Note: Descriptions are shown in the official language in which they were submitted.
1331182
-- 1 --
Optically Active Glycidyl Ethers Suitable for Use in the
PreParation of Liouid CrYstalline Com~ounds
This application has been divided out of Canadian
Patent Application Serial No. ~6,646 filed September 7,
1988.
This invention relates to novel optically active
glycidyl ethers suitable for use as intermediates in the
preparation of liquid crystalline compounds. The liquid
crystalline compounds that can be prepared from the
intermediates of the present invention have an optically
active y-lactone ring and are useful as elements in display
devices or as elements in opto-electronic devices. These
liquid crystalline compounds include not only compounds
which can exhibit the liquid crystal phase by themselves but
also compounds which do not exhibit the liquid crystal phase
alone but are useful as a component of liquid crystal
compositions.
Liquid crystals have been widely used as a
material for display devices, where a TN (Twisted Nematic)
type display system is usually employed. Advantages of such
a TN display system include less electric consumption, less
eye fatigue because it is a receptor type, and the like. On
the other hand, disadvantages of this system include a
driving force that is very weak because it is driven mainly
on the basis of anisotropy of dielectric constant and slow
response speed. Hence, this system cannot be applied to
devices which require a high response speed.
'T~'
, ~
.. . ~ ..... . , . ~ ,
,~
. ,.
1331~82 - ~
Liquid crystals having ferroelectricity were first
discovered by R.B. Meyer et al. in 1975 (cf. J. Physique,
36, L-69, 1975). This type of l;quid crystal is driven by a -~
comparatively large force derived from spontaneous
polarization, shows extremely high response speed and also
has, good memory. Because of these excellent properties, the
ferroelectric liquid crystal has been noted as a new type
of display element. In order to exhibit the ferro-
electricity, the liquid crystalline compounds should show a
chiral smectic C phase (SmC* phase) and thus should contain
at least one asymmetric carbon atom in the molecule. It is
also necessary to have a dipole moment in the direction
vertical to the long axis of the molecule.
A ferroelectric liquid crystal DOBAM8C synthesized
by Meyer et al. has the following formula~
CH3
C10H21o ~ CHIN ~ CH-CH-CO2CH2-~H-CH2CH3
and satisfies the above conditions, but it contains a Schiff
base and hence is chemically unstable and showsa low ; -~
spontaneous polarization as low as 3 x 10 9 C/cm2. Since
then, many ferroelectric liquid crystalline compounds have
been synthesized, but practically useful compounds having
sufficiently high response speeds have never been found.
Among the known ferroelectric liquid crystalline
1331182
compounds, DOBA-1-MBC which ha~ the asymmetric carbon atom
at the position nearer to the carbonyl group than in DO~AMBC
and has the following formula:
CH3
C1 OH21 0~cH=N~cH=cH-co2~H-c3H7
shows a spontaneous polarization of 5 x 10 8 C/cm2 which is
larger than that of DOBAMBC. It is assumed that this is
caused by the following difference. The
asymmetric carbon atoms and the dipole which are important :
factors for the appearance of ferroelectricity are
positioned close each other, and thereby, the free rotation
of the dipole moiety of the molecule is depressed and the
orientation of the dipole i9 increased. Thus, it is assumed
that the known ferroelectric liquid crystalline compounds
. .
cannot give satisfactory spontaneous polarization and high
response speed because the asymmetic carbon atom having an
inhibitory action of the free rotation of the molecule is .r~
present on the linear chain in the known ferroelectric
liquid crystalline compounds and hence the free rotation of
the molecule cannot be completely inhibited and the dipole
moiety cannot be fixed.
Under the circumstances, the present inventors have
studied intensively in an effort to determine a method of
inhibiting the free rotation of the dipole moiety in
conventional ferroelectric liquid crystalline compounds and
- ~3~11=82
-- 4 --
have found that the free rotation can be inhibited by
providing a compound wherein the asymmetric carbon atom is
contained in a 5-membered lactone ring, by which there can
be obtained a chemically stable-liquid crystalline compound
having ferroelectricity.
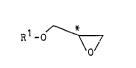
The present invention thus provides in one
embodiment an optically active glycidyl ether of the
formula~
R -O ~ (B)
O '
wherein Rl is a group selected from the group consisting of
R3~(0)n ~ ~ R3~(0)n ~ and
R3-()n ~ N
n and e are each independently O or l; R3 is an alkyl group
having 1 to 15 carbon atoms; and the symbol * denotes an
asymmetric carbon atom, which can be used to prepare liquid
crystalline compounds of the formula (A) as set out below.
In drawings which illustrate preferred embodiments
of the present invention as well as the invention set out in
the parent application:
Fig. 1 shows a graph of the relationship between
the relative dielectric constant and temperature in the q-
lactone derivative prepared in Example l.
1331182
- 4a -
Fig. 2 shows a graph of the relationship between
the relative dielectric constant and temperature in the
y-lactone derivative (2S, 4S) prepared in Example 32.
The liquid crystalli;~-compounds prepared using
the novel optically active glycidyl ethers of the
1331~8~
. 5
invention are compounds having an optically active q-lactone
ring and having the following formula~
R 1 _ 0~ R2
I ¦ (A)
~0 ,.
wherein R1 is a group selected from the group consisting of
R3~(0)n ~ ~ R3~(0)n ~ , and
R3~(0)n ~ \ ~ , n and e are each independently
O or 1, R3 is an alkyl group having 1 to 15 carbon atoms, R2
is a group of -(Co)m-R4, m is O or 1, and R4 is a hydrogen ~:
atom or an alkyl group having 1 to 15 carbon atoms, and * ~
denotes an asymmetric carbon atom. ~.
In the specification, the term "alkyl group" for R3 :
and R4 includes methyl, ethyl, n-propyl, n-butyl, n-pentyl,
n-hexyl, n-heptyl, n-octyl, n-nonyl, n-decyl, n-undecyl, n- :.
dodecyl, n-tridecyl, n-tetradecyl, n-pentadecyl, isopropyl,
t-butyl, 2-methylpropyl, 1-methylpropyl, 3-methylbutyl, 2-
methylbutyl, 1-methylbutyl, 4-methylpentyl, 3-methylpentyl,
2-methylpentyl, 1-methylpentyl, 5-methylhexyl, 4-methyl-
is hexyl, 3-methylhexyl, 2-methylhexyl, 1-methylhexyl, 6-
methylheptyl, 5-methylheptyl, 4-methylheptyl, 3-methyl-
heptyl, 2-methylheptyl, 1-methylheptyl, 7-methyloctyl, 6-
methyloctyl, 5-methyloctyl, 4-methyloctyl, 3-methyloctyl, 2-
methyloctyl, 1-methyloctyl, 8-methylnonyl, 7-methylnonyl, 6-
1331182
methylnonyl, 5-methylnonyl, 4-methylnonyl, 3-methylnonY1, 2-
I methylnonyl, 1-methylnonyl, 3,7-dimethyloctyl, 3,7,11-
¦ trimethyldodecyl, and the like.
The compounds of the parent application contain a
carbonyl group within a 5-membered ring and one or two -
asymmetric carbon atoms on the ring as a moiety having a
dipole moment as an origin of ferroelectricity, and hence,
the free rotation at this moiety is inhibited and thereby
the dipole moiety is directed to one direction, which is
effective for enlarging the spontaneous polarization and for
increa~ing the response speed. In the liquid crystalline
compounds (A) of this invention, when R2 is a hydrogen atom,
only one asymmetric carbon atom i5 contained, but when R2 is
a group other than hydrogen, two asymmetric carbon atoms are
lS contained in the ~-lactone ring and hence there are present
two kinds of diastereomer. These are all suitable for
inhibition of free rotation of the dipole moiety, and they
are used as a liquid crystal alone or in a mixture of two or
more thereof.
The compounds of the formula (A) can be prepared
by a process which comprises reacting an optically active
glycidyl ether of the formula:
R1_o ~ (B)
:, -:
wherein R1 and the symbol * are the same as R1 and * in the -;
formula (A), with a ~-ketoester or malonate of the formula,
respectively
1 3 3 1 1 8 2 ~ ~
,~`` ~`,
o o
. . :. ~ .
R4 ~ oR5 ~- (C) ~ ~ -~
: ~ ....
or
O O .
~:.
R50 ~ oR5 (D)
R4
wherein R4 is a hydrogen atom or an alkyl group having 1 to 15
carbon atoms and R5 is a lower alkyl group having 1 to 4
carbon atoms, in the presence of a base in an organic
solvent.
The starting optically active glycidyl ether (8)
can be prepared by a process a~ shown in the following
reaction scheme:
* ~
~ Cl
R10H - ~ Rl-
Base o
wherein Rl and the symbol * are the same as R1 and * in the
formula (A).
