Note: Descriptions are shown in the official language in which they were submitted.
- 1 ~ 3 2 ~
4-l6l?9/-
Process for the manufacture of novel substituted aminomethane-
di~hosphonic acids
,~ _
The invention relates to a process for the manufacture of
novel substituted aminomethanediphosphonic acids, especlally
of heteroarylaminomethanediphosphonic acids of the formula
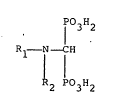
1 3 2
¦ Rl - N- CH (I)
R2 P03H2
in which R1 represents an optionally benzo- or
cyclohexeno-fused 5-membered heteroaryl radical that
contains, as hetero atom(s), either from 2 to 4 N-atoms
or 1 or 2 N-atoms as well as 1 O-atom or S-atom and that
is unsubstitutad or is C-substituted by lower alkyl;
by phenyl that is unsubstituted or ~s substituted by
lower alkyl, lower alkoxy and/or by halogen; by lower
alkoxy; by hydroxy; by di-lower alkylamlno; by lower
2 1 3 2 ~ . ~
alkylthio and/or by halogen; and/or that is N-substi-
tuted by lower alkyl; or by phenyl-lower alkyl that is
unsubstituted or is substituted by lower alkyl, lower
alkoxy and/or by halogen; and R2 represents hydrogen or
lower alkyl, with the proviso that R2 is other than
hydrogen when R1 represents a pyrazol-3-yl or isoxazol-
3-yl radical that is optionally substituted by alkyl
and/or by halogen, and of their sal~s.
Optionally benzo- or cyclohexeno-fused S-membered
heteroaryl radicals containing, as hetero atom(s),
either fro~ 2 to 4 N-atoms or 1 or 2 N-atoms as well as
1 O-atom or S-atom are, for example, imidazolyl, for
example imidazol-2-yl or -4-yl, thiazolyl, for example
thiazol-2-yl, or also thiazol-5-yl or -4-yl, oxazolyl,
for example oxazol-2-yl, or also oxazol-4 yl, triazolyl,
for example 4H-1,2,4-triazol-3-yl or 2H-1,2,3-triazol-
4-yl, tetrazolyl, for example tetrazol-5-yl, thiadia-
zolyl, for example 1,2,5-thiadiazol-3-yl, oxadiazolyl,
for example 1,3,4-oxadiazol-2-yl, benzimidazolyl, for
example benzimidazol-2-yl, benzoxazolyl, for example
benzoxazol-2-yl, or benzothiazolyl, for example
benzothiazol-2-yl. The radicals mentioned may contain
one or several identical or different, especially one or
two identical or different, substituents from among
those mentioned at the beginnin~. Radicals R1 having
substitutable N-atoms are preferably N-substituted as
indicated. Radicals R1 are, for example, 1-C1-C4-
alkylimidazol-2-yl radicals, such as 1-methylimidazol-
2-yl, 1-phenyl-C1-C4-alkylimidazol-2-yl radicals,
such as 1-benzylimidazol-2-yl, oxazol-2-yl, thiazol-2-
yl, 4- and 5-C1-C4-alkylthiazol-2-yl radicals, such as
4- or 5-methylthiazol-2-yl, 5-phenylthiazol-2-yl,
1,2,4-thiadiazol-5-yl, 3-phenyl-1,2,4-thiadiazol-5-yl,
1 3 2 ~ 3 ~ ~
1,3,4-thiadiazol-2-yl, 5-methyl-1,3,4-thiadiazol-2-yl,
benzoxazol-2-yl and benzothiazol-2-yl.
Hereinbelow, there is to be understood by lower
radicals and compounds, for example, those containing
up to and including 7, especially up to and including 4,
C-atoms. In addition, the general terms have, for
example, the following meanings:
- -~ Lower alkyl is, for example, C1-C4-alkyl, such as
methyl, ethyl, propyl or butyl, or also iso-, sec.- or
tert.-butyl, but may also be a C5-C7-alkyl group,
such as a pentyl, hexyl or heptyl group.
Phenyl-lower alkyl is, for example, phenyl-C1-C4-
alkyl, espe~ially 1-phenyl-C1-C4-alkyl, such as
benzyl.
Lower alkoxy is, for example, C1-C4-alkoxy, such
as methoxy, ethoxy, propoxy, isopropoxy, butoxy,
isobutoxy, sec.~butoxy or tert.-butoxy.
Di-lower alkylamino is, for example, di-C1-C4-
alkylamino, such as dimethylamino, diethylamino~
N-ethyl-N-methylamino, dipropylamino, N-methyl-N-propyl-
amino or dibutylamino.
Lower alkylthio is, for example, C1-C4-alkylthio,
such as methylthio, ethylthio, propylthio or butylthio,
or also iso-, sec.- or tert.-butylthio.
Halogen is, for example, halogen having an atomic
number of up to and including 35, such as fluorine,
chlorine or bromine.
Salts of compounds of the formula I are especially
the salts thereof with pharmaceutically acceptable
bases, such as non-toxic metal salts derived from metals
of groups Ia, Ib, IIa and IIb, for example alkali metal
salts, especially sodium or potassium salts, alkaline
earth metal salts, especially calcium or magnesium
salts, copper salts~ aluminium salts or zinc salts, or
ammonium salts with ammonia or organic amines or
quaternary ammonium bases, such as optionally
-- 4 --
'j 'J ~ ~!
C-hydroxylated aliphatic amines, especially mono-, di-
or tri-lower alkylamines, for example methyl-, ethyl-,
dimethyl- or diethyl-amine, mono-, di- or tri-(hydroxy-
lower alkyl)-amines, such as ethanol-, diethanol- or
triethanol-amine, tris(hydroxymethyl)amino-m~thane or
2-hydroxy-tert.-butylamine, or N-(hydroxy-lower alkyl)-
N,N-di-lower alkylamines or N-(polyhydroxy-lower alkyl)-
N-lower alkylamines, such as 2-(dimethylamino)-ethanol
or D-glucamine, or quaternary aliphatic ammonium
hydroxides, for example tetrabutylammonium hydroxide.
It should also be mentioned in this connection that
the compounds of the formula I may be in the form of
internal salts, for example of the formula
H PO3H~3
R - ~ - CH (I').
