,4-thiadiazol-
3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-propen-1-yl)-3-cephem-
4-carboxylate (IX-l) (E isomer,l.07 g, 1.5 mmole), 4-hydroxy-
methylpyridine (818 mg, 7.5 mmole) in CH3CN (4.5 ml) and CH30H (3
ml) was stirred at room temperature under N2 atmosphere for one
hour. After removal of ~he solvents by evaporation, the residual
oil was triturated with isopropyl ether, collected by filtration,
and washed with a mixture of isopropyl ether and methanol (3 : 1,
10 ml) to give 1.28 9 of the quaternized cephem ester as a yellow
powder. A solution of the ~uaternized ester (1.25 g) and sodium
bisulfite (600 mg) in B5 ~ HCOOH (10 ml) was stirred a~ room
temperature under N2 atmosphere for one hour. After the addition
of 85 % ~COOH (5 ml), the mixture was stirred under the same
conditions for an additional hour. Toluene was added and the
reaction mixture was evaporated azeotropically under reduced
pre sure. The residue was triturated with acetone to yield 1.17
g of the crude formate of the title compoundO A suspension of
this compound tl.l5 g) in water (100 ml) was filtered to remove
insolubles, which were washed wi~h water (10 ml x 2)o The
filtrate and the washes were combined and subjected to reverse
phase column chromatography. The column, which was packed with
the packing taken out of a prepPAK-500/C18 cartridge column
1L;276~29
68
(Waters) 60 ml), was developed with water, 5 % methanol and 10 %
methanol, successively. The fractions containing the desired
compound were combined, concentrated under reduced pressure, and
precipitated by the addition of acetone to give 100 mg of the
title compound (I-lL)as a pale yellow powder. To a suspension o,
the powder (90 mg) in methanol (9 ml) was added 1 M HCl in CH30H
(0.5 ml) and the mixture was stirred at room temperature and
concentrated in vacuo. To the concentrate was added isopropanol
to precipitate 77 mg of the hydrochloride of the title compound.
Pale yellow powder. M. p. >190C(dec.).
KBr
IR : vmax cm 1, 1775, 1670, 1635, 1530.
Phosphate buffer (pH7)
UV ~max nm (), 230 (22600), 264 (sh, 163~0)
MR : ~D20
ppm 3.83 (2H, br. 2-CH), 4.17 (3H, s,
OCH3~, 5.06 (2H, s, ~ CH2OH), 5.36 (lH, d,
J=4.5 Hz, 6-H), 5.41 (2H, d, J=7 Hz, CH=CH-CH2),
5.94 (lH, d, J=4.5 Hz, 7-H), 6.36 (lH, d-t,
J=16 and 7 Hz, CH=CHCH2), 7.13 (lH, d, J=16 Hz,
CH=CH-CH2), 8.08 and 8.83 (each 2H, d,
J=7 Hz, P~-H).
~L27~g
69
N C ~ CON~ ~ S ~
H2N ~ CC H ~ ~ CH=C~H2--~~ oNH2
I-2H ~E
7-[2-(5-Amino-1,2,4-thiadiazol-3-~1)-2-ethoxYiminoacetamido]-3-
(I-2H)
To a solution of 200 mg of 7-amino-3-[3-(4-carbamoyl-
pyridinio)- l-propen-l-yl~-3-cephem-4-carboxylate hydrochloride
(E isomer) in 5 ml of 50 ~ aqueous acetone was added portionwise
190 mg of 2-ethoxyimino-2-(5-amino-1,2,4-thiadiazol-3-yl)acetyl
chloride hydrochloride Lprepared according to the procedure
described in published Japan patent application (Rokai) 57-24389
(2/8/82)], and the mixture was adjusted ~o pH 6.5-7.0 with 2 N
Na~CO3 (about 1 ml). The reaction mixture was stirred at 10 C
~or an hour, acidi ied to pH 2 with 1 N HCl and evaporated in
vacuo. The residue was filtered and the filtrate was chroma-
tographed on a column of HP-2~, which was eluted with 500 ml of
water and 25 4 aqueous isopropanol, successively. ~ractions
containing the desired product were combined and evaporated under
reduced pressure. The oily residue was treated wi~h isopropanol
(20 ml) to give 263 mg ~93 ~) of the title compound (I 2H). M.
p. 170 C ~dec.~
To a stirred suspension of 225 mg ~0.40 mmole) of ~he
above zwitterion in 10 ml of methanol was added 1 ml of 1 N HCl
in CH30H and the mixture was stirred at room temperature for 30
minutes. The solution was filtered and concentrated under
* Trade ~ark
~,~
3L27~
reduced pressure. To the residue was added 15 ml of isopropyl
alcohol, and the resulting precipitate was collected by
filtration and dried in vacuo to give the title compound as its
hydrochloride. Yield 146 mg (57 %). M. p. 160 C (dec.).
Estimated purity 65 ~.
KBr
IR : vma~ cm 1, 3300, 1780, 1680, 1620.
~V : ~phosphate buffer tPH7) nm (E), 227
max (22300), 288 (22800).
NMR : ~D2 1.44 (3H, t, J=7 Hz, OCH2-CH3),
ppm 3.74 (2~, br. s, 2-H) 4.45 (2H, q, J-7 Hz,
OCH2-CH3), 5.36 (1~, d, J=4.5 Hz, 6-H), 5.46
(2H, d, J=7 Hz, 3-CH-CH-CH2),5.92 (lH, d, J=4.5
Hz, 7-H), 6.20 (lH, m, 3-C~=CH), 7.04 (lH, d,
J=16 Hz, 3-CH=CH), 8.43 (2H, d, J=7 H~,
Py-HA), 9.10 (2H, d, J=7 Hz, Py-HB).
Example 16
7-[2-(5-Amino-1,2,4-thiadlazol-3-~)-2-meth~ mlnoacetamido ? -3-
[3-(4-carbamoylpyridinio?-1-propen-1-yl]-3-cephem-4-ca_boxylate
(I-lH) (E isomer)
This Example shows the preparation of Compound I-lH via
the last few steps of ~eaction Scheme la or lb, wherein the
intermediate benzhydryl 7-[2-(5-amino-1,2,4-thiadia201-3-yl)-2-
methoxyiminoaceta~ido]-3-[3-(4-carbamoylpyridinio)-1-propen-1-
yl]-3-cephem-4-carboxylate formate (XXVII-lH) is isola~ed.
z9
71
A. Benzhydryl _ 12-~5-Amlno-1~2,4-thiadiazol-3-yl)-2-methoxy-
imlnoacetamido]-3-[3-(4-ca~ ~ o)~ ropen-1-~1]-3-
ce~hem-4-carboxylate Formate (E isomer) ~XXVII-lH)
A solution of XI~-lH ( ~ = ~ , E isomer) (34 g, 75%
pure) in a mixtuee of acetone and C~30H tl/l, 200 ml) was placed
on a column of Amberlite*IRA-410 (formate form 340 ml). The
column was eluted with the same solven~ system. The first
fraction (1 L) was evaporated to about 100 ml of the volume and
the brown residue was triturated wi~h isopropyl ether (400 ml).
The resulting powder was collected by filtration and dried under
vacuum to afford 29 9 (75~ pure by HPLC) of the title compound
XXVII-lH (E isomer) as a brown powder melting at >150C 5dec.).
IR : vKBr cm 1 3300, 1780~ 1680, 1630, 16GO.
max
UV ~EtOH nm (El% ) 282 (186).
max 1 cm
I~IR ~acetone-d6/CH30H~d4(1/1) 4.0 (3H, s, OCH3), 5.26r (lH~ d~
ppm J=4.5 Hz, 6-H), 5.43 (2H, d, J=7 Hz, CH2N ),
5.99 (lH, d, J=4.5 Hz, 7-~), 6.5 (lH, m,
3-CH=C~), 6.92 (lH, s, CHPh2~, 7.1 (lH, d,
J=16 Hz, 3-CH), 7.35 ~lOH, m, Ph-H), 8.36
(lH, s, HCOO), 8.46 & 9.12 (2H each, d,
J=8 Hz, Py-H).
B. 7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacet-
amido]-3-[3-(4-carbamoyl-1-pyridinio?-1-pro~en-1-yl]-3-cephem-4-
carboxYlate (I-lH) (E lsomer)
A mixture of XXVII-lH (E isomer) from Step A (29 g, 75%
pure) and 85% formic acid (290 ml1 was stirred for 2 hours at
room temperature. Evaporation of the mixture gave a brown oil
which was triturated with acetone (500 ml). The powder was
collected by filtration, washed with acetone (2 x 100 ml) and
* Trade Mark
~27~i~3Z~
. ., ~
72
dried in vacuo to give 24 g (50~ pure by HPLC) of the crude title
compound. The brown solid was treated twice with 2 N HCl ~l L
and 0.5 L). The aqueous extracts were combined and placed on a
column packed with Diaion*~P-20 ll.5 L). The column was washed
with water (8 L) and eluted with 30~ CH30H (5 L). The fraction
containing the desired produc~ was evaporated to about 30 ml.
The concentrate was treated with acetone (200 ml) to give a
precipitate, which was collected by filtration and dried in vacuo
to give lO.l 9 (85~ pure) of the title compound (zwitterion form)
as a yellow powder. To a suspension of this product in CH30H
(lOO ml) was added N HCl in CH30H (55 ml) at room temperature and
the mixture was stirred for 30 minutes. The resulting clear
solution was filtered to remove insolubles, concentrated to about
50 ml of the volume and precipitated with isopropanol (200 ml).
The resulting powder was collected, washed with isopropanol (50
ml) and dried _ vacuo to give 1005 g (85% pure) of the title
compound I-lH (E isomer) (HCl salt), melting at >l80C (dec.~.
Pale yellow powder.
Example 17
7-1~2-(5-Amino-l,2,4-thiadiazol-3-yl)~2-methoxyiminoaceta~ido]-3-
[3-(4-carbamoylpy~idinlo)-l-propen-l~yl]-3-cephem-4-carboxylate
(I-lH) (E isomer)
This example shows the preparation of Compound I-lH via
the last few steps of Reaction Scheme la or lb, wherein inter-
mediate XXVII-lH (the formate) is not isolated.
A solution of IX-l (E isomer) (27.6 9, 38.5 mmole) and
isonicotinamide (22.8 9, 187 mmole) in a mixture of CB3CN (120
ml) and C~30H (lOQ ml) was stirred at room temperature for l hour
under a nitrogen atmosphere. After evaporation of the organic
solvents, the oily residue was triturated with isopropyl ether to
give 50.5 g of a mixture of the quaternized salt and isonicotin-
amide. A solution of the mixture (50.3 g) and sodium bisulfite
~l6 g) in 85% HCOOH (160 ml) was stirred at room temperature for
40 minutes and subsequently at 40C for l hsur under N2. The
* Trade Mark
T ' , ~
~ ;Z!76i~2~
73
mixture was evaporated in vacuo. The residual oil was mixed wi~h
toluene (50 ml), evaporated azeotropically and triturated with
acetone (400 ml~ to give 27.8 g of the crude title compound.
This material was treated twice with 2 N HCl (1 L and 0.5 L).
The acid extrac~s were combined and placed on a column of HP-20
resin (1.5 L). The column was eluted with water (9L) and 30%
methanol (lOL). The fractions containing the desired compound
were combined and concentrated to give a yellow oil, which was
triturated with acetone (300 ml) to yield 9.35 g of the
zwitterion form of the title compound.
To a suspension of the product (9.3 g) in CH30H (180
ml) was added 1 N HC1 in CH30H (55 ml) to obtain a clear
solution. The solution was concentrated to about 100 ml and
diluted with isopropanol to precipitate 9.50 g (pueity 754) of
the title compound I-lH (E isomer) as its hydrochlorid.e. Pale
yellow amorphous powder. M.p. >195C (dec.).
.
Example 18
7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxy_minoacetamido]-3-
[3-(4-carbamoylpyridlnio)-1-propen-1-yl]-3-cephem-4-carboxylate
(I-lH) (E isomer?
Tkis example shows the preparation of Compound I-lH via
the last step (7-N-acylation) of Reaction Scheme lco
To an ice-chilled suspension of the 7-amino-cephem
hydrochloride XXII-H (E isomer) ~S.O g, 12.6 mmole) in 50~
aqueous acetone (100 ml) was added sodium bicarbonate in small
portions. The pH of the mixture was monitored by a pH meter
throughout the reaction. To the cold neutralized solution (pH
about 7) was added 2-~5-amino 1,2,4-thiadiazol-3-yl)-2-
methoxyiminoacetyl chloride hydrochloride (4.02 g, 15.6 mmole) in
small portions over a period of an hour, and the pH of the
reaction mixture was maintained in the range of 6.8-7.5 by
occasional additions of sodium bicarbonate. The reaction was
also monitored by tlc. After all of Compound XXII-H had been
. . ~ .
~76~Z~
.
74
consumed, the mixture was acidified to pH 3 by the addition of 2
N hydrochloric acid. The mixture was filtered, and the filtrate
was concentrated under reduced pressure. The residue was diluted
with acetone ( 4bo ml) to separate the precipitate, which was
collected by filtration to afford 9.5g g of the crude title
compound as a light yellow powder. Estimated purity 40% by HPLC.
A suspension of the crude product (9.S g) in 2 N hydrochloric
acid (150 ml) was filtered, and the filtrate was adsorbed on a
column of HP-20 resin (S00 ml~. After washing with water (1.5
L), the column was eluted with 25% aqueous isopropyl alcohol and
the eluate was collected in 100-ml fractions. The desired
fractions were pooled, acidified with 2 N hydrochloric acid (10
ml) and concentrated. The residual oil was triturated with
isopropyl alcohol (200 ml), and the precipitate was collected by
filtration. After drying over phosphorus pentoxide, 5.18 g of
the title compound I-lH (E isomer) hydrochloride was obtained as
a yellow amorphous powder. M.p. >19ODC (dec.). Estimated purity
75%.
Example 19
Purification and crystallization of Compound I-lH (E isomer)
Compound I-lH hydrochloride obtained in Example 16 was
a pale yellow amorphous powder of 85~ purity.
Procedure 1
Six grams of the 854 purity hydrochloride was dissolved
in 20 ml of H2O and filtered through a celite pad. The amber-
colored filtrate ~pH 2~ was passed through a reverse phase column
(the packing of prepPAK-500/C18 cartridge, Waters; 120 ml), which
was eluted with water. The eluate was collected in 120-ml
fractions with monitoring by ~PLC*. Fraction No. 3 through
fraction No. 5 were combined and concentrated to about 10 ml, and
precipitated by acetone (100 ml) to give 3.3 y of the zwitterion
form of I-lH (pale yellow amorphous powder; estimated purity
95~).
~2~6~2g
To a suspension of the 95~ purity powder (3.2 9) in
CH30H (32 ml) was added N HCl in CH30H (18 ml), and the mixture
was stirred at roo~ temperature until a clear solution was
obtained. The solution was filtered and the filtrate was
concentrated to about 10 ml. To the concentrate was added
isopropanol (100 ml) to separate a pale yellow precipitate, which
was collected by flltration, washed with isopropanol (5 ml) and
dried to yield 2.6 g of ~he HCl salt (amorphous powder; estimated
purity 95~).
A solution of the 95~ purity hydro~hloride (1 g) in
water (4 ml) was adjusted to pH 6.5 with NaHCO3 (200 mg) and
stirred for 30 minutes. The crystals which separated during
stirrins were collected by filtration, washed with water (2 x 5
ml)- and dried in vacuo to give 710 mg of I-lH (zwitterion form)
__
as pale yellow prisms. M.p. >185C (dec.)~ Microanalysis showed
it to be the trihydrate.
