Note: Descriptions are shown in the official language in which they were submitted.
1326489
FP-1662
~s is a divisi~ application of oqp~Y~s~ application serial
no. 561,556, filed Mbrch 16, 1988.
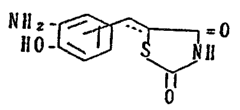
This invention relates to a novel benzoxazole
derivative, and processes for preparing same. More
S particularly, it relates to a benzoxazole derivative of the
formula:
R-Alk ~ ~ .0
~ (I)
wherein R is a substituted or unsubstituted phenyl group, a
substituted or unsub~tituted naphthyl group, a substituted or
unsubstituted cycloalkyl group or a substituted or
unsubstituted hetero cyclic group; Alk is single bond, a
fiubstituted or un~ubstituted lower alkylene group, a lower
alkenylene group or a lower alkynylene group; and the group~
is a group of the formula: -CH2- or -CH-, or a
pharmaceutically acceptable salt thereof.
--1--
1326489
A variety of biguanide derivatives and sulfonylurea
derivatives have been 50 far used for treatment of diabetes.
However, these anti-diabetic agents are still unsatisfactory
in that the biguanide and sulfonylurea derivatives cause side .
effects such a~ lactic acidosis and severe hypoglycemia,
respectively.
. The novel benzoxazole derivative (I) of the present
invention and a salt thereof are useful for therapeutic
treatment of diabetes because they elevate insulin
sensitivity and show potent hypoglycemic activity.
Examples of the benzoxazole derivative of the present
invention are those of the formula (I) in which R is phenyl
group, naphthyl group, cyclohexyl group, l,3-thiazol-4-yl
group, 1,3-oxazol-4-yl group, pyridyl group, benzoxazolyl
group, thienyl group, quinolyl group or benzofuranyl group
~these groups may optionally have a substituent or
substituents selected from a (lower alkoxy)carbonyl group, a
lower alkoxy group, a lower alkyl group, a trihalogeno-lower
alkyl group, a lower alkylthio group, a lower alkylsulfinyl
group, a lower alkylsulfonyl group, phenyl group, phenoxy
group, a phenyl-lower alkoxy group, hydroxy group, a halogen
atom, nitro group, amino group, a lower alk~noylamino group,
a di~lower alkyl)amino group, a cycloalkyl group, pyrrolidino
group, piperidino group, morphorino group and pyrrolyl
25 -group.~; and Alk is single bond, a straight or branched lower
alkylene group, a lower alkenylene group or a lower
1326~89
alkynylene group ~said lower alkylene group may optionally be
substituted with hydroxy group, oxygen atom, phenyl group or
a cycloalkyl group.~.
Among them, preferred examples of the benzoxazole
S derivative of the invention are those of the formula (I) in
which R is CD a phenyl group which may optionally have 1 to 3
substituent(s) selected from the above mentioned groups; ~ a
naphthyl group which may optionally be substitutea with a
lower alkoxy group, a lower al~yl group, a halogen atom or
10 . nitro group; C9 a cyclohexyl or pyridyl group which may
optionally be substituted with a lower alkyl group; @9a 1,3-
thiazol-4-yl or 1,3-oxazol-4-yl group which may optionally
have 1 to 2 substituent(s) selected from a lower alkyl group,
a lower alkyl~hio group, a cycloalkyl group and phenyl group;
0 a 1,3-benzoxazolyl group which may optionally be
substituted with phenyl group; 6~ thienyl group; ~ quinolyl
group; or ~D benzofuranyl group.
More preferred examples of the compound of the invention
are those of the formula (I) in which R is ~9 a phenyl group
which may optionally have 1 to 3 substituent(s) selected from
a lower alkoxy group, a lower alkyl group, a trihalogeno-
lower alkyl group, a lower alkylthio group, a lower
alkylsulfinyl group, a lower alkylsulfonyl group, phenyl
group, a halogen atom, nitro group, pyrrolidino group,
25 ..piperidino group, morphorino group, pyrrolyl group and a
di(lower alkyl)amino group; ~ a naphthyl group which may
optionally be substituted with a lower alkoxy group, a lower
-3-
i326489
alkyl group, a halogen atom or nitro group; C~ a pyridyl group
which may optionally be substituted with a lower alkyl group;
or ~D a 1,3-thiazol-4-yl or 1,3-oxazol-4-yl group which may
optionally have 1 to 2 substituent(s) selected from a lower
alXyl group, a lower alkylthio group, a cycloalkyl group and
phenyl group.
Other preferred examples of the benzoxazole derivative
of the invention are those of the formula (I) in which Alk ~s
a straight or branched lower alkylene group and/or 2,4-
dioxothiazolidin-5-yl(or ylidene)-methyl group is bound to
the benzoxazole ring at the 5- or 6-position thereof,
especially at the 5-position thereof.
In the above-mentioned examples of the benzoxazole
derivative (I), the lower alkoxy group, the lower alkyl
group, the lower alkanoyl group, the cycloalkyl group, the
lower alkylene group, the lower alkenylene group and the
lower alkynylene group include an alkoxy group of one to six
carbon atoms, an alkyl group of one to six carbon atoms, an
alkanoyl group of two to six carbon atoms, a cycloalkyl group
of three to nine carbon atoms, an alkylene group of one to
four carbon atoms, an alkenylene group of two to four carbon
atoms and an alkynylene group of two to four carbon atoms,
respectively. Preferred ex~mples of these group are an
alkyl group of one to four carbon atoms, an alkenyl, alkynyl
or alkanoyl group of two to five carbon atoms, and a
cycloalkyl group of f our to ~even carbon atoms.
1326~89
On the other hand, the benzoxazole derivatives (I) of
the present invention in which the 2,4-dioxothiazolidine
binds to the benzoxazole ring via methylene group (i.e., the
com2ound in which the group~C~is methylene group) may exist
S in the form of two optically active isomers due to the
asymmetric carbon atom thereof. On the other hand, the
benzoxazole derivative (I) of the present invention in which
the 2,4-dioxothiazolidine binds to ~enzoxazole ring via a
group of the formula: -CH= (i.e., the compound in which the
~0 group~is -CH=) may exist in the form of the two geometrical
isomers. The present invention includes within its scope
either one of these optical or geometrical isomers and a
mixture thereof.
According to the present invention, the compound ~I) can
be prepared by the step of:
(1) condensing a compound of the formula:
R-Alk -C-RI
Il (Il)
wherein Rl is hydroxy group, a lower alkoxy group or a
reactive residuel Y is oxygen ~tom or imino group, and R and
Alk are the same as defined above, or a salt thereof with a
dioxothiazolidine compound of the formula:
Nll~- ~ .0
~0
_5_
1~26~8~
wherein the symbol is the same as defined above, or a 6alt
thereof,
(2) dehydra~ing an amide of the formula:
5 R-AIk -CONH- ~ jO
wherein the symbols are the same as defined above, or a salt
thereof, or
~10 (3) dehydrogenating an azomethine compound of the formula:
R-Alk -CHjN- ~ i (V)
b
wherein the symbols are the same as defined above, or a salt
thereof.
