Note: Descriptions are shown in the official language in which they were submitted.
~3~ RAN 4007/50
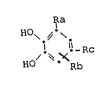
l~he catechol derivatives of the general formula
~ a
H0\ ~.
H0/ ~ X Rc la
Rb
lo
wherein Ra signifies nitro or cyano, Rb signifies
hydrogen or halogen, Rc signif i2S halogen, nitro,
cyano or the group ~(~)n (Q)m
~(~)n~Q~R , A signifies vinylene optionally
substituted by lower alkyl, n signifies the number 0
or 1, m signifies the number 0 or 1, Rl signifies
the group -CoR3, a carbocyclic, aromatic group or an
aro~atic or partially unsaturated, heterocyclic group
attached via a carbon atom, R~ signifies hydrogen or
an optionally substituted, saturated or partially
unsaturated, lower hydrocarbon residue, R3 signifies
hydroxy, amino, an optionally substituted, saturated
or partially unsaturated, lower hydrocarbon residue
attached via an oxygen atom or an imino or lower
alkylimino group or a saturated, N-containing hetero-
cyclic group attached via a ring nitrogen atom, Q
signifies the group -C0- or >C=N-(Z)p-R , Z
signifies an oxygen atom or an imino group, p
signifies the number 0 or 1 and X4 signifies30
hydrogen or a saturated or partially unsatura~ed,
lower hydrocarbon residue which is optionally
substituted and which is optionally attached via a
carbonyl group,
the ester and ether derivatives which are hydrolyza~le
3 under physiological conditions and the pharmaceutically
acceptable salts thereof possess valuable pharmacological
Mt/27,1.87 ~ ~
- ~ -
properties. In particular, these compounds inhibit the
enzyme ca~echol-O-methyltransferase (COMT), a soluble,
magnesium-dependent enzyme which catalyses the transfer-
ence of the methyl group of S-adenosylme~hionine to a
catechol substrate, whereby the corresponding methyl
ethers are formed. Suitable substrates which can be
~-methylated by COMT and which can thus be deactivated
are, for example, extraneuronal catecholamines and
exogenously-administered therapeutically active substances
having a catechol structure.
The compounds of formula Ia above can accordingly be
used in the prevention or control of illnesses in which a
deactivation of extraneuronal catecholamines by CO~T plays
a role, for example in the prevention or control of
depressions. In this case the compounds of formula Ia
above can be used as individual compounds or in combina-
tion with other therapeutically active substances which
favourably influence the course of the illness. The
compounds of formula Ia can, however, also be used as
comedications with other therapeutically active substances.
The compounds of formula Ia can, however, also be used
to improve the prevention or control of illnesses with
therapeutically active substances which have a Gatechol
structure. ~he treatment of Parkinson's disease and of
parkinsonism with L-dopa, a therapeutically active
substance having the catechol structure, can be mentioned
as an example. In such cases the compounds of formula Ia
can be used in the form of a comedication or as
combination preparations.
The field of diagnostics offers a further possibility
for the use of the compounds of formula Ia above. After
the administration of ~1 F']-6-fluoro-L-dopa,
[ F]-dopamine in the brain can be visualized with the
~3~
-- 3
aid of positron emission tomography. By adding a compound
of formula Ia above the COMT is inhibited and thus the
formation of ~ E`]-3-0-methyldopa is prevented. ln the
absence of a COMT-inhibitor, the [18~`]-3-0-methyldopa
would penetrate into the brain and lead to a greatly
increased background which would make the diagnosis very
much more difficult.
Formula Ia above embraces not only known, bu~ also
novel compounds. The compounds of formula la which are
known per se fall under the general formula
HO\ ~-
,I~YIi-RC~' la
wherein Ra signifies nitro or cyano, Xb signifies
hydrogen or halogen, ~c" signifies nitro, cyano or the
group -(A)n-COO~ or ~(~)n~Q~H, A signifies
vinylene optionally substituted by lower alkyl, n
signifies the number O or 1, Q signifies the group
-CO- or ~C=N-(Z)p-R4, Z signifies an oxygen atom
or an imino group, p signifies the number O or 1 and
R signifies hydrogen or a saturated or partially
unsaturated, lower hydrocarbon residue which is
optionally substituted and which is optionally
attached via a carbonyl group, with the proviso that
Ra signiies nitro when Rc" signifies cyano or nitro,
ester and ether derivatives which are hydrolyzable under
physiological conditions and pharmaceutically acceptable
salts theceof.
llhe novel compounds of formula la are the compounds of
the general formula
~2~
-- 4
~a
HO\ ~-
I Ib
-~herein Ra signifies nitro or cyano, Rh signifies
hydrogen or halogen, Rc' signifies nitro. cyano or the
grou~ -(A) n~ (Q)m~R or - (A) n~Q~R , A
signifies vinylene optionally subs~ituted by lower
alkyl, n signifies the number O or 1, m signifies the
number O or 1, R signifies the group -COR , a
carbocyclic, aromatic group or an aromatic or
partially unsaturated, heterocyclic group attached via
a carbon atom, R signifies an optionally substi-
tueed, satueated or partially unsaturated, lower
hydrocarbon residue, R 1 signifies hydroxy, amino,
an optionally substituted, saturated or partially
unsaturated, lower hydrocarbon residue attached via an
oxygen atom or an imino or lower alkylimino group or a
saturated, N-containing heterocyclic group attached
via a ring nitrogen atom, Q signifies the geoup
-CO- or >C=N-(Z)p-R , Z signifies an oxygen atom
or an imino group, p signifie~ the number O or 1 and
R4 signifies hydrogen or a saturated or partially
unsaturated, lower hydrocarbon residue which is
optionally substituted and which is optionally
attached via a carbonyl group, with the proviso that
Ra signifies cyano when Rc` signifies cyano or nitro
and R31 has a significance different from hydroxy
when m signifies ~he number O,
and the ester and ether derivatives thereof which are
hydrolyzable under physiological conditions and the
- 35 pharmaceutically acceptable salts thereof.
Objects of the present invention are: The above
~3~
-- 5
compounds of formula Ia and the mentioned derivatives
thereof for use as therapeutically active substances:
medicaments based on these compounds and derivatives the
manufacturs of such medicaments; the use of the compounds
and derivatives in question in the prevention or control
of illnesses; the use of the compounds ancl derivatives in
question for the manufacture of medicaments which in a
given case inhibit the enzyme COMT in the sense of a
desired side-effect; the compounds of formula Ib above and
the mentioned derivatives thereof per se; the manufacture
of these compounds and derivatives: as well as inter-
mediates for their manufacture.
The term l'lower" denotes residues and compounds with a
maximum of 7, preferably a maximum of ~, carbon atoms. The
term "alkyl", ta~en alone or in combinations such as
"alkyl group", "alkoxy", "alkylthio" and "alkylimino",
denotes straight-chain or branched, satura~ed hydrocarbon
residues such as methyl, ethyl, propyl, isopropyl,
n-butyl, s-butyl, i-butyl and t-butyl. The term "saturated
or partially unsaturated, lower hydrocarbon residue"
denotes open-chain and cyclic groups and combinations
thereof. Examples of saturated and partially unsaturated
lower hydrocarbon residues are: lower alkyl groups such as
those defined above; loweL alkenyl groups such as
Z-propenyl, 2-butenyl, 3-butenyl and 2-methyl-2-propenyl:
C3 7-cycloalkyl and C8 10-bicycloalkyl groups
optionally substituted by lower alkyl groups such as
cyclopropyl, cyclopentyl, 2-methylcyclopentyl, cyclohexyl
and 3-methylcyclohexyl; lower cycloalkenyl groups
optionally substituted by lower alkyl groups such as
3-cyclopentenyl, 1-methyl-3-cyclopentenyl and
3-cyclohexenyl: lower alkyl or alkenyl groups substi-
tuted by lowe~ cycloalkyl or cycloalkenyl groups such as
cyclopropylmethyl, cyclopropylethyl, cyclopentylmethyl,
cyclohexylmethyl, 2-cyclohexenylmethyl and 3-cyclopropyl-
~L3~
-- 6
-2-propenyl. The lower alkenyl groups preferably contain
2-4 carbon atoms: the cycloalkyl and cycloalkenyl geoups
preferably contain 3-6 carbon atoms.
The following come into consideration as substituents
for the above lower hydrocarbon residues: Hydroxy, cyano,
nitro, halogen, amino, lower alkylamino, di(lower alkyl)-
amino, lower alkoxy, lower alkoxycarbonyl, aryl,
arylaminocarbonyl, arylcarbonyl, arylcarbonylamino, lower
alkanoyloxy, lower alkanoyl, carbamoyl, mono- or di(lower
alkyl)carbamoyl, lower alkylenedioxy, trifluoromethyl,
carboxy, lower alkanoylami~o, lower alkoxycarbonylamino
a~d lower alkylthio. The saturated or partially
lS unsaturated, lower hydrocarbon residues are preferably
unsubstituted or mono- or disubstituted.
l~he term "aryl" denotes carbocyclic aromatic groups,
preferably mono- or bicyclic groups. Especially preferred
carbocyclic aromatic groups are ehenyl and naphthyl
groups, especially phenyl groues. These groups are
optionally substitued by: halogen, trifluoromethyl, nitro,
amino, mono- oe di(lower alkyl)amino, lower alkyl, lower
alkoxy, lower alkylthio, lower alkanoyl, lower alkoxy-
carbonyl, carboxy, hydroxy, cyano, lower alkanoyloxy,carbamoyl, mono- or di(lower alkyl)carbamoyl, lower
alkylenedioxy, lower alkanoylamino or lower
alkoxycarbonylamino. The carbocyclic aromatic groups are
preferably unsubstituted or mono- or disubstituted.
The term "aromatic or partially unsaturated, hetero-
cyclic group`' preferably denotes a mono-, di- or
tricyclic, aromatic or ~artially unsaturated, heterocyclic
group with up to 5 hetero atoms from the group consisting
of nitrogen, sulphur and oxygen. These heterocyclic groups
preferably contain 1-4 nitrogen atoms and/or an oxygen or
sulphur atom. They are preferably mono- or bicyclic. The
hetero atoms are preferably distributed on one or two
rings, whereby nitrogen atoms can simultaneously also be
components of 2 rings. The heterocyclic groups are
preferably aromatic. They can be substituted, in this case
they are preferably mono- , di- or trisubstituted. As
substituen~s ehere come into consideration: halogen,
trifluoromethyl, nitro, carboxy, amino, arylamino, lower
alkyl, lower alkoxy, hydroxy, lower alkoxycarbonyl, lower
alkanoyl, lower alkanoyloxy, oxo, lower alkylenedioxy,
mercapto, lower alkylthio, lower alkylamino, di(lower
alkyl3amino, C3 7-cycloalkylamino, C8 1O-bicycloalkyl-
amino, lower al~anoylamino, lower alkoxycarbonylamino,
carbamoyl, mono- or di(lower alkyl)carbamoyl, cyano, aryl,
aryl-lower alkyl, aryl-lower alkylamino, heteroaryl,
heteroaryl-lower alkyl, heteroarylamino and C3 7-
-cycloalkyl. The monocyclic heterocyclic groups are
pre~erably 5- or 6-membered and contain a maximum of four
hetero atoms. The bicyclic heterocyclic groups are
preferably 8- to 10-membered, with the individual rings
being preferably 5- or 6-membered.
~ 'he follo~ing are to be mentioned as examples of such
heterocyclic groups: Pyridyl, pyrazinyl, triazinyl,
thiadiazinyl, thiazolyl, oxazolyl, oxadiazolyl, pyrazolyl,
tetrazolyl, imidazolyl, thienyl, quinolinyl,
isoquinolinyl, dihydroisoquinolinyl, benzoxazinyl,
quinoxalinyl, benzopyranyl, benzimidazolyl, indolyl,
imidazothiazolyl, imidazothiadiazolyl, imidazopyridyl,
benzothiazinyl, benzoquinoxalinyl and imidazo-
benzothiazolyl.
The term ''heteroarylll denotes aromatic, heterocyclic
groups as defined above.
. .
The term "a saturated, N-contalnlng heterocycllc group
attached via a ring nitrogen atom~ preferably denotes a
l~Z~
3- to 7-membered, preferably 4- to 6-membered, saturated
N-heterocycle which, in addition to the said nitrogen
atom, can contain an oxygen, sulphur or nitrogen atom as a
second hetero atom. These saturated N-heCerocycles can be
mono- or disubstituted by: lower alkyl, hydroxy, lower
alkoxy, lower alkanoyloxy, lower hydeoxyalkyl, lower
alkoxyalkyl, lower alkanoyloxyalkyl, lower alkoxycarbonyl,
lower alkanoyl, carbamoyl, mono- or di(lower alkyl)-
carbamoyl, oxo and/or lower alkylenedioxy.
The following are to be mentioned as examples of suchN-containing heterocyclic groups: 4-morpholinyl,
l-pyrrolidinyl and l-azetidinyl.
The ester and ether derivatives which are hydrolyzable
under physiological conditions are preferably compounds of
Lormula la in which at least one of the two phenolic
hydroxy groups is acylated by a lower fatty acid or
etherified by a lower l-alkoxycarbonyloxy-l-alkyl, lower
l-alkanoyloxy-l-alkyl or by a lower 2-oxo-l-alkyl group.
The substituent Ra preferably signifies nitro. The
substituent Rb is preferably situated in the p-position to
the substituent Ra and preferably signifies hydrogen,
chlorine or fluorine, with hydrogen being especially
preferred. The substituent Rc` preferably signifies the
group -CO-R l in which R signifies an aromatic,
mononuclear carbocyclic group or an aromatic, mononuclear
heterocyclic group with 1-3 nitrogen atoms as the hetero
ring member(s) which is attached via a cacbon atom. In an
especially preferred embodiment Rll signifies a phenyl
group optionally mono- or disubstituted by halogen,
trifluoromethyl, cyano, hydroxy or lower alkyl or a
pyridyl group.
Particularly preferred compounds in the scope of the
~3~:~,24~L8
g
present invention are:
3,4-Dihydroxy-5-nitrobenzo~henone,
2'-fluoro-3,4-dihydroxy-5-nitrobenzophenone and
3,~-dihydroxy-5-nitrophenyl 4-pyridyl ketone.
The compounds of formula lb, ~he ester and ether
derivatives which are hydrolyzable under physiological
1 conditions and the pharmaceutically acceptable salts
~hereof can be manufactured in accordance with the
invention by
a) cleaving off the lower alkyl ether group(s) in a
compound of the general formula
~a
\ o ~ \
R~0/
wherein one of the symbols X and R' signifies lower
alkyl and the other signifies hydrogen or lower alkyl
and Ra, Rb and Rc' have the above signi~icance,
or
b) reacting a compound of the general formula
~a
H0\ ~-
t ll Ib
H0/ ~.~ (A)n-C0-C~2-X
Rb
wherein X signifies a leaving group and Ra, Rb, A and
n have the above significance,
with a thioamide, thiourea, thiocarboxylic acid hydrazide,
thiosemicarbazide, amidine, guanidine, amidrazone,
~lL3~
-- 10 --
aminoguanidine, cyclic amidine, l,Z-diamine, 1,2-amino-
thiol or a 1,2-aminoalcohol and, if desired, dehydrogen
ating the cyclocondensation product obtained, or
c) reacting a compound of the general formula
~a
~ A)n-CO-COOR~ lb2
wherein R" signifies lower alkyl and Ha, Rb, A and n
have the above significance,
with a 1,2-diamine, ~,2-aminothiol, 1,2~aminoalcohol,
semicarbazide, thiosemicarbazide, amidrazone or an amino-
guanidine and, if desired, dehydrogenating the cyclocon-
densation product obtained, or
d) reacting a compound o~ formula Ibl above with a
B-aminocarbonyl compound, or
e~ converting the carboxaldehyde group(s) in a compound
of the general formula
fH0 ~N fHo
H0\ ~-\ H0\ ~ \o
H0/ ~.X Rc ~ CH0 Ia2 or/l~ xll_CH0 lV
wherein Xc"' signifies nitro, cyano or the group
-(~) -R12 and R12 signifies the group -COX
a carbocyclic, aromatic group or an aromatic or
partially unsaturated heterocyclic group attached via
a carbon atom and Ra, Rb, A, n and X have the
above significance,
or in a di-0-lower alkanoyl derivative thereof into the
cyano group(s), or
f) reacting a di-0-lower alkanoyl derivative of a
carboxylic acid of che general formula
lo i~ k'~- (A)n-cooH /3~ kli- (A) -C0-COOH
wherein Xa, Rb, A and n have the above significance,
in the presence of a condensation agent or a reactive
derivative or a di-0-lower alkanoyl derivative of a
carboxylic acid of formula la3 or lb3 with a compound
of the general formula
Ho-R5 V or HN~6~7 VI
wherein R5 signifies an optionally substituted,
saturated or partially unsaturated, lower hydrocarbon
residue, R6 signifies hydrogen or lower alkyl and
R7 signi~ies hydrogen or an optionally substituted,
saturated or partially unsaturated, lower hydrocarbon
residue or R6 and R7 togather with ~he nitrogen
atom signify a saturated W-containing heterocyclic
group,
or
g) hydrolyzing a compound of formula Ib2 or a compound
of the general formula
8 ~a
R o\ ~
R80/ ~ c lb4
Rb
wherein R signifies lower alkanoyl and Ra, Rb and
~L3~
- 12 -
Rc' have th~ above significance,
or
h) eeacting a compound of the general Eormula
~ a
H0\ ~.\
lo Rb Ib5
wherein Ra and Rb have the above signi~icance and X"'
signifies hydrogen or lower alkyl,
or a di-0-lower alkanoyl derivative thereof in the
presence of a secondary amine with a compound of the
general formula
CH3Co-R23 VII
wherein R siynifies an o~tionally substituted,
saturated or partially unsaturated, lower hydrocarbon
residue,
or
i~ reacting a compound of the general ~ormula
~a
H0\ ~
H0/ ~.~ ( )n C0-CH2-COOR" Ib6
0
wherein Ra, Rb, A, n and R" have the above signifi-
cance,
with a hydrazine or an amidine, or
j) reacting a compound of formula Ib above in which m
signifies the number l and Q signifies the group -C0- with
a compound of the general formula
~3g~
- 13 -
H2N-(Z)p-R4 VIII
wherein Z, p and R4 have the above significance,
and, if desired,
k) converting a compound of formula Ib above into an
ester or ether derivative which is hydrolyzable under
physiological conditions or into a pharmaceutically
acceptable salt thereof.
In accordance with process variant a) the compounds of
formula lb can be manufactured by cleaving the ether
group(s) in a compound of formula lI. This ether cleavage
can be carried out according to methods which are known
per se and which are familiar to any person skilled in the
art. The ether cleavage can be carried out, Eor example,
by treatment with hydrogen bromide in a suitable solvent.
5uitable solvents are, ~or example, water, acetic acid and
mixtures thereof. The reaction is preferably carried out
at an elevated temperature, for example in a temperature
range of about 100C to the boiling temperature of the
reaction mixture. There are preferably used 48 percent
hydrobromic acid or mixtures thereof with acetic acid.
The ether cleavage can also be carried out by
treatment with boron tribromide in a suitable solvent at
temperatures of about -60C to about room temperature.
Suitable solvents aee especially halogenated lower hydro-
carbons such as met~lylene chloride, chloro~orm and thelike. Further suitable methods are: treatment with
2yridinium hydrochloride at temperatures of abouc 150C to
about 250C and treatment with sodium iodide/silicon
tetrachloride in an inert organic solvent at an elevated
temperature, for example at the re~lux temperature of the
reaction mixture. Suitable solvents for the latter process
are, for example, acetonitrile, aromatic hydrocarbons such
~L3~
- 14 -
as benzene or toluene, mixtures thereof and the like.
In accordance with process variant b) there can be
manufactured compounds of the general formula
~a
H0/ ~ ~Rb n Ib7
wherein Ra, Rb, A and n have the above significance
and Q signifies a group of the formula
/N-.~ N /NHRf ~N-N~ NHRf
15 ~ (a)~ (b), -S/ (c), \--S (d),
/Re /NHRf
N=~ /N=i /N=N\ /N=N\
.-~H (e), ~ H (f)~ --Re (g)r _~ -NHRf (h
/Rg /Rg
_0~ ~ Q4 (i), ~-~ Rh (k) or --~ /~-Rh (k'),
--\ /--Rh (1) or --~ /--Rh (m),
in which Re signifies hydrogen, C3 7-alkyl, C3 7-
-cycloalkyl, aryl, heteroaryl, aryl-lower alkyl or
heteroaryl-lower alkyl, Rf signifies hydrogen, aryl,
aryl~lower alkyl, lowec alkyl, lower alkoxycarbonyl,
he~eroaryl, heteroaryl-lower alkyl, C8 lo~bicyclo~
alkyl or C3 -cycloalkyl, Rg and Rh each signify
hydrogen, cyano, lower alkyl, C3 7-cycloalkyl, aryl,
aryl-lower alkyl, heteroaryl or heteroaryl-lower alkyl
~3~2~
- 15 -
or Rg and Rh ~ogether with the two carbon atoms to
which they are attached signify a carboxycyclic
aromatic group or an aromatic or partially
unsaturated, heteLocyclic group, the dotted line
signifies an optional bond and Q4 together with the
carbon a~om and the nitrogen a~om signify an aromatic
or partially unsaturated, heterocyclic group which
contains at least one nitrogen atom as a hetero riny
mPmber.
Suitable solvents for this process aspect are lower
alcohols such as ethanol, n-butanol, n-hexanol and
ethylene glycol, open-chain and cyclic ethers which can
contain free hydroxy groups such as tetrahydrofuran,
dioxan, t-butyl methyl ether, ethylene glycol dimethyl
ether, dieChylene glycol dimethyl ether, ethylene glycol
monomethyl ether and diethylene ylycol monomethyl ether,
acetonitrile, dimethylformamide, dimethylacetamide and
dimethyl sulphoxide. The desired reaction can also be
carried out withou~ a solvent by dry heating th~ reaction
partners. The reaction is preferably carried out at an
elevated temperature, for example in a range of about ~0C
to 150C, whereby it is preferably carried out at the
boiling temperature of the solvent insofar as it is
carried out in the presence of a solvent and the boiling
point lies in the previously men~ioned range.
In accordance wi~h process variant c) there can be
manufaccured compounds of the general focmula
~a
H0\ ~.\
H0 Rb lbB
in which Ra, Rb, A and n have the above significance
- 16 -
and Q~ signifies a gcoup of ~he formula
_ ~ /.-Rh (n~ -Rh ~o), - ~ Rh (P)~
~--N\ ~ ~ ~ -SH ~.-N/ (s) or
( )
~--N
in which Re, R~, Rg, Rh and the dotted line have the
above significance.
The reaction in accordance with process variant c) can
be carried out under the same reaction conditions as
process variant b).
. In accordance with process variant d) there can be
manufactu~ed compounds of the general formula
~a
UO/ ~`Y n Ib9
wherein Q signifies ~he group of the formula
:~3~
\ ~\ /Rg
/~ (u)
Re Rh
and Ra, Rb, Re, RX, Rg, Rh, ~, n and the dotted line
have the above significance.
0
~rocess variant d) can also be carried out under the
same reaction conditlons as process variant b~.
In accordance with process variant e) there can be
manufactured comeounds of formula lb in which Xa sigrLifies
cyano, Rc' signifies nitro, cyano or the group
~(~)n~R and R signifies the group -COR , a
carbocyclic, aromatic group or an aromatic or partially
unsaturated, heterocyclic group attached via a carbon atom
and Rb, ~, n and R3l have the above significance. 'rhe
conversion of the carboxaldehyde group(s) into the cyano
qroup(s) can be efected according to methods which are
known per se and which are familiar to any person skilled
in the art. For example, a compound of formula Ia2, III
or IV can be treated with hydroxylamine O-sulphonic acid
at an elevated temperature, with water being preferably
used as the solvent. ~he reaction can be carried out in a
temperature range of about 50C to about 100C.
ln accordance with process variant f) there can be
manufactured di-O-lower alkanoyl derivatives of compounds
of formula Ib in which Rc' signifies the group
-(A)n-(CO)m-COR and R signifies amino, an
optionally substituted, saturated or partially
unsatucated, lower hydrocarbon residue actached via an
oxygen atom or an imino or lower alkylimino group or a
saturated, N-containing heterocyclic group attached via a
~3~Z~
- 18 -
ring nitrogen atom and ~, n and m have the above signifi-
cance. This reaction can also `De carried out accocding to
methods which are known per se and which are familiar to
any person skilled in the art. Lower alkyl esters can be
manufactured, for examele, by treating the carboxylic acid
with the corresponding lower alcohol in the presence of an
acid, with the corresponding lower alcohol being
preferably used as the solvent. Suitab].e acids are, ~Of
example, mineral acids such as hydrogen chloride and
organic sulphonic acids such as p-toluenesulphonic acid.
