Note: Descriptions are shown in the official language in which they were submitted.
~40 ~2~L
Specification:
The synthesis of peptide aminoalkylamides and peptide
hydrazides by the solid-phase method
s
The introduction of an aminoalkylamide into the C-terminal
end of a biologically active peptide has in some cases had
beneficial effects on the metabolic stability and activity
(EP-A 179 332). The preparation of the peptides modified in
this way has made use of the classical coupling of frasments
in solution.
In the solid-phase synthesis of peptides (see Patchornik,
Cohen in Perspectives in Peptide Chemistry, pages 118 - 1Z8
(Karger, Basle 1981)) the reactive chains are often not
grafted directly onto the synthetic resin material, but
are bonded to the carrier material by what are called spacers
or links. The literature (for example Atherton, Sheppard
in Perspectives in Peptide Chemistry, pages 101 - 117 (Karger,
Basle 1981)) discloses, for example, reagents for introducing
such spacers (called "linkage agents") which have the formulae
YI, VII and VIII.
HOCH2 -~CH2- CH2- C02H HOCH~ OCH2 - CO2H HOCH2~ C02H
(VI) (VII) (VIII)
New linkage agents which allow direct construction, by solid
phase synthesis, of peptides modified by C-terminal amino-
alkylamide or hydrazide have been found.
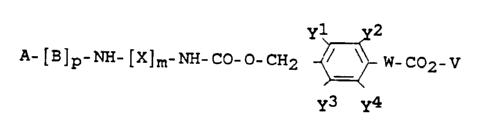
Thus the present invention relates to compounds of the tor-
mula I
yl y2
( I ) A- [B]p-NH- [X]m-NH-CO-O-CH2 _~_ W-C02-V
y3--y4
*
-- 2
in which 1 3 ~ O ~ 2 1
A denotes hydrogen or an amino protective group which
is labile to bases or labile to weak acids,
B represents identical or different amino acid resi-
dues,
X denotes (C1-C12)-alkylene or (C6-C10)-arYl-(c1-c12)
alkylene,
y1, y2~ y3 and Y4 are identical or different and denote
hydrogen, methyl, methoxy or nitro, at Least one of
these radicals denoting hydrogen,
V denotes hydrogen or a carboxyl protective group,
~ denotes -tCH2]n- or ~0-[CH2]n-,
m is 0 or 1,
n is an integer from 0 to 6, and
15 p is an integer from 0 to 5.
Preferred compounds of the formula I are those in which p is
0, 1 or 2, in particular 0, and/or in which m is 1.
X is preferably ~CCH2]q~~ it being possible for q to be 1 -
12, preferably 1 - 8.
Preferably at least 2, in particular at least 3, of the radi-
ls y1 y2 y3 and Y4 denote hydrogen-
25Protective groups which are labile to bases or labile to
weak acids are, in particular, urethane protective groups,
such as Fmoc, Ddz, Bpoc, Msc, Peoc, Pse and Tse, preferaDly
Fmoc (see, for example, Hubbuch, Kontakte (Merck) 1979, No.
30 3, pages 14 - 23).
B represents the residue of an amino acid, preferably of an
~-amino acid, which, if chiral, can be present in the D or
L form. Preference is given to residues of naturally occur-
35 ring amino acids, their enantiomers, homologs, derivativesand simple metabolites (see, for example, ~unsch et al.,
Houben-~eyl 15/1 and 2, Stuttgart, Thieme 1974). Thus, for
example, the following are suitable:
_ 3 - 134~'121
Aad, Abu, ~Abu, ABz, 2ABz, ~Aca, Ach, Acp, Adpd, Ahb, Aib,
B Aib, ALa, ~ Ala, a Ala, ALg, ALL, Ama, Amt, Ape, Apm, Apr,
Arg, Asn, Asp, Asu, Aze, Azi, Bai, Bph, Can, Cit, Cys, Cyta,
Daad, Dab, Dadd, Dap, Dapm, Dasu, Djen, Dpa, Dtc, Fel, Gln,
Glu, Gly, Guv, hCys, His, hSer, Hyl, Hyp, 3Hyp, Ile, Ise,
Iva, Kyn, Lant, Lcn, Leu, Lsg, Lys, ~ Lys, a Lys, Met, Mim,
Min, nArg, Nle, Nva, Oly, Orn, Pan, Pec, Pen, Phe, Phg, Pic,
Pro, a Pro, Pse, Pya, Pyr, Pza, Qin, Ros, Sar, Sec, Sem, Ser,
Thi, ~ Thi, Thr, Thy, Thx, Tia, Tle, Tly, Trp, Trta, Tyr, Val
and the residues of the corresponding enantiomeric D-amino
acids.