Among the compound (I) of the invention, an olefinic
compound of the formula:
~ N ~ CH. ~ 0
wherein the symbols are the same as defined above, may also
be prepared by reacting an aldehyde compound of the formula:
N~J,~_~CH0
R-Alk ~ ~ (U)
1326489
wherein the symbols are the same as defined above, or a salt
thereof with 2,4-dioxothiazolidine or a ~alt thereof.
On the other hand, the compound of the formula:
~-AIk~ )~;0 ( I -- ~ )
wherein the symbols are the same as defined above, may be
prepared by hydroly~is of an imino compound of the formula:
10 - -
R-Alk ~ ~ 0
wherein the symbols are the same as defined above, or a salt
thereof or reduction of the compound (I-b) or a salt thereof.
The starting compound tII) in which Y is oxygen atom,
and the starting compound (III) to (V) and (VII), compound
(I-b) and 2,4-dioxothiazolidine, may, if required, be used
for the above reactions in the form of a mineral acid salt
(e.g., hydrochloride), an alkali metal salt, an alkaline
earth metal salt. The starting compound (II) in which Y is
imino group and the starting compound (VI) may, if required,
be used for the above reaction in the form of a salt with a
mineral acid te.g., hydrochloric acid).
1326489
When the group Y is oxygen atom, the condensation of the
starting compound ~I) or a salt thereof and the
dioxothiazolidine compound (III) or a ~alt thereof can be
conducted in the pre~ence or absence of a condensing agent.
S Preferred examples of the reactive residue (Rl) include
halogen atoms such as chlorine or bromine atoms.
Trimethylsilyl polyphosphate, ethyl polyphosphate and the
like are preferably used as the condensing agent. If
required, the reaction may be carried out in an inert solvent
such as dichlorobenzene, dichloromethane, tetrahydrofuran,
benzene or toluene. On the other hand, when the group Y is
imino group, the condensation of the starting compound (II)
or a salt thereof and the compound (III) can be conducted in
a solvent. Dioxane, tetrahydrofuran, ethanol or methanol
are suitable as the solvent. Either one of these reactions
may be carried out at 40 to 260 C.
The dehydration of the amide (IV) or a salt thereof can
be conducted in the presence or absence of a dehydrating
agent or an acid. It is preferred to carry out the reaction
in an inert solvent such as those mentioned above; but, if
the reaction is carried out in the absence of the dehydrating
agent or an acid, it i9 not always necessary to use a
solvent. Preferred examples of the dehydrating agent and
acid include phosphoryl chloride, thionyl chloride,
- trimethylsilyl polyphosphate, ethyl polyphosphate, p-
toluenesulfonic acid and the like. It is preferred to carry
out the reaction at SO to 250 C.
- 132648~
The dehydrogenation of the szomethine compound (V~ or a
salt thereof can be conducted in the presence of a
dehydrogenating agent. Examples of such dehydrogenating
agent include lead tetraacetate, nickel peroxide and the
like. It is preferred to carry out the reaction at O to 100
C in a solvent such as dioxane, tetrahydrofuran, benzene,
toluene, water, ethyl acetate, acetic acid, pyridine and the
mixture thereof.
The reaction of aldehyde compound (YI) or a salt thereof
and 2,4-dioxothiazolidine or a salt thereof can be conducted
in the presence or absence of a base. Piperidine, pyridine,
a tri(lower alkyl)amine, sodium hydride, sodium methylate,
sodium ethylate, lithium diisopropyl amide, etc., can be used
as a base. The solvent which is used in the condensation of
the compound (II) in which Y is imino group can be used in
this reaction. It is preferred to carry it out at O to 150
C .
Hydrolysis of the imino compound (VII) or a salt thereof
can be conducted in an inert solvent according to a
conventional manner. For example, said hydrolysis is
preferably carried out by treating the compound (VII) with an
acid such as hydrochloric acid, hydrobromic acid, hydroiodic
acid, p-toluenesulfonic acid, trifluoroacetic acid,
methanefiulfonic acid and the like. It is also preferred to
carry it out at 50 to 150 C. On the other hand, reduction
of olefinic compound (I-b) or a salt thereof can be conducted
in the presence of a catalyst in hydrogen atomosphere.
-ga-
1326~89
Palladium carbo~, palladium, platinum oxide or Raney nickel
can be used as a catalyst. It is preferred to carry it out
at 10 to 80 C. In these hydrolysis and reduction, dioxane,
tetrahydrofuran, ethanol, methanol, ethylene glycol
monomethyl ether, acetic acid and the mixture thereof are
suitable as the solvent.
Concomitantly, the benzoxazole derivative (I) of the
invention in which Alk is a lower alkylene group substituted
with oxo group may be converted into the corresponding
benzoxazole derivative (I) in which Alk is a hydroxy-lower
alkylene group. ~his conversion is carried out by reduction
of the former derivative with a reducing agent such as sodium
borohydride at 0 to 100 C in an inert solvent such as
methanol, ethanol, tetrahydrofuran and the mixture thereof.
On the other hand, the benzoxazole derivative (I) in which R
is a lower alkylsulfinyl- or lower alkylsulfonylphenyl group
may be obtained by oxidation of the benzoxazole derivative
(I) in which R is a lower alkylthiophenyl group. Said
oxidation is preferably carried out by treatment with an
oxidative agent such as m-chloroperbenzoic acid, perbenzoic
acid or peracetic acid at -70 to 100 C in an inert solvent
such as lower alkanol, methylene chloride, chloroform,
tetrahydrofuran, dioxane, water and the mixture thereof.
Alternatively, the benzoxazole derivative (I) in which R is a
hydroxyphenyl group may be obtained by debenzylation of the
benzoxazole derivative (I) in which R i8 benzyloxyphenyl
group. Said debenzylation is preferably carried out by
-9b-
-` 1326489
treatment with an acid such as hydrochloric acid or
hydrobromic acid at O to 100 C in an inert ~olvent such as
acetic acid.
_ _ _ _
--9c--
1326489
The benzoxazole derivative (I) of the present invention
and a salt thereof exhibit potent hypoglycemic activity and
are useful for treatment and/or prophylaxis of diabetes,
especially for the treatment of patients with non-insulin
dependent diabetes. Such therapeutic effect of the compound
(I) is based on the elevation of insulin sensitivity in celle
and, unlike the known anti-diabetic agents, said compound is
advantageous in that it can be used as an anti-diabetic agent
7 without affecting patients of normal blood glucose level.
Moreover, the toxicity of the benzoxazole derivative (I) of
the present invention is low. For example, when 5-~(2,4-
dioxothiazolidin-S-yl)methyl~-2-t(2-phenylthiazol-4-
yl)methyl~benzoxazole at a dose of 100 mg/~g (CMC suspension)
was orally administered to mice, no mice died during a 72
hour-observation period.
