Note: Descriptions are shown in the official language in which they were submitted.
13296G9
4-16803/+
Araliphatylaminoalkanediphosphonic acids
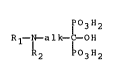
The invention relates to araliphatylaminoalkanediphosphonic acids of
formula
lpO3H2
R~ alk- -OH (I),
R2 03H2
wherein Rl is an aromatically substituted aliphatic radical, R2 is
hydrogen or a monovalent aliphatic radical and alk is a divalent ali-
phatic radical, and their salts, to processes for the preparation of the
compounds of the invention, to pharmaceutical compositions containing
them and to their use as active ingredients in medicaments.
Aromatically substituted aliphatic radicals Rl are, for example, lower
alkyl or lower alkenyl radicals that are substituted by at least one
aromatic or heteroaromatic radical. Aromatic and heteroaromatic substi-
tuents are, for example, 5- or 6-membered monocyclic aryl radicals, or
bicyclic aryl radicals composed of 5- or 6-membered rings, or 5- or
6-membered monocyclic heteroaryl radicals, or bicyclic heteroaryl
radicals composed of 5- or 6-membered rings, which heteroaryl radicals
contain as hetero atom(s) 1 or 2 N-atoms, 1 O-atom or S-atom, 1 N-atom
and 1 O-atom, or 1 N-atom and 1 S-atom, such as phenyl or, secondly,
naphthyl or pyrrolyl, thienyl, furyl, pyridyl, imidazolyl, pyrimidinyl or
quinolinyl, especially phenyl, thienyl, pyridyl or imidazolyl, each of
which aryl and heteroaryl radicals is unsubstituted or, especially, is
mono- or poly-substituted, preferably mono- or, secondly, di-substituted,
by lower alkyl, lower alkoxy, lower alkylthio and/or by halogen. The
radical Rl may be substituted by one or more than one such substituent
R3, for example by one substituent R3 or by two identical or different
substituents R3. These are preferably bonded by way of a carbon atom but
may also be bonded by way of an additional nitrogen atom which may be
132~6~9
present. If Rl has more than one such substituent, preferably two of
these are bonded to the same carbon atom of the aliphatic radical.
Preferred radicals Rl are those of formula
R~
/CH-alk'- (Ia~,
R4
wherein R3 is an aromatic radical RA, R4 is hydrogen or an aromatic
radical RB and alk' is lower alkylene, RA and RB each having one of the
meanings given for R and the sum of the carbon atoms of alk' preferably
not exceeding 6.
Monovalent aliphatic radicals are, for example, lower alkyl or lower
alkenyl radicals, and divalent aliphatic radicals are especially lower
alkylene radicals.
Hereinafter there are to be understood by lower radicals and compounds,for example, those radicals and compounds having up to and including 7,
especially up to and including 4, carbon atoms. The general terms also
have, for example, the following meanings:
Lower alkyl is, for example, Cl-C4alkyl, such as methyl, ethyl, propyl,isopropyl or butyl, and also isobutyl, secondary butyl or tertiary butyl,
but may also be a Cs-C7alkyl group, such as a pentyl, hexyl or heptyl group.
Lower alkoxy is, for example, Cl-C4alkoxy, such as methoxy, ethoxy,
propoxy, isopropoxy or butoxy, and also isobutoxy, secondary butoxy or
tertiary butoxy.
Lower alkylthio is, for example, C1-C4alkylthio, such as methylthio,
ethylthio, propylthio or butylthio, and also isobutylthio, secondary
butylthio or tert~ary butylthio.
Halogen is, for example, halogen having an atomic number of up to and
including 35, such as chlorine or fluorine, and also bromine.
_ 3 _ ~ ~ 2 9 6 0 9
Lower alkenyl is, for example, Cz-C4alkenyl, such as vinyl, allyl or
buten-2-yl, but may also be a Cs-C7alkenyl group, such as a pentenyl,
hexenyl or heptenyl group.
Lower alkylene alk is, for example, Cz-C4alkylene, especially straight-chained C2-C4alkylene, such as ~,~-C2-C4-alkylene, for example ethylene,
1,3-propylene or, secondly, 1,4-butylene.
.
Lower alkylene alk' is, for example, C2-C6alkylene, especially straight-
chained C2-C6alkylene, such as ~,~-C2-C6alkylene, for example ethylene,
1,3-propylene, 1,4-butylene, 1,5-pentylene or 1,6-hexylene, but may also
be 1,2-propylene, 1,2- or 1,3-butylene or 1,4-pentylene.
Salts of compounds of formula I are especially internal salts thereof and
salts thereof with pharmaceutically acceptable bases, such as non-toxic
metal salts derived from metals of groups Ia, Ib, IIa and IIb, for
example alkali metal salts, especially sodium or potassium salts,
alkaline earth metal salts, especially calcium or magnesium salts, copper
salts, aluminium salts or zinc salts, or ammonium salts with ammonia or
organic amines or quaternary ammonium bases, such as optionally
C-hydroxylated aliphatic amines, especially mono-, di- or tri-lower
alkylamines, for example methyl-, ethyl-, dimethyl- or diethyl-amine,
mono- di- or tri-(hydroxy-lower alkyl)-amines, such as ethanol-,
diethanol- o~ triethanol-amine, tris(hydroxymethyl)amino-methane or
2-hydroxy-tert.-butylamine, or N-(hydroxy-lower alkyl)-N,N-di-lower
alkylamines or N-(polyhydroxy-lower alkyl)-N-lower alkylamines, such as
2-(dimethylamino)-ethanol or D-glucamine, or quaternary aliphatic
ammonium hydroxides, for example tetrabutylammonium hydroxide. These
salts include both complete salts and partial salts, i.e. salts with 1,
2, 3 or 4, preferably 2, equivalents of base per mole of the acid of
formula I.
The compounds of formula I and their salts have valuable pharmacological
properties. In particular, they exhibit a pronounced regulatory action on
the calcium metabolism of warm-blooded animals. In particular? in rats,
_ 4 _ ~3~96~3
they effect a marked inhibition of bone resorption, which can be demon-
strated both in the test procedure according to Acta Endocrinol. 78,
613-24 (1975) by reference to the PTH-induced increase in the serum
calcium level after subcutaneous administration in doses of from approxi-
mately 0.01 to approximately 1.0 mglkg, and in the TPTX
(thyroparathyroidectomised) rat model by reference to the experimental
hypercalcaemia, induced by vitamin D3, after the administration of doses
of approximately from 0.001 to 1.0 mg s.c.. The tumour hypercalcaemia
induced by Walker-256-tumours is likewise inhibited after peroral
administration of from approximately 1.0 to approximately 100 mg/kg.