The reaction temperature preferably lies in a range of
room temperature to the boiling temperature of the chosen
solvent.
The remaining esters and the amides are preferably
manufactured starting from reactive carboxylic acid
derivatives. Suitable reactive carboxylic acid derivatives
are, for example, the corresponding ca~boxylic acid
halides, especially the carboxylic acid chlorides, corres-
ponding carboxylic acid anhydrides and mixed anhydrides
(e.g. with trifluoroacetic acid and organic sulphonic
acids such as mesitylenesulphonic acid and p-toluene-
sulphonic acid), corresponding carboxylic acid
imidazolides and the like. The reaction is conveniently
carried out in ~he presence of an acid-binding agent and
in an inert organic solvent. Suitable acid-binding agents
are, for example, tertiary amines such as triethylamine
and pyridine. In the manufacture of amides, excess amine
of formula Vl can also be used as the acid-binding agent.
Suitable solvents are, for example, open-chain and cyclic
ethers such as tetrahydrouran, diethyl ether, t-butyl
methyl ether, dioxan, ethylene glycol, dimethyl ether or
the like, halogenated hydrocarbons such as methylene
chloride, chloroform and 1,2-dichloroethane, acetonitcile
and dimethylformamide.
- 19
The hydrolysis of compounds of formula Ib~ to the
corresponding catechol derivatives in accordance with
process variant g) can also be carried out according to
methods which are known per se and which are familiar to
any person skilled in the art. The hydrolysis can be
carried out, for example, by treatment with an alkali
metal hydroxide such as sodium hydroxide or potassium
hydroxide in a suitable solvent. Suitable solvents are,
for example, lower alcohols such ~ethanol and water or
mixtures thereof. The hydrolysis can be carried out, for
example, in a temperature range of about 0C to the
boiling temperature of the solvent, whereby, however, it
is preferably carried out at room temperature.
In accordance with process variant h) there can be
manufactured compounds of the general formula
~lO\ ~-
~!~ k -C (R''')=CH_co-R23 lbl
wherein Ra, Rb, X"' and R have the above signifi-
cance,
and the corresponding di-0-lower alkanoyl derivatives
thereof. Cyclic amines such as pyrrolidine, piperidine,
morpholine and thiomocpholine are preferably used as the
secondary amine. Suitable solvents for this process are,
for example, open-chain and cyclic ethers such as tetra-
hydrofuran, diethyl ether, t-butyl methyl ether, dioxan,
ethylene glycol and dimethyl ether, halogenated hydro-
carbons such as methylene chloride, ch}oroform and
1,2-dichloroethane, acetonitrile and dimethylformamide.
The reaction temperature conveniently lies in a range of
about 0C to the boiling temperature of the chosen solvent
whereby the reaction i5 preferably carried ou~ at room
~3~
- - 20 -
temperature. In an especially preferred embodiment the
reaction is carried out in the presence of an acid,
preferably a carboxylic acid such as acetic acid.
In accordance with process variant i) there can be
manufactured compounds of the general formula
~a
lo ~o/ ~ k - (A)n~Q5 Ibll
wherein Q signifies the group
Rf /Re
(v) or _.// ~N (w)
\OH ~OH0
and ~a, Rb, Re, Rf, A and n have the above signifi-
cance.
Suitable solvents for this process are, for example, lower
alcohols such as methanol and ethanol, open-chain and
cyclic ethers such as tetrahydrofuran, diethyl ether,
t-butyl methyl ether, dioxan, ethylene glycol and dimethyl
ether, acetonitrile and dimethylformamide. The reaction is
pre~erably carried out at an elevated temperature, for
example in a range of about 50C to the boiling
temperature of the chosen solvent, whereby it is
preferably carried out at the boiling temperature of the
chosen solvent.
In accordance with process variant j) there can be
manufactured compounds of the general formula
- 21 -
HO\ ~-\ /Ri
~ ( A)n-C~ ) R4 Ibl2
wherein ~i signi~ies the group -~`OR , a
carbocyclic, aromatic group or an aromatic or
partially unsaturated, heterocyclic group attached via
a carbon atom or an optionally substituted, saturated
or partially unsaturated, lower hydrocarbon residue
and Ka, Rb, ~ , R4, A, Z, n and p have the above
significance.
This process can also be carried out accordiny to methods
which are known per se and which are familiar to any
person skilled in the art. Suitable solvents are, for
example, lower alcohols such as methanol and ethanol,
dimethylfocmamide and water. The rea~tion is con~eniently
carried out at room ~emperature.
In accordance with process variant k) the compounds of
formula Ib can be converted into ester or ether
derivatives which are hydroly~able under physiological
conditions. Suitable ester derivatives which are
hydrolyzable under physiological conditions are especially
the compounds of formula Ib in which at least one of the
two phenolic hydroxy groups is acylated by a lower fatty
acid. These can be manufactured according to methods which
are known per se and which are familiar to any person
skilled in the art. ln a preferred embodiment the
acylation is carried out with the corresponding lower
fatty acid anhydride in the presence of a catalytic amount
of a strong acid, with excess fatty acid anhydride being
preerably used as the solvent. Suitable acids are, for
example, sulehuric acid and organic sul~honic acids such
as p-toluenesulphonic acid.
~3~
- ~2 -
Suitable ether derivatives which are hydrolyzable
under physiological conditions are, for example, comeounds
of formula lb in which at least one oi the two phenolic
hydroxy groups is etheri~ied by a lower l-alkoxycarbon~l-
oxy-l-alkyl, lower l-alkanoyloxy-l-alkyl or by a lower
2-oxo-1-alkyl group. The etherification can be carried out
according to methods which are known per se and which are
familiar to any person skilled in the art. For example, a
compound of formula Ib can be reacted with a lower
l-alkoxycarbonyloxy-l-alkyl halide, a lower l-alkanoyloxy-
-l-alkyl halide or a lower 2-oxo-1-alkyl halide, with this
atherification being conveniently carried out in the
presence of a base. Rs halides there come into considera-
tion, in particular, the iodides. Suitable bases are, forexample, alkali metal hydroxides and alkali metal
carbonates such as sodium hydroxide and sodium carbonate.
In accordance with process variant k) compounds of
formula Ib above can also be converted into pharmaceut-
ically acceptable salts. As salts there come into
consideration, in particular, salts with pharmaceutically
acceptable bases. As examples of such salts there are to
be mentioned the alkali metal salts such as the sodium and
potassium salts. These salts can be manufactured according
to methods which are known per se and which are familiar
to any person skilled in the art.
The various compounds which are used as stacting
materials are known or can be prepared according to
methods known eer se. The following Examples contain
detailed information concerning the preparation of these
starting materials.
As mentioned earlier, the compounds of formula Ia
inhibit the enzyme COMT. This activity can be determined
quantitatively as follows: Rat liver homogenate is
~3~3~
- 23 -
incubated in the presence of a suitable substrate as
described in J. Neurochem. 38, 191-19S (1982~ and the COMT
activity is measured. In a second series of experiments
the incubation is carried out in the presence of a
compound of fo~mula Ia. The IC50 can then be calculated
from the difference of the CO~T activity which is
determined. IC50 is given in nmol/l and is that concen-
tration in the incubation mixture which is required to
reduce the COMT activity by 50%. The IC50 values for
some compounds of formula Ia are given in the following
Table. Moreover, this Table contains data concerning the
acute toxicity of these compounds (LD50 in mg/kg in the
case of single oral administration to mice).
L5 Compound of formula IaIC50 in nmol/1 50
mg/kg p.o.
3,4-Dihydroxy-5-nitrophenyl
2-pyridyl ketone 53.4 1250-2500
20 3,4-Dihydroxy-5-nitrophenyl
3-pyridyl ketone 47.0 1000-2000
3,4-Dihydroxy-5-nitrophenyl
4-pyridyl ketone 67.0 1000-2000
n-Butyl 3,4-dihydroxy-5-
25 -nitrobenzoate 20.0 312- 625
n-Butyl 3,4-dihydroxy-5-
-nitrocinnamate 25.9 2500-5000
2thyl 3,4-dihydroxy-5-
-nitrophenylglyoxylate 48~ 1250-2500
3~4-Dihydroxy-5-nitrobenzo-
phenone 48.0 500-1000
3,5-Dinitropyrocatechol 36.9 500-1000
2l-Fluoro-3,4-dihydroxy-S-
nitrobenzophenone 42.0 312- 625
The described substances can be used as medicaments,
e.g. in the form of pharmaceutical preparations for
~36~
-- 2g --
enteral or parenteral administration. The comeounds of
formula Ia can be administered, eor example, perorally,
e.g. in the form of tablets, coated tablets, dragees, hard
and soft gelatine capsules, solutions, emulsions or
suspensions, rectally, e.g. in the form of supeositories,
or parenterally, e.g. in the form of injection solutions.
-
The manufacture oE the pharmaceutical preparations can
be effected in a manner which is familiar to any personskilled in the art by bringing the compounds of formula
Ia, optionally in combination with other therapeutically
valuable substances, into a galenical administration form
together with suitable, non-toxic, inert, therapeutically
compatible solid or liquid carrier materials and, if
desired, the usual pharmaceutical adjuvants.
As carrier materials there are suitable not only
inorganic carrier materials, but also organic carrier
materials. Thus, for tablets, coated ~ablets, dragees and
hard gelatine capsules there can be used as carrier
materials, for example, lactose, maize starch or
derivatives thereof, talc, stearic acid or its salts.
Suitable carriers for soft gelatine capsules are, for
example, vegetable oils, waxes, fats and semi-solid and
liquid eolyols (depending on ~he nature of the active
substances no carriers are, however, required in the case
of so~t gelatine capsules). Suitable carrier materials for
the manufacture of solutions and syrups are, for example,
water, polyols, saccharose, invert sugar and glucose.
Suitable carrier materials for injection solutions are,
for example, water, alcohols, polyols, glycerine and
vegetable oils. Suitable carrier materials for supposi-
tories are, for example natural or hardened oils, waxes,
fats and semi-liguid or liquid polyols.
As pharmaceutical adjuvants there come into considera-
~3~
- 25 -
tion the usual preserving agen~s, solubilizers,
stabilizing agents, wetting agents, emulsifying agents,
flavour-improving agents such as sweetening agents and
S flavouring agents, colouring agents, salts for varying the
osmotic pressure, buffers, coating agents and antioxidants.
The dosage of the described substances can vary within
wide limits depending on the illness to be treated, the
age and the individual condition of the patient and on the
mode of administration and will, of coursa, be fitted to
the individual requireman~s in each particular case. In
the improvement of the treatment of Parkinson's disease
and of parkinsonism with L-dopa a daily dosage of 25 mg to
about 1000 mg, especially about 100 mg to about 300 mg,
comes into consideration. Depending on the dosage it is
convenient to admlnister the daily dosage in several
dosage units.
~`he pharmaceutical preparations in accordance with the
invention conveniently contain about 25 mg to about
300 mg, ereferably about 50 mg to about 1~0 mg, of a
compound of formula Ia or of an ester or ether derivative
which is hydrolyzable under physiological conditions or of
a pharmaceutically acceptable salt thereof.
The following Examples are intended to illustrate the
present invention in more detail, but are not intended to
limit its scope in any manner. All temperatures are given
in degrees Celsius.
~L~
a) 17.1 g (~6.7 mmol) of 4-hydroxy-3-methoxy-5-nitro-
benzaldehyde are treated with 170 ml of constant-boiling
hydrobromic acid and heated under reflux for 3.5 hours.
After cooling the separated precipitate is filtered off
13~r~ 8
under suction, washed twice with ice-water and taken up in
ethyl acetate. The oryanic phase is washed twic~ with
S0 ml oE sodium chloride solution each time, dried over
S magnesium sulphate and evaporated in a water-jet vacuum.
The crystals obtained are taken up in methylene chloride,
whereupon the solution is filtered over a ten-fold amount
of silica gel. The material obtained is crystalli2ed from
ethyl acetate/isopropyl ether. There i6 obtained
3,4-dihydroxy-5-nitrobenæaldehyde in the form of yellow
crystals of m.p. 142-143.
b) A solution of 1.7 g (15 mmol) of hydroxylamine o-
-sulphonic acid in 6 ml of water is added to a solution of
l.a3 g (10 mmol) of 3,4-dihydroxy-S-nitrobenzaldehyde in
25 ml of water, subsequently stirred a~ 65 for 3.5 hours,
cooled, the separated precipitate is Eiltered off under
suction and taken up in ethyl acetate. The organic phase
is dried over sodium sulphate and evaporated in a water-
-jet vacuum. The crystals obtained are cecrystallized from
ethyl acetate/n-hexane. There is ob~ained 3,4-dihydroxy-5-
-nitrobenzonitrile in the form of yellow crystals of m.p.
194-195.
Example 2
aa) 10 ml of tert.butyl lithium solution (1.4M in hexane)
are added dropwise at -70 within 10 min. to 4.1 g of
4-(benzyloxy)-3-methoxy-bromobenzene dissolved in 40 ml of
tetrahydrofuran. ~fter stiEring at -70O for 2 hrs. 1 ml of
pyridine-3-carbaldehyde is added within 5 min. ~he
reaction mixture is stirced at -700 for 1 hr. and at 0
f.or 2 hrs. and poured into 100 ml of lN hydrochloric acid.
The mixture is extracted three times with 50 ml of ether
each time. The combined ether phases are washed with
loO ml of lN hydrochloric acid and 20 ml of water. The
combined aqueous phases are made alkaline with aqueous
~3~
- 27 -
ammonia solution and extracted three times with 100 ml of
methylene chloride each time. The combined methylene
chloride phases are dried over sodium sulphate and
evaporated. There is obtained
alpha-~4-(benzyloxy)-3-methoxyphenyl]-3- -pyridinemethanol
as an oil.
ab) ln an analogous manner, using pyridine-4-carbaldehyde
there is obtained alpha-[4-(benzyloxy)-3-methoxyphenyl~-4-
~pyridinemethanol as an oil.
ba) 3.2 g of alpha-t4-(benzyloxy)-5-methoxyphenyl]-3-
pyridineme~hanol suspended in 50 ml of water are treated
with 2.5 g of potassium permanganate, whereupon the
mixture is stirred at 90 f or 30 min. Af ter adding a
further 1.0 g of potassium permanganate and stirring for a
further 30 min. at 90 the mixture is cooled to room
temperature and extracted twice with 1~0 ml of ethyl
acetate each time. The combined ethyl acetate phases are
washed with sodium chloride solution, dried over sodium
sulphate and evaporated. l`he thus-obtained residue is
chromatographed on 50 g of silica gel with ethyl acetate.
After recrystallization from methylene chloride/hexane
there is obtained 4-(benzyloxy)-3-methoxyphenyl 3-pyridyl
ketone of m.p. 76.
bb) ln an analogous manner, Erom alpha-[4-(benzyloxy)-3-
-methoxyphenyl)-4-pyridinemethanol there is obtained
4-(benzyloxy)-3-methoxyphenyl 4-pyridyl ketone of m.p.
8s-87O (methylene chloride/hexane).
ca) 50 ml of 33 percent hydrobromic acid in acetic acid
are added dropwise within 15 min. at 10 to 20 g of
4-(benzyloxy)-3-methoxyphenyl 3-pyridyl ketone dissolved
in 200 ml of methylene chloride. After stirring at 20 f or
3 hrs. the reaction mixture is poured into a mixture of
~3~
- 28 -
100 ml of conc. aqueous ammonia and ice. The pH is
adjusted to 6 by adding acetic acid. The methylene
chloride phase is separated; the aqueous phase is
extracted a further twice with 100 ml of methylene
chloride each time. The combined methylene chloride phases
are dried over sodium sulehate and evapora~ed. The residue
is recrystallized from methylene chloride/hexane. There is
obtained 4-hydroxy-3-methoxyphenyl 3-pyridyl ketone of
m.p. 150-151.
cb) In an analogous manner, from 4-(benzyloxy)-3-methoxy-
phenyl 4-pyridyl ketons there is obtained 4-hydroxy-3-
-methoxyphenyl 4-eyridyl ketone of m.p. 215-218
(acetonitrile).
da) 0.38 ml of 65 percent nitric acid is added dropwise at
room temeerature to l.lS g of 4-hydroxy-3-methoxyehenyl
3-pyridyl ketone dissolved in 15 ml o~ acetic acid. After
stirring for 2 hrs. the reaction mixture is poured into
120 ml of ice-water, whereupon the mixture is adjusted to
pH 5 with conc. ammonia and the precipitate formed is
filtered off. The thus-obtained residue is heated under
reflux in 20 ml of acetonitrile, whereupon it is again
filtered off. There is obtained 4-hydroxy-3-methoxy-5-
-nitrophenyl 3-pyridyl ketone as brown crystals of m.p.
193.
db) In an analogous mannec, from 4-hydroxy-3-methoxyphenyl
4-pyridyl ketone there is obtained 4-hydroxy-3-methoxy-5-
-nitroehenyl 4-pyridyl ketone of m.p. 240~.
e) 3.5 g o 4-hydroxy-3-methoxy-5-nitrophenyl 3-pyridyl
ketone dissolved in 70 ml of 48 percent aqueous hydro-
bromic acid are stirred at 100 for 18 hrs. The reaction
mixture is subsequently evaporated under reduced pressure.
The residue is recrystallized from water. There is
~3~
- 29 -
obtained 3,4-dihydroxy-5-nitroph~nyl 3-pyridyl ketone
hydrobromide of m.p. 2~50.
~) In an analogous manner, from 4-hydroxy-3-methoxy-5-
-nitrophenyl 4-pyridyl ketone there is obtained
3,4-dihydroxy-5-nitrophenyl 4-pyridyl k~etone of m.p. 246
(from water).
g) 13.2 g of 3,4-dihydroxy-5-nitrophenyl 4-pyridyl ketone
are suspended in 500 ml of methanol and treated while
stirring with 4.88 g of methanesulphonic acid. The
suspension is heated under reflux for 60 minutes. It is
subsequently cooled to 10, the crystals are fil~ered of f
under suction and washed twice with 30 ml of methanol each
time. There is obtained 3,4-dihydroxy-5-nitrophenyl
4-pyridyl ke~one methanesulphonate of m.p. 260~261 (dec.).
ExamPle
a) A solution of 2.6 g (12.2 mmol) of 4-hydroxy-3-
-methoxy-5-nitrobenzoic acid in Z6 ml of constant-boiling
hydrobromic acid is heated under reflux for 2 hours. After
cooling the solvent is distilled off in a water-jet
vacuum. The crystalline residue is recrystallized from
50 ml of water at boiling temperature. There is obtained
3,4-dihydroxy-5-nitrobenzoic acid in the form of yellow
crystals of m.p. 224-226.
b) 1.0 g (5 mmol) of 3,4-dihydroxy-5-nitrobenzoic acid is
treated with 20 ml of methanolic hydrochloric acid,
stirred at 45 for 3 hours and, after removing the
solven~, the residue is taken up in me~hylene chloride.
The organic phase is washed with sodium chloride solution,
dried over sodium sulphate and evaporated. The crystalline
product obtained i6 taken up in methylene chloride and
~3~
- 30 -
filtered over a ~en-fold amount of silica gel. The
material obtained is recrystallized from ethyl acetate/n-
-hexane. There is obtained methyl 3,4-dihydroxy-5-nitro-
benzoate in the form of yellow crystals of m.p. 144-1~5.
The following esters are obtained in an analogous
manner starting from 3,4-dihydroxy-5-nitrobenzoic acid:
c) Ethyl 3,4-dihydroxy-5-nitrobenzoa~e of m.p. 106-107
(from ethyl acetate/n-hexane),
d) n-butyl 3,4-dihydroxy-5-nitrobenzoate of m.p. 73-74
tfrom methylene chloride) and
e) n-hexyl 3,4-dihydroxy-5-nitrobenzoate of m.p. 44-45
~from isopropyl ether).
Example 4
a) 25 ml of 2M phenyl lithium solution (in benzene/e~her
(7:3)) are added dropwise within 15 min. to 10.0 g of
3,4-dimethoxy-5-nitrobenzaldehyde dissolved in 150 ml of
tetrahydrofuran and the mixture is stirred at 0 for 1 hr.
and at 20 ~or 2 hrs. The mixture is subsequently treated
with 150 ml of 2N sul~huric acid and extracted three times
with 150 ml of ether. The combined ether phases aee washed
with sodium chloride solution, dried over sodium sulphate
and evaporated. The thus-obtained residue is chromato-
graphed on 180 g of silica gel with methylene chloride.
There is obtained 3,4-dimethoxy-5-nitrobenzhydrol as an
amorphous solid.
b) 2.5 g of 3,4-dimethoxy-5-nitrobenzhydrol dissolved in
~0 ml oE methylene chloride are treated with 2.2 g of
pyridinium chlorochromate, whereupon the mixture is
stirred at room temperature for 2 hrs. The insoluble
consti~uents are subsequently filtered off. The filtrate
is evaporated and the residue is chromacographed on 60 g
of silica gel with methylene chloride. ~fter crystalliza-
~3~2~
- 31 -
tion from methylene chloride/hexane there is obtained
3,4-dimethoxy-S-nitrobenzophenone of m.p. 78-80O.
c~ 0.5 g of 3,~-dimethoxy-5-nitrobenzophenone is stirred
at 110 for 30 hrs. in a mixture of 4 ml of acetic acid
and ~ ml of 48 percent aqueous hydrobromic acid. The
reaction mixture is subsequently evaporated to dryness.
The residue is taken up in methylene chloride. It is
washed with water, dried over sodium sulphate and evapor-
ated. After recrystallization from methylene chloride/
hexane there is obtained 3,4-dihydroxy-~-nitrobenzophenone
of m.p. 132.
xamPle 5
a) 4.9 g t0.2 mol) of magnesium are suspended in 15 ml of
absolute ethanol and, after adding 1 ml of carbon tetra-
chloride, warmed until the reaction starts. A solution of
31.8 g (0.2 mol) of diethyl malonate in lg.9 ml of
absolute ethanol and 80 ml of absolute ~oluene is then
added dropwise while stirring so that the temperature lies
between 50 and 60. The reaction mixture is subsequently
stirred at this temperature for a further 1 hour,
whereupon it is cooled to -5 and a solution of 49.3 g
(0.2 mol) of 3,4-dimethoxy-5-nitrobenzoyl chloride (m.p.
82-85O) in 300 ml of absolu~e toluene and 50 ml of
absolute tetrahydrofuran is added dropwise so that the
temperature does not exceed -5. The mixture is
subsequently stirred at room temperature overnight. After
distillation of the solvent the residue is dissolved in
soo ml of ethyl acetate. The solution is treated while
stirring and cooling with ice with an ice-cold solution of
12 ml of concentrated sulphuric acid in 80 ml of water.
The organic phase is washed with sodium chloride solution,
dried over magnesium sulphate and evaporated. The oil
obtained is chromatographed on a ten-fold amount of silica
z~
~ 32 -
gel with methylene chloride. The crystalline material
obtained is recrystalli2ed from isopropyl ether. There is
obtained diethyl 3,4-dime~hoxy-~-nitrobenzoylmalonate in
the form of pale heige crystals of m.p. 70.
b) 19.0 g ~51.4 mmol) of diethyl 3,~-dimethoxy-5-nitro-
benzoylmalonate are dissolved in 100 ml of glacial acetic
acid, 5 drops of concentrated sulphuric acid are added
thereto and the mixture is heated under reflux for 16
hours. The acetic acid is distilled off at 60 in a water
-jet vacuum and the residue is treatad three times with
250 ml of toluene each time, whereby it is evaporated each
time. The crystalline residue is extracted with ethyl
acetate. The organic phase is washed with water, dried
over magnesium sulphate and evaporated. The crystals
obtained are chromatographed on a 30-fold amount of silica
gel with Coluene. The crystalline material obtained is
recrystallized Erom isopropyl ether. There is obtained
3,4-dimethoxy-5~-nitroacetophenone in the form of
yellowish crystals of m.p. 86-87.
c) 2 g (8.9 mmol) of 3`,4'-dimethoxy-5`-nitroacetophenone
are treated with 30 ml of constant-boiling hydrobromic
2S acid and stirred at 140 for Z.5 hours. After cooling the
mixture is poured into 200 ml of ice-water and extracted
three times with 100 ml of ethyl acetate each time. The
organic phase is washed twice with 25 ml of sodium
chloride solution each time, dried over sodium sulphate
and evaporated. The product obtained is filtered with
ethyl acetate over a 20-fold amount o~ silica gel. By
recrystallization of the material obtained from water
there is obtained 3',4~-dihydroxy-5'-nitroacetophenone in
the form of yellow crystals of m.p. 159-160~.
The same compound is obtained starting from
4'-hydroxy-3'-methoxy-5'-nitroacetophenone by treatment
~3~
- 33 -
with hydrobcomic acid at the boiling temperature.
