Note: Descriptions are shown in the official language in which they were submitted.
Z[)(32045
,
4- 17289/-~
Process for the preparation of optically active acvloxvazetidinones
The present invention relates to a process for the preparation of 3,4-trans-disubstituted
azetidinones of the formula
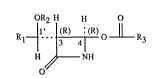
OR2 H H o (I)
R~ ~o ll R3
H .~ N~l
o
in which R1 is hydrogen or lower alkyl, R2 is hydrogen or a hydroxyl protective group R~'
and R3 is substituted or unsubstituted phenyl or lower alkyl.
The general definitions used within the scope of this invention preferably have the
following meanings:
Groups or compounds marked "lower" preferably contain up to and including 7, preferably
up to and including 4 carbon atoms.
As lower alkyl, Rl is preferably methyl and also ethyl, n-propyl, isopropyl, n-butyl,
isobutyl or tert-butyl. Rl is primarily methyl and also hydrogen.
Hydroxyl protective groups R2~, including their introduction and removal, ale described,
for example, in "Protective Groups in Organic Chemistry", Plenum Press, London and
New York 1973, and also in "Protective Gro~lps in Organic Chemistry", Wiley, New York
1974. A hydroxyl protective group is, for example, the acyl radical of a carboxylic acid,
for example lower alkanoyl which is substituted by halogen, such as fluorine or chlorine,
for example 2,2-dichloroacetyl or 2~2,2-trifluoroacetyl, or a benzoic acid which is unsub-
stituted or substituted, for example, by nitro, halogen, such as chlorine, or lower alkoxy,
such as methoxy, for example benzoyl or 4-nitrobenzoyl, 3,5-dinitrobenzoyl, 4-chloro-
~QC~4~
benzoyl or 4-methoxybellzoyl, and also the acyl radical of a carbonic acid lower alkyl or
lower alkenyl half-ester which is unsubstituted or substituted, for example, by halogen,
sllch as chlorine, or by phenyl which can contain substituents, suCIl as 4-nitrophenyl, for
example 2,2,2-trichloroethoxycarbonyl, allyloxycarbonyl, benzyloxycarbonyl or 4-nitro-
benzyloxycarbonyl, or the acyl radical of an organic sulfonic acid, for example lower
alkanesulfonyl, such as methanesulfonyl, or arylsulfonyl, such as toluenesulfonyl.
Hydroxyl protective groups are also 2-oxacycloalkyl or 2-thiacycloalkyl having 5 or 6 ring
atoms, for example 2-tetrahydrofuryl or 2-tetrahydropyranyl, and also a thia analogue
thereof, 1-lower alkyloxy-lower alkyl, for example methoxymethyl or 1-ethoxyethyl, or
silyl which is trisubstituted, in particular, by lower alkyl, aryl-lower alkyl and/or aryloxy,
for example tri-lower alkylsilyl, such as trimethylsilyl, triethylsilyl, dimethyl-(2-ethyl-2-
propyl)-silyl, diethyl-(2-ethyl-2-propyl)-silyl, dimethyl-(tert-butyl)-silyl or dimethyl-(2,3-
dimethyl-2-butyl)-silyl, diaryl-(lower alkyl)-silyl, for example diphenyl-(tert-butyl)-silyl,
aryl-(di-lower alkyl)-silyl, for example tert-butylmethylphenylsilyl, and di-lower alkyl-
(lower alkylphenoxy)-silyl, for example dimethyl-(2,4,6-tri-tert-butylphenoxy)-silyl. Pro-
tective groups R2' which are particularly preferred because of their stability to bases and
hydrolysis are trialkylsilyl, in particular trimethylsilyl, tert-butyldimethylsilyl or
dimethyl-(2,3-dimethyl-2-butyl)-silyl.
As substituted phenyl, R3 is phenyl which is substituted, preferably monosubstituted to
trisubstituted, for example by lower alkoxy, for example methoxy, lower alkyl, for
example methyl and/or halogen, for example chlorine, for example 4-methoxyphenyl,
4-nitrophenyl, 2-, 3- or 4-chlorophenyl or 4-tolyl. As lower alkyl, R3 is, for example,
ethyl, n-propyl, isopropyl, n-butyl, isobutyl or tert-butyl, especially methyl. R3 is
preferably phenyl and methyl.