Further, in adjuvant arthritis in rats in the test procedure according to
Newbould, Brit. J. Pharmacology 21, 127 (1963) and according to Kaibara
et al., J. Exp. Med. 159, 1388-96 (1984), they exhibit a marked
inhibition of the progression of chronic arthritic processes in doses of
approximately from 0.01 to 1.0 mg/kg s.c.. They are therefore eminently
suitable as active ingredients in medicaments for the treatment of
diseases that may be associated with calcium metabolism disorders, for
example inflammatory processes in joints and degenerative processes in
the articular cartilage, of osteoporosis, periodontitis,
hyperparathyroidism and of calcium deposits in blood vessels or on
prosthetic implants. A favourable effect is produced both in diseases in
which an anomalous deposition of sparingly soluble calcium salts is to be
observed, such as diseases from among the forms of arthritis, for example
~rechterew's disease, neuritis, bursitis, periodontitis and tendinitis,
fibrodysplasia, osteoarthrosis and arteriosclerosis, and in diseases in
which an anomalous degeneration of hard body tissue is the principal
symptom, such as hereditary hypophosphatasia, degenerative processes in
the articular cartilage, osteoporoses of various origins, Paget's disease
and osteodystrophia fibrosa, and also in tumour-induced osteolytic
processes.
The invention relates, for example, to compounds of formula I wherein R1,
R2 and alk have the meanings given at the beginning, with the proviso
that in compounds of formula I wherein R1 is monosubstituted by phenyl,
1329609
Rz is hydrogen or, if Rl contains 2 or 3 carbon atoms in the aliphatic
moiety, is an aliphatic radical containing at most 3 carbon atoms, and
their salts.
The invention relates especially to compounds of formula I wherein Rl is
a lower alkyl or lower alkenyl radical that is mono- or di-substituted by
a 6-membered monocyclic aryl radical or by a bicylic aryl radical com-
posed of 5- or 6-membered rings or by a 5- or 6-membered monocyclic
heteroaryl radical or by a bicyclic heteroaryl radical composed of 5- or
6-membered rings, which heteroaryl radicals contain as hetero atom(s) 1
or 2 N-atom(s), 1 O-atom or 1 S-atom, 1 N-atom and 1 O-atom, or 1 N-atom
and 1 S-atom, and which aryl and heteroaryl radicals are unsubstituted or
substituted by lower alkyl, lower alkoxy, lower alkylthio and/or by
halogen, R2 is hydrogen, lower alkyl or lower alkenyl, and alk is lower
alkylene, for example those wherein R2 is hydrogen or Cl-C3alkyl and R1
in the aliphatic moiety contains 2 or 3 carbon atoms if Rl is substitused
by ~nsubstituted phenyl, and their salts, especially their pharma-
ceutically acceptable salts.
The invention relates especially, for example, to compounds of formula I
wherein Rl is a lower alkyl radical that is mono- or di-substituted by a
6-membsred monocyclic aryl radical or by a bicyclic aryl radical composed
of 5- and/or 6-membered rings, which aryl radicals are unsubstituted or
substituted by lower alkyl, lower alkoxy, lower alkylthio and/or by
halogen, or is a lower alkyl radical that is mono-substituted by a 5- or
6-membered monocyclic heteroaryl radical that contains as hetero atom(s)
1 O-atom or S-atom or 1 or 2 N-atoms, R2 is hydrogen or a lower alkyl
radical, with the proviso that R2 is hydrogen if Rl is an unsubstituted
benzyl- or phenyl-C3-C7alkyl radical, or that R2 is hydrogen or lower
alkyl containing 1 or 2 carbon atoms if Rl is a phenethyl radical, and
alk is lower alkylene, and their salts.
The invention relates especially to compounds of formula I wherein Rl is
a radical of formula (Ia) wherein R3 is an aromatic radical RA, R4 is
hydrogen or an aromatic radical RB and alk' is lower alkylene, RA and RB
1329609
-- 6 --
each being a 5- or 6-membered monocyclic aryl radical or a bicyclic aryl
radical composed of 5- or 6-membered rings or a 5- or 6-membered mono-
cyclic heteroaryl radical or a bicyclic heteroaryl radical composed of 5-
or 6-membered rings, which heteroaryl radicals contain as hetero atom(s)
1 or 2 N-atom(s), 1 O-atom or S-atom, 1 N-atom and 1 O-atom or 1 N-atom
and 1 S-atom, and which aryl and heteroaryl radicals are unsubstituted or
substituted by lower alkyl, lower alkoxy, lower alkylthio and/or by
halogen, R2 is hydrogen, lower alkyl or lower alkenyl, and alk is lower
alkylene, for example those wherein alk' is Cz-C310wer alkylene and R2 is
hydrogen or C1-C3alkyl if R3 is unsubstituted phenyl and R4 is hydrogen,
and their salts, especially their pharmaceutically acceptable salts.
The invention relates more especially, on the one hand, to compounds offormula I wherein R1 is a radical of formula (Ia) wherein R3 is a phenyl,
thienyl, pyridyl, imidazolyl or naphthyl radical that is unsubstituted or
mono- or di-substituted by C1-C4alkyl, such as methyl, C1-C4alkoxy, such
as methoxy, C1-C4alkylthio, such as methylthio, and/or by halogen having
an atomic number of up to and including 35, such as fluorine or chlorine,
R4 is hydrogen or, if R3 is a phenyl radical that is unsubstituted or
mono- or di-substituted by C1-C4alkyl, such as methyl, C1-C4alkoxy, such
as methoxy, C1-C4alkylthio, such as methylthio, and/or by halogen having
an atomic number of up to and including 35, such as fluorine or chlorine,
R4 is likewise a phenyl radical that is unsubstituted or mono- or di-sub-
stituted by C1-C4alkyl, such as methyl, C1-C4alkoxy, such as methoxy,
C1-C4alkylthio, such as methylthio, and/or by halogen having an atomic
number of up to and including 35, such as fluorine or chlorine, alk' is
C1-Csalkylene, such as ethylene, 1,3-propylene or 1,4-butylene, Rz is
hydrogen, and alk is straight-chained C2-C4alkylene, such as ethylene or
1,3-propylene, and their salts, especially their pharmaceutically
acceptable salts.
