Note: Descriptions are shown in the official language in which they were submitted.
~30(~35
N-SUBSTITUTED ARYLCYCL0 .
BUTENYL-UNSATURATED CYCLIC IMIDES
This invention relates to N-substituted aryl-
cyclobuteno-unsaturated cyclic imides, and to novel
polyimides prepared from such compounds.
In recent years the search for high perfor-
mance materials, especially high temperature-reslstant
polymers, has gained momentum. In order for a material
to have stability at high temperatures, it must fulfill
several requirements including a high melting or
softening temperature, a high modulus or rigidity, a
resistance to solvent and chemical degradation, and
tou~hness. The intrinsic thermal and oxidative
stability of aromatic structures has long been
recognized, and a variety of polymers have been made in
which benzene rings are linked together by various
connecting groups. Among the more stable aroma.tic
polymers that fulfill the requirements of high
temperature resistance are the polybenzimidazoles, the
polybenzoxazoles and the polyimides. Of these
polymers, the polyimides have had the most interest.
2~
,~
28,913C-F -1-
~ 3~ i3S
--2--
The major difficulty encountered in the commercial
development o~ these materials is that they are usually
obtained in the form of a powder which cannot be
readily fabricated into useful objects.
The polyimides prepared from aliphatic diamines
and aromatic carboxylic acids are generally soluble and
thermoplastic. Aliphatic polyimides have been prepared
from bis(dienophiles) and a diene such as
cyclopentadiene. Such reactions often involve gas evo-
lution.
Aromatic polyimides, such as polypyromellit-
imides, have a spectrum of superior properties. Those
polyimides may be prepared by the reaction of an
aromatic dianhydride with an aromatic diamine to give a
soluble polyamic acid, which on cyclodehydration gives
the insoluble desired product.
High performance plastics reduce the weight of
mechanical components, and not just by virtue of their
densitieQ. Their high perPormance properties allow
greater design stresses, and often elements can be
downsized accordingly. In recent years, aromatic
polyimides have become widely accepted as premillm, high
performance engineering plastics~ These resins are
well-known for having excellent properties at elevated
temperatures (i.e., chemical resistance) but are also
costly. Historically, polyimide resins are difficult
to fabricate into objects other than fibers and films.
The most common methods of manufacturing parts having
the highest strength and temperature properties are hot
compression-molding, machining from hot-compression
molded or extruded rod, and direct forming (a process
similar to the powder-metallurgy processes). Given the
28,913C-F -2-
~30~)63~
_ 3 _ 64693-4007
synthetic and fabrication d~ficultie~, a new route to polyimides
is desirable.
A further problem with the preparatlon of certain
polyimide~ is the need for the use of catalysts, initiators or
curing agents. The presence of such ~ompounds often results in
the preparation of impure polymertc composition~. Further, the
presen~Q of ~uch compounds often results in undesirable properties
in such polymerlc compo~lt~ons. What are needed are monomers which
prepare polyimlde~ whereln the polymer~ can be easily proces~ed,
for exa~ple, fabr~cated into ugeful objects. What are further
needed are monomers which can be polymerlzed in a manner such that
no ga3 1~ evolved. What are further needed are monomers which can
be poly~er~zed wlthout t~e need or catalysts, curing agents or
initlators.
The inventlon 1~ a compound whlch comprlses an un-
satuxated cyclic lmide moiety and an aryl cyclobute:ne moiety,
wherein the cyclobutene moiety i~ fused to the aryl radlc~l, and
wherein the lmide nltrogen is connected to the aryl radical by a
brldglng m0~bar or a dlreGt bond.
Accordlng ~o one a~pect of the pre~ent lnven~ion
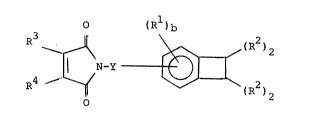
there i8 pro~lded a compou~d whlch corregpond8 to the formula
o ( R 1 ) ~
~(R2)2
wherein
- 3
~0~)~;35
- 3a - 64693-4007
Rl is separately in each o~curr~cea hydrocarbyl,
hydrocarbylthio, hydrocarbyloxy, electron-withdrawing or electron-
donating group;
R is separately in each occurrence hydrogen, cyano,
halo or an electron-donating group;
R3 is separately in each occurrence hydrogen, hydro-
carbyl, hydrocarbyloxy or hydrocarbylthio;
Y is a direct bond or a divalent organic radical; and
b is an integer of from 0 to 3, inclusive; with
the proviso that at least two of R are hydrogen and the further
proviso that the moieties Rl, R2 and R3 do not interfere with
polymerization of the compound.
According to a further aspect of the present inven-
tion there is provided a compound which corresponds to the formula
~ ~ _ y - Ar (C(R )2)2
wherein
Ar is a carbocyclic aromatic radical;
R is separately in each occurrence a hydrocarbyl,
hydrocarbyloxy, hydrocarbylthio, an electron-donating or electron-
withdrawing group;
R is separately in each occurrence hydrogen, cyano,
halo, or electron-donating group;
R3 is separately in each occurrence hydrogen, a hydro-
carbyl, hydrocarbyloxy or hydrocarbylthio group;
,, .
- 3a
()0635
- 3b - 64693-4007
Y is a direct bond or a divalent organic radical; and
a is an integer of from 0 to 3;
with the proviso that the two carbon atoms of the (C(R2)2)2 moiety
which are bound to Ar are bound to adjacent carbon atoms; the same
aromatic ring of Ar; with the further proviso that at least two
of R2 are hydrogen; and the further proviso that the moieties Rl,
R2 and R3 do not interfere with polymerization of the compound.
According to another aspect of the present invention
there is provided a monomer which corresponds to the formula
~ ~ )2
wherein
Rl is separately in each occurrence an electron-with-
drawing or electron-donating group;
R2 is separately in each occurrence hydrogen, a cy-
: ano, an alkoxy, a halo, or an alkyl group;
R3 is separately in each occurrence hydrogen, hydro-
carbyl, hydrocarbyloxy or hydrocarbylthio;
Y is a direct bond; and
b is an integer of from 0 to 3, inclusive with the
proviso that at least two of R2 are hydrogen with the further
proviso that Rl, R2 and R3 do not interfere with polymerization
: of the monomer.
Another aspect of this invention is a polyimide
polymeric composition which results from the polymerization of
3b
1300~35
- 3c - 64693-4007
one or more of the above-described compounds.
The novel compounds of this invention are easily
processable into useful articles. The polymerization of such
compounds does not result in the evolution of gaseous or volatile
by-products which can create problems in the eventual product
prepared.
- 3c
,, .
~30~3~
Furthermore, in order to prepare the polymers of these
monomers, there is no need for catalysts, initiators or
curing agents.
In general, the compounds of this invention
comprise unsaturated cyclic imides which are N-substi-
tuted with arylcyclobutene moieties. In such
arylcyclobutene moieties the cyclobutene ring is ~used
to the aromatic radical. The nitrogen atom of the
cyclic imide is connected to the aryl radical of the
arylcyclobutene moiety by a bridging member or a direct
bond. The cyclic imide can be substituted with
hydrocarbyl, hydrocarbyloxy or hydrocarbylthio
substituents. The aryl radical on the arylcyclobutene
moiety can be substituted with electron-withdrawing
groups, electron-donating groups, hydrocarbyl groups,
hydrocarbyloxy groups or hydrocarbylthio groups. The
cyclobutene ring may be sub~tituted with electron-
~ithdrawing groups or electron donating groups.
The cyclic imide can be any cyclic imide moietywhich contains olefinic unsaturation, and which may be
substituted in the manner described hereinbefore.
It is pre~erable that the olefinic unsaturation be
adjacent to one of the carbonyl moieties of the imide
functionality. In one preferred embodiment, the cyclic
imide is a 5-membered heterocycle, in particular, a
maleimide. Preferably, the substituents which may be
on the carbon atoms of the imide ring are C1-20 alkyl~
C1 20 alkoxy, C1_20 alkylthio, C6_20 aryl, C6_20
aryloxy~ C6-20 arylthio, C7_20 alkaryl, C7 20
alkaryloxy, C7_20 alkarylthio, C7_20 aralkyl, C7_20
aralkoxy or C7_20 aralkylthio. More preferred
28,913C-F -4-
~3~0~i35
-5--
substituents include Cl_20 alkyl, with C1_3 alkyl being
most preferred.
~he arylcyclobutene moiety can be any aro-
matic radical which has a cyclobutene ring fused to one
of the aromatic rings. The term "aryl" refers herein
to any aromatic radical. Aromatic as used herein
refers to carbocyclic or heterocyclic rings in which 4n
+ 2 delocalized n elections are contained in an orbital
ring. This property is also known as resonance
stablization or delocalization. Preferred carbocyclic
aromatic radicals include benzene, naphthalene, phenan-
threne, anthracene 7 a biaryl radical, or two or more
aromatic radica]s bridged by alkylene or cycloalkylene
moieties. More preferred carbocyclic aromatic radicals
include benzene, naphthalene, biphenyl, binaphthyl or a
diphenylalkylene or a diphenylcycloalkylene compound.
The most preferred carbocyclic aromatic radical is
benzene. Examples of preferred heterocyclic aromatic
compounds included pyrrole, furan, thiophene,
imidazole, oxazole, thiazole, pyrazole, pyridine, and
pyrimidine. More preferred heterocyclic aromatic
compounds are pyridine, furan, and thiophene, with
pyridine being most preferred. The carbocyclic aromatic
ring~ are pre~erred over the heterocyclic aromatic
rings.
The aryl radical can be substituted with
electron-withdrawing groups, electron-donating groups,
hydrocarbyloxy groups, hydrocarbyl groups or
hydrocarbylthio groups. Electron-withdrawing groups
re~er herein to cyano, carboxylate, hydrocarbyl-
carbonyloxy, nitro, halo9 hydrocarbylsulfinyl or
hydrocarbylsulfonyl groups. Electron-donating groups
refer herein to amino groups, hydroxy groups or alkyl
28,913C-F -5-
13~)0~35
--6--
groups. Preferred substituents on the aryl radical
include C1_20 alkyl, C1_20 alkoxy, C1_20 alkylthio, C6_
20 aryl, C6_20 aryloxy, C6_20 arylthio, C7_20 alkaryl,
C7_20 alkaryloxy, C7_20 alkarylthio, C7_20 aralkyl, C7_
20 aralkoxy, C7_20 aralkylthio, cyano, carboxylate,
hydrocar~ylcarbonyloxy, nitro, halo, hydrocarbyl-
sulfinyl, amino or hydrocarbylsulfonyl. More preferred
substituents on the aryl radical include C1_20 alkyl,
halo, nitro or cyano. The most pre~erred substituents
on the aryl moiety include Cl_3 alkyl, halo, nitro or
cyano.
The cyclobutene ring may be substituted with
electron-withdrawing groups or electron donating
groups, wherein electron-withdrawing groups and
electron-donating groups are described hereinbefore.
Preferred substituents on the cyclobutene ring are
cyano, carboxylate, hydrocarbylcarbonyloxy, nitro,
halo, hydrocarbylsulfonyl or hydrocarbylsulfinyl. More
preferred substituents include halo, nitro or cyano
groups; with cyano groups being most preferred.