Functional groups in the side chains of the said amino acid
residues can be in protected form. Suitable protective
groups are described in Hubbuch, Kontakte (Merck) 1979, No.
3, pages 14 - 23, and in Bullesbach, Kontakte (Merck) 198Q,
No. 1, pages 23 - 35. The preferred groups are those which
are stable to bases and weak acids and can be eliminated
using strong acids.
Alkylene can be straight-chain or branched. Examples of
definitions of (C6-C10)-aryl are phenyl, tolyl or naph-
thyl; phenyl is preferred.
A carboxyl protective group V is, for example, (C1-C6)-aLkyL
or (C7-C11)-araLkyL; preference is given to methyL, ethyL,
tert.butyL, benzyL and modified benzyl, such as p-chloro-,
p-bromo-, p-nitro- and p-methoxybenzyl and the nitrogen ana-
log picolyl. In the wider sense, such protective groups in-
clude activated ester groups such as ONSu, OBt, OObt or p-
nitrophenoxy.
The invention also relates to a process for the preparation
of the compounds of the formula I, which comprises
a) reaction of a compound of the formula II
Y ~ y2
R-co-o-cH2~/ ~rW-co2-v
~ ==~ (II)
y3 y4
~ 4 ~ 13~04~1
in which
R represents a leaving group which can be detached
nucleophilically,
V represents a carboxyl protective group, and
~, y1~ y2, y3 and Y4 are as defined i~ claim 1, wit~ a com-
pound of the formula III
A- [B]p-NH- [X]m~NH2 ( I I I )
in which A represents an amino protective group which
is labile to bases or labile to weak acids, and B, X,
p and m are as defined in claim 1, and elimination of,
where appropriate, one or both of the protective
groups A and/or V in the resulting protected compound
of the formula I, with the formation of the free NH2
and/or C02H group(s), the preferred processes being
those in which V is selectively eliminated, for exam-
ple by reductive cleavage with Zn/glacial acetic acid,
or
b) reaction of a compound of the formula I in which A
denotes hydrogen, and B, X, y1, y2~ y3, y4, V, W, m,
n and p are as defined in claim 1, with a compound Ot
the formula IV
A-[B]s p-OH ~ (IV)
in which A, B and p are as defined above, but A does
not denote hydrogen, or its active ester, halide or
azide, and, if V is not hydrogen, where appropriate
elimination of a carboxyl protective group V with the
formation of the carboxyl group.
A leaving group R which can be detached nucleophilically
is, for example, halogen, such as chlorine, bromine and
iodine, or activated aryloxy, such as p-nitrophenoxy.
The reaction of a compound of the formula II with a com-
pound of the formula III is preferably carried out in an
13~0~1
aprotic solvent such as, for example, THF, DMF, CHCl3 or
CH2Cl2, advantageously in the presence of a base such as,
for example, a tertiary amine, for example ethyl triiso-
propylamine, triethylamine or pyridine, the addition of
an acylation catalyst such as, for example, DMAP, HOObt
or HOBt having an advantageous effect, at a temperature
between 0~C and the boiling point of the reaction mixt~re,
preferably between 0~C and 40~C.