That is, a phenol derivative of the formula R1OH is
reacted with an optically active epichlorohydrin in the
. .. -
pre~ence of a base. The optically active epichlorohydrin is ;~
preferably used ln an amount of 1 to 10 equivalents to 1 equiv~
alent of the phenol derivative, and the base is preferably
:" - 81-331182
used in an amount of 1 to 5 equivalents to 1 equivalent of
the phenol derivative. The base includes alkali metal
hydroxides or alkoxides, e.g. sodium hydroxide, potassium
hydroxide, potassium t-butoxide~,-and the like. The above -
reaction may procee~ smoot~ly ~7ithouta catalyst, but may be
carried out in the presence of a catalyst. The catalYst
may include quaternary ammonium halides, e.g. benzyltri-
ethyl ammonium chloride, benzyltriethylammonium bromide,
henzyltrimethylammonium chloride, benzyltrimethylammonium
bromide, etc. and is used in an amount of 0.01 to 0.1
equivalents to l equivalent of the phenol derivative. An
excess amount of the optically active epichlorohydrin m~y
be used as the solvent, but there is preferably used a
suitable polar solvent, e.g. dimethylformamide, dimethyl-
sulfoxide, dimethylacetamide, acetonitrile, t-butyl
alcohol, and water. The reaction is usually carried out
at a temperature of 50 to 80 C for 0.5 to 3 hours.
Alternatively, the optically active glycidyl
ether tB) mav also be prepared by reacting the phenol
derivative of the formula RlOH with an optically active
epichlorohydrin in the presence of an amine (e.q.
morpholine, piperidine, pyridine, etc.) of O.l to 0.5
equivalents to 1 equivalent of the phenol derivative and -~
subjecting the resulting optically active chlorohydrin
derivative to a cyclization reaction with l to 5 equiva-
lents of a base, for example, an alkali metal hydro~ide, ; ~ ~
':
13311~2 .~
carbonate or alkoxide (e.g. sodlum hydroxide, potassium
hydroxide, potassium carbonate, sodium carbonate, potassium
t-butoxide, etc.). The latter process is carried out in two
steps but is advantageous in that the extraction of the
product can easily be done. This reaction is usually
carried out at a temperature of 50 to 80C for 3 to 24 ;
hours.
When a racemic epichlorohydrin is used in the above
reaction, there is obtained a glycidyl ether in the form of
a racemic mixture. The starting optically active epichloro-
hydrin can be prepared in a high purity by the processes as
described in-Japanese Patent First Publication (Kokai) Nos.
132196/1986 and 6697/1987 (as to R isomer) and by the
process as described in Japanese Patent Application No.
283393/1987 (as to S isomer).
The starting phenol derivative used for
the preparation of the compound (B) can be prepared by the
processes as shown in the following Reaction Schemes I to
VI, wherein R3 is the same as R3 in the formula (A), R3 is a
hydrogen atom or an alkyl group having a carbon atom one ;
smaller than that in R3, Ph means phenyl, and R' is a lower
alkyl group having 1 to 4 carbon atoms.
That is, 4-(4-trans-alkylcyclohexyl)phenols, 4-(4-
alkyloxyphenyl)phenols, and 4-(4-alkylphenyl)phenols are
prepared by the known processes as shown in Reaction
Schemes I, II and III, respectively.
., _ ....... . . . . .
-- 10 --
1331182
Reaction Scheme-I
R3 CO ~ Cl Q R3 CO
,l~ NH2N~2 H20
R3'C~2 ~
AlCl3 ~ CH3COCl
R3 CH2 ~ 1) H202; ~C02H 3~ ~ COCH3
2) H30
Reaction Scheme-II
.
C~I3C02~ ~ CH3Co2~3CocH3
~ ~30~ ~ ;
HO ~ COC~3 -- ;
~ R3X
R30-~3oE~ 1<) H202 ~ EIC02H R30~ COCH3
2) H30
Reaction Scheme-III -~
CH3C02 ~ AlCl3 CoR3
~ NH2NH2-H20
HO ~ CH2~3 ~:~
. 4-(5-Alkyl-2-pyrimidinyl) phenols and 4-(5-
alkyloxy-2-pyrimidinyl)phenols are prepared by the processes
13311~2
as shown in the followin~ Reaction Schemes IV and V,
respectively, which are disclosed in Japanese Patent First :~ .
Publication (Kokai) Nos. 189274/1986 published Au ~ t 22, 1986.
Reaction Scheme-IV
(CH3)2NCHO 3 /~caN(CH3)2
R CH2CH(OC2H5)2 > R C
POC13 \C~O
--~1) C2H5~H, HCl fi--~ /NH
HO~ CN ~ HO~ C/ HCl- ~
2) NH3~=~ ~NH2 ~ NaOC2~5
/ R3 ~ ~ ~ OH
/ N
H2 r Pd-C
C~Y)~}~
NaOC2Hs
P~3CH(co2c2H5~2 ~ 3~ N\~ / POC13
~ N ~ OH
o// H ~: :
Reaction Scheme-V -~
. .
R3~H + BrcH2cH~oc2H5)2 ~ R3ocH2cH(oc2Hs)2
~ (CH3)2NCHO - POC13 :~-
Ho~3~/NH E~Cl
N~ \NH2 :3 ~CHN(CH3)2
R30~ OH-C R O ~
\=N~ NaOC2H5 CHO
. Moreover, 4-[5-(4-alkyloxyphenyl~-2-pyrimidinyl]- :
25phenols and 4-[5-(4-alkylphenyl)-2-pyrimidinyl]phenols are
~ . ., ,.,,.. . .... ~.. ~ ...... . . . - .
- 12 -
1331182
prepared by the processes as shown in the following Reaction
Scheme-VI.
Reaction Scheme-VI
HO ~ CN HO ~ CH2CO2H R3 ~ COCE3
~ PhCH2Cl R'OH ~ H+ S ~ ~ ~
PhCH2O ~ CNHO ~ CH2CO2R' R ~ CH2CO2H
l) C2H5OH, HCl R3X ~ ~ R'OH, H+
2) NH3
~NH R3O ~ CH2C2R' R3 ~ CH2CO2R'
PhCH2O ~ C ~Cl :~
~=~ ~NH2 ~ / ,OR' -~-~
(E) \ / O=C :~
Rt ~ CH(cO2R )2
NaOC2H5
. -I (F) n = 0 ~ ::
\ f ~G) n = l .
R3-~o ~ \ ~ OCH2Ph
. O H ~ POCl3Cl
R3t O ~ ~ ~ OCH2Ph
H2 ~ Pd-C
R3t O ~ \ ~ OH
(H) n = 0 - :
(I) n = l
~ - 13
13311~2
According to the process of Reaction Scheme-VI,
Compound (E) is prepared by protecting the hydroxy group of p-
hydroxybenzonitrile with a benzyl group and converting the
cyano group thereof into an amidine hydrochloride in a usual
manner. Separately, p-hydroxyphenylacetic acid is esteri-
fied with a lower alcohol, and the pnenolic hydroxy group is
alkylated with an alkyl halide, alkyl p-toluenesulfonate or
alkyl methanesulfonate, followed by reaction with diethyl
carbonate in the presence of a base to give a diethyl malonate ~;
. .
derivative (G).
The amidine hydrochloride (E) is condensed with the
diethyl malonate derivative (G) in the presence of a base, for
example~alkali metal alkoxides (e.g. sodium ethoxide, sodium
methoxide, etc.), followed by reaction with phosphorus
oxychloride in the presence of a base, for example, organic amines
(e.g. N,N-diethylaniline, pyridine, 4-(N,N-dimethylamino)-
pyridine, etc.), and the resulting compound is reduced with
hydrogen gas in the presence of Pd-C catalyst to give the
de~ired 4-[5-(4-alkyloxyphenyl)-2-pyrimidinyl]phenol (I). -
In the above process, when a diethyl p-alkylphenyl-
malonate (F) i9 used instead of the diethyl malonate
derivative (G) and the compound (E) and the compound (F) are
reacted as in the reaction of the compound (E) and the
compound (G), there is prepared 4-~5-(4-alkylphenyl)-2-
pyrimidinyl]phenol (H).
The diethyl p-alkylphenylmalonate (F) can be
. ' . . ~ ' . ' ' ' ' '
- 14 -
1331182
prepared by subjecting a p-alkylacetophenone to a Willgerodt
reaction, esterifying the resulting phenylacetic acid
derivative with a lower alcohol, and condensing the
resultant product with diethyl carbonate.