The bridging member can be a divalent organic
radical which is bonded to the nitrogen of the cyclic
imide and the aryl radical o~ the arylcyclobutene
moiety. The divalent organic radical use~ul as a
bridging member is any divalent organic radical which
i9 capable of being bonded to both the nitrogen of a
cyclic imide and an aryl radical. The divalent organic
radical i5 preferably a hydrocarbylene, hydrocarbylene-
amido, hydrocarbylenecarbonyloxy, hydrocarbyleneoxy,
hydrocarbylenethio, hydrocarbylenesulfinyl or hydrocar-
bylenesulfonyl radical. More preferred divalent
organic radicals are alkylene, arylene 9 alkylene-
bridged polyarylene, cycloalkylene-bridged polyarylene,
28,913C-F -6-
` ~0~635
--7--
alkyleneamido, aryleneamido, alkylene-bridged
polyaryleneamido, cycloalkylene-bridged
polyaryleneamido, alkylenecarbonyloxy,
arylenecarbonyloxy, alkylene-bridged polyarylene-
carbonyloxy, cycloalkylene-bridged polyarylene-
carbonyloxy, alkyleneoxy, aryleneo~y, alkylene-bridged
polyaryleneoxy, cycloalkylene bridged polyaryleneoxy,
alkylenethio, arylenethio, alkylene-bridged
polyarylenethio, cycloalkylene-bridged polyarylenethio,
alkylenesulfinyl, arylenesulfinyl, alkylenebridged
polyarylenesulfinyl, cycloalkylene-bridged
polyarylenesulfinyl, alkylenesulfonyl, arylenesulfonyl,
alkylene-bridged polyarylenesulfonyl or cycloalkylene-
bridged polyarylenesulfonyl. Even more preferreddivalent organic radicals include alkylene, arylene,
alkylenecarbonyloxy, arylenecarbonyloxy, alkyleneamido,
aryleneamido, alkyleneoxy, aryleneoxy, alkylenethio or
arylenethio. Most preferred divalent organic radicals
include alkylene and aryiene radicals.
Preferably, the aryl moiety and cyclic imide
are connected by a direot bond or a bridging member
which compriqes an alkylene, arylene, alkylene-bridged
polyarylene or cycloalkylene-bridged polyarylene; and
more preferably a direct bond or a bridging me!mber
which comprises an alkylene or arylene moiety. Most
pre~erably the cyclic imide nitro~en and the aryl
radical are connected by a direct bond.
3o
Preferred N-substituted arylcyclobutenyl cyclic
imideq correspond to the ~ormula
28,913C-F -7-
" ~OO~i35
--8--
~G~
X ~I~Y-Ar/ ~ C(R2)2)2
\C/ (Rl)a
0
wherein
Ar is an aromatic radical;
Rl is separately in each occurrence a hydro-
carbyl, hydrocarbyloxy, hydrocarbylthio,
electron-donating or electron-withdrawing group;
R2 is separately in each occurrence hydrogen or
an electron-withdrawing group;
X is an alkenylene moiety which can be sub-
stituted with one or more hydrocarbyl, hydrocarbyloxy
or hydrocarbylthio groups;
Y is a direct bond or divalent organic moi-
ety; and
a is an integer of between 0 and 3.
More preferred N-substituted arylcyclobu
tenyl-unsaturated cyclic imides include those which
correspond to the formula
28,913C-F -8-
g
R3 ~
N-Y-Ar (C(R2)2)2
wherein
Ar is an aromatic radical;
R1 is separately in each occurrence a hydro-
carbyl, hydrocarbyloxy, hydrocarbylthio, an electron-
donating or electron-withdrawing group;
R2 is separately. in each occurrence hydrogen or
an electron-withdrawing group;
R3 is separately in each occurrence hydrogen, a
hydrocarbyl, hydrocarbyloxy or hydrocarbylthio group;
Y is a direct bond or a divalent organic
radical; and
a is an integer of between 0 and 3.
In an even more preferred embodiment, the N-
substituted arylcyclobutenyl-unsaturated cyclic imide
corresponds to the formula
28,913C-F -9-
~3~ 3S
--1 o--
9 ~ )2
wherein
R1 is separately in each occurrence a hydro-
carbyl, hydrocarbylthio, hydrocarbyloxy9 electron-
withclrawing or electron-donating group;
R2 i9 separately in each occurrence hydrogen or
an electron-withdrawing group;
R3 is separately in each occurrence hydrogen,
hydrocarbyl, hydrocarbyloxy or hydrocarbylthio;
Y is a direct bond or a divalent organic
radical; and
b is an integer of between 0 and 3, inclu~ive.
In the above formulas, R1 i9 preferably C1~2~
alkyl, C1_20 alkoxy, C1_20 alkylthio, C6_20 aryl, C6_20
aryloxy~ C6-20 arylthio, C7_20 alkaryl, C7 20
alkaryloxy, C7_20 alkarylthio, C7_20 aralkyl, C7_20
3 aralkoxy, C7_20 aralkylthio, cyano, carboxylate,
hydrocarbylcarbonyloxy, nitro, halo, hydrocarbylsul-
finyl, hydrocarbylsulfonyl or amino. R1 is more pref-
erably C1_20 alkyl, halo, nitro or cyano. Most prefer-
ably R1 is C1-3 alkyl, halo, nitro or cyano.
287913C-F -lQo
~ 306~5
"
R2 is preferably hydrogen, cyano, carboxylate,
hydrocarbylcarbonyloxy, nitro, halo, hydrocarbyl-
sul~onyl hydrocarbylsulfinyl, alkyl, amido,
hydrocarbyloxy. R2 is more preferably hydrogen, halo,
nitro or cyano. R2 is even more preferably hydrogen or
cyano and most preferably hydrogen.
R3 is preferably hydrogen, C1_20 alkyl7 C1_20
alkoxy, C1-20 alkylthio, C6_20 aryl, C6_20 aryl-oxy,
C6 20 arylthio, C7_20 alkaryl~ C7_20 alkaryloXY~ C7-20
alkarylthio, C7_20 aralkyl~ ~7-20 aralkoxy or 7-20
aralkylthio. R3 is more preferably hydrogen or C1_20
alkyl. R3 is even more preferably hydrogen or C1_3
alkyl and most preferably hydrogen.
In the above formulas, Y is preferably a direct
bond, a hydrocarbylene, hydrocarbyleneamido,
hydrocarbylenecarbonyloxy, hydrocarbyleneoxy, hydro-
carbyleneamino, hydrocarbylenecarbonyl, hydrocarbyl-
enethio, hydrocarbylenepolythio, hydrocarbylenesul-
finyl or hydrocarbylenesulfonyl. Y is more prefer-
ably a direct bond, alkylene, arylene, alkylene-bridged
polyarylene, cycloalkylene-bridged polyarylene,
alkyleneamido, aryleneamido, alkylenecarbonyloxy,
arylenecarbonyloxy, arylenecarbonyl, alkylenecarbonyl,
aryleneoxy, alkyleneoxy, aryleneamino, alkyleneamino,
alkylenethio, alkylenepolythio, arylenethio,
arylenepolythio, arylenesulfinyl, alkylenesulfinyl,
arylenesulfonyl or alkylenesulfonyl. Y is most
preferably a direct bond, alkylene or arylene.
In the formulas described hereinbefore, Ar is
preferably a benzene, naphthalene, phenanthrene,
anthracene or biaryl radical, or two or more aromatic
radicals bridged by alkylene moieties. Ar is more
28,913C-F -11-
0635
-12-
preferably benzene, naphthalene, biphenyl, binaphthyl
or a diphenylalkylene. Ar is more preferably a benzene
radical.
Hydrocarbyl means herein an organic radical
containing carbon and hydrogen a~oms. The term hydro-
carbyl includes the following organic radicals:
alkyl,alkenyl, alkynyl, cycloalkyl, cycloalkenyl, aryl,
aliphatic and cycloaliphatic aralkyl and alkaryl. Ali-
phatic re~ers herein to straight- and branched-, and
saturated and unsaturated, hydrocarbon chains, that is,
alkyl, alkenyl or alkynyl. Cycloaliphatic refers
herein to saturated and unsaturated cyclic
hydrocarbons, that is, cycloalkenyl and cycloalkyl.
The term aryl re~ers herein to biaryl, biphenylyl,
phenyl, naphthyl, phenanthranyl, anthranyl and two aryl
groups bridged by an alkylene group. Alkaryl refers
herein to an alkyl-, alkenyl- or alkynyl-substituted
aryl substituent wherein aryl i9 as de~ined
hereinbeYore. Aralkyl means herein an alkyl, alkenyl
or alkynyl group substituted with an aryl group,
wherein aryl is as defined hereinbefore. Cl_20 alkyl
includes straight- and branchad-chain methyl, ethyl,
propyl, butyl, pen~yl, hexyl, heptyl, octyl, nonyl,
decyl, undecyl, dodecyl, tridecyl, tetradecyl,
perltadecyl, hexadecyl, heptadecyl, octadecyl,-nonadecyl
ancl eicosyl groups. Cl_5 alkyl includes methyl, ethyl,
propyl, butyl and pentyl.
3o
Cycloalkyl re~ers to alkyl groups containing
one, two, three or more cyclic rings. Cycloalkenyl
re~'ers to mono-, di- and polycyclic groups containing
one or more double bonds. Cycloalkenyl also refers to
28,913C-F -12-
-
-13-
cycloalkenyl groups wherein two or more double bonds
are present.
Hydrocarbylene herein refers to a divalent
hydrocarbon radical and is analogous to the hydrocarbyl
radicals described hereinbefore with the single differ-
ence that the hydrocarbylene radical is divalent.
Hydrocarbyleneamido refers herein to a divalent
radical wherein a hydrocarbylene radical is
bonded to an amido group, and corresponds to the for-
mula
o
-R4-CN-
R5
wherein R4 is a hydrocarbylene radical and R5 is hydro-
gen or a hydrocarbyl radical.
Hydrocarbyleneoxy refers herein to a divalent
radical in which a hydrocarbylene radical is bonded to
a divalent oxygen atom and corresponds to the formula
-R4-o~ wherein R4 i9 as defined hereinbefore.
Hydrocarbylenecarbonyloxy refers to a hydro-
carbylene moiety which is bonded to a carbonyl moiety
which is further bonded to a divalent oxygen atom and
corresponds to the formula
R4-Co~
wherein R4 is as defined hereinbeforeO
28,913C-F -13-
0~3S
--1 4--
Hydrocarbylenethio refers herein to a radical
in which a hydrocarbylene radical is further bonded to
one or more sulfur moieties and corresponds to the
formula -R4-(S)p- wherein R4 is as hereinbefore
defined, and wherein p is between 1 and 3.
Hydrocarbyleneamino refers herein to a
hydrocarbylene radical bonded to an amino moiety and
generally corresponds to the formula
-R4-N-
R5
wherein R4 and R5 are as defined hereinbefore.
Hydrocarbylenesulfinyl refers herein to a
hydrocarbylene moiety bo~ded to a sulfinyl moiety and
generally corresonds to the formula
-R4-S-
wherein R4 is as hereinbefore defined.
Hydrocarbylenesulfonyl ~enerally corresponds to
a radical in which a hydrocarbylene radical is bonded
to a sul~onyl radical and corresponds to the formula
2~,913C-F -14-
~30()~i3S
--15--
-R4-S-
o
wherein R4 i~ as hereinbefore defined.
Wherein the bridging member is a hydrocarbyl-
eneamido, hydrocarbyleneoxy7 hydrocarbyleneamino,
hydrocarbylene~hio, hydrocarbylenecarbonyloxy moiety,
the amido, amino, oxy, thio, sul~inyl or sulfonyl
moiety is preferably bonded to the aryl portion of the
arylcyclobutene.
