Note: Descriptions are shown in the official language in which they were submitted.
~287~44
2~205-449
-- 1
BICYCLIC COMPOI~NDS, THEIR PRODUCTION AND USE
This invention relates to novel bicyclic compounds,
which are useful as pharmaceuticals, and a process ~or
producing the same.
As compounds having hypotensive effect due to
s inhibitory activity to angiotensin converting enzyme, there
are known various amino acid derivatives [e.g. Japanese
Patent Vnexamined Publication (Kokai) Nos. 77-116457,
77-136117, 79-12372, ~0-3~382 and 80-81~45, which corre-
spond to USP-4105776, USP-4053651, ~ritish Unexamined Pub.
No, 2~D5D~, European Unexaminad Pub. Nos. 9183 and 12401,
respectivelyl The compounds of the present invenkion are
different from these known compounds in skeletal structure,
and moreover have superior angiotensin converting enzyme
inhibitory and hypotensive activities.
Thus, the present invention provides novel bicyclic
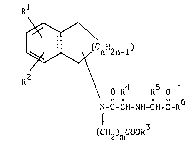
compounds represented by the formula:
Rl
~/\(C
2 0 2~\~< O R'~ t~5 O ~ I )
t~ N-C-C~I-N~I-C~I-C-R
~CH2 ) mCoOR3
wherein Rl and R2 are the same or different and each repre-
sents hydrogen, hydroxyl or lower alkoxy; ~3 is hydrogen, benzyl or
lower alkyl; R4 is hydrogen, lower alkyl, amino-lower-alkyl
r~1
1~8~444
- 2 - 24205-449
or acylamino-lower-alkyl; R5 i9 hydrogen, lower alkyl or
aralkyl which may be substituted; R6 is hydroxyl, lower
alkoxy, benzyloxy, amino or lower alkylamino; and _ and n
each means 1 or 2,
and pharmaceutically acceptable salts thereof.
Referring to the above formula (I), the lower alkoxy
group represented by Rl or R2 includes those containing
about 1-4 carbon atoms, such as methoxy, ethoxy, propoxy,
butoxy and isopropoxy. R and R I when they are adjacent,
may form a lower ~Cl 4) alkylenedioxy, such as methylene-
dioxy or ethylenedioxy.
The lower alkyl group represented by R3 includes
those containing about 1-4 carbon atoms, such as methyl,
ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl and
tert-butyl.
The lower alkyl group represented by R4 includes
lower alkyl ~roups similar to those represt~nted by R3.
The amino-lower-alkyl group represented by R4 includes
straight or branched ones containing about 1-4 carbon
atoms, such as aminomethyl, l-aminoethyl, 2-aminoethyl, 3-
aminopropyl, 4-aminobutyl and 3-amino-2-methylpropyl. The
acylamino-lower-al~yl group represented by R4 includes
those groups in which the amino group oE the above-
mentioned amino-lower-alkyl group is acylated with a
carboxylic acid- or carbonate ester-derived acyl group.
Said acyl group is, for example, C2 4 alkanoyl ~e.c~.
acetyl, propionyl), benzoyl, C2 ~ alkoxycarbonyl (e.9.
ethoxycarbonyl) or benzyloxycarbonyl~
ReferrintJ to R5, the lower ~lkyl ~roup represented
thereby includes lower alkyl groups similar to those repre-
sented by R3. The aralkyl group represented by R5
includes phenyl-lower (Cl 4)-alkyl containing about 7-10
carbon atoms, such as benzyl, phenethyl, 3-phenylpropyl,
~-methylbenzyl, ~-ethylbenzyl, ~-methylphenethyl, ~-
3~ methylphenethyl and ~-ethylphenethyl. The phenyl moiety
of said phenyl-lower-alkyl group may optionally have 1-3
,. 3L~ ~37L~fl~
substituents, such as halogen (e.g. fluorine, chlorine, bromine,
iodine), Cl 4 alkyl (e.g. methyl, ethyl, propyl, butyl), Cl 4
alkoxy (e.g. methoxy, ethoxy, propoxy, isopropoxy, butoxy, mekhyl-
enedioxy), amino, nitro, or hydroxyl. Examples of such substi-
tuted phenyl-lower-alkyl group are 2-(4-chlorophenyl)ethyl, 2-
(4-hydroxyphenyl)ethyl, 2-(4-methoxyphenyl)ethyl, 2-(3,4-di-
methoxyphenyl)ethyl, 2-(3,4,5-trimethoxyphenyl)ethyl, 2-(3,4-
methylenedioxyphenyl)-ethyl, 2-(p-tolyl)ethyl, 3,4-dimethoxy-
benzyl, 3,4-methylenedioxybenzyl, 3,4,5-trimetho~ybenzyl, 4-
ethylbenzyl and 4-chlorobenzyl.
Referring to R6, the lower alkoxy represented thereby
includes lower alkoxy groups containing about 1-4 carbon atoms,
such as methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy,
sec-butoxy and tert-butoxy, and the lower al~ylamino ~roup in-
cludes mono- or di-lower (Cl 4)-alkylamino groups, such as methyl-
amino, ethylamino, propylamino, butylamino, dimethylamino and
diethylamino.
The salts of compounds (I) include pharmaceutically
acceptable salts, ~or example, inor~anic acid addition salts,
such as hydrochloride, hydrobromide, sulfate, nitrate and phos-
phate; organic acid addition salts, such as acetate, tartrate,
citrate, Eumarate, maleate, toluenesulEonate and methanesulfo-
nate; metal salts, such as sodium, potassium, calcium and alum-
inum ~alt5; and 5alt5 with organic bases, such as -triethylamine,
guanidine, ammonium, hydrazine, quinine and cinchonine salts.
In the above-mentioned compounds (I), preferred em-
bodiments are those of the formula (I) ~herein Rl and R inde-
~--1
` ~2~7
-- 4 --
pendently represent hydrogen, hydroxyl or Cl_4 alkoxy, or Rl and
R2 jointly form Cl 4 alkylenedioxy; R is hydrogen or Cl 4 alkyl;
R4 iS hydrogen, Cl 4 alkyl or amino-Cl 4 alkyl which is unsub-
stituted or substituted by acyl of the class consisting of C2 4
alkanoyl, benzoyl, C2 4 alkoxycarbonyl or benzylo~ycarbonyl; R5
is hydrogen, Cl_4 alkyl or phenyl-Cl ~ alkyl which is unsubsti-
tuted or substituted by 1 to 3 members of halogen, Cl_4 alkyl,
Cl 4 alkoxy, amino, nitro and hydroxyl, R is hydroxyl, Cl 4
alkoxy, amino, or mono- or di-Cl_4 alkylamino; and m and _ each
means 1 or 2, and pharmaceutically acceptable salts thereof.
Among the compounds (I), further preferred are com-
pounds wherein Rl and R2 are each hydrogen, R3 is hydro~en, R4
is lower (Cl 4) alkyl or amino-lower (Cl 4)-alkyl, R is phen-
ethyl, R6 is lower(Cl 4) alkoxy or hydroxyl, and _ and n are
each 1, or compounds of the formula:
O R R5 O
-N-C-CH-NH-CH-C-R6 (I')
CH2COOH
wherein R4 is hydrogen or Cl_4 alkyl, R5 is hydrogen, Cl 4
alkyl or phenyl-Cl 4 alkyl, and R is hydroxyl or Cl_4 alkoxy,
and their pharmaceutically acceptable salts with a metal or an
orc~anic base and their pharmaceutically acceptable acid addi-
tion salts.
Reerrin~ to the above formula (~'), the groups in R4 ,
R and R correspond to those of R , R and R respectively.
A preferable specific embodiment in the present inven-
tion is N-(l-ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N-(indan-2-
-- 5 --
yl)glycine andits pharmaceutically acceptable salts.
The pr~sent invention also provides a process for pro-
ducing the compounds of formula (I) as defined before, and phar-
maceutically acceptable salts thereof. The process comprises:
a) subjecting a compound of the formula:
Rl
R2 ~ ~ O R4 (II)
.. .
7-C--CH--NH2
(CH2 ) mcooR3
wherein the symbols are each as above defined, and a compound
of the formula:
R5_C_C_R6
O O
wherein R5 and R6 are as above defined, to condensation under
reductive conditions; or
b) hydrolyzing an ester compound of formula (I) wherein
R3 is Cl 4 alkyl and/or R6 is Cl 4 alkoxy and all other symbols
are as defined above, to provide a compound of formula (I)
wherein R3 is hydrogen and/or R6 is hydroxyl and all other sym-
bols are as defined above; or
c) subjecting a benzyl ester of a compound oE Eormula (I)
wherein R3 is benzyl and/or R6 is benzyloxy and all other sym-
bols are as deEined above, to catalytic reduction to provide a
compound of formula (I) wherein R3 is hydrogen and~or R6 is
hydroxyl and all other symbols are as defined above; or
d) subjecting a compound of the formula:
lZ8~
-- 6
~,~
~ ( n 2n-1) (IV)
R2 \ O R Z R5 O
,. . . . ..
N-C-CH-M-CH-C-R
( CH2 ) mCoOR3
wherein Z is a protective group removable by hydrolysis or cata-
lytic reduction and the other symbols are as defined above, to
hydrolysis or catalytic reduction; or
e) reacting a compound of formula (II) with a compound of
the formula: ~5 O
.
X - CH - C - R6 (V)
where.in R5 and R6 are as defined above, and X is halo~en or a
group of the formula R7So2-o- in which R7 is Cl 4 alkyl, phenyl
or p~tolyl; and
f) if desired, converting the thus obtained compound of
the formula (I) into a pharmaceutically acceptable salt thereof.
Said reductive conditions employed in process alterna-
tive a) above include those reaction conditions used in cata-
lytic reduction using as a catalyst such metal as platinum, pal-
ladi~n, Raney nickel or rhodium or a mixture thereof with an
appropriate carrier; reduction with a metal hydride compound
such as lithium aliminum hydride, lithium borohydride, lithium
cyanoborohydride, sodium borohydride or sodium cyanoborohydride;
reduction with metallic sodium or metallic magnesium and an al-
~2~
-- 7 --
cohol; reduction with such a metal as iron or zinc and such an
acid as hydrochloric acid or acetic acid; electrolytic reduc-
tion and reduction with a reducing enzyme. The above reaction
is usually carried out in the presence of water or an organic
solvent (e.g. methanol, ethanol, ethyl ether, dioxane, methyl-
ene chloride, chloroform, benzene, toluene, acetic acid, dimethyl-
formamide, dimethylacetamide). The reaction temperature depends
on the means of reduction employed, but generally temperatures
ranging from -20C to +100C are preferred. The reaction can
proceed in a satisfactory manner at ordinary pressure, but the
reaction may also be carried out under pressure or under reduced
pressure according to circumstances.