,l
2 P3H2
The mentioned compounds can accordingly also be
converted, by treatment with a strongly protonic acid,
such as with a hydrohalic acid, sulphuric acid,
sulphonic acid, for example methane- or p-toluene-
sulphonic acid, or sulphamic acid, for example
N-cyclohexylsulphamic acid, into the corresponding acid
addition salts of the formula
11 ~ 2 ~
H 13H2
Rl - ~ - CH A~ (I"),
~2 P3H2
in which A~ represents the anion of the protonic acidO
The compounds of the formula I and their salts have
valuable pharmacological propertiesO In particular,
they exhibit a pronounced regulatory action on the
calcium metabolism of warm-blooded animals. In
particular, in rats, they bring about pronounced
inhibition of bone resorption, which can be demonstrated
both in the test procedure according to Acta Endocrinol.
78, 613-24 (1975) by reference to the PTH-induced
increase in the serum calcium level after subcutaneous
administration in doses of from approximately 0.01 to
approximately 1.0 mg/kg, and in the TPTX tthyropara-
thyroidectomised) rat model by reference to the
experimental hypercalcaemia, induced by vitamin D3,
after the administration of doses of approximately from
0.001 to 1.0 mg s.c.. The tumour hypercalcaemia induced
by Walker-256-tumours is likewise inhibitsd after
peroral administration of from approximately 1.0 to
approximately 100 mg/kg. Further, in adjuvant arthritis
in rats in the test procedure according to Newbould,
Brit. J. Pharmacology 21, 127 (1963) and according to
Kaibara et al., J. Exp. Med. 1~9, 1388-96 ~1984),
they exhibit a marked inhibition of the progression of
chronic arthrltic processes in doses of approximately
from 0.01 to 1.0 mg/kg s.c.. They are therefore
excellently suitable as active ingredients in
medicaments for the treatment of illnesses that can be
- 6 - 1 ~ 21~J~
attributed to calcium metabolism disorders, for example
inflammatory processes in joints and degenerative
processes in the arthrodial cartilage, of osteoporosis,
periodontitis, hyperparathyroidism and of calcium
deposits in blood vessels or on prosthetic implants.
favourable effect is produced both in illnesses in which
an anomalous deposition of sparingly soluble calcium
salts is to be observed, such as those from among the
forms of arthritis, for example Morbus Bechterew,
neuritis, bursitis, periodontitis and tendinitis,
fibrodysplasia, osteoarthrosis and of artereosclerosis,
and in those illnesses in which an anomalous degener-
ation of hard body tissue is well to the fore, such as
hereditary hypophosphatasis, degenerative processes in
the arthrodial cartilage, osteoporoses of various
origins, Morbus Paget and osteodystrophia fibros~, and
also in tumour-induced osteolytic processes.
The invention relates especially to the manufacture
of compounds of the formula I in which Rl represents an
imidazolyl, benzimidazolyl, 2H-1,2,3- or 4H-1,2,4-tri-
azolyl, tetrazolyl, oxazolyl, benzoxazolyl, oxadiazolyl,
thiazolyl, benzothiazolyl or thiadiazolyl radical that
is C-unRubstituted or C-mono- or C-di-substituted by
lower alkyl; by lower alkoxy; by phenyl that is
unsubstituted or is mono- or di-subsl:ituted by lower
alkyl, lower alkoxy and/or by halogen; by hydroxy; by
di-lower alkylamino; by lower alkylthio and/or by
halogen; and that is unsubstituted at a substitutable
N-atom which may optionally be present or preferably
N-mono- or N-di-substituted by lower alkyl or by
phenyl-lower alkyl that is unsubstituted or is mono- or
di-substituted by lower alkyl, lower alkoxy and/or by
halogen; and R2 represents hydrogen or lower alkyl, and
of their salts, especially their internal salts and
pharmaceutically acceptable salts with bases.
1324383
The invention relates especially, for example, to
the manufacture of compounds of the formula I in which
Rl represents an imidazolyl, benzimidazolyl, 2~ 1,2,3-
or 4H-1,2,4-triazolyl, tetrazolyl, oxazolyl, benzoxazolyl,
oxadiazolyl, thiazolyl, benzothiazolyl or thiadiazolyl
radical that is unsubstituted or is mono- or di-substi-
tuted by lower alkyl; by lower alkoxy; by phenyl that is
unsubstituted or is mono- or di-substituted by lower .
alkyl, lower alkoxy and/or by halogen; by hydroxy; by
di-lower alkylamino; by lower alkylthio and/or by halogen;
and R2 represents hydrogen or lower alkyl, and of their
salts, especially their internal salts and pharmaceutically
acceptable salts with bases.
The inv~ntion rel~tes espeeially to the manufacture
of compounds of the formula I in whieh Rl represents a
thiazolyl, such as thiazol-2-yl, radical, a benzothiazol-
2-yl radieal, a thiadiazolyl, sueh as 1,2,4-thiadiazol-
5-yl or 1,3,4-thiadiazol-2-yl, radieal, an oxazolyl, sueh
as oxazol-2-yl, radical or a benzoxazol-2-yl radieal eaeh
of which is unsubstituted or is C-substituted by Cl-C4-
alkyl, such as methyl, or by a phenyl radieal that is
unsubstituted or is mono- or di-substituted by Cl-C4-.
alkyl, such as methyl, Cl-C4-alkoxy, such as methoxy,
and/or by halogen, sueh as ehlorine; or represents an
imidazolyl, such as imidazol-2-yl or imidazol-4-yl,
radieal or a henzimidazol-2-yl radieal eaeh of whieh is
unsubstituted or is C-substituted by C1-C4-alkyl, sueh
as methyl, or by a phenyl radieal that is unsubstituted
or is mono- or di-substituted by C1-C4-alkyl, such as
methyl, C1-C4-alkoxy, sueh as methoxy, and/or by
halogen, such as chlorine, and/or each of which is
N-substituted by C1-C4-alkyl, sueh as methyl, sr by a
phenyl-C1-C4-alkyl radical, such as a benzyl radical,
that is unsubstituted or is mono- or di-substituted by
C1~C4-alkyl, sueh as methyl, C1-C4-alkoxy, sueh as
methoxy, and/or by halogen, such as chlorine; and ~2
1324383
represents hydrogen, and of their salts, especially their
internal salts and pharmaceutically acceptable salts with
bases.
