Note: Descriptions are shown in the official language in which they were submitted.
1339665
The present invention relates to compounds which can be used in
contrast ~edia for radiography.
Iodobenzene compounds containing several lodine atoms ln the
benzene nucleus, usually 3 iodine atoms per benzene nucleus, and various
S other substituents have been used for a long time as contrast mediu~.
These other substituents are pharmacologically acceptable groups which
enable the compounds to be administered to man and animals. Generally
speaking, these substituents are chosen, on the one hand, in order to
confer adequate solubillty in water on the compound so that they can be
administered in aqueous solution and, on the other, in order to confer
on these compounds sufficient tolerance for them to be tolerated by the
human organism.
For this purpose, non-ionic structures have been suggested, i.e.
iodobenzene derivatives possessing non-ionic substituents.
Thus, in the patent FR-A-2 053 037, carbamoyl iodobenzene compounds
containing a total of at least one N-hydroxy alkyl group and at least
two hydroxy groups were suggested.
A compound illustrative of this class is metrizamide which has,
however, proved to be of limited stability.
The present invention aims to provide novel non-ionic compounds
which are well tolerated by the human organism, very stable in aqueous
solution, which possess a high solubility in water and which exhibit low
viscosity in solution.
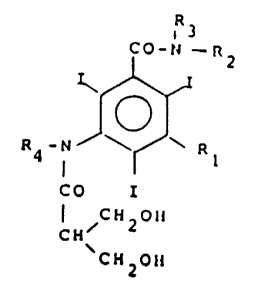
To this end, the subject of the present invention is compounds of
formula:
C0-N - R2
R - N ~ R
CO
CH 20H
'CH2~H ~L
in which
13~966lj
Rl is selected from a group of formula
- N - C0 - R5
- R6
in which R5 is selected from C1-C4 alkyl, C1-C4 hvdroxya1kyl or C1-C4
polyhydroxyalkyl, and
R6 is selected from hydrogen, Cl-C4 alkyl, C1-C4 hydroxyalkyl or Cl-C4
polyhydroxyalkyl,
and a group of formula
iR8
- C0 - N - R7
in which R7 is selected from C1-C4 hydroxyalkyl or C1-C4 polyhydroxyalkyl,and
R8 is selected from hydrogen or C1-C4 alkyl,
R2 is selected from hydrogen, Cl-C4 hydroxyalkyl or C1-C4 polyhydroxy-
alkyl,
R3 is selected from hydrogen or C1-C4 alkyl, and
R4 i8 selected from hydrogen, C1-C4 alkyl, C1-C4 hydroxyalkyl or C~-C4
polyhydroxyalkyl.
In the present invention by polyhydroxyalkyl group i8 meant a
linear or branched polyhydroxyalkyl group.
A preferred compound of formula I is the compound of formula I in
which
R1 = -C0-NH-CH2-CH20H, R2 = -CH2-CH20H, R3= H
R4 = - CH2-CHOH- CH20H.
~oreover, a preferred group of compounds of formula I is that
constituted by the compounds of the symmetrical diamino type, i.e. the
compounds of formula:
1339665
C O - N --R 2
I ~,~ I
131 I I
R4-N ~ jN-R 4
, C H 2 ~ H I ~ C H 2 ~ H
CH2~H \CH20H
The compounds of formula I can be prepared in a standard manner in
particular by acylation and/or alkylation reactions starting from known
compounds.
Thus, the compounds of the symmetrical diamino type (compounds of
formula II) can be prepared by
a) acylation of a diamino compound of formula:
R
13
CON - R'2
~\/~/ I
III
N H 2
R2' representing a R2 group, the hydroxy groups of which have been
protected
with an acid chloride of formula :
RCOCl IV
in which R represents
~ 2
a -CH group, the hydroxy groups of which are protected,
~ CH2-OH
b) alkylation of the compound obtained, if necessary, with an
1339665
alkylat~ng reagent of formula:
4Z V
in vhlch R'4 has the meanings given previously except hydrogen and Z
represents a labile group such as an atom of chlorine, bromine or
iodine, in the presence of a base such as sodium methylate, sodium
ethylate, sodium hydride or sodium hydroxide.
c) deprotection
Compounds of formula III are described in the French patent
application FR-A-2 614 299 , pub~shed October 25, 1988.
