Note: Descriptions are shown in the official language in which they were submitted.
--~ 1 336428
- 1 - T1011
SUBSTITUTED DIBENZOCYCLOHEPTENIMINES
This invention relates to a class of
dibenzocycloheptenimines, and in particular to
derivatives of 5-methyl-10,11-dihydro-5H-dibenzo[a,d]-
cyclohepten-5,10-imine substituted with a hydrocarbon
or hydrocarbyloxycarbonyl group. The compounds are
useful as anticonvulsant agents and in the treatment
of neurodegenerative diseases.
British Patent No. 2,004,872 (and US Patent
No. 4,399,141) describes 5-methyl-10,11-dihydro-5H-
dibenzo[a,d]cyclohepten-5,10-imine and derivatives
thereof as anticonvulsant agents. The use of those
compounds as non-competitive N-methyl-D-aspartate
(NMDA) receptor antagonists, and therefore their value
in the treatment of neurodegenerative diseases, is
disclosed in European patent application, publication
No. 230370.
It has now been found that derivatives
substituted with a hydrocarbon or
hydrocarbyloxycarbonyl group on the benzo ring are
also good anticonvulsants and NMDA receptor
antagonists. None of the prior documents discloses or
suggests hydrocarbon or hydrocarbyloxycarbonyl
substituents at that position. The compounds of this
invention are useful in the prevention and/or
treatment of neurodegeneration in pathological
conditions such as stroke, hypoglycaemia, cerebral
palsy, transient cerebral ischaemic attack, cerebral
ischaemia during cardiac pulmonary surgery or cardiac
l 336428
- 2 - T1011
arrest, perinatal asphyxia, epilepsy, Huntington's
chorea, Alzheimer's disease, Olivo-pontocerebellar
atrophy, anoxia such as from drowning, spinal cord
injury and poisoning by exogeneous NMDA poisons (e.g.
some forms of lathyrism).
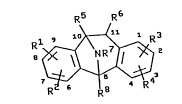
The present invention provides a compound of
formula (I):
R5 R6
7~ 4 R3
(I)
or a pharmaceutically acceptable salt thereof,
wherein one of Rl, R2, R3 and R4 represents a
hydrocarbon or hydrocarbyloxycarbonyl group and the
remaining groups Rl, R2, R3 and R4 independently
represent hydrogen, hydrocarbon,
hydrocarbyloxycarbonyl, halogen, hydroxy or Cl_6
alkoxy, or Rl and R2 or R3 and R4 may together
represent the residue of a carbocyclic ring; R5 and
R6 independently represent hydrogen, hydrocarbon,
hydroxy or fluoro; R7 represents hydrogen or Cl_3
alkyl; and R8 represents methyl or ethyl.
The term 'hydrocarbon' includes groups
having up to 18 carbon atoms, suitably up to 10
carbon atoms, conveniently up to 6 carbon atoms.
Suitable hydrocarbon groups include Cl_6 alkyl, C2_6
alkenyl, C2_6 alkynyl, C3_7 cycloalkyl, C3_7
cycloalkyl(Cl_6)alkyl, aryl, and aryl(Cl_6)alkyl.
1 336428
- 3 - T1011
The term 'hydrocarbyloxycarbonyl' as used
herein refers to a group of formula -CO2R in which R
represents a hydrocarbon group as defined above.
Suitable alkyl groups include straight and
branched chain alkyl groups containing from 1 to 6
carbon atoms, such as methyl, ethyl, propyl and
butyl. A particular alkyl group is methyl.
When used herein the term 'aryl' includes
optionally substituted phenyl and naphthyl.
Any of the hydrocarbon groups may be
substituted with a group selected from halogen, C1_6
alkyl, phenyl, C1_6 alkoxy, halo(C1_6)alkyl, hydroxy,
amino, nitro, cyano, carboxy, C1_6 alkoxycarbonyl,
C1_6 alkoxycarbonyl(C1_6)alkyl, C1_6 alkylcarbonyloxy
and C1_6 alkylcarbonyl, and groups of formula
- -CONRaRb and -NRa.CORb in which Ra and Rb
independently represent hydrogen or hydrocarbon.
Preferred substituent groups include halogen,
hydroxy, C1_6 alkyl, C1_6 alkoxy and C1_6
alkoxycarbonyl.
Suitable aryl groups include phenyl and
chlorophenyl.
Suitable aryl(C1_6)alkyl groups are benzyl
and chlorobenzyl.
Preferably R7 represents hydrogen and R8
represents methyl.
When R1 and R2 or R3 and R4 together
represent the residue of a carbocyclic ring, the ring
may be saturated or unsaturated. The R1/R2 or R3/R4
carbon chain may comprise from 2 to 6 carbon atoms,
preferably 3 or 4 carbon atoms, i.e. forming a fused
5- or 6-membered ring, such as benzo.
1 336428
-
- 4 - T1011
In a preferred subgroup of compounds of
formula (I), one of Rl to R4 represents Cl_6 alkyl,
hydroxy(C1_6)alkyl, phenyl or C1_6 alkoxycarbonyl,
and the remaining groups R1 to R4 represent hydrogen
or halogen, in particular chloro or bromo. It is
also preferred that R5 represents hydrogen, hydroxy
or methyl and R6 represents hydrogen or hydroxy, in
particular exo-hydroxy.
The hydrocarbon or hydrocarbyloxycarbonyl
group, R1 to R4, may be present at any position of
the benzo rings, suitably at positions 2, 3, 4, 6, 7
or 8, in particular at positions 2, 3, 7 or 8.
Hydrocarbon or hydrocarbyloxycarbonyl substitution at
positions 3 and 7 leads to preferred compounds.
Particular compounds of this invention
include:
5,7-dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine;
5,9-dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine;
3,5-dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine;
1,5-dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine;
5-methyl-3-phenyl-10,11-dihydro-5H-dibenzo[a,d]-
cyclohepten-5,10-imine;
7-methyl-12,13-dihydro-7H-benzo[4,5]cyclohepta[1,2-a]-
naphthalen-7,12-imine;
7-methyl-12,13-dihydro-7H-benzo[4,5]cyclohepta[1,2-a]-
naphthalen-7,13-imine;
2,5-dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine;
- 1 336428
- 5 - T1011
5,8-dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine;
3-methoxycarbonyl-5-methyl-10,11-dihydro-5H-dibenzo-
[a,d]cyclohepten-5,10-imine;
- 5 3-hydroxymethyl-5-methyl-10,11-dihydro-5H-dibenzo-
[a,d]cyclohepten-5,10-imine;
and salts thereof.
