Note: Descriptions are shown in the official language in which they were submitted.
~ 2~7332
RAN 4070/66
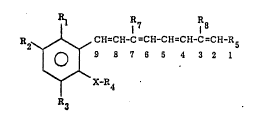
. The present invention relates to compounds of the
formula:
~7 R8
~0 ~2~ ~ ~H=CR-C=CH-CH=CH-C=CH-R5
3 ~ 7 6 S 4 3 2 l
X-R4
A3
L5
wherein n i~ an integer selected from 6 or 7; Rl is
hydrogen, loweralkyl, chlorine, fluorine or
t~i~luoromethyl: R2 is hydrogen, lowe~ alkoxy,
trifluoromethylloweralkoxy, hydro~y, lower alkyl,
chlorine, trifluoromethyl, or fluorine; R3 i8
hydrogen, loweralkyl, chlorine or fluorine; R4 is
alkyl contai~ing from 4 to lO carbon atoms or
-CH2~CH2)nCH20H; X is -CH o-,
Rlo
-CH-, ,-C=CH- ,-0-. or ~N~; R5 is -I-OR9;
Rlo Rlo Rlo
: and R7, R~ ~9 and Rlo are individually lower
alkyl or hydrogen:
and salts thereof where R9 is hydroge~ to a process for
their ~repacation and to ~harmaceutical compositions
containing them.
Figures 1, 2, 3, 4, 5, 6 and 7 represent~ s~hematic
~rocess stee~ for ~re~aring the compounds of formula I above.
Grn/11.6.85
~ r,~ j~
7332
-- 2
In the compound6 of this invention the term "halogen"
includes all four halogens, i.e. chlorine, bromine, iodine
and fluoeine with chlorine and bromine being preferred. The
term "lower alkyl" as used herein designates both straight
and beanched chain lower alkyl grou~ containing from 1 to 7
carbon atoms. Among the pre~e;cred lower alkyl groups are
methyl, ethyl, iso~ropyl, n-butyl, etc., with methyl and
ethyl being especially preferred. The term "lower alkoxy"
de~ignates lower alkoxy groups containing ~rom 1 to 7 carbon
atom~ such as methoxy, ethoxy, isopro~oxy, isobutyoxy, etc.
The term trifluoromethyl lower alkoxy de~ignates a
trifluoromethyl substituted lower alkoxy ~ubstituent where
lower alkoxy is defined as above. The term alkyliden~
designates a aliphatic saturated hydrocarbon group where the
teLminal carbon atoms i5 divalent.
The term ~larylll designates mononuclear aromatic
hydrocarbon groups which can be un~ubstituted or ~ubstitutad
in one or more po6itions with a lower alkyl groups, ~uch a~
phenyl or tolyl, etc. and polynuclear aromatic groups which
can be un6ubstituted or sub~tituted in ona or more po6itions
with a lower alkyl group and a napthyl, phenanthryl or
a~thryl. The preferred aryl group is phenyl.
In one of the embodiments of the com~ounds of Formula I,
O- and R7 and R8 are lower alkyl preferably
methyl. In another ereferred embodimen~, R4 is -(CH2)y H
with y being 6 to 9, more particularly 8 to 9 with 9 being
especially preferred. In this embodiment of the invention,
30 Rl, R2 and ~3 are ere~erably hydrogen or Rl and R3
can be hydrogen and R2 can be lower alkoxy such as methoxy
or ethoxy. On the other hand in ~his embodiment R2 and
R3 can be hydrogen with Rl being chlorine or fluorine or
Rl and R2 can be hydrogen with R3 being chlorine or
35 fluorine.
In still another embodiment of the compounds of formula
~.
.
'77332
-- 3
I~ R4 is -CH2~CH2)nCH~OH. In this embodiment of
the compounds of Eormula I, Rl, R2 and R3 are
hydrogen, or Rl i8 chlorine or f luorine with R2 and R3
being hydrogen. On the other hand in this embodiment of the
compounds of formula I, R1 and R3 are hydrogen and R2
is lower alkoxy preferably methoxy or ethoxy. Also
preferred are those compounds of this embodiment o~ the
compound of formula I where Rl and R2 are hydrogen and
R3 i8 chlorine or fluorine.
Also included in this invention are salts of the
compound of formula r above with pharmacautically
acceptable, non-toxic, inorganic or organic bases, e.g.
alkali metal and alkaline earth metal salts. Among the
preferred salt5 are the sodium, potassium, magnesium or
calcium salts, as well as salts with ammonia or suitable
non~toxic amines, such as lower alkyl amines, for example
triethylamine, hydroxy-lower alkylamines, for example
2-hydroxyethylamine, bis-~ hydroxyethyl~amine or
zo tris-~2-hydroxyethyl~-amine, cycloalkylamines, for example
dicyclohexylamine, or benzylamines, ~or example
N,N'-dibenzyl-ethylenediamine, and dibenzylamine. These
salts can be prepared by treating the compounds of formula
I, where Rg is hyd~ogen wi~h inorganic or organic ~ases by
conventional mea~s well known in the a~t.
The compounds of formula I as well a6 salts thereof are
effective as disease modi~iers Eor treating cheumatoid
arthritis as well as related disorder~ such as
30 osteo-arthritis.
The compounds of formula I can be utilized to treat
patients suffering from rheumatoid arthritis and related
disorders. In such ca~es, the compounds modify the effects
35 of these diseases by reducing destruction of ~he bone joints
- caused by this disease as well as reducing inflammation,
heat and pain of the bone joints which results from
.
~D,2t7''73~
-- 4
rheumatoid arthriti6 and related disorder6. The compounds
of formulae I and salts thereof are also useful fo~ treating
diseases resulting from immune hyperactivi~y such as trans-
plantation autoimmunity, autoimmune disease and graft versus
host di6ease. The unexpected lack of toxicity of the
compounds of this invention can be seen by the ~act that the
compound ~all-E)-9-[2-(nonyloxy)-phenyl]-3,7-dimethyl-
-2,4-6,8-nonatetraenoic acid has a LD50 in mice of grea~er
than 1,000 mg/kg both i.p. and p.o.
That the compounds of this invention are effec~ive
~nti-arthritic agents can be seen from the results obtained
when these compound~ are admini~tered to rats in accordance
with the chronlc adjuvant arthritis te~t system disclosed in
Billingham and Davies "Handbook of ~xperimental
Pharmacology" (editors J.R. Vane and S.H. Ferreira) Vol.
50/II, pg. 108-144, Springer-Verlag, Berlin, 1979).
~ ' '
In this procedure, adjuvant arthritls was induced by the
subplantar injection on day O of 0.05 ml o~ adjuvant ~a
suspension of heat-killed, dessicated Mvcobacterium
butYricum! 0.5% (w/v), in heavy mineral oil containing 0.2
digitonin] into the right hind paw of male Charles River
Lewis rats (120-140g) that were hou6ed individually and
z5 given food and water ad lib. Paw volumes (both hind paws)
were measured immediately after injection of the adjuvant.
Paw Volumes were also measured, to follow the development of
inflammation-induced ~welling in the ar~hritic paws, at
inteLvals of 3 to 7 days by immer~ion of the paw to the
level sf the lateral malleolus in a mercury plethysmograph.
Drugs were adminis~ered once a day tstarting on the day
~ of adjuvan~ in}ection) by incubation using Tween*80
;~ (polyoxyethylene ~orbitan mono-oleate~ at a do6e of 0.25
35 ml~100 g body weight as the vehicle. Arthritic control rats
received daily dose6 of the vehicle only. On day 23-25, the
rats were sacrificed, plasma collected and plasma fibrinogen
* trade mark.
:'
~2 ;~733r~d
-- 5
levels determined (ammonium sulfate turbidimetric method) as
described by Exner et al., Amer. J. Clin Path~ 71:521-527
(1979). Anti-inflammatory activity of the test drugs were
determined by comparing the extent of paw swelli~g (paw
volume on a particular day, i.e. day four to day
twenty-five, minus the paw volume on day 0) in drug-treated
arthritic rats with the extent of paw swelling in the
vehicle-treated arthritic rats. Drug induced decreases in
the level of plasma fibrinogen,, an acute phase protein tha~
0 i8 elevated in the plasma of rats with adjuvant-induced
arthritis, were also used to guantitate the
anti-inflammatory activity.
The results of various compounds of thi6 invention when
compared to Indomethacin and 13-cis-vitamin A acid are given
in the following table (TABLE I)~
,7--_~_ . _._ ~ __ __
. ~ - ~ 1 ^ 1 ^ 1 ~ 1 I ~r
.' _ _ , _ ~ .~. _
E ~ ~ C~ _ n ~ . ,.,
_ _ _ _ _ . _ _ __
: ,OE~ I ~, ~.S3 .cl~ e~ ~ 1~ ~t O,~ T
uo~ _ _ ,_ _ ~_ ~ ~ .......... _
_ ~ ~ ~o ~ ~ ~ ~7 q,7 ~ ~
m _ _ ~ _ : _ . . . . . _
:~ ~ OQ ~ O ~o I~ n .u~ ~n ~
~277;~3~
-- 7
In the above Table I the ~ercent reduc~ion in paw volume
demonstrates the effectiveness of the compounds o~ this
invention to reduce swelling caused by adjuvant arthritis.
As seen from the results in this Table, the compounds o~
this invention e~fectively reduce the swelling caused by the
adjuvant. Furthermore the compounds of this in~ention were
ef~ective in reducing the plasma fibrinogen generally
associated with rheumatoid arthritis. Furtharmore as seen
from the weight gain of the animals, the compounds of this
L0 invention at the dosage tested produced no ~ubstantial
reduction in the weight gain o~ the animals. This indicates
the lack of toxicity exhibited by the compounds of this
invention.
The compounds of formula I and their pharmaceutically
acceetable salts can be used in a variety of pharmaceutical
preparations. In these preparations, the~e compounds are
administrable in the fo~m of oral unit dosage forms such as
tablets, pills~ powders, ca~ules, as well a~ in such forms
as injectables, solu~iong, suppositories, emulsion6,
dispersions, and in other suitable forms. The pharmaceu-
tical pre~arations which contain the com~ounds of formula I
are conveniently formed by admixing with a non-toxic
~harmaceutical organic carrier or a non-toxic pharmaceu~ical
inorganic carrier. Typical of pharmaceuti- cally acceptable
carriers are, foc example, water, gelatin, lactose,
starche6, magnesium stearate, talc, vegetable oils,
polyalkylene glycols, ~etroleum jelly and other conventio-
nally em~loyed pharmaceutically acceptable carriers. The
pharmaceutical pce~arations may al60 contain non-toxic
auxiliary subs~ances such as emulsifying, preserving and
wetting agents and the like, as for exam~le, socbitan
monolaurate, triethanol amine oleate, polyoxyethylene
sorbitan, dioctyl sodium sulfosuccinata and the like.
The daily dose administered for the com~ounds will, of
course, vary with the ~articular novel compound employed,
~7733~
-- 8
the chosen eoute of administration and the size of ~he
recipient. The dosage administerQd is not subject to
definite bounds but it will usually be in ef~ective amoun~s
of the pharmacologically function of the compound~ of this
invention. Reeresentative of a typical method for
administering the comeounds of formula I or their 6alts is
by oral administration. By this route, the compounds o~
formula I or their salts can be administered at a dosage of
O.S mgtkg per day p.o. to lOO mg/kg per day p.o. Preferably
these com~ound8 can be administ:ered daily to ~atient~ i~
unit oral dosage forms at daily dosages of ~rom l to 30
mg/kg of body weight, with dosages o~ ~rom l to lO mg/kg
being especially ereferred.
The compound of Pormula I where X is -O- can be prepared
from compounds of the formula ~
R2~CH(:)
I O ~
~ OH
R3
wherein Rl, R2 and R3 are as above,
via the reaction scheme in Figure l.
2~
In the reaction scheme of Figure l, n. Rl, R2, R3,
R7 and R8 are as above and R'g is lower alkyl, Z i8 a
; leaving grou~; Y is aryl preferably phenyl; Z' is halo.
The compound of formula III is converted to the compound
of formula IV via reaction step (a) by reducing the aldehyde
group to the alcohol. This reaction i~ carried out
utillzing a conventional reducing agent which converts
aldehyde~ to alcohols. Any conventional reducing agent for
35 this pur~ose can be utilized in the reaction of step (a).
