Note: Descriptions are shown in the official language in which they were submitted.
~L~233~
K 4889
OLEFINIC BENZOCYC~OBUTENE POLYMERS AND P~CESSES
FOR TJ~E PREPAP~TIC~N THE~30F
The inventlon relates to polymers of a benzocyclobutene and to
processes for the preparation thereof. ~ore particularly, this
invention relates to novel polymers of certain olefinic benzocyclo-
butene monomers. Furthermore, the invention relates to a novel
benzocyclobutene derivative.
m e polymerization of -monoolefins to useful thermoplastic
polymers by catalysis employing coordination catalysts is well
known. Generally, the coordination catalysts comprise at least tw~
co~pounds, one being a compound of a transition metal of groups 4
to 8 of the Periodic Table of the Elements, referred to herein as
"procatalyst" and the other being an organcmetallic compound of a
metal of groups l to 3 of the Periodic Table of the Elements,
referred to herein as "cocatalyst". Such coordination catalyst
systems are often referred to as Ziegler catalysts. A group of
these catalysts which is stereoregulating in the production of
polymers of propylene and higher -monoolefins is often referred to
as Ziegler-Natta catalysts. In commercial Ziegler-Natta catalysts,
the procatalyst is generally an active form of titanium chloride
and the cocatalyst an aLkyl comFound of aluminium or an alkyl
halide compound of alum m ium. These catalysts may be further
modified by addition of compounds which increase the stereo-
regulating effect of the catalysts, referred to herein as
selectivity control agents.
Polymers of ethylene, propylene, l-butene and 4-methyl-l-
pentene produced by means of such coordination catalysts are
materials of conmerce, both as homcpolymers and copolymers.
A novel alphamonoolefin polymer has now been found.
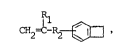
The invention provides a polymer of a benzocyclobutene of the
general formula (I):
CH2-C-R2 ~ (I)
i~;
1~93~90
where ~ represents a hydrogen atom or a methyl group and R2 a
CH2 +n group where n is 0 or an integer from l to 6.
The polymers according to the present invention are not
substituted at the cyclobutene ring. According to a preferred
embodiment they are solid homopolymers.
Further, the inventlon provides a process for the preparation
of a polymer of a benzocyclobutene, which process ccmprises homo-
polymerizing a benzocyclobutene of the general formula II):
Rl
CH2=C-R2 ~ (I)
where Rl represents a hydrogen atom or a methyl group and R2 a
-~-CH2-~-n group where n is 0 or an integer from l to 6, in the
presence of a polyolefin coordination catalyst.
The homopolymers according to the present invention are very
reactive at elevated temperature above about 200 C. Such a polymer
may be crosslinked and employed as a plastic in typical plastics
operations such as in making extruded or moulded products.
Alternatively, such a polymer may be functionalized, for example
with maleic anhydride, and employed as a polymer blend component,
for example with nylons. Fundamentally, nylons are condensation
products of hexamethylene diamine and adipic acid. Further, such
polymer can be an important intermediate that may be used as a
matrix with other polymerizable monomers.
One key aspect of the present invention involves the homo-
polymerization of the benzocyclobutene monomer of the general
formula I and the resulting polymer. Preferably, Rl represents a
hydrogen atom and n is 0, l or 20 Preferred homopolymers are:-
~onomer Hereinafter also referred to as
4-(3-butenyl)benzocyclobutene 4~BC
3-t3-butenyl)benzocyclobutene 3BBC
4-allyIbenzocyclobutene 4ABC
30 3-allylbenzocyclobutene 3ABC
4-vinylbenzocyclobutene 4VBC
3-vinylbenzocyclobutene 3VBC
4-isopropenylbenzocyclobutene 4IBC
3-isopropenylbenzocyclobutene 3IBC
1~93~ 90
According to another eDbxY1Lment the present invention provides
a solid copolymer of a benzocyclobutene of the general formula I
and of:-
(a) a C2-C8 alphamonoolefin,
(b) a monoalkenyl arene and which copolymer is a random copolymer,
or
(c) a monoalkenyl arene and which copolymer is an AB block copolymer,
AB~ block copolymer, (AB-t-mY block copolymer, (PE~-~n--Y-~-A)o
block copolymer or an ( ~ Y-~-B)p block copolymer or a
mixture thereof where each "A" represents a copolymer block of
a monoalkenyl arene monomer and a benzocyclobutene of the
general formula I in which n is zero, each "B" represents a
polymerized conjugated diene hydrocarbon block or a substantial-
ly completely hydrogenated conjugated diene polymer block and
each of "m", "n", "o" and "p" is an integer in the range of
from 1 to 30 and "Y" represents the residue of a multi-
functional coupling agent or multifunctional initiator.
The known crystalline olefin polymers, in their usual form,
have some outstanding good properties and some undesirable ones.
For example, desirable properties of highly crystalline poly-
propylene are high tensile strength and substantial hardness. One
disadvantage of the usual forms of highly crystalline polypropylene
is poor creep resistance. Poor creep resistance means that such
polymers are deficient for dimension stability. One means of
improving creep resistance is to crosslink the polymer by peroxide.
However, such crosslinking is useful only for ethylene polymers,
and not propylene polymers, l-butene polymers etc. Still further,
such crosslinking techniques are also not desirable because of
premature crosslinking, bubble formation, etc.
The novel solid copolymers of a benzocyclobutene of the
general formula I and a C2-C8-alphamonoolefin and which have been
mentioned hereinbefore possess a unique balance of properties along
with a unique curing or crosslinking approach.
The present invention also provides a process for the5 preparation of a solid copolymer, said process comprising
lZ93(~90
copolymerizing a C2-C8 alphamonoolefin and a benzocyclobutene of
the general formula (I)
Rll
CH2=C-R2~P (I)
where ~ represents a hydrogen atom or a methyl group and R2 a
-~-CH2 +n group where n is 0 or an integer frcm l to 6, in the
presence of a polyolefin coordination catalyst.
Peroxide crosslinking of polyethylene gives useful products
such as cable insulations, foams, abrasion resistant profiles,
coatings and bearings. These products can be prepared by a variety
of processes such as extrusion, injection moulding and sintering.
Some of the problems existing in the peroxide curing of poly-
ethylene are generation of bubbles from peroxide deccmposition;
interference of crosslinking reactions by some stabilizer additives
which are radical scavengers; and the inability to prevent curing
at temperatures below 170 C limits some of the processing
techniques to the lower melting LDPE only.
Peroxide crosslinking of substituted polyolefins are generally
not feasible because hydrogen abstraction takes place preferent-
ially at the tertiary carbons to give polymeric radicals which
preferentially undergo ~-scission than recombination.
~ CH2CH CE12CIH ~ CH2CH CH2 1 ~
R R R R
recombination
/ ~-scission
~ ~ ,
Rl
--CH2C CH2CIH __ --~ CH2~H
R R
_ CH2C ,~ +
R ~ CH2 C
R
1~3~ 9C~I
The present invention provides the following advantages over
peroxide crosslinking:-
l. It can be applied to substituted polyolefins such as poly-
propylene and poly~4-methyl-l-penter.e).
2. It is not affected by stabilizers which are radical
scavengers.
3. It eliminates mixing problems.
4. It eliminates the problem of bubble formation.
5. It can be applied to PE in processes which require no
premature crosslinking at temperatures below 170 C.
This invention is suitable for the homc- and co-polymerization
of C2-C8 alpha-monoolefins. Such olefins include ethylene, propylene,
l-butene, 4-methyl-l-pentene, l-hexene, l-heptene, l-octene and
mixtures thereof. Preferred olefins include ethylene, propylene,
l-butene and 4-methyl-l-pentene with propylene being the most
preferred alpha-monoolefin monomer.
A key aspect of the present invention involves the incorpora-
tion of the benzocyclobutene monomer of the general formula I in
the polymerization process and the product. Preferred are copolymers
of a C2-C~ alphamonoolefin and of:-
Monomer
4-(3-butenyl)benzocyclobutene,
3-(3-butenyl)benzocyclobutene,
4-vinylbenzocyclobutene,
3-vinyIbenzocyclobutene,
4-allylbenzocyclobutene,
3-allylbenzocyclobutene,
4-isopropenylbenzocyclobutene, or
3-isopropenylbenzocyclobutene.
of the above, the first six are preferred with 4BBC being most
preferred.
