Note: Descriptions are shown in the official language in which they were submitted.
I 3363~
Fusion proteins, a process for their preparation and their
use
In the preparation by genetic engineering methods of rela-
tively small eukaryotic proteins with a molecul~r ~eight
up to about 15000 Daltons the yield obtained in bacteria
is frequently only small. It is presumed that the proteins
which are formed are rapidly degraded by proteases intrin-
sic to the host. For this reason, proteins of this type
- are advantageously prepared as fusion proteins, in particu-
Lar having a portion of protein intrinsic to the host,
which is then cleaved off.
It has now been found that a segment composed of only about
70 amino acids of the D-protein from the trp operon of E.
coli is particularly suitable for the formation of fusion
proteins, specifically in the region of the sequence of
amino acids 23 to 93 (C. Yanofsky et al., NucLeic Acids Res.
9 (1981)6647), also caLled "D'-peptide" hereinafter. Be-
tween the carboxyl terminal end of this peptide and of the
amino acid sequence of the desired eukaryotic protein
there is a sequence comprising one or more genetically
codabLe Dmino acids which permits the desired protein to be
cleaved off chemicalLy or enzymatically. In preferred em-
bodiments of this invention, the amino terminal end ;s foL-
lowed by a short amino acid sequence composed of Lys-Ala,
optionally followed by a sequence of 1 to 10, in particular
1 to 3, other genetically codable amino acids, preferably
by two amino acids, in particular by Lys-Gly.
Hence the invention relates to a fusion protein of the general
formula
Met-Xn-D'-Y-Z
in which
n is zero or 1,
i,
~ - 2 - l 33~329
X is a sequence of 1 to 12 gen~t~cally codable amino aC~s,
preferably Lys-Ala,
D' is a sequence of about 70 amino acids in the region o~f the
sequence of amino acids 23-93 of the D-peptide in the trp
operon of E. coli,
Y denotes a sequence of one or more genetically codable
amino acids which permits the following amino acid
sequence Z to be cleaved off, and
z is a sequence of genetically codable amino acids
representing the desired final protein.
The present invention will now be described in association with
preferred embodiments with reference to accompanying drawings,
in which:
Figure 1 shows a process for preparing plasmid pH120/14, which
is suitable ~or the expression of a fusion protein having the
first three amino acids of the L-peptide.
Figure 2 shows a process for preparing plasmid pH106/4 coding
for proinsulin.
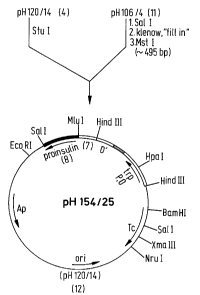
Figure 3 shows a process for preparing plasmid pH154/25, which
is suitable for the expression of a fusion protein under the
control of the trp operon, in which the amino acid sequence
Ala-Ser-Met-Thr-Arg is located after the L'- and D'-peptide and
is followed by the amino acid sequence of proinsulin.
Figure 4 shows a process for preparing plasmid pH254, which is
suitable for the expression of a fusion protein having the
amino acid sequence L', D~-proinsulin under the control of the
trp promoter, and a process for preparing plasmid pH255 which
is suitable for the insertion of a structural gene into one of
the restriction sites MluI, SalI and EcoRI.
Figure 5 shows a process for preparing plasmid pH256 which is
suitable for the insertion of structural genes into the EcoRI
site, and a process for preparing plasmid pH257.
1~
1 336329
~ - 2a
.,.
Figure 6 shows a process for ~ ~ ing plasmid pJ1~ -
coding for proinsulin.
Figure 7 shows a process for preparing plasmid pK150
coding for hirudin.
Figure 8 shows a process for preparing plasmid pK160,
which contains, immediately upstream of the trp-D sequence,
a multiple restriction enzyme recognition sequence which
embraces restriction sites for the enzymes XmaI, SmaI,
BamHI, XbaI, HincII, SalI, AccI, PstI and HindIII, and an
EcoRI restriction site downstream of the 3'-end of the
hirudin sequence.
Figure 9 shows a process for preparing plasmid pK170 which
contains a DNA sequence which codes for Met-Asp-Ser-Arg-
Gly-Ser-Pro-Gly-trp-D'-(hirudin) fused onto the trp
operator.
Figure 10 shows a process for preparing pK180 coding for
hirudin.
Figure 11 shows a process for preparing pH154/25 * coding
for proinsulin.
Figure 12 shows a process for preparing plasmid pint 13
coding for an amino ac`id sequence allowing chemical cleavage
by cyanogen bromide, n-bromosuccinimide or acids.
~ ~ - 2b '
1 336329
.. , , ;
Further aspects of the invention and preferred embodiments
are described here;nafter and defined in the patent claims.
