Note: Descriptions are shown in the official language in which they were submitted.
13~38~
-- 1 --
NOVEL beta-LACTAMS AND THEIR PRODUCTION
This application is a division of our prior
application Serial No. 498,692 filed on December 27, 1985.
The present invention relates to beta-lactams and
their production. More particularly, it relates to novel
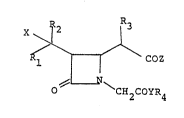
beta-lactam compounds of the formula:
~2 ~3
R~COZ ( I)
o CH2COYR4
wherein Rl and R2 are the same or different and each represent
a hydrogen atom or a lower alkyl group, R3 is a lower alkyl
group, R4 is a hydrogen atom, a carboxyl-protecting group or
a thiolcarboxyl-protecting group, X is a hydrogen atom, a
hydroxyl group or a protected hydroxyl group, Y is an oxygen
atom or a sulfur atom and COZ is a carboxyl group, an
activated or protected carboxyl group, a thiolcarboxyl group
or an activated or protected thiolcarboxyl group, and their
production.
Since the successful isolation from nature of the
antibiotic substance "thienamycin" [U.S. patent 3,950,357;
J.Am.Chem.Soc~, 100, 313 (1978)], various carbapenem compounds
have been reported. Among them, there are Xnown some carba-
penem compounds substituted with an alkyl group at the
l-position, and l-methylcarbapenem compounds are particularly
notable as they exert strong antimicrobial activity
against various microorganisms with excellent stability
in living bodies [EP-0071908A; ~eterocycles, 21, -
- 2 - 1~ ~33 8 Q
29 (1984)]. However, their known methods of preparation are
troublesome in that they require a lengthy series of reaction
steps. Further, such methods do not permit the stereospecific
formation of the l-methyl group.
As a result of extensive study, it has now been
found that the beta-lactam compounds of the formula (I)
according to the invention are valuable intermediates for the
production of l-alkylcarbapenem compounds having the following
fundamental skeleton:
-~3
,.~N ~'
O
COO~
whèrein R3 is as defined above. Particularly, they make it
possible to stereospecifically form an alkyl group at the
l-position.
In the definitions of the beta-lactam compounds of
the formula (I?, the term "lower" is intended to mean a group
15 having not more than 10 carbon atoms, preferably not more than
8 carbon atoms, more preferably not more than 5 carbon atoms.
For instance, the lower alkyl group represented by Rl, R2 or
R3 may be an alkyl group of 1 to 4 carbon atoms, e.g.,
methyl, ethyl, n-propyl or isopropyl.
The hydroxyl-protecting group (i.eO the group
protecting the hydroxyl group) in the protected hydroxyl group
~ 3 ~ 13 ~3 3 8 Q
may be lower alkoxycarbonyl for example Cl-C4 alkoxycarbonyl
(e.g. t-butyloxycarbonyl), halogenated lower alkoxycarbonyl
for example, halogenated (Cl-C3)alkoxycarbonyl (e.g. 2-iodoethyl-
oxycarbonyl, 2,2,2-trichloroethyloxycarbonyl), ar(lower)-
alkoxycarbonyl for example, phenyl(Cl-C4)alkoxycarbonyl op-
tionally bearing any substituent(s) on the benzene ring
(e.g. benzyloxycarbonyl, o-nitrobenzyloxycarbonyl, p-nitro-
benzyloxycarbonyl, p-methoxybenzyloxycarbonyl?, tri(lower)-
alkylsilyl for example, tri(Cl-C4)alkylsilyl te.g. trimethyl-
silyl, t-butyldimethylsilyl?, substituted methyl for example,
Cl-C4 alkoxymethyl (e.g. methoxymethyl), Cl-C4 alkoxy-
(Cl-C4~alkoxymethyl (e.g. 2-methoxyethoxymethyl), Cl-C4
alkylthiomethyl (e.g. methylthiomethyl), tetrahydropyranyl,
etc.
The carboxyl-protecting group (i.e. the group
protecting the carboxyl group) and the thiocarboxyl-
protecting group (i.e. the group protecting the thiolcarboxyl
group) may be conventional ones, and specific examples
include lower alkyl for example, Cl-C4 alkyl (e.g. methyl, ethyl,
isopropyl, t-butyl?, halogenated lower alkyl for example, halo-
genated Cl-C3 alkyl (e.g. 2-iodoethyl, 2,2,2-trichloro-
ethyl), lower alkoxymethyl for example, Cl-C4 alkoxymethyl (e.g.
methoxymethyl, ethoxymethyl, isobutoxymethyl), lower ali-
phatic acyloxymethyl for example, Cl-C5 alkanoyloxymethyl (e.g.
acetoxymethyl, propionyloxymethyl, butyryloxymethyl, piva-
loyloxymethyl?, lower alkoxycarbonyloxyethyl for example, 1-
(Cl-C4 alkoxycarbonyloxy)ethyl (e.g. l-methoxycarbonyloxy-
ethyl, l-ethoxycarbonyloxyethyl~,optionally substituted
- 4 - 1313380
lower alkenyl for example, C3-C10 alkenyl optionally sub-
stituted with Cl-C4 alkyl or phenyl (e.g. allyl, 2-methylallyl,
3-methylallyl, 3-phenylallyl), optionally substituted mono-
aryl(lower)alkyl for example, phenyl(Cl-C4)alkyl optionally
bearing any substituent(s) chosen from Cl-C4 alkoxy, nitro,
halogen and the like on the benzene ring (e.g. benzyl,
p-methoxybenzyl, 2,4-dimethoxybenzyl, o-nitrobenzyl, p-
nitrobenzyl, p-chlorobenzyl), optionally substituted diaryl-
(lower)alkyl for example, diphenyl(Cl-C4)alkyl optionally
bearing any substituent(s) chosen from Cl-C4 alkoxy and the
like on the benzene ring(s) (e.g. diphenylmethyl, di-p-anisyl-
methyl?, aryl for example, phenyl optionally substituted with
halogen, nitro, Cl-C4 alkoxy or the like (e.g. phenyl,
p-chlorophenyl, 2,4,5-trichlorophenyl, p-nitrophenyl,
o-nitrophenyl, p-methoxyphenyl), heteroaryl for example, pyridyl
or pyrimidyl optionally substituted with Cl-C4 alkyl (e.g.
2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 2-(4,6-dimethyl)
pyrimidyl?, phthalidyl, etc.
The carboxyl-activating group (i.e. the group
activating a carboxyl group) and the thiolcarboxyl-
activating group (i.e. the group activating a thiolcarboxyl
group may be the ones respectively derived from carboxyl and
thiolcarboxyl so as to enhance their reactivity, and their
examples include active ester, active acid anhydride, etc.
Specific examples of the symbol Z include halogen (e.g. chlo-
rine, bromine, iodine?, lower alkoxycarbonyloxy for example,
Cl-C5 alkoxycarbonyloxy (e.g. ethoxycarbonyloxy, isopropoxy-
carbonyloxy, sec-butoxycarbonyloxy?, lower alkanesulfonyloxy
13~3~80
-- 5 --
for example, Cl-C4 alkanesulfonyloxy (e.g. methane-sulfonyl-
oxy), arylsulfonyloxy for example, phenylsulfonyloxy
optionally bearing any substituent(s) on the benzene ring
(e.g. p-toluenesulfonyloxy), di(lower)alkylphosphoryloxy
for example, di(Cl-C4)alkylphosphoryloxy (e.g. dimethylphos-
phoryloxy, diethylphosphoryloxy?, diarylphosphoryloxy for
example, diphenylphosphoryloxy optionally bearing any substi-
tuent(s) on the benzene ring (e.g. diphenylphosphoryloxy?,
cyclic imidoxy for example, N-succinimidoxy or N-phthalimidoxy,
heteroaryl for example, imidazolyl or triazolyl, heterocyclo-
alkyl for example, 3-(2-thioxo)thiazolidinyl, etc.
Production of the beta-lactam compounds ~I)
according to the invention will be hereinafter explained in
detail.
lS Process A:-
The beta-lactam compound of the formula:
X IR2 R~
Rl ~ N \ COY R5 (I-l)
O . CH2COOR4
wherein Rl, R2, R3 and X are each as defined above, Y is an
oxygen atom or a sulfur atom, R4 is a protective group for the
carboxyl and R5 is a protective group for the carboxyl or
thiolcarboxyl is obtainable by reacting a compound of the
formula:
- 6 - ~313380
_ R2 R3
Rl n COY'R5 (II)
o~L~EI
wherein R1, R2, R3, R5, X and Y' are each as defined above
with a compound of the formula:
M-CH2COOR4 (III)
wherein R4 is as defined above and M is an activated
hydroxyl group, i.e. an active ester,in an inert solvent in
the presence of a base. If necessary, a phase transfer
catalyst may be used.
Examples of the inert solvent include aromatic
hydrocarbons (e.g. benzene, toluene), ethers (e.g.
tetrahydrofuran, dioxane, diethyl ether), halogenated
hydrocarbons (e.g. methylene chloride, dichloroethane,
chloroform), ketones (e.g. acetone, methyl isobutyl ketone),
acetonitrile, dimethylformamide, dimethylsulfoxide,
hexamethylphosphoric amide (HMPT), t-butanol, water, etc.
These may be used solely or in combination. As the base,
there may be used organic bases (e.g. 1,8-diaza-
bicyclo[5.4.0]undeca-7-ene (DBU), alkali metal hydrides
~e.g. sodium hydride, potassium hydride), metal salts of
amines (e.g. sodium amide, lithium diisopropylamide, lithium
bis(trimethylsilyl)amide), alkali metal hydroxides (e.g.
sodium hydroxide, potassium hydroxide~, alkali metal
carbonates ~e.g. sodium carbonate, potassium carbonate),
alkali metal alkoxides (e.g. potassium t-butoxide), etc. As
~ 7 - 13~338~
_ the phase transfer catalyst, there may be employed benzyl
triethyl ammonium chloride, tetra-n-butyl ammonium bromide,
tetraethyl ammonium bromide, etc.
The base or the phase transfer catalyst may be
used in such an amount that the reaction proceeds smoothly.
Occasional heating or cooling is desirable to accelerate or
control the reaction.
Still, preferred examples of the reactively
modified hydroxyl group represented by M are active esters
for example, suIfon~l esters-(e.g. mesylate,-tosylate) and
halogens (e.g. chlorine, bromine, iodine).
Process B:-
(1) ~he beta-lactam co~pound of the formula:
R2 R3
X>~ ~
Rl ~ COOH (I-2)
0~
CH2COOR4
wherein R1, R2, R3, R4 and X are each as defined above is
obtainable by subjecting the compound (I-1) wherein Y' is a
sulfur atom to selective hydrolysis. The selective hydroly-
sis may be carried out by a per se conventional procedure,
for instance, under basic conditions.-
` - 8 - 13133g~
(2) The beta-lactam compound of the formula:
R2 R3
>1,~
Rl l l COOR5 (I-3)
CH2COOR4
wherein R1, R2, R3, R4 and X are each as defined above and
R5 is a hydrogen atom or a protective group for the carboxyl
but at least one of R5 and R4 represents a hydrogen atom
can be prepared by eliminating at least one of the protec-
tive groups R4 and R5 from the compound ~I-l) wherein Y' is
an oxygen atom.
(a) When both of R5 and R4 in the compound (I-3)
represent hydrogen atoms, a protective group R4 may be
introduced therein. (b) When R5 and ~4 in the compound
(I-3) represent respectively a protective group and a
hydrogen atom, a protective group R4 may be introduced
therein, followed by selective elimination of the protective
group R5. In both cases, the beta-lactam compound (I-2) can
be obtained as the ultimate product.
Elimination of the protective group(s) R4 and/or
R5 may be accomplished by various per se conventional
procedures depending on the protectiYe ~roup,and those procedures
can be chosen from hydrolysis, catalytic reduction, treat-
ment with acids or bases, reduction, etc. For selectiveelimination of either one of the protective groups R4 and
R5, it is convenient to select an appropriate combination of
9 13~3~
R4 and R5 so as to make such selective elimination possible.
The introduction of the protective group R4' in the case (a)
as well as the introduction of the protective group R4 and
the subsequent elimination of the protective group R5 may be
5 accomplished by ~ se conventional procedures.
Process C:-
The beta-lactam compound of the formula:
R2 3
COOZ' (I-4)
C82COOR4
wherein R1, R2, ~3, R4 and X are each as defined above and
COZ' is an activated ox protected carboxyl group or an
10 activated or protected thiolcarboxyl group can be produced
by subjecting the compound (I-2) to any treatment for
converting the carboxyl group into an activated or protected
carboxyl or thiolcarboxyl group.
When the -COZ' group represents an activated
carboxyl group for example, an active ester or an active acid anhydride
or a protected thiolcarboxyl group, the symbol Z' may be any
one chosen from those as hereinabove exemplified for the
symbol Z. In case of the -COZ' group being a substituted
aryloxy group, for instance, it may be preferably p-nitro-
phenyloxy, o-nitrophenyloxy, 2,4,5-trichlorophenyloxy or the
like. In case of the -COZ' group being a heteroaryloxy
group, it is preferred to be o-pyridyloxy, p-pyridyloxy or
the like.
- lo ~3~338~
The above conversion may be accomplished by
various procedures, of which typical exzmples are shown
below:
(a) The compound (I-2) is reacted with a
halogenating agent (e.g. oxalyl chloride, thionyl chloride)
in the presence or absence of a base to obtain the corres-
ponding acid halide.
(b) The compound (I-2) is reacted with a chloro-
formic ester le.g. ethyl chloroformate) in the presence of a
base to obtain the corresponding mixed acid anhydride.
(c) The compound (I-2) is re~cted with l,l'-car-
bonyldiimidazole to obtain the corresponding acylimidazole
derivative.
(d) The compound (I-2) is reacted with thiazoli-
~ine-2-thione in the presence of a dehydrating agent (e.g.
dicyclohexylcarbodiimide) to obtain the corxesponding
acylthiazolidine-2-thion derivative.
(e1 The compound (I-2) is reacted with a thiol
pound for e~le, a substituted or unsubstituted thiophenol,
4,6-dimethyl-2-mercaptopyrimidine or 2-mercaptopyridine with
the aid of a dehydrating agent (e.g. dicyclohexylcarbodi-
imide), or is converted into its active esterfor e~ple, acid
halide, mixed acid anhydride or acylimidazole derivative,
followed by reacting with said thiol compound.
(f) ~he compound (I-23 is reacted with a hydroxyl
compound for ~mple~N-hydroxysucc~mide, N-hydroxyphthalimide,
a substituted or unsubstituted phenol or 2-pyrridone with the
aid of a dehydrating agent (e.g. dicyclohexylcarbodiimide),
13~ 338~
or is converted into its active ester for.e~le, acid halide,
mixed acid anhydride or acylimidazole derivative, followed
by reacting with said hydroxyl compound.
Process D:-
The beta-lactam compound of the formula:
R2 R3
X>~ ~
Rl r l COY~R5 (I-6~
0~ CH2COSR4
wherein Rl, R2, R3, R5, X and Y' are each as defined above
and R4 is a thiolcarboxyl-protecting group can be obtained
by reacting a compound of the formula:
R2 R3
R~COY ' R5 (I-5)
C~I2COOH
`- wherein Rl, R2, R3, R5, X and Y' are each as defined above
10 with a thiol of the formula:
, ~SR4
wherein R4 is as defined above.
The reaction may be carried out by a ~er se
conventional procedure for acylation of a thiol group.
- 12 - 1 3 ~ 3~ 80
Process E:-
The beta-lactam compound of the formula:
R2 R3
X~, I
R ~ ~ COZ' (I-7~
' O H2CSR4
wherein Rl, R2, R3, R4, X and Z' are each as defined above
can be produced from the corresponding compound (I-6) ac-
5 cording to the procedure as explained in Processes B and C.
Process F:-
The beta-lactam compound of the formula:
R2 R3
R~ COOH (I-2)
O CH2C~OR4
wherein Rl, R2, R3 and R4 are each as defined above and X'
is a hydrogen atom or a protected hydroxyl group can be
10 produced by reacting the compound of the formula:.
R2 R3
OH
1 ~ (IV~
O H
wherein Rl, R2, R3 and X' are each as defined above with an
acetic acid compound of the formula:
- 13 - 1~33~
M-C~2CR4 (III)
wherein R4 and M are each as defined above in an inert
solvent in the presence of a base and subjecting the
resulting product of the formula:
R2 R3
OH
Rl r l (v)
O CEI2COOR4
wherein Rl, R2, R3, R4' and X7 are each as defined above
5 to oxidation.
In the above process, the reaction at the first
step may be carried out substantially by the same process
as in Process A. The oxidation at the second step may be
effected by a per se conventional procedure for conversion
10 of a primary alcohol into the corresponding carboxylic acid,
for instance, by treatment with an oxidizing agent (e.g.
chromium (VI) oxide-sulfuric acid, chromium oxide-pyridine).
As stated above, the beta-lactam compounds lI) of
the invention are useful as intermediates for production
15 of l-alkylcarbapenem compounds. For instance, a compound of
the formula:
~ 2 13
R ~ COZ' (I-8)
0 ~2COYF~4
- 14 - 131~
wherein Rl, R2, R3, Y and Z' are each as defined above, X'
is a hydrogen atom or a protected hydroxyl group and R4 is a
carboxyl or thiolcarboxyl-protecting group, which covers the
compounds (I-4) and (I-7) as well as the corresponding
5 compounds derived therefrom, when the symbol X represents a
hydroxyl group, by protection of such hydroxyl group in a
per se conventional procedure, may be used as the starting
material, which is converted into the 1-alkylcarbapenem
compound (I) in various ways, of which typical examples are
10 shown below.
Procedure (1):-
COY4 ~ ~ ~
COYR"
(I-8) (VI) 4
wherein Rl, R2, R4, X', Y and Z' are each as defined above
and R3 is a hydrogen atom or a lower alkyl group.
The beta-lactam compound (I-8) is treated with a
15 base in an inert solvent to give the compound (VI). When R3
is a lower alkyl group, there can ~e obtained as the major
product the compound (VI) retaining the steric configuration
based on the asymmetric carbon atom at the 5-position
bonding to the 4-position of the beta-lactam ring in the
20 starting compound (I-8). As the inert solvent, there may
be used ethers (e.g. diethyl ether, tetrahydrofuran,
- - 15 ~ g0
dioxane, ethylene glycol dimethyl ether), aromatic hydro-
carbons (e.g. benzene, toluene), acetonitrile, dimethyl-
formamide, hexamethylphosphoric triamide (HMPT), t-butanol,
etc. These solvents may be used solely or in combination.
5 Preferred examples of the baseinclude me ~ salts of amines
(e.g. lithium diisopropylamide, lithium bis(trimethylsilyl)-
amide, sodium amide), metal salts of alcohols (e.g.
potassium t-butoxide), alkali metal hydrides (e.g. sodium
hydride, potassium hydride), sodium methylsulfinylmethide,
10 etc.