The process according to tlle invention comprises
(a) treating a compound of the formula
OR2 H H 0 (
R ~ ¦ (S) ~ (S) ¦¦ R
H .~N~
o C1~21r R4
~o~
-3-
in which R4 is substituted or unsubstituted phenyl or lower allcyl with a peracid, or
decarboxylating by acyloxylation a compound of the formula
OR2 H H (IIb~
Rl~ - ~ COOH
H.~ N\
O CH2COOH
with the introduction of the radicals R3-C(=O)- or R4-C(=O)-, (b) replacing the radical of
the formula R4-C(=O)- by hydrogen in a compound of the formula
OR2 H H O (III)
R1~---~o 1I R3
H.~ N\
o CH2--o ~r R4
obtainable as in (a), and (c) removing, in a compound of the forrnula
OR2 H rl O (IV)
Rl~- - ~O ~I R3
rl ~ N\
O CH2--OH
thus obtainable, the hydroxymethyl group in the 1-position, if appropriate after conversion
into a forrnyl group, and, if desired, in a compound of the forrnula I thus obtainable in
which R2 is a protective group R2', converting the protected hydroxyl group into the free
hydroxyl group, andJor, if desired, in a compound of the for nula I thus obtainable in
which R2 is hydrogen, converting the free hydroxyl group into a protected hydroxyl group.
Preferred starting materials are those of the formulae Ila or IIb in which Rl is hydrogen or
methyl, R2 is hydrogen or a hydroxyl protective group R2', such as dimethyltert-butylsilyl
or dimethyl-(2,2-dimethyl-2-butyl)-silyl, and R3 and R4, preferably both identical, are
phenyl or methyl.
~o~zo~
The treatment of the starting material of the form-lla IIa with a peracid is preferably
caTried out by the Baey~r-Villiger reaction, in which organic or inor~Tanic peracids or salts
thereof, such as alkali metal or alkaline earth metal salts, for example sodium, potassiu}n
or magnesium salts, can be employed. Organic peracids are, in particular, the corres-
ponding percarboxylic acids, inter alia lower alkanepercarboxylic acids which are unsub-
stituted or substiîuted, for example, by halogen, such as peracetic acid or trifluoroperacetic
acid or trihalogenoiminoperacetic acid, for example trichloroiminoperacetic acid or tri-
fluoroiminoperacetic acid, aromatic peracids which are unsubstituted or substituted, for
example, by nitro and/or halogen, such as perbenzoic acid, 4-nitroperbenzoic acid,
3-chloroperbenzoic acid, 3,5-dinitroperbenzoic acid or monoperphthalic acid, or a salt
thereof, for example magnesium monoperphthalate. Examples of inorganic peracids are
peroxosulfuric acids, such as peroxosulfuric acid. Peracetic acid, 3-chloroperbenzoic acid
and monperphthalic acid are preferred.
The peracids can also be formed in situ, for example from the corresponding acids or acid
salts thereof and hydrogen peroxide. The iminopercarboxylic acids in particular are
usually prepared in situ, for example from the corresponding nitriles and hydrogen
peroxide, preferably in the presence of a weakly acid buffer, for example potassium
hydrogen phosphate.
The reaction is usually carried out in a suitable solvent, such as a halogenated hydro-
carbon, for example methylene chloride or chloroform, an ester, such as ethyl acetate, an
acid, such as acetic acid, for example in the form of glacial acetic acid, or an alcohol, such
as a lower alkanol, for example ethanol, at temperatures between about 0 and abowt
100C, if appropriate in an inert gas atmosphere and if appropriate under presswre, for
example up to about 10 bar.
The acyloxylating decarboxylation whicll takes place with the introduction of thc acyl
groups R3-C(=O)- or R4-C(=O)- of a compoun~l of the formwla llb is carried out in a
manner known per se, for example by trcatment with lead(lV) acylates or by means of
anodic oxidation in the presence of a carboxylic acid. The acyl radicals in this lead(lV)
acylate correspond to the radicals R3-C(=O)- or R4-C(=O)-, and the carboxylic acid
present in the anodic oxidation corresponds to the acids of the formula R3-C(=O)-OH or
R4-C(=O)-OH; i.e in an intermediate product of the formula III which is prepared from a
starting material of the formula lIb, the radicals R3 and R4 are identical.