Compounds of the formula I (A = hydrogen) are reacted
with compounds of the formula IV, their active ester, ha-
lide or azide preferably in an organic solvent, such as
DMF, advantageously in the presence of a base such as,
for example, a tert.amine, at a temperature between -10~C
and the boiling point of the reaction mixture, preferably
at room temperature. Examples of suitable active esters
are the ONSu, OBt, OObt and p-nitrophenoxy compounds.
Preferred halogen derivatives are the chlorides. Pyridin-
ium perchlorate can be added to improve the solubility.
Compounds of the formula II are prepared by, for example,
reacting esters of the formula IX
yl y2
HO-CH2 ~ ~ W-C02-V (IX)
y3 y4
in which y1~ y2~ y3, y4, W and V are as defined above,
but V does not denote hydrogen, with phosgene or phosgene
derivatives such as, for example, nitrophenyl chloroform-
ate in an aprotic polar solvent, for example THF or DMF,
mixed with a tert. base, for example a tert. amine such
as pyridine, preferably in the ratio 1 : 1, at a tempera-
ture between -40~C and room temperature, preferably be-
tween -20~C and 0~C.
The invention also relates to the use of a compound of the
formula I, in which V denotes hydrogen and A does not
- 6 - 13404~1
denote hydrogen, in the solid-phase synthesis of compounds
of the formula V
P-NH- [X]m-NH2 (V)
s
in which P represents a peptide residue comprising
q < p+1 ~-amino acids, and X, m and p are as defined
above, and to a process for the preparation of a
peptide of the formula V, in which P, X, m and p are
as defined above, by solid-phase synthesis, which
comprises coupling a compound of the formula I, in
which A does not denote hydrogen, and V represents
hydrogen, to a resin, eliminating the protective
group A, stepwise coupling on q-p ~-amino acids
which are, where appropriate, in the form of their
activated derivatives and which have been temporarily
protected by amino protective groups which are labile
to bases or labile to weak acids and, after construction
is complete, liberating the peptide from the resin by
treatment with a moderately strong to strong acid, the
temporarily introduced side-chain protective groups being
eliminated again at the same time or, by suitable measures,
subsequent thereto.
If necessary to prevent side reactions or for the synthe-
sis of specific peptides, the functional groups in the
side chain of amino acids are additionally protected by
suitable protective groups (see, for example, T.W. Greene,
"Protective Groups in Organic Syntheses", New York, John
Wiley & Sons, 1981), those primarily used being Arg(Tos),
Arg(Mts), Arg(Mtr), Asp(OBzl), Asp(OBut), Cys(4-Me8zl),
Cys(Acm), Cys(SBut), Glu(OBzl), Glu(OBut), His(Tos),
His(Fmoc), His(Dnp), His(Trt), Lys(Cl-2), Lys(Boc), Met(0),
Ser(Bzl), Ser(But), Thr(Bzl), Thr(But).
The resins used as carrier material are commercially
available. BHA and MBHA resins are preferred.
The peptide of the formula V is then cleaved off by
treatment with the moderateLy strong to strong acids1 340 42
customarily used in peptide synthesis (for example
trifluoracetic acid and HF), there being cleavage of
the urethane protective group contained in the spacer.
It is possible to use as coupling reagent for the com-
pound of the formula I (V = H) and the other amino acid
derivatives all possible activating reagents used in pep-
tide synthesis, see, for example, Houben-Weyl, Methoden
der organischen Chemie (Methods of organic chemistry),
volume 15/2, but in particular carbodiimides such as, for
example, N,N'-dicyclohexylcarbodiimide, N,N'-diisopropyl-
carbodiimide or N-ethyl-N'-(3-dimethylaminopropyl)carbo-
diimide. This coupling can be carried out directly by
addition of the amino acid derivative with the activating
reagent and, where appropriate, an additive which supp-
resses racemization, such as, for example, 1-hydroxybenzo-
triazole (HOBt) (W. Konig, R Geiger, Chem. Ber. 102, 708
(1970)) or 3-hydroxy-4-oxo-3,4-dihydroxybenzotriazine
(HOOBt) (W. Konig, R. Geiger, Chem. Ber. 103, 2054 (1970))
to the resin, or the preactivation of the amino acid deri-
vative can be carried out separately as the symmetric an-
hydride or HOBt or HOObt ester, and the solution of the
activated species in a suitable solvent can be added to
the peptide-resin which is ready for coupling.