The desired compound (A) of this invention can be
prepared by reacting under reflux the compound (~) with 1 to ;~
5 equivalents of the compound (C) or the compound (D) in the
presence of 1 to 5 equivalents of a base in an organic
solvent for 1.5 to 24 hours. The base used therein includes ~ -~
alkali metal alkoxides (e.g. sodium methoxide, sodium
ethoxide, potassium t-butoxide, etc.), alkali metal hydrides
(e.s. sodium hydride, lithium hydride, etc.), and alkyl
alkali metals (e.g. n-butyllithium, etc.), and the organic
solvent includes alcohols (e.g. methanol, ethanol, t-butyl
alcohol, etc.), ethers (e.g. tetrahydrofuran, diethyl ether,
dimethoxyethane, diethylene glycol dimethyl ether, dioxane,
etc.), aprotic polar solvents (e.g. dimethylformamide,
dimethylsulfoxide, hexamethylphosphoric triamide, etc-.), and
a mixture of these solvents.
In the above process, when R4 in the compound (D) is
a hydrogen atom, the final compound prepared by the process
is mixed with an inorganic salt (1 to 10 equivalents) and
water under neutral condition and then is refluxed in a
polar solvent to give the desired compound (A). The solvent
u~ed therein includes polar solvents, e.g.
dimeth~lformamide, dimethylacetamide, dimethylsulfoxide,
;. -, . . . , , ~. ,~ i : .. ... ~ . ~ ,
: ~ -: . . :,:..... : : .: :: , .. . .. : : :
~31~82
- 15 -
hexamethylphosphoric triamide, diethylene glycol dimethyl
ether, dioxane, and the like. The inorganic salt includes
alkali metal or alkaline earth metal halides, e.g. lithium
chloride, sodium chloride, pot~ssium chloride, lithium
5 bromide, sodium bromide, potassium bromide, lithium iodide, -
sodium iodide, potassium iodide, magnesium chloride, calcium
chloride, strontium chloride, barium chloride, magnesium
bromide, calcium bromide, barium bromide, magnesium iodide,
calcium iodide, barium iodide, and the like. Water is
preferably used in an amount of 5 to 50 equivalents. The
reaction is completed in 1 to 15 hours.
The liquid crystalline compounds prepared using
the optically active glycidyl ethers of this invention may
be obtained in the form of a racemic mixture when a racemic
epichlorohydrin is used as the starting material, and the
racemic compounds may be added to other optically active
liquid crystalline compounds in order to regulate the
helical pitch thereof. The liquid crystalline compounds
have excellent heat stability and light stability, and the
optically active compounds have excellent properties as
ferroelectric liquid crystals. The liquid crystalline
compounds are also useful for the following utilities.
(1) Additives for TN (Twisted Nematic) type or
STN (Super Twisted Nematic) type liquid crystals in order to
inhibit occurrent of reverse domain.
(2) Display element utilizing cholesteric
nematic phase transfer effects (cf. J.J. Wysoki, A. Adams
and W. Haas; Phys. Rev. Lett., 20, 1024, 1968).
` 1331182
- 16 - -
(3) Display element utilizing White-Taylor type
guest host effects (cf. D.L. White and G.N. Taylor; J. Appl.
Phys., 45, 4718, 1974).
(4) Notch filter or band-pass filter utilizing -
.~ - .
5 selective scattering effects by fixing the cholesteric phase ~-
in matrix (cf. F.J. Kahn; Appl. Phys. Lett., 18, 231, 1971).
(5) Circularly polarized light beam splitter ~ ~
utilizing circularly polarized light characteristics of the -
cholesteric phase (cf. S.D. Jacob; SPIE. 37, 98, 1981).
This invention set out in the present application
as well as that set out in the parent application is
illustrated by the following Preparations and Examples, but
should not be construed to be limited thereto.
In the Examples, the positions of R and S in the
optically active compounds (A) are shown by the position
numbers in the following formula: ~
: '
R1 -0--~R2
O ~ (A)
,' '
The phase transfer temperature in the Examples was
measured by DSC (Differential Scanning Colorimetry) and with
a polarizing microscope. The symbols in the phase transfer
20 temperature mean as follows: v
C : Crystalline phase
SmA: Smectic A phase
SmC: Smectic C phase
','''''~
- 17
1 3 3 1 1 ~ 2
:~"
SmC*: Chiral smectic C phase
Sml: Non-identified ~mectic phase other than SmA,
SmC and SmC*.
N: Nematic phase - ;`
N*: Chiral nematic phase
I: Isotropic liquid
The chiral smectic C phase (SmC*) was further
confirmed by measuring the dielectric constant thereof.
Preparation o~ phenol derivatives
Preparation 1
Preparation of 4-[5-(4-n-octyloxyphenyl)-2
pyrimidinyl]phenol~
i) Preparation of 4-benzyloxyphenylamidine hydro-
chlor~de.
4-Cyanophenol (95.2 g), benzyl chloride (127 g)
and potassium carbonate (138 g) were refluxed in acetone
(160 ml for 5 hours. The product was separated by
filtration, concentrated under reduced pressure, and ~
; benzene thereto was added. The mixture was washed wit'n `
water, and benzene was distilled off under reduced
pressure to give 4-benzYloxybenzonitrile (141.38 g). The
, . . .
4-benzyloxvbenzonitrile (141 g) was dissolved in benzene
(338 ml), ethanol (270 ml) was added thereto and the - -
mixture was cooled to 0C. Into the resulting slurry
was bubbled hydrogen chloride gas (36 liters) with -
- 18 -
13311~2 : ~
stirring. Thereafter, the temperature was raised to 25C,
arld the mixture was allowed to stand for 2 days. The reaction
mixture was concentrated under reduced pressure to a 1/3
volume, and ether was added to ~he concentrated mixture . The
precipitated crystals were separated by suction filtration to
give an imide ester (183 g).
The above-obtained imide ester (183 g) was mixed with
ethanol (270 ml) to give a slurry, and a solution of ammonia ;~
(60.75 g) in ethanol (405 ml) was added thereto. After
allowing the mixture to stand at room temperature for 2 days,
the solvent was distilled off under reduced pressure to give
4-benzyloxyphenYlamidine h~drochloride (154.5 g). NMR
(DMSO-d6) ~: 5.19 (2H, s), 7.17 (2H, d, J=9.0 Hz), 7.35 (5H, ~
s) 7.86 (2H, d) ~ -
ii) Preparation of diethyl 4-n-octyloxyphenyl-
malonate:
4-Hydroxyphenylacetic acid (50.0 g) was dissolved in
ethanol (400 ml) and conc. sulfuric acid (0.5 ml) was added
thereto. The mixure was refluxed with stirring, and ethanol
was distilled off to give ethyl 4-hydroxyphenylacetate (60 g).
The ethyl 4-hydroxyphenylacetate (59 g) and sodium
ethoxide (22.4 g) were dissolved in ethanol (150 ml) and
n-octyl bromide (63.5 g) was added. The mixture was refluxed
for 3 ho~3rs, concentral:ed under reduced pressure, and ethYl
acetate was added there to to dissolve the oily
- 19 - ~: '
1 3 3 1 1 8 2
substance. The mixture was washed with water, dried over
anhydrous magnesium sulfate, distilled under reduced
pressure to remove ethyl acetate, and further distilled ;
under reduced pressure to give~ethyl -
4-n-octyloxyphenylacetate (79.6 g, b.p. 179C/0.1 mmHg).
The obtained ethyl 4-n-octyloxyphenylacetate (79
g), ethanol (140 ml), diethyl carbonate (300 ml) and sodium
ethoxide (19.3 g) were mixed, and the mixture was heated
with stirring while ethanol was distilled off. The reaction
10 mixture was transferred into ice water and acidified with
hYdrochloric acid. The organic layer was separated and the
solvent distilled off to give diethyl
4-n-octyloxvphenYlmalonate (91.6 g).
NMR (~DC13) ~: 0.5-2.0 (21H, m), 3.90 (2H, t, J=~.0 Hz),
~.16 (4H, a, J=7.2 Hz), a.52 (lH, s), 6.80 (2H, d, J=9.0
H~), 7.26 (2H, d, J=9.0 Hz) ~- -
iii) Preparation of 4-[5-(4-n-octyloxyphenyl)-2-
pyrimidinyl]phenol:
4-Benzyloxyphenylamidine hydrochloride (65.6 g) and
diethyl 4-n-octyloxyphenylmalonate (91.0 g) were dissolved in
methanol (500 ml) and thereto was added sodium methoxide (44.8
g). The mixture was refluxed with stirring for 9 hours.
After cooling, the reaction mixture was acidified with
sulfuric acid, and the precipitated crystals were separated by
suction filtration to give yellow crystals (77.7 g).
The ahove yellow crystals (77 g), phosphorus oxy-
chloride (310 ml) and N,N-diethvlaniline (4~.5 ml) were mixed
,.. ,.",., .,., .. ~., . - -.. -~
7 :
~ 1331182 :~
and refluxed with stirring for 26 hours. The excess
phosphorus oxychloride was d stilled off under reduced
pressure, the residue transferred into ice-water and
extracted with ether. The extract was washed with water and
distilled to remove ether to giv~ a crude product (70 g).