The other compounds of formula III can be prepared in an analogous
manner startlng in particular from an alkyl 3,5-dinitrobenzoate of
formula:
COOR'
¦ VI
,~,
02N N02
20in ~hich R' ~s a C1-C4 alkyl group ~uch as methyl.
Asymmetrlc dlamino compounds can also be prepared startlng from a
compound of formula VI by:
a) reaction vith an amine of formula:
NH-R2 VII
R3
80 as to produce a compound of formula:
30 CON - R2
~ VIII
02N N02
b) reduction by means of ammonium sulflde 80 as to give rise to a
compound of formula: 13 .3 9 6 ~ 5
l3
CON - R2
5 ~ ~ IX
H2 N02
c) acylatlon of the compound of formula IX by an acid chloride of
formula RCOCl ~IV) 80 afi to give rise to a compound of formula:
CON - R2
15 ~ ~ X
R-CO-HN N~2
20d~ reduction and iodination of the compound of formula X so as to
give rise to a compound of formula:
co l - R2
25CO IH2 XI
R
e) alkylation, lf required, of the compound of formuls XI ~ith an
alkylating reagent of formula V so as to give rise to a compound of
formula:
1~39665
C O - N - R 2
I ~ I X I I
4 1 1 2
COR
f) deprotection of the compound of formula XII
g) acylation of the deprotected compound obtained with an acid
chloride of formula:
C1-CO-R'5 XIII
R'5 representing a group R5, the hvdroxy groups of which are protected,
so as to give rise after deprotection to a compound of formula :
CO-N -~2
I ~ I
0 I XIV
. ~ NH-COR~
R4-1 1
cO
~CH20H
CH2~H
~0 the steps f and g may be carried out in the reverse order, and if
necessary
h) alkylation to give rise to a compound of formula I in which R6
has the meanings indicated except hydrogen.
Symmetrical compounds of the isophthalic type (compounds of
formula I in which Rl = -CO-N-R2
13396~5
can be prepared by
a) acylation of an amine of formula:
-
COCl
~ COCl XV
10with an acid chloride of formula RCOCl (IV) 80 as to give rise to a
compound of formula:
C OC 1
I / 1 I
~ O ~ XVI
R -CO-HN~ \~/ COCl
I
b) reaction of the compound of formula XVI with an amine of formula
H-N-R2 (VII) so as to give rise to a compound of formula:
R3
CO-N --R2
I
~ ~ XVII
R-CO-Ni~ ' CO-N--R2
r R3
then, if desired, either
c) alkylation of the compound of formula XVII with an alkylating
8 13396~
reagent of formula R'4Z such as that specified previously and finally
d) removal of the protecting groups from the -CH(CH20H)2 group, or
e) removal of the protecting groups fro~ the -CH(CH20H)2 group and,
if desired,
S f) alkylation of the deprotected compound with an alkylating
reagent R'4Z.
The asymmetric compounds of the lsophthalic type (compounds of
CO I R7 vith R7~R2 and/or R ~ R ) can be
R8
prepared by
a) acylation of an amine of formula
1 3
CO-N - R
lS ~ ~ I R7 XVIII
H2 CON - R
vith an acid chloride of formula RCOCl,
b) alkylation, lf desired, vith an alkylating reagent of formula
R ' 4Z and
c~ removal of the protecting groups from the -CH-(CH20H)2 group.
The compound of formula XVIII may be obtained as descrlbed in EP-O
2S OlS 867, published September 17, 1980.
As an alternative, the asymmetric compounds of the isophthalic type
can be prepared by
a1 acylation of an amine of formula:
COC 1
2 ~ CO~ -R~7 XIX
I R8
1333~65
g
in which R'7 represents a R7 group in which the hydroxy groups are
protected,
with an acld chloride of formula RCOCl ~IV) co as to give rise to a
compound of formula:
COC 1
~N / ~ CO-N -R'7 XX
CO I 8
R
b) reaction of the compound of formula XX with an amine of formula:
HN-R2 ~VII)
so as to give to a compound of formula:
R3
I ~ I
L~l xx~
~N~ CO-N-R'7
CO I R8
- R
then, if desired, either
c) alkylation of the compound of formula XXI with an alkylating
reagent of formula R'4Z such as that previously specified and finally
d) removal of the protecting groups from the -CH-~CH20H)2 group,
or
e) removal of the protectlng groups from the -CH-~CH20H)2 group,
and, if desired,
f) alkylation of the deprotected compound with an alkylating
3S reagent R'4Z.
