Note: Descriptions are shown in the official language in which they were submitted.
1333618
This patent application is a division of
application No. 514,378 filed on July 22, 1986.
Technical Field
This invention relates to synthetic intermediates
for preparing substituted di-t-butylphenols which are anti-
allergic agents.
Background of the Invention
The leukotrienes are a group of biologically
active mediators derived from arachidonic acid through the
action of lipoxygenase enzyme systems. There are two groups
of leukotrienes derived from the common unstable precursor
Leukotriene A4. The first of these are the peptido-lipid
leukotrienes, the most important being Leukotrienes C4 and
D4. These compounds collectively account for the
biologically active material known as the slow reacting
substance of anaphylaxis.
The leukotrienes are potent smooth muscle
contracting agents, particularly on respiratory smooth
muscle, but also on other tissues as well. In addition,
they promote mucous production, modulate vascular
permeability changes and are potent inflammatory mediators
in human skin. The most important compound in the second
group of leukotrienes, namely dihydroxy fatty acids, is
Leukotriene B4. This compound is a potent chemotactic agent
for neutrophils and eosinophils and, in addition, may
modulate a number of other functions of these cells. It
also affects other cell types such as lymphocytes and, for
example, may modulate the action of suppressor cells and
natural killer cells. When injected ln vivo, in addition to
promoting the accumulation of leukocytes, Leukotriene
B4 is also a potent hyperalgesic agent and can modulate
'~$
` -2- 1333618
-
vascular permeability changes through a neutrophil
dependent mechanism. Both groups of leukotrienes are
formed following oxygenation of arachidonic acid througl
the action of a lipoxygenase enzyme. See, for example,
D. M. Bailey et al., ~nn. Rpts. Med. C!lem., 17, 203 (1982).
RESPIRATORY CONDITIONS
Asthma. The leukotrienes are potent spasmogens
of human trachea, bronchus, and lung parenchyma, and when
administered to normal volunteers as aerosols are 3,800
times more potent than histamine at inducing a 50% decrease
in air flow at 30% of vital capacity. They mediate
increases in vascular permeability in animals and promote
mucous production in human bronchial explants. In
addition, Leukotriene B4 may also mediate mucous production
and could be an important mediator of neutrophil and
eosinophil accumulation in asthmatic lungs. Lipoxygenase
products are also thought to be regulators of mast cell
degranulation, and recent studies with human lung mast
cells have suggested that lipoxygenase inhibitors (but not
corticosteroids), may suppress antigen-induced mast cell
degranulation. In vitro studies have shown that antigen
challenge of human lung results in the release of
leukotrienes and that, in addition, purified human mast
cells can produce substantial amounts of leukotrienes.
There is, therefore good evidence that the leukotrienes are
important mediators of human asthma. Lipoxygenase
inhibitors would, therefore be a new class of drugs for the
treatment of asthma. See, for example, B. Samuelsson,
Science, 220, 568-575 ~1983).
SKIN DISEASES
Psoriasis. Psoriasis is a human skin disease
which affects between two and six percent of the
-3- 1333~18
population. There is no adequate therapy for psoriasis and
- related skin conditions. The evidence for leukotriene
involvement in these diseases is as follows. One of the
earliest events in the development of prepapillary lesions
is the recruitment of leukocytes to tlle skin site.
Injection of Leukotriene B4 into human skin results in a
pronounced neutrophil accumulation. There are gross
abnormalities in arachidonic acid metabolism in human
psoriatic skin. In particular, highly elevated levels of
free arachidonic acid can be measured as well as large
amoun'its of lipoxygenase products. Leukotriene B4 ha.s been
detected in psoriatic lesions, but not in non-involved
skin, in biologically significant amounts.
ALLERGIC CONDI~IONS
Leukotrienes can be measured in nasal washings
from patients with allergic rhinitis and are greatly
elevated following antigen challenge. Leukotrienes may
mediate this disease through their ability to regulate mast
cell degranulation, to modulate mucous production and
mucocillary clearance, and to mediate the accumulation of
inflammatory leukocytes.
Leukotrienes may also mediate other diseases.
These include atopic dermatitis, gouty arthritis, gall
bladder spasms and ulcerative colitis. In addition, they
may have a role in card;ovascular disease because
Leukotrienes C4 and D4 act as coronary and cerebral
arterial vasoconstrictors and these compounds may also have
negative inotropic effects on the myocardium. In addition,
the leukotrienes are important mediators of inflammatory
disease through their ability to modulate leukocyte ancl
lymphocyte function.
Many substituted di-t-hutylphenols are known.
Generally these compounds may he useful as antioxidants.
~_ _ 4 - 1 333 618
Some of these compounds are also known to be active
antiinflammatory agents.
Compounds wherein 2,6-di-t-butylphenol is
substituted in the 4 position by an unsubstituted phenyl or
certain simply-substituted phenyls are known
antiinflammatory agents. See, for example, U.S. Patent
4,172,151 and references cited therein. The compound 2,6-
di(tertiary-butyl)-4-(4'-carboxyphenylimino)-2,5-cyclohexan-
diene-l-one is disclosed in Chemical Abstracts 67:81701n.
No compounds wherein a 2,6-di-t-butylphenol is
substituted in the 4 position by an anilino group wherein
such anilino group is substituted by a moiety including
carboxy, tetrazolyl, N-methyl-tetrazolyl, or N-trifluoro-
methylsulfonyl are known.
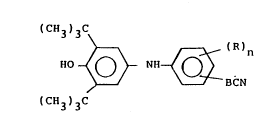
Summary of the Invention
This invention relates to certain compounds of
formula:
t 3)3C
HO ~ ~ (R~n
~ 3)3C
wherein R is hydrogen, lower alkyl, lower alkoxy, lower
alkylthio, hydroxy or halogen; n is 0, 1 or 2, with the
proviso that when n is 2, the R substituents combined
contain no more than 6 carbon atoms; and B is a carbon-
carbon bond, lower alkylene, lower alkenylene, lower
alkylene containing one ether or thioether link in the
alkylene chain, or -CNHCH2-.
The compounds of the invention are useful as
synthetic intermediates for preparing certain of the
_ 5 _ 1 33361 8
antiallergic compounds which are disclosed and claimed in
the canadian co-pending applications Nos. 514,386 and 514,378.
Moreover, it is believed that certain of the antiallergic
compounds disclosed in said copending application No.
514,386 are prodrugs of certain antiallergic compounds
disclosed herein. For example, N-(3-carboxyphenyl)-N-(3,5-
di-t-butyl-4-hydroxyphenyl)succinamic acid disclosed in
copending application 514,386 is believed to possibly be a
prodrug of 3-(3,5-di-t-butyl-4-hydroxyanilino)benzoic acid
which is disclosed herein.
Detailed Description of the Invention
The invention of the parent application No.
514,378 relates to compounds of formula (I):
3)3C R'
HO--~ N ~ (R)n
3)3C
wherein R is hydrogen, lower alkyl, lower alkoxy, lower
alkylthio, halogen (preferably chloro or fluoro), amino,
lower alkylamino, di(lower)alkylamino, lower acylamido or
hydroxy, and n is 0, 1 or 2 with the proviso that if n is 2,
then all R substituents combined contain no more than 6
carbon atoms; R' is hydrogen, lower alkyl, acetyl or
trifluoroacetyl; A is carboxyl, tetrazolyl, N-methyltetra-
zolyl or -CNHS02CF3; and when A is carboxyl, B is a carbon-
carbon bond, lower alkylene, lower alkenylene, lower
alkylene containing one ether or thioether link in the
1333618
alkylene chain, -CNHCH2-; when A is tetrazolyl or N-
R
methyltetrazolyl, B is a carbon-carbon bond, -CH2- or -CNH-;
1l
and when A is -CNHSO2CF3, B is a carbon-carbon bond; and
derivatives of compounds wherein A is carboxyl selected
from the group consisting of the lower alkyl esters,
(lower)alkylamino(lower)alkyl esters, pharmaceutically
acceptable (lower)alkylamino(lower)alkyl ester acid-addition
salts and pharmaceutically acceptable carboxylate salts, and
derivatives of compounds wherein B is tetrazolyl selected
from pharmaceutically acceptable alkali metal and alkaline
earth salts of the tetrazolyl moiety.
Presently preferred are compounds wherein the
group -B-COOH, -B-tetrazolyl or -B-N-methyltetrazolyl is
IR'
oriented para or meta to the -N- linking group.
Presently preferred compounds are those wherein A
is carboxyl.
Presently preferred as B is a carbon-carbon bond.
When B is alkylene it is preferably methylene. When B is
alkenylene it is preferably ethenylene.
