Note: Descriptions are shown in the official language in which they were submitted.
-- 1 --
_ el process for preparing 1,5-benzothiaze~ine derivatives
This invention relates to a novel process for
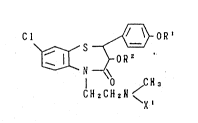
preparing l,5-benzothiazepine derivatives of the formula:
Cl ~ OR'
OR Z
O CH~
CHzCH2N /
X '
wherein Rl is a lower alkyl group, R is hydrogen atom
or a lower alkanoyl group, and Xl is hydrogen atom or a
lower alkyl group, or a salt thereof.
U.S~. Patent 3,562,257 discloses vario~s benzothiazepine
derivatives including~7-chloro-1,5-benzothiazepine deriv-
atives such as 2-(4-methoxyphenyl)-3-hydroxy(or acetoxy)-S-
t2-(dimethylamlno)ethyl]-7-chloro-2,3-dihydro-1~5-benzo-
thiazepin-4(5H)~-one. The aforesaid U.S. Patent also
discloses~ that these benzothiazepine derivatives have
antidepressive,~tranquilizing and/or coronary vasodilating
activities~
~As compared with these known compounds, the compounds
(I)~of the~ pr~esent~lnventi~on in which Xl is a lower
alkyl~group show stronger hypotensive and cerebral or
..
.
` :
- 2 -
coronary vasodilating activities and are more useful
for the treatment, amelioration and/or prophylaxis of
hypertension; cerebral diseases such as cerebral vasospasm
or cerebral infarction; and heart diseases such as angina
pectoris, arrhythmia or myocardial infarction. On the
other hand, the compounds (I) of the invention in which
X is a hydrogen atom show platelet aggregation-
inhibiting activity and are useful for the treatment,
amelioration and/or prophylaxis of thrombotic diseases
such as cerebral thrombosis (cerebral infarction), tran-
sient cerebral ischemia, coronary thrombosis (myocardial
infarction), pulmonary embolism, peripheral vascular embol-
ism, thromboangiitis and/or other thromboembolism (e.g.
the thromboembolism following heart valve replacement).
According to the present invention, the compounds (I)
can be prepared by:
i) alkylating a compound of the formula:
Cl ~ OH
OR3
I O CH3
CHzCH2N <
wherein R is a hydrogen atom or a lower alkanoyl group
~ and x2 is a hydrogen atom, a lower alkyl group or a
protecting group;
ii) when X is a protecting group, removing said
protecting group therefrom;
iii) when R3 is a lower alkanoyl group, optionally
removing said lower alkanoyl group therefrom and
iv) if required, further converting the product into a
salt thereof.
.
:
- 3 -
In the above-mentioned reactions, a wide variety of
protecting groups normally employed to protect amino
groups may be used as the protecting group X~. Examples
of such protecting groups include unsubstituted or sub-
stituted benzyloxycarbonyl groups e.g. benzyloxycarbonylor p-methoxybenzyloxycarbonyl groups; unsubstituted or
substituted lower alkoxycarbonyl groups e.g. tert.-
butoxycarbonyl, ~ -trichloroethoxycarbonyl or
iodoethoxycarbonyl groups; and unsubstituted or
substituted phenyl-lower alkyl groups e.g. benzyl,
p-methoxybenzyl or 3,4-dimethoxybenzyl groups.
The alkylation reaction of this invention can be
carried out by treating the compound (II) with a low~r
alkylating agent. Such an alkylating agent may be, for
example, a lower alkanol, a lower alkyl sulfate e.g.
dimethylsulfate, a lower alkyl ester of alkyl or aryl
sulfonate e.g. methyl tosylate, methyl methanesulfonate
and dimethyl 2-oxo-1,3-propane disulfonate, a lower alkyl
halide e.g. methyl iodide and methyl bromide, a lower
alkyl quaternary ammonium compound e.g. trimethylphenyl-
ammonium hydroxide, a lower alkyl sulfonium or sulfoxonium
compound e.g. trimethylsul~onium hydroxide, trimethyl-
sulfoxonium iodide, a lower alkyl selenonium compound e.g.
trimethylselenonium hydroxide, and a diazo lower alkane
e.g. diazomethane.
