Note: Descriptions are shown in the official language in which they were submitted.
The present invention rela-tes to a process for
preparing optically active alpha-arylalkanoic acids and in
particular, the presen-t invention concerns an overall enantio-
selective process for the preparation of optically active alpha-
aryl-alkanoic acids comprising two main steps: a stereoselective
halogenation of novel chiral (optlcally active) ketals and a
stereoselective rearrangement of the thus obtained products.
In particular the present invention relates to
optically active esters of ~ -arylalkanoic acids which are
intermediates in the process.
This, application is a devisional application of
copending application No. 478,585 filed April 4, 1985.
The alpha-arylalkanoic acids constitute a very large
class of compounds, of which many have assumed considerable
commercial importance in relatively recent years as anti-
inflammatory and analgesic drugs.
These include 2-(4-isobutylphenyl)-propionic acid known
as Ibuprofen, 2-(3-phenoxyphenyl)-propionic acid known as
Fenoprofen, 2-(2-fluoro-4-diphenyl)-propionic acid known as
Flurhiprofen, 2-[4-(2-thienylcarbonyl)-phenyl]-propionic acid
known as Suprofen, 2-(6-methoxy-2-naphthyl)-propionic acid, of
which the tS) isomer is known as Naproxen, and others.
Another group of alpha-arylalkanoic acids are well
known as intermediates in the preparation of pyrethroid
insecticides. These include 2-(4-chlorophenyl~-3-methyl-butyric
acid and 2-t4-difluoromethoxyphenyl)-3-methyl-butyric acid.
A number of the alpha-arlyalkanoic acids exist as a
mixture of optically active isomers.
.~3~8~
- 2 -
Very often, a decidedly higher biologlcal activity is associa-ted with
one enantiomer which thus is much more important than -the other from an
i.ndustrial vlewpolnt. ~~
A particularly lmportant example i9 Z-(6-methoxy-2-naphthyl)-propio-
6 nlc acid, of which the (Sj isorner (Naproxen3possesses pharmacological
properties whlch are decldedly better than those of the (R) lsomer
and of the raceme mixture, so that in practice i-t ls only the (S)
isomer ~hich ls used as pharmaceutlcal drug.
Of the many methods for synthesising alpha-arylalkanoic aclds which
have recently appeared in the literature, the most interesting are
those which use rearrangement of aryl-alkyl-ketals which are functio-
nalised on the alkyl position alpha to the ketal. These include the
~ethods:.:described in:European.,patent.ap~lications.34871.(Blaschim).,
35305 (Blaschim), 48136 (Sagami), 64394 (Syntex), 89711 (Blaschim),
and 101124 (Zambon), and in Italian patent applications 21841 A/8Z
(Blaschim and CNR), 22760 A/82 SZambon) and 19438 A/84 (Zambon), and
in the publication J. Chem. Soc., Perkin I, 11, 2575 (1982).
All these processes lead to racemicmixtures of the two optical
isomers .
Optically active alpha-arylalkanoic acids can be prepared by separating
..the enantiomer ~rom the racemicmixture obtained by using the afore-
~aid procedures (for example by using optically active bases), or
by applying some of said. rearrangements to optically
active ketal~, which have been previou~ly prepared and isolated,
as described for example in European patent applications 67698 ~Saga-
mi) and 81993 (Syntex).
However, the preparation o~ optically active ketals as described in
these European patent applications appears rather laborious and
costly, and also involves the preparations of intermediates by sophis
ticated methods with low yields, and are not suitable for indus-
trial preparation~
The ro~.olution Or alpha-arylalkanoic acids from the racemic mixture in
_ 3 _ ~ 3~
a conventional way,that is by using optically active bases has -the d~
common to all these processes: material costs, manufacturing labor and
equipment for -the recovery and racemi2ation of` the undesired optical
isomer.
Therefore, it is important to have a stereoselective process for pro-
ducing the desired isomer directly. Such a process obviates -the neces
sity of subsequently resolving the d- and l-isomers using optically
active bases, subh as cinchonidine, brucine, alpha-phenyiethylamine,
N-methyl-glucamine and the like.
The elimination of resolution steps results in a substantla~ saving,
both in material cost and manufacturing labor and equipment.
The savings can be particularly significant with regard to compounds
which are approved for pharmaceutical use as a substantialiy pure,
optically active isomer,such as S(~2-(6-methoxy-2-naphtyl)-propionic
acid (Naproxen) or a precursor thereof which may be easily converted
to this acid.
For the sake of clarity we will state hereinafter the meaning of some
terms used in the following specification:
"Chiral" refers to a chemical structure having at least an asymmetry
center. The con~iguration of an asymmetric carbon atom is classified
as "R" or "S" according to the Cahn -Ingold-Prelog method.
'IEnantiomer" or "enantiomorph" refers to a molecule whlch is non-
superimposable on its respective mirror image. A necessary and
sufficient condition for a molecule to show optical acti~ity (i.e.
to be an enantiomer) is that such a molecule not be superimposable
with its mirror image. This phenomenum usually occurs in organic chemi-
stry when a carbon atom is attached to four different atoms or chemi-
cal groups. "~nantiomer" and "optical isomer" are often used inter-
changeably in this context.
"Enantiomeric excess" or "e.a." refer~ to a definition; i.e. th~ per-
centage of the predominant enantlomer mlnus that Or the oth~r. rrhus,
a mlxture of 95% (~) isomer and S% ~-) isom~r wollld have a 90~
~ 4 ~ ~ 3 1 ~ ~
"Optical yield" or "optlcal purity" may be defined as enantlomeric
excess. However, strictly speaking, it refers to the measursd rokation
shown by the mixture which may or may not reflect the true proportions
of the enantiomers. In thi3 application the ~wo terms are used
interchangeably.
"Optically active" refers to a sy3tem or compound which rotate~ the
plane of polarized light.
"Epimers" are two dia~tereoisomers which have a different confi6ura-
tion at only one chiral center.
"Diastereoisomers" are stereoisomers that are not mirror images of
each other: they have the same con~iguratlon at at least one a~ymmetric
center and, at the same time, different configura-tion at at least one
asymm~tric center.
"Diastereotopic'~ refers to the case in which two atoms or groups in a
molecule e.gu CX2WY are in such a position that replacing each of
them by a group Z leads to diastereoisomers.
"Stereoselective synthesis" re~srs to ang reaction in which one among
a number of stereoisomers is formed exclusively or predominantly.
"Enantioselective synthesis" refers to any reaction ln which one of
two enantiomers i5 form0d exclusively or predominantly.
"Race~ization" refers to the conversion of the molecules of one
enantiomer into a racemic mixtur0 of both.
We ha~e now prepared new ketals of alkyl-aryl-ke-tones of formula
H ~¦ ¦~ COR2 (A)
O /O
Ar \ C _ CH - R
in which: X
Ar represents aryl, optionally subs-titu-ted;
R repre6ents linear or branched Cl-C4 alkyl;
~ 3 ~
Rl and ~2~ which can be equal to or different from each o-ther, re-
present a hydroxy, a O M , OR3 or NR4R5 group wher~ R3 is Cl-C24 alkyl,
C3-C6 cycloalkyl, phenyl or ben~yl; M is -the cation of an alkaline metal;
R~ and R5, which can be equal to or different from each other,re~resent
a hydrogen atom,a Ol-C~ alkyl,a C5-C6 cycloalkyl,or a -(CHz) -CH20H
group where n ls 1, 2 or 3 or R4 and R5 taken together con~titute a
(CH2)m- group where m i5 4 or 5 or a -CH2-C~2-R -CH -CH - group where
R7 is an oxygen atom,a NH group or a Cl-C4 N-alkyl group; X représent~ a
hydrogen,chlorine,bromine or iodine atom. The carbon atoms indicated
by an asterisk are both contemporaneously ln (R) or (S) configura-
tion. Thus the ketals of formula A are optically active.
The ketals of formula (A) have shown quite unexpected properties
which allow the realization of the new process according to
the present invention.
In fact, we have found that when ketals of formula A, in which X is
hydrogen, are reached with achiral halogenating reagents, a chemo- ,
selective halogenation occurs in high yield on the diastereotopic carbon
atom inthe alpha position with respect to the ketal group and in the
thus obtained alpha halogen ketals ~formula A; X = Cl, ~r, I) only one
of the e~imers is formed or s-trongly prevails over the other. It is
worth noting that the absolute configuration (R,R or S,S) of the chiral
centers already present on the starting ketals A (X=H) is untouched.
As far as we know, a ~tereoselectiYe halogenation in the alpha
position of a ketal has never been previously described.
Moreover, we have found that the ketals of formula A in which X = Cl,
8r, I provide in high yields alpha-arylalkanoic acids in which the
enantiomeric ratio reflects the epimeric ratio of the s-tarting
ketals or, depending on the rearrangement conditions,the acid enan-
tiomeric ratio is higher than the epimeric ratio of -the starting
ketals.
'rO our knowledge, it is the Eirst time that a rearrangment of ketals
ie described whlch gives rise to chemically pure alptla-arylalkanoic acids
- 6 - ~ 3 ~
having an enantiomeric excess higher than ths epimeric exces of the
starting ketals.
Thus the present invention provides an enantioselective
process for the preparation of alpha-aryl~alkclnoic acids by diastereo-
selec-tive halogenation, in the alpha position to the ketal group, of
optically active ketals of formula (A) wherein X = H and the enantio-
selec-tive rearrangement of the obtained halo-ketals into the correspond
ing alpha-arylalkanoic acids.
An enantioselective process for preparing optically active alpha-
arylalkanoic acids is completely new.
~ 3 ~
The arylalkanoic acids prepared according to the present invention
fall within the formula
' F~
Ar - CH - C00~ (I)
in which ~l is a Cl C4 alkyl; Ar is as heretofore defined and prefe-
rably a monocyclic, polycycli.c, or or~hocondensed polycyclic aro-
matic or heteroaromatic group having up to l2 carbon atorns in the
aromatic s~ystem' 5UCII as phenyl, diphenyl,naph~hyl,'hienyl,or pyrrolyl.
The possible substituents of these aromatic g~oups ccmprise one
or more halogen atoms, Cl-C4 alkyls, C3-C6 cycloalkyls, benzyl,
hydroxy, Cl-C4 alkoxy, Cl-C~ al~ylthio, Cl-C4 haloalk~l, Cl-C4
haloalkoxy, phenoxy, thienvlcarbonyl and benzoyl.
Specific examples of such substituted aryls are 4-isobutyi-phenyl,
3-phenoxy-phenyl, Z-fluoro-~-diphenyl, 4'-fluoro-4-diphenyl,
4-(2-thienyicarbonyl)-phenyl, 6-methoxy-2-naphthyl, 5-chloro-~-
methoxy-2-naphthyl and 5-bromo-6-methoxy-2`naphthyl, 4-chloro-
phenyl, 4-difluoromethoxy-phenyl, G-hydroxy-2-naphthyl, and
5-bromo-6- hydroxy-2-naphthyl.
The ketals of Eormula (A) which constitute the starting compounds
for the new process according to the present invention are prepa-
red by ke-talization of a ketone of formula
Ar - C - CH2 - R ~II)
O
(in which Ar and R have the aforesaid meanings) by means of
L(~)-tartaric acid ~2R, 3R-dihydroxy-butanedioic acid) or
D(-)~tartaric acid (25, 3S-dihydroxybutanedioic acid) or derivatives
thereof.
The ketones of formula II are products which are known or are easily
prepared by known methods, for example by Friedel-Crafts acylation.
The ketalization reaction is carried out according to conven-
tional methods, for example in the presence of an acid catalyst and
~ 3
an orthoester. Alterna-tively, the water formed during the react-
ion can be removed by azeotropic distillation, for example with
benzene, toluene, xylene, heptane or other su:i-table solvents.
The absolute configuration and the optical pur:Lty of -the ketals
.5 of formula A in which X is hydrogen are the same as those of thestar-ting diol (tartaric acid or derivative thereof). Thus, start-
ing from L(~)-tartaric acid, the obtained ketal of formula A has
both the carbon atoms marked by an asterisk in formula A hereabove
in the R configuration.
This reaction is particularly suitable for preparing compounds of
formula (A) ~n which Rl and R2 represent a OR3 group, by react-
ing the ketones of formula (II) with a tartaric acid ester.
The ketals of formula (A) in whlch Rl and R2 are other than OR3
are preferably prepared startin~ from these latter compounds by
suitable transformation of the OR3 group.
For example, starting from esters of formula (A) in which Rl and
R2 are OR3 groups, the corresponding mono-salts (for example
Rl = 0 M ~nd R2 = OR3~ can be prepared by partial saponification
with one equivalent of a base~for example alkaline hydroxide), and
from these the corresponding mono-acids (for example Rl = OH,
R2 = OR3) can be prepared by acidlfication.
Hydrolysis of the ester~ with two equivalents of an alkaline base
leads to the formation of the corresponding salts ~Rl = R2 = M )
which by acidification produce the free dicarboxylic acids (Rl =
R2 = OH) which are th~ starting compounds for preparing different
deriv~tives such as other mono or di-esters (Rl and/or R2 = OR3)
or mono or di-amideq (Rl and/or R2 = NR4R5).
. The amides can also be obtained directly from the esters of formu-
la (A) by treatment with a suitable amine of formula R4R5-N-H.
As stated heretofore, the compounds (A) wherein X = H are useful as the
starting compounds for preparing -the compounds of formula (A)
in which X represents a chlorine, bromine or iodlne atom.
9 ~ 3 ~
The compounds of formula (A) are halogenated byknown h~logenating agents
for example bromine, quaternary ammonlum perhalides, sul-
phuryl chloride, cupric chloride or bromide, N-bromo or N-chloro-
succinimide, N-chlo~o-phthalimide, pyridine or pyrrolidone per-
bromide or pyridine perchloride or the analogous iodides,hexachloro-
2,4-cyclohexadienone, iodine and iodlde chlorlde, or analogous
systems.
We have found that the halogenation of ketals having the carbon
atoms marked by an asterisk in formula A hereabove both in configu
ration R, that is ketals prepared from L(~)-tartaric acid or a de-
rivative thereof (i.e. the na-turally occurring tartaric acid),
give rise to the formation of a mixture of epimeric alpha-halo
ketals in which the epimer in which the carbon atom bonded to the
halogen is in the S configuration, strongly prevails. Since the con-
figuration of the carbon atoms marked by an asterisk in formula A
hereabove remains unchanged, the major epiMer of the alpha halo-
-icetals derived from the naturally occurring tartaric acid or a
derivative thereof, will be hereinafter referred to as RRS epimer
and the minor one as RRR epimer.
We have also found that starting from ketals derived from
D(-)-tartaric acid, the ma~or epimer has the carbon atom bonded
to the halogen akom in the R configuration.
From the above firldings it clearly results that the described
halogenation reaction is a new stereoselective reaction.
The ratio between the epimers RRS/RRR iS generally hlgher than
75:25 and in most of the cases is higher than 94:6. Depending on
the substrate and the reactlon conditions it is also possible
to obtain the RRS epimer as the only chemically pure alpha-halo-
gen-ketal, the other epimer RRR . present, if any, in an amount
lower than 1%.
Generally, the yields in alpha-halogen ketals are higher than
90%-
- 10 - ~L3~
The stereoselectivity of the halogenation reaction ~s only slightly
affected by the polarity of the solvent. a number of solvents
such as carbon te-tracllloride, 1,2-dichloroethane, chlorobenzene,
benzene, toluene, acetonitrile, cyclohexane, ethylacetate, carbon
disulphide, acetic acid and so on,may be used. Best results are obtained
by using solvents of` low polarity. The reaction may be carried out at
room temperature with satisfactory results. ~'he stereoselectivity of the
halogenation reaction increases by lowering the reaction temperature.
The reaction still occurs up to -70C.
Preferably', traces of a mineral acid are required to start-up
the halogenation reaction which i~ usually tsrminated in a few mi
nutes. As far as yields and stereoselectivity are concerned, the
preferred halogenation reation is the bromination. Said reaction
is preferably carried out with bromine as the halogenating agent,
at a temperature between -40and +20C in solvents such as carbon
tetrachloride, methylene chloride, 192-dichloro-ethane and carbon
disulphide.
The peculiar characteristics of the ketals of formula A and in
particular the shown high stereoselectivity in the halogenation
reaction,were completely unpredictable on the base of the present
knowledge o~ stereocontrolled reactions.
Independently from the aforesaid, the fact that the ketals of formula
(A) where X = halogen ~exist in the form of diastereoisomers easily
separable by known methods, for example by ~ractional crystalliza-
tion, is also important.
If required, it is therefore possible to separate the desired
isomer of the ketal of formula (A) and sub~ect this to rearrangemen-t
to obtain the alpha-arylalkalloic acid in the substantially pure
optically active form.
It is also important to note that tartaric acids and esters, in
particular L(~)-tartaric acid and the relative methyl and ethyl
esters, have a commercial cost which is competitlve with that o~
$ ~ ~ 7
the glycols described as ketali~ing agents in the processes of the
known art, and the preparation of the tartaric acid derivatives
(ester, amides, salts) certainly does not constitute a costly
process.
The possibili-ty of having groups of different nature in the ketals
of formula A, wi-th reference to the substituen-ts R1 and R2,'enables
-to vary the hydrophilic and lipophilic propert;ies of said ketals
within wide limits, from compounds containing polar grups (allcaline
salts, amides) to lipophilic compounds~esters of long-chain
alcohols~.
This wide possibility of choice allows to select the ketal of
formula A most suitable for -the experimental conditions
(solvents, temperature, catalysts) used in the various proces es
for the preparation,of alpha-arylalkanoic acids or their deriva- -
tives by rearrangement.
As far as the rearrangement of the ketal3 of formula A (in which
X=Cl, Br, I~ is concerned, we have found that the ketals having
the configuration RRS (wherein S is'the configuration of the
carbon atom bonded to the halogen atom) provide the S-enantiomer
of the c,orresponding alpha-arylalkanoic acid.
This is particularly important because (a) the S-enantiomer of
alpha-arylalkanoic acid is generally the biologically more active
isomer and the alpha-arylalkanoic acids present on the market in
optically active form are all of S-configuration and because (b)
the ketals of formula A having configuration RRS are selectively
obtalned by halogenation of the ketals o~ formula A, X=H in turn easily
prepare,d from the appropriate ketone and the naturally occurring
L(~)-tartaric acid (or a derivative thereof) which is a really
~nexpensive material.