1~33~90
The relative amount of olefinic benzocyclobutene monomer of
the general fonmula I depends upon the degree of "crosslinking"
ultimately desired. The following table shows the preferred ranges
in mol per cent:
PreferredMbre Preferred
Alphamonoolefin moncmer99.99 to 8599.9 to 95
Olefinic benzocyclobutene monomer0.01 to 15 0.1 to 5
IOTAL 100% 100~
According to a further embodiment the present invention
provides a solid substantially amorphous copolymer of a benzo-
cyclobutene of the general formula I, ethylene and propylene.
Coordination catal~sts are used to copolymerize ethylene with
many other ~-olefins to prepare high molecular weight, linear,
substantially crystalline polymers. When mixtures of olefins are
polymerized with certain of these coordination catalysts, e.g.
V~Cl3 and diethylaluminium chloride, amorphous, elastomeric
polymers are formed. Those copolymers made from ethylene and
propylene have found wide interest and commercial usefulness. I~lese
ethylene-propylene copolymers (termed "EPR") are saturated and,
thus, cannot be sulphur-cured but require a peroxide or other
special cure. R~ndom EPR's typically contain about 30 to 70 per
cent weight ethylene.
EPR's are desirable rubbers because these are prepared from
low-cost monomers and have good mechanical and elastic properties,
as well as outstanding resistance to ozone, heat and chemical
attack. One disadvantage of such polymers is their poor creep
resistance. Poor creep resistance means that such polymers are
deficient for applications under load. One means of improving creep
resistance of EPR's is to crosslink the polymer with peroxides.
~cwever, such crosslinking is not without its associated problems
such as unpleasant odours and difficult curing procedures,
interference by certain stabilizers which are radical inhibitors,
and rernoval of peroxide deccmposition products.
~Z~3~ 90
-- 7 --
The novel solid substantially amorphous copolymer mentioned
hereinbefore does not need to be crosslinked with peroxides, since
it possesses its cwn unique crosslinking capability.
Flo~her, the present invention also provides a process for the
preparation of such a solid, amorphous polymer, said process
comprising copolymerizing ethylene, propylene and an olefinic
benzocyclobutene monomer of the general formula I described
herinbefore in the presence of a polyolefin coordination catalyst.
The incorporation of latent curing sites into the amorphous
copolymers according to the invention eliminates difficult blending
procedures and unpleasant odours. The cured rub~er would contain no
residual unsaturation and thus should be more resistant to oxidative
and thermal degradation, as explained by H.J. Harwood in J. Testing
and Evaluation, 289 (1983).
The amorphous copolymers of the present invention may be used
to replace ccm~ercial EPR's in whole or in part, and are useful in
the standard uses of EPR, such as moulded and extruded goods (e.g.
hose, gaskets and belts. See generally, Elastomers, Synthetic,
Kirk-Othmer Encyclopedia of Chemical Technology, Volume 7, pages
20 686-692 (Interscience Publishers 1965).
The relative amount of benzocyclobutene of the general formula
I in the amorphous copolymer according to the invention depends
upon the degree of "crosslinking" ultimately desired. The following
table shows the preferred ranges (in mol per cent in the product):
PreferredMore Preferred
Ethylene monomer 20 to 80 40 to 75
Propylene monomer 80 to 20 60 to 30
Olefinic benzocyclobutene monomer0.1 to 20 0.1 to 10
TOTAL 100~ 100%
m e amorphous copolymers of the present invention have a
random structure and number average molecular weight of about
50,000 to about 300,000.
1Z93~J9O
As shown in Example 1 hereinafter, the thermal electrocyclic
ring-opening of such benzocyclobutene monomers is the key to their
particular usefulness in the present invention. Such monomers
should have very good stability up to at least 100 C ( t~ = 12
years at 100 C for benzocyclobutene) and high reactivity at
elevated temperature (t~ = 1.5 minutes at 250 C for benzocyclo-
butene).
A number of different coordination catalysts of the Ziegler-
Natta type are useful in the process of this invention. Broadly,
such catalysts comprise a pro-catalyst which is a solid compound of
a transition metal of group 4 to 8 of the Periodic Table of the
Elements and a cocatalyst which is an alkyl compound, including
alkyl halides and alkyl hydrides, of a metal of groups 1 to 3. It
is now well kncwn that only a limited number of these compounds are
practical for effectively converting a given monomer into a desired
polymer. In general, the same catalysts which are effective for the
polymerization of a particular monomer feed in a conventional
polymerization process are also effective for the same conversion
in the process of the present invention.
Ziegler-Natta coordination catalysts are discussed in detail
in the book "Ziegler-Natta Catalysts and Polymerizations" by John
Boor, Jr., Academic Press, 1979 and in numerous patent specifica-
tions and review articles, including those cited by ~cor.
More recently, catalysts having much higher activity have been
developed both for polymerization of ethylene to linear high
density polyethylene and for the stereoregular polymerization of
higher-~-monoolefins. The most active of these catalysts ccmprise
procatalysts composites of magnesium or manganese halide, titanium
halide and, in the case of stereoregulating catalysts, an electron
donor. The cocatalysts generally are aluminium trialkyls and, in
the case of a stereoregulating catalyst, a selectivity control
agent. Such catalysts are described, for example, in U.S. patent
specifications 4,113,654 and 4,265,785 and many other patent
specifications for ethylene polymerization and in U.S. patent
35 specifications 4,107,413 and 4,329,253 and European patent
~93~90
specifications 19,330 and 29,623 and many others for stereospecific
polymerization of a-monoolefins.
Suitable procatalysts for conversion of propylene and other
~- noolefins to isotactic polymers (which catalysts are useful
herein) are violet TiC13 and composites of titanium chloride,
magnesium chloride and an electron donor. Procatalysts of the type
of violet TiC13 are preferably employed with alkyl aluminium
halides, typically diethyl aluminium chloride (DEAC), as
cocatalyst. Procatalysts of the type of composites of titanium
halide, magnesium halide and electron donor are preferably employed
with trialkylaluminium, typically triethylaluminium (TEA) as
cocatalyst, and with an aromatic ester electron donor, such as
ethyl p-methoxybenzoate (p-ethyl anisate) or p-methyl toluate as
selectivity control agent.
Other catalysts may also be employed including halides or
alkoxyhalides of a transition metal such as zirconium, vanadium,
chromium and molybdenum. In the active catalyst the transition
metal is at a valence belcw its maximum. Among the halogens the
order of preference runs from chlorides to bromides to iodides to
fluorides.
Preferred catalysts for the preparation of the amorphous
copolymers according to the invention include halides or alkoxy-
halides of a transition metal such as zirconium, vanadium, chromium
and molybdenum. In the active catalyst the transition metal is at a
valence below its maximum. Among the halogens the order of prefer-
ence runs from chlorides to bromides to iodides to fluorides.
Preferred catalysts are V0C13 and diethylaluminium chloride.
See generally U.S. patent specifications 3,000,866, 3,063,973 and
3,093,621 for suitable catalysts and reaction conditions.
The benzocyclobutene monomer of the general formula I is
homopolymerized or copolymerized with a C2-C8 alpha-monoolefin, for
example with a mixture of ethylene and propylene, in a manner
similar or identical to that used in the polymerization and
copolymerization of alpha-monoolefin moncmers. These
lZ93C 90
-- 10 --
polymerizations may be carried out by any one of the conventional
techniques, such as gas phase polymerization or slurry
polymerization using liquid monomer or an inert hydrocarbon diluent
as liquid medium. Hydrogen may be used to control the molecular
weight of the polymer without detriment to the performance or the
stereospecific performance of the catalyst cc~positions.
Polymerization may be effected batchwise or continuously with
constant or intermittent supply of the catalyst to the
polymerization reactor or reactors.
Polymerization, as well as catalyst preparation, is carried
out in the absence of air and water or in the presence of only very
limited amounts of these, since otherwise the catalyst would be
deactivated. Desired polymerization temperatures are between 20 C
and 100 C, preferably between 40 C and 80 C.