Another ~ubject of the present invention i8 contrast ~iu~
lo 13396~
which contain at least one compound of formula I.
These contra8t me~i a are used in man and animals for
radiological purposes.
The preferred pharmaceutical form of the contrast materials
according to the invention consists of aqueous solutions of the
compounds.
The aqueous solutions usually contain a total of from S to 100 g of
compounds of formula I per 100 ml and the volume of such solution to be
injected usually varies from 1 to 1000 ml.
The aqueous solution of the compounds of formula I may also contain
certain additives such as:
- sodium chloride at concentrations included between 0.1 and 10 mM/1
- disodium EDTA at concentrations included between 0.1 and 2 mM /1
- sodium citrate at concentrations included between 0.1 and 10 mM /1
- heparin at doses included between 10 and 100 units per 100 ml of
solution.
These compounds may be administered by all routes conventionally
used for iodinated non-ionic contra8t medium Thus, they may be
administered by the enteral route or the parenteral route (intravenous
route, intra-arterial route, opacification of the cavities) and in
particular into the subarachnoid space.
An example of the composition according to the present invention
will be given below.
Composition
Composition of example 1 65 g
Water for injectable preparation
QS 100 ml
The following examples illustrate the preparation of the compounds
of formula r.
EXAMPLE 1
Preparation of 5-[3-hydroxy-2-(hydroxymethyl)-N-~2,3-
dihYdroxypropyl)propionamido]-N',N~-bis-(2-hYdroxYethyl)-2,4,6-triiodo-
isophthalamide.
a~ PreParation of 5-[2-isopropyl-1,3-d10xane-5-carboxamido~-2,4,6-
triiodo-isophthaloYl dichloride.
137 g of 5-amino-2,4,6-trilodoisophthaloyl chloride ~0.23 mole) are
1~396~
11
d1ssolved in 460 ml of DnAC to Yhlch are added 110 9 (0.57 mole) of 2-
1~opropyl-1,3-dioxane-S-carboxylic acid chloride. The reaction mixture
is stirred under argon at ambient temperature for 4 days. The D~AC
removed in a vacuum. The oil obtained 1s extracted vith 3 1 of ethyl
S acetate and vashed tvice vith l l of ice-cold vater. The organic phaie
lS dried and concentrated to dryness. The product is crystalli.ed from
200 ml of CH2C12. After filtration, 110 g of solid are obtalned:
Yield: 64%
TLC: SiO2 CH2C 2 Rf : 0.13
~60 F 254J 51~2 ether/petroleum ether S0/S0
Rf : 0.52
b) Preparation of 5-[2-isopropyl-1,3-dioxane-5-carboxamido~-N',N~-
bis-(2-hydroxyethyl)-2,4,6-tri1odo-isophthalamide.
130 9 of the product obtained in a (0.173 mole) are dissolved in a
solution of 750 ml of D~AC and 75 ml ~0.534 mole) of trlethylamine.
33.7 9 of ethanolamine ~0.552 mole) are added dropvi3e to the reaction
mixture. The reaction mixture i~ then stirred for 3 hours at ambient
temperature. The triethylamine hydrochloride is removed by filtration
and the ~nAC is removed in a vacuum. The oil obtained is crystallized
from 1 liter of vater. The product is filtered off and dried in a
vacuum.
Yield : 95~
TLC : SiO2 Rf : 0.25 CH2C12/methanol 9tl
~60F254) SiO2 Rf : 0.67 CH2C12~methanol 8/2
% I : 45.6 (found~ - 47.6 (theory)
Hypers11 C8 S~ 15 cm
HPLC purity : 97% 0.01 n NaH2PO4 = so
~ethanol = S0
c) Preparation of 5-[3-hYdroxY-2-hydroxymethyl-N-(2,3-
dihydroxypropyl)propionamido]-N',N~-bis-(2-hydroxyethyl)-2,4,6-triiodo-
isophthalamide.