3~ In order to conveniently transform the optically active ketals of
formula A tX=Cl, Br, I) it is necessary to use a rearrangement method
which provides optically active alpha-arylalkanoic acids having,an
enantiorner~ic ratio very close to that o~ the epimers in the starting
- 12 - ~ 4~ r,
ketals. This implies that the reaction has to b0 stereospecific and
that -the react.ion conditions are such that no racemization occurs in
the final products. We have found that the known methods provide alpha
arylalkanoic acids having enantiomeric ratio equal to or lower than the
epimeric ratio of the starting ketals. We have also found, and this is a
further ob~ect of th~ present invention,a n~w enantioselec~ive.rearran-
gement method'which overcomes.the above limits.
Such a prOCesG is here~ith defined as enantioselective in so far as
the enantiomeric composition ~ratio between enantiomers S and R)
of the alpha-arylalkanoic acids thus obtained~differs from the ep~-
meric composition of the ~tarting ketals of formula A and more
precisely and quite surprisingly corresponds to an increase in the
optical purity of the alpha-aryialkanoic acid with respect to
the epimeric composition of the starting ketals.
Thanks to this new, surprising rearrangement process, starting from
e.g. a mixture of epimeric ketal~.'of formula A (in which X=Cl, Br, I)
sufficiently enriched in the RRS epimer,it i~ possible to obtain
in a optically pure form the S-enantiomer of the corresponding
alpha-arylalkanoic acid.
It is worth noting that the yield of the ~ew rearrangement process is
- a~ high as 80-90%.
The enantioselective process object of the present invention essen-
tially consists in rearranging a ketal of formula A in which X is a
' chlorine, bromina or iodine atom, in aqueous medium at an acid pH,at a
temperature comprised between room temperature and 100C.
The above mentioned rearrangement conditions are particularly un-
expected and surprising in that it i8 well known that the treatment
of a ketal with water under acidic conditions i8 a general method
to convert ketals into the'corresponding ketones and the . -
alcohol or diol, Accordingly, the previously known alpha-haloal-
kyl-aryl ketals, under the above reaction conditlons, undergo a
fast hydrolysis providlng the corresponding alpha haloalkyl-aryl-
ketono ~ncl alcollol or dlol.
.,
- 13 ~
On the contrary, the ketals of formula A obJect of the present
invention, when treated in aqueous acid medium, provide in high
yield -the corresponding alpha-arylakanoic acids, ketones being pre-
sen-t, if any, in negligeable amounts.
The rearrangement process ob~ect of the invention i3 preferably
carried out by using ketals of formula A (ln which X-Cl, Dr, I) BO-
luble or at leas-t partlally soluble ln water ~nder the reac-tlon
conditions, i.e. the ke-tals of formula A in which R and/or R are
hydrophilic groups.
The rearrangement is preferably carried out by heating the ketaL
of formula A in water at a pH comprised betwaen 3.5 and 6.5.
The desired pH values may be maintained by adding a sultable amount
of a buffer.
The reaction duration depends mainly on the nature of the ke-tal
of formula A, and on the reaction temperature. Generally, a high
cohversion degree is reached after s~me hours.
Usually, the alpha-arylalkanoic acids are scarcely soluble in water,
therefore at the end of the reaction the optlcally active alpha-
arylalkanoic acid may be isola-ted by simple filtration. A pharmaceutical
product as pure as required by U.S.Pharmacopeia is obtained by sim~le
acid-base treatment of the product isolated by filtra-tion~ As far aR we
know,this is the first time ~h~t a rearrangement of halogenketals for the
preparation of alpha-aryialkanoic acids is carried out in water as the
ohly reaction solvent. The main advantage~ of the present rearrangement
process from an industrial point o~ view, may be summarized as follows:
(:a? the proce6s is enantioselective and provides alpha-arylalkanoic acids
in high yields and with an enantiomeric ratio higher than the epimeric
ratio of the starting ketals; ~b) the reaction solvent is water with the
consequent economic and safety advantages; (c) no metal catalyst is re-
quired and (d) the optically active alpha-arylalkanoic acid is separated
from the reaction mixture by simple filtration.
By considering the overall process for the preparation o~
optically active alpha-arylalkanoic acid~ according to the
present invention it may be said that it csnsists of two
quite new steps: the stereo-selective halogenation of a
ketal of formula A in which X is hydrogen and the
enantioselective rearrangement of the thus obtained ketal of
formula A in which X is a chlorine, bromine! or iodine atom.
More specifically the overall process for the selective
preparation of the S-enantiomer of an alpha-arylalkanoic acid
according to the present invention consists thus of two quite
new steps: the stereo-selective halogenation of the suitable
ketal of formula A in which X is hydrogen and in which the
carbon atoms marked by an asterisk are both in the R
con~iguration, to selectively obtain the epimer RRS of the
ketal of formula A in which X is a chlorine, bromine or
iodine atom and the enantioselective rearrangemnt of the thus
obtained ketal in water under acidic conditions.
Such a process is possible thanks to the unexpected
characteristics of the ketals of ~ormula A shown both in the
alpha halogenation step and in the a~ueous rearrangement
step.
The rearrangemnt method may be also performed in different
less advantageous manners depending on the starting ketal.
For exa~nple, the ketals of ~ormula (A) in which X is a iodine
atom, when Ar is the 5-methoxy-2-naphthyl group and R is a
methyl, can be rearranged according to the procedure given in
European patent application 89711, published September 28,
1983, or by oxidation as described in Italian patent
application 21841 A/82 open to public inspection December 11,
1983 (now Italian patent 1,~90,867).
- 14 -
,~
11 3 1 ~ ~
~ikewise, the ketals of formula (A) in which X is any halogen
atom can be rearranged in the presence of certain metal
salts, as described in European patent applications Nos.
34871 and 35305 and in J~ Chem. Soc., Perkin I, 11, 2575
(1982), or in a protic polar msdium in neutral or weakly
alkaline conditions, optionally in the
- 14a -
presence of an inert diluent, as described in Italian patent appli-
cation No. 22760 A/82 or in European patent application
lOL,124.
The latter aforesaid method has important advantages relative in
particular to its ease of industrial realizatiom and to the fact that
it does not require the presence of metal salts as catalysts.
The aforesaid rearrangement reactions lead in general to the forma-
tion of alpha-arylalkanoic acids in the form of their derivative,
in particular esters. These are then hydrolysed to the corresponding
free acids by conventional methods.
Of the optlcally active alpha-arylalkanoic acids, the most import-
ant from the pharmacological viewpoint is 2-(6-methoxy-2-naphthyl)-
propionic acid, of which the S(~isomer is known as Naproxen.
In a specific embodiment, the present invention relates to compounds
of formula
! Rl.C0 H
~ 0 tB)
ZO ' \ C
~ I H -CH3
Z-O
'
(in which R1, R2 and X have the meanings given for the formula (A),
Y represents a hydrogen atom or a chlorine or bromine atom and Z
represents a hydrogen atom, a methyl or an alkaline metal) and their
use in the preparation of Naproxen by rearrangement.
The carbon atom~indicated with an asterisk have R configuration and
when X is different from hydrogen, the carbon atom to which it is
bonded has S configuration.
A compound o~ formula (B) in which X represents a halogen atom and
_ 16 -
Z a methyl, may be rearranged in the presence of certain metal salts
such as Ag and Z~,or in a polar solvent under neutral or slightly
alkaline conditions.
Moreover a compound of formula (B) in which Z repressnts an alkaline
metal, may be rearranged in an aqueous or organic medium under neutral
or alkaline conditions.
In any case the preferred embodiment according to the present
invention is the rearrangement of the ketals of formula B (in
which X = Cl, Br, I) in water, under acidic conditions.
The rearrangement of the epimer RRS of the ketals of formula B leads to
S(+)-Naproxen or its direct precursors, for example containing Y
substituent.
In preparing Naproxen, it is necessary to eliminate the substituent
Y when this is a chlorine or bromine atom. This is done by hydrogeno-
lysis either on the alpha-arylalkanoic acid or on the ~elative ester.
The reaction involving rearrangement of the compounds of formula (A),
in particular when conducted in a medium free from alcohols and
glycols under mild conditions, can lead to the formation of new lnter-
mediate esters of formula
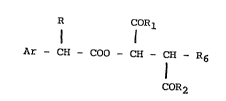
R COR
1 1
Ar - CH - GOO - CH - fH R6 (c)
COR2
(in which Ar, R, Rl and R2 have the meanings given for formula A)
and R6 i6 OH, Cl, Br or I. Depending on the reaction conditions,
R6 can also assume other meanings such as acetate, propionate or
benzoate.
Hydrolysis of the compounds of Eormula (C) then leads to the corres-
poding alpha-arylalkanoic acid~.
Likewise, the rearrangement of the compounds of formula (B), when
carried out in a medium free from alcohol9 and ghycols, can lead to
the pro~uction of intermedia-te esters of formula:
- 17 - ~ 3 ~
CH
, 3
C-O-CH-CH-R
z _o ~$~ 2
(in which R1, R2~ R6 and Y have the meanings given for formula (B),
and Z represent~ a hydrogen atom or a methyl), whlch on hydrolysis
form the alpha-arylalkanoic acid known as Naproxen or lt~ immediate
precur~ors. Again in this caF~e, in which the transformation of the halogen
ketals to aryl-alkanoic acids takes place in two stages, there i5 no
substantial racemisation, and thus the desired optically active
aryl-alkanoic acid is selectively and prevalently obtained.
The compounds of formula tc) are new compounds and are
provided by the present invention, in that they have
interesting properties which make them useful ~rom varlous aspects.
As already stated, the compounds of formula (C) form the Gorrespond-
ing alpha-arylalkanoic acids on hydrolysis.
Moreover, because of the presence of the two asymmetric carbon atoms in
the alcoholic moiety(the atoms to which the CORl and COR2 groups are
bonded respectively), the esters of formula (C) are use~ul for the
optical resolution of the alpha-arylalkanoic acids.
The resolution of an acid into its optical iæomers is generally
carried out by ~orming salts with an optically active base.
The us~ of the compounds (C) constitutes a new process for the reso-
lution of mlxtures of optically active alpha-arylalkanoic acids by
forming an ester with tartaric aGid or one of its derivatives,
instead of forming a salt with an optically active base.
The use of the compounds of formula (C) for resolving an alpha-aryl-
alkanoic acid i~ particularly advantageous when, by mean~ of the
aforesaid process for rearranging the ketal6 (A), es-ters of ~ormula
(C) are obtained enriched ln the desired isomer.
- 18 ~
I-t i5 evident -that the compounds of formula (C) are useful for
the optical resolu-tion of alpha-arylalkanoic acids independently
from the rnethod of preparation.
In thls respect, it is possible to prepare the compounds of formula
S (C) by es-t0rifying a racendc alpha-arylalkanoic acid ~or.one whlch
is already rich in one of the.two .enantiomers) independently from how
this ha3 been prepared.
The compounds o~ formula (D), whether prepared by rearrangement
of a compound of formula (B) or prepared by esterifying race~c .
2-(6-methoxy-2-naphthyl)-proplonic acid or one of ita immediate
precursors using tartaric acid or one of its derivatives, are
u~eful for separating, by means of crystallization, the
ester of formula (D) which on hydrolysis produces Naprox~n in a
substantially pure form.
A further unexpected property of the compounds of formula (C) i5
that they are in themselves pharmacological1y active compounds.
The compounds of formula (D) have proved particularly interesting~
The following tables give the data relative to the anti-inflammato-
ry and antipyretic activity of the compounds (D) in which:
1 2 3; .6 ; ; 3 (a)
R1 = Rz = OCH3`; R6 = ~; Y = Br; Z = CH3 ~b)
compared with ~aproxen and with 5-Br Naproxen. (c)
From these data it i3; evident that the n0w considered compounds,
although having a lesser activity than Naproxen, still have an
interesting activity which could find practical application in
human therapy under determined conditions,
~ 3 ~
-- 19 --
TABLE 1 - An-ti~inflammatory activity of -the derivatives (a)and
(b) with respect to Naproxen and 5-bromo-Naproxen(c)
by ora].admi.nistra-tion
CompoundDose Inhibi-tion ED50
_ ~M/kg/os (after 3 h1 % (L.C. 95%1
(a) 10 0 175
; 30 0 (110 ~ 280)
lO0 16
(b) 10 3 160
14 (100 - 250
.1~0 20
(c) 10 6
34
00 34 196
300 56 (120 - 304)
Naproxen 10 38
31
lO0 66 (19 - 49)
- ~
,
~L 3 ~ P~
,~, . .
~o, _.
In C~ ~
,-: ~ ~ -- ~ ~4 ~ 0 a) ~ 1` ~5 ~ 0 a)
~: :~ ~ o ~ r~ 0 _~ w ~o
o o o o o o o o . ~ o,
'.o c: td .~
n O . .
0 ~,1,_ . ; - .
. c: ~ ~ t~ _ r~ N CG N ID
r ~., C O O O ,~ 7 ~
O O C O O O Oo ~ ~ o ._ ~,
~ I I I .
t^d l . ' .
. . ' . . ..
~,~ . .
'~V ~2 : .............. ....... - ,' '.
n ~ .
~a ~ o G C O O O O o O C- ~ C O
,~Co; ~:: . ..
'`1 1 ' ~ '~ ' '
21
" , ''' .
:~, . J
. x~ _ .~J o al ~g
~ O OO' O O O ~
1~1 ~ ~ t
C _ . .'
C . .
~ . ~ ' . . .
Q~ ~ O U7 0 U~ ~ O ~
h ~rl _I O --~ O O ~
,~:, qJ ~ .1 1 .1 1 1 1 1
_. ~~ . . .
a u~ o
~ 5~J ID N ~ T~ O
_~ ~ O O O O O O ~
0~ ,~J 1.~ 1 1,,
E t:: . .
. _ ~
" C a :: o o o o o o o .
.
. .f~ . .' ."
''. 1 . ' ~ ' . . .
~ D Z
~ 3 ~
- 22 -
Some pratica:L examples of the process according to the present
invention are described hereinafter in order to illustrate the
invelltion bu-t wi.thout in any way limiting it.
ExamPle l
Preparation of the compound 2-ethyl-2-(6-m~thoxy-2-
naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxylic acid dimethyl
ester.
1-(6-methoxy-2-naphthyl)-propan-1-c~ne (46.5 g; 0.217
moles), L(+)tartaric acid dimethyl ester (300 g)~ trimethyl
orthoformate (94 g; 0.887 moles) are gradually heated up to
complete solution. Methane-sulphonic acid (1.48 g; 0.0154 moles~
is then added and the obtained solution is refluxed for 2 hours;
it is cooled at room temperature and the reaction mixture is
slowly added to a 10% solution of Na2CO3 (500 ml). It is
extracted with methylene chloride and the organic e~tracts are
1~ repeatedly washed with water.
The organic phase is dried on Na2SO~ and the solvent is
evaporated under reduced pressure.
The residue is crystallized from methanol (250 ml)~
The desired product is obtained (51.S8 g; 0.138 moles; yield
63.6%) having the following characer~stics:
m.p. = 73-74C
Ed ]D = ~33.04 (C = 1%, C~Cl3)
I.R. (Nu~ol a trademark): 1770,1740 cm~l (stretching C=O)
NMR ~CDC13 - TMS, 200 MHz) S(ppm): 0.94 ~t, 3H, J=7, 5 Hz);
2.08 (q, 2H, J=7.5 Hz); 3.46 (s, 3H); 3.84 (s, 3H); 3.90 (Sr 3H);
4.86 ~2H, ABq, ,~ )=10.80, J=6 Hz); 7.1--7.9 (m, 6H) .
Example 2
Preparation of the mixture of the diastereolsomers oE
2-(1-bromo-ethyl)-2-(6-methoxy-2-naphthyl)-1,3-dioxolane-
4(R~,5(R)-dicarboxylic acid dlmethylester.
3 ~ $ ~
To a solution of the compound obtained in Example 1
(37.4 g; 0.1 mole).
- 23a -
, ,
g ~ ~ r~
- 24 -
in 1,2-dichloro~thane (100 ml), tetra-n-~utylammonium perbromide
/ N(n.C4Hg)~ Br3 / (48.Z g; 0.1 mole) is added.
The reaction mlxture is Icept at 20C for 24 h and then slowly
added under stirring to a 10% solution of Na2C03 (200 ml). It
is extracted with toluene (2 x 200 ml) and the combined organic
ex~ract~ are washed with a 2% solution ofNal-lC03 ~3 x 100 ml).
The organic phase is ~ried on Na2S0~ and the solvent evapo-
rated under reduced pressure. The crude product obtained (48 g) is pu
rified by chromatography on a silica gel column (eluent hexene:
diethylether = 75:25) to give 13 g of the desired mix-ture of dia-
stereoisomers.
The ratio between the two diastereoisomers (1:2) determined
by H-NMR (200 MHz) is 7:3.
Diastereoisomer l (R~S)
H-NMR (CDCl3 - TMS), ~ (ppm): 1.68 (d, 3H, J=7.5 Hz); 3.54 (s,
3H); 3.90 (s, 3H); 4.08 (s, 3H); 4.48 ~q, 1H, J=7.5 Hz); 4.94
(ZH, ABq,~ ~ =26.8; J=7.2 Hz); 7.1-8.0 (6H,m).
Diastereoisomer 2 (RRR)
H-rlMR (CDC13 - TMS), ~ (ppm): 1.64 (d, 3H, J=7.5 Hz); 3.58 (s,
3H); 3.89 (~, 3H); 4.08 (s, 3H); 4.50 tq, lH, J=7.5 Hæ); 4.89
(2H, A~q,~ ~ = 36.3, J=6.3 Hz); 7.1-8.0 (6H,m).
EXAMPLE 3
Pr~ ~rO,,t~
of the 2(R)-hydroxy-3(R)-/ 2-(6-methoxy 2-naphthyl)-
propanoyl / _ butanedioic acid dimethyl ester.
A mixture of diastereoisomers 1:2 = 67:33, obtained according to
example Z (5 g; 0.011 moles) dissolved into CH2C12 t61 ml) and kept
at ~C under inert atmosphere is added with silvsr tetrafluoroborate
(2.33 g; 0.012 moles). The reaction mixture is kep-t at 0C for
- 25 - ~ 3~3'~ ~
30 minutes and then -the temperature is allo~ed to raise up to room
temperature.
The mixture i9 fil-tered and the precipitate washed with CH2C12. The
organic pha~:es are washed with water and dried 2 4
The solvent is evapo~ated under reduced pressure
to give a mixture oE diastereoisomeric esters (ratio
determined by NMP~, 200 MH~, A:B = 64:36).