The invention relates more especially, on the other hand, to compounds of
formula I wherein R1 is a radical of formula (Ia) wherein R3 is a phenyl,
naphthyl, thienyl, pyridyl or imidazolyl radical that is unsubstituted or
mono- or di-substituted by C1-C4alkyl, such as methyl, C1-C4alkoxy, such
_ 7 _ 1 3 2 9 6 ~ 9
as methoxy, C1-C4alkylthio, such as methylthio, and/or by halogen having
an atomic number of up to and including 35, such as fluorine or chlorine,
R4 is hydrogen or, if R3 is a phenyl radical that is unsubstituted or
mono-or di-substituted by C1-C4alkyl, such as methyl, C1-C4alkoxy, such
as methoxy, C1-C4alkylthio, such as methylthio, and/or by halogen having
an atomic number of up to and including 35, such as fluorine or chlorine,
R4 is likewise a phenyl radical that is unsubstituted or mono- or di-sub-
stituted by C1-C4alkyl, such as methyl, C1-C4alkoxy, such as methoxy,
C1-C4alkylthio, such as methylthio, and/or by halogen having an atomic
number of up to and including 35, such as fluorine or chlorine, alk' is
C1-Csalkylene, such as 1,3-propylene, 1,4-butylene or 1,5-pentylene, Rz
is C1-C4alkyl, such as methyl, or Cz-C4alkenyl, such as allyl, and alk is
straight-chained Cz-C4alkylene, such as ethylene or 1,3-propylene, with
the proviso that alk' is methylene and Rz is hydrogen or C1-Czalkyl if R3
is unsubstituted phenyl and R4 is hydrogen, and their salts, especially
their pharmaceutically acceptable salts.
The invention relates preferably to compounds of formula I wherein R1 is
a radical of formula (Ia) wherein R3 is phenyl that is unsubstituted or,
sscondly, mono- or di-substituted by C1-C4alkyl, such as methyl, C1-C4-
alkoxy, such as methoxy, or by halogen having an atomic number of up to
and including 35, such as chlorine or fluorine, R4 is hydrogen, alk' is
straight-chained, terminally bonded Cz-C4alkylene, such as ethylene or
1,3-propylene, Rz is hydrogen or C1-C4alkyl, such as methyl, and alk is
C2-C3alkylene, such as ethylene, for exampla those wherein alk' is C1-C2-
alkylene and R2 is hydrogen or C1-C3alkyl if R3 is unsubstituted phenyl,
and their salts, especially their pharmaceutically acceptable salts.
The invention relates most preferably to compounds of formula I whereinR1 is mono- or di-phenyl-Cz-Csalkyl that is unsubstituted or mono- or
di-substituted in the phenyl moiety by C1-C4alkyl, such as methyl, C1-C4-
alkoxy, such as methoxy, and/or by halogen having an atomic number of up
to and including 35, such as chlorine, or is imidazolyl-, thienyl- or
1329~0~
-- 8 --
pyridyl-C2-C6alkyl, Rz is hydrogen or C1-C4-alkyl, such as methyl, and
alk is Cz-C3alkylene, such as ethylene, with the proviso that R2 is
hydrogen if R1 is unsubstituted phenyl-C3-C6alkyl, or Rz is hydrogen or
C1-C2alkyl if R1 is an unsubstituted phenethyl radical, and their salts,
especially their pharmaceutically acceptable salts.
The invention relates most especially to compounds of formula I whereinR1 is a radical of formula (Ia) wherein R3 is phenyl that is unsubsti-
tuted or, secondly, mono- or di-substituted by C1-C4alkyl, such as
methyl, Cl-C4-alkoxy, such as methoxy, and/or by halogen having an atomic
number of up to and including 35, such as chlorine or fluorine, R4 is
hydrogen, alk' is straight-chained, terminally bonded C2-C4alkylene, such
as ethylene or 1,3-propylene, R2 is hydrogen, and alk is C2-C3alkylene,
such as ethylene, and their salts, especially their pharmaceutically
acceptable salts.
The invention rela~es specifically to the compounds of formula I
mentioned in the Examples and their salts, especially their internal
salts and pharmaceutically acceptable salts with bases.
The invention also relates to a process for the preparation of compounds
of formula I and their salts, which process is based on methods that are
known per se. This process comprises
a) in a compound of formula
~1
Rl -~ - alk - ~ - OH (II),
Ro X2
wherein Ro is a radical R2 or an amino-protecting group and Xl is a
functionally modified phosphono group and X2 is a free or functionally
modified phosphono group, converting functionally modified phosphono X
and, where appropriate, Xz into a free phosphono group, or
132g6~3
b) reacting compounds of formulae
~o3H2
Rl - Yl (III) and Y2 - alk - ~ - OH (IV),
03Hz
wherein one of the radicals Y1 and Y2 is a reactive esterified hydroxy
group and the other is a group of formula -N(Ro)-H wherein Ro is a
radical R2 or an amino-protecting group, or salts thsreof, with each
other, or
c) reacting a compound of formula
Rl - ~ - alk - X3 (V),
o
wherein Ro is a radical R2 or an amino-protecting group and X3 is
carboxy, carbamoyl or cyano, especially carboxy or cyano, with phos-
phorous acid and phosphorus trichloride, hydrolysing the primary product
and, in an intermediate of formula
~03H2
Rl - ~ - alk - - NH2 (VI)
Ro 03H2
obtained starting from compounds of formula V wherein X3 is cvano or
carbamoyl, or in a salt thereof, replacing the amino group by hydroxy by
treatment with nitrous acid, and in each case removing the amino-
protecting group if present and, if desired, converting a resulting
compound into a different compound of formula I and/or converting a
resulting free compound into a salt or a resulting salt in the free
compound or into a different salt.
