Note: Descriptions are shown in the official language in which they were submitted.
- 1 -
Brief Description of the Invention
A process is disclosed of the production of
non-conventional lobe oil base stocks or blending
stocks of very low pour point, pour point of about
-21'C or lower, preferably about -24'C or lower, said
pour points being achieved by conventional dewaxing
techniques without resort to deep dewaxing procedures,
and very high viscosity index (VI), VI's of about 130,
and higher, preferably 135 and higher by the isomeriza-
tion of waxes over isomerization catalysts in an
isomerization unit to a level of conversion such that
about 40o and less, preferably 15-350, most preferably
20-30o unconverted wax remains in the fraction of the
isomerate boiling in the lobe boiling range sent to the
dewaxing unit calculated as (unconverted wax)/(uncon-
verted wax + dewaxed oil)X100. For the purposes of
this application the amount of unconverted wax in the
370'C+ oil fraction is taken to be the amount of wax
removed or recovered from said oil fraction upon
dewaxing. The total product from the isomerization
(isom) unit is fractionated into a lobe oil fraction
boiling in the 330'C+ range, preferably in the 370'C+
range: This lobe oil fraction is solvent dewaxed
preferably using 20/80 mixture of MEK/MIBR and uncon-
verted wax is recycled to the isomerization unit.
Operating the isomerization unit at a level
of conversion such that the oil fraction sent to the
dewaxer Contains about 40 o and less wax, preferably 15-
......r... 1
~.3330~7
- 2 - '
35% wax, most preferably 20-30% unconverted wax goes
against the conventional wisdom of isomerization
operations. Lower levels of conversion, i.e. those
levels at which a substantial portion of wax remains
unconverted in the lobe oil fraction sent to the
dewaxer (and is subsequently recovered at the dewaxer
for recycle) are typically seen as favoring maximiza-
tion of lobe oil production since operation at lower
levels of conversion tend to favor the production of
lobe oil as compared to lower boiling fuels. The
amount of wax present in the oil sent to the dewaxer
normally should have no significant impact on the
dewaxability of the oil or the pour point which can be
achieved. There may be a point beyond which so much
wax is present as to be beyond the ability of the
dewaxer to handle the volume of waxy oil but this
tradionally is a materials handling problem and does
not affect the ability of the dewaxer to dewax oil to
the desired pour point using conventional dewaxing
techniques and temperatures. High levels of conversion
however tend to produce larger quantities of fuels.
It aas been discovered, that at low levels
of conversion difficulty is encountered in producing a
lobe oil having a pour point of at least -2loC from wax
isomerate. To produce a lobe oil fraction which can be
easily dewaxed to a pour point of at least -21°C it has
been found that the isomerizdtion c~s~:t should be run at
a level of wax conversion such that about 40g
and less preferably 15-350, most preferably 20-30o unconverted wax
is in the lobe fraction sent to the dewaxer.
Description of the Figures
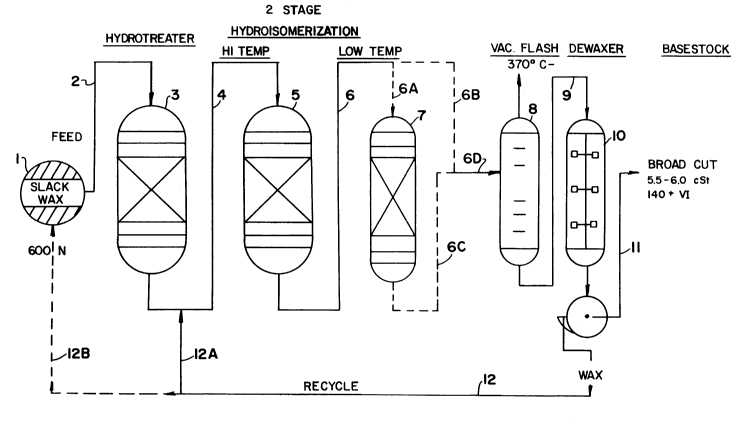
Figure 1 is a schematic of the step
sequences of the process of the present invention.
~,i~ ~ 333057
-3-
Figure 2 is a schematic of the step sequences of the process of the
present invention including the optional step of waxy fractionator bottoms
recycle.
Figure 3 illustrates the conversion behavior for three different Pt F/A1203
catalysts on a light slack wax (obtained from 600N raffinate) containing about
22% oil.
Detailed Description of the Invention
In Figure 3, the shape of the curves on the ternary diagram are a
measure of the selectivity -for converting wax into oil (e.g. 370°C+
oil) and
fuels (e.g. product boiling below 370°C-1. These curves were generated
by
running the catalysts on a 600N wax feed at conditions of 1000 psi H2, 0.9
V/V/hr, 5000 SCF/bbl, H2, and temperatures ranging from 280-360°C.
The most selective catalysts produce higher oil yields and less fuel at any
given residual wax level. Catalyst I (Catalyst 1 of Example 4 herein) produces
a maximum once through oil yield of almost 55 wt. % on feed. Catalysts II
(catalyst 8 of Example 5 herein) and III (comparison catalyst 1 of Example 5)
produce maximum once-through oil yields of about 50 and about 45 wt.%
respectively. Though the curves represent catalyst selectivity on a once
through operation, they are a good guide to performance in a recycle-to-
extinction process.
In principle a wax extinction process for maximizing tube yields would
involve operation at a very low severity i.e. where conversion to fuels is at
a
minimum. Under these circumstances the amount of
-4- ~~i333057
unconverted wax recycled to the isomerization reactor would be large and
differences in catalyst selectivity would be less important.
In practice however, it is not possible to operate in a low conversion
mode. Instead, the operating severity is governed by the need to make a low
pour ( <_ -21 °C pour point) oil. It has been discovered that low pours
cannot
be achieved from isomerates made at low conversion. This is unexpected since
with natural oils the amount of wax present did not effect the ability to
dewax
the oil to low target pour point. A critical determinant in reaching low pours
is
that the amount of wax remaining in the 370C + fraction obtained from
isomerization should not exceed 40% and for lower pour points may have to
be as little as 25%. To maximize yield in this situation the choice of
catalyst
becomes important.
As wax in 370C+ oil product declines from 50 to 25%, (Figure 3), the
ratio of oil to fuels decreases. This trend is much more pronounced with the
least selective catalyst III. This is also illustrated in the Table blow. All
yields
are based on a once through operation.
CataI~L
Wax in 25 40 50 25 40 50 25 40 50
oil product
Wax left 18.5 34 44.5 17 32 43 12 30 42
(% of feed)
Oil yield 54.5 50 44.5 49.5 48 43 36 45 42
(% of feed)
Fuels
Yield 27.0 16 10 33.5 20 14 52 25 16
( % of feed
O~r i 333057
-5-
The full recycle oil yields for catalysts I, II and III, in which wax is
recycled to extinction, can be predicted assuming the same conversion
selectivity applies for recycled wax. On this basis, the yield distinctions
between catalysts are even more pronounced.
Catalyst -I- -II- -III-
Wax in oil 25 40 50 25 40 50 25 40 50
(once-through)
Predicted extinction
recycle yield of
370C + oil 69 78 82 60 72 79 40 62 72
At a 25% wax in oil conversion level, Catalyst I is actually 70% more
selective for oil than Catalyst III in an extinction recycle process. Thus
small
differences in catalyst selectivity identified in once through operations can
translate into significant yield differences in a recycle process.
Another way to express the different performance of each catalyst is to
determine the reaction severity required to achieve a particular target oil
yield
in a full recycle operation. For the target of 70% oil yield shown in Figure 1
catalyst I converts much more wax into oil than does catalyst III (i.e. there
is
less unconverted wax remaining in catalyst I product). In this case, catalyst
III
cannot simultaneously meet a target yield of 70% oil and a target of _< -21
°C
pour point, since the amount of unreacted wax in oil exceeds 40%.