H-NMR (CDC13 - TMS), ~, (ppm- ):
Diastereoisomer A (RRS):
1.62 (d, 3H, J=8 ~Iz); 3.22 (s, 3H); 3.83 (s, 311); 3.92 (s, 3H),
3.21 (d, lH, J=7.2 Hz); 3.95 (q, lH, J=8 H~); 4.68 (dd, lH,
JCH oH=7-2 H7, JCH CH=2.47 Hz); 5.37 (d, lH, J =2.47 Hz); 7.1-7.8
(6H, aromatic protons).
Diastereoisomer B (RRR):
1.66 (d, 3H, J=8 Hz); 3.58 (5, 3H); 3.72 (s, 3H); 3.92 (s, 3H);
3.24 (d, lH, J=7.6 Hz); 3.97 (q, lH, J=8 Hz), 4.78 (dd, lH,
JCH-OH 7-6 Hz~ JCH-CH = 2-47 Hz); 5-45 (d, lH, J=2.47 Hz); 7.1~7.8 -
(6H, aromatic protons).
EXAMPLE 4
Preparation of 2-(6-methoxy-2-naphthyl) propionic acid.
A mixture of diastereoisomer esters A and B prepared as described
in Example 3 (ratio A:B = 62:38) (3.2 g) dimethoxyethane (24 ml~,
hydrochloric acid ~2 N (24 ml) is kept under stirring, at 95C
for~ Z.5 h. It is cooled to room te0perature, poured into water
and extracted with CH2C12.
The combined organic extracts are washed with a sa-turated
solution of sodium bicarbonate
The aqueous phase is acidified to give the 2-(6-methoxy-2-naphthyl)-
propionic acid (1.3 g).
1 3 ~
An analytically pure sample obtained by column
chromatography on silica gel (eluant hexene: diethylether=1:1),
Wit~l [d~]D = +12-9 (c = 1%, CHC13) i~ esterified with
diazomethane.
The obtained methyl ester is analyzed by lH-NMR (200
MHz) using an optically active shift agent (Europium (III) -
tris-[3-(eptafluoropropylhydroxymethylene)-d--camphorate] in
10 CDCl 3)-
The ~nantiomeric ratio is (~)S:(-)R=62:38.
Example 5
Preparation of the 2-(6-methoxy-2-naphthyl~-propionic
acid.
A mixtur~ of diastereoisomeric ketals prepared as
described in Example 2, in the ratio 1:2=67:33, is heated at
125C in ethylene glycol, in the presence of pstassium acetate
for 20 h. After the work up of the reaction mlxture, a mixture
of esters is obtained that are hydrolyzed as described in Example
4. The (~)~S)-2 (6-methoxy-2-naphthyl) propionic acid (Naproxen)
is obtained, with an optical purity of 40~; m.p. = 151-152C.
Example 6
Preparation of the diastereoisomeric mixture of the
compound 2-~1-bromoethyl)-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-
dioxolane-4(R), 5(R)-dicarboxylic acid dimethyl ester.
To a solution of 2-ethyl-2-(6-methoxy-2-naphthyl)-l~3
dioxolane-4tR), 5(R)-dicarboxylic acid dimethyl ester (3.74 g;
0.01 moles) in CC14 (70 mlJ kept at 0C under inert atmosphere, a
- 26 -
~ ~L 3 ~ 8 9 d~ 7
solution of bromine ~3.2 g; 0.0'2 moles) in CC1~ (7 ml) cooled at
0C is added dropwise in 1 h.
- '26a -
13 1 ~ ~ ~ 7
- 27 -
The mixture is Icept at 0C for -two hours, then pG~ed under vi
gorous stirring into an 10% a~ueous solution of Na2C03 (250 ml)
and extrac-ted wi-th C~I Cl (3 x 50 ml).
The comblned orgaIlic extracts are dried on Na2S04 and the
solvent evaporated under vacuum. The residue (5 g; 0.0093 moles;
yield 93%) consists of a mixture o-f the two diastereoisomers ide~-
tif`ied wi-th 3 and 4.
The ratio between the diastereoisomers 3:4 determined by HPLC
and H-NMR i5 95:5.
The major isomer has the ~ame configura-
tion (S) of the dias-tereoisomer l described in Example 2, refe-
rerring to the aliphatic carbon a-tom bonded to bromine.
Diastereoisomer 3 IRRS)
H-NMR (200 MHz) (CDC13 - TMS), ~ (ppm):1.66 (d, 3H, J=6.8 Hz);
3.52 (s, 3H); 3.88 (s, 3H); 4.05 (s, 3H); 4.46 (q, lH, J=6.8 Hz);
4.94 (2H ABq, J=6 Hz); 7.28-8.24 (5H, aromatic protons).
Diastereoisomer 4 (RRR)
H-NMR (200 MHz) (CDCl3 - TMS) ~ ~ppm): 1.63 (d, 3H, J=6.8 Hz);
3.56 (s, 3H); 3.87 (s, 3H); 4.05 (s, 3H); 4.48 (q, lH, J=6.8 Hz);
4.91 (2H, ABq, J=6 Hz); 7.28-8.24 (5H, aromatic protons).
The HPI.C analysis (high pressure liquid chromatography) has
been performed under the following conditions:
Hewlett Packard instru~ent mod. 1084/B with variable
wavelength W detector:
AnalYtical conditions:
- Column BRAWNLEE LABS RP8 (5 ~ ) spheri 250 mm x 4.6 mm
(internal ~iameter)
- Solven-t A: bidistilled water, flow 0.9 ml/min
- Solvent B: methanol, flow 1.1 ml/min
- Solvent A temperature: 60C
- Solvent B temperature: 40C
- .
- 28 - ~3
- Column temperature: 50C
- Wavelengttl (A ): 254 nanometers
- Injecl;ion: 10 ~ 1 of a solution containing 3 mg/ml o~ a sample
in acetonitrile.
Retention times:
Diastereoisomer 3 : 18.20 min
Diastereoisomer 4 : 19.90 min
= . = . = . _ . _
A mixture of diastereoisomers 3 and 4 in ratio 95:5 obtained as
above described is chromatographated on silica gel, by
using as eluent a mixture of diethylether : hexane = 3 : 7.
The collected fractions are separately analy~ed by HPLC.
The frac-tions containing the diastereoisomer 3 showing a diaste-
reoisomeric purity higher than 99%~are collected.
The solvent is evaporated under vacuum -to give the pure diaste-
reoisomer 3.
H-NMR (200 MHz) (CDC13-TMS) delta (ppm): 1.66 (d, 3H, J=7.S Hz);
3.52 (s, 3H); 3.83 (s, 3H); 4.05 (s, 3H); 4.46 (q, lH, J=7.5 Hz);
4.94 (2H, ABq, J=7.2 Hz); 7.28-8.24 (5H, aromatic protons).
EXAMPLE 7
Preparation of a mixture of diastereoisomers of the compound
2-~1-bromoethyl)-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-
4(R),5(R)-dicarboxylic acid dimethyl ester.
The reaction described in Example 6 has been repeated wi-th different
solvents and a-t different temperatures according to the following
procedure.
To a solution of 2-ethyl--2-(6-methoxy-2-naphthyl)-1,3-dioxolane-
-4~R), 5(~)-dLcarboxylic acid dimethyl ester (0.01 moles) Ln -the
- 29 - ~3~
solvent indicated in the following Table (70 ml), kep-t under
inert atmosphere at the temperature also indicated in -the Table, a
solutlon of bromine(o.o2 moles) in -the same solven-t (7.0 ml),
pre-coolecl to the temperature of the above mixture, is added.
The so obtained reaction rnix-ture is kept at -the ternperature indica-
ted to reach~asubstantially complete conversion. It is then worked
up as described in Example 6. The ratio between the diastereaiso-
mers 3 and 4 is indicated in the Table.
~ 30 - 1 3 ~
T A B L E
... . . _ .. .. .
Solven-t T Ratio
(C) diast. 3
dias-t. 4
Carbon te-trachloride 20 93/7
1 t 2-Dichloroethane 20 93/7
1,2-Dichloroethane O 91J9
1,2-Dichloroethane -30 92/8
1,1,2,Z-Tetrachloroethane 20 89/11
10 Chlorobenzene 20 90/10
Benzene 20 91/9
Benzene 0 92/8
Toluene 20 91/9
Ethylenglycoldimethylether 20 86/14
15 Acetonitrile 20 82/18
Cyclohexane 20 88/12
Orthodichlorobenzene 20 89.2/10.8
Sulfolane 27 78/22
Ethylacetate 20 91/9
Para-dichlorobenzene 60 87/13
Carbondisulfide 15 92.3/7.7
Acetic acid 15 89/11
Hexafluorobenzene 15 90.3/9.7
Molar yield 90-95%
- 31 ~ r~
EXAMPLE 8
Preparation of 2-(1-bromoethyl)-2-(5-bromo-6-methoxy-2-naphthyl)-
1,3-dioxolane-4(R), 5(R)-dicarboxylic acid dimethylester.
To a solution of 2-ethyl~2-(~methoxy-2-naphthyl)--1,3-dioxolane-
-4(R), 5(R)-dicarboxyllc acid dime-thyl ester (70 g; 0.187 moles)
in 1,2-dichloroethane (175 ml) kept at -30C, under inert atmosphe-
re and under stirring, a bromine solution (59.8 g; 0.374 moles)
in 1,2-dichloroethane (140 ml) is added in 2 h.
The reaction mixture is kept at -30C up to a complete conversion
of the starting product, then is added slowly dropwise to a 10~
solution of Na2C03 (1000 ml) under vigorous stirring.
The organic phase is separated, washed with water, dried
on Na2S04 and the solvent evaporated under vacuum. The mixture
of the two dias-tereoisomers 3:4 is obtained in a ratio 9:1.
The above ratio has been determined by HPLC and H-N~R.
EXAMPLE 9
Preparation of 2(R)-hydroxy-3(R)-L 2-(5-bromo-6-methoxy-2-naphthyl)-
propanoyL /-butanedioic acid dimethy}ester.
To a solution of 2-(1-bromoethyl)-2-(5-bromo-6-methoxy-2-naphtyl)-
1,3}dioxQ~ 4(R), S(R)-dicarboxylic acid dLme ~ 1 ester (2.66 g; 0. ~ moles;
ratio diastereoisomer 3 to diastereoisomer 4 = 85:15 determined
by HPLC) in 1,2-dichloroethane (20 ml), kept under stirring at
15C under inert atmosphere, sllver tetra~luoroborate~(l.l7 g 0.006
moles) is added.
The reaction mixture is kep-t at -15C for 2 h, then allo~ed -to reach
room temperature in about 1 h and filtered. The organic pha
se is washed with water, dried on Na2S04 and the solvent
evaporated under vacuum.
- 32 - ~ 31~ ~ ~ r~
The desired product is obtained (2.2 g; 0.0047 moles; yi.eld 94%)
as a mixture o.f -two diastereoisomers named C and D,
in a ratio C:D = 84:16 determined by H-NMR, 200 ME~z .
Il-NMR (CDC13 - TMS)
Diastereoisomer C (RRS) - The data are consisti.ng
~ith the given 5 tructure; ths da-ta which
refer to the aliphatic part are analogous
to those of the diastereoisomer A descri-
bed in Example 3.
Diastereoisomer D (RRR) - The dat,a . are quite consisting
with the given structure; the data which
refer to the aliphatic part are analogous
to those of diastereoisomer B described
in Exampl.e 3.
The diastereoisomer C has been separated in pure form by crystalli-
zation from methanol. M.p. = 124 - 126DC; /~ / D = + 60.2 (c = 1%
in CHC13).
EXAMPLE 10
.
Preparation of 5(~-2-(5-bromo-6-methoxy-2-naphthyl)-propionlc acid.
~a) a mixture of:
- 2(R~-hydroxy-3(R)-/2-(5-bromo-6-methoxy-Z-naphthyl-JPrPan~
butanedioic acid dimethyl ester (diastereoisomer C of Exam-
ple 9 ; 0.5 g; 1.065 mmoles)
- sodium hydroxide (0.170 g; 4.26 mmoles)
- water t2.5 ml)
- methanol (3.5 ml)
is kept under stirring at room temperature for 18 hours.
The mixture is diluted with water and extracted with dichloro
methane. The aqueous phase is acidified with conc. HCI and
extracted with dichloromethanc.
- 33 ~
The organic phase is then washed with water, dried and
the solvent evaporated under vacuum~ The 50 obtained crude acid
is purified by chromatography on silica gel (eluent hexene:
diethyle-ther = 8 : 2).
The S(~)-2-(5-bromo-6-mettloxy-2-naph-th~l)-propionic in
the pure form is obtained; m.p. = 15S-157"C;
/C~ / 578 ~ ~ 20.5 (c=0.5% in CIIC13). Star-ting
from this acid, by debromination according to -the method descri-
bed in the Belgian Patent 89Z.689, Naproxen is obtained ha~ing
the same optical purity of the starting 5-Bromo-derivative.
.
b) a mixture-of:
- 2(R~-hydroxy-3(R)-/ 2-(5-bromo-6-methoxy-2-naphthyl)-propa-
noyl /-butanedioic acid dimethyl ester
(diastereoisomer C obtained according to
Example 9 ; 0.2 g; 0.426 mmoles)
- 1,2-dimethoxy-ethane (3 ml)
- conc. HCl t3 ml)
is kep-t at 95C for 2 h. The reaction mixture is then cooled
to room temperature, diluted with water and extracted with
CH2Cl2.The organic phase is washed with wa-ter and extracted with
10% sodium bicarbonate.
The basic aqueous extract is acidified with conc.HCl and
extracted with CH2Cl2.
The organic extract is washed with water, dried on Na2S04
and the solvent evaporated under reduced pressure.
The optically pure S(~)-2-(5-bromo-6-methoxy-2-naphthyl)-propio-
nic acid is obtained:
/ ~ / 578 = ~ 4~-9 (c - 0.5% in C~IC13).
- 34 ~ 7
This acid i5 debromina-ted to give Naproxen hav ~ the same ~tiG~
purity, following the procedure described in the Belgian Pa-tent
892.689 /C~ / 20 = ~66(c = 1% in CHCl )-
EXAMPLF. 11
Preparation of the 2-(5-bromo- 6-meth~xy-2-naphthyl)
propionic acid.
. .
A mixture of 2-~1-bromoethyl)-2-t5-bromo-6-methoxy-2-naphthyl)-1,3-
dioxolane-4(R) 7 5(R)-dicarboxylic acid dimethyl ester prepared
according to Example 8 (2.66 g; 5 mmoles; diaster.3:diaster.4 =
9:1 as determined by HPLC), sodium bicarbonate (1.7 g; 20 mmole)
and water is refluxed for 22 h. The reaction mixture ls cooled
to room temperature and extracted with die-~lethe~. The aqueous
phase is acidified wi-th conc.HCl and the precipita-te filtered and
washed with water.
The so obtained crude acid (1.13 g) is purified on a sllica gel
column (eluent hex~qe:diethyletherin ratio 8:2).
The 2~(5-bromo-6-me-thoxy_2-naphthyl)-propionic acid (0.92 g; 3 mmoles;
yield 60%) is obtained - m.p. = 156-158C; / ~ / 578 = + 23.5
(c = 0.5% in CHC13).
EXAMPLE 12
Preparation of 2-ethyl-2-(6-metho~y-2-naphthyl)-1,3-dioxolane-4(R),
5(R)-dicarboxylic acid diethyl ester.
1-(6-methoxy-2~naphthyl)-propan-1-one (20.0 g; 0.093 moles), diethyl
ester of L(~)-tartaric acid (160 g) and triethyLorthoformate ~37 g;
0.25 moles) are slowly heated up to complete solution.
Methanesulphonic acid (0.68 g; 0.007 moles) is added and the ~;olution
i9 refluxed f'or } h.
- 35 ~
The reac-tion mix-ture is cooled to room temperature and added to
a 10% solution of Na2C03 (250 ml) under vigorous stirring.It is
extracted with Cll2C12 and the organic extracts are repeatedly
washed with water.
The organic phase is dried on NQ2SO4 and the solvent is eva-
portated under reduced pressure.
The crude product is grad~ally heated up to 180C (external bath)
under a pressure of 0.1 mmEIg.
The desired product is obtained (33.6 g~ 0.084 moles; yield 90%)
having the following characteristics:
/ ~ / 20 = + 20.59 (c = 1%, C~IC13)
I.R. (NEAT): 1770,1740 cm (stretching C=0)
H-NMR (CDC13 - TMS) S (ppm): 0.95 (t, 3H, J=6.4~);1.02 (t, 3H,
J=7.3Hz);1.3 (t, 3H, J=7.3Hz);2.08 (q, 2H, J=6. ~ );3.9 (s, 3H);
3.88 (dq, 2H, J=1 ~ , J=7.3Hz);4.30(q, 2H, J=7.3Hz);4.82 (ABq, 2H,
J=5.~);7-8 (6H, aromatic protons).
EXAMPLE 13
Preparation of the diastereoisomers mixture of 2-(1-bromoet~yl)-
-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R), 5(R)-
dicarboxylic acid diethyl ester.
To a solution of 2-ethyl 2-(6-methoxy-2-naphthyl)-1,3-dioxolane-
4(R), 5(R)-dicarboxylic acid diethyl ester (2 g; 0.005 moles) in
CC14 (35 ml) is added a solution of brune (1.6 g; 0.01 moles~ in
CC14 (3.5 ml),under inert atmosphere, at 20C.
The mixture is kept at 20C for -two hours and thenWD~ced up as de-
scribed in Example 6.
The desired diastereoisomericmixture is obtained (named as 5
and 6) in 93% yield.
The ratio between the diastereoisomers , determined by HPLC,ls
5:6 ~ 91.5:~.5.
- 36 - ~ 3 ~
The diastereoisomer 5 (which is the prevalent one) shows the same
configuration (S) af the dias-tereoisomer 1 (Example 2) and of
diastereoisomer 3 (Example 6) with respect to the aliphatic car-
bon atom bonded to bromine.
H-NMR (CDC13 - TMS) (200 MHz)
Diastereoiso~ 5 (~RS):S(ppm) 1.04 (t, 3H,J=~); 1.31 (t, 3H, J-~lz);
1.65 (d, 3H, J=6-~-~);3.92 (dq, 2H, J=ll.~lz, J=~lz);3.98 (s, 3H);
4.3 (q, 2H,J=~lZ); 4.48 (q, lH, J=6.8~);4.88 (ABq, 2H, J=6.~);
7.2-8.2 (5H, aromatic pro-tons).