Of course, it i~ advantageous if the undesired portion (in-
trinsic to the host) of the fusion protein is as small as
possible since then the cell produces only little "ballast"
and hence the yield of desired protein is high. Further-
more, when the undesired portion is cleaved off, fewer by-
products are produced, which fac;litates working up~ A
factor opposing this is that the (assumed) "protective
function" of the undesired portion is to be expected only
above a certain size. It has now emerged, surprisingly,
that the segment chosen according to the ;nvention from
the D-protein fulfils this task although it contains only
about 70 amino acids.
In many cases, especially in the preferred embodiment in
which X represents Lys-Ala or contains this sequence at
the ~-terminal end, the fusion protein formed is insoluble.
The latter can easiLy be separated from the solubLe pro-
teins, which great~y facilitates the working up and in-
creases the y;eld. The formation of an insolubLe fusion
prote;n is surpr;s;ng since, on the one hand, the bacterial
portion of only about 70 amino acids ;s quite small and,
on the other hand, it is a constituent of a protein which
is present in solution in the host cell~
"About 70 amino ac;ds in the region of the sequence of amino
_ 3 _ 1336329
acids 23 to 93 of the D-peptide" means that it is possible
to carry out, in a manner known per se, variations, that
is to say it is poss;ble for individual amino acids to be
deleted, replaced or exchanged without this significantly
changing the properties of the fusion proteins according
to the invention. The invention likewise relates to vari-
ations of this type.
The desired eukaryotic protein is preferably a biologicalLy
active protein, such as a hirudin, or a precursor of a pro-
tein of this type, such as human proinsulin.
The fusion protein is obtained by expression in a suitablesystem, and in the particularly preferred embodiment is,
after disruption of the host cells, isolated from the sedi-
ment in which it is concentrated owing to its sparing solu-
bility. Hence it is easy to separate it from the solubLeconstituents of the cell.
Suitable host cells are all those for which expression sys-
tems are known , that is to say mammalian cells and micro-
organisms, preferably bacteria, in particular E. coli
since, after all, the bacterial portion of the fusion
protein is a protein intrinsic to the host E. coli.
The DNA sequence which codes for the fusion protein accord-
ing to the invention is incorporated, in a known manner,
into a vector which ensures satisfactory expression in the
selected expression system.
In bacterial hosts, it is expedient to choose the promoter
and operator from the group comprising Lac, Tac, PL or
PR of the phage ~, hsp, omp or a synthetic promoter,
such as are described, for example, in German Offenlegungs-
schrift 3,430,683 (European Patent Application 0,173,149)~
A particularly suitable vector is one which contains the
following elements of the trp operon of E. coli: the pro-
1 336329
4moter, the operator and the ribosome binding site of the
L-peptide. It is particularly advantageous for the first
three amino acids of this L-peptide to follow in the cod-
ing region, and then to be followed by a short amino acid
sequence and the amino acids 23 to 93 of the D-protein in
the trp operon.
The intermediate sequence Y which makes it possible to
cleave off the desired polypeptide depends on the compo-
sition of this desired peptide: for example, if this con-
tains no methionine it is possible for Y to denote Metand then chemical cleavage with cyanogen bromide is car-
ried out. If there is cysteine at the carboxyl terminal
end in the connecting member Y, or if Y represents Cys,
then cysteine-specific enzymatic cleavage or chemical cleav-
age, for example after specific S-cyanylation, can be car-
ried out. If there is tryptophan at the carboxyl terminal
end of the bridging member r, or if Y represents Trp, then
chemical cleavage with N-bromosuccinimide can be carried
out. If Y represents Asp-Pro, then proteolytic cleavage
can be carried out in a manner known per se (D. Piszkie~icz
et al., Biochemical and Biophysical Research Communications
40 (1970) 1173-1178). The Asp-Pro bond can, as has been
found, be made even more acid-labile if Y is
(Asp)m-Pro or Glu-(Asp)m-Pro,
m denoting 1, 2 or 3. In these cases the cleavage products
obtained start at the N-terminal end with pro and terminate
at the C-terminal end with Asp.
Examples of enzymatic cleavages are likewise kno~n, it
also being possible to use mod;fied enzymes of improved
specificity (cf. C.S. Craik at al., Science 228 (1985)
291-297). If the desired eukaryotic peptide is human pro-
insulin it is possible to choose as the sequence Y a pep-
tide sequence in which an amino acid which can be cleaved
off by trypsin (Arg, Lys) is bonded to the N-terminal
amino acid (Phe) of the proinsulin, for example Ala-Ser-
` 5 1 33632q
Met-Thr-Arg, since then the arg;nine-specific cleavage can
be carried out ~ith the protease trypsin. If the des;red
protein does not contain the amino acid sequence Ile-Glu-
Gly-Arg, it is also possible to cleave ~ith factor Xa (Euro-
pean Patent Application 0,161,937).
It is also possible in the design of sequence Y-to take ac-
count of the synthetic c;rcumstances and to incorporate
suitable cleavage sites for restriction enzymes. The DNA
sequence corresponding to the amino acid sequence Y can
thus also assume the function of a Linker or adapter.