When a lower~alkanol is used as the alkylating agent,
the reaction is preferably carried out in the presence of
a dehydrating agent. Conventional dehydrating agents,
e.g. dicyclohexylcarbodiimide or a mixture of triphenyl-
phosphine and diazenedicarboxylic acid diethyl ester, maybe used for this purpose. The reaction is preferably
carried out at temperature of 0 to 40C.
When the above-mentioned lower alkyl sulfate, lower
alkyl ester of alkyl or aryl sulfonate or lower alkyl
halide is used as the alkylating agent, the reaction is
preferably carried out in the presence of a base. Examples
of the base include inorganic bases e.g. an alkali metal
p~
-- 4 --
oxide, an alkali metal carbonate, an alkali metal
bicarbonate, an alkali metal hydride or an alkali metal
alkoxide, and organic bases e.g. ethyl diisopropyl amine.
The reaction is preferably carried out at a temperature
between OC and the refluxing temperature, and especially
at 10 to 40C in an inert solvent (e.g., acetone,
acetonitrile, methanol, benzene, dioxane, tetrahydroEuran,
dimethylsulfoxide, dimethylformamide). The reaction is
also preferably carried out in the presence of a phase-
transfer catalyst e.g. tetrabutylammonium fluoride.
On the other hand, when the lower alkyl quaternary
ammonium compound, lower alkyl sulfonium or sulfoxonium
compound or lower alkyl selenonium compound is used as the
alkylating agent, the reaction is preferably carried out
at a temperature between 10C and the refluxing tempera-
ture, and especially at 50~100C, either in an inert
solvent or without a solvent. Toluene, dioxane, methanol,
dimethylformamide and dimethylsulfoxide are suitable
solvents.
When the diazo lower alkane is used as the alkylating
agent, the reaction is preferably carried out at a
temperature of 0 to 40C, and especially 20 to 30C, in
an inert solvent. Dioxane, tetrahydrofuran, methanol and
mixtures thereof are suitable as the solvent. When the
group R3 of the starting compound ~II) is a lower alkanoyl
group, the reaction is preferably carried out in the
presence of fluoroboric acid or silica gel.
When carrying out the above-mentioned alkylation
reaction, it is particularly preferred to use the lower
alkyl sulfate, lower alkyl sulfonate or lower alkyl halide
as the alkylating agent when the group x2 of the
~ 5 ~3~
starting compound (II) is a hydrogen atom and the group
R is the lower alkanoyl group; or alternatively it is
particularly preferable to use the lower alkyl quaternary
ammonium compound, lower alkyl sul~onium or sulfoxonium
compound, lower alkyl selenonium compound or diazo lower
alkane as the alkylating agent when both the groups x2
and R3 of the starting compound (II) are a hydrogen atom.
When the group x2 of the thus-obtained product is a
protecting group, removal of the protecting group may be
conducted in a conventional manner. For example, when
the protecting group is an unsubstituted or substituted
benzyloxycarbonyl or unsubstituted lower alkoxy carbonyl,
the group may be readily removed by treating the compound
with an acid e.g. hydrogen bromide, hydrogen chloride or
trifluoroacetic acid in a solvent. When the protecting
group is a substituted lower alkoxy carbonyl, the group may
be removed by treatment with zinc in a solvent. On the
other hand, when the protecting group is an unsubstituted
or substituted phenyl lower alkyl, the group may be
removed by first substituting it with a protecting group
(e.g., benzyloxycarbonyl group) which is readily removable
with an acid, and then treating the latter group in the
same manner as mentioned above. Substitution of the
unsubstituted or substituted phenyl lower alkyl with the
benzyloxycarbonyl group is pre~erably carried out by
treating the product (i.e., the compound in which the imino
group is protected by the unsubstituted or substituted
phenyl lower alkyl group) with benzyloxycarbonyl halide,
e.g. benzyloxycarbonyl chloride, at a temperature between
50 and 130C in a solvent.
When R3 is the lower alkanoyl group, the protecting
group and the lower alkanoyl group may be removed simul-
taneously, or the protecting group may be removed without
removal~ of the lower alkanoyl group. These reactions can
be controlled by changing the reaction conditions, e.g.
the amount of the reaction agents, the reaction time
and/or the reaction solvent used.
- 6 ~
If required, the compound (I) of the present invention
thus obtained may be converted into its acid addition
salts by treating it with an acid. Examples of the acid
addition salts include inorganic acid addition salts e.g.
hydrochloride, hydrobromide, hydroiodide, perchlorate,
sulfate or phosphate, organic acid addition salts e.g.
oxalate, maleate, fumarate or methanesulfonate, and so
forth.