The catalysts employed in the production of the subject
homopolymers and copolymers may be of sufficiently high activity
that no product deashing step is required. If catalyst residues are
to be deactivated and removed, this may be accomplished by
conventional means employed in cleanup of olefin polymers produced
over such catalysts, for example, by contact with an alcohol,
followed by extraction with water.
Over the years a large number and variety of important
polymers have been developed with styrene monomers. Amorphous
homopolymers of styrene prepared via free-radical polymerization
are still the st important polymers. Subsequently other forms of
polystyrene have been prepared. These include Ziegler-Natta
isotactic crystalline polystyrene and anionic polystyrene with an
MW/Mn in the range of 1.05-1.10 with controlled molecular weight.
Alkylated styrenes, such as alphamethylstyrene and paramethyl-
styrene have also been of interest for certain end uses. See, forexample, "Styrene Plastics", Kirk-Othmer Encyclopedia of Chemical
Technology, Volume 19, pages 85-134 (Interscience Publishers,
1969).
It has long been known that the brittle nature of polystyrene
can be overcome by incorporating a minor amount of rubber. m e
1~93~90
rubber is largely incompatible with the polymeric vinyl compounds
resulting in a two-phase system comprising a dispersed rubbery
phase and a poly( novinyl arcmatic) matrix. See, for example, U.S.
patent specification 4,309,515. m ese graft copolymers are cc~monly
termed high impact polystyrene or HIPS.
Acrylonitrile copolymers with styrene (SAN) are another large
group of styrene polymers. m ese copolymers are transparent, and,
in comparison to polystyrene, more solvent- and craze-resistant and
relatively tough. m ey also constitute the rigid matrix phase of
the ABS (acrylonitrile-butadiene-styrene) copolymers which are of
comm~n usage.
Styrene has long been copolymerized with butadiene to form
both rubbers (SBR) and toughened plastics.
Other copolymers with styrene include -methylstyrene
copolymers, p-methylstyrene copolymer (see e.g. U.S. patent
specification 4,230,836), methyl methacrylate copolymer, maleic
anhydride copolymer and ~any more.
The solid random copolymers of a benzocyclobutene of the
general formula I and a monoalkenylarene described hereinbefore
constitute a new set of polymers having improved properties. Said
copolymers of a benzocyclobutene of the general formula I and a
monoalkenylarene are prepared by polymerizing a monoalkenylarene
and a benzocyclobutene of the general formula I in the presence of
a catalyst at a temperature in the range of from -80 C to +150 C.
Said copolymer preferably has a content of the benzocyclobutene of
the general formula I in the range of from 0.01 mol per cent to 10
mol per cent.
Said random copolymers of a benzocyclobutene and a
monoalkenylarene, according to the present invention, may be
crosslinked at elevated temperatures (above about 200 C) resulting
in improved solvent resistance (insolubility in certain solvents)
and a higher glass transition temperature (Tg). Further, these
polymers of the present invention may be functionalized via a
Diels-Alder reaction, resulting in polymers having improved
interfacial adhesion.
lZ93(3 90
- 12 -
The monoalkenyl arene or moncvinyl aromatic compounds are
those having the vinyl radical, i.e. ethylenically unsaturated
radical, attached directly to a carbon atom of the aromatic
nucleus.
Styrene is the preferred monovinyl aromatic compound. Examples
of other compounds applicable herein are the alkyl and dialkyl
derivatives of styrene such as the dimethylstyrenes,
p-methylstyrene, ethylstyrenes, isopropylstyrenes r butyl-styrenes,
etc., the halogen derivatives of styrene, for example, chloro- and
dichlorostyrenes and the mono- and dibromostyrenes and
alkylhalostyrenes as well as mixtures of these compounds with
styrene or with each other. Alphamethylstyrene may be substituted
in a minor amount, for example 2 to 30% by weight, preferably from
about 5 to 25% in the total composition for a portion of the
monovinylaromatic monomer to improve properties of the
interpolymers such as heat distortion temperature.
The mixture of the monoalkenyl arene monomer and
benzocyclobutene monomer may be polymerized by themselves or with
other copolymerizable mono~ers. In general the polymerization
conditions appropriate to styrene will be appropriate herein, as
long as the polymerization temperature is maintained below 150 C
(in order not to prematurely "activate" the benzocyclobutene
monomer). Thus, polymerization may be effected under bulk
conditions or in solution, suspension or emulsion techniques
comparable to those used for styrene polymerization. The
polymerization catalysts may be of the free radical or anionic
types. Suitable free radical initiators include di-tertiary butyl
peroxide, azobis(isobutyronitrile), di-benzoyl peroxide, tertiary
butyl perbenzoate, di-cumyl peroxide and potassium persulphate.
Anionic initiators are generally of the formula RMy where R is
organo, mono- or polyvalent and may be alkyl, alkenyl, aryl,
aralkyl, and alkaryl, and may contain from 1 to about 50 carbon
atoms; and y is 1 to 4, and preferably 1 or 2. Such initiators as
methyl lithium, ethyl lithium, methyl sodium, propyl lithium,
n-butyl lithium, sec-butyl lithium, tert-butyl lithium, butyl
1~93C 90
- 13 -
sodium, lithium naphthalene, sodium naphthalene, potassium naphtha-
lene, cesi~lm naphthalene, phenyl sodium, phenyl li ~ ium, benzyl
lithium, cumyl sodium, cumyl potassium, methyl potassium, ethyl
potassium, and so forth may be used in this reaction. Branched
chain polymers may be obtained by using multifunctional initiators,
for example, 1,3,5-trilithiocyclohexane and 1,4,7,10-tetrapotassio-
decane. In the anionic polymerization each molecule of the
initiator starts one anionic polymer chain; multiple anions can
permit addition of secondary chains to the main chain. Stereo-
specific catalysts can also be used to advantage. Such catalystsare generally of the well ~nown Ziegler type, CQmpriSing a
transition metal of Group 4A, 5A, 6A, or 7, in a valence state
lower than its maximum in combination with an organometallic
compound of Group 2 or 3.
Among the reducible transitional metal compounds suitable for
the purposes of this invention are the heavy metal, inorganic
compounds such as halide, oxyhalides, complex halides, hydroxides,
and organic compounds such as alcoholates, acetates, benzoates, and
aoetyl acetonates, of the requisite ~etals. Such metals include
titanium, zirconium, hafnium, thorium, uranium, vanadium, niobium,
tantalum, chromium, molybdenum, tungsten and iron. The metal
halides, particularly the chlorides are generally preferred.
Titanium, zirconium and vanadium are the most active metals. The
following heavy metal compounds are readily reducible: titanium
tetrachloride, titanium tetrabromide, zirconium tetrachloride,
vanadium tetrachloride, and zirconium acetylacetonate.
m e reduction may be effected in any suitable manner, for
example, by reduction with hydrogen or aluminium. Titanium
tetrachloride can be readily reduced to titanium trichloride by
reduction with hydrogen, aluminium or titanium metal. Suitable
reduction methods are well known in the art and are described, for
example, in U.S. patent specification No. 3,362,940
The other component of the Ziegler catalyst system is at least
one organometallic compound of a metal of Groups 2 or 3. These
compounds will have at least one hydrocarbon radical, i.e., alkyl,
1~3C'90
cycloalkyl, aralkyl, aLkaryl, or aryl, attached to the metal
through a carbon atcm. The other substituents in the organometallic
compound can be hydrocarbon radicals, halogen radicals, alkoxy,
amino, hydrogen, etc., or combinations thereof. Non-limiting
examples of the organametallic ccmpounds are triethylaluminium,
tripropylaluminium, dipropylzinc, triisobutylaluminium, diethyl-
magnesium, diphenylaluminium chloride, cyclohexyl-ethylzinc,
diethylaluminium bramide, diethylaluminium chloride, diethyl-
aluminium iodide, ethylzinc chloride, propyl-magnesium chloride,
dipropylaluminium chloride, dioctylaluminium chloride, diisobutyl-
aluminium hydride, phenylaluminium dihydride, cyclohexylbromc-
aluminium hydride, dipropylaluminium hydride, propyl zinc hydride,
ethylmagnesium hydride, and methoxyaluminium diethyl. Mixtures of
two or more organometallic compounds can be used.
m e catalyst can be formed by methods well kncwn in the art.
Thus, for example, it can be made by charging the components
separately to the polymerization zone or they may be combined
immediately prior to entry into the zone.