The coupling and activation of the compound of the formula
I (V = H) and of the amino acid derivatives with one of
the abovementioned activating reagents can be carried out
in dimethylformamide or methylene chloride or a mixture
of the two. The activated amino acid derivative is nor-
mally used in a 1.5- to 4-fold excess. In cases where in-
complete coupling occurs, the coupling reaction is re-
peated, without previously carrying out the deblocking of
the ~-amino group of the peptide-resin which is neces-
sary for coupling the next amino acid in the sequence.
Successful completion of the coupling reaction can be
checked using the ninhydrin reaction as described, for
-
- 8 - 1340~421
example, by E. Kaiser et al. Anal. Biochem. 34, 595 (1970).
The synthesis can also be carried out automatically, for
example using an Applied Biosystems model 430A peptide
S synthesizer, it being possible to use either the synthe-
sis programs provided by the apparatus manufacturer or
those constructed by the user himself. The latter are
particularly employed when amino acid derivatives protec-
ted with the Fmoc group are used.
When the peptide amides are cleaved off the resin with
hydrogen fluoride and trifluoroacetic acid, it is custo-
mary to add substances as cation traps, such as phenol,
cresol, thiocresol, thioanisole, anisole, ethanedithiol,
dimethyl sulfide, ethyl methyl sulfide or a mixture of
two or more of these auxiliaries. In this connection,
the trifluoroacetic acid can also be used diluted by sui~-
able solvents such as, for example, methylene chloride.
2û Abbreviations used:
Fmoc 9-fluorenylmethyloxycarbonyl
Ddz a,~-dimethyl-3,5-dimethoxybenzyloxycarbonyl
Bpoc 2-[4-biphenylyl]-2-propyloxycarbonyl
Msc Methylsulfonylethyloxycarbonyl
Peoc pyridylethyloxycarbonyl
Pse phenylsulfonylethyloxycarbonyl
Tse tolylsulfonylethyloxycarbonyl
HONSu N-hydroxysuccinimide
HOBt 1-hydroxybenzotriazole
HOObt 3-hydroxy-4-oxo-3,4-dihydrobenzotriazine
THF tetrahydrofuran
DMF dimethylformamide
DMAP dimethylaminopyridine
The examples which follow serve to illustrate the present
invention without intending to confine it to them.
1340421
Example 1: Methyl 4- hydroxyethylphenoxyacetate
18.2 9 of 4-hydroxymethylphenoxyacetic acid are dissolved
together with 17.1 ml of N,N-diisopropylethylamine in
S 50 ml of DMF, and then 6.1 ml of methyl iodide are added
to the stirred solution. The mixture warms slightly dur-
ing this. The reaction is complete after 3 h. The sol-
vent is removed in vacuo. The residue is taken up in
ether, and the solution is extracted once with 0.5 N
hydrochloric acid. The aqueous phase is then extracted
three times with ether, and the combined ether phases are
washed with aqueous sodium bicarbonate solution and con-
centrated. The residue is dissolved in ethyl acetate and
filtered through a short silica gel column. The pale
yellowish oil which is obtained after concentration crys-
tallizes on being left to stand.
NMR and mass spectrum are consistent with the indicated
structure.