Diast~oiso~ 6(RRR)-S (ppm) 1.09 (t, 3H,J--7HZ~; 1-29 ~t, 3H~ J= ~ );
1.62 (d, 3H, J=6-8Hz);3.98 (s, 3H); 4.29 (q, 2H,J=7Hz); 4.85 (ABq,
2H, J=6~5~);7.2-8.2 (5H, aromatic protons).
HPLC analysis performed under essentially the same conditions as
described in Example 6, with the only difference that the percen-
tage oE the solvent B is 58% (total flow 2 ml/min).
Diastereoisomer 5: retention time 24.03 minutes
Diastereoisomer 6: retention time 25.00 minutes.
- 37 - ~ 3 ~ 7
.E~AMPL~ 14
Preparation of 2-ethyl-2-(6-methoxy-2-naphttlyl)-1,3-dioxolane-4(R),
5(R)-dicarboxylic acid
.5 A mixture of 2-e-thyl-2-(6-methoxy-2-naphthyl) 1,3-dioxolane-4(R),
5(R)-dicarboxylic acid dimethyl ester (~.68 g; 12.5 mmoles),
NaOH (1 g, 25 mmoles) and water (50 mL) is kept under stirring at
room temperature for 5 h.
The reaction mixture is filtered and the aqueous phase acidified
with conc. HCl to pH 1.
It is then extracted with die~wle~er and the~ombined organic ~:
extracts are washed with wa-ter and dried on Na2S04.
Evaporatian of the solvent under vacuum gives 2-ethyl-2-(-methoxy- .
2-naphthyl)~1,3-dioxolane-4(R), 5(R)-dicarboxylic acid (3.46 gj:10 mmoles);
yield 80%), m.p. = 100-102~C.
H NMR (200 MHz) (CDC13-TMS) delta (ppm): O.g2 It, 3~1, J = 7 H~);
2.07 (q, 2H, J = 7 Hz); 3.86 (s, 3H); 4.78 (2H, ABq, ~ ~ = 4.2;
J = 5.8 Hz); 7,0-8,0 (6H, aromatic protons).
A sample esterified with diazomethane in diethyle~ gives the starting
methyl ester with unchanged : HNMR, I.R., m.p.,
and i~7.
.
EXAMPLE 15
Preparation of 2-(1-bromoethyl)-2-(5-bromo-6-methoxy-2-naphthyl)-
1,3-dioxolane-4(R), 5(R)-dicarboxylic acid.
A mixture consisting of the two diastereoisomers of 2~ bromo-ethyl)-
2-¦5-bromo-6-me-thoxy-2-naphthyl)-1,3-dioxolane-4(R), 5(R)-dicarboxylic
acid dimethyl ester, in ratio 9:1 (6.65 g 12.5 mmole~), NaOH (1 g;
25 mmoles), dimethoxyethane (10 ml) and water (10 ml) is kept under
~tirring at room temperature for 2 h.
~ 3'3 ~ ~ 3~
The reaction mixture is diluted with water and extracted wi-th
diethylether.
The aqueous phase is then acidified to pil 1 with conc. HCl and
extracted with diethylether.
The combined organic extracts are washed with water and
dried on Na SO .
2 4
The evaporation of the solvent under vacuum leads to
the two diastereoisomers of 2-(1-bromoethyl)-2-(5-bromo-6-methoxy-
2-naphthyl)~1,3 dioxolane-4(R), 5(R~-dicarboxylic acid (5.8 g;
11.5 mmoles; yield 92%) named with the numbers 7 and 8.
The ratio between the diastereoisomers 7 and 8 , determined by
HNMR (200 MH~),is of 9:1.
Diastereoisomer 7 (RRS) (CDC13-TMS) delta (ppm): 1.60 (d, 3H, J = 7 Hz);
4.00 (s, 3H); 4.49 (q, lH, J = 7 Hz); 4.87 (2H, ABq,~ ~ = 18.9;
J = 6.5 Hz): 7.2-8.2 (5H, aromatic protons).
EXAMPLE 16
Preparation of Z-(l-bromoethyl)-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-
dioxolane-4(R), 5(R)-dicarboxylic acid.
A mixture of 2-/l(S)bromoethyl7-2-(5-bromo-6-methoxy-2-naphthyl)-
1,3-dioxolane-4(R), 5(R)-dicarboxyllc acid dimethyl ester~diastereoisomer
3 in the pure form; 6.65 g; 12.5 mmoles), NaOH (1 g; 25 mmoles),
dimethoxyethane (10 ml) and water (lO ml) is kept under stirring at room
temperature for 2 h.
The reaction mixture is diluted with wa-ter and ex-tracted with diethyl-
ether. Then the aqueous phase is acidified a-t pH 1 with conc. IICl and
extracted with diethylether.
The ~ombined organic extracts are washed with water and
dried on NazSO4.
E~x~a~rg of U~ solventunder vacuum gives 2-~l(S)-brollloe-thyl7~2-~5-bromo-
3L 3 ~ 7~
6-methoxy-2-naphthyl)-1,3 dioxolane-4(R), 5(R)~dicarboxylic
acid (diastereoisomer 7).
lH NMR (200 MHz) (CDC13-TMS) delta (ppm): 1.60 (d, 3H,
J = 7 Hz); 4.00 (s, 3H); 4.49 (~, lH, J = 7 Hz); 4.B7 (2H,
ABq,~ ~ ~ 18.9; J = 6 Hz); 7.2-8.2 ~5 H, aromatic protons).
Example_17
Preparation of 2(R)-hydroxy-3(R~-[2-(5 bromo-6-methoxy2-
naphthyl)-propanol~-butanedioic acid dimethyl ester.
To a mixture of the diastereoisomerd 3 and ~ in ratio
94:6 (determined by HPLC) (10~0 g, 0.0188 moles) in 1,2-
dichloroethane (75 ml) kept under stirring at +15C, under
inert atmosphere, a solution of silver tetrafluoborate
(4.4 g; 0.0226 moles) in 1,2-dichloroethane ~30 ml), is added
in 15 min~
The reaction mixture is kept at ~15C for 7 h, poured
slowly into cooled water (lO0 ml) in such a manner that the
temperture does not overcome +10C. The mixture is then
filtered on Celite (a trademark) and the filtrate washed with
CH2Cl2 (100 ml).
The organic phase is washed with water (2x200 ml) and
dried on Na2SO4. Evaporating of the solvent under reduced
pressure gives a residue (7.2 g; 0.0154 moles; yield 82%)
consisting of a mixture of diastereoisomeric esters (ratio
diast. C:3 ~ 91:9, determined by lH NMR analysis).
- 39 -
.
.
Example 18
Preparation of the compound 2-ethyl-2-(6-methoxy-2-
naphthyl)-1,3-dioxolane-4(R), 5(R)-dicarboxylic acid di-
isopropyl ester.l-(S-methoxy-2-naphthyl)-propan-1-one (10.3 g; 0.048 moles),
di-isopropyl ester L (+) tartaric acid (94 g) and
trimetlylorthoformate~
- 39a-
~ 40 - ~3 ~
(7.57 g; 0.071 moLes), are gradually hea-ted up to complete
solution.
It is ther~ added me~ncsulphonic acid tO.37 g; 0.0039 moles) and the
solution is refluxed for 2.5 h (temperature Or the solution 90C).
~5 The reac-tion mixture is cooled and slowly added t;o a 10% solution o~
Na2C03 (100 ml), under vigorous stirring.
It is extracted wi-th CH2Cl2 and the organic ex-tracts are washed with
water (100 ml).
The organic phase is dried on NazSO4 and the solvent is evaporated
under reduced pressure to give 94 g of crude product.
~hë: drude product
is then slowly heated up to 220C lexternal bath) at
0.2-0.3 mm/Hg. The residue is purified by cromatography
~n a silica ~el column (eluent hexene : diethyl- '
1~ ether = 85:15)2-ethyl-2(6-me-thoxy-2-naphthyl)-1,3-dioxolane-4(R), 5(R)
dicarboxylic acid diisopropylester (14.2 g; 0.033 moles; yield 69%)
was obtained.
I.R. (Neat): 1770, 1740 Gm ( stretching C = 0)
H NMR (CDCi3-TMS) (200 MHz~ delta (ppm): 0.95 (t, 3 H, J = 7.6 Hz);
0.96 (d, 3 H, J = 6.4 Hz); 1.05 (d, 3 H, J = 6.4 Hz); 1.29 (d, 6 H,
J = 6.4 Hz); 3.8 (s, 3 H); 4.75 (A~q, 2 H, J = 6.6 Hz); 4.79 (q, 1 H,
J = 6.4); 5.14 ~ept., 1 H, J = 6.4); 7-8 (m, 6 H).
EXAMPLE 19
Preparation of the diastereoisomer~ mixture of the 2-(1-bromoethyl)-
2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R), 5(R)-dicarboxylic
acid diisopropylester.
A s~lu;~ion of brom~e~l6 g; 0.01 moles)in CCl4(3.5 mL) is added
drogwise, at 15C, under inert atmosphere, in 1 h to a solution of
.2-e-thyl-2-(6-methoxy-2-naphthyl)-1,3-dioxolane- 4(Q), 5(R)-dicarboxylic
.
-
- 41 - ~ 3 ~ 7~
acid diisopropyl ester (2.15 g; 0.005 moles) in CCl4 (35 ml).
5he mixture is kept at 15C fO~ 2 h and then ~orked up as described
i~ example 6.
The desired diastereoisomeric mixture (isomers 9 and 10) is obtained
in a 94% yield.
The ratlo between the two dias-tereoisorners as detr-rmined by HPLC is
9:10 = 93.9:6.1- . lH NMR (CDC13-TMS) (200 MlIz)
Dias-tereoisomer 9 (RRS): delta (ppm): 0.96 (d, 3HI, J = 6.4 Hz);
1.06 (d, 3H, J = 6.4 Hz); 1.3 (d, 6 H, J = 6.4 H~); 1.67 (d, 3H,
J=~.2 Hz);3.98 (s, 3H); 4.47 ~q, lH, J = 7.2 Hz); 4.80(ABq, 2H, J = 6.6 H~);
4.80(m, lH, J = 6.4 Hz); 5.15 (m, lH, J = 6.4 Hz); 7.2-8.2 (5H, aromatic
protons).
Diastereoisomer 10 (RRS): delta (ppm): 0.96 (d, 3H, J = 6.4 Hzj;
1.06 (d, 3H, J = 6.4 Hz); 1.28 (d, 6H, J = 6.4 Hz); 1.63 (d, 3H, J=7.2 Hz~;
3.98 (s, 3H); 4.47 (q, lH, J - 7.2 Hz); 4.80(A~q, 2H, J = 6.6 Hz);
4.80~m, lH, J = 6.4 Hz); 5.15 (m, lH, J = 6.4 Hz); 7.2-8.2 (5H, aromatic
protons).
HPLC analysis performed as described in ex. 6I with the only difference
that the percentage of solvent B~is 62.5% ~to-tal flow 2 ml/min.)
Diastereoisomer 9:retention time 23.68 min.
Diastereoisomer lO:retention time 24.46 min.
EXAMPLE 20
Preparation of 2(R)hydroxy-3(R)-/2-(5-bromo-6-methoxy-2-naphthyl)-
prPanY17-butanedioic acid diisopropylester.
Following the procedure described in ex. 17 a mixture
of diastereoisomeric ketals 9 and 10 (ex. 19) in a ratio 9:10 = '~4:6
determined by HPLC (2.0 g; 3.4 mmoles), a residue is obtained (1.6 g)that
af~r purification by c~Y~at ~ aphy qn silir~a gel colwn (eluent hexene:dietkylether
~- 1:1) gives a mixture of diastereiosomers ester~ (E ~nd F) in ratio
- 42 - ~ 3 ~
~0:10 ~(determined by 11 NMR (200 MHz) analysis)
H-NMR (CDC13-TMS) (200 Mllz)
Diastereoisomer E (RRS): delta (ppm): 0.55 (d, 311, J = 6.12 I-lz);
1.02 (d, 3~l, J = 6.12 Hz); 1.24 (d, 3H, J = 6.12 Hz); 1.27 td. 3H,
J = 6.12 H~); 1.61 (d, 3H, J = 7 Hz); 3.17 (d, lZI, J = 6.8 Hz);
4.00 ~q, lH, J = 7 Hz); 4.02 (s, 3l1); ~.52 (ept, 1ll, J = 6.12 llz)
CH-C~I 2.2 Hz, JcH-oH = 6.8 Hz); S.13 (ep-t, lH, J = 6 12
Hz); 5.30 (d, lH, J - 2.2 Hz); 7.2-8.2 (5H, aromatic system).
Dias-tereoisomer F (RRR): delta (ppm): 0.95 (d, 3H, J = 6.1Z Hz);
1.12 (d, 3H, J = 6.12 Hz); 1.14 (d, 3H~ J = 6.1Z Hz); 1.19 (d, 3H,
J = 6.12 Hz); 1.62 (d, 3H, J = 7 Hz); 3.17 (d, lH, J = 6.8 Hz);
4.00 (q, lH, J = 7 Hz); 4.02 (s, 3H); 4.52 (ept, lH, J = 6.12 Hz);
4.62 (dd, lH, JC~I C~l = 2.2 Hz, JCH CH = 6.8 Hz); 5.13 (ept, lH, J = 6-12 Hz);
5.41 (d, lH, J = 2.2 Hz); 7.2-8.2 (5H, aroma-tic system).
EXAMPLE 21
Preparat~on of the 2-(5-bromo-6-methoxy-2-naphthyl)-propionic acid.
A mixture of dias-tereoisomers E and ~ (ex. 20) in a ratio E:F = 90:10
(0.35 g; 0.648 mmoles), dimethoxyethane (4.6 ml) and 12 N HCl (4.6 ml) is
kept at 88C under stirring `for 2 h. It is cooled to room temperature
and then it is worked upas described in ex. lO(b).
The so obtained crude pnx~ct ig eluted through a silica gel column
(eluent hexene:ethyl ether = 8:2), to give the 2-(5-bromo-6-methoxy-
2-naphthyl)propionic acid: m.p. = 148-151C; /d 7 = ~38
578
(c = 0.5% CHC13).
The methylester of the above acid ob-tained by esterification with
diazomethane , analyzed ~ H-NMR (200 M~) using
optically active shift ~agent (europium (III) tris-~-(eptafluoro-
propylhydroxymethylene)-d-camphorate/ in CDC13,shows a ratio between
,the enantiomers of S(~):R(-) = 90:10.
3 ~ ~ ~
EX~MPLE 22
Preparation of 2(~)hydroxy-3(R)-~2-(5-bromo-6-methoxy-2-naphthyl)-
propanoyl7-butanedioic acid diethylester.
Following the p ~ e~eas described in ex.].7 a mixture of diastereo-
isomer~ ketals 5 ~nd 6 ~ex. 13) h~ving a ratio 5:6 = 93:7, determined
by IIPLC,(2.41 g; 4.3 mmoles), a residue is obtained (1.95 g) tha-t by
elutlon through a silica gel column (eluent~hexane : diethylether = 1:1)
gives a mixture of diastereoisomericesters named as G and H
~1.77 g; 3.6 mmoles; yield 83%) in ratio G:H = 86:14 de-termined by
H-NMR, 200 MHz. H-NMR (CD~13-TMS) (200 MIIz):
Diastereoisomer G ~RRS): delta (ppm): 0;76 (t, 3H, J = 7.2 Hz~;
1.27 (t, 3H, J = 7.2 Hz); 1.58 (d, 3H, J = 7 Hz); 3.10(d, lH, J = 7.12 Hz);
3.58 (q di AB, 2H, J = 12 Hz, J = 7.2 Hz); 4 (q, lH, J = 7 Hz);
gem
4.01 (s, 3H); 4.27 (q, 2H, J = 7.2 Hz); 4.65 (dd, lH, JCH 0~l = 7.12 Hz~;
JCH OH = 2.4 Hz); 5.32 (d, lH, J = 2.4 Hz); 7.2-8.2 (5H, aromatic protons).
Diastereoisomer H (RRR): delta (ppm): 1.08 (t, 3H, J = 7.2 Hz);
1.14 (t, 3H, J = 7.2 Hz); 1.62 (d, 3H, J = 7 Hz); 3.1 (dl 1}l, J = 7-12 Hz);
3.58 (q di A8, 2H, Jgem = 12 Hz, J - 7.2 Hz); 4.pO (q, lH, J --7Hz) 4.01
(s, 3H); 4.27 (q1 2H, J = 7.2 Hz); 4.65 (dd, lH, JCH OH = 7.12 ~Z; JcH_cH
= 2.4 Hz), 5.44 (d, lH, J = 2.4 Hz); 7.2-8.2 ~5H, aromaticFrotons).
EXAMPLE 23
A mixture of diastereoisomeric es-ters G and H prepared as described in
ex. 22 (ratio G:H = 86:14) (0.64 g; 1.28 mmoles), dimethoxyethane (9 ml)
- 25 and 12 N HCl (9 ml) is kept at 95C (temperature of the bath) under
stirring for 1 h.
It is cooled to room temperature and then it is worked up as described
in ex. lQ(b). The so obtained crude acid is elu-ted through a silica gel
column (eluent hexane: diethyle:ther _ 1 : 1).
The 2-(5-bromo-6-methoxy-2-naphthyl)-propionic acid is obtained-
M.p. = 149-151C and /~7 = ~33.94 (c = 0.5%, CHC13)-
578
44 - ~ 3 ~ $ ~ 5 b
~A sample is esterified wi-th cliazome-thane and the ob-tained methylester
is analysed with H-NMR (200 M Hz) . using an optically active
shift ager~t ~europium (III) tris /3-(eptafl~loropropyL hyd~oxymethylene)
_d -camphorate~in CDC13
~5 The enantiomers ratio is S~ R(-) = 86:14.
EXAMPLE 24
Preparation of 2-ethyl-2-(6-methoxy-2-naph-thyl)-1.3-dioxolane-4(S), 5(S)-
dicarboxylic acid dimethylester.
1-(6-methoxy-2-naphthyl)-propan-1-one (20 g; 0.093 moles), dimethylester
of D(-)tartaric acid (129 g) and trimethyl orthoformate(29 g; 0.27 moles)
are gradually heated up to a complete solution. It is then added methane-
sulphonic acid (0.74 g; 7.7 mmoles) and the solution is refluxed (84C)
for 1 h; it is cooled to room temperature and the mixture is poured
slowly in a 10% solution of Na2C03 (250 ml) under vigorous stirring.