The benzoxazole derivative (I) can be used for
- pharmaceutical use either in the free form or in the form of
a salt. Suitable salts of the compound (I) for
pharmaceutical use include, for example, phamaceutically
acceptable salts such as an alkali metal salt (e.g., sodium
salt, potassium salt), an alkaline earth metal salt (e.g.,
calcium salt, magnesium salt), and acid addition salts
(hydrochloride or sulfate). Such salt may be obtained by
treating the compound (I) with a stoichiometrically equimoler
amount of the acid or base according to a conventional
manner.
--10--
~32~89
The compound (I) and a salt thereof may be administered
either orally or parenterally and may also be used in the
form of a pharmaceutical prepar~tion containing the same
compound in admixture with pharmaceutical excipients suitable
S for oral or parenteral administration. The pharmaceutical
preparation~ may be in solid form such as tablets, capsules
or suppositGries or in liquid form such as solutions,
suspensions or emulsions. Moreover, when administered
parenterally, the phamaceutical preparation may be used in
the form of injections.
The dose of the compound (I) or a salt thereof may vary
depending on the age, condition and body weight of patients,
the kind and severity of diseases to be treated and
administration route, etc, but may usually be about 0.005 to
lS about 100 mg/kg, preferably about 0.01 to about 10 mg/kg, per
day.
All of the starting compounds (III) to (VII~ of the
invention are novel. Among them, the dioxothiazolidine
compound (III) in which the group_q~ is methylene group can
be prepared by hydrolyzing a compound of the formula:
H o -@~7i
~H
and then nitrating the product with conc. niric acid,
followed by reduction with a reducing agent such as sodium
hypophosphite in the presence of a catalyst such as
, ~.
132648~
palladium-carbon at 0 to 100 C. On the other hand, the
dioxothiazolidine compound (III) in which the group,~is a
group of the formula: -CH= can be prepared by condensing 4-
hydroxy-3-nitrobenzaldehyde or 3-hydroxy-4-nitrobenzaldehyde
with 2,4-dioxothiazolidine in the presence of a base (e.g.,
piperidine), and then treating the product with a reducing
agent (e.g., sodium hypophosphite) in the presence of a
catalyst (e.g., palladium carbon). The amide (IV) can be
prepared by condensing the dioxothiazolidine compound (III)
.
and the compound (II) under a mild condition, e.g., in the
presence of a condensing agent such as
dicyclohexylcarbodiimide. The azomethine compound (V) can
be prepared by condensing the compound of the formula:
R-Alk-CIlO
wherein the symbols are the same as defined above, or a salt
thereof with the dioxothiazolidine compound (III) in the
presence or absence of a catalyst (e.g., hydrochloric acid).
Aldehyde csmpound (VI) can be prepared by dehydrating the
compound of the formula:
R-Alk - CONH- ~ CllO
HO- ~
-12a-
:~326~89
wherein the symbols are the same as defined above, or a salt
thereof according to the same condition of the-dehydration of
the compound (IV). Further, the imino compound (VI) or a
salt thereof can be prepared by diazotizing the aniline
S compound of the formula:
N NH2
~0 ~ ( ~ )
wherein the symbol~ are the same as defined above, or a salt
thereof` in the presence of hydrogen halide, reacting the
product with methyl acrylate in the presence of a copper
. _ _ . . . _ . r _ . _ _ . . . _ . _
.~
-12b-
1326489
catalyst (e.g., copper(I) oxide), and then reacting the
product with thiourea in the presence of a base such as
sodium acetate.
S Experiment
Genetically obese and diabetic mice, K~-AY (Tokyo
Laboratory Animals Science Corp., Tokyo, Japan; 1.5 ~o 11
months old), were used. Mice were divided into groups of 4
mice with roughly egual means in blood glucose level and body
weight after prefeeding powdered chow (CE-2, Clea Japan Inc.,
Tokyo, Japan). ~ice were fed ad libilum for 5 days the
powdered chow containing O.S mg% of test compound. After 5
days, blood was collected from the tail tip. Blood glucose
was enzymatically determined. Hypoglycemic activity of test
5 compound was calculated as f ollows;
blood glucose level of the medicated group
.( 1- - , ' ~xlOO
blood glucose level of the non-medicated group
The results are shown in Table 1.
-13-
-` 1326~89
Table 1
~ . ._ . __ _ _ _ .
Test compounds Hypoglycemic
activity
5-~(2,4-dioxothiazolidin-5-yl)methyl~-2- 63%
~(2-phenylthiazol-4-yl)methyl~benzoxazole
. __.
5-~(2,4-dioxothiazolidin-5-yl)methyl~-2- 52%
C(2-phenyloxazol-4-yl)methyl~ben20xazole
.
S-~(2,4-dioxothiazolidin-5-yl)methyl)-2- 49%
((S-methyl-2-cyclohexyloxazol-4-
yl)methyl3benzoxazole
.. _ .. .
S-~2,4-dioxothiazolidin-5-yl)methyl)-2- 58
~(S-methyl-2-phenyloxazol-4-
yl)methyl~benzoxazo1e
. .
-14-
- i326489
Example 1
(1) A mixture of 1.76 g of sodium nitrite in 5 ml of
water is added dropwise under ice-cooling to a mixture of
4.87 g of S-amino-2-phenylbenzoxazole, 6 ml of conc.
S hydrochloric acid and 50 ml of acetone. The mixture is
stirred at the same temperature for 10 minutes, and then 12.1
g of methyl acrylate are added thereto. l50 mg of copper(I)
oxide are added gradually to the mixture at 40 C. After
nitrogen gas evolution ceases, the mixture is kept at 35 C
for 20 minutes. Water is added thereto, and the aqueous
mixture is extracted with ethyl acetate. The extract is
washed with water, dried and evaporated to remove the
solvent. The residue is purified by silica gel column
chromatograghy (solvent; chloroform), whereby 5.13 g of
methyl 3-t2-phenylbenzoxazol-5-yl)-2-chloropropionate are
obtained as pale brown oil.
IR ~ axat(cm 1):1740
~2) A mixture of 5.13 g of the product obtained above,
2.30 g of thiourea, 1.50 g of sodium acetate and 35 ml of
ethylene glycol monomethyl ether is heated at 100 C for 8
hours. The solvent is distilled off, and water and n-hexane
are added to the residue. The precipitated crystals are
collected by filtration, washed, and dried, whereby 4.35 g of
.. . . .. . .. . . . . . .. . . .
5-t~2-imino-4-oxothiazolidin-S-yl)methyl~-2-phenylbenzoxazole
are otained as colorless powder.
-15-
- 1326~89
M.p. 281 to 283 C (decomp.)