Examples of preferred N-substituted benzocy-
clobutenyl maleimides include N-benzocyclobutenyl male-
imide, N-benzocyclobutenylmethyl maleimide, N-benzocy-
clobutenylethyl maleimide, N-benzocyclobutenylpropyl
maleimide, N-benzocyclobutenylbutyl maleimide, N-ben-
zocyclobutenylpentyl maleimide, N-benzocyclobutenyl-
hexyl maleimide, N-benzocyclobutenylphenyl maleimide,
N-benzocyclobutenylbiphenyl maleimide, N-benzocyclobu-
tenylamidomethyl maleimide, N-benzocyclobutenylamido-
ethyl ma.leimide, N-benzocyclobutenylamidopropyl male-
imide, N-benzocyclobutenylamidobutyl maleimide, N-ben-
zocyclobutenylamidopentyl maleimide, N-benzocyclobuten-
ylamidohexyl maleimide, N-benzocyclobutenylamidophenyl
maleimide, N-benzocyclobutenylamidobiphenyl maleimide,
: N-benzocyclobutenyloxycarbonylmethyl maleimide, N-
benzocyclobutenyloxycarbonylethyl maleimide 9 N-
benzocyclobutenyloxycarbonylpropyl maleimide, N-
benzocyclobutenyloxycarbonylbutyl maleimide, N-
; 35 benzocyclobutenyloxycarbonylpentyl maleimide, N-
benzocyclobutenyloxycarbonylhexyl maleimide, N-
28,913C-F -15-
~3~)~)635
.--16--
benzocyclobutenyloxycarbonylphenyl maleimide, N-
benzocyclobutenyloxycarbonylbiphenyl maleimide, N-ben-
zocyclobutenylthiomethyl maleimide, N-benzo-
cyclobutenylthioethyl maleimide, N-benzocyclo-
butenylthiopropyl male-imide, N-benzocyclo-
butenylthiobutyl maleimide, N-benzocyclo-
butenylthiopentyl maleimide, N-benzocyclo-
butenylthiohexyl maleimide, N-benzocyclo-
butenylthiophenyl maleimide, N-benzocyclo-
butenylthiobiphenyl maleimide, N-benzocyclo
butenyloxymethyl maleimide, N-benzocyclobutenyloxyethyl
maleimide, N-benzocyclobutenyloxypropyl maleimide, N-
benzocyclobutenyloxybutyl maleimide, N-benzocyclo-
butenyloxypentyl maleimide, N-benzocyclobutenyloxyhexyl
maleimide, N-benzocyclobutenyloxyphenyl maleimide, N-
benzocyclobutenyloxybiphenyl maleimide.
The arylcyclobutene moieties can be prepared by
several synthesis schemes.
In one synthesis scheme, an alkyl-substituted
aromatic compound which is further substituted with an
aryl deactivating substituent is chloroalkylated in a
position ortho to the alkyl group. In the preferred
embodiment wherein the aromatic compound is benzene,
the starting material corresponds to the following for-
mula
~3o
28,913C-F -16-
,
3 ~ ~ 6 3
-17-
.
(R2)2
lH
(R1
R9
wherein R1 and R2 are defined hereinbefore; R9 is any
aryl deactivating substituent; and c is an integer of
o, 1, 2, or 3. The alkyl N-substituted aromatic
compound is chloroalkylated by contacting the alkyl
aromatic compound with a chloroalkylating agent and
thionyl chloride in the presence of an iron chloride
cataly~t so a~:to result in a product which contains a
chloroalkyl group ortho to the alkyl substituent. In
the embodiment wherein the aromatic compound is a
benzene ring, the product corresponds to the formula
(R2)2
CH
(R~ C(R2)2-Cl
.
wherein R9 i9 a hydrocarbyloxycarbonyl, carboxamide,
hydrocarbylcarbonyl, carboxylate, halocarbonyl,
28,913C F -17-
3C~0635
--18--
nitrile, nitro, sulfone or sulfoxide group. R9 is more
preferably a halo or hydrocarbyloxycarbonyl group, with
hydrocarbyloxycarbonyl being the most preferred group.
Preferably c i9 0 or 1, most pre~erably 0.
In this process the chloroalkylating agent is
preferably chloromethyl methyl ether, although other
chloroalkylating agents such as bis(chloromethyl) ether
could be used. At least a 2:1 molar excess of the
chloroalkylating agent to the alkyl-substituted
aromatic compound is needed. It is preferable to use
between a 6:1 and 3:1 ratio of chloroalkylating agen~
to alkyl aromatic compound. The catalyst is ferric
chloride (FeCl3) while the cocatalyst is thionyl
chloride. The catalyst can be present in between 0.1
and 1.0 mole per mole of alkyl aromatic. More
preferably between 0.2 and 0.4 mole of catalyst are
present for each mole of alkyl aromatic compound.
Preferably between 0.1 and 1.0 mole of thionyl chloride
per mole of alkyl aromatic is used, more preferably
between 0.2 and 0.4 mole per mole of alkyl aromatic.
This process can be performed at a tempera-
ture of between 40C and 80C, preferably 40C and 60C.
Below 40C, the reaction rate is low. The boiling point
of some of the components of the reaction mixture
starts at 80C.
3~ This process can be done by contacting the
alkyl aromatic compound with the chloromethylating
agent, catalyst and cocatalyst in a suitable solvent.
Suitable solvents include chlorinated hydrocarbon
solvents. Thereafter the reaction mixture is heated to
the appropriate temperature.
28,913C F -18-
0 6~ 5
-19-
The product can be recovered by quenching the
reaction mixture with alcohols or water to inactivate
the chloroalkylating agents remaining, stripping off
the volatiles and washing out the catalyst with water.
The product thereafter is recovered by distillation.
..
The ortho chloroalkylated alkyl aromatic
compounds can be converted to aromatic compounds with
cyclobutene rings ~used thereto, by pyrolysis. This is
achieved by contacting the ortho chloroalkylated alkyl
aromatic compound with at least 2 times its weight of a
suitable diluent, and thereafter passing the mixture
through a reactor at a temperature of 550C or greater
and a pressure of between about atmospheric and 25 mm
of mercury. Suitable diluents are generally
substituted aromatic compounds which are inert to the
chloromethylated alkyl aromatic compound and are stable
at pyrolysis temperatures. Examples of suitable
diluents are benzene, toluene, xylenes, chlorobenzenes,
nitrobenzenes, methylbenzoates, phenyl acetate or
diphenyl acetate. Preferred diluents are the xylenes.
Preferable temperatures are between 700C and 750C.
Preferable pressures are between 35 and 25 mm o~
mercury. In a preferred embodiment, the reaction
mixture i9 pa~sed through a hot tube packed with an
inert material 5 for example, quartz chips or stainless
steel helices. The product can be recovered by
distillation. The product wherein the aromatic
3 compound is benzene corresponds to the formula
28,913C-F -19-
;35
.~ .
-20-
~ R2 ) 2
R9
wherein R1, R2, R9 and c are as hereinbefore defined.
In the preferred embodiment wherein R9 is a
hydrocarbyloxy carbonyl moiety, the hydrocarbyloxy
carbonyl moiety can be converted to a carboxylate moi-
ety by contacting the substituted (arylcyclobutene)
compound with at least a molar equivalent o~ alkali
metal hydroxide in an alkanol-water solvent system. In
the embodiment wherein the aromatic radical is benzene,
the product corresponds to the formula
~ ~2)2
HOC
o
Thereafter the carboxylate-substituted (aryl-
cyclobutene) compound can be converted to an acid chlo-
ride by contacting the carboxylate-substituted (arylcy-
28,913C-F -20-
,..
) i35
-21-
clobutene) compound with thionyl chloride and refluxing
at 70C to 80C. The acid halide-substituted
(arylcyclobutene) so formed can be used to prepare the
novel monomers of this invention, as described
hereinafter. In the embodiment wherein the aryl
radical is a benzene ring, the product ~orresponds to
the formula
(R1 )~R2)2
ClC
o
In an alternative synthesis, an aryl compound
with ortho dibromomethyl groups can be converted to a
1,2-diiodoarylcyclobutene, by contacting the aryl com-
pound substituted with ortho dibromomethyl moieties
with an alkali metal iodide in an alkanol solvent at
reflux ~o a~ to form the diiodoarylcyclobutenes. The
product can be recovered by filtering, evaporating the
filtrate and recrystallizing the product. In the
embodiment wherein the aryl radical is a benzene
radical, the starting material corresponds to the
formula
28,913C-F -21-
1~ 35
-22-
(R1)G ~ HBr2
~ CHBr2
and the iodobenzocyclobutene corresponds to the formula
( R 1 ) G ~ I
The 1,2-diiodoarylcyclobutenes can be converted
to arylcyclobutenes by dissolving the 1,2-diiodo-
arylcyclobutenes in an alcohol solvent, preferably
methanol or ethanol and contacting the solution with an
alkali metal hydroxide in the presence of a palladium-
on-carbon catalyst and H2 gas at a temperature of 20C
3 to 30C. In general, at least between about 2 and 4
moles of alkali metal hydroxide per mole of 1,2-
diiodoarylcyclobutene is used. Preferably, between 50
and 200 psi (344.7 kPa and 1379 kPa) of hydrogen gas is
u~ed. The arylcyclobutenes prepared in this manner can
be recovered by distillation. In the embodiment
28,913C-F -22-
~L30~3~3~i
--23--
wherein the aryl radical is a benzene radical, the
product corresponds to the formula
(R1 )~ )Ll
The arylcyclobutene is thereafter brominated.
In this process, the arylcyclobutene is dissolved in
acetic acid and contacted with a brominating agent of
pyridinium hydrobromide perbromide in the presence of
mercuric salts, for example, mercuric acetate, at a
temperature of between 20C and 50C. The brominated
product can be recovered by extraction and
distillation. In the embodiment wherein aryl radical
is benzene, the product corresponds to the formula
(R1)
Br
The brominated arylcyclobutene can there~after
be carbonylated to prepare a hydrocarbyloxy carbonyl-
substituted arylcyclobutene. This carbonylation is
achieved by dissolving the brominated arylcyclobutene
in an alkanol solvent~ and thereafter contacting the
solution with carbon monoxide under pressure in the
presence of a palladium catalyst, wherein the palladium
is in the zero valence state, in the further presence
of an acid acceptor under conditions such that the
28,913C-F -23-
:
` ~0~)~;35
-24-
brominated arylcyclobutene compound undergoes car-
bonylation. Preferred catalysts are palladium acetate
with a cocatalyst of triphenyl phosphine, palladium
triphenyl phosphine tetrakis, andbis(triphenyl
phosphine) palladium chloride complex. The acid
acceptor is generally a tertiary amine. In general,
the reaction vessel is pressurized with carbon monoxide
to a pressure of between atmospheric and 3000 psi
(20,684 kPa), preferred pressures are between 600 and
1000 psi (4136 and 6895 kPa).
This process is preferably run at a tempera-
ture of between 100C and 140C, most preferably between
120C and 130C. The hydrocarbyloxycarbonyl
arylcyclobutene can be recovered by filtering off the
catalyst, washing away the acid scavenger with a 10
percent strong acid solution, stripping off the solvent
and distilling the product to puri~y it. To prepare a
carboxamide-substituted arylcyclobutene, a primary or
secondary amine is substituted for the alcohol solvent.
In the embodiment wherein the aryl radical is a benzene
radical, the process corresponds to the following
equation:
(R1)C--~ +R100H + N(R6)3~R1)C~
wherein R1 and c are as hereinbe~ore de~ined and R6 and
R10 are hydrocarbyl moieties. The hydrocarbyloxy-
carbonyl-substituted or carboxamide-substituted
arylcyclobutenes can thereaYter be acidified and
28,913C-F -24-
3 5
-25-
converted to acid chlorides by the process described
hereinbefore.