The mixture is extracted with CH2C12 (250 ml) and the organic extracts
are washed with water.
The organic phase is dried ~on Na2S04 and the solvent i5 evaporated
under reduced pressure.
The c ~ e~roduct (40.3 g) is gradually heated up to 180C at 0.1-0.5 mm/Hg,
under stirring.
The residue (33.3 g) is crystallized from methanol (100 ml) thus obtain-
ing the desired product (23.7 g; 0.0635 moles ; yield 68%) with the
following characteristics:
m.p. 72-73C; /~ ~D = ~34-0 (c = 1%, CHC13)
I.R. (Nujol):1770, 1740 cm (stretchlng C = 0)
H-NMR (CDC13, TMS) (200 M Hz).
Thel data are identical to those of the compound 2-ethyl-2-(6-methoxy--
Z-naphthyl)-1.3-dioxolane--4(R),5(R)-dicarboxylic acid dimethyl ester,
descrlb~d in ex. 1.
~ 3 ~
- 45 -
,EXAMPLE Z5
.
Preparation of 2-(1-bromoethyl)-2-(5-bromo-6-me-thoxy-2-nQphthyl)-1,3-
dioxolane-4(S), 5(S)-dicarboxylic acid dimethyl ester.
By processing ~8 described in ex. 19 the 2-e-thyl-2-~6-methoxy-2-naph-thyl)-
1,3-dioxolane-4(S), 5(S)-dicarboxylic acid dimethyl ester (9.35 g;
0.025 moles) the desired mix-ture of diastereoisomers is obtained (identi-
fied as 3' and 4') in 93% yield.
The ratio between the diastereoisomers as determined by HPLC is 3':4' = 93:7.
The diastereoisomer 3', that is the prevailing one is the enantiomer of
the diastereoisomer 3 described in ex. 6.
H-NMR (CDC13-TMS) (200 MHz)
Diastereoisomer 3' (SSR):the data are identical to those of the diastereo-
isomer 3 described in ex. 6.
Diastereoisomer 4' (SSS): the data are identical to those of the diastereo-
isomer 4 described in ex. 6.
HPLC analysis performed as described in ex. 6.
Diastereoisomer 3': ret0ntion time 18.41 min.
Diastereoisomer 4': retention time 19.33 min.
EXAMPLE 26
-
Preparation of 2(S)-hydroxy-3(S)-/2-(5-bromo^6-methoxy-2-napthyl)-
propanoyl7-butanedioic acid dimethyl ester.
By processing as described in ex. 17, a diastereoisomericmixture of
2~ bromoethyl)-Z-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane~l(S),
S(S)-dicarboxylic acid dimethyl ester (compounds 3' and 4' of ex. 25 in
ratio 3l:41 = 93.7; 2.66 g; 5.0 mmoles) a mixture of the desired
diastereoisomers is obtained (1.98 g; 4.2 mmoles; yield 84.4%) identified
as compounds C' and D').
~The ratio determined by H-NMR (ZOO MHz) is C': D' = 85:15
- 46 -
~-NMR (CDC13-TMS) (200 Ml Iz )
Diastereoisomer C' (SSR): the data are identical to those of diastereoi-
somer C described in ex. 9.
Diastereolsomer D' (SSS): the data are identical to t,hose of diastereoi-
somer D described in ex. 9.
EXAMPLE 27
Preparation of R(-)-2-(5-bromo-6-methoxy-2-naphthyl)-propionic acid.
A mixture of diastereoisomers C' and D' prepared according to ex. 26
(ratio Ct D' = 85:15; 1.2 g; 2.56 mmoles), dime-thoxyethane (18 ml),
12 N HCl (18 ml) is kept at 88C under stirring for 1 h.
The reaction mixture is cooled to room tempera-ture and ls then~orked up
as described in ex. 10 (b).
The so obtained crude acid i8 eluted through a silica gel column
6eluent hexane: ,dieth,ylether 1:1~.
The 2-(5-bromo-6-methoxy-2-naphthyl) propionic acid is obtained..
M.p. = 146-148C; fc~7578 = ~33.39 (c = 0.5%; CHC13).
This acid is esterified with diazomethane and the obtained methylester
analyzed by H-NMR (200 MHz) using an optically active shift
agent ~europium (III)-tris/3-(eptafluoropropylhydroxymethylene)-d-
camphorate 7 in CDCl3.
The ratio between the enantiomers is R(-):S(+) = 85:15.
The methylester when crysta}lized from methanol and hydrolized with an
acid, leads to the R(-)-2-(5-bromo-6-methoxy-2-naphthyl)-propionic acid
in optically pure form.
EXAMPLE 28
Preparation of 2-ethyl-2-(6-methoxy-2-naphthyl)-1,3-dioxolane-4~R), 5(R~-
dicarboxylic acid dimethyl ester.
. .
- 47 ~ r~
1-(6-methoxy-2-naphthyl)-propan-1-one (465 g; 2.17 moles), dimethylester
of L(-~) tartaric acid (773 g; 4.34 moles) and trimethyl ~thoformate
(461 g; 4.3~ mole~) are gradually heated up to complete solutlon.
The solutlon is added with methanesulFhonic acid(15 g; 0.155 moles).
The reaction mixture is kept at 100C ~or 4 hours, distilling off
the volatile compoùnds (about 400 g).
It is cooled to 50C and poured slowly under stirring into a 10% aqueous
solution of NaHC03 (5 1). It is extracted with CH2C12 and the organic
extrac-t is washed with wa-ter and dried 2 4
By evaporating the sol~ent under reduced pressure, a residue ,
containing the desired product as determined by HPLC analysis (743 g;
yield 91.6%). is obtained.
An analitycally pure produc-t is ob,tained by crystalli~ing from 1.3 1 of
methanol (672 g; 1.8 moles ; yield 82.8%).
EXAMPLE 29
Preparation ,of the 2-ethyl-2-/4-(2-methylpropyl)-phenyl7-1,3-dioxolane-
4(R), 5(R)-dicarboxylic acid dimethylester.
A mixture of 1-/4~(2-methylpropyl)-phenyl7-propan-1-one (110 g; 0.58 moles),
dimethyl ester of L(+) tartaric acid (206 g; 1.16 moles) ana trimethyl
orthoform:ate (122.7 g; 1.16 moles) is gradually heated up to complete
solution (50C). The solution is added with methanesulpha'nic aci~3.9 g;
0.04 moles).
The reaction mixture is heated to 85~C and kept at this temperature for
2 h, then c~oled to room temperature and ,worked up as described in ex. 1.
Ihecn~b product !(210 g) is eluted through a silica gel column (eluent
hexane:~diethylether 8:Z) and the desired product is obtained (175.2 g;
0.501 moles; yield 86.5%) having the following charac-teristics:
I.R.~(Neat): 1730-1760 cm (stretching C = 0)
- 48 -
H-NMR (CDC13-TMS) (200 MHz) del-ta (ppm): 0 . 84 ( d , 6H , J = 6 . 4 Hz );
0.89 (t, 3~1, J = 7 . 5 HZ ); 1. 8 (-t-~:p-t~ , JCI-I C~-l -64 HZ ~ JCII-CH ~;
1.97 (q, 2~1, J = 7.5 ~Iz);
2.41 (d, 211, J = 7.1 Hz); 3.78 (9, 311); 3.a4 (s, 3H); 4.78 (AB, 2H,
5 J = 5.7 llz); 7-7.4 (AA'BB', "Il, ~romatic protons).
EXAMPLE 30
Preparation of dias-tereoisomers of the compound 2-(1-bromoethyl)-2-
/4-(2-methylpropyl)-phenyl7-1,3-dioxolane-4(R), 5(R)-dlcarboxylic acid
10 dimethyl ester.
To a solution in 1,2-dichloroethane (70 ml) of 2~ethyl-2-/4-t2-methylpropyl)-
phenyl7-1,3-dioxolane-4(R), 5(R)-dicarboxylic acid dime-thylester (7.0 g;
20 mmoles obtained according to ex. 29),deoxygenated and added with hydrobromic
15 acid (0.324 g; 4 mmoles), it is added dropwise in 1 h under inert
atmosphere at -~15C, a solution of bromine (3.20 g; 20 mmoles) in
1,2-dichloroethane (7.0 ml) previously deoxygenated.
The mixture is kept at 15C for an additional hour and then worl~ed up as
described in example 6.
20 The so obtained residue is eluted through a silica gel column (eluent
hexane: diethylether 8:2) to give ~ mixture of the desired diastereoi-
somers, identified as 11 and 12, in 77% yie~d.~ -
The ratio between the compounds 11 and 12 as determined by HPLC is 88:12
H-NMR (CDC13-TMS) (200 MHz):
25 Diastereisomer 11 (E~RS) : delta (ppm): 0.87 (d, 6H, J = 6.4 Hz);
1.61 (d, 3H, J = 7.1 Hz); 1.84 (t-ept, lH, JC~-~ CH- 6.4 I-lZt JCli CH = 7.1 Hz);
2,45 (d, 211, J = 7.1 Hz); 3.53 (s, 3H);
3.84 (8, 3H); 4.38 (q, lH, J = 7.1 Hz) 4.9 (AB, 2H, J - 5.g Hz);
7-7.4 (AA'BB', 4H, aromatiç prP~tOns).
30 ~iastereoisomer }2 (RRR): delta (ppm): 0.87 (d, 6H, J = 6.4 Hz);
~ 3 ~
~1.58 (d, 3H, J = 7.1 Hz); 1.87 ~t-ept, lH, JCI-~ C~l = 6.~ Hz,
Cll-CH2 .1 ~I~); 2.53 (d, 2H, J = 7.1 I~
3.6 (s, 3ll); 3.83 (5, 3~l); 4.41 (q, lH, J = 7.1 llz); 4.85 (Aa, 2H,
J = 6.5 1l~); 7-7.4 (~A'BB', 411, aromatic protons).
The ilPLC analysis has been performed under the following conditions:
I-lewlett PaCkard instrument mod. 1090 with a
variable wavelength UV de-tec-tor (mod. 1040 DAD).
Analytical conditions:
- column BROWNLEE LABS RPS (5 ~) spheri, 250 mm x 4.6 mm (in-ternal
diameter)
- solvent A: bidis-tilled water
- solvent B:, acetonitrile:methanol = 40:60
- flow: 2 ml~min.
- percentage solvent B: 54%
- column -temperature: 50C
- wavelength (~ ): 222 nanometers
- injection: 4 ~ of a solution containing 0.5 mg/ml of product in
aceton;trile:methanol 40:60
- retention times: diast. 11 = 22.61 min.
diast. 12 = 23.63 min.
- 50 -
EXAMPLE 31
Prepara-tion of 2(R)-hydroxy-3(R)- (2-/4-(2-me-thylpropyl)-phenyl /-
-propanoyl -butanedioic acid dimethyl ester.
Operating under analogous conditions to -those described in Example
17, ~fter work up o~ the reaction mixture, starting from a mixture
of diastereoisomers 11 and 12 (3.0 g; 7.0 mmole ) (ratio determined
by HPLC, 11:12 = 88:12), with a rea~ontlme of 6 hours at ~28C
the mixture of diastereoisomeric esters indicated herein as I
and J is obtainedO
H-NMR (CDCl3 - TMS) (200 MHz)
Diastereoisomer I (RRS): delta (ppm): 0.87 (d, 6H, J=6.4 H~);
1.485 (d, 3H, J=7.1 Hz); 1.8 (t-hept, lH, JCH CH =6.4 Hz,
JCH CH =7.1 Hz); Z.42 (d, 2H, J=7.1 Hz); 3.15 (d, lH, J=7.05 Hz);
3.32 (s, 3H); 3.78 (s, 3H); 3.8 (q, lH, J=7.1 Hz); 4.67 (dd, lH,
JcH_cH=2.3 Hz~ JCH-oH=7 05 Hz); 5.36 (d, lH, J=2.3 Hz); 7.02-7.16
(AA'BB', 4H, aromatic protons).
Diastereoisomer J (RRR): delta (ppm): 0.87 (d, ~H~ J=6.4 H~);
1.525 (d, 3H, J=7:1 Hz); 1.825 (t-hept, lH, JCH CH =6.4 H~,
JCH CH = 7.1 Hz); 2.43 (d, 2H, J=7.1 Hz); 3.15 (d, lH, J=7.05 Hz);
3.62 (s, 3H); 3.69 (s, 3H); 3.82 (q, lH, J=7.1 Hz); 4.73 (dd, lH,
JCH CH=2-3 Hz, JCH oH=7 05 Hz); 5.43 (d, lH, J=2.3 Hz); 7,04-7.2
(AA'BB', 4H, aromatic protons).
A E X f~ L~
25 ~ U~ 32
,.. , f
,~ ~ i Preparakion of 2-/ 4-(2-methylpropyl)-phenyll -propionic acid
(Ibuprofen).
Operating in a analogous manner to that described in Example lO(b),
crude 2-/ 4-(2-me-thylpropyl)-phenyl~ -propionic acid is obtained
from a mixture of dia~tereoisomeric ester 8 I and J, prep~red as
3 ~ 7
described in Example 31 (1.37 g; 3.74 mmoles). Af~er chromatogra-
phy on silica gel, the pure acid is ob-tained (0.7 ~)
/ ~ / D ~19 (C = 1%, 95% ethanol).
EXAMPLE 33
Prepara-tion of 2-(1-bromoethyl)-Z-~ 4-(2-methy:Lpropyl)-phényl_/-
1,3-dioxolane-4(R), 5(R)-dicarboxylic acid.
A solution of diastereolsomers 11 and 12 (see Example 30)(10.0 g;
0.0233 moles) in methylene chloride (20 ml) is added dropwise to
a solu-tion o~ sodium hydroxide ~l.B7 g; 0.0466 moles) in water
25 ml) and methanol (100 ml), kept under stirring at 20C.
The reaction mixture is kept under stirring at this temperature
for 1 hour . The solvent is evaporated under reduced pres-
sure. The residue is taken up in water (100 ml) and acidified to
pH lr with concentrated HCl.
It is extracted with diethyletller(3 x 50 ml). The organic phase
is extracted with a 10% sodium bicarbonate solution (3 x 50 ml).
The alkaline solution is acidified to pH 1,with concentrated HCl and
extract-ed with diethyle-ther (3 x 50 ml). The combined organic phases
are dried over sodium sulpha-te, and the solvent is evaporated under
reduced pressure to give the crude product (8.3 g; acidimetric
assay 92%; yield 81%).
HPLC analysis of a sample esterified with diazomethane æhows that
the ratio of the two diastereosiomers13 and 14 is 87:13.
H-NMR (CDCl3-TMS) delta (ppm)
Dia~tereoisomer 13 (RRS): delta (ppm): 0.87 ~d, 6H, J=6.4 Hz);
1.59 (d, 3H, J57.1 Hz); 1.95 (t- ept, lH, JCH CH =6.4 Hz,
CH CH =7 Hæ); 2.55 (d, 2H, J=7 Hz); 4.42 (q, lH, J=7.1 Hz);
~o 4.88 ~AB, 2H, J=6.4 Hz~; 7-7.4 (AA'B~f, 4H, aromatic protons~,
8.2 (9, 2H).
- 52 - 1 3i~
Diastereoisomer 14 (RRR): delta (ppm): 0.87 (d, 6H, J=6.4 Hz);
1.5~ (d, 311, J=7.1 ~1z); 1.95 (t- ept, l1-1, JC~1 C~1 = 6.4 11z,
JC~1 C11 = 7 11z); 2.55 (d, 211, J=7 1-1~); 4.42 (q, ~H, J=7.l ~1z);
4.8 ( AB, 211, J-.6.4 Hz); 7-7.44 ( AA ' BB ', ~H, aromatlc protons);
8.2 (s, 2H).
EXAM
Preparation of (+)-2~R)-hydroxy-3(R)-J 2(S)(6-methoxy-2-naphthyl)-
propanoyl / -butanedioic acid dimethyl ester.
A solution of triethylamine (4.45 g; 0.044 moles) in methylene
chloride (lO ml) is added dropwise in -a period of 5 minutes to a
mixture of 2(R),3(R)-dihydroxy-butanedioic acid dimethyl ester
(L(~)tartaric acid dimethyl ester )(44.5 g; 0.25 moles) and methylene
chloride (90 ml), cooled to -10C and kept under s-tirring, followed
by -the dropwise addition in a period of 20 minutes of a solution,
in mcthylene chloride (25 ml~ of S(~)2-(6-methoxy-2-naphthyl)-
-propionyl chloride (5.0 g; 0.020 moles) prepared as described in
Japanese patent applica tion 57jl45841 (C.A. 98, 72492h).
The reaction mixture is then poured into a 10% sodium bicarbonate so-
lution (200 ml), extracted with methylene chloride (lO0 ml), and the
organic phase washed with dilute hydrochloric acid and dried over
sodium sulphate. The residue (5.5 g~ is obtained by evaporating the
solvent under reduced pressure, and is crystallised from a mixture of
heptane and diethylether(l:l, 165 ml).
The desired product (diastereoisomer A, see Example 3) (2.75 g) is
obtained, having the following characteristics:
I.R. (C=5% in CHC13) 1750 cm
/ ~ / D = ~73 7 (C=1%, CHCl3)
M.P.= 77-79C
- 53 - ~ d~ r~
H-NMR (CDC13 TMS) (200 Ml-lz): delta (ppm): 1.58 (d, 3H, J=7.4 Hz);
3.07 (s, 311); 3.31 (d, lH, J=7.4 Hz); 3.79 (s, 3~l); 3.87 ~s, 3H);
3 96 (q 1~l J=7.4 Hz); 4.66 (dd, ~ Jc~l-ctl=2 3 ~ ' Cfl-OH
5.37 (d, lH, J=2.3 ~Iz); 7-7.8 (61-1, aromatic system).
A solution of bromine (0.410 g; 2.56 mmoles) in 1,2-dichloroethane
(3 ml) is added in 15 minu-tes -to a solution of the ester thus ob-
tained in 1,2 dichloroethane (10 ml), cooled to 0C.
The reaction mixture is kept at 0C for 1 hour, and is -then poured
into a 10% sodium bicarbonate solution (10 ml) and extracted with me-
thylene chloride (10 ml).
The comb~ed or~c phases are washed with wat~r (20 ml x 2), dried
over sodium sulphate, and the solvent evaporated under reduced pres-
sure.