To a suspens10n of 100 9 of the product obtained in b (0.125 mole)
in 350 ml of ethylene qlycol are added dropvise 125 ml (O.S mole) of 4 N
methylate at 60-C folloved by 65 Q (0.625 mole) of 1-chloro-2,3-
propanediol. After 1 hour at 60CC the mass of the reaction mixture has
increased. 100 ml (0.4 mole) of 4 N methylate and S5.2 Q (O.S mole) of
12 1~39~S
1-chloro-2,3-propanediol are added. The mixture is maintained overnight
at 600C. A further addition of 31 ml l0.125 mole) of 4 N methylate and
20.7 q of 1-chloro-2,3-propanediol is made. Stirring is maintained for
4 hours at 600C. The mineral salts are removed by filtration. The
ethylene glycol is evaporated in a vacuum.
The distillation residue is taken up in 800 ml of 10 N HC1 and the
solution is stirred overnight at ambient temperature. The reaction
mixture is concentrated to dryness and the residue is taken up in 300 ml
of ethanol. The mineral salts are removed by filtration. The ethanol
is evaporated in a vacuum and the residue is crystallized from 1 liter
of isopropyl alcohol. The precipitate is filtered off and purified by
HPLC (RP 18~ ~elution with water~.
Total yield ~alkylation-deprotection-purification) : 52%
1) TLC ~sllica 60F254) : CH2Cl2/methanol 7/3 Rf : 0.4
2) HPLC Hypers11 C8 5u 15 cm
Buffer 0-01 M NaH2P~4 ~- 97
methanol .. 3
Purity : 97t.
3) '~. I : 45.8 ~found) - 46.4 (theory)
4) NMR ~DMSO)
Poorly resolved multiplet centered at 3.5 ppm ~18H); multiplet
centered at 4.5 ppm ~OH) exchangeable with D20 ~6 H); broad peak at
8.4 ppm ~NH) exchangeable vith D20 ~2 H).
EXAMPLE 2 - Preparation of 5-qlycolamido-3-t3-hydroxy-2-
hydroxymethyl-N-(2,3-dihydroxypropyl)propionamido]-2,4,6-triiodo-N-
hydroxyethyl benzamide
a) PreParation of 3,5-dinitro-N-~2-hYdroxyethyl)benzamide
750 g ~3.32 moles) of methyl 3,5-dinitro benzoate are suspended in
2 liters of methanol in the presence of 222.7 g ~3.65 moles) of ethanol-
amine. The reaction mixture is refluxed for 48 hours until the ester
has disappeared. After 4 hours at room temperature, the crystalline
product is filtered off, vashed vith 500 cm of methylene chloride, then
dried in an oven at 600C in a vacuum for 4 hours. This procedure leads
to the recovery of 718 g of product in a yield of 85%
Melting point: 140~C.
TLC ~toluene/methyl ethyl ketone/formic acid (60/25/25) Rf : 0.5.
13 1339S6~
b) Preparation of 3-nitro 5-amino-N-(2-hydroxyethyl)benzamide.
To a suspension of 25.5 g (0.5 mole) of 3,5-dinitro-N-~2-hydroxy
ethyl)benzamide in 135 cm of water are added at 700C 12.25 g (0.18
mole) of ammonium sulfide. At the end of the addition the mixture is
homogenous but reprecipitation occurs after 1/2 hour at 700C. The
reaction mixture is allowed to cool to ambient temperature and stirring
is continued for 2 hours. The precipitate is filtered off, washed with
methanol ~70 cm ) then dried in an oven (600C).
Mass obtained : 15.1 g - yield 67%.
TLC (toluene/methyl ethyl ketone/formic acid 60/25/25). Rf : 0.3
H NMR (~MSO) : 3.4 ppm (multiplet; 4H,CH2 aliphatics); 4.65 ppm
(multiplet, H exchangeable with D20, NH2); 5.9 ppm (singlet, H
exchangeable with D20, OH); 7.4 - 7.7 ppm (2 multiplets; 3H, aromatic
protons); 8.6 ppm (multiplet, lH, NH).
c) Preparation of 3-nitro-5-t2-isopropyl-1,3-dioxane-5-
carboxamido]-N-hydroxyethyl benzamide
40 g (0.177 mole) of 3-nitro-5-amino-N-~2-hydroxyethyl)benzamide
are dissolved in 400 cm of DMAC. The addition of 74.9 g ~0.389 mole)
of 2-isopropyl-1,3-dioxane-5-carboxylic acid chloride in the presence of
triethylamine ~54.6 cm ) gives rise to an exothermic reaction.