Suitable acid addition salts of compounds of
this invention include pharmaceutically acceptable
inorganic salts such as the sulphate, nitrate,
phosphate, borate, hydrochloride and hydrobromide and
pharmaceutically acceptable organic acid addition
salts such as acetate, tartrate, maleate, citrate,
succinate, benzoate, ascorbate, methanesulphonate,
a-ketoglutarate~ ~-glycerophosphate, and
glucose-1-phosphate. Preferably the acid addition
salt is a hemisuccinate, hydrochloride,
~-ketoglutarate, ~-glycerophosphate or glucose-1-
phosphate, in particular the hydrochloride salt.
Also included within the scope of the
present invention are pharmaceutical compositions
comprising the imines of this invention. Preferably
these compositions are in unit dosage forms such as
tablets, pills, capsules, powders, granules, sterile
parenteral solutions or suspensions, or suppositories
for oral, parenteral or rectal administration. For
preparing solid compositions such as tablets, the
principal active ingredient is mixed with a
pharmaceutical carrier, e.g. conventional tableting
ingredients such as corn starch, lactose, sucrose,
sorbitol, talc, stearic acid, magnesium stearate,
dicalcium phosphate or gums, and other pharmaceutical
diluents, e.g. water, to form a solid preformulation
" 1 336428
- 6 - T1011
composition containing a homogeneous mixture of an
imine of the present invention, or a non-toxic
pharmaceutically acceptable salt thereof. When
referring to these preformulation compositions as
homogeneous, it is meant that the active ingredient
is dispersed evenly throughout the composition so
that the composition may be readily subdivided into
equally effective unit dosage forms such as tablets,
pills and capsules. This solid preformulation
composition is then subdivided into unit dosage forms
of the type described above containing from 0.1 to
about 500 mg of the active ingredient of the present
invention. The tablets or pills of the novel
composition can be coated or otherwise compounded to
lS provide a dosage form affording the advantage of
prolonged action. For example, the tablet or pill
can comprise an inner dosage and an outer dosage
component, the latter being in the form of an
envelope over the former. The two components can be
separated by an enteric layer which serves to resist
disintegration in the stomach and permits the inner
component to pass intact into the duodenum or to be
delayed in release. A variety of materials can be
used for such enteric layers or coatings, such
materials including a number of polymeric acids and
mixtures of polymeric acids with such materials as
shellac, cetyl alcohol and cellulose acetate.
The liquid forms in which the novel
compositions of the present invention may be
incorporated for administration orally or by
injection include aqueous solutions, suitably
flavoured syrups, aqueous or oil suspensions, and
flavoured emulsions with edible oils such as
1 336428
- 7 - T1011
cottonseed oil, sesame oil, coconut oil and peanut
oil, as well as elixirs and similar pharmaceutical
vehicles. Suitable dispersing or suspending agents
for aqueous suspensions include synthetic and natural
gums such as tragacanth, acacia, alginate, dextran,
sodium carboxymethylcellulose, methylcellulose,
polyvinyl-pyrrolidone, and gelatine.
The novel imines of this invention are
useful as anticonvulsants at a dosage level of from
about 0.01 to about 20 mg per kilogram of body
weight, preferably about 0.05 to 2 mg/kg of body
weight, on a regimen of 1 to 4 times a day.
In the treatment of neurodegeneration, a
suitable dosage level is about 0.01 to 50 mg/kg,
preferably about 0.05 to 10 mg/kg and especially
about 0.05 to 0.5 mg/kg/day. The compounds may be
administered on a regimen of 1 to 4 times per day.
The compounds of this invention wherein R5
is hydrogen or hydrocarbon and R6 is hydrogen may be
prepared by a process which comprises reducing the
N-hydroxy compound of formula (II):
Rls H
y~ 3 R
(II)
1 336428
- 8 - T1011
wherein R1 to R4 and R8 are as defined with respect
to formula (I) above and R15 represents hydrogen or
hydrocarbon; and, when R7 is other than hydrogen,
alkylation of the product. The preferred reducing
agent is nascent hydrogen generated by the action of
a metal, preferably zinc, with an acid, such as
acetic acid. Suitable conditions for the process are
a temperature of from 40 C to lOO C for 1 to about 10
hours.
The intermediate compounds of formula (II)
may be prepared by the cyclisation of a compound of
formula (III):
Rl5 H
R1 ~ NHOHR
(III)
wherein R1 to R4 and R8 are as defined with respect
to formula (I) above and R15 is as defined with
respect to formula (II) above. The conditions for
this reaction are elevated temperature or the
presence of a base and are generally as described in
US Patent No. 4477668. Both compounds (II) and (III)
are novel compounds and represent further aspects of
this invention. The compound (II) may also be
prepared by processes analogous to those described in
U.S. Patents Nos. 4399141 and 4232158.
1 336428
- 9 - T1011
The novel compounds of this invention
wherein R8 represents a group of formula -CH2R9, in
which R9 is hydrogen or methyl, may also be prepared
by ring closure of a 10-NHX-5-(=CHR9)-
10,11-dihydro-5H-dibenzo[a,d]cycloheptene (where X is
the group R7 or a protecting group, such as acetyl)
by treatment with a strong base such as an
organometallic reagent, for example n-butyllithium,
in an ethereal solvent such as tetrahydrofuran or
1,2-dimethoxyethane, at about O-C to about 30-C for
about 5 minutes to about 1 hour, followed if
necessary by removal of the protecting group.
The compounds of formula (I) in which at
least one of R1 to R4 represents a hydrocarbon group
may be prepared, where appropriate, by alkylation or
arylation, i.e. by reaction of a compound of formula
(IV):
R5 R6
R ~ R
(IV)
wherein R5 to R8 are as defined with respect to
formula (I) above, one of R11, R12, R13 and R14
represents halogen, and the remaining groups R11,
R12, R13 and R14 independently represent hydrogen,
hydrocarbon, halogen, hydroxy or C1_6 alkoxy; with a
reagent which replaces halogen by hydrocarbon. For
example, a Grignard reagent such as an alkyl- or
~ 336428
- 10 - T1011
arylmagnesium bromide may be employed in the presence
of a catalyst such as
1,3-bis(diphenylphosphino)propane nickeltII)
chloride. If the hydrocarbon substituent is phenyl,
a suitable reagent is PhB(OH)2/(PPh3)4Pd [phenyl
boronic acid/tetrakis(triphenylphosphine)palladium].