When R iS lower alkyl, lower alkoxy or lower
alkylthio, it is presently preferred to be methyl, methoxy,
or methylthio respectively. The presently preferred R group
is hydrogen.
By "lower" as used in connection with "alkyl" and
"alkylene" is meant that such groups contain one to about
four carbon atoms. Most preferred alkyl groups contain one
or two carbon atoms. By "lower" as used in connection with
"alkenylene" is meant that such groups contain two to about
four carbon atoms.
In the compounds of formula (I) wherein A is
~~ ~ 7 ~ 133 3 6 1 8
tetrazolyl, two tautomeric forms of tetrazolyl exist as is
known to those skilled in the art. Tautomerism does not
exist in tetrazolyl moieties where the tetrazolyl ring is
substituted on a nitrogen atom by methyl. Instead, two N-
methyl isomers are obtained, one in which the methyl group
is in the l-position, the other in which it is in the 2-
position. All such tautomers and isomers are within the
scope of the invention of the parent application.
It is well known to the art that pharmaceutically
acceptable salts such as alkali metal, alkaline earth,
aluminum and other metal and amine salts of pharmaceutically
active acids are the equivalents of the acids in terms of
activity, and in some cases may even offer advantages in
absorption, formulation and the like. Pharmaceutically-
acceptable carboxylate salts of the compounds of formula (I)
which contain carboxyl as A are prepared in an inert
atmosphere by reaction of the acid with a base and
subsequent evaporation to dryness, preferably under mild
conditions. The base may be organic, e.g., sodium methoxide
or an amine, or inorganic, e.g., sodium hydroxide.
Alternatively, the cation of a carboxylate salt, e.g.,
sodium, may be displaced by a second cation such as calcium
or magnesium when the salt of the second cation is more
insoluble in a selected solvent.
Other useful derivatives of the compounds of
formula (I) which contain carboxyl as A include alkyl
esters, alkylaminoalkyl esters, and salts of the latter. In
the ester derivatives, the hydrogen portion of the
carboxylic acid group is replaced with an alkyl or
substituted alkyl, preferably an alkylaminoalkyl group.
Esters of the compounds of formula (I) may be
obtained as intermediates during the preparation of the
- 8 - 133 3618
acidic compound. In some cases, the esters may be prepared
directly using standard synthetic methods. These esters may
exhibit antiallergic activity, but they are primarily of
interest as synthetic intermediates, although in some
instances hydrolyzable or salt-forming esters may be of
interest as therapeutic agents. Preferred esters are alkyl
esters and alkylaminoalkyl esters having one to four carbon
atoms in the alkyl group. Especially preferred are
alkylaminoalkyl esters such as the dimethylaminoethyl esters
which will form salts, e.g., hydrochlorides.
Ester derivatives may be obtained by alkylation of
an alkali metal salt of the compound in dimethylformamide
with an alkyl iodide or dialkylaminoalkylchloride, or by
starting with esters instead of acids in Scheme I, Step (1)
below.
Pharmaceutically acceptable alkali metal and
alkaline earth salts may also be prepared of compounds of
formula (I) wherein A is tetrazolyl by methods known to
those skilled in the art.
The preferred compounds of formula (I) are 4-(3,5-
- di-tertiary-butyl-4-hydroxyanilino)benzoic acid, 3-(3,5-di-
tertiary-butyl-4-hy~droxyanilino)benzoic acid, 5-C3-(3,5-di-
tertiary-butyl-4-hydroxyanilino)phenyl~-tetrazole, and 5-~4-
(3,5-di-tertiary-butyl-4-hydroxyanilino)phenyl]tetrazole.
Compounds of formula (I) may be prepared by the
method of Scheme I, wherein A, R, and B are as defined
above, and R' is hydrogen.
- 8a -
1333618
SCHEME I
(CH3)3c
~0 + NH2~
-A
(CH3)3c
II III \
(1) \~
3)3C
O=~
3 ) 3C
/
(2~/
3)3C
HO ~NH--~
B-A
( 3)3C V
-9- 1333618
-
The reaction of step (1) is a Lewis acid
catalyzed condensation of the known compound
2,6-di(tertiary-butyl)-p-benzoquinone (II) and a
substituted aromatic amine ( III ) . Suitable substituted
aromatic amines for preparing compounds of Formula I
wherein A is carboxyl are known compounds such as the
aminobenzoic acids, for example, 3- and 4-aminobenzoic
acid, the aminophenylacetic acids, aminophenylbutyric
acids, aminophenylthioacetic acids, aminophenyloxyacetic
acids, alkyl aminophenylacetates, aminophenylcinnamic
acids, and the like. Similarly, suitable tetrazolyl-
substituted aromatic amines for providing compounds of
Formula I wherein A is tetraæolyl are known such as 5-(3-
or 4-aminophenyl)tetrazoles.
Suitable Lewis acid catalysts include boron
trifluoride, tin tetrachloride, titanium tetrachloride and
the like.
The reaction of step (1) is carried out by
combining the reactants in an inert solvent such as an
ether, for example, tetrahydrofuran, and heating gently, if
necessary. The products of Formula IV are novel solids
which are readily isolated and may be recrystallized from
polar solvents.
The reaction of step (2) is a reduction of the
imino quinone system of the intermediate of Formula IV to
an amino phenol. It is readily accomplished using
catalytic reduction with hydrogen gas in an inert solvent
when A is carboxyl. It may be carried out under neutral
conditions or in the presence of base, for example, an
equimolar amount of base. Suitable catalysts include
platinum or palladium on charcoal. Chemical reduction can
also be carried out, for example, with sodium thiosulfite,
or zinc and acetic acid to provide compounds wherein A is
carboxyl or tetrazolyl. Chemical reduction is preferred
when B contains a double bond.
-lo- 1333618
-
Compounds of formula (I) wherein R' is alkyl
and A is carboxy are prepared from a compound of Formula v
(obtained above) by reacting the compound with an alkyl
halide, particularly an alkyl bromide or an alkyl iodide.
This reaction may be carried out in a solvent SUCll as
N,N-dimethylformamide, optionally in the presence of base.
When base is present, the carboxyl will generally become
esterified, and therefore subsequent hydrolysis by
conventional methods may be desired.
Compounds of formula (I) wherein R' is acetyl
or trifluoroacetyl and A is carboxy are prepared from a
compound of Formula V by reacting the compound with the
appropriate anhydride.
Compounds of Formula I wherein A iS
N-methyltetrazolyl are preferably prepare~ by alkylating
an alkali metal salt of the corresponding compound of
Formula I wherein A is tetrazolyl with methyl iodide.
o
Compounds of Formula IV wherein A is -IC-NHSO2CF3
may be prepared from the corresponding compound of Formula
IV wherein A is carboxy via reaction of that compound with
thionyl chloride and subsequent reaction of the resulting
acid chloride with sodium trifluoromethanesulfonamide.
Catalytic reduction provides compounds of Formula I wherein
o
A i S -CNHSO2CF 3 .
Compounds of Formula I wherein A is tetrazolyl
may also be prepared by the method of Scheme II wherein R,
n and B are as defined above and R' is hydrogen.
-11- 13~3618
-
SCEIEME II
(CH3)3C ~
O- ~ O + NH2 ~ (c~n
(CH3)3C
II VI
\ (1)
l 5 ( C 3 ) ~ ~ BCN
( C 3 ) 3
V I I
(~
b
2 0 ( CH 3 ) 3 C ( 4 ) ( Cli 3 ) 3~ ~( R ) ~1
(C 3)3 ( CH3 ) 3
VIII \ X
\~ (3)
(CH3)3
HO ~ Nl~ ~ B~R)n
(C 3)3 > N
HN
\ N
\ //
IX N
133361~
- 12 -
The reaction of step (1) of Scheme II is a Lewis
acid catalyzed condensation similar to step (1) of Scheme I
except that here an aminonitrile of formula (VI) is used in
place of the substituted aromatic amine used in step (1) of
Scheme I. Compounds of formula (VI) are known or may be
prepared by conventional methods. The reaction is conducted
as described in connection with step (1) of Scheme I. The
product of step (1) of Scheme II is a novel intermediate of
formula (VII).
The reaction of step (2) of Scheme II is a reduc-
tion of the type (and performed using the method of) step
(2) of Scheme I to provide a novel intermediate of formula
(VIII) which is the one forming the subject of the present
application.
In step (3), the intermediate of formula (VIII) of
the present invention is reacted with sodium azide in the
presence of ammonium chloride and lithium chloride. The
reaction is preferably conducted in N,N-dimethylformamide
and is conducted under a nitrogen atmosphere and accompanied
by heating.