As previously mentioned, the polymerization may be carried out
in bulk, in solution, in suspension or in emulsion. Solution
polymerization will generally employ inert hydrocarbon solvents
such as toluene, benzene, cyclohexane, or ethyl toluene. Suspension
polymerization is generally carried out in an aqueous medium
camprising water and suspending agents such as calcium phosphates,
polyvinyl alcohol, hydroxyethyl cellulose or sodium polyacrylates.
Suitable suspension polymerization techniques will be comparable to
those used with styrene nomer, which are well kncwn in the art
and described, for example, in U.S. patent specification No.
2,715,118. EmNlsion techniques also will be c~mparable to those
used for styrene, using an aqueous medium with the addition of
suitable surfactants. Catalysts will normally be of the
free-radical type, for example, a ccmbination of butyl peroxide and
tertiary butyl perbenzoate.
The polymerization conditions will generally be similar to
those used for styrene. Thus, temperatures will generally be in the
12~3~90
- 15 -
range of -80 to ~150 C, preferably 0 C to 150 C.
The polymerizable mixture may also be copolymerized with other
monamers. m e conditions for the copolymerization will, in general,
be similar to those used for polymerizing the mixture by itselfs
and for copolymerizing styrene. m us, initiators, temperatures,
pressures, solvents, and recovery processes will be simi]ar to
those previously described. The types of copolymer produced may
include random and graft copolymers. m e preparative procedures
will be those appropriate to the type of copolymer in question. In
general the monoalkenyl arene content will be above 60% weight for
such copolymers.
Random copolymers may be made with a wide range of comonamers
including other vinyl monomers such as alpha-methyl styrene,
acrylates including methyl acrylate, ethyl acrylate, methacrylates
including methyl methacrylate, acrylonitrile, olefins especially
diolefins such as butadiene, isoprene, chloroprene and mono olefins
such as ethylene and propylene.
One class of random copolymers are the randam copolymers with
butadiene. m ey may be produced by methods similar to those used in
the manufacture of GR-S synthetic rubber as described, for example,
in "Synthetic Rubber", Ed. Whitby et al, John Wiley, N.Y., 1954.
m e copolymers with acrylonitrile are another class of
copolymers. They have a low affinity for hydrocarbons, especially
as compared to the hamopolymer. The affinity for hydrocarbons can
therefore be controlled by copolymerizing the monamer mixture with
varying amounts of acrylonitrile. Generally, the amount of
acrylonitrile will be from 15 to 35%, preferably about 30%, by
weight, although lawer amounts, e.g., about 10% of the camonomer
will effect useful changes in the properties of the polymer.
Another class of copolymers are the high impact copolymers.
These are generally graft copolymers produced by grafting units
derived fram the polymerizable mixture onto a backbone polymer
which is generally of a rubbery nature. Suitable backbone polymers
include polybutadiene, poly~dimethyl butadiene), polyisoprene,
~35~90
- 16 -
polychloroprene and other synthetic rubbers such as the styrene-
butadiene rubbers (SBR), ethylene-propylene rubbers (EPR), ethylene-
propylene-diene elastomers ~EPDM), polyacrylates, nitrile rubbers
and copolymers with other arQmatic monomers including vinyl toluene.
The backbone will generally comprise 2 to 25 per cent by weight of
the high impact copolymer, preferably 3 to 10 per cent by weight.
Normal techniques, e.g. grafting, comparable to those used for
making high impact polystyrenes are useful; they are well known in
the art and referred to, for example, in U.S. patent specification
10 2,694,692 and British patent specification 1,054,301.
A key aspect of the present invention is the amount of benzo-
cyclobutene monomer incorporated in the polymer. The relative
amounts of benzocyclobutene moncmer and monoalkenyl arene mOnQmer
depend upon the desired crosslink density or functionality. m e
table below shows suitable ranges in mol per cent:
Preferred More Preferred
Benzocyclobutene monomer 0.01 to 200.1 to 10
Monoalkenyl arene nomer 99.99 to 8099.9 to 90
T~TAE 100 100
Block copolymers have been developed rapidly within the recent
past, the starting monamers usually being monoalkenyl arenes such
as styrene or alphamethyl styrene block polymerized with conjugated
dienes such as butadiene and isoprene. A typical block copolymer of
this type is represented by the structure
polystyrene-polybutadiene-polystyrene. When the monoalkenyl arene
blocks comprise less than about 55~ by weight of the block
copolymer, the product is essentially elastomeric. Moreover, due to
their peculiar set of physical properties they can be referred to
more properly as thermoplastic elastomers. By this is me~lt
polymers which in the melt state are processable in ordinary
thermoplastic processing equipment but in the solid state behave
like chemically vulcanized rubber without chemical vulcanization
having been effected. Polymers of this type are highly useful in
lZ93S,~90
- 17 -
that the vulcanization step is eliminated and, contrary to scrap
frcm vulcanized rubbers, the scrap fram the processing of thermc-
plastic elastomers can be recycled for further use.
These block copolymers have enjoyed broad cammercial success.
Nevertheless, Lmprovements in such polymers are desired. In
particular, for particular applications such polymers require
greater solvent resistance and higher use temperatures. Still
further, such polymers also need improved adhesion to polar
materials when used in certain blend campositions. What has now
been discovered is a novel unhydrogenated block copolymer that
overcomes these deficiencies.
m e oxidative stability of such block copolymers is improved
by selective hydrogenation of the diene blocks without
hydrogenation of the styrene blocks. Such block copolymers are
described in U.S. Reissue specification 27,145 and U.S. patent
specification 3,595,942.
These hydrogenated block copolymers have enjoyed broad
commercial success. Nevertheless, improvements in such polymers are
desired. In particular, for particular applications such particular
polymers require greater solvent resistance and higher use
temperature. Still further, such polymers also need imprcved
adhesion to polar materials when used in certain blend
campositions. ~hat has now been discovered is a novel partially
hydrogenated block copolymer that overcomes these deficiencies.
m e present invention, therefore, also pravides novel
copolymers of monoalkenyl arenes and/or conjugated dienes with a
benzocyclobutene derivative. In particular, the present invention
provides AB block copolymers, ABA block copolymers, (AE~-mY block
copolymers, (AB ~ A)o block copolymers and (AB~-nY-~B)p block
copolymers and mlxtures thereof where each "A" represents a
copolymer block of a monoalkenyl arene monomer and a benzocyclo-
butene monamer of the general formula II
CH2=C ~ (II)
1;~93~90
- 18 -
where R represents a hydrogen atom or a CH3 group, each "B"
represents a polymerized conjugated diene hydrocarbon block or a
substantially completely hydrogenated conjugated diene polymer
block and each of "m", "n", "o" and "p" is an integer in the range
of from 1 to 30 and "Y" represents the residue of a multifunctional
coupling agent or mlltifunctional initiatior.
The invention also relates to a process for preparing such
polymers.
The block copolymers of the present invention possess a number
of advantages c,ver prior art block copolymers. When the polymers of
the present invention are crosslinked at elevated temperatures, the
resulting polymers possess improved solvent resistance along with
higher use temperatures. In addition, it is possible to
functionalize such non-crosslinked polymers to obtain polymers
having improved adhesion to polar materials. Still further, such
hydrogenated polymers also possess improved oxidative stability.
The block copolymers of the present invention have idealized
structures as follows:
Structure ~YE~
AB 2 block copolymer
A~A linear block copolymer
(AB ~' radial block copolymer
(AB }nY~~A)o asymmetric radial block copolymer
(ABt-ny~B)p asymmetric radial block copolymer
~xtures of the above structures are also contemplated.