(3) 3.18 g of the product obtained above are dissolved
in 50 ml of ethylene glycol monomethyl ether, and 2.05 g of
toluenesulfonic acid monohydrate and 6 ml of water are added
thereto. After the mixture is refluxed for 1 hour and 45
minutes, the solvent is distilled off. Water is added to
~he residue, and the solution is extracted with ethyl
acetate. The extract is washed, dried, and evaporated to
remove the solvent. The residue i5 purified by 8ilica gel
~0 column chromatography (solvent; chloroform : methanol - 20 :
1), whereby 1.83 g of 5-~(2,4-dioxothiazolidin-5-yl)methyl~-
2-phenylbénzoxazole are obtained.
M.p. 192 to 194 C
Mass~m/e):3~4(M )
IR ~Nu~ol(TM) (Cm-1) 3180,1745,1680
Examples 2 to 4
The corresponding starting compounds are treated in the
same manner as described in Example 1 to give the compounds
shown in Table 2.
Table 2
11-11 k ~ ~NN r >Il - A l k ~ O
tum' )
.. ..
( I' )
-16-
1326~8~
(wherein the group,~is methylene group)
. __ _ ___ . _
Ex. Compound~I') Properties
.
No. R-Alk-
_
o~CHJ M.p. 107 to 110C
2 - ~1ass(m/e~:344(M+)
IR*:17?0, 1685
_ _ ._
M.p. 209 to 212C
3 Cl-~ Mass(m/e) :358,360(M )
. IR :1760, 1740, 1700, 1685
_ . .... ~
M.p. 219 to 222C
4 ~ CH=CH- Mass(m/e):350(M
. IR :1740, 1680
__ ._ . . . __
: IR 3maXl(cm 1) (same in the following Examples)
Example 5
~ 1) A solution containing 2. 38 g of 2-phenyl-4-
thiazoleacetyl chloride in 10 ml of tetrahydrofuran is added
dropwise at 0 ~C to the mixture of 3.10 g of 5-(3-amino-4-
S hydroxybenzyl)-2,4-dioxothiazolidine, 3.63 g of N,N-
dimethylaniline, 25 ml of tetrahydrofuran and 5 ml of
dimethylformamide, and the mixture is stirred at room
temperature for 20 minute~. After the reaction, the mixture
i~ poured into water, and extracted with ethyl acetate. The
10 extract is washed, dried and evaporated to remove the
--17--
1326489
solvent. The residue is crystallized with ethyl acetate,
whereby 3.35 g of N-~5-(2,4-dioxothiazolidin-5-yl)methyl-2-
hydroxyphenyl~-2-phenylthiazole-4-acetamide are obtained.
Yield 76%
M.p. 227 to 22a C ~decomp.)
Mass(m/e):4391M ), 202
IR ~maU~l(cm 1):1740, 1690, 1670
(2) l.S g of the product obtained above are adaed to a
trimethylsilyl polyphosphate solution prepared from 3.2 g of
phosphorus pentaoxide, 6.6 ml of hexamethyldisiloxane, and
12.5 ml of 1,2-dichloroethane. The mixture is heated at 100
C for 30 minutes. The reaction mixture is poured into
ice-water, and extracted with ethyl acetate. The extract is
dried, and evaporated to remove the solvent. The residue is
recrystallized from methanol, whereby 880 mg of S-t(2,4-
dioxothiazolidin-5-yl)methyl)-2-t(2-phenyl-1,3-thiazol-4-
yl)methyl~benzoxazole are obtained.
Yield 61~
M.p. 86 to 89 C
Mass(m/e):421tM ), 305
IR ~NUaxl(cm 1):1750, 1690
Example 6
(1) A mixture of 2.03 g of 2-phenyl-4-oxazoleacetlc
acid, 2.38 g of 5-~3-amino-4-hydroxybenzyl)-2,4-
dioxothiazolidine, 2.06 g of dicyclohexylcarbodiimide, 2 mlof dimethylformamide and 20 ml of tetrahydrofuran is stirred
-18-
1326489
at room temperature for 18 hours. After the reaction,insoluble materials are filtered off. The filtrate is
poured into ice-water, and the solution is extracted with
ethyl acetate. The extract is washed with water, dried and
evaporated to remove the solvent. The residue is purified
by silica gel column chromatography (solvent; chloroform :
methanol = 10 : 1), whereby 2.83 g of N-~5-(2,4-
dioxothiazolidin-5-yl)methyl-2-hydroxyphenyl3-2-
phenyloxazole-4-acetamide are obtained.
Yield 67%
M.p. 197.5 to l99.S C
Mass(m/e):423(M ), 186
IR ~ma~ ¦cm ):1740, 1690, 1670
(2) 635 mg of the product obained above are heated at
230 C for 40 minutes. After cooling, the reaction product
is purified by silica gel column chromatography (solvent;
chloroform : methanol = 10 : 1), and recrystallized from
acetonitrile, whereby 383 mg of 5-~(2,4-dioxothiazolidin-5-
yl)methyl~-2-~(2-phenyl-1,3-oxazol-4-yl)methyl~benzoxazole
are obtained.
Yield 63%
M.p. 174 to 177.5 C
Mass(m/e):405(MI), 289
IR vmUjl(cm 1) 1730, 1710
Examples 7 to 30
--19--
- i326~89
The corresponding starting compounds are treated in the
same manner as described in Example 5-(1) and (2) or 6-(1)
and (2) to give the compounds shown in Table 3.
Table 3
R-AIk-llR~ t NH2- ~
~) 1,. (m ) ~
R-Alk -CON~- ~ =O
NO- ~ ~ "~
I
'
R-Alk ~ O
~ (1' )
(wherein Y is oxygen atom, Rl is hydrogen atom, and the group,~
i8 methylene group~
Ex. Compound~I') Properties
No. R-Alk-
.
.. . .. ... . M.p. 167.5 to 168C . .
7 . Cll ~ ~ Mass(m/e):359(M~)
. . . IR :1740, 1700
_ _ . . .. __ . ....... _
--~0--
1326~89
N 1- M.p. 239 to 2420C
8 ~ S~ Mass(m/e):407(M ), 291
IR :1740, 1690
__ _ -- ----- . I
M.p. 158 to 159C
9 ~-CI12- Mass(m/e):338(M ), 222
. IR :1750, 1690
_ . , .__ ---I
. M.p. 177 to 180C
lo ~NlCII z- Mass(m/e):339(M ), 223
IR :1730, 1700
_ . ...
M.p. B6 to 89C
11 ~ Cllz- Mass(m/e):344(M ), 262, 228
IR :1750, 1690
OCIIJ M.p. 79.5 to 81.0C
12 ~ Cll 2 - Mass(m/e):368(M )
IR :1760, 1745, 1700
..
M.p. 217 to 218C
13 (Cll,) 2N-~CII2- Mass(m/e):381(M )
IR :1745, 1680
. : _.. ,.. ~ .. _
. M.p. 162 to 163C
14 (CzH~) zN-~CHz- Mass(m/e):409(M+)
. IR :1760, 1700
.. . ~
M.p. 229 to 232C
15 CN ~CH~ Mass(m/e):407(M+)
. IR :1750, 1690
M.p. 205 to 208C
16 ~ - ~CHZ- Mass(m/e1:421(M+)
__ IR :1750, 1690
-21-
1326~89
_ ..