Suitable amino-protecting groups Ro are, for example, ~-aryl-lower alkyl,
such as benzyl or ~-methoxybenzyl, a,~,~-triaryl-lower alkyl, such as
trityl, or tri-lower alkylsilyl, such as trimethylsilyl. ~-Aryl- and
~,,~-tri-aryl-lower alkyl can readily be removed by hydrogenolysis, and
tri-lower alkylsilyl and ~,,~-tri-aryl-lower alkyl can readily be
removed by hydrolysis. The removal of ~-aryl- and ~ -tricosyl-lower
alkyl Ro using hydrogenolysis is effected especially by reaction with
hydrogen in the presence of a hydrogenation catalyst, such as a nickel or
13296~9
-- 10 --
noble metal catalyst, for example palladium-on-carbon, preferably in a
lower alkanol under normal pressure and temperature conditions, for
example at approximately from 20C to 30C and from approximately
0.95 bar to approximately 1.3 bar.
Functionally modified phosphono groups that are to be converted into free
phosphono in accordance with process variant a) are, for example, in the
form of an ester, especially in the form of a diester of formula
-P(=O)(OR)2 (IIa) wherein OR is etherified hydroxy, for example lower
alkoxy, lower alkanoyloxy-lower alkoxy, such as C2-C7alkanoyloxy-C1-C4-
alkoxy, for example acetoxymethoxy or pivaloyloxymethoxy, or is a
phenoxy, -phenyl-lower alkoxy or silyloxy group each unsubstituted or
substituted by lower alkyl, lower alkoxy, halogen, trifluoromethyl and/or
by hydroxy, such as tri-lower alkylsilyloxy.
The conversion of functionally modified phosphono groups into free
phosphono groups is effected in customary manner, such as by hydrolysis,
for example in the presence of a mineral acid, such as hydrochloric or
sulfuric acid, at from approximately 80C to approximately 110C, for
example at boiling temperature, or by reaction with a tri-lower alkyl-
halosilane, for example trimethylchlorosilane or, especially, trimethyl-
iodosilane or trimethylbromosilane, preferably in methylene chloride, in
a temperature range of from approximately 0 to approximately 40C, and
by subsequent treatment with water. ~-Phenyl-lower alkyl esters can
furthermore be converted into compounds of formula I by hydrogenolysis,
for example reaction with hydrogen in the presence of a hydrogenation
catalyst, such as a nickel or noble metal catalyst, for example
palladium-on-carbon, preferably in a lower alkanol under normal pressure
and temperature conditions.
The starting materials of formula II can be prepared, for example, by
reacting a compound of formula
R1 - ~ - alk - COOH (IIb),
o
1329609
-- , 1 --
or preferably the anhydride or acid chloride thereof, with a corre-
sponding phosphorous acid triester of formula P(OR)3 (IIc), for example
at from 0C to approximately 60C, to give a compound of formula
,OR
Rl - ~ - alk - ~ - ~ - OR (IId),
and further reacting the latter with a phosphorous acid diester of
formula H-P(=O)(OR)z (IIe) or P(OH)(OR)z (IIf) in the presence of a di-
lower alkylamine, for example diethylamine, or in the presence of an
alkali metal lower alkanolate, for example sodium methanolate, to give
the corresponding compound of formula
R
Rl\` = ~ ~ OR
~N - alk - - OH (IIg).
Ro = ;R- OR
Starting materials of formula IIb can, if they are not known, be
prepared, for example, by reacting a corresponding compound of formula
Rl-N(Ro)-H (IIh),
wherein Ro is a group Rz or an amino-protecting group, with a compound of
formula
Y - alk - COOR (IIi),
wherein Y is halogen, such as bromine, or, for the preparation of
compounds IIb wherein alk is 1,2-lower alkylene, for example ethylene,
with a compound of formula
alko - COOR (IIj),
uherein alko is lower alk-l-enyl, hydrolysing the ester obtained in each
case to the acid and anhydridising or chlorinating the latter, for
example using phosphorus pentachloride, and, if desired, removing the
amino-protecting group if present.
13296~9
- 12 -
Reactive esters (III) and (IV) that are to be used in accordance with
process variant b) contain as the reactive esterified hydroxy group, for
example, a halogen atom, such as a chlorine, bromine or iodine atom, or a
sulfonyloxy group, such as an alkanesulfonyloxy group or an unsubstituted
or substituted benzenesulfonyloxy group, for example methanesulfonyloxy
or p-toluenesulfonyloxy.
The reaction with the reactive esters mentioned is effected, for example,
in the presence of a base, such as an alkali metal hydroxide or an
alkaline earth metal hydroxide, for example sodium hydroxide, or a
quaternary ammonium hydroxide, for example tetrabutylammonium hydroxide,
advantageously in the presence of a solvent or diluent, for example a
lower alkanol, a di-lower alkyl ketone or a cycloaliphatic ether, for
example isopropanol, methyl ethyl ketone, dioxane or tetrahydrofuran.
The starting materials of formula IV can be prepared, for example, by
reacting a compound of formula
Y2 - alk - COOH (IVa)
in customary manner, for example in chlorobenzene, with phosphorous acid
and phosphorus trichloride or with phosphoric acid and an excess of
phosphorus tribromide, and subsequently working up by hydrolysis.
The reaction of compounds of formula V with phosphorous acid and phos-
phorus trichloride in accordance with process variant c) is effected in
customary manner, the phosphorous acid component preferably being formed
in situ by reaction of excess phosphorus trichloride with water-
containing phosphoric acid, for example with commercially customary
approximately 75 % to approximately 95 %, preferably approximately 85 %,
phosphoric acid. The reaction is advantageously carried out while
heating, for example at from approximately 70 to approximately 120~C, in
~329609
- 13 -
a suitable solvent, such as tetrachloroethane, trichloroethane, chloro-
benzene, chlorotoluene or paraffin oil, and with working up being
effected by hydrolysis.
The treatment of intermediates of formula VI with nitrous acid is
effected in customary manner with the latter being freed in aqueous
solution from one of its salts, for example sodium nitrite, by acid
treatment, for example by the action of hydrochloric acid, during which a
corresponding, unstable diazonium salt, for example diazonium chloride,
is formed as intermediate, which diazonium salt, with the introduction of
the ?-hydroxy group, splits off nitrogen.
The starting materials of formula V can, if they are not known, be
prepared, for example, by reacting a corresponding compound of formula
R1-N(Ro)-H (IIh),
wherein Ro is a group Rz or an amino-protecting group, with a compound of
formula
Y - alk - COOR (IIi),
wherein Y is halogen, such as bromine, or, for the preparation of
compounds of formula V wherein alk is 1,2-lower alkylene, for example
ethylene, with a compound of formula
alko - X3 ( IIf),
wherein alko is a lower alk-l-enyl radical, and in each case removing the
amino-protecting group if present and, if desired, in each case hydro-
lysing the resulting primary product to the acid.