The reaction mixture is maintained under argon for 18 hours at
ambient temperature. The mixture is filtered and the filtrate is
diluted with vater and extracted with ethyl acetate. The residue
obtained after evaporation of the solvent is treated with potassium
carbonate (12 g) in 300 cm of methanol. After being stirred at ambient
temperature for 48 hours, the mixture is concentrated, then extracted
vith ethyl acetate. The crude product obtained after treatment is
recrystallized from a mixture of ether/ethyl acetate 80/20.
37.8 g of product are isolated in a yield of 56-/
TLC (ethyl acetate Rf : 0.48).
HPLC Hypersil C8 5u 15 cm.
Buffer: 0.01 M NaH2P04 50%
MeOH 50%
Purity: 94X.
d) Preparation of 5-amino-3-t2-isoProPy~ 3-dioxane-5
carboxamido]-2,4~6-triiodo-N-hydroxYethyl-benzamide.
14
A methanolic solution (1.4 1) of 40 g of 3-nitro-S-t2-isopropyl-
1,3-dioxane-S-carboxamido]-N-hydroxyethyl benzamide i8 stirred under an
atmosphere of hydrogen (S.10 Pa) for 5 hours at SO~C in the presence
of 4 g of palladlzed charcoal. The catalyst is then filtered off and
S the filtrate is evaporated under reduced pressure. The resulting
compound is suspended in 950 cm of water. The mixture is made
homogenous by the addition of 20 cm of 2 N hydrochloric acid. 63 cm
of iodine chloride (70/. in iodine) are then added dropwise with
vigorous stirring. After 24 hours at ambient temperature,the
precipitate is filtered off, washed with water, taken up in ether.
After drying, 32 g of product are obtained in a yield of 42%.
TLC (dichloromethane/methanol 90/10) Rf : 0.8.
e) Preparation of 5-amino-3-~N-(2,3-dihydroxypropyl)-2-isopropyl-
1,3-dioxane-5-carboxamido]-2-4,6-triiodo-N-hydroxyethyl benzamide.
lS To a solution of the compound obtained in d) 120g, 0.027 mole) in a
mixture of ethylene glycol-dimethylformamide v/v (160 ml) are added
dropwise 84 cm (0.337 mole) of 4N ~odium methylate. The mixture is
heated at 60~C for 1/2 hour and 36.1 cm (0.432 mole) of 1-chloro-2,3-
propanediol are added at thls temperature. The reaction mixture is
maintained at 60OC under nitrogen for 60 hours. The mineral salts are
removed by filtration. The ethylene glycol and the DMF are evaporated
in a vacuum. The crude product obtained is purified on silanized silica
(elution with water, followed by water~methanol 50/S0). 16.5 g of
product are isolated. Yield 76%.
TLC ~dichloromethane/methanol 80/20). Rf : 0.8.
f) Preparation of S-amino 3-[3-hydroxy-2(hydroxymethyl)-N-(2,3-
dihydroxypropyl)propionamido]-2,4,6-triiodo-N-hydroxyethyl benzamide.
16g ~0.02 mole) of the product obtained in e) are deprotected in
the presence of 80 cm of 10 N hydrochloric acid for 48 hours at ambient
temperature. After neutralization and evaporation under reduced
pressure, the residue is precipitated with a mixture of methanol-ether
~9/1), filtered off then purified by HPLC ~RP 18) ~elution with water
then with water/methanol 90/10).
4g of product are isolated in an overall yield ~deprotection,
purification) of 30%.
TLC ~dichloromethane/methanol 80/20). Rf : 0.25.
1 3 3 9 b 6 ~
HPLC Hypersil C8 5p 15 cm.
Buffer: 0.01 ~ NaH2P04 90X
MeOH lOX
Purity: 97Z.
g) Preparation of 5-N-qlycolamido-3-[3-hydroxy-2(hydroxymethyl)-N-
(2,3-dihydroxypropyl)proplonamldo]-2,4,6-triiodo-N-hydroxyethyl
benzamide.