In another preparation of an arylcyclobutene,
the reaction may ~ollow that reported by Skorcz and
Kaminski, Or~. Syn., 48, pages 53-56 (1968). In a
typical preparation, an alkyl cyanoacetate is added to
- a solution o~ sodium metal in ethanol followed by the
addition of an ortho-halomethylaryl halide. The alkyl
2-(o-halomethylaryl)cyanoacetate is isolated and
treated with aqueous sodium hydroxide. Subsequent
acidi~ication results in the cyanoacetic acid
derivative. That derivative is placed into N,N-
dimethylformamide and is refluxed to form the 3-(o- -
halomethylaryl)propionitrile derivative which is
isolated and added to a suspension of sodamide in
liquid ammonia. ~fter an appropriate reaction time,
ammonium nitrate is added and the ammonia allowed to
evaporate. The cyanoarylcyclobutene is isolated by
ether extraction and purified by ~ractional distilla-
tion under reduced pressure.
Substituted arylcyclobutenes can be prepared by
the same technique by using the appropriately substi-
tuted reactants, such as an alkyl or alkoxybenzyl
halide. Also substituents can result ~rom using an
alkyl haloacetate or a dialkylmalonate.
In another preparation based on the paper by
Matsura et al., Bull. ChemO Soc. Jap., 39, 1342 (1966),
o-a~inoaryl carboxylic acid is dissolved in ethanol and
hydrochloric acid added. Isoamylnitrite is slowly
added to the cold stirred solution and diethyl ether is
then added. The product, aryldiazonium-2-carboxylate
hydrochloride, is filtered. That product is placed in
28,913C-F -25-
3~;
- 26 - 64693-4007
a solvent, preferably ethylene dichloride, and acrylonitrile and
propylene oxide is added to the stirred mixture which is then
heated under nitrogen until the reaction is complete. After cool-
ing, the mixture is filtered and the product, l-cyanoarylcyclo-
butene, is isolated by fractionally distilling the filtrate under
reduced pressure.
Amounts of reactants, reaction parameters and other
details can be found in the cited article, the examples of this
application, or can be easily deduced therefrom.
In the next sequence of reactions, the cyanoarylcyclo-
butene or substituted derivative is nuclear substituted. In one
preparation, the cyanoarylcyclobutene is added slowly to a cold
solution of sodium nitrate in concentrated sulfuric acid to form
5-nitro-1-cyanoarylcyclobutene. That nitro compound is isolated,
dissolved in ethanol and reduced by hydrogenation over a palladium
on carbon catalyst. The isolated product is 5-amino-1-cyanoaryl-
cyclobutene. In the preferred embodiment where the aryl radical
is benzene, the product corresponds to the formula
N 2 ~\ ~ CN
(R )c ~ (R2)2
Cyclobutapyridines are prepared by the pyrolysis of
4-pyridyl propargyl ether at 550C. See J.M. Riemann et al.
Tetrahedron Letters, No. 22, pp. 1867-1870 (1977).
~ ;
0~ 5
Alternatively, a pyridine-4-carbonitrile with an alkyl
substituent on the carbon atom adjacent to the nitrile
iq reacted with sodium azide and ammonium chloride in
N,N-dimethylformamide to prepare a 5(alkyl-4-pyridyl)-
tetrazole. The 5(alkyl-4-pyridyl)tetrazole i9
pyrolyzed at about 600C to prepare a cyclobutapyridine.
See. W. D. Crow et al. Australian Journal of Chemistry
1741 et seq. (1975).
Amino cyclobutapyridines are prepared by
reacting a cyclobutapyridine with sodamide (NaNH2) in
N,N-dimethylaniline solvent at 110C. A hydroxycyclo-
butapyridine is prepared by reacting one mole of an
aminocyclobutapyridine with one mole of sodium nitrite
and two moles of sulfuric acid in water at 0C for a
period of time and thereafter warming to 50C. Halo-
substituted cyclobutapyridine i9 prepared by reacting a
hydroxypyridine in thionyl at reflux either neat or in
solution, for example, thionyl chloride or thionyl
bromide, in N,N-dimethylformamide solvent.
The N-substituted arylcyclobutenyl-unsaturated
cyclic imides of this invention wherein the bridging
member is a direct bond can be prepared by the fol-
lowing method. An unsaturated cyclic anhydride is
contacted ~ith an amine-substituted arylcyclobutene
under conditions qo as to form an N-arylcyclo-
butenylamido alkenoic acid. Such acid can thereafter
be dehydrated to cyclize ~he amido alkenoic acid into a
cyclic imide ring and form the N-sub-stituted
arylcyclobutenyl-unsaturated cyclic imide.
The formation of the arylcyclobutenyl amido
alkenoic acid is achieved by reacting an unsaturated
cyclic anhydride with an amine-substituted arylcyclo-
28,913C-F -27-
;35
-28-
butene. This reaction is exemplified in one preferred
embodiment wherein the anhydride is maleic anhydride
and the arylcyclobutene is 5-aminobenzocyclobutene, and
is illustrated by the following equation:
NH ~
O O
lC
The cyclic anhydride and amino~substituted
arylcyclobutene are contacted in a suitable solvent at
a temperature of between -40C and 100C. Suitable
solvents include aliphatic hydrocarbons, aromatic
hydrocarbons, ethers and halogenated hydrocarbons. It
is preferred to run the process under an inert
atmosphere. It is also preferred to use freshly
sublimed anhydride as any impurities in the anhydride
can result in very poor yields. It is also preferred
to use at least`a 5 percent excess of anhydride so as
to drive the reaction to completion with respect to the
amino-substituted arylcyclobutene compound.
Preferred temperatures are between 0C and 50C
with between 20C and 25C being most preferred.
The N-arylcyclobutenylamido alkenoic acid can
thereafter be dehydrated by one of two methods. In the
preferred embodiment, the N-arylcyclobutenylamido
alkenoic acid is contacted with a dehydrating agent in
28,913C-F -28-
j35
-29-
an aprotic reaction medium in the presence of a nickel
II salt catalyst. In general, the reaction medium is
an aprotic solvent and can include ketones, ethers,
amides or aliphatic halogenated hydrocarbons.
Preferred reaction media include the ketones, with
acetone being most preferred. The dehydrating agents
include anhydrides, carbodiimides and isocyanates; with
the anhydrides being preferred and acetic anhydride
being most preferred.
The catalyst is any nickel II salt with nickel
II acetate being most preferred. In general, between 1
and 5 percent of the catalyst is useful. It is
preferable to run this process in the presence of an
aprotic base such as a carbonate or tertiary amine,
preferably a tertiary amine. In general, between 20
and 200 mole percent of a tertiary amine is used, with
between 100 and 150 mole percent being preferred,
wherein mole percentages are based on the starting N-
arylcyclobutenylamido alkenoic acid. The mole ratio of
the dehydrating agent to the N-arylcyclobutenylamido
alkanoic acid is between 4:1 and 1:1, preferably
between 1.5:1 and 1:1.
It is preferred to run this process under an
inert atmosphere. Temperatures which are useful are
those at which the dehydration takes place. Preferable
temperatures are between -20C and 100C, with between
15C and 25C being most preferred.
In this reaction, the N-arylcyclobutenylamido
alkenoic acid is not ~oluble in the reaction medium but
the cyclic imide product is solubleO The reactant is
slurried in the reaction media and exposed to the
reaction conditions described. The completion of the
28,913C-F -29-
S
--30--
reaction is noted by dissolution of the reactants
indicating formation of products.
In an alternative procedure, the N-arylcyclo-
butenyl amido alkenoic acid can be dehydrated bydispersing the compound in a glacial acetic acid
reaction media in the presence of an alkali or alkaline
earth metal acetate salt, and heating the reaction
mixture to a temperature at which the dehydration takes
place to form the cyclic imide rings. Generally, a
sufficient amount of alkali or alkaline earth metal
acetate salt to cause complete dehydration is suitable.
Preferably, at least an equimolar amount of alkali or
alkaline earth metal acetate salt is used, most
preferably an excess of 5 mole percent. The process
can be run at any temperature at which the dehydration
takes place, preferable temperatures are between 50C
and 140C, with between 100C and 120C being most
preferred. Completion of the reaction is indicated by
dissolution of the product.
In both instances, the product can be reco~-
ered by washing with water and thereafter an aqueous
solution o~ an inorganic baqe.
To prepare an N-substituted arylcyclobu- tenyl
cyclic imide with a hydrocarbylene amido, hydro-
carbyleneoxy or hydrocarbyleneoxycarbonyl bridge, an
unsaturated cyclic anhydride is reacted with a hydro-
carbon substituted with amino and carboxyl moieties,
for the hydrocarbylene amido-bridged species, or a
hydrocarbon substituted with amino and hydroxyl moi-
eties, for the hydrocarbyleneoxy and hydrocarbylene-
28,913C-F -30-
~3~6~5
--3 1--
oxycarbonyl-bridged species, to prepare an amido alka-
noic acid wherein the amido nitrogen is substituted
with a carboxy-substituted hydrocarbyl or hydroxy-sub-
stituted hydrocarbyl moiety. This reaction can be per-
formed at a temperature of between -40C and 100C in a
suitable solvent. Suitable solvents include
aliphatichydrocarbons, aromatic hydrocarbons, ethers
and halogenated hydrocarbons. It is preferred to run
the process under an inert atmosphere. It is preferred
to use freshly sublimed anhydride as any impurities can
result in very poor yields. It is preferred to use at
least a 5 percent excess of anhydride so as to drive
the reaction to completion.
In the embodiment wherein the anhydride is
maleic anhydride, these reactions are exemplified by
the following equations
28,913C-F -31-
L3~ 5
-32-
O O ' '
R3 ~ R3 ~
O + NH2-R4-CoH~ ¦ NH2-R4-CoH
R3 ~ R3 ~ C /
O O
and
O O
R3 ~ R3 ~
\ I \~H-R4-oH
O + NH2-R4-oH D- l
1 R3 ~ R3 ~ C-OH
O
wherein R3 i9 as hereinbefore defined and R4 is a
hydrocarbylene radical.
The amido alkenoic acid can be dehydrated using
one of the two dehydration methods described
hereinbefore so as to prepare a N-substituted cyclic
imide wherein the substituent is a N-hydrocarbylcar-
bonyloxycarbonyl cyclic imide, or a N-hydrocarbylcar-
bonyloxy cyclic imide. In the embodiment wherein the
N-substituted amido alkenoic acid was derived ~rom
28,913C-F -32-
~L3t~6~5
-33-
maleic anhydride, this reaction is exempli~ied by the
~ollowing equations
O O
R3 ~ R3
~ , ' \ 0 00 ` / 00
\ ~- " " _Q~t ~ " ,.
~ NH-R4-coH ~ R5CoCR5 t-amine , ~j-R4-cocR5
R3 C-OH R3 O
o
and ,,
+H20 ~ R5-CoH
O O
R3 ~ ~
" " cat _ ,-
NH-R4-oH + R5CoCR5 t-amine N-R4-oCR5
, \ , \
R3 C-OH R3 ~ .
.
o
+H20 + R5-CoH
wherein R3 and R4 are as hereinbefore defined and R5 is
a hydrocarbyl moiety.
28,913C-F -33-
- 130~)635
.