The base is to be used in such an amount that the
reaction can proceed smoothly, and it may be usually from
1.5 to 3 equivalents to the starting compound (I-8~. The
reaction ~emperature may be accelerated or controlled by
1~ heating or cooling, and it may be normally from -75 ts 50C.
Reo~y-of the produced compound (VI) from the
reaction mixture may be accomplished by application of a per
se conventional procedure for post-treatment. However,
post-treatment of the reaction mixture should be
20 effected with special care, because, for instance, the alkyl
group at the l-position of the compound of the formula:
R2 R3
X~
Rl ¦ >=o ~VI-l)
0~ ~
COYR4
wherein Rl, R2, R3, R4, X' and Y are each as defined above
- 16 _ 1313~
may be epimerized on treatment with a base or during
concentration.
~ l-a) The thus produced compound (VI) wherein Y is
an oxygen atom can be converted into the corresponding
S l-alkylcarbapenem compound having an antimicrobial activity
according to the following route:
R2 R3 R2 R3
X~=o ~ Rl ~5--Ro
(VI I ) COOR4 (VII ~ ) COOR4
R2 R3
X~
~1 ~ ",/> S-~o
(VIII) COOH
wherein Rl, R2, ~3, R4 and X' are each as defined above and
X is a hydrogen atom or a hydroxyl group and Ro is an
organic group.
The compound (VI') is first converted into the
carbapenem compound (VII') by the procedure as described in
U.S. Patent 4,350,631, European Patent 54,917 or Japanese
Patent Publication (unexamined) No. 123182/82 or any similar
procedure thereto. Then, the resulting carbapenem compound
15 (VII') may be, if necessary, subjected to elimination of the
hydroxyl-protecting group, elimination of the carboxyl-
~L31338~
- 17 -
protecting group and/or elimination of the amino-protecting
group to give the carbapenem compound (VIII).
Eliminatlon of the protecting group may be
accomplished by a ~ se conventional procedure, althcugh it
~varies- with the kind of the protecting group. When, for
instance, the hydroxvl-protecting group and the nitrogen-
protecting group in the compound (VII) are haloaenated lower
alkoxycar~onyl or arllower)alkoxycarbonyl or the carboxy-
protecting group in the compound (VII) are halogenated lower
alkyl, ar(lower)alkyl or b~hydryl, it may be eliminated by
application of an appropriate reduction. Such reduction may
be effected using zinc with acetic acid, tetrahydrofuran or
methanol in case of the protecting group being halo(lower)-
al~oxycarbonyl or ~alo(lower)alkyl, or using a catalyst e.g.,
platinum or palladium-carbon in the case-where the protect~
group is ar(lower)alkyloxycarbonyl, ar(lower)alkyl or
benzhydryl.
When a catalyst as stated above is used, the
reduction is no~mally effected in an inert solvent chosen
from lower al]canols te.g. methanol, ethanol), ethers (e.g.
tetrahydrofuran, dioxane), organic acids (e.g. acetic acid),
water and buffers le.g. phosphate buffer, morpholinopropane-
sulfonate buffer), etc. These solvents may be used solely
or in combination. The reaction temperature may be usually
from 0 to 100C, preferably from 0 to 40C. The hydrogen
pressure may be ;atmospheric or elevated.
Still, such a protecting group as o-nitrobenzyl-
oxycarbonyl or o-nit-obenzyl may be eliminated also by
3 3 ~ ~
.
photo-reaction.
(1-b) The compound (VI~ wherein Y is a sulfur
atom can be converted into the corresponding carbapenem
compound according to the following route:
R2 R3 R2 R3
X~ I X'~ I
O ; Rl ~ SRo
(VI~) CSR4 / I COSR4
~ ~ (VII")
R2 R3 R2 R3
1 ~ ~ S~ ~ SRo
(VII') COOR4 (VIII~
wherei~ Rl, R2, R3, R4, R4, Ro, X' and X are each as
defined above.
The compound (VI") is first converted into the
carbapenem compound (VII n ) in the same manner as in (l-a).
Then, the carbapenem compound (VIIn) may be, if necessary,
subjected to elimination of various protecting groups. As
to a thioester group, the application of a p r ~e conven~
tional hydrolytic procedure can successfully accomplish its
elimination giving the carbapenem compound (VIII;. Instead
of application of the hydrolytic procedure, treatment with a
lS silyl compound e.g.,- silanol may be applied to the carba-
- 19 ~3~3~
penem compound (VII") so that the carbapenem compound (VIII)
can be obtained directly. Alternatively, the carbapenem
compound (VII n ) may be treated with an alcohol in the
presence of a silver salt te.g. silver trifluoroacetate) to
give the compound (VII'), which is then treated in the same
manner as in (l-a) to give the carbapenem compound (VIII).
In the substituent SRo of the carbapenem compounds
(VII') and (VII"), Ro may be any one as heretofore used in
connection with carbapenem compounds, and its examples
include substituted or unsubstituted alkyl or alkenyl having
1 to 10 carhon atoms; cycloalkyl, alkylcycloalkyl or cyclo-
alkylalkyl in which the cycloalkyl group has 3 to 6 carbon
atoms; aryl le.g. phenyl), aralkyl wherein the aryl group is
phenyl and the alkyl portion has 1 to 6 carbon atoms;
heteroaryl, heteroarylalkyl or heterocycloalkyl, etc. These
groups may optionally ~ear at least one substi-
tuent chosen from amino, mono-, di- or trialkylamino,
hydroxyl, alkoxy, mercapto, alkylt~io, arylthio (e.g.
phenylthio), sulfamoyl, amidino, guanidino, nitro, ~alo
(e.g. chloro, bromo, fluoro), cyano and carboxyl. In said
substituents having a hetero ring, the hetero atom(s) in the
hetero ring may be chosen from oxygen, nitrogen and sulfur,
and their number may be from 1 to 4. The alkyl moiety in
said subs~ituents may have 1 to 6 carbon atoms.
- 20 - ~3
Procedure (2):-
COZ; ~ ~Rl ~ 3
COYR4
(I-8) (VI-2)
R-A ~ R ~ 0-8 + R-Z' + A
COYR4
(VI-2)
~2 R3
3 ~
COY~4
(IX)
wherein R1, R2, R3, R4, X', Y and Z' are each as defined
above and L is an activated hydroxyl group e.g.,- an active
ester of hydroxyl, B is an alkali metal atom and R-A is an
alkylating or acylating agent.
For the direct production of the compound (IX) from
the compound ~I-8), the latter is treated as in Procedure
- 21 - 13~3~
(1). Without isolation of the product, the reaction mixture
is treated with an alkylating or acylating agent (e.g. iodo-
methane, iodopropane, allyl bromide, benzyl bromide, methyl
p-toluene sulfonate) so as to catch the residue of the
activating group e.g., an active ester residue, an active
acid anhydride residue or a thiol residue, followed by
treatment with a hydroxyl-activating agent e.g., an active
esterifying agent for hydroxyl to give the carbapenem
compound ~IX). The treatment with the alkylating or
acylating agent is preferably caxried out in the presence of
a base in an inert solvent.
~ s the active ester of hydroxyl represented by the
symbol L, there are exemplified substituted or unsubstituted
arylsulfonic esters (e.g. benzenesulfonic esters, p-toluene-
15 sulfonic esters, p-nitroben2enesulfonic esters, p-bromo-
benzenesulfonic esters3, lower alkanesulfonic esters (e.g.
methanesulfonic esters, ethanesulfonic esters), halo(lower)-
alkanesulfonic esters (e.g. trifluoromethanesulfonic
esters), diarylphosphoric esters (e.g. diphenylphosphoric
20 esters), halides (equal to esters with hydrogen halides)
(e.g. chlorides, bromides, iodides), etc. Preferred are
p-toluenesulfonic esters, methanesulfonic esters, diphenyl-
phosphoric esters, etc. Accordingly, any reagent which is
reacted with the compound (VI-2) to give the acti~e ester as
25 exemplified above may be used as the active esterifying
agent. Examples of the alkali metal atom represented by the
s~l B ~ lud~ li~um, sodium, potassium, etc. As the base,
there may be used the one as exemplified in Procedure (1)
i3~38~
- 22 -
:
for production of the compound (VI).
When the symbol R3 in the compound (I-8) is a
lower alkyl group, its treatment with a base in an inert
solvent affords the enolate salt (VI-2), which retains the
steric configuration on the basis of the asymmetric carbon
atom at the 5-position of the compound ~I-8). Even after
. conversion of the enolate (VI-2) into the compound (IX), the
steric configuration of the alkyl group represented by the
symbol R3 is unchanged. Thus, adoption of th~s Procedure
(2) gives the carbapenem compound (IX) without epimeriza-
tion. Still, the enolate (VI-2) in this case has a possi-
bility of taking a chelate structure of the formula:
R2 ~3
Rl~ _ O
o ~N ~= B
R4Y
..
wherein R1, R2, R3~ R4, X', Y and B are each as defined
above.
The active esterifying agent is to be used in an
amount sufficient to effect the reaction smoothly, and its
amount may be from 1 to 1.5 equivalents to the compound
(I-8). The reaction temperature may be usually from -78 -to
60C, prererably from -40 to 10C.
- 23 ~ 131 3 38 0
,
Procedure ( 3 ): -
~?~co; ~ o-~+3~)z6 1
: COYR4
(I-8) (VI-2)
R--A ~ o--9 + R-Z ~ +
COYR4
(VI-2 )
~L + R-Z' + A(~136)
COYR4
( IX)
Ro ~ i ~ S-Ro
COYR4
(VII )
- 24 - 13~3~8~
wherein Rl, R2, R3, R4, Ro, R-A, B, X', Y and Z' are each as
defined above.
When direct production of the carbapenem compound
- (VII) from the compound (I-8) is desired, the latter may be
converted into the carbapenem compound (IX) in the same
manner as Procedure (2). Without isolation of the compound
(IX), the reaction mixture is treated with a mercaptan
compound of the formula:
Ro~SH (X)
wherein Ro is as defined above in the presence of a base to
give the carbapenem compound (VII). The base to be used in
the treatment with the mercaptan compound (X) may be the
same as or different from that as used in the cyclization of
the compound (I-8) to the compound (IX). Likewise, the
inert solvent to be used in said treatment may be the same
as or different from that as used in said cyclization.
As the base, there may be used one chosen from
those as exemplified in Procedure (1). Other examples of a
suitable base include organic amines è.g., triethylamine,-
diisopropylethylamine, 4-dimethylaminopyridine, 1,8-diazabi-
cyclo[5.4.0]-7-undecene (DBU), 1,5-diazabicyclo[4.3.0~-
5-nonene (DBN) and 1,4-diazabicyclo[2.2.2]octane (DABCO).
Preferred examples of the solvent which is used to-ensure a
smooth reaction include acetoni-trile, dimethylform-
amide, dimethylsulfoxide, etc.
The base to be used together with the mercaptan
compound (X) may be employed in such an amount to assure
a` smooth reaction, and its amount may be
- 25 - 13~3~8~
in a large excess, preferably from 1 to 2 equivalents to the
compound (I-8). The mercaptan compound (X) and the base
may be introduced into the reaction system separately.
Alternatively, the salts formed between them may be added to
the reaction system.
Conversion of the beta-lactam compound (I-8) into
the carbapenem compound (IX) may be accomplished by carrying
out the reactions as above explained in order. When
desired, the carbapenem compound (IX) is subjected to
hydrolysis or elimination of the protecting group so that
the carbapenem compound (VII) can be-obtained. The
hydrolysis or ~he elimination of the protecting group may be
carried out in the same manner as in Procedure (l-a) (i.e.
conversion of the compound (VII') into the compound (VIII))
or (1-b) (i.e. conversion of the compound (VIIn) into the
compound (VIII)).
A typical example of the conversion from the beta-
lactam compound (I-8) into the l-beta-methylcarbapenem
compound is shown below:
- 26 - 13~3~
~Ri O jORi O
¦' ~LT~O-B + B(~3Z ~3
N~ //
O `CH2cOYR4 O
COYR4
(I-8 ) (VI-2 )
ORi O
R-A ~ ~ ~ -B + Q-Z' ~ A~
COYR4
(VI-2 )
ORl~ ORlo
H H H H
3 , ~ Ro
COYR4 COYR4
~IX ) (VII )
wherein R4, Y, Z', B, R-A, L and Ro are each as defined
above and Rlo is a hydroxyl-protecting ~roup.
Namely, 1) the beta-lactam compound (I-8 ) is
treated with a base in an inert solvent, 2) the residue Z'
is caught with an alkylating or acylating agent and then 3)
- 27 - 13~3~
the resulting product is treated with a hydroxyl-activating
group e.g., an active esterifying agent for hydroxyl.
When desired, 4J the resultant product is reacted with the
mercaptan compound (X) in the presence of a base or the salt
of the mercaptan compound (X) with the base. These reac-
tions may be carried out in a single re~ction vessel in
order to give the carbapenem compound IIX ) or (VII ).
Alternatively, the compound (I-8 ) may be
subjected to the reaction in 1), followed by post-treatment
to give the compound of the formula:
ORlo
(VI )
ro
O
COYR4
wherein R4, Rlo and Y are each as defined above. The beta-
methyl group at the l-position is apt to be epimerized when
the product is stored in a high concentration solution or in
a polar solvent e.g., acetonitrile. Thus, the stereo-
specific production of the compound (IX ) from the compound(VI ) as once isolated in a large scale would have a
technical problem. To the contrary, the conversion of not
the compound (VI ) but the compound (VI-2 n ~ into the
compound (IX ) is advantageous, because the production of
* *
the compound (IX ) or (VII ) can be accomplished without
epimerization of the methyl group at the l-be~a-position.
- 28 - 1313~8~
.
The compound lII) as the starting material in
production of the beta-lactam compound (I) according to this
invention can be produced in the manner as described in U.S.
Patent 4,350,631, European Patent 54917 or Japanese Patent
Publication (unexamined) No. 123182/82. For production of
the compound (II) wherein Y is a sulfur atom, introduction
of the -SR4 group may be carried out by a conventional
procedure as in the case of production of the compound
(I-6). While the starting compound (IV) can be produced in
the same manner as described in European Patent 10317 or
Japanese Patent Publication (unexamined) No. 8g285~80, it
may be produced according to the route as shown in the
following scheme:
R2 R2
X ~ COOH X ~ COOR4
J_~ ~ R~
O - R' O N R'
ll) (2)
Rl j~ ` R;7~\ D
o ~ R' O - R'
(3) (4)
- 29 _131~38~
~ ~ J
(5) (6)
X ~ ¦
O \ R' R'
(7) (8)
X~ 0
N
(IVa)
wherein Rl, R2, X and X' are each as defined above, R' is a
nitrogen-protecting group, A' is a halogen atom and P is a
hydroxyl-protecting group.
In the above scheme, the step A is directed to
conversion of the carboxylic acid (1) into the corresponding
ester (2). The conversion is usually carried out by appli-
cation of a per se conventional esterification procedure.
For instance, the carboxylic acid (1) obtained by the
process as described in Japanese Patent Publication (un-
examined) No. 96060/83 may be reacted with an alkyl halide
13~33~
- 30 -
in the presence of an acid-eliminating agent or with an
alkanol in the presence of a dehydrating agent to give the
ester (2).
At the step B, the ester (2) is reduced by treat-
ment with an organic metal compound e.g., magnesium halideor methyl lithium in an inert solvent, optionally followed
~y protection of a hydroxyl group in a ~er se conventional
procedure to give the corresponding alcohol (3).
At the step C, the alcohol (3) is dehydrated by
treatment with a dehydrating agent e.g., thionyl chloride
or tosyl chloride in the presence or absence of a base to
give the corresponding methylene compound (4).
At the step D, the methylene compound (4) is
halogenated by treatment with a halogenating agent e.g.,
molecular halogen or N-halogenosuccinimide in an inert
solvent to give the corresponding halogenated compound ~5).
At ~he step E, the halogenated compound (5) is
hydrolyzed by treatment with water in the presence of a low
atomic valency ion salt of a heavy metal ~e.g. copper,
silver) to give the corresponding hydroxyl compound (6).
At the step F, the hydroxyl compound (6) is
subjected to protection on the hydroxyl group to give the
corresponding protected hydroxyl compound (7).
At the step G, the protected hydroxyl compound (7)
is hydrogenated by catalytic hydrogenation to give the
corresponding methyl compound ~8).
At the step H, the methyl compound (8) is sub-
jected to elimination of the hydroxyl-protecting group
- 31 - 13~33~
. --
represented by the symbol P and elimination of the amino-
protecting group represented by the symbol R' simultaneously
or stepwise to give the corresponding free compound (IVa).
In the beta-lactam compound (I), the carbon atoms
at the 3- and 4-positions and the carbon atom bonding to the
beta-lactam ring and in the substituent attached to the
,, 4-position of such beta-lactam ring are all asymmetric
, - carbon atoms. Further, in the casewhere all of Rl, R2 and X
are different ~xm one another (e.g. Rl = methyl, R2 = hydrogen,
X = hydroxyl), the carbon atom bonding to the beta-lactam
ring and in the substituent attached to the 3-position of
such beta-lactam ring is an asymmetric carbon atom.
Accordingly, the beta-lactam compound of the formula (I~
covers optical isomers and stereo isomers due to said
asymmetric carbon atoms. Among those optical isomers and
stereo isomers, the compounds of the following formula:
H R3
J~
,. ~ COz
O CH2COYR4
wherei~ R3, R4 and Y and Z are each as defined aboveJare
particularly preferred in that they,have the same',configuration as
that of naturally occuring thienamycin at the carbon atom at
the 4-position.
Practical and presently preferred embodiments of
the invention are illustratively shown in the following
Examples and References Examples. This invention is,
- 32 - ~3~338~
however, not limited to these examples. In these examples,
the abbreviations have the following meanings: TBDMS,
t-butyldimethylsilyl; Ph, phenyl; tBu, t-butyl; PNB, p-
nitrobenzyl; PMB, p-methoxybenzyl; Im, l-imidazolyl; Bt,
l-benzotriazolyl; Ac, acetyl; PNZ, p-nitrobenzyloxycarbonyl;
DAM, di(p-anisyl)methyl; Z, benzyloxycarbonyl; Me, methyl.
13133~0
Exam~Le 1-1
OTBDMS OTBDMS
H H ~ ~ H H ~
COOcx2Ph ~L I COOC~I2Ph
N ~ - ~ ~ N
O H O ~ CH2COOtBu
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-~(lR)-l-benzyloxyczrbonylethyl]-
azetidin-2-one ~755 mg) in methylene chloride (10 ml), there
were added successively t-butyl bromoacetate (1.88 g), 50 %
S sodium hydroxide (620 mg) and triethylbenzylammonium
chloride (220 mg), followed by stirring at room temperature
for 2 hours. The reac~ion mixture was diluted with water
and diethyl ether. The aqueous layer was separated from the
organic layer and extracted twice with diethyl ether.