~m~
s
a) 100 g (0.8 mol? of guaiacol are dissolved in 136.~ g
(0.86 mol) of isobutyric anhydride, treated with 120 g
(0.88 mol) o~ anhydrous zinc chloride (whereby all passes
into solution), che reaction mixture is heated to 155 and
cooled after three minutes. The residue is firstly
subjected to a steam distillation in order to remove
readily volatile constituents and is then extracted three
times with 500 ml of ether each time. The organic phase is
washed twice with 250 ml of water each time, once with
150 ml of saturated bicarbonate solution and agdin with
250 ml of water, dried over sodium sulphate and evaporated
in a water-jet vacuum. The brown resin obtained is
distilled in a high vacuum. The distillate Oe b.p.
105-120 (~.67 Pa) is dissolved in ether, whereupon the
solution is treated with n-hexane until crystallization
begins. The crystals obtained are recrystallized from
ether/hexane. There is obtained 4~-hydroxy-3'-methoxy-2-
-methyl-propiophenone in the form of colourless crystals
of m.p. 86-87~.
b) 15.0 g (77.2 mmol) of 4`-hydroxy-3'-methoxy-2-methyl-
-propiophenone are dissolved in 300 ml o~ glacial acetic
acid and 7.65 ml of 50.5 percent nitric acid (11.2N) in
40 ml of glacial acetic acid are added dropwise thereto
while stiring within 15 minutes. After 15 minutes the
reaction mixture is poured into ice-water and the
separated crystals are ~iltered off under suction, washed
with water and dissolved in methylene chloride. The
solution is dried over sodium sulphate and evaporated. The
crude product obtained is taken up in methylene chloride
and ~iltered over 100 g of silica gel. The thus-obtained
crystals are recrystallized from methylene chloride~
~3~
- 34 -
.
n-hexane. There is ob~ained 4'-hydroxy-3'-methoxy-2-
-methyl-5l-nitropropiophenone in the form of yellow
crystals of m.p. 85-87O.
c) 8.0 g (33.4 mmol) of 4'-hydroxy-3'-methoxy-2-methyl-
-S~-nitropropiophenone are treaced with 64 g of pyridine
hydrochloride and stirred at 180 for 45 minutes. After
cooling the reaction mixture is poured into 500 ml of
ice-water, whereupon it is made acid with 20 ml of 3N
hydrochloric acid and extracted with methylene chloride.
The organic phase is washed with water, dried over sodium
sulphate and evaporated in a water-jet vacuum. After
crystallization from methylene chloride/n-hexane there is
obtained 3',4~-dihydroxy-2-methyl-S'-nitropropiophenone in
the form of yellow crystals of m.p. 98-9~.
ExamPle 7
.
a) 100 g (805.5 mmol) of guaiacol are dissolved in
136.4 g (86.2 mmol) of butyric anhydride, created with
120 g (880 mmol) of zinc chloride, heated for 3 minutes as
given in Example 6.a and then worked-up as described
there. The crude product obtained after high vacuum
distillation is chromatographed with toluene on 600 g of
silica gel. ~fter recrystallization from ether/n-hexane
there is obtained 4'-hydroxy-3'-methoxy-butyrophenone in
the form of colourless crystals of m.p. 40-41.
b) 6.5 ml of 11.2N nitric acid are added dropwise to a
solution of 12.7 g (65.4 mmol) of 4'-hydroxy-3`-methoxy-
butyrophenone in 250 ml of glacial acetic acid while
stirring within 10 minutes. The reaction mixture is
subsequently stirred for 15 minutes, poured into ice-
-wa~er, the separated precipitate is filtered of~ under
suction, washed with ice-watec and taken up in methylene
chloride. The methylene chloride solution is filtere~ over
~3~
- 35 -
50 g of silica gel. The mateLial obtained is recrystall-
ized from methylene chloride/n-hexane. There is obtained
4'-hydroxy-3l-methoxy-Sl-nitrobutyrophenone in the form of
yellow crystals of m.p~ g2-93~.
c) 10.2 g (42.6 mmol) of 4~-hydroxy-3l-methoxy-5l-nitro-
butyrophenone are treated with 80 g of pyridine hydro-
chloride and stirred at 200 for 40 minutes. After cooling
the reaction mixture is poured into 500 ml of ice-water.
The mixture is treated with 30 ml o~ 3N hydrochloric acid
and extracted with methylene chloride. The organic phase
is dried over sodium sulphate and evaporated. The crude
product obtained is chromatographed with methylene
chloride on 150 g of silica gel. The material obtained is
recrystallized from methylene chloride/n-hexane. There is
obtained 3',4'-dihydroxy-5'-nitrobutyrophenone in the form
of yellow crystals Oe m.e. 88-90.
ExamPle 8
a~ 2.25 g (10 mmol) of 3,4-dihydroxy-5-nitrocinnamic acid
are dissolved in 50 ml of methanol and hydrochloric acid
gas is introduced into this solution for 10 minutes. ~fter
1 hour 50 ml of isopropyl ether are added thersto, and the
separated precipitate is filtered off under suction and
washed with isopro~yl ether. ~fter recrystallization from
methanol/ether there is obtained methyl 3,4-dihydroxy-5-
-nitrocinnamate in the form of yellow crystals of m.p.
186-187.
b) In an analogous manner, from 3,4-dihydroxy-5-nitro-
cinnamic acid and butanolic hydrochloric acid solution
there is obtained n-butyl 3,4-dihydroxy-5-nitrocinnamate
in the form of yellowish crystals of m.p. 129-130.
'~3~
- 36 -
Exam~le 9
5.0 g (13.5 mmol) of diethyl 3,4-dimethoxy-S-nitxo-
benzoyl malonate are dissolved in 50 ml of absolute
methylene chloride. ~fter cooling to -20 a solution of
16.9 g (67.5 mmol) of boron tribromide in 30 ml of
methylene chlo~ide is added dropwise thereto while
stirring so that the temperature does not exceed -20. The
mix~ure is subsequently stirred at roorn temperature
overnight. After adding 80 ml of ethanol the mixture is
stirred at room temperature for 30 minutes and the solvent
is subsequently distilled off in a water-jet vacuum. The
residue is treated with water and extracted with methylene
chloride. The organi~ ~hase is washed with water, dried
over sodium sulphate and evaporated. The crude product
obtained is filtered with ethyl acetate over 50 g of
silica gel. The crystalline residue obtained is recryst~
allized from methylene chloride/n-hexane. There is
obtained ethyl 3,4-dihydroxy-5-nitro-benzoylacetate in the
form of yellow crystals of m.p. 141-142.
Example 10
a) ~ solution of 1.49 g (12.3 mmol~ of 2-phenylethylamine
in 100 ml of methylene chloride is treated with 1.26 g of
triethylamine. ~ solution of 3.0 q o~ 3,4-dimethoxy-S-
-nitrobenzoyl chloride in 100 ml of methylene chloride is
added dropwise thereto while stirring, whereupon the
mixture is stirred for a further 15 minutes. The organic
phase is then extracted twice wi~h 50 ml of ice-water each
time, dried over sodium sulphate and evaporated in a
water-jet vacuum. Ater recrystallization from methylene
chloride/n-hexane there is obtained 3,4-dimethoxy-5-nitro-
-N-phenethylbenzamide in the form of pale beige needles of
m.p. 121-122.
~3~ 3
b) 3.6 g (10.9 mmol) of 3,~-dimethoxy-5-ni~ro-N-
-phene~hylbenzamide are heated under reflux with 36 ml of
phosphorus oxychloride under a nitrogen atmosphere for 96
hours. After dis~illing off the excess phosphorus
oxychloride in a water-jet vacuum at 60 ~he residue is
treated three times with 100 ml of toluene each time, with
the solvent being distilled off each time. The residue is
taken up in methylene chloride. The organic phase is
washed with water, dried over sodium sulphate and
evaporated in a water-jet vacuum. The red resin obtained
i5 chromatographed on 120 g of silica gel with methylene
chloride/ethyl acetate (1:1). There is obtained 1-(3,4-
-dimethoxy-5-nitrophenyl)-3,4-dihydroisoquinoline in the
form of a yellow resin.
c) 1.4 g (4.5 mmol) of 1-(3,~-dimethoxy-5-nitrophenyl)-
-3,4-dihydroisoquinoline are treated with 15 ml o
constant-boiling hydrobromic acid and heated to boiling
under reflux under a nitrogen atmosphere for 1.5 hours.
After distilling off the hydrobromic acid in a water-jet
vacuum Che crystalline residue is recrystallized from
acetone. There is obtained 5-(3,4-dihydro-1-
-isoquinolinyl)-3-nitropycocatechol hydrobromide in the
form of yellow crystals of m.p. >250 (decomposition).
ExamPle 11
a) 5.0 g (27.7 mmol) of 4-hydroxy-5-methoxy-isophthal-
aldehyde are treated with 50 ml of constant-boiling
hydrobromic acid and heated to boiling under reflux and
while stirring under an argon atmosphere for 3 hours.
~fter cooling 50 ml o ice-water are added theeeto, and
the separated precipitate is ~iltered off under suction
and washed with water. The crude product is taken up in
ethyl acetate and filtered over 50 g of aluminium oxide
(activity grade 11). The cryscalline material obtained is
~3g~Z4~
- 38 -
recrystallized from ethyl acetate/n-hexane. There is
obtained 4,5-dihydroxyisophthalaldehyde in the form of
slightly orange crystals of m.p. 201-20Z.
s
b) A solution of 3.27 g (2~.9 mmol) of hydroxylamine o-
-sulphonic acid in 12 ml of water is added dcopwise at 30
while stirring to a solution of 2.0 g (12.0 mmol) of
4,5-dihydroxyisophthalaldehyde in 20 ml of water,
whereupon the mixture is held at 65 for 10 hours. After
cooling the sepacated precipitate is filtered off under
suction, washed with water and taken up in ethyl acetate.
The organic phase is washed with water, dried over sodium
sul~hate and eva~orated in a water-jet vacuum. ~fter
recrystallization from ethyl acetate there is obtained
4,S-dihydroxyisophthalonitrile in the form of yellow
crystals which decompose above 300~.
Example 12
a) ~ solution of 38 g of ~uming nitric acid (96%) in
S0 ml of glacial acetic acid is added dropwise while
stirrin~ and within 30 minutes at 20-25 to a solution of
112.5 g of 2-bromo-4'-hydroxy-3'-methoxyacetophenone in
560 ml of glacial acetic acid. Yellow-brown crystals
thereby separate. After 90 minutes the reaction mixture is
poured on to 300 g of ice. The crystals are filterad off
under suction, washed with 1000 ml of water and dissolved
in 1000 ml of methylene chloride. The organic phase is
washed with saturated sodium chloride solution, dried over
sodium sulphate, filtered and ehe filtrate is evaporated
at 50 in a water-jet vacuum until crystallization begins.
The crystallizate, cooled to room temperature, is filtered
off under suction and washed with a small amount o~
methylene chloride. There is obtained 2-bromo-4~-hydroxy-
-3'-metboxy-SI-nitroacetophenone of m.p. 14/-149.
~3~
- 39 -
b) Method A~
ba) A suspension of 580.1 mg of 2-bromo-4~-hydroxy-3~-
S -methoxy-5`-nitroacetophenone in 10 ml of ethanol is
treated with 443.8 mg of selenium dioxide and heated under
reflux for 71 hours. Thereafter, the selenium is filtered
off and the Eiltrate is evaporated. The residue is
dissolved in methylene chloride, washed with saturated
sodium chloride solution, dried over sodium sulehate,
filtered and evaporated. There is obtained eth~l
4-hydroxy-3-methoxy-5-nitrophenylglyoxylate of m.p.
165-167 (from ethanol).
In an analogous manner:
bb) From 2-bromo-~'-hydroxy-3~-methoxy-5~-nitroaceto-
phenone and n-butanol there is obtained n-butyl ~-hydroxy-
-3-methoxy-S-nitro~henylglyoxylace of m.p. 105-107 (from
ethanol) and
bc) from 2-bromo-4l-hydroxy-3l-methoxy-5'-nitroaceto-
phenone and n-hexanol there is obtained hexyl 4-hydroxy-3-
-methoxy-S-nitrophenylglyoxylate of m.p. 103-105 (from
n-hexanol~petroleum ether).
c) Method B:
ca) ~ suspension of 29.01 g of 2-bromo-4'-hydroxy-3'-
-methoxy-5l-nitroacetophenone in 300 ml of tert.butanol is
treated with 27.74 g of selenium dioxide and heated to
boiling under re~lux for 1~3 hours. The hot reaction
mixture is suction filtered through a filter aid Oe
diatomaceous earth while rinsing with methylene chloride.
l'he filtrate is evaporated and the residue is suspended in
lS0 ml o~ hot methylene chloride. The crystalline
precipitate is filtered of~ under suction and washed with
- 40 -
a small amount of methylene chloride. There is obtained
4-hydroxy-3-methoxy-5-nitrophenylglyoxylic acid of m.p.
169-171.
s
2.42 g of 4-hydLoxy-3-methoxy-5-nitrophenylglyoxylic
acid are dissolved in 25 ml of dry N,N-dimethylformamide,
treated at room temperature with 50 mg of 4-dimethylamino-
pyridine and 920 mg of dry methanol, subsequently cooled
to 0 with an ice-bath and 27 g of N,N-dicyclohexyl-
carbodiimide are added thereto. After 10 minutes the
ice-bath is removed and the reaction mixture is stirred
for a further one hour at room temperature. The mixture is
subsequently evaporated. The residue is dissolved in ethyl
acetate, whereupon insoluble urea is filtered off, the
filtrate is washed four times with water, dried over
sodium sulphate, filtered and evaporated. There i6
obtained methyl 4-hydroxy-3-methoxy-5-nitrophenyl-
glyoxylate of m.p. 15S-157 (from methylene chloride/
ether).
In a~ analogous manner:
cb) E'rom 4-hydroxy-3-methoxy-5-nitrophenylglyoxylic acid
and ethanol there is obtained ethyl 4-hydroxy-3-methoxy-5-
-nitrophenylglyoxlate of m.p. 165-167 (from ethanol) and
cc) ~'rom 4-hydroxy-3-methoxy-5-nitrophenylglyoxylic acid
and isopropanol there is obtained i-propyl 4-hydroxy-3-
-methoxy-5-nitrophenylglyoxylate of m.p. 99-101 (from
isopropanol ) .
d) ~ suspension of 17.2 g of ethyl 4-hydroxy-3-methoxy-5-
-nitrophenylglyoxylate in 100 ml of dry acetonitrile and
100 ml of dry toluene is treated with 10.53 g of sodium
iodide and 11.9 g of silicon tetrachloride and heated
under re~lux for 47 hours. The reaction mixture is ~hen
~L3~
- 41 -
evaporated and the residue is distilled six times with
200 ml of toluene each time. The residue obtained is
par~itioned between water and ether and filtered through a
filter aid of diatomaceous earth. The ethereal phase is
washed with four times with saturated sodium chloride
solution, dried over sodium sulphate, filtered and
evaporated. The oily residue is treated three times with
ether and active carbon. ~here is obtained ethyl
3,4-dihydroxy-5-nitrophenylglyoxylate of m.p. 77-79O (from
ether/n-hexane).
In an analogous manner:
e) From methyl 4-hydroxy-3-methoxy-5-nitrophenyl-
glyoxylate there is obtained methyl 3,4-dihydroxy-5-nitro-
phenylglyoxylate as a yellow distillate at 145-150 and
10.67 Pa,
f) ~rom isopropyl 4-hydroxy-3-methoxy-5-nitrophenyl-
glyoxylate there is obtained isopropyl 3,4-dihydroxy-5-
-~itrophenylglyoxylate as a yellow distillate at 155-160
and 12.0 Pa,
g) from n-butyl 4-hydroxy-3-methoxy-5-nitroehenyl-
glyoxylate there is obtained n-butyl 3,4-dihydroxy-5-
-nitrophenylglyoxylate as a yellow distillate at 160-165
and 10.67 Pa and
h) from n-hexyl 4-hydroxy-3-methoxy-5-nitrophenyl-
glyoxylate there is obtained hexyl 3,4-dihydroxy-5-nitro-
phenylglyoxylate as a yellow distillate at 165-170 and
12.0 Pa.
Example 13
236.1 mg of n-hexyl 3,4-dihydroxy-nitrophenyl-
~3~2~
- - 42 -
glyoxylate are dissolved in L5 ml of ethanol and treated
with 7.59 ml of O.lN sodium hydroxide solution. ~fter one
hour the reddish-yellow solution is evaporated. The
resulting sodium salt of the n-hexyl 3 t ~-dihydroxy-5-
-nitrophenylglyoxylate is crystallized from water and has
a m.p. of ~ 3000.
~xample 14
A solution of 3.18 g of n-hexyl 3,4-dihydroxy-5-nitro-
phenylglyoxylate in 47.5 ml o~ ethanol is trea~ed with
1.11 g of 0-methylhydroxylamine hydrochloride, 1.95 g of
sodium acetate and 2.5 ml of water and heated to boiling
under reflux for 5 hours. ~herea~ter, the reaction mixture
is evaporated and the residue is treated with ether and
water. The organic phase is washed with saturated sodium
chloride solution, dried over sodium sulphate, filteLed
and evaporated. The residue is chromatographed on silica
gel with methylene chloride. There is obtained a 7:3
mixture of n-hexyl E- and Z-3,4-dihydroxy-5-nitrophenyl-
glyoxylate 0-methyl oxime as a reddish oil; 80 M~l~ NMR
spectrum (C~C13): signal for 0-methyl at 3.96 and
4.05 ppm.
ExamPle 15
A solution of 5.1 g of ethyl 3,4-dihydroxy-5-nitro-
phenylglyoxylate in dry methylene chloride is treated
dropwise at -10 within 15 minutes with 2S g of boron
tribromide. The mixture is then stirred at -10 for one
hour and subsequently at room temperature for 17 hours.
Thereafter, the reaction mixture is evaporated, the
residue is treated cautiously with water and stirred at
50 for a further 30 minutes. After cooling to room
temperature the flocculent precipitate is filtered off
under suction. The aqueous phase is acidified with 10 ml
~3~2~
- 43 -
of lN hydrochloric acid, extracted four times with e~her,
the combined organic extracts are washed four times with
saturated sodium chloride solution, dried over sodium
sulphate, filtered and evaporated. The crude product is
filtered three times in succession in e~her through a
filter aid of diatomaceous earth. There is obtained
3,4-dihydroxy-5-nitrophenylglyoxylic acid of m.p. l72-174
(from isopropyl ether~.0
xample 16
a) A mixture of 3.93 g of n-hexyl 3,4-dihydroxy 5-nitro-
phenylglyoxylate and I.38 g of 2-aminophenol is melted at
120 while stirring. The melt crystallizes after 5
minutes. ~fter Z hours it i6 cooled and recrystallized
~rom me~hanol. There is obtained 3-(3,4-dihydroxy-5-nitro-
phenyl)-2H-l,4-benzoxazin-2-one of m.p. 202-204.
In an analogous manner:
b) From n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
2-amino-p-cresol there is obtained 3-(3,4-dihydroxy-5-
-nitrophenyl)-6-methyl-2H-1,4-benzoxazin-2-one of m.p.
233-235 (from methanol/methylene chloride),
c) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
2 amino-4-propylphenol there is obtained 3-(3,4-dihydroxy-
-5-nitrophenyl)-6-propyl-2~1-l,4-benzoxazin-2-one of m.p.
200-202 (from methanol),
d) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
3-amino-4-hydroxybenæoic acid there is obtained 3-(3,4-
-dihydroxy-S-nitrophenyl)-2-oxo-2H-l,4-benzoxazine-~-
-carboxylic acid of m.p. 286-~87 (from acetone/petroleum
ether),
~3~
- 44 -
e) from n-hexyl 3,4-dihydroxy 5-nitrophenylglyoxylata and
2-amino-4-chlorophenol there is obtained 6-chloro-3-
-(3,4-dihydroxy-5-nitrophenyl)-2H-1,4-benzoxazin-2-one of
m.p. 241-243 (from methanol),
f) Erom n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
2-amino-4,6-dichlorophenol there is obt:ained 6,8-dichloro-
-3-(3,4-dihydroxy-5-nitrophenyl)-2H-l,~-benzoxazin-2-one
of m.p. 237-239 (from ethanol/ether) and
g) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
2-amino-5-nitrophenol there is obtained 3-(3,4-dihydroxy-
-5-nitrophenyl)-7-nitro-2H-1,4-benzoxazin-2-one of m.p.
253-255 (from acetonitrile/ethanol).
Exam~le 17
a) A mixture of 396.0 mg of n-hexyl 3,4-dihydroxy-5-
-nitrophenylglyoxylate and 137.6 mg of l,2-phenylene-
diamine is heated to 12~ for 60 minutes. Thereafter, the
mixture is suspended in methanol, filtered off under
suction and recrystallized from N,N-dimethylforinamide/
water. There is obtained 3-(3,4-dihydroxy-5-nitrophenyl)-
-2(1H)-quinoxalinone of m.p. >300.
In an analogous manner:
b) From n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
N-methyl-1,2-phenylene diamine there is obtained l-methyl-
-3-(3,4-dihydroxy-5-nitrophenyl)-2(1H)-quinoxalinone of
m.p. 271-273 (from methanol),
c) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
N-propyl-1,2-phenylene diamine there is obtained l-propyl-
-3-(3,4-dihydroxy-5-nitrophenyl)-2(1~)-quinoxalinone of
m.p. 183-lB5 (from methanol),
~ J~
- 45 -
d~ from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
4,5-dimethyl-1,2-phenylenediamine there is obtained
3-(3,4-dihydroxy-5-nitrophenyl)-6,7-dimethyl-2(1}1)-
-quinoxalinone of m.p. >300 (feom N,N-dimethylformamide/
water),
e) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxyla~e and
4,5-dichloro-1,2-phenylenediamine there is obtained
6,7-dichloro-3-(3,4 dihydroxy-5-nitrophenyl)-2(lH)-
-quinoxalinone of m.p. >300 ~from N,N-dimethylformamide/
water),
f) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
3-chloro-5-trifluoromethyl-1,2-phenylenediamine there is
obtained a 1:1 mixture of ~(and 5)-chloro-3-(3,4-
-dihydroxy-5-nitrophenyl)-6 (and ~)-tri~luoromethyl-2(1H)-
-quinoxalinone of m.p. >300 (from N,N-dimethylformamide/
wate r ) , .
g) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
4-methoxy-1,2-phenylenediamine there is obtained a 1:1
mixture of 3-(3,4-dihydroxy-5-nitrophenyl)-6(~nd
7)-methoxy-2(1H)-quinoxalinone of m.p. >300 (from
ethanol/ether),
h) from n-hexyl 3,4-dihydroxy-5-nitrophenylglyoxylate and
4-nitro-1,2-phenylenediamine there is obtained 1:1 mixture
of 3-(3,4-dihydroxy-5-nitrophenyl)-6(and 7)-nitro-2(1H)-
-quinoxalinone of m.p. >300 (from N,N-dimethylfoemamide/
water) and
i) from n-hexyl 3,~-dihydroxy-5-nitrophenylglyoxylate and
N-hexyl-1,2-phenylenediamine ~here is obtained
l-hexyl-3-(3,4-dihydroxy-5-nitrophenyl)-2(lH)-quinoxalinone
of m.p. 152-154 (from methanol~.
~3V;~
- 46 -
Example 18
A solution of 1.07 g of n-hexyl 3,4-dihydroxy-5-nitro-
phenylglyoxylate and 696.3 mg of 2,3-diaminonaphthalene in
3 ml of l-hexanol is heated ~o boilinq under reflux for 3
hours. The reaction mixture is then cooled and diluted
with methanol. The crude product is filtered off under
suction and recrystallized from N,N-dimethylformamide/
water. There is obtained 3-(3,4-dihydroxy-5-nitrophenyl)-
benzo[g]quinoxalin-2(1H)-one of m.p. >300.
ExamPle 19
A suspension of 2.05 g of n-hexyl 3,4-dihydroxy-5-
-nitroehenylglyoxylate and 600.8 mg of thiosemicarba2ide
is stirred intensively at 90 for 30 minutes. The mixture
is then cooled to 40 and treated with a solution of
870.1 mg of sodium hydroxide in 15 ml of water. The
solution is subsequently heated to 90 for 30 minutes,
cooled to room temperature and treated dropwise with 2 ml
of conc. hydrochloric acid. The crystallized-out eroduct
is filtered off under suction and then dissolve~ in ethyl
acetate. The solution is washed with saturated sodium
chloride solution, dried over sodium sulphate, filtered
and evaporated. There is obtained
6-(3,4-dihydroxy-5-nitrophenyl)-3 -mercapto-1,2,4-triazin-
-5(4~1)-one of m.p. 282-284 (from ethanol).