The protective group represented by Z in formula (IV)
and removable by hydrolysis includes all sorts o~ acyl ~roups
and trityl. In particular, such ~roups as benzyloxycarbonyl,
tertbutoxy carbonyl, trifluoroacetyl and trityl are advanta-
geous for a reaction under relatively mild conditions. The pro-
tective group represented by Z and removable by catalytic re-
duction includes benzyl, dipnenylmethyl and benzyloxycarbonyl,
among others. The hydrolysis reaction in this method is carried
out in water or an organic solvent such as methanol, ethanol,
dioxane, ~yridine, acetic acid, acetone or methylene chloride,
or a mixture thereof. For acceleratin~ the reaction, an acid
(e.cJ. hydroch1oric, hydrobromic, hydroiodic, hydrofluoric, sul-
furic, methanesulfonic, p-toluenesulfonic or trifluoroacetic
acid) or a base (e.g. sodium hydroxide, potassium hydroxide,
~L2~
- 7a -
potassium carbonate, sodium hydrogen carbonate, sodium acetate,
triethylamine) may be added to the reaction system. The re-
action temperature is usually in the range of about -10C to
+150C. The catalytic reduction in this method is carried out
in water or an organic solvent such as methanol, ethanol, di-
oxane or tetrahydrofuran, or a mixture thereof, in the presence
of an appropriate catalyst such as platinum or palladium-carbon.
This reaction is carried out at ordinary pressure or under pres-
sure up to about 150 kg/cm2 and at ordinary temperature or a
temperature up to 150C. Generally, the reaction can proceed
sufficiently smoothly at ordinary temperature and at ordinary
pressure.
The reaction of the compound (II) with the compound
(V) is preferably carried out by maintaining both the reactants
in an apporpriate solvent at a temperature within the range of
about -10C to about +150C. For accelerating the reaction,
such a base as potassium carbonate, sodium hydroxide, sodium
hydrogen carbonate, pyridine or triethylamine may be made to
coexist in the reaction system as a deacidifying agen-t.
The starting material of process alternative (b) may
be prepared by the process alternative (a), (d) or (e) mentioned
above usin~ the startin~ materials wherein R is Cl_~ alkyl and/or
R6 i~ alkoxy.
The starting material of process alternative (c) may
be prepared by a process corresponding to the process of al-
ternative (a), (d) or (e) mentioned above, but using starting
materials wherein R3 is benzyl and/or R6 is benzyloxy.
s
3 ~
- 7b -
When a compound of formula (I) wherein R3 is hydrogen
and R is methoxy or ethoxy is desired, it is convenient to em-
ploy process alternative a) or e) using starting materials where-
in R3 is tert-butyl and R is methoxy, ethoxy or propoxy and
then partial hydrolysis according to process alternative b) to
convert the tert-butoxy ester to the free carboxyl group while
retaining the methoxy, ethoxy or propoxy group.
The object compounds (I) of the present invention pro-
duced in this way can be isolated from the reaction mixture by
usual means of separation and purification, such as extraction,
concentration, neutralization, filtration, recrystallization,
column chromatography and/or thin layer chromatography.
A preferred process for producing the pre:Eerred com-
pound of formula (I') or a pharmaceutically acceptable acid addi-
tion salt thereof or a pharmaceutically acceptable salt thereof
with a metal or an organic base comprises:
i) subjecting a compound of the formula
~ O R4 (II')
C 2
4'
wherein R is as defined above, and
2~ R is hydrocJ~n, Cl ~ alkyl or b~nzyl an~ a compound of the
formula:
R5 - C - C - R
O O
wherein R is as defined above, and
R6 is hydroxyl, Cl 4 alkoxy or benzyloxy, to condensation
under reductive conditions; or
- 7c -
ii) subjecting a compound of the formula:
R Z R O
N-C-CH-N-CH-C-R6 (IV')
CH2COOR
wherein Z is a protective group removable by hydrolysis or cata-
lytic reduction; and the other symbols are as defined above to
hydrolysis or catalytic reduction: or
iii) reacting a compound of formula (II') as defined above
with a compound of the formula:
R O
X - CH - C - R6
wherein X is halogen or a group of the formula R7So2-o- in which
0 R is Cl 4 alkyl, phenyl or p-tolyl, and
RS and R6 are as defined above; and
iv) la] when in the starting materials R is Cl 4 alkyl or
benzyl, subjecting the thus obtained compound to hydrolysis so
as to convert the Cl 4 alkyl group into hydrogen, or to cata-
lytic reduction so as to convert the benzyl group into hydrogen;
[b~ or when in the starting materials R is benzyloxy, sub~ec-
ting the thus obtained compound to catalytic reduction so as to
convert the benzyloxy gr-oup into hydroxyl; [c] or when in the
starting materials R6 is Cl ~ alkoxy and if a compound o:E for-
mula (I') wherein R is hydroxyl is required, subjecting thethus obtained compound to hydrolysis so as to convert the Cl 4
alkoxy group into hydroxyl; and if desired, converting the thus
obtained compound of formula (I') into a pharmaceutically accept-
. .
- 7d -
able acid addition salt thereof or into a pharmaceutically accep-
table salt thereo~ with a metal or an organic base.
Depending on the presence or absence of the substi-
tuents represented by R and R5, there may exist two to eight
steric isomers of a compound (I). These individual isomers and
mixtures thereof naturally fall within the scope of the present
invention. Such isomers, if desired, can be prepared individ-
ually. For example, a single optical isomer of (I) can be ob-
tained by carrying out the above reaction using a single isomer
of the starting compound (II) or (IV). When the product is a
mixture of two or more isomers, they can be separated into in-
dividual isomers by a usual separation technique, such as salt
formation using an optically active acid (e.g. camphorsulfonic,
tataric or dibenzoyltartaric acid) or an optically active base
(e.g. cinchonine, cinchonidine,
" ~2~37~4
quinine, quinidine, alphamethylbenzylamine, dehydro-
abietylamine), a variety of chromatographic techniques or
fractional crystallization. For ~hose compounds (I)
wherein R4 and R5 are each other than hydrogen, the isomers
respectively having an S configuration generally have more
preferable physiological activities as compared with the
corresponding compounds having an R configuration.
The compounds of the present invention, i.e., the
bicyclic compounds represented by formula (I) and phar-
maceutically acceptable salts thereof, exhibit inhibitoryactivities on angiotensin converting enzyme, bradykinin
decomposing enzyme (kininase) and enkephalinase in animals,
in particular, mammals (e.g. human, dog, cat, rabbit,
guinea pig, rat) and therefore are useful as drugs for
diagnosis, prevention or treatment of hypertension and as
analgesic and analgesic-activity-potentiating agents. The
compounds of the present invention are of low toxicity,
well absorbed even on oral administration and highly stable.
Therefore, when they are used as the above-mentioned drugs,
they can safely be administered orally or parenterally,
per se or in admixture with suitable pharmaceutically
acceptable carriers, excipients or diluents in various
pharmaceutical formulations such as powders, granules,
tablets, capsules, injectable solutions, etc. While the
dosage level generally varies depending upon the conditions
of the diseases to be treated as well as the administration
route used, for example, in the treatment o hypertension
in adult human, the compounds may be administered orally
at a single dose of about 0.02-20 mg/k~, preferably about
0.2-2 mg/k~, or intravenously at about 0.002-0.2 mg/kg,
preferably about 0.02-0.2 mg/kg, about 2 to 5 times per
day according to the conditions.
The starting compounds (II~ of this invention can
easily be prepared, for example, by the process shown by
the following reaction scheme:
~2~4
g
Rl ` Rl
~ n 2n-1~ 2 2 m , ~ ~ CnH2n 1)
R NH
(~ 2)m
(VI) (VIII)
R4 R1
C -NHCHCOOH (IX) ~ ~ H2
bz (CnH2n-1) , (II)
/ \ O R4
R2 N-C-CH-NH-Cb
1 COOR3
(CH2)m
~t)
In the above ormulas, Rl, ~2, R3, R4, m and n are as above
2C defined, and Cbz is benzyloxycarbonyl.
The process for preparing (II) as shown by the above
reaction scheme is now illustrated in more detail. Firstly,
in the step (VI) -~ (VIII), (VI) and (VII) are reacted in
an appropriate solvent to yield a Schiff base, which is
then subjected to reduction. An organic solvent such as
methanol, ethanol, dioxane, methylene chloride, chloroform,
benzene or toluene is used as the solvent,and the reaction
is conducted generally at a temperature within the range
of about -10C to +150C. For advantageous progress of
the reaction, a catalyst, such as sulfuric acid or p-
toluenesulfonic acid, or a dehydrating agent, such as
anhydrous sodium sulfate, anhydrous magnesium sulfate or
calcium chloride, may be added to the reaction mixture.
It is also possible to make the reaction proceed advan-
tageously by using a water separating device (trap). TheSchiff base obtained is, either in the form of a reaction
12~79~ ~4
-- 10 --
mixture or after isolation in a usual manner, again added
to a solvent and subjected to reduction. The means of
reduction include catalytic reduction using platinum or
palladium-carbon, for instance, as a catalyst, and a method
using such a reducing agent as sodium borohydride or sodium
cyanoborohydride.
It is also possible to make the Schiff base formation
and reduction proceed simultaneously be allowing such a
reducing agent to coexist in the reaction mixture of (VI)
and (VI~) from the beginning. In the step (VIII) (X),
(VIII) is reacted with (IX) or a carboxyl-derived functional
derivative thereof. The carboxyl-derived functional
derivative of compound (~ ) incl~udes among others acid
halides, such as acid chloride and acid bromide; acid
anhydrides obtainable by removing one mole of water from
two moles of (~ ); mixed anhydrides formed by substitution
of the hydrogen atom of the carboxyl group in (~ ) by
ethoxycarbonyl, isobutyloxycarbonyl, benzyloxycarbonyl,
etc.; and reactive esters of (IX) derived from l-hydroxy-
benzotriazole, N-hydroxyphthalimide, N-hydroxysuccinimide,
etc. Generally, the reaction is carried out in an appro-
priate solvent, which may be of any kind so long as it
does not disturb the reaction. When (IX) is used as it is
without converting it to a functional derivative, the
reaction is preferably carried out in the presence of a
dehydrating agent such as dicyclohexylcarbodiimide or
carbonyldiimidazole. When an acid halide is used as the
functional derivative, the reaction may also be carried
out in the presence of a base such as pyridine, picoline,
triethylamine, sodium hydroxide, sodium hydrogen carbonate
or sodium carbonate. aenerally, the reaction temperature
is within the range of about -20C to about +150C. In
most cases, however, the reaction can proceed in a
satisfactory manner at ordinary temperature. The step
(X) ~ (II) consists in removal of the N-protecting group
by catalytic reduction. The catalytic reduction is carried
~2~37~
11 --
out in water or an organic solvent, such as methanol,
ethanol, dioxane, tetrahydrofuran or acetic acid, or a
mixture thereof, in the presence of an appropriate catalyst,
such as platinum, palladium-carbon or Raney nickel. This
reaction is carried out at ordinary pressure to about 150
kg/cm2 and at ordinary temperature to 150~C. Generally,
the reaction can proceed in a satisfactory manner at
ordinary temperature.