4S
Suitable lead(lV) acylates are the corresponding lower alkanoates wllich are unsubstituted
or substitutcd, for examyle by halogen, such as fluorine, for example lead tetraacetate,
lead tetrapropionate or lead tetra-(trifluoroacetate), or benzoates which are unsubstituted
or substituted, for example, by lower alkyl, such as methyl, lower alkoxy, such as
Methoxy, and/or halogen, such as fluorine or chlorine, for example lead tetrabenzoate.
These reagents are used in a customary manner, for example in the presence of a suitable
solvent, such as an inert, polar solvent, for example tetrahydrofuran, dioxane, acetic acid,
dimethylformamide, N-methylpyrTolidone or hexamethylphosphoric triamide, or a mixture
of solvents, if necessary with the addition of an alkali metal acylate, for example sodium
acetate or potassium acetate, and/or an amine, for example pyridine or lutidine, with
cooling or heating, for example at temperatures from about 20C to about 80C and/or in
an inert gas atmosphere, for example an argon atmosphere.
The anodic oxidation is usually carTied out in a mono-cell or in a divided cell having a
mechanical diaphragm and being composed, for example, of porous clay, glass or
polyvinyl chloride, or having an ion exchanger diaphragm, for example a diaphragm
obtainable under the trade name Nafion~), and electrodes made of noble metal, such as
platinum, titanium alloys, for example titanium-iridium or titanium-tantalum, and also
nickel, lead dioxide, glassy carbon and/or graphite. The reaction is carried out in the
presence of an acylate donor, such as an acetate donor, for example in acetic acid or in a
mixture of acetic acid and an inert organic solvent, such as acetonitrile, dioxane or
dimethylformamide, with the addition of an amine, such as a tri-lower alkylamine, for
example triethylamine or tri-n-butylamine, or pyridine, and/or an alkali metal acylate, for
example sodium acetate or potassium acetate, and, if desired, an additional so-called
conductive salt which increases the conductivity, for example a lithium, sodium,potassium or tetraalkylammonium salt, for example the colTesponding tetrafluoborate.
Suitable curTent densities for tl-e electrolysis are betwcen about 10 alld about 400 mA/cm2,
for example about 40 mA/cm2. Thc rcaction temperature is between about 0 and about
50C, preferably between room tempcr.ltllre and about 30C.
Both the reaction with a lead(IV) acyl.lte an(l the anodic oxidation in the presence of an
acylate donor preferably result in a compound of the formula III in which the
1-hydroxyalkyl group at the C(3) ring carbon atom ~nd the acyloxy group at the C(4) ring
carbon atom are arTanged in the trans-position to one another, i.e. result in a diastereomer
having the (3R,4R) or the (3S,4S) configuration
2~2~4~
The selective removal of the acyl group R4-C(=O)- in an intermediate of the formula III
with the formation of the hydroxyl group is preferably carried out by means of enzymatic
ester cleavage. Known esterases which are particularly suitable for cleaving esterified
primary hydroxyl groups are used in this reaction. Thus, for example, for removing an
acetyl group in an interrnediate of the formula III in which R4 is methyl, it is preferred to
use the esterase from Nocardia mediterranei, an organism used for the preparation of
rifamycin (Schar el al., Appl. Microbiol. Biotechnol. Volume 27, page 451 (1988)). A
benzoyloxy group in an intermediate of the formula III in which R4 is phenyl is cleaved,
for example, by means of cholesterol esterase (Sigma C9530 from porcine pancreas,
which is commercially available).
The reaction is carried out in a manner known per se, preferably under neutral conditions,
for example in a suitable aqueous phosphate buffer solution, and at temperatures from
about 10C to about 30C, usually at room temperature.
The removal of the hydroxymethyl group which is a substituent on the ring nitrogen atom
can be effected in a manner known per se, for example by hydrolysis or by treatment with
an acid reagent, preferably after prior oxidation of the hydroxymethyl group to the formyl
group.