The product was recrystallized from ether to give a compound
(21 g) of the following ~ormula:
n-C8H1 70~N>~-OCH2Ph '
(Ph: phenyl)
NMR (CDC13) ~: 0.4-2.1 (15H, m), 3.99 (2H, t, J=6.o Hz),
5.09 (2H, s), 6.7-7.5 (11H, m), 8.38 (2H, d, J-9.0 Hz)
The colorless crystals obtained above (19.8 g),
ethanol (757 ml), magnesium oxide (11.4 g), water (57 ml)
and 10 ~ Pd-C (4 g)were heated with stirring at 60OC under
hydrogen atmosphere until a theoretical amount of hydrogen
was absorbed. The reaction mixture was filtered with suction,
and the filtrate was concentrated to give the desired 4-~5-
(4-n-octyloxyphenyl)-2-pyrimidinyl]phenol (7.7 g), m.p.
137C.
NMR (CDC13) ~: 0.5-2.1 (15H, m), 4.00 (2H, t, J-6.o Hz),
6.92 (2H, d, J-9.0 Hz), 7.01 (2H, d, JD9.0 Hz), 7.50 (2H, d,
J~9.0 Hz), 8.30 (2H, d, J-9.0 Hz), 8.94 (2H, s)
~Preparation o~ the compounds (B)]:
The starting optically active epichlorohydrins were
prepared by the processes as disclosed in Japanese Patent ~-
: ~
1331182 - ~
First Publication (Kokai) Nos. 132196/1986 and 6697/1987 and
in Japanese Patent Application No. 283393/1987. These were
R-(-)- and S-(l)-epichlorohydrins which have a chemical
purity of 98.5 % or more (measured by gas chromatographic
analysis) and an optical purity of 99 % or more [the
specific rotation, [a]D = -34.0, +34.0, c = 1.2, methanol,
respectively].
Preparation 2
To a mixture of the above R-(-)-epichlorohydrin ~;
(5.55 g), 4-(trans-4-n-pentylcyclohexyl)phenol (2.46 g) of
the following formula:
n C5H~1 ~ OH
and benzyltriethylammonium chloride (0.04 g) was added
dropwise aqueous sodium hydroxide (NaOH 0.45 g, water 15 ml)
with stirring at 600C over a period of 20 minutes, and the
mixture was further refluxed for one hour. The reaction
mixture was cooled to room temperature and extracted twice
with ether. The extract was washed once with a saturated
saline solution and distilled under reduced pressure to
remove the solvent. The residue was purified by silica gel
chromatography to give (S)-2,3-epoxypropyl 4-(trans-4-n-
pentylcyclohexyl)phenyl ether (1.8 g) of the following
formula:
n C5H11 ~
I - 22 -
13311~2
[a]2D5- +4.44 (c - 1.36, CH2Cl2)
NMR (CDC13) ~: 0.45-2.50 (21~- m), 2.50-3.00 (2H, m), 3.15- -
3.50 (1H, m), 3.70-4.30 (2H, m), 6.79 (2H, d, J=9.0 Hz),
7.09 (2H, d, J=9.0 Hz)
Preparation 3
~ The starting phenol derivative (2.50 g) of the
¦ following formula:
n-CgH17~0H
and the same R-(-)-epichlorohydrin (4.25 g) and benzyl-
10 triethylammonium chloride (20 mg) as used in Preparation 2
were dissolved in dimethylformamide (3 ml) and thereto was
added dropwise 24 wt.~ aqueous sodLum hydroxide t1.2 equi-
valent) at 60oC. After reacting at the same temperature for
40 minutes, the reaction mixture was cooled to room
15 temperature and extracted with ether. The extract was
distilled under reduced pressure to remove the solvent. The
residue was p~rified by silica gel chromatograrhy to give an S
isomer of glycidyl ether (1.62 g) of the following formula:
n-CgH17 ~
m.p. 90C
[]D5- ~4 44 (C ~ 1.01, CH2Cl2)
NMR (CDCl3) ~: 0.50-3.00 (19H, m), 3.10-3.50 (1H, m), 3.80- -~
4.30 (2H, m), 6.75-7.60 (8H, m)
:~?. ~
~ .-. . . : ~ ~ : :.:. :,: - : ~ .
- 23 -
1331182 ~
Preparation 4
The qtarting phenol derivative (10.0 g) of the
following formula:
n-C8H170~0H
and the same R-(-)-epichlorohydrin (18.6 g) as used in
Preparation 2, piperidine (367 ml) and dimethylformamide (1
ml)were mixed and stirred at 60OC for 10 hours. The
reaction mixture was distilled under reduced pressure to
remove the solvent and acetone ~5 ml) was added thereto.
Further, a 24 wt.~ aqueous sodium hydroxide (1.2 equivalent)
was added dropwise with stirring at room temperature, and the
mixture was reacted for 30 minutes. The reaction mixture was
adjusted to pH 7 with 2N hydrochloric acid and extracted
with ethyl acetate. The extract ~Tas dried over anhydrous
magnesium sulfate and distilled under reduced pressure to
remove the solvent. The residue was purified by silica gel
chromatography to give S isomer of glycidyl ether (1.58 g)
of the following formula~
n-CgH170~0/?~7
m.p. 131C
~]D ~ +3-3 (c - 0.55, CH2C12)
NMR (CDC13) ~: 0.70-2.20 (17H, m), 2.55-3.00 (2H, m), 3.15-
3.45 (lH, m), 3.75-4.20 (2H, m), 6.89 (2H, d, J~9.0 Hz),
6.92 (2H, d, J~8.4 Hz), 7.43 (4H, d, J-9.0 Hz) -
,. ~
-- 24 --
13311~2
Preparation 5
A phenol derivative (5.28 g) of the following
formula:
n C3H7 O~OH~
S-(I)-epichlorohydrin (11.55 g), potassium t-butoxide (3.00
g) and t-butyl alcohol (45 ml) were mixed and the mixture
stirred at 600C for 3 hours. The reaction mixture was
distilled under reduced pressure to remove the solvent and ~ ~
the residue extracted with chloroform. The extract was ~-
distilled under reduced pressure to remove the solvent. The
residue ~7as purified by silica gel chromatography to give R
isomer of glycidyl ether (5.82 g) of the following formula:
n-c3H7 0~ /~
[~]3D ~ ~5-71 (c ~ 1.66, CH2Cl2)
NMR (CDCl3) ~: 0.60-2.50 (17H, m), 2.60-2.95 (2H, m), 3.15-
3.60 (1H, m), 3.80-4.30 (2H, m), 6.76 (2H, d, Js8.4 Hz),
7.07 (2H, d, J~8.4 Hz)
Preparation 6
In the same manner as described in Preparation 5
except that a compound of the following formula: -~ ~ -
n-C1 2H25~0H
was used as the starting phenol derivative, there was prepared ~ ~
R i~omer of glycidyl ether of the following formula: ~-
n-C12H25~ ~7
1331182
m.p. 91C
[a]D ~ ~3-59 (c 3 1.07, CH2Cl2)
NMR (CDCl3) ~: 0.85-2.93 (27H, m), 3.34-3.40 (1H, m), 3.97-
4.27 (2H, m), 6.94-7.53 (8H, m)
Preparation 7
A mixture of the starting pnenol derivative (10 g)
of the following formula:
n-C8H17 ~ \ ~ OH,
the same R-(-)-epichlorohydrin (16.07 g) as used in
Preparation 2, 20 wt.% aqueous sodium hydroxide (7.33 g) and
dimethylformamide (20 ml) was heated with stirring at 60-70C
for one hour. The reaction mixture was cooled and thereto was
added water. The mixture was extracted with chloroform to
obtain a crude product (11.67 g). The crude product was
purified by silica gel chromatography to give an S isomer of
glycidyl ether (9.07 g) of the following formula:
n-C8H170 ~ N ~ O
m.p. 74C -~-
~a]D ~ +1.660 (c 3 1.02, CH2Cl2)
NMR (CDCl3) ~: 0.5-2.2 (15H, m), 2.6-3.0 (2H, m), 3.1-3.7
(1H, m), 3.8-4.4 (4H, m), 6.95 (2H, d, J=9.0 Hz), 8.26 (2H, ~ r
d, J-9.0 Hz), 8.36 (2H, s)
Preparation 8
A mixture of the starting phenol derivative (7.44
g) of the following formula:
~ ,.. .. . ... .. . .. ;.~ . . . .. . . . . . . . .