_6 _ f~_~i_~;~=;
The wax which is isomerized may come from any
of a number of sources. Synthetic waxes from Fischer-
Tropsch processes may be used, as may be waxes recover-
ed from the solvent or autorefrigerative dewaxing of
conventional hydrocarbon oils as well as mixtures of
these waxes. Waxes from dewaxing conventional hydro-
carbon oils are commonly called slack waxes and usually
contain an appreciable amount of oil. The oil content
of these slack waxes can range anywhere from 0 to 45%
or more, usually 5 to 30% oil. For the purposes of
this application, the waxes are divided into two
categories: (1) light paraffinic waxes boiling in the
range about 300-580'C and (2) heavy micro waxes having
a substantial fraction (>50%) boiling above 600'C.
Isomerization is conducted over a catalyst
containing a hydrogenating metal component typically
one from Group VI or Group VIII or mixtures thereof,
preferably Group VIII, more preferably noble Group VIII
most preferably platinum on a halogenated refractory
metal oxide support. The catalyst typically contains
from 0.1-5.0 wt.% metal, preferably 0.1 to 1.0 wt.%
metal, most preferably 0.2-0.6 wt.% metal. The refrac-
tory metal oxide support is typically a transition e.g.
gamma or eta alumina and the halogen is most usually
fluorine.
~ J .~~ 5 l~ "~-
A preferred catalyst con-
tains a hydrogenation metal component which is a Group
VIII metal or mixtures thereof, preferably noble Group
VIII metal, most preferably platinum on a fluorided
alumina or material containing alumina, preferably
alumina or material consisting predominantly (i:e.
>50%) of alumina, most preferably gamma or eta alumina
wherein said catalyst in its as introduced to waxy feed
form is characterized by possessing (1) a hydrate level
of 60 or less, preferably 10 to 60 determined as the
relative amount of hydrate represented by a peak in the
X-ray diffraction (XRD) pattern at 20 - 5.66~r when a
hydrate level of 100 corresponds to the XRD peak height
exhibited by a standard material constituting 0.6 wt%
Pt on 150 m2/g ~ alumina containing 7.2 wt% F wherein
the fluorine has been deposited using an aqueous
solution containing a high concentration of HF, i.e. 10
wt% HF and greater, preferably 10 to 15 wt% HF and the
material dried at 150'C for 16 hrs: (2) a surface
nitrogen content N/A1 ratio of 0.01 or less, preferably
0.007 or less, most preferably 0.004 or less as deter-
mined by X-ray photoelectron spectroscopy (XPS): (3) a
bulk fluorine concentration of about 2 to 10 wt% and
(4) a surface fluorine present in a layer extending
frog the surface of the particle (e. g. 1/16 inch
extrudates) to a depth of 1/100 inch, of less than 3
wt%, preferably less than 1 wt%, most preferably less
than 0.5 wt% fluorine in that zone provided that the
surface fluoride concentration is less than the bulk
fluoride concentration.
The fluoride content of the catalyst can be
determined in a number of ways.
' 8 ' t J-) i ' l
J li ~:i ~
One technique analyzes the fluorided catalyst
using oxygen combustion methodology which is well
established in the literature. Approximately 8-l0 mgs
of sample is mixed with 0.1 g benzoic acid and 1.2 gms
of mineral oil in a stainless steel combustion capsule
which is mounted in a 300 mL. Parr oxygen combustion
bomb. The "sample" is purged of air and subsequently
combusted under 30 Atms of pure oxygen. Combustion
products are collected in 5 mL. of deionized water.
Once the reaction has gone to completion (about 15
minutes), the absorbing solution is quantitatively
transferred and made to fixed volume.
Fluoride concentration of the sample is
determined by ion chromatography analysis of the
combustion product solution. Calibration curves are
prepared by combusting several concentrations of
ethanolic KF standards (in the same manner as the
sample) to obtain a 0-10 ppm calibration range.
Fluoride concentration of the catalyst is calculated on
an ignition-loss-free-basis by comparison of the sample
solution response to that of the calibration curve.
Ignition loss is determined on a separate sample heated
to 800 degrees F for at least 2 hours. Ion chromato-
graphic analysis uses standard anion conditions.
Another procedure employs the use of fluoride
distillation with a titrimetric finish. Fluorides are
converted into fluorosilicic acid (H2SiF6) by reaction
with quartz in phosphoric acid medium, and distilled as
such using super heated steam. This is the Willard-
Winter-Tananaev distillation. It should be noted that
the use of super heated, dry (rather than wet) steam is
crucial in obtaining accurate results. Using a wet
steam generator yielded results 10-20% lower. The
collected fluorosilicic acid is titrated with standard-
ized sodium hydroxide solution. A correction has to be
' . _ 9 _ ~ j~j.J~
made for the phosphoric acid which is also transferred
by the steam. Fluoride data are reported on an igni-
tion-loss-free-basis after determination of ignition
loss on a sample heated to 400 degree C for 1 hour.
~ p~erred catalyst is a catalyst prepared by a
process involving depositing a hydrogenation metal on
an alumina or material containing alumina support,
calcining said metal loaded support typically at
between 350 to 500'C, preferably about 450 to 500'C for
about 1 to 5 hrs, preferably about 1 to 3 hrs and
fluoriding said metal loaded support using a high pH
fluorine source solution to a bulk fluorine level of about
8 wt~ or less , ( l . a . 2 .to 8 wt=°s) preferably about 7~ wt~ or
less,
. said high pH source solution being at a pH of 3.5 to
4.5-and preferably being a mixture of NH4F and HF
followed by rapid drying/heating in a thin bed or
rotary kiln to insure thorough even heating in air,
oxygen containing atmosphere or an inert atmosphere to
a temperature between about 350 to 450'C in about 3
hours or less, preferably 375 to 400'C and holding at
the final temperature, if necessary, for a time suffi-
cient to reduce.the hydrate and nitrogen content to the
aforesaid levels, e.g. holding for 1 to 5 hours or
using a low pH fluorine source solution having a pH of
less than 3.5 to a bulk fluorine level of about 10 wt~ or less;
( l . a . 2~ to 10 wtg ) preferably about- 8 wt~ or less followed by
drying/heating in a thin bed or rotary kiln to a
temperature of about 350 to 450'C, preferably 375 to
425'C and holding, if desired, at that temperature for
1 to 5 hours, in air, an oxygen containing atmosphere,
or inert atmosphere. The alumina or=alumina containing
support material is preferably in the form of extru-
dates and are preferably at least about 1/32 inch
across the longest cross sectional dimension. If the
catalyst is first charged to a unit, heating a dense
l~ ~ .~ .~e ,~ i.i '_;: 7
bed charge of catalyst will be for a longer period,
longer than 5 hours, preferably longer than 10 hours
and preferably at temperatures of 400 to 450'C.
The above catalysts typically contain from
0.1 to 5.0 wt% metal, preferably 0.1 to 1.0 wt% metal,
most preferably 0.2 to 0.6 wt% metal.
The dried/heated catalyst has a surface
nitrogen content N/Al of 0.01 or less by X-ray photo-
electron spectroscopy (XPS), preferably an N/A1 of
0.007 or less, most preferably an N/A1 of 0.004 or less
by XPS.
The catalyst, following the above recited
heating step, can be charged to the isomerization
reactor and brought quickly up to operating conditions.
Alternatively following the above recited heating step
the catalyst prepared using the pH 3.5 - 4.5 solution
technique can be activated preferably in pure or plant
hydrogen (60-70 vol% H2) at 350 to 450°C, care being
taken to employ short activation times, from 1 to 24
hours, preferably 2 to 10 hours being sufficient. Long
activation times (in excess of 24 hours) have been
found to be detrimental to catalyst performance. By
way of comparison, catalysts made using solutions of pH
less than 3.5 can be activated in pure or plant
hydrogen at 350 to 500'C for from 1 to 48 hours or
longer. In fact, if catalysts prepared using solutions
of pH 3.5 or less are not heated first, then it is
preferred that they be subsequently activated at more
severe conditions, i.e. for longer times and/or at
higher temperatures. On the other hand, if they are
heated first, then moderate activation procedures
similar to those employed with catalysts made from the
higher pH solution treatment will suffice.