The invention will be further illustrated in more
detail by the following reference examples, embodiment
examples, test examples and dosage form examples, which,
however, are by no means limitative of the present invention.
Reference Exa~ple 1
Glycine ethyl ester hydrochloride (20 g) is dissolved
in a solution of 10 g of 2-indanone in 200 ml of methanol,
and then 5.0 g of sodil~ cyanoborohydride is added portion-
wise to the solution with ice cooling and stirring. After
stirring at room temperature for 2 hours, the reac~ion
mixture is poured into 500 ml of ice water, and the whole
mixture is made alkaline with sodium hydrogen carbonate
and extracted with 300 ml of ethyl acetate. The extract
is washed with water and dried, the ethyl acetate is
distilled off under reduced pressure, 10 ml of 20~ ethanolic
hydrochloric acid and 200 ml of ethyl ether are added to
the residue, and the mixture is allowed to stand at room
temperature. The resulting crystalline precipitate is
collected by filtration and dried to give 11 c3 of N-(indan-
2-yl)glycine ethyl ester hydrochloride as colorless
needles melting at 165-167C.
Reference Example 2
_ _ _ . _
To a solution of 5 g of 2-indanone in 150 ml of
methanol is added 15 g of glycine benzyl ester para-
toluenesulfonate, and then 5 g of sodium cyanoborohydride
is added portionwise with ice cooling and stirring.
Thereafter,the mixture is treated in the same manner as
in Reference Example 1 to give 6.5 g of N-(indan-2-yl)-
874~L
- 12 -
glycine benzyl ester hydrochloride as colorless prisms
melting at 186-189C.
Reference Example 3
2-Indanone (40 g) is dissolved in 300 ml of methanol,
78 g of glycine tert-butyl ester phosphite and 150 g of
water are added, and then 23 g of sodium cyano~orohydride
is added over 15 minutes with ice cooling and stirring.
The resulting mixture is ~urther stirred at room temperature
for 4 hours. To the reaction mixture, 400 ml of 20~
phosphoric acid is added portionwise over an hour, 200 ml
of water is then added, the mixture is stirred for 30
minutes and then extracted with 800 ml of ethyl ether, and
the aqueous layer is adjusted to pH 10 with 20~ sodium
hydroxide and extracted with 4 portions (500 ml in total)
of chloroform. The extract is dried over anhydrous sodium
sulfate and then distilled under reduced pressure. 50 ml
of ethanol and then 150 ml of water are added to the oil
obtained, and the mixture is cooled. The crystalline
precipitate is collected by filtration and recrystallized
twice from aqueous ethanol to give 47 g of (indan-2-yl)-
glycine tert-butyl ester as colorless prisms melting at
54-SSC.
Reference Example 4
A solution of 22.3 g of N-carbobenzoxy-L-alanine and
14 ml of triethylamine in 200 ml of tetrahydrofuran is
cooled to -10C, and 13.1 ml of isobutyl chlorocarbonate
is added dropwise in portions with stirrin~. ~fter stirring
for 30 minutes, a solution of 2~.1 g of N-(indan-2-yl)-
c31ycine ethyl ester hydrochloride and 14 ml of triethyl-
amine in 200 ml o chloroform is added dropwise at -10C to -5C.
A~ter standincJ o~ernight at room temperature, the reaction
mixture is washed in sequence with water, aqueous sodium
hydrogen carbonate, 10~ hydrochloric acid and water, and
dried over anhydrous sodium sulfate. The solvent is
distilled off under reduced pressure, the residue is dis-
solved in 100 ml of methanol, 75 ml of 2N sodium hydroxide
:1~74 ~4
- 13 -
is added, and the mixture is stirred at room temperature
for 2 hours. Then, the mi~ture is made acidic with 10%
hydrochloric acid to separate the resulting oil, which is
extracted with 500 ml of ethyl acetate. The extract
washed with water and dried, and the solvent is distilled
off under reduced pressure to give 25 g of N-carbobenzoxy-
L-alanyl-N-(indan-2-yl)glycine as an oil. This is
dissolved in 50~ ethanol and subjected to catalytic reduc-
tion in the presence of 4 g of 10% palladium-carbon. When
the hydrogen absorption has ceased, the catalyst is
filtered off, and the filtrate is concentrated under
reduced pressure. Addition of 50 ml of methanol to the
residue yields 11 g of L-alanyl-N-(indan-2-yl)glycine as
colorless needles melting at 18Q-182C.
Reference Example 5
N-Carbobenzoxy-L-alanine (21.8 g) and 12.3 ml of
triethylamine are dissolved in 200 ml of tetrahydrofuran,
and 8.5 g of ethyl chlorocarbonate is added dropwise at
-15C with stirring. After the dropping, stirring is
continued for 15 minutes, and then a solution of 22 g of
(indan-2-yl)glycine *ert-butyl ester in 100 ml of chloro-
form is added dropwise at -10C or below. After stirring
at room temperature for an hour, the reaction mixture is
poured into 500 ml of water, and the chloroform layer is
separated and the chloroform is distilled off. The residue
is dissolved in 300 ml of ethyl acetate, the solution is
washed with two 50-ml portions of lN a~ueous sodium
hydroxide, one 50-ml portion oE water, ~wo 50-ml portions
of 20% aqueous phosphoric acid and one 50-ml portion of
water, in that order, and then dried over anhydrous
magnesium sulfate, and the solvent is distilled off to
give 35 g of N-carbobenzoxy-L-alanyl-N-(indan-2-yl)glycine
tertbutyl ester as an oil. This is dissolved in 300 ml of
methanol and, aEter addition of 7 g of oxalic acid and
3.5 g of 10% palladium-carbon (containing 50~ water), is
subjected to catalytic reduction at ordinary temperature
- 14 -
and ordinary pressure. After the reaction, the catalyst
is filtered off, the filtrate is distilled off under
reduced pressure, and 500 ml of ethanol is added to the
residue. On cooling, a precipitate forms, which is
collected by filtration and dried to give 21.8 g of L-
alanyl-N-(indan~2-yl)glycine tert-butyl ester oxalate
melting at 138-141C.
r~D + 20.4 (c=l, methanol)
Reference Example 6
N-(Indan-2-yl)glycine benzyl ester hydrochloride
(6 g) is added to a mixture of 300 ml of ethyl acetate and
200 ml of S~ aqueous potassium carbonate, followed by
vigorous stirring. To the resulting solution is added
dropwise 6 ml of chloroacetyl chloride over 30 minutes
with ice cooling. Thereafter, stirring is continued for
an hour. The ethyl acetate layer is then separated, washed
with water and dried over anhydrous magnesium sulfate, and
the solvent is distilled off under reduced pressure.
Addition of ether to the residue gives 6 ~ of N-chloro-
acetyl-N-(indan-2-yl)glycine benzyl ester as colorless
scales melting at 99.5-100.5C.
~eference Example 7
N-Chloroacetyl-N-(indan-2-yl)glycine benzyl ester
(3 g) and 2 g of N-benzylglycine ethyl ester are dissolved
in 50 ml of methyl ethyl ketone, 10 g of potassium
carbonate is added, and the mixture is refluxed for 24
hours with stirring. After cooling, the insoluble matter
is filtered off, and the filtrate is distilled under
reduced pressur~ to give an oil. This is purified by
silica gel column chromato~raphy to give 3 g of N-ethoxy-
carbonylmethyl-N-benzyl~lycyl-N-~indan-2-yl)glycine benzyl
ester as an oil.
Infrared (IR) Absorption Spectrum vmaexatcm 1 1730, 1640
Reference Example 8
3~ By reacting 7 g of N-(indan-2-yl)glycine tert-butyl
ester with 8.5 g of N-(carbobenzoxy)-L-leucine and treating
12~
- 15 -
the reaction mixture as in Reference Example 5, there is
obtained 3.5 g of L-leucyl-N-~indan-2-yl)glycine tert-butyl
ester as colorless amorphous powder.
Reference Example 9
By reacting 8.2 g of N-(indan-2-yl)glycine tert-butyl
ester with 13 g of N~-tert-butoxycarbonyl-NE-carbobenzoxy-
L-lysine as in Reference Example 5, there is obtained 14 g
of Natert-butoxycarbonyl-N~-carbobenzoxy-L-lysyl-N-(indan-
2-yl)glycine tert-butyl ester as a colorless oil.
NMR~CDC13) ~: 1.40, 1.45(18H), 5.05(2H)/ 7,0-7.3(9H)
IR Spectrum vmax cm : 1700, 1640
~]D ~ 14.3 (c=0.9 methanol)
~eference Example 10
Na-tert-Butoxycarbonyl-N~-carbobenzoxy-L-lysyl-N-
(indan-2-yl)glycine tert-butyl ester (5 g) obtained by the
procedure of Reference Example 9 is dissolved in 100 ml of
lN solution of hydrogen chloride in 0thyl acetate, and the
solution is allowed to stand at room temperature for 6
hours. On adding 500 ml of petroleum ether to the reaction
mixture, an oily substance separates, which is isolated.
The solution layer is concentrated under reduced pressure
and again subjected to a similar treatment with hydrogen
chloride. The oil fractions obtained are combined, dis-
solved in 100 ml of ethyl acetate, and washed with 100 ml
of lN aqueous sodium hydroxide and with water. The
organic layer is dried over anhydrous ma~nesium sulfate
and, after addition of 1 ~ of oxalic acid, the solvent is
distilled o~f under reduced pressure. Addition of a mix-
ture oE ethcr ~nd petroleum ether to the residue yields
2.~ g oE N~-carbobenzoxy-L-lysyl-N-(indan-2-yl)glycine
tert-butyl ester oxalate as colorless powder.