` - 25 - ~ ~
1331182
n-CgH170 ~ N ~ OH
as prepared in Preparation 1, the same R-(-)-epichlorohydrin
(9.16 g) as used in Preparation 2, 50 wt.% aqueous sodium
hydroxide (1.74 g) and dimethylformamide (77 ml) was stirred
at 60-700C for 3 hours. The reaction mixture was cooled and
wa~er was added. The mixture was extracted with
dichloromethane. The extracted product was purified by
silica gel chromatography to give an S isomer of glycidyl ether
(6.90 g) of the following formula:
n-CgH1 70~N>~ ~7
m.p. 198C ;~
~a]D ~ +0.95 (c = 1.04, CH2Cl2)
NMR (CDC13) ~: 0.6-2.1 (15H, m), 2.6-3.0 (2H, m), 3.2-3.5
(lH, m), 3.8-4.5 (2H, m), 6.99 (4H, d, J-9.0 Hz), 7.50 (2H,
d, J-9.0 Hz), 8.40 (2H, d, J-9.0 Hz), 8.90 (2H, s) -~;
Preparation 9
The starting phenol derivative (1.01 g) of the
following formula:
n-CgH17 ~ N ~ OH,
the same R-(-)-epichlorohydrin (2.01 g) as used in
Preparation 2 and benzyltriethylammonium chloride (16 mg)
were mixed and heated at 70C, and a 24 wt.% aqueous sodium
hydroxide (650 mg)was dropwise added thereto. The mixture was
stirred at 70C for 2 hours. The reaction mixture was cooled
~ ~ - 27
1331182
to room temperature and extracted three times with
chloroform. The.extract was dried over anhydrous magnesium
sulfate and distilled under reduced pressure to remove the
solvent. The residue was recrystallized from hexane to give an
S isomer of glycLdyl ether (380 mg) of the following
formula:
n-c8H17 ~ N
¦ m-p- 65C
1 10 ~]D = ~1.90 (c = 0.46, CH2C12)
NMR (CDCl3) C: 0.6-3.0 (19H, m), 3.2-3.6 (lH, m), 3.9-4.5
(2H, m), 6.99 (2H, d, J~9.0 Hz), 8.36 (2H, d, J-9.0 Hz),
8.55 (2H, s)
Preparation 10
-. :
A mixture of the starting phenol derivative (3.12
g) of the following formula:
n-C10H21 ~ \ ~ OH,
the same R-(-)-epichlorohydrin (4.627 g) as used in
Preparation 2, 50 wt.% aqueous sodium hydroxide (0.88 g) and
dimethylformamide (30 ml) was heated with stirring at 60C
~or 2.5 hourc. The reaction mixture was cooled and distilled
under reduced pre~sure to remove the solvent. The product
was purified by silica gel chromatography to give an ~ isomer of
glycidyl ether (2.96 g) of the following formula:
n-C10H21 ~ N ~ ~
- 28 - ~
1331~82
m.p. 650C
[a]D7= +2.47 (c = 1.02, CH2Cl2)
NMR (CDC13) ~: 0.6-2.0 (19H, m), 2.4-3.0 (4H, m), 3.2-3.5
(lH, m), 3.8-4.5 (2H, m), 6.98 (2H, d, J=9.0 Hz), 8.33 (2H,
d, J=9.0 Hz), 8.53 (2H, s)
Preparations 11-12
In the same manner as described in Preparations 2-
10, there were prepared optically active glycidyl ethers as
shown in Table 1, wherein R3, n, X and the symbol * are of
the following formula:
3 ~
R ~(0)n~X~0 ~0/ ~-
Table 1
~ ,: ~:;
No. R3 n - [a]D
_
r-~ r~~~ +4.780 (c = 1.08,
11 n-C6H13 0 ~ S CH2Cl2, 30C)
_ _
r~ +3.860 (c = 1.06,
12 n-CgH19 0 ~ S CH2Cl2, 31C)
Preparation of Compound (A)
Example 1
A dispersion of 50 wt.% sodium hydride (224 mg) in
m~neral oil was washed twice with dry ether and dry tetra-
hydrofuran (lO ~l) was added thereto. To the mixture was added
dropwise methyl 3-oxododecanate (1.07 g) with stirring at
40OC. After stirring the mixture for 5 minutes, (S)-2,3-
, , .. , ~ ~ . .... . . ... . . . . .
`~ - 29 -
1331182
epoxypropyl 4-(trans-4-n-pentylcyclohexyl)phenyl ether (1.41
g) as prepared in Preparation 2 was added dropwise to the
reaction mixture, and the mixture was refluxed for 20
hours. The reaCtion mixture was cooled to room temperature
and 4N hydrochloric acid was dropwise added thereto until the
pH was adjusted to l. The mixture was extracted twice with
ether, and the extract was washed once with a saturated saline
solution and distilled under reduced pressure to remove the sol-
vent. The residue was purified by silica gel chromatography to
give y-lactone derivatives (A) (430 m~ as a mixture of (2S,4S)
isomer : (2R, 4S) isomer = 50 : 50) of the following
f ormulae:
(2R, 4S) isomer:
n C5H11 ~ ~ C0-C9H19-n 1
(2S, 4S) isomer:
n~C5H11 ~ 0 ~ Co-c9H1g-n
H o H
NMR (CDCl3) ~: 0.87-1.86 (39H, m), 2.26-3.06 (3H, m), 3.73-
4.21 (3H, m), 4.85-4.90 (1H, m), 6.82 (2H, d, J-8.54 HZ),
7.12 (2H, d, J-8.55 Hz)
IR (KBr): 1778, 1720 cm~1
~a]D9~ +18.1 (c - 1.06, CHCl3)
23C 91C 100C
25 C e~ SmC* SmA ~ - I
>
24C 92C 101C
13311~2
The Y-lactone derivative~ prepared in the above
Example 1 were sealed in a cell made of glass wherein a
polyethylene terephthalate fiIm (thicl<ness 50 llm) was used
as a spacer. The cell was charged with an alternating current
of 70 Hz, and the relative dielectric constant was measured
by a bridge method. The results are shown in the accompanying ~ ;~
Fig. 1. It is clear from the test results that these
:
compounds have ferroelectric properties.
Example 2
In the same manner as described in Example 1 except
that methyl 3-oxononanate (1.14 g) was used instead of methyl
3-oxododecanate, it was reacted with (S)-2,3-epoxypropyl 4-
(trans-4-n-pentylcyclohexyl)phenyl ether as prepared in
Preparation 2 to give Y-lactone derivatives (A) (970 mg, as
a mixture of (2S, 4S) isomer: (2R, 4S) isomer ~ 50 : 50) of
the following formulae:
(2R, 4S) isomer:
A ~j\ /y~, C~C6H13-n
n-C5H11 ~ H 0- ~0
(2S, 4S) isomer:
A fi~ /~ CO-C6H1 3-n
n~c5H11 V~ H 0
NMR (CDCl3) ~: 0.87-1.88 (33H, m), 2.20-3.09 (3H, m), 3.72-
4.21 (3H, m), 4.77-4.99 (lH, m), 6.81 (2H, d, J-8.55 Hz),
7.10 (2H, d, J~8.55 Hz)
IR (KBr): 1762, 1716 cm
,
. 31 - .
1331182
29
~]D = +13-3 (c = 1.09, CHCl3)
10C 79C - 820C
C ~ S~C* ~ SmA
20C 80C 830C
Example 3 ~ : .
A dispersion of 50 wt.~ sodiu~ hydride (224 mg) in
mineral oil was washed twice with dry ether and dry tetrahydro~
furan (lO ml) was added thereto. To the suspension was
,; ,.,,...,~
added dropwise dimethyl n-butylmalonate (130 mg) with
stirring at 40C. After stirring the mixture for 5 minutes,
(S)-2,3-epoxypropyl 4-(trans-4-n-pentylcyclohexyl)phenyl
ether (1.41 g) as prepared in Preparation 2 was added :: ,.
dropwise to the mixture, and the mixture was refluxed with
stirring for 20 hours. The reaction mixture was cooled to
room temperature and 4N hydrochloric acid was added dropwise
thereto until the;pH was adjusted to l. The ~ixture was
extracted twice with ether, and the extract was washed once
with a saturated saline solution and distilled under reduced
pressure.to.remove,the.solvent. The residue was purified by
silica gel chromatography to give y-lactone derivatives, (2S,
4S) isomer and (2R, '4S) isomer -(50.mg. and 40 mg, respectively)
of the follo~in~ 'f'ormu-lae~
(2S, 4S) isomer~
~ fi~ /~\~c4H
n C5H11 ~ ~ 0 ` ~ H
' ;~
:: ~
:
- 32 -
1 3 3 1 1 ~ 2
Phase transfer temperature~
C ~ I
840C - --
[~]D - +33.45 (C = 0.658, CH2Cl2)
NMR (CDCl3) ~: 0.88-1.98 (30H, m), 2.38-2.67 (3H, m), 4.07-
4.13 (2H, m), 4.67-4.73 (1H, m), 6.83 (2H, d, J-8.3 HZ),
7.12 (2H, d, J=8.3 HZ)
IR (KBr): 1762 cm~
Elementary analysis for C26H4003:
Calcd. (%): C,77.95; H,10.07
Found (%): C,77.91; H,10.12
(2R, 4S) isomer:
n-C5H11 ~ ~ C4H9-n
Phase transfer temperature:
` C ~ I
850C
[a]D ~ +20.37 ~c - 1.05, CH2Cl2)
NMR (CDCl3) ~: 0.70-2.95 (33H, m), 4.00-4.25 (2H, m), 4.50-
4.95 (lH, m), 6.77 (2H, d, J-8.4 HZ), 7.11 (2H, d, J-8.4 HZ) -
IR (KBr): 1762 cm
Example 4
Dry l,2-dimethoxyethane (3 ml) was added to a disper-
sion of 50 wt.% sodium hydride (163 mg) in mineral oil, and
a solution of dimethyl n-heptylmalonate (716 mg) in 1,2-
dimethoxyethane (3 ml) was added thereto dropwise with
stirring at room temperature over a period of 10 minutes.