- In carrying out this reaction it is generally pre~ereed to
utilize an alkali metal borohydride such as sodium
~LZ7733~
g
borohydride as the reducing agent. Any of the conditions
conventional in such reduction ceactions can be utilized to
carry out the reaction of step (a). If R2 is hydroxy it
- is generally preferred to protect the hydroxy designated by
R2 during the reduction of the compound of formula III and
its subsequent conversion to the compound of formula II-~.
Any conventional hydrolizable hydroxy erotecting group such
as a lower alkanoyl group may be utilized to protect the
hydrox~ group when R2 i8 hydroxy. This e~ter protecting
L0 group can be cleaved by con~entional ester hydrolysi~ after
the focmation of the Wittig salts o~ formulae XIII and XVI
or after the formation of the ether of formula X.
The com~ound oi i'ormula IV is converted to the compound
of formula VI via reaction step (b) by treating the compound
of formula IV with a triarylphosphine hydrohalide. In this
manner the phosphoniu~ ~alt of formula VI ifi produced. ~ny
conven~ional method o~ reacting an allylic alcohol with a
triarylphosphine hydrohalide can be used to carry out this
~, 20 reaction~ The phosphonium salt o~ formula V~ is reacted via
a Wittig reaction with the compound of ~ormula VII in ~tep
(c) to ~orm the compound of formula VIII. ~ny of the
conditions conventionally used in Wittig Leactions can be
utilized to carry out the reaction of ~tep (c~.
2s
On the other hand the compound of formula III may be
directly conver~ed to ~he compound of formula VIII via ~he
~eaction with the phosphonium salt of the compound of
formula IX as in reaction step (e). The Leaction of the
phosphonium salt of formula I~ with the compound of formula
III to produce the compound of formula VIII is carried out
utilizing the same conditions as described in connection
with reaction step (c).
The compou~d of formula VIII is conveLted to the
compound of formula X by etherifying or alkylating the
compound of f.ormula VIII with a compou~d of formula V as in
~;27733?,
-- 10 --
reaction step (d). In the compound of formula V, Z can be
any conventional leaving group such as mesyloxy, tosyloxy OL
a halide. ~ny conventional method of etherification of a
hydroxy group though reaction with a halide or a leaving
group can be utilized to carry out ~he reaction of step (d).
In accordance with another embodiment of thi~ invention
~he compound of formula X, ~here when R2 iB hyd~oxy, the
hydroxy group is protected ~ia a hydrolizable ester, can be
~o produced from the com~ound of the ~ormula III by alkylation
or etherification of the compound o~ formula III with the
compound of formula V to produce the comeound o~ XI. This
reaction i8 carried by alkylating the compound of formula
III with the compound of formula V as in step ~d). In the
reaction of step6 (f) and (d) where R4 iB a hydroxy alkyl
group, the hydroxy contained in a~ need not be protected.
This is true since under the conditions used in this
reaction s~ep, the compound of formula V will react with
either the compound of formula III or the compound of
formula VIII to produce the compound of formula XI or the
compound o~ ~ormula X without the neces6ity for protecting
~ ~he hydroxy grou~ contained on the ~lkyl chain. Alkylation
:~ or etharifi~ation will occur direc~ly with the ~h~nyl
- hydroxy moiety o~ either the compound of formula III or the
compound of formula VIII and there will be little, if any
~ reaction with the hydroxy group contained on the alkyl chain
:~ designated by R4. The compound of formula XI is converted
to the compound of formula ~II, via reaction step (g) by
reduction. The same;conditions described in connection with
30 reaction ste~ (aj can be utilized to convert the com~ound o
~: ~ormula XI to the compound of formula XII~
:;
The compound of formula ~II is converted, via reaction
step (h), to the compound of formula XIII by treating the
35 compound of formula XII with a triaryIphosphine hydro-
;- halide in ~he manner described hereinbefore in connection
with step (bj. The compound of formula XIII is converted to
~Z7733~,
- 11
the compound of formula X by reacting the compound of
formula XIII with the com~ound of formula VII via reaction
step (i). This reaction step is carried out in the same
manner as described hereinbefore in connection with reaction
step (c).
In accordance with another embodiment of this invention
the compound of formula X is p~oduced by first convert~ng
the compound of formula XI to the compound of formula XIV.
L0 The compound of formula XI is converted to the compound of
formula XIV by aldol condensation with the compound of
formula XX. Any conventional method o~ aldol condensation
can be utilized to react the compound of formula XI with the
compound of formula XX to form the compound of formula XIV.
L5 In the next ~tep the compound of fo~mula XIV is condensed
via either a Grignard reaction with vinyl magnesium halide
or a lithium condeFLsation reaction with vinyl lithium to
produce the compound of formula XV. The reac~ion of step
(k) can be carried ou~ by utilizing any of the conditions
conventional in lithium condensation6 or Grignard
condensation reactions. The compound of formula XV is
converted to the compound of formula XVI by reacting the
compound of formula XV with a triarylpho&phine hydrohalide
~: in the manner described hereinbefore in connection with the
:~ 25 reaction of step ~b~. The compound of formula XVI i6
thereafter converted to the compound of formula ~, via
reaction step (m). by reaction with the compound of formula
XVIl. The reaction of step (m~ is carried out utilizing a
standard ~ittig reaction as described in connection with the
30 reaction o~ step (c). The compound of formula XVI by the
reaction with ~he compound of formula XVII produces the
com2ound of formula X. The compound of formula X can be
converted to the free acid i.e. the compound of formula I
where R5 is COOH by ester hydrolysis. Any co~entional
35 method of ester hydrolysis will produce the compound of the
formula I where R5 is COOH.
~2~33~
~ 12 -
The compounds of formula I where X is -N- a~e prepared
Rlo
from the compounds oE the formula
R
R2 ~ C1~20T~
L J XXII
y~ NH2
R3
wherein Rl, R~ and R3 are as above
via the reaction scheme in Figure 2.
L5In Figur~ 2- Rl, R2, R3. ~4~ 7 8 9
Zl and Y are a~ above. In Figure 2, R13 is the same as
R4 with one less carbon than the alkyl group contained ~y
R4. Therefore R13 is an alkyl group containing from 3
o 9 carbon atoms or -C~2(CH2)mCH~OH where m is a
~o number one less than n, i.e. m is an integer of from 5 to
S. In ~igure 2, Rlo i~ hydrogen or lower ~Ikyl containing
from 1 to 7 carbon atoms and Rlo' is a lower alkyl group
containing from 1 to 7 carbon atoms. In this embodiment
Rlo" i~ a lower alkyl group having one carbon atom le s
zs the alkyl group de~ignatad by Rlo'.
In Figure 2 the compound of formula XXII is reacted with
acid ~hloride of formula XIX where Z' is halogen tG produce
the compound of formula XXIII ~hich is later conv~rted
either to the compound of formula ~XVI or the compound o~
; ~XXI. ~here R13 in the compound of for~ula XIX is
: -CH2-(CH2)mCH20H where m is an in~eger of from 5 to
6 carbon atoms, the pesence of the hydLoxy group on the
substituent of R13 will not effect the reaction to produce
the compound of formula XXVI or the compound of formula
XXXI. It has been found that this hydroxy groue remains
una~fected throughout the series of reaction~ set forth in
- :
r~
~ 13 --
Figure 2.
On the other hand this hydroxy group can be erotected by
mean~ of forming a hydrolyzable ether functional gcoup which
protects the hydroxy group throughout these reactions. ~ny
conventional ether protecting grou~ can be utilized to
protect the hydroxy group throughout these reactions. Among
the preferred ether pro~ecting groups, are included:
tetrahydropyranyloxy, t-butoxy, triloweralkylsilyloxy i.e.
trimethylsllyloxy, etc. Any conven~ional ether erotacting
group can be utili~ed to protect the terminal hydroxy groue
which may be present as Rl3. On the other hand, the
reactions set forth in Figure 2 can be carried out without
any protection of the terminal hydroxy gcoup.
In the first ste~ of the reaction, the compound of
~ormula XXII is reacted wi~h the compound of formula XIX to
p}oduce the compound of formula ~XIII. Any conventional
~ method of condensing an amine with an acid halide can be
; 20 utili2ed to carry out this reaction. In the next step the
compound of formula XXIII is converted to the compound o~
formula XXIV, via reaction step (n), by treating the
compound of formula XXIII with a reducing agent. ~ny
conventional alkali metal aluminum hydride reducing agent
can be utilized to caIry out this reaction with the
preferred reducing agent being lithium alum;num hydride.
Any of the conditions conventional in reducing with an
alkali metal aluminum hydride reducing agent can be utilized
to cacry out this reaction.
0
The compound of ~ocmula XXIV can be converted to ~he
com~ound of formula XXVI via the intermedia~e XXV. In the
first step of this procedure, step (o), the compound of
formula XXIV is converted into the phosphonium salt of
35 formula XXV by reac~ion wi~h a ~ciacylehosphine hydro-
halide as described hereinbefore in connection with the
reaction of s~e~ (b). The compound of formula XXV is
.
~ , .
127733,?~
- 14 -
converted to the compound of formula XXVI Yia reaction step
(p), by reaction with the aldehyde of formula VII. (See
Figure 1). The reaction of step (p) to produce the com~ound
of formula XXVI is carried by a Wittig reaction in ~he same
manner as described in reaction step (c) hereinbefore. The
compound of formula XXVI where R9 is lower alkyl can, if
desired, be converted into the Eree acid by conventional
ba~ic hydroly6is. Any conventional method of basic
hydroly~i~ to hydrolize esters can be utilized to convert
lo the compound of formula ~XVI to the free acid. On the other
hand, the compound of formula XXVI where R4 contain~ a
terminal hydroxy group etherified with a conventional ether
~rotecting group can be converted into the free alcohol by
acid hydrolysis. Any of the conventional methods of
L5 hydrolyzing ethers can be utilized to carry out this
reaction. Tho ether hydroly6is of the com~ound of formula
XXVI can be carried out either befoLe or after the acid
hydrolysis used to hydrolize the ester protecting Rg'. On
the other hand if R4 in the compound o~ formula X in
zo Figure 1 contains an etheri~iPd hydroxy group, the compound
o~ formula X can be hydrolyzed in the same manner ~s is the
compound Oe formula XXVI.
On thç other hand, the compound of formula XXIV can be
zs converted to th~ tertiary amine compound of formula XXXI.
In this reaction, the com~ound oE formula XXIV is reacted,
via reaction step (q), with the acid halide of formula
~IX-A, to produce the compound of formula XXVII in the same
manner de~cribed hareinbefore in connection with the
30 reaction to convert the compound of formula XXII to the
compound of formula XXIII.
The compound o~ formula XXVII i5 converted to the
formula XXIX, via reaction ste~ (r), by treating the
35 comeound of Eormula XXVII with a lithium aluminum hydride
reducing agent as described in connection with reaction o~
~;~77~33%
- 15 -
step (n).
In the next step of this reaction scheme, the compound
of formula XXIX is converted to the compound of formula XXX,
via reaction step ~5), by treating with a triarylpho6~hine
hydrohalide. This reaction is carried out in the same
manner as described in connection with reaction step (b)
described hereinbefore. The compound of formula XXX is
converted to the compound of formula XXXI, via reaction ste~
(t), by reaction wîth the compound of foemula VII (Figure
1). In carrying out the reaction of ~tep (t) a Wittig
reaction iæ utilized. The reaction of stey (t) can be
carried out in the same manner as described hereinbefore in
connection with the reaction of step (c). The compound of
; LS formula XXXI where Rg' is a lower alkyl grsup can be
converted, if desired, to the corresponding compound of the
formula XXXI containing the free acid group by basic
hydrolysis. 0~ the other hand, if R4 contains a terminal
hydroxy group protected through the Pormation of a
; 20 hydrolizable ethe~, the compound of formula XXXI can be
converted to the corresponding compound where R4 is a free
hydroxy group by conventional ether hydrolysis. This ether
hydroly~is can be carried out before or after hydrolysis of
the e~er group denoted by R
; In the reac~ion scheme of Figure 2, when R2 i8
h~droxy, it i8 preferred that this hydroxy group be
~ protected via the formation of an ester protecting group.