The compounds of formula (I) in which at
least one of R1 to R4 represents a
hydrocarbyloxycarbonyl group may conveniently be
prepared by metalation of a compound of formula (IV)
as defined above with, for example, n-butyllithium;
treatment of the resulting anion in situ with carbon
dioxide; and subsequent esterification of the
resulting carboxylic acid by standard means, such as
by treatment with boron trifluoride etherate in the
presence of a suitable alcohol.
Where appropriate, a compound of formula (I)
initially obtained may subsequently be elaborated
into a further compound of formula (I) as defined
above. For example, a compound of formula (I)
wherein one or more of R1 to R4 is/are hydroxymethyl
may conveniently be obtained from the corresponding
compound of formula (I) wherein one or more of R1 to
R4 is/are hydrocarbyloxycarbonyl by treatment of the
latter with a suitable reducing agent, e.g. lithium
aluminium hydride.
The compounds of formula (I) in which R5
represents hydroxy may be prepared by methods
analogous to those described in EP-A-0264183. A
further method is to treat the N-hydroxy compound of
formula (II) above, in which R15 is hydrogen, with
manganese acetate, and subsequently to reduce the
1 336428
- 11 - T1011
product thereby obtained with, for example, zinc in
acetic acid. This method may be illustrated by the
following scheme:
S
When it is desired that R7 should be other than
hydrogen, the resulting product can subsequently be
alkylated.
The compounds of formula (I) in which R6
represents hydroxy may be prepared by reducing an
11-oxo compound, in which the nitrogen atom may be
protected. The reduction may be effected by
treatment with, for example, diisobutylaluminium
hydride (Dibal-H) in an ethereal solvent such as
ether, tetrahydrofuran or 1,2-dimethoxyethane at
about -90 to -60-C, preferably about -78 C, for about
1 to 3 hours.
This process produces a mixture of endo and
exo hydroxy derivatives which, after deprotection,
may be separated by conventional techniques such as
preparative chromatography. Alternatively, in order
to obtain the 11-hydroxy compound selectively as its
desired exo or endo isomer, the 11-oxo compound may
be reduced with a chiral reducing agent such as D- or
L-Selectride. Thus, it will be appreciated that a
mixture of the exo and e _ isomers can be converted
1 336428
- 12 - T1011
into a single desired exo or endo isomer by oxidation
to the 11-oxo compound and subsequent application of
the foregoing selective reduction technique. This
approach can equally be adopted for the conversion of
- S a single exo or endo isomer into the opposite isomer.
Further illustrative methods of preparing
the exo and endo isomers of the 11-hydroxy compound
selectively are described in EP-A-0264183.
The 10-fluoro derivative of compound (I) may
be prepared by treating the corresponding 10-hydroxy
compound with diethylaminosulphur trifluoride (DAST)
in an inert organic solvent such as a chlorinated
hydrocarbon such as chloroform or methylene
dichloride at about 15 to 30-C for about 30 minutes
to 2 hours.
The isomeric 11-fluoro compound may be
prepared by treating the aziridine of formula (V):
R1 ~.8 RR3
(V)
wherein R1 to R5 and R8 are as defined with respect
to formula (I) above; with hydrogen fluoride-pyridine
(HF-70%; pyridine-30%) at about -90 to -60C,
preferably about -78 C, followed by spontaneous
warming to ambient temperature (15 to 30C) for about
12 to 36 hours.
1 336428
- 13 - T1011
During any of the above reactions it may be
necessary to protect any reactive groups on any of
the molecules concerned with conventional protecting
groups which may subsequently be removed using
methods known from the art.
The compounds useful in this invention bind
with a high affinity and in a reversible and
saturable manner to membranes from rat brain cortex.
In addition these compounds potently and selectively
block responses to NMDA in a brain slice from rat
cortex, and antagonise NMDA-induced seizures in the
mouse.
Binding Studies
The compounds of the invention were tested
for their ability to displace a standard compound
from rat brain. The standard compound employed is
S-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine, hereinafter referred to as MK-801.
Binding of [3H]-MK-801 to rat brain in vitro
- 20 was conducted in a crude synaptosomal membrane
fraction (P2) prepared from rat cerebral cortex
according to a modified method of Hulme et al.,
Molecular Pharmacology, 1978, 14, 737-750. Compounds
of the invention displaced the [3H]-MK-801 binding in
a concentration-dependent manner. The concentrations
of the compounds of accompanying Examples 1 to 12
required to displace 50% of specific [3H]-MK-801
binding (ICso) were below 10 ~M in each case.
1 336428
- 14 - T1011
Cortical Slice Studies
The effects of compounds of the invention on
responses to NMDA were assessed using the rat
cortical slice as described by Wong et al, Proc.
Natl. Acad. Sci. USA, 1986, 83, 7104. The apparent
equilibrium constant (Kb) was calculated from the
righthand shift in the NMDA concentration-response
curve produced by the compound under test. The
compounds of accompanying Examples 1 to 6, 8 to 10
and 12 were tested and their Kb values were found to
be below 5 ~M in each case.
Antagonism of NMDLA-induced seizures
Compounds of the invention were examined for
their ability to antagonise tonic seizures induced by
N-methyl-DL-aspartic acid (NMDLA). Groups of 8 male
Swiss-Webster mice (25-30 g) were injected
intravenously with the test compound at various
doses, 15 min before s.c. administration of NMDLA
(500 mg/kg). Animals were observed for the following
30 min and the number of mice protected from tonic
extension of the forelimbs noted. ED50 values for
the antagonism of the NMDLA induced tonic seizures
were determined using probit analysis. The compounds
of accompanying Examples 1, 2, 5, 6 and 12 were
tested and their ED50 values were found to be below
10 mg/kg in each case.
The following Examples illustrate the
preparation of compounds of this invention:
1 336428
- 15 - T1011
EXAMPLE 1
5,7-Dimethyl-10,11-dihydro-5H-dibenzo[a,d]
cyclohepten-5,10-imine.
Step A: Preparation of 2-(2-(4-methylphenyl)ethyl)
benzoic acid.
1,5-Diazabicyclo[4.3.0]non-5-ene (29.lg) was
added to a solution of 2-(carbomethoxy)benzyl
triphenylphosphonium bromide (lOOg) and
p-tolualdehyde (26.7g) in acetonitrile (250ml). The
mixture was refluxed for 10 minutes, the solvents
removed in vacuo and the residue dissolved in
chloroform (200ml), washed with lN aqueous
hydrochloric acid (lOOml) and water (lOOml), then
dried over sodium sulphate, filtered and evaporated.