In step (4), the intermediate of formula (VIII) of
the present invention is hydrolyzed, in an inert atmosphere,
by known means such as with sodium hydroxide in aqueous
ethanol, to provide compounds of formula X.
Compounds of formula (I) wherein R' is alkyl and A
is carboxy, tetrazolyl or N-methyltetrazolyl may be prepared
by alkylating the intermediate of formula (VIII) of the
present invention by conventional methods prior to
conducting step (3) or (4).
Again, as in Scheme 1, compounds of formula (I)
wherein R, is acetyl or trifluoroacetyl may be prepared from
the compounds of formula (IX) and (X) by reacting the
compound with an appropriate anhydride as discussed
previously.
Similarly, N-methyltetrazolyl derivatives may be
~ - 12a - 1333618
prepared as described in connection with Scheme I above.
The anti-allergic activity of the compounds of
formula (I) may be demonstrated via a variety of biological
assays including in vitro assays for measuring inhibition of
lipoxygenase activity and leukotriene synthesis, and in vivo
assays for inhibiting bronchoconstriction.
-13- 1 3 3 3 61 8
More specifically, a suitable assay for
demonstrating inhibition of lipoxygenase activity by the
compounds of Formula I utilizes lipoxygenase isolated from
mammalian lung tissue, for example, the lung tissue of
guinea pigs. An example of such an assay is that described
by Ben Aæiz et al., ~nal. ~iochem. 34, ~ ~l970). The
inhibition of lipoxygenase activity is measured by a rapid
and sensitive spectrophotometric technique. The compounds
of Formula (I) exhibit an IC50 (the
concentration at which 50% of the enzymatic activity is
inhibited) of less than about 100 micromoles per liter.
Preferred compounds exhihit an IC50 of less than about 50
micromoles per liter. Most preferred compounds exhibit an
IC50 of less than about 10 micromoles per liter.
The activity of the compounds of For~ula I may
also be demonstrated in a more specific test for
leukotriene biosynthesis inhibition. This test utilizes
the cell free leukotriene biosynthesis system of M.
Steinhoff et al., Biochim. Biophys. Acta. 68, 28 (1980),
which consists of homogenized rat basophil leukemia cells.
Leukotriene synthesis is initiated by the addition of
arachidonate. Solutions are centrifuged and supernatants
assayed using a radioimmunoassay developed as described by
Aeringhaus et al., FEBS Letter 146, 111-114. Drugs are
dissolved in ethanol or dimethyl sulfoxide and preincubated
for five minutes. Phenidone is used as a positive control.
The compounds of Formula I exhibit an IC50 of 100
micromoles per liter or less. Preferred compounds exhibit
an IC50 less than 25 micromoles per liter, and most
preferred compounds exhibit an IC50 of less than 10
micromoles per liter.
The compounds of Formula I are relatively
inactive as inhibitors of cyclooxygenase. This is
important in order for there to be good in v vo
antiallergic activity. A convenient in vitr_ method for
-14- 1333618
-
measuring cyclooxygenase activity is an assay wherein the
amount of thromboxane s2 production is measured-in a whole
blood clotting assay. The thromboxane s2 production is
measured by a radioimmunoassay as described by Patrono, et
al, Thromb. Res. 17, 317 (19~0). The compounds of Formula
I do not show appreciable activity at concentrations of 100
micromoles per liter.
The _ vivo test used to demonstrate
anti-allergic activity may be any of those known to those
skilled in the art. Preferably, bronchoconstriction in
sensitized guinea pigs is measured upon antigen challenge.
This test is described in hroad terms by Piechuta, et al.,
Immunology, 38, 385 (1979), and more specifically by
Hammerbeck and Swingle, Int. Archs. Allergy Appl. Immun.
_ , 84-90 (1984). It is used in a modified form as
follows: Male Hartley guinea pigs (250-600 g) are dosed
with a compound of Formula I in an amount generally about 1
to 40 mg/kg. Fifteen minutes later the animals are aerosol
challenged with eithee water or ovalbumin at a
concentration of 10 mg per ml. The animals are then placed
under an inverted dessicator jar (18 x 14 cm) with a
constant flow of air coming into the chamber from a
compressed-air source to prevent hypoxia. Air flow leaving
the chamber and fluctuations due to respiration are
monitored through a separate outlet with a Fleisch No. 0000
pneumotachograph (available from Beckman Instruments, Inc.,
Schiller Park, Illinois) coupled to a Beckman Type R
dynograph (available from Beckman Instruments).
Aerosolization through a third outlet is made via a No. 40
DeVilbiss nebulizer (available from The DeVilbiss Company,
Somerset, PA) for 90 seconds at 150 mm Hg. The
characteristic respiratory patterns observed are summations
of two air exchange processes occurring simultaneously in
the chamber. One exchange process is due to inspiration
3S and expiration of air into and out of the animal, while the
other exchange process is due to the air flow into and out
of the chamber due to respiratory movements. The tracing
-15- 1333618
obtained is the mechanical representation of the summation
of those flows. Superimposed on the tracings was a
characteristic spiking ('notching'), which appears to be
due to an exaggerated expiratory movement, the frequency of
which correlates with the severity of the
bronchoconstrictive reaction. The frequency of notching
for 15 minute periods beginninq 4 minutes after the
beginning of the aerosol challenge is used for comparing
various treatments. Effects are considered significant if
the t value achieves p<0.05. Compounds of Formula I
exhibit an intraperitoneal ED40 of 100 mg per kg or less
when tested in the above model. Preferred compounds
exhibit an ED40 of 20 mg per kg or less. Most preferred
compounds of the invention exhibit an ED40 of 10 mg per kg
or less and are effective orally.
The imine intermediates of Formula IV are also
active as an antiallergic agent and are believed to be
reduce in vivo to the corresponding compounds of Formula I.
Specifically,
4-amino-3-(2,6-di-t-butycyclohexadienon-4-ylideneamino)benz
oic acid, 4-(2,6-ditertiary-butylcyclohexadienon-4-
ylideneamino)benzoic acid, 4-(2,6-di-tertiary-
butylcyclohexadienon-4-ylideneamino)hippuric acid,
4-(2,6-ditertiary-butylcyclohexadienon-4-ylideneamino)
cinnamic acid, 4-acetamido-3-(2,6-di-tertiary-
butylcyclohexadienon-4-ylideneamino)henzoic acid, and
3-(2,6-di-tertiary-butylcyclohexadienon-4-ylideneamino)
benzoic acid have been found to exhibit useful activity in
the above described in vivo assay involving
bronchoconstriction. The last compound mentioned above,
when administered in vivo to a dog, has been found to be
converted to the compound 3-(3,5-di-tertiary-butyl-4-
hydroxyanilino)benzoic acid.
One of the preferred compound.s of Formula
(I), namely
` -16- 1333618
,
3-(3,5-di-tertiary-butyl-4-hydroxyanilino)benzoic acid has
been found to be active as a bronchodilator in the small
airways of the guinea pig as determined using the method
described in L. Diamond et al., J. ~ppl. Physiol.:
Respirat. Environ. Exercise Physiol., ~3 (6), 942-9~n
(1977).
Thus, compounds of Formula I are antiallergic
agents exhibiting _ vivo activity in mammals. The
p h a r m a c e u t i c a l c o m p o s i t i o n s w i l l
contain sufficient compound of Formula I in a dosage form
suitable for inhibiting the mammalian biosynthesis of
leukotrienes, or for the treatment desired. The effective
concentration of the Formula I compound in the composition
will vary as required by the mode of administration, dosage
form, and pharmacological effect and level desired.
For treating pulmonary conditions such as asthma,
the mode of administration may be oral, parenteral, by
inhalation, by suppository and the like. Suitable oral
dosage forms are tablets, elixirs, emulsions, solutions,
capsules, including delayed or sustained release dosage
forms. Dosage forms for administration by inhalation
include aerosols and sprays which may be administered in
metered doses if desired.
For treating other allergies or allergic
reactions, the compound of Formula I may be administered by
any conventional mode, for example, orally, parenterally,
topically, subcutaneously, by inhalation and the like. The
oral and parenteral dosage forms are as described for
pulmonary treatment. The topical application dosage forms
include ointments, sprays, controlled release patches,
powders, solutions and the like.
For treating inflammation, the mode of
administration may be oral, parenteral, by suppository and
the like. The various dosage forms are as described above.
For treating skin diseases such as psoriasis,
atopic dermatitis and the like, oral, topical or parenteral
administration is useful. For topical application to the
-17- 1333618
-
diseased area, salves, patches, controlled release patches,
emulsions, etc. are convenient dosage forms.
For treating cardiovascular conditions any
suitable mode of administration may be used.