Since the above-mentioned reactions of the invention
can be carried out without racemization, the compound (I)
in an optically active form can be readily obtained by the
use of an optically active isomer of the comp~nd (II) as
the starting compound.
The starting compound (II) may be prepared, for
lS example, by: reacting 3-(4-benzyloxyphenyl)glycidic acid
methyl ester with 5-chloro-2-aminothiophenol to give 2-(4-
benzyloxyphenyl)-3-hydroxy-8-chloro-2,3-dihydro-1,5-benzo-
thiazepin-4(5H)-one or a salt thereof; condensing the
product with a compound of the formula:
" , CH3
Y CH2CH2N (III)
X2
wherein Y is a halogen atom and x2 is the same as
defined above, or a salt thereof, to give a compound of
the formula:
Cl ~ ~ O-CH2 ~
OH (IV)
I O CH
CH2CHzN /
~X 2
.
- 7 ~
wherein x2 is the same as defined above; removing the
benzyl group Erom the compound (IV); and if required,
converting the hydroxy gro~p of the thus-obtained product
at the 3-position of the benzothiazepine ring into a lower
alkanoyloxy group before or after removal of the benzyl
~roup.
The compound ~I) and the starting compound (II) can
exist in the form of two diastereoisomers (e.g., cis and
trans) or four optical isomers (e.g., (+)-cis, (-)-cis,
(+)-trans, and (-)-trans) due to the two asymmetric carbon
atoms involved therein, and all of these isomers and
mixtures thereof are included within the scope o~ the
invention. Among these isomers, however, the cis isomer
o~ the compound (I) is pre~erred for medicinal use.
Presently preferred embodiments of the invention are
described in detail in the following Examples which should
not be construed as limiting.
Example 1
690 mg of (+)-cis-2-(4-hydroxyphenyl)-3-hydroxy-5-
[2-(N,N-dimethylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one were dissolved in 20 ml of
tetrahydrofuran. Sodium hydride (78 mg, 60% oily) was
added thereto at room temperature and the mixture was
stirred ~or 30 minutes at the same temperature. A
solution of 245 mg of dimethyl sul~ate in 10 ml of
dimethylformamide was added thereto, and the mixture was
stirred ~or 1 hour at room temperature. The reaction
mixture was poured into water and extracted with ethyl
acetate. The extract was washed with water, dried and
evaporated to remove the solvent. The residue was
recrystallized from a mixture of ethyl acetate and
n-hexane. The product was 509 mg of (~)-cis-2-~4-
methoxyphenyl)-3-hydroxy-5-12-(N,N-dimethylamino)ethyl]-
8-chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one.
M.p. 122-124C (decomp.)
[~] D0 +144.6 (C=0.85, methanol)
- 8
Examples 2 to 4
The corresponding starting compounds were treated in
the same manner as described in Example 1 to gi~e the
compounds shown in Table 1.
Table 1
Cl S ~ OH Cl S ~ OR'
OR3 ~ ~ oR2
I O CH3 ~ O CH3
CHzCH2N / CHzCHzl~ /
(Il) \XZ ( I ) ~X'
(R', X' and XZ- -CH3, R3= RZ)
Ex. Compound ~I) Optlcal Properties
No. R activity
.
2: -H cis-(-) M.p. 121-123C (decomp.)
: ~20 -142.70(c=l 04
_ D methanol)
3 -COCH3 ~ cis-~+) Maleate(recrystallized ~rom
~ etha.nol-ether)
: : M.p. 158-160C
_ ~ ~ ( )2~75 4 (C=1.0,methanol) ~
4 -COCH3 cis-~ Hydrochloride :
:: ~ M~.p. 128-132C(decomp.)
; ~ : I : t~)D -93.3(C=0.872,
ethanol)
:
:
:
Example 5
A 210 mg amount of (~)-cis-2-(4-hydroxyphenyl)-3-
acetoxy~5-[2-(N-methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one, 250 mg of potassium carbonate
and 140 mg of dimethylsulfate were dissolved in 5 ml of
methanol, and the solution ~7as stirred at room temperature
overnight. Insoluble materials were filtered of, and
the filtrate was evaporated to remove the solvent. The
residue was purified by silica gel column chromatography
(solvent; benzene : ethyl acetate = 4 : 1), converted into
the maleate and recrystallized from a mixture of ethanol
and ether. The product was 212 mg of (~)-cis-2-(4-
methoxyphenyl)-3 acetoxy-5-[2-(N,N-dimethylamino)ethyl]-
8-chloro-2,3-dihydo-1,5-benzothiazepin-4(5H)-one maleate.