Example 2: Fmoc-NH-(cH2)4-NH-co-o-cH2 ~ 0-CH2-COOCH3
9.8 9 of methyl 4-hydroxymethylphenoxyacetate are dis-
solved in 200 ml of dry CH2Cl2, and then 10.1 9 of p-nitro-
phenyl chloroformate and 7 ml of triethylamine are added.The mixture is boiled under reflux for about 6 h, until
the precursor has completely reacted. Then a suspension
of 15.5 9 of Fmoc-NH-(CH2)4-NH2 (prepared by reaction
of Boc-NH-(CH2)4-NH2 with Fmoc-ONSu followed by elimin-
ation of Boc) in 100 ml of dry CH2Cl2 and a further 7 mlof triethylamine are added, and the mixture is boiled
under reflux. After the reaction is complete, the solvent
is removed in vacuo, and the residue is digested with
ether and filtered off ~ith suction. The residue on the
filter is washed with aqueous 1 N Na2CO3 solution and then
with hot water, and is dried under high vacuum in a desic-
cator.
Melting point 122-124~C, NMR and mass spectrum are
-
consistent with the indicated structure. 134G~21
Exa-Ple 3: H2N-(CH2)4-NH-CO-0 CH2 ~ 0-CH2-COOH
5.2 9 of the ester obtained as in Example Z are suspended
in 100 ml of methanol, and 6 equivalents of an aqueous
1 N NaOH solution are added. After the reaction is com-
plete, the pH is adjusted to 3 with aqueous 1 N HCl, and
the methanol is removed in vacuo. The precipitate is fil-
tered off with suction, washed with a little H20, and thendigested in ether and again filtered with suction.
Melting point starts at 196~C (decomposition), NMR and mass
spectrum are consistent with the indicated structure.
Exa-ple 4: Fmoc-Phe-NH-(CH2)4-NH-CO-O-CH2 ~ 0-CH2-COOH
1.5 9 of the product obtained as in Example 3 are suspen-
ded in 50 ml of dry DMF. Then, successively, 0.9 9 of
ZO pyridinium perchlorate (to improve the solubility) and
2.6 9 of Fmoc-Phe-OObt and 0.5 ml of triethylamine are
added. The mixture is stirred at room temperature. After
the reaction is complete, the solvent is removed in vacuo,
and the residue is partitioned between ethyl acetate and
HzO. The aqueous phase is extracted once more with ethyl
acetate, and the combined organic phases are dried and
concentrated. The residue is digested with a little CHCl3
and is filtered off with suction. The residue on the fil-
ter is washed with a little ether and is dried.
Melting point starts at 140~C (decomposition), NMR and mass
spectrum are consistent with the indicated structure.
Exa-ple 5: Fmoc-Phe-NH-(CH2)4-NH-CO-O-CH2 ~ 0-CH2-CO-
(4-eethylbenzhydryla-ine resin)
1.4 9 of the Fmoc-phenylalanine spacer acid obtained as
in Example 4 are dissolved together with 350 mg of HOBt
in 40 ml dry DMF, and the solution is added to 3.66 9 of
4-methylbenzhydrylamine resin (Nova Biochem, loading
0.4 mmol/g). Then 0.6 ml of diisopropylcarbodiimide 3s 21
added, and the reaction is allowed to go to completion,
mixing continuously. After the reaction is complete, the
product is filtered off with suction, washed with DMF,
isopropanoL, CHzClz and tert.-butyl methyl ether and is
dried under high vacuum. Loading according to elemental
analysis (nitrogen determination): 0.3 mmol/g.
Example 6: Synthesis of [des-Tyr24, des-Arg23]-r-
atriopeptinIII-(4-amino)butyla-ide
The peptide synthesis is carried out on 1 9 of the above-
mentioned resin using OOBt esters of Fmoc-amino acids with
an Applied Biosystems model 430A automatic peptide synthe-
sizer and synthesis programs modified by ourselves.
For this, 1 mmol of each of the appropriate amino acidderivatives is weighed into the cartridges supplied by
the manufacturer, Fmoc-Arg(Mtr)-OH, Fmoc-Asn-OH and Fmoc-
Gln-OH are weighed together with 1.5 mmol of HOBt into
the cartridges. These amino acids are preactivated direct-
ly in the cartridges by dissolving in 4 ml of DMF and
adding 2 ml of a 0.55 M solution of diisopropylcarbodi-
imide in DMF. The HOObt esters are dissolved in 6 ml of
DMF and then pumped, in the same way as the amino acids
arginine, asparagine and glutamine which are preactivated
in situ, onto the resin which has previously been deblocked
with 20% piperidine in DMF. The amino acids which are
activated in situ are coupled twice.