IR : vKBr cm 1 1780, 1695, 1660, 1630, 1610.
max
UV ~phOsphate buffer (pH7) nm (E) 227 (22000), 290 (23000)-
max
N~ DMSo-d6+D2O
ppm 3.45 (2H, br, s, 2-H), 3.9 (3H, s, OCH3),
4.99 (lH~ d, J=4.5 Hz, 6-H), 5.16 (2H, d,
J=7 Hz, CH2N ), 5.61 (lH, d, J=4.5 Hz, 7-H),
5.8 (lH, d-t, J-16 h 7 Hz, 3-CH=CH), 6.93
(lH, d, J=16 Hz, 3-CH), 8.18 & 8.89 ~each
2H, d, J=7 Hz, Py-H).
Anal. Calc d for C21H20N8O652 2 ~ ; ,
N, 18.72; S, 10.71.
Found: C, 42.41; H, 4.35;
N, 18.86; S, ll.G0.
* Column, Lichrosorb RP-18, 4x300 mm: Mobile phase, 0.01 M
76
phosphate buffer (pH 7.2)~CH30H = 85/15: Detection, uv
(254 nm);
Procedure 2
Once crystalline I-lH had been obtained from Procedure
1, it was possible to obtain the crystalline zwitterion form of
I-lH directly from the crude I-lH hydrochIoride by seeding with a
few crystals of the pure I-lH.
A solution of the 85~ pure hydrochloride (250 mg) in
water (1 ml) was treated with charcoal. The solu~ion was
adjusted to p~ 6.5 with NaHC03 (60 mg) and decolorized with
charcoal. The filtrate was seeded with a few pieces of the
crystals obtained from Procedure 1 and stirred overnight at room
temperature. The separated crystals were collected by
filtration, washed with water (2 x 2 ml) and dried under reduced
pressure to give 170 mg (80% recovery) of pale yellow prisms of
I-lH (zwitterion form), melting at >185C (dec.), which was
identical with that obtained by Procedure 1, (as shown by IR, W,
NMR).
The crystalline zwitterion form of Compound I-lH was
slightly soluble in water (6 m~/ml in saline at 23C).
6~
77
Example.Z0
N CONH ~ 5
H2~ 1 ~ OCH3 ~ N ~ CH-C~-CHz-
I-lR ~E
7-[2-(5-Amino-1,2,4- hiadiazol=3-yl)-2-methoxyiminoacetamido]-3-
[3-(3-hydroxymethylpyrldinio)-1-1~r~ enyl]-3-cephem-4-carboxylate
(I-lK) (E isomer)
A. DiphenylmethYl 7-[2-(5-Am no=1,2,4-thiadiazol-3-yl)-2-
ethoxyiminoacetamido]-3~[3-~3-hydroY.ymethylpyridinio)-l-
prope~yl~-3-cephem-4-carboxylate iodide (E-isomer) (XII-lK)
To a solution of IX-l (E-isomer, 1.79 g, 2.5 mmoles) in
2.5 ml of CH30~ and 7.5 ml of CH3CN was added 3-hydroxymethyl-
pyridine (545 mg, 5 mmoles), and the mixture was stirred at room
te~.perature for 3 hours. The reaction mixture was poured into
ethyl acetate (100 ml) with vigorous stirringO The resulting
precipitate was collected by filtration, washed with a small
volume of ethyl acetate and dried to give 2.06 g (100%) of the
title compound XII~lK as a tan powder. Mp. 170-180C (dec.).
IR : vmax (KBr) in cm 1 1780, 1725, 1675, 1615, 1530, 1385,
1225, 1040, 750, 700.
UV : ~ax (CzH50H) in nm ~E ) Z90 ~196).
7~
7B
NMR : ~ (DMSO + D20) in ppm 3.7 (2H, br.s, 2-H), 3.91 (3H, s,
OCH3), 4.70 (2H, s, Py C~2-OH),
5.28 ~2H, m, CH2 N+), 5.23 (lH, d,
- J-5Hz, 6-H), 5.90 (lH, d, J=5Hz,
7-H), 6.34 (lH, m, 3-CH=C~), 6.86
(lH, d, J=16H~, 3-CH)l 6.89 (1~,
s, CHPh2), 7.35 (lOH, m, Ph-H),
7.9-8.9 (4H~ m, Py-H).
B. 7-[2-(5 Amino-1,2,4-thiadiazol-3-yl)-2-methoxyimino-
acetamido]-3-[3-(3-hydr~ymethylpyridinio)-1-pr~enyl]-3-
cephem-4-carboxylate (I-lK) (E isomer)
A mixture of XII-lK ~E isomer, 2.~ g, 2.4 ~noles) and
sodium bisulfite (1 g) in 85% ~COOH (10 ml) was stirred for 2
hours at room temperature. The reaction mixture was concentrated
to ca. 5 ml under reduced pressure~ The oily residue was poured
into acetone (100 ml) with vigorous stirring. The precipitate
was collected by filtration, washed with a small amount of
acetone and dried to give 1.1 g of a tan powder, which was
purified by column chromatography [using the packing o~ a
PrepPAK-500/C18 cartridge (Waters)] to give 283 mg (22%) of I-lK
as an amorphous powder. The powder was crystallized from 4N
H2S04 and acetone to give 144 mg of the title compound I-l~ as
colorless needles. Mp. 185~188~C (dec.).
IR : vmax (X~r) in cm 1 1775, 1680sh, 1660, 1630, 1225, 1045,
B50.
UV : ~ma~ (Phosphate buffer, pH 7) in nm (El~ ) 236.5 (283);
292.5 (330).
~ ,~,.
, .
, ~76gZ~ I
79
NMR : ~ (D2O) in ppm 3.75 (2~, s, 2-H), 4.18 ~3H, s, OCH3),
4.97 (2H, s, Py-CH2OH), 5.35 (lH, d,
J=4Hz, 6-H), 5.43 (2H, d, J=6.5HZ,
CH2-N ), 5.92 (1~, d, J=4Hz, 7-H), 6.18
(1~, d-t, J=16Hz, J=6.5Hz, 3-CH=C9-),
6.97 (lH, d, J=16Hz, '-CH), 8.13 (lH,
d-d, J=8Hz, J=6Hz, Py-H), 8.60 (lH, d,
J=~Hz, Py-H), 8.84 (lH, d, J=6Hz, Py-H),
8.90 (1~, s, Py-H).
Example ~1
~ CONH ~ ~ ~
H2N S ~ oc~3 CH=CH-~2- ~ 0NHCH3
I-lM *E
7-[2-(5-Amino-1,2l4-thiadiazol-3-yl)-2-(Z)-methoxyimino-
acetamido]-3-[3-(4-N-meth~tlcarbamoYlPYridinio)-l-propenyl]-3-
cephem-4-carboxylate (I-lM) (E isomer)
A mixture of diphenylmethyl 7-[~-(5-amino-1,2,4-
thiadiazol 3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-1-propenyl)-
3-cephem-4-carboxylate tIX-l) ~E isomer, 450 mg, 0.62 mmole) and
4-N-methylcarbamoylpyridine [prepared according to the procedure
of M. Samejima, Yakugaku 2asshi, 80, 1706 (1960)] (215 mg, 1.58
mmoles) in acetonitrile (2 ml) was stirred under nitrosen
atmosphere for 5 hours at room temperature. The mixture was
evaporated under reduced pressuxe and the residue was triturated
with ether to give 530 mg o the guaternary salt. A mixture of
the solid and sodium bisulfite (150 mg) in 85~ formic acid (2 ml)
was stirred for 4 hours and then heated at 40C for 30 minutes.
The mixture was evaporated under reduced pressure. The residue
was triturated with acetone and the crude product was collected
by filtration. The crude product was chromatographed on a column
of HP-20 (1.5 x 18 cm) and the column was eluted with water and
30% aqueous methanol. The methanolic eluate was evaporated under
~2~Z9
~o
redueed pressure and ~he residue was freeze-dried to give 140 mg
of an amorphous powder, which was further purified by HPLC
(Column: Lichrosorb RP-18, Solvent: 15~ CH3OH) and the eluate
of HPLC was freeze-dried ~o give 60 mg (18%) of the title product
I-lM. Mp. 180-183C (dec.). Estimated purity: 80~.
IR : ~max (KBr) in cm 1 17~0, 1660, 1600.
UV : ~max (Phosphate buffer, pH 7) in nm (~) 230 (22100), 286
(22100).
NMR : ~ (D2O) in ppm 3.08 (3H, s, CONHCH3), 3.72 (2H, s, 2-H),
4.16 (3H, s, OCH3), 5.35 (lH, d, J=4.5Hz,
6-~), 5.95 (lH, d, J=4.5Hz, 7-H), 7.00
~lH, d, J=16Hz, 3-CH), 8.35 (2~, d,
J-6Hz, pyridine~H), 9.05 (2H, d, J=16Hz,
pyridine-H).
Example 22
N ~ N CONH ~ S ~
H2N S ~ OC~3 ~ ~ CH-CH-CH2- ~ OOH
I-lN *E/Z = 7/l
7-12-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-
~3-(4-carboxypyridini_)-1-propenyl]-3-cephem-4-carboxylate
(I-lN)
To a stirred suspension of isonicotinic acid (340 mg,
2.8 ~moles) in dry DMF (3.5 ml) was added N,O-bis(trimethyl-
silyl)acetamide (0.7 ml, 2.8 mmoles) under nitrogen atmosphere.
To the resulting clear solution was added diphenylmethyl t-[2-
(5-amino-1,2,4-thiadiazol-3-yl3-2-methoxyiminoacetamido]-3-~3-
iodo-l-propenyl)-3-cephem-4-carboxylate (IX-l) (E isomer, 720 mg,
l mmole) in one portion, and the red solution was stirred for 1.5
hours at room temperature. The reaction mixture was added
~ , .
,.. .
~` ~
~27~9
81
dropwise to a stirred saturated sodium chloride solution (50 ml)
containing sodium thiosulfate (150 mg). The yellow precipitate
was collected by filtration, washed with water, and dried to
obtain 722 mg of a pale yellow powder. The powder (700 mg) and
sodium bisulfite (70 mg~ were dissolved in 85~ formic acid (S
ml), and the solution was allowed to stand at room temperature
for 1.5 hours. The mixture was suspended in toluene (50 ml) and
concentrated. T~e residue was triturated with acetone (70 ml),
and the precipitate was isolated by filtration to afford 421 mg
of a yellow powder. This crude powder (400 mg) was suspended in
water (2 ml) and to the suspension was added sodium bicarbonate.
The resulting dark solution was adsorbed on a column of the
packing (50 ml) of a PrepPAK/C18 cartridge (Water's Sys~em 500)r
and the column was eluted by water (200 ml). The eluent was
fractionated into 10 fractions (20 ml of each), and the desired
fractions (Fractions Nos. 4-7) were combined, acidified to pH 3
with 2N hydrochloric acid, and concentrated. The residue was
triturated with acetone (30 ml) and the precipitate was collected
by filtration to give 201 mg (37~ of the title compound I-lN as
a yellow powder. E/Z = 7/1; 80~ pure. Mp. >lB9C (dec.).
XR : v~ax (KBr) in cm 1 1770, 1665, 1600.
UV : ~max (Phosphate buffer, pH 7) in nm () 227 (22500),
290 (22100).
NMR : ~ (D2O ~ NaHCO3) in ppm 3.7 (2H, br.s), 4.15 (3H, s),
5.32 (lH, d, J=4Hz), 5.39 (2H,
d, J=6Hz), 6.14 (lH, d-t,
J=15.5 and 6Hz), 7.03 (lH, d,
J=15.5Hz), 8.31 (2H, d, J=7Hz),
8.94 ~2H, d, J=7Hz).
~27~
82
Examp_e 23
S
N ~T--C ~ CONH
H2NJ~S~ ~OC~13 ~N ~C~I-CH-CH;2-~
coo~3 )~
I-lO *E
?-[2-(5-~mino-1,2,4-thiadiazol-3-yl)-2-(Z)-methoxyimino-
acetamido]-3-[3-(2,3~c~cLopentenopyridinio~-l-propenyl]-3-
cephem-4-carboxylate (I-lO) (E isomer)
A mixture of diphenylmethyl 7-[2-(5-amino-1,2,4-
thiadia~ol-3 yl)-2-methoxyiminoacetamido~-3-(3-iodo~l-propenyl)-
3-cephem-4-carboxylate (IX-l) (E isomer, 450 mg, 0.62 mmole) and
2,3-cyclopentenopyridine (217 mg~ 1.83 mmole) in acetonitrile (2
ml) was stirred under nitrogen atmosphere for 4 hours at room
temperature. After evaporation under reduced pressure, the
mixture was triturated with ether to give 560 mg of the
quaternary salt. A mixture of the solid and 85% formic acid (2
ml) was stirred under nitrogen for 3 hours at room temperature
and then ~eated at 40C for 30 minutes. The mixture was
evaporated under reduced pressure and trituration of the residue
afforded 391 mg oX the crude product, which was purified by
chromatography on a column of HP-20 (1O5 x 18 cm). The column
was eluted with water and 30~ aqueous methanol. Evaporation of
the methanolic eluate under reduced pressure, followed by
freeze-drying afforded 160 mg of an amorphous powder, which was
further purified by HPLC (Column: Lichrosorb, Solvent: 10~
CH30H). The eluate of HPLC was freeze-dried to give 5~ mg (15~)
of the title product I-lO. Mp. ~190C (dec.). Estimated purity:
75%.
IR : vmax (KBr) in cm 1 1765, 1670, 1600.
W : ~max (Phosphate buffer, pH 7) in nm (~) 235 (20000), 283
(25000).
~,~
1~76~;ZY) I
~3
MR : ~ (D2O ~ NaHCO3) in ppm 2.2-2.6 (2H, m, -CH2-), 3.1-3.6
(4H, m, -CH2-)~ 3.72 (2H, s,
2-H~, 4.17 (3H, OCH3), 5.33 (lH,
d, J=4.5~z, 6-H), 5.90 (lH, d,
J=~.5Hz, 7-H), 6.75 (1~, d,
J=16Hz, 3-CH), 7.65-8.2 (3H, m,
pyridine-H).
~ ple 24
N - CONH ~ ~
2 ~ O ~ ~ C~=C~-CN2--~ OOH
I-2N
7-[2-(5-Amino-1~2,4-th_ diazol-3-yl)-2-(z)-ethoxyimino-
acetamido]-3-[3-(4~carboxY~Yridinio)-l-~roPenYl]-3-cephem-4
carboxylate (I-2N, E isomer)
and
7-[2-(5-Amino-1~2,4_thiadiazol-3-yl)-2-(Z)-ethoxyimino-
acetamido~-3-[3-(4-carboxypyridinio)-1-~ropenyl]-3-cephem-4-
carboxYlate ~I~2N, Z isomer)
To a chilled mixture of BSA (1.0 ml, 4.12 mmoles) and
isonicotinic acid (506 mg, 4.12 mmoles) was added IX-2 (from
Preparation No. 21~ (1.0 g, 1.37 ~moles), and the mixture was
stirred under nitrogen at room te~perature for 2 hours. The
mixture was poured into 10~ Na2S2O3 (20 ml) to precipitate 1.3 9
of the quaternary salt, which was collected by filtration, washed
with water and driedO A mixture of the solid and sodium
bisulfite (0.3 g) in formic acid (98%, 5 ml) was heated at 40C
for 1 hour and evaporated under reduced pressure. The residue
was triturated with acetone and iltered to give 900 mg of the
crude product (E-propenyl isomer:Z-propenyl isomer = 2:1).