The physico-chemical properties of this product were
identical to those of the product prepared in Example 3.
Example 6
(1) (-)-cis-2-(4-hydroxyphenyl)-3-hydroxy-5-[2-
(N-benzyl-N-methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin~4(5H)-one was treated in ~he same manner as
described in Example 1 to give (-)-cis-2-(4-methoxyphenyl)-
3-hydroxy-5-[2-(N-benzyl-N-methylamino)ethyl]-8-chloro-2,3-
dihydro-1,5-benzothiazepin-4(5H)-one.
Perl_hlorate (recrystallized from ethanol)
M.p. 161-163C (decomp.)
~] D -76.4 (C=0.589, methanol)
(2) 4.3 g of the product obtained in paragraph (1) were
dissolved in 50 ml of benzene and reflux~d. A solution of
4.55 g of benzyloxycarbonyl chloride in 10 ml of benzene
was added dropwise thereto for 15 minutes. The solution
was refluxed for 1 hour, cooled to room temperature, and
evaporated to remove the solvent. To the residue were
added 30 ml of ethanol and 50 ml of 5% aqueous sodium
hydroxide, and the mixture was stirred for 2 hours at room
temperature. The reaction mixture was diluted with water
and extracted with chloroform. The extract was washed
with water, dried and evaporated to remove the solvent.
The product was 4.79 g of (-)-cis-2-(4-methoxyphenyl)-3-
hydroxy-5-[2-(N-benzyloxycarbonyl-N-methylamino)ethyl]-8-
chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one.
IRv CHC13 (cm~l): 3510, 1695, 1660
[~] D -133.3 (C=0.582, methanol)
(3) 1.07 g of the product obtained in paragraph (2)
were dissolved in 2 ml of benzene, and 1.7 ml of a 25%
hydrogen bromide-acetic acid solution were added thereto.
The mixture was stirred for 2 hours at room temperature.
Ether was added thereto, and the precipitates were
collected by filtration and washed with ether. To the
precipitates were added water and benzene, and the mixture
was made alkaline with potassium carbonate. The benzene
layer was washed with water, dried, and evapora~ed to
remove the solvent. Ether was added to the residue. The
precipitates were collected by filtration, and recrystal-
lized from a mixture of ethyl acetate and n-hexane. The
product was 0.47 g of (-)-cis-2-(4-methoxyphenyl)-3-
hydroxy-5-[2-(N-methylamino)ethyl]-8-chloro-2,3-dihydro-
1,5-benzothiazepin-4(5H)-one.
M.p. 142-145C
[~] D -147.7 (C=0.814, chloroform)
Fumarate (recrystallized from ethanol)
M.p. 164-167C(decomp.)
Examples 7 to 9
(1) The corresponding starting compounds were treated
in the same manner as described in Example 6-(1) to give
the compounds shown in Table 2.
Table 2
Cl : ~30~1 Cl ~OR~
\~S~ ~S
O R 3~ O R ~
O ~CH~ - - > l \o CH3
CH2:CHzN \ CHzCH z N <
( V )
~R~= -CH~, ~XZ= -CHzCt,Hs)
`' ~ :
.
~ -- ~- - - ---~
Ex. Compound (V) Optical Properties
No. R3 activity
. _ _ _ . _ _
7 -H cis-(+) Perchlora~e(recrystallized
from ethanol)
M.p. 161-163C(decomp.)
methanol)
_ ~ . ,
8 -COCH3 cis-(-) Oxalate(recrystallized
from ethanol)
M.p. 192-194C(decomp.)
[~] D -96.5(C=1.0,
dimethylformamide)
.~ _ . ~ _ .
9 -COCH3 cis-(+) Oxalate(recrystallized
from ethanol)
M.p~ 191-194C (decomp.)
[a] D ~96.8(C=0.73,
_ _ dimethylformamide)
(2) The products (V~ obtained above were treated in
the same manner~as described in Example 6-(2) to give the
compounds shown~in Table 3.