Elemental analysis for C29H39N3O5-C2H2O4-H2O
Calcd.: C, 60.28; EE, 7.02; N, 6.80
Found : C, 59.83; H, 7.01; N, 6.41
~a]23-5+ 17.5 (c=l, methanol)
Reference Example 11
- 16 -
In a solution of 13 g of 2-indanone in 100 ml of
methanol, there is dissolved 13 g of ~-alanine ethyl ester
hydrochloride, and, with ice cooling and stirring, 6.5 g
of sodium cyanoborohydride is added portionwise. After
standing at room temperature overnight r the mixture is
poured into ~00 ml of ice water, made alkaline with sodium
hydrogen carbona~e and extracted with 200 ml of chloroform.
The extract is washed with water, and dried, followed by
distilling orf chloroform under reduced pressure. To the
residue, 10 ml of 20~ ethanolic hydrochloric acid and 50 ml
of ethyl ether are added successively, and the mixture is
allowed to stand at room temperature~ The resulting pre-
cipitate is collected by filtration and dried to give 10 g
of N-(indan-2-yl)-~-alanine ethyl ester hydrochloride as
colorless scales meltin~ at 150-151C.
Reference Example 12
By reacting 7 g oE N-(indan-2-yl)-~-alanine ethyl
ester hydrochloride with 7 g of N-carbobenzoxy-L-alanine
and treating the reaction mixture as in Reerence Example 4,
there is obtained ~ g of L-alanyl-N-tindan-2-yl)-~-alanine.
Melting point 205-206C.
[~]23 5+ 1~ (c=0.8, lN HCl)
Reference Example 13
In a solution of 20 g o~ 1,2,3,4-tetrahydro-2-
naphthalenone in 200 ml of methanol, there is dissolved 23 g
of glycine ethyl ester hydrochloride, and 9.0 ~ of sodium
cyanoborohydride is added portionwise with ice cooling and
stirrin~. ~fter stirring at room temperature for 2 hours,
the reaction mixture is poured into 500 ml oE ice water,
and the whole mixture is made alkaline with sodium hydrogen
carbonate and extracted with 500 ml of ethyl acetate. The
extract is washed with water and dried, the ethyl acetate
is distilled off under reduced pressure, 10 ml of 20~
ethanolic hydrochloric acid and 200 ml of ethyl ether are
added to the residue, and the mixture is allowed to stand
at room temperature. The crystalline precipitate is
~;~B7~
- 17 -
collected by filtration and dried to give 25 g of N-(1,2,3,
4-tetrahydronaphthalen-2-yl)glycine ethyl ester hydrochloride
as colorless needles melting at 198~200C.
Reference Example 14
By reacting 13.5 g of N-(1,2,3,4-tetrahydronaphthalen-
2-yl)glycine ethyl ester hydrochloride with 11.6 g of N-
carbobenzoxy-L-alanine and treating the reaction mixture as
i~ Reference Example 4, there is obtained 7.5 g of L-alanyl-
N-(1,2,3,4-tetrahydronaphthalen-2-yl)glycine as colorless
amorphous powder.
IR spectrum vmai cm : 1720, 1640
Reference Example 15
By reacting 13 g of (indan-l-yl)glycine ethyl ester
hydrochloride with 11.6 g of N-carbobenzoxy-L-alanine and
treating the reaction mixture as in Reference Example 4,
there is obtained 8.0 g of L-alanyl-N-(indan-l-yl)glycine
as colorless amorphous powder.
IR spectrum vmaXlcm 1 1730, 1640
Reference Example 16
_
By reaciing 10 g of (5-~enzyloxyindan-1-yl)glycine
ethyl ester with 8 ~ of N-carbobenzoxy-L-alanine and treat-
ing the reaction mixture as in Reference Example ~, there
is obtained 5.2 g of L-alanyl-N-(5-hydroxyindan-1-yl)-
glycine as colorless amorphous powder.
Reference Example 17
5,6-Dimethoxy-l-indamine hydrochloride (11 y) is sus-
pended in 200 ml of methyl ethyl ketone, then 6.~ ~ o~
potassium carbonate, 2.0 g of potassium iodide and 8.2 g of
tert-butyl chlorocarbonate are added, and the mixture is
refluxed for 8 hours. The reaction mixture is poured into
500 ml of water and extracted with 200 ml of ethyl acetate,
the extract is washed with water and dried, and the ethyl
acetate is distilled off under reduced pressure. The
residue is subjected to silica gel column chromatography
and eluted with hexane-acetone (7:3) to give 6.0 g of N-
(5,6-dimethoxyindan-1-yl)glycine tert-butyl ester as an oil.
12~7~44
- 18 -
This is dissolved in 50 ml of ethyl ether, 2.0 g of oxalic
acid is added, and the mixture is allowed to stand at room
temperature. There is thus obtained 7.3 g of N-(5,6-
dimethoxyindan-l-yl)glycine tert-butyl ester oxalate as
colorless needles melting at 158-160C.
Reference Example 18
By reacting 7 g of N-(5,6-dimethoxyindan-1-yl)glycine
tert-butyl ester oxalate with 4.8 g of N-carbobenzoxy-L-
alanine and treating the reaction mixture as in ~eference
Example 5, there is o~tained 4 g of L-alanyl-N-(5,6-dimethoxy-
indan-l-yl)glycine tert-butyl ester oxalate as colorless
amorphous powder.
Reference Example 19
A mixture of 143 g of ethyl 3-phenylpropionate, 234 g
of ethyl oxalate and 154 ml of 28~ sodium ethoxide solution
in ethanol is heated on a water bath at a bath temperature
of 60-70C for 1.5 hours, while distilling off the ethanol
under reduced pressure. To the resultin~ red syrupy residue
is added 1.3 liters of 15 v/v ~ sulfuric acid~ The mixture
is boiled under reflux with stirrin~ or lS hours, and the
oil layer is separated, neutralized with 10~ sodium hydroxide
and extracted with ethyl acetate. The aqueous layer is made
acidic with diluted sulfuric acid. The resulting oil is
extracted with ethyl acetate, washed with water and dried.
Removal of the ethyl acetate by distillation under reduced
pressure gives 130 g of 2-oxo-4-phenylbutyric acic as an
oil.
Re _rence Example 20
2-Oxo-4-phenylbutyric acid (130 ~) is added to a mix-
ture of 650 ml of ethanol and 13 ml of concentrated suluric
acid, ~nd the whole mixture is refluxed for S hours. The
reaction mixture is concentrated to approximately half the
original volume, and then diluted with 500 ml of water.
The resulting oil is collected and, the aqueous layer is
3~ extracted with ethyl acetate. The extract and the oil are
combined and dried, and the solvent is distilled off under
- 19 -
reduced pressure. The residue is distilled under reduced
pressure to give 113 g of ethyl 2-oxo-4-phenylbutyrate as
a colorless oil boiling at 135-141C/3 mmHg.
Reference Examples 21-27
Compounds shown in Table 1 can be pre~pared from the
respectively corresponding starting compounds by a similar
manner to Reference Examples 19 and 20.
Table 1
R~ ~ CH2CH2COCOOR"
No. R' Boiling Point
.
1521 H 3 108-112C/0.5 mmHg
22 H ( 2)2 3 105-118C/l mmHg
_
2023 H -CH ~CH3 132-135C/3 mmHg
24 H -(CH2)3CH3 145-150C~4 mmHg
2525 H -CH2-CH ~CH3 120-132C/0.5 mmHg
26 Cl -CH2CH3 125-135C/l mmHg
27 CH3 C 2CH3 120-130C/l mmHg
Reerence Example 28
Piperidine ~7.5 ml) is added to a solution of 99.6 g
of veratrum aldehyde and 124.8 g of malonic acid in 240 ml
of pyridine, and the mixture is heated at 80-85DC for an
hour and further at 110-115C for 3 hours. After cooling,
the reaction mixture is poured into a large amount of water,
and the resulting crystalline precipitate is collected by
7~
- 20 -
filtration. The crystals are dissolved in diluted aqueous
sodium hydroxide. The solution is made acidic with hydro-
chloric acid to give 70 g of 3,4-dimethoxycinnamic acid as
needles melting at 182-183C. 35 g of these crystals are
dissolved in S00 ml of ethanol, and the solution is
saturated with gaseous hydrogen chloride and allowed to
stand at room temperature overnight. The crystals obtained
by distilling off the ethanol are dissolved in ethyl acetate.
The solution is washed with diluted aqueous sodium hydrogen
carbonate and with water, and dried. The crystals obtained
by distilling off the ethyl acetate are recrystallized from
ethanol to give 65 g of ethyl 3,4-dimethoxycinnamate as
scales melting at 53-55C. 34 g of these crystals are
dissolved in 300 ml of ethanol, 10 g of 5~ palladium-carbon
is added to the solution, and the mixture is shaken in a
hydrogen atmosphere at room temperature. After 3 hours,
the catalyst is filtered off, and the filtrate is concen-
trated to give 34 g of ethyl 3,4-dimethoxyphenylpropionate
as an oil. A solution of 34.1 g of this oil in 43 g of
diethyl oxalate is added to a sodium ethoxide solution
(prepared from 3.8 g of metallic sodium and 150 ml of
ethanol) at 60C with stirring. After completion of the
addition, stirring is continued at 70-75C for further 3
hours. The ethanol is distilled off under reduced pressure,
200 ml of water is added to the residue, and the mixture is
washed with ethyl acetate. The aqueous layer is made acidic
with hydrochloric acid and then extracted with ethyl acetate.
The ethyl acetate layer is washed with water, dried and
concentrated to give 27 g of ethyl 2-oxo-3-ethoxycarbonyl-
4-t3,4-dimethoxyphenyl)butyrate as an oil. 26 g of this
oil is dissolved in a mixture of 80 ml of dimethyl sulfoxide
and 8 ml of water. To the solution is added 6.3 g of sodium
chloride, and the mixture is stirred at 120C for 2 hours.
After cooling, a large amount of water is added, and the
3~ mixture is extracted with ethyl acetate. The extract is
washed with water, dried and concentrated to give crystals.
-
~8~
- 21 -
Recrystallization from ether yields 15 g of ethyl 2-oxo-4-
~3,4-dimethoxyphenyl)butyrate ad pillars melting at 85-87C.
Reference Example _
Proceeding as in Reference Example 28, there is
obtained ethyl 4-(p-benzyloxyphenyl)-2-oxobutyrate as a
yellow oil from the corresponding starting material.