Separation of the isomers was carried out by HPLC (Column:
~2~ 91
. 84
Lichrosorb, Solvent: 15% CH30H). The faster moving fractions of
HPLC were colle~ted, evaporated under rsduced pressure and
~reeze-dried to give the E-propenyl isomer of I-2N (44 mg, yield
6%). The slower moving fractions gave the Z-propenyl isomer of
I-2N (32 mg, yield 4%~ in a similar procedure.
I-2N, ~ isomer
Mp.: >2D0C (dec.).
IR : vmax (KBr) in cm 1 1765, 1660, 1620, 1380.
UV : ~max (Water) in nm (E) 228 (22200), 291 (23600).
N~ (D2O) in ppm 1.45 (3H, t, J=6Hz, CH2CH3), 3.72 (2H, s,
2-H), 4.45 (2H, q, CH2CH3), 5.4.0 (lH, d,
J=4Hz, 6-H), 5.90 (lH, d, J=4Hz, 7-H),
. 7.05 (lH, d~ J=15Hz, 3-CH), 8.30 ~2H, d,
J=6Hz, Py-H), 8.95 (2H, d, J=6Hzr Py-H).
I-2N, Z isomer
Mp.: >200C (dec.).
I-R : vmax (KBr) in cm 1 1760, 1660(sh), 1620, 1370.
UV : )~max (Phosphate buffer, pH 7) in nm (E) 225 (22400), 275
(sh, 16000).
NMR : ~ (D~O) in ppm 1.45 (3H, t, J=7Hz, CH2CH3), 3.50 (lH, d,
J=17Hz, 2-H), 3.75 (lH, d, J-17Hz, 2-H),
5.38 (lH, d, J-4Hz, 6-H), 5.95 (lH, d,
J=4Hz, 7 H), 6.62 (lH, d, J=llHz, 3-CH),
8.35 (2H, d, J=6Hz, Py-H), 8.92 (2H, d,
J=6Hz, Py-H).
- ~2~ 9
Example 25
S
CONH
H2N ~ S ~ \O ~ N ~ CH-C~-CH2-N ~ CONH2
~H2-CEI=C~2 COO~
I-3~ *E
7-[2-(Amino-1,2,4-thiadiazol-3-yl)-2-~prol?en-3-yloxyimino)-
acetamid ~ io)-l-pr~p~enyl]-3-ce~h_m-4-
carboxylate (I-3~ (E isomer)
To a solution of 35 mg (0.08 mole) of 7-amino-3-13-(4-
carbamoylpyridinio)-l~(E)-propenyl]-3-cephem-4-carboxylate
hydrochloride in 2 ml of 50% aqueous acetone was added 52 mg of
2-[5-amino-1,2,4-thiadiaæol-3-yl)]-2-(propen-3-yloxyimino)acetyl
chloride ~.ydrochloride (from Preparation No. 25~ and ~he mixture
was adjusted ~o pH 6O5-7~0 with 21~ Na2CO3. The mix~ure was
stirred at room temperature for 1 hour, acidified to pE3 2 with lN
HCl and concentrated under reduced pressure. The residue was
chromatographed on a column of HP-20 resin which was eluted with
300 ml of water and 30~ CH3OH-H2O. Fractions containing the
product were combined and evaporated under reduced pressure. The
residue, 73 mg, was purified by a column of reverse phase carrier
which was taken out of a PrepPAK-500/C18 cartridge (Waters, 30
ml). The column was eluted with water, 5~ CH30EI, 10% CH30H and
20~ CH30H, successively. Fractions containing the product were
combined and lyop~ilized to afford 26 mg (62~) of the title
product I-3H. Mp. 160C (dec.).
IR : vmax (K~r) in cm 3400, 1765, 1680, 1605, 1400.
V : AmaX (Phosphate buffer, pH 7) in nm (~) 226 (24600), 288
(22800).
~7~
86
NMR : ~ (D20) in ppm 3.75 (2H, s, 2 H), 5.41 ~lH, d, J=SHz,
6-H), 5.50 (4H9 m, CH2N & CH=CH2), 5.~8
~lE, d, J=SHz, 7-H), 6.20 (lH/ m,
3-CH=CH), 7.09 (lH, d, J=17~z, 3-CH),
8.50 (2H, d, J=7Hz, Py-H), 9.16 (2H,
d, J-7Hz, Py-H).
Example 26
N C ~ CONH ~ ~
~2N 1 ~ ~H C ~C~ ~ N ~ CH--CH-CH2- ~ CONH2
I-4H *E
7-[2-(5-Amino-1~2~4-thiadiazol-3-yl)-2-prO~argylOXyiminO-
acetamido~-3-~3-(4-carbamo,ylpyridinio)-1-propenyl]-3-ce~hem-4-
carboxylate (I-4H) (E isomer)
To a solution of 86 mg (0.19 mmole) of 7-amino-3-[3-
(4-carbamoylpyridinio)-1-(E)-propenyl]-3-cephem-4-carboxylate
hydrochloride (XXII-H) in 2 ml of 50% aqueous acetone was added
63 mg of 2-propargyloxyimino-2-~5-amino-1,2,4-thiadiazol-3-yl)-
acetyl chloride hydrochloride (from Preparation No. 26). The
suspension was maintained at pH 6.5-7.0 with 2N Na2C03 and then
stirred at rosm temperature for 1 hour. The reaction mixture was
acidified to pH 2 with lN HCl and concentrated in vacuo. The
residue was diluted with 30 ml of water, neutralized with NaHC03
and filtered. The filtrate was transferred on the top of a
column which was packed with reverse phase carrier (30 ml) taken
from a PrepPAK-500/Cl8 cartridge (Waters). The column was elu~ed
with water, 5~ CH30H, 10% CH30H and 20~ CH30H, successively.
Fractions containing the product were combined and lyophilized to
afford 13 mg (12%~ of the title product I-4H. Estimated purity
7096~ ~p~ 160Co
IR : vmax (KBr~ in cm l 3400, 2120, 1765~ 1680~ 1610
B7
UV : ~max (Phosphate buffer, pH 7) in nm (~) 229 (24000),
288 (21200).
NMR : ~ (D2O) in ppm 3.78 (2H, s, 2-H), 5.15 (2H, d, J=lHz,
-CH -C - CH), 5.40 (lH, d, J=5~z, 6-H),
--2
5.50 (2H, ~, CH-N ), 5.98 (lH, d, J=5Hz,
7-H), 6.20 ~lH, m, 3-CH=CH), 7.05 (1~, d,
J=17Hz, 3-CH), 8.50 (2H, d, J=7Hz, Py-H),
9.16 (2H, d, J-7Hz, Py-H).
.
Example 27
S
~ CONH ~
H2N S ~ ~ ~ N ~ CH=CH-CH2-N ~ CONH2
I-5H *E
7-[2-(5-Amino-1,2,4-thiadiazol-3 yl)-2-cyclopentyloxyimino-
acetamido]-3-[3-(4-carbamoylp~ridinio)-1-propenyl]-3-cephem-4-
carboxylate (I-5H) ~E i~omer)
To a stirred solution of 139 mg (0.31 mmole) of 7-amino-
3-[3-(4-carbamoylpyridinio)-1-propenyl]-3-cephem-4-carboxylate
hydrochloride in 3.5 ml of 50% aqueous acetone in an ice-cooling
~ath was added portionwise 120 mg (0.44 mmole) of 2-(S-amino-
1,2,4-thiadiazol-3-yl)-2-cyclopentyloxyiminoacetyl chloride
hydrochloride (from Preparation No. 27). The mixture was
adjusted to pH 6.5-7.0 with 2N Na2CO3 (0.9 ml) and stirred for 1
hour at 10C. The reaction mixture was acidified to pH 2 with lN
HCl and evaporated under reduc~ed pressure. The residue was
chromatographed on a column of HP-20 resin ~20 ml) and was eluted
with 300 ml of water and 30~ CH3OH-H2O, successively. Fractions
containing the product were combined and concentrated in vacuo.
The residue was treated with 60 ml of acetone to give 111 mg
( R3%) of the title compound I-5H. Mp. 160C (dec.). Estimated
purity 70%.
B~ 1276~32 !3
.
IR : vmax (K8r) cm 1 8400, 1770, 1680, 1605, 1530.
UV : ~'ma~ (Phosphate buffer, pH 7) in nm (~) 224 (23300),
286 (24600)~
NMR: ô (DMSO-d6) in ppm 1.70 (8H, br.s, H~ ), 4.68 (lH,
br.s, )~ )/ 5O05 (lH, d, J=5Hz~
6 H), 5.30 (2H, m, C~2N ), 5.67 ~
d-d, J=5Hz ~ 7Hz, 7-H), 6.20 (lH, m,
3-CH=CH), 7.08 (lH, d, J=17~z, 3-CH),
R.34 (2H, d, J=7Hz, Py-Hl, 9.11 (2H,
d, J=7Hz, Py-H), 9.38 (lH, d, J=7Hz,
7-NH).
H2N ~ OC 3 ~ * (~)
CO~
I-lP *E
ï-[2-(5-Amino-1,2,4-thiadiazol-3-ylJ-2-methoxyiminoacetamido]-3-
[3-(3-carboxymethylpyridinio)-1-propenyl]-3-cephem-4-carboxylate
(I-lP) (E isomer)
A. Diphenylmethyl 7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-
methoxyiminoacetamldo~-3-[3-(3-carboxymethylp ridinio)-l-
proPenyl]-3-cephem-4-carboxylate (XII lP, iodide, E isomer)
To a suspension of 3-carboxymethylpyridine hydro-
chloride (0.89 g, 5 mmoles) in 10 ml of CH2C12 was added
N,O-bis(trimethylsilyl)acetamide (4.97 ml, 18 mmoles), and the
mixture was stirred at room temperature until a clear solution
was obtained. To the solution was added IX-l (1.79 g, 2.5
mmoles), and the mixture was allowed to stand at room
. ~
1~ 7~ 9 !
8~
temperature. After 3 hours, 3 ml of CH3OH was added to the
cooled mixture and the solution was evaporated 1n vacuo to give
an oil which was triturated with ethyl acetate to afford 2.28 g
of the title compound XII-lP as a tan powder. Mp. 161C (dec.).
R : vmax (XBr) ln cm 1 1780, 1720, 1675, 1630, 1530, 1385,
1225, 1045, 755, 700.
UV ~max (C2HsoH) in nm (El% ) 295 (188).
1 cm
NMR : ~ (DMSO + D2O) in ppm 3.70 (2H, br.s, 2-~), 3.90 (5H,
s, OCH3 & Py-CH2CO), 5.25 (3H, m,
-CH2N & 6-H), 5.92 (lH, d,
J=4.5Hz, 7-H), 6.35 (lH, m,
3-CH=CH-), 6.90 (lH, d, J=16~,
3-CH), 6.92 (lH, s, CHPh2), 7.35
(lOH, m, Ph-H), 8.8-9.0 (4H, m,
P H)
B. 7-[2-(5-Amino-1,2~4-thiadiazol-3-yl)-2 methoxyimino-
acetamido]-3-[3-~3-carboxymethylpyridinio)-1 Pro~enyl]-3-
cephem-4~carb_xylate (I-lP) (E isomer)
A mixture of XII-lP ~iodide) (2.28 g) and sodium
bisulfite (1.1 g) in 85% HCOOH (10 ml) was stirred at room
temperature ~or 2 hours. The reaction mixture was concentrated
to ca. 5 ml under reduced pressure. The oily residue was
triturated with acetone (100 ml) to give 1.22 9 of the crude
pro~uctt which was purified by column chromatography (HP-20, 420
ml) to afford 533 mg of the title compound I-lP (38%, from IX-l)
as a pale yellow amorphous powder. Mp. 165~C (dec.).
IR : vmax (KBr) in cm 1 1770, 1670, 1600, 1530, 1385, 1140,
1040.
Zg
9o
UV : ~max (Phosphate buffer, pH 6.2) in nm (El% ) 234 (374),
1 cm 277sh (390)~
290 (402).
MR : ~ (D2O + NaHCO3) in ppm 3.78 (2H, s, 2-H), 3.92 (2H, s,
Py-CH2CO), 4.22 (3H, s, OCH3),
5~40 (lH, d, J=4~z, 6-H~, 5.44
(2H, d, J=6.5Hz, -CH2-N ), 5.97
(lH, d, J=4Hz, 7-H), 6.20 (lH,
d-t, J=16 ~ 6.5Hz, 3-CH~CH),
7.08 (1~, d, J=16Hz, 3-CH), 8.11
(lH/ d-d, J-8 & 7~z, Py H5), 8.53
(lH, d, J=8Hz, Py-H4), 8~82 (lH,
d, J=7Hz, Py-H6), 8.86 (lH, s,
Py-H2).
Example 29
N ~ ~ CONH
~2N ~ S ~ H3 ~ ~ H-CH-C~2-N ~ S-CH2C~
I-lQ *E
7.-[2-(5-Amino-1,2,4-thiadiazol-3-Yl)-2-methoxyiminoacetamido]-3-
[3-(4-carboxymeth~lthio~yridinio)-1-propenyl~-3-cephem-4-
carboxylate (I-lQ) (E isomer)
A. Diphenylmethyl 7-~2-(5-Amino-1/2,4-thiadiazol-3-yl)-2-
methoxyiminoacetamido~-3-[3-(4-carboxymethylthiopyridinio)-
l-propenyl]-3-cephem-4-carboxylate ~XII-lQ, iodide,_E
isomer)
To a suspension of 4-carboxymethylthiopyridine (0.88 9,
5 mmoles) in 10 ml of CH2C12 was added N,O-bis(trimethylsilyl)-
acetamide (5 ml, 18 mmoles), and the mixture was stirred at room
temperature until a clear solution was obtained. To the solution
was added IX-l (E isomer 1.79 g, 2.5 mmoles), and the mixture was
~L~7~Z9
91
allowed to stand at room temperature. After 3 hoursl 3 ml of
CH30}~ was added to the cold mixture and the solution was
evaporated in vacuo to give oily residue which was triturated
with ethyl acetate to give 2.43 g of the title compound XII-lQ
(iodide) as a tan powder. Mp. 155C (dec.).
IR : vmax (KBr) in cm 1 1789, 1720, 1670, 1625, 1525, 1385,
122~, 1115, 1040, 755, 700.
max (C2HsoH) in nm (E ) 312 (299).
NIIR: ~ (DMSO-d6 ~ D2O) in ppm 3.70 (2H, br.s, 2-H), 3.93
(3H, s, OCH3), 5.07 (2H, m,
CH2-N ), 5.23 (lH, d, J=5Hz,
6-H), 5.90 (lH, d, J=5Hz, 7-H),
6.29 (lH, m, 3-CH~CH), 6.87 (lH,
d, J=16Hz, 3-CH), 6.91 (lH, s,
CHPh2), 7.35 (10H, m, Ph-H),
7.88 & 8.58 (each 2~, d, J=6Hz,
Py-H).
B. 7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyimino-
acetamido]-3-[3-(4-carboxymethylthiopyridinio)-1-
propenyl]-3-cephem-4-carboxylate (I-lQ) (E isomer)
A mixture of XII-lQ (iodide, 2.43 g~ and sodium
bisulfite (1.1 g) in 85% HCOOH (10 ml) was stirred at room
temperature for 2 hours. The reaction mixture was concentrated
to ca. 5 ml under reduced pressure. The oily residue was
tri~urated with acetone (100 ml)r filtered and dried to give a
crude product (1.39 ~), which was purified by column
chrohlatography (HP-20, 20 ml) to afford 577 mg of 'che title
compound I-lQ (39~ from IX-l) as a pale yellow amorphous powder.