The fusion protein according to the invention is advanta-
geously expressed under the control of the trp operon of
E. coli. A DNA segment containing the promoter and operator
of the trp operon is no~ commercialLy available. The ex-
pression of proteins under the control of the trp operonhas been described many times, for example in European Appli-
cation 0,036,776. The induction of the trp operon can
be effected by the absence of L-tryptophan and/or the pre-
sence of indolyl-3-acrylic acid in the medium.
Under the control of the trp operator there is first trans-
cri~?tion of the ~ regian coding for the L-peptide. ~his L-peptide
which is composed ~f 14 amino acids, contains L-tryptophan in
each of the positians 10 and 11. The rate of protein ~ynthesis of the L-peptide
determines ~hether the downstream structural genes are like-
~ise translated or ~hether protein synthesis is term;nated.~hen there is a deficiency of L-tryptophan there no~ takes
pLace slo~ synthesis of the L-peptide as a result of the
~ow concentration of the tRNA for L-tryptophan, and the
follo~ing proteins are synthesized. In contrast, ~hen there
are high concentrations of L-tryptophan the corresponding
segment of mRNA is rapidly read and termination of protein
biosynthesis takes place, since the mRNA assumes a terminator-
like structure (C. Yanofsky et al., loc. cit.).
The frequency of translation of a mRNA is great~y influenced
1 336329
- 6 -
by the nature of the nucleotides-in the vicinity of the start
codon. Thus, on expression of a fusion protein with the aid of
the trp operon, it appears favorable to insert the nucleotides
for the first few amino acids of the L-peptide for the start of
the structural gene of the fusion protein. In the preferred
embodiment of the invention using the trp system the
nucleotides of the first three amino acids of the L-peptide
(called L'-peptide hereinafter) were chosen as codons for the
N-t~rm; n~ 1 amino acids of the fusion protein.
Hence the invention also relates to vectors, preferably
plasmids, for the expression of fusion proteins, the DNA of the
vectors having the following feature from the 5'-end (in
suitable order and in phase): a promoter, an operator, a
ribosome binding site and the structural gene for the fusion
~5 protein, the latter containing amino acid sequence I (appendix)
upstream of the sequence of the desired protein. Upstream of
the structural gene, or as the first triplet of the structural
gene, there is located the start codon (ATG) and optionally
further codons for other amino acids which are arranged between
the start codon and the D'-sequence or between the D~-sequence
and the gene for the desired protein. The choice of the DNA
se~uence upstream of the structural gene depends on the amino
acid composition of the desired protein, in order to make it
possible to cleave the desired protein off from the fusion
protein.
It may prove advantageous in the expression of the fusion
protein according to the invention to modify individual
triplets of the first few amino acids downstream of the ATG
start codon in order to prevent any base-pairing at the level
of the mRNA. Modifications of this type are, just as are
modifications, deletions or additions of individual amino acids
in the D'-protein, familiar to those skilled in the art, and
the invention likewise relates to them.
1 33632q
- ~ - 7 -
Since relatively small plasmids confer several advantages,
a preferred embodiment of the invention comprises the e~i-
~ mination of a DNA segment with the structural gene for tetra-
cycline resistance from pLasmids derived from pBR 322. It
is advantageous to delete the segment from the HindIII
restriction site at position 29 to the PvuII restriction
site at position 2066. It is particularly advantageous to
delete a DNA segment which is even somewhat larger from
the plasmids according to the invention, by making use of
the PvuII restriction site at the start (in the direction
of reading) of the trp operon (which is located in a non-
essential part). It is thus possible to carry out direct
ligation of the resulting large fragment with the two PvuII
restriction sites. The resulting plasmid, which has been
shortened by about 2 kbp, effects an increase in expres-
sion, and this is possibly attributable to an increased
copy number in the host cell.
The invention is illustrated in detail in the examples which
follow.
Example 1
a) Chromosomal E. coli DNA is cut with Hinf I, and the 492 bp
fragment which contains the promoter, operator, the struc-
tural gene of the L-peptide, the attenuator and the codons
for the first six amino acids of the trp-E structural gene
from the trp operon is isolated. This fragment is filled
in with deoxynucleotide triphosphates with the aid of
Klenow polymerase, linked at both ends to an oligonucleo-
tide which contains a recognition site for HindIII and is
then cut with HindlII~ The HindIII fragment thus obtained
is Ligated into the HindIII restriction site of pBR 322.
This results in the plasmid ptrpE2-1 (J.C. Edmann et al.,
Nature 291 (1981) 503-506). This is converted into the
plasmid ptrpL1 as described.