In addition to the common dosage forms listed
above, the compounds of Formula I may also be administered
for various utilities and indications or for inhibiting
leukotriene synthesis by conventional controlled release
means and/or delivery devices.
In preparing suitable dosage forms, conventional
compounding procedures and ingredients, for example,
diluents, carriers, etc. may be used. Examples of suitable
solid carriers are lactose, terra alba, sucrose, talc,
gelatin, agar, pectin, acacia, magnesium stearate, stearic
acid, and the like. Examples of suitable liquid carriers
are syrup, peanut oil, olive oil, water, and the like.
Similarly, the carrier or diluent can include any time
delay material well known to the art, such as glyceryl
monostearate or glyceryl distearate, these being useful
alone or, for example, in combination with wax.
The following Examples are provided to illustrate
the invention of the parent application, and the present
invention, but are not intended to limit the invention.
EXAMPLE 1 - Preparation of 4-(3,5-Di-tertiary-butyl-4-
hydroxyanilino)benæoic ~cid
Step A
A mixture of 22 g (0.10 mole) of
2,6-di(tertiary-butyl)-p-benzoquinone, 13.7g ~0.10 mole) of
4-aminobenzoic acid, 175 ml of tetrahydrofuran and 1 ml of
boron trifluoride: diethyl ether complex was heated on a
steam bath for 1.25 hours. The mixture was allowed to cool
to about 20C over 16 hours under a nitrogen atmosphere.
Evaporation provided a solid which was washed with hexane
and recrystallized from ethanol to provide orange solid
2,6-di(tertiary-butyl)-4-(4'-carboxyphenylimino)-2,5-
cyclohexadien-l-one, m.p. 305-309C. Analysis: Calculated
for C21H25NO3: ~C, 74.3; ~H, 7.4; %N, 4.1; Found: ~C, 74.2;
%H, 7.4; %N, 4.1.
-18-
1333618
Step B
To a solution of 5.~ g (0.0147 mole) of
2,6-di(tertiary-butyl)-4-(4'-carboxyphenylimino)-2,5-
cyclohexadien-l-one in 300 ml of ethanol was added 0.25g of
5 percent palladium on charcoal. It was subjected to
hydrogenation in a Paar apparatus and filtered. This
solvent was removed by evaporation under vacuum and the
residue was recrystallized from a 5:2 (v/v) ethanol-water
mixture to provide light-orange crystals of
4-(3,5-di-tertiary-butyl-4-hydroxyanilino)ben7Oic acid,
m.p. 241-243C. Analysis: Calculated for C211i27NO3: ~C,
73.9; %H, 8.0; ~N, 4.1; Found: ~C, 73.9; %H, 7.9; ~N, 3.8.
EXAMPLE 2
Using the method of Example 1, 2,6-di(tertiary-
butyl)-p-benzoquinone was reacted with 3-aminobenzoic acid
to provide red-orange crystals of 2,6-di(tertiary-butyl)-
4-(3'-carboxyphenylimino)-2,5-cyclohexadien-1-one, m.p.
230-231C. Analysis: Calculated for C21H25NO3: %C, 74.3;
~H, 7.4; %N, 4.1; Found: %C, 74.1; %H, 7.6; ~N, 3.7.
EXAMPLE 3-6
Using the general method of Example 1, the
aminobenzene starting materials of Formula III, shown in
Table I below were reacted with 2,6-di(tertiary-butyl)-p-
benzoquinone to provide the imine products indicated in
Table I.
1333618
T~l)le I
Ex~mpleStarting M~teri~l ~r~ lct ~r Formu]a IV
No.of Formula III (r~ ). in `C)
34-aminopllenylacetic 0- ~ -N ~ ) C~2C
acid (~ll3)3C ~~-
(186~
4 2 ~ ( 3)3 NI~CII~COOH
(239.5-240)
~ COOH ((H3)3 ~ ~ OH
H2N OH ( 3)3
(225.5-226)
( Cl~3 ) 3C~ - COO~I
6H2N ~ COOH O ~ N
3 ~ ( 3)3 CH3
(226.5-228)
EXUUMPLE 7
To a mixture of 200 ml of ethanol and 23.8g
(0.0701 mole) of 2,6-di(tertiary-butyl)-4-(4'carboxyl-
phenylimino)-2,5-cyclohexadien-1-one was added 2.9 g (0.072
mole) of sodium hydroxide in 20 ml of water. To this
mixture was added l.Og of 10~ palladium on charcoal,
followed by the addition of 50 ml of water. The mixture
was reduced by agitating on a Paar apparatus for about 16
hours. Celite*was added to the mixture, and the mixture
was filtered through a bed of celite*. The mixture was
acidified with 6N hydrochloric acid, and the resulting
yellow solid precipitate was collected by filtration to
provide 4-[3,5-di(tertiary-butyl)-4-hydroxyanilinolbenzoic
acid, m.p. 241-242C.
* Trade-mark
-20- 1333618
EX~MPLE 8
To a mixture of 200 ml of ethanol and 25.0g
(0.0736 mole) of 2,6-di(tertiary-butyl)-4-
(4'-carboxyphenylimino)-2,5-cyclohexadien-1-one and 12 g
(0.087 mole) of potassium carbonate warmed on a steam bath
was added l.Og of palladium on charcoal. The mixture was
reduced using a Paar apparatus for 2 hours. The mixture
was diluted with 300 ml of water, filtered through celite,
and the filtrate acidified with 6N hydrochloric acid. The
yellow solid precipitate was collected by filtration to
provide 4-[3,5-di(tertiary-butyl)-4-hydroxyanilino]benzoic
acid, m.p. 241-242C.
EXAMPLES 9-13
Using the general method of Example 7 or 8 the
imine intermediates obtained in Example 2-6 were reduced to
provide compounds of Formula I shown in Table II below:
~1 1333618
o
r ~ ~, a, '~
~a ~ W X ~X ~1 X
~X
U'~
C ,~, ~O o
a ~ O
o
m o=u
o ~
H u u m
~ Uo= U ~) ~ o
s ~ ~ o~ ~ U~
Z
u~ u~,
m ~ ~ ~ ~ ~ m ~ ~ ~ r $~
O $ ~ ~ m ~ $ $ u
q~
o ~
H
W X ~ ~" X ~"
r . O
O ~ ,_~
r~
-22-
1333618
EX~MPLE 14
A mixture of a solution of 6.3 g (0.018 mole) of-
4-[3,5-di-(tertiary-butyl)-4-hydroxyanilinolphenylacetic
acid in 10 ml of N,N-dimethylformamide and 5 g (0.036 mole)
of potassium carbonate was heated on a steam bath until gas
evolution ceased. The mixture was allowed to cool to
ambient temperature, and 5 milliliters of methyl iodide
were then added. The mixture was heated at its boiling
temperature and 5 ml aliquots of methyl iodide were added
at 20, 45 and 60 minutes. After the mixture had
evaporated, the residue was taken up in water and 2N sodium
hydroxide solution was added. The mixture was warmed, and
the insoluble residue of methyl 4-[3,5-di(tertiary-butyl)-
-4-hydroxy-N-methyl-anilino] phenylacetate was separated by
filtration. The residue was suspended and partially
dissolved in 50 ml of methanol, and 10 ml of 2. 5N sodium
hydroxide solution was added. The mixture was stirred for
about 16 hours, diluted with 300 ml of water and 300 ml of
diethyl ether, and was then further diluted with about 100
ml of hexane. The mixture was acidified with dilute
hydrochloric acid and the aqueous phase was discarded. The
organic phase was washed first with 10~ sodium bicarbonate
solution and then with 5~ sodium carbonate solution. The
product remained in the organic phase as the sodium salt.
The organic phase was acidified with 10~ aqueous
hydrochloric acid and washed with sodium chloride solution,
followed by drying. Evaporation provided a residue which
was extracted with 20 ml of boiling benzene. Hexane (6 ml)
was added, and the light yellow solid was recrystallized
first from 80% aqueous ethanol, and then from benzene to
provide fine yellow needles of 4-l3,5-di(tertiary-butyl)-
4-hydroxy-N-methylanilino]phenylacetic acid, m.p.
181-182.5C. Analysis: Calculated for C23H31NO3; %C, 74.8;
%H, 8.5; %N, 3.8; Found: %C, 74.8; %H, 8.6; %N, 3.6.