Mp. 188C (dec.).
IR : vll~ax (KBr) in cm 1765, 1670, 1625~ 1530, 1380, 1110,
1035.
92
UV : AmaX (Phosphate buffer, pH 6.2) in nm (El% ) 234 (459),
l cm 310 (678)-
MR : ~ (D20 ~ NaHC03) in pp~ 3.79 (2H, br.s, 2-H), 4.10 (2H,
s, S-CH2), 4.23 ~3H, s, OCH~),
5.25 (2H, d, J=6.5Hz, CH2-N ),
5.39 (lH, d, J=4.0Hz, 6-H), 5.97
(lH, d, J=4Hz, 7-H), 6.18 (lH,
d-t, J=15.5Hz & 6.5Hz, 3-CH=CH),
7.05 (lH, d, J=lS.5Hz, 3-CH),
7.84 & 8.55 (each 2H, d, J=7Hz,
Py~
E~le 30
N -- CONH~
H2N1 5~0CH O~N~CH CH--C~2\--N~
*E/Z -- 7/1
7-[2-(5 Amino-1,2,4-thiadiazol-3-Yl)-2-methoxyiminoacetamido]-
3-[3~ methYlpyrrolidinio~ propenyl]-3-cephem-4-carboxylate
sulfate (I-lA, sulfate)
A. ~iphenylmethyl 7-12-(5-Amino-lL2,4-thiadiazol-3 yl)-2-
~ethoxyiminoacetamido]-3-[3-(1-methylpyrrolidinio)-1-
propenyl]-3-cephem-4-carboxylate (XII-lA, iodide~
To a cold solution of diphenylmethyl 7-[2-(5-amino-
1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-(3-iodo-
propenyl)-3~cephem-4-carboxylate (IX-l) (from Preparation No. 14
(21.5 g, 30 mmoles) in ethyl acetate (300 ml) was added dropwise
a solution of l-methylpyrrolidine (2.55 g, 30 mmoles) in ethyl
acetate (30 ml) over a period of 1 hour at -5 to 0C, with
stirring. After stirring for an additional lO minutes, the
resulting precipitate was collected by filtration and washed with
~z~s~
93
chloroform (200 ml) to give 23.0 g ~95.8%) of the title compound
(IX-lA, iodide), melting at >175C (dec.).
IR : vmax (KBr) in cm 1 3300, 1780, 1730, 1685, 1615.
max (C2H5oH) in nm (E ~ 218 (435), 295 (188~.
B. Diphenylmethyl 7-[2-(5-Amino-1,2,4-thiadiazol-3-~_)-2-
methoxyiminoacetamido]-3-[3-(1-methylpyrrolidinio)-1-
p~ropenyl]-3-cephem-4-carboxylate_(XII-lA, chloride)
The compound (XII-lA, iodide) (23 g, 28.7 mmoles) was
dissolved in a mixture of acetone and methanol (1:1, 230 ml~ and
applied on an Amberlite IRA-410 (chloride form, 230 mll column
which was pretreated with the same mixed solvent. The column was
developed with the solvent and the fractions containing the
desired compound were combined and concentrated to an oily
residue, which was triturated with ethyl acetate (300 ml) to
yield 17.9 9 (87.7~) of the ~itle compound (XII-lA, chloride),
melting at l90~C Idec.).
I~ : vmax (KBr) in cm 1 3380, 1780, 1680, 1620.
1%
max (C2H5H) in nm (E ) 220 (369), 290 (232).
6~Z~'
94
. 7~ (5-Amino-lr2~4-thiadia~ol~3-y~ S~
acetamido~-3 [3~ methylpyrrolidinio)-1-proPenyl]-3-
-
ce~hem-4-carboxylate su fa~e (I-lA, sulfate)
A mixture of the compound (XII-lA, chloride) (17.8 g,
25 mmoles) in 85% formic acid (178 ml) was stirred at room
temperature for 2 hours under a nitrogen atmosphere. The mixture
was evaporated in vacuo and the oily residue was triturated with
acetone to give 9.80 g of crude I-lA. Concentration of the
filtrate and the acetone washings yielded additional 2.95 g of
crude I-lA. Two crops of the crude material were combined and
extracted with 2N ~Cl (1 L and O.S L). The combined extracts
were adsorbed on a Diaion HP-20 resin (1.5 L column), which was
eluted with water and 30% aqueous methanol. The desired
fractions were collected and evaporated ln vacuo to an oily
residue, which was tri~urated with isopropanol (200 ml) and
acetone (200 ml), successively, to yield 7.09 g of a light yellow
powder. This material (6.80 g) was dissolved in water (20 ml)
and then subjected to column chromatography over the packing of
PrepPAK-500~C18 cartridge (90 ml), using water and 10~ aqueous
methanol as an eluent. The eluate was collected in 20-ml
fractions with monitoring by HPLC. [Column, Nucleocil
SSC-ODS-262~ 8 x 100 mm; Mobile phase, O.OlM phosphate buffer (pH
7.2)/CH30H = 90:10; Detection, UV (254 nm)]. Fraction No. 4
through Fraction No. 10 were combined, evaporated under reduced
pressure and lyophilized to give 2. 28 9 of a yellow powder (E/Z =
7/1, 70% pure) [Crop 1~. Fraction No. 11 through Fraction No. 85
were worked up in the same manner as described above to give 3.27
g of yellow powder (E/Z = 5/lr 70% pure) [Crop 2]. A portion of
rop 1 (1.0 g~ was purified by rechromatography on the packing of
PrepPAK-500/C18 cartridge (90 ml). The column was eluted with
water and S~ aqueous methanol, successively. The eluate
containing the desired compound was concentrated and lyophilized
to yield 638 mg (E/Z = 7/1, 80~ pure) of yellow powder. Another
portion of Crop 1 tl.l4 g) was worked up the same way to give 880
mg (E/Z = 7/1, 80% pure) of yellow powder. The two purified
samples were combined and a portion (1.4S g) dissolved in lN
sulfuric acid (S ml). The solution was diluted with acetone (315
~Z7~i~2~
g5
ml), with stirring. The creamy precipitate was collected by
filtration to obtain 1.48 g of the title compound (I-lA, sulfate)
(E/Z = 7/1, 80~ pure), melting at >185C (dec.).
IR : vmax (KBr) in cm 1 3380, 3000, 1765, 1675, 1630, 1535,
139~, lllS.
UV : ~max (Phospha~e buffer, pH 7) in nm (~) 236 (19900),
291.5 (22500).
~ H
NMR : o (D20 + NaHC03) in ppm 2.36 (4H, br., N ~ ), 3.15
(3H, s, CH3+N ~ ), 3.62 (SH,
br., 2-H and N ~ ), 3. 83 (lH,
b~., 2-H), 4.13 (2H, d, J=8Hz,
CH2N ), 4.22 (3H, s, OCH3), 5. 39
(lH, d, J=4.5Hz, 6-H), 5.96 (lH,
d, J=4.5Hz, 7-H), 6.00 (lH, m,
3-CH=CH), 6.67 (1/8H, d, J=lOHz,
3-CH, cis), 7.04 (7/8H, d,
J=16Hz, 3-CH, trans).
9~9
- 96
Example 31
~ CON~ ~ ~
H2N S ~ \ OC~ o ~ ~ C~=CH-CH2-N(CH3)3
CO
I-lD *E/Z = 10/1
7-[2-(5-Amino-1~2,4-thiadiazol-3-yl)-2-methoxyiminoacetamidoi-
3-[3 trimethylammonio-1-propen~l]-3-cephem-4-carboxylate ~I-lD)
A. DiPhenylmethyl 7-[2-(S amino-1,2,4-thiadiazol-3-yl)-2-
metboxyiminoacetamido]-3-(3-t~ o-1-propenyl)-
3-cephem-4-carboxylate (XII-lD, iodide)
To a solution of 13.0 g (19 mmoles) of diph~nylmethyl
7-[2-~5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetamido]-3-
~3-iodopropenyl)-3-cephem-4-carboxylate (IX-l, from Preparation
No. 10) in 38 ml of dry ethyl acetate was added 1.75 ml (19.1
- mmoles) of l.lN trimethylamine in ethyl acetate at -5C, and the
mixture was stirred for 1 hour at -5C. The resulting
precipitate was filtered off, washed well with CHC13 and dried to
give 12.5 g (88%) of the title compound (XII-lD) as the iodide.
IR : vmax (KBr) in cm 1 3300, 1765, 1720, 1665.
~ax (C2~50H) in nm (~) 300 (18400)
B. ~ ylme~yl 7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-
_inoacetamido]-3-(3-trimeth~ammonio-1-propeny~-
3-cephem-4-carboxylate (XII-lD, chloride)
The iodide (XII-lD, 12.5 g) was dissolved in 60 ml of
C~30H-acetone (1:1~ and passed through a column of ion-exchange
resin [IRA-410 (Cl ), 125 ml]. The colum~ was eluted with 300 ml
of CH30H-acetone (1:1), and the eluate was evaporated in vacuo
and triturated with 300 ml of isopropyl-ether to afford 10.4 9
(91~) of the quaternary salt (XII-lD, chloride).
6~29 i
97
IR : vmax (KBr) in cm 1 8300, 1765, 1710, 1665.
UV : Amax (C2H5OH) in nm (e) 298 (15100).
C. 7-[2-(5-Amino-1,2,_-thiadiazol-3-yl)-2-methoxyimino-
acetamido]-3-~3-trimethylammonio-1-propenyl]-3-cephem-
4-carboxylate (I-lD, sulfate, E isomer)
A solution of 10.4 g (16.0 mmoles) of XII-lD (chloride)
in 20.8 ml of 85% formic acid was allowed to stand for 3 hours at
room temperature and concentrated in vacuo. The residue was
treated with 210 ml of isopropanol and the precipitate was
filtered offO The solid (10.1 g) was triturated with 210 ml of
water and neutralized with sodium bicarbonate. The suspension
was filtered off and ~he filtrate was chromatographed on a column
of HP-20 (300 ml) which was eluted with water (1000 ml), 10~
CH30H (200 ml) and 30~ CH30H (150 ml), successively. Fractions
containing the desired product were combined and concentrated
under reduced pressure. The residue was purified by reverse
phase chromatography. The column was packed with a packing
obtained from a PrepPAX-500/C18 cartridge (Waters, 200 ml).
Elution with water (600 ml) and 30% CH30H (200 ml), successively,
followed by concentration of fractions containing the desired
product gave 2.52 g (18%) of the title compound. A solution of
the zwitterionic product (1.5 g) in lN H2SO4 (5 ml) was added
portionwise to 300 ml of acetone and the resulting precipitate
was filtered and dried. Yield o I-lD sulfate was 1.42 5 (80~).
The ratio of E/Z was approximately 10/1 based on HPLC.
IR : vmax (KBr) in cm 1 3380, 1765, 1665.
V : ~max (Phosphate buffer, pH 7) in nm (E) 237 (19500),
293 (22400).
6~
98
NMR : ~ ~D2O) in ppm 3.25 (9H, s~ N+-CH3), 3.94 (2H, s,
2-H), 4.14 (2H, d, J=7Hz, CH2N~), 4.23
(3H, s, O-CH3), 5.42 ~lH, d, J=4.5Hz,
6-H), 6.00 (lH, d, J=4.5Hz, 7-H), 6.23
(lH, d-t~ J=7 & 16Hz, 3-CH=CH), 7.23
(lH, d, J=16Hz, 3-CH).
NC----CON~I~
H2N~3 ~ ~ `CH--C~_CH2_N~CONH2
cod9
~ E
7-[2-(S-Amino-1,2,~-thiadia~g~ 3~ ~L ~acLe~8~amido]-
3-~3-(4-carbamoylpyridinio)-1-propenyl]-3-cephem-4-carboxylate
(I-lH, E isomer)
To a mixture of 7-amino-3-[3-(4-carbamoylpyridinio)-
l-(E)-propenyl]-3-cephem-4-carboxylic acid hydrochloride (XXII-~,
397 mg, 1 mmole) and NaHC03 (168 mg, 2 mmoles) in aqueous DMF
(water, 3.5 ml and DMF, 7.5 ml) was added benzotriazol-1-yl~2-
(5-amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetate (479 mg,
1.5 mmoles) (from Preparation No. 28). The mixture was stirred
at room temperature for 3.5 hours. The reaction mixture was
adjusted to pH 3-9 with 3N HCl and diluted with ?00 ml of acetone
to give a precipitate, which was collected by filtration. The
crude product was dissolved in a small volume of aqueous THF and
the solution was adjusted to pH 6.8 with NaHCO3, treated with
decolorizing carbon, concentrated to ca. 1 ml and seeded with a
few pieces of crystalline I-lH. After stirring overnight, the
crystalline precipitate was collected by filtration to afford the
title compound I-lH (zwitterion form). Yield 83 mg (16%). Mp.
>185C (dec.). Physico-chemical data of this product were
identical to those of the compound in Example 10.
.
6~29
Preparation No. 1
Diphenylmethyl-7-~2-(5-Amlno-l~2~4-thiadiazol-3-~l)-2
methoxyiminoacetamldo]-3-chloromethyl-3-cephem-4-
carboxylate (IV-l)
To a ~tirred suspension of 2-(5-amino-1,2,4-thiadiazol-
3-yl)-2-methoxyiminoacetic acid (III-l) (2.1 g, 10 mmole) in dry
CH2C12 (50 ml) was added PC15 (2.09 9, 10 mmole) at -30C, and
the mixture was stirred for 20 minutes at -15 to -20 C. To the
above acid chloride solution was added a solution of diphenyl-
methyl 7-amino-3-chloromethyl-3-cephem-4-carboxylate
hydrochloride (II) (4.5 g, 10 mmole~ in C~2C12 (50 ml) containing
N,O-bis-(trime~hylsilyl3acetamide (10 g, 50 mmole) at -30 DC.
After stirring at -10 C for 1 hour, the mixture was c,oncentrated
to,remove the CH2Cl~ and diluted with ethyl acetate (200 ml).
The mixture was washed with 10 ~ aqueous NaHC03 (2 x 40 ml), H20
(2 x 20 ml) and brine (10 ml), successively, and dried over
MgS04. The solvent waC evaporated ~n vacuo and the resulting
oily residue tlO g) was dissolved in CHC13 (20 ml) and chromato-
graphed on a silica gel (Wak~ gel C-200, 100 g containing 10 ml
of 1/1.5 M pH 7 phosphate buffer) using 1 - 3 ~ CH30H-CHC13.
Fractions containing the title compound were evapora~ed to give
5.7 g (95 %) of IV-l as a yellow amorphous powder. M. p. >140 DC
(dec.).
IR : vKBrcm 1 3300, 1780, 1720, 1680, 1620.
max
V ~EtHnm () 245 (1800), 280 ~9900).
max
NMR : ~DMSO d6 3.S3 (2~, AB~, 2~ 3.94 (3H, s, OCH3), 4.42
ppm (2H, s, 3 CH2~, 5.22 (1~, d, J~4.5, 6-H), 5.32
(lH, d-d, J=4.5 ~ 6, 7-H), 6.93 (lH, s, CHPh2),
7.36 (lOH, m, Ph-H), 8.1 (2H, br~s, NH2), 9.58
~lH, d, J-6, 7-NH).