A~ter stirring the mixture for 5 minutes, a solution o~ (S)-
1~311~
2,3-epoxypropyl 4-ttrans-4-n-pentylcyclohexyl)phenyl ether
(940 mg) as prepared in Preparation 2 in 1,2-dimethoxyethane
(4 ml) was added dropwise to the mixture over a period of lO -
minutes, and the mixture was refluxed with stirring for 2.5 ,~
hours. The reaction mixture was cooled to room temperature
and 4N hydrochloric acid was dropwise added thereto until the
pH was adjusted to l. The mixture was extracted twice with
ether, and the extract was washed once with a saturated saline
solution and distillsd under reduced ~ressure to remove the
solvent. The residue was purified by silica gel chromatography
to give a y-lactone derivative, (2S, 4S) isomer (13 mg) of the
following formula:
(2S, 4S) isomer:
n-C5H1 1 0~o~/~C7H1 5-n
Phàse transfer temperature:
1 1 0C .~: :. "
[o~]D5~ +27.61 (c - 0.039, CH2C12)
NMR (CDC13) ~: 0.78-2.82 (39H, m), 3.97-4.19 (2H, m), 4.40-
4.82 (1H, m), 6.77 (2H, d, J--8.4 Hz), 7.08 (2H, d, J-8.4 Hz)
IR (K8r): 1758 cm 1 ~ `
Example 5 .~
: ,:-::
The optically active glycidyl ether prepared in
Preparation 2, i.e. (S)-2,3-epoxypropyl 4-(trans-4-n-pentyl-
cyclohexyl)phenyl ether (370 mg), potassium t-butoxide (151
mg), dimethyl methylmalonate (357 mg) and t-butyl alcohol (3
ml) were mixed, and the mixture refluxed with stirring for
- 34 - - ~ ~
1331182 ~
8 hours. The reaction mixture was cooled to room temperature
and 4N hydrochloric acid was added dropwise thereto until the pH
was adjusted to l. The mixture was extracted twice with ether,
and the extract was washed once with a saturated saline solution and
distilled under reduced pressure to remove the solvent. The
residue was purified by silica gel chromatography to give
Y-lactone derivatives, (2S, 4S) isomer and (2R, 4S) isomer
(60 mg and 50 mg, respectively) of the following formulae:
(2S, 4S) isomer:
10n-C5H11 {~o~ ~CH3
Phase transfer temperature: 18C 54C
C SmA c
- 101C >
~]D ~ +14.03 (c = 0.493, CH2Cl2)
NMR (CDCl3) ~: 0.88 (3H, t, J~7.0 Hz), 0.97-1.84 (21H, m),
2.39 (lH, t, J-12.2 Hz), 2.49-2.56 (lH, m), 2.69-2.76 (1H,
m), 4.04-4.12 (2H, m), 4.65-4.71 (1H, m), 6.83 (2H, d, J-8.7
Hz), 7.11 (2H, d, J-8.7 Hz)
IR (KBr): 1760 cm~1
MS m/e (relative intensity, %): 359 [(M+1)+, 26],
358 [M+, 100]
Theoretical weight as C23H3403:
Calcd.: 358.2509
Found: 358.2537
. 35 ~
1331182
,, .. .~.
(2R, 4S) isomer:
n~CsH11 ~ 0 ~ CH3
Phase transfer temperature:
C > I
101C
[~]D = +20.25 (c - 0.490, CH2Cl2)
NM~ (CDC13) ~: 0.89 (3H, t, J=6.8 Hz), 0.97-1.40 (17H, m),
1.84 (3H, d, J-10.7 Hz), 2.02-2.10 (lH, m), 2.39 (1H, t,
JA12.2 HZ), 2.45-2.51 (1H, m), 2.87-2.93 (1H, m), 4.01-4.12
(2H, m), 4.76-5.03 (1H, m), 6.79 (2H, d, J=8.6 HZ), 7.11
(2H, d, Ja8.6 Hz)
IR (KBr): 1760 cm~1
MS m/e (relative intensity, %): 359 ~(M+1)+, 26] `
358 ~M+, 100] ~;-
Example 6
The optically active glycidyl ether prepared in ;~
Preparation 5, i.e. (R)-2,3-epoxypropyl 4-(trans-4-n-propyl-
cyclohexyl)phenyl ether (416 mg), potassium t-butoxide (188
mg), dimethyl methylmalonate (443 mg) and t-butyl alcohol
(2.5 ml) were mixed, and the mixture was refluxed with
5tirring for 2 hour~. The reaction mixture was cooled to
room temperature and 4N hydrochloric acid was added dropwise
thereto until the pH was adjusted to l. The mixture was ex- ~-~
tracted three times with chloroform, and the extract was washed :~
once with a saturated saline solution and distilled under reduced
pressure to remove the solve~t. The residue-was purified by
.
: .. ~. ,...... ~ , , . : : : : -
- 36 -
1331182
silica gel chromatography to give Y-lactone derivatives,
(2R, 4R) isomer (77 mg) and (2S, 4R) isomer (86 mg) of the
following formulae~
(2R, 4R) isomer:
n-c3H7 0~ /y~CH3
~0 ~- :
Phase transfer temperature: 11C 17C
C~ SmA _ I :
117C
~]D ~ -16.820 (c - 0.98, CH2Cl2)
NMR (CDCl3) ~: 0.6-3.0 (23H, m), 4.0-4.2 (2H, m), 4.4-4.95
(1H, m), 6.76 (2H, d, J=8.0 Hz), 7.10 (2H, d, J=~8.0 Hz)
IR (KBr): 1762 cm~1
(2S, 4R) isomer: --
n-c3H7O~o ~CH3
H ~ H
Phase transfer temperature: 52C
C ~
139C
" 0 - .
[a]D ~ -27.82 (c - 1.03, CH2Cl2)
NMR (CDCl3) ~: 0.65-3.0 (23H, m), 4.0-4.2 (2H, m), 4.6-5.0
(1H, m), 6.76 (2H, d, J-8.0 Hz), 7.10 (2H, d, J-8.0 Hz)
IR (KBr): 1762 cm~1
Examples 7 to 11
In the same manner as described in Examples 1 to 6,
there were prepared optically active ~-lactone derivatives as ~:
shown .in Table 2, wherein R3. R4, n, m, and symbols 2* and
4* are of the following.formula: ~ -
13311~2
R3~o~ocH2~CO~R4
o
Table 2
No. R3 n 2' 4~ m R4 C Sm1 SmC* SmA N* I
7 n~C3H7 o S R 0 n-C9H19 . ~ , -~
11 1. R R 11 11 , __~ _ _ _ _ , .