-23- 1333618
EXAMPLE 15
A solution of 3.4g (0.010 mole) of
4-(3,5-di-tertiary-butyl-4-hydroxyanilino)benzoic acid
(prepared in Example 1) in 35 ml of N,N-dimethylformamide
and 3.5 ml of methyl iodide was heated at 95C for about 4~
hours under nitrogen. The reaction mixture was poured into
cold water and the resulting solid was collected and then
taken up in chloroform. The chloroform solution was
filtered, washed with water, dried with magnesium sulfate
and evaporated to give a tan solid. This material was
recrystallized first from benzene and then from a mixture
of ethanol and water to give 1.5 g of white crystalline
4-(3~5-di-tertiary-butyl-4-hydroxy-N-methylanilino)benzoic
acid, m.p. 240-244C. Analysis: Calculated for C22H29NO3:
%C, 74.3; %H, 8.2; %N, 3.9. Found: %C, 74.4; %H, 8.3; %N,
3.5.
EXAMPLE 16
A mixture of 2.0 g (0,.00589 mole) of
2,6-di(tertiary-butyl)-4-(4-carboxyphenylimino)-2,5-
cyclohexadien-1-one and 2.5 g of sodium thiosulfite in 25
ml of lN sodium hydroxide solution and a few ml of diethyl
ether were stirred at 20C. ~fter one hour of stirring the
mixture was heated on a steam bath while adding 1.5 g of
sodium thiosulfite and enough sodium hydroxide to make the
solution alkaline. After one hour the solution was
acidified with 6N hydrochloric acid. The precipitate
separated by filtration to provide a light orange solid was
about 50~ 3-[3,5-di(tertiary-butyl)-4-hydroxyanilino]-
benzoic acid according to thin layer chromatographic andinfrared spectral analyses.
EXAMPLES 17 - 19
Using the general method of Example 1, the
aminobenzene starting materials of Formula III below were
reacted with 2,6-di(tertiary-butyl)-p-benzoquinone to
provide the imine products indicated in T~BLE III.
-29-
1333618
TA~LE III
Starting Product of Melting
Example Material of Formula IV Point
Number Formula III (m p. in C in ac
S CO2H tC113)3C CO21l
17 H2N- ~ O S O ~ > N- ~ ~ (241-245)
Cl~3 (C 3)3C Cl~3
CO2H (CH3 ~C CO2H
18 2 ~OE~ O~N~~ - OH ( 249-259)
(C 3)3C
CO2H (CH3)3C CO2H
19 H2N~Cl ~N~ Cl ( 195-199)
(C 3)3C
EXAMPLE 20
To a mixture of 225 ml ethanol and 13.6 g (0.038
mole) of 2,6-di(tertiary-butyl~-4-(5'-carboxy-2'-methyl-
phenylimino)-2,5-cyclohexadiene-1-one (from Example 18) was
added 1 9 of 5% palladium on charcoal (50~ water wet). The
mixture was reduced by agitating on a Paar apparatus for 2
hours. The mixture was filtered through celite to remove
the catalyst and the filtrate was concentrated on a rotary
evaporator to give 13.0 g of a light orange solid, m.p.
229-233aC. This material was recrystallized from aqueous
ethanol to give 10.8 9 orange crystalline solid m.p.
-25- 1333618
234-239C. This material was in turn recrystallized from
benzene to give 9.6 g pale orange solid
3-(3,5-di-tertiary-butyl-4-hydroxyanilino)-4-methylbenzoic
acid, m.p. 234-239C. Analysis: Calculated for C22H29NO3:
S %C 74.3; ~H 8.2, 6N 3.9. Found: ~C, 74.3; ~11, 8.2; ~N 3.8.
EXAMPLE 21 - 22
Using the general method of Example 20, the imine
intermediates obtained in Examples 18 and 19 were reduced
to provide compounds of Formula I shown in TABLE IV below:
TABLE IV
Example
Number Product of Formula I (m.p. in C)
(CH3)3/ CO2H
21 HO ~ NH ~ -Cl (200.5-201.5)
(CH3)3c
(CH3)3c CO2~-l
22 HO- ~ NH- ~ OH (234-238)
(C 3)3C
EXAMPLE 23
A mixture of 22.0 g (0.10 mole) of
2,6-di(tertiary-butyl)-p-benzoquinone, 19.0 g (0.105 mole)
of (4-aminophenyl)thioacetic acid, 100 ml of
tetrahydrofuran and 1 ml of boron trifluoride: diethyl
ether complex was heated at gentle reflux, with stirring,
for 1.5 hours. The reaction mixture was concentrated under
a nitrogen gas flow to a volume of 75 ml. The concentrate
was diluted with 250 ml ethanol and 1 g of palladium on
-26- 1333618
charcoal was added. This mixture was hydrogenated on a
Paar apparatus for 12 hours, then filtered through celite
to remove the catalyst. The filtrate was concentrated to
give 33.7 g of an oil. The oil was taken up in diethyl
ether. This solution was washed with dilute (about 10~)
hydrochloric acid, then dried and evaporated to give 18.4 g
of a gummy solid. This material was recrystallized from
5:6 benzene:hexane to give 6.3 g of pink solid
4-(3,5-di-tertiary-butyl-4-hydroxyanilino)phenylthioacetic
acid, m.p. 136.5-137.5C. Analysis: Calculated for
C22H29NO3S: ~C, 68.2; %H, 7.5; %N, 3.6. Found: %C, 68.3;
%H, 7.7; %N, 3.3.
EXAMPLE 24
A mixture of 22.5 g (0.105 mole) of
2,6-di-tertiary-butyl-p-benzoquinone, 11.8 g (0.10 mole) of
anthranilonitrile, 50 ml of tetrahydrofuran and 1 ml of
boron trifluoride:diethyl ether complex was heated at
gentle reflux for 2 hours. Heating was continued for an
additional 2 hours under a stream of nitrogen gas to
concentrate the mixture. The concentrated mixture was
diluted with 50 ml of ethanol, warmed to effect complete
dissolution, and then allowed to cool. The precipitate was
collected, rinsed with cold 4:1 methanol:water, and oven
dried to give 23.6 g of orange crystals of
2,6-di-tertiary-butyl-4-(2'-cyanophenylimino)-2,5-
cyclohexadien-1-one, m.p. 109-110.5C.
To a mixture of 15.0 g (0.0468 mole) of
2,6-di-tertiary-butyl-4-(2'-cyanophenylimino)-2,5-
cyclohexadien-1-one (obtained above) and 200 ml of ethanol
was added 1 g of palladium on charcoal. The mixture was
hydrogenated on a Paar apparatus for 10 minutes. The
solvent was decanted off, then the residual solid was
dissolved in chlo-roform. The chloroform solution was mixed
with ethanol, filtered, and concentrated to give 14.6 g of
tan solid, m.p. 170-173C. ~ portion (1.9 g) of this
material was recrystalli%ed from ethanol to give pale
-27- I 333618
orange prisms of the novel compound 2-(3,5-di-tertiary-
butyl-4-hydroxyanilino)benzonitrile, m.p. 171.5-172.5C.
Analysis: Calculated for C21H26N2O: ~C 78.2; ~1~ 8.1; ~N
8.7. Eound: %C 78.3; ~H 8.3; ~N 8.6.
A mixture of 5.~ g (0.018 mole) 2-(3,5-di-
tertiary-butyl-4-hydroxyanilillo)ben7.onittile, 40 g of 50%
sodium hydroxide and 100 ml of ethanol was heated at gentle
reflux for 3 hours. The reaction mixture was poured onto a
mixture of 100 ml of 6N hydrochloric acid and ice. The
precipitate was collected and dried in a vacuum oven to
give 5.8 g of yellow solid, m.p. 217-219C. This material
was recrystallized from a mixture of ethanol and water to
give 4.9 g of light orange needles of N-(3,5-di-tertiary-
butyl-4-hydroxyphenyl)anthranilic acid, m.p. 220.5-221.5C.
Analysis: Calculated for C21tl27NO3: %C 73.9; ~H 8.0; ~,N
4.1; Found: %C 74.1; ~H 8.1; %N 4Ø
EXAMPLES 25-26
Using the general method of Example 1 the
starting materials of Formula VI shown in TABLE V below
were reacted with 2,6-di(tertiary-butyl)-p-ben~oquinone to
provide the imine products indicated in TABLE V.
Starting
Example Material of
Number Formula VI Product of Formula I (m.p. in C)
CN (CH3~ C CN
NH2 ~ N ~ (102.5-103)
(C113)3C
(CH3)3c
26 NH~ ~N ~ N ~ CN (140-142)
(C1~3)3C
-2~- 1333618
-
EXAMPLE 27
Using the method of Example 20, ~.0 g of
2,6-di-t-butyl-4-(3'-cyanophenylimino)-2,5-cyclohexadien-1-
one (from Example 26) was converted to 3-(3,5-di-t-butyl-
4-hydroxyanilino)benzonitrile, m.p. 150-153C.