Compounds of formula I obtained by the process of the invention or by
another method that is known per se can be converted into other compounds
of formula I in a manner known per se.
- -
_ 14 _ 1 3 2 9 6 ~ 3
For example, in a resulting compound of formula I wherein Rz is hydrogen,
by reaction with a lower alkanal under reducing conditions, for example
with formaldehyde and formic acid, or, secondly, with a reactive ester of
a lower alkanol or lower alkenol in customary manner, preferably in the
presence of a basic condensing agent, such as an alkali metal lower
alkanolate, hydrogen can be replaced by a lower alkyl or lower alkenyl
radical R2, respectively.
Furthermore, non-aromatic double bonds present in R1 and/or R2 can be
reduced to single bonds in customary manner by hydrogenolysis, for
example reaction with hydrogen in the presence of a hydrogenation
catalyst, such as a nickel or noble metal catalyst, for example
palladium-on-carbon, preferably in a lower alkanol under normal pressure
and temperature conditions.
The aromatic substituent(s) of Rl can also be substituted. For example,
halogen can be introduced by reaction with a customary nuclear
halogenating agent, for example with chlorine or bromine in the presence
of a Lewis acid, such as iron(III) chloride.
Depending on the starting materials and procedures chosen, the novel
compounds may be obtained in the form of one of the possible isomers or
as a mixture thereof, for example depending on the number of asymmetric
carbon atoms, they may be obtained in the form of pure optical isomers,
such as antipodes, or in the form of isomeric mixtures, such as
racemates, diastereoisomeric mixtures or mixtures of racemates.
Resulting diastereoisomeric mixtures and mixtures of racemates can be
separated on the basis of the physico-chemical differences between the
components into the pure isomers, diastereoisomers or racemates in known
manner, for example by chromatography and/or fractional crystallisation.
- 15 - 1329609
Resulting racemates can furthermore be resolved into the optical
antipodes by known methods, for example by recrystallisation from an
optically active solvent, with the aid of microorganisms, or by reaction
of an acid end product with an optically active base that forms salts
with the racemic acid and by separation of the salts obtained in that
manner, for example on the basis of their differing solubilities, into
the diastereoisomers from which the antipodes can be freed by the action
of suitable agents. Advantageously, the more active of the two antipodes
is isolated.
Resulting free compounds of formula I, including their internal salts of
formula I, can be converted into salts with bases by partial or complete
neutralisation with one of the bases mentioned at the beginning. Acid
addition salts also can be converted in an analogous manner into the
corresponding free compounds or internal salts thereof.
Conversely, resulting free compounds of formula I can be converted intoacid addition salts by treatment with a protonic acid.
Resulting salts can be converted in a manner known per se into other
salts having a lower proportion of cations (partial salts) or into the
free compounds, for example by treatment with an acid reagent, such as a
mineral acid. Resulting free compounds can be converted by treatment with
a base, for example alkali hydroxide solution, into salts and/or
resulting salts having a lower proportion of cations (partial salts) can
be converted in the same manner into salts having a higher proportion of
cations, for example complete salts.
The compounds, including their salts, may also be obtained in the form of
their hydrates or may include the solvent used for crystallisation.
Owing to the close relationship between the novel compounds in free form
and in the form of their salts, throughout this specification there is to
be understood by the free rompounds or their salts, where appropriate and
expedient, also the corresponding salts or free compounds, respectively.
1329609
- 16 -
The invention relates also to those embodiments of the process in which a
compound obtainable as an intermediate at any stage of the process is
used as starting material and the remaining steps are carried out, or a
starting material is used in the form of a salt and/or racemate or
antipode or, especially, is formed under the reaction conditions.
The starting materials used in the present invention are preferably those
which result in the compounds described at the beginning as being espec-
ially valuable. The invention relates also to novel starting materials
and to processes for the preparation thereof.
The pharmaceutical preparations according to the invention, which contain
compounds of formula I or pharmaceutically acceptable salts thereof, are
for enteral, such as oral or rectal, and parenteral administration and
contain the pharmacologically active ingredient on its own or together
with a pharmaceutically acceptable carrier.
The novel pharmaceutical preparations contain, for example, from approxi-
mately 10 % to approximately ôO %, preferably from approximately 20 % to
approximately 60 %, active ingredient. Pharmaceutical preparations
accorting to the invention for enteral and parenteral administration are,
for example, those in dosage unit form, such as dragées, tablets,
capsules or suppositories, and also ampoules. These are prepared in a
manner known per se, for example by means of conventional mixing,
granulating, confectioning, dissolving or lyophilising processes. For
example, pharmaceutical preparations for oral administration can be
obtained by combining the active ingredient with solid carrisrs, if
desired granulating a resulting mixture and processing the mixture or
granulate, if desired or necessary after the addition of suitable
adjuncts, into tablets or dragée cores.
Suitable carriers are especially fillers, such as sugars, for example
lactose, saccharose, mannitol or sorbitol, and cellulose preparations,
and also binders, such as starch pastes using, for example, corn, wheat,
- 17 - 1 329 6 ~
rice or potato starch, gelatine, tragacanth, methylcellulose and/or poly
vinylpyrrolidone, and/or, if desired, disintegrators, such as the above-
mentioned starches, also carboxymethyl starch, crosslinked polyvinyl-
pyrrolidone, agar, alginic acid or a salt thereof, such as sodium
alginate. Adjuncts are especially flow-regulating and lubricating agents,
for example silica, talc, stearic acid and/or polyethylene glycol. Dragee
cores are provided with suitable coatings which may be resistant to
gastric juices, there being used, inter alia, concentrated sugar
solutions which may contain gum arabic, talc, polyvinylpyrrolidone, poly-
ethylene glycol and/or titanium dioxide, or lacquer solutions in suitable
organic solvents or solvent mixtures, or, for the preparation of coatings
that are resistant to gastric juices, solutions of suitable cellulose
preparations, such as acetylcellulose phthalate or hydroxypropylmethyl-
cellulose phthalate. Colourings or pigments may be added to the tablets
or dragée coatings, for example for identification purposes or to
indicate different doses of active ingredient.