The residue (1.14 g) is crystallized from methanol. (+)-2(R)-hydroxy-
3(R)~2-(S)-(5-bromo-6-methoxy-2-naphthyl)-propanoyl ~butanedioic acid
dimethyl ester is obtained (0.889 g; 1.9 mmoles; yield 74%); M.P.
124-126~C; / ~ / D = ~61.4 (C = 1%; CHC13).
The chemical-physical data (M.P., / ~ ~ D and H-NMR-200 MHz)
are equal to those of the diastereoisomer ester C described in Exam-
ple 9. When treated with palladium-on-carbon and hydro~en at atmo-
spheric pressure and room -temperature in the presence of triethyl-
amine, the product produces the diastereoisomer A.
EXAMPLE 35
Preparatio~ of the mixture of diastereoisomers 7 and 8 of 2-(1-bromo-
ethyl)-2-(5~bromo-6-methoxy-2-naphthyl)-1,3 dioxolane-4(R),5(R)-di-
carboxylic acid.
A solution of bromine (171 g; 1.68 moles) in carbon tetrachloride
(360 ml) is added dropwise in 1 hour to a solution of 2-ethyl-2-
(6~me-thoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxylic acid di-
methyl ester (200 g; 0.534 moles) in carbon -tetractlloride (2000 ml)
kept under an inert atmosphere at 0C.
- 54 ~ 8~
The reac-tion mixture is kept at O~C for 2 hours, and worked as
described in Example 6.
The crude product-thus ob-tained (351 g) is dissolved in methanol
(2000 ml), and a solution of sodlum hydroxide (38.4 g; 0.96 moles)
in water (384 ml) is added dropwise to the resultant solution at am-
bient temperature in 1 hour. The reaction mixture i8 kep-t at ambient
-temperature under stirring for 20 hours . The methanol is
evaporated under vacuum, maintaining -the initial volume of the
solution by adding water.
The pH of the aqueous solution obtained is adjusted to 7 wi-th dilute
hydrochloric abid. The solution is then extrac-ted with methylene chlo-
ride and the aqueous solution is acidified with concentrated HCl to
pH 1.
It is extracted with diethylether(3 x 250 ml) and the combined organic
phases are washed with water and dried over sodium sulphate. The sol-
vent is evapora-ted under vacuum to give a residue that is crystalli-
sed from methylene chloride.
A mixture of the two diastereoisomers 7 and 8 of
2-(1-bromoethyl~-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),
5(R)-dicarboxylic acid is obtained (205 g; 0.407 moles; yield 76%) in
the ratio of 7:8 = 94:6.
EXAMPLE 36
A mixture of the two diastereoisomers 3 and 4 of 2-(1-bromoethyl)-2-
(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R), 5(R)-dicarboxylic
aci~ dimethyl ester in the ratio 3:4 = 9:1 (1 g; 1.87 mmoles),
zinc bromide (0.84 g; 3.75 mmoles) and 1,2-dichloroethane (12 ml) is
heated at reflux (83C),under stirring~ and under nitrogen,for
66 hours.
The reaction mixture is cooled to ambient temperature, and water (5 ml)
is added. The phase are separated and the organic phase is dried over
80dium sulphate.
- 55 - ~3~
The solvent i5 evaporated under vacuum to giv~ a residue (0.9 g) to
which dioxane (10 ml) and concentrated HCl (~ ml) are add~d. The
mixture is heated to 70C under,stirring, for 2 hours, is then diluted
with water (10 ml) and extracted with diethyle-ther (3 x 20 ml).
The combined organic ex-trac-ts are washed with water and dried over so-
dium sulphate. Evaporatlon of the solvent under vacuwn give5 a re~idue
which by chromatography on silica gel (eluent hexane:ethyl ether =
7:3) gives 2-(5-bromo-ô-methoxy-2-naphthyl)-propionic acid (0.28 g;
0.9 mmoles; yield 48%);
M.P. 166-167C
2~
/ ~/ D = +15^44 (C=0.5, CHC13).
The ratio of the enantiomeric acids S(+)/R(-) is 65:35.
ESEMPI0 37
Preparation of 2-(S)-(5-bromo-6-methoxy-2-naphthyl)-propionic acid
methyl ester from 2-(1-~S)-bromoethyl)-2-(5-bromo-6-methoxy-2-naph-
thyl)-1,3-dioxolane-4(R),5(R)-dicarboxylic acid dimethyl eæter.
A mixture of pure 2-(1-(S)-bromoethyl)-2-(5-bromo-6-methoxy-2-naph-
thyl)-1,3-dioxolane-4(R),5(R)~dicarboxylic acid dimethyl ester (1.03 g,
1.93 mmol), silver trifluoromethanesulfonate (0.6 g, 2.31 mmol) and
methanol (5 ml) i5 heated at reflux for 7 hours. The reaction mixture -
is cooled at room temperat~re, filtered, poured into water, and extrac-
ted with dichloromethane. The combined organic extracts are ~ashed
with water, dried (Na2S04), and-filtered.
Evaporation of the solvent under reduced pressure gives the optically
pure 2-(S)-(5-bromo-6-methoxy-2~naphthyl)-propionic acid methyl eæter.
M.P. 94-95C
L ~ - / D ~ + 52(c=0.5, CHCl3)
The product is found to be optically pure by H-NMR (200 MHz) analy-
sys, carried out in CDCl3 using an optically activc shif-ting agent
(Europium (III) Tris-~ 3-(eptafluoropropylhydroxyme-thylen~)-d-aampho-
rate.1
~6 - 3 ~
EXAMPLE 38
Bromination of 2-ethyl-2-(6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),
5(R)-dic~rboxylic acid.
~romine (0.32 g; 2 mmol) is added dropwise, in 5 minutes at 15C
and under argon, to a suspension of 2-eth~1-2--(6-methoxy-2-naph-
thyl)-l,3-dioxolane-4(R),5~R)-dicarboxylic acid (0.346 g, 1 mmol).
The reaction mixture is heated at 40~C and kept at 40C for 12 hours;
then it is poured into a 10% aqueous solution of sodium bicarbonate
and extracted with diethylether. The aqueous phase is acidified to
pH = 1 with conc. HCl and extracted with diethylether. The combined
organic extracts are washed with water, dried (Na2S04), and filte-
red. Evaporation of the solvent under reduced pressure gives a react-
ion crude which, after purification leads to a diastereOisomeric
mixture of 2-(1-bromoethyl)-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-
dioxolane-4(R),5(R)-dicarboxylic acid in ratio 7:8 = 81:19 (deter-
mined by H-NMR).
H-NMR (90 MHz, Acetone-d6-TMS) ~ (ppm):
Diastereoisomer 7 (RRS):
1.70 ~3H, d, J=6.8 Hz); 4.03 (3H,s); 4.66 (lH, q, J-6.8 Hz);
4.95 (2H, A~q,4 ~ = 15.31, J=6.9 Hz); 7.45-8.18 (5H, m).
Diastereoisomer 8 (RRR):
1.70 (3H, d, J=6.8 Hz3; 4.03 (3H, s); 4.66 (lH, q, J=6.8 Hz);
4.95 (2H, ABq,A~=14.46, J=6.6 Hz); 7.45-8,18 (5H, m).
The diastereoisomeric ratio is confirmed analyzing by H-NMR and
HPLC the product obtained by esterificatibn with diazomethane.
- 57 _
EXAMPLE 3g
Preparation of the diastereoisomeric mixture of 2-(1-iodoethyl)-2-
(6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5~R)-dicarboxylic acid
dimethyl ester,
A solution of 2-ethyl-2-(6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),
5(R)-dicarboxylic acid dimethyl ester (0.935 ~, 2.5 mmol) and of
iodine monochloride (0.81 g, 5 mmol) in dichloromethane (5 ml) is
kept under nitrogen and at 15~C for 24 hours. The reaction mixture
is poured into a 10% aqueous solution of sodium bicarbonate, ancl
extracted with additional dichloromethane. The combined organic
extracts are ~ashed with a 5% aqueous solution of sodium thiosulphate,
with water, dried (Na2S04), filtered, and concentrated in vacuo.
Purification of the residue by column chromatography (silical gel,
eluent hexane:diethyl ether = 7:3) gives the diastereoisomeric
mixture of 2-(1- iod~ethyl)-2-(6-methoxy 2-naphthyl)-1,3-dioxolane-
4(R),5(R)-dicarboxylic acid dimethyl ester 15 and 16 in ratio l5:16=
60:40 (determined by H-NMR).
H-NMR (200 MHz, CDC13-TMS) ~ (ppm):
Diastereoisomer 15 (RRS)
1.80 (3H, d, J-7 Hz); 3.44 t3H, s1; 3.84 (3H, s); 3.90 (3H, s);
4.58 (lH, q, J=7 Hz); 4.95 (2H, ABq,4 ~ = 20.70, J=6 Hz); 7.8-3.0
(6H, m).
Diastereoisomer 16 (RRR)
1.80 (3H, d, J=7 Hz); 3.58 (3H, 8); 3.84 ~3H, s); 3.90 (3H, s);
4.58 (lH, q, J=7 Hz); 4.87 (2H, ABq,~ ~=46.04, J=6.8 Hz); 7.8-8.0
(6H, m).
~ - 58
~3~3~ ~-7
EX~MPL,E 40
Preparation of 2-(6-methoxy-2-naphthyl)-propionic acid from a
diastereoisomeric mixture of 2-(1-iodoethyl)-2-(6-methoxy-2-naphtyl)-
-1,3-dioxolane-4(~),5(R)-dicarboxylic acid dlmethyl es-ter.
Silver trif]uoromethanesulfonate (1.2 g, 4.8 rnmol) is added, under
argon and stirring, at 15C to solution of a diastereoisomeric
mixture of 2-(1-iodoethyl)-2-(6-me-thoxy-2-naphthyl)-1,3-dioxolane-
4(R),5(R)-dicarboxylic acid dimethyl ester in ratio 60:40 (1.~ g,
3.2 mmol) in 1,2-dichloroethane (20 ml). The reaction mixture is
kept in the dark at 15C for 3 hours; then it is filtered, poured
into water. The organic layer is separated, washed with water,
dried (Na2S04), filtered and concentrated in vacuo.
The residue is dissolved into dioxane (5 ml) and conc. HCl (5 ml) is
added. The mixture is heated at 70C for 2 hours coolsd at room
temperature, poured into water, and extracted with die-thyl ether.
The combined organic extracts are washed with water and back-extracted
with a 2% aqueous solution of sodium bicarbonate. The aqueous phase
is acidified with conc. HCl and extracted with diethyl ether. The
combined organic extracts are washed with water, dried (Na2S04),
filtered. Evaporation of the solvent under reduced pressure gives
the 2-(6-methoxy-2-naphthyl)-propionic acid.
M.p. = 154-155C.
/ ~ 7D = -~ 6.02 (c = 1,CHC13)
HPLC analysis, carried out as described in J. Pharm. Sci. 68, 112
(1979) and H-NMR (200 MH~) analysis carried out on the methyl ester
in CDCl3 using an optically active shifting agent (Europium (III)
Tris-/3(eptafluoropropylhydroxymethylen)-d-camphorate7) shows an
enantiomeric ratio S(+) : R(-) = 55 : 45.
_ 59 _
~ 3 ~
EXAMPI.E 41
Preparation of 2-ethyl-2-(6-hydroxy-Z-naphtyl)-1,3-~dioxolane-4(R),5~R)-
dicarboxylic acid dimethyl ester.
A mixture of 1-(6-hydroxy-2-naphtyl)-propan-1-one (Z5 g, 0.125 mol),
2(R), 3(R)-dihydroxyb~ltalledioic acid dlmetyl este~ (178 g, 1 mol),
trimethyl orthoformate (54 g, 0.51 mol), and of methane~ulphonic acid
(0.84 g, 0.08a mol) is heated, under argon and under stirring,
at 70C for 4 hours.
The reaction mixture is cooled at room temperature, poured into a
10% aqueous solution of sodium carbonate (409 ml), and extracted
with diethylether ~4 x 50 ml). The combined organic extracts are
washed with water (3 x 150 ml?, dried (Na2S04), filtered, and
concentrated in vacuo.
Purification of the crude by column chromatography (silica gel,eluent
hexane : diethyl~ther = 1 : 1) gives the pure 2-ethyl-2-~6-hydroxy-
-2-naphtyl)-1,3-dioxolane-4(R),5(R)-dicarboxylic acid dimethylester
(17 g) as an oil.
H-NMR(90 MHz, CDC13-TMS) ~tppm):
1.93 (3H, t, J = 6.5 Hz); 2,10 (2H, q, J = 6.5 Hz); 3.43 ~3H, s);
3.80 (3H, s); 4.83 (2H,ABq,a~ = 6.7, J = 6 Hz); 6.00 (lH, s, OH);
7.07-7.85 (6H, m).
EXAMPLE 42
Preparation of the diastereoisomeric mixture of 2-(1-bromoethyl)-2-
-(5-bromo-6-hydroxy-2-naphthylj-1,3-dioxolane-4(R),5~R)-dlcarboxylic
acid dimethyl ester.
A solution of bromine (5.12 g, 32 mmol) in carbon tetrachloride (5 ml)
is added dropwise in 10 minutes, under argon and at 15C, to a
solution of 2-ethyl-2-(6-hydroxy-2-naphthyl)-1,3-dioxolan~-4(R),5(R)-
-~ - 60 -
:L 3 ~
dicarboxylic acid dimethyl ester (6 g, 16 mmol) in carbon tetrachloride
(60 ml). The reaction mixture is kept at 15C for 2 hours and poured
into a 5% aqueous solution of sodium thiosulfate (200 ml).
The org~nlc layer is separated, washed with water, dried (Na2S04),
filtered, and concentrated in vacuo.
Purification of the reaction crude by column chromato~raphy (silica .
gel, hexane : diethyl ether = 1 : 1) gives a diastereoisomeric mixture
of 2~ bromoethyl)~2-(5-bromo-6-hydroxy-2-naphthyl)-1,3-dioxolane-
-4(R),5(R)-dicarboxylic acid dimethyl ester (8 g, 15 mmol; yield 93%)
as a solid.
Ratio diaster~oisomers 17 : 18 = 90 : 10 (determined by H-NMR and
HPLC).
m.p. 116-117C.
H-N~R(200MHz, CDC13TMS) ~ (ppm):
diastereoisomer 17 (RRS)
1.66 (3H, d, J = 7 Hz); 3.52 (3H, s); 3.88 (3H, s); 4.48 (lH, q, J=7
H~):4.96 (2H, ABq,a ~ = 27.80, J =6.1 Hz); 7.2-8.0 (5H, m).
diastereoisomer 18 (RRR)-
1.6Z (3H, d, J = 7 Hz); 3.56 (3H, s); 3.87 (3H, s); 4.48 (lH, q, J = 7
Hz); 4.90 (2H, ABq,~ 35.44, J=6.3 Hz); 7.2-8.0 (5H, m).
Tbe diastereoisomeric ratio 17 (RRS) : 18 (RRR) = 90 : 10 is confirmed
by converting the product in the diastereoisomeric mixture of 2-(1-
bromoethyl)-2-(5-bromo-6-methoxy-2~napnthy~-1,3-dioxolane-4(R),5(R)-
-dicarbox~lic acid dimethyl ester 3 and 4 following the present
procedure:
a mixture of the product (0.52 g, 1 mmol), potassium carbonate (1.38 g,
10 mmol), me-thyl iodide (0.426 g, 3 mmol), and of acetone (10 ml) iS
kept under stirring at room -temperature for 4 hours.
The reaction mixutre si fil-tered and concentrated in vacuo. The residue,
so obtained, is a diastereoisomeric mixture of 2~(1-bromoethyl)-2-(5-
bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dlcarboxyllc acid
- 61 - 1 3 3 ~
dimethyl e~ter in ratio 3 (RRS):4(RRR~ = 90 : 10 (determined by
H-NMIl and HPLC).
EX~MPL~ 43
Preparation of 2-(5-bromo-6-hydroxy-2-naph-thyl)-propionic acid.
A mixture of the dias-tereoisomers 17 and 18 in the ratio 90:10 (see
Example 42) (0.57 g; 11 mmoles), sodium hydroxide (0.132 g; 33 mmoles)
and water (20 ml~ is heated to 60~C for 2 hours. The reaction mixtu-
re is cooled to room temperature, acidified to pH 1 ~ith concen-
trated HCl and extracted with diethylether.
The combined organic phases are washed with water, dried over sodium
sulphate and concentrated under vacuum. The residuè thus obtained
$s purified by chromatography on silica gel, to give pure
2-(5-bromo-6-hydroxy-2~naphthyl)-propionic acid.
On the basis of H-NMR analysis as described in Example 4, the ratio
of the S to R enantiomer is 90:10.
EXAMPLE 44
Preparation of 2-(1-bromoethyl)-2-(5-bromo-6-hydroxy-2-naphthyl)-
-1,3-dioxolane-4(R~,5(R)-dicarboxylic acid.
A mixture of the diastereoisomers 17 and 18 in the ratio 90:10 (see
Example 42) (5.6 g; 0.0108 moles), water (52 ml), methanol (30 ml)
and an aqueous 10% (w/v) sodium hydroxyde ~olution (11.5 ml) is kept
under s~irring at room temperature for 6 hours.
The reaction mixture is then acidified with concentrated ~ICl to pH 1
and extracted with diethylether. The combined organic extracts are washed with
.
wa-ter and dried over sodium sulphate.
Evaporation of the solvent under vacuum gives the diastereoisomers 19
and 20 (4.8 g; 0.0098 moles; yield 90%) in the ratio 19:20 = 92:8
6 2 ~ ~ 5
H-NMR (90 MHz., CDCl3-TMS~ S (ppm)
Diastereoisomer 19 (RRS):
1.66 (d, 3M, J= 7 ~3z); 4.63 (q, 11-1, J=7 Hz); 4.93 (2H~ ABq,a ~ =16.42,
J=6.5 Hz); 7.23-8.15 (m, 511); 8.27 (1l1, broad)
EXAMPLE 45
Preparation of 2-(5-bromo-6-hydroxy-2-naphthyl~-propionic acid
A mixture of the diastereoisomers 2-(1-bromoethyl)-2-(5-bromo-6-
hydroxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxylic acid 19 and
20 (1.76 g; 3.6 mmoles) in the ratio 19:20 = 92:8 (s~e Example 44),
sodium bicarbonate (2.4 g; 2~ mmoles) and water (50 ml) is heated
under reflux, under stirring , for 4 hours. The reaction mixture,
cooled to ambient temperature, is acidified to pH l with 6 N HCl
and extracted with diethylether. The combined organic phases are washed
with water and dried over sodium sulphate, Evaporating the solvent
under vacuum gives a crude product to which dimethoxyethane (17 ml)
and 12 N HCl (17 ml) are added. The reaction mixture is heatad under
reflux, under stirring for 2 hours, cooled and extracted with diethyl-
ether.:The combined organic phases are washed with water and dried
over sodium sulphate. Evaporation of the solvent under vacuum gi~es a
residue which is chromatographed over silica gel (eluent diethylether-
hexane 7:3). In this manner the pure acid is obtained /c~7D = ~42.3
(C_l in acetone). A samples is esterified with diazomethane. The me-
thyl ester is analysed by H-NMR (200 MHz) using an opti-
cally active shift agent. The ratio of the enantiomeric acids
(~)S/(-)~ is 98:2.