The "A" blocks are copolymer blocks of a monoalkenyl arene
moncmer and a benzocyclobutene monomer of the for~,ula
CH2=C ~1
where R is H or CH3. When R is H, the benzocyclobutene monomers are
4-vinylbenzocyclobutene or 3-vinylbenzocyclobutene. When R is CH3,
the benzocyclobutene monomers are 4-isopropenylbenzocyclobutene or
lZ93~90
-- 19 --
3-isopropenylbenzocyclobutene. The preferred benzocyclobutene
monomer is 4-vinylbenzocyclobutene. Preferably the monoalkenyl
arene is styrene. Other useful monoalkenyl arenes include
alphamethyl styrene, tert-butyl styrene, paramethyl styrene and the
other ring alkylated styrenes as well as mixtures of the same.
m e relath~e amour.ts of benzocyclobutene mon er and
monoalkenyl arene mon er in the A blocks depend upon the desired
functionality or degree of crosslink. The table below shcws
suitbable ranges in mol per cent:
PreferredMore Preferred
Benzocyclobutene mono~er0.01 to 200.1 to 10
Monoalkenyl arene monomer99.99 to 8099.9 to 90
TOTAL 100 100
m e B blocks are polymer blocks of conjugated dienes or of
substantially c~l~letely hydrogenated conjugated dienes. Preferred
dienes include butadiene and isoprene. A much preferred diene is
butadiene. Mixtures of conjugated dienes may also be employed.
The Y moiety stands for the residue of a multifunctional
coupling agent. Linear polymers tA~A) are formed by employing
coupling agents having two reactive sites or by sequential
polymerization. One type of coupling agent employed in the forming
linear polymers is a dihalo alkane such as dibromoethane. See
British patent specification 1,014,999. Another coupling agent
employed in making linear polymers is phenyl benzoate as disclosed
in U.S. patent specification 3,766,301. Radial polymers are formed
by employing coupling agents having more than two reactive sites.
Examples of such coupling agents include among others: SiC14--U.S.
patent specification 3,244,664; Polyepoxides, polyisocyanates,
polyimines, polyaldehydes, polyketones, polyanhydrides, polyesters,
polyhalides--U.S. patent specification 3,281,383; Diesters--U.S.
patent specification 3,594,452; Methoxy silanes--U.S. patent
specification 3,880,954; Divinyl benzene--U.S. patent specification
3,985,830; 1,3,5-benzenetricarboxylic acid trichloride--U.S. patent
lZ~:t3(~ 90
- 20 -
specification 4,104,332; and glycidoxy-methoxy silanes--U.S. patent
specification 4,185,042.
m e linear and radial block polymers may also be formed by
sequential polymerization using multi-functional initiators having
at least 2 reactive carbon-lithium bonds. The dilithium initiators
are represented by the formula LiRLi. Examples of these dilithium
initiators are 1,1,6,6-tetraphenyl-1,5-hexadiene,
1,3-divinylbenzene, 1,3-bis(1-methylvinyl)benzene,
1,4-bis(2-phenylvinyl)benzene, 1,3-bis(1-phenylvinyl)benzene,
1,4-bis(1-phenylvinyl)benzene, 4,4'-bis(1-phenylvinyl)biphenyl,
2,7-diphenyl-1,7-octadiene, 2,7-di-4-tolyl-1,7-octadiene,
1,2-bis(4-(1-phenylvinyl)phenyl)-ethane, and
1,4-bis(4-(1-phenylvinyl)phenyl)butane. Initiators with more than
two lithium-carbon bonds can be formed by the reaction of RLi and
DVB.
The letters "m", "n", "o" and "p" stand for the relative
number of arms in each polymer molecule. Accordingly, m, n, o and p
are integers when referring to a single polymer molecule. However,
a polymer mass will generally contain molecules of varying
functionality. When referring to the polymer (AEt-mY, it is
preferred that m be 1 to 15, preferable 2 to 8. When referring to
the polymers (ABt-nY-~A)o and (AB~-nY-~A)p, it is preferred that the
sum of n + o be greater than 3, preferably 3 to 15 and that the sum
of n + p be greater than 3, preferably 3 to 15. Accordingly n is
preferably 2 to 8 for both polymers.
m e block copolymers of the present invention are produced by
anionic polymerization employing an organomonolithium initiator.
(The following description refers only to mono-lithium initiators,
though it is appreciated, as stated above, that multi-functional
initiators may also be used.) The first step of the process
involves contacting the monoalkenyl arene monomer, benzocyclobutene
monomer and the organomonolithium compound (initiator) in the
presence of an inert diluent therein forming a living polymer
compound having the simplified structure A-Li. The monoalkenyl
arene is preferably styrene. The inert diluent may be an aromatic
~L29~3a90
- 21 -
or naphtenic hydrocarbon, e.g., benzene or cyclohexane, which may
be modified by the presence of an alkene or alkane such as pentenes
or pentanes. Specific examples of suitable diluents include
n-pentane, n-hexane, 2,2,4-trimethylpentane, cyclohexane, toluene,
benzene, xylene and the like. The organomonolithium compounds
(initiators) that are reacted with the polymerizable additive in
step one of this invention are represented by the formula R1Li;
wherein R1 is an aliphatic, cycloaliphatic, or aromatic radical, or
combinations thereof, preferably containing from 2 to 20 carbon
atoms per molecule. Exemplary of these organomonolithium compounds
are ethyllithium, n-propyllithium, isopropyllithium, n-butyl-
lithium, sec-butyllithium, tertoctyllithium, n-decyllithium,
n-eicosyllithium, phenyllithium, 2-naphthyllitium, 4-butylphenyl-
lithium, 4-tolyllithium, 4-phenylbutyllithium, cyclohexyllithium,
3,5-di-n-heptylcyclohexyllithium, 4-cyclopentylbutyllithium, and
the like. The alkyllithium compounds are preferred for employment
according to this invention, especially those wherein the alkyl
group contains from 3 to 10 carbon atoms. A much preferred
initiator is sec-butyllithium. See U.S. patent specification
20 3,231,635. The concentration of the initiator can be regulated to
control molecular weight. Generally, the initiator concentration is
in the range of about 0.25 to 50 millimol per 100 g of monomer
although both higher and lower initiator levels can be used if
desired. The required initiator level frequently depends upon the
solubility of the initiator in the hydrocarbon diluent. These
polymerization reactions are usually carried out at a temperature
in the range of -75 to +150 C and at pressures which are
sufficient to maintain the reaction mixture in the liquid phase.
Next, the living polymer in solution is contacted with a
conjugated diene. Preferred dienes include butadiene and isoprene.
A much preferred diene is butadiene. The resulting living polymer
has a simplified structure A--B--Li. The predomunantly cis-1,4
microstructure of the polybutadiene blocks obtained from polymer-
ization in cyclohexane can be modified to a random mixture of 1,4-
and 1,2-structures by the addition of a small a~ount of ether
lZ'~3~30
m~difiers such as diethyl ether or tetrahydrofuran.
The B-Li polymer arms may be formed in a separate reactor
employing an inert solvent, organ~monolithium initiator and
conjugated diene monomer. In an alternative en~xYlDment, the B-Li
S arms may be formed in the same reactor as the AE-Li polymer arms.
In that case, after the A-Li arms are formed, additional initiator
is added. Then the conjugated diene monomer is added. In this
alternative e~bodiment, the B arms and the B portion of the AB arms
will necessarily be similar in com,position and molecular weight.
The molecular weights of the living polymer arms (A and B) may
vary between wide limits. Suitable number average molecular weights
are:
Preferred More Pre erred
A 300 to 30,000 3,000 to 20,000
B 15,000 to 100,000 25,000 to 60,000
m e living AE-Li and B-Li or A-Li polymer arms are then
reacted with a multifunctional coupling agent. Exemplary coupling
agents are listed above. The AB and ABA polymers do not require use
of coupling agents.
The coupling agent should be added to the living polymer after
the polymerization of the monomers is substantially complete, i.e,
the agent should only be added after substantially all of the
monomer has been converted to living polymers.
The amount of coupling agent added depends upon the structure
of the coupling agent and on the desired number of arms, and the
choice is within the skill of the average polymers chemist.
The coupling reaction step may be carried out in the same
solvent as for the polymerization reaction step. A list of suitable
solvents is given hereinbefore. The coupling reaction step
temperature may also vary between wide limits, for example, from 0
to 150 C, preferably fram 20 to 120 C. m e reaction may also take
place in an inert atmosphere, for example, nitrogen and under
pressure, for example, a pressure of frcm 0.5 to 10 bar.
Hydrogenation of the block copolymers is preferably effected
by use of a catalyst ocmprising the reaction products of an
aluminium alkyl ccmpound with nickel or cobalt carboxylates or
1~3~ ~0
23 63293-2704
alkoxides under such conditions as to substantially completely
hydrogenate at least 80% of the aliphatic double bonds while
hydrogenating no more than about 25% of the alkenyl arene aromatic
double bonds. Preferred block copolymers are those where at least
99% of the aliphatic double bonds are hydrogenated while less than
5% of the aromatic double bonds are hydrogenated.