Other orally administrable pharmaceutical preparations are dry-filled
capsules consisting of gelatine, and also soft sealed capsules consisting
of gelatine and a plasticiser, such as glycerol or sorbitol. The dry-
filled capsulss may contain the active ingredient in the form of a
granulate, for example in admixture with fillers, such as lactose,
binders, such as starches, and/or glidants, such as talc, and, if
desired, stabilisers. In soft capsules, the active ingredient is prefer-
ably dissolved or suspended in suitable liquids, such as fatty oils,
paraffin oil or liquid polyethylene glycols, to which stabilisers may
also be added.
Suitable rectally administrable pharmaceutical preparations are, for
-example, suppositories that consist of a combination of the active
ingredient with a suppository base material. Suitable suppository base
materials are, for example, natural or synthetic triglycerides, paraffin
hydrocarbons, polyethylene glycols or higher alkanols. It is also
possible to use gelatine rectal capsules that contain a combination of
1329609
- 18 -
the active ingredient with a base material; suitable base materials are,
for example, liquid triglycerides, polyethylene glycols or paraffin
hydrocarbons.
For parenteral administration there are suitable, especially, aqueous
solutions of an active ingredient in water-soluble form, for example in
the form of a water-soluble salt, or suspensions of the active
ingredient, such as correponding oily injection suspensions, in which
suitable lipophilic solvents or vehicles, such as fatty oils, for example
sesame oil, or synthetic fatty acid esters, for example ethyl oleate or
triglycerides, are used, or aqueous injection suspensions that contain
viscosity-increasing substances, for example sodium carboxymethyl-
cellulose, sorbitol and/or dextran and, if desired, also stabilisers.
The present invention relates also to the use of compounds of formula Iand their salts, preferably for the treatment of diseases that are
attributable to calcium metabolism disorders, for example of the
rheumatic type, and especially of osteoporoses.
Dosages under 0.01 mg/kg body weight have only a negligible effect on
pathological calcification or the degeneration of hard tissue. At dosages
a-Dove 100 mg/kg body wsight, toxic side-effects may occur in long-term
use. The compounds of formula I and their salts can be administered both
orally and, in the form of a hypertonic solution, subcutaneously, intra-
muscularly or intravenously. The preferred daily doses are in the range
of approximately from 0.1 to 5 mg/kg in the case of oral administration,
in the range of approximately from 0.1 to 1 mg/kg in the case of sub-
cutaneous and intramuscular administration, and in the range of approxi-
mately from 0.01 to 2 mg/kg, for example approximately from 0.013 to
0.67 mg/kg, in the case of intravenous administration.
The dosage of the compounds used is, however, variable and depends on the
particular conditions, such as the nature and severity of the disease,
the duration of treatment and on the particular compound. Single doses
contain, for example, from 0.01 to 10 mg, dosage unit forms for
1329609
- 19 -
parenteral, such as intravenous, administration contain, for example,
from 0.01 to 0.1 mg, preferably from 0.02 to 0.08 mg, and oral dosage
unit forms contain, for example, from 0.2 to 2.5 mg, preferably from 0.3
to 1.5 mg, per kg of body weight. The preferred single dosage for oral
administration is from 10 to 100 mg and for intravenous administration
from 0.5 to 5 mg, and can be adminis.ered up to 4 times per day. The
higher dosages in the case of oral administration are necessary owing to
the limited resorption. In the case of long-term treatments, the
initially higher dosage can normally be changed to lower dosages while
still maintaining the desired effect.
The following Examples illustrate the invention described above; they are
not intended, however, to limit the scope thereof in any way. Tempera-
tures are given in degrees Celsius.
Example 1: (0.1 mol) of 3-[N-(3-phenylpropyl)-N-methyl- amino]-propionic
acid hydrochloride is heated at 100~ under reflux with 13.4 ml of 85 %
phosphoric acid and 50 ml of chlorobenzene while stirring. Then, at 100,
27 ml of phosphorus trichloride are added dropwise, gas evolution taking
place. A thick mass separates from the reaction mixture over the course
of 30 minutes. The mixture is heated at 100 for a further 3 hours and
the supernatant chlorobenzene is then removed by decanting. The viscous
mass which remains is refluxed with 100 ml of 9N hydrochloric acid for
3 hours while stirring. The mixture is filtered while hot, with the
addition of carbon, and the filtrate is diluted with acetone, whereupon
3-[N-(3-phenylpropyl)-N-methyl-amino]-1-hydroxy-propane-1,1-diphosphonic
acid separates out in crystalline form; m.p. 219 (decomposition).
The 3-[N-(3-phenylpropyl)-N-methyl-amino]-propionic acid hydrochloride
used as starting material can be prepared as follows:
(0.15 mol) of N-(3-phenylpropyl)-N-methyl-amine are introduced into 50 ml
of diethyl ether, and 15.1 g of ethyl acrylate are gradually added
thereto while stirring. With a slight increase in tempera~ure, a clear
- 20 - ~3296~9
solution forms. After standing overnight at room temperature, the ether
is removed by distillation. The oil which remains is crude 3-[N-(3-phen-
ylpropyl)-N-methyl-amino]-propionic acid ethyl ester.
The resulting ester is refluxed with 600 ml of 4N hydrochloric acid for24 hours. The mixture is then completely concentrated by evaporation
under reduced pressure and the crystalline residue is triturated with
acetone. After the crystals have been filtered with suction, washed and
dried, 3-[N-(3-phenylpropyl)-N-methyl-amino]-propionic acid hydrochloride
is obtained.
Example 2: In a manner analogous to that described in Example 1,
3-(3-phenylpropylamino)-1-hydroxy-propane-1,1-diphosphonic acid,
m.p. 219 (decomp.),
3-(3-phenylprop-2-ylamino)-1-hydroxy-propane-1,1-diphosphonic acid,
m.p. 208-210 (decomp.),
3-[N-(3-phenylpropyl)-N-ethyl-amino]-l-hydroxy-propane-1,1-diphosphonic
acid, m.p. 195-197 (decomp.),
3-(4-phenylbutylamino)-1-hydroxy-propane-1,1-diphosphonic acid,
m.p. 191-193 (decomp.)
ant
3-[3-(pyrid-3-yl)propylamino]-1-hydroxy-propane-1,1-diphosphonic acid
can be obtained starting from or via
3-(3-phenylpropylamino)-propionic acid hydrochloride (m.p. 219,
decomp.),
3-(3-phenylprop-2-ylamino)-propionic acid hydrochloride (m.p. 126-127),
3-[N-(3-phenylpropyl)-N-ethyl-amino]-propionic acid hydrochloride, oil,
3-(4-phenylbutylamino)-propionic acid hydrochloride, m.p. 135-137 and
3-[3-(pyrid-3-yl)propylamino]-propionic acid hydrochloride, respectively.