The extracts were cor~ined with the organic layer, washed
with water twice and with brine three times, dried over
sodium sulfate and evaporated. The residue was purified by
silica gel chromatography to give (3S,4S)-3-[(lR)-l-t-butyl-
di~ethylsilyloxyethyl]-4-~(lR)-l-benzyloxycarbonylethyl]-
lS l-(t-butyloxycarbonylmethyl)azetidin-2-one.
IR vmaxat (cm 1): 1755, 1730, 1450, 1400, 1380,
1360, 1242, 1~20, 1150, 830, 765, 740, 685.
NMR ~ (CDCl3): 0.04 (3H, s), 0.07 (3H, s), 0.85
(9H, s), 1.23 (3H, d, J = 6.3 Hz), 1.24 (3H, d, J = 6.9 ~z),
1.44 (9H, s~, 2.90 (1~, qd, J = 6.9 and 3.6 H,), 2.99 (;H,
dd, J = 2.0 and 6.6 Hz), 3.83 (2H, m), 5.10 (2H, s), 7.35
(SH, s).
- 34 _
131338
;
Example 1-2
OTBDMS OTBDMS
~ H H I ~ H H
/\ ~\ ~\
~ ¦ COOCH2Ph ~ I COOH
O CH2COOtBt O ~CH2COOt~3u
A solution of (3S,4S)-3-~(lR)-1-t-butyldimethyl-
silyloxyethyl]-4-[(lR)-1-benzyloxycarbonylethyl]-1-(t-
- butyloxycarbonylmethyl)azetidin-2-one (0.45 g) in 99.5 %
ethanoi (6 ml) was subjected to hydrogenation at room
tempera~ure in the presence of 10 % palladium-carbon (90 mg)
under atmospheric pressure, followed by filtration to remove
the catalyst. The filtrate was evaporated to give (35,4S)-
3-[(lR)-1-t-butyldimethylsilyloxyethyl]-4-[(lR)-l-carboxy-
ethyl]-l-(t-butyloxycarbonylmethyl)azetidin-2-one.
10IR ~naxt (cm 1~: 1760, 1740, 1730, 1455, 1360,
1245, 1224, 1150, 830, 770, 745.
NMR ~ (CDC13): 0.06 (3H, s), 0.08 (3H, s), 0.87
(9H, s), 1.24 (3H, d, J = 6.3 Hz), 1.25 (3H, d, J = 7.3 Hz),
1.48 (9H, s), 2.94 tlH, ~d, J = 7.1 and 3.0 ~z), 3.04 (lH,
15dd, J = 2.3 and 5.5 Hz), 3.98 (2H, m), 4.00 (lH, m), 4.21
(lH, m).
Example 1-3(1)
OTBDMS OTBDMS
~ H H 7 ~ H H ~
~ COOH ~ COSPh
0~ CH2COOtBu 0 ~CH2COOtBu
A mlxture of (3S,4S)-3-[(lR)-1-t-but~ldimethyl-
silvloxyethyl]~ (lR)-l-carboxyethyll -l-(t-butyloxy-
~33~
carhonylmethyl)azetidin-2-one (1.29 g) and N,N'-carbonyl-
diimidazole (604 mg) in dry acetonitrile (25 ml) was stirred
at room temperature for 1 hour. To the mixture, there were
aaded successively a solution of thiophenol (410 mg) in dry
acetonitrile (6 ml) and a solution of triethylamine (377 mg)
in dry acetonitrile (6 ml). After stirring at room temper-
ature for 0.5 hour, the reaction mixture was diluted with
ethyl acetate and dilute hydrochloric acid. The aqueous
layer was separated from the organic layer and extracted
with ethyl acetate three times. The extracts were combined
with the organic layer, washed with brine twice, dried
over sodium sulfate and evaporated. The residue was
purified by silica gel chromatography to gi~e (3S,4S)-3-
[(lR)-l-t-bu~yldimethylsilyloxyethyl]-~-[(lR~-l-phenylthio-
carbonylethyl]-1-(t-butyloxycarbonylmethyl)azetidin-2-one.
IR vmaxt (cm )~: 1760, 1740, 1705, 1367, 1250,
1227, 835, 770, 740.
~ xample 1-3(2)
OTBDMS OTBDMS
H H ~ ~ H H
~-~COO~ ~/~/\COS~C
O CH2COOtBu O ~CH2COOtBu
In the same manner as in Example 1-3(1) but
replacing thiophenol by p-chlorothiophenol, there was
obtained (3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxy-
ethyl]-4-[(lR)-l-p-chlorophenylthiocarbonylethyl]-l-tt-
butoxycarbonylmethyl)azetidin-2-one.
IR ~ma~ (cm ): 1760, 1740, 1705, 1480, 1365,
131~3~9
1260, 1230, 1155, 1095, 838, 775.
N~ ~ (CDC13): 0.10 (6H, s), 0.89 (9H, s), 1.26
t3H, d, J = 6.3 Hz), 1.31 (3H, d, J = 6.9 Hz), 1.43 (9H, s),
3.02 tlII, dd, J = 2.3 and 6.9 ~~), 3.14 (lH, qd, J = 3.3 and
6.9 Hz), 3.92 (2~, m), 7.34 (4H, m).
ExamDle 1-3(3)
OTBDMS OT~DMS
r H H ~ ~ H H ~ ~1
COOH ~ COO ~ Cl
O CH2COOtBu ~CH2COOtBu
To a solution of (3S,4S)-3-[tlR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-carboxyethyl]-l-(t-butyloxy-
carbonylmethyl)azetidin-2-one (100 mg) and 2,4,5-trichloro-
phenol (33 mg) in dry tetrahydrofuran (4 ml), there was
added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydro-
chloride (96 mg) under ice-ccoling, followed by stirring
overnight. The reaction mixture was diluted with diethyl
ether and ~ater. The organic layer was separated from the
aqueous layer, washed with brine, dried over sodium sulfate
and distilled ofr to remove the solvent to give (35,4S)-3-
~ )-1-t-butyldimethylsilyloxyethyl]-4-[(lR)-1-~2,4,5-tri-
chlorophenyloxy)carbonylethyl]~ t-butyloxycarbonylmethyl)-
azetidin-2-one.
IR ~maxat (cm 1): 1760, 1740 (sh), 1455, 1362,
1250, 1225.
~ 3~
Exam~le 1-3(43
OTBDMS OTBDMS
COOH COCl
O CH2COOtBu O CX2COOtBu
To a solution of (3S,4S)-3-[(lR)-1-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-carboxyethyl]-1-(t-butyloxy-
carbonylmethyl)azetldin-2-one (98 mg) in dry methylene
. chloride (1 ml), there was added a solution of thionyl
chloride (34 mg) in dry methylene chloride (0.5 ml) at room
temperature. The resultant mixture was stirred at the same
temperature for l hour and then boiled under reflux for 3
hours. After removal of the solvent, the residue was
dissolved in dry toluene, again distilled off to remo~e the
solvent and dried in vacuo to give (35,4S)-3-[(lR)-l-t-
butyldimethylsilyloxyethyl]-4-~(lR)-1-chlorocarbonylethyl]-
l-(t-butyloxycarbonylmethyl)azetidin-2-one.
IR ~naxt (cm 1): 1800 (sh), 1740, 1450, 1360,
1244, 1220, 930, 825, 770.
- 15Example 1-3(5)
OTBD~IS OTBDMS
COOH ~ COOBt
O CH2COOtBu O C~12COOtBu
In the same manner as in Example 1-3(3), (3S,4S)-
3-[(lR)-1-t-butyldimethylsilyloxyethvl]-4-[(lR~ carbo~y-
ethyl]-l-(t-butyloxycarbonylmethyl)azetidin-2-one (104 mg)
was treated with l-o~yben-triazole (5, mg) and 1-ethyl-3-
8 ~
(3-dimethylaminopropyl)carbodiimide hydrochloride (93 mg) to
give (3S,4S)-3-~(lR)-l-t-butyldimethylsilyloxyethyl3-4-
[(lR)-1-(1-benzotriazolyloxy)carbonylethyl]-1-(t-butyloxy-
carbonylmethyl~azetidin-2-one
IR vmeaat (cm 1): 1740 (sh), 1730, 1450, 1360,
1240, 1220, 822.
Exam~le 1-3(6)
OTBDMS OTBDMS
~COOEI ~\cos~1
O CH2COOtBu O ' H2COOtBU
In the same manner as in Example 1-3(3), (3S,4S)-
3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-[(lR)-l-carboxy-
ethyl]-l-(t-butyloxycarbonylmethyl)azetidin-2-one ~100 mg~
10 was treated with 2-mercaptopyridine (35 mg) and 1-ethyl-3-
(~-dimethylaminopropyl)carbodii~ide hydrochloride (100 mg)
to gi~e (3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyll-4-
[(lR)-1-(2-pyridylthio)carbonylethyl]-1-(t-butyloxycarbonyl-
methyl)azetidin-2-one.
15IR ~max (cm ): 1755, 1690, 1360, 1247, 1220,
1142, 830, 770.
Example 1-4
OTBDMS ~ OTBDMS
COSPh ~ COSPh
O H O CH2COOtBu
To a suspension of sodium hydride (31 mg) in dry
dimethylformamide (4.3 ml), there ~-ere added successi~ely
- - 39 -
~ 3 ~
t-butyl alpha-bromoacetate !835 mg) and (35,4S)-3-[(lR)-1-t-
butyldimethylsilyloxyethyl~-4-[(lR)-1-phenylthiocarbonyl-
ethyl]azetidin-2-one (0.42 g), and the resultant mixture was
stirred at room temperature for 1 hour under a nitrogen
stream. The reaction mixture was diluted with diethyl ether
and adjusted to pH 6.86 with a phosphoric acid buffer
solution. The aqueous layer was separated from the organic
layer and e~tracted with diethyl ether three times. The
extracts were combined with the organic layer, washed three
times with brine, dried over sodium sulfate and evaporated.
The residue was puriried by silica gel chromatography to
give (3S,4S)-3-[(lR)-l-t-butyldi~ethylsilyloxyethyl]-4-
[(lR)-l-phenylthiocarbonylethyl]-l-(t-butyloxycarbonyl'-
methyl)azetidin-2-one.
lhe IR spectrum of the product thus obtained was
identical with that of the product in Example 1-3(1).
Example 1-5
OTBDMS OT9DMS
H H ~ ~ H H
COS ~ Cl ~ COS ~ Cl
O'd O H2COOtBu
In the same manner as in Example 1-1, there was
obtained (3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-
[(lR)-l-p-chlorophenylthiocarbonylethyl]-l-t-butyloxy-
carbonylmethylazetidin-2-one from (3S,4S)-3-[(lR)-l-t-butyl-
dimethylsilyloxye~hyl]-4-[(lRJ-l-p-chlorophenylthiocarbonyl-
ethyl]azetidin-2-one.
- 40 -
131~3~
-
The IR spectrum and NMR spec~rum of the product
thus obtained were identical with those OL the product in
Example 1-3(2).
Example 2-1
OZ OZ
/ ~ \`COOCH2Ph ~ COOCH2Ph
0 H O CH2~0OtBu
To a solution of ~3S,4S)-3-[(lR)-l-t-benzyloxy-
S carbonyloxyethyl]-4-~(lR)-l-benzyloxycarbonylethyl]-
azetidin-2-one (71.94 g) in dry dimethylformamide (700 ml),
there were added successively t-~utyl bromoacetate (68.25 g)
and sodium hydride (9.24 g, S0 ~ oil suspension) with
ice-cooling, followed by stirring for 1 hour. The reaction
mixture was diluted with a 10 % aqueous ammonium chloride
solution (S00 ml), stirred for 30 minutes and extracted with
toluene (2 liters). The extract was washed with brine,
dried over sodium sulfate and evaporated. The residue was
purified by ~ilica gel chromatography to give (3S,4S)-3-
[(lR)-l-benzyloxycarbonyloxyethyl]-4-~(lR~ benzyloxy-
carbonylethyl]-l-(t-butyloxyca~bonylmethyl)azetidin-2-one.
IR vnaext (cm 1): 1765 (sh), 1740, 1455, 1370,
1268, 1160.
NMR ~ (CDCl3): 1.18 (3~, a, J = 6.9 Hz), 1.45
20 (3H, d, J = 6.3 Hz), 1.45 (9H, s), 2.86 ~lH, m), 3.26 (lH,
dd, J = 2.0 and 9.0 Hz), 3.55 (lH, d, J = 18 Hz), 4.04 (lH,
dd, J = 2.0 and 4.6 Hz), 4.10 (lH, d, J = 18 Hz).
~31~
ExamPle 2-2
OZ OE~
OOCH2Ph ~ COOH
O ~ N \ CH2COOtBu o ~ L ~ ~ CH2COOtBu
A solution of (3S,45)-3-[(lR)-l-benzyloxy-
carbonyloxyethyl]-4-ttlR)-l-benzyloxycarbonylethyl]-l-(t-
butyloxycarbonylmethyl)azetidin-2-one (81.50 g) in ethanol
(800 ml) was subjected to hydrogenation at room temperature
in the presen~e of 10 % palladium-carbon (8.15 g) under
~tmcspheric pressure, followed by filtration to remove the
catalyst. The filtrate and the washings were combined and
evaporated to give (3S,4S)-3-[(1~)-1-hydroxyethyl]-4-[(lR)-
l-carboxyethyl]-l-(t-butyloxycarbonylmethyl)azetidin-2-one.
IR ~n-eat (cm 1): ~740, 1720, 1440, 1360.
NMR ~ (CDC13): 1.28 (3~, d, J = 6.9 Hz), 1.33
(3H, d, J = 6.6 Hz), 2.84 tlH, m), 3.09 (lH, dd, J = 2.0 and
6.6 Hz), 3.76 (lH, d, J = 1~ Hz), 4.03 ~lH, dd, J = 2.0 and
5.3 Hz).
Example 2-3
OH OH
COO~ ~COSPh
O CH2COOtBu O CH2COOtBu
In the same manner as in Example 1-3~1), there was
obtained (3S,4S)-3-[(lR)-l-hydroxyethyl]-4-[[lR)-l-phenylthio-
Carbonylet~yl]-l-(t-butyloxycarbonylmethyl)azetidin-2-one
from (3S,4S)-3-t(lR)-l-hydroxyethyl]-4-[(lR)-l-carboxy-
- -42 -
13~3~
.
ethyl]-l-(t-butyloxycarbonylmethyl)zzetidin-2-one.
IR Vma~ (cm ~: 1745, 1725, 1290, }230, 1140 ,
950, 750.
Example 3-1
0~ OZ
- ~ H H ~ ~ H H e
~a) / ~ O~
O H 0 CH2COGtBu
OZ
(b) ~COOH
O ~H2COOtBu
(a) To a solution of (3S,4R)-3-~(lR)-l-benzyloxy-
carbonyloxyethyll-4-[(lR)-l-hydroxymethylethyl]-2-azeti-
dinone (30.7 g) in acetone (300 ml), there were added
t-butyl bromoacetate (33.0 g) and potassium carbonate (27.6
-- g), followed by stirring under reflux for 17 hours. The
reaction mixture was cooled down to room temperature and
filtered to remove insoluble materials. The filtrate
contained (3S,4R)-3-[(lR)-l-benzyloxycarbonyloxyethyl]-
4-[(lR)-l-hydro~ymethylethyl]-l-t-butoxycarbonylmethyl-
2-azetidinone.
IR ~maxat (cm 1): 1735, 1360, 1250, 1150, 1030,
15 955).
(b) The filtrate was diluted with water (15 ml)
and treated with the ~ones reagent, which was prepared from
chromium trioxide (16.92 g), 98 ~ sulfuric acid (26.52 g)
~ 43 ~ i 3 ~ 3 ~ ~ a
and wate- (~9.2 g), while ice-cooling for l hour. The
reaction mixture was quenched with isopropanol and diluted
with ethyl acetate (l liter) and water (300 ml). The
organic layer was washed with brine (300 ml x 4), dried over
magnesium sulfate (50 g) and evaporated in vacuo to give an
oily residue, which was purified by silica gel chromato-
graphy to give (35,4S)-3-[(lR)-1-ben~yloxycarbonyloxy-
ethyl]-4-[(lR)-l-carboxyethyl]-1-t-butoxycarbonylmethyl-
2-azetidinone (23.73 5, 54.5 %).
IR ~maxt (cm l): 1735, 1450, 1365, 1250, 1150,
1040.
NMR ~ tCDC13): l.l9 (3P., d, J = 6.9 ~z), 1.46
(9~, s), 3.26 (lH, dd, J = 2.3 and 8.6 Hz), 4.06 ~1~, dd, J
= 2.3 and 4.0 Hz), 7.36 (5H, s).
lS Examsle 3-2
OZ OZ
H
COOH ~~OSPh
O CEI2COOtBu O C~I2COOtBu
To a solution of (3S,4S)-3-[(lR)-l-benzyloxy-
carbonyloxyethyl]-4-[(lR)-1-carboxyethyl]-1-t-butoxy-
carbonylmethyl-2-azetidinone (23.75 g) in dry acetonitrile
(237 ml), there was added N,N'-carbonyldiimidazcle (10.SS g)
under ice-cooling, followed ~y stirring for 0.5 hour.
Thiophenol (7.21 g) and triethylamine (6.62 g) were then
added thereto under ice-cooling, followed by stirring for
2.5 hours. The reaction mi~ture was diluted with ethyl
44 -
1~33~
acetate (500 ml) and washed with lN hydrochloric acid (200
ml). The aqueous layer was separated from the organic layer
and extracted with ethyl acetate ~200 ml x 2). The extract
was combined with the organic layer, washed with brine (300
ml x 3), dried over magnesium sulfate and-e~aporated in
vacuo to give an oily residue, which was puriried by silica
gel chromatography to obtain (3S,4S)-3-[(lR)-1-benzyloxy-
carbonyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-l-t-
butoxycarbonylmethyl-2-azetidinone (19.46 g, 67.64 %).
IR ~max (cm ): 1760, 1735, 1700, 1440, 1365,
1255, 1150, 1045, 950, 740.
NMR ~ (CDCl3): 1.25 (3H, d, J = 6.9 Hz), 1.44
(9H, s), 1.47 (3H, d, J = 6.2 Hz), 3.12 (lH, m)~ 4.13 (1~,
dd, J = 2.6 and 4.5 ~z), 7.36 (lOH, m).
Exam~le 4-1
OTBDMS ~ OTBDMS
H H ; ~ H H
\ COOCH2Ph ~ COOCH2Ph
O CH2COOtl3u
In the same manner as in Example 1-1, a solution
of (3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-[(lS)-
l-benzyloxycarbonylethyl]azetidin-2-one (3.50 g) in methy-
lene chloride (45 ml) was treated with t-butyl alpha-bromo-
20 acetate (8.73 g), 50 % aqueous sodium hydroxide solution
(2.86 g) and triethylbenzylammonium chloride (1.02 g) to
give (3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-~-
[(lS)-l-benzyloxycarbonylethyl]-l-(t-butyloxymethyl)azeti-
~ ~5 ~ 13133~
din-2-one.