EXAMPLE 28
Eight g (0.025 mole) of
3-(3,5-di-t-butyl-4-hydroxyanilino)benzonitrile (from
Example 27), 4.9 g (0.075 mole) of sodium azide, 4.0 g
(0.075 mole) of ammonium chloride, 1.06 g (0.025 mole) of
lithium chloride and 60 ml of N,N-dimethylformamide were
combined under a nitrogen atmosphere and heated at 110C
for 48 hours. The reaction mixture was poured into cold 6N
hydrochloric acid and a gummy solid precipitated out. The
supernatant was decanted off and the residue dissolved in
ethanol. The ethanol solution was diluted with water and a
pink solid was collected. This material was recrystallized
from a mixture of ethanol and water to give 5.97 g of
5-[3-(3,5-di-t-butyl-4-hydroxyanilino)phenyl]tetrazole,
m.p. 231-233C. Analysis: Calculated for C21H27NSO: %C,
69.0; ~H, 7.4; %N, 19.2; Found: %C, 6n.4; ~H, 7.6; %N,
18.8.
EX~MPLE 29
Using the method of Example 20, 10 g of
2,6-di-t-butyl-4-(4'-cyanophenylimino-2,5-cyclohexadien-1-
one (from Example 27) was hydrogenated to give
4-(3,5-di-t-butyl-4-hydroxyanilino)benzonitrile.
EXAMPLE 30
Seven g (0.0205 mole) of 4-(3,5-di-t-butyl-4-
hydroxyanilino)benzonitrile (from Example 29), 4.01 g
(0.0615 mole) of sodium azide, 3.29 g (0.0615 mole) of
ammonium chloride and 50 ml of N,N-dimethylformamide were
combined and heated first at 105C for 20 hours and then at
150C for 9 hours. The reaction mixture was diluted with
-29- 1333618
-
diethyl ether and water and acidified with 6N ~ICl. The
ether phase was washed with sodium chloride solution, dried
over sodium sulfate, and concentrated to an oil under a
stream of nitrogen. The crude product was treated with
chloroform and hexane to convert it from an oil to a solid.
The solid was recrystalli7ed from a mixture of ethanol and
water to give 2.6 g of light orange needles of 5-[4-(3,5-
di-t-butyl-4-hydroxyanilino)phenylltetrazole, m.p.
22~-225C. ~nalysis: Calculated for C211~27N5O C2l~5OII:
%C, 67.1; %H, 8.1; ~N, 17.0; ~ound: ~C, 67.1; ~H, 8.2; ~N,
16.9.
EX~MPLE 31
Preparation of N,N-Dimethyl-2-aminoethyl
3-(3,5-Di-tertiary-butyl-4-hydroxyanilino)benzoate
Under a nitrogen atmosphere, 6.0 g (0.0176 mole)
of 3-(3,5-di-tertiary-butyl-4-hydroxyanilino)benzoic acid
(from Example 13) and 2.53 g (0.0176 mole) of
2-dimethylaminoethyl chloride hydrochloride were dissolved
in 17 ml of N,N-dimethylformamide. To this mixture was
added 4.9 ml (0.035 mole) of triethylamine and the reaction
waS heated at 100C for 25 hours. The reaction temperature
was then raised to 120C and heating was continued for an
additional 28 hours. The reaction mixture was diluted with
10 ml of diethyl ether, then filtered to remove
triethylamine hydrochloride. The filtrate was diluted with
additional diethyl ether and then shaken with cold 10%
hydrochloric acid. A middle layer contained most of the
product hydrochloride. It was diluted with water and
ether, and basified with solid sodium carbonate. The ether
extract was washed with saturated aqueous sodium chloride
and then concentrated to give 1.7 g of a tan solid, m.p.
119-120C. This material was recrystallized first from 20
ml of cyclohexane and then from a mixture of 10 ml of
ethanol and 3 ml of water to give 1.48 g of light yellow
prisms of N,N-dimethyl-2-aminoethyl 3-(3,5-di-t-butyl-4-
hydroxyanilino)benzoate m.p. 123-124C. Analysis:
-30- 1 3 3 3 6 18
Calculated for C25H36N23 %C~ 72-8; ~H~ 8-8; ~N~ 6-8-
Found: %C, 73.0; %H, 8.8; ~N, 6.7.
EXAMPLE 32
Preparation of N-Acetyl-3-(3,5-di-t-butyl-4-
hydroxyanilino)benzoic Acid.
A mixture of 5.6 g of 3-(3,5-di-t-butyl-~-
hydroxyanilino)benzoic acid (from Example 13) and 15 ml of
acetic anhydride was heated under a nitrogen atmosphere at
70-120C for about 90 minutes. The mixture was cooled to
80C, 5 drops of pyridine were added and the reaction was
reheated to 120C for 10 minutes. An additional 0.25 ml of
pyridine was added at about 80C, followed by the gradual
addition of 10 ml of water to hydrolyze the excess acetic
anhydride and the mixed anhydride of the product. I~eating
was continued until a precipitate formed. The reaction
mixture was allowed to cool to room temperature, and the
precipitate was then collected, rinsed with a cold mixture
of methanol and water, and dried to give 4.3 g of off-white
solid m.p. 204-205.5C. This material was recrystallized
first from a mixture of 30 ml of ethanol and 5 ml of water,
and then from a mixture of 30 ml of isopropanol and 5 ml of
hexane to give 3.3 g of white solid N-acetyl-3-(3,5-di-t-
butyl-4-hydroxyanilino)benzoic acid m.p. 214.5-215.5C.
Analysis: Calculated for: C23H29NO4 1/2(CH3)2CHOH: %C,
71.2; %H, 8.0; %N 3.4. Found: %C, 71.4; ?~H, 8.1; %N, 3.2.
EXAMPLE 33
Preparation of N-Trifluoroacetyl-3-(3,5-di-t-
butyl-4-hydroxyanilino)benzoic Acid.
2.96 g of 3-(3,5-~i-t-butyl-4-hydroxyanilino)-
benzoic acid (from Example 13) was slurried in 10 ml of
trifluoroacetic anhydride. After several minutes, the
reaction boiled (40C) and then became clear. The reaction
mixture was poured into a mixture of ice and water and the
resulting solid was collected and dried. This material was
recrystallized from a mixture of qO ml of ethanol and 12 ml
-31- 1 33361 8
of water to give 3.07 g of white crystalline N-trifluoro-
acetyl-3-(3,5-di-t-butyl-4-hydroxyanilino)-ben7Oic acid
m.p. 181C. Analysis: Calculated for C23H26F3NO~: ~C,
63.1; %H, 6.0; %N, 3.2. Found: ~C, 63.0; ~H, 6.4; ~sN, 2.8.
s
EX~MPLE 34
A mixture of 22.0 g (0.10 mole) of
2,6-di-t-butyl-p-benzoquinone, 15.2 g (0.10 mole) of
3,4-diaminobenzoic acid, 50 ml of tetrahydrofuran and 1 ml
of boron trifluoride etherate was heated at about 55C for
45 minutes. The reaction mixture was diluted with 100 ml
of ethanol and 40 ml of water and was then allowed to stand
overnight at 25C. The resulting precipitate was collected
and dried to give 31.8 g of deep red solid 4-amino-3-(3,5-
di-t-butylcyclohexadienon-4-ylideneaminoJben~oic acid, m.p.
253-253.5C. Analysis: Calculated for C21H26N2O3: %C,
71.2; ~H, 7.4, ~N, 7.9. Found: ~C, 71.0; ~il, 7.5; ~N, 7.8.
A mixture of 21.1 g of 4-amino-3-(3,5-di-t-butyl-
cyclohexadienon-4-ylideneamino)- benzoic acid, 50 ml of
ethanol, 20 ml of water, 2.4 g of sodium hydroxide and 0.03
g of 5~ palladium on charcoal catalyst was placed on a Paar
appartus. After 16 hours the hydrogen uptake was complete.
Under a nitrogen atmosphere, the reaction was filtered into
13 ml of 6N hydrochloric acid. The filtrate was diluted
with water and additional hydrochloric acid. The resulting
precipitate was collected, rinsed with a cold mixture of
methanol and water and dried to give 15.5 g of a lavender
solid, m.p. 261.5-262C. Two g of this material was
stirred with 150 ml of warm ethyl acetate, and was then
filtered. The filtrate was diluted with 25 ml of hexane.
The resulting precipitate was collected and dried to give
0.3 g of light purple crystalline 4-amino-3-(3,5-di-t-
butyl-4-hydroxyanilino)benzoic acid m.p. 261-261.5~C.