5.5 g of O-acetylated glycolic acid chloride (0.04 mole) are added
dropwise at ambient temperature to a solution of 3 g of the compound
obtained in step f (0.004 mole) in 30 cm of anhydrous DnAC. The reac-
- tion mixture is heated at 400C for 12 hours, then poured into 250 cm of
ice-cold water. The precipitate obtained is filtered off then extracted
with ethyl acetate. After treatment followed by evaporation, the
product obtained dissolved in 50 cm of methanol is deprotected in the
pre6ence of 10 cm of 1 N sodium hydroxide. The solution is stirred at
ambient temperature for 14 hours, then desalted by successive passages
through H IIRN77) and OH (IRN78) resins. After evaporation to
dryness, the residue is taken up in ethyl ether, filtered then dried.
Mass obtained: 1.5 g. Overall yield: 47%.
Purity in iodine: 99%.
TLC lethyl acetate/methanol/ammonia 60/40/1).
Rf : 0.25.
HPLC Hypersil C8 5u 15 cm.
Buffer: 0.01 M NaH2P04 90%
Me OH 10%
Purity : 89%
EXAMPLE 3 - Preparation of 3,5-bis-(3-hYdroxY-2-hydroxymethYl -
propionamido)-2,4,6-triiodo-N-(2,3-dihydroxYpropyl)benzamide
a) Preparation of 3,5-diamino-2,4,6-triiodo-N-(2,3-
diacetoxypropyl)benzamide
301.5 g (0.5 mole) of 3,5-diamino-2,3,6-triiodo-N-~2,3-
dihydroxypropyl)benzamide are suspended in 1 1 of anhydrous pyridine
cooled to 15~C. After the addition of 2450 ml of acetic anhydride, the
solution i8 stirred for 18 h at ambient temperature, then poured into
3S acidulated water. After extraction with ethyl acetate, drying of the
organic phase and evaporation, 270 g of product are obtained in a yield
13~9~
of 78.5%.
Purity in iodine : 98.3~/
TLC toluene/methylethylketone/HCOOH 60/25/35. Rf: 0.70.
b) Preparation of 3,5-bi6~2-isopropyl-1,3-dioxane-5-
carboxamido)2,4,6-triiodo-N-~2,3-diacetoxypropyl~benzamide
114.5 g (0.166 mole) of the compound obtained in a) are di~solved
in 350 ml of anhydrous DMAC. The addition of 128 g ~0.66 mole) of 2-
isopropyl-1,3-dioxane-5-carboxylic acid chloride is carried out at OoC.
After being stirred overnight the reaction mass i8 poured into a mixture
of ice-water. The precipitate is filtered off, washed with vater then
dried in a vacuum at 504C.
c) Preparation of 3,5-bis~2-isopropyl-1,3-dioxane-5-carboxamido)-
2,4,6-triiodo-N(2,3-dihydroxypropyl) benzamide
175 g of the compound obtained in b) suspended in 2.5 l of methanol
are stirred at ambient temperature in the presence of 45 g of potassium
carbonate overnight. After evaporation of the reaction mixture, the
product crystallizes from vater. After filtration and drying, the
crystals obtained in 85% yield are used directly in the next step.
d) Preparation of 3,5-bis~3-hydroxy-2-hydroxymethyl propionamido)-
2,4,6-triiodo-N-(2,3-dihydroxypropyl~benzamide.
The compound obtained in c) is dissolved in 2 l of 5 N HCl at 50oC.
After being stirred for 18 h, the suspension obtained is filtered. The
filtrate is concentrated in a vacuum and the residue is taken up in
isopropanol.
108 g of crystalline product are obtained in 2 crops in a yield of 94%.
TLC SiO2 8utanol 60, water 25, CH3COOH 11 : Rf : 0.2.
The product is purified by preparative HPLC on SiO2 RP18 15,25 u vith
water as eluant in a yield of 47%.
Purity in iodine: 99.6%.
HPLC purity: 99.1% (Hypersil C8 5u 15 cm 0.01 M NaH2P04 95, MeOH 5).
H NMR 200 MHz (DMS~)
8.5 ppm ~m,lH exchangeable with D20, O-CONH)
9.9 ppm ~t,2H exchangeable with D20, O-NH-CO)
4.6 ppm ~m,6H exchangeable, OH)
3-4 ppm ~m, 13H, CH)
2.7 ppm ~m, 2H, NH-CH2).