The latter reaction can be carried out, for example, by treating a compound of the formula
IV with a suitable oxidizing metal compound, such as an appropriate chromiurn(VI)
compound, for example chromium(VI) oxide, or an analogous oxidizing agent, preferably
under the conditions of the Killiani oxidation, for example using an approximately 8 N
solution of chromium trioxide in aqueous sulfuric acid.
The removal of the N-hydroxymethyl or, in palticular, the N-formyl group can preferably
be carried out under acid conditions, using primarily inorganic acids, such as mineral
acids, for example hydrochloric acid or sulfuric acid, usually in the form of dilute,
aqueous solutions, and also organic carboxylic or sulfonic acids, for example p-toluene-
sulfonic acid. The N-formyl group, which can, for example, also be removed by treatment
with an acid aluminium oxide, for example under the conditions of chromatography, can
also be removed under mild basic conditions, for example in the presence of an alkali
metal hydroxide, carbonate or bicarbonate or alkaline earth metal hydroxide, carbonate or
bicarbonate, which is usually present in aqueous solution.
4S
The hydrolytic removal of the N-hydroxymethyl or N-formyl group is usually carried out
at room temperature or at a slightly elevated temperature, for example between about
lS~C and about 50C, it being possible, in a given case, additionally to use organic
solvents, such as suitable alcohols, for example water-soluble lower alkanols, or ether
compounds.
In a compound of the formula I which can be obtained in accordance with the invention,
and also in an intermediate at any stage of the process according to the invention, the
removal of a protective group R2 can be effected in a manner known per se, for example
by solvolysis, such as acidolysis, reduction or oxidation.
An acyl group R2 can be removed under acid or mild alkaline conditions, depending on
the meaning of R2, for example by treatment with acids, such as formic or trifluoroacetic
acid, or with bases, such as an alkali metal hydroxide, carbonate or bicarbonate, for
example sodium bicarbonate, a bicyclic amidine, such as 1,5-diazabicyclo[5.4.0]undec-
5-ene, or an alkali metal halide.
Certain acyl radicals of half-esters of carbonic acid, such as halogeno-lower alkoxy-
carbonyl and benzyloxycarbonyl which is unsubstituted or substituted by nitro, can be re-
moved and replaced by hydrogen by reduction, for example by means of catalytic hydro-
genation in the presence of a suitable hydrogenation catalyst, for example platinum oxide
or palladium, or by tretment with chemical reducing agents, such as zinc in the presence of
aqueous acetic acid.
The removal of a lower alkenyloxycarbonyl group in which lower alkenyl is especially
allyl can preferably be effected by treatment with a lower alkenyl group acceptor in the
presence of a suitable catalyst, swch as a mctallic phosphine compound, for example
tetrakistriphenylphosphine palladium, if appropriate in the presence of triphenylphosphine.
Examples of suitable acceptors for lower alkenyl groups, such as the allyl group, are
amines, in particular sterically hindered amines, for example tert-butylamine, and also
tri-lower alkylamines, for example triethylamine, morpholine or thiomorpholine, and also
aliphatic or cycloaliphatic ~-dicarbonyl compounds, for example acetylacetone, ethyl
acetoacetate or, preferably, dimedone, and also lower alkanecarboxylic acids, for example
acetic acid or propionic acid. The reaction is usually carried out by treating the compound
of the formula I having a suitably protected hydroxyl group with 1.5 to 10 molar
Z~ 4S
- 8 -
equivalents of the lower alkenyl group acceptor in the presence of 2 to 10 mol %, in
particulLIr S to 8 mol % (relative to the st~uting compound), of the catalyst, in particular
tetrakistriphenylphosphine palladium catalyst, if appropriate in the presence of up to 50
mol % of triphenylphosphine, in an inert solvent, such as an ether, for example dioxane or
especially tetrahydrofuran, a halogenated hydrocarbon, for example methylene chloride, a
lower alkanol, for example ethanol, an ester, for example ethyl acetate, or a mixture of
such solvents, at temperatures from about 0C to about 40C, preferably at about 2V to
about 2~C, and, if necessary, in an inert gas atmosphere, such as an atmosphere of
nitrogen or argon.