EXAMPLE 46
A solution of silver tetrafluoroborats (0.6 g; 3.08 mmoles) in l,2-
dichloroethane t4 ml) is added dropwise to a mixture of 2-(1-bromo-
ethyl)-2-(5-bromo-6-methoxy-2-rlaphthyl)-1,3-dioxolarle-4(R),5(~)-
- 63 - ~ 3 ~
-dicarboxylic acid dime-thyl ester (dias-tereoisomer 3 : diastereoiso-
mer 4 = 94:6, ratio determined by HPLC) (1.33 g; 2.5 mmoles) and
1,2-dichloroethane (10 mL) kept under stirring at ~15~C.
After 73 hours the reac-tion mixture is poured into water (20 ml)
and filtered through celite, -the filtrate beix~g washed ~rith methylene
chloride (10 ml~.
The organic phase is washed with wa-ter (2 x 20 ml) and dried over
sodium sulphate.
Evaporation of the solvent under reduced pressure gives a residue
(0.95 g) in which -the diastereoisomers C and D of the ester are pre-
sent in the ratio C:D = 79:21, determined by H-NMR analysis at
60 MHz.
In an analogous test carried out in parallel, in which water (0.1 g;
6 mmoles) was added to the reaction mixture before adding the sodium
tetrafluoroborate, the ratio of the diastereoisomers,after 73 hours.
is C:D = 94:6;
EXAMPLE 47
Preparation of 1-(4-chlorophenyl)-3-methyl-butan-1-one
3-methyl-butyrryl chloride (12~3.6 g; 1.07 moles) is added in lS
minutes to a suspension of aluminum chloride (153.8 g; 1.15 moles)
in methylene chloride (200 ml) cooled to -5C and kept under stirring
~in an inert atmosphere.
At the end oP the addition, the mixture is heated to +20C
ahd chloroben~ene (100 g; 0.89 moles) is added in 15 ~inutes. The
reaction mixture is heated to +45C for 7 hours, then cooled to
ambiente temperature and poured under stirring. into concentrated HC1
(200 ml) and ice (1500 g).
The aqueous phase is extracted with me-thylene chloride (3 x 300 ml).
The organic extracts are washed with a 1% sodium hydroxide solution
~ - 64 - ~ 3 ~
(3 x 700 ml) and with water (3 x 700 ml~.
After drying over sodium sulphate, the organic solvent is evaporated
under reduced pressure to give a residue (161 g) which, after crystal-
lization from n~hexane (100 ml) provides 1-(4-c:hlorophenyl)-3-methyl-
bu-tan-l-one (121.5 g; 0.62 moles; yield 69.4%).
M.P. = 39-40
I.R. (Nu~ol) = 1680-1700 cm (stretching c=o)
H-NMR ~CDCl3-TMS) (90 MHz): ~ (ppm):
0.97 (d, 6H, J=6,7 Hz); 2.27 (m, lH, JC~ CH = 6.7 Hz); 2.77 (~art AB of
an ABX system, 2H~; 7.3-7.9 (AA~BB~, 4H aromatic protons)
EXAMPLE 48
.
Preparation of 2-(4-chlorophenyl)-2-(2-methylpropyl)-1,3-dioxolane-
4(R),5(R)-dicarboxylic acid dimethyl ester.
A mixture of 1-(4-chlorophenyl)-3-methyl-butan-1-one (40.0 g; 0.204
moles), 2(R),3(R)-dihydroxy-butanedioic acid dimethyl ester (72.4 g;
0.407 moles) and trimethyl orthoformate (43.1 g; 0.406 moles) is
heated gradually untill a complete solution (60DC~. ~ethanesulphonic
acid (1.4 g; 0.015 moles) is added to the solution, which is then
heated to 75~C.
After a reaction time of 3 hours, the mixture is cooled to ambient
temperature a~d poured into a 10% sodium bicarbonate solution (250 ml)
under vigorous stirring. The aqueous phase is extracted with
methylene chloride (2 x 250 ml) and the o~ganic extracts washed with
water (Z x 400 ml). After drying the organic phase over sodium sul-
phase, the solvent is evaporated under reduced pressure.
The residue obtained (68.7 g) is chromatographed ov0r silica gel
(eluent hexane:diethylether _ 8:2).
2-(4-chlorophenyl)-2-(2-methylpropyl)-1,3-dioxolane-4(R),5(~)-
dicarboxylic acid dimethyl ester (41 g; 0.115 moles; yield 56,4%)
i9 obtained.
- 65 -
~ 3 ~ 7 li
M.P. = 40C
/ ~ / = +21.6 (c = 1%; C~IC13)
I.R. (Nujol) = 1770-1740 cm (stretching c=0)
H-NMR (200 Ml-l~) (CDC13-TMS): S (ppm):
0.87 (d, 6H, J=6.9 llz); 1.67 (m, lfl, JCI~ C~l = 6-9 ~Iz); 1-86 (part
AB of an A~X system, 2ll); 3.55 (8, 3H); 3.8~ (s, 3H);~4.7a (ABq,
2H, J=6.0 Hz); 7.2-7.4 (AA'BB', 4H aromatic protons).
EXAMPLE_49
Preparation of 2-(1-bromo-2-methylpropyl~-2-(4-chlorophenyl)-1,3-
dioxolane-4(R),5(R)-dicarboxylic acid dimethyl es-ter.
A solution of bromine (8.06 g; 0.05 moles~ in 1,2--dichloroethane
(18 ml) is added in 1 hour and 15 minutes to a solution of
2-(4-chlorophenyl)-2-(2-methylpropyl)-1,3-dioxolane-4(R),5~R)-
dicarboxylic acid dimethyl ester (18.0 g; 0.05 moles) in 1,2-
dichLoroethane (180 ml~, to which methanesulphonic acid (3.6 g;
0.038 moles) had been previously added, the reaction mixture being
kept under stirring in an inert atmosphere at +15~C. After 1
hours at 15C, the mixture is poured into a 10% sodium carbonate solution
(400 ml) under vigorous stirring. and extracted with methy- .
lene chloride (2 x 250 ml).
The organic phase is washed with water (2 x 400 ml) and dried over
sodium sulphate.
After evaporating the solvent under reduced pressure, a residue
(20.5 g) is obtained which contains the two diastereoisomers of the
2-(1-bromo-2-methylpropyl)-2-(4-chlorophenyl)-1,3-dioxolane-4(R),
5(R)-dicarboxylic acid dimethyl ester, here indicated as 21 and 22,
ln the ratio 21:22 = 97:3 (ratio determined by H-NMR (300 MHz)
analysis and Confirmed by HPLC analysis).
- 66 - ~ 7
By crystallization from n-hexane (60 ml), the diastereoisomer 21 is
obtained (13.6 g; 0.031 moles; yield 62.5%), and is found to be pure on
H-NMR analysis (300 Milz).
H-NMR (300 MHz ) ( CDC13-TMS )
Diastereoisomer 21 (RRS):
0.93 (d, 31-l, J_6.9 Hz); 0.98 (d, 3H, J=6.6 ll~); 1.70 (m, 1~l,
CH-CH H~, JCH-C~ =6-6 Hz~ Jc~l_cH =6 9 Hz); 3.59 (s, 3~); 3.85
(s, 3H), 4.28 (d, 1l~, J-1.8 Hz); 4.87 (ABq, 2H, J=6.2 Hz); 7.3-7.5
(AA'BB', 4H aromatic protons).
The HPLC analysis was performed under the following conditions:
Hewlet-t Packard instrument mod. 1090 with U.V. variable
waveleng-th U.V. detector.(mod. 1040 DAD).
Analytical conditions:
- Brownlee column LABS RP 8 (5 ~ )balls; 250 ml x 4.6 mm (inner
diameter)
- Solvent A: bidls-tilled water
- Solvent B: methanol
- Flow: 1.7 ml/min
- Percentage solvent B: 63%
~ Column temperature: 40C
- Wavelength ( ~ ~: 230 nanometer
- Injection 5 ~ of a solution containing 0.5 mg~ml of product in me-
thanol
- Retention times:
Diastereoisomer 21 = 11.71 minutes
Diastereoisomer 22 = 12.85 minutes
~.~ 3 ~ . 7
-67 -
Example 50
Preparation of 2(R)-hydroxy-3(R)-l2(S)-(4-chlorophenyl)-3-methylbut
anoy~7-butane oic acid dime-thyl ester
A solution of silver tetrafluoroborate (1.6 g, 8.2 mmol) in
1,2-dichloroethane (15 ml) was added in 20 minutes to a mixture of
2-(1-bromo--2-methylpropyl)-2-(4-chlorophenyl)-1,3-dioxolane-4(R),5(
R)-dicarboxylic acid dimethyl ester (diastereoisomer 21) (3 g, 6.9
mmol), w~ter (0.2 g)and of 1,2-dichloroethane (18 ml) at 20~C. The
reaction mixture was heated at 50C for 7 hours, ccoled at 20C
and poured in water (50 ml). The mixture was filtered on celite
and the precipitate washed with dichloromethane (30 ml).
The organic phase was separeted, washed with ~ater~ dried
over sodiu~ sulfate, and concentrated in vacuo. Purification of
the reaction crude (2.3 g) by column chromatography (silica gel;
eluent hexane:diethylether = 1:1) gave the pure diastereoisomer
2(R)-hydroxy-3(R)-~2-(S)-(4-chlorophenyl)-3-methylbutanoy~-bu-ta-
nedioic acid dimethyl ester K (1.95 g, 5.2 mmol; yield 75.9%).
H-NMR (300 MHz, CDCl3-TMS) delta (ppm):
Cll-C~I 6.9 Hz); 1.06(d, 3H, J=6.2 Hz), 2.33(m lH
JCH Cll=10-6 ~Iz~ JCI~ C~l -6-9 Hz~ JC~ Cll =6.2 Hz); 3.22(d, 1~l,
JCH C~l =6.9S Hz); 3.24(d, lH, J=10.6 Hz); 3~30(s, 3H); 3.77(s,
3H); 4.63(dd~ lH~ JCH CH=2 6 ~Iz); 5.36(d~ lH~ J =2.6 Hz);
7.21-7.23(AA'88', 4H, aromatic protons).
pl~
-- 68 _
Example 51
Prepara-tion of 2(R)-hydroxy-3(R)-L2(S)-(4-chlorophen~1)-3-methyl-
bu~tanoyl~-butandioic acid.
A mixture of 2(R)-hydroxy--3(R)-~ (S)-(4-chlorophenyl)-3-methylbu-
tanoyl/-butanedioic acid dimethyl ester (dias-tereoisomer K) (1 g,
2~6 mmol), 1,2-dimethoxyethane tl3-3 ml) and of conc HCl (18.3 ml)
was heated, under stirring, at 70C for 1 hour. THe reaction
mixture was cooled at room temperature, poured into water (50 ml)
and extracted with dichloromethane (2 x 50 ml). The organic phase
was extracted with a 10% aqueous solution of sodium bicarbonate (4
x 50 ml). The aqueous phase was acidified with conc HCl to pH
and extracted with dichloromethane (3 x 50 ml). The combined
organic phase was washed with water, anhydrified over sodium
sulfate, filtered, and concentrated in vacuo.
Crystallization of the residue (0.8 g) gave the pure 2~R)-
hydroxy-3(R)-~2(S)-(4-chlorophenyl)-3-methylbutanoyl/-butanedioic
acid ~0.4 g) (diastereisomer L).
bl.p. = 173-175C
H-NMR ~300 MHz, CDC13-TMS) delta (ppm): Diastereoisomer L (RRS)
0.56(d, 3H, J=6.7 Hz); 0.94(d, 3H, J=6.5 Hz); 2.20(m, lH,
CH-CH3 6 Hz~ JCH-CH =6-5 Hz~ JcH_c~-~=10 4 Hz); 3.16(d, lH,
J=10.4 Hz); 4.65(d, lH, JC~ C~l~2.1 Hz); 5.33(d, lH, J=2-1 llz);
7.00-7.27(AA'BB', 4H, aromatic protons).
1 _ 69
H-NMR analysis carried ou-t on the corresponding dirnethyl ester,
obtained by reactlon with dia~omethane, showed only the presence
the diastereoisomer K (~RS).
Example 52
Preparation of (~)-2(S?-(4-chlorophenyl)-3-methvlbutanoic acid.
A mixture of the diastereoisomer K (0.9 g, 2.3 mmol), 1,4-dioxane
~ (16 ml) and of conc HCl (16 ml) was heated, under stirring, at
90C for 18 hours~ The reaction mixture was cooled at room
temperature, diluted with water (30 ml), . and extracted with
dichloromethane ~3 x 20 ml~). The organic phase was extracted with
a 10% aqueous solu-tion of sodium bicarbonate (5 x 10 ml). The
aqueous phase was acidified with conc HCl to pH 1 and extracted
with dichloromethane (5 x 10 ml). The combined organic phase was
washed with water, dried . ~ over sodium sulfate, and con-
centrated in vacuo.
Purification of the reaction crude (0.25 g) by column chroma-
tography (silica gel; eluente hexane:di~thylether= 80:20) gave
pure 2(S)-(4-chlorophenyl)-3-methylbutanoic acid (0.2 g).
/ ~ /D =~38.6 (c=1%, chloroform)
Example 53
Preparation of 2-(l(S)-bromo-2-meth lpropyl?-2-(4-chlorophenyl)-1,3
, . ~ . . Y
-dioxolane-4(R),5(R)-dicarboxylic acid
_ 70 _
A solution of the diastercoisomer 21 (10 g, 23 mmol) in dichlometha-
ne (lO ml)was added dropwise in 15 minute3 at 20C to a solution
of sodium hydroxyde (2 g, 50.6 mmol) in water (25 ml) and methanol
(100 ml). The reaction mixture wa~ kept at 20C for 1 hour and
the solvent removed under reduced pressure. Water (100 ml) was
added. The solution, so obtained, was acidified with conc HCl -to
pH 1 and extracted with diethyle-ther (3 x 75 ml). The organic
phase was extracted with a l~fi aqueous solution of sodium
bicarbonate (3 x 75 ml). The aqueous phase was acidified with conc
HCl to pH 1 and extracted with diethylether (3 x 75 ml). The
combined organic extracts were washed with water and anhydrified
over sodium sul~ate. Evaporation of the solvent under reduced
pressure gave the Z-(l(S)-bromo-2-methylpropyl)-2-(4-chlorophenyl)
-1,3-dioxolane-4(R),5(R)-dicarboxylic acid (diastereoisomer 23)
(7.2 g,l9.8 mmol; yield 86%).
H-NMR (300 MHz, CDCl3-TMS) delta tppm): diastereoisomer 23(RRS)
0.92(d, 3H, J=6.6 Hz); 0.98(d, 3H, J=6.2 Hz); 1.58(m, lH,
CH-CH Hz~ JcH-cH =~-6 Hz~ JCH CH =6-2 Hz); 4-37(d, lH, J=1.8
Hz); 4.86(ABq, 2H, ~=6.2 Hz); 7.36-7.46(AA'BB', 4H, aromatic
protons).
The presence of one diastereoisomer was con~irmed by HPLC analysis
carried out on a sample o~ the corresponding dimethyl ester
(diastereoisomer 21) obtained by reaction with diazomethane.
Example 54
Preparation of 2-ethyl-2-(6-methoxy-2-naehthyl)-1,3-dioxolane-4(R),
5(R)-dicarboxylic acid N,N!N',N'-tetraethyl amide.
~3~g~7
_ 71 -
A mixture of 2-ethyl-2-(6-methoxy-2-naphthyl)-1,3-dioxolane-4(~),
5(R)-dicarboxylic acid dimethyl ester (9.36 g, 25 mmol),
diethylamine (25 ml) and of water (20 ml)was kept, under stirring,
at room temperature for 15 hours. The solvents were removed by
evaporation.a-t room temperature under reduced pressure.
Diethylether (50 ml) was added to the residue and the mixture was
refluxed for 1 hour; then it was cooled at room temperature,
filtered and the filtrate was dried under reduced pressure.
2-ethyl-2-(6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxyl
ic acid N,N,N',N'-tetraethyl amide (11 g, 24 mmol; yield 96%) was
so obtained.
M.p.=108-112C
H-NMR 1200 MHz, CDCl3-TMS) delta (ppm): 0.83(t, 3H, J=7 Hz);
l.ll(t, 12H, J=7 Hz); 2.00(q, 2H, J=7 Hz); 2.79(q, 8H, J=7 Hz);
3.83(s, 3H); 4.32(2H, ABq, ~ ~ =17.8, J=8 Hz); 6.9-7.8(6H,
aromatlc protons).
IR (Nujol): 1605, 1630 (stretching C=0)
Exarnple 55
Preparation of 2-(1-bromoethy1)-2~(5-bromo-6-methoxy-2-naphthyl)-
1,3-dioxolane-4(R),5~R_-dicarbox~lic acid N,N,NI,N'-tetraethyl
amlde.