NMR Spectrum (CDC13) ~: 1.3(t,3H), 2.7-3.3(m,4H), 4.3
~ q,2H), 5.0(s,2H), 6.7-7.4(m,9H)
In the above NMR data, s means a singlet, d a doublet, t
a triplet, q a quartet, m a multiplet, and Ph a phenyl
group. (Hereinafter the same shall apply.)
Reference Example 30
2-Oxo-4-phenylbutyric acid t9 g) is dissolved in 100 ml
of benzene, and 10 g of phosphorus pentachloride is added
1~ thereto portionwise with ice cooling~ After stirring at
room temperature for an hour, a solution of 10 g of butyl-
amine in 20 ml of tetrahydrofuran is added dropwise to the
benzene solution. The reaction mixture is stirred at room
temperature for 30 minutes and poured into 100 ml of ice
water. The mixture is extracted with ethyl acetate, the
extract is washed with water and dried, and the ethyl
acetate is distilled off under reduced pressure. The
residue is subjected to silica gel column chromatography by
the use of hexane-acetone (7:3) as the eluent to give 6.0 g
of N-butyl-2-oxo-4-phenylbutylamide as a slightly yellow
oil.
NMR Spectrurn (CDC13) ~: 0.90~3~l,mlCEl3), 1.20-1.50(4H,
m), 2.70-3.40t5H,m), 7.20(5EI,s,Ph)
Example 1
To a solution of 1.0 q of I,-alanyl-N-(indan-2-yl)-
glycine and 6.0 ~ of ethyl 2-oxo-4-phenylbutyrate in 200 ml
of ethanol, there is added 8 g of molecular sieve, and the
mixture is stirred at room temperature for an hour. Then,
1.0 ~ of sodium cyanoborohydride is added. After standing
overnight, the reaction mixture is concentrated under reduced
pressure, the residue is adjusted to pH 9.0 with 10~ sodium
- 2~ -
hydroxide, and the insoluble matter is removed by extraction
with ethyl ether. The aqueous solution is adjusted to pH 4
with 10~ hydrochloric acid and extracted with two 200-ml
portions of ethyl acetate. The extract is washed with
water and dried over sodium sulfate, and the solvent is
distilled oEf under reduced pressure. The residue is dis-
solved in 2 ml of 20% ethanolic hydrochloric acid, 100 ml
of ethyl ether is tnen added, and the mixture is allowed to
stand at room temperature to give 0.4 g of N (1-ethoxy-
carbonyl-3-phenylpropyl)-L-alanyl-N-(indan-2-yl)glycine
hydrochloride melting at 168-170C.
Example 2
L-Alanyl-N-(indan-2-yl)glycine tert-butyl ester oxalate
(21 g) is dissolved in 200 ml of ethanol. To the solution
4.1 g of sodium acetate, 10 ml of acetic acid, 25 g of
ethyl 2-oxo-4-phenylbutyrate and 25 g of molecular sieve 3
are added in sequence. Thereafter, 30 g of Raney nickel
suspended in 100 ml of ethanol is added with ethanol, and
catalytic reduction is carried out under ordinary temperature
and ordinary pressure. When the absorption of hydro~en has
~easecl, the supernatant is separated by decantation, and the
precipitate is washed two or three times with ethanol. The
supernatant and the washings are combined and concentrated
under reduced pressure. The residue is dissolved in 500 ml
of ethyl acetate, and the solution is washed with aqueous
sodium hydrogen carbonate and filtered with 30 ~ of
diatomaceous earth. The ethyl acetate layer is separated
from the filtrate, washed with water and dried over anhydrous
ma~nesium sulfate, and the solvent is distilled off under
reduced pressure to gi~e 24 g of N~ ethoxycarbonyl-3-
phenylpropyl)-L-alanyl-N-(indan-2-yl)glycine tert-butyl
ester as a slightly yellow, viscous oil.
IR Spectrum ~Neatcm 1 1730(ester), 1640(amide)
NMR Spectrum (CDC13) ~: 1.27(3H,t,CH3), 1.40(9H,s,CH3 x 3),
1.8-2.2(3H,m,CH3), 2.6-4.5-(lOH,m), 3.8-3.9(2H,m,CH2),
4.2(2H,q,CH2), 4.9(1H,t,CH), 7.1-7.4(9H,m,Ph)
` ~
_ 23 -
Example 3
The N-(l-ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N-
(indan-2-yl)glycine tert-butyl ester obtained in Example 2
is subjected to column chromatography using 700 g of silica
gel and eluted with benzene, benzene-acetone (10:1 to 4:1)
and methanol-benzene (1:9) to give two fractions. Each
fraction is further subjected to column chromatography using
400 g of silica gel and purified by the above procedure.
The first fraction gives 2 g of N-[l-(R)-ethoxycarbonyl-3-
phenylpropyl]-L-alanyl-N-(indan-2-yl)glycine tert-butyl
ester as a colorless viscous oil.
[a]D ~ 16.~ (c=l, methanol)
On the other hand, the second fraction gives 16.5 g
of N-~ 1- (S) -ethoxycarbonyl-3-phenylpropyl]-L-alanyl-N-
(indan-2-yl)glycine tert-butyl ester as a colorless viscous
oil.
[~]D ~ 12.6 (c=l, methanol).
Example 4
5 g of N-~l-(S)-ethoxycarbonyl-3-phenylpropyl]-L-
alanyl-N-(indan-2-yl)~lycine ter~-butyl ester obtained in
Example 3 is dissolved in 5 ml of acetic acid, 20 ml o 25
hydrobromic acid in acetic acid is added to the solution,
and the mixture is shaken for 10 minutes. The crystals
which precipitate on addition of 300 ml of ethyl ether are
collected by filtration to give 5 g of N-~l-(S)-ethoxy-
carbonyl-3-phenylpropyl]-L-alanyl-N-(indan-2-yl)glycine
hydrobromide as colorless crystals meltin~ at 180-183C.
[~]20+ 15.6 (c=l.~, methanol)
Example 5
By using N-~l-(R)-ethoxycarbonyl-3-phenylpropyl]-L-
alanyl-N-(illdan-2-yl)~lycine tert-butyl ester obtained in
Example 3 and treating as in Example 4, there can be
obtained N-[l-(R)-ethoxycarbonyl-3-phenylpropyl]-L-alanyl-
N-(indan-2-yl)glycine hydrobromide as colorless crystals `
melting at 150-155C.
[~]D ~ 20.2 (c=l, methanol)
- . ::. . .
- 24 -
Example 6
To a mixture of 500 ml of ethyl acetate, 33 g of
sodium hydrogen carbonate and 500 ml of water is added
16.2 g of N-[l-(S)-ethoxycarbonyl-3-phenylpropyl]-L-alanyl-
N-(indan-2-yl)glycine hydrobromide prepared by the procedure
of Example 4. After stirring to complete dissolution, the
solution was adjusted to pH 4 with lN hydrochloric acid.
The ethyl acetate layer is separated, washed with water,
dried and, after addition of 20 ml of 7N ethanolic hydro-
chloric acid, concentrated under reduced pressure. To theresidue are added 250 ml of ethyl ether and 250 ml of
petroleum ether, and the resulting precipitate is collected
by filtration to give 11 g of N-[l-(S)-ethoxycarbonyl-3-
phenylpropyl]-L-alanyl-N-(indan-2-yl)glycine hydrochloride
as colorless crystals.
Recrystallization from a mixture of acetone and lN
hydrochloric acid affords colorless plates melting at 166-
170C with decomposition.
[~]D ~ 18.5 (c=l, methanol)
IR Spectrum vNUa]lcm 1: 1740(COOC2H5), 1705(C~O~l), 16~0
(CO-N)
Example 7
L~Alanyl-N-(indan-2-yl)glycine tert-butyl ester
oxalate (4.1 g ) is dissolved in 40 ml of ethanol, then
0.85 g of sodium acetate, 2 ml of acetic acid, 5 g of
propyl 2-oxo-4-phenylbutyrate and 5 g of molecular sieve
3A are added, thereafter a suspension of 6 ~ of Raney
nickel in 20 ml of ethanol is added, and catalytic reduction
is carried out at ordinary temperature and ordinary pressure~
When the hydrogen absorption has ceased, the s~lpernatant is
separated by ~ecantation. The precipitate is washed two
or three times with ethanol. The supernatant and the
washings are combined, and the solvent is distilled off
under reduced pressure. The residue is shaken with 100 ml
3; of ethyl acetate and aqueous sodium hydrogen carbonate.
Filtration with 10 g of diatomaceous earth, separation of
... .
87~4
- 25 -
the ethyl acetate layer of the filtrate, washing with water,
drying and removal of solvent by distillation give N-(1-
propoxycarbonyl-3-phenylpropyl)-L-alanyl-N-(indan-2-yl)-
glycine ter~-butyl ester as a slightly yellow, viscous oil.
To this are added 3 ml of acetic acid and 12 ml of 25% hydro-
bromic acid solution in acetic acid, and the mixture is
shaken occasionally for 15 minutes so as to make the reac-
tion proceed. On adding 150 ml of ethyl ether, crystalline
precipitate forms, which is collected by filtration to give
3.4 g of N-(l-propoxycarbonyl-3-phenylpropyl)-L-alanyl-N-
(indan-2-yl)glycine hydrobromide as colorless crystals.
They are added to a mixture o~ 70 ml of water and 70 ml of
ethyl acetate, and the mixture is stirred, neutralized with
sodium hydrogen carbonate and then adjusted to pH 3-4 with
10~ hydrochloric acid. The ethyl acetate layer is qeparated,
dried over anhydrous magnesium sulfate, and made acidic with
ethanolic hydrochloric acid. The solvent is distilled o~f,
and 150 ml of ethyl ether is added to the residue. A~ter
allowing the mixture to stand for 10 minutes, the resultin~
crystalline precipitate is collected by filtration to give
1.7 g of N-(l-propoxycarbonyl-3-phenylpropyl)-~-alanyl-N-
(indan-2-yl)glycine hydrochloride melting at 147-150C with
decomposition.
IR Spectrum vmUajlcm 1 1740(ester), 1710(COOH),
1640(CO-N)
E~ample 8
The same procedure as Exan~ple 7, exceptin~ -the use of
butyl 2-oxo-4-phenylbutyrate in place of propyl 2-oxo-4-
phenylbutyrate, c3ives N-(l-butoxycarbonyl-3-phenylpropyl)-
~-~Lanyl-N-~indan-2-yl)c31ycine hydrochloride mel-ting at
154-156C with decomposition.