Example 3: 9.3 g (26.3 mmol) of 3-(3-phenylpropylamino)-1-hydroxy-
propane-1,1-diphosphonic acid are refluxed for 36 hours with 6.1 ml of
formic acid and 4.2 ml of a 38 % formaldehyde solution in water. The
reaction solution is concentrated by evaporation under reduced pressure
- 21 - 1 3 2 9 6 ~ 9
and the residue is diluted with acetone, yielding 3-[N-(3-phenylpropyl)-
N-methyl-amino]-1-hydroxy-propane-1,1-diphosphonic acid in the form of
colourless crystals of m.p. 219 (decomp.).
Example 4: In a manner analogous to that described in Example 1, 3-[4,4-
di(~-fluorophenyl)butylamino]-1-hydroxy-propane-1,1-diphosphonic acid,
m.p. 220-222 (decomp.) is obtained starting from 3-[4,4-di(~-fluoro-
phenyl)butylamino]-propionic acid ethyl ester via 3-[4,4-di(~-fluoro
phenyl)butylamino]-propionic acid hydrochloride, m.p. 165-166.
The starting material can be prepared, for example, in the following
manner:
26.1 g (0.1 mol) of 4,4-di(p-fluorophenyl)butylamine and lô.1 g of
3-bromopropionic acid ethyl ester are refluxed with 21.0 g of potassium
carbonate in 200 ml of 2-butanone for 24 hours while stirring. The
reaction mixture is filtered and the filtrate is concentrated by evapor-
ation under reduced pressure, yielding crude 3-[4,4-di(p-fluorophenyl)-
butylamino]-propionic acid ethyl ester in the form of an oil.
In an analogous manner, 4-[N-(2-phenethyl)-N-methyl-amino]-1-hydroxy-
butane-l,l-diphosphonic acid is obtained starting from N-(2-phenethyl)-N-
methyl-amine and 4-bromobutyric acid ethyl ester vla 4-[N-(2-phenethyl)-
N-methyl-amino]-butyric acid ethyl ester and 4-[N-(2-phenethyl)-N-methyl-
amino]butyric acid hydrochloride.
Example 5: In a manner analogous to that described in Example 1, 3-~N-[3-
imidazol-4-yl)propyl]-N-methyl-amino}-1-hydroxy-propane-1,1-diphosphonic
acid is obtained starting from N-[3-(imidazol-4-yl)propyl]-N-methyl-amine
via 3-~N-[3-(imidazol-4-yl)propyl]-N-methyl-amino~-propionic acid ethyl
ester and 3-~N-[3-(imidazol-4-yl)propyl]-N-methyl-amino}-propionic acid
hydrochloride.
The starting material can be prepared, for example, in the following
manner:
- 22 _ 1 3 2 9 ~ ~ 9
16.8 g (0.12 mol) of 3-[imidazol-4(5)-yl]propionic acid are refluxed with
13.1 ml of thionyl chloride for 2 hours. After removing excess thionyl
chloride by distillation, 3-[imidazol-4(5)-yl]propionic acid chloride
hydrochloride remains as a semi-solid mass (yield 100 %).
23.4 g (0.12 mol) of 3-[imidaæol-4(5)-yl]propionic acid chloride hydro-
chloride are dissolved in 80 ml of dimethylformamide and the solution is
cooled to -10. Methylamine gas is passed into the solution for 2 hours
until there has been a weight increase cf 15 g. After standing overnight
at room temperature, the reaction mixture is concentrated to dryness by
evaporation under reduced p}essure. The residue is chromatographed on
220 g of silica gel with the eluant chloroform/methanol/conc. ammonia
(80:20:1). The fractions containing the product yield, from tetrahydro-
furan, colourless crystals of 3-[imidazol-4(5)-yl]propionic acid
(N-methyl)amide, m.p. 168-170. 10.9 g (0.0718 mol) of 3-[imidazol-4(5)-
yl]propionic acid ~N-methyl)amide are added in portions, while stirring,
to a suspension of 2.8 g of lithium aluminium hydride in 200 ml of tetra-
hydrofuran and the reaction mixture is then refluxed for 30 hours. The
reaction mixture is then cooled to 0, and 3 ml of water, 2.2 ml of lON
sodium hydroxide solution and 8.2 ml of water are added dropwise in
succession. The inorganic precipitate is filtered off and the filtrate is
concentrated by evaporation in vacuo. The oil which remains is crude
N-[imidazol-4(5)-yl-propyl]-N-methylamine.
Example 6: In a manner analogous to that described in Example 1,
3-[4-(4-methoxyphenyl)butylamino]-1-hydroxy-propane-1,1-diphosphonic
acid, m.p. 160-164 (decomp.);
3-[3-(4-methoxyphenyl)propyl-N-methyl-amino]-1-hydroxy-propane-1,1-di-
phosphonic acid, m.p. 154-156 (decomp.)