~1 3 ~
A mixture of the two diastereoisomers of 2~ bromoethyl)-
-2-(S-bromo-6-methoxy-2-naphtllyl)-1,3-dioxolane-4(R),5(R)-dicarboxy
lic acid dime-thyl ester 3 and 4 in ratio 3:4=9:1 (6.65
g, 12.5 mmol), diethylamine (27.5 ml) and of water (20 ml) ~as
kept, under s-tirring, a-t room temperature for 15 hours. The
solvents were removed under reduced pressure. Diethylether (50 ml)
was added to the residue~ The insoluble was filtered, washed with
diethylether, and dried under reduced pressure. The diaste-
reoisomeric mixture of 2-(1-bromoethyl)-2-(5-bromo-6-
-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarbaxylic acid
N,N,N',N'-tetraethyl amide 24 and 25 (6.75 g, 11 mmol; yield 88%),
in ratio 24:25=9:1 (determined by H-NMR, 200 MHz)
H-NMR (200 MH~, CDCl3-TMS) delta (ppm) :
diastereoisomer 24 ~RRS): 1.06(t, 12H, J=7 ~z); 1.69(d, 3H, J=7
Hz); 2.76(q, 8H, J=8 Hz); 4.00(s, 3H); 4.55(2H, ABq, ~ ~ =35.1,
J=8 Hz); 4.54(q, 2H, J=7 Hz); 7.2-8.2(5H, aromatic p~otons).
Example 56
Preparation of ?- (l-bromoethyl)-2-(5-bromo-6~methoxy-Z-naphthyl)-
1,3-dioxolane-4(R),S(R)-dicarboxylic acid disodium salt.
A mixture of the two diastereoisomers of 2-(1-bromoethyl)-
-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxy
lic acid dimethyl es-ter 3 and 4 in ratio 3.4=9:1 (6.65 g, 12.5
mmol~, sodium hydroxyde (1 g, 25 mmol), dimethoxyethane (10 ml),
and of water (10 ml) was kept, under stirring, at room temperature
for 2 hours. The reaction mixture was diluted wi-th ~a-ter and
ex-tracted with diethylether. The aqueous phase was concentrated
under reduced pressure to give the diastereoisom~ric mix-ture of
2-(1-bromoethyl)-2-(5-bromo-6-me-thoxy-2-naphthyl)-
73
-1,3-dioxolane-4(R),5tR)-dicarboxylic acid disodium salt 26 and 27
(11.5 mmol; yield 92%) in ratio 26:27=9:1 (de-termined by H-NMR
200 MHz).
Example S7
Preparation of (~)-2(S)-(5-br ~ -2-naphthyl)-propio iC
acid from a diastereoisomeric mixture o~ 2-(1-bromo_hyl)-2-(5-
bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxylic
acid 7 and 8 in ratio 7:8=93:7
. _ . . _
A mix-ture of the t~lo diastereoisomers of 2-(1-bromoethyl)-
-2-(5-bromo-6-methoxy-2-naphtyl)-1,3-dioxolane-4(R),5(R)-dicarboxyl
ic acid 7 and 8 in ratio 7:8=93:7 (9.3 g, 18.45 mmol) and of an
aqueous solution (110 ml) prepared by dissolving K2HP04 ~26.1 g)
and KH2P04 (5.7 g) in water ~384 ml) ~as heated, under stirring,
at 100C for 21 hours. The reaction mixture was cooled at room
temperature (pH 4.2), acidlfied with conc HCl to pH 1, and
extracted with diethylether (3 x 100 ml). The combined organic
extracts were washed with water and dried - over sodium
sulfate. Evaporation of the solvent under reduced pressure gave a
residue that on the basis of the GLC analysis carried out on a
sample treated with diazomethane was constituted of 2-(5-bro-
mo-6-methoxy-2-naphthyl)-propionic acid (4.33 g, 14.02 mmol; yie~
76%) and of the starting diastereoisomer 7 (1.3 g).
Purification by column chromatography of the reaction crude
(sllica gel; eluent hexane: diethylether = 7:3) gave the pure
(*)-2(S)-(5-bromo-6-methoxy-2-naphthyl)~propionic acid (4.22 g,
13~66 mmol; yield 74%) in 97% enantiomeric excess.
M.p. =168-170C
/ ~ 7D =~40.8 (c=0.5%, chloroform)
~ i 3
_ 74_
HPLC analysis, carried out as described in J.Pharm.Sci. 68, 112
(1979), showed ~n en~n-tiomeric ratio S(+):R(-)=98.5:1.5.
The er~antiomeric ratio was confirmed by H-NMR 200 MHz analysis
carried out in CDC13 using an optically active shifting agent
(europium (III) tris-L3-(eptafluoropropylhydroxymethylene)-
-d-camphorate~) on the corresponding me-thyl ester obtanined by
treating a sample of acid with diazomethane.
Example 58
A mixture of the two diastereoisomers of 2-(1-bromoethyl)-
-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxy
lic acid 7 and 8 in ratio 7:8=93:7 (2.27 g, 4.5 mmol) and of an
aqueous solu-tion (31.5 ml) prepared dissolving K2HP04 (26.1 g) and
KH2P04 (5.7 g) in water (384 ml) was heated, under stirring, at
100C for 42 hours. The reaction mixture was cooled at room
temperature (pH 4.2) and worked up as described in example 57.
(+)-2(S)-(5-bromo-6-methoxy-2-naphthyl)-propionic acid (1.32 g,
4.2 mmol; yield 93%) was obtained in g7% enantiomeric excess.
The enantiomeric ratio S(~):R(-)=98.5:1.5 was confirmed by HPLC
and by H-NMR analysis carried out as described in example 57.
Example 59
Preparation of the pure 2-(l(S)-bromoethyl)-
-2-(5-bromo~6-methoxy-2-naphthyl)-1,3-dioxola~e-4(R),5(R)-dicarboxy
lic acid (diastereoisomer 7).
_ __ ._
A mixture of the two diastereoisomers of 2-(1-bromoethyl)-
2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxyl
ic acid 7 and 8 in ratio 7(RRS):8(RRR)=g4:6 (134.42 g, 0.266 mol)
and of an aqueous solution (1726 ml) prepared dissolving K2HP04
(174 g) and KH2P04 (38 g) in water (2000 ml) was heated, under
stirring, at 90C for 14 hours. The reaction mixture was cooled at
room temperature (acidic pH), acidified with conc HCl -to pH 1, and
e~tracted with d.iethylether (3 x 150 ml). The combined organic
ex-tracts were wa~hed with water and anhydrified over sodium
sulfate. Evaporation of -the solven-t under reduced pressure gave a
residue that wa~ dried under vacuo at 80C for 12 hours. A
solution of me-thanesulfonic acid ( 1 ml) in methanol (2000 ml) was
added to the resldue (118 g)so obtained. The solution was heated
at reflux for 2 hours, cooled at room temperature, neutralized
with sodium bicarbonate.The solvent was removed under reduced
pressure and water (1000 ml) was added to the residue. The
solution was extracted with diethylether ~2 x S00 ml). The
c¢mbined organic extracts were washed with water, - dried over
sodium sulfate, and concentrated in vacuo. Purification of the
residue by column chromatography (silica gel; eluent hexa-
ne:diethyle-ther = 8:2) gave the pure 2-(l(S)-bromoethyl)-2-
-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxylic
acid dimethyl ester 3 (56 g, 0.105 mol~.
A solution of sodium hydroxyde (5.32 g, 0.133 mol) in water (70
ml) was added dropwise in 1 hour, under stirring, to a solution of
the diastereoiæomer 3 (35.4 g, 0.0665 mol) in methanol (250 ml) at
20~C. The reaction mixture was kept at 20C for 2 hours; then
methanol was removed under reduced pressure mantaining the initial
volume of the solution by addition of water~ The aqueous solution,
so obtained, was extracted with dichloromethane, acidified with
conc HCl to pH 1, and ex-tracted with diethylether (3 x 100 ml).
The combined organic extracts were washed with water, anhydrified
over sodium sulfate, filtered, and concentrated in vacuo.
~ ~?3 ~ 3~ r~i
Crystallization of the residue from dichloromethane gave the pure
2-(l(S)-bromoe-thyl)-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolarle-
4(R),5(R)-dicarboxylic acid (diastereoisomer 7).
M.p.=184-186C
- 20
H-NMR (200 MHz, esadeuteroacetone-TMS) delta (ppm): 1.68(d, 3H,
J=7 Hz); 4.03(s, 3H); 4.66(q, lH, J=7 Hz); 4.95(2H, ABq,
=3~.67 Hz, J=6.5 Hz); 7.46-8.18(m, 5H, aromaticprotons).
Example 60
Preparation of (+?-2(S)-~4-(2-methylpropyl ~ henyl~propionic_acid
A mixture of the two dias-tereoisomers of 2-(1-bromoethyl)-
-2r4-(2-methylpropyl)-phenyl/-1,3-dioxolane-4(R),51R)-dicarboxylic
acid 13 and 14 in ratio 13:14=87:13 (3.29 g, 8.2 mmol) was added
to an aqueous solution (49 ml) of K2HP04 (4.26-g)and KH2P04 (0.93
g). The solution (pH 6.6) was heated, under stirring, at 100C for
68 hours. The reaction mixture was cooled at room temperature (pH
5.5), diluted with water (100 ml), acidified with con HCl to pH 1,
and extracted with diethylether (3 x 40 ml). The organic phase was
then extracted with a I0% aqueous solution of sodium bicarbonate
(6 x 40 ml?. The combined aqueous extracts were acidified with
conc HCl to pH 1 and extracted with diethylether (3 x 50 ml). THe
combine organic extracts were washed with water, drie~ over
sodium sulfate, and concentrated in vacuo. Purification by column
chromatography (silica gel; eluen-t hexane:diethylether =8:2) gave
the pure 2~4-(2-methylpropyl)-phenyl/-propionic acid (0.28 8)-
/ ~ / 2=+47.90 (c=1%, ethanol 95%)
Example 61
d ~J
- 77 -
A mix-ture of the two diastereoisomer of 2-(1-bromoethyl)-
-2-r4--(2-methylpropyl)-phenyl7-1,3-dioxolane-4(R),5(R)-dicarboxylic
acid 13 and 14 in ratio 13:14=87:13 (3.29 g, 8.2 mmol) was added
to an aqueous solution tll5 ml)of KH2P04 (L6.4 g) and NaOH (0.82
g). The solution (pH 5) was heated, under stirring, at 100C for
90 hours.
The reaction mixture was cooled at room temperature ~pH 3.5~ and
worked up as described in example 60.
Pure 2-r4-~2-methylpropyl)-phenyl7-propionic acid (0.66 g) was
obtained.
/ ~ /D =+48.8 (c=1%, ethanol 95Yo)
Example 62
A mixture of the two diastereoisomers of 2-(1-bromoethyl)-
-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4( R ), 5 ( R ) -dicarboxy
lic acid 7 and 8 in ratio 7:8=94:6 (2.52 g, 5 mmol)was added to
an aqueous solution (70 ml) of KH2P04 tlO g) and NaOH ~1.4 g). The
solution (pH 6) was heated at 90C for 50 hours. The reaction
mixture was cooled at room temperature (pH 6.0) and worked up as
described in example 57.
Pure (+)-2(S)-(5-bromo-6-methoxy-2-naphthyl)-propionic acid (1.3
g, 4.2 mmol; yield 84%) was obtained in 90% enantiomeric excess.
M.p. ~ 16a-170~C
/ ~ ? D =*37.85 (c=0.5%, chloroform)
The enantiomeric ratio S(+):R(-)=95:5 was confirmed by HPLC and by
H~NMR analysia carried out as described in example 57.
Example 63
The pure diastereoisomer 2-(l(S)-bromoethyl)-2-(5-bromo-
-6-methoxy-2-na~hthyl)-1,3-dioxolane-4tR),5tR) dicarboxylic acid 7
t2-52 g, 5 mmol) was added to an aqueoU solution t70 ml) of
~ 3 ~ d
- 78 -
KH2P04 (10 g~ and NaOi~ (1.4 g). The solution (pH 6) was heated at
90C for 50 hours. The reaction mixture was cooled at room
temperature (pH 5.9) and worked up as described in example 57.
Pure (~)-2(S)-(5-bromo-6-methoxy-2-naphthyl)-propionic ~cid (l.OZ
g, 3.3 mmol; yield 66%)was obtained in 9E3% enantiomeric excess.
M.p.=168-170C
L~1D =+40 74 (c=0.5%, chloroform~
The enantiomeric ratio S(~):R(-)=99:1 was confirmed by HPLC and by
H-NMR carried out as described in example 57.
Example 64
Comparative example at pH higher than 7.
The pure diastereoisomer 7(RRS) (2.52 g, 5 mmol)was added to an
aqueous solution (70 ml)of KH2P04 (10 g) and rlaOH (2.5 g).
The solution (pH 7.2)was heated at 90C for 50 hours. The reaction
mixture was cooled at room temperature (pH 7.0)and worked up as
described in example 57.
Pure ~)-2(S)-(5-bromo-6-methoxy-2-naphthyl)-propionic acid (0.88
g, 2.85 mmol; yield 57%) was obtained in 78% enatiomeric excess.
M.p.=166-168C
L~ 7D =~32.58 (c=0.5%, chloroform)
The enantiomeric ratio S(+):R~-)=89:11 was confirmed by HPLC and
by H-NMR as described in example 57.
Example 65
Comparative example a-t pH higher then 7.5.
The pure diastereoisomer 7(RRS) ~2.52 g, 5 mmol) was added to an
aqueous solution (70 ml) of KH2P04 (10 g) and ~aOH (3 g).
The solution (pH 7.65)was hea-ted at 90C for 50 hours. The
reaction mixture was cooled at room temperature (pH 7.5)and worked
up as described in example 57.
~$~
- 79 -
Pure (~)-2(S)-(5-bromo~6-methoxy-2-naphU~ propionic acid (1.03
g, 3.33 mmol; yield 67%) was obtained in 74% enatiomeric excess.
M.p.=164-168C
~ ~ 7D =-~31.20 (c=0.5%, chloroform)
The enarltiomeric rat~o S(~):R(-)=~7:13 was confirmed by HPLC and
by H-NMR as described in example 57.
Example 66
A mixture of the two diastereoisomers 7(RRS) and 8(RRR)in ratio
7:8=94:6 (2.52 g, 5 mmol) was added to an aqueous solution (70 ml)
of KH2P04 (10 g) a~d NaOH (0.5 g).
The solution (pH 5.1)was heated at 90C for 52 hours. The reaction
mix-ture was cooled at room temperature (p~l 4.2)and worked up as
described in example 57.
Optically pure (-~)-2(S)-(5-bromo-6-methoxy-2-naphtyl)-propionic
acid (1.27 g, 4.11 mmol; yield 82%) was obtained.
M.p.=167-169C
rO~J D =t4Z.2~ (c=0.5%, chloroform)
The optical purity was confirmed by HPLC and by H-NMR as
described in example 57.
Example 67
The pure diastereoisomer 7(RRS) (2.52 g, 5 mmol) was added to an
aqueous solution (70 ml) of KH2P04 (10 g) and NaOH (0.5 g).
The solution (pH 5.15)was heated at 90C for 52 hours. The
reaction mixture was cooled at room temperature (pH 4.2)and worked
up as described in example 57.
Optically pure (~)-2(S)-(5-bromo-6-methoxy-Z-naphthyl)-propionlc
acid (1.30 g, 4.20 mmol; yield 84%) was obtained.
M.p.=168-170C
~ 42.2 (c=0.5%, chloroform)
~ 3 ~ L ~
- 80 ~
The optical purity was confirmed by HPLC and by H-NMR as
described in example 57.
~xample 68
The pure diastereoisomer 7(RRS~ (2.52 g, 5 mmol) was added to an
aqueous solution (35 ml) prepared dissolving KH2P04 (26.1 g) and
Kll2P04 (5.7 g) in water (384 ml).
The solution was heated at 100C for 45 hours. The reaction
mixture was cooled at room temperature (pH 4.1)and worked up as
described in example 57.
Optically pure (-~)-2(S~-(5-bromo-6-methoxy-2-naphtyl)-propionic
acid (1.3 g, 4.2 mmol; yield 84%) was obtained.
M.p.=168-170C
/ ~ ~D =+42.23 (c=0.5%, chloroform)
The optical purity was con~irmed by HPLC and by H-NMR as
described in example 57.
Example 69
A mixture of the two diastereoisomers 7(RRS) and 8(RRR)in ratio
7:8=93'7 (2.52 g, 5 mmol) was added to an aqueou~ solution (70 ml)
of KHzP04 (10 g).
The solution (pH 4.2)was heated at 90C for 50 hours. The ~eaction
mixture was cooled a-t room temperature (pH 3.2)and worked up as
described in example 57.
Pure (+)-2(S)-(5-bromo-6-methoxy-2-naphtyl)~propionic acid (0.65
g, 2.10 mmol; yield 42%) was obtained in 94% enantiomeric ~xcess.
M.p.=164 165C
r~ lD =+40.08 (c=0.5%, chloroform)
The enantiomeric ratio S(~):R(-)=97:3 was confirmed by HPLC and by
H-NMR as described in example 57.
Example 70
~ A :~
- 81 -
A solutlon of the two dlastereoisorners of 2-(l~bromoe-thyl)-
-2-(5-bromo-6-methoxy-2-naphthyl)-1,3-di oxol ane-4(R),5(R)-dicarboxy
lic acid N,N,N',N'-tetraethyl amide 24(~RS) and 25(~RR)in ratio
24:25=9:1 (2.93 g, 5 mmol) in water (70 ml)was heated a-t 90C for
50 hours. The reaction mixture was coolecl at room temperature (pH
5.6)and worked up as described in example 57.
Pure (+)-2(S)-(5-bromo-6-methoxy-2-naphtyl)~propionic acid (0.58
g) was obtained in 98% enan-tiomeric excess.
~.p.=164-165C
f ~ ~D =-~41.74 ~c=0.5%, chloroform)
The enantiomeric ra-tio S(+):R(-)=99:1 was confirmed by HPLC and by
H-NMR as described in example 57.
Example 71
A mixture of the two diastereoisomers of 2-~1-bromoethyl)-2-
-(5-bromo-6-methoxy-2-naphthyl)-1,3-dioxolane-4(R),5(R)-dicarboxyli
c acid N,N,N'!N'-tetrethyl amide 24(RRS) and 25(RRR)in ratio
24:25=9:1 (2.93 g, 5 mmol) was added to an aqueous solution (70
ml) of KH2P04 (10 g) and NaOH (0.5 g).
The solution (pH 5.7)was heated at 90C for 50 hours. The reaction
mixture was cooled at room temperature (pH 4.2)and worked up as
described in example 57.
Pure (+)-2(S)-t5-bromo-6-methoxy-2-naphtyl)-propionic acid (0.54
g) was obtained in 98% enantio0eric excess.
M.p.al66-168C
~ ~D =~41.86 (c=0.5%, chloroform)
The enantiomeric ratio St+):R(-)=99:1 was confirmed by HPLC and by
H-NMR as described in example 57.