A 2-oxacycloalkyl or 2-thiacycloalkyl group R2 can be removed by treatment with an
acid, such as a mineral acid, for example hydrochloric or sulfuric acid.
A silyl group R2 which is substituted as indicated can be removed in the presence of
suitable, preferably polar, solvents, such as solvents containing hydroxyl groups, if
appropriate under acid conditions or, in particular, by treatment with a salt of hydrofluoric
acid which affords fluoride anions, such as an alkali metal fluoride, for example sodium
fluoride, if appropriate in the presence of a macrocyclic polyether ("crown ether"), or with
the fluoride of an organic quaternary base, such as a tetra-lower alkylammonium fluoride,
for example tetrabutylammonium fluoride, if appropriate in the presence of an acid
reagent.
In a compound of the formula I obtainLlble in accordLmce with the invention, and also in an
intermediate at any stage of the process, in which R2 is hydrogen, the free hydroxyl group
can be protected in a manner known per se, it being possible, if necessary, to protect
temporarily an unsubstituted ring nitrogen atom. The protective group can be introduced,
for example, by treatment with a suitable halogenosilane, for example a tri-lower alkyl
halogeno silane, such as trimethylchlorosilane or dimethyltert-butylchlorosilane, or a
suitable silazane compound, such as hexamethyl(lisilazane, or by treatment with a reactive
derivative of an acid whose acyl radical acts as a hydroxyl protective group, for example
by reaction with an anhydride, for example trifluoroacetic anhydride, a mixed anhydride,
for example an acid halide, such as 2,2-dichloroacetyl chloride, or a mixed anhydride of a
carbonic acid half-ester, for example 2,2,2-trichloroethyl, allyl, phenyl or p-nitrophenyl
chloroformate, or by reaction with 1,2-dihydrofuran or 1,2-dihydropyran in the presence of
an acid, for example p-toluenesulfonic acid.
2~ 5
The present invention preferably relates to a process for the preparation of compounds of
the formula I in which Rl is hydrogen or, in particular, methyl, R2 is hydrogen or a
hydroxyl protective group R2, such as the acyl radical of a carbonic acid half-ester, for
example 2,2,2-trichloroethoxycarbonyl, allyloxycarbonyl, benzyloxycarbonyl or p-nitro-
benzyloxycarbonyl, the acyl radical of a substituted or unsubstituted benzoic acid, for
example 4-nitrobenzoyl or 3,5-dinitrobenzoyl, or, in particular, tri-lower alkylsilyl, for
example trimethylsilyl, tert-butyldimethylsilyl, or dimethyl-(2,3-dimethyl-2-butyl)-silyl,
and R3 is phenyl or methyl, and in which the 1'-C atom of the side chain can have the S
configuration or preferably the R configuration, if Rl is methyl.
The invention relates primarily to a process for the preparation of
(3R,4R, l 'R)-4-acetoxy-3-( 1 '-hydroxyethyl)-azetidin-2-one or
(3R,4R,l'R)-4-benzoyloxy-3-(1'-hydroxyethyl)-azetidin-2-one and their 1'-O-tri-lower
alkylsilyl derivatives, such as 1'-O-tert-butyldimethylsilyl or
1'-O-dimethyl-(2,3-dimethyl-2-butyl)-silyl derivatives.
Compounds of the formula I, particularly those in which Rl is hydrogen or methyl, R2 is
hydrogen or a hydroxyl protective group R2 and R3 is methyl or phenyl are valuable
intermediates which can be processed further in a manner known per se to give penem or
carbapenem compounds which have an antibiotic activity. Further processing is carried
out analogously to that of known azetidinone compounds of the formula I in which R3 is
methyl, for example as described by T. Hayashi et al., Chem. Pharm. Bull. 29, 3158, 1981,
or in European Published Patent Applications No. 42,026 (for penems) or No. 78,026 (for
carbapenems).
The starting materials and intermediates used in the process according to the invention are
in part novel. They can be prepared in a manner known per se.