Analysis: Calculated for C21H28N2O3: ~C, 70.8; ~H, 7.9; %N,
7.9. Found: %C, 70.8; ~H, 7.9; ~N, 7.6.
` -32- 1333618
EXAMPLE 35
Under a nitrogen atmosphere, a suspension of 2.0g
of 5-[4-(3,5-di-tertiary-butyl-4-hydroxyanilino)phenylJ-
tetrazole, prepared in Example 30, and 2.0g of potassium
carbonate in 4 ml of N,N-dimethylformamide was warmed to
obtain a deep red solution of the potassium salt.
Approximately 4g of methyl iodide was added and the
reaction was warmed for several minutes until the color
lightened. An additional 4g of methyl iodide was added and
the reaction mixture was heated at a gentle reflux for
about five minutes. The reaction mixture was cooled,
diluted with diethyl ether, and poured into dilute
hydrochloric acid. The ether phase was washed with water
and brine, dried with sodium sulfate, and evaporated to
- 15 give 1.889 of a red-brown solid. This material was
recrystallized from a mixture of benzene and hexane to give
0.53g of light yellow-tan Material #1, m.p. about 205C. A
solid precipitated from the mother liquor of Material ~1.
It was collected to give 0.73g of pink Material #2, m.p.
172-175.5C. Material #1 was recrystallized from a mixture
of 20 ml of ethanol and 5 ml of water to give 0.34g of
light brown granules, m.p. 218-221C. Material #2 was
stirred with 50 ml of ethanol, then filtered to remove some
undissolved material. The filtrate was diluted with 20 ml
of water to give 0.56g of pale pink leaflets, m.p.
175.5-177C. By proton NMR analysis, Material #1 is
believed to be the 1-methyltetrazole and Material #2 the
2-methyltetrazole. The delta-values are 4.15 and 4.36 ppm,
respectively, for the two products.
EX~MPLE 36
Using the method of Example 1, 4-aminobenzyl-
cyanide was reacted with 2,6-di-tertiary-butyl-p-benzo-
quinone to give 2,6-di-tertiary-butyl-4-(4'-cyano-
methylphenylimino)-2,5-cyclohexadiene-1-one, m.p.
126.5-127.5C. Following the reduction method of Example
1, Step B, the corresponding anilino phenylacetonitrile,
-33- 1333618
m.p. 146-147C, was obtained. This was converted to
5-14-(3,5-di-_-butyl-4-hydroxyanilino)benzylltetrazole,
m.p. 212-214C (dec), following the method of Example 30,
but carried out at 110C for 48 hours.
EXAMPLE 37
Example 36 was rerun using tin tetrachloride
instead of boron trifluoride as the catalyst. Thin layer
chromatography using two different systems showed that the
reaction mixture contained the desired 2,6-di-tertiary-
butyl-4-(4'-cyanomethylphenylimino)-2,5-cyclohexadien-1-
one.
EXAMPLE 38
Example 36 was rerun using titanium tetrachloride
instead of boron trifluoride as the catalyst. Thin layer
chromatography using two different systems showed that the
reaction mixture contained the desired 2,6-di-tertiary-
butyl-4-(4'-cyanomethylphenylimino)-2,5-cyclohexadiene-1-
one.
EXAMPLE 39
A mixture of 1.70g (0.005 mole) of
3-(3,5-di-tertiary-butyl-4-hydroxyanilino)benzoic acid
(prepared in Example 13) and 50 ml of hot isopropyl alcohol
was filtered to remove a small amount of insoluble
material. The resulting solution was deoxygenated with a
stream of nitrogen gas. Uner a nitrogen atmosphere, a
solution of 0.44g (0.005 mole) of morpholine in 1.5 ml of
isopropyl alcohol was added with rapid stirring.
Evaporation provided a solid which was recrystallized from
a mixture of isopropyl alcohol and isopropyl ether to give
solid morpholinium-3-(3,5-di-tertiary-butyl-4-hydroxy-
anilino)benzoate, m.p. 147-150C. Analysis: Calculated for
C21H27NO3C4HgNO: ~C, 70.1; %H, 8.5; %N, 6.5; Found: %C,
69.8; %H, 8.5; %N, 6.4.
-34- 1333618
EXAMPLE 40
A suspension of 6.68g (0.0196 mole) of
2,6-di-tertiary-butyl-4-~3'-carboxypllenylimino)-2,5-cyclo-
hexadien-l-one (prepared in Example 2) in 20 ml of benzene
and 3 ml of thionyl chloride was heated at reflux with
stirring until gas evolution had ceased. Evaporation
provided an oil which was diluted witll a small amount of
tetrahydrofuran and added dropwise to a suspension of 3.79
of anhydrous 5-aminotetrazole in 25 ml of tetrahydrofuran
containing 1.5 ml of pyridine. The reaction mixture was
allowed to stand at room temperature under a nitrogen
atmosphere for 16 hours. The reaction mixture was diluted
to a volume of 500 ml with diethyl ether, and was then
filtered. The filter cake was rinsed with diethyl ether
and resuspended in 300 ml of diethyl ether and filtered
again. The combined filtrates were evaporated to give 4.7g
of an orange solid, m.p. 241-244C. One g of this material
was recrystallized from ethanol to provide 0.3g of orange
3-(2,6-di-tertiary-butylcyclohexadienon-4-ylideneamino)-N-
(S-tetrazolyl)benæamide, m.p. 271.5C (dec.). Analysis:
Calculated for C22H26N6O2
Found: %C, 65.1; %H, 6.4; %N, 20.5.
A of mixture of 3.lg 3-(2,6-di-tertiary-butyl-
cyclohexadienon-4-ylideneamino)-N-(5-tetrazolyl)benzamide~
200 ml of tetrahydrofuran, and 0.7g of palladium on
charcoal catalyst was placed on a Paar apparatus.
Hydrogenation was complete after two hours. Under a
nitrogen atmosphere, the reaction was filtered to remove
the catalyst. The filtrate was evaporated to give 4.39 of
a sticky yellow solid which was recrystallied from a
mixture of 130 ml acetic acid and 20 ml of water to give
1.6g of yellow solid 3-(3,5-di-tertiary-butyl-4-hydroxy-
anilino)-N1-(5-tetrazolyl)benzamide, m.p. 282-283C.
Analysis: Calculated for C2zH2~N6O2: %C, 64.7; ~11, 6.9; %N,
20.6; Found: %C, 64.6; ~H, 6.8; ~N, 20.2.
~ -35- 1333618
EXAMPLE 41
A mixture of 5.51g (0.025 mole) of 2,6-di-
tertiary-butyl-p-benzoquinone, 4.54 g (0.027 mole) of
3-(4-aminophenyl)propionic acid, 50 ml of tetrahydrofuran
and 0.25 ml of boron trifluoride etherate was heated on a
steam cone under a slow stream of nitrogen for two hours.
The resulting solid was triturated with hexane, collected,
rinsed with hexane and recrystallized from a mixture of
ethyl acetate and hexane to give 5.lg yellow 3-[4-(2,6-di-
tertiary-butylcyclohexadienon-4-ylideneamino)phenyll-
propionic acid, m.p. 165-167C. Analysis: Calculated for
C23H29NO3: %C, 75.2; %H, ~.0; hN, 3.~; Found ~C, 74.8; %I~,
7.9; %N, 3.7.
A mixture of 4.0 g of 3-14-(2,6-di-tertiary-
butylcyclohexadienon-4-ylideneamino)phenyl~propionic acid,
200 ml of ethanol of O.lg of 10~ palladium on charcoal
catalyst was placed on a Paar apparatus. Hydrogen uptake
was complete after 30 minutes. Under a nitrogen
atmosphere, the reaction mixture was filtered to remove
catalyst. The filtrate was evaporated to give an oil which
was coevaporated with hexane to remove all traces of
ethahol and then triturated with hexane to give a light
orange crystalline solid. This solid was recrystallized
from a mixture of ethanol and water to give 3.lg
3-[N-(3,5-di-tertiary-butyl-4-hydroxyphenyl)-4-amino-
phenyl]propionic acid, m.p. 140-142C. Analysis:
Calculated for C23H31NO3: %C, 74.8; %1~, 8.5; %N, 3-8;
Found: ~C, 74.7; %~I, 8.3; ~N, 3.9.
EXAMPLE 42
A mixture of 22.0g (0.10 mole) of
2,6-di-tertiary-butyl-p-benzoquinone, lO.9g (0.01 mole) of
m-aminophenol, 50ml of tetrahydrofuran and 0.5 ml of boron
trifluoride etherate was stirred at room temperature for
about one hour. The reaction mixture was diluted with
diethyl ether, and the ether solution was extracted with
10% hydrochloric acid and dried over magnesium sulfate.