IR vmax (cm ): 1760 (sh), 1735, 1455, 1362,
1245, 1222, 835, 773.
Example 4-2
OTBDMS ; OTBDMS
~--COOC~2Ph ~ oo~
O CH2COOtBu O CH2COOtBu
In the same manner as in ~xample 1-2, (3S,4S)-3-
[~lR)-1-t-butyldimethylsilyloxyethyl]-4-[(lS)-l-benzyloxy-
carbonylethyl]-l-(t-butyloxycarbonylmethyl)azetidin-2-one
wa~ subjected to hydrogenation to give (3S,4S)-3-~(lR)-1-
t-butyldimethylsilyloxyethyl]-4-~(lSJ-l-carboxyethyl]~
(t-butyloxycarbonylmethyl)azetidin-2-one.
10IR ~NUaxol (cm 1): 3300 (broad), 1760 (sh), 1742,
1690, 990, 9~0, 825, 767.
Example 4-3(1)
OTBDMS ~ OTBDMS
H EI ~ ~ H H
COOH ~ COSPh
O `~CH2COOtBu o ~CH2COOtBu
In the same manner as in Example 1-3(1) but
replacing the starting material by (3S,4S)-3-[(lR)-l-t-
butyldimethylsilyloxyethyl]-4-[(lS)-l-carboxyethyl]-l-t-
butoxycarbonylmethyl-2-azetidinone, there was obtained
(3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-[(lS)-
l-phenylthlocarbon~lethyl]-1-t-butoxycarbonylmethyl-2-
13~-3~?J~J~
- azetiainone.
IR ~naext (cm 1): 1760, 1740 (sh!, 1700, 1365,
1250, 1225, 950, 830, 773, 740, 680.
Example 4-3(2~
OTBDMS _ OTBDMS
--`coo~ ~\Cos-<~N~C~3
CH2COOtBu ~ X2COOtBu 3
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lS)-l-carboxyethyl]-1-t-butyloxyca-
rbonylmethylazetidin-2-one (lG0 mg) in dry methylene
' - chloride ( ml), there was added oxalyl chloride (37 mg),
and the resultant mixture was stirred at room temperature
for 2 hours in the presence of a catalytic amount or
dimethylformamide. 4,6-Dimethyl-2-mercaptopyrimidine 150
mg) and 4-dimethylaminopyridine (44 mg) were added thereto,
Collowed by stirring. The reaction mixture was diluted with
methylene chloride, washed with dilute sulfuric acid, water
and sodium bicarbonate and brine successively, dried over
- 15 sodium sulfate and distilled off to remove the solventO The
residue was purified by silica gel chromatography to give
(3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-[(lS)-
1-(4,6-dimethylpyrimidin)-2-ylthiocarbonylethyl]-1-t-butyl-
oxycarbonylmethyl)azetidin-2-one.
IR ~ma~ tcm ): 1760, 1740 (sh), 1580, 1362,
1247, 830, 770.
- 47 - 1~3~
~xamPle 4-3(3)
OTBDMS _ OTBDMS ~ O
COOH ~ <OO-
O CH2COOtBu O CH2COOtBu O
To a solution of (3S,4S)-3-[(lR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lS)-l-caxboxyethyl]-l-(t-butyloxy-
- carbonylmethyl)azetidin-2-one (100 mg) and N-hydroxysuccin-
imide (33 mg) in dry dimethylformamide (0.3 ml), there was
added N,N'-dicyclohexylcarbodiimlde (74 mg), followed by
stirring at 0 to 5C overnight. The reaction mixture was
diluted with ethyl acetate, followed by addition of sulfuric
acid. The a~ueous layer was seprated from the organic layer
and extracted with ethyl acetate twice. - The extracts
were combined with the organic layer, wasned with water four
times and brine, dried over sodium sulfa~e and distilled off
to remove the solvent. The residue was purified by silica
gel chroma~ography to give 135,4S)-3-[ (lR)-1-t-butyldi-
methylsilyloxyethyl]-4-{(lS)-l succinimidoxycarbonylethyl]
1-tt-butyloxycarbonylmethyL~azetidin-2-one.
IR ~meaxt (cm lJ: 1805, 1760 (sh), 1740, 1455,
1360/ 1200, 11~5, 1055, 830, 762, 745.
13~338~
Example 4-3(4)
OTBD~fS OTBD~S
r H H ~ ~ H H
~f \ COOH ~ ~ COIm
0 ~ ~ CH2COOtBu O CH2COOtBu
A solution of (3S,4S)-3-[(lR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lS)-l-carboxyethyl]-l-(t-butyloxy-
carbonylmethyl)azetidin-2-one (42 mg) and N,N'-carbonyl-
diimidazole (19 mg) in dry acetonitrile (0.9 ml) was stirred
at room temperature for 1 hour and distilled off to remove
the solvent. The residue was purified by silica gel
chromatography to give (3S,4S)-3-[(lR)-l-t-butylaimethyl-
silyloxyethyl]-4-[(ls)-l-(l-imidazolyl)carbony~lethyi3
(t-butyloxycarbonylmethyl)azetidin-2-one.
IR ~meaxt (cm 1): 1760 (sh), 1735, 1382, 1360,
1225, 114S, 935, 827, 745.
Example 5-1
OTBDMS OTBDMS
r ~ H ~ H H
--`COOCH2Ph COOCH2Ph
O ~ H O ~ CH2COOMe
To a solution of (3S,4S)-3-[(lR)-l-t-butyl-
dimethylsilyloxyethyl]-4-~(lS)-l-benzylo~ycarbonylethyl~-
azetidin-2-one ~1.56 g) in methylene chloride (20 ml), there
were added successi~ely methyl alpha-bromoacetate (916 mg),
50 % aqueous sodium hydroxide solution (1.28 g) and
triethylbenzylam~onium chloride (455 mg), followed by
stirring at room temperature for 2 hours. The reaction
~ 49 ~ ~3~ 33~ ~
mixture was diluted with water. The aqueous layer was
separated from the organic layer and extracted twice
with diethyl ether. The extracts were combined with the
organic layer, washed successively with watex twice and
brine three times, dried over sodium sulfate and evaporated.
The residue was purified by silica gel chromatography to
give (3S,45)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-
~(lS)-l-benzyloxycarbonylethyl]-l-(methoxycarbonyl~ethyl)-
azetidin-2-one.
10- IR vmaxt (cm 1): 1760, 1740, 1460, 1407, 1360,
1250, 1~15, 1180, 1140, 835, ~70, 745.
MMR ~ (CDC13): 0.07 (3H, s), 0.08 (3~, s), 0.87
(9H, s), 1.24 (3H, d, J = 6.3 Hz), 1.25 (3H, d, J = 7.2 Hz),
2.77 (lH, m), 3.63 (3H, s), 3.93 (2H, s), 3.96 (lH, dd, J =
2.0 and 9.6 Hz), 4.19 (lH, m), 5.09 (2H, s), 7.36 (SH, s).
Example 5-2
OT~DMS ` OTBDMS
H H _ ~ H H
COOCH2Ph ~ COOH
~ N~ ~ ~I N~
o CH2COOMe ~ CH2COOMe
A solution of t3S,4S)-3-[(lR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lS)-1-benzyloxycarbonylethyl~-1-(methox-
carbonylmethyl)azetidin-2-one (400 mg) in 99.S % ethanol (6
ml) was subjected to hydrogenation at room temp~rature in
the presence of 10 % palladium-carbon (80 mg) under
atmospheric pressure, followed by filtration to remove the
catalyst. The filtrate was evaporated to give (3S,4S)-3-
[(lR)-l-t-butyldimethylsil~loxyethyl]-4-[(lS)-l-carboxy-
3 8 ~
ethyl]-1-(meth~xycarbonylmethyl)azetidin-2-one.
IR ~maex (cm ): 1740, 1705, 1435, 1240, 1215,
1135, 830, 770.
Example 5-3
OTBDMS OTBD~IS
H H - Y H H
COOH ~ OSPh
o~cH2cooMe ~CH2COOMe
In the same manner as in Example 1-3(1) but
replacing the starting material by (3S,4S)-3-[(lR)-l-t-
butyldimethylsilyloxyethyl]-4-[(lS)-l-carboxyethyl]-1-
methoxycarbonylmethyl-2-azetidinone, there was obtained
-(3S,4S)-3-[(lR)-1-t-b~tyldimethylsilyloxyethyl]-4-[(lS)-
: 1-phenylthiocarbonyiethyll-1-methoxycarbonylmethyl--2-
azetidinone.
IR ~meax (cm 1): 1750, 1695, 1437, 1405, 1247,1202, 950, 830, 770, 740.
NMR ~ (CDC13): 0.07 (3H, s), 0.09 (3H, s), 0.87
(9H, s), 2.88 llH, dd, J = 2.3 and 6.6 Hz), 3.03 (lH, m),
15 3.70 (3H, s), 4.02 (lH, dd, J = 2.0 and 9.2 ~z), 4.19 (lH,
m), 7.41 (SH, m).
- 51 - i 3 ~ a
~'
Example 6-1
OTBDMS OH
> ~ COSPh
O H2COOtBu O CHzCOOH
OTBDMS
COSPh
(b)
O CH2COOH
(a) To a solution of (3S,4S)-3-[(lR)-l-t-butyl-
dimethylsilyloxyethyll-4-[(lR)-1-phenylthiocarbonylethyl]-1-
(t-butyloxycarbonylmethyl)azetidin-2-one (190 mg) in
methanol (4.5 ml) was added 6N hydrochloric acia (1.5 ml),
rollowed by s~irring at room temperature for 15 minutes.
The reaction mixture was diluted with chloroform and brine.
The aqueous layer was separated from the organic layer and~
extracted with chloroform twice.-~ -. The extracts were
combined with the organic layer, dried over sodium sulfate
10 and evaporated. The residue was dissolved in trifluoro-
acetic acid (1.0 ml) and anisole (0.1 ml). After stirring
at room temperature for 25 minutes, the solvent was evapo-
rated and removed azeotropically with dry toluene twice -
to give (3S,4S)-3-[(lR)-l-hydroxyethyl]-4-[(lR)-l-phenyl-
15 thiocarbonylethyl]-1-(carboxymethyl)azetidin-2-one as a
crude product.
- 52 ~ 13~ 3~$~
(b) A mixture of the cruae product as obtained
above, t-butyldimethylchlorosilane (246 mg) and imidazole
(151 mg) in dry dimethylformamide (2 ml) was stirred over-
night. The resulting mixture was poured into water and
extracted with ethyl acetate three times. The organic layer
was washed successively with dilute sulfuxic acid, water
(five times) and brine ~ twice), dried over sodium.
sulfate and evapora~ed. The residue was purified by silic~
gel chromatography to give (3S,4Si-3-[(lR)-l-t-butyl-
dimethylsilyloxyethyl]-4-[(lR)-1-phenylthiocarbonylethyl~-1-
(car~oxymethyl)azetidin-2-one.
IR ~meaxt (cm 1): 3350 (broad), 1760 (sh), 1737,
1700, 1245, 1140, 830, 767, 742.
xample 6-2
OT8DM~ OTBDMS
H H ~ ~ H H
COSPh ~ COSPh
O ~CH2COOH ~CH2COSPh
A mixture of (3S,4S)-3-[(lR)-l-t-butyldimethyl-
silyloxyethyl~-4-[(lR)-1-phenylthiocarbonylethyl]-l-
(carboxymethyl)azetidin-2-one (154 mg) and N,N'-carbonyl-~
diimidazole (66 mg) in dry acetonitrile (2.9 ml) was stirred
at room temperature for l hour. Thiophenol (56 mg) in dry
acetonitrile (1 ml) and triethylamine (52 mg) in dry
acetonitrile (0.5 ml) were added thereto, followed by
stirring at room temperature for 30 minutes. The reaction
mixture was diluted with ethyl acetate, poured into dilute
sulfuric acid anà e~.tracted with ethyl acetate three times.
_ 53 - ~3~
The organic layer was washed successively with dilute
sulfuric acid ana brine ( twice), drled over sodium
suirate and e~aporated. The residue was purified by silica
gel chromatography to give (35,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-1-
(phenylthiocarbon~lmethyl)azetidin-2-one.
IR vmaxt (cm 1): i760, 1700, 1478, 1440, 1250,
1140, 835, 773, 7~0, 682.
NMR C (CDC13): 0.08 (3H, s), 0.10 (3~, s), 0.89
(9H, s), 1~27 (3H, d, J = 6.3 Hz), 1.32 (3H, d, J - 7.3 Hz),
3.11 (lH, dd, J - 2.0 and 6.9 Hz), 3.20 (lH, m), 4.24 (lH,
m), 4.30 (2H, m), 7.41 (lOH, s).
Exam~le 7-1
OTEDMS ~ OTBDMS
H ~ ~ H H
COSPh ~ C~SPh
~ >
O' CH2COOtBu O -CH2cH
In the same manner as in Example 6-1, there was
obtained (3S,45)-3-[(lR)-1-t-butyldimethylsilyloxyethyl]-4-
[(lS)-1-phenylthiocarbonylethyl]-1-(carboxymethyl)azetidin-
2-one from (35,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-
[(lS)-l-ph~nylthiocarbonylethyl]-l-(t-butyloxycarbonyl-
methyl)azetidin-2-one.
IR vmaxt (cm 1): 3350 (broad), 1760 (sh), 1740,
1250, 940, 825, 770, 740, 680.
_ 54 ~ 33 ga
Example 7-2
OTBDMS OTBD~IS
H H ~ ~ H ~ `
OSPh ~ `COSPh
O CH2COOH o ~C~I2COSPh
In the same manner as in Example 6-2 but replacing
(3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-~(lR)-l-
phenylthiocarbonylethyl]-l-(carboxymethyl)a~etidin-2-one by
(3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-[(lS)-1-
phenylthiocarbonylethyl]-1-(carboxymethyl)azetidin-2-one,
there was obtained (3S,4S)-3-[(lR)-l-t-butyldimethylsilyl-
cxyethyl]-4-~(lS)-l-phenylthiocarbonylethyl]-l-(phenylthio-
carbonylmethyl)azetidin-2-one.
IR vneat (cm 1): 1760, 1705, 1480, 1442, 1250,
g55, 8~5, 742, 682.
~xam~le 8-1
OTBDMS OTBDMS
r H H ~ ~ ~ H
OOPMB ~ COOP~
H O CH2COOPNB
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-1-p-methoxybenzyloxycarbonyl-
ethyl]azetidin-2-one (1.12 g) in methylene chloride (14 ml),
there were successively added p-nitrobenzyl alpha-bromo-
15 acetate (1.09 g), 50 % aqueous sodium hydroxide solution(0.85 g~ and triethylbenzylammonium chloride (303 mg),
followed by stirring at room temperature for 30 minutes.
The reaction mixture was diluted with water and e:ctracted
~ 55 ~ 1~33~)~
with a mixture of diethyl ether and methylene chloride (3 :
1) three times. The organic layer was washed successively
with water twice and brine tnree times, dried over
sodium sulfate and evaporated. The residue was purified by
silica gel chromatography to give (3S,4S)-3-[(lR)-l t-
butyldimethylsilyloxyethyl]-4-[(lR)-1-p-methoxybenzyloxy-
carbonylethyl]-l-(p-nitrobenzyloxycarbonylmetAyl)azetidin-
2-one.
IR vmax (cm ): 1760, 1742, 1607, 1515, 1458,
1342, 1241, 1170, 830, 747.
NMR ~ (CDC13): OoOl (3H, s), 0.05 (3H, s), 0.83
(9H, s), 2.86 (lH, qd, J = 7.2 and 3.0 Hz), 3.0C (lE, dd, J
= 2.3 and 6.6 Hz), 3.80 (3H, s), 5.01 (2H, m), 5.20 (2H, s),
6.88 (2H, d, J = 8.6 Hz), 7.49 (2H, d, J = 8.9 Hz), 8.22
(2H, d, J = 8.6 ~z).
Example 8-2
OTBDMS
~ H H r
/~ooP~8
. .>
0~ ~CH2COOPNB
OTBDMS OH
H H ¦ r H H
COOH ~ COOH
O ~ CH2COOPNB O ~ CH2COOPNB
To a solution of (3S,45)-3-[(lR~-1-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-p-methoxybenzyloxycarbonyl-
ethyl]-l-(p-nitrobenzyloxycarbonylmethyl)azetidin-2-one (142
13~3P~
mg) in dry methylene chloride, there was BF3-Et2O complex
~163 mg) under ice-cooling, followed by st-rring at room
temperature. The reaction mixture was poured into 2 cold
aqueous sodium bicarbonate solution, acidified with dilute
hydrochloric acid and extracted with ethyl acetate three
times. The organic layer was washed successively with
dilute hydrochloric acid and brine, dried over sodium
sulfate and evaporated. The residue was puriied by silica
gel chromatography ~o give (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-carboxyethyl]-l-(p-nitro-
benzyloxycarbonylmethyl)azetidin-2-one ~Compound A3 and
(3S,45)-3-[(lR)-l-hydroxyethyl]-4-[~lR)-l-carboxy-
ethyl]-l-(p-nitrobenzyLoxycarbonylmethyl)azetidin-2-one
(Compound B).
Compound A:-
IR ~neaat (cm 1): 3100 (broad), 1760, 1730, 1520,
1342, 1245, 1180, 830, 770.
~ MR ~ (CDCl3): 0.03 (3H, s), 0.07 (3H, s), 0.85
(9H, s), 1.25 (3H, d, J = 6.3 Hz), 1.26 (3E, a, J = 7.3 Hz),
20 2.91 (lH, qd, J - 3.0 and 7.3 Hz), 3.05 ~lH, dd, J = 2.3 and
5.9 Hz), 4.13 (2H, m), 5.26 (2H, s), 7.52 (2H, d, J = 8.9
Hz), 8.23 (2H, d, J = 8.9 Hz).
Compound B:-
IR ~maxt (cm 1): 3430 (broad), 1760, 1735, 1705,
25 1520, 1345, 1180, 7~5.