The invention relates more especially to the
manufacture of compounds of the formula I in which R1
represents a thiazolyl, such as thiazol-2-yl or thiazol-
4-yl, radical that is unsubstituted or is.substituted by
Cl-C4-alkyl, suc~ as methyl, by Cl-C4-alkoxy, such as
methoxy, by phenyl, by hydroxy, by di-Cl-C4-alkylamino,
such as dimethylamino or diethylamino, by Cl-C4-alkylthio,
such as methylthio, or by halogen having an atomic
number of up to and including 35, such as chlorine, and
R2 represents hydrogen, and of their salts, especially
their internal salts and pharmaceutically acceptable
salts with bases.
The invention relates even more especially to the
manufacture of compounds of the formula I in which Rl
represents a thiazolyl, such as thiazol-2-yl, radical,
a l-Cl-C4-alkyl-, such as 1-methyl-imidazol-2-yl or -4-yl,
radical, or a phenyl-Cl-C4-alkyl-, such as benzyl-
imidazol-2-yl or -4-yl, radical each of which is
unsubstituted or is C-substituted by Cl-C4-alkyl, such as
methyl, by Cl-C4-alkoxy, such as methoxy, by phenyl, by
hydroxy, by di-Cl-C4-alkylamino, such as dimethylamino or
diethylamino, by C1-C4-alkylthio, such as methylthio,
or by halogen having an atomic number of up to and
including 35, such as chlorine, and R2 represents
hydrogen, and of their salts, especially their internal
salts and pharmaceutically acceptable salts with bases.
The invention relates most especially to the
manufacture of compounds of the formula I in which Rl
represents a thiazol-2-yl radical that is unsubstituted
or is mono- or disubstituted, especially in the 4- and/or
5-position, by Cl-C4-alkyl, such as methyl, or by phenyl,
o~ represents an imidazol-2-yl or benzimidazol-2-yl
rad~cal that is unsubstituted or is mono-substituted in
1324383
g
the l-position by Cl-C4-alkyl, such as methyl, or by
phenyl-Cl-C4-alkyl, such as benzyl, respectively, or
represents an unsubstituted benzoxazol-2-yl or benzo-
thiazol-2-yl radical, and R2 represents hydrogen, and of
their salts, especially their internal salts and
pharmaceutically acceptable salts with bases.
The invention relates specifically to the manufacture
of the compounds of the formula I mentioned in the
Examples and of heir salts, especially their internal
salts and pharmaceutically acceptable salts with bases.
The invention process for the manufacture of
compounds of the formula I and to their salts is based on
methods that are known per se and is characterised in that
a3 in a compound of the formula
Rl - N - 1 - H (II)
2 2
which is optionally intermediately protected at a
substitutable N-atom of the radical R1 and in which X1
represents a functionally modified phosphono group X and
X2 represents phosphono or similarly represents a
functionally modified phosphono group X, the group(s) X
is(are) converted into free phosphono, or
b) a compound of the formula
132~383
- 10 -
R~ CH=O . ( III ),
R2
which is optionally intermediately protected at a
substitutable N-atom of the radical R1, is reacted
first with phosphorus trioxide and then with water,
and, if desired, in each case, a resulting compound is
converted into a different compound of the formula I
and/or a resulting free compound is converted into a
salt or a resulting sait is converted into the free
compound or into a different salt.
Functionally modified phosphono groups X that are
to be converted into free phosphono according to
process variant a) are, for example, in the form of an
ester, especially in the form of a diester of the
formula
-P(=O)(OR)2 (IIa)
in which OR represents, for example, lower alkoxy, or a
phenoxy group that is optionally substituted by lower
alkyl, lower alkoxy, halogen, trifluoromethyl and/or by
hydroxy.
The conversion of a functionally modified phosphono
group into a free phosphono group is effected in
customary manner by hydrolysis, for example in the
presence of a mineral acid~ such as hydrochloric or
hydrobromic acid or sulphuric acid, or by reaction with
a tri-lower alkyl-halosilane, for example with
trimethylchlorosilane or, especially, trimethyliodo-
sllane or trimethylbromosilane, preferably while
cooling, for example in a temperature range of from
132~383
approximately 0 to approximately 25C.
The starting materials of the formula II can be
manufactured, for example, by condensing a compound of
the formula
R1-N(R2)-H (IIb; R2=H)
with at least the equimolar amount of an orthoformic
acid triester of the formula
H-C(OR)3 (IIc~
in which OR represents, for example, lower alkoxy, or a
phenoxy group that is optionally substituted by lower
alkyl, lower alkoxy, halogen, trifluoromethyl and/or by
hydroxy, there probably being formed initially a
corresponding compound of the formula
R1-NH-CH(OR)2 (IId1) or R1-N=CH-OR (IId2),
and by further reactinq the condensation product with at
least double the molar amount of a phosphorous acid
diester, for example of the formula
H-p(oR)2 ~IXe),
and, if desired, lower alkylating the resulting compound
(II, R2=~l) to form the corresponding compound (II; R2=
lower alkyl)~
In intermediates II in which the radical R1 is
N-substituted by lower alkyl or by phenyl-lower alkyl
that is unsubstituted or is substituted by lower alkyl,
lower alkoxy and/or by halogen, the N-substituent can
be removed, lower al~yl being removed, for example, by
treatment with a haloformic acid ester, such as a
- 12 - 1324383
bromoformic or chloroformic acid lower alkyl ester, and
subsequent hydrolysis of the resulting carbamate, and
~-phenyl-lower alkyl radicals being remo~ed, for
example, by hydrogenolysis, for example by treatment
with hydrogen in the presence of a hydrogenation
catalyst, for example palladium-on-carbon and/or
platinum oxide, or by reduction with a metal, for
example by treatment with an alkali metal in ammoniaO
It is also possible, however, to reac~ the starting
material IIb in a manner known Per se with the
phosphorous acid diester IIe in the presence of an
orthoformic acid triester IIc without isolating the
intermediate stage. Thus, according to an especially
preferred embodiment, the corresponding compound IIb is
reacted at boiling heat in the presence of at least the
equimolar amount of an orthoformic acid triester IIc
with at least double the molar amount of the phosphorous
acid diester IIe without isolating the intermediate
stage, ~or example of the formula IId1 or IId2, and the
primary product II is hydrolysed by treatment with
aqueous hydrochloric acid at boiling heat.