- o
t 336329
- 8 -
By use of the synthetic oligonucleotides (N1) and (N2)
5' CGA ~AA TGA AAG CAA AGG 3' (N1)
5' CCT TTG CTT TCA TTG T 3' (N2)
which hybridize to th~ double-strand~d ~ligo--
nucleotide tN3)
5' CGA CAA TGA AAG CAA AGG 3'
3' T GTT ACT TTC GTT TCC 5'
the DNA sequence for the first three amino acids of the
L-peptide is incorporated, and a restriction site (StuI)
for the insertion of further DNA is formed, in the ClaI
site of the plasmid ptrpL1 (Figure 1). The plasmid
ptrpL1 is reacted with the enzyme CLaI in accordance
with the manufacturer's instructions, and the mixture
is extracted with phenol and the DNA is precipitated
with ethanol. The opened plasmid is reacted with al-
kaline phosphatase from E. coli to remove the phosphate
groups at the 5'-ends. The synthetic nucleotides are
phosphorylated at the 5'-ends and are inserted, using
T4 DNA ligase, into the opened pLasmid which has been
treated with phosphatase. After the li~ase reaction
is complete, transformation into E. coli 294 and seLec-
tion of the transformants by Amp resistance and the pre-
sence of a StuI restriction site are carried out.
About 80Z of the resulting clones had the expected re-
striction site ; the nucLeotide sequence depicted in
Figure 1 was confirmed by sequence analysis. The plasmid
pH120/14 which contains downstream of the ribosome bind-
ing site for the L-peptide the nucleotide triplets for
the first three amino acids of the L-peptide (L'-peptide),
followed by a StuI site which in turn permits the in-
sertion of further DNA and thus aLlows the formation of
fusion proteins having the first three amino acids of
- 9 _ 1 336 32q
the L-peptide, is obtained.
b) The example of the oligonucleot;de (N1) employed above
;s used below to ;llustrate the chem;cal synthes;s of
such oligonucleot;des:
The method of M.J. Ga;t et al., NucLe;c Acids Research
8 (1980) 1081-1096 is used to bond covalently the nu-
cleos;de at the 3'-end, that ;s to say guanos;ne in the
present case, to a glass bead support (CPG ~=controlled
pore glass) LCAA (=long-chain alkylam;ne) supplied by
Pierce) via the 3'-hydroxyl group. This entails the
guanosine being reacted as the N-2-;sobutyryl 3'-0-
succ;nyl 5'-d;methoxytrityl ether ~;th the modified sup-
port in the presence of N,N'-dicyclohexylcarbodiimide
and 4-dimethylaminopyr;dine, there being acylation of
the amino radical of the long-chain amine on the support
by the free carboxy~ group of the succinyl radical.
In the subsequent steps ;n the synthesis the base com-
ponent is used as the dialkylamide or chlor;de of the
mono-methyl ester of the 5'-0-d;methoxytr;tylnucLeoside-
3'-phosphorous acid, the adenine being in the form of
the N6-benzoyl compound, the cytosine being in the
form of the N4-benzoyl compound, the guanine being in
the form of the N2-isobutyryL compound, and the
thymine, ~hich contains no am;no group, being without
a protective group.
40 mg of the support containing 1 ~mol of bound guano-
s;ne are treated success;vely ~;th the follow;ng agents:
a) Methylene chlor;de,
b) 10% tr;chloroacetic acid ;n methylene chloride,
c) methanol,
d) tetrahydrofuran,
e) acetonitriLe,
f) 15 ~mol of the appropriate nucleoside phosphite and
-
1 336329
- 10 -
70 ~mol of tetrazole in 0.3 ml of anhydrous aceto-
nitriLe ~5 minutes),
9) 20% acetic anhydride in tetrahydrofuran containing
4QX lutidine and 10~ dimethylaminopyridine (2 minutes),
S h) tetrahydrofuran,
i) tetrahydrofuran containing 20X water and 40X lutidine,
j) 3% iodine in collidine/water/tetrahydrofuran in the
ratio by vo~ume 5:4:1,
k) tetrahydrofuran and
L) methanol.
The term "phosphite" in this context is defined as the
monomethyl ester of the deoxyribose-3'-monophosphorous
acid, the third valency being saturated by chlorine or
a tertiary amino group, for example a diisopropylamino
radical. The yields from the indiv;dual steps in the
synthesis can be determined after each detritylation
reaction b) by spectrophotometry by measurement of the
absorption of the dimethoxytrityl cation at a wavelength
of 496 nm.
Once the synthesis of the oligonucleotide is complete,
the methyl phosphate protective groups on the oligomer
are cleaved off by use of p-thiocresol and triethylamine.
The oligonucleotide is then detached from the solid sup-
port by treatment with ammonia for 3 hours. Treatment
of the oligomers with concentrated ammonia for 2 to 3
days quantitatively cleaves off the amino protective
groups on the bases. The crude product thus obtained
is purified by high pressure liquid chromatography
(HPLC) or by polyacrylamide gel electrophoresis.