M.p. 213 to 215 C
17 0 N- ~ -CNz- Mass(m/e):423(M )
~ *
IR :1760, 1740, 1700
M.p. 161 to 171C
18 (CN~) 2N-~ Mass(m/e):381~M+)
IR :1745, 1705
. - . . .
pale yellow powder
19 ~ ~ CH 2 - Mass(m/e):421(M+)
IR :1750, 1700
_
M.p. 183 to 186C
20 ~OCN s Mass(m/e):418(M )
. IR :1750, 1700
. , . ._ . _
CNz- M.p. 201 to 202 C
21 ~CN3 Mass(m/e):402(M )
IR :1735, 1700
_ _ . ~---- --''---I
M.p. 153 to 155C
22 CH~OCO-~CHz- Mass(m/e):396(M )
IR :1740, 1725, 1705
._
M.p. 165 to 167C
C2Hs~' +
23 ~N~H~- Mass(m/e):367(M )
IR :1770, 1740, 1700
- . . .
M.p. 180 to 183C
24 ~ C - C- Mass~m/e):348~M~)
IR :2220, 1750, 1690
. .__
. M.p. 129 to 132C
ZS C~ C~ Masstm/e):402, 400(M+)
.
-22-
1326~89
CHJ M.p. 154 to 156C
26 E~ I Mass(m/e):402, 400(M )
CHI IR :175S, 1730, 1690, 1670
_ , . - ------I
N0~ M.p. 183 to 184C
27 ~ CHz- Mass(m/e):433(M )
IR :1745, 1705
__ . . . I
M.p. 158 to 160C
28 ~ O ~ CN 2 - Mass(m/e):378(M )
IR :1750, 1730, 1690
M.p. 251.5 to 254C
29 ~ C~z- Mass(m/e):389(M )
IR :1740, 1695
_ . .. _
M.p. 208 to 210C
~ ~ N ~ CHz- Mass(m/e):455(M )
IR :1740, 1700
. . . __
ExamJele 31
A mixture of 4.0 g of phosphorus pentaoxide, 10 ml of
hexamethyldisiloxane, and 20 ml of 1,2-dichlorobenzene is
refluxed for 5 minutes to give a trimethylsilyl polyphosphate
solution. Then, 1.52 g of 2-phenyl-S-methyl-4-oxazoleacetic
acid and 2.17 g of 5-(3-amino-4-hydroxybenzyl)-2,4-
dioxothiazolidine are added thereto, and the mixture is
heated at 150 C for 2 hours. The reaction mixture is
poured into ice-water, and extracted with ethyl acetate.
The extract is washed with water, dried and evaporated to ~
remove the solvent. The residue is purified by silica gel
-23-
1326~89
column chromatography (solvent; chloroform : methanol = 100 :1), and recrystallized from a mixture of ethyl acetate and
n-hexane, whereby 1.99 g of 5-~(2,4-dioxothiazolidin-5-
yl)methyl)-2-t(2-phenyl-5-methyl-1,3-oxazol-4-
yl)methyl~benzoxazole are obtained.
Yield 68%
M.p. 175 to 178 C
Mass(m/e):419(M ), 348, 303
IR vNUa~l(cm 1):1745, 1700, 1640
Examples 32 to 72
The corresponding starting compounds are treated in the
same manner as described in Example 31 to give the compounds
shown in Table 4.
Table 4
R-Alk-CR~ + NH2-~ =0
ll H0- ~ ~ ~ H
m ) ~
R-Alk ~ ~ H
(I' )~
1326~89
(part l)(wherein Y is oxygen atom, R is hydrogen atom, and
the group,~is methylene group)
Ex. Compound(I'~ Properties
No. R-Alk-
. _ _ _
~ ~ M.p. 86 to 89C
32 ~ S Mass(m/e):427(M ), 372, 359
IR :1770, 1690
N~ M.p. 146 to 148C
33 ~O~CH, Mass(m/e):425(M )
t IIJ *
~_, _ IR :1760, 1680
N~ ~ - M.p. 288 to 289C
34 ~ ~ Mass(m/e):480(M ), 458
IR sl665, 1565
_
N M.p. 174.5 to 176C
35 CN ~ Mass(m/e):391(M )
IR :1740, 1700
. __ - -- ..
N M.p. 177 to 179C
36 CNs~ Is Mass(m/e):357(M )
IR :1770, 1740, 1700
. _
M.p. 183 to 186C
37 F - ~ CH ~ - Mass(m/e):356(M~)
IR :1740, 1690
_ . _ _
M.p. 143 to 146C
38 ~CH~O-~CNr- Mass(m/e):368(M+)
IR :1760, 1740, 1710
1326~89
_
M.p. 180 to 183 C
39 CH~- ~ - Cll 2 - Mass(m/e):352(M )
IR :1760, 1740, 1710
_ . .. ~
M.p. 173 to 174C
CF3- ~ -CH2- Mass(m/e):406(M )
IR :1750, 173Q, 1690
_ ._ ._
M.p. 156.5 to 159.5C
41 Ci-~-CNz- - Mass(m/e):406, 408, 410(M )
C IR :1740, 1700
..~
M.p. 139 to 141C
42 ~CH z - . Mass(m/e):344(M )
S *
IR :1750, 1690
. .
M.p. 118 to 121C
43 NO2- ~ -CHz- Mass(m/e):383(M )
IR :1720, 1700
M.p. 75 to 78C
44 Cl- ~ CH2CN2- Mass(m/e):386, 388(M )
IR :1750, 1690
Cl M.p. 139 to 141 C
~ CH z - . Mass(m/e):372, 374(M )
IR :1740, 1700
. _
M.p. 170.5 to 172-C
46 ~CHz- Mass~m/e):372, 374(M )
IR :1745, 1700
. ... -- .... _
M.p. 194 to 197C
47 ~ ~ c b 2 - I R 1745, 1680
.
-26-
~32~89
CHz- M.p. 150 to 152.5C
48 ~ Mass(m/e):388(M )
IR :1745, 1690
M.p. 208.5 to 210.SC
49~ CH 2 - Mass(m/e):388(M )
IR :1745, 1680
.. __ I
colorless powder
50Cl- ~ CU(Clls)^ Mass(m/e):388, 386(M )
IR :1750, 1690
. .
~ colorless powder
51 ~ Cll- Mass(m/e):420(M )
IR :1750, 1695
. . ___ , ~
~ colorless powder
52 ~ ~ Mass(m/e):414(M )
IR :1750, 1690
... __ I
~ M.p. 175 to 178-C
53~ æ Cllz- Masstm/e):428(M )
IR :1740, 1690
._ -. . .. _ ----- --.