-34-
The N-hydrocarbylcarbonyloxycarbonyl cyclic
imide is converted to a hydrocarbylene amido-bridged N-
substituted arylcyclobutenyl cyclic imide by reacting
the N-hydrocarbylcarbonyloxycarbonyl cyclic imide with
an amino-substituted arylcyclobutene in the presence of
a tertiary amine. ThiS process can be accomplished~by
contacting the starting reactants in a chlorinated ali-
phatic hydrocarbon solvent at 0C with agitation under
an inert atmosphere. This process iS exemplified by
the following equation
~ N-~4-CoCRS + ~ ~ (R6)3-N~
lt
1' R3. ~
~ N-R4-CN/ ~ ~ ~ +R5CoH-N-(R6)3
2( 0
wherein R4 and R5 are as herelnbefore defined, and R6
is a hydrocarbyl radicalO
28,913C-F -34-
- 13~06~S
To prepare a hydrocarbyleneoxy or hydrocar-
byleneoxycarbonyl-bridged N-substituted arylcyclo-
butenyl cyclic imide, the N-hydrocarbylcarbonyl-
oxyhydrocarbyl cyclic imide is hydrolyzed to prepare a
N-hydroxyhydrocarbyl cyclic imidec The hydrolysis is
usually run in an aqueous/alkanol solvent system in the
presence of an acid or base catalyst at between room
temperature and reflux of the solvent mixture (20C to
60C). This reaction is exemplified by the following
equation
~ W-R~-oC~5 +~2 ~ N-R4-o~ + ~5CoH
wherein R3, R4 and R5 are as hereinbefore defined.
To prepare the hydrocarbyleneoxycarbonyl-
bridged N-substituted arylcyclobutenyl cyclic imides,
the N~hydroxyhydrocarbyl cyclic imide is reacted with a
chlorocarbonyl-substituted arylcyclobutene. In prac-
tice, the N-hydroxyhydrocarbyl cyclic imide is dis-
solved in a chlorinated aliphatic hydrocarbon solvent
to which is added a tertiary amine, which functions as
an acid acceptor, and thereafter the chlorocarbonyl-
substituted arylcyclobutene in a chlorinated aliphatic
hydrocarbon is added slowly to the mixture. This is
2~ preferably done at 0C in an inert atmosphere. It is
28,913C-F -35-
~00~35
-36 -
preferred to stir the reaction mixture for a period of
time at 0C after the addition is complete. This
reaction is exemplified by the following equation
R3 ~ 0
\~ N-R4 OH + ClC~
R3 ~ ~ ¦ I (R6)3-N- -
R3 ~
~ N-R4-oC - ~ + (R6)3-N HCl
1( 0
wherein R3, R4 and R6 are as hereinbefore defined.
The hydrocarbyleneoxy-bridged N-substituted
arylcyclobutenyl cyclic imides can be prepared from th-e
N-hydroxyhydrocarbyl cyclic imide in the following man-
ner. The N-hydroxyhydrocarbyl cyclic imide is reacted
with p-toluene sulfonyl chloride and pyridine to pre-
pare a cyclic imido hydrocarbyl p-toluene sulfonate.
Either excess pyridine or methylene chloride are used
as the solvent. The reactants are contacted in equi-
molar amounts, unless pyridine is the solvent, at a
temperature of between 0C and 25C. This reaction is
exemplified by the following equation
28,913C-F -36-
` ~3()0~5
-37
~ N-~4-oH + CH3 ~ 502Cl + ~ N ~
R3
o
o
~ N-R4-o-so2 ~ CH3 + ~ N-HCl
R3
O
wherein R3 and ~ are as hereinbefore defined.
The ayclic imido hydrocarbyl p-toluene sul-
fonate is contacted with a hydroxy-substituted arylcy-
clobutene in the presence of a four to five molarexcess of an alkali metal carbonate (~uch as potassium
carbonate) based on the sulfonate, in a N,N-dimethyl
formamide solvent, to prepare a hydrocarbyloxy-bridged
N-substituted arylcyclobutenyl cyclic imide. This
reaction takes place at temperatures of between 20C and
140C. This process is exemplified by the ~ollowing
equation
28,913C-F -37-
1:~00~35
-38-
N-R4-0-502 ~ H0 ~ ¦ + 4-5 E2C03
o
o
~ ~W-F~4-o~=1 0 s +H2
wherein R3 and R4 are as hereinbefore defined.
The hydrocarbylene amino-bridged N-substi-
tuted arylcyclobutenyl cyclic imides can be prepared by
the ~ollowing procedure. An amino-substituted aryl-
cyclobutene is reacted with about an equimolar amount
of a hydrocarbon substituted with aldehyde and nitro
moieties; in the presence o~ between about 0.3 to 1.5
moles of sodium cyanoborohydride in a methanolic sol-
vent at 20C to 25C. The product is nitrohydrocarbyl
amino-substituted arylcyclobutene. The process can be
exemplified by the following equation
28,913C-F -38-
6 3 5
-39-
No2-R4-CH + NHR7 ~ NaBH3CN
.
No2-R4-CH2NR7 ~
wherein R4 15 as hereinbefore defined and R7 is hydro-
gen or a hydrocarbyl moiety. The nitro moiety on the
nitrohydrocarbyl amino-substituted arylcyclobutene is
reduced to an amine moiety by contacting with an excess
of metallic zinc in a concentrated hydrochloric acid
solution at between 20C and reflux. The product
corresponds to the formula
NH2-R4-CH2NR7
wherein R4 is as hereinbefore defined. The aminohy-
drocarbyl amino-substituted arylcyclobutene is there-
after reacted with an unsaturated cyclic anhydride to
prepare a hydrocarbylene amino-bridged N-arylcyclo-
28,913C-F -39-
~30~
,
-40-
butenyl amido alkenoic acid. The conditions for this
reaction are as described hereinbefore for the reaction
of an amino-substituted arylcyclobutene and a cyclic
anhydride. This reaction i~ exemplified by the
following equation
~ "0 + NH2-R4-CH2NR'
~ ~NH-R4-;H2NR7
R3 C-OH
"
o
The hydrocarbylene amino-bridged N-aryl
cyclobutenyl amido alkenoic acid is thereafter dehy-
drated by one of the methods described hereinbefore to
prepare the hydrocarbylene amino~bridged N-substi-
tuted arylcyclobutenyl cyclic imide. This product
corresponds to the formula
28,913C-F -40
6~5
"
-41-
~ N-~4-CH2N~7 ~ O ~
wherein R4 and R7 are as hereinbefore defined.
A hydrocarbylene-bridged N-substituted aryl-
cyclobutenyl cyclic imide can be prepared by the
following procedure. A carboxy-substituted or
carboxyhydrocarbyl-substituted arylcyclobutene is
reduced to a hydroxyhydrocarbyl-substituted
arylcyclobutene by reacting the starting material with
about a 3:1 molar excess of diborane in an ether or
cyclic ether solvent at between 0C to 20C. This
process is exemplified by the following equation
15 0
HOCR8 ~ ¦ + B2H6 ~ HOCH2R8 ~ 1
wherein R8 is a direct bond or a hydrocarbylene moi-
ety. The hydroxyhydrocarbyl-substituted arylcyclobu-
tene is reacted with a slight excess of thionyl chlo-
ride to prepare a chlorohydrocarbyl~substituted aryl-
28,913C-F -41-
6:~$
-42-
cyclobutene. The reactants are usually contacted neat
or in a methylene chloride solvent at 2 temperature of
between 0C and 50C. An example of the product
corresponds to the formula
Cl-CH2-R8
~1 .
The chlorohydrocarbyl-substituted arylcyclobutene is
thereafter reacted with about an equimolar amount of
potassium phthalamide to prepare an N-arylcyclobutenyl-
hydrocarbyl phthalamide. The reactants are generally
contacted neat at temperatures of between 100C and
200C. This reaction is exemplified by the following
equation
ClCH2-R8~ ~= ~
________~ ~ ~ CH2-N~ ~ ~ KCl
0
28,913C-F -42-
~oo~s
-43-
wherein R8 is as hereinbefore defined. The N-aryl-
cyclobutenylhydrocarbyl phthalamide is reacted with
about one equivalent of hydrazine hydrate to prepare an
aminohydrocarbyl-substituted benzocyclobutene. The
reactants are contacted in an alkanol solvent at the
reflux of the solvent. The product corresponds to .the
formula
NH2-CH2-~1
wherein R8 is as hereinbefore defined. The aminohy-
drocarbyl-substituted benzocyclobutene is thereafter
reacted with an unsaturated cyclic anhydride to prepare
an N-hydrocarbylarylcyclobutenyl amido alkenoic acid
under the conditions described hereinbefore. This
process is exemplified b~ the following equation
~ O + N112CH2R8~ ~ NHR~
wherein R3 and R8 are as hereinbefore defined. The N-
hydrocarbylarylcyclobutenyl amido alkenoic acid is then
dehydrated to form a cyclic imide ring thus preparing
an N-hydrocarbylarylcyclobutenyl cyclic imide. This
process i~ performed using one of the two dehydration
processes described hereinbefore.
28,913C-F -43-
3 0 ~ ~ ~ 5
-44-
To prepare a mercaptoarylcyclobutene, an
arylcyclobutene sulfonic acid and equimolar.amounts of
sodium hydroxide are contacted in aqueous solution at
20C-25C to prepare sodium arylcyclobutene sulfonate.
The sodium arylcyclobutene sulfonate is dried at 100C,
and thereafter contacted in neat Porm with 0.48 ~ole of
phosphorous pentachloride at 170C to 180C to prepare
an arylcyclobutene sulfonyl chloride. The arylcyclo-
butene sulfonyl chloride is reduced with zinc, 4.9
moles, in the presence of 6.8 moles of concentrated
sulfuric acid at 0C to prepare the
mercaptoarylcyclobutene.
To prepare the alkylenethio-bridged N-sub-
stituted arylcyclobutenyl-unsaturated cyclic imide,
equimolar amounts of a mercapto arylcyclobutene, sodium
hydroxide and a dihaloalkane are contacted in an alka-
nol solvent at between 0C and 50C. The product is a
haloalkyl-substituted arylcyclobutenyl sulfide. This
reaction is exemplified by the following equation
HS - ~ + NaOH + R4X2 ~X-R4-S - ~ + HX
wherein X is halogen and R4 is a divalent alkane radi-
cal. Two moles of the haloalkyl-substituted arylcyclo-
butenyl sulfide is contacted with 0.8 moles of
potassium phthalimide and 0.4 mole of potassium
carbonate. The reactants are contacted neat at a tem-
perature of 190C to prepare an n-phthalimidoalkyl
28,913C-F -44-
~ 3 0 ~
-45-
arylcyclobutenyl sulfide. This process is exem-
plified in one preferred embodimen~ by the following
equation
X-R4-S - ~ + ~ N-K + K2C3 `~
o
O
N-R4-S ~ ~ ~ K2C03 + KX
O
The phthalimidoalkyl arylcyclobutenyl sulfide
i9 contacted with a hydrazine hydrate in a mole ratio
of 1 to 1.25, respectively, in an alkanol solvent at
re~lux to prepare an aminoalkyl arylcyclobutenyl
sulfide. In one preferred embodiment, the product
corresponds to the formula
H2N-R4-S~ +
.
28,913C-F -45-
-46-
The aminoalkylarylcyclobutenyl sulfide is then
reacted with an unsaturated cyclic anhydride to
prepare a thioalkylene-bridged N-aryl cyclobutenyl
minoalkenoic acid. This is achieved under conditions
described hereinbefore. Thereafter, the alkylenethio
N-arylcyclobutenyl amidoalkenoic acid can be dehydrated
by one of the methods described hereinbefore to prepare
an alkylenethiobridged N-substituted arylcyclobutenyl
cyclic imide.
To prepare arylenethio-bridged N-arylcyclo-
butenyl cyclic imide, equimolar amounts of a mercapto
arylcyclobutene, sodium hydroxide and a halonitro-sub-
stituted aromatic compound are contacted in an alkanol
solvent under reflux to prepare a nitroaryl arylcyclo-
butenyl sulfide. The nitro group on the nitroarylarylcyclobutenyl sulfide is reduced by contacting one
mole of such compound with about two moles of tin and
about six moles of concentrated hydrochloric acid to
prepare an aminoaryl arylcyclobutenyl sulfide.