; The e~ter protecting group can be removed after the
~ 30 formation o~ the Wittig ~alts oE formula XXX.
.
The compounds oE formula I where X is -CH-
P~10
are prepared from a compound of the formula:
~27733~'?J
- L6 -
I
R2 ~ zH XXXV
~l
- r 17 Rlo
R3 0
wherein Z" i~ ether bromo or iodo: Rl, R2, and R3
are as above: and Rlo is hydrogen or lower alkyl
lo as set forth in Figure 3. In Figure 3, Rl, R2, R3,
4~ R7~ R3, ~ 9~ Rlo~ ~13.~ Y~ Z', and Z" are a~
above.
In Figure 3, the compound of formula XXXV is ~irst
reacted with the compound of formula ~X%IV via reactio~ ste~
(u) to eroduce the compound of formula XXXVI. This reaction
is carried out via a Wittig reaction. In the compound of
formula XXXVI R13 can be, if desired, -CH2-(CH2)m -
~:~ 0H where the free hydroxy grou~ can, i~ desired, be
protected through the formation of any of the aforementionedco~ventional ether group~. On the other hand it has been~
found that thi~ OH group can be a P~ee hydrsxy group a~d
~:~ need not be protected by means o~ an ether protecting
group. In carrying out ~he reactions o~ Figure 3 this free
. ~ 25 hydroxy grou2 i$ not af~ected by the reaction~ which convert
: ~he compound of formula XXXVI to the compound of ~ormula
X~XXI. However for best yield~ it is generally preferred to
~rotect this hydroxy group via ~he formation o~ a
hydrolizable ether.
T~e reaction of stee (u) is carried out via a Wittig
. ~ reaction between the compound~ of formula XXXV and the
compound o~ foemula XXXIV utilizing the same reaction
conditions as described hereinbelow in connection with
35 reaction ste~ (c).
The comDDund of formula XXXVI ~n be converted to the
:
~ ' .
~t7733~
- 17 -
compound of formula XXXVII via a reaction step (v) by
hydrogenation. Any conventional method of hydLogenation can
be utilized to carry out this reaction. ~mong the
conventional method of hydrogenation are included treating
the compound of formula XXXVI, in an inert organic solvent
medium, with hydrogen gas in the presence of a catalyst.
~ny conventional hydrogenation catalyst can be utilized in
carrying out this reaction. Among the preferred catalysts
are included ealladi1lm. In carrying out this reaction any
~o conventional inert organic solvent can be utilized.
Furthermore, any of the conditions conventional in catalytic
hydrogenation can be utilized in reaction step (v~.
In the next ste~ o~ this reaction, the compound o~
formula XXXVII is converted to the compound of ~ormula XXXIX
via reaction step (w), by treating com~ound o~ formula
XXXVII with formaldehyde or a formaldehyde libera~ing
compound. In carrying out this reaction the co~pound o~
formula XXXVII is first metalated with an alkali metal alkyl
zo e.g. n-butyllithium. Generally this reaction is carried out
in an inert organic solvent such a6 an ether solvent. ~mong
the ~referred solvents are diethyl ether and
tetrahydrofuran. In carrying out this reaction temperature
and pressure are not critical. This reaction can be carried
out at room tem~erature and atmospheric pressure. If
desired, higher and lower tem~eratures can be utilized.
~fter treating the compound of formula XXXVIII with an
al~ali metal alkyl, formaldehyde or a ~ormaldehyde
liberating compound is added to the reaction medium. ~ny
co~ventional compound capable of l~berating ~ormaldehyde
such as paraformaldehyde can be utilized in carrying out
this reaction. This reaction i8 carried out in the same
reaction medium and utilizing ~he same conditions as the
metalation of the compound o formula XXXVIII was carried
out.
The com~ound of formula XXXIX is converted to the
~ll27733?,,
- 18 -
compound of formula XXXX, via reaction step (x), by treating
the compound of foemula XXXIX with triarylphosphine
hydrohalide. This reaction is carried out in the same
manner as described in connection with reaction step (b) as
described hereinbefore. The compound of formula XXXX is
converted, via reaction step (y), to the compound of formula
XXXXI by reac~ion with the compound of formula VII. (See
Figure 1). This reaction of step (y) is carried out via a
Wittig reaction utilizing the same conditions described in
L0 connection with reaction step (c). The compound of formula
XXXXI may be converted to corresponding compound containing
the free carboxyl group instead of Rg'. This reaction is
carried out by conventional ester hydrolysis in the manner
hereinbefore described. Any conventional method of ester
hydrolysis can be utilized. If R4 contains terminal
hydroxy group protected through the use of an ether
protecting group, this ether group can be hydrolixed to
yield the ~ree hydroxy group by conventional ether
hydrolys;6 ~uch as by utilizing an aqueous inorganic acid.
Any conventional method of ether hydrolysis can be
utilized. The protected ether hyd~oxy group can be
hydrolized either prior to or after hyd~olysis of the ester
grou~ to form the free acid of the compound of focmula XXXXI.
If it is desi~ed to produce compounds of the formulae I
and where X is -C=CH-, the com~ound of ~ormula XXXV in
~0
Figu~e 3 is reacted, via reaction step (u), with the
comeound of formula XXXIV where R13 is R4 to produce the
0 compound of formula XXXVI where R13 is R4. This
co~pound of formula XXXVI where ~4 is R13 ls then
subjected to the same series of reactions as the compound of
formula XXXVII, i.e. the reaction steps (w), ~x) and ~y), to
produce the compounds of formulae I where X is
35 -C=C~-.
Rlo
1277;33?~
-- 19 ~
In the reaction scheme in Figure 3 where R2 in the
compound of formula XXXV is a hydroxy group, it is preferred
to protect this hydroxy group via esterifica~ion with a
lower alkanoic acid. This ester protecting group can be
cleaved after formation of the Wittig salt of formula XXXX.
The compound of formula I and II where X is -CH-0
~ 10
can be erepared from a compoun-l of the formula
~0
~-F~Z'
3 ~
l ~ L
~3
wherein Rl, R2, R3 and Z" as above
by the reaction scheme of Figure 4. In Figure 4, Rl,
~ R R R R R ' Y d Zll b
and ~15 is R4 wherein a hydroxy group contained in R15
is protected in the form of a hydrolizable ether group such
a~ tetrahydropyranyl as well a6 the ether groups mentioned
hereinbefore.
: 25 The compound of ~ormula L is co~verted to the compound
of formula LI by reaction with the alkali metal alkoxide of
formula L-A. This reaction i8 carried out by reacting the
: compound sf formula L with the compound of formula L-A
utilizing the conditions conventional in reacting an alkali
30 metal alkoxide with a halide.
:
In the ste~, the compound of formula LI is converted to
the compound of formula LII by first treating the compound
of formula L[ with an alkyl lithium such as n-butyl lithium
to metalate the compound of formula 1I. The metalated
compound of formula LI i6 thereafter reacted with
; formaldehyde or a formaldehyde liberating compound. In
773~
- 20 -
converting the compound of formula LI to the compound of
formula LII, the same reaction conditions as described in
connection with reaction step (w) are used in this
conver~ion. The compound of formula LII is converted to ~he
phosphonium salt of ~ormula LIII treating the compound of
formula LII with a triarylphosl?hine hydrohalide in the
mannar set for~h in reaction step (b) above. The
phosphonium salt of formula LIII is eeacted via a Wittig
reaction with the compound of formula VII (see Figure 1) to
L0 form a compound of formula LIV. This reaction to form the
compound o~ fo~mula LIV i~ carriad out in the same manner as
described in connection with step (c~ hereinbafore.
When R15 in the compound of formula LIV contains a
protected hydroxy substituant said substituent can be
hydrolized to form the free hydroxy compound by conventional
methods for hydrolizing easily remo~able ether groups. Any
conventional method for hydrolizing ether protecting groups
can be utilized. The conditions conventional for
zo hydrolizing ether protacting groups will not affect the
other ether group contained within the compound of formula
LIV. The com2ound o~ ~ormula LIV can be convarted to the
~rea acid by conventional ester hydrolysis.
Z5 If R2 in the compounds of formulae L, LI, LII and LIII
is hydroxy, it is peeferable that the hydroxy group be
protec~ed via a hydrolyæable ester grou~ such as lower
alkanoylvxy. The hydrolyzable ester ~rotecting group can be
cleaved after forming the Wittig salt of formula LIII.
If desired, the double bonds within the comeound of
formula I at positions 2-3, 4-5, 6-7 and 8-9 can be either
in the ci~ or trans configuration. On the other hand, these
compounds can be a mixture of the various cis and trans
35 isomers. In the compou~d of formula VII, the double bonds
contained therein can be either in the cis or trans
con~iguratioll de~ending upon tùe desired ~tereo
~;277331~:
- Zl -
configuration of the double bonds within the compounds of
formula I. The Wittig reaction carried out in producing the
compounds of formula I and II such as in steps ~c), (e),
(i), etc. produces the double bond at the 8-9 po6ition as a
mixture o~ the 8-9 cis and trans isomers. These cis and
tran~ i~omers can be separated by conventional means such as
fractional crys~allization, etc.
In addition, where the compound6 o~ formula I have a
L0 double bond in the trans configuration at the 2-3 position,
this isomer can be converted to the corresponding Ci8 double
bond with conventional methods of isomeriæation known in the
art. bmong these ~rocedures are included treating the
compound of either formula I with iodine in an inert organic
L5 solvent. I60merization with iodine produces the compound o~
formula I with a 2-3 double bond in the cis position.
The comeounds of ormula I include all of its geometric
isomers including mixtures of these geometric isomer5.
The eompound o~ formula XI where Rl is fluoro is a new
compound and can be prepared from a comeound of the formula
~ F
~ 25 ~ ~ LV
where R2 and R3 are above
via the reaction scheme given of Figure 5. In Figure 5,
R2. R3 and R4 are as above.
In Figure 5, ~he compound o~ formula LV i6 alkylated by
reaction with an allyl bromide. If the compound where R2
~ i6 hydroxy is desired, the compound of formula LV where the
:
~7733~
- 22 -
hydroxy group designated by R2 is protected by
esterification is used as the starting material, i.e. the
compound of formula LV where R2 is a protected hyroxy
group. Any conventional method of alkylating a hydroxy
group with an allyl bromide can be utilized to carry out the
reaction o~ converting the com~?ound o LV to the compound of
formula LVI. The compound of ormula LVI i8 rearranged to
the compound of formula LVII by heating the compound of
formula LVI to a temperature from 190 degrees to 230 degrees
L0 centigrade. This rearrangement can take place without the
use of any solvent or in the ~resence of a high boiliny
hydrocarbon solvent. If R3 is hydrogen, the com~ound of
fo~mula LVII i8 formed as a mixture with the isomer of the
compound of formula LVII where the allyl group i6 para to
the ~luorine substituent on the benzyl ring. This isomer
can be separated or utilized in the subsequent reactions and
separated from the reaction mixture at a later ~tage.
The compound of formula LVII i8 thereafter converted to
zo the compound of formula LVIII by reaction with the compound
of formula V (Figure 1) as set orth in reaction s~ep (d)
hereinbefore. In the next step o~ this reaction scheme, ~he
compound of formula LVIII is converted to the compound o~
formula LIX by isomerization with a strong base such as an
alkali metal al~o~ide in the presence of an inert organic
sol~ent preferably pota~sium tertiary butoxida in dimethyl
sulfoxide. The compound of formula LIX i5 converted to the
compound of formula LX by treating the compound of formula
LIX with ozone gas. In carrying out this reaction,
30 temperature of from minu~ 70 de~rees centigrade to minus 20
degrees centigrade are utilized. Furthermore thi~ reaction
is carried out in an iner~ organic solvent. Any
conventional inert organic solvent can be utilized,
~re~erably halogenated hydrocarbons such as methylene
35 chloride.
A compound of formula LX is the com~ound of focmula XI
~7733,~
- 23 -
wherein Rl is fluoro. This compound can be converted to
the compound of formula X in accordance with the reaction
scheme set forth in Figure 1.
Where R2 is hydroxy in the compound of formula III
there are two hydroxy groups. Therefore, it i8 generally
preferred to prepare the compound of formula XI where R2
is a protected hydroxy group from a compound of the formula
LXI as shown in Figure 6. In this manner, compounds of
L0 formula I can be prepared where X i8 -0-; R2 iS hydroxy
and R3 and R4 are as above. In Fig. 6, Rl and R15
taken with its attached oxygen atom, is hydroxy protected by
a conventional hydrol~zable protecting group preferably a
lower alkanoyl.