* Trade Mark
100
Preparation No. 2
Diphenylmethyl 7-[2-(5-Amlno-1,2~4-thiadiazol-~-yl)-2-
methoxyiminoacetamido]-3-iodomethyl-3-cephem-4 carboxylate (V-l)
A mixture of IV-l from Preparation No. 1 (5.7 g, 9.5
mmole) and NaI (4.3 g 29 mmole) in dry acetone (50 ml) was
stirred for 5 minutes at room temperature. The mixture was
concentrated under reduced pressure and the resulting oil was
shaken with a mixture of ethyl acetate (100 ml) and H20 (10 ml).
The organic layer was separated and washed with 10 % w/v sodium
thiosulfate and brine, successively. After drying, the ethyl
acetate was removed in vacuo to give 6.1 9 (93 %) of the title
compound (V-l) as a yellow amorphous powder melting at > 120 C
(dec.).
IR : vK rcm 1 8300, 1780, 1725, 1680, 1620.
max
UV : ~E Hnm (~) 245 (17000), 282 (12000).
max
NMR : ~DMSO d6 3.72 (2H, ABq, 2-H), 3.94 ~3H, s, OCH3~, 4.23
ppm (2H, s, 3-CH2), 5.21 (1~, d, J=4.5, 6-H), 5.89
(lH, d-d, J=4.5 & 6, 7-H), 6.94 (lH, s, CHPh2),
7.35 (lOH, m, Ph-H), 8.12 (2H, br-s, NH2), 9.65
(lH, d, J=6, 7-NH).
Preparation No. 3
Diphenylmethyl 7-[2-(5-Amino-1,2,4-thiadiazol-~y~-2-
methoxyiminoacetamido]-3~triphenylphosphoniomethyl-3-cephem-4-
ca_boxylate iodide (VI-lJ
A mixture of Y-l from Preparation No. 2 (690 mg, 1
mmole) and triphenylphosphine (786 mg, 3 mmole) in ethyl acetate
(20 ml) was stirred overnight at room temperature. The solid
which separated was collected, washed with ethyl acetate (2 x 10
10 1 ~76q3~9
ml) and dried to give 950 mg (100 ~) of the phosphonium iodide
VI-1. M. p. 186 C (dec.).
IR : vKBrcm 1 3300, 1780, 1710, 1680, 1610.
max
UV : ~E Hnm (e) 268 (15000~, 275 (13000), 300 (7300).
max
NMR : ~DMSO d6 3.52 (2H, br-s, 2H), 3.94 (3H, s, OCH3), 5.34
ppm (lH, d, J~4.5, 6-H), 5.9 (lH, m, 7-H), 6.3 (lH,
s), 7~3 (lOH, m, Ph-H), 7.8 (lSH, m, Ph-H).
~ ~ e ~ 3 ~
Diphenylmethyl 7-[2-(5-Amino-1,2,4-thiadiazol-3 yl)-2-methoxy-
iminoacetamido]-3-~(tri~henylphosphoranylidene~methyl]-3-
ce~hem-4-carboxylate (VII-l)
.
A mixture of VI-1 from Preparation No. 3 (952 mg, 1
mmole), Amberlite IRA-410 (OH form, 500 mg) and N NaO~ (4 ml) in
CH2C12 (10 ml) was stirred for 1 hour at room temperature. The
mixture was filtered and the separated organic layer was dried
over MgSO4 and concentrated under diminished pressure. The
resul~ing oil was triturated with ethyl acetate and the resulting
yellow precipitate was collected by filtration to give 740 mg
(90%) of the title compound VII-l. M.p. ~180C (dec.).
IR : vKBr c~ 1 3400, 1750, 1630
max
UV : ~EtOH nm ~) 268 (1200D), 276 (10000), 384 (23000).
max
~6~9
102
Pre~aration No. 5
Diphen~methyl 7-[2-tS-Amino-1,2,4-'thiadiazol-3~1)-2-methoxy-
iminoacetamido]-3-(3-chloro-1-propen-1-yl)-3-cephem-4-carboxYlate
(VIII-l)
To a solution of VII-l from Preparation No. 4 (6.9 g,
8.4 mmole) were added MgS04 (3 g) and 40~ chloroacetaldehyde (810
mg, 8.4 mmole). The mix~ure was stirred for 1.5 hours at room
temperature and ~hen filtered. The filtrate was eluted on silica
gel (Wakogel C-200, 100 g containing 10 ml of 1/1.5 M phosphate
buffer) column by using CHC13, and CHC13 containing CH30H.
Fractions containing the desired product (0.5 - 1% CH30H-CHC13)
were evaporated in vacuo to give 1.6 9 (30~) of the title
compound VIII-l as a yellow amorphous powder, which was a mixture
of the Z and E isomers ~ith respect to the chloroprope~yl moiety
(Z/E=2/1, by nmr). M.p. ~130~C (dec.).
IR: vKBr cm 1 3300, 1780, 1725, 1680, 1620.
max
UV : ~E OH nm ( ~ ) 240 (20000), 286 (12000).
max
NMR ~DMSO-d6~D20 3.56 ~ 3.8 (m, 2-H), 3.94 (3H, s, OCH3),
ppm 4.16 (d, J=7.5, CH2Cl), 5.26 (lH, d,
J=4.5, 6-H), 5.87 (lH, d, J=4.5, 7-H),
6.28 (2~3H, d, J=ll, 3-CH cis-H), 6.72
(1/3H, d, J=16, 3-CH trans-H), 6.81
(2/3H, s, CHPh2~, 6.92 (1/3H, s, CHPh2),
7.4 (lOH, m, Ph-H).
103
P~epara~ion No. 6
Diphenvlmethyl 7-~enzylideneamino-3-[(tri~henylphosphoranyli-
dene)methyl~-3-ce~hem-4-carbox~late (XVI)
To a solution of diphenylmethyl 7-kenzylideneamino-3-
[(triphenylphosphonio)methyl]-3-cephem-4-carboxylate iodide (XV)
[prepared according to the procedure of Japan published patent
application (Kokai) 56-86187 (7/31/81)] (60 g, 70 mmole) in
CH2C12 (350 ml) were added N NaOH (140 ml) and Amberlite IRA-410
(OH form, 35 g) at 5C. The mixture was stirred for 1 hour at
5C and filtered. The organic layer was separated, dried over
MgSO4, concentrated to ca. 100 ml of volume and precipitated with
ethyl acetate (500 ml). The resulting yellow solid was collected
~y filtration and dried in vacuo to afford 48 9 (94~) of the
title compound XVI, melting at 195~8C (dec.).
IR : vKBr cm 1 1770 1620
max
Preparat on No 7
~iP~ylmeth~ 7-Benzylideneamino-3-(3-chloro-l-pro~en-l-yl)-3
cephem-4-carboxylate (XVII)
To a stirred solution of XVI from Preparation No. 6
(2.9 g, 4 mmole) in a mixture of CH2C12 (40 ml) and H2O (10 nl),
was added anhydrous chloroacetaldehyde (800 mg) at room tempera-
ture. ~o the mixture was added additional 800 mg of chloro-
acetaldehyde in three portions over a period of 1 hour, while the
p~ of the mixture was kept between 6 to 9 by addition of N NaOH.
After 15 minutes, the aqueous layer was removed and the organic
layer was dried over MgSO4. Evaporation of the solvent gave a
red oil which was dissolved in a mixture of ethyl aceta~e and
isopropyl ether ~1/2, 80 ml~. The solution was washed with
saturated aqueous NaHCO3 (l0 ml) and H2O (10 ml), successively.
After drying over MgSO4, removal of the solvent afforded 3.3 g of
yellow oil. A solution of the oil in CH2C12 (50 ml) was filtered
~ Z76~2~
10~
with aid of silica gel (12 9, Wakogel C 200) containing 1/1.5 M
phosphate buffer (1.2 ml, pH 6.4) and the silica gel was washed
with CH2C12 (50 ml). The filtrate and washing were combined and
evaporated to dryness. The residue was triturated with n-hexane
to give 1.7 g (80~) of ~:he title compound (XVII) as a yellow
powder. The nmr spectrum indicated that the ch.loropropenyl
moiety had the Z configuration. M.p. >50C (dec.).
IR: vKBr cm 1 3400, 1775, 1720, 1630.
max
UV:~E OH nm (E) 253 (11000), 258 (llOûO), 265 (looao), 273
max(8300), 281 (7000), 290 (6300).
Nr~ DMSO d6 3.63 (2H, br-s, 2~H), 4.0 (2H, m, CH2-Cl), 5.42
ppm (2H, m, 6-H & 3-CH=CH), 5.72 ~lH, d, J=A.5, 7-H),
6.27 (lH, d, J=ll, 3-CH), 6.85 (lH, s, C~lPh2),
7.33 (lOH, m, Ph-H).
Preparation of anhydrous chloroac_taldehyde
Anhydrous calcium chloride was added to a chilled solution
of 50~ aqueous chloroacetaldehyde (50 ml), with stirring, to
separate it into two layers. The chloroacetaldehyde hydrate
layer (1) (the upper layer) was separated and diluted with CHC13
(100 ml), mixed with MgS04 (20 g), heated to reflux for 5 min~
utes, and filtered. The solvent and water were removed azeo-
tropically (b.p. 56-64C) (2), and the residue was distilled to
give anhydrous chloroacetaldehyde (3), b.p. 70-82C/760 mm.
IR vfilm cm 1 1720.
max
(1) R.P. Kurkjy, E. V. Brown, J. Amer. Chem. Soc., 74,
5778 (1952).
(2) S. Trippett, D. M. Walker, J. Chem. Soc., 1961 1266.
31L;2~69~9
105
(3) H. G. House, V. X. Jones, G. A. Frank, J. Org. Chem.,
29, 3327 (1964).
Preparation No. 8
Diphenylmethx~_7-Amino-3-(3-chloro-1-proeen-1-yl)-3-cephem-4-
carboxylate (XYIII)
A solution of XVII from Preparation No. 7 (180 mg, 0.34
mmole) in ethyl acetate (10 ml) was added to a solution of Girard
Reagent T [(carboxymethyl)trimethylammonium chloride hydrazide]
(251 mg, 1.5 mmole) in CH30H (10 ml) containing acetic acid (0.25
ml), at 5C. After stirring for 30 minutes at 5C, the mixture
was concentrated to remove the CH30~ and then ethyl acetate (20
ml) was added. The ethyl acetate solution was washed with H20 (2
x 5 ml), saturated aqueous NaHC03 (5 ml) and brine (5 ml),
successively and dried over MgS04. Evaporation of the solvent
gave 145 mg (97%) of the title compound XVIII (Z isomer) as a
yellow powder. M.p. >100C (dec.).
IR : vK r cm 1 3400, 1770, 1720.
max
UV : ~E nm (E) 252 (3700), 258 (3800), 260 (4000), 274
max (4000), 285 (4000).
Preparation No. 9
Di~enylme~yl 7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxy-
iminoacetamido]-3-(3-chloro-1-Propen-l-yl)-3-cephem-4-
sarboxylate (VIII-l?
A mixture of 2-(S-amino-1,2,4-thiadiazol-3-yl)-2-
methoxyiminoacetic acid (III-l) (10.1 g, 50 mmole) and PC15 (10.4
g, 50 mmole) in dry CH2C12 (100 ml) was stirred at -7 to -15C
for 2 hours. The clear solution was poured into n-hexane (500
~1) to give a precipitate. The organic layer was discarded by
~276~Z9
106
decantatlon and the remaining solid was triturated with n-hexane
(100 ml). The yellow precipitate was collected by filtration and
dried 1n vacuo to give 12.5 9 (99~) of the acid chloride, melting
at 80C (deo.).
IR : v i cm 1 1770
max
The acid chloride (25 mg, 0.1 mmoleJ was added to a
solution of XVIII (Z isomer) from Preparation No. 8 (44 mg, 0.1
mmole) in dry CH2C12 (5 ml) at room temperature, with stirring.
After 30 minutes, ~he mixture was concentrated under reduced
pressure and diluted with ethyl acetate ~20 ml) and saturated
aqueous NaHC03 (5 ml). The organic layer was washed with sat-
urated aqueous NaHC03 (S ml), brine (5 ml), 10% ~Cl (5 ml) and
brine (5 ml). The solvent was dried over MgS04 and then
evaporated to dryness to give the product as a yellow foam. The
foam was purified by silica gel (Wakogel C-200, 1 g, containing
0.1 ml of 1/1.5 M phosphate buffer pH 6.4) column chromatography
by elution with CH2C12-CH30H (100 : 1), to give 31 mg (50%) of
the ti~le compound VIII-l (Z isomer) as a yellow powder. M.p.
>150C (dec.).
IR : vKBr cm 1 3400, 1775, 1720, 1675, 1630.
. max
UV : ~EtOH nm () 240 (17000), 280 (10000).
max
NMR : ~DMSO ~6 3.6 (2H, m, 2-H), 3.92 (3H, s, O-CH3), 4.0 (2H,
ppm m, CH2ClJ, 5.27 (2H, m, 6-H & 3-CH=CH), 5.83 (lH,
d-d, J=4.5 ~ 10, 7-H), 6.25 (lH, d, J=ll, 3-CH),
6.83 ~lH,s, CHPh2), 7.33 (lOH, m, Ph-H), 8.0 (2~,
br-s, NH2), 9.57 (lH, d, J=10, 7-N~).
92~
107
Preparation No. 10
Diphenylmethyl 7-[2-(5-Amino-lJ2,4-thiadia2ol-3~ -2-methoxy-
iminoacetamido] ~ ephem-4-carboxylate
(IX-l)
A solution of VIII-l from Preparation No. 5 (Z/E=2/1,
480 mg, 0.77 mmole) in dry acetone (10 ml) containing NaI (346
mg, 2.3 mmole) was stirred for 30 minutes at ambient temperature.
The reaction mixture was evaporated under reduced pressure. The
resulting oil was partitioned between ethyl acetate (50 ml) and
wa~er (10 ml). The upper layer was washed with 10~ w/v aqueous
sodium thiosulfate solution (10 ml) and brine (10 ml)
successively, and dried over MgS04. Evaporation of the solvent
gave 540 mg (98~) of the title compound IX-l (Z/E=l/l) as a
reddish amorphous solidt melting at >120C (dec.).
IR : vKBr cm 1 3300, 1780, 1720, 1680, 1620.
max
~EtOH n~ (~) 240 (21000), 290 (12000)
max
NMR : ~DMSO D20 3.67 (2H, m, 2-H), 5.29 (lH, d, J=4.5, 6-H),
ppm 5.95 (lH, d, J=4.5, 7-H), 6.27 (1/2H, d,
- J=ll, 3-CH cis), 6.72 (1/2H, d, J=16, 3-CH
trans), fi.87 h 6.96 (each 1/2H, s, CHPh2),
7.4 (lOH, m, Ph-H).
7~t~3 Z9
108
Preparation No. 11
Diphenylmethyl 7 [2-(5-Amlno-1,2,4-thiadiazol~ oxy-
iminoacetamido]-3-(3-iodo-1-propen-1-yl)-3-cephem-4-carboxylate
(IX-l)
.
A mixture of VIII-l (Z isomer) from Preparation No. 9
(5.6 g, 9 mmole) and NaI (4 g, 27 mmole) in dry acetone (100 ml)
was stirred for 1.5 hours at room temperature. The mixture was
evaporated and the resulting oil was diluted with ethyl acetate
(90 ml). The ethyl acetate layer was washed with 10% w/v aqueous
sodium thiosulfate solution (10 ml) and H2O (10 ml). Removal of
tke dried (MgsO4) solvent gave a yellow oil, which was solidified
by trituration with isopropyl ether. Filtration of the
precipitate gave 4.3 g (67%) of the title compound IX-l as the E
isomer. M.p. >165C (dec.).