_ 11 _ ! ~~ ) .~ r i ;..i
A typical activation profile shows a period
of 2 hours to go from room temperature to 100'C with
the catalyst being held at 100'C for 0 to 2 hours then
the temperature is raised from 100 to about 350 over a
period of 1 to 3 hours with a hold at the final temper-
ature of from 1-4 hours. Alternatively the catalyst
can be activated by heating from room temperature to
the final temperature of 350-450'C over a period of 2-7
hours with a hold at the final temperature of 0-4
hours. Similarly activation can be accomplished by
going from room temperature to the final temperature of
350-450°C in 1 hour.
It is possible to dispense with a separate
activation procedure entirely, (provided the catalyst
has first been heated in air). In these instances, the
calcined catalyst is simply charged to the reactor,
heated to just above the melting point of the wax feed,
feed and hydrogen introduced onto the catalyst, and
thereafter the unit brought quickly up to operation
conditions.
Another preferred catalyst comprises a hydrogenating
metal on fluorided alumina or material containing
alumina support made by depositing the hydrogenation
-12 - ~ ~, ~~ 333057
metal on the support and fluoriding said metal loaded support using acidic
fluorine sources such as HF by any convenient technique such as spraying,
soaking, incipient wetness, etc. to deposit between 2-10%F preferably 2-8%F.
Following halogenation the catalyst is dried, typically at 120°C
and then
crushed to expose inner surfaces, the crushed catalyst and is double sized to
remove fines and uncrushed particles. This sieved catalyst is 1 /32 inch and
less and typically from 1 /64 to 1 /32 inch in size across its largest cross-
sectional dimension.
The starting particle or extrudate may be of any physical configuration.
Thus particles such as cylinders, trilobes or quadri lobes may be used.
Extrudates of any diameter may be utilized and can be anywhere from 1 /32 of
an inch to many inches in length, the length dimension being set solely by
handling considerations. It is preferred that following sizing the particle
have
a length smaller than the initial extrudate diameter.
Following deposition of the hydrogenation metal and the fluoriding of the
particle or extrudate, the particle or extrudate is crushed or fractured to
expose
inner surfaces.
The crushing is conducted to an extent appropriate to the particle or
extrudate with which one is starting. Thus, an extrudate which is 1 food long
and 1 /16 inch in diameter would be sized into pieces which range anywhere
from 1 /64 to 1 /32 inch across its longest cross-sectional dimension.
Similarly,
if the extrudate is only 1 /16 inch to begin with it will be enough simply to
break
it in half, into two 1 /32 inch pieces, for example.
~,~ 1333057
-13-
Alternatively, one can take a metal loaded support particle which is
already about 1 /32 inch in size or smaller and fluoride it as described above
using HF.
Generally, therefore, the sized material will range in size between about
1 /64 to 1 /32 inch in size.
The uncalcined sized catalyst is activated in a hydrogen atmosphere such
as pure hydrogen or plant hydrogen containing 60 to 70 vol% hydrogen by
heating to 350 to 500°C, preferably 350 to 450°C for from 1 to
48 hours or
longer. The hydrogen activation profiles described above may similarly be
employed here.
~~i333057
- 14-
This sized catalyst is unexpectedly superior for wax isomerization as
compared to the uncrushed particle or extrudate starting material. It has also
been discovered that 370°C+ oil products made using the sized catalyst
as
compared to the uncrushed or extrudate material starting with wax possessing
about 5-10% oil exhibit higher VI's than do 370°C+ oil products made
starting
with wax possessing 0% oil (on the one hand) and about 20% oil (on the
other). Therefore, to produce products having the highest VI one would
isomerize wax having from 5-15% oil, preferably 7-10% oil using the "sized"
catalyst produced using HF.
As one would expect isomerization catalysts are susceptible to
deactivation by the presence of heteroatom compounds (i.e. N or S
compounds) in the wax feed so care must be exercised to remove such
heteroatom materials from the wax feed charges. When dealing with high
purity waxes such as synthetic Fischer-Tropsch waxes such precautions may
not be necessary. In such cases subjecting such waxes to very mild hydro-
treating may be sufficient to insure protection for the isomerization
catalyst.
On the other hand waxes obtained from natural petroleum sources contain
quantities of heteroatom compounds as well as appreciable quantities of oil
which contain heteroatom compounds. In such instances the slack waxes
should be hydro-treated to reduce the level of heteroatoms compounds to levels
commonly accepted in the industry as tolerable
~Gi533057
-15-
for feeds to be exposed to isomerization catalysts. Such levels will typically
be
a N content of about 1 to 5 ppm and a sulfur content of about 1 to 20 ppm,
preferably 2 ppm or less nitrogen and 5 ppm or less sulfur. Similarly such
slack
waxes should be deoiled prior to hydrotreating to an oil content in the range
of
0-35% oil, preferably 5-25% oil. The hydrotreating step will employ typical
hydrotreating catalyst such as Co/Mo, Ni/Mo, or Ni/Co/Mo on alumina under
standard, commercially accepted conditions, e.g., temperature of 280 to
400°C, space velocity of 0.1 to 2.0 V/V/hr, pressure of from 500 to
3000 psig
H2 and hydrogen gas rates of from 500 to 5000 SCF/b.
In the present invention isomerization of waxes over the above
particularly recited isomerization catalysts is conducted to a level of
conversion
which optimizes the conversion of wax to lube range materials while minimizing
production of fuels range materials (i.e. 370°C- products) yet
producing an
overall tube oil product which does not contain more unconverted wax than can
be efficiently handled by the solvent dewaxing unit i.e. 25-40% wax to the
dewaxer.
Isomerization is conducted under conditions of temperatures between
about 270 to 400°C, preferably 300-360°C, pressures of 500 to
3000 psi H2,
preferably 1000-1500 psi H2, hydrogen gas rates of 1000 to 10,000 SCF/bbl,
and a space velocity in the range 0.1-10 v/v/hr, preferably 1-2 v/v/hr.
~,A r 333057
- 16-
Following isomerization the isomerate is fractionated into a tubes cut and
fuels cut, the tubes cut being identified as that fraction boiling in the
330°C+
range, preferably the 370°C+ range or even higher. This tubes fraction
is then
dewaxed to a pour point of about -21 °C or lower. Dewaxing is
accomplished
by techniques which permit the recovery of unconverted wax, since in the
process of the present invention this unconverted wax is recycled to the
isomerization unit. It is preferred that this recycle wax be recycled to the
main
wax reservoir and be passed through the hydrotreating unit to remove any
quantities of entrained dewaxing solvent which solvent could be detrimental to
the isomerization catalyst. Alternatively, a separate stripper can be used to
remove entrained dewaxing solvent or other contaminants. Since the
unconverted wax is to be recycled dewaxing procedures which destroy the wax
such as catalytic dewaxing are not recommended. Solvent dewaxing is utilized
and employs typical dewaxing solvents. Solvent dewaxing utilizes typical
dewaxing solvents such as C3-Cg ketones (e.g. methyl ethyl ketone, methyl
isobutyl ketone and mixtures thereof), Cg C,o aromatic hydrocarbons (e.g.
toluene) mixtures of ketones and aromatics (e.g. MEK/toluene),
autorefrigerative solvents such as liquified, normally gaseous C2-CQ
hydrocarbons such as propane, propylene, butane, butylene and mixtures
thereof, etc. at filter temperature of -25 to -30°C. The preferred
solvent to
dewax the isomerate especially isomerates derived from the heavier waxes (e.g.
bright stock waxes) under miscible conditions and thereby produce the highest
yield of dewaxed oil at a high filter rate is a mixture of MEK/MIBK (20/80
v/v)
used at a temperature in the range -25 to -30°C. .Pour points lower
than -21 °C
can be achieved using lower filter temperatures and other ratios of said
solvents
but a penalty is paid because
133307
- 17 -
the solvent-feed systems becomes immiscible, causing
lower dewaxed oil yields and lower filter rates.
Further, when dewaxing isomerate made from a microwax,
e.g. Bright Stock slack wax it is preferred that the
fraction of the isomerate which is sent to the dewaxer
is the "broad heart cut" identified as the fraction
boiling between about 330 to 600'C, preferably about
370-580'C. After such fractionation the fraction sent
to the dewaxer has about 40 o and less unconverted wax _ The
heavy bottoms fraction boiling above about 580 to 600'C
contains appreciable wax and can be recycled to the
isomerization unit directly. However if any hydro-
treating or deoiling is deemed necessary or desirable
then the fractionation bottoms are reisomerized by
being first sent to the fresh feed reservoir and
combined with the wax therein.
r' :_
f,~~;_
_ 18
It has also been found that prior to fractio-
nation of the isomerate into various cuts and dewaxing
said cuts the total liquid product (TLP) from the
isomerization unit can be advantageously treated in a
second stage at mild conditions using the isomerization
catalyst or simply noble Group VIII on refractory metal
oxide catalyst to reduce PNA and other contaminants in
the isomerate and thus yield an oil of improved day-
light stability.