The other oligonucLeotides are also synthesized entirely
correspondingly.
c) The plasmid ptrpES-1 (R. A. Hallewell et al., Gene 9
(1980) 27-47) is reacted with the restriction enzymes
HindIII and Sa~I in accordance with the manufacturer's
~ 1 33632~
instructions, and the DNA fragment of about ~20 bp is
removed. The synthetic oligonucleotides ~N4) and (N5)
5' AGC TTC CAT GAC GCG T 3' (N4)
5' ACG CGT CAT GGA 3' (N5)
.
S are phosphorylated, incubated together at 37C and,
by use of DNA ligase, added onto the blunt-ended DNA
for proinsuLin t~. ~etekam et al., Gene 19 (1982) 179-
183). After reaction with HindIII and SalI, the pro-
insulin DNA ~hich has now been extended is covalently
incorporated into the opened plasmid using the enzyme
T4 DNA ligase (Figure 2), this producing the plasmid
pH106/4.
The plasmid pH106/4 is first reacted once more ~ith
SalI, the overlapping ends are filled in with KLenow
polymerase to give blunt ends and the product is then
incubated with the enzyme MstI. A DNA fragment of
about S00 bp which contains the entire part coding for
proinsuLin and a segment of about 210 bp of the D-
protein from the trp operon of E. coLi is isoLated.
The DNA fragment is bLunt-ended and is inserted into
the StuI site of the plasmid pH120/14, thus producing
the plasmid pH154/25 (Figure 3). This is suitabLe for
the expression of a fusion protein under the contro~
of the trp operon, in which the amino ac;d sequence
Ala-Ser-Met-Thr-Arg is Located after the L'- and D'-
peptide and is foLLowed by the amino acid sequence of
proinsuLin.
Example 2
The plasmid pH154/25 (Figure 3) is reacted with the re-
striction en2ymes BamHI and XmaIII. The protruding ends
are filled in w;th KLenow poLymerase and linked using T4
DNA ligase. This results in the plasm;d pH254 (F;gure 4)
1 3~6329
- 12 -
which is suitable for the expression of a fusion protein
having the amino acid sequence L', D'-proinsulin under the
control of the trp promoter~ The plasmid is somewhat smal-
ler than pH154/25, which may be an advantage.
Example 3
lncubation of the plasmid pH254 (Example 2; Figure 4) with
the restriction enzymes MluI and SalI is carried out to
liberate a DNA segment of 280 bp, and this is removed. The
remainder of the plasmid is converted with Klenow poly-
merase into the blunt-ended form and is covalently cyclized
again with DNA ligase. This results in the plasmid pH255
(Figure 4) which is suitable for the insertion of a struc-
tural gene into one of the restriction sites MluI, SalI
and EcoRI. The formation of a fusion protein with the L',
D'-protein is carried out under inducing conditions. Of
course, it is possible to insert further restriction sites
into the plasmid pH255 by suitable linkers.
Example 4
The plasmid pH154/25 (Figure 3) is incubated with the en-
zymes MluI and EcoRI, and the liberated DNA fragment (about
300 bp) is removed. The remainder of the plasmid is filled
in with Klenow polymerase. Ring closure is effected by
the action of DNA ligase. The resulting pLasmid pH256
(Figure 5) can be used for insertion of structural genes
into the EcoRI site.
Example 5
Deletion of a 600 bp fragment from the plasmid pH256
(Example 4; Figure 5) using the restriction enzymes BamHI
and NruI results in the plasmid pH257 (Figure 5). For this
purpose, pH256 is first incubated with BamHI and blunt ends
are generated with Klenow polymerase. After incubation
with NruI and removal of the 600 bp fragment the formation
~ 1 336329
- 13 -
of pH257 is effected after incubation ~ith DNA ligase.
Example 6
Insertion of the lac repressor (P.J. Farabaugh, Nature 274,
(1978) 765-769) into the plasmid pKK 177-3 (A~ann et al.,
S Gene 25 (1983) 167) resu~ts in the p~asmid pJF1~8. This
is reacted with EcoRI and SalI, and the remainder of the
plasmid is isolated.
A fragment about 495 bp in size is obtained from the plas-
mid pH106/4 (Figure 2) by the action of SalI and incubation
~ith MstI.
The oligonucleotides $N6) and (N7) obtained by synthesis
5' ACG AAT TCA TGA AAG CAA AGG 3' (N6)
5' CCT TTG CTT TCA TGA ATT CGT 3' (N7)
are phosphorylated and, using DNA ligase, added onto the
blunt-ended DNA fragment. Reaction ~ith EcoRI and Sa~I
liberates oYerlappin~ ends which permit ~igation into the
opened plasmid pJF118.
After transformation into E. coli 294 of the hybrid plasmid
which has thus been obtsined, the correct clones are selec-
ted on the basis of the size of the restriction fragments.This plasmid is called pJ120 (Figure 6).