IR : vRBr cm 1 3400, 17B0, 1725, 1680, 1610.
max
UV : ~E OH nm t~) 240 (18000), 297 (11000).
max
NMR : ~D SO 6 2 3.90 (3H, s, OCH3), 5.25 (lH, m, 6-H), 5.95
ppm (1~, m, 7-H), 6.72 (d, J=16, 3-CH trans), 6.96
(lH, s, CH-Ph2), 7.4 (lOH, m, Ph-H).
'~_
Benzhydryl 7-Amino-3-[3-cbloro-1-propen-1-yl~-3-cephem-4
carboxylate (Z-isomer) ~XVIII)
Compound XVIII is the common intermediate u~ilized in
Reaction Schemes lb and lc.
A. Benzhydryl 7-Benzylideneamino-3-triphenylphosphoniomethyl-3-
cephem-4-carboxylate chloride (XV)
769291
109
To a suspension of benzhydryl 7-amino-3-chloromethyl-3-
cephem 4-carboxylate hydrochloride (II hydrochloride) (200 g,
0.44 mole) in CH2C12 (940 ml~ was added 1 N NaOH (440 ml) at room
temperature. ~he mixture was shaken for 10 minutes and the
organic layer was separated. To the organic layer were added
MgSO4 (75 g) and benzaldehyde (51 g, 0.48 mole) and the mixture
was allowed to stand for 3 hours. The reaction mixture was
filtered and the insolubles were washed with CH2C12 (200 ml). To
the combined filtrate and washings was added triphenylphosphine
~126 g, 0.48 mole). The mixture was concentrated to about 400 ml
and allowed to stand for 4 days. The resulting viscou~ oil was
di~uted with ethyl acetate (1 L) and triturated to separate the
title compound XV a pale yellow crystalline powder which was
collected by filtration and dried in vacuo. Yield 322 g (96~).
M.p. 185-190C (dec.).
IR : v cm 1780, 1720, 1630.
max
.
UV ~CH2C12 nm (e) 260 (24100).
max
B. Benzhydryl 7-~enzylideneamino-3-[~triphenyl~hosphoran-
ylidene)methyl]-3-cephem-4-carbox~_ate (XVI)
A mixture of XV (322 g, 0.42 mole) and 5 N Na2CO3 (252
ml) in CH2Cl~ (1.6 L) was stirred vigorously for 15 minutes at
room temperature. The organic layer was separated, dried over
MgSO4 and concentrated to about 500 ml of volume. The
concentrate was added to acetone (1 L), with stirring, to give a
light yellow crystalline powder which wa~ collected by filtration
to yield 237 g (78%) of XVI, melting at 195-198C (dec.).
~769~9
110
IR vXBr cm~l 1770 1620
max
UV : ~CH2C12 nm (E) 2S4 (23000), 389 (22000).
max
NMR : ~CDC13 2.56 & 3.16 (2H, ABq), 5.00 (lH, d, J=4 Hz), 5.23
ppm (lH, d, J=4 ~z), 5.47 (lH, d, J=22 ~z), 6.95 (lH,
s), 7.2-7.8 (30H, m), 8.55 (lH, s~.
C. Benzb~dryl ?~Amino-3-[chloro-1-propen-1-yl]-3-cephem-4-
carboxylate hydrochloride (Z isomer) (XVIII Hydrochloride)
To a refluxing solution of XVI (214 g, 0.294 mole) and
N,O-bis-(trimethylsilyl)acetamide (40 ml, 0.15 mole) in dry
CH2C12 (2.9 L) was added dropwise, with stirring, a 50% solution
of chloroacetaldehyde (93 g, 0.59 mole) in CHC13 over a period of
15 minutes. After standing for 30 minutes, the mixture was
concentrated to dryness. To the residual oil were added CH2C12
(1.5 L), Girard Reagent T (99 g, 0.59 mole) and 10% aqueous HCl
(300 ml), and the mixture was stirred for 1 hour at roo~ tempera-
ture. The organic layer was washed with water (200 ml) and a
saturated NaCl solution (200 ml), dried over MgSO4, tre~ted with
charcoal (5 g) and filtered. The filtrate was cooled to -10C
and treated with 1 N HCl in CH30H (300 ml). The mixture was
stirred for 30 minutes at room temperature and concentrated ~o
about 300 ml. The concentrate was diluted with ethyl acetate
(400 ml) and seeded with a few crystals of XVIII hydrochloride.
After 2 hours the separated crystals were collected by
filtration, washed with ethyl acetate (200 ml) and dried in vacuo
to give ~4 g (53%) of the title compound XVIII as its
bydrochloride, melting at ~185C (dec.). Pale yellow needles.
~2~6~9
111
IR : vKB~ cm 1 28309 1780, 1720.
max
UV ~EtOH nm (~) 286 (8800).
max
NMR : ~DMSO d6 3.73 (2H, br, s, 2-H), 3.97 (2H, m, C~2Cl), 5.22
ppm (lH, d, J=4.5 Hz, 6-H), 5.37 (lH, d, J=4.5 Hz,
7-H), 5.77 (lH, m, 3-CH=CH), 6.45 (lH, d, J=ll
Hz, 3-CH), 6.88 (lH, s, CHPh2), 7.33 (lOH, br,
s, Ph-H~.
Anal- Calc'd for C23H21N23SCl HCl C, 57-87; H, 4-65;
N, 5.87; S, 6.72;
C1, 14.85.
Found: C, 57.62; H, 4.53;
N, 5.70; S, 6.64;
C1, 14.89.
Preparation No. 13
Benzhydry~ 5~Amino-1,2,4-thiadiazol-3-yl)-2-methoxyimino-
acetamido]-3-[3-chloro-1-Pro~e -~yl]-3-cephem-4-carboxylate
(Z isomer) (VIII-l)
To a stirred solution of XVIII (Z isome}) (20 g, 42
mmole) in CH2C12 (420 ml) containing N,O-bis(trimethylsilyl)-
acetamide (34 ml, 125 mmole) was added 2-(5-amino-1,2,4-
thiadiazol-3-yl)-2-methoxyiminoacetyl chloride hydrochloride
(15.2 9, 59 mmole) in three portions over a period of 30 minutes
at -10 to 0C. The mixture was stirred for 30 minutes at 0-5C
and concentrated under reduced pressure. The residual brown oil
was dissolved in ethyl acetate (420 ml) and the solution was
washed successively with saturated aqueous NaHC03 (3 x 15 ml),
saturated aqueous NaCl (15 ml), 10% HCl (15 ml) and saturated
aqueous NaCl (15 ml), and concentrated to about 50 ml of the
volume. To the concentrate was added n-hep~ane !200 ml) to give
~7~i9~
il2
28.5 g t90~ pure) of the title compound VIII-l (Z-isomer) as a
colorless powder. M.p. >150C (dec.)O
IR : vKBr cm 1 3400, 1780, 1720, 1680, 1620.
max
UV : ~E QH nm ~E) 240 (20000), 283 (12000).
max
NMR ~acetne-d6 3.6 (2H, m, 2-H), 3.95 (3H, s, OCH3~,
ppm (2H, m, CH2Cl), 5.32 (1~, d, J=4.5 ~z,
6-H), 5.62 (1~, m, 3-CH=CH), 6.03 (1~, d,
J=4.5 Hz, 7-~), 6.32 ~lH, d, J=11 Hz, 3;CH),
6.87 (lH, s, CHPh2), 7.33 (lOH, br, s, Ph-H).
Preparation No. 14
Benzhydryl 7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyimino-
acetamido]-3-[3-iodo-1-propen-1-yl]-3-cephem-4-carboxylate
(E isomer) (IX-l)
A mixture of VIII-l (Z isomer) (28.5 g, 90% pure) and
sodium iodide (19 g) in dry acetone (420 ml) was stirred for 10
minutes at room temperature and allowed to stand at 5C for 2
~ours. The mixture was concentrated under reduced pressure. To
the residue were added ethyl acetate (420 ml) and 10~ w/v aqueous
sodium thiosulfate solution (30 ml), and the mixture was shaken .
The organic layer was washed with water (30 ml), dried over MgS04
and evaporated to about 50 ml of volume. The concentrate was
diluted with n-heptane (200 ml) to yield 30.5 g (9S~ pure) of the
title compound IX-l (E isomer~ as a yellow powder, melting at
> 12 0 C ( dec . ) .
~. 27 ~ 9
113
IR : vKBr cm 1 3400, 1780, 1725, 1680, 1620.
max
UV ~EtOH nm () 306 (15000).
max
NMR ~acetone-d6
ppm 3.71 (2H, m, 2-H), 3.97 (3H, s, OCH3), 4.0
(2H, d, J=8 Hz, CH2I), 5.26 (lH, d, J-4.5 Hz~
6-H), 6.03 (lH, d-d, J=4.5 & 8 Hz, changed to
doublet J=4O5 Hz by D2O, 7-H), 6.32 ~lH, d-t,
J=15 & 8 Hz, 3-CH=CH), 6.79 (lH, d, J=15 Hz,
3-CH~, 6.98 (lH, s, CHPh2), 7.35 (10~, ~,
Ph-H), 7.63 (2B, br, s, disappeared by D2O7
NH2), 8.52 (lH, d, J=8 Hz, disappeared by
.
Preparation No. 15
Benzhydry~7-E2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxvlmino-
acetamido]-3-~3-(4-carbamoyl-1-pyr1dinio)-l-propen-1-ylJ-3-
cephem-4-carboxylate Iodide (E isomer) (XII-lH)
To a suspension of IX-1 (E isomer) (30.5 g) and
isonicotinamide (26 g, 212 mmole) in CH3CN (120 ml) was added
CH30H ~100 ml) until the mixture became clear. The solution was
stirred for 2 hours under nitrogen atmosphere at room temperature
and concentrated ~o about 100 ml under reduced pressure. The
residual semi-solid was triturated with isopropyl ether (200 ml).
The solvent was removed by decantation and the residual yellow
powder was washed with a mixture of isopropyl ether and CH30H
(3/1, 120 ml). The powder was collected by filtration and dried
in vacuo to give 36 g (75% pure estimated by HPLC) of the title
compound XII-lH (E isomer) as a light yellow powder mel~ing at
>150C (dec.).
~ ~ ,.
'~.276~
114
IR : ~ cm 1 3300, 1780, 1720, 1680, 1620.
max
UV : AEtOH nm ~El%H ) 282 (170)
max 1 cm
N~ ~DMSO-d6
ppm 3.72 (2H, m, 2-H), 3.90 (3H, s, OCH3), 5.25 (3H,
m, 6-~ & CH2N ), 5.9 (lH, d-d, J=4.5 & 8 Hz,
changed to a doublet J=4.5 ~z by D20 addition,
7-H), 6.35 (lH, m, 3-CH=CH), 6.89 (lH, s,
CHPh2), 6.9 (lH, d, J=16 Hz, 3-CH), 7.35 (lOH,
m, Ph-H), 8~06 (2H, br, ~, disappeared by D20,
~H2), 8.21 (2H, br, s, disappeared by D20
addition, NH2), 8.36 & 9.07 (each 2H, d, J=6 ~z,
Py-H), 9.57 ~lH, d, J=8 Hz, disappeared by D20
addition, 7-NH).
Preparation No. 16
Benzkydryl 7-Benzylideneamino-3-[3-chloro-1-~ropen-1-yl]-3-
cephem-4-carboxYlate (XVII) (Z isomer)
To an ice-cooled mixture of the crystalline 7-amino-
cephem intermediate XVIII (Z isomer) (13.4 9, 28 mmole) and
benzaldehyde (3.3 g, 31 mmole) in ethyl acetate (150 ml) was
added dropwise 0.5 N sodium hydroxide (56 ml, 28 mmole) over a
period of 20 minutes~ to maintain the temperature of the reaction
mixture below 10C. The mixture was stirred with cooling for
another 15 minutes, and the organic layer was separated, washed
with sa~urated aqueous sodium bicarbonate (100 ml x 2) and dried
over magnesium sulfate. To the dried solution was added a small
amount o charcoal and the mixture was filtered. The filtrate
was concentrated to dryness. The residual oil was dissolved in
carbon tetrachloride ~50 ml), and concentrated again. This
procedure was repeated 3 times, and the mixture was monitored by
reverse pha~e tlc to confirm that all of the starting 7-amino-
.
" ~'Z~7Ç~29
115
cephalosporin was converted to the Schiff's base. Removing thesolvent ln vacuo gave 16.45 g of the title compound XVII (Z
isomer) as a pale yellow powder (estimated purity 85~; M.p. 74C
(dec.), which was ussd for the next step without purification.
IR : v r cm 1 1780, 1725, 1635.
max
UV : ~CH2C12 nm (El% ) 257 (
max 1 cm
NMR : ~CDC13 6.18 (lH, d, J-ll Hz).
ppm
Preparation No. 17
Benzhydryl 7-Benzylideneamino-3-[3-(4-carbamoyl-1-pyridinio)-1-
propen-l-yl]-3-cephem-4-carboxylate Iodide (XXI-~) (E isomer)
To a chilled mixture of the 3-chloropropenylcephem XVII
(Z isomer3 (16.4 g) in acetone (5 ml), was added dropwise a
solution of sodium iodide (6.3 g, 42 mmole) in acetone (~0 ml)
over 10 minutes under nitrogen atmosphere, and the mixture was
stirred at room temperature. The reaction was monitored by the
ratio of uv absorption ~El ~ (255 nm)/El % (320 nm)]. When
1 cm 1 cm
the ratio reached below 1.30 (after 45 minutes), the mixture was
diluted with carbon tetrachloride (400 ml), and allowed to stand
at room tempera~ure. When the ratio came to below 1.10 (after 3
hours), the mixture was concentrated to a half its volume. The
concentrate was treated with a small amount of charcoal and
diatomaceous earth, and filtered. The filter cake was washed
with a 1:1 mixture (100 ml) of methylene chloride and carbon
tet~achloride. To the combined solution of the filtrate and
washings, was added a solution of isonicotinamide (3.5 g, 28.7
mmole) in dimethylformamide (20 ml) and the mixture was concen-
trated under reduced pressure. The concentrate was allowed to
stand at room temperature for 1.5 hours and washed with isopropyl
.
116
e~her (100 ml x 3). The residual brown semi-solid was dissolved
in methylene chloride ~50 ml) and the solution was added
dropwise, with stirring, to ethyl acetate (1.5 L). ~he resulting
precipitate was collected by filtration and washed with ethyl
acetate (200 ml). After drying over phosphorous pentoxide in
vacuo, 17 g of the title compound XXI-H (E isomer) was obtained.
Yellow amorphous powder. M.p. 150-155C (dec.). Estimated
purity 80% by nmr.
IR : v cm 1775, 1725, 1690, 1635.
max
~C12 nm (El% ) 258 (335)~ 298 (255)-
max 1 cm
NMR ~DMSO-d6
ppm 3.4-3.8 (2H, br.), 5.35 (2H, br.), 5.41 (lH, d,
J=4 Hz), 5.73 (lH, d, J=4 Hz), 6.93 (lH, s),
6.97 (lH, d, J=16 Hz), 7O3-7.5 (15H, br. s),
8.40 (2H, d, J=6.5 Hz), 9.15 (2H, d, J=6.5 Hz).