Thus, for example, compounds of the formul~ II (a and b) can be obtained by d) cyclizing
the carbanion of a compound of the formula
2~ 45
- 10-
H o (V)
3 2 11
Rl--CH--C(R) C112--C--R3
0~ N
O CH2--C--R4
o
in which R3' has the meaning of R3 or is etherified hydroxyl, and R4' has the meaning of
R4 or is etherified hydroxyl, R3' and R4' either having the meanings of R3 and R4,
respectively, or both being etherified hydroxyl, and in which, if Rl is lower alkyl, the 3-C
atom has the R configuration or the S configuration, or the carbanion of a compound of the
formula
\ 3 2 / (VI)
~ CH--C(R) CH2--C--R3
R20 l I l
/~ N
CH2--C--R4
in which R2 is a hydroxyl protective group which cannot be removed under the conditions
of the cyclization process and X is a nucleofugic leaving group and, if Rl is lower alkyl,
the 3-C atom has the R configuration or the S configuration, or e) by isomerizing a
cis-compound of the formula
loR2 11 o (VII)
R~l Cllllll~llllll C-- R3
¦ (S) (R)
H
~ N~
C112--C--R~,
o
and, if desired, converting a compound of the formula II obtainable in accordance with the
invention into another compound of the formula II.
3~
In a starting material of the formula VI a hydroxyl protective group R2 which cannot be
removed under the conditions of the cyclization process is one of the acyl groups men-
tioned under R~2 of a substituted carboxylic or sulfonic acid, for example 2,2,2-trifluoro-
acetyl, and also the acyl radical of a carbonic acid half-ester, for example p-nitrobenzyl-
oxycarbonyl, or 2-tetrahydrofuryl or 2-tetrahydropyranyl, and also 1-lower alkoxy-lower
alkyl, for example methoxymethyl or 1-ethoxyethyl, or substituted or unsubstituted aroyl,
for example 4-nitrobenzoyl or 3,5-dinitrobenzoyl. A suitable nucleofugic leaving group X
is preferably reactive esteri~led hydroxyl, for example halogen, for example chlorine,
bromine or iodine, lower alkanoyloxy, such as acetoxy, arylsulfonyloxy, for example
phenylsulfonyloxy or p-toluenesulfonyloxy, or lower alkylsulfonyloxy, for example
methanesulfonyloxy.
An etherified hydroxyl group R3 or R4 is, in particular, a hydroxyl group which is
etherified with an aliphatic, aromatic or araliphatic radical, such as lower alkoxy, for
example methoxy, ethoxy, n-propoxy, isopropoxy or primarily tert-butoxy, aryloxy, for
example phenoxy, or aryl-lower alkoxy, for example benzyloxy.
In carbanions of compounds of the formula V or VI the negative charge on the carbon
atom is located in the a-position relative to the ~roup of the formula -CO- R3; i.e. the
corresponding compound has the partial formula ~N--C~ --CO--R3 . Carbanion com-
pounds of this type are prepared in situ by treating a compound of the formula V or VI in a
suitable solvent with a base which forms carbanions, in the course of which the cyclization
of process d) takes place. Bases of this type are, inter alia, alkali metal bases, for example
sodium hydroxide or carbonate, potassium hydroxide or carbonate or lithium hydroxide or
carbonate, or organic alkali metal compounds, for example lower alkyllithium, such as
n-butyllithium or tert-hutyllithium, alkali metal amides, for example lithium diisopropyl-
amide or di-tert-butylamide, and especially alkali metal amides in which two hydrogell
atoms have been replaced by org.lnic silyl grollps, for examplc trimethylsilyl ~roups, for
example lithium bis-(trimethylsilyl)-amicle, sodium bis-(trimethylsilyl)-amide or
potassium bis-(trimethylsilyl)-amide, inter alia lithium hexamethyldisihlzide, and also
suitable organic nitrogen bases, such as cyclic amidines, for example 1,5-diazabicyclo-
[4.3.0]non-5-ene or 1,5-diazabicycloLS.4.0]undec-5-ene, and also fluorine compounds
which release fluoride ions in an aprotic, organic solvent and which are particularly suit-
able for the preparation of carbanions of compounds of the formula V, preferably tetra-
lower alkylammonium fluorides, especially tetra-n-butylamonium fluoride.