Clls colorless powder
54Cl- ~ l cU2 , Mass(m/e):416, 414(M )
llls IR :1750, 1695
__ . .. _ _ . _ ._
M.p. 137 to 140C
~C112- ~ Mass(m/e):368(M+)
CllsO IR :174~, 1705
M.p. 165 to 168C
56 C211~0-~Clli- Mass(m/e)s382(M )
_ IR sl745, 1705
-27-
- 1326~9
M.p. 155 to 157C
S7C411~0- ~ -CHz- Mass(m/e):410(M+)
IR :1750, 1690
._
M.p. 122 to 123C
58 ~ 0- ~ -CHz- Mass(m/e):430(M )
IR :1745, 1690
M.p~ 138 to 141C
59 ~ - CHsO~ ~ CHz- Mass(m/e~:444(M )
IR :1745, 1685
_ _
r-\ M.p. 163 to 166C
CH O- ~ -CHz- Mass(m/e):398(M+)
IR :1745, 1700
CHJO ~ M.p. 190 to 193C
61 CHaO (/\~CH 2 - Mass(m/e):428(M )
~= *
CHJ0 IR :1750, 1700
. .. ~
M.p. 183 to 185C
62 ~N- ~ CHz- Mass(m/e):403(M )
IR :1750, 1700
. .__ .
N(C t Hs)z foam
. 63 ~CH2- Mass~m/e):409(M )
. IR :1750, 1700
. .
CRs- M.p. 203 to 205.5.C
64 ~ Mas~(m/e):418(M )
H~ IR :1750, 1690
CHs~ M.p. 225.5 to 227.5C
~ Mass(m/e):402(M )
CHa _ IR :1745, 1685
~ -28-
- 1326489
. . .
~ CI~z- ~1.p. 214 to 216C
66 ~ Mass(m/e~:424, 422(M )
Cl IR :1750, 1680
. .
M.p. 144 to 147C
67 CH3S- ~ -Cllz- Mass(m/e):384(M )
IR :1740, 1700
: ....
M.p. 159 to 162C
68 (CH3)3C- ~ C~2- Mass(m/e):394(M )
__ IR :1760, 1740, 1700
M.p. 188 to 189C
69 ~ C0-Mass(m/e):352(M )
IR :1740, 1710, 1680
. .... _
(part 2)(wherein Y is oxygen atom, Rl is hydrogen atom, and
the group~is methine group)
.. _ . - ._
Ex. Compound(I') Properties
No. R-Al~-
..... _
N ~ M.p. 261.5 to 263C
~ 0 ~CH Mass(m/e):417(M )
IR :1740, 1720, 1700
N~ M.p. 221 to 222.5C
71 ~ S ~ Mass(m/e):419(M )
~ IR :1740, 1705
. . __ .. ... __
M.p. 218 to 219.5-C
72 C I - ~ CH 2 - Mass(m~e):372, 370~M+)
~ .. . .. _ _ ,
note): ~): sodium salt
-29-
1326489
Example 73
A mixture of 500 mg of 5-(3-amino-4-hydroxybenzyl)-2,4-
dioxothiazolidine, 405 mg of 2-phenyl-4-formylthiazole and 30
ml of ethanol is refluxed for 20 minutes, and the solvent i8
distilled off. 840 Mg of 4-(2,4-dioxothiazolidin-5-
yl)methyl-2-~(phenylthiazol-4-yl)methylidene)aminophenol
obtained as a crude product are dissolved in 40 ml of
benzene, and 1.26 g of lead tetraacetate are added thereto.
After the mixture is stirred at room temperature for lS
minutes, the solvent is distilled off. Ethyl acetate and
water are added to the residue, and the ethyl acetate layer
is washed with water, dried, and evaporated to remove the
solvent. The residue is purified by silica gel column
chromatography (solvent; chloroform : methanol = lO0 : 1),
15 whereby 400 mg of 5-t(2,4-dioxothiazolidin-5-yl)me~hyl~-2-
(2-phenyl-1,3-thiazol-4-yl)benzoxazole are obtained. The
Mass and IR data of this product are identical with those of
the product obtained in Example 8.
Example 74
(1) 9.47 g of N-(5-amino-2-hydroxyphenyl)-2-(4-
chlorophenyl)acetamide are treated in the same manner as
described in Example 1-(1) to (3) to give 5.64 g of N-tS-
(2,4-dioxothiazolidin-5-yl ? methyl-2-hydroxyphenyl)-2-(4-
chlorophenyl)acetamide as pale yellow powder.
M.p. 207 to 209 C
Mass(m/e):392, 390(M~)
-30-
~326~89
IR vma~l(cm ):1750, 1720
(2) 5.G4 g of the product obained above are heated at
220 C for 50 minutes. After cooling, the products are
purified by silica gel column chromatography (solvent;
S chloroform : methanol = 50 : 1), and recrystallized from
ether, whereby 4.0 g of 5-~(2,4-dioxothiazolidin-5-
yl)methyl)-2-(4-chlorobenzyl)benzoxazole are obtained.
M.p. 169.5 to 170.5 C
Mass(m/e):374, 372(M )
IR vma~ (cm 1):1750, 1690
Example 75
N-(5-amino-2-hydroxyphenyl)-4-hydroxy-3,5~di(tert.-
butyl)benzamide is treated in the same manner as described in
Example 74 to-give 5-((2,4-dioxothiazolidin-5-yl)methyl)-2-
~4-hydroxy-3,5-di(tert.-butyl)phenyl)benzoxazole.
M.p. 213 to 216 ~C
Mass(m/e):452(M+)
IR vmaXl(cm 1):1750, 1700
Exam~le 76
N-(4-amino-2-hydroxyphenyl)-2-(4-chlorophenyl)acetamide
i~ treated in the same manner as described in Example 74 to
give 6-~(2,4-dioxothiazolidin-5-yl)methyl)-2-~4-
chlorobenzyl~benzoxazole.
M.p. 219.5 to 220.5 C
Mass(m/e):374, 372(M+)
-31-
1326489
IR vma~l(cm 1~ :1745, 1695
Example 77
3.48 g of 5-t(2,4-dioxothiazolidin-5-yl~meth~l)-2-(4-
nitrobenzyl)benzoxazole are dissolved in a mixture of 70 ml
of tetrahydrofuran and 70 ml of methanol, and 2.5 g of 10%
palladium-carbon are added thereto. The mixture is
subjected to catalytic hydrogenation in hydrogen gas
atmosphere under atmospheric pressure. Insoluble materials
are filtered off, and the filtrate is condensed. The
residue is purified by silica gel column chromatography
(solvent: chloroform : methanol = 20 : 1) and recrystallized
from ethyl acetate, whereby 1.86 g of 5-~(2,4-
dioxothiazolidin-5-yl)methyl~-2-(4-aminobenzyl)benzoxazole
are obtained.
M.p. 180 to 183 C
Mass(m/e):353(M )
IR vma~l(cm ):1735, 1700
Example 78
A mixture of 0.99 g of 5-~(2,4-dioxothiazolidin-5-
yl)methyl)-2-(4-aminobenzyl)benzoxazole, 2 ml of acetic
anhydride and 10 ml of pyridine i8 stirred at room
temperature overnight. 10~ Hydrochloric acid i8 added
thereto, snd the solution i8 extracted with ethyl acetate.