~ Table 3
Cl ~ ~ OR'
- \~ S ~ \=/:
OR3
N :~
b: ~C It 3
c N 2 c a ~ly \CH
(V-a)
~S
N
O C I 1 3
CH,CHzN
(Y-b)
, ~ ~
( R; ~ P ~ - C II 3~
`: :
- 12 ~ 3~
,~ . . .
Ex. Compound (V-b) Optical Properties
No. R3 activity
... _ .....
7 -H cis--(+) [~,] 20 ~132.6 (C=0.550,
methanol )
8 -COCH3*) cis- (-) IR~mCaHxcl3 (cm~l) :1740,1685
[c~ ] 20 -115. 4 (C=l . O,
. D methanol)
9 -COCH3 * ) c i s- (+ ) [cl ] 20 +115 . 5 (C= 1- O
D methanol)
~note: *): After th~ ~ reaction, the residue was not
treated with ethanol-aqueous sodium hydroxide.
(3) The products (V-b) obtained above were treated in
the same manner as described in Example 6- (3) to give the
compounds shown in Table 4.
: Table 4
Cl~ ~ ,~OR':
\~ ~OR3
N ~\
1 O CH3
CHzCH~N /
~\C O O C H, ~
C i ~ O R '
o R 2
> I O CH3
C H ~ C R ~ N <
R~ C N 3, ~ R Z = - R, R ' ~ - H o r - C O C H 3 )
: :
.
:
- 1 3 - ~' .3~
.
Ex . Compound (I-a)
._._ _ . . . , .. __
No. Optical activity Properties
. . _._ ._ _
7 cis-(+) Hydrochloride 1/2 hydrate
(recrystallized from the
mixture of ethanol and ether)
M.p. 137-140C
[a] 20 +8304tC=0.415,methanol)
. _
8 cis-(-) identical with those of
the product prepared in
Example 6-(3)
. ..
9 cis-(+) identical with those of
the product prepared in
Example 7-~3)
.
Example 10
450 mg of (-)-cis-2-(4-hydroxyphenyl)-3-hydroxy-5-
[2-~N-benzyl-N-methylamino)ethyl]-8-chloro-2,3-dihydro-
1,5-benzothiazepin-4(5H)-one were dissolved in 20 ml of
S toIuene, and a solution of 490 mg of benzyloxycarbonyl
chloride in 10 ml of toluene was added thereto at 80C.
The mixture was stirred at 80C for 5 hours. After the
reaction, the solvent was distilled off. To the residue
were added 10 ml of 5% aqueous sodium hydroxide and
10 ml of methanol, and the mixture was stirred at room
temperature for 2 hours. The mixture was extracted with
ethyl acetate. The extract was washed with water, dried,
and the solvent was evaporated off. Then the residue was
purified by silica~gel column chromatography (solvent;
chloroform : methanol = 9 : 1) to give 410 mg of (-)-cis-2-
(4-hydroxyphenyl)-3-hydroxy-S-~2-(N-benzyloxycarbonyl-N-
methylamino)~ethyl]-8-chloro-2,3-dihydro-1,5-benzothiazepin-
415H)~-one.
: :
- 14 - ~ ~q~
t2) A mixture of 410 mg of the product obtained in
paragraph (1), 340 mg of potassium carbonate, 170 mg o~
methyl iodide and 10 ml of methanol was stirred at room
temperature for 40 hours. After the reaction, the mixture
was evaporated to remove the solvent, and the residue
was dissolved in ethyl acetate. The solution was washed
with 10% aqueous sodium hydroxide and water, dried, and
evaporated to remove the solvent. The product was 315 mg
of (-)-cis-2-(4-methoxyphenyl)-3-hydroxy 5-[2-(N-benzyloxy-
carbonyl-N-methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one.
The physico-chemical properties of this product were
identical to those of the product prepared in Example
6-(2)-
(3) The product obtained in paragraph (2) was treated
in the same manner as described in Example 6-(3), whereby
(-)-cis-2-(4-methoxyphenyl)-3-hydroxy-5-[2-(N-methylamino)
ethyl]-8~chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one
was obtained.
Example 11
(1) (-)-cis-2-(4-hydroxyphenyl)-3-acetoxy-5-[2-
(N-benzyl-N-methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one was treated in the same manner as
described in Example 8-(2) to give (-)-cis-2-(4-hydroxy-
phenyl)-3-acetoxy-5-[2-(N-benzyloxycarbonyl-N~methylamino)
ethyl]-8-chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one.