~Qlal2U4~
- 12-
lf a suitable silyl reagellt, for example lithiurn hexamethyldisilazide, is used for the
cyclization of a compound of the formula V or VI, under certain circumstances, for
example if the reaction is carried out carefully, a compound of the formula Ila in which R2
is a silyl protective group, for example trimethylsilyl, can be obtained directly.
Examples of solvents suitable for the formation of carbanions are halogenated or non-
halogenated hydrocarbons, such as benzene, toluene, methylene chloride or chloroform,
ethers, such as dioxane, tetrahydrofuran or dimethoxyethane, amides, such as dimethyl-
formamide or hexamethylphosphoric triamide, sulfoxides, such as dimethyl sulfoxide, or
nitriles, such as acetonitrile, or mixtures thereof. The reaction can also be carried out
under phase transfer conditions, for example in a system consisting of methylene chloride
and 50 % aqueous sodium hydroxide solution in the presence of a phase transfer catalyst,
such as benzyltriethylammonium chloride or tetrabutylammonium bisulfate. The reaction
temperature, which depends, inter alia, on the choice of the base used, is between about
-80C and about 80C, the reaction being preferably carried out under an inert gas
atmosphere, for example an atmosphere of argon or nitrogen.
Under the conditions of the process, an inversion of the configuration at the 2-C atom of
the starting material takes place, so that a 3S compound of the formula IIa is formed from
a 2R compound of the formula V or VI; the configuration of the 1'-C atom in the side
chain, if Rl is lower alkyl, remains unchanged. For example, 3S azetidinones of the for-
mula IIa are obtained from R-glycidamides of the formula V, or (3S,l'R)-3-hydroxyethyl-
azetidinones of the formula IIa are obtained from (2R,3R)-2,3-epoxybutyramides of the
formula V.
The isomerization of a compound of the formula VII (process variant e) can be carried out
in a suitable solvent or solvent mixture in the presence of a base. Suitable solvents and
bases, and also the reaction conditions, are the sarne as those which can be used for the
cyclization process d). It is preferred to employ aprotic dipoLu solvents, such as dirnethyl-
formamide, N-methylpyrrolidone or dimethyl sulfoxide, and, as bases, in particular
potassium carbonate or 1,5-diazabicyclo[5.4.0~undec-5-ene, and it is also possible to carry
out the reaction under the phase transfer conditions described.
Starting materials of the formula Ilb can be obtained in a manner known per se, for
example by solvolysis, from diesters of the formula IIb obtainable in accordance with the
Q~
- 13-
invention. The corresponding diester compounds can be converted into the free
dicarboxylic acid compounds, for example by means of acid or basic hydrolysis, a di-tert-
bu~yl ester ( R3 = R4~ = tert-butoxy) can, for example, be converted by treatment with
dilute, for example 1 N, sodium hydroxide solution into a lower alkanol, for example
methanol or ethanol, or with trifluoroacetic acid with the application of heat.
The cis-compounds of the formula VII can be formed as byp}oducts in the course of the
cyclization reaction (d) and can be separated off from the products of the des*ed con-
figuration in a customary manner, for example by means of chromatography.
Compounds of the forrnula V can be obtained in a manner known per se, for example by
reacting an epoxy acid compound of the formula
H (VIII)
Rl--CH--C
\ o / C
// ~
0 0~1
or a suitable salt thereof with an amine compound of the formula
(IX)
Il
~ CH2--C-- R3
HN
CH2--C--R4
o
The reaction can be carried out under conditions which are analogolls to those of the
process described in detail bclow for the preparation of compounds of the formula VIa.
The compounds of the formula VIII are known or c~m be prepared by processes known per
se.
The starting materials of the formula V are preferably prepared in situ under the
abovementioned carbanion-forming conditions and with a Walden inversion at the 2-C
- 14-
atom from compounds of the formula
Rl~ 3 H X O (VIa),
~ CH 2 C(S) CH2--C--R3
R20 D~ N
o CH2--C-- R4
o
in which R~'is hydrogen or a hydroxyl protective gTOup which can be removed under the
conditions of epoxide forrnation and in which, if Rl is lower alkyl, the 3-C atom has the R
configuration or the S configuration, the elements of a compound of the formula R~ -X
being removed.