After the synthesis is complete, the peptide butylamide
is cleaved off the resin, simultaneously removing the
side-chain protective groups with trifluoroacetic acid
which contains th-ioanisole and m-cresol as cation traps.
The residue obtained after removal of the trifluoroacetic
acid in vacuo is subjected to digestion with ethyl ace-
tate and centrifugation several times. The remaining
crude peptide is treated with tributylphosphine in tri-
fluoroethanol to remove the cysteine protective group.
- 12 - 1340421
After the solvent has been removed, the residue is again
digested with ethyl acetate and centrifuged. The reduced
crude peptide is immediately oxidized with iodine in 80%
strength aqueous acetic acid, the excess I2 is removed with
ascorbic acid, and the reaction mixture is concentrated
to a small volume and then salt is removed on ~ Sephadex
G25 with aqueous 1 N acetic acid. The fractions contain-
ing the pure peptide are combined and freeze-dried.
According to amino acid analysis, the amino acid composi-
tion of the peptide corresponds to the indicated formula.
Example 7: Phenacyl 4-hydroxymethylphenoxyacetate
182 9 of 4-hydroxymethylphenoxyacetic acid and 199 9 of
~-bromoacetophenone are dissolved in 600 ml of dry DMF,
and then, at 0~C, 138 ml of triethylamine are rapidly
added dropwise. The mixture is allowed to reach room
temperature, and is stirred overnight. The DMF solution
is poured into 3.5 l of water, and the aqueous phase is
extracted with ethyl acetate. The organic phase is washed
with water, dried over sodium sulfate and concentrated.
The product precipitates out on evaporation. It is fil-
tered off with suction, washed with ethyl acetate/n-hexane
1:1 and dried under high vacuum.
Melting point: 94-95~C, NMR is consistent with the indi-
cated structure.
Example 8: O2N ~ O-CO-O-CH2 ~ O-CH2-CO2-CH2-CO
30 9 of phenacyl 4-hydroxymethylphenoxyacetate are dis-
solved, under protective gas, in 500 ml of a 1:1 mixture
of THF and pyridine, and the solution is cooled to -20~C.
Then 21 9 of p-nitrophenyl chloroformate dissolved in
100 ml of THF are added dropwise. After the mixture has
been stirred at this temperature for 30 minutes, it is
allowed to warm to 0~C and stirred into 1 l of a half-
saturated aqueous NaCl solution at 0~C, and the mixture is
then stirred for 30 minutes. The precipitate is filtered
1~0421
- 13 -
off with suction, washed with ice-water and, after drying,
triturated with n-hexane.
Melting point: 142-145~C, NMR is consistent with the indi-
cated structure.
ExaepLe 9:
oc Phe- NH- ( CH2 ) 8- NH- CO- O- CH2 ~ O- CH2- C02- CH2- CO
9.3 9 of the compound prepared in Example 8, 12.25 9 of
Fmoc-Phe-NH-(CH2)g-NH2 trifluoroacetate and 3.26 9 of HOObt
are placed as the solid substances in a flask, and then
a mixture of 2.58 9 of ethyl diisopropylamine in 100 ml
of dry DMF is poured over. The mixture is then stirred
at 40~C for 3.5 hours and then stirred into 500 ml of hal~-
saturated aqueous NaCl solution. The precipitate whichseparates out is filtered off with suction, washed with
ice-water and, after drying, triturated with ether/ethyl
acetate.
Melting point: 147-150~C, NMR and MS are consistent
with the indicated formula.
The following compounds (Examples 10 to 14) are prepared
in analogy to Example 9:
Exa~ple 10:
Fmoc- Phe- NH- ( CH2 ) 4- NH- CO- O- CH2 ~ O- CH2- C~2 - CH2- CO ~
Melting point 144-147~C, NMR and MS correspond to the indi-
cated formula.