The reaction of compounds III with phosphorus
trioxide according to process variant b) is preferably
effected with the latter bein~ formed in situ, for
example by reacting phosphorus trichloride and
phosphorous acid at elevated temperature, for example at
approximately from 50 to 65C, adding the reactant III,
heating further and working up the primary product, a
1:1 adduct of the aldehyde of the formula
R1-~-CH=O (III)
with phosphorus trioxide of hitherto-unknown structure,
by hydrolysis, preferably by treatment with water.
In a modification o~ this 2referred embodiment of
- 13 - 1324383
process variant b), orthophosphoric acid is reacted, at
approximately from 50C to 70C, with an approximately
1.1- to approximately 2-fold, preferably approximately
1.5-fold, excess of phosphorus trichloride, the reactant
III is added, the whole is heated for a prolonged period
at approximately from 50C to 70C, diluted with 80%
phosphoric acid and worked up by hydrolysis.
- Starting materials III can be manufactured in
customary manner, for example by reacting an amine of
the formula
1-N(R2)-H (IIb; R=H)
with formic acid or a functional carboxy derivative
thereof, for example with a formic acid ester of the
formula
H-COOR (IIe)
in which OR represents, for example, a phenoxy group
that is optionally substituted by lower alkyl, lower
alkoxy, halogen, trifluoromethyl and/or by hydroxy, or
with formamide.
For the intermediate protection of a substitutable
N-atom of the radical R1 the customary N-protecting
groups and methods of introducing and removing them are
suitable, for example 2,2,2-trihaloethoxycarbonyl
radicals, such as 2,2,2-triiodo-, 2,2,2-tribromo- or
2,2,2-trichloro-ethoxycarbonyl radicals, which can be
removed, for example, by treatment with zinc in acetic
acid, a-phenyl-lower alkoxycarbonyl radicals, such as
benzyloxycarbonyl, which can be removed, for example, by
catalytic hydrogenation, and lower alkanesulphonyl
groups, such as methanesulphonyl, which can be removed,
for example, by treatment with bis(2-methoxyethoxy)_
sodium aluminium hydride, and also, however, ~-phenyl-
- 14 - 132~383
alkyl or alkyl groups, the removal of which is dealt
with hereinafter.
Cornpounds of the formula I obtained in accordance
with the process of the invention or by another process
that is known Per se can be converted into other
compounds of the formula I in a manner known Per se.
For example, lower alkyl R2 can be introduced into
compounds of the formula I in which R2 represents
hydroqen by reaction with a reactive ester, such as a
hydrohalic acid ester or an organic sulphonic acid
ester, of a lower alkanol. It is also possible,
however, to introduce an aliphatic radical, for example
methyl, by reaction with an aliphatic aldehyde, for
example with formaldehyde and formic acid.
It is also possible in compounds of the formula I
in which the radical R1 is N-substituted by lower alkyl
or by phenyl-lower alkyl that is unsubstituted or is
substituted by lower alkyl, lower alkoxy and/or by
halogen, to remove the N-substituent, lower alkyl being
removed, for example, by treatment with a haloformic
acid ester, such as a bromoformic or chloroformic acid
lower alkyl ester, and subsequent hydrolysis of the
resulting carbamate, and ~-phenyl-lower alkyl radicals
being removed, for example, by hydrogenolysis, for
example by treatment with hydrogen in the presence of a
hydrogenation catalyst, for example palladium-on-carbon
and/or platinum oxide~ or by reduction with a metal, for
example by treatment with an alkali metal in ammonia.
Resulting free compounds of the formula I,
including their internal salts of the formula I', can be
converted into salts with bases by partial or complete
neutralisation with one of the bases mentioned at the
be~inning. Acid addition salts of the formula I" also
can be converted in an analogous manner into the
corresponding free compounds of the formula I or internal
salts of the formula I'.
1324383
- 15 -
Conversely, resulting free compounds of the formula
I can be converted into acid addition salts of the
formula I" by treatment with one of the protonic acids
mentioned at the beginning.
Resulting salts can be converted into the free
compounds in a manner known ~ se, for example by
treatment with an acid reagent~ such as a mineral acid,
or, as the case may be, with a base, for example alkali
liquor.
The compounds, including their salts, may also be
obtained in the form of their hydrates or may include
the solvent used for crystallisation.
Owing to the close relationship between the novel
compounds in free form and in the form of their salts,
hereinbefore and hereinafter there is to be understood
by the free compounds or their salts, where appropriate
and expedient, optionally also the corresponding salts
or free compounds, respectively.
The invention relates also to those embodiments of
the process according to which a compound obtainable as
an intermediate at any stage of the process is used as
starting material and the remaining steps are carried
out or a starting material in the form of a salt and/or
racemate or antipode is used or especLally is formed
under the reaction conditions.
The starting materials that are used in the process
of the present invention are preferably those which
result in the compounds described at the beginning as
being especially valuable. The invention relates also
to novel starting materials and processes for the
manufacture thereof~
1324383
- 16 -
The invention relates also to pharmaceutical
compositions containing, as the active ingredient, a known
compound of the formula I, wherein R2 represents a pyrazol-3-
yl or isoxazol-3-yl radical that is unsubstituted or mono- or
disubstituted by lower alkyl and/or halogen and R2 denotes
hydrogen, specifically l-(isoxazol-3-ylamino)methane-1,1-
diphosphonic acid, l-(4-methylisoxazol-3-ylamino)methane-1,1-
diphosphonic acid, l-(5-methylisoxazol-3-ylamino)methane-1,1-
diphosphonic acid, l-(pyrazol-3-ylamino)methane~ di-
phosphonic acid, l-t4-methylpyrazol-3-ylamino)methane-1,1-
diphosphonic acid or l-(5-methylpyrazol-3-ylamino)methane-1,1-
diphosphonic acid or a pharmaceutically acceptable salt
thereof, to the use of the active ingredient as a medicament
and to a method of treatment of illnesses associated with
calcium metabolism disorders.