In that embodiment the total isomerate is passed
over a charge of the isomerization catalyst or over just
noble Gp VIII on e.g. transition alumina. Mild conditions
are used, e.g. a temperature in the range of about 170-270°C,
preferably about 180 to 220°C, at pressures of about 300 to
1500 psi H2. Preferably 500 to 1000 psi H2, a hydrogen gas
rate of about 500 to 10,000 SCF/bbl, preferably 1000 to
5000 SCF/bbl and a flow velocity of about 0.25 to 10
v/v/hr., preferably about 1-4 v/v/hr. Temperatures at
the high end of the range should be employed only when
similarly employing pressures at the high end of their
recited range. Temperatures in excess of those recited
may be employed if pressures in excess of 1500 psi are
used, but such high pressures may not be practical or
economic.
The total isomerate can be treated under
these mild conditions in a separate, dedicated unit or
the TLP from the isomerization reactor can be stored in
tankage and subsequently passed through the aforemen-
tioned isomerization reactor under said mild condi-
tions. It has been found to be unnecessary to frac-
tionate the 1st stage product prior to this mild 2nd
~G~333057
-19-
stage treatment. Subjecting the whole product to this mild second stage
treatment produces an oil product which upon subsequent fractionation and
dewaxing yields a base oil exhibiting a high level of daylight stability and
oxidation stability. These base oils can be subjected to subsequent
hydrofinishing using conventional catalysts such as KF-840 or HDN-30 (e.g.
Co/Mo or Ni/Mo on alumina) at conventional conditions to remove undesirable
process impurities to further improve product quality.
Figures 1 and 2 present schematic representations of preferred
embodiments of the wax isomerization process.
In Figure 1, slack wax feed, derived from, for example a lighter oil such
as 600N oil or lighter is fed from reservoir (1 ) to a hydrotreater (3) via
line 2
wherein heteroatom compounds are removed from the wax. This hydrotreated
slack wax is then fed via line 4 to the isomerization unit (5) after which the
total liquid product is fed either directly via lines 6, 6B and 6D to the
separation
tower (unit 8) for fractionation into a tubes fraction boiling above about
370°C+ and a light fraction boiling below about 370°C- or, in
the alternative
the TLP from the isomerization unit is fed first via lines 6 and 6A to a low
temperature, mild condition second stage treating unit (unit 7) wherein the
TLP
is contacted with the isomerization catalyst or simply a noble Group VIII
metal
on alumina catalyst to produce a stream which is then sent via lines 6C and 6D
to the fractionation tower (unit 8). In either case the tube steam boiling in
the
370°C+ range is then forwarded via line 9 to the. solvent dewaxer (unit
10) for
the separation of waxy constituents therefrom, the dewaxed oil fraction being
recovered via
~~~ i 333057
-20-
line-11 and if necessary forwarded to other conventional treatment processes
normally employed on base stock or blending stock oils. The recovered wax
is recycled either directly via line 12 and 12A to the slack wax stream being
fed
to the isomerization unit or it is recycled to the wax reservoir (1 ) via line
12B
for passage through the hydrotreater prior to being recycled to the
isomerization
unit.
In Figure 2 the wax processing stream is much like that of Figure 1, the
main differences being that Figure 2 represents the scheme for handling
heavier
slack wax feeds, such as a wax feed derived from Bright Stock oil. In such a
case the wax from reservoir 1 is fed via line 2 to the hydrotreater (3) prior
to
being sent via line 4 to the isomerization unit (unit 5) after which it is
either fed
via lines 6 and 6A to a low temperature mild condition second stage treating
unit (unit 7) wherein it is contacted with a further charge of isomerization
catalyst or simply noble Group VIII metal on alumina and fed via lines 6C and
6D to the fractionator tower (unit 8), or fed directly via lines 6, 6B and 6D
to
the fractionation tower (unit 8). In the fractionation tower the isomerate
made
using the heavy wax is fractionated into a light fraction boiling in the
370°C-
(a fuels cut) a tube cut boiling in the 370°C+ range and a bottoms
fraction
boiling in the 580°C+ range. The tubes fraction, a broad cut boiling in
the
370°C to 580°C range is sent via line 9 to the dewaxer (unit 10)
as previously
described. The 580°C+ bottoms fraction contains appreciable wax and is
recycled via line 13, 13A, 13B and 4 to the isomerization unit (5). This
bottoms fraction optionally can be combined via line 13 and 13C with the wax
in line 12 recovered from the dewaxing unit (10) in which case this total
recycled
.~1~ ~ 533057
-21 -
stream can be fed directly to the isomerization unit via lines 12A, 13B and 4
or it can be sent to the wax reservoir (1 ) via lines 12B for treatment in the
hydrotreater prior to being fed to the isomerization unit.
The invention will be better understood by reference to the following
examples which either demonstrate the invention or are offered for comparison
purposes.
EXAMPLES
Examale 1
Catalyst 1
A synthetic hydrocarbon synthesis wax (a Fischer-Tropsch wax,
characterized as being 100% 370°C+ material possessing a melting point
in
the range 104 to 110°C, a mean carbon number (from viscosity data) of
about
65 carbons, a boiling range of about 450-650°C (initial to 70 LV% off
by GCD)
and a kinematic viscosity of 9.69, was isomerized over a 14/35 meshed
platinum on fluorided alumina catalyst made by first fluoriding a platinum
loaded
1 /16" alumina extrudate (0.6 wt. % platinum) using a 11.6 wt% aqueous HF
solution (by soaking) after which the fluorided metal loaded extrudate was
washed with 10 fold excess water and dried at 150C in vac. oven. The metal
loaded fluorided extrudate was not calcined. It was crushed to produce
particles of about 1 /30" (meshed to 14/35). Catalyst 1 had a fluorine content
of 8.3 wt%.
The sized catalyst, Catalyst 1, was activated by heating to 450°C
in 50
psi flowing H2 in the following manner: room temperature to 100°C in 2
hours,
hold at 100°C for 1 hour; heat from 100°C to 450°C in 3
hours, hold at
450°C for 1 hour.
~3330~7
- 22 -
U
0
o
r~ W
z
00 -. w
v1 N x O b
W o c9 a O u~ o o ao ~ o
0 o c~
0
~n
~n
O H ~O O N N r1 u1 c"1
C~ M
O
I
o x ao E~ o I rmo ~
mn ~ w
o t
~
~ -- W M
oo
ao
cn x >,
o~ x
~
3
H J'
(x ~' ~ O
O
O
I~ .-. O ,'7
tf7
x o g ao vo r~ M ~ o~ u~ co
0 c~ W cn
o
c~
d
o O c~ W o .-i .-I a
r1 r~ ~o
o
I
I
o a o M t~ I
~
~mn
N
I
,~ M
O
U
H
H
W
a~ x
r1
z --
N
H cn x
x s~ -
U O ~ m
~ o o ~ o
O - .s~ \ .~ - U
x ~o w w in >', m - o +~ 0 0
E s~ ~ U h N C N o sy o 0
t O c~t~ U1 O >'I r-I~ d' r~ b
.r: r-I
fx U x ~ v 1--IO -rlp u U ~r u O
.I
~ ~ ~ O v O
r1 ?~ ~ - +~ S. H ~ ~ O
N ~ .t
U O m +~ +~ N b s.~ u1 A, D t!~ U7 ~
~ 'O +
U1 O 11-~ ~ a +~ >~ O U U U :~ O
O U
H ri U S-i U O t1 r1 1.i H U
f~.t o
W +~ ~ O +~ s~ ~ U ~ ~ -~ O ~ ~ O
I o
td C1 r1 O 'b N ?i O +~ ?, >, tx
~ U u1 U 9r t~
C9 N ~ O rtf -r1 O U s~ ~ . U r-I M
o o En +~ +s +~
z .a ~ > a~ ~n s~ a s~ ~ o .a ro o .~I -~I x
a o .~I -~I
H s~ ~ sa s~ w .~Ia ~I m a~ w <n m a~ ~a
o ~ s'a ~
e~ b x a~ o x I o 0 0 3
~ +~ 0
a~
~ d U CL ~ ~ t<:~ ~ U r0 f.1 I-1
O ~1 U U U
3 o sa ~ m a x o 3 ~ ~ N 3 a ~ cn v~ w
a~ ~ ~ l ~ m
w m a~ a. o ro r-I~ o .~I -.~a~ o o .~I +
ro a~ +~ -~I .~ -~I
-~I
D H m b~EnEi U3 31 U A ViOW ~ O W W~~~ 3
OAi333057
-23-
It is apparent that at low levels of conversion, where large quantities of
unconverted was remain in the 370°C+ oil to the dewaxer, it is not
possible to
achieve a low pour (i.e. about -21 °C) using typical dewaxing solvents
under
standard conditions (i.e. filter temperature of -30°C). Lower pour
point could
be achieved if one were to go to extremely low filter temperature such as
-40°C, but this puts strains on the refrigeration capability of the
plant as well
as possibly being beyond the metallurgical limitations of most plants.