Theaminoaryl arylcyclobutenyl sulfide is thereafter
contacted with a an unsaturated cyclic anhydride in
equimolar amounts in methylene chloride at a
temperature of 0C to 25C to prepare an arylenethio-
bridged N-arylcyclobutenyl amidoalkenoic acid. The
arylenethio-bridged N-arylcyclobutenyl amidoalkenoic
acid is dehydrated using procedures described
hereinbefore to prepare an arylenethio-bridged N-
arylcyclobutenyl cyclic imide.
The hydrocarbylenethio-bridged N-arylcy-
clobutenyl cyclic imides can be contacted with equi-
molar amounts of peracetic acid in an ethyl acetate
solvent at between 0C to 20C to prepare a
hydrocarbylenesulfinyl-bridged N-arylcyclobutenyl
28,913C-F -46-
3 ~ S
-47-
cyclic imide. The hydrocarbylenethio-bridged N-aryl-
cyclobutenyl cyclic imide can be contacted with 2 moles
of peracetic acid for each mole of the bridged cyclic
imide in ethyl acetate solvent at 0C to 20C to prepare
a hydrocarbylenesulfonyl-bridged N-arylcyclobutenyl
cyclic imide.
To prepare the various bridged N-arylcyclo-
butenyl cyclic imides wherein the aryl moiety is a
heterocycle, the appropriately substituted hetero-
cyclic N-arylcyclobutenyl cyclic imide is reacted in
the manner described herein to get the appropriately
desired compound.
The compounds of this invention are unique in
several respects. They have intramolecular diene and
dienophile functionality. They are thermally stable
for long periods at elevated temperatures, up to 100C.
They are readily polymerizable. The compounds of this
invention are useful in the preparation of polyimides
by polymerization of one or more of the compounds of
this invention. It is believed that the polymerization
takes place by a Diel~-Alder reaction wherein the
unsaturation on the cyclic imide acts as a dienophile
while the cyclobutene ring forms a diene which reacts
wlth the dienophile to form the polymeric compositions.
The polymers of this invention are prepared by
heating the compounds described hereinbefore to a tem-
perature o~ 170C or greater. Preferable temperatures
for polymerization are 200C or greater. In general 7 it
is preferable to run the polymerization at a
temperature of between 170C and 300C, with between
200C and 300C being most preferred.
28,913C-F -47-
:~30~i35
-48-
Wherein the N-substituted arylcyclobutenyl
cyclic imides correspond to the formula
R3
\ (R ~
, ¦ N-Y-A~,C(R2)2)2
R3 ~
wherein Ar, p~1, R2, R3, Y and a are as described here-
inbefore; it is believed that the polymeric composition
contains units which correspond to the formula
(R2)2
(R1)a ¦ R3
\ / ~ C \
Ar~ N-Y-
\C /~C'/
~
(R2)2
It is further believed that in one preferred
embodiment the polymers derived from monomers of such a
28,913C-F -48-
3 ~ ~ 6~ 5
-49-
formula result in the preparation of polymers which
correspond generally to the ~ormula
(R2)2
_y ~ N-Y - - Ar ~ C(~2)2~2
R3 n ¦ 0 c
O \ (R2)2
wherein Ar, R1, R2, R3, Y and a are as described here-
inbefore and c is a real number of 2 or greater, and
most preferably 20 or greater.
In another preferred embodiment, the poly~
meric composition is the polymer of one or more com-
pounds which corresponds to the formula
Q
ll (R1)b
R ¦
O
wherein R1, R2, R3, Y and b are as hereinbefore
de~ined. In this embodiment, lt is believed that the
28,913C-F -49-
.
r 1300~;~35
-50-
polymer prepared contains units which correspond to the
formula
\-Y
(R )2
wherein Rl, R2, R3 J Y and b are as hereinbefore
defined.
In one preferred embodiment wherein the com-
pound or compounds polymerized corresponds to said
formula, it is believed that the polymer prepared
corresponds to the formula
~ O
R3 R ,, d (R2)2
o
: wherein Rl, R2, R3, Y and b are as hereinbefore
defined, and d is a real number of 2 or greater. d is
preferably 20 or greater.
28,913C-F -50-
~C16~S
The novel N-substituted arylcyclobutenyl-
unsaturated cyclic imide compounds of this invention
are useful in the preparation of polymeric
compositions. In general, these polymeric compositions
are prepared by contacting these N-substituted
arylcyclobutenyl-unsaturated cyclic imide compounds and
heating them to the polymerization temperature of the
particular monomer used. The polymerization is an
addition polymerization wherein no volatiles are
generated. Furthermore, no cataIyst initiator or
curing agents are necessary for the polymerization to
take place. It is believed that the polymerization
takes place when the cyclobutene ring undergoes
transformation to prepare an aryl radical with two
olefinic unsaturated moieties ortho to one another
wherein the olefinic unsaturated moieties thereafter
undergo reaction with the unsaturated cyclic imide
moieties. It is to be noted that the temperature at
which polymerization is initiated is dependent upon the
nature of sub~tituents on the cyclobutene ring. In
general, wherein the cyclobutene ring is unsubstituted,
the polymerization is initiated at 200C. Wherein the
cyclobutene ring is substituted with an electron-
donating substituent, the polymerization temperature is
generally lowered, the higher the ability of the
substituent to donate electrons, the lower the
polymerization initiation temperature is.
The method of polymerization of the N-sub-
stituted arylcyclobutenyl unsaturated cyclic imide
monomers has a significant effect on the nature and
properties of the polymeric composition prepared. In
one embodiment, the N-substituted arylcyclobutenyl-
unsaturated cyclic imide monomers of this invention can
28,913C-F -51-
o~s
-52-
be melt polymerized. The melt polymerization of N-
substituted arylcyclobutenyl-unsaturated cyclic imide
monomers allows their use in the preparation of solid
parts, as coatings, in composites, as adhesives and as
fibers.
In one embodiment of the melt polymerization,
the monomers are heated to the temperature at which it
melts, preferably this is a temperature of between 80C
and 100C, and thereafter poured or injected into a
mold. Thereafter, pressure may be applied on the
melted monomer in the mold. &enerally, pressures of
between atmospheric and 2000 psi (13,789 kPa) are
suitable. Thereafter, the monomer is heated to a
temperature at which the monomers undergo
polymerization. This is preferably a temperature of
between 200C and 300C, more preferably between 200C
and 250C for between 10 minutes and 3 hours. Upon
cooling, the polymerized composition can be removed
~rom the mold.
Polymers prepared in this manner can subse-
quently be thermally treated at temperatures above 200C
to raise the modulus and lower the coefficient of
expansion of such polymeric compositions.
In general, the polymers prepared by this
method are insoluble in that they swell but do not dis-
solve, are thermally stable at 200C, have a good modu-
lus, a low water pickup and are reasonably hard.
In another embodiment, the N-substituted
arylcyclobutenyl-unsaturated cyclic imide monomers of
this invention can be used to prepare coatings and
films. In such embodiments, the monomers are dissolved
28,913C-F -52-
0t)~i35
in a suitable solvent and coated onto the substrate of
- choice, and thereafter the coated substrate is exposed
to temperatures at which the monomers undergo polymeri-
zation over a period of time sufficient for the
polymerization to go to completion. Under preferable
conditions, temperatures of above 200C for between 1
and 5 hours are used. Suitable solvents are those
which volatilize away at temperatures below the
polymerization temperature. Preferred solvents are
cyclic and aliphatic ethers, lower alkanols, amides,
and chlorinated hydrocarbon solvents. It is preferable
to saturate the solvent with the monomer, a 20 to 30
weight percenk concentration of monomer in the solvent
is preferred.
The N-substituted arylcyclobutenyl-unsatu-
rated cyclic imide monomers may be combined with the
powder-form or fibrous fillers or reinforcing materials
either before or after heat treatment. For example, it
is possible to impregnate powder-form or fibrous
fillers or reinforcing materials such as quartz sand or
glass cloths, with the N-substituted arylcyclobutenyl-
unsaturated cyclic imide monomers, optionally in
~olution.
Suitable fillers and reinforcing materials are,
generally, in any powder form and/or fibrous prod-
ucts, for example, of the type commonly used in the
production of moldings based on unsaturated polyester
resins or epoxide resins. Examples o~ products such as
these are, primarily, granular fillers such as quart~
powder,ground shale, asbestos powder, powdered
corundum, chalk, iron powder, aluminum powder, sand,
gravel and other fillers of this kind, also inorganic
or organic fibers, more especially glass fibers in the
28,913C-F -53-
~ ~0~;35
, --
-54-
usual textile forms of fibers, filaments ro~Jings,
yarns, nonwovens, mats and cloths, etc. In this
connection, amino silane-based finishes have proven to
be particularly effective. It is also possible to use
corresponding textile structures of organic, preferably
synthetic fibers (polyamides, polyesters) or on the
basis of quartz, carbon, metals, etc., as well as
monocrystals (whiskers).
The end products combined with fillers or
reinforcing materials may be used in particular in ves-
sel and pipe construction by the winding technique, in
electrical engineering, in mold construction and tool
making and also in the construction of heavily stressed
components, in the lightweight construction of vehicles
in aeronautical and astronautical engineering.
In another embodiment, the N-substituted aryl-
cyclobutenyl-unsaturated cyclic imide monomers can be
used as adhesives. In such embodiment, one of the sub-
strates to be joined is contacted with some form of themonomers, for example, the monomer in a powdered form.
Thereafter, the second substrate to be adhesivated is
contacted with the substrate previously contacted with
the monomer where the monomer was contacted with the
first substrate. Thereafter, pressure of at least 1
psi is applied and the monomers and substrates are
raised to a temperature at which the monomer undergoes
polymerization.
In one embodiment, the N-substituted aryl-
cyclobutenyl-unsaturated cyclic imide monomers can be
formed into a prepolymer which thereafter can be
polymerized. To form the prepolymer, the N-substituted
arylcyclobutenyl-unsaturated cyclic imide monomers are
28,913C-F -54-
~L~0~63S
--55--
contacted in an inert atmosphere or under vacuum and
heated to a stage at which the polymerization mixture
is sufficiently viscous enough to be moldable in
conventional molding equipment. In general, the
monomers can be contacted at a temperature of 190C to
220C for between about 1 and 10 minutes. Thereafter,
the prepolymer can be used in various techniques to
prepare the polymeric compositions of this invention.
In one preferred embodiment, the prepolymer is cooled
to form a powder which can be used to form compression
molded articles, as an adhesive, and in many other
uses. In another embodiment, a prepolymer of the N-
substituted arylcyclobutenyl-unsaturated cyclic imide
monomers can be prepared by precipitation
polymerization. In particular, the technique involves
heating such ~onomers in a solvent to prepare a low
molecular weight prepolymer that contains unreacted
arylcyclobutene rings. A solvent is used which
dissolves the monomer but not the prepolymer. As the
prepolymer forms, it precipitates and is removed. The
prepolymer can be fabricated in a hot compression mold
which reacts out the remaining arylcyclobutene rings to
give a thermoset polymer. The product is a fine white
powder.
Preferable solvents are nonpolar solvents, such
as aromatic hydrocarbons, aliphatic hydrocarbons,
28,913C-F -55-
63S
aliphatic chlorinated hydrocarbons, aromatic chlori-
nated hydrocarbon solvents, biphenols, naphthalenes or
polychlorinated biphenols. The polymerization can take
place at temperatures generally of between 200C and
240C for period~ of between 1 and 5 hours. In general,
the monomer can be dissolved up to saturation in the
solvent used. A 20 to 30 percent by weight solution of
the monomer in the solvent is preferred.