Exa-ple 11:
Fmoc- Al a- NH- ( CH2 ) 8- NH- CO- O- CH2 ~ O- CH2 - C02- CH2 - Co~3
Melting point 179-181~C, NMR and MS correspond to the indi-
cated formula.
- 14 -
Exa~ple 12: 134~1
E~oc- NH- ( CH2 ) 8- NH- CO- O- CH2 ~ O- CH2- C02- CH2- CO ~=~
S Melting point 144-145~C, NMR and MS correspond to the indi-
cated formula.
Formu~a 13:
1 ~ E moc-NH- ( CH2 ) 6-NH- CO- O- CH2 ~ O- CH2- C~2- CH2- CO~
Melting point 172-175~C, NMR corresponds to the indicated
formula.
ExampLe 14:
Fmoc-NEI-(CH2)4-NH-CO-O-CH2 ~ O-CH2-C02-CH2-CO ~ )
Melting point 165-166~C, NMR corresponds to the indicated
formuLa.
Exa~pLe 15:
E'moc- Phe- NH- ( CH2 ) 8- NH- CO- O- CH2 ~ ~- CH2- C02H
8 . 4 9 Fmoc- Phe- NH- ( CH2 ) 8- NH- CO- O- CH2 ~ O- CH2 - C~2 - CH2 - CO
are suspended in a mixture of 150 ml of glacial acetic
acid and 50 ml of dichloromethane, and 12 9 of zinc pow-
der which has previously been activated by washing with
1 N HCl and dry ethanol are added in portions. After a
few minutes, the suspension becomes more viscous and dif-
ficult to stir, while there is slight evolution of heat.
Hence a further 80 ml of glacial acetic acid and 50 ml of
dichloromethane are added, and stirring is continued over-
night. The mixture is then filtered with suction through
a filter with a clarifying layer, washing with glacial
acetic acid and dichloromethane. The filtrate is
-
- 15 - 13~0~21
concentrated, and the oil which remains as residue is
- taken up in a little dichloromethane and stirred with
ethyl acetate and ether. The precipitated product is
filtered off with suction and dried under high vacuum.
Melting point: decomposition above 160~C, NMR and MS are
consistent with the indicated formula.
In addition, the compounds of Examples 16 to 18 are pre-
pared by the method described in Example 15:
1 0
Exacple 16:
E~oc- Phe- NH- ( CH2 ) 4- NH- CO- O- CH2~ O- CH2- C~2H
Melting point: decomposition above 150~C, NMR and MS are
consistent with the indicated formula.
Exa~pLe 17:
2 0 Fmoc- Phe- NH- ( CH2 ) 8- NH- CO- O- CH2 ~o- CH2 - C~2H
Melting point: decomposition above 160~C, NMR and MS are
consistent with the indicated formula.
25 Exanple 18:
Fmoc- NH- ( CH2 ) 8- NH- CO- O- CH2-~30- CH2- C02H
Melting point: decomposition above 154~C, NMR and MS are
consistent with the indicated formula.
Exa~pLe 19:
Fmoc- NH- ( CH2 ) 6- NH- CO- O- CH2 ~ O- CH2 - C02CH3
The synthesis is carried out in analogy to Example 2.
Melting point: 115-118~C, NMR and MS are consistent wi~h
the indicated formula.
,, . .. . . . ~
- 16 -
ExampLe 20: 1340421
NH2- (CH2)6-NH-CO-O-cH2~0-cH2-co2H
was prepared by the method described in Example 3.
Melting point: 184-187~C decomposition, NMR and MS are con-
sistent with the indicated formula.
Exa~pLe 21:
1 0
Fmoc- Phe- NH- ( CH2 ) 6- NH- CO- O- CH2~0- CH2 - C02H
The synthesis is carried out in analogy to Example 4.
Melting point: deco~position above 120~C, NMR and MS are
consistent with the indicated formula.