Operating
at higher levels of conversion (e.g. 30% wax in the 370°C+ fraction to
the
dewaxer) is seen to facilitate achieving a low pour point while still being
within
the typical operating parameters of standard dewaxing plants.
Exams Ip a 2
Catalyst 1
Slack wax from 600N oil was isomerized over Catalyst 1 described in
Example 1 to three levels of conversion.
The slack wax was first hydrotreated over HDN-30 catalyst (a
conventional Ni/Mo on alumina catalyst) at 350°C, 1.0 v/v/hr., 1500
SCF/BBL,
H2, 1000 psi (H2). The catalyst had been on stream for 1447-1577 hours. The
hydrotreated slack wax had sulfur and nitrogen contents of less than 1 ppm
and contained about 23% oil.
~~i333057
- 24 -
TABLE 2
DEWAXING OF ISOMERATES DERIVED FROM
600N SLACK WAX (370°C+)
Isomerization Conditions
Pressure, psi 1000 1000 1000
Space Velocity (v/v/hr) 0.9 0.9 0.9
Gas treat rate
(SCF/bbl, HZ) 5000 5000 5000
Temp. C 318 324 327
Conversion Level (Low) (Medium) (High)
Wt%370C- 11.8 20 25.8
Dewaxer, Feed Cloud, C 60 54 49
Dewaxing Conditions (Batch Conditions)
Solvent: 100% MIBK
Dilution Solvent/Feed/v/v 5.1 3.5 3.4
Filter Temperature, °C -25 -25 -25
Viscosity,CS @100°C 5.63 5.03 4.61
Dewaxed Oil Properties
Pour Point, C -14 -19 -23
Pour-Filter TC 11 6 2
Viscosity, cSt @40C 27.6 22.8 20.7
Viscosity, cSt @100C 5.63 5.03 4.61
Viscosity Index 149 147 144
Wt. % Wax recovered from
370C + oil fraction 56 39 30
From this it is seen that even for isomerates obtained by isomerizing waxes
from a natural petroleum source, the ability to dewax the isomerate to the
desired
low pour point of at least about -21 °C is dependent upon the level of
conversion.
Low conversion levels produce isomerate which cannot be dewaxed to a low
target pour using conventional dewaxing solvents under typical dewaxing filter
temperature conditions.
~A1333057
-25-
EXAMPLE 3 (Comparative)
It has been discovered that waxy isomerates behave differently than
waxy conventional oils when being dewaxed. With waxy conventional oils the
wax content of the oil (usually a solvent extracted distillate) has virtually
no
impact on the pour point of the dewaxed oil nor on the ease with which that
pour point can be achieved. In Table 3 below two typical oils, 150 neutrals
having viscosities of about 5.4 cSt 0100°C, viscosities very similar to
those
of the isomerates described in the present text, were solvent dewaxed using
ketone solvents. The difference between the two natural oil stocks is wax
content; one stock from a South Louisiana crude contains about 9-10% wax,
the other stock from a North Louisiana crude contains about 19-22% wax.
Both stocks were processed under nearly identical conditions as shown in the
Table. Despite the differences in wax content the pour points of the dewaxed
oils obtained by dewaxing under nearly identical conditions were identical.
Both natural oil stocks were dewaxed in a dewaxing plant employing MEK/MIBK
under DILCHILL conditions as described in U.S. Patent 3,773,650 to a
temperature of -6°C. Further chilling to the filtration temperature was
done
employing laboratory scraped surface chilling apparatus. While feed filter
rates
and wax cake liquids/solids differed, both oils could be dewaxed to about the
same pour point using nearly identical dewaxing conditions.
This is to be compared with the results obtained in the prior example
wherein dewaxing isomerate of different wax contents under nearly identical
dewaxing conditions gave dewaxed oils of different pour points, thus showing
the unexpected effect that the wax content of the isomerate has on dewaxing
performance.
- 26 -
~,~ 1333057
Z (n (n C7
T
m
~ c cc
Q
~ W ~ ~ ~ m
N
r r
7C ~ _ co
O o
'
~ csio - o
~
O
( ~ ~ T
n
O O N N
~ 0
r
+
fD
D ~ 7
..
C
O c
~ n
3 7. CO CD n m
.
~ O O
~ m
~ _ '-t
O a
,. N O CD N
.t
-
O
''
'
O
D
fn CD X O
.
3
0 0
O o - 7C
c~
0
'*. ~ ~ .P o
0 o
O r+ W N 3~ C
O
O
~ pp '+ Q rrtO
r+ r+ o ~ _
+ n (D
r ~ ~
W
r"
O O
O
X ~ ~ o ~
.
-~ o~ a n n~
-+ C O O ~ T Cn
C
-p
O
C
m
_ ~ ao
o~ oo < m
n m
O
N
_
O ~G
.
h. c
C O N N O O
Q' 00 Cf1~ 7
X
n' a~ ~ < m
0 0
W
~la r 333057
-27-
EXAMPLE 4
Catalysts 2 to 7
In the following runs the isomerate was made from slack wax obtained by
solvent dewaxing a 600N oil. The slack wax was hydrotreated over HDN-30
catalyst at 350°C, 1.0 v/v/hr. 1500 SCF/bbl, H2, 1000 psi H2 or over KF-
840 at
340°C, 0.5 v/v/hr., 1000 psi, 1500 SCF/-bbl. These hydrotreated waxes
had oil
contents ranging from 21 to 23%, S ranging from 3 to 10 (ppm), N <_ 1- (ppm).
This wax feed was contacted with platinum on fluorided alumina produced in
the following way.
Catalyst 2 One sixteenth inch alumina extrudates impregnated with platinum
were
obtained from the commercial supplier containing 0.6 wt. % platinum and 1
chlorine on the extrudate. The metal loaded extrudate was then fluorided using
a 10
fold excess 11.6 wt% aqueous HF by immersion for 16 hrs. at ambient
temperature.
The resulting catalyst was washed with 2 fold excess H20 and dried at
150°C in
vacuum for 16 hrs. The fluoride content was 8.0 wt. %. The sample of Catalyst
2
as charged to the 200 cc unit was activated in 300 psi H2 at 6.3 SCF H2/hr as
follows: heat from room temperature to 100°C at 35°C/hr; hold at
100°C for 6 hrs;
heat from 100°C to 250°C at 10°C/hr; hold at 250°C
for 12 hrs; heat to 400°C
at 10°C/hr; hold at 400°C for 3 hrs. The sample of Catalyst 2 as
charged to the
3600 cc unit was activated as follows: at 300 psi H2 at 11 SCF H2/hour per
pound
of catalyst, heat from room temperature to 100°C at 10°C/hour;
hold at 100°C for
24 hours; heat from 100°C to 250°C at 10°C per hour; hold
at 250°C for 15
hours; then at 22 SCF H2/hour per pound of catalyst, heat from 250 to
400°C in 31
hours; hold at 400°C for 3 hours.