The expression of the fusion protein is carried out in shaken
flasks as follo~s:
An overnight culture in LB medium (J.H. Miller, Experi~ents
2~ in Molecular Genetics, Cold Spring Harbor Laboratory, 1972),
containing 50 ~g/ml ampicillin, of E. coli 294-transformants
which contain the plasmid pJ120 is used to set up a fresh
culture in the ratio of about 1:100, and the growth
1 336329
- - 14 -
is follo~ed by measurement of the OD. ~hen the OD is O.S,
isopropyl B-D-galactopyranoside (IPTG) is added to the
culture in an amount such that its concentration is 1 mM,
and the bacteria are removed by centrifugation after 150
to 180 minutes. The bacteria are boiled for 5 minutes in
a buffer mixture (7M urea, 0.1X SDS, 0.1 M sodium phosphate,
pH 7.0), and samples are applied to a SDS gel e~ectrophoresis
plate. After the electrophoresis, the bacteria which con-
tain the plasmid pJ120 provide a protein band vhich corres-
ponds to the size of the expected fusion protein and whichreacts ~ith antibodies against insulin. After the fusion
protein has been isolated the expected proinsulin derivative
can be liberated by cleavage with cyanogen
bromide. After disruption of the bacteria (French Press;
(R)Dyno-mill ) and centrifugation, the L', D'-proinsu-
lin fusion protein is located in the sediment, so that con-
siderable amounts of the other proteins can nou be removed
~ith the supernatant.
The stated induction conditions apply to shake cultures;
for Larger fermentations it is advantageous to modify the OD
values accordingly and, ~here appropriate, to vary the IPTG
concentrations slightly.
Example 7
An overnight culture in LB medium containing SO ~g/ml ampi-
cillin is prepared from E. co~i 294 transformants which
contain the plasmid pH154/2S (Figure 3) and the next morn-
ing ;s diluted in the ratio of about 1:100 in M9 medium
(J. H. Miller, Loc. cit.) containing 2000 ~g/ml casamino
acids and 1 ~g/ml thiamine. ~hen the OD = O.S indolyl-3-
acryl;c acid is added so that the final concentration is15 ~g/ml. After incubation for 2 to 3 hours, the bacteria
are removed by centr;fugation. SDS gel electrophoresis shows
a very pronounced protein band ~hich is at the place ex-
pected for the fus;on protein and which reacts ~ith anti-
bodies against insulin. After disruption of the bacteriaand centrifugation, the L', D'-proinsu~in fusion protein
1 336329
- ~ - 15 -
is located in the sediment so that, again, considerable
amounts of the other proteins are no~ removed with the
supernatant.
In the present case too, the stated induction conditions
apply to shake cultures. Fermentations in larger volumes
require altered concentrations of casamino acids or addi-
tion of L-tryptophan.
Example 8
The plasmid pH154/25 (Figure 3) is opened ~ith EcoRI, and
the protruding DNA single strands are filled in ~ith Klenow
polymerase. The DNA thus obtained is incubated ~ith the
enzyme MluI, and the DNA coding for insulin is cut out of
the plasmid. Separation by gel electrophoresis is carried
out to remove this fragment from the remainder of the plas-
mid, and the remainder of the plasmid is isoLated.
The plasmid sho~n in Figure 3 of German Offenlegungsschrift3,429,430 (European Patent Application A1 0,171,024) is
reacted with the restriction enzymes AccI and SaLI, and
the DNA fragment containing the hirudin sequence is removed.
After the protruding ends of the SalI restriction site have
been filled in with Klenow polymerase, the DNA segment is
ligated ~ith the synthetic DNA of the formula (N8)
Met Thr
5' CCC ACG CGT ATG ACG T 3'
3' GGG TGC GCA TAC TGC ATA 5' (N8)
The ligation product is incubated with MluI. After the en-
zyme has been inactivated at 65C, the DNA mixture is treated
~ith bovine alkaline phosphatase at 37C for one hour.
This is followed by removal of the phosphatase and the re-
striction enzyme from the mixture by extraction with phenol,and the DNA is purified by ethanol precipitation. The DNA
~hich has thus been treated is inserted using T4 ligase
-
~ 1 336329
- 16 -
;nto the opened remainder of the plasmid pH154/25, this
resulting in the plasmid pK150 which has been characterized
by restriction anaLysis and DNA sequencing by the method
of Maxam and Gilbert (Figure 7).
Example 9
E. coli 29~ bacter;a which contain the plasmid pK150 (Fi-
gure 7) are cultured in LB medium containing 30 to 50 ~g/ml
ampicillin at 37C overnight. The culture is diLuted
in the ratio of 1:100 ~ith M9 medium which contains 2000
~g/ml casamino acids and 1 ~g/ml thiamine, and the mixture
is incubated at 37C, mixing continuously. ~hen the
OD600= 0.5 or 1, indolyl-3-acrylic acid is added to a final
concentration of 15 ~/ml, and the mixture is incubated
for 2 to 3 hours or 16 hours respect;vely. The bacteria
are then removed by centrifugation and disrupted in 0.1 M
sodium phosphate buffer tpH 6.5) under pressure. The spar-
ingly soluble proteins are removed by centrifugation and
analyzed by SDS polyacryLamide gel electrophoresis. It
emerges that cells whose trp operon has been induced con-
tain in the region below 20,000 Daltons but above14,000 Da~-
tons a new protein ~hich is not found in non-induced cells.