~xample 20
aaa) A suspension of 2.9 g of 2-bromo-4'-hydroxy-3`-
-methoxy-5`-nitroacetophenone and 761.2 mg of thiourea in
170 ml of ethanol is treated at 60 with 820.3 mg of
sodium acetate and stirred for 6 hours. The reaction
mixture is then evaporated, the residue is treated with
170 ml of water and heated to 60 for 30 minutes. After
~3~Z~
- 47 _
cooling the product is filtered off under suction and
washed with water. There is obtained 4-(2-amino-4-
-thiazolyl)-2-methoxy-6-nitrophenol of m.p. 248-250 (from
ethanol).
In an analogous manner:
aab) From 2-bromo-4'-hydroxy-3'-methoxy-5'-nitroaceto-
phenone and ~-phenylthiourea there is obtained
4-(2-anilino-4-thiazolyl)-2-methoxy-6-nitrophenol of m.p.
185-187 (from ether).
aba) S0 ml of 1,2-dichloroethane and 3.56 g of calclum
carbonate are added to a solu~ion of 4.88 g of 6-amino-4'-
-(trifluoromethyl)-hexanilide hydrochloride in 70 ml Oe
water. The suspension is treated within 60 minutes while
stirring at room temperature with a solution of ~.6 g of
thiophosgene in 1.7 ml of toluene. ~fter 16 hours the
precipitate is filtered off under suction and the organic
phase is washed with lN hydrochloric acid and water. After
drying over sodium sulphate and filtration the filtrate is
evaporated. There is obtained crude 4~-(trifluoromethyl)-
hexananilide-6-isothiocyanate.
abb) A solution of 5.2 g of crude 4~-(trifluoro-
methyl)hexananilide-6-isothiocyanate in 60 ml o ethanol
is treated with 100 ml of conc. ammonia solution. ~fter 30
minutes the reaction mixture is evaporated. The residue is
dissolved in ethyl acetate, washed with water, dried over
sodium sul~hate, filtered and evaporated. There is
obtained 1-~5-[(a,a,-trifluoro-p-tolyl)caebamoyl~-
pentyl]-2-thiourea of m.p. 140-142 (~rom ethanol).
abc) R suspension of 666.7 mg of 1-~5-~a,a,a-
-trifluoro-p-tolyl)carbamoyl]pentyl]-2-thiourea and
580.2 mg of 2-bromo-4~-hydroxy-3~-methoxy-5~-nitroaceto-
3~3~
- 48 -
phenone in 50 ml of ethanol is treated at 60 with
164.2 mg of sodium acetate. A reddish-yellow solution
thereby results. After 90 minutes the reaction mixture is
evaporated, the residue is treated with water, the
precipitate is filtered off under suction and washed four
times with 10 ml of water each time. There is obtained
6-tt4-(4-hydroxy-3-methoxy-5-nitrophenyl)-2 -thiazôlyl]-
amino]-4~-(trifluoromethyl)hexananilide of m.p. 160-162
(from ethanol).
ac) A solution of 29.0 g of 2-bromo-4~-hydroxy-3'-methoxy-
-5'-nitroacetophenone and 13.5 g of 2-aminoacetophenone in
250 ml of dry N,N-dimethylformamide is stirred at 90 for
16 hours. The reaction mixture is then evaporated to about
two thirds and poured on to ice. The separated crystals
are filtered off under suction and dissolved in methylene
chloride. The solution is washed with saturated sodium
chloride solution, dried over sodium sulphate, filtered
and evaporated. There is obtained 2-(4-hydroxy-3-methoxy-
-5-rlitrobenzoyl)-3-methylindole of ra.p. 195-197 (from
methylene chloride/methanol).
ada) ~ suspension of 14.5 g of 2-bromo-4l-hydroxy-3'-
-methoxy-5'-nitroacetophenone and 5.41 g of 1,2-phenylene-
diamine in 350 ml of methanol is treated with 4.92 g of
sodium acetate and the mixture is heated to boiling under
reflux for 22 hours. The reaction mixtura is then evapor-
ated, the residue is dissolved in methylene chloride, this
solution is washed ~our times with water, dried over
sodium sulphate, filtered and evaporated. There is
o~tained 2-(4-hydroxy-3-methoxy-5-nitrophenyl)quinoxaline
of m.p. 195-197 (from methylene chloride/methanol).
In an analogous manner:
adb) ~`rom 2-bromo-4'-hydroxy-3'-methoxy-5'-nitroaceto-
~3~'~J2~
- 49 -
phenone and 4,5-dimethyl-1,2-phenylenediamine there is
obtained 6,7-dimethyl-Z-(4-hydroxy-3-methoxy-5 -nitro-
phenyl)quinoxaline of m.p. 207-20g (from methylene
chloride/methanol).
b) A suspension o~ 267.3 mg of 4-(2-amino-4-thiazolyl)-2-
-methoxy-6-nitrophenol in 10 ml of dry methylene chloride
is trea~ed at -20 with 1.25 g of boron tribromide. ~fter
the addition the mixture is stirred at -20 for a further
one hour and at room temperature without cooling for 18
hours. The reaction mixture is then evaporated, the
residue is treated cautiously with water and stirred at
50 for 30 minutes. ~fter cooling to room temperature the
mixture is suction filtered and the crude product is
filtered off and washed with a small amount of water.
There is obtained 5-(2-amino-4-thiazolyl)-3-nitropyro-
catechol hydrobromide oE m.p. 244-246 (Erom methanol).
In an analogous manner:
~) From 4-(2-anilino-4-thiazolyl)-2-methoxy-6-nitrophenol
there is obtained 5-(2-anilino-4-thiazolyl)-3-nitropyro-
catechol of m.p. 202-204 (from methanol),
d) from 6-[4-(4-hydroxy-3-methoxy-5-nitrophenyl)-2-
-thiazolyl)amino]-4'-(trifluoromethyl)hexananilide there
is obtained 6-[[4-(3,4-dihydroxy-5-nitrophenyl)-2-
-thiazolyl]amino]-4'-(trifluoromethyl)hexananilide of m.p.
214-216 (from methanol),
e) from 2-(4-hydroxy-3-methoxy-5-nitrobenzoyl)-3-
-methylindole there is obtained 5-bromo-2-(3,4-dihydroxy-
-5-nitrobenzoyl)-3-me~hylindole of m.p. 265-267 (from
3~ methanol),
~) from 2-(4-hydroxy-3-methoxy-5-nitrophenyl)quinoxaline
~3~æf~
-- so
there is obtained 2-(3,4-dihydroxy-5-nitrophenyl)-
quinoxaline of m.p. 241-243 (from methanol) and
g) from 6,7-dimethyl-2-(4-hydroxy-3-methoxy-5-nitro-
phenyl)quinoxaline there is obtained 6,7-dimethyl-2-(3,4-
-dihydroxy-5-nitrophenyl)quinoxaline of m.p. 274-276
(from methanol).
ExamPle 21
A mixture of 3.12 g of 2-bromo-3`,4l-dihydroxy-5'-
-nitroacetophenone and 849.3 mg of thioacetamide are
heated to boiling under reflux for 18 hours in 40 ml of
ethanol. After cooling the ccystals are ~iltered off under
suction and recrystallized from methanol/isopcopanol.
There is obtained ~-(2-methyl-4-thiazolyl)-3-nitropyro-
catechol hydrobromide of m.p. 280-2/32.
ExamPle-22
A solution of 1.38 g of 2-bromo-3~,4~-dihydroxy-5~-
-nitroacetophenone and 461 mg of thiosemicarbazide in
20 ml of n-butanol is heated to boiling under reflux for
60 minutes. After cooling to room temperature the crystals
are filtered off under suction and recrystallized from
n-butanol. There is obtained 5-(2-amino-6~-1,3,4-
-thiadiazin-5-yl)-3-nitropyrocatechol hydrobromide of m.p.
265-267~.
ExamplP 23
A solution o~ 1.38 g of 2-bromo-3l,4'-dihydLoxy-5'-
-nitroacetophenone and 505.7 mg of Z-amino-1,3,4-
-thiadiazole in 25 ml of n-butanol is heated to boiling
under re~lux for 7 hours. The reaction mixt-ure is then
cooled to room temperature and the separated crystals are
~3~Z~
- 51 -
filtered off under suction. There is obtained 5-(imidazo-
~2,1-b]-1,3,4-thiadiazol-6-yl)-3-nitropyrocatechol of m.p.
278-2800 (from methanol).
ExamPle 24
A mixture of 2.78 g of 2-bromo-3',4'-dihydroxy-5'-
-nitroacetophenone, l.ol g of 2-aminothiazole and 40 ml of
ethanol is heated to boiling under reflux for 23 hours.
The reaction mixture is then cooled to room tem~erature
and the crystals are filtered off under suction. There is
obtained 5-~imidazot2,1-b]thiazol-6-yl)-3-nitropy~o-
catechol hydrobromide of m.p. 286-288 (from methanol).
Example 25
A mixture of 1.95 g of Z-bromo-3',4'-dihydroxy-5'-
-nitroacetophenone, 1.06 g o~ 2-aminobenzothiazole and
50 ml of ethanol is heated to boiling under reflux for 17
hours. The rsaction mixture is then cooled to room
temperature, whereupon the ceystals are filtered off under
suction. There is obtained S-(imidazo[2,1-b]benzothiazol-
-2-yl)-3-nitropyrocatechol of m.p. 303-30~ (from
N,N-dimethylformamide/methanol).
ExamPle 26
~ mixture of 2.78 g o~ 2-bromo-3~,4'-dihydroxy~
-nitroacetophenone, 1.51 g of 2-aminothiophenol and 50 ml
of ethanol is heated to boiling under reflux for one hour.
The reaction mixture is then cooled to room temperature,
whereupon the crystals are filtered off under suction.
There is obtained 5-(2~1-1,4-benzothiazin-3-yl)-3-nitro-
pyrocatechol of m.p. 302-3040 ~from N,N-dimethyl-
formamidetmethanol).
~3~
A mixture of 987.5 mg of 2-bromo-3l,4~-dihydroxy-5~-
-nitroacetophenone and 1.01 g of 2-aminopyridine is melted
at 110. After 30 minutes the melt is t:reated with 15 ml
of ethanol and heated to boiling under reflux ~or 3 hours.
The reaction mixture is then cooled to room temperature
and the crystals are filtered off under suc~ion. There is
obtained 5-(imida20~1,2-a]eyridin-3-yl)-3-nitropyro-
catechol of m.p. 250-252 (from N,N-dimethylformamide/-
methanol).
ExamP~~ 28
a) 8.9 g of l,l'-carbonyldiimidazole are added to a
solution o~ 10.7 g of q-hydroxy-3-methoxy-5-nitrobenzoic
acid in 500 ml o dry tetrahydrofuran and the reaction
mixture is subsequently heated to 65-70 for 5 hours. It
is then cooled to room temperature and a solution of
21.6 g of 6-aminohexyl-t-butylcarbamate in 50 ml of dry
tetrahydrofuran is added dropwise thereto within 15
minutes. The reaction mixture is ~hen heated to 65-70 for
18 hours, evaporated, the residue is suspended in ethyl
acetate, suction filtered and the suction ~iltered
material is chromatographed on silica gel with
aceto~e/methylene chloride (3:1). There is obtained
~6-t4-hydroxy-5-nitro-m-anisamido)hexyl]-t-butylcarbamate
of m.p. 145-147 (from isopropanol).
b) 5.8 ml of hydrobromic acid in glacial acetic acid
(~33 percent) are added at room temperature to a
solution of 4.1 g of ~6-~4-hydroxy-5-nitro-m-anisamido)-
hexyl]-t-butylcarbamate in 80 ml of glacial acetic acid.
~`he mixture is stirred for 2 hours, the separated crystals
are filtered off under suction and washed WiCh ether.
l`here is obtained N-(6-aminohexyl)-4-hydroxy-S-nitro-m- -
~3~
- 53 -
-anisamide hydrobromide of m.p. 207-209~ (from
isopropanol).
c) 1.25 ~ of boron teibromide are added -20 to a
suspension of 392.3 mg of N-(6-aminohexyl)-4-hydroxy-5-
-ni~ro-m-anisamide hydrobromide in 25 ml of dry methylene
chloride. After the addition the mixture i~ stirred at
20 ~or one hour and subsequently at room temperature for
17 hours. The reaction mixture is then evaporated, the
residue is treated with 10 ml of water and stirred at room
temperature ~or one hour. After evaporating the water the
residue is chromatographed on Sephadex LH 20 with
toluene/ethanol (1:1). There is obtained N-6-aminohexyl-
-3,4-dihydrox~-5-nitrobenzamide hydrobromide of m.p.
205-207 (feom ethanol).
aa) ~ solution of 4.93 g of 5-nitrovanillin and 2.7 g of
1,2-phenylenediamine in 45 ml o~ methanol and 15 ml of
nitrobenzene is heated to boiling under reflux. After 15
minutes crystals begin to separate from the red solution.
After 18 hours the reaction mixture is cooled to room
temperature and dilu~ed with 60 ml of methanol. The
crystals are filtered off under suction and washed with
methanol. There i6 obtained 4-(2-benzimidazolyl)-2-
methoxy-6-ni~rophenol of m.p. 198-200 (from
N,N-di~ethylformamide/methanol).
ln an analogous manner:
ab) From 5-nitrovanillin and 4,5-dichloro-1,2-phenylene-
diamine there is obtained 4-(5,6-dichloro-2-benz-
imidazolyl)-2-methoxy-6-nitrophenol of m.p. 258-260 ~from
N,N-dimethylformamide/ether).
*Trade Mark
~L3~Z~
- 54 -
b) A suspension of 860.1 mg of 4-t2-benzimidazolyl)-2-
-methoxy-6-nitrophenol in 10 ml of glacial acetic acid and
10 ml of 48 percent hydrobromic acid is heated to boiling
under reflux for 72 hours. The mixture is then evaporated
and the residue is treated four times with 50 ml of
toluene each time, which is again distilled off each time.
There is obtained 5-(2-benzimidazolyl)-3-nitropyrocatechol
of m.p. >300 (from acetone/water).
In an analogous manner:
c~ From 4-(5~6-dichloro-2-benzimidazolyl)-2-methoxy-6
-nitrophenol there is obtained 5-(5,6-dichloro-2-
-benzimidazolyl)-3-nitropyrocatechol of m.p. 282-284
(from acetone/water).
Example 30
A suspension of 29.0 g of 2-bromo-4'-hydroxy-3'-
-methoxy-5~-nitroacetophenone in 700 ml of dry methylene
chloride is treated at -20 within 30 minutes with a
solution of 125.3 g of boron tribromide in 300 ml of dry
methylene chloride. ~fter the addition the mixture is
stirred at -20 for a further one hour and at room
temperature for 16 hours, then evaporated, the residue is
treated cautiously with water while cooling with ice and
stirred at 50 for 30 minutes. After cooling the mixture
is extracted with ether, the ethereal phase is washed with
water, dried over sodium sulphate, filtered and evapor-
ated. There is obtained 2-bromo-3`,4'-dihydroxy-5'-nitro-
acetophenone of m.p. 138-140 (from methylene chloride).
Exam~le 31
a) A solution of 3.6 g of ethyl 3,4-dihydroxy-5-nitro-
phenylglyoxylate in 30 ml of acetic anhydride is hea~ed at
~3~
- 55 -
110 for 30 minutes in the presence of a catalytic amount
of conc. sulphuric acid, cooled to room temperature, the
reaction mixture is poured into 150 ml of water and
stirred for 60 minutes. The mixture is extracted with
ether, washed with saturated sodium ch:Loride solution, the
combined organic ex~racts are dried sodium sulphate,
filtered and evaporated. ~here is obtained ethyl
3,4-diacetoxy-5-nitrophenylglyoxylate of m.p. 87-89 (from
ether/petroleum ether).
In an analogous manner:
b) From 3-~3,4-dihydroxy-5-nitrophenyl)-2~ 1,4-
-benzoxazin-2-one there is obtained 3-(3,4-diacetoxy-5-
-nitrophenyl)-2~1-1,4-benzoxazin-2-one of m.p. 186-188
(from methylene chloride/methanol),
c) from 3-(3,4-dihydroxy-5-niCrophenyl)-2(lH)-
-quinoxalinone there is obtained 3-(3,4-diacetoxy-5-nitro-
phenyl)-2(1H)-quinoxalinone of m.p. Z41-243 (from
methylene chloride/methanol),
d) from 3,4-dihydroxy-5-ni~robenzophenone there is
obtained 3,4-diacetoxy-5-nitrobenzophenone of m.p.
141-143 (from methylene chloride/ether),
e) from 2~-fluoro-3,4-dihydroxy-5-nitrobenzophenone there
is obtained 3,4-diacetoxy-2~-fluoro-5-nitrobenzophenone of
m.p. 122-124 (from ether) and
f~ from 3,4-dihydroxy-5-nitrophenyl 4-eyridyl ketone
there is obtained 3,4-diacetoxy-s-nitrophenyl 4-pyridyl
ketone of m.p. 14~-150 (from methylene chloride/ether).
- 56 -
Example 32
a) A soluCion of 2.0 g of 3,5-dinitropyrocatechol in
25 ml of propionic anhydride is heated at 110 for 18
hours in the presence of a catalytic amount of conc.
sulphuric acid, the excess anhydride is distilled off at
70 in a high vacuum ~1.33 Pa), the residue is dissolved
in methylene chloride, washed with water, dried over
sodium sulphate, filtered and evaporated. There is
obtained l,2-dipropionyloxy-3,5-dinitrobenzene of m.p.
74-76 (from methylene chloride/petroleum ether).
In an analogous manner:
b) ~rom 3-(3,4-dihydroxy-S-nitrophenyl)-6-methyl-2H-1,4-
-benzoxazin--2-one there is obtalned 3-(3,4-dipropionyloxy-
-5-nitrophenyl)-6-methyl-2H-1,4-benzoxazin-2-one of m.p.
158-160 (from methylene chloride),
c) from 6-chloro-3-(3,4-dihydeoxy-5-nitrophenyl)-2H-1,4-
-benzoxazin-2-one there is obtained 5-(6-chloro-2-oxo-2~1-
-1,4-benzoxazin-3-yl)-3-nitro-o-phenylene-dipropionate o~
m.p. 148-150 (from methylene chloride/ether),
d) from 3-(3,4-dihydroxy-5-nitrophenyl)-7-nitro-ZH-1,4-
-benzoxazin-2-one there is obtained 3-nitro-5-(7-nitro-2-
-oxo-2H-1,4-ben20xazin-3-yl)-o-phenylene-dipropionate of
m.p. 177-179 (from methanol),
e) ~rom 3-(3,4-dihydroxy-5-nitrophenyl)-2--oxo-2H-1,4-
-benzoxazine-6-carboxylic acid there is obtained 3-~3,4-
-bis(propionyloxy)--5-nitrophenyl]-2-oxo-2H-1,4-benzoxazine-
-6-carboxylic acid of m.p. 192-194 (from methylene
chloride),
f) from 3-nitro-5-(2-quinoxaiinyl)pyrocatechol there is
- 57 -
obtained 3-nitro-5-(2-quinoxalinyl)-o-phenylene-
-dip~opionate of m.p. 152-154 (from methylene chloride/-
ether,
g) from 5-(2-benzimidazolyl)-3-nitropyrocatechol there is
obtained S-t2-ben2imidaæolyl)-3-nitro-o-2henylene-
-dipropionate of m.p. 179-181 tfrom ether),
h) from 6,7-dichloro-3-~3,4-dihydroxy-5-nitrophenyl)-
-2(lH)-quinoxalinone there is obtained 5-(6,7-dichloro-
-3,4-dihydro-3-oxo-2-quinoxalinyl)-3-nitro-o-phenylene-
-dipropionate of m.p. 260-~62 ~from methylene chloride),
i) from S-bromo-2-(3,4-dihydroxy-5-nitrobenzoyl)-3-
-methylindole there is obtained 5-bromo-2-(3,~-
-dipropionyloxy-5-nitrobenzoyl)-3-methylindole of m.p.
196-198 (from ether) and
j) from 6-(3,4-dihydroxy-5-nitrophenyl)-3-mercapto-1,~,4-
-~riazin-5~4H)-one there is obtained 3-ni~ro-5-~2,3,4,5-
-tetrahydro-5-oxo-3-thioxo-as-triazin-6-yl)-o-phenylene-
-dipropionate of m.p. 237-239~ tfrom ether).
Example 33
a) ~ solution of 1.09 g of ethyl 3,4-dihydroxy-5-nitro-
ehenylglyoxylate in 6 ml of isobutyric anhydride is heated
at 110 for 17 hours in the presence of a catalytic amount
o~ conc. sulphuric acid. The reaction mixture is then
treated ten times with 10 ml of toluene each ti~e, whereby
it is evaporated each time at 80 and 18.7 mbar. The oily
residue is distilled in a bulb-tube at l75-lao~ and
8.0 Pa. There is obtained ethyl 3,4-diisobutyryloxy-5-
-nitrophenylglyoxylate.
ln an analogous manner:
~31~
- 58 -
b) From 3,5-dinitropyrocatechol there is obtained 1,2-
-diisobutyryloxy-3,5-dinitrobenzene of m.p. 7)3-80 (from
ether)~
s
c) feom 3-(3,~-dihydroxy-5-nitrophenyl)-6-methyl-2~-1,4-
-benzoxazin-2-one there is obtained 3-(3,4-diisobutyryl-
oxy-5-nitrophenyl)-6-methyl-2H-1,4-benzoxazin-2-one of
m.p. ~2-144 (from ether),
d) from 2-(3,4-dihydroxy-5-nitrophenyl)quinoxaline there
is obtained 2-(3,4-diisobutyryloxy-5-nitrophenyl)-
quinoxaline of m.p. 155-157 (from ether),
e) from 1-methyl-3-(3,4-dihydroxy-S-nitrophenyl)-2(1H)-
-quinoxalinone there is obtained l-methyl-3-(3,4-
-diisobutyryloxy--5-nitrophenyl)-2(1~1)-quinoxalinone of
m.p. 138-140 (from ether),
f) from 5-(imidazo~2,1-b]-1,3,4-thiadiazol-6-yl)-3-nitro-
pyrocatechol there is obtained 5-(imidazo[Z,l-b]-1,3,4-
-thiadiazol-6-yl)-3-nitro-o-phenylene diisobutyrate of
m.p. 169-171 (from methylene chloride),
g) from 2-bromo-3',4'-dihydroxy-5'-nitroacetophenone
there is obtained 5-(bromoacetyl)-3-nitro-o-phenylene
diisobutyrate of m.p. 56-58 (from methylene chloride/
hexane),
h) from 5-(2-amino-4-thiazolyl)-3-nitropyrocatechol
hydeobromide theLe is obtained 3-nitro-5-(Z-isobutyramido-
-4-thiazolyl)-o-phenylene diisobu~yrate of m.p. 157-159
(from methylene chloride/ether),
i) from 5-(2-amino-6~1-1,3,4-thiadiazin-5-yl)-3-nitro-
pyrocatechol hydrobromide theee is obtained 3-nitro-5-(2-
-isobutyramido-4-isobutyryl-1j3,4-thiadiazin-5-yl)-o-
~3~
- 59 -
-phenylene diisobutyrate of m.e. 177-179~ (from ether),
j) from 3-(3,4-dihydroxy-5-nitrophenyl)-6--propyl-2H-1,4-
-ben70xazin-2-one there is obtained 3-nitro-5--~2-oxo-6-
-propyl-2H-1,4-benzoxazin-3-yl)-o-phenylene diisobutyrate
of m.p. 131-133 (from methanol),
k) from S-(imidazotl,2-a3pyridin-3-yl)-3-nitropyro-
catechol there is obtained 5-timidazo[l,2-a]pyridin-3-yl)-
-3-nitro-o-phenylene diisobutyrate of m.p. 137-139 (~rom
ether),
1) from 5-(imidazo~2,1-b]benzothiazol-2-yl)-3-nitropyro-
catechol there is obtained 5-(imidazot2,1-b]ben20thiazol-
-2-yl)-3-nitro-o--phenylene diisobutyrate of m.p. 197-lg9
(from methylene chloride/ether and
m) from 3,4~dihydroxy-5-nitrophenyl 2-pyridyl ketone
hydrobromide there is obtained 3-nitro-5-(2-pyridyl-
carbonyl)-o-phenylene diisobutyrate of m.p. 83-85 (fro~
ether/hexane).
xamPle 34
a) A solution o~ 613.0 mg of ethyl 3,4-dihydroxy-5-nitro-
phenylglyoxylate in 4 ml of pivaloyl anhydride is heated
to 100 for 17 hours in the presence of a catalytic a~ount
of conc. sulphuric acid, the cooled solution i5 diluted
with ether, washed with saturated sodium solution, dried
over sodium sulphate, filtered and evaporated. The residue
is treated ten times with 10 ml of toluene, whereby it is
evaporated again each time. ~ter bulb-tube distillation
(air-bath) at 175-180 and 4.0 Pa there is obtained ethyl
3,4-dipivaloxyloxy-5-nitrophenylglyoxylate.