In Figure 6, the compound of Formula LXI is converted to
the compound of formula LXII utilizing the same reaction as
described in ~onnection with the conver6ion of a compound of
~ormula LV ~o a compound o~ the Formula LVI. (See Fig. 8)
The compound of Formula LXII is next converted to a com~ound
of Formula LXIII by utilizing the same procedure described
in connection with the conversion of the compound of Formula
LVI to LVII. In the next 6tep6 the compound o Formula
LXIII is converted to the compound LXIV ~y the ~ame
zs procedure described in connection with step (g') in Figure 5
and then to the compound of Formula ~XV by the procedure
described in connection with step (h') in ~igure 5. The
conver~ion of bromobenzene compound of Formula LXV to the
~henol compound of Formula LXVI takes place by erocedures
30 well known in the art such as disclosed by Kidwell and
Darling, Tetrahedron Letters, (1966) pgs. 531-535.
: In ~he next step of preparing the in~ermediate of
formula XI where R2 is a protected hydroxy group, i.e. the
35 compound of Formula XI~ he hydroxy group is protec~ed on
the compound of formula LXVI through esterification with any
conventional hydrolyzable ester group to form the compound
~.~Z7733.~,
- 24 -
of Formula LXVII where Rl5 taken together is attached
oxygen forms a hydrolyzable ester group. Any conventional
method of esterifying a hydroxy groups with an organic acid
such as a lower alkanoic acid containing from 1 to 7 carbon
atoms can be used to prepare the compound of Formula LXVII.
The com~ound of Formula XI-~ is formed from the compound of
Formula LXVII by the reaction described hereinbefore with
reseect to the conversion of a compound of the Formula LIX
to LX (See Pig. 5).
In carrying out the convQrsion of a compound of Formula
XI-A, to a compound of Formula XVII, as in Figure 1, it ;8
generally prefe~red to hydrolyzs the ester substituent which
forms R2 aftec the formation of the Wittig salt of Formula
XIII or Formula XVI.
In accordance with another embodiment of this invention
the compound of formula XI where Rl is CF3 (the compound
of formula XI-B) can be formed by the reaction outlined in
2Q Figure 7 from a compound of Formula LXX~ In the fir6t ~te~
of this reaction the compound of Formula LXX is converted to
a compound of Formula LXXI utili~ing the ~ame procedule
dascribed hereinbefore in connection with the reaction. via
step (d) where the compound of Formula VIII is reacted with
a compound of the Formula V to produce a compound of the
Formula X. In this reaction where R2 i5 OH, alkylation
~ occurs very slowly on the hydroxy group ortho to the CF3
; group. Therefore, erotection of this group may no~ be
neces6ary since alkylation eroceeds ~refeeably with the meta
0 h~droxy ~roup. Any mixtures of alkylated products obtained
from this rea~tion can be separated by conventional
separation procedures. The compound of formula LX~I is
converted to the compound of formula XII-B by conventional
procedures of formylating a benzene Ling such as by
35 treatment with an alkyl lithium and dimethylformamide.
The following examples are illus~rative but limitative
~2>7733?J
- 25 -
o~ the invention. In the exam~les the ether is diethylether
and the solvents were removed in vacuo.
Example 1
[[(2-tNonyloxy~DhenYllmethYlltriPhenYlPhosPhonium bromide
2-Hydroxybenzaldehyde (110 g). was alkylated by mixing
this compound with l-bromononane (180 g), anhydrous
potassium carbonate and dimethylformamide (800 mL). This
mixture was heated at 80C ~or 14 hours. Hexane and watec
were then added and the hexane extract was concentrated and
the residue was distilled to yîeld the
2-nonyloxybenzaldehyde ~210 g). be 121C (0.3mm Hg).
solution of 2-nonyloxybenzaldehyde prepared above (100 g) in
ethanol (1000 mL) at 10C was reduced by treating wi~h an
L5 excess of sodium borohydride (6 g) and after 6tirring the
mixture for a further 15-20 min. at room te~perature, the
compound 2-nonyloxybenzylalcohol was isolated by extraction
into hexane. Removal of the hexane in vacuo yielded the
crude 2-nonyloxybenzylalcohol (98 g). The resulting
2-nonyloxybenzylalcohol was added to a mixture of
triphenylphosphine hydrobromide (144 g) in acetonitrile t500
mL) and the resultant solution wa~ heated at reflux for 14
hours. Removal of the solvents in vacuo and crystallization
of the re~idue from a tetrahydrofuran/ethyl ether mixture
gave the pure ~2-tnonylox~)phenylJmethyl]triphen
phosphonium bromide (208 g3.
~(2-HYdroxYPhenyl?methylLtriPhen~l~--sphonium bromide
2-Hydroxybenzyl ~lcohol was treated with t~iphenyl-
phosphine hydrobromide in acetonitrile as described in
Example 1 to yield [(2-Hydroxyphenyl)methyl~riphenyl-
phosphonium bromide.
ExamPle 3
ALL(EL-9-(2-Hydroxyphen~1)-3,7-dime hYl-2,4,6,8-nona-
tetraenoic ac_d ethyl ester
~.2~7733~
- 26 -
A 601ution of ~t2-hydroxyphenyl)methyl]triphenyl-
phosphonium bromide (1 mol~ in tetrahydrofuran was converted
to the ylide at -35 with a solution of n-butyllithium in
hexane (2.1 mol equiv.) and then exposed to 7-formyl-3-
5 methyl-2,4,6-octatrienoic acid ethyl ester (1 mol) and then
stirred at -70 for a fuether 15 min. Isolation of the
organic products with a hexane~ethyl acetate mixture (4:1
parts by volume~ and dilute mineral acid (2M aqueous HCl)
gave the pure all(E)-9-(2-hydroxyphenyl)-3,7-dimethyl-
2,4,6,8-nonatetraenoic acid eth~l eeter (90% yield) a~ter
chromatography followed by crystallization from a
dichloromethane/hexane mixture.
Example 4
ALL(E~-9=~2-HvdroxvphenrlL-3~7-dimethyl-2~ J8-nona
tetraenoic acid ethyl_ester
mixture of 2-hydroxybenzaldenyde (0.5 mol), (7-
carboxy-2,6-dimethyl-2,4,6-heptatrien-1-yl)triphenylphos-
phonium bromide (0.6 mol) in 1,2-epoxybutane (750 mL) was
heated at re1ux for 30 ~in. cooled, poured into an
ether/hexane mixture (1:1 ~arts by volume) fileered and
concentrated. The residue was then crystallized from a
hexane/ether mixture to yield all(E)-9-(2-hydroxyphenyl)-3,
7-dimathyl-2,4,6,~-nonatetraenoic acid ethyl ester (38
yield), mp 143-1~5.
Example 5
E)-9-~2~-(Nonyloxy)phenY11-3,7-di~ yl-2 4,6~8-nona-
tetraenoic acid
A solution of ~[2-(nonyloxy)ehenyl]methrl]triphenyl-
~hosphonium bromide (150 g3 in tetrahyd~ofuran (1100 mL) was
cooled to -50C to yield a fine suspension of the solid
8alt. To this mixture was added a solution o~
n-butyllithium in hexane (180 mL, a 1.6 molaL) to yield a
solution of the ylide. The mixture was then stirred a
further 15 min at -40C, cooled to -70~ and tLeated with
7-formyl-3-methyl-2,4,6-octatrienoic acid ethyl ester (65 g)
, , .
Z7;733~?J
- 27 _
dissolved in tetrahyd~ofuran (250 mL). Addition o~ hexane
and aqueous methanol (40~) to the reaction mixture followed
by concentration of the hexane extract yielded
All(E)-9-[2-(nonyloxy)phenyl]-3,7-dimethyl-2,4,6,8-nona-
tetraenoic acid ethyl ester (64 g, 58% yield), mp52-53C. This ester was then hydrolized by foeming a
solution of ~his ester (70 g) in ethanol (1000 mL). This
solution was trea~ed with aqueous potassium hydroxide (80 g
in 400 m1 water) and heated at reflux for 1 h. Watar and
L0 aqueous mineral acid was then added and the solids were
extracted into chloroform. Concentration of this o~ganic
extract and crystallization of the residue from an ethyl
acetate hexane mixture yielded All(E)-9-E2-(nonyloxy)phenyl]-
3,7-dimethyl-2,4,6,B-nonatetLaenoic acid (38 g), mp
lOZ-1~3C.
ExamPle_6
(All_E) 2~2-(NonYloxY)~henYl~,7-dimethvl-2,4,6l8-nona-
tetrae~oic acid
; 20 A mixture o~ sodium hydride (24 g, 50~ by weight in
mineral oil) and dimethylformamide (1000 mL) at 10C was
treated with All(E~-9-t2-hydroxyphenyl3-3,7-dimethyl-2,4,
6,8-nonatetraenoic acid ethyl ester (0.4 equiv.). The
~esulting mixture was then stirLed at room temperature until
all hydrogen evolution had stoeped to produce the sodium
salt o~ All(E)-9-(2-hydroxyphenyl)-3,7-dimethyl-2,4,6,8-
nonatetraenoic acid ethyl ester. ~ solution of l-nonyl
tosylate (O.S equiv.) in dimethyl~ormamide (200 mL) was ~hen
added to this salt solution and the reaction mixture ~as
stirred at 45 for 14 h. Hexane/water was then carefully
added and the hexane extract was concentLated and the
residue was purified by chromatography over silica geI.
Crystalli2ation from hexane then yielded the puee
All(E)-9-~2-(nonyloxy)ehenyl]-3,7-dimethyl-2,4,6,8-nona-
tetraenoic acid ethyl ester. Hydrolysis of this ester as in
Examele 5 gave (All-E)-9-[2-(nonyloxy)phenyl]-3,7-dimethyl-
:~2773~
- 28 -
2,4,6~8-nonatetraenoic acid.
~ xample 7
~ll-E2-3,7-DimethYl-9-r2-t~2 2-dimethYl-octYl)oxYlPhen
s 2,4,5,8-nonatetraenoic acid
2-Hydroxybenzaldehyde was condensed with
2,2-dimethyl-1-iodo octane to yield 7- (2,2-dimethyl
octyloxy) benzaldehyde which was reduced to 2-(2,2-dimethyl,
octyloxy)benzyl alcohol and ~hen converted to
10 ~2-(Z,2-dimethyloctyloxy~ehenyllmethyl]t~iphenylphosphonium
bromide a~ in Example 1. Condensatîon of this phosphonium
bromide with 7-formyl-3-methyl-2,4,6-octatrienoic acid ethyl
ester as described in Example 3 followed by hydrolysis, as
in Example 5, gave the (All-E)-3,7-Dimethyl-9-~2-t(2,2-
L5 dimathyl-octyl)oxy]phenylJ-2,4,5,8-non-atetraenoic acid mp
113-117 (from dichloromethane/hexane mixture).
Examele 8
(All-E?-3,7-Dimethyl-,9-[2-L~octylox~)-methYllDhenyll-Z,4L6,8-
nonatetraenoic a~id
Lithium octanoate, prepared from oc~anol and
~-butyllithium, in a mixture of tetrahydrofuran/hexane
dimethyl foemamide was condensed with 2-bromobenzyl bromide
to yield 2(octyloxy)methylbromobenzene. This material was
treated with n-but~llithium in ether/hexane mixtu~e and
subsequently treated with para~ormaldehyde to yield
2-(octyloxy)methylbenzyl alcohol. This material was then
treated wi~h ~riphenyl phosphonine bromide to yield
~[Z-[(octyloxy~-methylJphenyl~me~hyl3triphenyl pho~phonium
bromide. Co~dansation of this material with
7-formyl-3-methyl-2,4,6-octatrienoic acid ethyl estee, as in
Example 3, ~ollowed by hydrolysis, as in Example 5, gave the
(~ll-E)-3,7-Dimethyl-9-~2-~(octyloxy)-methyl~phenyl]-2,4,6,8-
nonatetraenoic acid, mp 120-121 (from dichloro
35 methane/hexane mixture).