~ 3 ~
Example 9-1
OTBDMS OTBDMS
'I .H li ~ 11 .H H
/~COS~Cl ~ I COS~Cl
O ~ CH2COOtBU O ~H2COOH
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
- methylsilyloxyethyl]~4-[(lR)-l-p-cnlorophenylthiocarbonyl-
ethyl]-l-(t-butyloxycarbonylmethyl)azetidin-2-one (200 mg)
in dry methylene chloride (l.S ml), there was added BF3-ET2O
comple~ (263 mg), followed by stirring at room temperature
for 1 hour. After evaporation or the solvent, the residue
wzs dissolved in methanol (O.S ml), diluted with brine and
extracted with ethyl acetate. The organic layer was washed
with brine, dried over sodium sulfate and evaporated to give
(3S,4S)-3-[(lR)-l-hydroxyethyl~-4~[(lR)-l-p-chlorophenyl-
thiocarbonylethyl]-l-(carboxymethyl)azetidin-2-one as a
crude product. The crude product, t-butyldimethylsilyl
chloride (246 mg) and imidazole (151 mg) were dissolved in
dry dimethylformamide (2.5 ml) and allowed to stand at room
temperature overnight. The reaction mixture was poured into
cold brine, adjusted with lM potassium hydrogensulfate to pH
2 and e~tracted with diethyl ether three times. The organic
layer was washed with brine twice, dried over sodium
sulfate and evaporated. The residue was purified by silica
gel chromatography to give (3S,4S)-3-[~lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-p-chlorophenylthiocarbonyl-
- ethyl]-l-(carboxymethyl)azetidin-2-one.
IR vmax (cm ): 3300 (broad), 1760, 1740, 1700,
13~3~
1480, 1382, 1250, 1140, 1087, 830, 775.
Example 9-2
OTBDMS OH
COS ~ Cl ~ COS ~ Cl
O ~CH2COOH \CH2COOPNB
To a mixture of (3S,4S)-3-~(lR)-l-t-butyldimethyl-
silyloxyethyl)-4-[(lR)-l-p-chlorophenylthiocarbonylethyl3-1-
carboxymethylazetidin-2-one (70 mg) and p-nitrobenzyl
alcohol (24 ml) in ethyl acetate (0.3 ml), there was added
5 a solution of N,N'-dicyclohexylcarbodiimide (30 mg) in dry
ethyl acetate (0.2 ml), and the resultant mixture was
stlrred at 5 to 10C overnight. The precipitated N,N'-di-
cyclohexylurea was collected by filtration and washed with
ethyl acetate. The washing was combined with the filtrate,
washed with water and brine, dried over sodium sulfate and
distilled off to remove the solvent. The residue was
purified by silica gel chromatography to give (35,45)-3-
[(lR)-l-t-butyldimethylsilyloxyethyl)-4-[(lR)-l-p-ch~oro-
phenylthiocarbonylethyl]-l-(l-nitrobenzyloxycarbonyLmethyl)-
15 azetidin-2-one.
IR vmax (cm ): 1760, 1750, 1700 t 1602, 1520,
1478, 1343, 1250, 1180, 1090, 835, 775, 742.
~ R ~ (CDC13): 0.07 (3E~, s), 0.08 (3H, s), 0.88
(9H, s), 1.27 (3H, d, J = 6.3 Hz), 1.31 (3H, d, J = 7.3 Hz),
20 3.01 (lEi, dd, J = 2.6 and 7.1 Hz), 3.14 (lH, qd, J = 2.6 and
7.3 Hz), 4.12 (2H, m), 4.17 (2H, m), 5.20 (2H, m), 7.34 (4EI,
m), 7.44 (2H, a, J = 8.6 Hz), 8.17 (2H, d, J = 8.9 Hz).
~ 59 ~ ~3133~Q
ExamDle 10-1
OH ~
COSPh ~ COSPh
~ .. >
0~ CH2COOtl~u O CH2COOH
OH
~ ~ H r
(b) ~ COSPh
--~,
O C~I2COOPNB
(a) (3S,45)-3-[(iR)~ ydroxyethyl]-4-[(lR)-l-
phenylthiocarbonylethyl]-l-(t-butyloxycarbonylmethyl)a~eti-
din-2-one (72.0 g) was dissolved in trifluoroacetîc acid
(500 ml) under ice-cooling, followed by stirring for 2
5 hours. The reaction mixture was evaporated in vacuo below
50C. The residue was then dissolved in toluene (250 ml)
and evaporated off to remove the solvent.
(b) To a solution of the residue in dry aceto-
nitrile (720 ml), there were added triethylamine (43.25 g)
10 and p-nitrobenzvl bromide ~92.33 g), followed by stirring at
room temperature for 1 hour. The reaction mixture was
diluted with ethyl acetate ~1.5 liters), washed with a 20
aqueous sodium chloride solution several times, dried over
sodium sulfate and evaporated. The residue was purified by
15 silica gel chromatography to give (3S,4S)-3-[(lR)-l-hydroxy-
ethyl]-4-[(lR)-l-phenylthiocarbonylethylJ-l-(p-nitrobenzyl-
oxycarbonylmethyl~azetidin-2-one.
IR VCaHC13 (cm 1): 1740, 1680, 1600, 1515, 1360,
13~33~
- 60 -
- 1250, ll&0, 950, 740.
Example 10-2
Oz OH
r H H ~ ~ H ~ r
~ COSPh ~ COSPh
O CH2COOtBu O ~ \C 2COO}i
To a solution of (35,4S1-3-[(lR)-l-benzyloxy-
carbonyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-1-t-
butoxycarbonylmethylazetidin-2-one (8.5 g) in 1,2-dichloro-
ethane (185 ml), there was dropwise added a solution of
` boron tribromide (26.4 g) in 1,2-dichloroethane (100 ml) at
-10~C for 20 minutes, followed by stirring at the same
temperature for 1 hour. Sodium bicarbonate (40 g) and
ice-water (600 g) were added thereto while stirring, and the
resultant mixture was diluted with ethyl acetate (200 ml).
The aqueous layer was acidified with 2N hydrochloric acid
(200 ml), extracted with ethyl acetate and combined with the
organic layer. The combined orsanic layer was washed with
- brine (200 ml x 3), dried over magnesium sulfate and evapo-
rated in vacuo to give (3S,4S)-3-[(lR)-1-hydroxyethyl]-
4-[(lR)-l-phenylthiocarbonylethyl]-1-carboxymethylazetidin-
2-one (10.4 g, 87.9 ~.).
IR ~ma~ (cm ): 1740, 1710, 1690, 1210, 1130,
1070, 940, 740.
- 61 _ ~ 3
Example 10-3
OH OTBDMS
H H ~ ~ H H
COSPh ~ COSPh
0 ~ ~CH2COOPNB O . ~ H2COOPNB
To a solution of (3S,4S)-3-[(lR~-hydroxyethyl]-4-
[(lR)-1-phenylthiocarbonylethyl]-1-(p-nitrobenzyloxy-
carbonylmethyl]azetidin-2-one (52.36 g) in dry dimethylform-
amide (262 ml), there were added imidazole (16.6 g) and
t-butyldimethylchlorosilane (23.38 g), followed by stirring
at room temperature ~or 5 hours. The reaction mixture was
diluted with ethyl acetate (1 iiter), washed with a 20 %
aqueous sodium chloride solution. The aqueous layer was
separated from the organic layer and extracted with ethyl
acetate (500 ml). The extract was combined with the organic
layer, washed with a 20 % aqueous sodium chloride solution
twice, , dr.ied over sodium sulfa~e and evaporated. The
residue was purified by silica gel chromatography to give
(3S,4S)-3-[(lR)-1-t-butyldimethylsilyloxyethyl]-4-
lS [(lR)-1-phenylthiocarbonylethyl~ (p-nitrobenzyloxy-
carbonylmethyl]azetidin-2-one.
IR ~ma~ (cm ): 1755, 1690, 1600, 1515, 1340,
1250, 1180, 835.
NMR ~ (CDC13): 0.08 (3H, s), 0.09 (3H, s), 0.89
(9H, s), 1.28 (3H, d, J = 6.0 HzJ, 1.32 (3H, d, J = 7.3 HZ),
3.01 (lH, dd, J = 2.3 and 7.3 Hz), 3.16 (lH, dd, J = 2.3 and
7.3 Hz), 3.96 (lH, d, J = 17.8 HzJ, 4.17 (2H, m), 4.31 (lH,
- 62 - 13 ~ 3
d, J = 17.8 Hz), 5.20 (2H, ABq, J = 13.5 Hz~, 7.25 - 7.45
(5H), 8.12 (2H, d, J = 8.9 Hz).
Exam~le 10-4
OH Me sio
r H H ~ 3
COSPh ~ COSPh
Q ~ ~ CH2COOPNB O ~ CH2ccopNB
In the same manner as in Example lG-3, there was
obtained (3S,4S)-3-[(lR)-l-trimethylsilyloxyethyl]-4-[(lR)-
l-phenylthiocarbonylethyl]-l-(p-nitrobenzyloxy~arbonyl-
methyl]azetidin-2-one from (3S,4S)-3-[(lR)-l-hydroxyethyl]-
4-t(lR)-l-phenylthiocarbonylethyl]-l-(p-nitrobenzyloxy-
carbonylmethyl]azetidin-2-one.
IR ~maxt (~m 1) 1760, 1695, 1600, 1520, 1440,
101340, 1250, 1180, 950, 840, 740.
NMR ~ (CDC13): 0.13 (9H, s), 3.04 (lH, dd, J =
2.3 and 7.6 Hz), 3.15 (lH, aq, J = 2.3 and 7.0 Hz), 3.92
(lH, d, J = 1~.1 Hz), 4.38 (lH, d, J = 18.1 Hz), 5.21 (2H,
ABq, J = 13.5 Hz), 8.12 (2H, d, J = 8.9 Hz).
15Reference ExamPle 1-1
OTBDMS OTBDMS
COOH / ~ COSPh
O ~ H ~ ~H
A mixture of (3S,4S)-3-[llR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lR)-l-carboxyethyll azetidin-2-one (301
mg) and N,N'-carbonyldiimidazole (194 mg~ in dry aceto-
- 63 ~
nitrile (8.6 ml) was stirred at room temperature for l hour.
To the reaction mixture, there were successively added
thiophenol (132 mg) in dry acetonitrile (2 ml) and triethyl-
amine (121 mg) in dry acetonitrile (2 ml), followed by
stir_ing at room temperature for 30 minutes. The resulting
mixture was diluted with ethyl acetate and washed with
brine. The aqueous layer was separated from the organic
layer and extracted with ethyl acetate twice. The
extracts were combined wtth the organic layer, washed wiih
brine, dried over sodium sulfate and distilled cff to remove
the solYent. The residue was purified by silica gel chro-
matography to aive (3S,4S)-3-~(lR)-1-t-butyldimethylsilyl-
oxyethyl]-4-~(lR)-phenylthiocarbonylethyl]azetidin-2-one.
IR vaeat (cm l): 3200 (broad), 1760, 1700, 1370,
15 1250, 1;40, 955, 830, 773, 740, 680.
Reference Example 1-2
OTBDMS OTBDMS
~COO~ ,~COS~Cl
A mixture of (3S,4S)-3-[(lR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lR)-l-carboxyethyl]azetidin-2-one (400
mg) and Nr~'-carbonyldiimidazole (259 mg) in dry aceto-
20 nitrile (11 ml) was stirred at room temperature for 1 hour.To the resulting mixture, there were successively added
p-chlorothiophenol (231 mg) in dry acetonitrile (3.2 ml) and
triethvlamine (162 mg) in dry acetonitrile (2.3 ml), fol-
lowed by stirring at-room temperature .or 30 minutes. The
- 64 - ~3~3~8~
reaction mixture was diluted with ethyl acetate and washed
with brine. The aqueous layer was separzted from the
organic layer and extracted with ethyl acetate twice. ~~
The extracts were combined with the organic layer, washed
with dilute hydrochloric acid, brine, aqueous sodium
bicarbonate solution and brine, dried over sodium sulfate
and distilled off to remove the solvent. The residue was
purified by silica gel chromatography to give (3S,4S)-3-
[(lR)-l-t-butyldimethylsilyloxyethyl]-4-[(lR)-l-p-chloro-
phenylthiocarbonylethyl]azetidin-2-one.
IR vmaext (cm 1): 3250 (broad), 1770, 1750, 1700,
1478, 1247, 1140, 1090, 820, 770.
NMR ~ (CDC13): 0.07 (6H, s), 0.88 (9H, s), 1.18
(3H, d, J = 6.3 Hz), 1.33 (3~, d, J = 6.9 ~z), 2.97 (lH, m),
3.02 (lH, m), 3.93 (lH, dd, J = 2.0 and 5.3 Hz), 4.22 (lH,
m), 5.86 (lH, broad, s), 7.36 (4~, m).
Reference Exam~le 1-3
OTBDMS CTBDMS
, ~ ~ COOPMB
A mixture of (3S,4S) 3-[(lR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lR)-1-carboxyethyl] azetidin-2-one (1.00
g), triethylamine (369 mg) and p-methoxybenzyl chloride (779
mg) in dry dimethylformamide (1 ml) was stirred at 70C for
2 hours and 40 minutes. The reaction mixture was poured
into ice-water, acidified with dilute hydrochloric acid to
pH 2 to 3 and extracted with diethyl ether three times. The
- 65 ~ 13~
organic layer was washed with cold lN aqueous sodium hydro-
xide solution, water and brine in this order, dried over
sodium sulfate and distilled off to remove the solvent. The
residue was purified by silica gel chromatography to give
(3S,4S)-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-4-[(lR)-l-
p-methoxybenzyloxycarbonylethyl]azetidin-2-one.
IR ~meaxt (cm 1): 3225 (broad), 1760, 1740, 1605,
1505, 1458, 1240, 1160, 1030, 950, 825, 767.
NMR ~ ~CDC13): 0.05 (6H, s), 0.86 (9~, s), 1.13
(3H, d, ~ = 6 Hz), 1.21 (3~, d, J = 7 Hz), 2.70 (1~, m),
2.95 (lH, dd, J = 2 and 4 Hz1, 3.81 (3H, s), 3.89 (1~, dd, J
= 2 and 5 Hz), 4.16 (lH, m), 5.05 (2H, s), 5.96 (lH, bro2d,
s), 6.87 (2H, d, J = 9 Hz), 7.27 (2H, d, J = 9 Hz).
Reference Example 2-1
OH OH
H K ~ H H
COOH ~ COOMe
O DAM O DAM
To a solution of (3S,4S)-4-carboxy-3-(1-(R)-
- hydroxyethyl)-1-di(p-anisyl)methyl-2-azetidinone (34 g) in
methanol (310 ml), there was added 98 ~ sulfuric acid (2.9
g). The resultant mixture was heated at 65C for 3 hours,
cooled down to 40C, neutralized with 8 % aqueous sodium
hydroxide solution (15 ml) and concentrated to make a one
third volume. The concentrate was diluted with 1,2-di-
chloroethane (105 ml) and washed with water. The aqueous
layer was separated from the organic layer and extracted
with 1,2-dichloroethane ~105 ml). The extract was combined
131338~
with the organic layer, washed with water and dried over
anhydrous sodium sulfate. After filtration, the filtrate
was concentrated in vacuo to give (3S,45)-4-methoxycarbonyl-
3-(1-(R)-hydroxyethyl)-l-di~p-anisyl)methyl-2-azetidinone.
m.p., 102 - 104C.
Reference E:cample 2-2
OH H H ¦ OH
COOMe
O DAM O \DAM
To a solution of (3S,4S~-4-methoxycarbonyl-3-(1-
(R)-hydroxyethyl)-1-di(p-anisyl)methyl-2-a-zetidinone (32.5
g) in dry tetrahydrofuran (310 ml), there was added dropwise
a lM susper.sion of methyl magnesium bromide in tetrahydro-
furan (370 g) at 0 - 5C, and the suspension was stirred at
the same temperature as above for 1 hour. 20 ~ Hydrochloric
acid (350 ml) was poured into the suspension at 20 - 25C,
and the resultant mixture was stirred for 1 hour, followed
by extraction with ethyl acetate (110 ml). The aqueous
layer was ree~tracted with ethyl acetate (110 ml). The
extracts were combined together, washed successively with
brine, a saturated sodium bicarbonate solution and water and
dried over anhydrous sodium sulfate. After filtration, the
filtrate was concentrated in vacuo to give (3S,4S)-4-(1-
hydroxy-l-methylethyl)-3~ (R)-hydroxyethyl)-1-di(p-
anisyl)methyl-2-azetidinone. m.p., 154 - 156C.
- 67 - ~3 ~3 ~ a
Reference Example 2-3
~,ON ,~J~J~ OH
O DAM O DA~
(3S,4S)-4~ Hydroxy-1-methylethyl)-3-(1-(R)-
hydrcxyethyl)-1-di(p-anisyl)methyl-2-azetidinone (26 g) and
4-dimethylaminopyridine (16 g) were dissolved in dry
dichloromethane (200 ml). Benzyl chloroformate (20 g) was
added dropwise thereto over a period of 1 hour with ice-
cooling, and the resultant mixture was stirred for 2 hours
and warmed to room temperature, followed by stirring at the
same temperature as above for 10 hours. 5 ~ Hydrochloric
acid llOO ml) was poured into the reaction mixture with ice-
cooling, and the resulting mixture was stirred for 0.5 hourand allowed to stand. The organic layer was washed suc-
cessively with water, a saturated sodium bicarbonate solu-
tion and brine and dried over anhydrous sodium sulfate.
After filtration, the filtrate was concentrated in vacuo to
give (3S,4S)-4-(1-hydroxy-1-methylethyl)-3-(1-(~ enzyloxy-
carbonyloxyethyl)-l-di(p-anisyl)methyl-2-azetidinone.
IR ~maexat (cm 1): 3450, 1750, 1615, 1515, 1250,
1180, 1030.
- NMR ~ (CDC13): 1.13 (6H, s), 1.38 (3H, d, J = 6
Hz), 3.70 (3H, s), 3.75 (3H, s), 5.10 (2H, s), 5.55 (lH,
bs), 7 . 29 ~5H, S) .
- 68 ~ 13~3~9
Reference Example 2-4
O \DAM ~ DAM
A solution of (3S,4S)-4-(1-hydroxy-1-methyl-
ethyl)-3-(1-(R)-benzyloxycar~onyloxyethyl)-l-di(p-anisyl)-
methyl-2-azetidinone (30 g) in dry toluene (350 ml) was
treated with thionyl chloride (9.0 g) at 20 - 30C for 5
S hours in the presence of pyridine (10 ml). Water (100 ml)
was added to quench the reaction Gi 10 - 25C. The organic
layer was separated, washed with water and dried over
anhydrous sodium sulfate. After filtration, the filtrate
was concentrated in vacuo to give an oily residue, which was
crystallized from a mixture of cyclohexane and ethyl acetate
to yield (3S,4S)-4~ methylethenyl)-3-(1-~R)-benzyloxy-
carbonyloxyethyl)-1-di(p-anisyl)methyl-2-azetidinone. m.p.,
117 - 118C.