8 n-C5H11 _ R S _ C2H5 , __~ _ _ _ _ ,
11 tl S S 1. 11 ~
_ _ .~ -:
9 n~C5H11 11 R S ll n-C11H23 122
11 11 S S 11 ll ~ ,,
, '
10 n-CgH19 1t R S ll CH3 , __~, - _ _ _ , ~
ll 11 S S ll 11 ~ --3' -- -- --` _ 1~
. :: ~ ~
11 n-C9H19 n R S n n-C5H11 , __~ _ _ _ _ ,
__ S S _ ~ - - _ _ ,
Example 12
The R isomer o~ glycidyl ether prepared in Prepara- : :~
tion 5 (380 mg), dimethyl malonate (274 mg), potassium t-
butoxide (163 mg) and t-butyl alcohol (2 ml) were mixed, and
1331182
the mixture was refluxed with stirring for 2 hours. The
reaction mixture was cooled to room temperature and 4N hydro-
chloric acid was d_opwise added thereto until the pH was
adjusted to l. The mixture was extracted three times with
chloroform, the extract washed with a saturated saline solution,
dried over anhydrous magnesium sulfate and distilled under re-
duced pressure to rera~ve the sol~ent. The residue was purified by
silica gel chromatography to give 4R isomer of methoxy- -
carbonyl-Y-lactone derivative (220 mg) of the following ~;
formula~
n~C3H7 O~o--~C02cH3
O
IR (KBr): 1781, 1744 cm~1
The above Y-lactone derivative (200 mg), magnesium
cP.loride (232 mg), dimethylacetamide (1.5 ml) and water (0.5
ml) were mixed and the mixture was refluxed with stirring fox
10 hours. The reaction mixture was cooled to room tempera- -~
ture and extracted twice with chloroform. The extract was
washed with a saturated saline solution, dried over
anhydrous magnesium sulfate and distilled under reduced ~-
pressure to remove the solvent. The residue was purified by
sllica gel chromatography to give 4R isomer of Y-lactone
de~ivative (145 mg) of the following formula:
- n-C3H7 O~(~ -~co
: ~
., ,, . :,: :. . - .~ . -
- 39 -
133~1~2
Phase transfer temperature:
C > I
74C
[a]3D = -18.640 (c z 1.27, CH2Cl2) ;
NMR (CDC13) ~: 0.65-3.45 (21H, m), 3.90-4.30 (2H, m), 4.55- ~ ;
5.00 (1H, m), 6.77 (2H, d, J=9.0 Hz), 7.11 (2H, d, J=9.0 Hz)
IR (KBr): 1778 cm 1 ~ .
Example 13
The S isomer of glycidyl ether prepared in . :;~
Preparation 3 (370 mg), diethyl n-propylmalonata (442 mg), ..
potassium t-butoxide (134 mg) and t-butyl alcohol (3 ml) were
mixed and the mixture was refluxed with stirring for lO hours.
The reaction mixture was cooled to room temperature and 4N
hydrochloric acid was dropwise added thereto until the pH was
adjusted to l. The mixture was washed with water and methanol to
lS give white crystals.. TIle Froduct was separated and purified by ~.
silica gel chromatography to give Y-lactone derivatives,
(2S, 4S) isomer (240 mg) and (2R, 4S) isomer (140 mg) of the .~.
following formulae:
(2S, 4S) isomer: .:. .
n- C8H1 7 ~ /;~K3 K7 ~n
Phase transfer temperature: . :
C > I
115C
~a]D ~ +32.67 (c ~ 1.081, CH2Cl2)
'` 25 NMR (CDC13) ~: 0.70-3.00 (27H, m), 4.00-4.25 (2H, m), 4.40- ~
4.85 (1H, m), 6.60-7.60 (8H, m) .,
IR (KBr): 1762 cm~1
Y.' , .,~. : , ,. ~,. ~: . . - ,.
-
i,~
j:", ~ . " ~, ;.. ,.: :: :;~ :
I` - 40 -
1~311~2
(2R, 4S) isomer:
n-CgH17 ~ 0 ~ C3H7-n
Phase transfer temperature:
C ~ I
117C
[~]D = +22.50 (c = 0.504, CH2Cl2)
NMR (CDCl3) ~: 0.70-3.00 (27H, m), 4.00-4.25 (2H, m), 4.50-
5.00 (1H, m), 6.60-7.60 (8H, m)
IR (KBr): 1762 cm~1
Example 14
- The S isomer of glycidyl ether prepared in
Preparation 4 (260 mg), dimethyl n-octylmalonate (269 mg),
potassium t-butoxide (90 mg) and t-butyl alcohol (2 ml) were
mixed and the mixture was refluxed with stirring for 13
hours. After the reaCtion, the reaction mixture was treated
in the same manner as described in Example 13 to give white
crystals. The product was separated and purified by silica
gel chromatography to give a Y-lactone derivative, (2S, 4S)
isomer (43 mg) of the following formula:
n-C8H17 ~ 0 ~ C8H17-n
Phase transfer temperature:
C ~ I
139C
[~]3 - +28.59 (c - 0.674, CH2Cl2) -
NMR (CDCl3) ~: 0.70-2.95 (37H, m), 3.80-4.20 (4H, m), 4.45-
4.90 (1H, m), 6.90 (4H, d, J-9.0 HZ), 7.42 (4H, d, J-9.0 Hz)
~ ::
,'t
. .~
~'.~': :. ' : ~ ': `:. :' ` '''
i ~ r
- 41 -
'' ' ' ' ' " '
1 3 3 1 1 8 2
~.
IR (KBr): 1760 cm~
Example.15 .
In the same manner as described in Example 13 -
except that the R isomer of glycidyl ether prepared in ~.
Preparation 6 was used as the optically active glycidyl ethex .~
and dimethyl n-butylmalonate was used instead of dimethyl n- -:
propylmalonate, there were prepared ~-lactone derivatives,
(2R, 4R) isomer and (2S, 4R) isomer of the following
formulae: ;~
t2R, 4R) isomer: :-~
n-C12H25 ~ 0 ~ C4H9-
Phase transfer temperature:
C ~ I .
130C ~:
[~]3D ~ -28.560 (c = 1.06, CH2Cl2) :~
NMR (CDCl3) ~: 0.85-2.69 (37H, m), 4.15-4.18 (2H, m), 4.71-
4.77 (1H, m), 6.95-7.53 (8H, m) :~
IR (KBr): 1764 cm~1 .
(2S, 4R) isomer: -
n-C12H25 ~ ~ C4H9~n
Phase transfer temperature: :
C ~ I
128OC .
~]3D ~ -22.98 (c ~ 1.07, CH2Cl2)
NMR (CDC13) ~: 0.85-2.85 (37H, m), 4.08-4.21 (2H, m), 4.81-
4.86 (1H, m), 6.93-7.52 (8H, m)
IR (KBr): 1760 cm~1
'~ .
~5 .. ~
- 42 -
1331182
Examples 16 to 22 :~
In the same manner as described in Examples 13 and
14, there were prepared optically active ~-lactone
derivatives as shown in Table 3, wherein R3, R4, n, m, and
symbols 2* and 4* are of the following formula~
R3~(O)n ~ OCH2 ~ (Co)m-R4
~0
Table 3
No ¦ n 2* 4* m R4 C Sm1 SmC* SmA N* I
_
16 n-C6H13 O R S O CH3 . ___~ - _ _ _ ,
15 1~ ll S S 1~ 1- , ___~ - - _ _ ,
17n-C6H13--R S _ n-C6H13. ___~ - - _ .
~- S S ll ~ ~_ _ _ _,
_ . 140
n-C6H13 ,~ R S " n-C10H21 . __~ - - _ _ ,
18 1~ .~ S S 1- 1- , 5 _ _ _ _ ,
,
19 n-C8H17 " R S n CH3 , __~ _ _ _ _ ,
.. ~ S S - - " ~ .
_ _ 127 :~
- to be continued - ~:
t ~
: :~
43 _ ~ ~
1331182 ;`:
Table 3 (continued)
Ex.R3 n 2* 4* m~4 C - Sml SmC* SmA N* I
No. . ~ ~:
_
20n-C8H17 O R S On-C9H19 . ~ _ _ _ _ ,
ll .. S S ,- " .~_ _ _ _ ,
i: '
21n-C8H17 ll R S "n-C12H25 . _~ _ _ _ _ , ;:~ :
ll ll S S ll ." .~"_ _ _ _,
_ .
22n-C8H17 1 R S llCH3 ~ _ _ _ _ ,
ll ll S S ll ll . _ ~ _ _ _ _,
_ 119
Example 23
lS In the same manner as described in Example 12
except that the S isomer of glycidyl ether prepared in
Preparation 11 (365 mg), dimethyl malonate (232 mg) and
potassium t-butoxida (138 mg) were used, there was prepared 4S
isomer of 2-(methoxycarbonyl)-Y-lactone derivative t226 mg)
of the following formula: ~ .
n C6H13~ ~,c02CH3 ~ :
H o
IR (KBr): 1740, 1768 cm~1
The Y-lactone derlvative of the above formula was
hydrolyzed and decarboxylated in the same manner as :
described in Example 12, to prepare 4S isomer of ?f_ ~
~ 44 - .:
1 3 3 1 1 ~ 2
lactone derivative (145 mg) of the following formula~
n-C6H13 ~ ~ 0
Phase transfer temperature:
C ~ I
138C -:
~]3 , +19.16 (c = 1.03, CH2Cl2)
NMR (CDCl3) ~: 0.80-1.75 (llH, m), 2.15-2.85 (6H, m), 4.05- ; :: .