[a] D -106.0(C=O.S0, chloro~orm)
(2) The product obtained in paragraph (1) was treated
in the same manner as described in Example 10-(2) to give
(-)-cis-2-(4-methoxyphenyl)-3-acetoxy~5-[2-(N-benzyloxy-
carbonyl-N-methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one.
(3) 37.5 g of the product obtained in paragraph (2)
and 75 ml of 25% hydrogen bromide-acetic acid and 95 ml
of dichloromethane were mixed under ice-cooling, and the
mixture was stirred at room temperature for 3 hours.
.
After the reaction, the dichloromethane was distilled off,
and ether was added to the residue. After the precip-
itates were washed with ether, the precipitates were
dissolved in water. The solution was neutralized wi'ch
potassium carbonate, and extracted with ether. The
extract was washed with water, dried and evaporated to
remove the solvent. The residue was conver'ced into the
oxalate, and recrystallized from methanol. The product
was 18.7 9 of (-)-cis-2-(4-methoxyphenyl)-3-acetoxy-5-~2-
(N-methylamino)ethyl]-8-chloro-2,3-dihydro-1,5~benzothiaze-
pin-4(5H)-one oxalate.
M.p. 174--176C
[~] 20 -74.2(C=0.814, methanol)
Example 12
645 mg of trimethylammonium tosylate were added to
sodium methoxide (prepared from 46 mg of sodium metal and
4.5 ml of methanol), and the mixture was stirred at room
temperature for 3 hours. Insoluble materials were filtered
off, and the filtrate was added to 20 ml of a toluene
solution containing 570 mg of (-)-cis-2-(4-hydroxyphenyl)-
3-hydroxy-5-[2- (N-methylamino)ethyl]-8-chloro-2,3-dihydro-
1,5-benzothiazepin-4(5H)-one. After the methanol was
distilled off, the solution was refluxed for 2 hours.
After the reaction, the mixture was evaporated ~o remove
the solvent. The residue was purified by silica gel
column chromatography (solvent: chloroform: ethanol =
95: 5) and recrystallized from the mixture of ethyl
acetate and n-hexane. The product was 275 mg of (-)-cis-2-
(4-methoxyphenyl)-3-hydroxy-5-[2- (N-methylamino)ethyl]-8-
chloro-2~3-dihydro-1,5-benæothiazepin-4(5H)-one.
The physico-chemical properties of this product were
identical to those of the product prepared in Example
6-(3)
- 16 -
Preparation of Starting Compounds
Preparation 1
(1~ A mixture of (+)-trans-3-(4-benzyloxyphenyl)
glycidic acid methyl ester and 5-chloro-2~aminothiophenol
was heated at 160C for 16 hours. Then (S)-N-(2-naphtha-
lenesulfonyl)pyrrolidine-2-carbonyl chloride and pyridine
were added to the reaction solution~ and the solution was
stirred. After the reaction, the product was separated by
silica gel column chromatography (solvent; benzene : ethyl
acetate = 4 : 1) into the (+)-isomer and (-)-isomer
thereof. After the separation, potassium carbonate, water
and methanol were added to each of the isomers, and the
mixtures were stirred at room temperature overnight.
Recrystallization of each product from ethyl acetate gave
(+)- and (-)-isomers of cis-2-(4-benzyloxyphenyl)-3-hydroxy
-8-chloro-2,3-dihydro-1,5-benzothiazepin-4-(5H)-one 1/5
ethyl acetate, respectively. (~)-isomer 1/5 ethyl acetate:
[~] 20 -74.2(C=l~00, dimethylformamide)
(-)-isomer 1/5 ethyl acetate:
[~] D -75.0(C=l.00, dimethylformamide)
(2) A mixture of (+)-cis-2-(4-benzyloxyphenyl)-3-
hydroxy-8-chloro-2,3-dihydro-1,5-benzothiazepin-4-(5H)-one,
2-(N-benzyl-N-methylamino)ethyl chloride hydrochloride,
potassium carbonate, acetone, ethyl acetate and water was
refluxed. After the reaction, insoluble materials were
filtered off, and the filtrate was evaporated to remove
the solvent. The residue was converted into oxalate and
recrystallized from a mixture of methanol and ether to
give (+)-cis-2-(4-benzyloxyphenyl)-3-hydroxy-5-[2-(N-
benzyl-N-methylamino)ethyll-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one oxalate.