In an analogous manner:
~3~ ~21'~
- 60 -
b) E~rom 3-(3,~-dihydroxy-5-nitrophenyl)-6-methyl-2~-1,4-
-benzoxa~in-2-one there is obtained 3-~3,4-dipivaloyloxy-
-5-nitrophenyl)-6-methyl-2H-1,4-benzoxazin-2-one of m.p.
180-192 (from ether),
c) from 3,4-dihydroxy-5-nitrobenzophenone there is
obtained 3,4-dieivaloyloxy-5-nitrobenzophenone o~ m.p.
101-103 (from t-butyl methyl ether) and
d) from Z'-fluoro-3,4-dihydroxy-5-nitrobenzophenone there
is obtained 3,4-dipivaloyloxy-2~-fluorobenzoehenone of
m.p. 74-76 (from low-boiling petroleum ether).
Example 35
A solution of 1.7 g of 3-(3,4-dihydroxy-~-nitro-
phenyl)-2(1H)-quinoxalinone in 17 ml of oenanthic
anhydride is heated to 110 for 17 hours in the presence
of a catalytic amounC of conc. sulphuric acid, the excess
anhydride is then distilled off in a high vacuum, the
residue is dissolved in methylene chloride, the organic
solution is washed with water, dried over sodium sulphate,
filtered and evaporated. There is obtained 5-(1,2-dihydro-
-2-oxo-3-quinoxalinyl)-3-nitro-o-phenylene diheptanoate of
m.p. 186-188 (from methylene chlorideJether).
ExamEle 36
a) 1.26 g of sodium acetate are added to a solution of
1.35 g of 2-bromo-3`,~'-dihydroxy-5'-nitroacetophenone in
~5 ml of alcohol and heated to ~oiling under reflux. After
6 hours the reaction mixture is filtered off from
separated sodium bromide and evaporated. The residue is
dissolved in ethyl aceta~e. The solution is washed with
saturated sodium chloride solution, dried over sodium
sulphate, filtered and evaporated at 50. There is
13~2~
obtained (3,4-dihydLoxy--5-niteobenzoyl)methyl acetate of
m.p. 166-168 (from ethyl acetate/ether).
In an analogous manner:
b) from 2-bromo-3',4l-dihydroxy-5l-ni~roacetophenone and
sodium isobutyrate there is obtained (~3,4-dihydroxy-5-
-nitrobenzoyl~methyl isobutyrate of m.p. 120-122 (from
ethyl acetate/ether) and
c) from Z-bromo-3',4'-dihydroxy-5~-nitroacetophenone and
sodium 3,4~dihydroxy-5-nitrophenylglyoxylate there is
obtained 3,4-dihydroxy-5-nitrophenacyl ~3,4-dihydroxy-5-
-nitrobenzoyl)formate of m.p. 224-226 (from methanol~
ethyl ace~ate).
Example 37
A suspension of 2.91 g of 2-bromo-3',4l-dihydroxy-5'-
-nitroacetophenone is treated with 1.31 g of thionicotin-
amide in 50 ml of alcohol and heated to boiling under
reflux for 2 hours. After cooling to room temperature the
crystals are filtered off under suction and recrystallized
from N,N-dimethylformamide/alcohol. There is obtained
3-nitro-5-[2-~3-pyridyl)-4-thiazolyl~pyrocatechol of m.p.
279-28lo .
Example 38
A solution of 5.52 g of 2-bromo-3',4'-dihydroxy-5'-
-nitroacetophenone and 2.7 g of 2-aminoacetophenone in
100 ~1 of dry N,N-dimethylformamide is stirred at 90 for
24 hours. The reaction mixture is evaporated, the residue
is dissolved in ethyl acetate, washed with water, dried
over sodium sulphate, filtered and evaporated. There is
obtained Z-(3,4-dihydroxy-5-nitrobenzoyl)-3-methylindole
_ 6Z -
of m.p. 212-214 (from n-butanol).
ExamPle 39
A suspension of 7.32 g of 2-bromo-3~,4~-dihydroxy-5~-
-nitroacetophenone is treated with 4.06 g of 1-~3-
-pyridinyl)-2-thiourea in 100 ml of n-butanol and heated
to boiling under reflux for 3 hours. ~fter cooling to room
temperature the crystals are filtered off under suction
and recrystallized from n-butanol. There is obtained
3-nitro-5-[2-(3-pyridylamino)-~-thiazolyl]pyrocatechol
hydrobromide of m.p. ~300.
Example 40
A suspension of 6.35 g of 2-bromo-3~,4'-dihydroxy-5l-
-nitroacetoehenone is treated with 4.68 g of 1-(3-
-guinolinyl)-2-thiourea in 150 ml of n-butanol and heated
to boiling under reflux for 3 hours. After cooling to room
temperature the crystals are filtered off under suction
and recrystallized from n-butanol. There is obtained
3-nitro-5-~2-(3-quinolinylamino)-4-thiazolyl]pyrocatechol
hydrobromide of m.p. >300.
ExamPle 41
A suspension of 8.28 g of 2-bromo-3~,4~-dihydroxy-5~-
-nitroacetophenone is treated with 6.37 g of rac-1-(2-exo-
-bornyl)-2-thiourea in 100 ml of n-butanol and heated to
boiling under reflux for 3 hours. ~fter cooling to room
temperature the crystals are filtered off under suction
and recrystallized ~rom n-butanol. There is obtained
rac-3-nitro-5-~2-(2-exo-bornylamino)-4-thiazolyl]-pyro-
catechol hydrob~omide of m.e. 262-264.
- 63 -
ExamPle 42
0.875 ml of pyrrolidine in 35 ml of tetrahydrofuran is
treated at 50 with 0.605 ml of acetic acid and
subsequently wi~h 1.73 g of 3,4-dihydroxy-5-nitrobenæ-
aldehyde and 2.87 g of 6-oxo-4~-(trifluoromethyl)-
~ heptananilide and stirred at 23 under argon for 56 hours.
The residue ob~ained after evaporati~g the reaction
mixture is partitioned between ethyl acetate and lN sodium
hydroxide solution. The combined sodium hydroxide extracts
are made acid with conc. hydrochloric acid, extracted with
e~hyl acetate, the combined ethyl acetate extracts are
washed with saturated sodium chloride solution, dried over
sodium sulphate, evaporated and ehe residue is chromato-
graphed on 120 g of silica gel with methylene chloride/
methanol (91:9). After recrystallization from ethyl
acetate/petroleum ether there is obtained (E)-8-(3,~-
-dihydroxy-5-nitrophenyl)-6-oxo-4'-(trifluoromethyl)-7-
-octenanilide of m.p. 194-197.
ExamPle 43
a) 26.0 g of 2-chloro-3-hydroxy-p-anisaldehyde are
dissolved in 400 ml of acetic anhydride and S ~1 of
pyridine. The solution is stirred at 80 for 8 hours,
subsequently evaporated, the residue is partitioned
between ice-water and methylene chloride, the organic
phase is dried over sodium sulphate, evapor- ated and the
residue is recrystalli~ed from methylene
chloeide/petroleum ether. There is obtained 2-chloro-3-
-formyl-6-methoxyphenyl acetate of m.p. 43-$0.
b) 38 g of 2-chloro-3-formyl-6-methoxyphenyl acetate are
introduced portionwise a~ -so to -10 within 15 minutes
into 150 ml of 98 ~ercent nitric acid. After stirring at
-5 ~or 30 minutes the eeaction mixture is poured into
~L3~2~
- 64 -
1.5 1 of ice-water and extracted three times with 500 ml
Oe methylene chloride. The combined organic phases are
washed with ice-water, dried over sodium sul~hate and
evaporated. The residue is crystallized from ether. There
is obtained 2-chloro-3-formyl-6-methoxy-5-nitrophenyl
acetate of m.p. 84-85O.
c) 3s.a g of 2-chloro-3-focmyl-6-methoxy-5-nitrophenyl
acetate are dissolved in 300 ml of methanol. After adding
145 ml of lN sodium hydroxide solution the mixture is
stirred at 23 for 1 hour. Af~er evaporation of the
methanol the residue is diluted with ice-water, made acid
with ZN hydrochloric acid and extracted twice with 400 ml
of ethyl acetate each time. Tha organic phases are washed
with saturated sodium chloride solution, dried over sodium
sulphate and evaporated. The residue is recrystallized
f rom methylene chloride/petroleum ether. There is obtained
2-chloro-3-hydroxy-5-nitro-p-anisaldehyde of m.p. 130.
d) 1.3 g of 2-chloro-3-hydroxy-5-nitro-p-anisaldehyde are
dissolved in 80 ml of methylene chloride, treated with
0.82 ml of boron tribromide, stirred at 23 for 18 hours,
the reaction mixture is treated with 5 ml of methanol
while cooling with ice, evaporated, the residue is dried
in a high vacuum, digested in water, filtered and
recrystallized f rom acetonitrile. There is obtained
2-chloro-3,4-dihydroxy-5-nitrobenzaldehyde of m.p.
193-195.
Example 44
a) ~ mixture of 30 g of 2-chloro-3-hydroxy-p-anisaldehyde
and 230 ml of ethanol is stirred at 70 for 4 hours in the
presence of 12.3 g of hydroxylamine hydrochloride, the
reaction mixture is subsequently evaporated, the residue
is dried in a high vacuum and recrystallized from
:~3~
- 65 -
methanol/water. There is obtained 2-chloro-3-hydroxy-p-
-anisaldehyde oxime of m.p. 174-176.
b) 21 g of 2-chloro-3-hydroxy-p-anisaldehyde oxime are
heated under reflux for 20 hours together with 400 ml oE
acetic anhydride. Thereupon, the reaction mixture is
evaporated, the residue is treated with 300 ml of
ice-water, stirred for 1 hour, decanted, the thus-cooled
residue is partitioned between methylene chloride and
water, the organic phase is dried over sodium sulphate,
evaporated, the residue is dried in a high vacuum,
chromatographed on 200 g of silica gel with methylene
chloride and recrystallized ~rom methylene chloride/
petroleum ether. There is obtained 2-chloro-3-cyano-6-
-methoxyphenyl acetate o m.p. 97-99.
c) In analogy to Examples 43b and 43c, ~rom 2-chloro-3-
-cyano-6-methoxyphenyl acetate there is obtained 2-chloro-
-3-hydroxy-5-nitro-p-anisonitrile of m.p. 157-159
(methylene chloride/hexane~.
d) ~n analogy to Example 43d, from 2-chloro-3-hydroxy-5-
-nitro-p-anisonitrile there is obtained 2-chloro-3,4-
-dihydroxy-S-nitro-benzonitrile of m.p. 180 (aceto-
nitrile).
Example 45
a) 5.5 g of a-chloro-2-fluoro-3,4-dimethoxytoluene and
4.05 g of potassium acetate are stirred at 80 for 25
hours in 50 ml of dimethylformamide. The reaction mixture
is subsequently poured into 150 ml of ice-water and
extracted with ether. The ether phases are washed with
sodium chloride solu~ion, dried over sodium sulphate and
evaporated. 2-Fluoro-3,4-dimethoxybenzyl acetate is
obtained as an oil.
~3~
- 66 -
b) 5.4 g of 2-fluoro-3,4-dimethoxybenzyl acetate arP
heated to 80 for 1.5 hours together with 50 ml of
methanol and 50 ml of lN sodium hydroxida solution. After
evaporation of the methanol the residue is extrac~ed with
methylene chloride. The combined extracts are washed with
water, dried over sodium sulphate and evaporated. The
residue is chromatographed on 80 g of silica gel with
methylene chloride/methanol (95:5). 2-Fluoro-3,4-
-dimethoxybenzyl alcohol is obtained as an oil.
c) 3.0 g of 2-fluoro-3,4-dimethoxybenzyl alcohol and
5.0 g of manganese dioxide are hea~ed under reflux for 1
hour togeCher with 50 ml of benzene. The insoluble
constituents are subsequently filtered of while washing
with methylene chloride. The iltrate is evaporated and
the residue is recrystallized from methylene chloride/
hexane. There is obtained 2-fluoro-3,4-dimethoxybenz-
aldehyde cf m.p. 52-54.
d) 4.4 g of 2-fluoro-3,4-dimethoxybenzaldehyde and 1.83 g
of hydroxylamine hydrochloride are heated under reflux for
5 hours together with 30 ml of ethanol. The reaction
mixture is subsequently evaporated and the residue, dried
in a high vacuum at 23, is introduced into 40 ml of
phosphorus oxychloride. ~fter stirring at 23 for 2.5
hours the reaction mixture is evaporated, the residue is
treated with ice-water, the precipitate which thereby
forms is filtered off, washed with water, taken up in
methylene chloride, the methylene chloride solution is
dried over sodium sulphate and evaporated. By recrystall-
ization of the residue from methylene chloride/hexane
there is obtained 2-~luoro-3,4-dimethoxybenzonitrile of
m.p. 64-65.
e) 2.0 g of 2~fluoro-3,4-dimethoxyben20nitrile are
dissolved in 60 ml of methylene chloride, treated with
~.3~ ,P4:~.8
- 67 -
1.1 ml of boron tribromide, stirred at 23 for 1 hour,
subsequently treated with a further 1.0 ml of boron
tribromide and stirred at 23 for a fuether 80 minutes.
5 Thereupon, the reaction mixture is poured into 100 ml of
ice-cold saturated sodium hydrogen carbonate solution,
whereupon ehe mixture is extracted twice with 300 ml of
ether, the combined ether phases are washed twice with
sodium chloride solution, dried over sodium sulphate,
evaporated and the residue is chromatographed on S0 g of
silica gel with methylene chloride and methylene
chloride/methanol (97:3). There is obtained 2-fluoro-3-
-hydroxy-p-anisonitrile of m.p. 198-200.
f) 0.9 g of 2-fluoro-3-hydroxy-p-anisonitrile are
dissolved in 10 ml o~ acetic anhydride and 0.5 ml of
pyridine, whereupon the mixture is stirred at 120 for 2
hours. The reaction mixture is subsequently evaporated and
the residue is eartitioned between ice-water and methylene
chloride. The organic phase is dried over sodium sulphate
and evaporated, and the residue is recrystallized from
methylene chloride/hexane. There is obtained 3-cyano-2-
-fluoro-6-methoxyphenyl acetate of m.p. 90-91.
g) 0.8 g of 3-cyano-2-fluoro-6-methoxyphenyl acetate are
introduced in three portions at -15 into 5 ml of 96
percent nitric acid, whereupon the mix~ure is stirred at
-10 for 1 hour. Thereupon, the reaction mixture is poured
on to 50 g of ice. The mixture is extracted twice with
70 ml of ethyl acetate each time. The combined organic
phases are washed with saturated sodium chloride solution,
dried over sodium sulphate and evaporated. The thus-
-obtained Lesidue is dissolved in S0 ml of lN sodium
carbonate solution and 50 ml of methanol, whereupon the
solution is stirred at 23 for 1 hour and the methanol is
distilled off. The residal aqueous phase is adjusted to
pH 2 at 0 with conc. hydrochloric acid and extracted
- 68 -
twice with 100 ml of ethyl acetate. The combined oLganic
phases are washed with satura~ed sodium chloride solution,
dried over sodium sulphate and evaporated, and the residue
is chromatographed on 45 g of silica gel with methylene
chloride/methanol (97:3). After recrystallization from
ether~hexane there is obtained 2-fluoro-3-hydroxy-5-nitro-
-p-anisonitrile of m.p. 112-113.
h) 550 mg of 2-fluoro-3-hydroxy-5-nitro-p-anisonitrile
are dissolved in 30 ml of methylene chloride, whereupon
the solution is treated with 0.~4 ml of boron tribromide.
~fter stirring at 23 ~or 6 hours a further 0.1 ml of
boron tribromide are added thereto, whereupon the mixture
is stirred at 23 for a further 1.5 hours. Thereupon, 3 ml
of ethanol are added thereto at -15. The reaction mixture
is evaporated and the residue is dried in a high vacuum at
23~. By recrystallization from ether/hexane there is
obtained 2-fluoro-3,~-dihydroxy-5-nitrobenzonitrile of
m.p. 154~.
Example 46
a) 9.5 g of 3,4-dimethoxy-5-nitrobenzoic acid are
suspended in 9~ ml of thionyl chloride. The suspension is
stirred at 8Q for 1 hour. By two-fold evaporation with
the addition of absolute toluene there are obtained lo g
of 3,4-dimethoxy-5-nitrobenzoyl chloride which is
dissolved in 100 ml of tetrahydrofuran. This solution is
added dropwise to 300 ml of 28 percent aqueous ammonia.
The mixture is subsequently stirred at 40 for 2 hours and
cooled to 5. The crystalline precipitate is filtered o~f.
There is obtained 3,4-dimethoxy-5-nitrobenzamide of m.p.
182-185.
b) 2.46 g of chlorine are introduced while cooling with
ice into a mixture of 8.0 g of sodium hydroxide, 50 ml of
- 69 -
water and 30 g of ice. Thereupon, 6.5 g of 3,~-dimethoxy-
-5-nitrobenzamide suspended in 5 ml of tetrahydrofuran are
slowly added thereto. The reaction mixture is heated to
700 within 30 minutes, stirred at 70 for 1 hour, cooled
to 5 and the separated crystals are filtered off. There
is obtained 3,4-dimethoxy-5-nitroaniline of m.p. 129-131.
By extraction of the filtrate with ethyl acetate, drying
the extract over sodium sulphate, concentration and
crystalliæation of the residue with the addition oE ether
there can be obtained a further portion of 3,4-dimethoxy-
-s-nitroaniline.
c) 10 g of finely powdered 3,4-dimethoxy-5-nitroaniline
are suspended in 15 ml of 12N hydrochloric acid and 40 ml
of water, whereupon the suspension is stirring at 30 for
1 hour. Thereupon, 3.8 g o~ sodium nitrite dissolved in
20 ml o water are added dropwise thereto at -5 within 15
minutes. The solution is stirred at -5 for 30 minutes and
the cold diazonium salt solution is added dropwise within
45 minutes to 100 ml of pyridine of 40. The mixture is
subsequently stirred at 70 for 1 hour. The reaction
~nixture is evaporated and the residue is taken up in
300 ml of ethyl acetate. It is extracted three times with
200 ml of 2N hydrochloric acid each time. The acidic-
-aqueous phase is adjusted to pH 9 with conc. ammonia and
extracted with methylene chloride. The combined methylene
chloride extracts are dried over sodium sulphate and
evapoeated, and the residue is chromatographed on 250 g of
silica gel with e~hyl acetate. After crystallizaCion from
methylene chloride/hexane there is obtained 2-(3,4-
-dimethoxy-5-nitrophenyl)pyridine of m.p. 89-90.
d) 1.5 g of 2-(3,4-dimethoxy-5-nitrophenyl)eyridine are
dissolved in 30 ml o~ 48 percent aqueous hydrobromic acid.
l`he reaction mixture is stirred at 100 for 16 hours and
at 23 for 18 hours. The precipitate which thereby ~orms
- 70 -
is filtered off and recrys~allized from methanol/ether.
There is obtained 3-nitro-5-(2-pyridyl)pyrocatechol
hydrobromide of m.R. 239-240.
ExamPle 47
a) 2.76 ml of 2-bromopylidine dissolved in 30 ml of
absolute tetrahydrofuran are treated -60 within 30
minutes with 19.2 ml of 1.6M n-butyl lithium solution (in
hexane), whereupon ~he mix~ure is stirred at -60 for 30
minutes. 6.0 g of 3,4-dimethoxy-5-nitrobenzaldehyde
dissolved in 50 ml of tetrahydrofuran are then added
dropwise thereto at -40 within 30 minutes. The reaction
mixture is warmed to 0 within 2 hours, poured into
ice-water and extracted with ethyl acetate. The combined
extracts are dried over sodium sulphate and evaporated,
and the residue is chromatographed on 200 g of silica gel
with ethyl acetate. There is obtained -(3,4-dimethoxy-
-5-nitrophenyl)-2-eyridinemethanol as a brown oil.
b) 7 g of manganese dioxide and added portionwise to
4~0 g of a-(3,4-dimethoxy-5-nitrophenyl)-2-pyridine-
methanol dissolved in lOo ml of acetone while constantly
heating under reflux over a period of 2.5 hours.
Thereupon, the mixture is heated under reflux for a
further 2 hours. The manganese salts are subsequently
filtered off and the residue obtained after evaporation is
recrystallized from ether/hexane. There is obtained
3,4-dimethoxy-5-nitrophenyl 2-eyridyl ketone of m.p. 113.
c~ 1.5 g of 3,4-dim~thoxy-5-nitrophenyl-2-eyridyl ketone
dissolved in 30 ml of 4~ percent hydrobromic acid are
stirred at 100 for 18 hours. Thereupon. the reaction
mixture is evaporated and the residue is recrystallized
from acetonitrile/methanol. There is obtained
3,4-dihydroxy-5-nitrophenyl 2-pyridyl ketone hydrobromide
2~
- 71 -
of m.p. 213.
Example ~8
a) 11.3 g of tin dichloride dihydra~e are added to 2.z5 g
of 3',4'~dimethoxy-5'-nitroacetophenone dissolved in 50 ml
of ethanol, whereupon the mixture is stirred at 75O for 30
minutes. Thereupon, the reaction mixture is poured on to
100 g of ice, neutralized with about 300 ml of saturated
sodium hydrogen carbonate solution and treated with 150 ml
of methylene chloride. The mixture is filtered and the
methylene chloride phase is separated. This is dried over
sodium sulphate and evaporated, and the residue is
recrystallized from ether/pe~roleum ether~ There i6
obtained S'-amino-3',4'-dimethoxyacetophenone Oe m.p.
63-65.
b) A solution of 5.2 g of sodium nitrite in 20 ml of
water is added dropwise at o within 20 minutes to 1~.0 g
of 5'-amino-3l,4'-dimethoxyacetophenone dissolved in
155 ml of lN hydrochloric acid. ~fter stirring at -2~ for
30 minutes the cold diazonium salt solution is added
dropwise within 30 minutes at 5-10 to a solution of 8.7 g
of copper~I) cyanide and 5.45 g of potassium cyanide in
60 ml of water. ~fter completion o~ the addition 200 ml of
methylene chloride are added, and the reaction mixture is
stirred at 23 for 3 hours and then filtered~ The organic
phase is separated, washed with water, dried over sodium
sulphate and evaporated. The residue is recrystallized
from methylene chloride/petroleum ether. There is obtained
5'-cyano-3l,4`-dimethoxyacetophenone of m.p. 125-126.
c) 3.75 g of aluminium powder and 28.5 g of iodine are
heated under reflux for 2 hours in 160 ml of absolute
benzene. 3.0 g of 5'-cyano-3`,4'-dimethoxyacetophenone and
0.5 g of tetra-n-butylammonium iodide are then added at
~L3~ 3
20, whereupon the mixture is heated under reflux for 1
hour. Thereupon, the mixture is treated at 20 with 50 g
of ice and filtered. The residue is washed with ethyl
acetate. The phases are separated and the aqueous phase is
extracted a further twice with ethyl acetate. The combined
organic phases are washed with 20 percent sodium thio-
sulphate solu~ion, dried over sodium sulphate and evapor-
ated. The thus-obtained residue is dissolved in 20 ml of
acetic anhydride and 0.5 ml of ~yridine and stirred at
12Q for 6 hours. The mixture is subsequen~ly evapordted
and the residue is partitioned between methylene chloride
and ice-water. The methylene chloride phase is dried over
sodium sulphate and evaporated, and the residue is
chromatographed on 100 g of silica gel with methylene
chloride. After recrystallization from ether there is
obtained 5'-cyano-3',4'-diacetoxyacetophenone of m.p.
76-79.
ExamPle 49
0.33 g of 5'-cyano-3',4'-diacetoxyacetophenone
dissolved in 3.3 ml of me~hanol is treated with 2.7 ml of
l.ON sodium hydroxide solution and the reaction mixture is
stirred at 23 for 45 minutes. The mixture is subsequently
acidified with 2N hydrochloric acid, diluted with 5 ml of
saturated sodium chloride solution and extracted twice
with 30 ml of ethyl acetate each time. The combined ethyl
acetate phases are dried over sodium sulphate and evapor-
3Q ated. The residue is recrystallized from toluene~aceto-
nitrile. There is obtained 5'-cyano-3',4'-dihydroxyaceto-
phenone as brownish crystals which decompose above 215.
a) 3.4~ g of phenyltrimethylammonium bromide dibromide
dissolved in 30 ml of tetcahydrofuran are added dropwise
~3~2~
- 73 -
at room temperature within 45 minutes to 1.9 g of
5~-cyano-3~,4~-dimethoxyacetophenone dissolved in 30 ml of
tetrahydrofuean, whereupon the mixture is stirred for 30
minutes. Thereupon, the reaction mixture is poured into
120 ml of ice-water and extracted three times with 70 ml
of methylene chloride. The combined methylene chloride
phases are washed with 2N sulphuric acid, dried over
sodium sulphats and evaporated. The residue is chromato-
graphed on Z0 g of silica gel with methylene chloride.After recrystallization from methylene chloride/hexane
there is obtained 5-(bromoacetyl)-2,3-dimethoxybenzo-
niteile of m.p. 138-141.
b) 1.45 g of 5-(bromoacetyl)-2,3-dimethoxybenzonitrile
and 1.12 g of selenium dioxide are stirred at 120 or 18
hours in 10 ml of n-hexanol. After cooling to room temper-
ature the mixture is diluted with 20 ml of methylene
chloride and filtered. The filtrate is washed with water,
dried over sodium sulphate and evaporated. The residue is
chromatographed on 70 g of silica gel with hexane/ether
(2:1). There is obtained hexyl ~3-cyano-4,5-dimethoxy-
phenyl)glyoxylate as an oil.
c) 4.8 ml of boron tribromide dissolved in 20 ml of
methylene chloride are added dropwise while cooling with
ice within 20 minutes to ~.0 g of hexyl ~3-cyano-4,5-
~dimethoxyphenyl)glyoxylate dissolved in 100 ml of
methylene chloeide and the reaction mixture is stirred at
room temperature for 18 hours. 40 ml of methanol are
subsequently added dropwise thereto at -60O, the mixture
is stirred at room temperature for 1 hour and evaporated.