Preparation No. 18
7-Amino-3-[3-(4-carbamoyl-1-pyridinio)-1-propen-1-yl]-3-c~phem-4-
carbox~late (XXII-H) (E isomer)
To a suspension of the quaternized cephem XXI-H (17 g)
in 85% formic acid (25 ml) was added dropwise concentrated
hydrochloric acid (5 ml), and the mixture was stirred at room
temperature for l.S hours and treated with a small amount of
charcoal. The mixture was filtered and washed with 854 formic
acid (5 ml). The filtrate was combined with the wash and poured
into acetone (1 L), with stirring. The resulting precipitate was
collected by filtration to give 9.52 g of yellow-colored crude
product. To a suspension of the crude material (9.5 9) in water
(50 ml) was added a small amount of charcoal, and the mixture was
filtered. The filtrate was added dropwise, with stirring, to
isopropyl alcohol (700 ml). The resulting precipitate was
~ ~ ~6$~9
117
collected by filtration, washed with a small a~ount of rnethanol
(30 ml), and dried to give 7.58 g of the title compound XXII-H (E
isomer) as the hydrochloride. Light yellow powder. ~stimated
purity 85% ~y VV. M.p. 173-188C (dec.).
IR : vKBr cm 1 1795, 1680, 1620, 1575, 1540.
max
UV ~Phosphate buffer (pH 7) nm (El ~ ) 294 (457)-
max 1 cm
N~ D2o+D~l
ppm 3.82 (2~, s), 5.17 (lH, d, J=5 H2), 5.33 (2H,
d, J=7 Hz), 5.43 (lH, d, J=S Hz), 6.37 (lH,
d-t, J=16 & 7 Hz), 7.23 (lH, d, J=16 Hz), 8.34
(2H, d, J=7 Hz), 9.00 (2H, d, J=7 Hz).
PreParation No._l9
2 (5-Amino-1,2,4-thiadiazol~x1)-2-methoxyiminoacetyl Chloride
Hy~ochloride ~ l as its acid chloride hydrochloride)
A. 2-Cyano-2-methoxyiminoacetamide
To a stirred mixture of ~-cyanoacetamide (252 g, 3
mole) and sodium nitrite (414 g, 6 mole) in water (600 ml) was
added acetic acid (371 ml, 10 mole) at 5-10C over 1.5 hours.
The mixture was allowed to stir for another 1.5 hours and
adjusted to pH 8.5 with 6 N NaOH. To the mixture was added
dimethyl sulfate (568 ml, 6 mole) at 15-20C and the mixture was
stirred at 45C for 1.5 hours. The reaction mixture was adjusted
to pH 8.5 with 6 N NaO~ and allowed to stand at 5C overnight to
separate the precipitate, which was collected by filtration,
washed with cold water and air-dried to give 292 g (77%) of ~he
title compound as brown needles melting at 170-172C.
118
IR : vKB~ cm 1 3400, 3180, 1720(sh), 1715, 1690,
max1615, 1570.
UV : ~H2O nm (~) 238.5 (8290), 268 (sh, 3870~.
max
NMR: ~DMSO d6 4.20 (3H, s, OCH ), 7.85 (28, br. NH2).
ppm 3
Anal. Calc'd. for C4H5N3O2: C, 37.80; H, 3.97; N, 33.~
Found: C, 37.43: H, 3.75; N, 32.51.
B. 2-Methoxyiminopro~anedinitrile
A stirred mixture of 2-cyano-2-methoxyiminoacetamide
(88.9 g, 0.7 mole), sodium chloride t70 g) and phosphorus oxy-
chloride (97 ml, 1.05 mole) in dry 1,2-dichloroethane (350 ml)
was refluxed for 16 hours. ~he insolubles were filtered off
through a dicalite pad and washed with dichloroethane. The
filtrate and t~e wash were combined, and poured into stirred
ice-water (1.5 L) to decompose the excess of phosphorus
oxychloride. The organic phase was washed with 10~ NaHCO3 (500
ml), water (500 ml x 3) and a saturated NaCl solution (500 ml),
and dried over MgSO4. The filtrate was distilled under
diminished pressure to give 61.5 g (81%) of the title compound
boiling at 62C/24 mm Hg. (Lit., b.p. 47-48C/12 mm Hg).
IR vLiquid Film cm~l 3020, 2960, 2245, 2020, 1530, 1455,
max1080.
NMR: ~CD 3
ppm 4.35 (3H, s, OCH3).
.: . . . . .
~.2~;~6 ~G3~2~
119
C. 2-Cyano~2-~ethox~iminoacetamidinium Acetate
To a solution of ammonium chloride (28.4 g 0.53 mole)
in 28~ aqueous ammonia (355 ml) and ethanol (180 ml) was added
dropwise a solution of 2-methoxyiminopropanedinitrile (58.0 g,
0.53 mole) in ethanol (120 ml) at -15 to -10C over a period of
30 minutes, with stirring. The mixture was stirred at -10C
overnight and then at ambient temperature (20-25C) for one day.
T~e reaction mixture was partitioned between water (350 ml) and
CH2C12 (350 ml~, and the aqueous phase was saturated with sodium
chloride, and extracted again with CH2C12 (300 ml~. The organic
extracts were combined, dried over MgSO4 and evaporated in vacuo.
A solution of the residue in ethyl acetate (1.6 L) was adjusted
to pH 3-4 with acetic acid to precipitate the title compound as
crystals, which were collected by filtration and washed with
ethyl acetate. Yield 67.6 g (69%). M.p. 152-4C (dec.). [Lit.,
m.p. 150-155C (dec.)~.
IR : vK cm 1 3160, 2900, 2360, 2235, 2000, 1665, 1555,
max 1495, 1415.
UV : ~E nm (~) 243 (8500), 265 (sh, 5380), 305 (sh, 1400).
max
NMR ~DMSO-d6
ppm 1.88 (3H, s, CH3COOH), 4.15 (3H, s, OCH3),
7.60 (4H, br.).
Anal. Calcld for C4H~N40-CH3COOH: C, 38.71; H, 5.41; N, 30O09
Found: C, 38.71; H, 5.59; N, 29.51.
D. 2-(5-Amino-1,2,4-thiadiazol-3-~1)-2-methoxyimino-
acetonitrile
To a su~pension of 2-cyano-2-methoxyiminoacetamidinium
ace~ate (125 9, 0.672 mole) in CH30H (1.25 L) were added dropwise
triethylamine (234 ml, 1.68 mole) at -10C, and subsequently Br2
. ~
120
(41.6 ml, 0.806 mole) ovee 20 minutes at -15 to -10C, and the
mixture was stirred for 20 minutes. To the mixture was added
dropwise a solution of KSCN (78.3 9, 0.806 mole) in CH30H (550
ml) over 1 hour at -15 to -10C. After stirring at 0-5C for 1
hour, the mixture was poured into ice-water (12 L) to form a
crystalline precipitate, which was collected by filtration,
washed with water and air dried to give 120 g ~98%) of the title
compound. M.p. 263-5C (dec.). The m.p. o the compound
prepared by us is higher by about 60C than that given in the
literature* [m.p. 210-15C (dec.)~, but our spectral and
microanalytical data are consistent for the structure.
IR : vK~r cm 1 3435, 3260, 3120, 2960, 2245, 2020, 1630,
max 1545, 1455, 1415.
UV : ~E OH nm ~E~ 248 (13300), 310 (3470).
max
Nl~ ~DMSO~d6
ppm 4.21 (3H, s, OCH3), 8.30 (2H, br. NH2).
nal. Calc'd for C5H5N50S: C, 32.?8; H, 2.75; N, 38.23; S, 17.50
Found: C, 32.76; H, 2.51; N, 38.02;
S, 17.50.
E. 2 (5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxYiminoacetic Acid
(III-l)
A mixture of 2-(5-amino-1,2,4-thiadiazol-3-yl-2-
methoxyiminoacetonitrile (18.3 g, 0.1 mole) in 4 N NaOH (250 ml)
was heated at 50-55C with stirring for 3 hours. The reaction
mixture was adjusted to pH 1 with H3PO4, and washed with ethyl
acetate (100 ml), saturated with NaCl, and extracted three times
with a mixture of ethyl acetate and tetrahydrofuran (3 : 1, 300
ml x 2, and 200 ml x 1). The extracts were combined, dried over
MgSO4 and concentrated under reduced pr-essure. The residue was
triturated with isopropyl ether to afford pale yellow crystals of
121
the title acid. Yield 16.8 g (83~). M~po 184-5C (dec.).
~Lit.*, m.p. 180-182C (dec.)].
IR O vKBr cm 1 3460, 3260, 3140, 1725, 1620, 1605, 1545.
max
UV : ~2 nm (E) 234 (13200), 288 (sh, 3620).
max
F. 2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-methoxyiminoacetyl
Chloride H~drochloride
To a suspension of 2-(5-amino-1,2,~-thiadiazol-3-yl)-2-
methoxyiminoacetic acid (III-l) (40.4 g, 0.2 mole) in dry CH2C12
(400 ml~ was added PC15 (41.6 g, 0.2 mole) in one portion at
-S0C. The mixture was stirred for 4 hours at 20 to -5C, and
poured into a mixture of n-heptane and isopropyl ether (2 : 1, 2
L). The yellow precipitate was collected by filtration, washed
with the same solvent mixture, and dried with KOH under reduced
pressure to give 46.0 g (90%) of the title acid chloride.
IR vNujol cm-l 1775
max
*Japan Kokai 57-158769 published September 30, l9B2, to Fujisawa
(Brit. appl., 3/6/81)
~L2~6~9
122
.
1 ~ C - CONH ~
H~N S ~ CH=CH-C~2-Cl
C2H5 COOCH (Ph ) 2
VIII-2 *Z
Diphenylmethyl 7-[2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-(Z)-
ethoxyiminoacetamido]-3-[3-chloro-l-propenyl]-3-cephem-4
carboxylate (VIII-2, Z isomer)
To a mixture of N,O-bis(trimethylsilyl)acetamide (2.3
ml, 9 m~oles) and crystalline diphenylmethyl 7-amino-3-[3
chloro-l-(Z)-propen-l-yl]-3-cephem-4-carboxylate hydrochloride
(XVIII) (1.338 g, 2.8 mmoles) (from Preparation No. 12) in
methylene chloride (10 ml) was added 2-(5-amino-1,2,4-
thiadiazol-3-yl)-2-(Z)-ethoxyiminoacetyl chloride hydrochloride
(800 mg, 2.95 mmoles) portionwise, with stirring, at -10C and
the mixture was allowed to stand at 0C for 2 hours. The mixture
was diluted with ethyl acetate (200 ml), washed with water and
evaporated under reduced pressure. Trituration of the residue
with isopropyl ether afforded the title product VIII-2 as an
amorphous powder. Yield 1.70 9 (95%). Mp. >150DC ~dec.).
IR : vmax (KBr) in cm 1 8300, 1780, 1720, 1690, 1380, 1220.
max (C2H5OH) in nm (E) 285 (llooo)
N~ (DMSO-d6) in ppm 1.26 (3H, t, J=7Hz, CH~CH3), 4.25
(2H, ~, J=7Hz, CH2CH3), 5.90 (lH,
d-d, J=4 & 8Hz, 7-H), 6.26 (lH, d,
J=llHz, 3-CH), 6.85 (lH, s, CHPh2),
9.53 (lH, d, J=8Hz, 7-NH3.
~:7~2~
123
Preparation No. 21
,. . .
~ CONH~
H2 S ~ ~ CH=C~--CH2
c~5 COocHlph)2
IX-2 *A mixture of E and Z isomers
DiPhenylmethyl- 7-[2-(5-Amino-1!2,4-thiadiazol-3~ 2-1Z)-ethoxY-
iminoacetamido]-3-[3-iodo-1-propenyl]-3-c~phem-4-carboxylate
(IX-2)
A mixture of VIII~2 (1.9b g, 3 mmoles) (from
Preparation No. 20) and sodium iodide (1.4 g, 9 mmoles) in
acetone (20 ml) was stirred for 10 minutes at room temperature
and then allowed to stand at 5C for 3 hours. The mixture was
evaporated under reduced pressure, diluted with ethyl acetate
(100 ml), washed with 10~ sodium thiosulate and water, and
evaporated under reduced pressure. Trituration of the residue
with isopropyl ether gave 1.82 g (84~) of the title product IX-2
as a light brown amorphous powder.
R : vmax (KBr) in cm 3290, 177û, 1720, 1670, 1530, 1370,
1220.
nmax (C2H5H) in nm (E ) 304 (199).
7~g2~ l
124
Preparation No. 22
~ r.
~CH=N~ ~ ~3
--CH=CH--C:H2 _~:ONH2
CoocH(ph~2
XXI~ dide *E
Diphenxlmethyl 7-Benzylideneamino-3-~(E)-3-(4-carbamoAy~-
~y~ri io)-l-propenyl]-3-ce~em-4-carbo~ylate (XXI-H iodide) (E
isomer)
To a chilled solution of the 3-chloropropenylcephem
(XVII, Z isomer, 42.8 g, 90 mmoles) (from Preparation No. 16) in
dry DMF (80 ml), was added KI (20 g, 120 mmoles) in one portion,
and the mixture was stirred at room temperature. The reaction
was monitored by the ratio of UV absorption [El% (255 nm)/
1 cm
El% (320 nm)]. When the ratio became below 1.10 (after 45
1 cm
minutes), the mixture was diluted with 800 ml of methylene
chloride~ treated with active carbon (4 g), and filtered. The
filter cake was washed with 100 ml of CH2C12. To the combined
filtrate and washings was added isonicotinamide (14.64 g), and
the mix~ure was concentrated under reduced pressure. The
concentrate was kept at room temperature for 1.5 hours and washed
with a mixture of toluene and n-heptane (1:1, 600 ml). ~he
residual brown semi-solid was dissolved in CH2C12 (100 ml) and
the solution was added dropwise to ethyl acetate (3 L) with
vigorous stirring. After drying over P2O5 in vacuo, 57.37 g
(88~ of the quaternized title product XXI-H was obtained as the
iodide. Yellow amorphous powder. Mp. 150-155C (dec.). This
product was identical to that obtained by iodination with NaI
(Preparation No. 17).
,
~76~
125
Preparat on No. 23
HCl-~2N ~ S ~
N ~ C~CH-CH2-Cl
~OOCH(Ph)2
XVIII *Z
Diphenylmeth ~ _7-Amino-3-(3-chloro-1-propen~l)-3-cephem-4
A 25~ solution of chloroacetaldehyde (69 g, 0.22
mmoles) in CHC13 was added to a solution of XVI (80 9, 0.11 mole)
in CH2C12 ll.l L3 containing N,O-bis(trimethylsilyl)acetamide
(16.2 ml, 0.06 mole) at -10C in one portion, and the mixture was
allowed to stand overnight at 5C. The mixture was concentrated
to ca. 0.3 L, diluted with a mixed solvent of ethyl acetate and
isopropyl ether (1/2, 0.6 L), treated with silica gel (Wakogel
C-100, 60 g) and filtered through a dicalite pad. The filter
cake was washed with the same solvent system (0.2 L). The
combined filtrate and washing were concentrated to ca. 0.2 L,
treated with Girard Reagent T (60 g, 0.26 mole) and 4N ~Cl (220
ml), and seeded with a few crystals of XVIII hydrochloride.
~fter stirring for 3 hours, the resulting crystals were collected
by filtration, washed with water (0.5 L) and ethyl acetate (0.5
L) and dried in vacuQ to give 37 g (70~3 of the title compound
XVIII hydrochloride, melting at >185C (dec.3. Pale yellow
needles. This product was identical to that obtained in
Preparation No. 12.
.
g
126
Preparation No. 24
S
HCl H2~ ~ ~
N ~ CH-CH-CH2~Cl
COOCH(Ph)2
XVIlI *Z
Diphenylme~yl 7-Amino-3-(3-chloro-1-propenyl) 3-cephem-4-
carboxylate hydrochloride (Z_isomer) (XVIII, Hydrochloride)
To a solution of chloroacetaldehyde (25% solution in
CHC13, 628 mg, 2 mmoles) in CH2C12 (10 ml) were added N,O-bis~
(trimethylsilyl)acetamide (0.135 ml, 0.5 mmole) and XVI (728 mg,
1 mmole), successively, at 5C. The mixture was allowed to stand
overnight at 5C. The mixture was evaporated and diluted with a
mixture of ethyl acetate and isopropyl ether (1/2, 10 ml~.