The residue was refluxed overnight in water (250ml)
and methanol (lOOml) containing potassium hydroxide
(56g), then cooled, diluted with water (250ml) and
washed with chloroform (3 x 250ml). The aqueous
solution was acidified to pH 2 with conc.
hydrochloric acid, and extracted with chloroform (3 x
200ml). The extract was washed, dried and evaporated
to afford 2-carboxy-4'-methyl stilbene (44.2g) as a
cis- trans mixture. This mixture was hydrogenated in
dimethylformamide (50ml) at 50 psi in the presence of
10% palladium on charcoal catalyst (2g). The
catalyst was removed by filtration and solvent
evaporated. The residue was dissolved in ether,
washed with water, dried and filtered to afford,
after evaporation and hexane trituration,
2-(2-(4-methylphenyl)ethyl)benzoic acid (40.0g), m.p.
80-2~C.
Step B: Preparation of 3-methyl-10,11-dihydro-5H-
dibenzo[a,d]cyclohepten-5-one.
1 336428
- 16 - T1011
The acid from Step A (37.5g) in sulfolane (200ml)
was heated to 110C and polyphosphoric acid (150ml)
added. After 1.5hr at 110C the mixture was poured
into water (500ml), extracted with hexane (3 x 200ml)
and the extract washed, dried, diluted with an equal
volume of ether and percolated through silica
(200g). The eluate was evaporated to afford 3-methyl-
10,11-dihydro-SH-dibenzota,d]cyclohepten-5-one
(30.5g) as an oil. 60MHz NMR in deuterochloroform
2.35 (CH3), 3.20 ppm [(CHz)2].
Step C: Preparation of 3-methyl-5H-dibenzo[a,d]
cyclohepten-5-one.
The ketone from Step B (30.25g) and
N-bromosuccinimide (29.lg) were refluxed in carbon
tetrachloride (300ml) for 5 hrs. The solution was
cooled, filtered and evaporated and the residue
dissolved in dimethylformamide (lOOml) and 1,5-
diazabicyclo[4.3.0]non-5-ene (22g) added. The
solution was heated to 80 for 20 mins, then poured
into water (300ml) and extracted with ether (3 x
300ml). The extract was washed with dilute aqueous
hydrochloric acid and water, then dried and
evaporated. Crystallisation from acetone/hexane gave
pure 3-methyl-5H-dibenzo[a,d]cyclohepten-5-one
(22.2g), m.p. 81-83C.
Step D: Preparation of 3,5-dimethyl-5H-dibenzo
[a,d]cyclohepten-5-ol.
To a solution of the ketone from Step C (22g) in
dry tetrahydrofuran (400ml) at 0C under nitrogen was
added a solution of methyl lithium in diethyl ether
(lOOml of 1.4M). The solution was stirred for 1 hr
at 0C, then water was cautiously added and
`` 1 336428
- 17 - T1011
solvents evaporated to give an aqueous mixture which
was extracted with ether (2 x 150ml). The combined
extracts were washed with water, dried and
evaporated. Crystallisation from ether/hexane gave
3,5-dimethyl-5H-dibenzota,d]cyclohepten-5-ol
(20.18g), m.p. 102-3C.
Step E: Preparation of 3,5-dimethyl-5H-dibenzocy-
clohepten-5-hydroxylamine.
To a refluxing, vigorously stirred suspension of
the carbinol from step D (Sg), anhydrous sodium
acetate (17.37g) and hydroxylamine hydrochloride
(14.6g) in dichloromethane (50ml) was added dropwise,
dichloroacetic acid (15ml in 50ml dichloromethane).
The reaction was followed by TLC on silica in 1:1
ethyl acetate/hexane. The mixture was cooled, washed
with 2N aqueous sodium hydroxide, (Z x 50ml), water
(50ml), then dried and evaporated. Trituration with
1:1 ether/hexane gave 3,5-dimethyl-5H-dibenzo[a,d]
cyclohepten-5-hydroxylamine (4.92g), m.p. 142-4C.
Step F: Preparation of a mixture of 3- and
7-methyl-5-methyl-12-hydroxy-10,11-dihydro-5H-dibenzo
[a,d]cyclohepten-5,10-imines.
To xylene (50ml) at reflux was added, over a 20
minute period, a solution of the hydroxylamine from
Step E (lg) in xylene (lOOml) under nitrogen. The
solution was refluxed a further 10 minutes, then
cooled, evaporated on silica using 2 : 3 ethyl
acetate/hexane eluant to give the product mixture
(520mg), as a buff solid, m.p. 125-30C. Two pairs
of atropisomers in the ratio 2:1 were evident in the
360 MHz H nmr spectrum.
1 3364~8
- 18 - T1011
Step G: Preparation of a mixture of 3- and 7-
methyl-5-methyl-10,11-dihydro-(5H)-dibenzo[a,d]
cycloheptenimines.
To a solution of the mixture from Step F (500mg)
in glacial acetic acid (5ml) was added zinc powder
(600mg) and the mixture stirred at 65C for 3 hrs. A
further 200mg zinc was added and the mixture heated a
further 1 hr. The mixture was filtered, the cake
washed with acetic acid and solvents evaporated. The
residue was partitioned between 2N aqueous sodium
hydroxide and ether, the organic layer separated,
washed with water, dried and evaporated then
chromatographed on silica using chloroform eluant to
afford a mixture of 3- and 7- methyl compounds as an
oil (270mg). Rf 0.31 on silica in 5%
methanol/chloroform.
- Step H: Preparation and separation of a mixture
of 3- and 7-methyl-5-methyl-12-tert-butoxycarbonyl-
10,11-dihydro-(5H)-dibenzo[a,d]cyclohepten-5,10-imines.
To a solution of the mixture of 3- and 7-methyl
compounds from Step G (250mg) in dichloromethane
(2ml) was added t-butylpyrocarbonate (327mg) and
triethylamine (152mg), and the solution stirred at
room temperature for 0.5hr. The solution was
evaporated to dryness, and the residue partitioned
between ether and water. The water was discarded and
the ether layer dried and evaporated to give an oil
which was chromatographed on a Lobar silica column
using 1% ethyl acetate/hexane as eluent. Fractions
were analysed by HPLC using 2% ethyl acetate/hexane.