O~i i 533057
-28-
Catalyst 3 was prepared using 1 /16 inch alumina extrudates impregnated with
0.6
wt% platinum and containing 1.0% chlorine as received from the commercial
supplier. The metal loaded extrudate was then fluorided using 5:1 volume
excess
of 11.6 wt% aqueous HF by immersion for 6 hours at ambient temperature
( ~ 25 °C). The resulting material when washed with two-fold excess H20
and dried
at about 120°C for 16 hrs was designated Catalyst 3. The bulk fluorine
content
was 7.2 wt%. Catalyst 3 was activated in atmospheric pressure H2 by heating
from
room temperature to 343°C in 4 hours followed by a hold at 343°C
for 2 hours.
Catal~ si t 4 is the same as catalyst 3 in all respects except that prior to
the hydrogen
activation step the material was heated at 400°C in air for 3 hours.
Catalyst 5 One sixteenth inch alumina extrudates impregnated with platinum
were
obtained from a commercial supplier containing 0.6 wt. % platinum and 1 %
chlorine.
The metal loaded extrudate was fluorided using a solution of HN4F/HF at pH 4.2
by
soaking. The soaked material was washed, then dried/heated for 2 hours at
400°C
in air. Fluorine content was found to be 7.0 wt%, and the surface N/AI = .0037
by X-ray photo spectroscopy. Catalyst 5 was activated by heating in 50 psi
flowing
H2 as follows: room temperature to 100°C in 2 hrs., hold for 1 hr.,
100°C to 450°C
in 3 hrs., hold for 4 hrs. For the sample of catalyst 5 charged to the small
unit (b)
used in the reported in Table 4, the final activation condition was
400°C for 0.75
hours.
~~i333057
-29-
Catalyst 6 was prepared by meshing the dried/heated form of Catalyst 5 to a
particle size of 1 /30" (14/35 mesh). After meshing to a particle size of 1
/30" (14/35
mesh), Catalyst 6 was activated in flowing hydrogen by heating from room
temperature to 100°C over a 2 hour period, holding at 100°C for
1 hour, heating
from 100 to 450°C over a 3 hour period, holding at 450°C for 1
hour. Activation
pressure was 50 PSI.
Catalyst 7 1 /16" A1203 extrudates were impregnated with chloroplatinic acid
to a
level of 0.26% pt. The extrudates were then sized and screened to 1 /30" mesh
and
subsequently fluorided using a 10 fold excess of 11.6 wt% aqueous HF by
immersion for 4 hrs at ambient temp. The resulting catalyst was washed in a 30
fold excess of H20 and dried at 130°C for 16 hrs. The catalyst was not
calcined.
The fluorine content was found to be 8.5 wt%. Activation procedure was the
same
as employed for Catalyst 1 (See Example 1 ).
Table 4 presents comparisons of these catalysts on slack wax from 600N oil.
Conditions are recited under which the catalysts were run. Dewaxed oil yields
were
determined by using the test method ASTM D-3235 on the 370°C+ fraction.
This example demonstrates that Catalyst 1 is unexpectedly superior to the
extrudate form of the HF treated catalyst (Catalyst 2), even when Catalyst 2
is run
at high mass velocity.
The importance of using the low pH halogenation media is also demonstrated,
compare Catalyst 4 with Catalyst 6, when each was run in a small unit in the
down
flow mode, clearly, sizing down the particles
O~i533057
-30-
does not always improve selectivity; it is only an advantage if fluoriding was
originally performed at low pH (e.g. <4) using for example HF. The performance
of
Catgalyst 7 of Table 4 also illustrates that the catalyst can be sized before
fluoriding.
Good selectivity again results when the low pH fluoriding media is used.
Table 4 also demonstrates the importance of the catalyst having a hydrate
level
of 60 or less. Catalyst 3 possesses a hydrate level of about 66 and is seen to
be
inferior to catalyst 4 which is identical except that the hydrate level is
lower (57).
Catalyst 4 produces a higher yield of 370°C+ oil than does
Catalyst 3.
'~ ( ~ b 1
- 31 -
o C~c-~H ~ s zxz ~n~n
~ W ~, fD x r C
0 n
..: rt .~ O ~ N -. \'~G ' ~-. I
.., ~ ~ t fD ~ \ OJpa ,
N ~n .. y
~c
~
~ o_ rr .-. .., C N o y c, b ~ o ~
or a w ~ o~ a. rrrt ~;
. r b rt
" p ~' U7 ~-t fD, n r. F
'~ N ~ r~
~
( ~ rt ~ ~ *~
~ n 4i .. rtd T r-. r-.
c ~
a ~ rr < ...rt ~
n ~ re ~
n ,~
~
p "~ \ fp \ N ~ ~ (~a
fp fD
~G
N C7 ~ rD ~ < --. 0iW - ~D ~ ~
n W a rt
y ~o ~ c ~ \ ~o '*n ro r
~ o ~C
-n . n .-. ~ ~o
-, ~
,~
~-..~p < p ~ ? ~. rt
rt cD <
rt
.
W
.
~w n . Q
w..
c ~ n. + _ _ ~ c < ~ ~ ~an
<
y ~ N V i n T rt ~ ~ n
o
~ . , , o ~. j
c o ...
N ~ I? o o I
c _ ~ ~ i o
'-
,.. ' rt !
... N
rt N ~
r.-~
O ,D a O O O
rt . W ~
~
N ~ .
. O . p
o~ N
, C 0 ~
C O O O ~D O O r
v '
~
O
7
,..
rt
N ~1
N N O O O
w
p
O ~
O O O ~ 'r
O ~
.r
N V1 V1 I--,
. o
~O ~" O H., W
O W
p 01N
O . 01
O O O O O ~ '" "~3
W 0
b
N as U'1 r C'
O 1- O ~7
W
O O O O ~ N ~ N ,a.
O O O ~ 0~I
CO 0
0
I
w ' cr O r
a ~ o
ow
o ~ o . ~..- c ~,
o ~. . ;,n ~
u,ow w
a o 0o rp --I
o
a o 00
r r
f..i ~' N
Vi
O
~I ~! O ' ~G
w
O is v7
.-
O :n tJm~ tJ~ s -~ ;
O O o .
~
v O O ,p
O
N V1 W w
O O O O
W
O
OD O O t0 O
O
N
O CD O O O
W
~," C'U1
O . O Op
N
'~
v O O ~O O O
O
W W V1 hr
J ~ O O O
W
_
~"~ O O ~D O O ~ 01
O o
N U~ r
O O O
W
_
r"'' O ~O O ~ v
O ~
~~1333057
-32-
Example 5
Catalysts 8 and 9 and Comparison Catalysts 1,2,3 and 4
In these Examples the hydrotreated 600N slack waxes are those previously
described in Example 4. Following isomerization in an upflow once through mode
of operation the isomerate was fractionated to obtain the 370°C+ tube
fraction.
Dewaxed oil yields were determined using the ASTM Test D-3235 method on
the 370°C+ fraction.
In this Example a series of catalysts was prepared using the NH4F/HF
fluoriding
procedures described above. Examples of superior catalysts made using the
NH4F/HF fluoriding procedures were seen to have surface fluorine content in
the low
recited desirable range. Results for these catalysts are shown in Table 5.
Less
satisfactory catalysts made using NH4F/HF treatment are shown in Table 6.
These
catalysts all contained high levels of surface fluorine resulting from initial
excessive
loading of bulk fluorine when using pH 4 or greater. In the case of comparison
Catalyst 3, while the bulk fluorine level is within the desired range and
surface
fluorine was initially low in the as charged catalyst, the excessively severe
activation
conditions employed subsequently increased the surface fluorine level of the
catalyst. This we believe is the reason for its poorer selectivity. All
catalysts were
dried and heated as reported in Tables 5 and 6.
-33- O~i333057
Table 5
Examples of Good Catalysts
in the Process of the Invention
Catalyst 8 9 9
Catalyst Charge (cc) 50 50 200
Method of fluoride treat NH4F/HF NH4F/HF NH4F/HF
Drying conditions C 400 400 400
(muffle) rotary
kiln
Catalyst Inspections
N/AI by XPS 0.0037 0.0021 0.0021
Hydrate level 29 24 24
F. (wt%) (bulk) 6.9 7.0 7.0
F wt% (surface) 1.7 2.0 2.0
Hydrogen Activation Times,
hrs.