Insolubles were removed by filtration and the filtrate was
concentrated to ca. 5 ml. The concentrate was treated with 4N
HCl (2 ml3, seeded with XVIII hydrochloride and stirred for 1
hour at room temperature. The crystals were collected by
filtration, washed ~ith ethyl acetate (10 ml) and water ~10 ml)
and dried in vacuo to give 384 mg (80%~ of the title compound
XVIII hydrochloride, melting at >185C (dec.). Pale yellow
needles. This product was identical to that obtained by
Preparation No~ 12.
PreParation No. 25
2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-(propen-3-yloxvlmlno)-
acetyl chloride hvdrochloride (III-3 as its_acid chloride
hydrochlor de
A. Methyl 2-(5-t-butoxycarbonylamino-1,2,4-thiadlazol-3-yl)-
2-(propen-3~ yimino)acetate
A mixture of 635 mg (3.37 mmoles) of N-(propen-3-
yloxy)phthalimide ~prepared according to the procedure of E.
.
.~7
127
,~
Grochosaki & J. Jurczak, Synthesis 1976 682] and 175 mg (3.35
mmoles~ of hydrazine hydrate in 5 ml of C2H50H was stirred for 1
hour at room temperature. ~he resulting precipitate was filtered
off and the filtrate and washings were combined. To the solution
was added 967 mg ~3.37 mmoles) of methyl 2-~5-t-butoxycarbonyl-
amino-1,2,4-thiadiazol-3-yl)-2-oxoacetate, and the mixture was
allowed to s~and for 1 hour at room temperature and concentrated
by a rotary evaporator. The residue was purified by silica gel
chromatography. The column was eluted with n-hexane/ethyl
acetate (401) and fractions containing the major product were
combined and evaporated under reduced pressure. Yield 514 mg
(46%). Mp. 83-86C.
IR : vmax tRBr) in cm 1 3100, 1745, 1710, 1610.
max (C2H50H) in nm (E) 223 (9700) ~42 (10000)
N~ (CDC13) in ppm 1.55 (9H, s, BOC-H), 4.40 (2H, d,
J=5~z, 0-CH2~l 5.21 (2H, m, CH2=CH),
5.90 (lH, m, -CH=CH2), 9.50 51H, br.s,
NH).
B. 2 (5-t-Butoxycarbonylamino-1,2,4-thiadiazol-3-yl)-2-
, ~
(propen-3-yloxyimino)acetic acidl'
A solution of 770 mg (2.3 mmoles) of methyl 2-(5-t-
butoxycarbonylamino-1,2,4-thiadiaæol-3-yl)-2-(propen-3-yloxy-
imino)acetate and 3.5 ml of 2N NaOH solution (7.0 mmoles) in 15
ml o~ CH30H was refluxed for 30 minutes. The reac~ion mixture
was concentrated in vacuo and dilut0d with 10 ml of ethyl
acetate-H20 (1:1). The water layer was separated, acidified to
pH 2 with 6N HCl and extracted with ethyl acetate (10 ml x 2).
The ethyl acetate solution was dried over MgS04 and concentrated
by a rotary evap~rhtor to afford 596 mg (81%) of the title
compound. ~p. 134-135C (litl): mp. 135-136C.
IR : vmax (Nujol*) in cm 1 3150, 1745, 1710, 1550.
* Trade Mark
'~7''
.... ~
~ 69~9
128
UV: )~max (C2H50H) in nm (E) 223 (11000), 242 (113t)0).
N~ (DMSO-d6) in ppm 1.55 (9H, s, BOC-H), 4.77 (2H, d,
J=SHz, ~CH2), 5.22 (2Hr m,
CH2=CH), 6.0 (lH, m, CH=CH2).
1)I. Csendes, et al., J. Antibiotics, 36, 1020 (1983).
C. 2-(5-Am_n_1,2,4-thiadiazol-3-~-2-(prol?en-3-Yloxyimino)-
acetic acid (III-3)1)
A solution of 570 mg (1.74 mmoles) of 2-(5-t-butoxy-
carbonylamino-1,2,4-thiadiazol-3-yl~-2-(propen-3-yloxyimino)-
acetic acid in 6 ml of trifluoroacetic acid was allowed to stand
for 1 hour at ambient temperature. Evaporation followed by
trituration with 30 ml of isopropyl ether gave 376 mg (95%) of
the title compound. Mp. 109C ~dec.).
IR : v~l~ax (Nujol) in cm 1 3180, 1710, 1545, 1460.
max (C2H5OH) in nm (f) 245 (13500)
NMR: ~ (DMSO-à6) in ppm 4.77 (2H, d, J=5Hz, ~CH2), 5.20
(2E~, m, CH=CH), 6.0 (lH, m, CH=CH2).
1) Japan Kokai 57-112396 (7/13/82, Fujisawa) Brit. applO 7935538
(10/12/79).
D. 2-(5~Amino-1,2,4-thiadiazol-3-yl)-2-(propen-3-yloxyimino)-
acetyl chloride hydrochloride
A solution of 350 rng (1.54 mmoles) of III-3 and 410 mg
(1.97 mmoles) of phosphorous pentachloride in.dichloromethane (5
ml) was stirred for 1 hour at 25C. The reaction mixture was
poured into 60 ml of n-hexane and the precipitate was iltered
off. ~ield 323 mg~
IR : vmaX (Nujol) in cm 1 1755.
.. ~ .
~L276'~Z9
129
Preparation No. 26
2-(5-Amino-1,2,4-~hiadiazol-3-vl)-2-propargy~oxyiminoacetyl
chloride hYdrochloride (III-4 as its acid chloride hydrochloride)
A. ~5ethyl 2-(S-t-Butoxycarbonylamino-1,2,4-thiadiazol-3-yl)-
2-propargyloxyiminoacetate
A suspension of 870 mg (4.32 mmoles) of N-propargyl-
oxyphthalimidel) and 290 mg (4.0 mmoles) of hydrazine hydrate in
5 ml of ethanol was stirred at 25C for 1 hour and filtered. To
the combined filtrate and washings was added 1.0 g (3.86 mmoles)
of methyl 2-(S-t-butoxycarbonylamino 1,2,4-thiadiazol-~-yl)-2-
oxoacetate2). The solu~ion was allowed to stand for 1 hour and
concentrated under reduced pressure. Purification by silica gel
chromatography followed by evaporation afforded 319 mg (27%) of
the title product. Mp. 72-75C.
IR : vmax (KBr) in cm 1 3200, 2380, 1745, 1710, i61b.
: ~m,x (C2H5OH) in nm (E~ 235 (12200)
DMSO-d6) in ppm 1.56 (9H, 5, BOC-H), 3.55 (lH, t,
J=2Hz, C ~C~), 4.85 (2H, d, J=2Hz,
-CH2-C_ CH), 8.9 (lH, br.s, NH).
1) Commercially available, Aldrich.
2) I. Csendes et al., J. Antibiotics 36, 1020 (1983).
B. 2-(S-t-Butoxycarbonylamino-1,2~4-thiadiazol-3-yl)-2-
propargyloxyiminoacetic acid
A solution of 490 mg (1.4 mmoles) of methyl 2-(5-t-
butoxycarbonylamino-1,2,4-thiadiazol-3-yl)-2-propargyloxyimino-
acetate and 2.2 ml of 2N aqueous NaOH solution (4.4 mmoles) in 14
ml of CH3OH was refluxed for 30 minutes. The reaction mix~ure
was concentrated under reduced pressure and 10 ml of ethyl
716929
130
acetate~H2O (1:1) was added to the solution. The separated water
layer was acidified to pH 2 with 6N HCl and extracted with ethyl
acetate (2 x lO ml). Drying over MgSO4 followed by evaporation
of the organic layer gave 149 mg (89~) of the title product. Mp.
135C (dec.).
IR : vmax (Nujol) in cm 1 3350, 1720, 1670, 1550.
max (C2H5OH) in nm (~) 233 (11500)
N~ (DMSO-d6) in ppm 1.55 (9H, s, BOC-H), 3.55 (iH, t,
JG2Hz, C~CH), 4.89 (2H, d, J=2Hz,
CH2C _ CH), 9.0 (lH, s, NH).
C. 2-(5-Amino-1,2,4-th adiazol-3-yl)-2-proparg~loxyimino=
acetic acid (III-4)
A solution o~ 410 mg (1.26 mmoles) of 2-(5-t-butoxy-
carbonylamino-1,2,4-thiadiazol-3-yl)-2-propargyloxyiminoacetic
acid in S ml of trifluoroacetic acid was allowed to stand for 1
hour at 25~C. Evapo~ation followed by trituration of the residue
with 25 ml of isopropyl ether gave 204 mg (72%) of the title
compound. Mp. 156-158C (dec.).
IR : vmax lNuiol) in cm 1 3300, 2480, 1730, 1610.
~ax (C2H5OH) in nm (~) 234 (12000)
MR : ~ (DMSO-d6) in ppm 3.52 (lH, t, J=2Hz, C~H), 4.86
(2H, d, J=2Hz, CH2-C _CH), 8.10 (2H,
br.s, NH2).
131
3) Japan Kokai 57-112396 ~7/13/82, Fujisawa) Brit. appl. 7935538
(10/12/79).
D.
acetyl chloride ~drochloride
A mixture of 175 mg ~0.07 mmole) of III 4 and 182 mg
(D.88 mmole) of phosphorous pentachloride in dichloromethane ~2
ml) was stirred for 1 hour at -5C. The reaction mixture was
poured into 30 ml of n-hexane and the precipitate was filtered
off. Yield 65 mg (34%).
IR : vmax (~ujol) in cm 1 1770.
PreParation No. 27
2-(5-Amino~1,2,4-thiadiazol 3-yl)-2-cyclopentyloximinoacetyl
chloride hydrochloride (III-5 as its acid chloride hydrochloride)
.
A. Methyl 2-(5-t-butox carbonylamino-1,2,4-thiadiazol-3-yl)-2-
cyclopen~xyiminoacetate
A suspension of 860 mg (3.7 mmoles) of N-(cyclo-
pentyloxy)phthalimidel) and 185 mg (3.7 mmoles) of hydrazine
hydrate in S ml o~ C2H5OH was stirred for 1 hour at ambient
temperature and filtered. The filtrate and washings were
combined and added to 1.06 g (3.7 mmoles) of methyl 2-(5-t-
butoxycarbonylamino-1,2,4-thiadiazol-3-yl)-2-oxoacetate2). The
solution was allowed to stand for 1 hour at room temperature and
concentrated in vacuo. The residue was purified by silica gel
column chromatography. Elution with n-hexane-ethyl acetate (4:1)
followed by evaporation gave the title product. Yield 906 mg
(81%). Mp. 115-118C.
IR : ~max (KBr) in cm 3200, 1745~ 1710, 1550.
UV : ~ax (C2H5OH~ in nm (~) 217 (1800), 252 (7600).
;9~ :
132
NMR : ~ (CDC13) in ppm 1.51 (9H, s, BOC H), 1.60 (8H, br.s,
H ~ ), 3.88 (3H, s, OCH3), 4.90 (lH,
br.s, ~ ), 8.70 (lH, br.s, I~H).
1) U.S. Patent 3,371,778 (7/27/76; Glaxo), Brit. appl. 49255
(10~25/72)o
2) I. Csendes et _ ., J. Antibiotics 36, 1020 (1983~.
B. 2-(5-t-Butoxycarbonylamino-1,2,4-thiadiaæol-3-yl)-2-cYclo-
pentyloxx_minoacetic acid
A solution of 500 mg (1.34 mmoles) of methyl 2-(5-t-
butoxycarbonylamino-1,2,4-thiadiazol-3-yl)-2-cyclopentyloxy-
iminoacetate and 2N NaO~ solution ~2 ml, 4 mmoles ) in 15 ml of
CH30H was refluxed for 30 minutes. The reaction mixture was
evaporated and 10 ~1 of ethyl acetate-H2O (1:1) wa~ added to the
solution. The water layer was separated, acidified to pH 2 with
6N HC1 and extracted with ethyl acetate (10 ml x 2~. The organic
layer was washed with brine, dried over MgSO4 and concentrated
under reduced pressure to give 377 mg (78~) of the title
compound. Mp. 185DC (dec.).
IR : vmax ( KBr ) in cm 3160, 1710, 1550.
max (C2H5OH) in nm (e) 238 (13300)
NMR : ~ (DMSO) in ppm 1.51 (9H, s, BOC-H), 1.70 (8~, br.s.,
H ~ ), 4.82 (lH, m, ~ ).
C. 2-!5-Amino-1,2,4-thiadiazol-3-yl)-2-cyclopentyloxyimino-
acetic acid (III-5, Z isomer)
_ _ __
A solution of 348 mg ~0.37 mmoles) of 2-(5-t-butoxy-
carbonylamlno-1,2,4-thiadiazol-3-yl)-2-cyclopentyloxyiminoacetic
~ ~17~
133
acid in 2 ml of trifluoroacetic acid was allowed to stand for 1
hour at room temperature. The reaction mixture was concentrated
under reduced pressure. The residue was triturated with 5 ml of
isopropyl ether and 10 ml of hexane to give 215 mg (86%~ of the
title compound. Mp. 162-165C (dec.) [lit3): mp. 160-165C
(dec.)].
IR : vmax (Nujol) in cm 1 3290, 3200, lilO, 1615, 1600.
max (C2H50H) in nm (E) 238 (13300)
N~ (DMSO-d6) in ppm 1.17-2.10 (8H, m), 4.60-4.98 (lH,
m), 8.22 (2H, S).
3) Japan Rokai 57-15R769 (9/30/82, Fujisawa) Brit. appl. 8107134
(3/6/81) .
D. 2-(5-Amino-1,2,4-thiadiazol-3-yl)-2-cy~lopentyloxy-lmin
acetyl chloride hydrochloride
A solution of 190 mg (O. 74 mmole) of III-S and 219 mg
(1.0 mmole) of phosphorous pentachloride in dichloromethane (5
ml) was stirred for 1 hour at room temperature. The reaction
mixture was poured into 50 ml of n-hexane. The resulting
precipitate was collected by filtration. Yield 122 mg (60~).
IR : vmax (Nujol) in cm 1 1760.
Preparation No. 28
Benzotriazol-l-yl-2-~5-amino-1,2,4-thiadiazol-3-yl)-2-methoxy-
iminoacetate
A mixture of l-hydroxybenzotriazole (207 g, 20 mmoles)
and dicyclohexylcarbodiimide (4.12 g, 20 mmoles) in 65 ml of DMF
was stirred at room temperature. After 15 minutes, IIX-l (4.04
g, 20 mmoles~ was added to the stirring mixture at 0C, and
stirring was continued for 3 hours. The reaction mix~ure was
~769i~g
134
filtered to remove the insoluble urea, and the filter cake was
washed with a small volume of DMF. The filtrate and washings
were combined and poured into 800 ml of ice water. The
precipitate was collected by filtration to give 5.24 g (82%) of
the title compound as a light grey powder. Mp. 189-192C (dec. ) .
IR : vmax (KBr) in cm 1 1815, 1620, 1540, 1415, 1090, 1060,
1005, 945, 865, 740,
max (C2~5O~) in nm (El ) 246 (580), 283sh (228).