The extract i8 washed with water, dried and evaporated to
-32-
>
132~89
remove the solvent. The residue is recrystallized from
ethyl acetate, whereby 0.56 g of 5-~(2,4-dioxothiazolidin-5-
yl)methyl~-2-(4-acetamidobenzyl)benzoxazole is obtained.
M.p. 233 to 236 C
Mass(m/e):395(M )
IR vmaUxl(cm 1):1745, 1690
Example 79
A mixture of 1.0 g of 5-~(2,4-dioxothiazolidin-S-
yl)methyl~-2-(4-methylthiobenzyl)benzoxazole, 0.58 g of 80%
m-chloroperbenzoic acid and 25 ml of methylene chloride is
stirred at room temperature for 10 minutes. The solvent is
distilled off, and ethyl a~etate is added to the residue.
The ethyl acetate solution is washed, dried and evaporated to
remove the solvent. The residue is purified by silica gel
column chromatography (solvent; chloroform : methanol = 20 :
1), whereby 0.67 g of 5-~(2,4-dioxothiazolidin-5-yl)methyl~-
2-(4-methylsulfinylbenzyl)benzoxazole are obtained as
colorless foam.
Yield 64 %
Mass(m/e):400~M~)
IR vmUa~l~cm 1):1750, 1690
Exam~le 80
A mixture of 1.2 g of 5-~(2,4-dioxothiazolidin-5-
yl)methyl~-2-(4-methylthiobenzyl)benzoxazole, 2.1 g of 80%
m-chloroperbenzoic acid and 30 ml of methylene chloride is
stirred at room temperature for 20 minutes. The reaction
-33-
~` 1326489
mixture is treated in the same manner as described in Example79, and recrystallized from a mixture of tetrahydrofuran and
n-hexane, whereby 1.2 g of 5-~(2,4-dioxothiazolidin-5-
yl)methyl)-2-(4-methylsulfonylbenzyl)benzoxazole are obtained
S as colorless powder.
Yield 69 %
M.p. 168 to 169 C
Example 81
1.3 g of S-t(2,4-dioxothiazolidin-S-yl)methyl)-2-
(benzoyl)benzoxazole are dissolved in a mixture of 30 ml ofmethanol and 6 ml of tetrahydrofuran, and 0.14 g of sodium
borohydride is added thereto. After the mixture is stirred
at room temperature for 5 minutes, water is added thereto.
The solution is extracted with ethyl acetate, and the extract
is evaporated to remove the solvent. The residue is
crystallized with ether, whereby 0.64 g of 5-~(2,4-
dioxothiazolidin-5-yl)methyl)-2-(a-hydroxybenzyl)benzoxazole
i5 obtained as colorless crystal.
M.p. 210 to 212 C (decomp.)
Example 82
A mixture of O.9S g of 5-((2,4-dioxothiazolidin-S-
yl)methyl~-2-(4-benzyloxybenzyl)benzoxazole, 10 ml of a 25 ~
hydrogen bromide solution in acetic acid and 10 ml of acetic
acid is stirred at room temperature overnight. Ethyl
acetate and water are added to the reaction mixture. The
ethyl acetate layer is washed with water, dried and
evaporated to remove the solvent. The residue is
-34-
1326~89
crystallized with n-hexane, and recrystallized from a mixture
of ethyl acetate and n-hexane, whereby 0.42 g of 5-~2,4-
dioxothiazolidin-5-yl)methyl)-2-(4-hydroxybenzyl)benzoxazole
is obtained as colorless needles.
S Yield 55 %
M.p. 201 to 204 C
Mass(m/e):354(M )
IR vNU~l(cm~l):3340 1730 1690
Example 83
(1) 5-(3-amino-4-hydroxybenzyl)-2,4-dioxothiazolidine
and 4-(N,N-dimethylamino)phenylacetyl chloride, prepared from
4-(N,N-dimethylamino)phenylacetic acid and oxalyl chloride,
are treated in the same manner as described in Example 5-(1)
to give N-~5-(2,4-dioxothiazolidin-5-ylImethyl-2-
lS hydroxyphenyl)-2-(4-dimethylaminophenyl)acetamide.
M.p. 215.5 to 217.5 C
~ 2) A mixture of 6.0 g of the product obtained above,
0.3 g of p-toluenesulfonic acid monohydrate and 35 ml of
diethylaniline is refluxed for l.S hours. After cooling,
the precipitated crystals are collected by filtration. The
crystals are washed and recrystallized from a mixture of
tetrahydrofuran and methanol, whereby 4.9 g of 5-~2,4-
dioxothiazolidin-5-yl)methyl~-2-(4-
dimethylaminobenzyl)benzoxazole are obtained.
Yoeld 78 ~
M.p. 217 to 218 C
1326489
~Preparation of Starting Compounds)
Preparation 1
- (1) A solution of 6.16 g of 2-amino-4-nitrophenol and
4.77 g of benzaldehyde in ethanol is refluxed. The reaction
solution is condensed and cooled. The crystalline
precipitates are collected by filtration to give 2-
benzylideneamino-4-nitrophenol. 15.5 g of lead tetraacetate
are added to a benzene suspension of the product obtained
above, and the mixture is stirred. After the reaction,
insoluble materials are filtered off. The filtrate is
washed, and condensed. The residue is purified by silica
gel column chromatography and recrystallized from ethanol,
whereby 5.08 g of 5-nitro-2-phenylbenzoxazole are obtained.
M.p. 169 to 171.5 C
(2) 3 g of 10% palladium-carbon are added to an acetic
acid solution of 8.85 g of the product obtained above, and
the mixture is subjected to catalytic hydrogenation in
hydrogen gas atmosphere. Insoluble materials are filtered
off, and the filtrate is condensed. The residue is
recrystallized from a mixture of chloroform and n-hexane,
whereby 7.02 g of 5-amino-2-phenylbenzoxazole are obtained.-
M.p. 150.5 to 153 C
Preparation 2
(1) 16.0 g of ~l-methylcyclohexyl)carbonyl chloride are
added dropwise to a tetrahydrofuran solution of 14.7 g of 2-
amino-4-nitrophenol and 12.1 g of N,N-dimethylaniline under
ice-cooling, and the mixture is stirred at room temperature.
-36-
- 1326489
10% Hydrochloric acid is added to the reaction mixture, and
the crystals are collected by filtration. The crystals are
added to 260 ml.of thionyl chloride, then the mixture is
refluxed. Thionyl chloride which remains unreacted is
distilled off, and the residue is purified by silica gel
column chromatography, whereby 21.7 g of 2~
methylcyclohexyl)-5-nitrobenzoxazole are obtained.
M.p. 57 to 59 C
(2) The product obtained above is treated in the same
manner as described in Preparation 1-(2) to give 5-amino-2-
(l-methylcyclohexyl)benzoxazole as a colorless oil.