The residue is taken up in methanol. It is heated under
reflux or 10 minutes, evaporated to dryness and dried in
a high vacuum. The thus-obtained crude product is recryst-
allized rom acetonitrile. There is obtained methyl
(3-cyano-4,5-dihydroxyphenyl)glyoxylate of m.p. 252.
~3~32~
- 74 -
ExamPle 51
1.075 g of methyl (3-cyano-4,5-dihydroxyphenyl)-
glyoxylate and 0.60 g of ~-amino-p-cresol are stirred at
120 for 70 minutes in 2 ml of dimethylformamide. The
~ixture is subsequently cooled to room temperature and
diluted with 15 ml of wat~r. The precipitate is filtered
off and dried in a water-jet vacuum at 80 for 6 hours.
A~ter recrystallization from acetonitrile there is
obtained 2,3-dihydroxy-5-(6-methyl-2-oxo-2H-1,4-
-benzoxazin-3-yl)benzonitrile of m.p. 27~-280.
Example 52
a) ~ solution of 2.5 g ~10.2 mmol) of 3,4-dimethoxy-5-
-nitrobenzoyl chloride in 10 ml o absolute tetrahydro-
~uran is treated with 1.1 ml o~ ethyl isocyanoacetate and
subsequently with a solution of 3.0 ml of triethylamine in
10 ml of tetrahydrofuran and stirred at room temperature
for 48 hours. ~fter distillation of the solvent the
residue is ex~racted with ethyl acetate/water. The crude
product obtained after evaporation is chromatographed on a
20-fold amount of silica gel with ethyl acetate. ~fter
recrystallization from ethyl acetate/hexane there is
obtained ethyl S-(3,4-dimethoxy-5-nitroehenyl)-4-oxazole-
carboxylate in the form of yellow crystals of m.p.
109-110.
b) 1.0 g (3.1 mmol) of ethyl 5-(3,4-dimethoxy-5-nitro-
phenyl)-4-oxazolecarboxylate is treated with lo ml of
constant-boiling hydrobromic acid and stirred at 140 for
2 hours. After distillation of the excess hydrobromic acid
the yellow residue is recrystallized from ethanol/acetone.
There is obtained 2-amino-3l,4)-dihydroxy-5~-nitroaceto-
phenone hydrobromide in the form of yello~ crystals of
m.p. >250 (dec.).
~3~
- - 75 -
Example 53
a) 10 g (34 mmol) of ethyl (3,4-dimethoxy-5-nitro-
benzoyl)acetate are suspended in 100 ml of ethanol,
treated with 1.7 g (37 mmol) of methylhydrazine and heated
under reflux for 16 hours. After distillation of about
5Q ml of ethanol the mixture is cooled to 0 and the
separated precipitate is filtered off under suction. After
recrystallization from ethanol there is obtained 3-(3,4-
-dimethoxy-5-nitrophenyl)-1-methylpyrazol-5-ol in the form
of yellow crystals of m.p. 200-202.
b) 2.0 g (7.Z mmol) of 3-(3,4-dimethoxy-5-nitrophenyl)-1-
lS -methylpyrazol-5-ol are æuspended in 100 ml of methylene
chloride. After cooling to -40 a solution of 4.9 ml of
boron tribromide in 60 ml of methylene chloride is added
dropwise thereto within one hour. ~he mixture is
subsequently stirred at room temperature for 16 hours,
cooled to -20 and treated within 30 minutes with 100 ml
of ethanol. ~fter stiering at room temperature for one
hour the solvent is distilled off in a water-jet vacuum at
40. The residue is ~reated three times with a mixture of
100 ml of ethanol/toluene each time, with the solvent
being distilled off each time. The residue is
recrystallized from ethanol. There is obtained
5-(5-hydroxy-1-methyl- pyrazol-3-yl)-3-nitropyrocatechol
hydrobromide in the form of yellow crystals of m.p. >250.
Example 54
a) 16.8 g ~0.7 g-atom) of magnesium are treated with
lS ml ol ethanol and, after adding Z ml of carbon tetra-
chloride, the reaction is initiated by heating. A solution
of 130.3 g of tert.butyl ethyl malonate in 70 ml of
ethanol and 600 ml of absolute ether is added dropwise
thereto while stirring within about 30 minutes so that the
~L3~2~
- 76 -
reaction proceeds at ~he reflux temperature. The mixture
is subsequently stirred at 50 for a further 3 hours and
the solvent i5 distilled off at 40 in a water-jet vacuum.
The residue is dissolved in 900 ml of tetrahydrofuran. To
this solution is added dropwise while stirring at 50O a
solution of 170 g ~0.7 mol) of 3,4-dimethoxy-5-nitro-
benzoyl chloride in 700 ml of absolute tetrahydrofuran and
the mixture is stiLred at the reflux temperature for one
hour. The solvent is distilled off at 40 in a water-jet
vacuum. The residue is treated with 1 1 o~ ether. 260 ml
of 3N sulphuric acid are added thereto while cooling and
stirring and the mixture is stirred for 30 minutes. The
aqueous phase is extracted twice wi~h 600 ml of ether each
time. The organic phase is washed neutral with water,
dried over sodium sulphate and evapora~.ed. The ~rown oil
obtained is filtered over 1 kg of silica gel with toluene.
The resulting mixture, consistiny of ethyl tert.butyl
(3,4-dimethoxy-5-nitrobenzoyl)malonate and ethyl
(3,4-dimethoxy-5-nitrobenzoyl)acetate, is dissolved in
600 ml of methylene chloride and treated while stirring
within about 30 minutes wi~h 193 ml of trifluoroacetic
acid. The mixture is subsequently s~irred at 40 for 2
hours and then evaporated at 40 in a water-jet vacuum.
The oil obtained is extracted with ether~water. The
organic phase is dried over sodiuln sulphate and evapor-
ated. ~fter dissolution in diisopropyl ethar/hexane and
cooling the separated crystals are filtered of e under
suction and recrystallized from diisopropyl ether. There
is obtained ethyl (3,4-dimethoxy-5-nitrobenzoyl)acetate in
the form of slightly yellowish crystals of m.p. 67-68.
b) 10.0 g (33.6 mmol) of ethyl (3,4-dimethoxy-5-nitro-
benzoyl)acetate are eeacted with 4.0 g (37 mmol) of
phenylhydrazine in analogy to Example 53a. A~ter recryst-
allization from methylene chloride/ethanol there is
obCained 3-(3,4-dimethoxy-5-nitrophenyl)-1-phenyl-2-
3 77~
-pyrazolin-5-one in the form of yellow crystals of m.p.
190-19Z.
c) In analogy to Example 53b, with bocon tribromide there
is obtained therefrom 5~(5-hydroxy-1-phenylpyrazol-3-yl)-
-3-nitropyrocatechol hydrobromide in the form of yellow
crystals of m.p. >220 (dec.).
ExamPle 55
20.1 g of diethyl (3,4-dimethoxy-5-nitrobenzoyl)-
malonate are dissolved in 200 ml of methylene chloride,
whereupon the solution is cooled to -20 and at this
temperature there is added dropwise while stirring within
15 minuCes a solution of 68.1 g of boron tribromide in
120 ml of methylene chloride. The mixture is subsequently
stirred at room temperature overnight. After cooling to
-Z0 the mixture is treated with 300 ml of ethanol and
stirred at room temperature for 30 minutes. The solvent is
distilled off in a water-jet vacuum at 40. The residue is
treated with 300 ml of ice-water and methylene chloride.
The organic phase is washed with water, dried over sodium
sulphate and evaporated. The crude product obtained is
chromatogra~hed on a 10-fold amount of silica qel with
toluene. ~fter crysCallization from methylene chloride/
hexane there is obtained ethyl ~3,4-dihydroxy-5-nitro-
ben~oyl)acetate in the form of y~llow crystals of m.p.
136-137.0
xam~le_56
2.0 g ~7.4 mmol) of ethyl 13,4-dihydroxy-5-nitro-
benzoyl)acetate are suspended in 50 ml of ethanol. After
treatment with 0.4 g of hydrazine hydrate the mixture is
held at the reflux temperature for 16 hours. After
distillation of the solvent the residue is held at the
iL3~
- 78 -
boiling temperature for S minutes with 50 ml of ethyl
acetate. The separated precipitate is filtered of under
suction and the filtrate is concentrated ~o 10 ml. The
crystals, which separate in the cold, are filtered off
under suction. There is obtained 5-(5-hydroxypy~azol-3-
-yl)-3-nitropyrocatechol in form of orange crystals of
m.p. 22~a ~dec.).
ExamPle 57
7.3 g (36.66 mmol) of 3,4-dihydroxy-5-nitrobenzoic
acid are treated with 30 ml of acetic anhydride, whereupon
the mixture is held at the reflux temperature for 8 hours.
The reaction mixture is poured on to ice. The separated
precipitate is ~iltered off under suction, washed with
water and taken up in methylene chloride. The organic
phase is dried over sodium sulphate and evaporated. ~he
residue obtained is recrystallized from methylene
chloride/n-hexane in the cold. There is obtained
3,~-diacetoxy-5-nitrobenzoic acid in the form of
colourless crystals of m.p. 126-127.
Example 58
a) g.7 g (32.2 mmol) o~ 3,4-diacetoxy-5-nitrobenzoic acid
are teeated with 12.5 ml of thionyl chloride, whereupon
the mixture is stirred at 100 for 1.5 hours. ~fter
distillation of the excess thionyl chloride the 5-(chloro-
carbonyl)-2-nitro-o-phenylene diacetate is dis~illed, b.p.
160 ~26.7 Pa).
b) 3.2 g (10.6 mmol) of 5-(chlorocarbonyl)-2-nitro-o-
-phenylene diacetate are dissolved in 50 ml of dimethyl-
formamide. The ice-cold solution is trea~ed while stirring
with a solution of 2.2 ml of diethylamine in 20 ml of
dimethylformamide. The mixture is subsequently stirred at
~3~
79 -
room temperature for one hour, whereupon the solvent is
distilled off in a water-jet vacuum at 50. The residue
obtained is treated with water and methylene chlocide. The
organic phase is dried over sodium sulphate and evapor-
ated. The yellow resin obtained is crystallized from
methylene chloride~ether. There is obtained N,N-diethyl-
-3,4 dihydroxy-5-nitrobenzamide in the form of yellow
crystals of m.p. 145-146.
Example 59
3.2 g (10.6 mmol) of 5-(chloeocarbonyl)-2-nitro-o-
-phenylenediacetate are dissolved in S0 ml of dimethyl-
formamide, whereupon the solution is treated at 0-5 while
~tirring and within 40 minutes with a solution of 3.02 ml
of Z, 2-diethylaminoethylamine in 20 ml of dimethyl-
formamide. The mixture is subsequently stirred at room
temperature for 30 minutes. The solvent is distilled off
in a water-jet vacuum at 60. The residue is extracted
twice with 20 ml of ethanol each time, dissolved in hot
ethanol and treated with an excess of ethanolic
hydrochloric acid, whereupon the mixture is evaporated.
Af~er recrystallization from ethanol/ethyl acetate there
is obtained ~-~2-(diethylamino)ethyl]-3,4-dihydroxy-5-
-nitrobenzamide hydrochloride in the form of yellow
crystals of m.p. 139 (dec.).
~xamPle 60
a) 10.0 g (47.4 mmol) of 2,3-dimethoxy-~-nitrobenz-
aldehyde are trea~ed with 50 ml of glacial acetic acid and
50 ml of constant-boiling hydrobromic acid and held at the
reflux temperature for 7 hours. After treatment with ice
the separated precipicate is filtered off under suction.
washed with water and taken up in ethyl acetate. The
organic phase is dri~d over sodium sulphate and evapor-
~3~
- 8~ -
ated. The crude product obtained is filtered over silica
gel with ethyl acetate. After crystalliza~ion from ethyl
acetate/hexane there is obtained 2,3-dihydroxy-5-nitro-
benzaldehyde in the form of brownish crystals of m.p.
2Z6-2280.
b) 4.5 g of 2,3-dihydroxy-5-nitrobenzaldehyde are
suspended in 75 ml of water. After treatment with 4.2 g of
hydroxylamine o-sulphonic acid the mixture is stirred at
65 for 16 hours. After cooling the separated crystals are
filtered off under suction and wa~hed with water. The
filtrate is extracted with ethyl acetate. The crystals and
the dried organic phase are combined, whereupon the
mixture is evaporated and the residue is recrystallized
from diisopropyl ether. There is obtained 2,3-dihydroxy-5-
-nitrobenzonicrile in the form of yellow crystals of m.p.
ls6-lsao .
ExamPle 61
a) 4.0 g (19.2 mmol) of 3,4-dimethoxy-5-nitro-benzo-
nitrile are dissolved in 50 ml of dimethylformamide,
whereupon ~he solution is treated with 1.66 g of ammonium
chloride and 2.02 g of sodium azide and stirred at 125
for 31 hours. ~fter in each case 8 and 15 hours the same
amounts of ammonium chloride and sodium azide are added
thereto. After cooling the mixture is poured on to ice.
l~he separated precieitate is filtered off under suction,
washed with water and dried. There is obtained 2-methoxy--
-6-nitro-4-(lH-tetrazol-5-yl)phenol in the form of orange
crystals of m.p. ~240 (dec.).
b) 4.0 g (16.9 mmol) of 2-methoxy-6-nitro-4-(lH-tetrazol-
-5-yl)phenol are treated with 40 ml of constant-boiling
hydrobromic acid, whereupon the mixture is stirred at 140
for a hours under a nitrogen atmostphere. After cooling
:3L 3 ~ -!t ~
the mixture is poured on to ice. The separated precipitate
is filtered off under suction and recrystallized from
ether. There is obtained 3 nitro-5~ -tetrazol-5-yl)-
pyrocatechol in the form of orange crystals of m.p. >240(dec.).
xam~le ~2
A eotal o 7.2 g (40 mmol) of 3,4-dihydroxy-5-nitro-
benzonitrile are introduced portionwise into 130 ml of
conc. sulphuric ~id whil~ stirring within 10 minutes,
whereupon he mix~ure is 6tirred at 50 ~or ~ hours. The
reaction mixture is poured into B00 ml of ice-water. The
separated precipitate is filtered off under suction,
washed with water and taken up in ethyl acetate. The
organic ~hase is dried over sodium sulphate and evapor-
ated. ~fter recrystallization from acetone/ethyl acetate
there is obtained 3,4-dihydroxy-5-nitrobenzamide in ~he
form of orange cr~stals of m.p. 235-236.
ExamDle 63
a) A solution of 11.25 g ~82.6 mmol) of 2,hydeoxyaceto-
phenone in 100 ml of absolute dimethylformamide is added
dropwise under an argon atmosphere within 15 minutes to a
suspension o 3.6 g (B2.5 mmol) of a 55 percent sodium
hydride dispersion in 50 ml of absolute dimethylformamide
and the mixture is stirred at room temperature for one
hour. Aft~r cooling to 0 a solution of 20.3 g (82.6 mmol)
of 3,4-dimethoxy-5-nitrobenzoyl chloride in 100 ml of
absolute dimethylfoLmamide is added dropwise thereto
within 20 minutes and the mixture is tirred at room
temperature overnigh~. The reaction mixture is poured into
ice-w~ter, whereupon the mixture is ~xcracCed twice with
250 ml o ethyl acetate each time. The organic phase is
extracted twice with ~00 ml of sodium chloride solution
i~
- 8Z -
each time, dried over sodium sulphate and evaporated. The
brown oil obtained is heated in 100 ml of toluene. The
separated precipitate is filtered off under suction and
the filtrate is chromatographed on a 30-fold amounc of
silica gel with toluene/ethyl acetate (4~ fter
recrystallization from ethyl acetate/hexane there is
obtained o-acetylphenyl 3,4-dimethoxy-5-nitrobenzoate in
the form of yellowish crystals of m.p. 108-109.
b) 10.0 g ~29 mmol) of o-acecylphenyl 3,4-dimethoxy-5-
-nitrobenzoate are dissolved in 50 ml of pyridine. After
treatment with 8.12 g (144 mmol) of powdered and dried
potassum hydroxide the mixture is stirred at 80~ for 5
minutes. After cooling the mixture is poured on to ice.
The aqueous solution is made acid by treatment with 3N
hydrochloric acid. The separated precipitate is filtered
off under suction. After recrystallization from ethyl
acetatethexane theLe is obtained l-(o-hydroxyphenyl)-3-
-(3,4-dimethoxy-5-nitrophenyl)-1,3-propanedione in the
form of yellowish crystals of m.p. 188-189.
c) A solution of 1.82 g (0.7 ml) of boron tribromide in
20 ml of methylene chloride is added dropwise within about
20 minutes to a solu~ion of 500 mg (1.45 mmol) of
l-(o-hydroxyphenyl)-3-(3,4-dimethoxy-5-nitrophenyl)-1,3-
-propanedione in 50 ml of methylene chloride while
stirring and under an argon atmosphere at -20, whereupon
the mixture is stirred at room temperature overnight.
After cooling to -20 the mix~ure is treated dropwise with
25 ml of ethanol and evaporated at 40 in a water-jet
vacuum. ~Eter recrystallization from ethanol there is
obtained l-(3,4-dihydroxy-5-nitrophenyl)-3-
-(o-hydroxyphenyl)-1,3-propanedione in the form of yellow
crystals o~ m.p. 251-25Z.
3~s
- ~3 -
Exam~le 64
a) A solution of 2.0 g (5.79 mmol) of o-acetylphenyl
3,4-dimethoxy-5-nitrobenzoate in 12.5 ml of glacial acetic
acid is treated with 0.94 g of sodium acetate and held at
the reflux tem~erature for 4 hours. ~fter cooling the
mixture is poured into ice-water. The separated crys~als
are filtered off under suction. After recrystallization
from ethyl acetate/hexane there is obtained 2-(3,4-
-dimethoxy-5-nitrophenyl)-4H-l-benzopyran-~-one in the
form of colourless crystals of m.p. 216-217.
b) A solution of 10 ml of boron tribromide in 50 ml of
methylene chloride are added dropwise within 30 minutes at
-10 to a solution of 1.0 g (2.9 mmol) o~ 2-(3,4-
-dimethoxy-5-nitrophenyl)~4H-l-benzopyran-4-one in 100 ml
of methylene chloride under an argon atmosphere, whereupon
the mixture is stirred at room temperature overnight.
After cooling to -20 20 ml of ethanol are added dropwise
there~o. The mixture is then evaporated in a water-jet
vacuum. The yellow residue obtained is extracted with
water/ethyl acetate. The organic phase is washed with
water, dried over sodium sulphate and evaporated. ~fter
recrystallization from ethanol/ethyl acetate there is
obtained 2-~3,4-dihydroxy-5-nitrophenyl)-4H-l-benzopyran-
-4-one in the form of yellow crystals of m.p. >240 ~dec.).
Example 65
a) 92 ml of n-butyl lithium solution (1.6M in hexane) are
added dropwise at -70 within 20 minutes to 33.0 g of
4-bromobenzotrifluoride (dissolved in 150 ml of tetra-
hydrofuran). After stirring at -70 for 45 minutes 36 g of
3-methoxy-4-benzyloxybenzaldehyde (dissolved in 100 ml of
tetrahydrofuran) are added droewise thereto at between
-70 and -~0. The reaction mixcure is s~irred at -70 for
~3~
- 84 -
2 hours and ac 0 for 1 hour, poured into a mixture of ice
and 100 ml of 2N sulphuric acid and exr,racted twice with
500 ml of ether. The combined ether phases are washed with
S saturated sodium chloride solution, dried over sodium
sulphate and evaporated. There is obtained 4-~benzyloxy)-
-3-methoxy-4l-(trifluoromethyl)benzhydrol which can be
used directly in ~he subsequent reaction step.
b) 52.6 g of 4-(benzyloxy)-3-methoxy-4'-(trifluoro-
methyl)benzhydrol (dissolved in 500 ml of methylene
chloride) are treated within 10 minutes at 20 with 30.6 g
of pyridinium chlorochromate and stirred at 20 for 2
hours. The precipitate formed is subsequently filtered of
and washed with methylene chlo~ide. The fil~rate is
evaporated and the residue is chromatographed on 150 g of
silica gel wi~h me~hylene chloride. After recrystalliza-
tion from methylene chloride/hexane there is obtained
4-(benzyloxy)-3-methoxy-4'-(tri~luoromethyl)benzophenone
of meleing point 101.
c~ 70 ml of 33 percent hydrobromic acid in acetic acid
are added within 15 minutes at 10 to 20 g of
4-(benzyloxy)-3-methoxy-4'-(trifluolomethyl)benzophenone
(dissolved in 150 ml Oe methylene chloride). After
stirring at Z0 for 1.5 hours the reaction mixture is
poured into 600 ml of ice-water; the methylene chloride
phase is separated and the aqueous phase is extracted a
further twice w.th 100 ml of methylene chloride. The
combined methylene chloride phases are washed with 600 ml
of water, dried over sodium sulphate and evaporated. ~he
residue is chromatographed on 150 g of silica gel with
methylene chloride. ~ftec recrystallization from methylene
chloride/hexane there is obtained 4-hydroxy-3-methoxy-4`-
-(trifluoromethyl)benzoehenone of melting point 97.
d) 3.2 ml of 65 percent nitric acid are added droewise
~L3~
- 85 -
within 10 minutes at 20 to 1~.8 g of 4-hydroxy-3-methoxy-
-4l-(trifluoromethyl)benzophenone (dissolved in 160 ml of
acetic acid). Af~er stirring for 1.5 hours the reaction
mixture is poured into 600 ml of ice-water, and the
precipitate formed is filtered off, washed with water and
dissolved in methylene chloride. The methylene chloride
solution is dried over sodium sulphate and evaporated, and
the residue is recrys~allized from methylene chloride/
hexane. There is obtained 4-hydroxy-3-methoxy-5-nitro-4'-
-(trifluoromethyl)ben20phenone of mel~ing point 172.
e) 2.0 g of 4-hydroxy-3-methoxy-5-ni~ro-4'-(trifluoro-
methyl)benzophenone (dissolved in 20 ml of 33 percen~
hydrobromic acid in glacial acetic acid) are stirred at
90 for 18 hours. Thereupon, 2Q ml of 4a percent aqueous
hydrobromic acid are added thereto, whereupon the mixture
is stirred at 110 for a further 18 hours. The reaction
~ixture is subsequently evaporated under reduced pressure
and the residue is recrystallized from water. There is
obtained 3,4-dihydroxy-5-nitro-4'-(trifluoromethyl)benzo-
phenone of melting point 116-118.
Example 66
a) 18.7 g of 4-hydroxy-3-methoxy-5-nitro-4~-(trifluoro-
methyl)benzophenone ~dissolved in 250 ml of tetrahydro-
furan) are treated at room temperature with 27.5 ml of 2N
potassium hydroxide solution, whereupon the mixture is
evaporated. The residue is treated with 200 ml of toluene,
whereupon the mixture is again evaporated. Thereupon, the
mixture is heated with 400 ml of toluene ~or 4 hours with
the separation of water and under reflux. 100 ml of
toluene are distilled off and 10 ml of dimethylformamide
and 20 ml of dimethyl sulphate (freshly distilled) are
added, whereupon the mixture is heated under reflux for 5
hours. 300 ml of lW sodium hydroxide solution are
- 86 --
subsequently added at 20. The reaction mixture is stirredfor 30 minutes and treated with 200 ml of ether. The
organic phase is separated; the aqueous phase is extrac~ed
a further twice with 100 ml of ether; the combined ether
phases are dried over sodium sulphate and evaporated, and
the residue is chromatographed on 70 g oE silica gel with
methylene chloride. After recrystallization from methylene
chloride/hexane there is obtained 3,4-dimethoxy-5-nitro-
-4~-(trifluoromethyl)benzophenone of melting point 115.
b) 49.5 g of tin dichloride dihydrate are added to 16.0 g
of 3,4-dimethoxy-5-nitro-4'-(trifluoromethyl)benzophenone
(dissolved in 300 ml of ethanol), whereupon the mixture is
stirred at 75 for 30 minutes. Thereupon, the reaction
mixture is poured into 800 ml of ice-water. It is
neutcalized with 28 percènt sodium hydroxide solution and
extracted three times with 600 ml of methylene chloride.