:
;
3LZ~7~3~i~
- 29 -
Exam~le 9.
(All-E)-9-r2-chloro-6-(nQnyloxy)PhenY11-3,7-dimethyl-2,4,6,
8-nonatet~aenoic acid
_
2-chloro-6-hyd~oxy-benzaldehyde was alkylated with
l-b~omononane as in Example 1 to give 2-chloro-6-nonyloxy
benzaldehyde. Reduction with sodium borohydroxide as in
Example 1 gave 2-chloro-6-nonyloxy benzyl alcohol which on
treatment with triphenylphosphine hydrobcomide in
acetonitrile as in Example 1 yielded ~[2-chloro-6-nonyl-oxy]-
L0 phenyl]methyl~triphenyl phosphonium bromide. Condensationwith 7-formyl-3-methyl-2,4,6-octatrienoic acid ethyl ester
as described in Example 3 produced (All E)-9~~2-chloro-6-
(nonyloxy)phenyl]-3,7-dimethyl-2,4,6,8-nonatetraenoic acid
ethyl ester. The ester was subjected to hydrolysis, as
~s in Example 5, to ~roduce (All-E)-9-~2-chloro-6-~nonyloxy)~
phenyl]-3,7-dimethyl-2,4,6,8-nonatetraenoic acid mp
129-131 (from ethyl acetate~hexane mixtu~e).
'
Exam~le 10
(All-E)-9-(5-Methoxy~2-nonYloxyehenyl)-3~7-dimethvl-?~4~6
8-nonatetraenoic acid
5-Methoxy-2-hydroxybenzaldehyde was alkylated with
nonylbromide and reduced with sodium borohydride, as in
Example 1, to yield 5-methoxy-2-nonyloxy-benzyl alcohol
which on exposu~e to triphenylphosphine hydrobromide gave
~5-methoxy-2-nonyloxyphenyl)methyl]~riphenyl phosphonium
bromide. Condensation of this material with 7-formyl-3-
methyl-2,4,6-oc~atrienoic acid ethyl ester, as in Example 3
followed by hydrolysis, as in Example 5, gave (All-E)-
9-(5-Methoxy-2-nonyloxyphenyl)-3,7-dimethyl-2,4,6,8-nona-
tetraenoic acid mp 125-126 (from methanol).
ExamPle 11
All(E)-g-~2-(8-H~droxyoctYl)oxy]pheny~-3L7-dimethyl-2~4~6
8-nonatetcaenoic acid
The sodium æalt of All(E)-9-(2-hydroxyphenyl)-3,7-
dime~hyl-2,4,6,3-nonatetraenoic acid ethyl ester in
~t7733;~
- 30 -
dimethylformamide ~repared as described in Example 6 was
treated with l,8-dihydroxyoctane monotosylate as desceibed
previously in Example 6 and gave ~ll(E)-9-~2-~8-hydroxy-
o~tyl)oxy]phenyl-3,7-diethyl-2,4,6,8-nonatetraenoic acid
ethyl ester after chromatog~aphy over 6ilica gel.
Hydrolysis as in Example 5 yielded
All(E)-9-[2-[(8-hydroxy-octyl)oxy]phenyl]-3,7-dimethyl-
phenyl]-3,7-dimethyl-Z,4,6,8-nonatetraenoic acid, mp
122-123 (from ethyl acetate).
L0
ExamPle 12
~ll(E)-rS-(2-NonyloxyphenYl)-3-methyl-2,4-1?entadienYlltri-
PhenYlPhosphonium bromide
2-(nonyloxy)benzaldehyde (62 g) di6solved in acetone
(500 mL) was treated with aqueous sodium hydroxide (100 mL,
lM) at coom temperature for 18 h. Brine and ethyl
acetate/hexane (1:1 parts by volume) wa~ then added.
Concentration of the organic phase followed by cystalliza-
tion from ~exane gave 4-(2-nonyloxyphenyl)-3-~utene-2-one
(53 g).
A solution of 4-(2-nonyloxyphe~yl)-3-butene-2-one (58 g)
in tetrahydrofuran (200 mL) was added to a solution of
vinylmagnesium bromide in tetrahydrofuran (200 mL, 1.6M
diluted to lL with more tetrahydro~uran) at -30C. ~fter
com~lete addition, the mixture was stirred at 0 C for 30
min, quenched wi~h saturated aqueou~ ammonium chloride (100
mL) and ether (2L) and filtered free o~ solids.
Concentration of the organic extract and purifica~ion by
30 chromatography over silica gel yielded (E)-5-(2-nonyloxy-
phenyl)-3-hydro~y-3-methyl-1,4-pentadiene (40 g) as an oil.
~ solution of (E)-5-(2-nonyloxyphenyl)-3-hydroxy-3-
me~hyl-1,4-~entadiene (66 g) in acetonitrile (250 mL) was
35 added to a slurry of triphenylphosphine hydrobromide (66 g)
in more acetoni~rile (300 mL) at 10C. After warming to
room temperature, ~he mixture was stirred at ~his
33.V~,
- 31 -
temperatuee ~or 2h to yield a solution. This solution was
then extracted with hexane (2 x 250 mL) and the acetonitrile
layer was concentrated (ca. 400 mL) and cooled to -10.
The solids were ~iltered o~f, washed with acetonitrile,
hexane, and dried to give pure All(E)-[5~(2-nonyloxyphenyl)-
3-methyl-Z,4-pentadienyl]triphenylphosphonium bromide (21 g).
Exam~
L0 Starting with All(E)-[$-(2-nonyloxyphenyl)~3-methyl
2,4-pentadienyl]triphenylphosphonium bromide and utilizing
the proceduce of Examele 5, the ylide was reacted with
3-~ormyl-2-butenoic acid ethyl ester to yield ~ll(E)-9-(2-
nonyloxyphenyl)-3,7-dimethyl-2,4,6,8-nonatetraenoic acid
after purification by chromatography over Bilica gel and
hydrolysis with aqueous ethanolic potas6ium hydroxide
; solution as in Example 5.
Example 14
(Z,EI~E3-9-~2=(NonYloxy)phenyll-3,7-dimethyl-2,4,6~8-non-
~ ~ll(E)-9-t2-~nonyloxy)phenyl]-3,7-dimethyl 2,4,6,8-
;~ nonatetraenoic acid ethyl ester S10 g) was dissolved in
~b hexane (200 mL) containing iodine (0.5 g) and ctirred at
room temperature for 30 min. ~he he~ane was washed ~ree o~
iodine with an aqueous sodium thiosulfate solution ~10~ by
weight), dLied and concentra~ed to give a mixture of double
bond isomers. Separation, by chro~atography on silica gel,
yielded pure (Z~E,E,E)-9-~2-(nonyloxy)phenyl]-3,7-dimethyl-
30 2,4,6,8-nonatetraenoic acid ethyl ester (1.5 g~. Hydrolysis
with aqueous ethanolic potassium hydroxide at re~lux gave
the pure ~Z,E,~,E)-9-~2-(nonyloxy)phenyl]-3,7-
dimethyl-~,4,6,8-nonatetraenoic acid: mp 135-136C.
, .
~7733~
_ 32 -
ExamPle 15
(E,E,E,Z)-9-[2-(NonYloxY)PhenY11-3,7-dimethyl-2,4,6,8-nona-
tetraenoic acid
The mother liquor material resulting from the
crystallization of All(E)-9-~2--(nonyloxy)phenyl]-3,7-
dimethyl-2,4,6,8-nonatetraenoic acid ethyl ester in Example
5 was a mixture containing various isomers. Pu~ification by
chromatography yielded an 80% pure ethyl ester which after
hydroly6is as in Example 5 yielded pure
(E,E,E,Z)-9~2(nonyloxy)phenyl~-3,7-dimethyl-2,4,6,~-
nonatetraenoic acld: mp 105-109.
Example 16
2-DecYl-l-bromgben ene
Nonylmethyltriphenyl phosphonium bromide (0.1 mol) in
tetrahydrofuran (200 mL) was converted to the ylide with
n-Butyllithium (0.1 mol equiv: 1.6M in hexane) at -10C.
2-Bromobenzaldehyde (0.09 ~ol) was then added in
tetrahydLofuran (25 mL) and after the mixture had been
stirred for a further 30 min at 0C hexane and aqueous
me~hanol (40:60) was added. The hexane extract was
conce~tLa~ed and the re6idue was di~tilled to yield
2-(1-decenyl)-l~bromobenzene (90%).
This material was dissolved in hexane containing a
palladium on carbon catalyst (10%) and hydrogenated at eoom
temperature and pres ure until the olefinic link was
saturated. The ~olids wera filtered o~f and removal of the
hexane and distillation o~ the residue gave pure
2-decyl-1-b~omobenzene (80~): bp 120 (.001 mm Hg).
~3
?-Decvl-l-hydroxymethylbenzene
2-Decyl-L-bromobenzene (0.1 mol) dissolved in ether (lS0
mL) was treated with n-butyllithium (0.11 eq. 1.6M in
hexane) and the mixture was stirred at room temperature for
~Z~33 ~
- 33 -
2 hours.
Dry paraformaldehyde (0.2 mol eq) was then added and the
mixture was stir~ed for a further 18h at room temperature.
Water and move ether wa~ then added and the ether
extract~ were dried and concentrated. The residue after
chromatography yielded pure 2-decyl-1-hydroxy methyl benzene
(75% yield).
L0
Example 18
All(El-9-(DecYlPhenyl)-3,7-dimethy~-214~6~8-nonatetraenoic
acid
2-Decyl-l-hydroxymethylbenzene was converted to the
L5 ~hosphonium salt with triphenylphosphonium hydrobromide in
acetonitrile by the ~rocedure of Example 1. This salt was
then ex~osed to n-butyllithium in tetrahydrofuran as be~ore
and then treated with 7-formyl-3-methyl-2,4,6-he~ta-
trienoic acid methyl e~tar as be~ore.
Purification of the crude condensation product by
chromatography on silica gel ~ollowed by basic hydrolysi6
yielded pure All(E)-9-(decyl~henyl)-3,7-dimethyl-2,4,6,8-
nonatetraenoic acid: mp 107-108 tfrom hexane-ether).
Example L9
~ll(E~-9-L~octYlaminophenYl)-3,7-dimethvl-2,4,6,8-nona-
tetraenoic ac d ethvl ester
2-Aminobenzyl alcohol ~1 mol) was treated with
30 octanoylchloride (2.2 mol) in a mixture of
dichloromethane-triethylamine at 0C. After 30 min at
10C the mixture was washed with water and ~he ether was
distilled off. The crude residue was dissoIved in
tetrahydrofuran ~2000 ml), treated wi~h aqueous sodium
35 hydroxide (lN, 1500 mL) and stirred a~ room temperature for
3h.
31 2~7~3~
- 34 -
Addition of water and ether yielded the crude
hydroxymethyl octylamide. Purification by chromatography
yielded the pure octyamide (~5%).
This material (100 g~ was dis~olved in tetrahydrofuran
(500 mL) and added to a slurry of Lithium aluminum hydride
(2 mol equiv) in tetrahydrofuran ~1000 mL). Th~ mixture
was then heated at reflux for 8h cooled to 0 C and
quenched with aqueous sodium ~ulfate solution (100 mL).
~0
The solids were filtered off, the solvents were removed
in vacuo and the residue wa~ puri~ied by chromatography on
silica gel to yield ~ure 2-hydroxymethyl-N-octylamaline
; (75g)-
~5
This material was dis601ved in acetonitrile (300 mL)
containing triphenyl pho~2hine hydrobromide (1.1 eq) and the
mixture was heated at ceflux for 24h and then concentrated.
The residue was digested with ether to give the phosphonium
salt as a white solid.
This material was converted to the coLresponding ylide
with n-butyllithium (1.5 mol eq) and stirred at 0 for
lh. Excess 7-formyl-3-methyl-2,4,6-heptatrienoic acid
~ zs ethyl ester (1.6 mol eq) was then added in tetrahydrofuran
-~ and the mixture was stirred at ~0C for lh.
Addition of hexane and aqueou~ methanol (2:3) and
removal of the hexane in vacuo gave the crude coupled
0 product. Purification by chromatography on silica gel and
crystallization from hexane gave pure ALL(E)-9-(2-octyl
amino phenyl)-3,7-dimethyl-2.4,6,8-nonatetraenoic acid ethyl
ester (25%): mp 38-40C.