The pharmaceutical preparations, which contain
compounds of the formula I or pharmaceutically
acceptable salts thereof, are for enteral, such
as oral or rectal, and parenteral administration
and contain the pharmacological active
ingredient on its own or together with a pharmaceuti-
cally acceptable carrier. The dosage of the active
ingredient depends upon the species of warm-blooded
animal, its age and individual condition and also on the
mode of administration. In a normal case, the estimated
approximate daily dose for a warm-blooded animal of
approximately 75 kg body weight is approximately from 20
to 1000 mg, preferably approximately from 30 to 300 mg,
~n the case of oral administration and approximately
from 1 to 25 mg, preferably approximately from 1 to
10 mg, in the case of intravenous administration, the
dose advantageously being dlvided into several equal
partial doses.
132~383
- 17 -
The pharmaceutical preparations contain, fGr
example, from approximately 10~ to approximately 80%,
preferably from approximately 20% to approximately 60%,
active ingredient. Pharmaceutical preparations
~ccording to the invention for enteral and parenteral
administration are, for example, those in dosage unit
form, such as dragées, tablets, capsules or supposi-
tories, and also ampoules. These are prepared in a
manner known Per se, for example by means of
conventional mixing, granulating, confectioning,
dissolving or lyophilising processes. For example,
pharmaceutical preparations ~or oral administration can
be obtained by combining the active ingredient with
solid carriers, if desired granulating a resulting
mixture and, if desired or necessary, processing the
mixture or granulate, after the addition of suitable
ad~uncts, into tablets or dragée cores.
Suitable carriers are especially fillers, such as
sugars, for example lactose, saccharose, mannitol or
sorbitol, cellulose preparations and/or calcium
phosphates, for example tricalcium phosphate or calcium
hydrogen phosphate, and binders, such as starch pastes
using, for example, corn, wheat, rlce or potato starch,
. . ~ .,
gelatine, tragacanth, methylcellulose and/or polyvlnyl-
pyrrolidone, and/or, if desired, disintegrators, such as
the above-mentioned starches, carboxymethyl starch,
crosslinked polyvinylpyrrolidone, agar, alginic acid or
a salt thereof, such as sodium alginate. Ad~uncts are
especlally flow-regulating and lubricating agents, for
example silica, talc, stearic acid or salts thereof,
- 18 - 1324383
such as magnesium or calcium stearate, and/or poly-
ethylene ~lycol. Dragée cores are provided with
suitable coatings which may be resistant to gastric
juices, there being used, nter alia, concentrated
sugar solutions which may conta~n gum arabic, talc,
polyvinylpyrrolidone, polyethylene glycol and/or
titanium dioxide, lacquer solutions in suitable organic
solvents or solvent mixtures, or, for the preparation of
coatings that are resistant to gastric juices, solutions
of suitable cellulose preparations, such as acetyl-
cellulose phthalate or hydroxypropylmethylcellulose
phthalate. Colourings or pigments may be added to the
tablets or dragée coatings, for example for iden~ifi-
cation purposes or to indicate different doses of active
ingredient.
Other orally administrable pharmaceutical
preparations are dry-filled capsules consisting of
gelatine, and also soft sealed capsules consisting of
gelatine and a plasticiser, such as glycerol or
sorbitol. The dry-filled capsules may contain the
active ingredient in the form of a granulate, for
example in admixture with fillers, such as lactose,
binders, such as starches, and/or glidants, such as talc
or magnesium stearate, and, if desired, stabilisers. In
soft capsules, the active ingredient is preferably
d~ssol~ed or suspended in suitable liquids, such as
fatty oils, paraffin oil or liquid polyethylene glycols,
it being possible also to add stabilisers.
Suitable rectally administrable pharmaceutical
1324383
-- 19 --
preparations are, for example, suppositories that
consist of a combination of the active ingredient with a
suppository base material. Suitable suppository base
materials are, for example, natural or synthetic
triglycerides, paraffin hydrocarbons, polyethylene
glycols or higher alkanols. It is also possible to use
gelatine rectal capsules that contain a combination of
the active ingredient with a base material; suitable
base materials are, for example, liquid triglycerides,
polyethylene glycols or paraffin hydrocarbons.
For parenteral administration there are suitable,
especially, aqueous solutions of an active ingredient in
water-soluble form, for example in the form of a
water-soluble salt, or suspensions of the active
ingredient, such as corresponding oily injection
suspensions in which suitable lipophllic solvents or
vehicles, such as fatty oils, for example sesame oil, or
synthetic fatty acid esters, for example ethyl oleate or
triglycerides, are used, or aqueous injection
suspensions that contain vicosity-increasing substances,
for example sodium carboxymethylcellulose, sorbitol
and/or dextran and, if desired, also stabilisers.
The present invention relates also to the use of
the compounds of the formula I and their salts,
preferably for the treatment of illnesses that can be
attributed to calcium metabolism disorders, for example
of the rheumatic type, and especially of osteoporoses.
Dosages under 0~01 mg/kg body weight have only a
negligible effect on pathological calcification or
the degeneration of hard tissue. At dosages above
100 mg/kg body weight, toxic side-effects may occur in
long-term use. The compounds of the formula I and their
salts can be adm~nistered both orally and, in the form
of a hypertonic solution, subcutaneously, intramuscul-
arly or intravenously. The preferred daily doses are in
the range of approximately from 0.1 to 5 mg/kg in the
- 20 - 1324383
case of oral administration, in the range of approxi-
mately from 0.1 to 1 mg/kg in the case of subcutaneous
and intramuscular administration and in the range of
approximately from 0.01 to 2 mg/kg in the case of
intravenous administration.
The dosage of the compounds used is, however,
variable and depends on the particular conditions, such
as nature and severity of the illness, duration of
treatment and on the particular compound. Single doses
contain, for example, from 0.01 to 10 mg, dosage unit
forms for parenteral, such as intravenous, admini-
stration contain, for example, from 0.01 to 0.1 mg,
preferably C.02 to 0.08 mg, and oral dosage unit forms
contain, for example, from 0.2 to 2.5 mg, preferably
from ~.3 to 1.5 mg, per kg of body weight. The
preferred individual dosage for oral administration is
from 10 to 100 mg and for intravenous administration
from 0.5 to 5 mg and can be administered in up to 4
single doses per day. The higher dosages in the case of
oral administration are necessary owing to the limited
resorption. In the case of long-term treatments, the
initially higher dosage can normally be converted to low
dosages while still maintaining the desired effect.