Reference Example 2-5
OZ oZ
Cl
\
O DAM o ~AM
(35,4S)-4-(1-Methylethenyl)-3-(1-(R)-benzyloxy-
carbonyloxyethyl)-1-di(p-anisyl)methyl-2-azetidinone (200 g)
was dissolved in ethyl acetate (3 liters), and a solution o~
chlorine in carbon tetrachloride (3.85 %, 870 g) W25 added
1 3 ~ $ 0
dropwise thereto at room temperature over a period of 15
minutes, followed by stirring for 1 hour. Water (1 liter)
and then lO % aqueous sodium thiosulfate solution (50 ml)
were poured into the reaction mixture, which was stirred for
0.5 hour and allowed to stand. The organic layer was washed
successively with a saturated sodium bicarbonate solution
and brine and dried over anhydrous sodium sulfate. After
filtration, the filtrate was concentrated in vacuo to give
(3S,4S)-4-(1-chloromethylethenyl)-3-(1-(R)-benzyloxycar-
bonyloxyethyl)-1-di(p-anisyl)methyl-2-azetidinone. m.p., 84
- 85C-
Reference Example 2-6
OZ OZ
Cl ~ H
N ~
0~ ~ ~ 0~ ~ DA~
To a solution of (3S,4S)-4-(1-chloromethyl-
ethenyl)-3-(l-(R)-ben~yloxycarbonyloxyethyl)-l-di(p-
anisyl)methyl-2-azetidinone (20 g) in dimethylsulfoxide (160
ml), there were successively added water (40 ml), cuprous
oxice (6.76 g) and p-toluenesulfonic acid (7.6 g), and the
resultant mixture was warmed to 50 to 55C and stirred for 2
hours at the same temperature. After cooling down to room
temperature, l % aqueous phosphoric acid (90 mi) and ethyl
acetate (200 ml) were poured into the reaction mixture,
followed by stirring for 0.5 hour. An insoluble material
was removed by filtration over celite and washed 3 times
with ethyl acetate (20 ml). The filtrate and the washings
~ 70 - ~3~3~
were combined together, and the aqueous layer was separated
from the organic layer and extracted with ethyl acetate (200
ml). The organic layer ana the extract were combined
together, washed with brine and dried over anhydrous sodium
sulfate. After filtration, the filtrate was concentrated in
vacuo, and the concentrate was crystallized from a mixture
of toluene and n-hexane (1 : 1) to give crystals of (3s,4S)-
4-(1-hydroxymethylethenyl)-3-(1-(R)-benzyloxycarbonyloxy-
ethyl)-l-di(p-anisyl)methyl-2-azetidinone. m.p., 118 -
120C.
Reference Exam~le 2-7
OZ Og
OH ~ OTB~MS
O ~DAM DA~
A solution of (3S,4S)-4-(1-hydroxymethyl-
ethenyl)-3-(1-(R)-~enzyloxycarbonyloxyethyl)-l-di~p-
anisyl)methyl-2-azetidinone (20 g) and imidazole (5.6 g) in
dry dimethylformamide (45 ml) was treated with t-butyl-
dimethylchlorosilane (6.77 g) at room temperature for 2
hours. The reaction mixture was diluted with cold water
(200 ml) and ethyl acetate (150 ml). The aqueous layer was
extracted with ethyl acetate (150 ml). The combined
extracts were washed successively with 5 % hydrochloric acid
solution (80 ml x 2) and brine (80 ml), and dried over
anhydrous sodium sulfate. After filtration, the filtrate
was concentrated in vacuo, and the concentrate was
crystallized from isopropanol to give crvstals of (35,4S)-
- 71 - ~3~3~
.
4~ t-butyldimethylsilyloxymethylethenyl)-3-(1-(R)-
benzyloxycarbonyloxyethyl)-1-di(p-anisyl)methyl-2-a~eti-
dinone. m.p., 90 - 92C.
Reference Example 2-8
OZ oZ
H H ¦¦ ~ H H r
/\L~9TBDMs ~,: TBDMS
O DAM O ~DAM
To a solution of (3S,4S~-4-(1-t-butyldimethvl-
silyloxymethylethenyl)-3-(1-(R)-benzyloxycar~onyloxyethyl)-
l-di(p-anisyl)methyl-2-azetidinone (20 g) in acetonitrile
(200 ml), there were added 5 % platinium on activated carbon
(4.0 g) and water ~4 ml) under nitrogen atmosphere. The
mixture was stirred at 10C in a hydrogen gas flow until 2.2
10 e~uivalents of hydrogen had been taken up. The catalyst was
removed by filtration and washed with ethyl acetate. The
filtrate and the washings were combined together and con~en-
trated in vacuo to give (35,4S)-4-(1-t-butyldimethylsilyl-
oxymethylethyl)-3-(1-(R)-benzylocycarbonyloxyethyl)-l-di-
(p-anisyi)methyl-2-azetidinone as an oil.
H gh performance liquid chromatography (HPLC)
[Lichrosorb RP-18], eluting with 85 % acetonitrile/water (1
ml/min~ and NM~ spectra indicated that this product was a
micture of 4-(1-(R)-t-butyldimethylsilyloxyethyl) compound
20 and the corresponding (S)-compound in a ratio of 7.7 : 1.
The above mixture was recrystallized from a mixture of
n-he:cane and ethyl acetate (10 : 1) to yield the (R)-
- 72 - ~3~33~
compound. m.p., 78 - 81C.
NMR ~ (CDC13): 0.01 (6H, s), 0.87 (9H, s), 1.40
(3H, d, J = 6 ~z), 3.31 ~lH, dd, J = 2.2 and 7.0 Hz), 3.44
(2H, d, J = 5.3 Hz), 3.73 (3H, s), 3.76 ~3H, s), 5.07 (lH,
m), 5.17 (2~, s), 7.38 (5~, s~.
Reference Example 2-9
OZ OZ
r ~ H ~ r H ~ ~
OTBDMS ~ OH
~ AM O H
To a solution of (3S,4R)-4-(1-(R)-t-butyldimethyl-
silyloxymethylethyl)-3-(1-(R)-benzyloxycarbonyloxyethyl)-l-
ditp-anisyl)methyl-2-azetidinone-(20 g) in dry dichloro-
methane (200 ml), there were added 1,3-dimethoxybenzene (7.8
g) and boron trifluoride etherate (23 g) at 10 - 20C, and
the resultant mixture was stirred at room temperature for 3
hours, followed by heating under reflux for 3 - 5 hours.
The reaction mixture was cooled down ~o 10 - 15C, washed
successively with brine (200 ml x 2), 2.5 ~ aqueous sodium
bicarbonate solution (200 ml) and brine (200 ml) and dried
over anhydrous sodium sulfate. After filtration, the
filtrate was concentrated in vacuo to give an oily residue,
which was purified by silica gel chromatography -to yield
(3S,4S)-4-(1-(R)-hydroxymethylethyl)-3-(1-(R)-benzyloxy-
carbonyloxyethyl)-2-azetidinone.
IR vmax (cm ): 3350, 1750, 1740, 1455, 1382,
1260, 1030.
_ 73 _ ~ ~ ~338~
NMR ~ (CDC13): 0.95 (3H, d, J = 7.0 Hz), 1-48
(3H, d, J = 6.5 Hz~, 3.14 (lH, dd, J = 2 and 9 ~z), 3.55
(lH, d, J = 2 Hz), 5.15 (2H, s), 6.05 tlH, broad, s), 7.37
(SH, s).
Reference Example 2-10
OZ OZ
~ -~ ~ COO~
A solution of (3S,4S)-4-(1-(R)-hydroxymethyl-
ethyl)-3-(1-(R)-benzyloxycarbonyloxyethyl)-2-azetidinone
(6.1 g) in acetone (60 ml) was treated with the Jones
reagent, prepared from chromium trioxide (2.7S g), 98 ~
sulfuric acid (4.4 g) and water (8.1 ml), at 10 - 20C for 1
hour. The reaction mixture was quenched with isopropanol
(0.5 ml) at 10 - 20C for 15 minutes, diluted with ethyl
acetate (122 ml) and washed with water (135 ml). The
aqueous layer was separated from the organic layer and
extracted with ethyl acetate 161 ml). The ethyl acetate
e~tracts and the organic layer were combined together and
extracted with 5 % aqueous sodium bicarbonate solution 130
ml). The extract was washed with dichloromethane (60 ml)
and acidified with 10 % hydrochloric acid solution (20 ml)
with ice-cooling. The acidic solution was extracted -twice
with dichloromethane (60 ml). The extracts were washed with
brine and dried over anhydrous sodium sulfate. After
.iltration, the filtrate was concentrated in vacuo to give
(35,4S)-4-(1-(R)-carboxyethyl)-3-(1-(R)-benzyloxycarbonyl-
- 74 - i3~3~3
oxyethyl)-2-azetidinone.
IR vmax (cm ): 3270, 1740, 1460, 1385, 1270,
750.
NMR ~ (CDC13): 1.19 (3E, a, J = 7.0 Hz), 1.40
(3H, a, J = 6.2 Hz), 2.67 (lH, m), 3.22 ~lH, broad, d, J =
7.5 Hz), 3.84 (lH, broad, d, J = 5.5 Hz), 5.14 (2H, s), 6.57
(lH, broad, s), 7.35 (5H, s), 7.63 (lH, broad, s).
Reference Example 2-11
' OZ OZ
~\COOI~ \COOCH2Ph
To a solution of 4-(1-(R)-carboxyethyl)-3-(1-
(lR)-benzyloxyca~x~yloxyethyl)azetidin-2-one (51.69 g) in acetone (510
ml), there were added anhydr~ potassium carbor.ate ~89.0 g)
and benzyl bromide (30.3 g), follcwed by stirring at 60C
or 1.5 hours. The reaction mixture was cooled to room
temperature and filtered off to remove insoluble materials.
~he filtrate and the washinss were combined and evaporated.
The residue was purified by silica gel chromatography to
gi~e (3S,4S)-3-[tlR)-l-benzyloxycarbonyloxyethyl]-4-[(lRJ-l-
benzyloxycarbonylethyl~azetidin-2-one.
IR ~maexat (cm 1): 1760 (sh), 1735, 1450, 1380,
1260, 1155.
NMR ~ (CDC13): 1.22 (3H, d, J = 6.9 Hz), 1.39
(3H, d, J = 6.3 Hz), 2.71 (lH, q, J = 6.9 Hz), 3.19 (lH, dd,
J = 2.0 and 7.9 Hzl, 3.83 (lH, da, J = 2.0 and 6.3 Hz), 5.92
(lH, s).
~ ~c?~3~
Reference Example 3-1
OTBDMS
/~~'-~
~L ~ ~ '
COSPh
OTBDMS OTBDMS
H H ~ ~ EI H 7
~OP (OPh) 2 ;~SPh
COSPh COSPh
To a solution of (4R,SR,6S,8R)-4-methyl-6-(1-
t-butyldimethylsilyloxyethyl)-l-aza~icyclo[3.2.0]hept-3,7-
dione-2-carbo~ylic acid phenylthioester (about 0.22 mmol)
containing thiophenol in dry acetonitrile (0.8 ml), there
were added a solution of diisopropylethylamine (S9 mg) in
dry acetonitrile (0.5 ml) and a solution of diphenyl
chlorophosphate (1~4 mg) in dry acetonitrile (0.5 mg) under
a nitrogen stream while ice-cooling, followed by stirring
for 1 hour. The reaction mixture was diluted with diethyl
ether and a phosphate buffer solution (pH, 6.86). The
aqueous layer was separated from the organic layer and
extracted with diethyl ether two times. The extracts were
combined with the organic layer, washed successively with a
0.1M potassium dihydrogen phosphate solution (three times),
15 water (two times) and brine, dried over sodium sulfate and
evaporated. The residue was purified by silica gel
- 76 ~ ~ 313~ ~ ~
chromatogrphy to give (4R,5R,6S,8R)-3-(diphenylphosphoryl-
oxy)-4-methyl-6-[1-t-butyldimethylsilyloxyethyl]-1-azabi-
cyclo[3.2.0]hept-2-en-7-one-2-carboxylic acid phenylthio-
ester (Compound A) and (4R,SS,6S,8R)-3-phenylthio-4-methyl-
5 6-(1-t-butyldimethylsilyloxyethyl)-l-azabicyclo[3.2.0]hept-
2-en-7-one-2-carboxylic acid phenylthioester (Compound B).
Compound A:-
IR vmaxa~ (cm 1): 1778, 1673, 1607, 1582, 1487,
1198, 1182, 1002, 962, 935, 767, 740, 680.
NNR C (CDC13): 0.09 (3H, s), 0.11 (3H, s), 0.91
(9H, s), 1.21 (3H, d, J = 7.3 Hz), 1.22 (3F, d, J = 6.0 Hz),
3.29 (lH, dd, J = 3.0 and 5.0 Hz), 3.52 (lH, m), 4.28 (2~,
m).
Comoound B:-
IR VCmaHxcl3 (cm 1): 1780, 1660 (sh), 1647, 1520,
1478, 1285, 1260, 1115, 1018, 945, 8~7.
NMR ~ (CDC13): 0.09 (3H, s), 0.13 (3H, s), 0.92
(9H, s), 0.95 (3H, d, J = 7.6 Hz), 1.17 (3H, d, J = 6.3 Hz),
3.07 (lH, m), 3.20 (lH, dd, J = 2.6 and 4.3 Hz), 4.31 (lH,
dd, J = 2.8 and 9.8 Hz), ~ 4.3 (lH, m), 7.3 - 7.6 (lOH, m).
Re.erence Example 3-2(1)
OTBDMS OTBDMS
P(OPh)2 ~ S ~ NHAc
COSPh COSPh
To a solution of (4R,SR,6S,8R)-3-(diphenylphos-
phoryloxy)-4-methyl-6-(1-t-butyldimethylsilyloxyethyl)-1-
- 77 - ~31~3$~
azabicyclo[3.2.0]hept-2-en-7-one-2-carboxylic acid phenyl-
thioes,er (10 mg) in dry acetonitrile (0.1 ml), there were
added a solution of diisopropylethylamine (5.2 ml) in dry
acetonitrile (0.2 ml) and a solution of N-acetylcysteamine
(4.8 mg) in dry acetonitrile (0.2 ml) at -30C under a
nitrogen strea~. The reaction mixture was warmed sradually
to -20C and diluted with diethyl ether and a phosphate
~uffer solution (pH, 6.86). The aqueous layer was separated
from the organic layer and ext~acted with diethyl ethe- two
10 times. The extracts were combined with the organic layer,
washed successively with a phosphate buffer solution (pH,
6.86), a O.lM potassium dihydrogen phosphate solution and
brine, dried over soaium sulfate and evaporated. The
residue was purified by sil:ica gel chromatography to give
(4R,5S,6S,8R)-3-(2-acetaminoethylthio~-4-methyl-6-(1-t-
butyldimethylsilyloxyethyl)-1-azabicyclo[3.2.0]hept-2-en-7-
one-2-carboxylic acid phenylthioester.
IR VmaCX13 (cm 1): 3460, 1765, 1665, 1250, 1102,
830.
NMR ~ (CDC13): 0.11 (3H, s), 0.14 (3~, s), 0.94
(9H, s), 1.25 (3H, d, J = 7.3 Hz), 1.25 (3H, d, J = 6.3 Hz),
1.97 (3H, s), 5.9 (lH, broad s), 7.3 - 7.6 (SE, m).
3 ~ ~
Reference Exam~le 3-2t2)
OTBDMS
H H
i ~ ~ OPtOPh)2
COSPh
- OTBDMS
H H
COSPh
In the same manner as in Reference Example ~-2(1),
(4R,5R,6S,8R)-3-(diphenylphosphoryloxy)-4-methyl-6-(1-
t-butyldimethylsilyloxyethyl)-l-azabicyclo[3.2.0]hept-
2-en-7-one-2-carboxylic acid phenylthio ester (6 mg), diiso-
5 propylamine (1.4 mg) and (2S,4S)-l-p-nitrobenzyloxy-
carbonyl-4-mercaptopyrrolidine (4 mg) were treated to give
(4R,55,65,8R,2'S,4'S)-3-[(1-p-nitrobenzyloxycarbonyl-2-
dimethylaminocarbonylpyrrolidin)-4-ylthio3-4-methyl-6-(1-t-
butyldimethylsilyloxyethyl)-l-azabicyclo[3.2.03hept-2-en-7-
10 one-2-carboxylic acid phenylthio ester.
IR VmHX13 (cm 1): 1767, 1700, 1650, 1518, 1340,
1100 .
Reference Example 3-2(3)
OT~DMS OTBDMS
P~OPh~2 ~ - SCHzPh
los Ph COSPh
- 79 -
In the same manner as in Reference Example 3-2(1),
(4R,5R,6S,8R)-3-(diphenylphosphoryloxy)-4-methyl-6-(1-
t-butyldimethylsilyloxyethyl)-l-azabicyclo[3.2.0]hept-
2-en-7-one-2-carboxylic acid phenylthio ester (20 mg),
diisopropylethylamine t5.2 mg) and benzylmercaptan (5 mg)
were treated to give (4R,5S,6S,8R)-3-benzylthio-4-methyl-
6-(1-t-butyldimethylsilyloxyethyl)-1-azabicyclo~3.2.0~-
hept-2-en-7-one-2-carboxylic acid phenylthio ester.
IR VmaX13 (cm ): 1770, 1660 (sh), 1640, 1298,
1266, 1250, 1142, 1102, 835.
NMR ~ (CDC13): 0.10 (3H, s), 0.13 (3H, s), 0.92
(9H, s), 3.24 (lH, dd, J = 2.6 and 5.3 Hz), 3.37 (lH, m),
4.09 (lH, m), 4.2 - 4.4 ~2H, m), 7.30 (5H, s), 7.3 - 7.6
(5H, m).
Reference Example 4
Me3SiO
r H H
_ COUMe2
COOPNB
QH
H H
/ ~ C ~;--C Me2
COOPNB
To a solution of (4R,5S,6S,8~,2'S,4'5)-p-nitro-
benzyl-3-[4-(1-p-nitrobenzyloxycarbonyl-2-dimethylamino-
- 8~ - 13~
carbonylpyrrolidinyl)thio]-4-methyl-6-(1-trimethylsilyloxy-
ethyl)-l-azabicyclo[3.2.0]hept-2-en-7-one-2-carboxylate (1.0
g) in dry tetrahydrofuran (10 ml), there was added a
phosphate buffer solution (pH 3; 8 ml), and the resultant
mixture was vigorously st rred at room temperature for 2.5
hours. The reaction mixture was diluted With ethyl aCetate
(50 ml), washed With brine~ dried over magnesium sulfate and
evaporated in vacuo to give (4R,SS, 6S,8R,2'S,4'S)-p-nitro-
benzyl-3-[4~ p-nitrobenzyloxycarbonyl-2-dimethylamino-
carbonylpyrrolidinyl)thio]-4-methyl-6-(1-hydroxyethyl)-1-
azabicyclo[3.2.0]hept-2-en-7-one-2-carboxylate,
IR ~meaxt (cm 1): 1760~ 1705~ 1645~ 1520~ 1402
1342, 1135, 1110.