~2~7~3~
_ 35 -
Example 20
2-Fluoro-6-nonyloxybenz~l alcohol
A solution of 3-fluoro phenol (100 g) in dimethyl-
formamide (1000 mL) containing potassium carbona~e (165 g)
was treated with allyl bromide (lL5 g) and heated at 80
for 18 hours.
Water and hexane were then added and the hexane
extract was washed with aqueou~; sodium hydroxide solution
L0 (5%). saturated brine solution and concentrated to yield the
allyl ether (155 g). This material (134 g) was heated at
220 for 16 hours to yield a mixture of 3-fluoro-2-(2-
butenyl)phenol and 5 fluoro-2-(Z-butenyl)~henol. This
mixture was di~solved in dimethyl formamide (2000 mL)
containing l-bromononane (170 g) and ~otassium carbomate
(150 g) and heated at 80 for 16 hours. Dilution with
water and extraction with hexane yielded a mixture of
~roducts on concentration. Distillation gave a mixture of
3-~(2-fluoro-6-nonyloxy)phenyl]-butene and 3-~(3-fluoro-
2-nonyloxy)phenyl]-butene (186 g) bp. 120-125 @ O.lmm.
This mixture of isomers (185 g) in dimethyl~ulfoxide
(1000 mL) containing potassium tert-butoxide (1.5 g) was
left at room temperature for 6 hours. Addition of water and
extraction ~ith hexane gave the mixture of
1-~(2-fluoro-6-nonyloxy)phenyl]-butene and 1-~(3-fluoro-
2-nonyloxy)phenyl]butPne.
This mixture of isomers (175 g) was dissolved in a
mixture of dichloromethane and methanol (9:1, Z000 mL) and
exposed to a stream of ozone at -40 f or 8h. ~fter this
time the reaction mixture ~as pouLed into a mixture of
water, hexane and dimethylsulfide ~100 mL) and s~irrad at
room temperature for L hour.
The hexane extract was washed (water), dried rMgS04),
treated with more dimethyl sulfide (50 mL) and left at room
.
733~
- 36 -
temeerature for 16 hours. `
Removal of the solvents yielded the mixture of aldehydes
2-fluoro-6-nonyloxybenzaldehyde and 4-fluoro-2-
nonyloxy-benzaldehyde (155 g).
This mixture of aldehydes (150 g) in ethanol (2000 mL)
was exposed to sodium borohydride (15 g) at 5 and then
stirred at room temperature for 30 min. ~ater (1500 mL),
brine (500 mL) were then added and the mixture of alcohols
was extracted into hexane. Remo~al of the solvents and
chromatography of the residue over sili~a gel (5~
ethylacetate-hexane mixture) yielded pure Z-~luoro-6-
nonyloxy-benzyl alcohol (76 g).
Example 21
(~ll-E~-9-2-Fluoro-6-tnonyloxY~PhenYll-3,7=dimethyl-2,4,6
e~e~ id
A mixture of 2-fluoro-6-nonyloxyben2yl alcohol (19 g)
and triphenylphosphine hydrobromide~(26 ~) in acetonitrile
(250 mL) was heated at reflux for 14 hour6 and then
concentrated to drynes~ to yield ~[(2-~luoro-6-
~ nonyloxy)phenyl]methyl]triphenyl phosphonium bromide (42
:~ g). Thi~ phosphonium salt was dissolved in tetrahydrofuran
(600 mL) cooled to -50 and treated with n-butyllithium
(45 mL, 1.6M in hexane). After stirring a further 15 min at
-50 7-formyl-3-methyl-2,4,6-octatrienoic acid ethyl ester
(8.4 g) was added and the reaction mixture was warmed ~o
room temperature and stirred for a further 15 min. Hexane
~ 30 wa~ then added and the mixture wa~ washed with water, 40%
:~ aqueous methanol and dried (MgS04). Concentration of the
hexa~e extract and purification by chromatography (5%
ether-hexane) ga~e the pure trans i60mer (11 g).
Cry tallization from hexane-ethyl acetate gave
(A11-E)-9-~2-fluoro 6-(nonyloxy)-phenyl]-3,7-dimethyl-204,6,8-
.
~.
733;2
- 37 -
nonate~raenoic acid ethyl ester (9.5 g).
A solution of the ester ~6.5 g) in ethanol (150 mL) was
treated with a solution of potassium hydroxide (7 g) in
water (40 mL) and heated at reflux for 1 hour. ~he cooled
reaction mixture was ~oured into cold aqueous hydrochloric
acid and the acid was extracted into chloroform. Removal of
the solvents and crystallization from hexane-ethyl acetate
gave pure (all-~)-9-~2-fluoro-6-(nonyloxy)phenyl]-3,7-
dimeth~l-2,4,6,8-nonatetraenoic acid mp 107-109.
:
: 25
`
~ 27733;~
- 38 -
Exam~le 2 ?
CAPSULE FORMUL~TIONS:
5 Item Inqredients mq/caPsule ~g~3p~ule mq/capsule
1. (All E)-9- 15 30 60
[2-(nonyloxy)
~henyl]-3,7-
dimethyl-2,4~6,
8-nonatetraenoic
acid
~5 2. Lactose 239 224 194
3. Starch 30 30 30
.
~. . .
4. Talc 15 15 15
~ 20
-~ 5. Magnesium
____ _____ _____
Capsule fill weight 300mg 300 mg 300 ~g
Z5
PROCEDURE:
1) Mix items 1-3 in a suitable mixer.
2) Add talc and magnesium stearate and mix for a short
period of time.
3~ Encapsulate on an appropria~e encaesulation machine.
~D ~ ~
~.~77~3~
- 39 ~
ExamPle 23
Capsules are prepa~ed by the procedure of Example
22 except that the active ingredient (item 1) wa~ (All
E)-9-[2-fluoro-6[nonyloxy)~henyl]-3,7-d}methyl-3,4,6,8-nona-
tetraenoic acid.
ExamPle 24
~o TABLET FORMULATION (Wet geanulation)
Item Inaredients mq~tablet mq~tablet mq~_ablet
: 1. (All E)-9- 100 250 500
L5 [2-(nonylo~y)
phenyl]-3,7-
dimethyl-20g,
6,8-nonatetrae-
noic acid
20 2. Lactose 98.5 147.5 170
3. Polyvinyl 15 30 40
pyrrolidone (PVP)
4. Modified sta~ch 15 30 40
5. Corn starch 15 30 40
25 6. Magnesium 1.5 2.5 5
: ~ stearate
Weight of tablet 245 mg 490 mg 7g5 mg
: 30
.,~
: 3s
'~
, .
:. ,
~ ~773~`~
-- ~o --
Procedure:
1. Mix i~ems 1, 2. 4 and 5 in a suitable mixer, granulate
with PVP and dissolve in water/alcohol. Dry the
gra~ulation. Mill the dry granulation through a
suitable mill.
2. Add magnesium stearate and compre66 on a suitable ~ress.
ExamPle 25
~0
Tablet are prepared in the same manner as Exam~le 24
except that the active inyredient (item 1) was (~11
E)-9-[2-fluoro-6(nonyloxy)~henyl]-3,7-dimethyl-2,4,6,8-nona-
tetraenoic acid.
Exam~le 26
T~BLET FORMULATIONS: (DiLect Compre~sion)
20 Item Ingredientmq~tablet mg~ mq/tablet
1. ~All E)-9- 15 30 60
~2-(nonyloyl~
phenyl]-3,7-
dimethyl-2,4,
6,8-nonate~rae-
noic acid
2. Lactose 207 192 162
3. Avicel~ 45 45 45
4. Direct 30 30 30
Compression StaLch
5. Magnesium 3 3 3
; 35 ---- -___ ____
~eight o~ tablet 300 mg300 mg 300 mg
* trade mark.
~'73~
- 41 -
PROCEDURE:
1. Mix Item 1 with equal amount oP lacto~e. Mix well.
2. Mix with Item 3, 4, and remaining amount of Item 2. Mix
well.
3. Add magnesium stearate and mix for 3 minutes.
4. Compre6s on a 6uitable punch.
Exam~
LO CAPSULE FORMUL~TION8~
Item Inqrsdients m~/caPsule m~/capsule mq/caP~ule
1. (~11 E)-(3,7- 15 30 60
~s dimethyl)-9-
~2~(8-hydroxy-
octyl)oxy]phenyl]-
Z,4,6,8-nonatetraenoic]
acid.
20 2. Lactose Z39 224 194
3. Starch 30 30 30
4. Talc 15 15 15
5. Magnesium
,
25 Capsule fill weight 300 mg 300 mg 300 mg
PROCEDU~E:
1) Mix items 1-3 in a suitable mixer.
30 2) Add talc and magnesium stearate and mix for a short
period of time.
3) Encapsulate on an appropriate encapsulation machine.
~1 27733~
- 42 -
Example 28
TABLET FORMULATIONS (Wet granulation)
Item Inqredient mq/tablet ma/tablet ma/tablet
1. (All E)-3,7- 100 250 500
~ - dimethyl-9-
- [2,~(octyloxy)
methyl~phenyl]-
2,4,6,8-nona-
tetraenoic acid
2. Lactose 98.5 147.5 liO
3. Polyvinyl 15 30 40
L5 pyrrolidone
4. Modified starch 15 30 40
5. Co~n starch 15 30 40
6. Magnesium 1.5 2.5 5
- stearate
~~~~~ ~~~~-~ ~~~~~
Weight of tablet 245 mg490 mg 795 mg
Procedure:
1. Mix items 1, 2, 4 and 5 in a suitable mixer, granulate
with PVP and dissolve in water/alcohol. Dry the
granulation. Mill the dry granulation th~ough a
suitable mill.
2. Add magnesium stearate and compress on a suitable press.
PhosPhonium bromide
mixture of a,~.~-tri~luoro-m-cresol (51 g)~
l-bromononane ~70 g), potassium carbonate (100 g) in
dimethylformamide was heated a~ 85C for 48 h. Addition
of water and hexane gave pure (3-trifluoromethyl) phenyl
.
~Z7733.~
- 43 -
nonyl ether (89 g): b.p. lL5C at 0.1 mmHg. This product
(89 g) in ether (1.5 L) at -20 C was mixed with
n-butyllithium (1.5 M in hexane: 233 mL) and then sti~red
for 2h at room temperature. This mixture was then cooled to
-40, treated with an excess o~ dry dimethyl~ormamide (40
m~) in ether (100 mL), warmed to 0 and then treated with
water. Extraction with hexane and chromatography on si.lica
(5% ether-hexane) gave (2-trifluoromethyl-6-nonyloxy)-
benxaldehyde (35 g). Reduction of this ~roduct with
L0 ~odiumborohydride i~ ethanol by the pcocedure set ~orth in
Example 1 gave (2-trifluoromethyl-6-nonyloxy)benzenemethanol
(32 g) after cheomatography over 8ilic~. This material (31
g) was converted into Ct2-trifluoromethyl-6(nonyloxy)phenyl]-
methyl]triphenyl~hosphoniu~ beomide by reaction with
triphenylphosphine hydrobromide by the procedure given in
Example 1.
Example 30
(All-E)-9-L~=(Tri~luoromethyl)-6-(nonYIoxy)ph ~ ll-3~7
dimethyl-2,4,6,8-nonatetraenoic acid
~ 2-trifluoromethyl 6-(nonyloxy)phenyl]methyl]triphanyl-
phosphonium bromide (97 mmol) in tetrahydro~uran (600 mL)
wa~ converted by ~eaction with 7-formyl-3-methyl-2,4,6-
octatrienoic acid ethyl ester to (All-E)-9-~2-(trifluoro-
methyl)-6-(nonyloxy)phenyl~-3,7-dimethyl-2,4,6,8-nona-
tetraenoic acid e~hyl ester by the procedure given in
Example 3. Purification by chromatography and
crystallization from hexane gave the pure ethyl ester
(41%). Hydrolysis (5.2 g) a~ in example 5 gave eure
(All-E~-9-~2-(t~ifluoromethyl)-6-(nonyloxy)phenyl]-3~7-
dimethyl-2,4,6,8-nonatetraenoic acid (3 g): mp 135-136
(from ethyl acetate hexane).