In another embodiment, the N-substituted
arylcyclobutenyl-unsaturated cyclic imide monomers can
be polymerized by solution polymerization techniques.
In this embodiment, the monomers are dissolved in
dipolar aprotic solvents with boiling points above the
polymerization temperature of the monomers. It is
preferable that the solvents have a boiling point of
above 200C and more preferable that the solvents have a
boiling point of above 250C. Examples of preferred
dipolar aprotic solvents include amides and sulfones.
It is necessary to add to the solution lithium salts
which solubilize the monomer in the solvents,
preferably between 5 and 20 weight percent based on the
monomer. A preferred lithium salt is lithium chloride.
The polymerization takes place by heating the
polymerization solution to a temperature at which the
monomer undergoes polymerization, preferably above
200C. The polymerization time is generally between
about 1 and 10 hours. The polymer can be recovered by
3 adding water to precipitate the polymer from the
reaction solution and thereafter stripping off the sol-
vent. The polymers prepared with this method can be
used in compression moldings or to prepare coatings.
It is often desirable to process these polymers under
elevated temperatures.
28,913C-F -56-
?0~3S
--57--
In another embodiment, t-he monomers of this
invention which undergo polymerization at a temperature
which is below the melting point of the monomer can be
polymerized in a solid state polymerization. In this
method, the monomers are heated to a temperature at
which polymerization takes place. Polymers prepared in
this method can be useful in the preparation of
bearings, seals and other parts by powder metallurgy
techniques.
The following examples are included to illus-
trate the invention, and do not limit the scope of the
invention or the claims. Unless otherwise specified,
all parts and percentages are by weight.
Example 1
(a) Preparation of ~thyl
2-(o-Chlorobenz~l) Cyanoacetate
Into a 3-liter, three-necked flask equipped
with a mechanical stirrer, reflux condenser, addition
funnel and nitrogen inlet was placed a solution of
35.64 g (1.55 moles) of sodium metal in 1050 mm of
absolute 2B ethanol. The solution was stirred under
nitrogen and cooled to 0C in an ice bath and 763.56 g
(6.75 moles) of ethyl cyanoacetate was added dropwise
over a period of 15 minutes. To this white suspension
was added 241.56 g (1.5 moles) of o-chlorobenzyl
chloride dropwise over 1 hour. After the addition was
complete, the ice bath was removed and the mixture was
slowly heated under nitrogen to reflux and held there
~or 3 hours. The resulting pink-colored mixture was
allowed to cool under nitrogen overnight at room
temperature. About 1 liter of ethanol was distilled
from the reaction mixture and 1.5 liters of water were
28,913C-F -57-
0~;35
-58--
added. The organic layer was taken up in three 400-ml
portions of methylene chloride, and the solutions were
combined and washed once with 150 ml of water. The
methylene chloride solution was dried over anhydrous
5 magnesium sulfate, ~iltered and evaporated on a rotary
evaporator~ The residual liquid was distilled under
reduced pressure through an insulated 12-inch (30.5 cm)
Vigreux column. A forerun of ethyl cyanoacetate
(boiling point 55C-60C/-0.3 mm Hg) comes over first
10 followed by pure ethyl 2-(o-chlorobenzyl) cyanoacetate.
The infrared, 'H and 13C nuclear magnetic resonance
were used to establish the structure. The yield was 68
percent of product having a boiling point of 130C-
135C/0.3 mm Hg.
(b) Preparation of 2-(o-Chloro-
benz~,rl)C~anoacetic Acid
In a 2-liter, three-necked flask equipped with
20 a mechanical stirrer, addition funnel and nitrogen
inlet was placed 243 g (1.02 moles) of ethyl 2-
(o-chlorobenzyl)cyanoacetate. A solution of 54.52 g
(1.363 moles) of qodium hydroxide pellets and 545 ml of
water was added over a period of 15 minutes while
25 stirring under nitrogen. Initially, the solution
turned cloudy and then became clear. The resulting
mixture was stirred for 5 hours at room temperature
under nitrogen. Water (445 ml) was added and the
mixture was cooled in an ice bath. Acidifying to pH 1
3 with 4 N hydrochloric acid gave a fine white
precipitate that was filtered and washed with water
until neutral to litmus. The product was dried in a
vacuum oven at 60C overnight to yield 20 g (97 percent)
35 of white powder. This material was recrystallized from
toluene to give pure white crystals of 2-(o-chloro-
28,913C-F -58-
o~s
-59-
benzyl)cyanoacetic acid identified by infrared, 'H and
13C nuclear magnetic resonance. The yield was 94
percent of product having a melting point of 132C-
134C
(c) Preparation of 3-(0-
chlorophenYl) propionitrile
Into a 1-liter, three-necked flask equipped
with a mechanical stirrer, reflux condenser and
nitrogen inlet was placed 138.5 g (0.66 mole) of 2-(o-
chlorobenzyl)cyanoacetic acid and Z20 ml of dry N,N-
dimethylformamide. The mixture was stirred and slowly
heated under nitrogen to reflux and held there for 6
15 hours. The resulting yellow mixture was allowed to
cool under nitrogen overnight at room temperature. A
precipitate (approximately 0.5 g) that formed was
filtered off and the filtrate was poured into 1 liter
of water. The organic layer was taken up in three 330-
ml portions of ethyl ether/hexane (1:1 ~/v), and thesolutions were combined and washed once with 150 ml of
water. The ethyl ether/hexane solution was dried over
anhydrous magnesium sulfate, filtared and evaporated on
a rotary evaporator. The residual liquid was distilled
under reduced pressure through an insulated 12-inch
(30.5 cm) Vigreux column with the product being
collected at 82C-85C/0.3 mm Hg as a colorless liquid
identified by infrared, 'H and 13C nuclear magnetic
resonance. Th~ yield was 94.7 percent.
(d) Preparation of 1-C~anobenzoc~obutene
A 3-liter, three-necked flask equipped with a
dry ice condenser, mechanical stirrer and Claisen
adapter fitted with an ammonia gas inlet and nitrogen
28,913C-F -59-
~L~30~35
-60-
inlet was rinsed with acetone, dried in an oven at
125C, and heated with an air gun while flushing with
nitrogen. The apparatus was cooled in a dry ice-
acetone bath and the condenser was filled with a dry
ice-acetone mixture. Ammonia gas flow was initiated
and 600 ml was condensed out. The ammonia inlet tube
was replaced by a stopper, and 0.4 g of powdered iron
(III) nitrate was added. Sodium metal, 51.52 g (2.24
moles) was added in small portions over 1 hour. After
all the sodium was added, the dry ice bath was removed
and cooling was left to the dry ice condenser.
Complete conversion of the sodium/ammonia solution to
sodamide was indicated by a color change from deep blue
to gray. Next, 92.82 g (0.56 mole) of 3-(o-chloro-
phenyl)propionitrile was added over a period of 10
minutes. The last traces of the nitrile were washed
into the flask with small amounts of anhydrous ethyl
ether. The dark green reaction mixture was stirred
vigorously for 3 hours and then was treated with
134.4 g (1.68 moles) of solid ammonium nitrate. The
ammonia was allowed to evaporate overnight at room
temperature. Water (420 ml) was cautiously added to
the residue. The organic layer was taken up in two
224-ml portions of chloroform, and the solutions were
combined and washed twice with 140 ml of aqueous 5
percent hydrochloric acid and once with 140 ml of
water. The chloro~orm solution was dried over
anhydrous magnesium sulfate, filtered, and evaporated
on a rotary evaporator. The residual liquid was
distilled under reduced pressure through an insulated
12-inch (30.5 cm) Vigreux column. The product was
collected at 59C-~9C/0.2 mm Hg. The infrared, 'H and
28,913C-F -~0-
-6 1- ~3~0635
13C nuclear magnetic resonance were run to identify the
product. The yield was 50 percent.
(e) Preparation of 5-nitro-
-1-c~anobenzoc~clobutene
Into a 500-ml, three-necked flask equipped with
an addition funnel, thermometer and nitrogen inlet was
placed 14.1 g (0.17 mole) of sodium nitrate and 135 ml
of concentrated sulfuric acid. The mixture was stirred
under nitrogen while cooling to -5C (calcium
chloride/ice) and 19.5 g (0.16 mole) of 1-cyano-
benzocyclobutene was added dropwise at such a rate as
to keep the reaction temperature below 2C. The
reaction mixture was then stirred under nitrogen at 0C-
5C for 0.5 hour, poured onto 1050 g of ice, andextracted with four 300-ml portions of methylene
chloride. The methylene chloride solutions were
combined, washed with four 150-ml portions of 10
percent sodium bicarbonate, once with 300 ml of water,
and dried over anhydrous magnesium sulfate. The
methylene chloride solution was filtered and evaporated
on a rotary evaporator to give 26.9 g of residue which
was recrystallized from absolute 2B ethanol to give
pure 5-nitro-1-cyanobenzocyclobutene identified by
infrared~ 'H and 13C nuclear magnetic resonance. The
melting point is 110C-112C and the yield was 64.1
percent.
(f) Preparation of 5-Amino-
-1`Cyanobenzoc~clobutene
Into a 1-liter, three-necked flask equipped
with a gas dipersion tube, reflux condenser~ rubber
septum and nitrogen inlet was placed 7 g (0.04 mole) of
5-nitro-1-cyanobenzocyclobutene and 400 ml of absolute
2B ethanol. The mixture was stirred under nitrogen and
28,913C-F -61-
0 6
-62-
heat was applied to dissolve the solid. After adding
2.4 ml of glacial acetic acid and 1.6 g of 5 percent
palladium on carbon, hydrogen flow was initiated and
the mixture was hydrogenated at atmospheric pressure
and ambient temperature. The hydrogenation was
followed by thin-layer chromatography (silica gel; 70
percent toluene, 25 percent ethyl acetate, 5 percent
triethylamine as eluent) and this showed the reaction
was essentially complete in 1 hour. After 3 hours, the
hydrogen flow was stopped and the system was purged
with nitrogen for l5 minutes to remove excess hydrogen
gas. The catalyst was removed by filtration using
Celite and quickly quenched in water. The filtrate was
evaporated to dryness on a rotary evaporator and the
residue was treated with aqueous 10 percent sodium
hydroxide. The aqueous solution was extracted with
three lO0-ml portions of ethyl ether, and the solutions
were combined and washed once with lO0 ml of water.
The ethyl ether solution was dried over anhydrous
potasqium carbonate, filtered and evaporated on a
rotary evaporator to give an amber-colored oil that
solidified on standing. The product was pumped under
vacuum overnight to remove the last traces of ethyl
ether and stored under nitrogen. The infrared, 'H and
13C nuclear magnetic resonance were run. The yield was
86.4 percent.
(g) Preparation of N-[5-(1-Cyano-
benzocyclobutenyl)]maleamic Acid
Into a 250-ml, three-necked flask equipped with
a mechanical stirrer, addition funnel, reflux con-
denser, thermometer and nitrogen inlet was placed 4.9 g
(0.05 mole) of freshly sublimed maleic anhydride and 50
ml of dry chloroform. The mixkure was stirred under
28,913C-F -62-
63 ~00635
nitrogen while cooling to 15C in an ice bath and a
solution of 7 g (0.05 mole) of 5-amino-1-cyanobenzocy-
clobutene in 50 ml of dry chloroform was added dropwise
at such a ~ate as to keep the reaction mixture below
20C. The reaction was maintained below 20C and
stirred under nitrogen for.1 hour after addition was
complete. The solid N-[5-~1-cyanobenzocyclo-
butenyl)]maleamic acid was filtered off, washed with
cold chloroform, then with hot ethyl acetate/2B ethanol
(absolute; 1:1 v/v), and dried overnight in a vacuum
oven at 60C~ The infrared, 'H and 13C nuclear magnetic
resonance, and carbon, hydrogen, nitrogen analyses were
run.