The following Examples illustrate the invention
described above; they are not intended, however, to
limit the scope thereof in any way. Temperatures are
given in degrees Celsius.
- 21 - ~32 4383
Example 1: 6.57 g (17 mmol) of 1~(thiazol-2-ylamino)-
methane-1,1-dipho~phonic acid tetraethyl ester are
dissolved in 70 ml of N hydrochloric acid and heated
under reflux for 6 hours. In the course of the
reaction, the product separates out in the form of a
fine white precipita.e. After cooling to room temper-
ature, filtration is carried out and the product is
washed with aqueous methanol. 4.33 g (93% of the
theoretical yield) of 1-(thiazol-2-ylamino)methane-
1,1-diphosphonic acid of m.p. 275 Idecomposition) are
obtained.
The starting material can be prepared, for example,
in the following manner:
A mixture consisting of 10.0 g (0.1 mol) of
2-aminothiazole, 20.0 ml (0.12 mol) of orthoformic acid
triethyl ester and 26.6 ml (0.2 mol) of diethyl
phosphite is heated under reflux for 1 hour. The
ethanol liberated is distilled off, the internal
temperature gradually increasing to approximately 150.
The residue is taken up in chloroform and filtered over
silica gel. The crude product is purified by column
chromatography (silica gel/ethyl acetate). 4.37 g (11%
of the theoretlcal yield) of 1-(thiazol-2-ylamino)-
methane-1,1-diphosphonic acid tetraethyl ester of m.p.
1~3-104 are o~ained.
ExamPle 2: In a manner analogous to that described
in Example 1 it is also possible to prepare 1-(oxazol-
2-ylamino)methane-1,1-diphosphonic acid of m.p. 245
(decomposition) and 1-(benzoxazol-2-ylamino)methane-
1,1-diphosphonic acid of m.p. 270 (decomposition).
Example 3: 4.36 g (10 mmol) of 1-~benzothiazol-2-
ylamlno)methane-1,1-diphosphonic acid tetraethyl ester
are heated in 40 ml of N hydrochloric acid at 110-120
for 6 hours. In the course of the reaction, the product
- 22 - 132~383
separates out in the form of a white precipitate. After
cooling to room temperature, filtration is carried out
and the product is washed with aqueous methanol. 3.09 g
~95% of the theoretical yield) of 1-(benzothiazol-~-yl-
amino1methane-1,1-diphosphonic acid of m.p. 290
(decomposition) are obtained.
The starting material can be prepared, for example,
in the following manner:
A mixture consisting of 3.0 g (20 mmol) of 2-amino-
benzothiazole, 4.0 ml (24 mmol) of orthoformic acid
triethyl ester and 5.3 ml (40 mmol) of diethyl phosphite
is heated at 120-125 for 5 hours~ The yellow precipi-
tate which separates out at the beginning of the
reaction gradually goes into solution again. The
ethanol liberated is distilled off during the reaction.
The crude product, which solidifies on being left to
stand, is purified by column chromatography (silica
gel/ethyl acetate/methanol). 5.62 g (64% of the
theoretical yield) of 1-(benzothiazol-2-ylamino)methane-
1,1-diphosphonic acid tetraethyl ester of m.p. 165-167
are obtained.
ExamPle 4: 1.30 g (3.2 mmol) of 1-(4-methylthia-
zol-2-ylamino)methane-1,1-diphosphonic acid tetraethyl
ester are heated in 20 ml of 1N hydrochloric acid at
100 for 20 hours. After cooling, 20 ml of methanol are
added. During subsequent stirring, the product
separates out in the form of fine white crystals. The
filtrate is subsequently washed with methanol and
petroleum ether. Yield: 615 mg (67% of the theoretical
yield) of 1-(4-methylthiazol-2-ylamino)methane-1,1-
diphosphonic acid of m.p. ~94 (decomposition).
The starting material can be prepared, for example,
in the following manner:
A mixture conslsting of 2.33 g (20 mmol) of
2-amino-4-methylthiazole, 4.0 ml (24 mmol) of
- 23 - 13243~3
orthoformic acid triethyl ester and 5.3 ml (40 mmol) of
diethyl phosphite is heated at 120-125 for 4 hours.
The ethanol liberated is distilled off. The residue is
purified by column chromatography (silica gel/ethyl
acetate/methanol). 1.32 g (17% of the theoretical
yield) of 1-(4-methylthiazol-2-ylamino)methane-1,1-
diphosphonic acid tetraethyl ester are obtained in the
form of a viscous oil.
Example 5: 1.97 g (4.9 mmol) of 1-(5-methylthiazol-
2-ylamino)methane-1,1-diphosphonic acid tetraethyl ester
are heated under reflux in 20 ml of N hydrochloric acid
for 6 hours. Upon cooling and leaving the reaction
mixture to stand at room temperature, the product
crystallises~ It is filtered and washed with acetone
and petroleum ether. Yield: 0.64 g (45% of the
theoretical yield) of 1-(5-methylthiazol-2-ylamino)-
methane-1,1-diphosphonic acid of m.p. 208 IdecomPo-
sition).
The starting material can be prepared, for example,
in the following manner:
A mixture consisting of 1.14 g (10 mmol) of
2-amino-5-methylthiazole, 2.0 ml (12 mmol) of
orthoformic acid triethyl ester and 2.65 ml (20 mmol) of
diethyl phosphite is heated at 120-125 for 4~ hours.
The ethanol liberated is distilled off. The residue is
purified by column chromatography ~silica gel/ethyl
acetate/methanol). 1.97 g ~49% of the theoretical
yield) of 1-(5-methylthiazol-2-ylamino~methane-1,1-
diphosphonic acid tetraethyl ester are obtained in the
form of a viscous oil.
ExamPle 6: 4.02 g (8.7 mmol) of 1-(5-phenylthiazol-
2-ylamino)methane-1,1-diphosphonic acid are heated under
reflux in 30 ml of N hydrochloric acid for 18 hours.
After cooling to room temperature, a small quantity of
-24 - 1324383
methanol is added and the whole is filtered. The
filtrate is heated under reflux in methanol for 1 hour,
filtered while hot and washed twice with hot methanol.