M.p. 108-115C
[ ] D +85.8(C=l.00, methar.ol)
-- 17
(3) The product obtained in paragraph (2) (free base)
was dissolved in pyridine, a solution of acetyl chloride
in dichloromethane was added thereto, and the mixture was
stirred. After the reaction, the mixture was evaporated
to remove the solvent, and the residue was dissolved
in ethyl acetate. The solution was washed with water J
5% aqueous sodium bicarbonate and water, successively.
Then, the solution was dried and evaporated to remove the
solvent. The residue was converted into the oxalate, and
recrystalIized from the mixture of methanol and ether.
The product was (+)-cis-2-(4-benzyloxyphenyl)-3-acetoxy-
5-[2- (N-benzyl-N-methylamino)ethyl]-8-chloro-2,3-dihydro-
1,5-benzothiazepin-4(5H)-one oxalate.
M.p. 173-175C
[ ] D +78.4(C=l.00, methanol)
(-)-cis isomer oxalate
M.p. 172-175C
(4) The product obtained in paragraph (3) (free base)
was dissolved in toluene, and a solution of benzyloxy-
20 carbonyl chloride in toluene was added dropwise thereto.The mixture was stirred under heating. After the reaction,
the solvent was distilled off. The residue was purified
by silica gel column chromatography (solvent; chloroform:
ethyl acetate = 95: 5), and recrystallized from a mixtu~e
25 of ethyl acetate and n-hexane. The product was (+)-cis-
2- (4-benzyloxyphenyl)-3-acetoxy-5-[2- (N-benzyloxycarbonyl-
N-me'chylamino)ethyl]-8-chloro-2,3-dihydro-1,5-benzothiazepin
-4(5H)-one.
[ ] D +79.0(C=l.00, chloroform)
30 (-)-isomer
[c~] 20 -79.2(C=l.00, chloroform)
- 18 -
(5) The product obtained in paragraph (4~ ~7as
dissolved in e~hyl acetate, and thereto was added 25%
hydrogen bromide-acetic acid under cooling. ~fter the
reaction, the solvent was distilled off. The residue ~as
washed with ether, neutralized with conc. aqueous ammonia
to give the corresponding free base, and extracted with
chloroform. The extract was washed with water, dried and
evaporated to remove the solvent. The residue was purified
by silica gel column chromatography and converted into the
oxalate. The product was (+)-cis-2-(4-hydroxyphenyl)-3-
acetoxy-5-[2-(N-methylamino)ethyl]-8-chloro-2,3-dihydro-
1,5-benzothiazepin-4(5H)-one oxalate.
.p. 186-189C
[a] ~ +81.2(C=0.50, methanol)
(-)-cis-isomer
M.p. 187-189C
[~] D -81.6(C=0.50, methanol)
Preparation 2
(1) (+)-cis-2-(4-benzyloxyphenyl)-3-hydroxy-5-[2-
(N-benzyl-N-methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one was treated in the same manner
as described in Preparation 1-(4) to give (~)-cis-2-(4-
benzyloxyphenyl)-3-hydroxy-5-[2-(N-benzyloxycarbonyl-N-
methylamino)ethyl]-8-chloro-2,3-dihydro-1,5-benzothiazepin-
4(5H)-one.
M.p. 94-96C
[~] 20 ~126.0(C=l. 00, chloroform)
(-)-cis-isomer
M.p. 93-95C
la] D0 -125.6(C=l.OOj chloroform)
-- 19 --
(2) The product obtained in paragraph (1) was treated
in the same manner as described in Preparation 1-(5). The
free base obtained was converted to the corresponding
2-(4-hydroxy-benzoyl)benzoate, and recrystallized from
aqueous ethanol to give (+)-cis-2-(4-hydroxyphenyl)-3-
hydroxy-5-[2~(N-methylamino)ethyl]-8-chloro-2,3-dihydro-
1,5-benzothiazepin-4(5H)-one 2-(4-hydroxybenzoyl) benzoate
3/2 hydrate.
M.p. 124-127C
[~j] D0 ~198.1(C=0.120, methanol)
(-)-cis-isomer 2-(4-hydroxy-benzoyl)benzoate 3/2 hydrate
M.p. 124-127C
[~i] D -197.5(C=0.120, methanol)
Preparation 3
lS (1) (+)cis-2-(4-benzyloxyphenyl)-3-hydroxy-a-chloro-
2,3-dihydro-1,5-benzothiazepin-4(5H)-one and 2-(N,N-
dimethylamino)ethyl chloride hydrochloride were treated in
the same manner as described in Preparation 1-(2) to give
(~)-cis-2-(4-benzyloxyphenyl)-3-hydroxy-5-[2-(N,N-dimethyl-
amino)ethyl]-8-chloro-2,3-dihydro-1,5-benzothiazepin-4
(SH)-one oxalate.