After the fusion protein has been isolated and reacted with
cyanogen bromide hirudin is liberated.
Example 10
The constructs described hereinafter permit the introduc-
tion, upstream of the 5'-end of the trp-D sequence, of DNA
sequences which contain as man~ recognition sites for
various restriction enzymes as possible in order
to incorporate the trp-D sequence into
as many as possible of the wide variety of prokaryoctic
expression systems.
The plasmids pUC12 and pUC13 (Pharmacia P-L Biochemicals,
5401 St. Goar: The Molecular Biology Catalogue 1983, Ap-
~ - 17 - 1 336329
pendix, p. 89) contain ~ polylinker se~uence, it being the
intention to ;nsert in the pl~sm;d pUC13, bet~een the restric-
tion cleavage sites for XmaI ~nd SacI, the Mstl-HindIII trp
fr~gment from the pl~smid p106/4 (Figure 2) ~hich is fused
vith the HindIII-hirudin-SacI fragment from the plasmid
pK150 ~Figure 7).
For this purpose, the DNA of the pl~snid pUC13 is first
treated ~ith the restriction enzyme XmaI. The ends of the
~inearized plasmid are fil~ed in by ~eans of the Kleno~
polymerase reaction. After ethanol precipitation, the DNA
is treated ~ith the enzy~e SacI and is ~g~in precipitated
fro0 the reaction mixture ~ith ethanol. The DNA is no~
reacted in ~n aqueous ligation ~ixture Yith the MstI-
HindIlI trp-D fragment isol~ted from plasmid pH106/4 and
~ith the HindIII-Sac~ hirudin fra~ment isolated from the
plas~id pK150, ~nd T4 DNA ligase.
The plasmid pK160 thus obtained no~ cont~ins, i~mediately
upstream of the trp-D sequence, aultiple restriction en-
zyme recognition sequence ~hich e~br~ces restriction sites
for the enz~nes XmaI, S~aI, BamHI, XbaI, HincII, SalI, AccI,
PstI ~nd HindIII. Furthermore, ~n EcoRI restriction site
is generated do~nstre-m of the 3'-end of the hirudin se-
~uence in this construction (Figure 8).
Example 11
The plasmid pH131/5 is prepared as follows:
The plas~id ptrpL1
is opened with ClaI and ligated with the synthe-
tically prepared, self-comPlement~ry oligonucleotide (N9)
5' pCGACCATGGT 3'; ~N9)
.
~he plasmid pH131/5 (Figure 9) thus obtained is opened at the
restriction site, vhich has been introduced in this 0anner,
- ~ ~ 336329
_ - 18 -
~ for the restriction enzyme NcoI, and the resulting pro-
truding single-stranded ends are filled in by means of the
- KLenov polymerase reaction. The linearized, blunt-ended
DNA is now cut w;th the enzyme EcoRI, and the lar~er of
the two resuLting DNA sequences is separated from the smal-
ler sequence by ethanol precipitation. The rema;nder of
the pLasm;d DNA of the plasmid pH131/5 thus obtained is
now Ligated with a fragment from pK160 coding for trp-D'-
hirudin by reaction vith T4 ligase. This fragment is
cleaved out of the p~asm;d pK160 by opening the plasm;d
w;th HincII and EcoRI. The fragment is removed from the
remainder of the plasmid by gel eLectrophoresis, and ;s
then eluted from the gel material. The Ligation product
;s transformed ;nto E. coLi K12. The clones containing
plasmid DNA are ;solated and characterized by restriction
anaLysis and DNA sequence analysis. The plasmid pK170
thus obtained contains fused onto the trp operator a DNA
sequence which codes for Met-Asp-Ser-Arg-GLy-Ser-Pro-Gly-
trp-D'-(hirudin~ (Figure 9).
ExampLe 12
The pLasm;d pJF118 (Example 6) is opened with EcoRI, and
the protruding DNA ends are converted into bLunt ends by
means of the KLenow polymerase reaction. The DNA thus
treated is then cut with the enzyme SalI, and the short
EcoRI-SaLI fragment is removed by gel eLectrophoresis.
The pLasmid pK 170 (Exsmple 11) is cleaved with NcoI, and
the protruding ends are converted into bLunt ends using
Klenov polymerase. The plasmid DNA is removed from the
reaction mixture by ethanol precipitat;on and ;s treated
with the enzymes HindIII and BamHI. Two of the resuLting
fragments are ;solated, namely the NcoI (w;th fiLled-in
end)-trp-D'-HindIII fragment and the H;ndIII-h;rud;n-~amHI
fragment (German Offenlegungsschr;ft 3,429,430). The two
fragments are isolated after separation by geL eLectro-
phoresis.