4.30 (2H, m), 4.75-4.95 (1H, m), 6.85-7.60 (8H, m) :.
IR (KBr): 1764 cm~1 -
Example 24
The S isomer of glycidyl ether prepared in
Preparation 8 (518 mg), dimethyl n-octylmalonate (1170 mg)
and potassium t-butoxide (269 mg) were dissolved in dimethyl-
formamide (5 ml) and t-butyl alcohol (5 ml), and the mixture
.
was heated with stirring at 90C for 5 hours. After the
reaction, the reaction mixture was treated in the same manner
as described in Example 13 to give Y-lactone derivatlves
(742 mg) of the following formulae. The product was a : :
mixture of diastereomers and was. purified by silica gel
chromatography to give (2S, 4S) isomer and (2R, 4S) isomer.
(2S, 4S) iso~er:
n-C8H17 ~ \ ~ ~ 8H17-n
Phase transfer temperature: 152C 1850C
C ' ~ SmC* ~ I . .
161C 186C
, ~
,,~.~ . . . : -: :
~ 45
1331182 ~ ~
[C']D = +19.45 (c = O.613, CH2Cl2)
NMR (CDCl3) ~: 0.4-3.0 (35H, ~), 3.7-4.3 (4H, m), 4.71 (1H, -
m), 7.00 (4H, d, J=9.0 HZ), 7.50 (2H, d, J=9.0 HZ), 8.39
(2H, d, J-9.0 HZ), 8.89 (2H, s)
IR (nujol): 1778 cm 1 ~- ;
(2R, 4S) isomer:
n-C8H1 70~{N\)~O/~C8H1 7-
Phase transfer temperature: 11 2C 198C
c c SmC* ~
126 C 199 C
~ a ] D ~ ' 7 - 09 ( c ~ 0.115, CH2C l 2) ~ :
NMR (CDCl3) ~: 0.4-3.0 (35H, m), 3.7-4.3 (4H, m), 4.82 (lH,
m), 7.00 (4H, d, J39.0 Hz), 7.50 (2H, d, J~9.0 HZ), 8.39
(2H, d, J-9.0 HZ), 8.85 (2H, s)
IR (nujol): 1778 cm~1
Example 25 ;
The S isomer of glycidyl ether prepared in
Preparation 7 (1.0 g), dimethyl n-butylmalonate (1.056 g)
and pota~sium t-butoxide (63 mg) were dissolved in dimethyl- -
formamide (10 ml) and t-butyl alcohol (10 ml), and the :~
mixture was heated with stirring at 90C for 2 hours. After :
I the reaction, the reaction mixture was treated in-the same
manner as described in Example 13 to give Y-lactone
deriva~ives (626 mg). The product was a mixture of
~" , ~
l ~J;
46 :
1331182
diastereomers and was purified.by si~ic~ gel ch~amatosra~hy ~ :
to give (2S~ 4S).isomer and (2R~ 4S) isomer.
,_ _
(2S~ 4S) isomer:
n-C8H170 ~ \ ~ 0 ~ 4H9 -n .
Phase transfer temperature:
C ~ I
130C
[a]D - +41~04 (c a 0~137~ CH2Cl2)
NMR (CDC13) ~ 0~4~3~1 (27H~ m), 3~9~4~3 (4H~ m), 4~66 (1H~
lOm), 6~92 (2H~ d, J=9~0 Hz)~ 8~25 (2H~ d, J-9~0 Hz), 8~35
(2H~ s)
IR (nujol): 1776 cm~1
(2R, 4S) isomer:
~ N~ ~ 0"~ "~ ~ C4H9-n
n-C8H170 ~ N ~ H 0 ~ H
~. ~
Phase transfer temperature: :
C - ~ I
108C
~a]D7~ l25~02 (c ~ 0~23~ CH2Cl2)
NMR (CDCl3) ~ 0~4~3~1 (27H~ m), 3~9~4~3 (4H~ m), 4~77 (1H~
20m), 6~92 (2H~ d, Ja9~0 HZ)~ 8~25 (2H~ d, J~9~0 Hz)~ 8~35
(2H~ s)
IR (nujol): 1776 cm~
Example 26
. . In the same manner as described in Example 25
25except that the S lsomer of glycidyl ether prepared in
Preparation 10 was used as the optically active glycidyl
:
.,
-~ _ 47 _ ~
~3311~2
:
ether and dimethyl n-dodecylmalonate was used instead of ~ .
dimethyl n-butylmalonate, there were prepared Y-lactone
derivatives, (2S, 4S) isomer and (2R, 4S) isomer. :~
(2S, 4S) isomer:
n-c1oH21~p~>~o/~c12H2s-n
Phase transfer temperature:
C > I
[a]D = +26.01 (c = 1.062, CH2Cl2)
NMR (CDCl3) ~: 0.5-2.9 (49H, m), 4.19 (2H, m), 4.82 (1H, m), .
6.95 (2H, d, J=9.0 Hz), 8.32 (2H, d, J=9.0 Hz), 8.52 (2H, s) ~
IR (nujol): 1778 cm 1 ~ :
(2R, 4S) isomer:
10 21 {-N ~ ~ C12H25-
Phase transfer temperature: ;
C ~ I
89C
[a]3D ~ +17.12 (c - 0.398, CH2Cl2) -
NMR (CDCl3) ~: 0.5-2.9 (49H, m), 4.19 (2H, m), 4.81 (1H, m),
6.95 (2H, d, J-9.0 Hz), 8.32 (2H, d, J=9.0 Hz), 8.52 (2H, s)
20 IR (nujol): 1778 cm~1 ~ ;
Example 27
The S isomer of glycidyl ether prepared in :~
Preparation 9 (320 mg), dimethyl n-hexylmalonate (406 mg) :~
and potassium t-butoxide (116 mg) were dissolved in t-butyl
alcohol (3.5 ml), and the mixture was refluxed with stirring
-~" ;~ " ~ "~ ~ ~c ~
~ - 48 -
13311~2
for 6 hours. After the reaction, the reaCtion miXtUre was
treated in the 9ame manner as described in Example 13 to
give a mixture of diastereomers of Y-lactone derivative (270
mg, (2S, 4S)~(2R, 4S) - 9/1).
(2S, 4S) isomer:
n-C8H17 ~ \ ~ 0 ~ 6H13-n
(2R, 4S) isomer:
n~C3H17 { ~ ~ 0 ~ C6H13-n
Physical properties of the mixture:
Phase transfer temperature:
C ~ I
1160C
[~]D ~ +37 93 (c ~ 1.024, CH2Cl2)
NMR (CDCl3) ~: 0.50-2.80 (33H, m), 4.10-4.25 t2H, m),
4.45-4.85 (1H, m), 6.95 (2H, d, J-9.0 HZ), 8.34 (2H, d,
J-9.0 HZ), 8.52 (2H, s)
IR (nu jol ): 1778 cm~1
Examples 28 to 33
In the same manner as described in Examples 24 to
27~ there were prepared optically active y-lactone
derivatives as shown in Table 4, wherein R3, R4, n, e, m and
the symbolq 2* and 4* are of the follow~ng formula:
R3~(0)n ~ N\ ~ OCH2 ~ (Co)m-R4
N 0 ~
'.
,.. ,.. :'.` ~"` ''
1331182
~ . ~ ~ ~ ' ; ~
.'.~' l ~ ~.;
~ ~ ~ ~ !~ ~
L'~ ~_~ ~
~ ~ = e u :: . ~ ~:
_
er E ~ = = - = = = =
~ u~ tn tn u~ u~ u~ . ~
~`1 t~ u~ ~: tn ~ u~ ~
o - _ - _ _ _ _ ''; " ;'-'
rl ~ ~ ~
~r tD _ ID ~ _' C.C~ . . ~ =
. . . r C
,yO :~ ~ ~ '. ~'~
1331182 ~ :
_ , . .
~ . . . . . .,
E ., . l l
~ .
e ~ ~ . ~ ~.
. . l l
~ ~ lo ~ ;s~
_~
~ l ~
. ..
u~ e ~ = = = . ~.
~ ~ ~n ~o ~n ~n -,:
.~ ~: ~n c: ~n :
~ . ., ~.
U _ _
~ I~ :C~' ,~ .
~ ~ = - ~ =_ ~ .
l l . .. ,,~
. . .
X ~ ~ .~
~ Z r~ ~ ,."
` 1331~2
The Y-lactone derivative (2S, 4S) prepared in
Example 32 was sealed in a cell made of glass (thickness of - :
spacer: 22 ~m). The cell was char;3e;l wi~ll an ~l~ernating
current (70 Hz, 1V), and the relative dielectric constant
wa~ measured by a bridge method. The results are shown in
accompanying Fig. 2. It is clear from the test results that ~:
the compound has ferroelectric properties.
.
.
,.
:,