RT. to final temp 7 4 7
Time at T 2 ~ 2 2
Final T, C 343 343 350
Hydrogen Activation Pressure
ambient ambient 50 psi
Isomerization Conditions
Temp. C 310 312 309
LHSV (v/v/h) 0.45 0.45 1.0
Press. PSI H2 1000 1000 1000
Gas rate 5000 5000 5000
(SCF/B, H2)
Max 370C+ oil 50(1 ) 49.8 49.3
Dewaxed oil yield, (wt% on
feed)
Conversion to
370C(wt% on feed) 28 24.5 35.2
(1 ) Interpolated data
~,~ i 533057
* cnI T! '*. _ 'n ~ o
C 0 70 T z Q.
W = ' ' ~
~ ~
O v
G)
~
r
~
vo nv ~ =m o a~G ~ 3 _.,~~~~ ,~ o.<m
m D
~ o oQg TN v, cn~ ~ -~ ~ c~ ,rto-Q-~ g ~ y
a a
O
r+ y o -, fn [ O p ~ ~ ~ ~ o o -
W ~ a O o ~
o
cn o
o ~ m ~ p ~ ~ \ \~ ~ o.~<
< ~ rt
m o ~ ~ o ~ c n ~
Q ~ ~
a m
m
. 3 ~ ~
~
Q o a
-
z ~ .
< * ~ ~
*
x m
. . o
o ~
c ~ :
m ? c) -~
a
N N
O
CD
O
-,
m
O
n
n
.
O 1 1 ~ _ ~ N
N N Ut - C3 (3 W t ~ ~ ~ ,a O
o ~ O O o W ~ o ~ N O ~
co ~ N ~
~
oo~o ~ cooooo? o ~ _ 3
~' ~'
000
0
a . ~ o
a m
0 0
x
o
~
~
n m
-c W
Z o a
W N C31 (f1
O ~ ~ O O -~ N /~ cn
~ O W O
cri
cG O ~ N ~ a~ ~ T N .
~ O ~ ~
O O O O O .
W TI O C7
C
O
3 v
n
r
+
_
G
r_+
O
C7
W ~ Z ~ O
W N O O .P ~ = 0
W ~ ~ '' ~ O O W N V O 7r ~ p
~ (") W p
U1
r+ a
N 00 p p O ~ p * O ~ T W
0 d
~ ~
O O ~ 0 ~ C '
. = 7
.
N T tn
G
n
W O
~
O 00 ~ /~ ~ O ~ ~, _ .a
O ~"~ O O ~ N N V
_, O
W 0
0
O
O
- 35 - i ~ ~ ~)"~.__' ~
EXAMPLE 6
The presence of oil in the wax has been found
to produce an enhanced VI product as compared to oil
free wax when isomerization is performed utilizing the
preferred "sized" catalyst made employing IiF. The
amount of oil in the wax, however, must fall within a
particular range as previously described, if this
enhanced VI phenomenon is to be obtained.
A meshed platinum on fluorided alumina
catalyst (Catalyst 1 from Example 1) was used to
isomerize a slack wax obtained from 600N oil. The wax
samples had oil contents of <1%, about 7% and about
23%. The wax containing less than about 1% oil was
made by recrystallizing a 600N slack wax by warm-up
deoiling then hydrotreating. This 1% oil wax has 99%
saturates, 0.8% aromatics and 0.2% polar compounds (as
determined by silica gel separation). It had an
initial boiling point of 382°C and a 99% off boiling
point of 588°C, as determined by GCD. Subsequently,
isomerized products were dewaxed to between -18 to
-21°C pour. Fractionation of the products showed that
at the higher viscosity range the isomerate made from
wax possessing about 7% oil exhibited an unexpected VI
enhancement as compared to the other wax samples having
<1% and 23% oil. This is to be compared with the
results obtained using an extrudate Pt/FA1203 catalyst.
Comparison Catalyst 4 was used to isomerize
slack waxes obtained from 600N oil, which slack waxes
contained <1%, 10.9% and 22% oil under conditions
selected to achieve the levels of conversion indicated
in Table 7. Comparing the results obtained using
Catalyst 1 with those obtained using Comparison Cata-
lyst 4 one sees that isomerization utilizing the meshed
catalyst (Catalyst 1) exhibits an unexpected VI
a :~ ~~.~ l~~
- 36 -
enhancement when the wax feed employed contains about
7% oil.
From the above it is clear that the sized
catalyst is preferred for use in the isomerization
process described herein. Reference to Figure 3 shows
that Catalyst 1 has the highest selectivity for oil
production making it a preferred catalyst (Catalyst I
of the Figure) .
- 37 -
_- 13~3fl~'~
C C ~f1 f~ I~ N t~~, C~ t0
Lj1 M r~ ~ Q. t-r1 P1
r-1 ~ nr r...yi n-1 rr rr r~
U
O
~~ O
C~~ O
c ~~ oG m op op
O lC O~
O tD
U1 . C' Q' er ~ ~ ~ v v
a N ' Q ~' ~ c
~
O
-l r..
C
E
U
C
c
.
.o
c
U
cm o
n
C ch
~.
O O ac oo M ~
> ~ C n O
C,
M Q' N . t0 OC m
CaTj d ~
x
~ r-!N M N N
v N
m
E y >
3
;J C
O O
=
G. U
O
x
Qr
c x
V1
7, ~C
3
w
,~
O w
it
~o O
G~
U
y
E ~
d
r e'
C y ~ N1 t~1 ra
p N
x C V N N V
tl~
r.1
W O N
G7
U
r
."
O
it
N U
7,
O
O
N
cp ri
U
~d
Ll~
O
U
-38- ~~ 1333~J57
Example 7
Slack wax from Bright Stock containing 15% oil was hydrotreated over
Cyanamid's HDN-30 catalyst at 399°C, 0.5 v/v/h, 1000 psi H2 and
1500
SCF/B, H2, yielding a hydrotreated slack wax with the following properties:
Wax Oil content: 22.8 wt%
Sulfur = 3ppm
Nitrogen = < 1 ppm
Distillation Data
GCD % off at °C. ibp,255
363
436
481
515
541
564
590
656
The hydrotreated slack wax was then isomerized over Catalyst 1 described
in Example 1 to produce the following isomerate products:
-39- ~G~333057
Isomerization Conditions: Run 1 Run 2
Temperature, C 332 332
Pressure psi H2 1000 1000
Gas rate SCF/B, HZ 5000 5000
LHSV (v/v/h) 0.9 0.9
Isomerate Product A B
Max 370°C+ 54.6 54.9
Dewaxed oil yield (wt% on feed)
(by ASTM D3235 method)
Conversion to
370°C-,(wt% on feed) 28.4 27.6
The isomerate products A and B made from the Bright Stock slack wax
were fractionated into a broad heart cut (from product A) and a narrow cut
(from product B) and dewaxed using MEK/MIBK under conventional dilution
chilling dewaxing conditions. This was a DILCHILL dewaxing operation run at
150 cm/sec. agitation top speed (2 inch agitator) at an outlet temp. of -
13°C.
Indirect chilling was then employed to get down to the filter temperature.
From
review of the data presented in Tables 8 and 8A it is apparent that
fractionating
the isomerate into a heart cut boiling between 370-582°C not only
facilitated
dewaxing the oil to the target pour point but permitted the dewaxing to be
more efficient (i.e. higher filter rates) then awith the narrow fracton.
Higher
yields of oil were obtained at good dewaxed oil filter rates on the broad
heart
cut as compared to narrow cut or 370°C+ topped fractions dewaxed under
the
same conditions. (Compare runs 1 and 2 Table ~8 with runs A, B and I, Table
8A). This shows the advantage of dewaxing the heart cut when dealing with
isomerate obtained from very heavy, high boiling wax fractions since operating
on the heart cut
~,1~ i 333057
-40-
permits dewaxing to be conducted under miscible conditions. Only when
dealing with a broad heart cut can low pour points, high yields and good
filter
rates be simultaneously achieved.