Isomers A (20mg) and B (9Omg) were collected.
1 336428
- 19 - T1011
Step I: Preparation of 5,7-dimethyl-10,11-dihydro-
SH-dibenzo[a,d]cyclohepten-5,10-imine hydrochloride.
To a solution of isomer B (9Omg) from step G, in
dry ethyl acetate (lml) was added hydrogen chloride
gas in ethyl acetate (5.SM, lml) and the solution
stirred for 0.5hr. Evaporation and crystallisation
from methanol/ether gave 5,7-dimethyl-10,11-dihydro-
5H-dibenzo[a~d]cyclohepten-5~10-imine hydrochloride
(32mg), mp 275-80C. Contamination with 3-isomer <
5% by HPLC. The structure was confirmed as the
7-methyl isomer by H nmr spectroscopy.
EXAMPLE 2
3,5-Dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclo-
hepten-5,10-imine.
To a solution of isomer A (20mg) from Example 1
(H) above, in dry ethyl acetate (0.5ml) was added
hydrogen chloride gas in ethyl acetate (S.SM, O.Sml)
and the solution stirred for O.Shr. Evaporation and
crystallisation from methanol/diethylether gave 3,5-
dimethyl-SH-dibenzo[a,d]cyclohepten-S,10-imine
hydrochloride (9.2mq). Contamination with 7-isomer <
5% by HPLCi The structure was confirmed as the 3
isomer by H nmr spectroscopy.
EXAMPLE 3
S,9-Dimethyl-10,11-dihydro-SH-dibenzo[a,d]cYclo-
hepten-S,10-imine.
- 1 336428
- 20 - T1011
Step A: Preparation of 2-t2-(2-methylphenyl)ethyl]
benzoic acid.
Prepared from o-tolualdehyde as described in
Example lA (61t yield), m.p. 122-3C (from
S ether/hexane).
Step B: Preparation of l-methyl-10,11-dihydro-5H-
dibenzota,d]cyclohepten-S-one.
Prepared from the above compound as described in
Example lB (65t yield), vmax (NU3O1~ 1660cm
(c~o) -
Step C: Preparation of l-methyl-5H-dibenzota,d]
cyclohepten-S-one.
Prepared from the above compound as described in
Example lC (20t yield), mp. 77-80C (from
acetone/hexane).
Step D: Preparation of 1,5-dimethyl-5H-dibenzo
ta,d]cyclohepten-5-ol.
Prepared from the above compound as described in
( example lD (60t yield), m.p. 107-9 (from ethyl
acetate/hexane).
Step E: Preparation of 1,5-dimethyl-5-hydroxy-
lamino-SH-dibenzota,d]cycloheptene.
Prepared from the above carbinol as in Example
lE. Yield 76%, m.p. 127-30C.
Step F: Preparation of a mixture of 1,5-dimethyl-
12-hydroxy-10,11-dihydro-SH-dibenzota,dlcyclohepten-
A
1 336428
- 21 - T1011
5,10-imine and S,9-dimethyl-12-hydroxy-10,11-dihydro-
SHdibenzota,d]cyclohepten-5,10-imine.
Prepared from the above hydroxylamine as in
Example lF, to give an orange solid in S0~ yield.
Two atropisomers of each compound were present as
adjudged by 360MHZ nmr: ~ (CDC13) 2.40 (0.3H),
2.55 (0.3H), 2.68 (O.lSH), and 2.85 ~O.lSH) (CH of
CH2, J=18Hz).
Step G: Preparation of a mixture of 1,5-dimethyl-
10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine
and S,9-dimethyl-10,11-dihydro-5H-dibenzo[a,d]
cyclohepten-5,10-imine.
To the mixture from step F (800mg) in glacial
acetic acid (6ml) was added powdered zinc (800mg) and
- the mixture heated at 60 for 2hr, cooled, filtered
and the cake washed with ether and water. Aqueous
sodium hydroxide was added to the washings to pH 14,
the ether layer separated, washed with brine, dried
and evaporated. The residue was purified by column
chromatography to give the product mixture as an oil
(200mg), which was used as such for step H.
Step H: Preparation of 1,5-dimethyl-10,11-dihydro-
12-tert-butoxycaloryl-5H-dibenzo[a,d]cyclohepten-S,10,
imine and S,9-dimethyl-10,11-dihydro 12-tert-butoxy
carbonyl-SH-dibenzo[a,d]cyclohepten 5,10 imine.
The mixture from step G was treated with
tert-butyl pyrocarbonate and the products separated
as described in Example l(H), to give the isomeric
products A (105mg) and B (27mg) as oils.
1 336428
- 22 - T1011
Isomer A: nmr ~(CDC13) 2.59 (lH, d, J=14.4
Hz, CHAHB); 3.61 (lH, dd CHAHB); 5.38 (lH, dd
CH).
Isomer B: nmr ~(CDC13) 2.40 (lH, d, J=18Hz,
CHAHB); 3.25 (lH, dd, CHAHB); 5.39 (lH, d,
J=8.6Hz, CH).
Step I: Preparation of 1,5-dimethyl-10,11-dihydro-
5H-dibenzo[a,d]cyclohepten-5,10-imine hydrochloride.
Isomer A from step H above (105mg) was treated
with dry HCl as in Example l(I) to give 1,5-dimethyl-
10,11-dihydro-5H-dibenzo[a,d]cycloheptene-5,10-imine
hydrochloride (80mg, 88%), m.p. 275-278C.
EXAMPLE 4
1,5-Dimethyl-10,11-dihydro-5H-dibenzo-[a,d]-
cyclohepten-5,10-imine.
Prepared from isomer B (Example (3H))as described
in Example l(I).m.p. > 325C (dec).
EXAMPLE 5
3,5-Dimethyl-10,11-dihydro-5H-dibenzo-[a,d]-
cyclohepten-5,10-imine hydrochloride.
A solution of 412 mg (1.37 mmol) 3-bromo
-10,11-dihydro-5-methyl-5H-dibenzo[a,d]-cyclohepten-5,
10-imine in 40 ml of ethyl ether was treated with 350
mg (0.64 mmol) of 1,3-bis(diphenylphosphino)propane
nickel (II) chloride under an inert atomosphere. To
the mixture was added a solution of methyl magnesium
bromide in ether (3.0 M, 1.4 ml) and after the
initial exothermic reaction had subsided, the mixture
was heated at 40 for 18 hours. TLC indicated
incomplete reaction and an additional 3.0 ml of the
1 336428
- 23 - T1011
Grignard reagent (3.0 M) was added portionwise and
the mixture heated at reflux for 6 hours. Thereafter
the reaction was cooled and quenched by the addition
of 100 ml each of water and ether, the mixture
transferred to a separatory funnel with an additional
300 ml of water and 200 ml of ammonium hydroxide.