Example 31
(All-E~-9-~2=~hexyloxY~henyll-3~7-dimeth~1-2~4~6~8-nona-
tetra no acid
t[2-(hexyloxy)phenyl]methyl]triphenylphos~honium
~L2773~
- 4~ -
bromide, prepared by the procedure of Example 1 by reacting
2-hydroxybenzaldehyde and l-bromohexane, was converted to
(All-E)-9-~2-(hexyloxy)phenyl]-3,7-diethyl-2,4,6,3-nona-
tetraenoic acid, m~ 137-138 (fLom ethanol) by the
procedure of Exam~le 3.
Exam~
r ~ 2-(Non~loxY)-5-(h~droxv)Dheny-llmethylltriphenvlphosphonium
bromide
A solution of 4-bromophenol (1 mol) in tetrahydro~ura~
(500 mL) was added to a Rlurry of sodium hydride (1.17 mol)
in dimethylformamide (1.2 L) at 25 C. ~fter complete
~eaction allylchloride (1.32 mol) wa~ added and after
stirring for a ~uLther 3 h at 45 the product was isolated
L5 with water and hexane. Di~tillation gave allyl-(4-bromo-
phenyl)ether. bp. 65-67 at 0.1 mm (82%). This material
was heated at 195 with dimethylanaline for 4 hours and
~hen di tilled to yield 2-allyl-4-bromophenol (0.81 mol). A
solution of this material (0.81 mol) in tetrahydrofuran (200
mL) was added to a mixture of l-bromononane (0.8 mol),
sodium hydride (0.92 mol) potassium iodide (1 g) in
dimethylformamide (lL) at 2SC. After hydrogen evaluation
was complete the mixture was heated at 50 for 14 h,
cooled, added to an exces~ of water and extracted with
hexane. Distillation furnished nonyl-(2-allyl-4-bromo-
phenyl)ether (256 g): b.p. 147-156 at 0.1 mm. This
material (255 g) in dimethylsulfoxide (1 L) and
tetrahydrofuran (0.5 L) was heated at 35-40 with
~otassium tert. bu~oxide (2 g) for 2 h and then quenched
30 with acetic acid (5 mL) and water. Isolation o~ the
reaction products with hexane yielded eure
l-r2-(nonyloxy)-5-(bromo)~henyl]eropene (234 g~: b.p.
145-155 at 0.1 mm. ~ solution of the above material
(0.56 mol~ in tetrahydrofuran (600 mL) was converted to the
35 Grignard reagent wi~h magnesium (1 mol) at 55C for 3 h.
After complete reaction ~he mixture was cooled to 0C and
~reated with trimethylborate (0.75 mol) in ether (200 mL).
~ ;~t7~73~
- 45 -
Af~er stirring for a further 30 min. at 25 C the mixture
was cooled to 0 and exposed to a mixture of ammonium
chloride (10%) and hydro~en peroxide (10%, 500 mL) and
stirred ~or a further lh at 25 C. Addition of water and
hexane gave the crude material after removal of the hexane
in vacuo. The crude product was passed through a plug of
silica gel to yield para [2-(1-propenyl 4-(nonyloxy)-
phenyl]phenol (73 g). Acetylation of this material (0.8 g)
with acetylchloride and trieth~ylamine in dichloromethane
lo gave the [2-(l-proeenyl)-4-(nol~yloxy)-l-(acetoxy)]benzene
(89%). This material (99 g) was dissolved in a mixture of
methanol (150 mL) and dichloromethane (1.5 L) and treated
with ozone at -40C until all the starting material had
been consumed. Dimethylsulfide (50 mL) and water (500 mL)
~5 were then added and after vigorous stirring for 30 min. a~
the organic phase was dried (MgS04~ and concentrated
to yield [2-(nonyloxy)-5-(acetoxy)~benxaldehyde (83 g).
Reduction of this material (80 g) with sodium borohydride (6
g) in ethanol (~Lj at 20 C for 2 h gave the crude
~2~(nonylo~y)-5-(acetoxy)]benzanemethanol which was
immediately exposed to aqueous potassium hydroxide (300 mL,
40%) in ethanol (lL) for 30 min at 60C. Acidification
with aqueous acid (6 Molar hydrogen chloride) and extraction
with chloroform yielded the crude p~oduct on concentration.
25 Digestion of the residue with hexane gave ~ure
~3-(hydroxymethyl)-4-~onyloxy)]phenol ~63 g) as a solid. A
solution of this matecial (62 g) in a mixture of
acetonitrile (0.5 L) and trihenylphosphine hydrobromide (86
g) was heated at reflux f OL L4 h and concen~rated to dryness
30 at 50C ts yield [[2-(nonylsxy)-5~h~droxy)phenyl]methyl]-
triphenylphosphonium bromide as a glass.
ExamPle 33
(~ll-E)-9-~S-Hydroxv-2-(nonYloxy)phenyl]-3,7-dimethYl-2,4,6,8-
35 nsnatetraensic acid
The ~2 ~nonyloxy)-5-~hydroxy)ehenyl]methyl~triphenyl-
phosphonium bromide (0.23 mol) in tetrahydrsfuran (1.5 L) at
33~
- ~6 -
-70C was treated with n-butyllithium ~1.6M in hexane: 315
mL) and then treated with ethyl 8-formyl-3,7-dimethyl-2,4,6-
oc~atrienoate (59 g) in tetrahydrofuran-. The mixture was
then warmed to -15C acidified with acetic acid and
extracted into ether and aqueous methanol (40%).
Purificatio~ by chromatography over silica gel gave pure
(All-E)-9-[5-hydroxy-2(nonyloxy)phenyl]-3,7-dimethyl-
2,4,6,8-nonatetraenoic acid ethyl ester. Hydrolysis of this
ester (6 g) by the procadure given in Example S gave
L0 (All-E)-9-~5-hydroxy-2(nonyloxy)phenyl]-3,7-dimethyl-2,4,6,8-
nonatetraenoic acid (3.5 g): mp 170-173 (from
ethylacetate).
Example 34
~All-E)-9-[2-(nonYloxY)-5-(2,2,2-triflUoroethoxy)phenvll-3~7
dimethYl-2 4,6,8-nona~etraenoic acid
(All-E~-9-~5-hydroxy-2-(nonyloxy)phenyl]-3,7-dimethyl-2,4,
6,8-nonatetraenoic acid ethyl ester (4.4 g) was heated at
90 C for 72 h with eota~sium carbonate (7 g),
2,2,2-tri~luoroethyl-p-toluensulphonate (6 g) in
dimethylformamide (Z00 mL~. ~ork up with water and hexane
followed by purification over silica gave the pure ethyl
ester (0.75 g). Hydroly~is o~ this ester ~0.9 g) by ~he
procedure of Example 5 gave pure (All-E)-9~2-(nonyloxy)-
5(2,~,2-trifluoroethoxy)phenyl]-3,7-dimethyl-2,4,6,8-nonatetra
enoic acid (0.6 g) after crystalli~ation from a mixture o~
tetrahydro~uran and hexane: mp 121C.
Ex ple 35
(z2-~[2-tl-Decenyl)phenyllmethyll-triphenylphosp4onium
bromide and ~E)-~r2(1-Decenyl)PhenYllmethylltriphenY
phosPhonium bromide
An (E,Z) mixture of 2-(1-decenyl)-1-bromobenzene as
prepared in Example 16 (1:4) was con~erted to a (E,Z)
35 mixture of 2-(1-decenyl)-1-hydroxymethyl benzene by the
: procedure of Example 17. This mixture was separated by
chromatography on silica gel to yield ths pure (E) and (Z)
~27'7332
- 47 _
alcohols. Reaction of each of these isomers with
triphenylphosphine hydrobcomide as in exam~le 1 gave the
corresponding phosphonium salts i.e. (Z)~2-(1-decenyl)-
~henyl]methyl]-triphenylphosphonium bromide and
(E~2-(1-decenyl)phenyl]methyl]triphenylphosphonium bromide.
ExamPle 36
tAll-E)-9-~2-(1-DecenVl~PhenYl.1-3.7-dimethYl-2,4,6,,8-nona-
tetraenoic acid
L0 The (E)-~2-(1-decenyl)phenyl]methyl]-triphenyl-
phosphonium bromide was converted into the ethyl ester of
(All E)-9-[2-(decenyl)phenyl]-3,7-dimethyl-2,4,6,8-nona-
tetraenoic acid by the procedure given in Example 1.
Hydrolysis with base as in Exam~le 1 and crystallization of
the crude acid ~rom acetonitrile gave tAll-E)-9-[2-tl-
decenyl)phenyl]-3,7- dimethyl-2,4,6,8-nonatetraenoic acid,
mp 10S-107.
ExamPle 37
(E~E~E~E~)-9-c2-(l-pecenyl)phenyl]-3~7-dimethvl-2L~ 8
nonatetraenoic acid
The title com~ound was pre~ared in the same manner as in
Example 36 employing the (Z)-C[2-~l-decenyl)phenyl~methyl~-
; triphenyl phosphonium bromide. Hydrolysis of the ethyl
- zs ester and crystallization of the crude acid from ether
yielded ~ure (E,E,E,E,Z~-9-~2-(1-decenyl)ehenyl]-3,7
~; ~ dimethyl-2,4,6,8-nonatetLaenoic acid, mp 103-105.
In the following examples, Compound A is All(E)-9-
[2(nonyloxy)phenyl]-3,7-dimethyl-2,4,6,8-nonatetraenoic
acid. In the following examples, Compound ~ was tested by
various ~ests for its anti-inflammatory activi~y in animal
models of inflammation and in certain chronic models foc
adjuvant arthritis.
In all tests, Compound A and the other retinoids tested
concurrently were formulated in arachis oil containing 0.05%
`- ~.27~
- 48 -
propylgallate as anti-oxidant. The dose volumes used were 5
ml.kg for rats and 10 ml.kg for mice. Controls were
dosed with the appropriate volume of arachis oil vehicle.
ExamP le 38
Effect of ComPound A on delaYed hYpersensitivity to
methYlated bovine serum albumin (MBSA)
LO Animals Male and femall~ MFI mice substrain E33.
Initial weight approximately 25 gm.
Materials Methylated bovine seeum albumin (MBSA)
(Sigma) Freunds comelete adjuvant (Difco)
L5
Method Grou~s of 10 mice were sensitized (day 0) by
injecting intradermally a~ two abdominal.
sites 0.05 ml. of a water i~ oil emulsion
of MBSA and Freunds complete adjuvant. On
day 9 the mice were challenged by injecting
20 1 of a 1~ MBS~ solution to one pa~ and
20 1 of water into the contralateral paw.
Paw volumes were measured 24 hours later by
mercury displacement plethy~mography. The
mean percentage increase in eaw volume of the
MBSA-challenged paw compared with the water
challenged paw was calculated for each
treatmen~ group. Dosing with vehicle a~d
~etinoid commenced on day O and finished on
day 9.
Results The result~ are given in the following table
(Table II~
.
.
~m33z
- 49
Table II The effects of Compound ~ in the M~3S~
delaYed hYpersensitivit~ test
Treatment Dose % increase %reduction Mean body
mg.kg paw volume ~cf. arachis weight change
~ oil control~ (q)
Arachis Oil lO9 + 11 M 3.8
LO E' 0.2
E~retinate lO 59 + 9** 46 M -0.8
F -2.0
Compound A lO 102 + lZ 6 M 3.3
F -0.2
15 COmpound A 30 50 + 7*** 5 M 2.8
0.~
Com~ound A lOO 40 ~ 5*** 63 M 3.3
- F -0.2
20 . _ _ _
:~;
Each group consisted of 4 male and 6 female mice (separately
~ caged). Drugs were dosed orally at a dose volume of 10
:~ z~ ml.kg l tlO doses).
ns. Not significant **p < O.Ol ***p <0.001 com~ared
with ~ehicle control u6ing Student's two-tailed t ~est.
.
- 50 -
Example 39
Effect of ComPond A on developinq ad3uvant arthritis in ~he
rat.
Animals AHH/R Female rats (PVG derived with an
initial weight range of 110 to 1~0 g.
were used.
Materials Adjuvant for injection. An homogenized
suspension of hea~ killed M. tuberculosis
L0 (Human strains C, DT and PN), 5 mg.M1 L
in liquid paraffin wa~ prepared.