~nal~sis Calculated Found
carbon 64.46 63.80
hydrogen 4.16 4.L~4
nitro~en 11.57 11.36
The yield was 11.32 g equal to 94.25 percent and the
melting point is 190C-192C.
(h) Preparation oE N-[5~ Cyano-
benzocyclobuten~l)]maleimide
Into a 250-ml, three-necked flask equipped with
a mechanical stirrer, reflux condenser, thermometer and
nitrogen inlet was placed 11 g (0.045 mole) of N-[5- (1-
cyanobenzocyclobutenyl)]maleamic acid, 2.4 g (0.03 mole)
of anhydrous sodium acetate, and 45.94 g (0.765 mole)
of fresh glacial acetic acid. The mixture was stirred
and slowly heated under nitrogen until a clear yellow
solution results (117C-118C). ~fter 5 minutes
28,913C-F -63-
64 ~ ~ ~
the heat was removed and the reaction mixture was
allowed to cool under nitrogen overnight at room
temperature. It was then slowly poured into a
vigorously stirred slurry of ice and water (120 g
total), and the resulting yellow precipitate filtered,
washed with water until neutral to litmus, and
transferred to a 500-ml beaker containing 150 ml of
aqueous saturated sodium bicarbonate. This mixture was
stirred for 10 minutes, then 150 ml of chloroform was
added and stirred for an additional 10 minutes. The
organic layer was taken up in three 50-ml portions of
chloroform, and the solutions were combined and washed
once with 150 ml of water. The chloroform solution was
dried over anhydrous magnesium sulfate, filtered and
evaporated on a rotary evaporator to give a viscous
yellow oil. The product was pumped under vacuum over-
night to give a yellow solid that was purified by col-
umn chromatography on silica gel using 70 percent tol-
uene/30 percent ethyl acetate as the eluent. Theinfrared, 'H and 13C nuclear magnetic resonance, and
carbon, hydrogen, nitrogen analyses were run.
AnalYsisCalculated Found
carbon 69.60 69.30
hydrogen3.60 3.70
nitrogen12.50 12.34
The yield was 5.7 g equal to 56.5 percent. The melting
point is 55C-60C.
28,913C-F -64-
1~0(~63S
--65--
Example 2
In a one-liter, one-necked, flask dispersed
with nitrogen was placed 10 g (0.413 mole) of maleamic
acid, 8.57 g (.0840 mole) of acetic anhydride, 0.2314 g
(0.0013 mole) of nickel (II) acetate, 429 ml of acetone
and 8.6 ml (6.24 g) of triethylamine. This solution
was stirred under nitrogen ~or 64 hours. Stirring was
stopped and the solution was poured into 300 ml of
water saturated with sodium carbonate. Chloroform (150
ml) was added to extract the organic layer. The sodium
carbonate came out of solution, collecting in the
bottom of the separatory funnel. Extra water was added
to the contents in the funnel to redissolve the sodium
carbonate. The organic layer was then extracted and
two 150-ml chloroform extractions were performed. The
three chloroform extractions were combined and washed
with 300 ml of water, dried over magnesium sulfate,
filtered and rotovaped. ~ yellowish-brown oil was
obtained and dried under vacuum for 16 hours to remove
remaining chloroform. Using a column packed with
silica gel and a solvent of 70 percent toluene, 30
percent ethylacetate, the product was chromatographed.
The combined ~amples containing the product (found by
TLC) were rotovaped and pumped under vacuum for 16
hours. Yield was 8.97 g oY product.
Example 3 - Preparation of Poly-N-[5-(1-cyano-
benzocyclobutenyl)]maleimide
Into a 25-ml, two-necked flask equipped with a
reflux condenser, nitrogen inlet and magnetic stir bar
was placed 0~5 g (2.2 mmole) of N-[5-(1-cyanobenzocyclo-
butenyl)]maleimide and 15 ml of mesitylene. The mi~ture
was purged with nitrogen and heated with stirring.
Initially, all of the maleimide derivative dissolved to
28,913C-F -65-
-66- ~ 635
give a clear yellow solution. Upon reaching reflux,
the soluti~n became cloudy and a beige powder
precipitated. After 2 hours o~ reflux, the reaction
was cooled and the precipitated polymer was filtered
off and washed free of residual mesitylene with
chloroform and dried. The yield was quantitative.
Example 4
Into a 25-ml, one-necked, round-bottomed flask
equipped with a nitrogen inlet was placed 0.1 g (0.446
mmole) of N-[5~ cyanobenzocyclobutenyl)]maleimide.
The flask was purged with nitrogen and immersed in an
oil bath. The bath temperature was raised to 200C over
1 hour. After heating at 200C for 20 minutes, the
melted monomer solidified to a pale yellow transparent
solid. The flask was cooled and the polymer removed
by breaking it up with a spatula. The yield was
quantitative.
- Preparation of 4-Nitrophenyl
-4-Benzocyclobutenyl Ketone
Into a lO0 ml roundbottom one neck flask
equipped with a magnetic stirring bar, a reflux
condensor and a nitrogen inlet was placed
benzocyclobutene lOg(96.15 mmol) and 4-nitrobenzoyl
chloride ll.9g(64.2mmol). To the flask was added Fe203
(0.65mmol, lmol %). The flask was heated to l50C under
3 a nitrogen atmosphere with vigorous stirring overnight.
The mixture was cooled to room temperature, mixed with
chloroform (120 ml) and transferred to a separatory
funnel. The dark brown solution was washed with 10
percent aqueous sodium bicarbonate (50 ml) twice, water
(50 ml) and brine (50 ml) each once. The solution was
28,913C-F -66-
-67- ~3(:1~)G3S
dried over magnesium sulfate overnight and filtered
through celite. The volatiles were removed on a
rotovap to give a deep red-black viscous liquid. The
liquid was contacted with 100 ml of n-hexane and heated
to boiling. The hot n-hexane was decanted away from
the undissolved brown liquid. The hexane treatment was-
repeated three more times. The resultant yellow n-
hexane layers were combined and slowly cooled to room
temperature. An off-white solid (rosettes)
precipitated out of solution. The solid was isolated
by suction filtration. The filtrate was concentrated
to one half of its volume and allowed to stand. The
solid recovered had a weight of 4.8 grams (a 30 percent
yield). NMR and IR spectra were taken of the solid and
the spectra agreed with the structure of 4-nitrophenyl-
4-benzocyclobutenyl ketone.
Example 6 - Preparation of 4-Aminophenyl-4-Benzocyclo-
buten~l Ketone
Into a 250 ml three neck round bottom flask
equippe~ with a magnetic stirring bar, a reflux
condensor with a nitrogen inlet and a thermometer and
an equilibrating addition funnel was charged loO g
t3.95 mmol) of 4-nitrophenyl-4-benzocyclobutenyl
ketone, 4.46 g (19.75 mmol) of (SnCl2-2H20) and 100 ml
of ethanol. Under a nitrogen atmosphere, the mixture
was heated to 60C. To the mixture, in a slow dropwise
manner was added sodium borohydride, 75 mg (1.975 mmol)
in 20 ml of ethanol, over a period o~ 20 minutes.
After the addition, the temperature of the mixture was
maintained at 60C for 30 minutes. The mixture was
cooled to 10C and 80ml of water previously chilled was
added. Concentrated sodium hydroxide was added until
the pH was 7 (4 M, NaOH, 6 ml). The mixture was
28,913C-F -67-
1~0063S
-68-
trans~erred to a 500 ml round-bottom flask and the
ethanol was removed on a rotovap. Thereafter, 100 ml
of water was added to the off-while slurry and the
aqueous phase was extracted with diethyl ether four
times with 100 ml aliquots. The ether extracts were
dried over sodium sulfate overnight. The sodium
sulfa~e was filtered from the diethyl ether extracts
and the diethyl ether was removed on a rotovap. A
bright orange solid (0.87g) was obtained. The solid
was recrystallized using carbon tetrachloride (200 ml)
and decolorizing charcoal. The first crop of crystals
was a pale yellow solid 0.54 g (61.3%). The carbon
tetrachloride was concentrated to 50 ml and left
standing overnight. NMR and IR indicate the product
was 4-aminophenyl-4-benzocyclobutenyl ketone.
Example 7 - The Preparation of
0 "
1~--C ~ / C c`~
o
Into a 100 ml round bottom flask equipped with
a magnetic stirring bar and a nitrogbn inlet was placed
l.Og(4.482 mmol) of 4-aminophenyl-4-benzocyclobutenyl
ketone and 20 ml of acetone. To the solution was added
4.40g(4.482 mmol) of maleic anhydride in several
portions over two minutes, with stirring. The solution
was stirred at room temperature ~or two days. A white
solid precipitated. The white solid was belleved to be
28,913C F -68-
69~ 63S
o o
~ ~
H02C
The white solid and the solution described above are
stirred with 4.9mg(8.955 mmol, 0.85ml) of acetic
anhydride, 35 mg(0.142 mmol) of h~drated sodium acetate
(four waters of hydration~ and 19 drops of triethyl
amine. As the triethyl amine was added, the solution
turned yellow. Stirring at room temperature was
continued overnight. The reaction mixture was an
orange hazy mixture (fine solid was present). The
reaction mixture was poured into 80 ml of vigorously
stirred aqueou~ sodium bicarbonate. An orange solid
precipitated from the solution. To the solution was
added 100 ml of chloroform, and the solution was
transferred to a separatory funnel. The layers were
separated and the aqueous phase was extracted with 100
ml of chloroform. The chloroform extracts were
combined and washed with 50 ml of water, and then 50 ml
of brine. The solution was dried over magnesium
sulfate and the suction filtered through celite. The
solvent was removed by a vacuum to give 1.20g of an
orange viscous syrup. The product was recrystalized
using ethanol and decolorizing charcoal. The mixture
3 was gravity filtered to give a pale yellow solution.
The solution was concentrated to one-half its volume
and placed in ice. A white solid precipitated. The
solution was allowed to stand overnight. The solid was
isolated and dried in air for 1 hour. The weight of
the product was 0.49g and has a melting point of 138-
139C. Another 0.39g of product was further isolated to
28,913C-F -69-
_70_ ~ S
give a total yield of 0.88g. The product was examined
by IR and NMR and shows agreement with the following
structure,
1 0 \~
o
The DSC of the product indicated a melting point of
147C and an exotherm was observed at 260.4C. The
energy of the exotherm was 347 J/g. A rescan of the
DSC showed no melting point or exotherm but did exhibit
a T2 at 260.4C.
Example 8 - Polymerization of 4-(N-maleimido)phenyl-
-4-Benzoc~clobuten~l Ketone
Into a tube was placed 146 mg of 4-(N-
maleimido)-phenyl-4-benzocyclobutenyl ketone and
ni~rogen. The tube was placed into a Wood's Metal bath
at 150C. The temperature was controlled and monitored
with a heater and a thermocouple. The following
sequence of times and temperatures were applied ~o the
tube:
28,913C-F -70-
~ -71- ~300~3S
150C 30 min.
180C 30 min.
210C 30 min.
235C 1 hour
260C 1 hour
270C 1 hour
At the end of the sequence, the heating was stopped and
the tube and thermocouple were removed from the heating
bath. The tube was allowed to cool overnight.
Thepolymer was an amber color with small voids trapped
in the matrix. The polymer was physically broken into
small pieces. A TGA was run on one of the pieces, 0.05
wt % loss occured at 327.78C and 5~ weight loss at
464.39C.
3o
28,913C-F -71-