The mixture was shaken with 600 ml more ether and the
layers allowed to separate. The ether layer was
removed and dried (sodum sulfate), then concentrated
under vacuum to afford 159 mg of a mixture which was
partially purified by flash chromatography (ethyl
acetate). Additional purification of the 3-methyl
compound was achieved by preparative HPLC using a
Waters PrepPak C-18 column. Homogeneous fractions
were combined and concentrated to dryness, the
material was flashed once more (chloroform, up to 5%
methanol) and the hydrochloride salt of the title
compound was prepared using ethanolic HCl:
C17H17N.HClØ4 H2O, 99.1% by HPLC.
Anal. Calcd.: N, 5.02; C, 73.18: H, 6.60
Found : N, 5.03: C, 73.48: H, 6.63
EXAMPLE 6
5-Methyl-3-phenyl-10,11-dihydro-5H-dibenzo-[a,d]-
cyclohepten-5,10-imine hydrochloride.
A mixture of 100 mg (0.33 mmol) of 3-bromo
-10,11-dihydro-5-methyl-5H-dibenzota,d]-cyclohepten-
5,10-imine, 20 mg (0.017 mmol) of tetrakis(triphenyl-
phosphine)palladium (0), 0.14 mL of triethylamine, 60
mg (0.5 mmol) of phenylboronic acid and 3 mL of
N,N-dimethylformamide was warmed to 100C under
nitrogen atmosphere for 12 h. The mixture was
`:- 1 336428
- 24 - T1011
concentrated by short path distillation under reduced
pressure (0.05 mm, bath temp 35C) to dryness. The
nearly black residue was taken up in 10 mL of dilute
aqueous ammonia (50 mL)) and extracted with three 50
mL portions of ethyl acetate. The combined extracts
were washed with 10 mL of dilute aqueous ammonia,
dried over magnesium sulfate, and concentrated to
dryness. The residue was partially purified by flash
chromatography with ethyl acetate as eluant, then
appropriate fractions combined and further purified
by preparative HPLC using a Whatman ODS-3 preparative
column, eluting with 3~ methanol, 57% water and 40%
acetonitrile (isochratic). The homogeneous fractions
(by HPLC) were combined, concentrated to dryness, and
the residue partitioned between 50 mL of 0.5 N sodium
hydroxide and 50 mL of chloroform. The residue from
concentration of the chloroform extracts was
dissolved in ether and acidified with 1 mL of 5.12 N
ethanolic hydrochloric acid. The white precipitate
Z0 (56 mg) was collected and dried to give the title
compound. C22H19N.HC1Ø5H20; 99.95% by HPLC
Anal. Calcd.: N, 4.09: C, 77.06: H, 6.17
Found : N, 4.38: C, 76.88; H, 6.21
EXAMPLE 7
7-Methyl-12,13-dihydro-7H-benzo[4,5]cyclo-
hepta[l,2-a]naphthalen-7,12-imine
Step A: Preparation of 7-hydroxy-7-methyl-7H-
benzo[4,5]cyclohepta[1,2-a]naphthalene.
To a solution of 7H-benzo[4,5]cyclohepta[1,2-a]
naphthalen-7-one (1.75g, prepared by the method of
`- 1 336428
- 25 - T1011
Bergmann et al, J. Orq. Chem., 1963, 28, 3341) in dry
tetrahydrofuran (40ml) at 0C under nitrogen was
added a solution of methyl lithium in diethyl ether
(8ml of 1.4M). After 90 minutes at 0C the reaction
was quenched by the dropwise addition of water (20ml)
and the reaction mixture extracted into diethyl ether
(2 x 75ml), dried (Na2SO4), filtered and the
solvents removed under reduced pressure to give a
light brown oil (1.9g).
Step B: Preparation of 7-hydroxylamino-7-methyl-
7H-benzo[4,5]cyclohepta[1,2-a]naphthalene
Sodium acetate (5.7g) and dichloroacetic acid
(8.5ml) were stirred together in dichloromethane
(8.5ml) at 0C for 5 minutes then hydroxylamine
hydrochloride (4.8g) added. After lh at room
temperature more dichloromethane (17ml) was added and
after a further 30 minutes, the product from Example
7, Step A (1.9g) was added. After 1 h at room
temperature the reaction mixture was poured into
ice-water (200ml) and washed with ammonia solution
(70ml). The organic layer was dried (Na2SO4),
filtered and concentrated in vacuo to give the
required product (2.04g) as a colourless form.
Spectral data were consistent with the proposed
structure.
Step C: Preparation of 12-hydroxy-7-methyl-12,13-
dihydro-7H-benzo[4,5]cyclohepta[1,2-a]naphthalen-
7,12-imine and lZ-hydroxy-7-methyl-12,13-dihydro-7H-
benzo[4,5]cyclohepta[1,2-a]naphthalen-7-13-imine.
1 336428
- 26 - T1011
To xylene (lOOml) at reflux was added, over a 20
minute period, the hydroxylamine product from Example
7B (1.9g) in xylene (lOOml) under nitrogen. After 30
minutes at reflux the reaction mixture was allowed to
cool and the solvent removed under high vacuum to
leave a brown residue. Trituration with ethyl
acetate and filtration gave a white solid which was
washed with methanol and dried in the vacuum oven to
give, as a mixture of atropisomers, 12-hydroxy-7-
methyl-12,13-dihydro-7H-benzo[4,5]cyclohepta[1,2-a]
naphthalen-7,12-imine (0.72g, 40%). The filtrate was
concentrated in vacuo and the residue obtained,
purified by chromatography on silica gel with 25%
ethyl acetate in hexane as eluent to give, as a white
solid, 12-hydroxy-7-methyl-12,13-dihydro-7H-benzo[4,5]
cyclohepta[l,2-a]naphthalen-7,13-imine (0.41g, 26%).
Step D: Preparation of 7-methyl-12,13-dihydro-7H-
benzo[4,5]cyclohepta[1,2-a]naphthalen-7,12-imine
hydrochloride.