Yield: 2.90 g (95% of the theoretical yield) of 1-(5-
phenylth~azol-2-ylamino)methane-1,1-diphosphonic acid of
m.p. 290 (decomposition).
The starting material can be prepared, for example,
in the following manner:
A mixture consisting of 2.93 g (16.6 mmol) of
2-amino-5-phenylthiazole, 3.3 ml (l9.9 mmol) of
orthoformic acid triethyl ester and 4.4 ml (33.5 mmol)
of diethyl phosphite is heated first for 2 hours at 120
and then for 2 hours at 130. The ethanol liberated is
distilled off in the course of the reaction. The
product, which solidifies upon cooling, is purified by
chromatography (silica gel/ethyl acetate/methanol).
4.12 g (54% of the theoretical yield) of 1-(5-phenyl-
thiazol-2-ylamino)methane-1,1-diphosphonic acid
tetraethyl ester of m.p. 151-153 are obtained.
Example 7: 2.5 g (5.96 mmol) of 1-(1-benzimidazol-
2-ylamino)methane~ diphosphonic acid tetraethyl ester
are dissolved in 25 ml of 1N hydrochloric acid and
heated at 100-110 for 26 hours~ In the course of the
reaction, the product separates out in the form of a
fine white precipitate. It is filtered while hot and
washed with water and then with methanol. 0.23 g (13%
of the theoretical yield) of 1~ benzimidazol-2-yl-
amino)methane-1,1-diphosphonic acid of m.p. 265
(decomposition) is obtained.
The starting material can be prepared, for example,
in the following manner:
6.66 g (50 mmol) of 2-aminobenzimidazole, 10.0 ml
(60 mmol) of orthoformic acid triethyl ester and 13.3 ml
(101 mmol) of diethyl phosphite are mixed together and
then stirred for 2 hours at 125-130~ until no more
- 25 - 132~383
ethanol is distilled off. The residue is purified by
column chromatography (silica gel/ethyl acetate/-
methanol, 9:1). 2.89 g (14% of the theoretical yield)
of 1~ benzimidazol-2-ylamino)methane-1,1-diphosphonic
acid tetraethyl ester of m.p~ 169-170 are obtained.
Example 8: 7 g of phosphorus trichloride are mixed
with 4.0 g of phosphorous acid and the mixture is
heated, while stirring, at 60 for 1 hour. 6.12 g of
N-(thiazol-2-yl)formamide are added thereto and the
mixture is heated for a further 6 hours at approximately
60. The mixture is then stirred with 30 ml of water,
filtered with suction, subsequently washed with aqueous
methanol and dried u~der reduced pressure. 2.0 g of
1-(thiazol-2-ylamino)methane-1,1-diphosphonic acid of
m.p. 275 (decomposition) are obtained.
ExamPle 9: 2.0 g (20.4 mmol) of crystalline
orthophosphoric acid are stirred with 3.5 g (25.5 mmol)
of phosphorus trichloride for 1 hour at 55-60~. 4.08 g
~20.0 mmol) of N-(4-phenylthiazol-2-yl)formamide are
then added thereto. The reaction mixture is left to
stand for approximately 24 hours at 60. For dilution,
10 ml of 80% phosphoric acid are added thereto and the
whole is left to stand overnight at room temperature.
It is then heated again to 60-70 and a further 1.37 g
(10 mmol) of phosphorus trichloride are added thereto,
the whole is further stirred for 2 hours at 60-70,
30 ml of water and 20 ml of acetone are added and the
whole is stirred for 2 hours at 60 to complete the
reaction. The reaction mixture is allowed to cool to
room temperature, and the fine, pale yellow precipitate
is filtered off and washed with water/acetone 3:2~ The
residue is purified by being boiled once with water/-
acetone 1:1 and twice with methanol. 18Q mg (2.6% of
the theoretical yield) of 1-(4-phenylthiazol-2-yl-
_ 26 - 13243~3
amino)methane~ diphosphonic acid of m.p. 238
tdecomposition) are obtained.
The starting material can be prepared, for example,
in the following manner:
13.22 g (75 mmol) of 2-amino 4-phenylthiazole are
heated at 110 for 5 hours with 40 ml sf formic acid.
The reaction mixture is cooled to room temperature and
poured onto ice. The white precipitate which separates
out is filtered and washed with ice-water. The product
is purified by means of petroleum ether. 7.01 g (45.8%
of the theoretical yield) of 4-(phenylthiazol-2-yl-
aminc3formamide of m.p. 161-164 are obtained.
.
Example 10: In a manner known Per se, for example
as described in Examples 1 to 7, it is also possible to
prepare the following:
1-(imidazol-2-ylamino)methane-1,1-diphosphonic acid,
1-(imidazol-4-ylamino)methane-1,1-diphosphonic acid,
1-(1-methylimidazol-2-ylamino)methane-1,1-diphosphonic
acid,
1-(tetrazol-5-ylamino)methane-1,1-diphosphonic acid,
1-(oxazol-2-ylamino)methane-1,1-diphosphonic acid,
1-(1,3,4-thiadiazol-2-ylamino)methane-1,1-diphosphonic
acid,
1-(5-methyl-1,3,4-thiadiazol-2-ylamino)methane-1,1-
diphosphonic acid, m.p. 260 (decomposition),
1-(3-phenyl-1,2,4-thiadiaæol-5-ylamino)methane~
diphosphonic acid, m.p. 198.
ExamPle 11. Tablets, each containing 50 mg of active
ingredient, for example 1-(thiazol-2-ylamino)methane-
1,1-diphosphonic acid or a salt, for example the sodium
salt, thereof, can be prepared in the following manner:
1324383
- 27 -
Constituents (for 1000 tablets)
active ingredient 50.0 g
lactose 50.7 g
wheat starch 7.5 g
polyethylene glycol 6000 5.0 g
talc 5 0 g
magnesium stearate 1.8 g
demineralised water q.s.