Hydrogenation temperatures must be maintained below
about 150C, preferably between about 0C and about 150C. See
generally U.S. Reissue specification 27,145 and U.S. patent
specification 3,595,942, which show the various conditions of
hydrogenation.
Then the product is typically recovered such as by
coagulation utilizing hot water or steam or both.
A key aspect of -the block copolymers according to the
present invention is that the end product contains randomly
distributed benzocyclobutene structures in the styrene end blocks.
A schematic structure for an ABA block copolymer is shown below,
where the monoalkenyl arene block is made from styrene (S) and the
diene block is made from butadiene (B):
( S )- ( B ) ( S )
Accordingly, when such a polymer is moulded at
temperatures above about 200C (or otherwise heated above such
temperatures), a cross]inked elastomer is obtained.
4-(3-Butenyl)benzocyclobutene (4BBC) is believed to be a
novel compound. This novel compound may be prepared by reacting a
4-halomethylbenzocyclobutene (halo being fluoro, chloro, bromo or
iodo) with metallic magnesium and reacting the 4-
iZ93~}~30
23a 63293-2704
halomagnesiummethylbenzocyclobutene thus formed with an allyl
halide, forming 4BBC and a magnesium halide. The 4-
halomethylbenzocyclobutene is preferably 4-
chloromethylbenzocyclobutene and the halide is preferably allyl
bromide.
The following Illustrative Experiment and Examples
further illustrate the invention.
~93~90
- 24 -
Illustrative Experiment
A key aspect of the present invention deals with the ring-
opening of the benzocyclobutene monomers to o-quinodimethanes. In
this Illustrative Experiment, half-life values for the parent
benzocyclobutene are calculated and summarized in the follcwing
Table 1, based on activation parameters reported in W.R. Roth et
al, Chem. Ber. 111 (1978) 3892-3903. m e results suggest that
reactive oligomers and polymers containing benzocyclobutenes which
are not substituted at the cyclobutene ring would have long shelf-
life and good reactivity at 200-250 C.
TABLE 1
k
Benzocyclobutene -------> _-quinodimethane
T (C) k (sec 1) t (h)
2.5 x 10 15 -J~----lo
100 1.7 x 10 9 l.l x 105
150 9.6 x 10 7 2 x 102
200 1.4 x lO 4 1.4
250 7.8 x lO 3 2.5 x lO 2
Example 1
With the exception of polyethylene and EPDM elastomer, it is
difficult to crosslink or introduce functional groups into poly-
olefins. By incorporating benzocyclobutene into polyolefins and
using its thermal reactivity to form carbon-carbon bonds, it is
possible to make new products such as crosslinkable polypropylene,
thermDformable polypropylene, and high temperature ethylene
propylene elastomers. This embodiment describes the preparation of
reactive polyolefins via Ziegler-Natta polymerization using
4-methyl-l-pentene (4MP1) and 4-(3-butenyl)benzocyclobutene (4BBC)
as model compounds.
4BBC was prepared from 4-chloromethyIbenzocyclobutene in a
two-step process by reacting the latter compound with metallic
magnesium, forming a substituent -CH2MgCl and reacting this sub,
~,'Z93(~90
- 25 -
stituent with allyl bromide, forming 4BBC and M~BrCl. This two-step
process had an cverall yield of 60% and is represented by the
follcwing equation:
ClCH2 CH2=CHCH2CH2
4BBC
The structure of 4BBC was confirmed by H and 13C ~MR. GC analysis
showed it to be >99% pure.
The homo- and copolymerizations of 4MP1 (4-methyl-1-pentene)
and 4BBC were carried out in 2,2,4-trimethylpentane (isooctane) at
50 C for 24 h using Stauffer TiC13.AA catalyst. The results
summarized in Table 2 showed that the presence of benzocyclobutene
had no deleterious effects on either polymer yields or molecular
weights. The high yields of polymers regardless of monomer com-
positions suggest that 4BBC is not a polymerization inhibitor and
should be readily copolymerizable with a wide variety of olefins.
The molecular weights as measured by gel permeation chrcmatography
(GPC) and intrinsic viscosities were high. m e GPC weight-average
molecular weight of a copolymer containing 6% 4BEC was estimated to
be about 1.33 million using polypropylene calibration. This value
is higher than those of commercial polyolefins since no molecular
weight control agents was used in the polymerization.
13C analysis of 4BBC homopolymer shcws twelve carbon
resonances whose chemical shifts are consistent with the 4BBC
repeating unit. This confirms that benzocyclobutene is stable under
ordinary Ziegler-Natta polymerization conditions. The chemical
shifts of the backbone carbons of 4BBC homopolymer are ~ 31.9 and
39.4 ppm, whereas the values of corresponding carbons in the
products from copolymerization are ~ 32.6 and 41.6 ppm. The
difference suggests that copolymerization produced random
copolymers rather than block polymers or mixtures of two homo-
lZ~3C9O
- 26 -
polymers. The nearly identical mono~er and copolymer compositions
shcwn in Table 2 are also consistent with a random copolymerization
process. m e formation of copolymer was also supported by the
formation of crosslinked copolymers containing as low as 0.5 %~l
of 4BBC upon ccmpression moulding. Homopolymer of 4MP1 moulded
under identical conditions remained soluble in decalin.
Copolymerization with 4BBC has been shcwn in this embcdiment
to be a feasible method of producing reactive olefins containing
benzocyclobutene.
TP~IE 2
Polymerization of 4MPl and 4BBCa)
Polymer Monomer Composition Polymer % 4BBC )
No 4MPl (%) 4BBC (%) Cocatalyst Yield (%) in Copolymer
1 0 100 ~E~ 86 100
2 100 0 DEAC 94 0
394.4 5.6 DE~C 93 6
498.7 1.3 DEAC 96 1.5
599.5 0.5 DEAC 93 <1
a) TiC13/aluminium alkyl = 3; monomer/Ti = 200 in all cases except
in the case of 100% 4BBC where monomer/Ti = 50.
b) Determined by 13C ~DR.
Example 2
Isotactic poly(4-methyl-1-pentene) containing benzocyclobutene
prepared by copolymerization of 4-methyl-1-pentene (4MPl) and 4-(3-
butenyl)benzocyclobutene (4BBC) is discussed in this example 2.
The copolymer is crystalline and can be crosslinked by comr
pression m~ulding. Since the performance of crystalline polymers
are greatly affected by morphology which in turn is determined by
thermal properties and history, it is important to understand how
crosslinking affects the properties and structures of polyolefins.
This e:}x~l~ment describes the effect of crosslinking on the
crystallinity, Tm, ar.d Tg of reactive poly(4MPl).
1~93C~91)
- 27 -
m e effect of crosslinking on the melting behaviour of a
series of copolymers containing up to 5.6 %mol of 4BBC was studied
by differential scanning calorimetry. m e samples were heated to
300 C, held at 300 C for 10 min to ensure cc)mplete reaction of
benzocyclobutene, quench cooled, and reheated to 300 C. The
results in Table 3 shcw that crosslinking caused decreases in
crystallinity, ranging from about 35% reduction in 0.5% 4BBC
copolymer to more than 90% in 5.6% cc)polymer. Control experiments
shc~ed that noncrosslinkable poly(4MPl) and a copolymer containing
4-phenyl-1-butene (4PBl) did not become less crystalline under
identical heat treatment.
m e differential scanning calorimetric (DSC) melting
transitions of the crosslinked copolymers were broad and each
polymer contained a relatively sharp and higher melting cc~ponent
whose TmaX was essentially identical to that of the as prepared
polymer.