12-Hydroxy-7-methyl-12,13-dihydro-7H-benzo[4,5]
cyclohepta[l,2-a]naphthalen-7,12-imine (0.25g) and
zinc dust (0.3g) were heated together at 65C in
glacial acetic acid (3ml) for 14h under an atmosphere
of nitrogen. After this time the reaction mixture
was filtered and the solvent removed in vacuo to
leave a light brown oil. This was dissolved in ether
(40ml) and washed with lN sodium hydroxide solution
(2 x 25ml) then water (1 x 25ml), dried (Na2S04),
filtered and concentrated under reduced pressure.
The residue was dissolved in a 5 molar solution of
hydrogen chloride in ethyl acetate then
reconcentrated and dried under high vacuum to give
` - 1 33642a
- 27 - T1011
7-Methyl-12,13-dihydro-7H-benzo[4,5]cyclohepta[1,2-a]
naphthalen-7,12-imine hydrochloride (130mg), m.p. >
250C (dec.). p.m.r. (free base) (360MHz, DMS0-d6)
~ 2-03 (3H, s, CH3), 3.09 (lH, d, J = 16.9Hz,
H-ll endo), 3.77 (lH, dd, J = 16.9 and 5.6Hz, H-ll
exo), 4.91 (lH, d, J = 5.6Hz, H-10), 7.08 (3H, m,
aromatics), 7.30-7.45 (3H, m, aromatics), 7.50 (lH,
d, J = 8.6Hz), 7.63 (lH, d, J = 8.6Hz), 7.72 (lH, d,
J = 7.9Hz) and 7.81 (lH, d, J = 7.9Hz). COSY spectra
showed that H-6, H-7 and H-8 are in the 3 proton
multiplet centred at ~ 7.08, H-9 is at ~ 7.33 and
H-4 is at ~ 7.50. Irradiation of the methyl group
(~ 2.03) produced a n.O.e. to H-4 (~ 7.50) and
H-6 (~ 7.08) while irradiation of H-10 gave a
n.O.e. to H-9 (~ 7.33). Found: C, 73.81; H, 5.78;
20 17 ' ~ 2 requires: C, 73.72; H,
6.19; N, 4.30~.
EXAMPLE 8
7-Methyl-12,13-dihydro-7H-benzo[4,5]cyclohepta
rl,2-a]naphthalen-7,13-imine hydrochloride.
12-Hydroxy-7-methyl-12,13-dihydro-7H-benzo[4,5]
cyclohepta[l,2-a]naphthalen-7,13-imine (Example 7,
Step C, 300mg) and zinc dust (370mg) were heated
together in glacial acetic acid (4ml) at 65C for 14h
under an atmosphere of nitrogen. After this time the
reaction mixture was filtered and the solvent removed
in vacuo to leave a residue which was dissolved in
ether (50ml) and washed with lM sodium hydroxide
solution (1 x 25ml), water (1 x 25ml) then dried
(Na2S04), filtered and concentrated under
vacuum. The white foam obtained was dissolved in a
5M solution of
1 336428
- 28 - T1011
hydrogen chloride in ethyl acetate and the solvent
removed in vacuo to leave as a white foam
7-methyl-12,13-dihydro-7H-benzo r 4,5]cyclohepta[1,2-a]
naphthalen-7,13-imine hydrochloride (0.19Sg), mp
208-210C. p.m.r. (free base) (360MHz, DMSO-d6)
~ 2.02 (3H, s, CH3), 2.85 (lH, d, J = 16.8Hz,
H-ll endo), 3.54 (lH, dd, J = 16.8 and 5.4Hz, H-ll
exo), 5.28 (lH, d, J = 5.4Hz, CH), 6.88 (lH, m), 7.05
(2H, m), 7.28 (2H, m), 7.40 (lH, dd, J = 8.2, 8.0Hz),
7.64 (lH, dd, J = 8.3, 8.0Hz), 7.81 (lH, d, J =
8.2Hz) and 7.86 (lH, d, J = 8.3Hz). COSY spectra
showed H-l to be at ~ 6.88 and H-16 to be at ~
7.86. Irradiation of H-10 (~ 5.28) gave a n.O.e.
to H-16 (~ 7.86).
EXAMPLE 9
2,5-Dimethyl-10,11-dihydro-5H-dibenzo[a,d]
cyclohepten-S,10-imine
2-Methyl-5H-dibenzo[a,d]cyclohepten-5-one
(prepared by the method of Dunn et al, J. Med. Chem.,
1977, 20, 1557) was converted as described in Example
1, Steps D-H, to a mixture of 2- and 8-methyl-5-
methyl-12-tert-butoxycarbonyl-10,11-dihydro-5H-dibenzo
[a,d]cyclohepten-5,10-imines. Separation of the
regioisomers was performed by reverse phase
chromatography, using a C-18 column, eluting with
CH3CN/H20 (65:35). The 2-methyl regioisomer was
deprotected as described in Example lI to give 2,5-
dimethyl-10~11-dihydro-5H-dibenzo[a~d]cyclohepten-
5,10-imine hydrochloride, m.p. 185-195C.
(360MHz, CDC13) 2.29 (3H, s, CH3), 2.33 (3H, s,
CH3), 2.88 (lH, d, CHAHB), 3,99 (lH, dd,
- ` 1 336428
- 29 - T1011
CHAHB), 5.25 (lH, d, CH) and 6.94-7.26 (7H, m,
ArH). Found: C, 66.55; H, 6.16; N, 4.49.
C17H17N.2HCl requires: C, 66.24; H, 6.21; N,
4.54%. m/e 235 (M+). The assigned regiochemistry
was confirmed by n.O.e. studies.
EXAMPLE 10
5,8-Dimethyl-10,11-dihydro-5H-dibenzo[a,d]
cyclohepten-5,10-imine
5,8-Dimethyl-12-tert-butoxycarbonyl-10,11-dihydro-
5H-dibenzo[a,d]cyclohepten-5,10-imine (separated from
the 2-methyl regioisomer as described in Example 9)
was deprotected as described in Example lH to give
5,8-dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine, m.p. 190-195C. ~ (360MHz, CDC13)
2.20 (3H, s, CH3), 2.31 (3H, s, CH3), 2.85 (lH,
d, CHAHB), 3.94 (lH, dd, CHA_B)~ 5-28 (lH, d,
CH) and 6.81-7.36 (7H, m, ArH). m/e 235 (M+). The
assigned regiochemistry was confirmed by n.O.e.
studies.