Method Rats were randomly 8plit into groups of
five and adjuvant arthritis was induced
L5 by the sub-plantar injection of 0.1 ml of
adjuvant suspension into the right hind
paw of each rat. Tes~ compounds were
administered by intubation each morning
commencing the day of adjuvant
injection. Two group~ of control rats
WeLe dosed with the vehicle as was a
group of three normal rats included for
comparative purposes. Dosing wa~ carried
out daily until the end of the te~t on
day 15 exce~t foL the fir~t weekend (day~
S ~ 6). Tre~ment groups are shown in
Table III and include etretinate as
~tandard re~inoid.
~;2 77;~2
- 51 ~
Measurements of right hind paw volume
were made initially and on days 2 and 4
after and adjufant injection (pLimary
phase). Right and left hind paw volume
were then measured. on day 8 and every
two or three days until the end of the
experiment on day 15 (secondary ~hase).
At this time the mobility of each ankle
joint and the
incidence and severity of seaondary
lesions on nose, ears, ~orepaws, left
hind paw and tail were also a6sessed in
terms of degrees o~ flexion ~ossible and
L5 by using an arbitrary scoring system,
respectively.
Asse6smen~ of refiults
The time course curves for the injec~ed
paws were in~egrated from days 0 to 4 to
reflect primary swelling and ~rom days 8
to 15 (secondary swelling). The secondary
swelling in the non-injected paw was
; 25 integrated similarly f rom days 8 to 15
Calculations were carried out using a
specific computer program which computed
mean + se for each lntegrated area. The
significance of differences from controls
was determined by Student's t tast (2
ta;led) and percentage reductions from
control areas were calculated.
Percen~age improvements in joint mobility
and percentage redu~tions in lesion score
were also determined. In the lattar case
the Wilcoxo~ rank ~um test (2 tailed) was
used ~o express the difference ~rom the
.
- 52 -
control score using raw data. Mean body
weiyht change in each group was recoeded.
Results The results are given in the following
table (Tablle III)
~ LO
: ~5
,
~ 25
; 30
:~ :
~ 35
: ~ :
.
12~332
-- 53 --
5 ~ ~
~ ~`' l l ~0 ~D O
~ . O ~ ~ ~ ------ b
~ 5 H . 5 = ~ _ __ u~ _ ~
. 3 ~0 . . _ ~O N ~
2 0 L ~ ~-- . * . O
Cl O O ~ l l t ~ ~1 1~ L V
`' i~ OU~- _ _ _ _ ~0~
~ ~ ~ ~~O ~D ~ ~ O ~
2 5 ~ . : G ~1 _ _ _ ~ D O
o O .. . _ ~.'
~:1 .' o!C ___ r~ ~ r/ 1: ~ -
3 0 ' ~--L~ '1:1 -- ~ ~ ~'
~ ,~V ~ E _ v v
~L2~'733~
- 54 -
Example 40
E~fect of Compound A on established t~Pe II collaqen
arthritis
Animals Male and female Alderley Park Strain 1
rats.
Materials Type 2 collagen (prepared ~rom bovine
nasal septum cartilage), Freunds
incomplete adjuvant ~Di~co).
Method Rats were sen6itized to type 2 collagen by
injecting them intradermally with 1 ml.
lS of a water in oil emulsion consisting of
equal parts of a 1 mg.ml~l solution of
type 2 collagen in 0.45M NaCl, 0.02M
Tris, pH 7.4 and Freunds incomplete
adjuvant. ~ats developing arthritis were
allocated on day 15 post sensitization to
a control arachis oil treated group (6
male, 4 female~ or to the Compound A
treated group (6 male, 5 female). Hind
paw volume measurement6 were ~aken to
ensure even dis~ribu~ion of rats between
groups. An overnight collection of urine
was made on days 15/16 and dosing
commenced on day 16. These urine sam~les
were analyzed for glycosaminoglycans
(GAG3. Compound A was dosed at 100
mg.kg 1 p.O. On days 19/20 a second
overnight collection of urine was made.
These urine samples were analyzed ~or
glycosaminoglycans (GAG). on day 20 a
second hind paw measurement was taken.
Rats were then anaesthetised with sodium
' ' .
1~77~32
~ 55 -
: pentobarbitone, bled, killed and X-ray~
; taken o~ hind and forepaws. Rats were
do~ed on days 16-19 inclu6ive (4 do~e~).
;
5 Results The results are ~iven in the following table
tTable IV).
L0
:
; L5
',
~ ~ :
` ~: :
~ 35 ~ ~
.
~: ' -' :-'` ' ' '
. ' . - ' .
~.27733%
- 56 -
+ I + I
~: S 3 ~ ~
~ ~ ~1 +1 0
L 0 h
~, : ~ +1 ~
:. v 3
L5 hc
~ ~ o
G
0 3
ZO _, U,~ ~ _ o ~ o
~ O
C
~ 6
Z5
0 3 ~
4.~ ~ ~ 6
O
u ~ ~9 o
~ 4 Ei
O
`: : 35
~: :
:, .
.
3L~Z~733~:
- 57 -
Example 41
Effect of Compound A on non-immune in1_mmation
Animals Female Alderley Park Strain 1 rats
weighing 170-205 g. at the start of the
experiment were used.
Materials Lambda -carrageenan. Pre~ared as a
solution in saline and sterilized by
LO autoclaving.
Method Compound ~ was administered orally to
grou~s of 8 rats once daily for 10 day~
at doses o~ 10, 30 and 100 mg.kg 1.
lS Control animals received the vehicle.
one hour after the last dose the animals
wece anesthetised with methohexitone
(Brietal, 50 mg.kg ~) and 0.2 ml of 1%
}ambda carrageenan was injected into the
~leural cavity. Four hours later the
animals were killed with an overdose of
pentobarbitone (Sagatal), the pleural
exudate was collected and the pleural
cavity was washed out with 2 ml o~
phosphate-buffered saline (PBS-A, Oxoid~.
The exuda~0 volume was recorded and cell
count were detarmined using an automatic
cell counter (Coulter). ~i~ferential
cell coun~s were ~erformed on exudate
0 smear~ stained with Giem6a stain in order
to determine separately the numbers o~
polymor~honucIear leucocytes (PMN) and
mononuclear cells (M~).
Immedia~ely aftsr recovery of the pleural
exudates the tibiae of the animals wece
~27733~
- 58 -
excised and theiL breaking strains were
determined.
The body weights of the animals were
recorded daily. Stati~tical analyses
were perfor~med using Student's two-tailed
t te~t.
10 Resul~s The results are given in the following
table (Tablle V). In the following table
: ~he dose iB in mg ~er kg ~er day.
:: ~5
.
' .
~ :
2~
:: 30
, ~
.
~ 35
'
:;
~ .
: :
~ : ' ,
: .
Z7
1 73~:
59
~_ ~ s l o
m ~ ~ . . .
r~ U~ ~ ~n ~n u
:~ 0 ~ ~ ^ ~Z; -l Z ~ Z ~
_ ~ ~ r O, O, O, O
~ 1~ _ ~ _ ~ 1 ~O I ~ ~ ~1
': _ ~ r~ ~ ~
L0 ~ ~ ~ ~ NO) Z 1:~ r~ ~ _~ ~1
,- c O ,,, O O 'r O~ ~ O ul ~1
E~ ~ _1 I I a: +1 r~ ~1 r~ ~1 O
_ . _ ~ . 3
',' l ~ ~ 0 l ~ C
`:: 1.5 ~, ' ~0 c~ U . .~, ~d 01
I e~ c~ ~ Z~ r ~ ~o ~ .C
~ -'~1 I -'+1 +l
`~ ~ O : _ _ __ '
~' ,CI g . ~11 1~ ~:
20 ~ ~ ~u c ~ u ~ ' = ~
:: O : O X 11 Z
C E~ ~ ~ -~ ~ r~ ~ O ~ r1
~ : N + I N ~ ~ _1 + ¦ O ~ U
__ _ ~ S
~: ` S ~1 C~ 0 0~ N _~
o b3 3~ ;: la ~ II ~D t-~
~ ~:; ~ ~ ~0 0~~ 00 O ;~
. : :
. ~ ~ d ~1 tJI ~ ~1 O
~: : : Cl 0 ~It~ O E o V l I
~ 8~ : ~ ~; ~o u
: ~
: : ~ : :
; -~ ~o,~ ¢~C: ~:: ~
: ~ ~ ~ o ~ ~ C:
: : U ~ 3 ' ~ : a. :
3 5 ~ ~ ~ o U, o: u z
: .:
, ` ~ ~ : ,: :
~ ~ .
"~
`:-.;, , ` . ,
i :, : .
.~
:
~: ~ " .:
`: : . . `
~::
~27733~
- 60 -
Example 42
Effect of ComPound A on the impreqnate,d sPonqe qranuloma
test in_the rat.
5 Animals ~HH/R female rats (PVG derived) with an
initial weight range of 120-140 g. were
used.
Materials Sponge prep,aration. Pelle~s (6.5 mm
lo diameter) w~ere punched from cellulose
sponge cloth ("~ettex") and 0.1 ml. of a
suspension containi~g 0.5 mg.ml~l of
heat killed M. tuberculosis (human
strains C, DT and PN) in ~terile saline
L5 wa~ applied to each pellet. The eellets
were dried, weighed and autoclaved.
-Method Rats were randomly divided into groups of
` ' five and daily dosinq with test compounds
zo was commenced. After the fifth dose the
rats were anaesthetised with Sagatal (45
mg. kg 1 i . p . ) the back~ were shaved and
two pellets weLe implanted subcutaneously
(one each side) into each rat through a
small dor~al midline incision. The
incision was closed and the rats allowed
to recover from the anaesthetic.
Seven days after implantation the rats
0 were killed and the pellets were removed,
; dissected free of extraneous tissue and
weighed. Each pellet wa~ then placed in
a 4 ml aliquot of distilled water,
chopped with fine scissors and
sonicated. ~fter centrifugation ~he
Na and K content of the supeLnatant
:~2~7332
- 61 -
was determined by flame photometry. In
addition the adrenal and thumus glands
from each rat were dis6ected out and
weighed and the lower hind limbs were
removed for measurement o~ tibial bone
breaking strain. Body weights we~e also
recorded throughout the teæt period~
Results The re~ultE, are given in the ~ollowing
~able (Table VI).
As essment of results
The mean + S~ for each of the parameters was calculated and
L5 differences from the control values were determined by
Student's t test (2-tailed). Percentage reductions of
granuloma weight, Na and K~ content and percentage
changes of adrenal and thymus, weight and tibial breaking
strain were de~ermined.
~ .
.
~ '
.
lZ~7~
v r~ O ~ ~1 + I O
~S ~ c 1 ~ ~ ~
0 3 ~ ~
OO~ a~ ~ ~ ~ .
30 ~ o,~ '' '' '' ` :
~V ~o j~ V
:
~7733Z
- 63 -
~nimals Male Lewis rats from Charle~ River were
used for these experiments.
Materials Heat-killed, dessicated Mycobacteriam
butvricum.
Method Adjuvant art:hritis was induced by the
injection of 0.1 ml of adjuvant ~a
su6pension of hea~-killed, dessicated
MYcobacterillm but~ricum, 0.5% CW/V) in
heavy mineral oil containing 0.2~
digitonin] into the base of the tail.
The arthritis wag allowed to develop for
21 days and then the volume of both hind
t5 paws were measured using a mercury
plethysmograph. The rats were divided
: into groups of 8 with equal mean paw
volumes and then the rats were treated
with Compound A, indomethacin (as a
: 20 control drug), or vehicle for 7 days at
: the end of the treatment period, the
: volumes of both hind paws were again
measured to assess antii~flammatory
effect~. Body weight changes were also
followed and, at the end of the
ex~eriment, ela6ma wa6 collected for
determina~ion of plasma fibrinogen ~Exner
et. al., Amer. J. Clin. Path, 71:
521-527).
Results: The results are given in the following
Table (Table VII).
. .
~ ,
-- 64 --
10 p
a
O O O
20 ~ ~ 3 ~
2 5 3 c ~ u ~ v
~ ~-1 q~ ~ 3 O ~ 3 ~ O L, ~
~ 30 ~ ô
- o ~ o u 3 g Z '-'
~: ~ .¢ ~ v