Dynamic Mechanical analysis shc~ed that crosslinking caused
increases in Tg from 52 C for the homopolymer to 54.5 C for 0.5%
4BBC copolymer, 58 C for 1.3~ and 68.5 C for 5.6%. The possibility
that the increase in Tg was due to cc~positional change was elimi-
nated by the decrease in Tg from 52 C for the homopolymer to
46.5 C for the noncrosslinked 1.3% 4PBl copolymer.
Contrary to a styrene-divinyl benzene system, the increase in
Tg was nonlinear with 4BBC concentration. m e crosslinking
efficiency, as measured by Tg elevation for every mmol increase in
4BBC/g polymer, c~ecreased with increasing 4BBC concentration
(Table 4). A possible explanation is that higher 4BBC concentration
gives higher crosslink density which leads to higher number of
isolated benzocyclobutene and consequently lower crosslink
efficiency. Alternatively, the lc~er crosslink efficiency at higher
4BBC concentration may be the result of an opposite effect of
cc ~ ositional change on Tg sin oe Tg drops frc~ 52 C for poly-(4MPl)
to 46.5 C for the noncrosslinked 1.3% 4PBl cc~polymer.
lZ~ 95~
- 28 ~
TABLE 3
Thermal Transitional Properties of 4~1 Copolymers
Polymer Composition ~H (cal/g3
%4BBC %4PBl ~ ~ 1st heat 2nd heat ~ )b)
0 0 244.510.4 10.2 52.0
0 1.3 238.38.4 9.6 46.5
0.5 0 238.39.9 6.5 54.5
1.3 0 236.5lO.0 3.6 58.0
5.6 0 232.1c) c) 68.5
a) DSC first heat data obtained at a heating rate of 10 C/min.
b) Dynamic mechanical data measured at 11 Hz.
c) 1st heat peak too broad and 2nd heat peak too small to give
accurate numbers.
TAB~E 4
Crosslink Efficiencies
-
Polymer Reactive Group ~Tg/~mmol Reactive Group
polystyrene 0-14% DVB 65 C )
poly(4MP1) 0.5% 4BBC 40 C
poly(4MP1) 1.3% 4BBC 38 C
poly(4MP1) 5.6% 4BBC 26 C
d) T.G. Fox and S. Losheak, J. Poly. Sci., 1955, 15, 371
Example 3
To determine if the BBC crosslinking process is affected by
the presence of stabilizers which scavange free radicals, equi-
valent amounts (1 mol per BCB) of Cyanox 2246 (5.5 %w)
l~t'~ t~ -
1293(~90
- 29 -
OH OH
tert.-C4Hg l l tert.-C4Hg
CH2 ~ Cyanox 2246
CH3 C~3
and Galvanoxyl (6.5 %w)
tert--c4H9 tert.-C4Hg
. O ~CH ~0 Galvanoxyl
tert.-c4H9 tert.-C4H9
were blended with poly(4-methyl-1-pentene) (containing 1.3 %m
4BBC), respectively, and the effects of the additives were studied
by extraction and DSC. Compression moulding at 250 ~C gave cross-
linked samples while under identical conditions P4MPI (1.3 %m 4PBI)
gave soluble film. Extraction with hot 1,1,2,2-tetrachloroethane
gave 95% gel in the case of Cyanox 2246 and 97% gel in the case of
,~ Galvanoxyl~ DSC study shcwed that the presence of Cyanox 2246 had
no effect on the crosslinking process as evidenced by the almost
identical reduction in ~Hf after crosslinking with or without
Cyanox 2246.
~Hf (cal/g)
1st Heat 2nd Heat
P4MPI (1.3 ~m 4BBC) 9.55 2.27
P4MPI (1.3 %m 4BBC) +
5.5 %w Cyanox 2246 9.43 2.45
The lack of effect by the stabilizers on crosslinXing suggests
the BCB crosslinking does not involve free radical inter~ediates as
in the case of peroxide crosslinking and consequently can be used
with substituted polyolefins in the presence of radical scavengers.
Example 4
With the exception of polyethylene and EPDM elastomer, it is
?~ I f C~
J~Z93~
- 30 -
difficult to crosslink or introduce functional groups into
polyolefins. By incorporating benzocyclobutene into polyolefins and
using its thermal reactivity to form carbon-carbon bonds, it is
possible to make new products such as high temperature ethylene-
propylene elastomers. This ~ntodLment describes the preparation ofreactive polyolefins via ~iegler-Natta polymerization using
ethylene/propylene and 4-13-butenyl)benzocyclobutene (4BBC) as
model compounds.
4BBC was prepared as described in example 1.
In a 2-litre resin kettle, 1 litre of toluene was rapidly
stirred and sparged with purified nitrogen for 20 minutes at 20 C.
Streams of propylene and ethylene were introduced. Feed rates of
0.5, 1.0 and 2.0 litre/min for ethylene, propylene and nitrogen,
respectively, were maintained throughout the reaction. After 20
minutes, 1 g of 4-(3-butenyl)benzocyclobutene, 0.173 g (1 mmol) of
vanadium(V) trichloride oxide in 5 ml of toluene, and 2.47 g (10
mmol) of triethyldialuminium trichloride in 10 ml of toluene were
added in rapid succession to give a deep purple colour solution
which gradually faded to a very pale purple colour. After 10
minutes, four additional portions of 0.5 g of 4-(3-butenyl)benzo-
cyclobutene, 0.2 mmol of vanadium(V) trichloride oxide in 1 ml of
toluene, and 2 mmol of triethyldialuminium trichloride in 2 ml of
toluene were added in 10-min intervals. The polymerization was
allowed to proceed another 20 minutes after the final addition;
then 20 ml of 2-propanol was added to deactivate the catalyst. The
polymer was precipitated from methanol, washed three times in a
Waring blender with methanol, and dried in vacuo at 65 C to give
12.5 g of a rubbery polymer. lH NMR analysis of the polymer in
1,1,2,2-tetrachloroethane shcwed the characteristic resonance of
benzocyclobutene at ~ 3,09 ppm. m e composition of the terpolymer
was estimated by ~R to contain approximately 0.15 %m
4-(3-butenyl)benzocyclobutene, 74 %m ethylene, and 25.7 ~m
propylene. The terpolymer is schematically illustrated as follows:
CH3 ~
1~3C~90
Example 5
_ - Preparation of 4-vinyIbenzocyclobutene
A solution of 4-chloromethylbenzocyclobutene (24.4 g, 160
mmol) and triphenylphosphine (41.9 g, 160 mmol) in 120 ml of
chloroform was heated at reflux for 24 h. Addition of diethyl ether
foll~ed by filtration gave triphenyl(4-benzocyclobutenyl)methyl
phosphonium chloride as a white powder: lH NMR (CDCl3) ~ 3.03 (m,
4H), 5.36 (d, 2H) 6.82 (m, 3H), 7.6-7.8 (m, 15H). To a solution of
the phosphonium salt in 500 ml of 37% formaldehyde in water was
added dropwise 75 ml of 50% aqueous sodium hydroxlde. The mixture
was stirred at ambient temperature for 2 h and then extracted with
diethyl ether. The ether extract was washed with brine and dried
over m gnesium sulphate. Fractional distillation gave 14.5 g of 90%
pure 4-vinylbenzocyclobutene: bp 63-66 C ~8 mbar); 1H NMR (CDCl3~
15 ~ 3.11 (s, 4H), 5.11 (d, lH), 5.63 (d, lH), 6.66 (dd, lH), 6.95 (d,
lH), 7.10 (s, lH), 7.18 (d, lH); 3C NMR (CDCl3) ~ 29.29, 29.44,
112.27, 119.87, 122.52, 125.70, 136.72, 137.97, 146.66, 146.01.