Mass(m/e):230(M )
Preparations 3 and 4
The corresponding starting compounds are treated in the
same manner as described in Preparation 1 or 2 to give the
compounds shown in Table 5.
Table 5
N NHr
~0
.
25 Pr. Compound(V m ') Properties
No. R Alk-
__
. -37-
- 1326489
¦ 3 ¦ ~ ¦M.P. 177 to 180C
. ~
4 ~ -CH=CII- M.p. 148 to 150.5C
Preparation 5
(1) A mixture of 8.55 g of 4-chlorophenylacetic acid and
35 ml of thionyl chloride is heated. After the reaction,
thionyl chloride is distilled off. The residue is added to
a tetrahydrofuran solution of 7.70 g of 2-amino-4-nitrophenol
and 6.65 g of N,N-dimethylaniline, and the mixture is stirred
at room temperature. The solvent is distilled off, and
diluted hydrochloric acid is added to the residue. Then,
the crystals are collectecd by filtration, whereby 14.2 g of
N-(5-nitro-2-hydroxyphenyl)-2-(4-chlorophenyl)acetamide are
obtained.
M.p. 250 to 252 C (decomp.)
(2) 13.93 g of the product obtained above are added to a
ethanol-ethyl acetate suspension of tin(II) chloride, and the
mixture is heated. After the reaction, the solvent i8
distilled off, and the residue is neutralized with sodium
hydroxide. The mixture is extracted with ethyl acetate.
The extract is evaporated to remove the solvent, and the
residue i~ recrystallized from ethyl acetate, whereby 9.86 g
of N-(s-amino-2-hydroxyphenyl)-2-(4-chlorophenyl)acetamide
are obtained.
-38-
~326489
M.p. 164 to 167 C
Preparation 6
4-hydroxy-3,5-di(tert.-butyl)benzoic acid and 2-amino-
4-nitrophenol are treated in the same manner as described in
5 Preparation 5-(1) and Preparation 1-(2) to give N-(5-amino-
2-hydroxyphenyl~-4-hydroxy-3,5-di(tert.-butyl)benzamide are
obtained.
M.p. 222 to 225 C (decomp.)
Preparation 7
(1) 4-aminophenol is treated in the same manner as
described in Example 1-(1) to (3) to give 5-(4-
hydroxybenzyl)-2,4-dioxothiazolidine.
M.p. 157 to 158.5 C
(2) 5.19 g of the product obtained above are added to 50
15 ml of 7096 nitric acid under ice-cooling, and the mixture is
stirred for 5 minutes. The reaction mixture is poured into
ice-water, and extracted with ethyl acetate. The extract is
washed with water, dried, and evaporated to remove the
solvent. The residue is recrystallized from a mixture of
20 ethyl acetate and n-hexane, whereby 4.85 g of 5-~4-hydroxy-
3-nitrobenzyl)-2,4-dioxothiazolidine are obtained.
Yield 78 %
M.p. 141 to 143.5 C
(3) 10% palladium-carbon, 4M-aqueous sodium
25 hypophosphite solution and water are added to a
dimethylformamide solution of 1.0 g of the product obtained
above, and thq mixture is stirred at room temperature.
--39--
- 1326489
After the reaction, insoluble materials are filtered off.
Water is added to the filtrate, and the solution is extracted
with ethyl acetate. The extract is evaporated to remove the
solvent, and the residue is recrystallized from
isopropylalcohol, whereby 7S5 mg of S-(3-amino-4-
hydroxybenzyl)-2,4-dioxothiazolidine are obtained.
M.p. 215 to 217.5C (decomp.)
Preparation 8
(1) A mixture of 18.0 g of 4-hydroxy-3-
nitrobenzaldehyde, 12.74 g of 2,4-dioxothiazolidine, 2.2 ml
of piperidine and 180 ml of dioxane is refluxed for 13 hours.
After cooling, 100 ml of water and 10 ml of 10% hydrochloric
acid are added thereto. The crystalline precipitates are
collected by filtration, dried, and recrystallized from a
mixture of tetrahydrofuran and n-hexane, whereby 14.8 g of
5-(4-hydroxy-3-nitrobenzylidene)-2,4-dioxothiazolidine are
obtained.
Yield 52 %
M.p. 256.5 to 258 C
(2) 12.85 g of the product obtained above are treated in
the same manner as de~cribed in Preparation 7-~3) and the
filtrate is poured into water. The crystalline precipitates
are collected by filtration, whereby 10.86 g of 5-(3-amino-
4-hydroxybenzylidene)-2,4-dioxothiazolidine are obtained.
Yield 95 ~
M.p. 260.5 to 261.5 C
Preparation 9
.
1326~89
(1) A solution of 13.3 g of L-4-hydroxy-3-nitro-
phenylalanine and 35 g of potassium bromide in 170 ml of an
aqueous 3N-sulfuric acid is cooled in ice-bath. Then, a
solution of 4.95 g of sodium nitrite in 10 m of water is
added dropwise to the mixture for 30 minutes~ After the
reacion at the same temperature for 10 minutes, the mixture
is extracted with e~hyl acetate. The ethyl acetate layer is
washed with water, dried and evaporated to remove the
solvent, whereby 15.2 g of 2-bromo-3-(4-hydroxy-3-
nitrophenyl)propionic acid are obtained as brown solid.
(2) A mixture of 15.1 g of the product obtained above,6.14 g of thiourea, 5.51 g of sodium acetate and 150 ml of
ethanol is refluxed for 3 hours. The solvent is distilled
off, and water is added to the residue. The precipitates
are collected by filtration, washed, and dried, whereby 12.2
g of 5-~4-hydroxy-3-nitrobenzyl)-2-imino-4-oxothiazolidine
are obtained as yellow powder.
M.p. 221 to 223 C (decomp.)
(3) A mixture of 12.1 g of the product obtained above,
15.6 ml of conc. hydrochloric acid, 125 ml of ethylene glycol
monomethyl ether and 12.5 ml of water is refluxed for 4
hours. The reaction mixture is condensed and the residue is
extracted with ethyl acetate. The ethyl acetate layer is
washed with water, dried, and evaporated to remove the
solvent. The residue is recrystallized from a mixture of
. .
- , .
1326489
ethyl acetate and n-hexane, whereby 10.4 g of 5-(4-hydroxy-
3-nitrobenzyl)-2,4-dioxothiazolidine are obtained as yellow
powder.
M.p. 141 to 143.S C
S (4) 10.3 g of the product obtained above are dissolved
in 170 ml of a mixture of tetrahydrofuran and methanol, and
2.5 g of lOg palladium-carbon are added thereto. The
mixtutre is subjected to catalytic hydrogenation at
atmospheric pressure. The catalyst is filtered off, and
filtrate is condensed. Isopropyl alcohol is added to the
residue, whereby 8.77 g of 5-(3-amino-4-hydroxybenzyl)-2,4-
dioxothiazolidine are obtained as pale yellow powder.
.p. 215 to 217.5 C
-42-