-- 1 33632~
- - 19 -
In addition, the BamHI-SalI-HirudinII fragment shown in
Figure 2 of German Offenlegungsschrift 3,429,430 is iso-
lated. In a ligation reaction the four fragments, namely
the remainder of the plasmid pJF 118, the NcoI-trp-D'-
HindIII fragment, the HindIII-hirudin-BamHI fragment and
the hirudinII fragment, are now reacted together and the
resuLting plasmid pK 180 (Figure 10) is transformed into
E. col; K12-W 3110. Correct plasmids are shown by it be;ng
possible to detect an EcoRI-trp-D'-hirudin-SalI fragment
in the pLasmid DNA. The trp-D'-hirudin sequence is now
attached to the tac promoter. The fusion protein is ex-
pressed as in Example 6.
Example 13
The plasmids derived from pBR 322, such as pH120/14 (Ex-
ample 1, Figure 1), pH154/25 (ExampLe 1, Figure 3), pH256
(Example 4, Figure 5), pK150 (Example 8, Figure 7) and
pK170 (Example 11, Figure 9), have - on the figures in the
clockwise direction - between the start codon of the fusion
protein and the next HindIII site (corresponding to HindIII
at position 29 in pBR322) an additional PvuII site ;n the
region of the fragment which contains the trp promoter and
operator, but outside the promoter region.
It has now been found that by deletion of the DNA segment
which is bounded by the PvuII site described and the PvuII
site which corresponds to position 2066 in pBR322, the yield
of a cloned protein (or fusion protein) is dis~inctly in-
creased~
The shortening of the p~asmid pH154/25 to give pH154/25*
is described by way of example hereinafter, it being pos-
s;ble for this to be effected correspondingly for the other
plasmids mentioned above (the shortened plasmids likewise
being identified by 3n asterisk):
pH154/25 is reacted with PvulI ~in accordance with the manu-
-
1 33632~
- 20 -
facturer's instructions), resulting ;n three fragments:
t
Fragment 1: From the PvuII restriction site of the proin-
sulin gene to the PvuII restriction site cor-
respond;ng to position 2066 in pBR322,
Fragment 2: from the PvuII restriction site near the trp
promoter to the PvuII site of the proinsulin
gene
and
Fragment 3: from the PvuII site near the trp promoter
fragment to the PvuII site corresponding to
position 2066 in pBR322.
The fragments can be separated by electrophoresis on agarose
and then isolated (Maniatis eb al., Molecular Cloning, Cold
Spring Harbor, 1982).
The fragments 1 and 2 are joined under blunt end conditions
using the enzyme T4 DNA ligase. Transformation into E. coli
294 is followed by testing for those colonies ~hich contain
a plasmid ~ith the complete proinsulin sequence and thus
have the fragments in the desired order. The pLasmid
pH154/25* is depicted in Figure 11.
A distinct increase in the proportion of fusion protein is
observed on expression, ~hich is carried out as described
in the preceding examples.
Example 14
The plasmid pH 154/25* ~Example 13, Figure 11) is digested
~ith HindIII and SalI, and the small fragment (having the
proinsulin sequence) is separated off by gel eLectrophore-
sis. The large fragment is isolated and ligated ~ith the
synthetic DNA (N10)
(Ala) Trp Glu Asp Pro Met Ile Glu (Gly) (Arg)
A GCT TGG GAG GAT CCT ATG ATC GAG GG (N10)
ACC CTC CTA GGA TAC TAG CTC CCA GCT
1 336329
Z1 -
The plasmid pInt13 (Figure 12) is produced.
The DNA (N10) codes for an amino acid sequence which con-
tains several cleavage sites for chemical cleavage:
a) Met for cyanogen bro-ide,
b) Trp for N-bromosuccinimide (NBS or BSI)
c) Asp-Pro for proteolytic cleavage, the upstream Glu ad-
ditionally weakening the Asp-Pro bond to~ards the action
of acids.
The introduction of this HindlII-Sa~l-linker (N10) into the
reading frame of a coded polypeptide thus allo~s the options
which have been mentioned for chemical cleavage off, de-
pending on the amino acid sequence of the desired prote;n
and on its sensitivity to the cleaving agents.
The figures are not to scale~
' ~ - 22 - 1 336329
Amino ac;d sequence I
23)
Ser Asn Gly His Asn Val Val Ile
Tyr Arg Asn His Ile Pro Ala Gln
Thr Leu Ile Glu Arg Leù A~a Thr
Met Ser Asn Pro Val Leu Met Leu
Ser Pro Gly Pro Gly VaL Pro Ser
Glu Ala Gly Cys Met Pro Glu Leu
Leu Thr Arg Leu Arg Gly Lys Leu
Pro ILe Ile Gly ILe Cys Leu Gly
(93)
His Gln ALa Ile Val Glu ALa