EXAMPLE 11
3-Methoxycarbonyl-5-methyl-10,11-dihydro-5H-
Z5 dibenzo[a,d]cyclohepten-5,10-imine
Step A: Preparation of 3-carboxy-5-methyl-10,11-
dihydro-5H-dibenzo[a,d]cyclohepten-5,10-
imine
In a 3 neck flask fitted with a thermometer,
magnetic stirrer, rubber septum and a gas inlet tube
connected to a bubbler was dissolved 500 mg (1.66
mmole) of 3-bromo-5-methyl-10,11-dihydro-5H-dibenzo-
1 336428
- 30 - T1011
[a,d]cyclohepten-5,10-imine in 10 ml of THF,
previously distilled over sodium benzophenone ketyl.
The solution, stirred at -78C under nitrogen
atmosphere, was treated with 2.45 ml (4.15 meqs) of a
1.7M solution of tert-butyllithium in pentane. After
stirring at -78C for 30 minutes, the nitrogen was
replaced by carbon dioxide generated from dry ice and
sublimed, passing through a Drierite column en route
to the reaction vessel. After a few minutes a
copious white precipitate had formed and the cooling
bath was removed. Stirring under C02 was continued
at ambient temperature for two hours, then 1 ml of
water was added and the THF removed in vacuo. The
residue was slurried in water (20 ml) until nearly a
clear solution, filtered, and any undissolved solids
washed with a small amount of water. The pH of the
clear filtrate was carefully adjusted to 6.5 by
addition of glacial acetic acid. The white solid
which precipitated was washed with water, then with
ether/acetonitrile mixtures to give the title
compound, 280 mg (64%). PMR (d6-DMSO) ~ 1.87 (s,
3H, CH3), 2.66 (d, lH, J = 17Hz, C-ll exo H), 3.39
(dd, lH, J = 17 and 5.6Hz, C-ll endo H), 4.61 (d, lH,
J = 5.3 Hz, C-10 H), 7.00-7.17 (m, 4H, aryl), 7.33
(d, lH, J = 6Hz, Hl), 7.64 (dd, lH, J = 7.8 and
1.5Hz, H-2) and 7.83 (s, lH, H-4).
Step B: Preparation of 3-methoxycarbonyl-S-
methyl-10,11-dihydro-SH-dibenzo[a,d]cyclohepten-5,10-
imine.
The 3-carboxy compound from Step A (265 mg, 1
mmole) was suspended in 25 ml abs. CH30H, the
mixture treated with 0.6 ml (5 mmol) boron
1 336428
- 31 - T1011
trifluoride etherate, and the resulting solution was
stirred at reflux for 18 hours. Removal of the
solvent left a residue which was extracted with
ether-saturated sodium bicarbonate. The combined
ether extracts were washed (H2O, brine) and dried
(Na2SO4) to afford 240 mg of 3-methoxycarbonyl-5-
methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-
imine. PMR (CDC13) ~ 1.99 (s, 3H, CH3), 2.63
(broad s, lH, NH), 2.80 (d, lH, J = 17Hz, C-ll-exo
H), 3.50 (dd, lH, J = 17, 5.4Hz, C-ll endo-H), 3.90
(s, 3H, OCH3), 4.72 (d, lH, J = 5.4Hz, C-10 H),
7.01-7.33 (m, 5H, aryl), 7.77 (dd, lH, J = 8 and
1.6Hz, H-2) and 7.95 (d, lH, J = 1.7Hz, H-4).
EXAMPLE 12
3-Hydroxymethyl-5-methyl-10,11-dihydro-5H-dibenzo-
ra,d]cyclohepten-5,10-imine
A solution of 3-methoxycarbonyl-5-methyl-10,11-
dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (279mg,
1.0mmol) in a mixture of ether (20 ml) and dry THF
(10 ml) was added dropwise with stirring, under
nitorgen, to a suspension containing 87 mg (2.3mmole)
of lithium aluminium hydride in 25 ml dry ether at
40C. When the addition was complete the mixture was
heated at 55C for 2 hours, then stirred at 25C for
18 hours. TLC (SiO2, CHC13:CH30H:NH40H
90:10:1) indicated the reaction was complete. The
mixture was chilled in an ice-bath and quenched by
the dropwise addition of lOml of saturated aqueous
sodium potassium tartrate. After stirring for
several hours the mixture was filtered and the while
solid washed with water, then ether to give 3-
1 336428
- 32 - T1011
hydroxymethyl-5-methyl-10,11-dihydeo-SH-dibenzo[a,d]
cyclohepten-5,10-imine (195 mg, 74%), mp 215-217C.
Additional product could be obtained from the ether
wash. The title compound had satisfactory PMR and
high resolution spectra; for C17H17NOØ5H2O.
Anal. Calc'd: N, 5.38; C, 78.43; H, 6.97
Found : N, 5.28; C, 78.53; H, 6.87
EXAMPLE 13
Tablet Preparation
Tablets containing 1.0, 2.0, 25.0, 26.0, 50.0 and
100.0 mg, respectively, of the following compounds
are prepared as illustrated below:
5,7-Dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine
3,5-Dimethyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-
5,10-imine
3-Hydroxymethyl-5-methyl-10,11-dihydro-5H-dibenzo[a,d]
cyclohepten-5,10-imine
3-Methoxycarbonyl-5-methyl-10,11-dihydro-5H-dibenzo-
[a,d]cyclohepten-5,10-imine
TABLE FOR DOSES CONTAINING FROM
1-25 MG OF THE ACTIVE COMPOUND
Amount-mg
Active Compound 1.02.0 25.0
Microcrystalline cellulose 49.25 48.75 37.25
Modified food corn starch49.2548.75 37.25
Magnesium stearate 0.50 0.50 0.50
- 1 336428
- 33 - T1011
TABLE FOR DOSES CONTAINING FROM
26-100 MG OF THE ACTIVE COMPOUND
Amount-mg
Active Compound 26.050.0100.0
Microcrystalline cellulose 5Z.0100.0200.0
Modified food corn starch 2.214.25 8.5
Magnesium stearate 0.390.75 1.5
All of the active compound, lactose, and a
portion of the corn starch are mixed and granulated
to a 10% corn starch paste. The resulting
granulation is sieved, dried and blended with the
remainder of the corn starch and the magnesium
stearate. The resulting granulation is then
compressed into tablets containing 1.0 mg, 2.0 mg,
25.0 mg, 26.0 mg, 50.0 mg and 100.0 mg of active
ingredient per tablet.