IR Spectrum vmUax lcm 1 1740(ester), 1700(COOH),
1640(CO-N)
Example 9
The same procedure as Example 7, excepting the use of
isopropyl 2-oxo-4-phenylbutyrate in place of propyl 2-oxo-
~Z87~
4-phenyl-butyrate, gives N-~l-isopropoxycarbonyl-3-
phenylpropyl)-L-alanyl-N-(indan-2-yl)glycine hydrochloride
melting at 150-153C with decomposition.
Example 10
The same procedure as Example 7, excepting the use of
isobutyl 2-oxo-4-phenylbutyrate in place of propyl 2-oxo-
4-phenyl~utyrate, gives N-(l-isobutoxycarbonyl-3-phenyl-
propyl)-L-alanyl-N-(indan-2-yl)glycine hydrochloride melting
at 148-149C with decomposition.
Example 11
The same procedure as Example 7 using methyl 2-
oxo-4-phenylbutyrate in place of propyl 2-oxo-4-phenyl-
butyrate and methanol as the solvent in place o~ ethanol,
gives N-(l-methoxycarbonyl-3-phellylpropyl)-L-alanyl-N-
(indan-2-yl)glycine hydrochloride as colorless needles
melting at 163-165C with decomposition.
IR Spectrum ~NaXolcm 1: 1750(COOCH3), 1705~COOH),
1640 (CO-N)
Example 12
The same procedure as Example 7, excepting the use of
ethyl 4-(4-chloropllenyl)-2-oxobutyrate in place of propyl
2-oxo-4-phenylbutyrate, gives N-~ l-ethoxycarbonyl-3-(4-
chlorophenyl)propyl~-L-alanyl-N-(indan-2-yl)glycine hydro-
chloride.
Melting point 163-168C (decomposition)
[a]D + 28.1 (c=l, methanol)
IR Spectrum v aUxlcm 1 1740, 1710, 1640
Example 13
The same procedure as Example 7, excepting the use of
ethyl 2-oxo-4-(p-tolyl)butyrate in place oE propyl 2-oxo-
4-phenylbutyrate, gives N-~ l-ethoxycarbonyl-3-(p-tolyl)-
propyl]-L-aLanyl-N-(indan-2-yl)glycine hydrochloride.
Melting point 160-163C (decomposition)
[~]D + 22.2 (c=l, methanol)
IR Spectrum vmaXlcm 1 1740, 1710, 1640
Example 14
374~4
_ 27 -
To 100 ml of ethanol, there are added 3 g of ~-alanyl-
N-(indan-2-yl)glycine tert-butyl ester oxalate, 0.75 g of
sodium acetate, 1.5 g of acetic acid, 5 g of molecular
sieve 3A and 5 g of ethyl 4-(4-benzyloxyphenyl)-2-oxobutyrate,
and catalytic reduction is carried out using Raney nickel
as catalyst. When the hydrogen absorption has ceased, the
catalyst is removed, and catalytic reduction is further
carried out usi-ng palladium-carbon as catalyst. The catalyst
is filtered off and the solvent is distilled off under
reduced pressure to give N-[l-ethoxycarbonyl-3-~p-hydroxy-
phenyl)propyl]-L-alanyl-N-(indan-2-yl)glycine tert-butyl
ester as oil. This oil is converted by the reaction pro-
cedure as used in Example 4 to N-[l-ethoxycarbonyl-3-(4-
hydroxyphenyl)propyl]-L-alanyl-N-(indan-~-yl)glycine hydro-
bromide, which is further converted to the hydrochloride bythe procedure of Example 6. There is thus obtained 0.65 c~
of N-[l-ethoxycarbonyl-3-(4-hydroxyphenyl)propyl]-L-alanyl-
N-(indan-2-yl)glycine hydrochloride as colorless crystals
melting at 126-130C with decomposition.
[a]D + 17.7 ~c=l, methanol)
Mass Spectrum m/e: 450(M -HCl-H2O), 335, 334, 331, 330,
284, 215, 214, 169, 168, 133, 129, 12Q, 117, 116, 107
Example 15
L-Alanyl-N-(indan-2-yl)glycine (3.5 g) is suspended
in a mixture of 30 ml of ethanol and 20 ml of water, and
to the suspension is added 15 g of ethyl 2-oxo-4-(3,4-
dimethoxyphenyl)butyrate With stirrin~ at room temperature,
a solution of 1.4 ~ of sodium cyanoborohydride in 10 ml of
ethanol is added dropwise over about 3 hours. After stirring
for an hour, the reaction mixture is concentrated, water is
added, and the insoluble matter is removed by extraction
with ether. On adjusting the aqueous layer to pH 4 with
diluted hydrochloric acid, an oil separates, which is
extracted with ethyl acetate. The ethyl acetate layer is
3~ washed with water, dried and concentrated under reduced
pressure. bn adding first 2 ml of 4N hydrochloric acid-
~2~7~4~
- 28 -
ethanol solution and then 300 ml of ether to the residue,
an oily substance separates. The ether is removed by
decantation, and the oil is further washed with ether by the
same procedure, whereby the oil turns into powder. This
powder is crystallized by treatment with dichloromethane-
ether and recrystallized from the same mixed sol~ent to
gi~e 0.4 g o~ N-[l-(S)-ethoxycarbonyl-3-(3,4-dimethoxy-
phenyl)propyl]-L-alanyl-N-(indan-2-yl)glycine hydrochloride
as crystals melting at 138-140C with decomposition.
~]22+ 34~ (c=0.5, ethanol)
Example 16
L-Alanyl-N-(indan-2-yl)glycine (1.0 g) and 6.0 g of
phenylpyruvic acid are dissolved in 50 ml of 70% aqueous
methanol, and the p~ is adjusted to 7.0 with aqueous
potassium hydroxide. Sodium cyanoborohydride (1.0 g ) is
added to this solution. After allowing the mixture to
stand at room temperature overnight, the solvent is dis-
tilled off under reduced pressure, the residue is dissolved
in 2 ml of water, made adsorbed on a Dowex 50 (H ) [trade
mark of ion-exchange resin~ column and eluted ~ith 2%
pyridine. The solvent is distilled off under reduced
pressure, the residue is purified by silica gel column
chromatography using acetonitrile~methanol ~4:1) as the
developer to give 0.3 g of N-(l-carboxy-2-phenylethyll-L-
2~ alanyl-N-(indan-2-yl)glycine as colorless powder.
Elemental Analysis for C23H26N205 HCl
Calcd.: C, 61.8~; H, 6.09; N, 6.27
Found : C, 61.43; H, 6.08; N, 6.61
NMR Spectrum (D20) S: 1.20-1.60(3~1,C~13), 2.80-4.Q2(lOH),
4.90-5.30(1~), 7.10-8.40(10~1,Ph)
Mass Spec~rum m/e: 392(M-~I20~
Example 17
By using 1.0 g of L-alanyl-N-(indan-2-yl)glycine,
6.0 g of 2-oxo-butyric acid and 1.0 g of sodium cyano-
borohydride in a similar procedure to that of Example 1,
there can be obtained 0.4 g of N-(l-carboxypropyl)-L-alanyl-
~;Z8~
29 -
N-(indan-2-yl)glycine as colorless powder.
Elemental analysis for C18H24N2O5
Calcd.: C, 62.05; H, 6.94; N, 8.04
Found : C, 61.97; H, 7.58; N, 7.46
NMR Spectrum (D2O) ~: 1.00(3H,J=6Hz,CH3), 1.25-1.50(3H,
-CH3), 3.00-3.85(10H), 5.10-5.20~1H), 7.28(4H,Ph)
Mass Spectrum m/e: 330(M-H2O)
Example 18
N-(N'-Ethoxycarbonylmethyl-N'-benzylglycyl)-N-(indan-
2-yl)glycine benzyl ester (3 g) is dissolved in 100 ml of
ethanol, and catalytic reduction is carried out at ordinary
temperature and pressure using 5% palladium-carbon as
catalyst. After absorption of 2 equivalents of hydrogen,
the reaction mixture is filtered to remove the catalyst.
1~ The ethanol is distilled off under reduced pressure to ~ive
an oily substance. Addition of ethanolic hydrochloric acid
with 100 ml of ether to the oily substance gives N-(N'-
ethoxycarbonylmethylglycyl)-N-(indan-2-yl)glycine hydro-
chloride as colorless powder.
Elemental analysis for C17H22N2O5 HCl~ H2O
Calcd.: C, 53.75; H, 6.37; ~, 7.38
Found : C, 53.63; H, 6.87; N, 7.18
Mass Spectrum m/e: 316(M-3/2H20-HCl)
Example 19
2~ N-(l-(S)-Ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N-
(indan-2-yl)glycine hydrochloride (1.2 ~) is dissolved in
30 ml of methanol. To the solution is added 5 ml of 2N
aqueous sodiu~ hydroxide. After stirrin~ at room tempera-
ture overni~ht, the reaction mixture is concentrated under
reduced pressure, and 30 ml of water is added. On adjusting
the pE~ to S-6 with diluted hydrochloric acid, an oil
separates, which is extracted with ethyl acetate. The ethyl
acetate layer is washed with water and dried, and the
solvent is distilled off under reduced pressure. Methanol
(S ml) is added to the residue, and the solution is allowed
to stand. There is obtained 0.6 g of N-(l-(S)-carboxyl-3-
~2E~7~
- 30 -
phenylpropyL)~L-alanyl-N-(indan-2-yl)glycine as crystals
melting at 140-142C.
[~]D + 26~ (c=0.6, 1~ ~Cl)
Example 20
N-[l-Ethoxycarbonyl-3-(3,4-dimethoxyphenyl)propyl]-
L-alanyl-N-(indan-2-yl)glycine hydrochloride (1.1 g) is
dissolved in 30 ml of methanol. To the solution is added
5 ml of 2N aqueous sodium hydroxide. After stirring for 4
hours, the reaction mixture is concentrated, and 30 ml of
water is added. On adjusting the pH to about 5 with diluted
hydrochloric acid, an oily substance separates, which is
extracted with ethyl acetate. The extract is washed with
water, dried and concentrated. Methanol (4 ml) is added to
the residue, and the mixture is allowed to stand. There is
15 obtained 60 mg of N-[l-carboxy-3-(3,4-dimethoxyphenyl)-
propyl]-L-alanyl-N-(indan-2-yl)glycine as crystals melting
at 160-165C with decomposition.