B - Free radical copolymerization of styrene and
4-vinylbenzocyclobutene
A mixture of styrene (5 g, 48 mmol), 4-vinylbenzocyclobutene
(0.1 g, 0.77 mmol) prepared as described in section A of this
Example and benzoyl peroxide (0.11 g) was heated under inert
atmosphere for 2 days at 50 C and 1 day at 60 C. The resulting
solid plug was dissolved in toluene and reprecipitated from
methanol. The precipitate was collected by vacuum filtration and
dried at 70 C in vacuo to give 4.56 g of a white powder. NMR
analysis of the product showed the presence of characteristic 13C
and 1H resonances of benzocyclobutene at ~ 29.24 and 3.1, respect-
ively. The product was estimated to contain 1-2 %m of 4-vinylbenzo-
cyclobutene based on N~DR integration. Compression moulding of thecopolymer at 150 C gave a film which was soluble in toluene,
whereas a filn of the copolymer prepared by moulding at 200 C for
17 h was insoluble in toluene, methylene chloride or tetrahydro-
furan.5 Example 6 - Anionic copolymerization of 4-vinylbenzocyclobutene
and styrene _
lZ93G9O
To a solution of 4-vinyIbenzocyclobutene (0.45 g, 3.5 mmol),
prepared as described in section A of Example 5 styrene (26.7 g,
257 mmol), 200 ~1 of 1-n-butoxy-2-t-butoxyethane in 233 g of
cyclohexane was added 0.84 mmol of s-~utyl lithium. After heating
at 50 C for 2 h under an inert atmosphere, the polymerization was
terminated by the addition of 3 ml of 1 %w Ionol ("Ionol" is a
. trade ~wY} for 2,6-ditert.-butyl-4-methylphenol~ in methanol. The
product was isolated by precipitation from methanol and dried in
vacuo at 80 C to give 26 g of a white, brittle solid. The polymer
was shown by lH NMR to contain 1.2 %m of 4-vinyIbenzocyclobutene
based on the benzocyclobutene resonance at ~ 3.08. Compression
moulding at 250 C for 10 min gave a film which was insoluble in
either toluene or tetrahydrofuran. Under identical conditions,
homopolystyrene gave soluble films.
Example 7 - Preparation of styrene-butadiene triblock polymers with
4-vinylbenzocyclobutene in the styrene block
To a solution of styrene (9.73 g, 93.6 mmol),
4-vinylbenzocyclobutene, prepared as described in Example 5 (164
mg, 1.26 mmol), and 25 ~l of 1-n-butoxy-2-t-butoxyethane in 233 g
of cyclohexane was added 1.3 mmol of s-butyl lithium. After the
mixture was heated at 50 C for 30 min under an inert atmosphere,
butadiene (25.6 g, 474 mmol) was added and the heating was
continued for an additional 2.5 h. The polymerization was
terminated by the addition of 0.5 mmol of methyl benzoate. GPC
analysis shcwed the product to be a mixture of polystyrene (7.9%,
MW 7,500), styrene-butadiene diblock (35.4%, MW 28,000),
styrene-butadiene-styrene triblock (54.4%, MW 58,000), and
styrene-butadiene multiblock (2.3%, MW 106,000). lH NMR showed the
product to contain 28 %w styrene, 1.2 %m 4-vinylbenzocyclobutene in
the styrene block, and 40 %m vinyl in the butadiene block.
Example 8
Various styrene-butadiene-styrene block copolymers with
4-vinyIbenzocyclobutene (VBC) in the styrene blocks were prepared
in a manner similar to that used in Example 7.
Reactive styrene-butadiene-styrene block polymers containing
VBC were prepared using styrene monomer containinq 1.3 ~m of V~C.
lZ931~90
The polymerizations were carried out in glass bottles at 50 C in
cyclohexane using s-BuLi as initiator and the results are
summarized in Table 5 hereinafter. Styrene-butadiene-styrene
triblock polymers were prepared by sequential anionic
polymerization of styrene and butadiene using twice the theoretical
amounts of BuLi followed by coupling the living diblock poly~ers
with methyl benzoate. Gæc analysis shcwed the products to be
mixtures of diblock and triblock polymer whose experimental
molecular weights were in good agreement with those calculated
based on zero consumption of BuLi by impurities. The coupling
efficiencies based on methyl benzoate were generally in the range
of 70-80~ (see Table 6 hereinafter).
The presence of benzocyclobutene in the products can be
readily confirmed by the magnetic resonance of the ethylene protons
in benzocyclobutene at ~ 3.1 ppm. Quantitative lH NMR showed the
styrene-butadiene-styrene triblock polymer to contain 1.2 %m VBC
based on styrene. This value agrees well with that of 1.3 %m VBC in
styrene monomer.
TABLE 5
Comparison of Calculated and GPC Mblecular Weights
BD
BuLi + SM +VBC - ---~ S Li -------> SB Li --~----> SBBS
SBBS MW (X103)
No Calculated Found
1 5.1-25.2-5.1 4.5-23.4-4.5
2 5.1-26.8-5.1 4.6-25.5-4.6
3 7.6-37.8-7.6 7.1-40.7-7.1
4 7.6-39.4-7.6 7.5-43.1-7.5
5.0-25.4-5.0 4.5-24.8-4.5
6 7.6-40.0-7.6 7.3-44.4-7.3
~3~9V
- 34 -
TABLE 6
%SBBS
No. Calculated Found Coupling Efficiency (%)
1 78 S8 73.4
2 79 50 63.3
3 77 62 80.5
4 77 54 70.1
79 61 77.2
6 77 62 80.5
Example 9 - Hydrogenation of styrene-butadiene
triblock polymer containing 4-vinyl-
benzocyclobutene in the styrene block
Hydrogenation of styrene-butadiene block copolymers (similar
to those prepared in Example 7) containing 1.3 %mol of VBC (based
on styrene) were carried out in a 300 ml autoclave using a
hydrogenation catalyst derived from nickel octoate and triethyl
aluminium. Conversions were determined by NMR and ozonolysis to be
> 98%. The VBC contents in the polymers before and after
hydrogenation were estimated from the characteristic proton
resonance of benzocyclobutene at ~ 3.1 ppm. While it is difficult
to determine the VBC content with high degree of accuracy due to
the low levels, the results in Table 7 suggest that most of the
benzocyclobutenes survived the hydrogenation intact and the
proposed method of manufacturing reactive hydrogenated block
copolymer via VBC is technically feasible.
To evaluate the effect of benzocyclobutene coupling on the
mechanical properties, a hydrogenated polymer (molecular weight,
7500S-42000EB-7500S; 60% triblock; 1.5-1.6 VBC/triblock chain) was
compression moulded at 150 C and 250 C, respectively. At 150 C,
little or no coupling occurred and the resulting film was
completely soluble in toluene. The 250 C film disintegrated in
toluene to give highly swollen gels as a result of the expected
coupling of benzocyclobutenes. The mechanical data in Table 8 shcw
3~90
that the higher moulding temperature gives higher modulus and lower
rate of stress relaxation. Since Shell KR~TON G~Rubber 1652, a
polyrner with simil æ molecular weight (7~00S-37500EB-7500S),
exhibits the opposite effect of temperature on modulus and stress
relaxation, the observed changes in properties may be attributed to
benzocyclobutene coupling ("KRATON G" is a trade name for a
partially hydrogenated styrene-ethylene/butylene-styrene block
copolymer).
The present example suggests the possibility that the
resistance to stress relaxation by low molecular weight poly~ers
such as KG-1652 can be improved by the incorporation of small
amounts of benzocyclobutene (<two per chain). One possible
mechanism is that the increase of molecular weight would lead to
better phase segregation and consequently improved properties.
TABLE 7
Hydrogenation ) of VBC~ dified S-B-S Block Polymers )
Catalyst Concentration Temperature % BBC )
No. (ppm Ni) (C) Before After
1 776 50-58 1.1 0.9
4 900 50-59 1.1 1.4
4 1300 53-66 1.1
200 56-64 0.8 1.4
400 56-92 0.8 0.9
6 900 50-59 0.9
a) Hydrogenations were carried out in cyclohexane, 15 %w polymer,
37.9-41.4 bar H2, and 1-3 h reaction time.
b) All polymer samples were prepared with the same moncmer
compositions - 1.2 ~m VBC based on styrene.
c) Estimated by H NMR analysis.
~ T~a~.? ~
1'~.93(~90
TABLE 8
Mechanical Properties of S-EB-S-VBC and KG-1652a)
Stress (MPa) % Elong. Stress
Mouldingd) Modulus 100% At At Rel,e)
Sample Temp.(C)(MPa) Elong. Break Break Slope
S-EE-S- ~ ) 1504.21 1.28 20.24 905 -0.086
S-EB-S- ~ ) 25017.60 2.93 12.75 795 -0.077
KG-1652C) 15049.83 4.10 30.9 581 -0.068
KG,1652C) 2501.10 1.88 18.12 579 -0.070
a) 0.05 cm thick microtensile specimens, strain rate = 2.54 cmJmin.
b) 7500S-42000EB-7500S (~60~ triblock).
c) 7500S-37500EB-7500S.
d) 10 min at 250 C; 20 min at 150 C.
e) At 150% elongation.