M.p. 148-151C (decomp.)
[ ] D ~94.0(C=l.00, methanol~
(-)-cis-isomer oxalate
M.p. 149-152C (decomp.)
[ ] D -94.2(C=l.00, methanol)
(2) A mixture of the product obtained in paragraph (1)
(free base), acetic anhydride and pyridine was stirred
under heating. After the reaction,ithe solvent was dis-
tilled off, and the residue was converted into the oxalate,and recrystallized from a mixture of methanol and ether.
- 20
The product was (+)cis-2-(4-benzyloxyphenyl~-3-acetoxy-5-
[2- (N,N-dimethylamino)ethyl]-8-chloro-2,3-dihydro-1,5-
benzothiazepin-4(5H)-one 1/2 oxalate.
M.p. 168-172C tdecomP )
[a ] 20 ~78.4(C=l.00, methanol)
(-)-cis-isomer 1/2 oxalate
M.p. 168-171C (decomp.)
[~] D -78.8(C=l.00, methanol)
(3) The product obtained in paragraph (2) (free base)
was dissolved in acetic acid, and 25~6 hydrogen bromide-
acetic acid was added thereto under cooling and the
mixture was stirred. After the reaction, the solvent
was distilled off. the residue was washed with ether,
neutralized with conc. aqueous ammonia to give the free
base, and extracted with chloroform. The extract was
washed with water, dried, and evaporated to remove the
solvent. The residue was converted into the oxalate and
recrystallized from ethanol. The product was (~)cis~2~
(4-hydroxyphenyl)-3-acetoxy-5-[2- (N,N-dimethylamino)ethyl~-
~ 8-chloro-2,3-dihydro-1,5-benzothiazepin-4(5H)-one oxalate
1 hydrate.
M~p. 142-149C
[] 20 ~ +79.2(C=O.S0, methanol)
ois-isomer oxala~e 1 hydrate
M.p. 143-149C
[a] 20 -78.~3(C=O.S0, methanol)
.
Preparations 4 to 6
The corresponding starting compounds are treated in
the ~same~manner~ as described in Preparation 3-(3~ to give
the compounds shown in Table 5.
,
-- 21 --
Table 5
Cl ~30-CH
k~ S---,
N ~
O CH3
CHzCHzN <
(IV) X2
Cl ~OH
\,~S~ .
O R 3
O C H 3
: : : : CHzCHzN /
~ ~ . ( Il ) \X2
~ ~ :
: : : ::
: ~ .
- 22 -
,( Pr. Compo~ Ind ~II) Optical Properties
No. R3 x2 activity
2-(4-hydroxybenzoyl)benzoat~
(recrystallized from a
ciS-(~) mixture o acetone and
isopropyl ether)
M.p. 165-170C (dec.)
[~]D +79.8~C=1.00,methanol)
4 -H -CH3 _
2-(4-hydroxybenzoyl)benzoate
(recrystallized from a
cis-(-) mixture of acetone and
isopropyl ether)
M.p. 166-170C (dec.)
~ a] 2-80.1(C=1.00lmethanol)
_
Oxalate (recrystallized from
. cis-(+) a mixture of ethanol and
ether)
M.p. 130-135C (dec.)
a] D+97.6(c=0.50,methanol)
5 -H -C~2C6H5
Oxalate (recrystallized from
cis-(-) a mixture of ethanol and
ether)
: M.p. 130-135C (dec.)
. ~ [a]D -97.5(C=0.50,methanol)
_ : _
1/2 oxalate 1 hydrate
. (recrystallized from a
: mixture of acetone and
: ciS-(+) ether)
M.p 103-113C (dec.)
: . ~ [a]D +78.4(C=1.00,methanol)
6 -COCH3 CH2 6 5 _ _ .
1/2 oxalate 1 hydrate
: (recrystallized from a
cis-(-) mixture of acetone and
: : ether)
: ~ M.p. 103-114C (dec.)
_ _ ~ ~ ~ [~]D -78.8(C=1.00,methanol)