' - 133~0~'~
C7 < p p p -~ C a. ~ ~ m ~ p 70
~ T 0 (n cn 10
< ~,
fJl~ N' fD (D ? N N ~p ~ r+ fD C O
O (D 3 ' O O O
O - -
O
o t0 X X . fD ~ C7 TI ~ ~ X
fOn n X O O ~ ~
~
o _ ~ (D N .~ ~ N ~ - ~p
~ ~ ~ .+, ~ 7 r! .-r
'+ rM Waa 7
~ N ~
~ a ~3
~
n ~ Ov ~ w~o v m
~ - ~ m ~~
~ a
~
~ ~ O Q . ~ C)
t~ ~ O O m' 7o m o N
O - ~ p \ O cD
- N X ~
x c) T ~ ~ ?
C)
N p ~ ~ ~ Q m c m < ~: o
O ~
m '< ~ o- s ~ ~ a < o. c7
X
'O-'. 0~7~~coo pWW o
o <
O
O r+ ;.+ ~ n
tn Q. .~ N N
+
~ 0 3
~r p
0
~ T
~ ~
N ~ N V N D
N -r
V O V Cf7 N W . . N . V W n C31
CT1 m
~
W Ci~ 00 V 0~ ~ C
- O
W T O
Z m D
7
o
V o ~ w cJC ~ N ~ co w ~ cN m C
N W ~
~
cn c N S N w m
r
N goo o~oo~ cao~ O
O
a ~
O
O
~ D
~ p
o ~ W
N N ~ N O V N W d W N ~ V
N O V Cf1 N W V W V ~ 00 ~ ~ O
Cf7 P ~
. ,
W V O CJ1 y ~ p
P Ui ~ ~. N ~' ~ 0 ~
. ~ ~ N a ~ (7
Z 7C
-~
w p
_ 3 ~ ,.. O
N W P N ? V N CTl -r 3 GJ N C7 ~ C
~
OD N O fJi ~ W U1 U7 07 -~ O W vu ,~ C m
~ CT1 ~ ~ ~
-~ ~ O N O CT7 V ~ -~ ~ ~ D
Vl CC
m m Z
0o
7
C
w
3
_ N V N ~ ~ ~, GJ W m
W W ~ N
N W ~ N ~ O U7 C31 O O O ~ n U1
00 O W CJ7 00 CJ1 00 ~: W 0
V V 00
~ 07
N ~ N O
p m
V N W cN'1 N
O O V Vi N 00 (D ~ W ' 00 W ~ U7 O
C31 .
~
W V W V O
W ~ m _
OW
- 42 - 1..~3~~~~
C) G 0 0 ~ s ~ a. ~ ~ ~ ~ ~ ~'
o ~ m
G
a O O C O
- O
p X O c < < ~ 7
n x n ~
Q ~
d O v N 07 ~ - S _ p>
' ~ x x x ~. (D -. ~ <D (D x
O n n TI ~ -~ ~
o T 7 ~
N. t
M
.f ~ ~ .~+ fD ~ CD ~ ~ ~ ~ r p
n< fD y+, O O r ',
< a ~
aa
n ~ , ~ ~ ,~ v m
o~ ~ ~a ~ 3 ~o~
ca ~ ~ ~y<
oa' ~ o' m
000
oo~ a~ ' co ,.+, c~
a . ~ ~~ a m
- .- x ~
p a o o
rt = N
~ p ~
x ~ ~ - ~
T ~ f ~
C7
N p ~ ~ n O ~ ~ a ~ o C Q p
~
y< ~ a ~ ~ o \ ~ m ~ ~ o C7
~
o ~ o W o TI t.'n,
~ a' a c~
m a ~ m
~ N Q
G
G
W
T
~Z
cnoc.~sico~N~ ~cNOOO~cnw~ ~ cNSm~~ y
~a ~
o
0o w m ~ TO
W
W
m
D y
Z70
~ ~ rw ~ cwc ~ ~ mC m
ap ~ ~ cNC N ~
w y
N
n .p W w
c . 'T~ 00
O~
W ~
n
00 W Ut W O CO OD
W Cs1
W ~
O W
_
O
~D
G)
0
.a
V N N 00 ~ ~ O O N O W W y Cft W
CD ~ ~
Cn O W .P -~ 00 00 07 n CO V ~ ~ ~ ~-I
W V ~' O W N ~ O
fD
O (7
Q
O~
O ~ N ~ ~ ~C
Ui OD 00 N Cf1 . . . . W W n C31 ~ O
00 w
W V O W
V _ O
m ~ Z
m
w
W
-~ C31 W ~ N (31
CS1
Cf1 N CO N CJ1 . ~ ~ . Cf1 W ~ Cf1
W ~ w
W O 00 W O ~' C31 ~
W
O ~
~ N ~ W W V N N n. n N
~ ~
'
00 O 00 W CT1 N ~ ~ C31 ~
O ~ N 00 .P
O CO 00 N tD CD ~' ~ N ~ W
W
7C O
0
n
-43-
~,b i 333057
Example 8
Slack wax derived from a 600N oil was hydrotreated over KF-840, a Ni/Mo
on alumina hydrotreating catalyst at 370°C, 0.33 LHSV, 1500 SCF HZ/bbl,
1000 psi H2. The hydrotreated wax had a sulfur content of 6 wppm, a nitrogen
content of < 1 wppm, an oil content of 18.7 wt%, an initial boiling point of
233°C and a 95% off boiling point of 338°C.
The slack wax was isomerized over Catalyst 2 in three runs at high mass
velocity as described in Table 9.
Table 9
Run 1 Run 2 Run 3
Pressure (psi) 1200 1200 1200
LHSV 1.0 1.0 1.0
gas rate SCF/bb,H2 2500 2500 2500
Temp C 329 328.9 327.1
Yield (wt%)
370C- 37.5 37.8 22.0
Max 370C+ Oil * 49.8 50.5 52.5
residual wax 12.7 11.8 25.5
Oil yield determined
using ASTM D-3235
test method
Isomerate from these three runs was combined to produce a feed to the
dewaxer having a 370°C~ wt% on feed of 26.6 The feed was fractionated
into
a 370°C+ fraction and 420°C+ fraction and dewaxed under
simulated
DILCHILL conditions in the laboratory using the procedure described in Example
7. DILCHILL dewaxing was
- . ~,~ ~ 333057
performed using two different solvent systems on the two above described
fractions. The results are presented in Table 10, below:
- ~,~ ; 333057
O ' ~< < C7 ~ O T! < n C) v,
C7 ~ ' (n
v>' v~' o ~ ~ in' 0 0
~ o ~ r+ ~ 0
~ X 'o o a ~
W a ~
co ~ - ~ ~ o
~
X T cn cn . T X ~ N
a n O . C7
~
N O n ~
O
W Q ~ '< ~G ,-+ Q' ~ ,<
N o N n C
~ o
O ~ ~ O x
~ C7
n
r ~
Q Q ~ f~
~ (~
a -i
~
T '~ fD Q D o
~ ~ o
c
I X ~ ~ 3 c~ <
o c~ ~
~
c
a o < o
n
n ~ <
a
a ~ ~ n
C)
r+
o r
n
Z
r
N o ~
m
gy
N O ~ . p V W O V p ~ -O X
.p .P V CT1 ~
N (
(D 00 V Cfl ~ N
(~
W N ~ cn ca
O
~ O
W
< ~ O O
n
O ~ (D
m ~ c
a ~ . . . cn ~ O
~ o
_ _ _ _
N V ~P ~ N Cfl O C31
~ 00 -~ O Cp
W
V W ~ N .p N
~
(D N
X
O
(D
N N N W U1
N
N (J7 CTI W O W ~ O C71 ~ ~p
Ci1 ~ V N
00
CO 00 W O CT~ N O cn
G7i W
W
N
O
0
n
rn
. .
_ _
N O ~ ~ N Cfl C4 CT1
C11 00 ~ O N ~
00 O
V ~ V V N O
CO C
N (D
O
CD
~~1533057
From this it can be seen that to achieve extremely low pour points, it is
preferred to use MEK/MIBK as the dewaxing solvent.