Preparation: ~ll the solid ingredients are first
forced through a sieve of 0.6 mm mesh width. Then the
active ingredient, the lactose, the talc, the magnesium
stearate and half of the starch are mixed. The other
half of the starch is suspended in 40 ml of water and
this suspension is added to a boiling solution of the
polyethylene glycol in 100 ml of water. The resulting
paste is added to the pulverulent substances and the
mixture is granulated, if necessary with the addition
of water. The granulate is dried overnight at 35,
forced through a sieve of l.2 mm mesh width and com-
pressed to form tablets of approximately 6 mm diameter
which are concave on both sides.
ExamPle 12: Tablets, each containing 100 mg of active
ingredient, for example 1-(thiazol-2-ylamino)methane-
1,1-diphosphonic acid or a salt, for example the sodium
salt, thereof, can be prepared in the following manner:
Constituents (for 1000 tablets)
active ingredient 100.0 g
lactose 100.0 g
wheat starch 47.0 g
magnesium stearate 3.0 g
Preparation: All the solid ingredients are first
forced through a sieve of 0.6 mm mesh width~ Then the
active ingredient, the lactose, the magnesium
. ,
13243~3
- 28 -
stearate and half of the starch are mixed. The other
half of the starch is suspended in 40 ml of water and
t~is suspension is added to 100 ml of boiling water.
The resulting paste is added to the pulverulent sub-
stances and the mixture is granulated, if necessary with
the addition of water. The granulate is dried overnight
at 35~ forced through a sieve of 1.2 mm mesh width and
compressed to form tablets of approximately 6 mm diameter
which are concave on both sides.
Example 13: In a manner analogous to that described
in Examples 11 and 12, it is also possible to prepare
tablets each containing 100 mg or 50 mg of another of
the compounds of the formula I mentioned in Examples 1
to 10, which compounds may also be in the form of salts
with bases, for example in the form of the disodium salt.
Example 14: Tablets for chewing, each containing
75 mg of active ingredient, for example 1-(thiazol-2-
ylamino)me~hane-1,1-diphosphonic acid or a salt, for
example the sodium salt, thereof, can be prepared, for
example, in the following manner:
Com~osition: (for 1000 tablets)
active ingredient 75.0 g
mannitol 230~0 g
lactose 150.0 g
talc 21.0 g
glycine 12.5 g
stearic acid 10.0 g
saccharin 1.5 g
5% gelatine solution q.s.
Preparation: All the solid ingredients are first
forced through a sieve of 0.25 mm mesh width. The
mannitol and the lactose a e mixed, granulated with the
. ,
~32~ 83
-29 -
addition of gelatine solution, forced thxough a sieve of
2 mm mesh width, dried at 50 and again forced through a
sieve of 1.7 mm mesh width. The active ingredient, the
glycine and the saccharin are carefully mixed, the
mannitol, the lactose granulate, the stearic acid and
the talc are added and the whole is mixed thoroughly and
compressed to form tablets of approximately 10 mm
diameter which are concave on both sides and have a
breaking groove on the upper side.
In an analogous manner, it is also possible to
prepare tablets each containing 75 mg of another of the
compounds of the formula I mentioned in Examples 1 to 9,
which compounds may also be in the form of salts with
bases, for example in the form of the disodium salt.
Example 15: Tablets, each containing 10 mg of active
ingredient, for example 1-(thiazol-2-ylamino)methane-
1,1-diphosphonic acid or a salt, for example the sodium
salt, thereof, can be prepared in the following manner:
Composition (for 1000 tablets)
active ingredient 10.0 g
lactose 328.5 g
corn starch 17.5 g
polyethylene glycol 6000 S.0 g
talc 25.0 g
magnesium stearate 4.0 g
demineralised water q.s.
Preparation: The solid ingredients are first forced
through a sieve of 0.6 mm mesh widtho Then the active
ingredient, lactose, talc, magnesium stearate and half
of the starch are intimately mixed. The other half of
the starch is suspended in 65 ml of water and this
suspension is added to a boiling solution of the
polyethylene glycol in 260 ml of water. The resulting
- 30 ~ ~32~383
paste is added to the pulverulent substances, and the
whole is mixed and granulated, if necessary with ~he
addition of water. The granulate is dried overnight at
35, forced through a sieve of 1.2 mm mesh width and
compressed to form tablets of approximately 10 mm
diameter which are concave on both sides and have a
breaking notch on the upper side.
In an analogous manner, it is also possible to
prepare tablets each containing 10 mg of another
compound of the formula I according to Examples 1 to 9,
which compound may also be in the form of a salt with
a base, for example in the form of the disodium salt.
Example 16: Gelatine dry-filled capsules, each
containing 100 mg of active ingredient, for example
1-(thiazol-2-ylamino~methane-1,1-diphosphonic acid or a
salt, for example the sodium salt, thereof, can be
prepared in the following manner:
Composition (for 1000 capsules)
active ingredient 350.0 g
microcrystalline cellulose 30.0 g
sodium lauryl sulphate 2.0 g
magnesium stearate 8.0 g
The sodium lauryl sulphate is sieved into the
active ingredient (lyophilised) through a sieve of
0.2 mm mesh width and the two components are intimately
mixed for 10 minutes. The microcrystalline cellulose is
then added through a sieve of 0.9 mm mesh width and the
whole is again intimately mixed for 10 minutes.
Finally, the magnesium stearate is added through a sieve
of 0.8 mm mesh width and, after mixing for a further
3 minutes, the mixture is introduced in portions of
390 mg each into slze 0 (elongated) gelatine dry-fill
capsules.
~ 31 - 1324383
In an analogous manner, it is also possible to
prepare capsules each containing 100 mg of another
compound of the formula I according to Examples 1 to 9,
which compound may also be in the form of a salt with
a base, for example in the form of the disodium salt.
Example_17: A 0.2% injection or infusion solution
can be prepared, for example, in the following manner:
active ingredient, for example 1-(thiazol-2-ylamino)-
methane-1,1-diphosphonic acid or a salt, for example the
sodium salt, thereof 5.0 g
sodium chloride 22.5 g
phosphate buffer pH 7.4 300.0 g
demineralised water to 2500.0 ml
The active ingredient is dissolved in 1000 ml of
water and filtered through a microfilter. The buffer
solution is added and the whole is made up to 2500 ml
with water. To prepare dosage unit forms, portions of
1.0 or 2.5 ml each are introduced into glass ampoules
(each containing respectively 2.0 or 5.0 mg of active
ingredient).