3-[3-(4-chlorophenyl)propyl-N-methyl-amino]-1-hydroxy-propane-1,1-di-
phosphonic acid, m.p. 162-166 (decomp.);
3-[3-(3-methylphenyl)propyl-N-methyl-amino]-1-hydroxy-propane-1,1-di-
phosphonic ~cid, m.p. 193-195 (decomp.);
- 23 - 13296~
3-[3-(imidazol-4(5)-yl)propyl-N-methyl-amino]-1-hydroxy-propane-1,1-di-
phosphonic acid, m.p. 130-136 (decomp.);
3-[3-(pyrid-2-yl)propyl-N-methyl-amino]-l-hydroxy-propane-l,l-di-
phosphonic acid, m.p. 142-147 (decomp.);
3-[N-(2-phenethyl)-N-methyl-amino]-l-hydroxy-propane-l,l-diphosphonic
acid, m.p. 187-188 (decomp.); and
3-[N-(2-phenethyl)amino]-1-hydroxy-propane-1,1-diphosphonic acid,
m.p. 205-206 (decomp.)
can be prepared starting from or via
3-[4-(4-methoxyphenyl)butylamino]propionic acid hydrochloride,
m.p. 150-152;
3-[3-(4-methoxyphenyl)propyl-N-methyl-amino]propionic acid hydrochloride,
m.p. 108-110;
3-[3-(4-chlorophenyl)propyl-N-methyl-amino]propionic acid hydrochloride,
m.p. 128-130;
3-[3-(3-methylphenyl)propyl-N-methyl-amino]propionic acid hydrochloride,
m.p. 97-98;
3-[3-(imidazol-4(5)-yl)propyl-N-methyl-amino]propionic acid dihydro-
chloride;
3-[3-(pyrid-2-yl)propyl-N-methyl-amino]propionic acid dihydrochloride,
m.p. 157-160 (decomp.);
3-[N-(2-phenethyl)-N-methyl-amino]propionic acid hydrochloride,
m.p. 144-145 (decomp.); and
3-[N-(2-phenethyl)amino]propionic acid hydrochloride, m.p. 150-152
(decomp.)
respectively.
Example 7: Tablets, each containing 75 mg of active ingredient, for
example 3-[N-(3-phenylpropyl)-N-methyl-amino]-l-hydroxy-propane-l,l-
diphosphonic acid or a salt thereof, for example the disodium salt, can
be prepared in the following manner:
13296~9
- 24 -
Constituents (for 1000 tablets)
active ingredient 75.0 g
lactose 263.5 g
corn starch 22.5 g
polyethylene glycol 60005.0 g
talcum 15.0 g
magnesium stearate 4.0 g
demineralised water q.s.
Preparation: The solid ingredients are first forced through a sieve of
0.6 mm mesh width. Then, the active ingredisnt~ lactose, talcum,
magnesium stearate and half of the starch are homogeneously mixed. The
other half of the starch is suspended in 65 ml of water and this
suspension is added to a boiling solution of the polyethylene glycol in
260 ml of water. The resulting paste is added to the pulverulent
substances, and the whole is mixed and granulated, if necessary with the
addition of water. The granulate is dried overnight at 35, forced
through a sieve of 1.2 mm mesh width and compressed into tablets of
approximately 10 mm diameter which are concave on both sides and have a
breaking notch on the upper side.
E~ampls 8: Tablets, each containing 10 mg of active ingredient, for
example 3-[N-(3-phenylpropyl)-N-methyl-amino]-l-hydroxy-propane-l,l-
diphosphonic acid or a salt thereof, for example the disodium salt, can
be prepared in the following manner:
Constituents (for 1000 tablets)
active ingredient 10.0 g
lactose 328.5 g
corn starch 17.5 g
polyethylene glycol 60005.0 g
talcum 25.0 g
magnesium stearate 4.0 g
demineralised water q.s.
- 25 - 1 3 2 9 6 0 9
Preparation: The solid ingredients are first forced through a sieve of
0.6 mm mesh width. Then, the active ingredient, lactose, talcum,
magnesium stearate and half of the starch are homogeneously mixed. The
other half of the starch is suspended in 65 ml of water and this
suspension is added to a boiling solution of the polyethylene glycol in
260 ml of water. The resulting paste is added to the pulverulent
substances, and the whole is mixed and granulated, if necessary with the
addition of water. The granulate is dried overnight at 35, forced
through a sieve of 1.2 mm mesh width and compressed into tablets of
approximately 10 mm diameter which are concave on both sides and have a
breaking notch on the upper side.
~xample 9: Gelatine dry-filled capsules, each containing 100 mg of active
ingredient, for example 3-[N-(3-phenylpropyl)-N-methyl-amino]-1-hydroxy-
propane-1,1-diphosphonic acid or a salt thereof, for example thc disodium
salt, can be prepared in the following manner:
Constituents (for 1000 capsules)
active ingredient 350.0 g
microcrystalline cellulose 30.0 g
sodium lauryl sulfate 2.0 g
magnesium stearate 8.0 g
The sodium lauryl sulfate is added to the active ingredient (lyophilised)
through a sieve of mesh width 0.2 mm and the two components are inti-
mately mixed for 10 minutes. The microcrystalline cellulose is then added
through a sieve of mesh width 0.9 mm and the mixture is again intimately
mixed for 10 minutes. Finally, the magnesium stearate is added through a
sieve of mesh width 0.8 mm and, after mixing for a further 3 minutes, the
mixture is introduced into size 0 (elongated) gelatine dry-filled
capsules in portions of 390 mg.
`~ 1329609
- 26 -
Example 10: A 0.2 X lnjection or infusion solution can be ~rep4r~d, for
cxampl-, in the following m~nnnr:
acti~e in~radient, for ox~ple 3-[N-(3-phenylpropy~ methyl-a~in
hydroxy-propane~ diphosphonic acid or a salt thereof,
for exam~le tho tlsodium salt 5.0 g
~odium chloride 22 . 5 g
phosphato buffer pH ~ 7.4 300.0 g
de~inoraliset watorad 2500.0 ml
Ihe actl~e ingredi0nt is dissolved in 1000 ml of water and filtered
thsough n microfllter. The buffer solucion is added and then watHr is
added to give ~ volume of 2500 ml. For the preparation Df do~age unlt
orms, portions of 1.0 or 2.5 ml are introtuced lnto glass ampoule~
(containin~ 2.Q or 5.0 mg of ac~ive ingredient, respectivelY)-
~3t~ele_~1 1,83 g of 3-(4-phenvlbutylamino)-1-hydroxy-propane~
dipho6phonic acit aro dissolved in 10 ml of lN sodium hydroxide solution.
Ihe resulclng solution i5 concencraced by evaporacion under reduced
pressure. ~he product i5 precipitaced by addi~ion of methanol allowed to
crystallised and isolated by suction-filtration. Disodiu~-3-(4-phffnyl-
bu~ylamlno)-l-hydro~y-propane-l~l-diphosphonate of m.p. 313-316
(decomp.) ls thus obtained-
In an analogous manner, disodiu~l-3-~-(3-phenylpropyl)-N~mechyl~amino]-
1-hydroxy-propane-1,1-diphosphonate of m.p. 321-325 is obtainod.