NMR ~ (CDC13): 1.30 (3~, d, J = 7.0 HZ)~ 1.35
~3H~ d~ J = 6,5 HZ)~ 2.99 (3H~ 5)~ 3.02 (3H~ d~ J = 15 ~Z)~
5,21 (2H~ 5)~ 5.20 and 5.43 (2H, ABq, J = 14 HZ), 7.51 (2H~
d, J = 8,5 HZ)~ 7.64 (2H, d, J = 8.5 HZ), 8.20 (4H, d, J =
8.5 HZ).
Re~erence E~.amPle 5-1
OTBDMS ~OTBDMS
Ph ~ SPh
COSPh COOPWB
(4R,5S,6S,8R)-3-Phenylthio-4-methvl-6-[(lR)-1-t-
butyldimethylsilyloxyethyl~-1-azabicyclo[3.2.0]hept-2-en-7-
one-2-carboxylic acid phenylthio ester tl7 mg) and p-nitro-
benzyl alcohol (24 mg) were dissolved in dry methylene
- 81 - 13~
chloride (0.6 ml). In the dar~, silver trifluoroacetate (7
mg) and 1,8-diazabicyclo~5.4.0]-7-undecene (5 mg) in dry
methylene cnloride (0.3 ml) were added tnereto successively
at room tamperature. The resultant mixture was stirred for
4.3 hours and diluted with a 0.lM phosphate buffer solution
tpH 7; 3 ml) and methylene chloride. After removal of any
insoluble materials by filtration, the filtrate was
extracted with methylene chloride twice. - The or~anic
layer was washed successively with a 2.5 % sodium dihydrogen
phosphate solution and brine, dried over sodium sulfate and
magnesium sulfate and evaporated. The residue was purified
by silica gel chromatography to give (4R,SS,6S,8R)-3-phenyl-
thio-4-methyl-6-[(lR)-l-t-butyldimethylsilyloxyethyl]-l-
azabicyclo[3.2.0]hept-2-en-7-one-2-carboxylic acid p-nitro-
15 benzyl ester.
IR and NMR spectra of this compound were identicalto those of ~he compound as in Example 11-4.
Reference ExampLe 5-2(1)
OTBDMS OH
~Ph 5 ~SPh
COSPh COSPh
To a solution of (4R,5S,6S,8R)-3-phenylthio-4-
methyl-6-[(lR)-l-t-butyldimethylsilyloxyethyl]-l-az2bicyclo-
[3.2.0]hept-2-en-7-one-2-carboxylic acid phenylthio ester
~20mg) in dry tetrahydrofuran (0.3 ml), there was dropwise added
a 0.27 M solution of tetra-n-butylammonium fluoride (0.14
-,82 - i3~ a
ml) in tetrahydrofuran under a nitrogen stream with ice-
cooling, followed by stirring for 1 hour. The reaction
mi~ture was diluted with a phosphate buffer solution (pH 7)
and extracted with methylene chloride three times. The
organic layer was washed with brine, dried over sodium
sulfate ana evaporated in vacuo to give an oily residue,
whicn was purified by silica gel chromatography to give
(4R,SS,6S,8R)-3-phenylthio-4-methyl-6-[(lR)-l-hydroxy-
ethyl]-l-azabicyclot3.2.0~hept-2-en-7-one-2-carboxylic acid
phenylthio ester.
IR VmaHCxl3 (cm 1): 3600 (broad), 1775, 1660 (sh),
1645, 1300, 1273.
NMR ~ (CDC13): 0.98 (3H, d, J = 7.3 Hz), 1.35
(3H, d, J = 6.3 Hz), 3.11 (lH, m), 3.23 (lH, dd, J = 2.3 and
6.9 Hz), 4.27 (lH, dd, J = 2.6 and 9.2 Hz), - 4.3 (lH, m),
7.3 - 7.6 (lOH, m).
Reference Example 5-2(2~
OH OH
J---,~, /r" ~J\
SPh ~ ~ SPh
COSPh COOH
To a mixture of (4R,5S,6S,8R)-3-phenylthio-4-
methyl-6-(1-hydroxyethyl)-1-azabicyclo[3.2.0~hept-2-en-7-
one-2-carboxylic acid phenylthio ester (3 mg) and trimethyl-
silanol (14 mg) in dry toluene (0.1 ml), there were added
silver trifluoroacetate (1.6 mg~ and a solution of 1,8-
diazabicyclo[5.4.0]-7-uncecene (1.1 mg) in dry toluene (0.1
~3~3~
ml) at room temperature, followed by stirring at 80C for 15
minutes. The reaction mixture was cooled down to room
temperature and ailuted with a 0.lM phosphate buffer solu-
tion (pH 7; 1 ml) and methylene chloride. After removal of
insoluble materials by filtration, the filtrate was ex-
tracted with methylene chloride twice. The aqueous
layer was stirred under reduced pressure to remove any
organic solvent. By identification of HPLC (Lichrosorb
RP-18; ~leOH (pH 7.0 - 7.2)/0.005 M phosphate buffer (3 : 7);
0.8 ml/minute) and TLC (silica gel; CHC13/~leOH/acetic acid
(200 : 50 : 1), (4R,SS,6S,8R)-3-phenvlthio-4-methyl-6-
(l-hydroxyethyl)-1-azabicyclo[3.2.0]hept-2-en-7-one-2-
carboxylic acid, i.e. the compound in Reference Example
6-(2), was found to be present in the aqueous layer.
Reference Example 6-1
OH
J"~o
COOPNB
OH OH
OP(OPh~2 ~ SPh
COOPNB COOPNB
To a solution of (4R,5R,6S,8R)-4-methyl-6-(1-
hydroxyethyl)-1-azabicyclo[3.2.0~hept-3,7-dione-2-carboxylic
acid p-nitrobenzyl ester (100 mg) in dry acetonitrile (1
t 3 '?L 3 ~ ~ ~7
- 84 -
ml), there were added a solution of diphenylchloro~hosphate
(67 mg) in dry acetonitrile (0.5 ml) and a solution of
diisopropylethyLamine (32 mg) in dry acetonitrile (O.S ml)
under a nitrogen stream while ice-cooling. The resultant
mixture was stirred at the same temperature for 30 minutes.
The reaction mixture was cooled down to -30C and then
thiophenol (37 mg) and diisopropylethylamine (44 mg) in dry
acetonitrile (0.4 ml) were added thereto successively. The
resulting mixture was stirred at the same temperature for 25
minutes and further for lS minutes under ice-cooling. The
mixture was diluted with ethyl acetate, washed successively
with brine, an aqueous solution of potassium dihydrogen
phosphate and brine, dried over a mixture or magnesium
sulfate and potassium carbonate and evaporated. The residue
W2S purified by silica gel chromatography to give
(4R,55,65,8R)-3-phenylthio-4-methyl-6-(1-hydroxyethyl)-
l-azabicyclo[3.2.0]hept-2-en-7-one-2-carboxylic acid p-
nitrobenzyl ester.
IR ~maxt (cm ): 3480 (broad), 1764, 1707, 1520,
1342, 1215, 1140.
NMR C (CDC13): 0.97 ~3H, d, J = 7.3 Hz), 1.31
(3H, d, J = 6.3 Hz), 3.10 (lH, m), 3.21 (lH, dd, J = 2.8 and
6.8 Hz), 4.18 (lH, dd, J = 2.8 and 9.4 Hz), 4.23 (lH, m),
5.42 (2H, m), 7.3 - 7.6 (5H, m), 7.69 (2H, d, J = 8.9 Hz),
8.24 (2H, d, J = 8.9 Hz).
- 85 - ~ ~3
Reference Exam~le 6-2
OH OH
,l""~ J-~
L ~ ~ Ph ~ SPh
COOPNB CGOH
To solution of (4R,5S,6S,8R)-3-phenylthio-4-
methyl-6-(1-hydroxyethyl)-1-azabicyclo~3.2.0]hept-2-en-7-
- one-2-carboxylic acid p-nitrobenzyl ester (37 mg) in tetra~
hydrofuran (2 ml~, a morpholinopropanesulfonic acid buffer
solution (pH 7.0; 2 ml) was added, and the resultant mixture
was subjected to hydrogenation at room temperature in the
presence of 10 % palladium-carbon ~56 mg~ under atmospheric
pressure for 4.5 hours. After filtration, the filtrate was
stirred under reduced pressure to remove tetrahydrofuran.
The filtrate was washed with methylene chloride and stirred
again under reduced pressure to remove any organic solvent.
The residue was purified by polymer chromatography (CHP-
20P), and the fractions eluted with 2 % and 5 % tetrahydro-
furan-water gave (4R,5S,6S,8R)-3-phenylthio-4-methyl-6-(1-
hydroxyethyl)-l-azabicyclo[3.2~o]hept-2-en-7-one-2
carboxyllc acid.
UV AmaX (nm): 306.
IR ~mBr (cm 1): 3425 (broad), 1745, 1595, 1400.
NMR ~ (D2O)(ppm): 0.90 (3H, d, J = 7.3 Hz), 1.18
(3H, d, J = 6.3 Hz), 3.00 (lH, m), 3.32 (lH, dd, J = 2.6 and
5.9 Hz), 4.09 (lH, dd, J = 2.6 and 9.2 Hz), 4.16 (lH, m),
7.3 - 7.6 (SH, m).
-86- t~ a
Example 11~
OTBDMS OTBD~IS
~/~OSPh
/
~CH2COOPNB O --~
COOPNB
A solution or (3S,4S)-3-[(lR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lR)-1-phenylthiocarbonylethyl]-1-p-nitro-
benzyloxycarbonylmethyl-2-azetidinone (70 mg) in dry toluene
(0.5 ml) was dropwise added to a supension of sodium hydride
(12.5 mg; 50 % in oil) in dry tetrahydrofuran (0.1 ml) under
ice-cooling, and the resultant mixture was stirred for 30
minutes. p-Toluenesulfonic acid monohydrate (57 mg) was
added thereto, ar.d the resulting mixture was further stirred
for 10 minutes. The reaction mixture was diluted with cold
ethyl acetate (20 ml), washed with brine, dried over magne-
sium sulfate and e~Japorated to give (4R,5R,6S,8R)-p-nitro-
benzyl-4-methyl-6-(1-t-bu~yldime~hylsilyloxyethyl)-1-aza-
bicyclo[3.2.0]hept-3,7-dione-2-carboxylate as an oil.
IR vmax (cm 1): 1760, 1605, 1520, 1460, 1350,
1250, 1220, 1110, 1045, 835, 780, 738.
N~IR ~ (CDC13): 1.21 (3H, d, J = 7.6 ~z), 1.29
(3H, d, J = 6.3 Hz), 2.80 (lH, m), 3.22 (lH, dd, J = 2.3 and
6.3 Hz), 4.18 (lH, dd, J = 2.3 Hz), 4.29 (lH, m), 4.72 (lH,
s), 5.30 (2H, ABq, J = 13.2 Hz), 7.54 (2H, d, J = 8.9 Hz),
8.24 (2H, d, J = 8.9 Hz).
Example 11-1(2)
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
~3~3~
methylsilyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-l-p-
nitrobenzyloxycarbonylmethyl-2-azetidinone (60 mg) in a
mi~:ture of benzene-d6 ana tetrahydrofuran-d8 (4 : 1) (0.6
ml), there was added sodium hydride (11 mg; 50 % in oil)
5 under ice-cooling, followed by stirring for l hour. The N~lR
spectrum of the reaction mixture was measured and shown
below in contrast to that as in Example 11-1(1).
N~ spectra data:-
Example No. Solvent 5-H 6-H
lO 11-1(2) C6D6-THFd8 3.08, d 3.93, d
(4 : 1) J = 6.3 Hz J = 8.9 Hz
11-1(1) C6D6-THFd8 2.58, dd 4.08, dd
(4 : 1) J = 2.6 and J = 2.6 and
5.3 Hz 7.9 Hz
15 Example 11-1-(3)
In the same manner as in Example 11-1(2) but
replacing the solvent by a mixture of benzene-d6 and di-
methylsulfoxide-d6 (9 : 1), the N~IR spectrum of the reaction
mixture was measured and shown below in contrast to that as
in E~ample 11-1(1).
NM~ spectra data:-
Example No. Solvent 5-H 6-H
11-1(3) C6D6-DMSOd6 3.16, d 4.00, d
(9 : 1) J = 7.3 Hz J = 7.6 Hz
11-1(1) C6D6-DMSOd6 3.08, dd 4.10, dd
J = 2.6 and J = 2.6 and
S.0 Hz 7.9 Hz
- 88 -
~3~
E~ample 11-2
OTBDMS OTBDMS
f H H ~ ~ H H
~ COSPh ~ 11
~ OP(OPh)2
O CH2COOPNB O --~
COOPN~
A solution of (3S,4S)-3-[(lR)-l-t-butyldimethyl-
silyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-l-p-nitro-
benzyloxycarbonylmethyl-2-azetidinone (117 mg) in a mixture
of dry toluene znd dry tetrahydrofuran (1 : 1) (1.2 ml) was
dropwise added to a suspension of sodium hydride (22 ml; 50
% in oil) in a mi~ture of dry toluene and dry tetrahydro-
furan (1 ~ 0.2 ml) at -20C, followed by stirrins at the
same temperature for 1 hour. A 2M solution (0.1 ml) of
iodomethane in tetrahydrofuran was added thereto, and
stirring was continued for 30 minutes. A solution of
diphenylchlorophosphate (56 ml) in dry toluene (0.1 ml) was
added to the mixture at the same temperature, followed by
stirring for 1.5 hours~ The resultant mixture was diluted
with ethyl acetate (20 ml), washed with brine, dried over a
mixture of magnesium sulfate and potassium carbonate (10 :
1) and evaporated in vacuo to give an oily residue, which
was purified by silica gel chromatography to obtain
(4R,5R,6S,8R)-p-nitrobenzyl-3-diphenylphosphoryloxy-4-
methyl-6-(1-t-butyldimethylsilylo~yethyl)-1-azabicyclo-
[3.2.0]hept-2-en-7-one-2-carboxylate (115 mg).
IR vmaexat (cm ): 1775, 1725, 1630, 1585, 1518,
1482, 1340, 1285, 1185, 1160, 9,8, 825, 770.
- 89 -
~,3~a
NMR ~ (CDC13): 0.06 (3H, s), 0.07 (3H, s), 0.86
(9H, s), 1.20 (3H, d, J = 7.9 Hz), 1.23 ~3H, d, J = 6.3 Hz),
3.29 (lH, dd, J = 3.0 and 6.0 H~), 3.43 (lH, m), 4.22 (2H,
m), 5.28 (2H, ABq, J = 13.5 Hz), 7.56 (2H, d, J = 8.9 Hz),
8.14 (2H, d, J = 8.9 Hz).
ExamPle 11-8
.
OTBDMS
~ H H
- ~ COSPh
O \ CH2COOPNB
OTBDMS
~ H H r
\ ~ ~ PNZ
COOPNB
A solution of (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-l-
p-nitrobenzyloxycarbonylmethyl-2-azetidinone (415 mg) in a
mixture of dry toluene and dry tetrahydrofuran (4 : 1) (4
ml) was dropwise added to a suspension of sodium hydride t75
ml; 50 ~ in oilj in a mixture of dry toluene and dry tetra-
hydrofuran (4 : 1) (0.75 ml) at -20C, followed by stirring
at the same temperature for 1 hour. A 0.5 M solution (1.49
ml) of iodomethane in tetrahydroruran was added thereto, and
stirring was continued for 30 minutes. A solution of
diphenyl chlorophosphate (218.5 mg) in dry toluene (2.2 ml~
~ 3~ ~2Q
-- so --
was added to the mixture at the same temperature, followed
by stirring for 2 hours. Thereafter, (2S,4S)-1-p-nitroben-
zyloxycarbonyl-2-dimethylaminocarbonyl-4-mercaptopyrrolidine
(237.5 mg) and sodium hydride (32.3 mg; 50 % in oil) were
added theretoi and stirring was continued for 2 hours. The
resultant mixture was diluted with ethyl acetate (50 ml),
washed with brine, dried over magnesium sulfate and e~apo-
rated in vacuo to give an oily residue, which was purified
by silica gel chromatography to obtain (4R,SS,6S,8R,2'S,4'S)-
p-nitrobenzyl-3-[4-(1-p-nitrobenzyloxycarbonyl-2-dimethyl-
aminocarbonylpyrrolidinyl)thio]-4-methyl-6-(1-t-butyldi-
methylsilyloxyethyl)-1-azabicyclo[3.2.0]hept-2-en-7-one-2-
carboxylate (329 mg).
IR vmax (cm ): 1775, 1715, 1660, 1610, 1525,
1400, 1345, 1210, 1140, 1110, ~35, 755.
Example 11-4
OTBDMS
r H H
~ COSPh
'J~
\ CH2COOPNB
OTBDMS
H H
~ 1l
P(oph)2
COOPI~B
+
131~
-- 91 --
.r . OTBDMS
r H H
SPh
COOPNB
A solution of (3S,4S)-3-[(lP~-l-t-butyldimethyl-
silyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-l-p-nitro-
benzyloxycarbonylmethyl-2-azetidinone (69 mg) in dry toluene
(0.6 ml) was dropwise acded to a suspension of sodium
hydride (12.5 ml; 50 % in oil) in dry tetrahydrofuran under
ice-cooling, and the suspension was stirred for 30 minutes.
Diphenyl chlorophosphate (67 mg) was added thereto under
ice-cooling, followed by stirring ~or 1 hour. The resultant
mixture was diluted with ethvl acetate (10 ml), washed with
brine, dried over a mixture of magnesium sulfate and potas-
sium carbonate (10 : 1) and evaporated in vacuo to give an
oily residue, which was purified by silica gel chromato-
graphy to obtain (4R,SS,6S,8R)-p-nitrobenzyl-3-phenylthio-4-
methyl-6-(1-t-butyldimethylsilyloxyethyl)-1-azabicyclo-
[3.2.0]hept-2-en-7-one-2-carboxylate (37 mg) (Compound A)
and (4R,5R,6S,8R)-p-nltrobenzyl-3-diphenylphosphoryloxy)-4-
methyl-6-(1-t-butyldimethylsilyloxyethyl)-1-azabicyclo-
[3.2.0]hept-2-en-7-one-2-carboxylate (34 mg) (Compound B).
Compound A:-
IR vmax (cm ): 1765, 1707, 1522, 1378, 1350,
1340, 1140.
~'MR ~ (CDC13): 0.06 (6H, s), 0.84 (9H, s), 0.95
(3H, d, J = 7.3 Hz), 1.17 (3H, a, J = 6.3 Hz~, 3.06 (lH, m),
- 92 -
: 3.19 (lH, dd, J = 2.9 and 5.0 Hz), 4.22 (2~, m), 5.40 (2H,
ABq, J = 1~.9 Hz), 7.3 - 7.6 (SH, m), 7.69 (2H, d, J = 8.9
Hz), 8.23 12H, d, J = 8.9 Hz).