Example 21
I.-Leucyl-N-(indan-2-yl)glycine tert-butyl ester
20 (3.5 g) is dissolved in 50 ml of ethanol, and 3.5 g of ethyl
2-oxo-4-phellylbutyrate, 1.5 g of sodium acetate, 3.5 g of
acetic acid, 7.0 g of molecular sieve 3A and 5.0 g of Raney
nickel are added, and catalytic reduction is carried out
under ordinary temperature and pressure. Af~ter absorption
of the theoretical amount of hydrogen, the catalyst is
filtered off, and the filtrate is concentrated ul~der reduced
pressure. To the residue, 100 ml oE water and 2 g of sodium
hydrogen carbonate are added, and the mixture is extracted
with 200 ml of ethyl acetate. ~fter washing the extract
~0 with water and dryin~, the ethyl acetate is distilled off
under reduced pressure, and 20 ml of acetic acid and 5 ml
of 25% hydrobromic acid solution in acetic acid are added
to the residue. After stirring at room temperature for 10
minutes, 100 ml of ether is added, and the resulting oil
3~ layer is separated and washed wi-th ethyl ether to give N-
(l-ethoxycarbonyl-3-phenylpropyl)-L-leucyl-N-(indan-2-yl)-
`` ~LZ~37~4
- 31 -
glycine hydrobromide. This is suspended in 10 ml of water,
the suspension is made alkaline with sodium hydrogen
carbonate, and the insoluble matter is extracted with ethyl
ether. The aqueous layer is adjusted to pH 4.0 with 10%
hydrochloric acid, and extracted with 200 ml of ethyl
acetate. The extract is washed with water and dried over
magnesium sulfate, 1 ml of ethanolic hydrochloric acid is
added, and the ethyl acetate is distilled off under reduced
pressure. On adding 200 ml of ethyl ether to the residue
and allowing the mixture to stand at room temperature, there
is yielded 0.8 g ~f N-(l-ethoxycarbonyl-3-phenylpropyl)-L-
leucyl-N-(indan-2-yl)glycine hydrochloride as colorless
amorphous powder.
NMR Specerum (d6-DMSO) ~: 0.93(6~,d,J=3Hz,CH(CH3)2),
1.30(3H,t,J=7Hz,CH3), 2.90-3.20(4H,m,CH~), 3.9~(2H,
s,CH2), 4.00-4.35(3H,m), 7.20(4H,s,Ph), 7.30(5H,s,Ph)
Mass Spectrum m/e: 476, 431, 361, 315, 171
Example 22
N~-Carbobenzoxy-L-lysyl-N-(indan-2-yl)glycine tert-
butyl ester oxalate (3.5 g) is dissolved in 20 ml of
methanol, and 1 g of sodium acetate, 1.2 g of acetic acid,
8 g of molecular sieve and 15 g of ethyl 2-oxo-4-phenyl-
butyrate are added. To this mixture is added dropwise with
stirring a solution of 3.3 g of sodium cyanoborohydride in
30 ml of methanol over 2 hours. 3 g of sodium cyanoboro-
hydride is further added, and the mixture is stirred for 3
hours. After addition of 200 ml of 25~ aqueous phosphric
acid, the reaction mixture is extracted with two 200-ml
portions of ethyl acetate. The extract is washed with 0.5N
a~ueous sodium hydroxide and with water, and dried over
anhy~rous magnesium sulfate. The ethyl acetate is distilled
off under reduced pressure, and the oil thus obtained is
separated and purified by silica gel column chromatography
(acetone-benzene (1:12 to 1:5)). The first fraction gives
1.1 g of N -[l-(R)-ethoxycarbonyl-3-phenylpropyl]-N -
carbobenzoxy-L-lysyl-N-(indan-2-yl)glycine tert-butyl ester
~28744~
- 32 -
as an oil.
Elemental analysis for C41H53N3O7
Calcd.: C, 70.36; H, 7.63; N, 6.00
Found : C, 69.89; H, 7.64; N, 5.76
[~]24 = _4~5O (c=l, methanol)
The second ~raction gives 1 g of Na-[l-(S)-ethoxy-
carbonyl-3-phenylpropyl]-N-carbobenzoxy-L-lysyl -N- ( indan-
2-yl)glycine tert-butyl ester as an oil.
Elemental analysis for C41H53N3O7
Calcd.: C, 70.36; H, 7.63; N, 6.00
Found : C, 70.28 H, 7.51; N, 5.93
~]D = -8.2 (c=l, methanol)
IR Spectrum v eaxtcm 1 3300(NH), 1720(C=0), 1630(C=0)
Example 23
N~-[l-(R)-Ethoxycarbonyl-3-phenylpropyl]-N-carbo-
benzoxy-L-lysyl-N-(indan-2-yl)glycine tert-butyl ester
(1 g) is dissolved in 2 ml of acetic acid. ~o the solution,
8 ml of 25% hydrobromic acid solution in acetic acid is
added, and the mixture is allowed to stand at room tempera-
ture for 30 minutes. On adding 200 ml of ether and 100 ml
of petroleum et;ler to the reaction mixture, a crystalline
precipitate forms. This is collected by filtration and
recrystallized from a mixture of methanol and ethyl ehter
to give 0.8 g of N~-[l-(R~-ethoxycarbonyl-3-phenylpropyl]-
L-lysyl-N-(indan-2-yl)glycine dihydrobromide melting at
128-133C with decomposition.
[a~24 = -11.6 (c=l, methanol)
Example 24
By a procedure similar to that described in ~xample
23, ~there is obtained 0.6 g of N~-[l-(S)-ethoxycarbonyl-3-
phenylpropyl]-L-lysyl-N-tindan-2-yl)glycine dihydrobromide
from 0.9 g of N -[l-~S)-ethoxycarbonyl-3-phenylpropyl]-N -
carbobenzoxy-L-lysyl-N-(indan-2-yl)glycine tert-butyl ester.
Melting point: 160-165C (decomposition)
[ ]24 + 18 0 ~c=l, methanol)
Example 25
~ 3 Z87~44~
- 33 -
By reacting L-alanyl-N-(indan-2-yl)glycine tert-butyl
ester oxalate (2.0 g) with N-butyl-2-oxo-4-phenylbutyrylamide
(4.0 g) in a similar manner to that of Example 2, there is
obtained N-(l-butylaminocarbonyl-3-phenylpropyl)-L-alanyl-
N-(indan-2-yl)glycine tert-butyl ester (0.5 g) as colorless
oil .
NMR Spectrum (~DC13): 1.90(3H,m,CH3), 1.40-1.55(16H,m),
2.60-3.49(8H,m,CH2), 3.70-3.95(2H,m), 7.20(4H,s,Ph),
7.30(5H,s,Ph)
Example 26
Use of N-(l-butylaminocarbonyl-3-phenylpropyl)-L-
alanyl-N-(indan-2-yl)glycine tert-butyl ester (0.5 g) as a
reactant in a similar manner to that of Example 4, gives
N-(l-butylaminocarbonyl-3-phenylpropyl)-L-alanyl-N-(indan-
2-yl)glycine hydrobromide as colorless amorphous powder.
NMR Spectrum (d6-DMSO) ~: 0.8-1.0(3H,m,CH3), 1.2-1.55
(7H,m,CH3+CH2x2), 1.90-2.25(2H,m,C~2), 2.40-2.60(2H,
m,CH2), 2.95-3.30(6H,m,CH2), 4.80-5.0(1H,m,CH), 7.10-
7.30~9H,m,Ph)
Example 27
L-Alanyl-N-(indan-2-yl)-~-alanine (2.0 g) is dissolved
in a mixture of 20 ml of water and 100 ml of ethanol, 10 g
of ethyl 2-oxo-4-phenylbutyrate is added, and a solution of
0 94 g of sodium cyanoborohydride in 20 ml of ethanol is
added dropwise over 2 hours. The mixture is stirred at room
temperature for 3 hours and then adjusted to p~l 4.0 with
10% hydrochLoric acid, and the e~hanol is distilled off
under reduced pressure. The residue is extracted with 200 ml
of ethyl acetate, washed with water and dried, and the ethyl
acetate is distilled off under reduced pressure. The
residue is dissolved in 2 ml of 20~ ethanolic hydrochloric
acid, 100 ml of ethyl ether is added, and the mixture is
allowed to stand at room temperature. Thus is obtained
1.2 g of N-(l-ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N-
(indan-2-yl~-~-alanine hydrochloride as colorless amorphous
powder.
. . . . . .
lZ8~444
-- 34 -
NMR Spectrum (d6-DMSO) ~: 1.30(3H,t,J=7.5Hz), 1.40(3H,
d,J=4.5Hz), 2.65-3.00(4H,m,CH2), 3.20-3.40(4H,m,CH2),
4.25(211,q,J=7.5Hz,CH2), 7.20(4H,s,Ph), 7.30(5H,s,Ph)
Example 28
L-Alanyl-N-(1,2,3,4-tetrahydrophthalen-2-yl)glycine
(3.0 g) is dissolved in 200 ml of ethanol, 15 g of ethyl 2-
oxo-4-phenylbutyrate and 12 g of molecular sieve 3A are
added, and the mixture is stirred at room temperature for
30 minutes. Thereafter, a solution of 1.3 g of sodium
cyanoborohydride in 50 ml of ethanol is added dropwise over
6 hours, and stirring is further continued for 2 hours. The
insoluble matter is filtered off, and the filtrate is con-
centrated to (lryness under reduced pressure. Water (50 ml)
is added to the residue, the mixture is made alkaline with
sodium hydrogen carbonate, and the insoluble matter is
removed by extraction with ethyl ether. The aqueous layer
is adjusted to pH 4.0 with 10% hydrochloric acid and then
extracted with 2ûO ml of ethyl acetate. The extract is
washed with water and dried, 1 ml of 20~ alcoholic hydro-
chloric acid is added, and the ethyl acetate is distilled
off under reduced pressure. Ethyl ether (100 ml) is added
to the residue, and the mixture is allowed to stand at
room temperature to give 1.5 g of N-(l-ethoxycarbonyl-3-
phenylpropyl)-L-alanyl-N-(1,2,3,4-tetrahydronaphthalen-2-yl)-
glycine hydrochloride as colorless amorphous powder.
NMR Spectrum (d6-DMSO) ~: 1.30(3H,t,J=7Hz,C~13), 1.40
(3H,d,J=6Hz,CH3), 1.70--2.30(5H,m,CH2), 2.80-3.05(7H,
m,CH2), 4.û0(2H,s,CH2), 4 30(2EI,q,J=7Hz,CH2), 7.10
(4H,s,Ph), 7.30(5H,s,Ph)
Mass Spectrum m/e: 448(M-HCl-H20), 344, 319, 168
Example 29
By reacting 3 g of L-alanyl-N-(indan-l-yl) glycine
with 15 g o~ ethyl 2-oxo-4-phenylbutylate and treating the
reaction mixture as in Example 28, there is obtained 1.2 g
3; of N-(l-ethoxycarbonyl-3-phenylpropyl]-L-alanyl-N-(indan-l-
yl)glycine hydrochloride as colorless amorphous powder.