~ 36 1333618
Evaporation gave an orange-red solid. This material was
dissolved in a mixture of diethyl ether and methylene
chloride. The solution was filtered, and was then
evaporated to give 29.1g orange-red solid. This material
was recrystallized from a mixture of 50 ml of benzene and
100 ml of hexane to give 17.0g of orange-red crystals, m.p.
162-169C. This material (2.5g) was recrystallized first
from a mixture of 15 ml benzene and 10 ml of hexane and
then from a mixture of 10 ml of isopropanol and 7 ml of
water to give 1.3g of orange crystalline 3-(2,6-di-
tertiary-butylchclohexadienon-4-ylideneamino)phenol, m.p.
169-169.5C. Analysis: Calculated for C20H25NO2: %C, 77.1;
~H, 8.1; ~H, 4.5; Found: %C, 77.2; 6~1, 8.0; fiN, 4.6.
1.36g (0.027 mole) of 50~ sodium hydride was
added in portions to a solution of 7.06g (0.023 mole) of
3-(2,6-di-tertiary-butylcyclohexadienon-4-ylidenamino)-
phenol in a mixture of 50 ml 1,2-dimethoxyethane and 10 ml
of dimethylacetamide. Three ml (0.027 mole) of ethyl
bromoacetate was then added in portions. The reaction
mixture was stirred at room temperature for about one hour,
and a solution of 1.3g of sodium hydroxide in 12 ml of
water wa$ added. After about 30 minutes the reaction
mixture was acidified with hydrochloric acid and was
extracted with diethyl ether. The ether extract was washed
with a saturated sodium chloride solution and evaporated to
give an orange solid. This solid was recrystallized from a
mixture of benzene and hexane to give 6.2g of an orange
solid, m.p. 161-162C. One g of this material was
recrystallized from a mixture of 10 ml of ethanol and 5 ml
of water to give 0.8g of orange crystalline 3-(2,6-di-
tertiary-butylcyclohexadienon-4-ylideneamino)phenoxyacetic
acid, m.p. 163-165C. Analysis: Calculated for C22H27NO4:
%C, 71.5; %H, 7.4; %N, 3.8; Found: ~C, 71.8; %H, 7.2; %N,
3.7.
A mixture of 5.0g of 3-(2,6-di-tertiary-butyl-
cyclohexadienon-4-ylideneamino)phenoxyacetic acid, 250 ml
of etllanol and 10 mg of 5~ palladium on charcoal catalyst
-37- 1333618
was placed on a Paar apparatus. Hydrogenation was complete
after five hours. The reaction mixture was filtered to
remove the catalyst and the filtrate was evaporated to give
a thick brown oil. The oil was dissolved in 20 ml of
benzene, filtered, diluted with 20 ml of c~clohexane and
10 ml of hexane, and chilled to give l.qg oF light tan
crystalline 3-(3,5-di-tertiary-butyl-4-hydroxyanilino)-
phenoxyacetic acid, m.p. 127-127.5C. Analysis: Calculated
for C22H29NO4; %C, 71.1; %E~, 7.9; %N, 3.8; Found: %C, 70.3;
%H, 7.6; %N, 3.7.
EXAMPLE 43
A suspension of 6.0g (0.027 mole) of
2,6-di-tertiary-butyl-p-benæoquinone, 3.3g (0.020 mole) of
p-aminocinnamic acid, 15 ml of tetrahydrofuran and 0.3 ml
of boron trifluoride etherate was heated at reflux for one
hour. The reaction mixture was dissolved in a minimum
amount of methylene chloride, and was diluted to a final
volume of 700 ml with diethyl ether. The ether solution
was washed first with cold 10~ hydrochloric acid and then
with brine, and was then dried over magnesium sulfate and
evaporated almost to dryness. The residue was diluted with
300 ml of hexane and evaporated almost to dryness before
being diluted with 500 ml of warm hexane. The mixture was
allowed to cool to room temperature before being filtered
to give 5.8g of a bright red powder. One g of thls
material was recrystallized from a mixture of 15 ml of
benzene and 3 ml of hexane to give 0.5g of red crystalline
4-(2,6-di-tertiary-butylcyclohexadienon-4-ylideneamino)-
cinnamic acid, m.p. 216-217.5C. Analysis: Calculated for
C23H27NO3; %C, 75.6; %H, 7.4; ~N, 3.~; Found: ~C, 75.9; ~H,
7.5; %N, 3.8.
A mixture of 2.0g of 4-(2,6-di-tertiary-butyl-
cyclohexadienon-4-ylideneamino)cinnamic acid, 100 ml of
methanol, 0.5 ml of concentrated hydrochloric acid and 2g
of zinc powder was stirred for ten minutes. The mixture
was filtered and the filtrate was evaporated to give a
' -38- 1333618
-
yellow gummy solid. This material was recrystallized from
a mixture of 15 ml of benzene, 4 ml of llexane and 2 ml of
cyclohexane to give 0.4 g of yellow granular 4-(3,5-di-
tertiary-butyl-4-hydroxyanilino)cinnamic acid, m.p.
199-200C. Analysis: Calculated for C23il29NO32/3C6H6: 6C,
77.4; %H, 7.9; %N, 3.3; Found: ~C, 77.2; ~H, 7.8; ~N, 3.3.
EXAMPL~ 44
A suspension of 5.0g (0.0147 mole) of
2,6-di-tertiary-butyl-4-(3'-carboxyphenylimino)-2,5-cyclo-
hexadien-1-one (prepared in Example 2) in 15 ml of benzene
and 2.5 ml of thionyl chloride was heated at reflux until
gas evolution ceased. The solution was evaporated, and was
evaporated twice more following additions of benzene. The
resulting acid chloride was added dropwise to a solution of
5.5g sodium trifluoromethanesulfonamide in 25 ml of
1,2-dimethoxyethane. The solvent was evaporated with a
stream of nitrogen to give a yellow solid. This solid was
stirred with 200 ml of tetrahydrofuran and was then
filtered to remove insoluble material. The filtrate was
hydrogenated for 16 hours on a Paar apparatus using 0.5g of
5% palladium on charcoal as the catalyst. The catalyst was
removed by filtration and the filtrate was evaporated to
give a dark brown oil. The oil was dissolved in 25 ml of
water. This solution was added to a mixture of 5.0 ml of
10% hydrochloric acid, water and ice to give 6.7 g of a
white solid. This solid was recrystallized from a mixture
of 70 ml of ethanol and 20 ml of water to give 3g of white
crystalline N-[3-(3,5-di-tertiary-butyl-4-hydroxyanilino)-
benzoyl]trifluoromethanesulfonamide, m.p. 234-234.5C.
Analysis: Calculated for C221i27F3N2O4S: ~C, 55.9; ~, 5.8;
%N, 5.9; Found: ~C, 56.1; %H, 5.8; ~N, 5.9.
EXAMPLE 45
A mixture of 13.2g (0.10 mole) of
3-amino-4-hydroxybenzoic acid, 22.0g (0.10 mole) of
2,6-di-tertiaey-butyl-p-benzoquinone, 25 ml of
' _39_ 1333618
-
tetrahydrofuran and 1 ml of boron trifluoride etherate was
heated at a gentle reflux for about 20 minutes by which
time a thick precipitate had formed. The reaction mixture
was diluted with 50 ml of ethanol and filtered to obtain
20.9g of an orange solid, m.p. 24~-250C. Six g of this
material was recrystallized from a mixture of 250 ml of
ethanol and 70 ml of water to give 3.4g of red granular
3-(2,6-di-tertiary-butylcyclohexadienon-4-ylideneamino)-4-
hydroxybenzoic acid, m.p. 275-276C. Analysis: Calculated
for C21H25NO4: %C, 71.0; %H, 7.1; %N, 4.0; Found: %C, 71.4;
~H, 7.1 %N, 3.8.
A mixture of 5.0g of 3-(2,6-di-tertiary-butyl-
cyclohexadienon-4-ylideneamino)-4-hydroxybenzoic acid,
0.05g of 5% palaldium on charcoal catalyst, 250ml of
ethanol and 50 ml of tetrahydrofuran was placed on a Paar
apparatus. Hydrogen uptake was complete in about 10
minutes. The reaction mixture was filtered to remove
catalyst. The filtrate was evaporated to qive a tan solid
which was recrystallized from a mixture of 40 ml of ethanol
and 15 ml of water to give 3.2g of reddish tan granular
3-(3,5-di-tertiary-butyl-4-hydroxyanilino)-4-hydroxybenzoic
acid, m.p. 254.5-255~C. (dec). Analysis: Calculated for
C21H27NO4: %C, 70.6; %H, 7.6; %N, 3.9; Found: ~C, 71.0; %t3,
7.6; %N, 4.1.