Compound B:-
The IR and N~5R spectra data were identical to
those of the compound ob~ained as in Exmaple 11-2.
Exam~le 12
OTBDMS
~ON ~ N
\ ~H2COOPNB
OTBDMS
CONMe2
~ ~ - PNZ
COOPNB
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-imidazolylcarbonylethyl]-l-
p-nitrobenzyloxycarbonylmethyl-2-azetidinone (52 mg) in a
mixture of dry toluene and dry tetrahydrofuran (4 : 1) (0.5
ml), there were added sodium hydride (10 mg; 50 % in oil)
and dry dimethylformamide (0.05 ml~ under ice-eooling,
follo~ed by stirring for 1 hour. Diphenyl chlorophosphate
(60 mg) was added thereto, and stirring was continued for 2
hours. To the resultant mixture, (2S,4S)-l-p-nitrobenzyl-
oxycarbonyl-2-dimethylaminocarbonyl-4-mercaptopyrrolidine
_ 93 ~ 33~
,
(35.3 mg) and sodium hydride (5 mg; 50 ~ in oil) were added
under ice-cooling, followed by stirring for 1.5 hours. The
reaction mixture was diluted with ethyl acetate (10 ml),
washed with brine, dried over magnesium sulfate and evapo-
rated in vacuo to give an oily residue, which was purifiedby thin layer chromatography on silica gel to obtain
(4R,SS,6S,8R,2'S,4'S)-p-nitrobenzyl-3-[4-(1-p-nitroben~yl-
oxycarbonyl-2-dimethylaminocarbonylpyrrolidinyl)thio]-4-
methyl-6-(1-t-butyldimethylsilyloxyethyl)-1-azabicyclo-
[3.2.0]hept-2-en-7-one-2-carboxylate.
The IR and NMR spectra data of this compound were
identical to those of the compound as in Example 11-3.
ExamDle 13
3SiO
,-~~
. ~ I COSPh
\ CH2COOPNB
~e3SiO
~ H H r
_/xcoz e2
COOPNB
In the same manner as in Example 11-3 but replac-
ing the starting material by (3S,4S)-3-[(lR)-l-trimethyl-
silyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl3-1-p~nitro-
ben~yloxycarbonylmethyl-2-azetidinone, there was obtained
~94~ 3~3~3~
r- (4R,5S,6S,8R,2'S,4'S)-p-nitrcbenzyl-3-[4-tl-p-nitrobenzyl-
oxycarbonyl-2-aimethylaminocarbonylpyrrolldinyl)thio]-4-
methyl-6-(1-trimethylsilyloxyethyl)-1-azabicyclo[3.2.0]hept-
2-en-7-one-2-carboxylate.
IR ~max (cm ): 1765, 1705, 1650, 1600, 1512,
1395, 1335, 1200, 1130, 1100, 840, 7~0.
Example 14-1
OTBD~IS OTBDMS
r H H r ~ ~ H
COSPh
O ~ CH2COSPh O ~ /
COSPh
To a solution of (3S,4S)-3-[-(lR)-1-t-butyldi-
methylsilyloxyethyl]-4-[(lRl-l-phenylthiocar~onylethyl]-l-
(phenylthiocarbonylmethyl)azetidin-2-one (20 mg) in dry
dimethylformamide (0.2 ml), there was added sodium hydride
(2.6 mg), followed by stirring at room temperature for 20
minutes. The reaction mixture was diluted with diethyl
ether, followed by addition of a phosphate buffer solution
- (pH, 6.86). The aqueous layer was separated from the
organic layer and extracted with diethyl ether twice. -
The extracts were combined with the organic layer, washed
successively with water three times and brine twice,.-
dried over sodium sulfate and distilled orf to remove the
solvent. The residue was puriried by silica ael chromato-
20 graphy to give (4R,5R,6S,8R)-4-methyl-6-(1-t-butyldimethyl-
silyloxyethyl)-l-azabicyclo[3.2.0]hept-3,7-dione-~-
carboxylic acid phenylthio ester.
- 95 - 1 3 ~ 3 ~ a
IR ~meaxt (cm 1): 1780 ~sh), 1760, 1750 (sh),
1710, 1250, 1140, 1062, 830, 775, 742.
Example 14-2
OTBDMS QTBDMS
~' H H ~ r H H Y
/~\COSPh ~
O~\ CH2COSPh o~ ~P (OPh) 2
COSPh
To a solution or (3S,4S)-3-[(lR)-1-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-1-
phenylthiocarbonylmethyl-2-azetidinone (60 mg) in dry
acetonitrile (1 ml), there was added sodium hydride (13 mg;
S0 % in oil) under ice-cooling, followed by stirring for 15
minutes. A solution of dinhenyl chlorophosphate (57.5 mg)
in dry acetonitrile (0.3 ml) was added there~o, and stirring
was continued for 1.5 hours while ice-cooling. The reaction
mixture was diluted with ethyl acetate ~10 ml), washed with
brine, dried over a mixture of masnesium sulfate and
potassium carbonate (10 : 1) and evaporated in vacuo to give
an oily residue, which was purified by thin layer chromato-
graphy on silica gel to obtain (4R,5S,65,8R)-3-diphenylphos-
phoryloxy 4-methyl-6-(1-t-butyldime~hylsilyloxyethyl)-1-
azabicyclo[3.2.0]hept-2-en-7-one-2-carboxylic acid phenyl-
thio ester (55 mg).
The IR and NMR spectra data of this compound were
identical to those or the compound as in Reference Example
3-1.
- 96 - ~L3~3~
E:camPle 15
OTBD~.S ~ OTBDMS
OSPh /'~\~ \=
O \CH2COSPh
COSPh
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilylo,~yethyl]-4-[(lS)-1-phenylthiocarbonylethyl~
phenylthiocarbonylmethyl-2-azetidinone (20 mg) in dry
hexamethylphosphoramide (~lPA) and tetrahydrofuran (1 : 100)
(0.2 ml), there was added a 0.5 M solution t0.3 ml) of
lithium bis(trimethylsilyl)amide in tetrahydrofuran unaer a
nitrogen stream at -30C, followed by stirrina at 0 to 5C
for 25 minutes. The reaction mixture was diluted with
diethyl ether and a phosphate buffer solution (pH, 6.86).
The aqueous layer was separated from the organic layer and
extracted with diethyl ether two times. The exiracts were
combined with the organic layer, washed with brine three
times, dried over sodium sulfate and evaporated. The
residue was purified by silica gel chromatography to give
(45,5R,6S,8R)-4-methyl-6-(1-t-butyldimehtylsilyloxyethyl)-1-
azabicyclot3.2.0]hept-3,7-dione-2-carboxylic acid phenylthio
ester.
IR vmaxt (cm 1): 1760, 1700, 1435, 1367, 1247,
827, 765, 740, 680.
1 3 ~
Example 16-1
OTBDMS OTBDMS
~CH\2C~OO~ B~I 0~--~=
COOtBu
mO a solution of (35,4S)-3-[(lR)-1-t-butyldi-
methylsilyloxyethyl]-4-[(lR)-l-phenylthiocarbonylethyl]-l-
(t-butyloxycarbonylmethyl)-2-azetidinone (65 mg) in dry
tetrahydrofuran (1.0 ml), there was added a 0.1 ~ solution
(3.84 ml) of lithium bis(trimethylsilyl)amide in tetra-
hydrofuran at -70C under a nitrogen strezm. After elevat-
ing the temperature to 0 to 5C, the reaction mixture was
quenched with a phosphate buffer solution (pH, 8.0) and
extracted with diethyl ether twice. - The organic layer
0 was washed with a phosphate buffer solution (pH, 8.0) twice
dried over sodium sulfate and distilled off to remove
the solvent to give (4R,5R,6S,8R)-t-butyl-4-methyl-6-(1-t-
butyldimethylsilyloxyethyl)-1-azabicyclo[3.2.0]hept-3,7-
dione-2-carboxylate.
NMR ~ (CDC13): 0.10 (6H, s), 0.89 (9H, s), 1.18
(3H, d, J = ?.9 Hz), 1.27 (3H, d, J = 6.6 Hz), 1.46 (9H, s),
2.76 (lH, m), 3.18 (lH, dd, J = 2.5 and 5.8 Hz), 4.22 (lH,
dd, J = 2.5 and 8.1 Hz), 4058 (lH, s).
- 98 ~ 1313~
ExamPle 16-2
COSPh ¦
O\C~2COOtBu ' O
~OOtBu
OTBDMS
~ O
> ~ ~ /~ o~(OPh)2
COOtBu
To a solution of (3S,4S)-4-[(lR)-1-phenylthio-
carbonylethyl]-3-[(lR)-l-t-butyldimethylsilyloxyethyl]-
l-(t-butyloxycarbonylmethyl)-2-azetidinone (101 mg) in dry
tetrahydrofuran (2 ml), there was added a 0.1 M solution (5
5 ml) of lithium diisopropylamide in tetrahydrofuran under a
nitrogen stream at -50C, followed by warming gradually to
0C for 2 hours. A solution of diphenyl chlorophosphate
(120 ml) in dry acetonitrile (7 ml) was added thereto under
ice-cooling, followed by stirring for 2 hours. The reaction
10 mi;~t~re was poured into cold diethyl ether (20 ml) and a .
phosphate buffer solution (pH, 6.86; 20 ml). The organic
layer was washed with brine, dried over magnesium sulfate
and evaporated in vacuo to remove the solvent. The residue
was purified by thin layer chromatography on silica gel to
15 give (4R,iR,6S,8R)-3-(diphenylphosphoryloxy)-4-methyl-6-(1-
t-butyldimethylsilyloxyethyl)-l-azabicyclo[3.2.0]hept-2-en-
- 99 - 13
'
7-one-2-carboxylic acid t-butyl ester (83.5 mg).
IR vnaat (cm 1): 1780, 1718, 1635, 1585, 1482,
1360, 12~5, 1185, 115&, 960, 940, 830, 765.
NRM ~ (CDC13): 1.17 (3~, d, J = 7.6 Hz), 1.21
5 (3H, d, J - 6.3 Hz), 3.22 (lH, dd, J = 3.0 and 6.0 Hz), 3.42
(lH, m), 4.12 (lH, dd, J = 3.0 and 10.0 Hz), 4.21 (lH, m).
Example 16-3
OTBDMS OTBDMS
o~C:~2COOt~u ~ '~
COOtBu
Compound (A)
In the same manner as in Example 16-1 but using
the starting material as shown in the table below, there was
10 produced a mixture of (4~,5R,6S,8R)-t-butyl-4-methyl-6-(1-
t-butyldimethylsilyloxyethyl)-l-a7abicyclo[3.2.0]hept-3,7-
dione-2-carboxylate and (4S,5R,6S,8R)-t-butyl-4-methyl-6-
(l-t-butyldi~ethylsilyloxyethyl)-l-azabicyclo[3.2.0]hept-
3,7-dione-2-carboxylate (Compound (A)).
- 100 - ~ 3~ 33~
No Starting material Reaction condition
_z
1 1 -Cl LiN[si(C~3)3]2/THF;
-70C > 0 to 5C
C ~ I (same as above)
3 -OBt ¦ (same as above)
4 ~ (same as above)
The IR and NMR spectra data of the product were
identical to those of the compound as in Example 16-1 and
Example 17-2
ExamPle 17-1(1)
OTBDMS OTBDMS
H ` r H H
o ~ CH\2~00Me O ~
COOMe
To a solution of (3S,4S)-3-[(lR)-l-t-butyldi-
methylsilyloxyethyl]-4-[(lS)-l-phenylthiocarbonylethyl]-1-
(methoxycarbonylmethyl)a~etidin-2-one (50 mg) in dry he~a-
methylphosphoramide and tetrahydrofuran (1 100) ~0 6 ml)
was added a 1 M solution (0 44 ml) of lithium bis(trimethyl-
silyl)amide in tetrahydrofuran at -30C under a nitrogen
stream, followed by stirring at room temperature After
- 101~
disappearance of the starting material, the reaction mixture
was diluted with a phosphate buffer solution (pH, 6.86) and
diethyl ether under ice-cooling. The aqueous layer was
separated from the organic layer and extracted with diethyl
ether ~wice. The extracts were combined with the
organic layer, washed with brine five times, dried over
sodium sulfate and evaporated. The residue was purified by
silicz gel chromatography to give (4S,5R,6S,8R)-methyl-4-
methyl-6~ t-butyldimehtylsilyloxyethyl)-1-azabicyclo-
[3.2.0]hept-3,7-dione-2-carboxylate.
IR vmax 3 (cm ): 1770, 1760, 1740, 1435, 1245,
1120, 825.
NMR 8 (CDC13): 0.10 (6H, s), 0.90 (9H, s), 1.27
(3H, d, J = 7.0 Hz), 1.30 (3H, d, J = 6.2 Hz), 2.29 (lH, m),
15 3.14 (lH, dd, J = 1.5 and 5.7 Hz), 3.77 (3H, s), 4.31 (lH,
m), 4.71 (lH, s).
Example 17-1(2)
OTBDMS OTBDMS
~ COSPh
O \ CH2COOMe O N ~
C~OMe
To a solution of (3S,4S)-3-[(lR) 1-t-butyldi-
methylsilyloxyethyl]-4-[(lS)-l-phenylthiocarbonylethyl]-l-
(methoxycarbonylmethyl)azetidin-2-one (15 mg) in dry hexa-
20 methylphosphoramide and tetrahydrofuran (1 : 100) ~0.2 ml),there was added a 2.6 M sodium methylsulfinylmethide
solution (0.05 ml), prepared from sodium hydride and di-
~3~3~
methylsulfoxide, at -20 to -25C under a nitrogen stream.
A.ter stirring at 0 to 5C ror 30 minutes and at room
temperature for 20 minutes, the reaction mixture was diluted
with a phosphate buffer solution (pH, 6.86) and diethyl
ether at 0 to 5C. The aqueous layer was separated and
extracted with diethyl ether. The extract was combined with
the organic layer, washed with water three times, dried over
sodium sulfate and evaporated. The residue was purif_ed by
silica gel chromatography to give (4S,SS,65,8R)-methyl-~-
methyl-6-~1-t-butyldimethylsilyloxyethyl)-1-azabicyclo-
[3.2.0]hept-3,7-dione-2-carboxylate.
The IR and NMR spectra data of this compound were
identical to those of the compound as obtained in Example
17-1(1).
ExamPle 17-1(3)
OTBDMS _ OTBDMS
H H ~ Y H H
~ '- ~ COSPh ~ h ` -
O CH2COOMe O
COOMe
To a solution of (35,45)-3-[(lR)-1-t-butyldi-
methylsilyloxyethyl]-4-[(lS)-l-phenylthiocarbonylethyl]-l-
(methoxycarbonylmethyl)azetidin-2-one (15 mg) in dry
hexamethylphosphoramide and tetrahydrofuran (1 : 100) (0.2
ml), there was added potassium t-butoxide (15 mg) at 0 to
5C, followed by stirring at room temperature for 30
minutes. The reaction mixture was diluted with a phosphate
burfer solution (pH, 6.86) and diethyl ether a~ 0 to 5C.
- 103 - 13~33~
The aqueous layer was separated from the organic layer and
extracted with diethyl ether. The e:ctract was combined with
the organic layer, washed successively with water two times
and brine, dried over sodium sulfate and evaporated. The
residue was purified by silica gel chromatography to give
(4S,SR,6S,8R)-methyl-4-methyl-6-(1-t-butyldimethylsilyloxye-
thyl)-l-azabicyclo[3.2.0]hept-3,7-dione-2-carboxylate.
The IR and NMR spectra data of this compound were
identical to those of the compound as in Example 17-l(l).
Example 17-2
OTBDMS OTBDMS
H H ~ r H H
COOR
Compound (B)
In the same manner as in Example 17-1(1) but
using the starting material as shown in the table below,
there was obtained (4S,SR,6S,8R)-4-methyl-6-(1-t-butyl-
dimethylsilyloxyethyl)-l-azabicyclo[3.2.0]hept-3,7-dione-2-
carboxylic acid ester (Compounds (B)).
No. -R Reaction condition
._
l -PNB LiN[Si(CH )3]2/
HMPA-THF ~l : 100);
-30C ~ room
temperature
.
2 ¦ -tBu (same as above)
HMPA: Hexamethylphosphoric triamide
- 104 -
l~t3~
.
~4S,5R,6S,8R)-p-Nitrobenzyl-4-methyl-6-[1-t-butyl-
dimethylsilyloxyethyl]-1-azabicyclo[3.2.0]hept-3,7-dione-2-
carboxylate (Compound (B): R = -PNB) -
IR VmaX13 (cm 1): 1780~ 1760~ 1720~ 1520~ 1345
1245, 1178, 835.
N~ ~ (CDC13): 0.08 (3E, S)~ 0.10 (3H~ s)~ 0.88
(9Hr S)~ 1.26 (3H, d, J = 6.8 HZ)~ 1.30 (3H~ d, J = 6.2 HZ)~
2.28 (lH, m), 3.17 (lH, dd, J = 2 and 7 H~), 3.67 (lH, dd, J
= 2 and 8 Hz), 4.30 (lH, m), 4.80 (lH, S)~ 5.29 (2H, S),
7.53 (2H~ d, J = 9 HZ)~ 8.24 (2H~ d~ J = 9 HZ).
(4S,5R,6S,8R)-t-Butyl-4-methyl-6-[1-t-butyl-
dimethylsilyloxyethyl]-l-azabicfClo[3.2.0]hept-3~7-dione-2-
carboxylate (Compound (B): R = -tBU):-
IR ~mHaC13 (cm 1): 1760~ 1730~ 1360~ 1142~ 825.
NRM ~ ~CDC13): 0.10 (6H~ S)~ ~.90 (9H~ 5)~ 1.27
(3H~ d~ J = 6.9 HZ)~ 1.30 (3~ d~ J = 5.9 HZ)~ 1.46 (5H~ S)~
2.24 (lH, m), 3.12 (lH, dd, J = 2.0 and 6.3 HZ), 3.66 (lH,
dd, J = 1.9 and 8.1 HZ)~ 4.29 (lH~ m), 4.57 (lH~ S).
Example 17-3
OTBDMS ` OTBDMS
O ~ OOtBU ~
COOtBU
Compound (C)
In the same manner as in Example 14-1 but using
the starting material as shown in the table below, there was
obtained (4S,5R,65,8R)-t-butyl-4-methyl-6-(1-t-butyldimethyl-
- 10-5 - I3~3~
silyloxyethyl)-l-azabicyclo[3.2.0]hept-3,7-dione-2-
carbo~ylate (Compound (C)).
No. ¦ Starting m2terial Reaction condition
I -Z
1 ¦ -SPh NaH/D~F;
l room temperature
Z ~ ~ ¦ (s~me es above)
The IR and NMR spectra data of the product were
identical to those of the compound as in Example 17-2.