. .
lZ~79~4~
NMR Spectrum (d6-DMSO) ~: 1.20-1.45(3H,m,CH3), 1.55-
1.65(3H,m,CH3), 2.70-3.05(4H,m,CH2), 4;00~2H,s,C~2),
4.40(2H,q,J=7HzrCH2), 4.80-5.00(1H,m,-CH-), 7.30(9H,
s,Ph)
Mass Spectrum m/e: 434(M-HCl-H2O), 258, 244, 180
Example 30
By reacting 3 g of L-alanyl-N-(5-hydroxyindan-1-yl)-
glycine with 15 g of ethyl 2-oxo-4-phenylbutylate and treat-
ing the reaction mixture as in Example 28, there is obtained
0.8 g of N-(l-ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N-
(5-hydroxyindan-1-yl)glycine hydrochloride as colorless
amorphous powder.
NMR Spectrum (d6-DMSO) ~: 1.10-1.70(6H,m,C~3x2), 1.75-
3.30(8H,m,CH2x4), 3.70-4.30(5H,mrCH2x2+CH), 4.60-
5.20(2H,m,CHx2), 6.55-7.30(8H,m,Ph)
Example 31
L-Alanyl-N-(5,6-dimethoxyindan-1-yl)glycine tert-butyl
ester (4.0 g) is dissolved in 80 ml of ethanol, and 0~77 g
of sodium acetate, 4.0 g o acetic acid, 4.0 ~ of ethyl
20 2-oxo-4-phenyl-butyrate, 12 g o~ molecular sieve 3A and
6.0 g o~ Raney nickel are added. Thereafter, the reaction
and treatment are carried out as in Example 2 to give 2.0 g
of N-(l-ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N-(5,6-
dimethoxyindan-l-yl)glycine tert-butyl ester as slightly
yellow oil.
NMR Spectrum (CDC13) ~: 1.20-1.50(15H,m,CH3xS), 1.90-
2.20(2H,m,C~I~), 2.50(2H,s,CH2), 2.60-2.95(4H,m,CH2),
3.80(3H,s,OCH3), 3.90(3H,s,oCH3), 4 20(2H,q,J=7.0Hz,
CH2~, 5.~0-5.60(1H,m,CH), 6.80-6.90~2H,m,Ph), 7.2S
(5H,s,Ph)
Mass Spectrum m/e = 568(M~), 495, 361, 308, 234
Example 32
N-(l-Ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N~(5,6-
dimethoxyindan-1-yl)glycine tert-butyl ester (2.0 g) is
35 dissolved in 10 ml of acetic acid, and 2 ml of 25~ hydro-
bromic acid in acetic acid is added dropwise with ice cooling.
lZ~'~444
- 36 -
After allowing the mixture to stand at room temperature for
5 minutes, 50 ml of ethyl ether is added to the reaction
mixture, whereupon amorphous powder precipitates. This is
collected by filtration and dissolved in 10 ml of water.
The solution is made alkaline with sodium hydrogen carbonake,
and the insoluble matter is removed by extraction with
ethyl ether. The aqueous layer is adjusted to pH 4.0 with
10~ hydrochloric acid and extracted with 100 ml of chloro-
form. Tne extract is washed with water and dried, 1 ml of
20~ ethanolic hydrochloric acid is added, and the chloro-
form is distilled off under reduced pressure. The residue
is dissolved in 2 ml of ethanol. On adding 20 ml of ethyl
ether, colorless amorphous powder precipitates. Collection
of this precipitate by filtration gives 0.2 g of N-(l-
ethoxycarbonyl-3-phenylpropyl)-L-alanyl-N-(5,6-dimethoxy-
indan-l-yl)~lycine hydrochloride as eolorless powder.
NMR Speetrum (d6-DMSO) ~: 1.25(3H,t,J=8Hz,CH3), 1.40
(3H, d, J=4.5Hz,CH3~, 3.75(3H,s,OCH3), 3.80(3lt,s,OCH3),
6.80(1H,s,Ph), 6.90(1H,s,Ph), 7~25(5H,s,Ph)
Example 33
By the similar procedure to that in Example 32, N-(l-
ethoxycarbonyl-3-pnenylpropyl)glycyl-N-(indan-2-yl)glycine
hydrochloride is obtained from N-(l-ethoxycarbonyl-3-phenyl-
propyl)glycyl-N-(indan-2-yl)glyeine tert-butyl ester as
2~ colorless amorphous powder.
NMR Speetrum (d6-DMSO) ~: 1.30(3H,t,J=6Hz,CH3), 2.25(2H,
m,C112), 2.70(2~1,m,C~12), 2.~0-3.20(4H,m,CH2), ~.00-
4.60(5~1,m), 4.30-5.10(1~,m), 7.30(4H,s), 7.35(5H,s,Ph)
Experiment l
~0 Inhibitions of An~iotensin I Convertin~ Enzyme (ACE)
by the Compounds of this Invention.
Experimental Method
The experiment was conducted in aecordance with a
modifieation of the method described by Cushman et al.
3~ [Biochemical Pharmacology, Vol. 20, 1637(1971)]. That is,
using hippuryl-L-histidyl-L-leucine(HHL) as the substrate,
121~74 ~4
- 37 -
the ACE inhibitory activity was determined in terms of
percent inhibition on the amount of hippuric acid produced
by ACE when the present compound was added. A solution of
the compound of the present invention dissolved in a 0.02
to 2~ dimethyl sul~oxide-100 mM potassium phosphate buffer
solution ~pH 8.3, containing 300 mM sodium chloride) was
added to 100 ~1 of ACE (protein concentration, 20 mg/ml)
and 100 ~1 of 1.25 InM HHL. In this experiment, a potassium
phosphate buffer solution containing dimethyl sulfoxide at
a concentration equal to that of the test solution was used
as a control. After incubating the solution at 37C for
one hour, 150 ~1 of lN hydrochloric acid was added to the
solution to termina~e the reaction. After 1 ml of ethyl
acetate was added, the solution was centrifuged at 3000
r.p.m. for 10 minutes. A O.S ml aliquot was separated from
the ethyl acetate layer and dried at a temperature below
50C under nitrogen gas streams. The residue was mixed
with 5 ml of 1 M aqueous sodium chloride and the mixture
was subjected to colorimetry at a wavelen~th o~ 228 nm.
Test Result
The test results obtained with respect to the com-
pounds of Examples 1, 7, 8, 9 and 19 are shown in Table 2
below.
Table 2
2~
,
Example No. Concentration Inhibitory Activity
of Tested on ACE
Compound (~M) (~)
__ . _ _
;30 ~' 11_ _
~ _ _ I _ 72-
~Z~
- 38 -
Experiment 2
Effect of Present Compounds against Hypertensive
Activity of Angiotensin I
Experimental Method
Male rats (Sprague-Dawley) weighing 250 g to 350 g
which were fed under free access to drinkinu- water and
feeds were ~sed as experimental animals. The rats were
anesthetized with intraperitoneal administration of
pentobarbital sodium (50 mg/kg) on the day before the
test day and a polye~hylene tube was inserted into each of
the femoral artery for measurement of blood pressure and
the femoral vein for injection of angiotensin I and II,
and then the tubes were fixed.
On the test day, an average blood pressure in the
control phase was recorded on an electric hemodynamometer
(MP-4T model manufactured by Nippon Koden, Japan) and
thereafter angiotensin I and then angiotensin II were
injected through the femoral vein at a dose of 300 ng/kg
and 100 ng/kg respectively, to measure the hypertensive
activity. Then, 13.8 ~M/kg of the compound of this inven-
tion was administered orally as an aqueous solution or an
aqueous gum arabic suspension, and 20, 60 and 120 minutes
after the administration, angiotensin I and II were injected
repeatedly to trace hypertensive reactions. In calculating
the percent inhibition to the hypertensive activity of
angiotensin I, the percent inhibitory value was`corrected
based on the variation with time in the hypertensive
reaction by angiotensin II.
Test Result
The test results obtained with respec-t to the com-
pounds of Examples 1, 7, 9 and 15 are shown in Table 3
below
~Z8744~
- 39 -
Table 3
, Percent Inhibition (%) against
Hypertensive Reaction by
Example No. of Angiotensin I
Tested Compound _
After After After
_ 20 min. 60 min. 120 rnin.
1 93 88 77
7 84 91 96
9 88 71 51
84 80 65
Preparation Example
The compounds (I) of the present invention are used,
for example, for the treatment of hypertension in the
following examples of formulation.
1. Tablets
(1) N~ Ethoxycarbonyl-3-phenylpropyl)-
L-alanyl-N-~indan-2-yl)glycine
hydrochloride 10 g
t2) Lactose ~0 g
(3) Corn Starch 29 g
(4) Magnesium Stearate 1 g
130 g
for 1000 tablets
The above ingredients (1), (2) and 17 g of corn starch
are blended, and granulated using a paste prepared from 7 g
of corn starch. Five grams of corn starch and the ingredient
(4) are added to the resulting granules and the mixture is
compressed by a tabletting machine to prepare 1000 tablets
having a diameter 7 mm each containing 10 mg of the active
ingredient ~1).
2. Capsules
(1) N-(l-Butoxycarbonyl-2-phenylethyl)-L-
:~8~4~
- 40 -
alanyl-N-(indan-2-yl?glycine
hydrochloride 10 g
(2) Lactose 135 g
(3) Cellulose Fine Powder 70 g
(4) Magnesium Stearate 5 g
220 g
for 1000 capsules
All of the above components are blended and
encapsulated into Gelatin Capsule No. 3 (IX Japanese
Pharmacopoiea) to prepare 1000 capsules each containing
10 mg of the active component (1).
3. Injectable Solution
(1) N-~l-(S)-Carboxy-3-phenylpropyl)-L-
alanyl-N-~indan-2-yl)glycine
sodium salt 10 g
(2) Sodium Chloride 9 g
~3) Chlorobutanol 5 g
(4) Sodium Bicarbonate 1 g
All o~ the above ingredients are dissolved in 1000 ml
of distilled water and charged into 1000 brown ampules
each containing 1 ml o~ the solution. The ampules are
replaced with nitrogen gas and sealed. The entire pre-
paration steps are conducted under sterile conditions.