17 l ~ 3~
EXAMPLE 4 - Preparation of 5-~3-hydroxy-2-~hydroxYmethyl)-N-~2-
hydroxYethyl)propionamido]-N-~2-hYdroxyethyl)-N'-~2,3-dihydroxypropyl)-
2,4,6-triiodo-isophthalamide.
a) Preparation of 5-~2-isoProPYl-1,3-dioxane-5-carboxamido)-2,4,6-
triiodo-3-N'-~2-acetoxyethYl) carbamoyl-benzoyl chloride.
5.36 g of 2-isopropyl-1,3-dioxane-S-carboxylic acid ~0.0308 mole)
are dissolved in 18 ml of DMAC. The reaction mixture is cooled to 5~C
and 2.55 ml ~0.0350 mole) of SOC12 are added dropwise such that the
temperature remains below 15~C. When the addition is complete, the
reaction mixture is left for 3 hours at ambient temperature.
Then 6.0 g ~0.00906 mole) of 5-amino-2,4,6-triiodo-3-~N-2-
acetoxyethyl) carbamoyl-benzoyl chloride are added. The reaction
mixture is maintained under argon for 4 days at ambient temperature.
The DMAC is removed in a vacuum. The oil obtained is taken up in
ethyl acetate; the organic phase is washed with water, dried and concen-
trated to dryness. The product is crystallized from 100 ml of ether.
After being filtered off and dried, 1.8 g of product are obtained in a
yield of 24%.
TLC ~silica 60F 254):
ethyl acetate/petroleum ether 80/20 - Rf = 0.83.
b) PreParation of 5-~2-isopropyl-1,3-dioxane-5-carboxamido)-2,4,6-
triiodo-N-~2-acetoxyethyl)-N'-~2,3-dihydroxypropyl)isophthalamide
1 g ~0.00122 mole) of the product obtained in a is dissolved in 100
ml of DMAC, then 0.26 ml of triethylamine ~0.00189 mole) are added.
0.18 g ~0.00196 mole) of 3-amino-1,2-propanediol are added dropwi~e to
the reaction mixture. After the addition is complete, the reaction
mixture is stirred under argon at ambient temperature for 24 hours.
The triethylamine hydrochloride is filtered off, then the DhAC is
evaporated. The oil thus obtained is crystallized from 20 ml of ether.
After being filtered off and dried, 0.8 g of product are obtained
in a yield of 75.5%.
TLC ~silica 60F254) : CHCl 3/ MeOH / NH ~H 53 / 30 / 10
Rf = 0.77.
c) Preparation of 5-[3-hydroxy-2-~hydroxYmethyl)-N-~2-
hydroxyethyl)propionamido]-N-~2-hydroxYethYl)-N'-~2,3-dihYdroxypropyl)-
2,4,6-triiodoisophthalamide
18 1 3 3 9 ~ b 5
0.4 9 (0.000458 mole) of the product obtalned in b are dissolved in
0.7 ml of ethylene glycol and 0.69 ml (0.00275 mole) of a 4 N solution
of sodium methylate. To this solution is added 0.18 ml ~0.00275 mole)
of chloroethanol. The reaction mixture is heated at 400C for 5 hours.
0.34 ml of 4 N 60dium methylate and 0.1 ml of chloroethanol are added.
The mixture is maintalned at 400C overnight.
The pH of the reaction mixture is brought to 7.00 by the additlon
of dilute hydrochloric acid.
The ethylene glycol is evaporated in a vacuum.
The residue after distillation is taken up in 6 ml of vater and 5
ml of concentrated hydrochloric acid, then gtirred overnight at ambient
temperature.
The reaction mixture is concentrated then purified by preparative
HPLC (RP 18, elution Yith vater). After evaporation and drying, 0.1 9
of product is obtained in an overall yield ~alkylation - purification~
of 27~/
TLC ~silica 60F254) : CH2C12/methanol/7/3 - Rf = 0.33.
HPLC : column of HypersiI C8 5~ 25 cm
Buffer: 0.01 n HaH2P04 / heOH : 95/5
Purity : 95%
TM
H HhR ~Bruker - 200 nHz) ln DnSO : in conformity vlth the expected
structure.
'~