The combined methylene chloride phases are washed with
water, dried over sodium sulphate and evaporated. ~fter
recrystallization from methylene chloride/hexane there is
obtained 5-amino-3,4-dimethoxy-4~-(trifluoromethyl)benzo-
phenone of melting point 95-96.
c) 3.25 g of 5-amino-3,4-dimethoxy-4'-(trifluoromethyl)-
benzophenone (dissolved in 50 ml of acetone) are evapor-
ated under reduced pressure after the addition of 15 ml of
2N sulphuric acid. The thus-obtained residue is suspended
in 20 ml of acetic acid, diluted with 100 ml of water and
treated at 5 with a solution of 700 mg of sodium nitrite
in 10 ml of water. It is stirred at 5 for one hour.
Thereupon, the diazonium salt solution is filtered, added
at 5 to a solution of 2.0 g of sodium cyanide and 1.0 g
of copper(I) cyanide in 20 ml of water and stirred at 5
for 1 hour. Thereupon, 200 ml of methylene chloride are
added thereto. Insoluble constituents are filtered off.
The phases are separated; the aqueous phase is extracted a
~3~
- 87 -
further twice with 100 ml of methylene chloride. The
combined methylene chloride phases are washed with water,
dried over sodium sulphate and evaporat:ed. The residue is
chromatographed on 30 g of silica gel with methylene
chloride. ~fter recrystallization from methylene
chloride/hexane there is obtained 5-cyano-3,4-dimethoxy-
-~-(trifluoromethyl)benzophenone of melting point 130.
d) 1.5 g of 5-cyano-3,4-dimethoxy-4'-(trifluoromethyl)-
benzophenone (dissolved in 75 ml of methylene chloride)
are treated at 5 with 2.18 ml of boron tribromide,
whereupon the mixtuee is stirred at room temperature for
18 hours. The reaction mixture is subsequently diluted
with 50 ml of methylene chloride. The mixture is heated
under reflux for a further 4 hours, treated at -70 with
15 ml o~ methanol, stirred at room temperature for 2
hours, evaporated, the residue is dried in vacuo and
partitioned between ethyl acetate and ice-water. The
aqueous phase is extracted a further twice with ethyl
acetate. The combined ethyl acetate phases are dried over
sodium sulphate and evaporated. The thus-obtained material
is stirred at 130 for 6 hours with 10 ml of acetic
anhydride and 1 ml of pyridine. The mixture is evaporated
and the residue is partitioned between ice-water and
methylene chloride. The methylene chloride phase is dried
over sodium sulphate and evaporated, and the residue is
chromatographed on 30 g of silica gel with methylene
chloride. The thus-obtained diacetate is dissolved in
10 ml of methanol. The solution is treated with 4.2 ml of
lN sodium hydroxide solution, stirred at 0 for 1 hour,
neutralized with acetic acid, evaporated and partitioned
between ethyl acetate and saturated sodium chlocide
solution. The ethyl acetate phases are dried over sodium
sulphate and evaporated, and the residue is recrystallized
from methylene chloride. There is obtained 5-cyano-3,4-
-dihydroxy-~'-(trifluoromethyl)benzophenone of melting
~3~2'~18
- ~8 -
point 204-206~.
ln an analogous manner:
al) E~rom ~-fluoro-4-hydroxy-3-methoxy-5-nitrobenzophenone
there is obtained 3,4-dimethoxy-2'-fluoro-5-nitrobenzo-
phenone of m.p. 86-88O (from ether/low-boiling petroleum
ether),
bl) from 3,4-dimethoxy-2'-~luoro-5-llitrobenzophenone there
is obtained 5-amino-3,4-dimethoxy-2l-fluorobenzophenone of
m.p. 93-95O (from ether/low-boiling petroleum ether),
cl) from S-amino-3,4-dimethoxy-Z`-~luorobenzophenone there
is obtained 5-benzoyl-2,3-dimethoxy-Z'-fluorobenzonitrile
of m.p. 13Z-134 tfrom methylene chloride/low-boiling
petroleum ether) and
2 dl) from 5-benzoyl-2,3-dimethoxy-2~-fluorobenzonitrile
there is obtained 5-benzoyl-Z,~dihydroxy-2`-fluorobenzo-
nitrile of m.p. Z28~Z30 (from ether/low-boiling petroleum
ether).
In an analogous manner:
bZ) ~rom 3,4 dimethoxy-5-nitrobenzophenone there is
obtained 5-amino-3,4-dimethoxybenzophenone as an amorphous
solid,
c2) from 5-amino-3l4-dimethoxybenzophenone there is
obtained 5-benzoyl-2,3-dimethoxybenzonitrile of m.p.
9B-100 (from etherthexane) and
d2) from 5-benzoyl-2,3-dimethoxybenzonitrile there is
obtained 5-benzoyl-2,3 dihydroxybenzonitrile of m.p.
212-214 (from ethyl acetate/ether).
~3~2~
- 89 -
ExamPle 67
a) A solution of 16.0 g (65.14 mmol) of 3,4-dimethoxy-5-
-nitrobenzoyl chloride in ~0 ml of pyridine is added
dropwise to a solution of 2.68 g (32.64 mmol) o~ l-methyl-
imidazole and 6.6 g (65.14 mmol) of triethylamine in 80 ml
of pyridine, whereupon the mixture is stirred at 60 for 3
hours. After treatment with 1.70 ml of 3N sodium hydroxide
solution the mixture is stirred for a further 1 hour and
subsequently poured into ice-water. The separated grey
crystals are filtered off under suction and taken up in
ethyl acetate, wheLeupon the organic phase is dried o~er
magnesium sulphate and the solvent is distilled of e . After
recrystallization from ethyl acetate/hexane there is
obtained 3,4-dimethoxy-S-nitrophenyl (l-methylimidazol-2-
-yl) ketone in the form of colourless crystals of m.p.
144-145.
b) 5.0 g tl7.17 mmol) of 3,4-dimethoxy-5-nitrophenyl
(l-methylimidazol-2-yl) ketone are treated with 50 ml of
hydrobromic acid (48%), whereupon the mixture is stirred
under reflux ~emperature for 2 hours. After cooling the
separated precipitate is filtered off under suction,
washed with ice water and recrystallized from ethanol.
There is obtained 3,4-dihydroxy-5-nitrophenyl (l-methyl-
imidazol-2-yl) ketone hydrobromide in ~he form of yellow
crystals of decomposition point >240.
In analogy to Example 67, from 3,4-dimethoxy-5-nitro-
benzoyl chloride and l-benzylimidazole there is obtained
l-benzylimidazol-2-yl (3,4-dimethoxy-5-nitrophenyl) ketone
in the form of colourless crystals of m.p. 134-135 tfrom
methylene chloride/hexane) and therefrom with hydrogen
bromide there is obtained l-benzylimidazol-2-yl
~3l~
90 --
(3,4-dihydroxy-5-nitrophenyl) ketone hydrobromide as
yellow crystals of m.p. 218-219 (dec.).
Example 69
a) A solution of 10.0 g (53.13 mmol) of l-~(benzyloxy)-
methyl]imidazole in 50 ml of acetonitrile is added
dropwise within 10 minutes while cooling with ice to a
solution of 13.0 g (53.13 mmol) of 3,4-dimethoxy-5-nitro-
benzoyl chloride and 5.4 g (53.13 mmol) of triethylamine
in 80 ml of acetonitrile so that the temperature does not
exceed 25. The mixture is subsequently stirred for a
further 3 hours, whereupon the solvent is distilled off.
After treatment with water the mixture is extracted with
ethyl acetate. After two-fold washing with water the
organic phase is dried over sodium sulphate and evapor-
ated. The resulting oil is chromatographed on 600 g of
silica gel with toluene/ethyl acetate (95:5), There is
obtained l-~(benzyloxy)methyl]imidazol-2-yl
(3,4-dimethoxy-5-nitrophenyl) ketone as a yellowish oil.
b) ln analogy to Example 67b) there is obtained after
treatment with hydrobromic acid 3,4-dihydroxy-5-nitro-
phenyl (imidazol-2-yl) ketone hydrobromide as yellow
crystals of m.p. 247-248.
Example 70
a) A solution of 25.0 g (120.16 mmol) of 3-bromoquinoline
is dissolved in 200 ml of dry ether and cooled to -60~. At
this temperature there are added dropwise within 15
minutes 75.1 ml ~120.16 mmol) of a 1.6 molar solution o~
n-butylliehium, whereupon the mixture is stirred for lo
minutes. A solution of 26.5 g (109.2 mmol) of vanillin
benzyl ether in 250 ml of dry ether is added dropwise
thereto at -60O, the mixture is subsequently stirred at
Z ~ ~ 8
- 91 -
room temperature for 3 hours, poured into about l.S 1 o~
ice-water and extracted three times with 600 ml of ethyl
acetate each time. The organic phase is washed with water,
dried over sodium sulphate and evaporated. The resulting
oil is chromatographed on 1 kg of silica gel with
methylene chloride/ethyl acetate (1:1). The crystalline
crude product obtained is recrystallized from e~hyl
acetate. There is obtained -~4-(benzyloxy)-3-methoxy-
phenyl]-3-quinolinemethanol in the form of colourless
crystals of m.p. 124-125.
b) A solution of 6.2 g (16.7 mmol) o ~-t4-(benzyloxy)-
-3-methoxyphenyl]-3-quinolinemethanol in 200 ml of
methylene chloride is treated with 62 g of manganese
dioxide and ~tirred at the reflux temperature 2 hours.
After cooling the precipitate is filtered off under
suction and washed with methylene chloride. The filtrate
is evaporated and the oil obtained is dissolved in hot
ether, whereupon the solution is treated with a small
amount of pentane. The separated crystals are filtered off
under suction. There is obtained 4-(benzyloxy)-3-methoxy-
phenyl (3-quinolinyl) ketone in the form of colourless
crystals of m.p. 110-111.
c) 11.0 g (29.7~ mmol) of 4-(benzyloxy)-3-methoxyphenyl
(3-quinolinyl) ketone are treated with 50 ml of
trifluoroacetic acid, whereupon the mixture is stirred at
room temperature for 2 hours. After distillation of the
trifluoroacetic acid the residue is treated twice wi~h
50 ml of ethanol each time and the solvent is distilled
off each time. The oil obtained crystallizes upon
treatment with ethanol. After recrystallisation from
ethanol there is obtained 4-hydroxy-3-methoxyphenyl
(3-quinolinyl) ketone in the form of yellowish crystals of
m.p. 196-197.
d) A solution of 1.91 ml o~ 65 percent nitric acid in
~3~
20 ml of glacial aceeic acid is added dropwise to a
solution of 5.5 g (19.69 mmol) of 4-hydroxy-3-methoxy-
æhenyl t3-quinolinyl~ ketone in 300 ml of glacial acetic
acid a~ 150 and the mixture is then stirred at this
temperature for a further 2 hours. The mixture i6 then
poured into ice-water and extracted three times wi~h
300 ml of ethyl acetate each time. The organic phase is
washed five times with 100 ml of water each time, dried
over sodium sulphate and evaporated. After treatment of
the residue with et~yl acetate there i~ obtained
4-hydroxy-3-methoxy-5-nitrophenyl (3-quinolinyl) ketone in
the form of yellow crystals of m.p. 220-221 (dec.).
e) 820 mg (2.53 mmol) of 4-hydroxy-3-methoxy-5-nitro-
phenyl (3-quinolinyl) ketone are treated with 50 ml of 4
percent hydrobLomic acid and held at the reflux temper-
ature or 3 hours. A~ter distillation of the hydrobromic
acid at 50 the residue is treat~d with 70 ml o~ hot water
and the insoluble constituent is filtered off under
suction. There is obtained 3,4-dihydroxy-5-nitrophenyl
(3-quinolinyl) ketone hydrobromide in the form of yellow
crystals of m.p. 270 (dec.).
Exam~le 71
In analogy to Example 70 there is obtained
a-~4-(benzyloxy)-3-methoxyphenyl]-4-quinolinemethanol as
colourless crystals of m.p. 117-118 (ethyl acetate),
there~rom with manganese dioxide there is obtained
4-(benzyloxy)-3-methoxyphenyl (4-isoquinolinyl) ketone as
colourless crystals of m.p. 126.5-127.5, therefrom with
trifluoroacetic acid there is obtained 4-hydroxy-3-
-methoxyphenyl (4-isoquinolinyl) ketone as yellow crystals
of m.p. 197.5-198.5 and therefrom by nitration wi~h
nitric acid in glacial acetic acid and subsequent
treatment with hydrobromic acid there is obtained
Z 4 ~ 8
- 93 -
3,~-dihydroxy-5-nitrophenyl (4-isoquinolinyl) ketone
hydrobromide as yellow crystals of m.p. 256 (dec.).
Exam~le 72
In analogy to Example 70 there is obtained
a-~4-(benzyloxy)-3-methoxyphenyl]-2-naphthalenemethanol
as colourless crystals (ethyl acetate/hexane) of m.p.
113-114, therefrom with manganese dioxide there is
obtained 4-(benzyloxy)-3-methoxyphenyl (2-naphthyl) ketone
as colourless crystals (ethyl acetate/hexane) of m.p.
104-105, therefrom by treatment with trifluoroacetic acid
and nitration with nitric acid in glacial acetic acid
there is obtained ~-hydroxy-3-methoxy-5-nitrophenyl
(2-naphthyl) ketone as yellow crystals of m.p. 187-1~
and ~here~rom by treatment with hydrobromic acid there is
obtained 3,4-dihydroxy-5-nitrophenyl (2-naphthyl) ketone
as yellow crystals of m.p. 184-185.
ExamPle 73
a) A solution of 20.0 g (100.5 mmol) of 3-phenylpropyl
bromide in 300 ml of ether is treated at -60 while
stirring within 15 minutes with a solution of 71.8 ml
(100.5 mmol) of tert.butyllithium (1.4 molar) in pentane.
After 10 minutes there is added dropwise at this temper-
ature within 15 minutes a solution of ~2.14 g (91.36 mmol)
of vanillin benzyl ether in 200 ml of ether, whereupon the
3 mixture is stirred ~or a further 2 hours. The mixture is
poured into 1 1 of ice-water and extracted three times
with 500 ml of ethyl acetate each time. The organic phase
is washed twice with 200 ml of water each time, dried over
sodium sulphate and evaporated. The oil obtained is
chromatographed on 1 kg of silica gel with methylene
chloride. The crystals obtained are recrystallized from
ether/pentane. There is obtained 4-(benzyloxy)-3-methoxy-
~3~
- 94 -
--(3-phenylpropyl)benzyl alcohol in the form of
colourless crystals of m.p. 71-73.
b) A solution of 9.0 g (24.83 mmol) of ~-(benzyloxy)-3-
-methoxy-~(3-phenylpropyl)ben2yl alcohol in 250 ml of
methylene chloride is treated with so g of manganese
dioxide and held at the reflux temperature for 2 hours.
~fter cooling the precipitate is filtered off under
suction and washed with methylene chloride. The filtra~e
is evaporated and the residue is recrystallized from e~hyl
acetate/e~her. There is obtained 4`-(benzyloxy)-3'-
-methoxy-4-~henylbutyrophenone in the form of colourless
crystals of m.p. 81-82.
c) A soluCion of 7.0 g (19.42 mmol) of 4'-(benzyloxy)-3~-
-methoxy-4-phenylbutyrophenone in 40 ml of 33 percen~
hydrobromic acid in glacial acetic acid is stirred at room
temperature for S hours and subsequently poured into
500 ml of ice-water. The solution is adjusted to pH 6.0 by
the addition of conc. ammonia and extracted three times
with 250 ml of ethyl acetate each time. The organic phase
is washed three times with 50 ml of water each time, dried
over sodium sulphate and evaporated. The oil obtained is
dis olved in 20 ml of ether, whereupon the solution is
treated with hexane until it becomes turbid and is left to
crystallize out. There is obtained colourless 4l-hydroxy-
-3'-methoxy-4-pheny}butyrophenone of m.p. 91-92.
d) A solution of 0.79 ml of 65 percent ni~ric acid in
25 ml of glacial acetic acid is added dropwise to a
solution of 2.2 g (8.14 mmol) of 4~-hydroxy-3~-methoxy-4-
-phenylbutyrophenone in 25 ml of glacial acetic acid and
the mixture is stirred at room temperature for a further 2
hours. The mixture is poured into 150 ml of ice-water and,
after treatment with 20 ml of 3N hydrochloric acid,
extracted three times with 75 ml of ethyl acetate each
~L3~
_ 9~ _
time. The organic phase is washed with water, dried over
sodium sulphate and evaporated. The crude product obtained
is taken up in ethyl acetate and filtered over 75 g of
silica gel. After recrystallization from acetonitrile
there is obtained 4~-hydroxy-3`-methoxy-5'-nitro-4-phenyl-
butyrophenone in the form of yellow crystals of m.p.
120-121~.
e) 1.0 g (3.2 mmol) of 4'-hydroxy-3'-methoxy-5'-nitro-4-
-phenylbutyrophenone is held at 200 for 1 hour with 8 g
of pyridine hydrochloride. The reaction mixture is poured
while still warm into ice-water and extracted with ethyl
acetate. The organic phase is washed with lN hydrochloric
acid and subsequently with water, dried over sodium
sulphate and evaporated. The dark residue obtained is
chromatographed on a 30-fold amount of silica gel with
ethyl acetate. P`rom methylene chloride/hexane there is
obtained 3',4'-dihydroxy-5'-nitro-4-phenylbutyrophenone in
the form of yellow crystals of m.p. 118-119.
Exam~le 74
a) A solution of 14.68 g (225.4 mmol) oE potassium
cyanide in 20 ml of water is added to a solution of 10.0 g
(47.4 mmol) of 3,4-dimethoxy-5-nitrobenzaldehyde in 100 ml
of dioxan. 18.81 ml ~1~0.76 mmol) of 37 percent hydro-
chloric acid are now added dropwise thereto within 30
minutes while stirring vigourously. AEter the addition of
3 lZ0 ml of ether the excess hydrogen cyanide gas is driven
off by passing argon through the mixture. The reaction
mixture is filtered over a siliceous earth filter aid, and
the organic phase is washed with water dried over sodium
sulphate and evaporated. The a-hydroxy-3,4-dimethoxy-
phenylacetonitrile (yellowish oil) which is formed is
dissolved in 200 ml of ether, whereupon the solution is
treated with 20 ml of ethanol, cooled to 0 and
~3i~
- 96 -
hydrochloric acid gas is passed in for 30 minutes. ~f~er 3
hours the separated colourless precipitate is filtered off
under suction and recrystallized from ethanol~ether. 'rhere
S is obtained ethyl ~3,4-dimethoxy-5-nitroehenyl)hydroxy-
acetimidate hydrochloride.
b) 19.7 g (61.46 mmol) o~ ethyl (3,~-dimethoxy-5-nitro-
phenyl)hydroxyacetimidate are dissolved in 500 ml of
ethanol, 6.73 g (61.46 mmol) of o-phenylenediamine are
added, the mixture is stirred at room temperature for 2
hours and subsequently held at the reflux temperature
overnight. ~fter distillation of the solvent the residue
is treated with 50 ml of water, made alkaline with sodium
carbonate solution and extracted twice with 250 ml of
methylene chloride each time. The organic phase is washed
with water, dried over sodium sulphate and evaporated. The
orange residue obtained is chromatographed on 400 g of
silica gel with methylene chloride/ethyl acetate (1:1).
From ether/hexane there is obtained -(3,4-dimethoxy-5-
-nitrophenyl)-2-benzimidazolemethanol in the form of
yellowish crystals o~ m.p. 50 (dec.).
c) 13.2 g (40.1 mmol) of -(3,4-dimethoxy-5-nitro-
phenyl)-2-benzimidazolemethanol are dissolved in 200 ml of
methylene chloride and, a~ter treatment with 130 g of
manganese dioxide, the mixture is stirred at the reflux
temperature for 2 hours. After filtration the solvent is
distilled off. There is obtained 2-benzimidazolyl
~3,~-dimethoxy-5-nitrophenyl) ketone in the form of
yellowish crystals of m.p. Z12-213.
d) l.o g (3.05 mmol) of 2-benzimidazolyl (3,q-dimethoxy-
-5-nitrophenyl) ketone and 8.0 g o-f pycidine hydrochloride
are held at 200O for 60 minutes. The dark solution is
poured while still warm into ice-water and extracted three
times with 100 ml of ethyl acetate each time. The organic
~3~
phase is washed with water, dried over sodium sulphate and
evaporated. After recrystallization from e~hyl acetate/
hexane there is obtained 2-benzimidazolyl (3,4-dihydroxy-
-5-nitrophenyl) ketone in the form of yellow crystals of
m.p, 249-250O.
. Example 75
a) 30.0 g (132 mmol) of 3,4-dimethoxy-5-nitrobenzoic acid
are dissolved in 250 ml of tetrahydrofuran and, after the
addition of 21.85 g (135 mmol) of l,l~-carbonyl-
diimidazole, the mixture is stirred at the reflux temper-
acure for 2 hours. The mixture is poured into 300 ml of
ice-water and the precipitated crystals are filteLed off
under suction after stirring for 30 minutes. The crystals
are taken up in methylene chloride, whereupon the organic
phase is washed with water, dried over sodium sulphate and
evaporated. After ccystallization from methylene
2 chloride~hexane there is obtained 1-(3,4-dimethoxy-5-
-nitrobenzoyl)imidazole in the foLm of colourless crystals
of m.p. 136-137.
b) 10.0 g (36.1 mmol) of 1-(3,4-dimethoxy-5-nitro-
benzoyl)imidazole in 50 ml of dimethylformamide are
treated with 6.95 g (93.8 mmol) o~ acetamidoxime,
whereupon the mixture is stirred at 70O for 1 hour. After
cooling the mixture is poured into 500 ml of ice-water and
stirred for 30 minutes. The separated crystals are
filtered of under suction and washed with water. After
crystallization from ethyl acetate there is obtained
N'-t(3,4-dimethoxy-S-nitrobenzoyl)oxy]acetamidine in the
form of colourless crystals of m.p. 165-166.
c) 2.0 g ~7.2 mmol) of N~-~(3,~-dimethoxy-5-nitro-
benzoyl)oxy]acetamidine aee held at reflux temperature for
1 hour in 20 ml of glacial acetic acid. After distillation
~3~
- 98 -
of the acetic acid the crystalline residue is recrys~all-
ized from ethec/hexane. There is obtained 5-(3,4-
-dimethoxy-S-nitrophenyl)-3-methyl-1,2,4-oxadiazole in the
form of colourless crystals of m.p. 111.
d) 2.5 g (9.43 mmol) of 5-(3,4-dimethoxy-5 nitrophenyl)-
-3-methyl-1,2,4-oxadiazole are dissolved in 70 ml of
methylene chloeide. After cooling to -60 there is added
lo dropwise thereto within 20 minutes while stirring a
~olution of 23.62 g (94.3 mmol) of boron tribromide in
50 ml of methylene chloride, whereupon the mixture is held
a~ the reflux temperature for 48 hours. ~fter cooling to
-60 the mixture is treated wieh 60 ml of ethanol and
subsequently stirred at room temperature for 30 minutes.
The yellow solution is evaporated to dryness, whereueon
the residue is treated three times with 100 ml of
toluene/ethanol ~1:1) each time and the solvent is
distilled off each time. ~fter crystallization from
ethanol ~here is obtained 5-(3-methyl-1,2,4-oxadiazol-5-
-yl)-3-nitropyrocatechol in the form of yellow crystals of
m.p. 201-202.
Example 76
a) 143.8 ml of n-butyllithium solution (1.53M in hexane)
are added dropwise at -70 within 30 minutes to 35.~ g of
l-bromo-2-fluorobenzene (dissolved in 600 ml of tetra-
hydrofuran). After stirring at -70 for 60 minutes 48.5 g
of 3-methoxy-4-benzyloxybenzaldehyde (dissolved in 450 ml
of tetrahydrofuran) are added dropwise thereto during 30
minutes. The reaction mixture is stirred at -70 for 2
hours and at 0 for 30 minutes, poured into a mixture of
ice and 150 ml of 2W sulphuric acid and extracted three
times with 500 ml of ether. The combined ether phases are
washed wi~h saturated sodium chloride solution, dried over
sodium sulphate and evaporated. There is obtained
~3~2~8
_ 99 _
4-(benzyloxy)-2'-fluoro-3'-methoxybenzhydrol as a
yellowish oil which can be used directly in the subsequent
reaction step.
In an analogous manner:
al) Fro~ 3-methoxy-4-benzyloxyben2aldehyde and 1-bromo-3-
-fluorobenzene there is obtained 4-(benzyloxy)-3'-
-fluoro-3-methoxybenzhydrol as an oil;
a2) from 3-methoxy-4-benzyloxybenzaldehyde and 1-bromo-4-
-fluorobenzene ~here is obtained 4-(benzyloxy)-4'-~luoro-
-3-methoxybenzhydrol as an oil;
a3) from 3-methoxy-4-benzyloxybenzaldehyde and l-bromo-
-2,6-di~luorobenzene there is obtained 4-(benzyloxy)-
-2',6~-difluoro-3-methoxybenzhydrol as an oil;
a4) fro~ 3-methoxy-4-benzyloxybenzaldehyde and 1-bromo-2-
-chlorobenzene there is obtained 4-(benzyloxy)-2~-chloro-
-3-methoxybenzhydrol as an oil;
aS) from 3-methoxy-4-benzyloxybenzaldehyde and 1-bromo-3-
-chlorobenzene there is obtained 4-(benzyloxy)-3'-chloro-
-3-methoxybenzhydrol as an oil;
a6) from 3-methoxy-4-benzyloxyben2aldehyde and 1-bromo-4-
~chlorobenzene there is obtained 4-(benzyloxy)-4~-chloro-
3 -3-methoxybenzhydrol as an oil:
a7) from 4-benzyloxy-3-methoxybenzaldehyde and
2-bromotoluene there is obtained 4-(benzyloxy)-3-methoxy-
-2'-methylbenzhydrol as an oil:
a8) from 3-methoxy-4-benzyloxybenzaldehyde and
4-bromotoluene ~here is obtained 4-(benzyloxy)-3-methoxy-
- 100 -
-4'-methylbenzhydrol as an oil:
a9) from 3-me~hoxy-4 benzyloxybenzaldehyde and
l-bromobenzonitrile there is obtained 4-(benzyloxy)-2'-
-cyano-3-methoxybenzhydrol as an oil and
alO) from 3-methoxy-4-benzyloxybenzaldehyde and
l-bromo-2-trifluoromethylbenzene there is obtained
4-(benzyloxy)-3-methoxy-2l-(trifluoromethyl)benzhydrol as
an oil.
b) 69.8 g of 4-(benzyloxy)-2~-fluoro-3-methoxybenzhydrol
(dissolved in 600 ml of methylene chloride) are trea~ed
within 30 minutes at 20 with 45.3 g of pyridinium
chlorochroma~e and stirred at 20 for 3 hours. The
precipitate Eormed is subsequently filtered off and washed
with methylene chloride. The filtrate i6 evaporated and
the residue is filtered on 100 g of silica gel wi~h ether.
After recrystallization from ether there is obtained
4-(benzyloxy)-2'-fluoro-3-methoxybenzophenone of m.p.
118-120.
In an analogous manner:
bl) From 4-(benzyloxy)-3`-fluoro-3-methoxybenzhydrol ~here
is obtained 4-(benzyloxy)-3'-fluoro-3-methoxybenzophenone
as an amorphous solid;
b2) from 4-(benzyloxy)-4~-fluoro-3-methoxybenzhydrol there
is obtained 4-tbenzYloxY)-4l-fluoro-3-methoxYbenzoPhenone
of m.p. 99-101 (Erom ether/hexane);
b3) from 4-(benzyloxy)-2',6'-difluoro-3-methoxybenzhydrol
3S there is obtained 4-(benzyloxy)-2~,6~-difluoro-3-methoxy-
benzophenone of m.p. 139-141 (from methylene
chloride/ether);
~a3~
- 101 --
b4) from 4-(benzyloxy)-2~-chloro-3~methoxybenzhydrol there
is obtained 4-(benzyloxy)-2~-chloro-3-methoxybenzophenone
of m.p. 128-130 (from ether);
s
b5) from 4-(benzyloxy)-31-chloro-3-methoxybenzhydrol there
is obtained 4-(benzyloxy)-3l-chloro-3-methoxybenzophenone
as an amorphous solid:
b6) from 4-(benzyloxy)-4~-chloro-3-methoxybenzhydrol there
is obtained 4-~benzy}oxy)-4'-chloro-3-methoxybenzophenone
of m.p. 106-108 (from methylene chloride/hexane):
b7) from 4-(benzyloxy)-3-methoxy-2~-methylbenzhydrol there
is obtained 4-(benzyloxy)-3-methoxy-2~-methylbenzophenone
of m.p. 86-88 (from isopropyl ether):
b8) from 4-(benzyloxy)-3-methoxy-4'-methylbenzhydrol there
is obtained 4-(benzyloxy~ 3-methoxy-4'-methylbenzophenone
of m.p. 79-81 (from ether/hexane);
b9) from 4-(benzyloxy)-2~-cyano-3-methoxybenzhydrol there
is obtained 4-~benzyloxy)-2'-cyano-3-methoxybenzophenone
as an amorphous solid and
blO) from 4-(benzyloxy)-3-methoxy-2~-(trifluoro-
methyl)benzhydrol there is obtained 4-(benzyloxy)-3-
-methoxy-2'-(trifluoromethyl)benzophenone of m. e . 103-105
(from ether).
c) 170 ml of 33 percent hydrobromic acid in glacial
acetic acid are added at 20-25 within 20 minutes to
42.4 g o~ 4-(benzyloxy)-2~-fluoro-3-methoxybenzophenone
(dissolved in 450 ml of methylene chloride). After
stirring at 20 for 1.5 hours the reaction mixture is
poured into 750 ml of ice-water: the methylene chloride
phase is separated and the aqueous phase is extracted a
~3~
- 102 -
further twice with 200 ml of methylene chloride. The
combined methylPne chloride phases are washed with 1200 ml
of water, dried over sodium sulphate and evaporated. In
order ~o remove the resulting benzyl bromide, the oily
residue is treated with hexane and decanted off. There is
obtained 2'~fluoro-4-hydroxy-3-methoxybenzophenona as a
yellowish oil which can be used directly in the subsequent
reaction step.
In an analogous manner:
cl) From 4-(benzyloxy)-3'-fluoro-3-methoxybenzophenone
there is obtained 3~-fluoro-4-hydroxy-3-methoxybenzo-
phenone of m.p. 133-135 (from methylene chloride/low-
-boiling petroleum ether);
c2) from 4-(benzyloxy)-~'-fluoro-3-methoxybenzophenone
there is obtainçd 4'-fluoro-4-hydroxy-3-methoxybenzo-
phenone of m.p. 139-141 (from etheL)
c3) from 4-(benzyloxy)-2',6'-difluoro-3-methoxybenzo-
phenone there is obtained 2~,6~-difluoro-4-hydroxy-3-
-methoxybenzophenone of m.p. 130~132 (from methylene
chloride/low-boiling petroleum ether);
c4) from 4-(benzyloxy)-2~-chloro-3-methoxybenzophenone
there is obtained 2'-chloro-4-hydroxy-3-methoxybenzo-
phenone as an amorphous solid;
c5) from 4-(benzyloxy)-3~-chloro-3-methoxybenzophenone
there is obtained 3'-chloro-4-hydroxy-3-methoxybenzo-
phenone of m.p. 136-13B (from methylene chloride):
c6) from 4-(benzyloxy)-4`-chloro-3-methoxybenzophenone
there is obtained 4~-chloro-4-hydroxy-3-methoxybenzo-
phenone of m.p. 114-116 (from methylene chloride/low-
~3~P~
-- 103 -
-boiling petroleum ether);
c7) from 4-(benzyloxy)-3-methoxy-2'-methylbenzophenone
there is obtained ~-hydroxy-3-methoxy-2l-methylbenzo-
phenone of m.p. 103-105 (from iso~ropyl ether);
c8) from ~-(benzyloxy)-3-methoxy-4~-methylbenzophenone
there is obtained ~-hydeoxy-3-methoxy-4~-methylbenzo-
phenone of m.p. 103-105 (from ethar/low-boiling petroleum
ether);
c9) from 4-(benzyloxy)-2`-cyano-3-methoxybenzophenone
there is obtained 2'-cyano-4-hydroxy-3-methoxybenzophenona
of m.p. 124-126 (from ether/n-hexane~ and
clO) ~rom ~-(benzyloxy)-3-methoxy-2~-(tri~luoro-
methyl)benzophenone there ls obtained 4-hydroxy-3-methoxy-
-2'-ttrifluoromethyl)benzophenone o~ m.p. 115-117 ~from
ether).
d) 7.8 ml of 65 percent nicric acid are added dropwise at
Z0 within 20 minutes to 29.4 g of 2~-fluoro-4-hydroxy-3-
-methoxybenzophenone (dissolved in 450 ml of acetic acid).
~fter stirring for 1.~ hours the reaction mixture is
poured into 2 1 o~ ice-watPr and ~he precipitate formed is
filtered off, washed with water an~ dissolved in methylene
chloride. 'rhe methylene chloride solution is washed with
water, dried over sodium sulphate and evaporated. The
residue is recrystallized rom methanol. There is obtained
~-fluoro-4-hydroxy-3-methoxy-5-nitrobenzophenone of m.p.
127-129.
In an analogous manner:
dl) From 3~-fluoro 4-hydroxy-3-methoxybenzophenone thera
is obtained 3'-fluoro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone of m.p. 163-170 (from methanol);
~L3~
- 104 _
d2) from 4'-fluoro-4-hydroxy-3-methoxybenzophenone there
is obtained 4'-~luoro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone of m.p. 126-128 (from ether):
d3) from 2l,6~-difluoro-4-hydroxy-3-methoxybenzophenone
there is obtained 2',6'-difluoro-4-hydroxy-3-methoxy-5-
-nitrobenzophenone of m.p. 147-149 (from methanol):
d4) from 2'-chloro-4-hydroxy-3-methoxybenzophenone there
is obtained 2~-chloro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone of m.p. 123 125 ~from ether);
d5) from 3'-chloro-4-hydroxy-3-methoxybenzophenone there
is obtained 3'-chloro-4-hydroxy-3-methoxy-5-nitrobenæo-
phenone o~ m.p. 152-154 (from methanol);
d6) from 4'-chloro-4-hydroxy-3-methoxybenzophenone there
is obtained ~'-chloro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone of m.p. 129-131 (from methylene chloride/low-
-boiling petroleum ether);
d7) from 4-hydroxy-3-methoxy-2'-methylbenzophenone there
is obtained 4-hydroxy-3-methoxy-2l-methyl-5-nitrobenzo-
phenone of m.p. 125-127~ (from ethanol);
d8~ from 4-hydroxy-3-methoxy-4'-methylbenzophenone there
is obtained 4-hydroxy-3-methoxy-4'-methyl-5-nitrobenzo-
phenone of m.p. 137-139 (from methylene chloride/ether);
d9) from 2~-cyano-4-hydroxy-3-methoxybenzophenone there is
obtained 2~-cyano-4 hydroxy-3-methoxy-s-nierobenzophenone
of m.p. 163-164 (from methanol);
dlO) from 4-hydroxy-3-methoxy-2'-(trifluoromethyl)-
benzophenone there is obtained 4-hydroxy-3-methoxy-5-
-nitro-2~-(trifluoromethyl)benzophenone of m.p. 138-140
~3~
- 105 _
(from methylene chloride/low-boiling petroleum ether):
dll) from 4-hydroxy-3,4~-dimethoxybenzophenone there
is obtained 4-hydroxy-3~,4`-dimethoxy-5-nitrobenzophenone
of m.p. 134-136 (from methanol~ and
dl2) from 4-hydroxy-3,3~,4'-tri~ethoxybenzo~henone
there is obtained 4-hydroxy-5-nitro-3,3`,4'-trimethoxy-
benzophenone of m.p. 178-180 (from methanol).
e) 24.8 g of 2'-fluoro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone (dissolved in ~20 ml of glacial acetic acid,
100 ml of 33 percent hydrobromic acid in glacial acetic
acid and 69 ml of 48 percent aqueous hydrobromic acid) are
boiled under reflux for 4 hours. The reaction mixture is
subsequently evaporated under reduced pressure and the
residue is distilled with toluene. The residue i~
dissolved in methylene chloride, washed with water, dried
over sodium sulphate, fil~ered and evaporated. The product
is crystallized from methylene chloride/low-boiling
petroleum ether. There is obtained 2'-fluoro-3,4-
-dihydroxy-5-nitrobenzophenone of m.p. 169-171.
In an analogous ~anner:
el) Prom 3'-fluoro-4-hydroxy-3-methoxy-5-nitrobenzophenone
there is obtained 3'-fluoro-3,~-dihydroxy-5-nitrobenzo-
phenone of m.p. 124-126 (from methylene chloride);
3~
e2) ~rom 4'-fluoro-4-hydroxy-3-methoxy-5-nitrobenzophenone
there is obtained 4'-fluoro-3,4-dihydroxy-5-niero~enzo-
phenone of m.p. 171-173 (from methylene chloride);
e3) from 2',6'-difluoro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone there is obtained 2',6'-difluoro-3,4-dihydroxy-5-
-nitrobenzophenone of m.p. 194-196 (from methanol);
~3~
- 106 -
e4) from 2'-chloro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone there is obtained 2'-chloro-3,~-dihydroxy-S-nitro-
benzo~henone of m.p. 129-131 (from methylene chloride/
low-boiling petroleum ether);
e5) from 3'-chloro-4-hydroxy-3-methoxy-5-nitrobenzo-
phenone there is obtained 3l-chloro-3,4-dihydroxy-5-ni~ro-
benzophenone of m.p. 143-145 (from methylene chloride/
low-boiling petroleum ether);
e6) ~rom 4'-chloro-4-hydroxy-3-methoxybenzophenone there
is obtained 4~-chloro-3,4-dihydroxybenzophenone o m.p.
174-176 (from methylene chloride):
e7) from 4--hydroxy-3-methoxy-2`-me~hyl-5-nitrobenzo-
p~lenone there is obtained 3,4-dihydroxy-2'-methyL-5-nitro-
benzophenone of m.p. 164-166 (from methy]ene chloride);
e8) from 4-hydroxy-3-methoxy-4'-methyl-5-nitrobenzo-
phenone there i5 obtained 3,4-dihydroxy-4l-methyl-5-nitro-
benzophenone o-f m.p. 146-148 (from methylene chloride);
e9) from 2`-cyano-4-hydroxy-3-methoxy-5-nitrobenzophenone
there is obtained 2'-cyano-3,4-dihydroxy-5-nitrobenzo-
phenone of m.p. 159-161 (from methanol~:
e}0) from ~-hydroxy-3-methoxy-5-nitro-2~ (trifluoro-
methyl)benzophenone there is obtained 3,4-dihydroxy-~-
-nitro-2'-(trifluoromethyl)benzophenone of m.p. 146-148
(from methanol);
ell) from 4-hydroxy-3,4'-dimethoxy-5-nitrobenzophenone
there is obtained 5-nitro-3,4,4l-trihydroxybenzophenone of
m.p. 212-214 (from methanol/methylene chloride) and
el2) from 4-hydroxy-5-nitro-3,3~,4~-trimethoxybenzo-
:~3~J;Z~
- 107 -
phenone there is obtained 5-nitro-3,3~,4,4l-tetrahydroxy-
benzophenone of m.p~ 222-224 (from ether).
ExamPle 77
A suspension of 13.8 g of 2-bromo-3',4'-dihydroxy-S'-
-nitroacetophenone is treated with 9.0 g of l-(phenethyl)-
-2-thiourea in 150 ml of n-butanol and the mixture is
heated to boiling under reflux for 3 hours. After cooling
to room ~emperature ~he crystals are filtered off under
suction and crystallized from n-butanol. There is obtained
'~-nitro-5-t2-(phenethylamino~-4-thiazolyl]pyrocatechol
hydrobromide of m.p. 249-251.
ExamPle 78
In analogy to Example 38, from 2-bromo-3',4'-
-dihydroxy-5l-nitroacetophenone and 2-aminobenzophenone
there is obtained 2-(3,4-dihydroxy-S-nitrobenzoyl)-3-
-phenylindole of m.2. ls6-l98o (from isopropanol).
Example 79
A suspension of 8.3 g of 2-bromo-3',~'-dihydroxy-5'-
-nitroacetophenone is treated with l-(l-adamantyl)-2-
-thiourea in 90 ml of n-butanol and the mixture is heated
to boiling under reflux for 4 hours. After cooling to room
temperature the crystals are filtered of f under suction
and recrystallized from n-butanol. There is obtained
5-[2-(1-adamantylamino)-5-thiazolyl]-3-nitropyrocatechol
hydrobromide of ~. p. 245-247~ .
Example 80
A suspension of 2.6 g of (3,4-dihydroxy-5-nitro-
banzoyl)methyl acetate in 20 ml of ethanol and 20 ml of lN
- 108 -
hydrochloric acid is heated to boiling under reflux for 5
hours. The reaction mixture is then evaporated, the
residue is distilled with toluene and then recrystallized
from ethanol. There is obtained 2,3`,4`-trihydroxy-5~-
-nitroacetophenone of m.p. 208-210.
Example ~1
A solution of 4.0 g of n-hexyl 3,~-dihydroxy-5-nitro
phenylglyoxylate and 1.5 g of diaminomaleonitrile in 35 ml
o~ ethanol is heated to boiling under reflux for 24 hours.
The alcohol is then distilled off, the residue is
dissolved in ether, washed with water, dried over sodium
sulphate, filtered and evaporated. There i~ obCained
6-hydroxy-5-(3,4-dihydroxy-5-nitrophenyl)-2,3-pyrazine-
dicarbonitrile of m.p. >300 (from ether/methylene
chloride).
~xample 82
a) 4.2 g of 5-~bromoacetyl)-2,3-dimethoxybenzonitrile
dissolved in 150 ml of methylene chloride are treated with
8.9 ml of boron tribromide. The reaction mixture is
stirred at 20 for 18 hours. IC is subsequently poured
into 22U ml of saturated sodium hydrogen carbonate
solution and 100 g of ice, adjusted to pH 6 with glacial
acetic acid and extracted with ethyl acetate. The organic
phase is washed with water, dried over sodium sulphate and
evaporated. There is obtained 5-(bromoacetyl)-2,3-
-dihydroxybenzonitrile as an amorphous solid.
b) 3.8 g of 5-~bromoacetyl)-2,3-dihydroxybenzonitrile
dissolved in 25 ml of N,N-dimethylformamide are treated
with N-phenylthiourea and stirred at 100~ for 5 hours.
Thereafter, the solvent is removed by evaporation. The
residue is treated with 200 ml of lN sodium carbonate
~ 3~
- 109 _
solution and extracted three times with 100 ml of
methylene chloride each time. The combined organic phases
are washed with water, dried over sodium sulphate and
evaporated. The residue is chromatographed on 30 g of
silica gel with ethyl acetate. The thus-obCained crude
product is treated with 40 ml of lN hydrochloric acid,
evaporated and crystallized from acetone. There is
obtained 5-(2-anilino-4-thiazolyl)-2,3-dihydroxybenzo-
nitrile hydrochloride of m.p. 245-247.
Exam~lè 83
a) 25 ml of tert.-butyllithium solution (1.4M in hexane)
are added dropwise at -70 within 10 minutes to 10 g of
4-(benzyloxy)-3-methoxy-bromobenzene dissolved in 100 ml
of tetrahydrofuran. After stirring at -70 for 1 hour 5 g
of quinoline-4-carbaldehyde dissolved in 50 ml of tetra-
hydrofuran are added dropwise within 30 minutes. The
reaction mixture is stirred at -40 for 1 hour and a~ -5
for 1 hour, poured into Z00 ml of water and adjusted to
p~ 4 with glacial acetic acid. The mixture is extracted
three times with 50 ml of ether each time. The combined
ether phases are washed with water, dried over sodium
sulphate and evaporated. There is obtained a-~4-(benzyl-
oxy)-3-methoxyphenyl]-4-quinolinemethanol as an amorphous
solid.
b) 9.4 g of a-~4-~benzyloxy)-3-methoxyphenyl]-4-
-quinolinemethanol dissolved in 200 ml of methylene
chloride are treated with 6.5 g of pyridinium chloro-
cheomate, whereupon the mixture is stirred at room
temperature for 3 hours. The insoluble constituents are
subsequently filtered off. The ~iltrate is eva~orated and
the residue is chromatographed on 150 g of silica gel with
ethyl acetate. There is thus obtained 4-~benzyloxy)-3-
-methoxyphenyl 4-quinolyl ketone as an amorphous solid.
~3~32~
-- 110 --
c) 15 ml of 33 percent hydrobromic acid in glacial acetic
acid are added dropwise within 5 minutes at room
temperature to 7.5 g of 4-(benzyloxy)-3-methoxyphenyl
4-quinolyl ketone dissolved in 150 ml oE methylene
chloride. After stirring at 20 for 4.5 hours the reaction
mixture is poured portionwise into 250 ml of saturated
sodium bicarbonate solution. The methylene chloride phase
is separated: the aqueous phase is extracted a further
twice with 100 ml of methylene chloride each time. The
combined methylene chloride phases are dried over sodium
sulphate and evaporated. The residue i6 recrystallized
from methylene chlcride/hexane. There is obtained
4-hydroxy-3-methoxyphenyl 4-quinolyl ketone of m.p.
190-192.
d) 0.37 ml of 65 percent nitric acid i6 added dropwise at
room temperdture to 1.3 g of 4-hydroxy-3-methoxyphenyl
4-quinolyl ketone. After stirring for 3 hours the reaction
mixture is poured into ice-water, adjusted to pH 6 with
conc. ammonia and the precipi~ate formed is filtered off.
The thus-obtained residue is heated under reflux in 20 ml
of acetonitrile, whereupon the crystals are filtered off
at 0. There is obtained 4-hydroxy-3-methoxy-5-nitrophenyl
4-quinolyl ketone of m.p. 246-248.
e) 1 g of 4-hydroxy-3-methoxy-5-nitrophenyl 4-quinolyl
ketone dissolved in 30 ml of 48 percent aqueous hydro-
bromic acid is stirred at 100 for 18 hours. After cooling
to room temperature the reaction mixture is diluted with
30 ml of water and the precipitate is filtered off under
suction. There is obtained 3,4-dihydroxy-5-nitrophenyl
4-quinolyl ketone hydrobromide of m.p. 273-275 (from
acetonitrile).
~3~
Example 84
a) 18.8 ml of n-butyllithium solution (1.6M in hexane)
are added dropwise at -50 within 10 minutes to 4.03 g of
thiophene dissolved in 40 ml of tetrahydrofuran. After
stirring at -~0 for 30 minutes 6.3 g of 3,4-dimethoxy-5-
-nitrobenzaldehyde dissolved in 100 ml of tetrahydrofuLan
are added dropwise within 30 minutes. The reaction mixture
is stirred at -50 for 1 hour at 0 for 30 minutes and
poured into lOo ml of 2N sulphuric acid. The mixture is
extracted three times with 100 ml of ether each time; the
combined ether phases are washed with sodium chloride
solution, dried over sodium sulphate, filtered and
evaporated. There is obtained a-(3,4-dimethoxy-5-
-nitrophenyl)-2-thiophenemethanol of m.p. 79-81 (from
methylene chloride/hexane).
b) 9.9 g of a-~3,4-dimethoxy-5-nitrophenyl)-2-
-thiophenemethanol dissolved in 30C ml of acetone are
treated with 90 g of manganese dioxide and heated under
reflux for 4 hours. The manganese dioxide is removed by
- suction filtration and the filtrate is evaporated. There
is obtained 3,4-dimethoxy-5-nitrophenyl 2-thienyl ketone
of m.p. 102-104 tfrom methylene chloride/hexane).
c) 2 g of 3,4-dimethoxy-5-nitrophenyl 2-thienyl ketone
are stirred at 100 for 8 hours in a mixture of 20 ml of
30-33 percent hydrobromic acid in glacial acetic acid and
20 ml of 48 percent aqueous hydrobromic acid. The reaction
mixture is subsequently evaporated to dryness. The residue
is taken up in ethyl acetate, washed with water, dried
over sodium sulphate and filtered, and the ~iltrate is
evaporated. ~fter recrystallization from ethyl acetate~
hexane there is obtained 3,4-dihydroxy-5-nitrophenyl
2-thienyl ketone of m.p. 155-157.
~3~
~ 112 -
ExamPle A
The interlocking gelatine capsules of the following
composition can be manufactured in a manner known per se:
Inqredients _ount in ma/capsules
~-Dopa 100
Benserazide hydrochloride 29.3
3,4-Dihydroxy-5-nitrophenyl
4-pyridyl ketone 25
Gelatine
Mg stearate
Avicel 93-7
Mannitol 100
Capsule fill weight 350 mg
*Trade Mark
A