In a compound of the formula VIa a hydroxyl protective gTOUp R~' which can be removed
under the conditions of epoxide formation is a tri-lower alkylsilyl group, for example
dimethyltert-butylsilyl or trimethylsilyl. X is preferably halogen, for example chlorine or
bromine, lower alkanesulfonyloxy, for example methanesulfonyloxy, or arylsulfonyloxy,
for example p-toluenesulfonyloxy.
The removal of R~'-xin which R~" iS hydrogen is effected in the course of the tTeatment
with one of the carbanion-forming bases mentioned, for example sodium hydroxide or
carbonate or potassium hydroxide or carbonate, preferably with an amidine, for example
1,5-diazabicyclo[5.4.0]undec-5-ene or with a tetra-lower alkylammonium fluoride, for
example dehydrated tetra-ll-butylammonium fluoride, for example under the reaction
conditions described by Djerassi et al., "Steroid Reactions", page 606 (Holden Day, San
Francisco, 1963). If R~' iS, for example, a tri-lower alkylsilyl group which can be
removed under the conditions of epoxide formation, it is preferred to use a tetra-lower
alkylammonium fluoride.
The reaction of a compound of tlle formula VIa to give an epoxy compound of the formula
V is preferably carried out in an inert solvent or solvent mixture, usually anhydrous, if
necessary with cooling or heating, for example within a temperature range from about
-40C to about +100C, preferably from about -10C to about +50C, and/or under an inert
gas atmosphere, for example an atmosphere of nitrogen.
2[11~2
- 15 -
In the course of this process thc conf igur;ltion at the 2-C atom is inverted, whereas the
configuration of the 3-C atom, if Rl is lower alkyl, is retained. Thus the (2R,3R) com-
pounds of the formula V are formed from 2S-halogeno-3R-hydroxy compounds of the
formula Vla and the (2R,3S) compounds of the formula V are formed from 2S-halogeno-
3S-hydroxy compounds.
After ;t has been prepared, the epoxide of the formula V can, if desired, be isolated by the
removal of R~'-x from a compound of the formula YIa by employing the amidine used as
the reagent in approximately equimolar amounts.
The above compounds of the formula VIa can be prepared, for example, by reacting a
compound of the formula
OR2' H X (X)
21~
R ~--C--C(S)
// \
O OH
or a reactive functional derivative thereof with an amine of the formula IX. This reaction
can be carried out in a manner known per se, for example by subjecting the two reactants
to a condensation reaction with one another in the presence of a condensing agent, and de-
hydrating the free acid, for example in the presence of carbodiimides, such as diethyl-
carbodiimide, diisopropylcarbodiimide or dicyclohexylcarbodiimide, or di-(N-hetero-
cyclyl)-carbonyl compounds, such .IS carbonyl(liimid.lzole, or 1,2-oxazolium compounds,
such as 2-ethyl-5-phenyl-1,2-oxazolium 3-sulfonate or 2-tert-butyl-5-methyl-1,2-oxazolium perchlorate, which are uscd in the prcsence of an inert solvent, such as di-
methylformamide, if necessary at a slightly redllce(l or elevaled temperature. Reactive
functional derivatives of Ll carboxylic acid of tlle formula X are primarily an}ly(lrides, in
particular mixed anhydrides, such as the corresponding carboxylic acid chloride or
bromide, which are preferably formed in situ by reacting a salt, for example the dicyclo-
hexylamine salt of epoxy acids of the formula VIII with suitable halogenating agents, such
as phosphorus oxychloride or thionyl chloride, in particular with oxalyl chloride, and
which are, if appropriate, reacted with the amine of the formula IX in the presence of a
base, for example N-methylmorpholine.
2~20~5
- 16-
The comyounds of the formula VI can be prepared analogously, starting from compounds
of the formula IX and from compounds of the formula
OR2 H (Xa)
-~x
Rl--C C
H C
// \
O OH
Compounds of the formula X and Xa in which X is halogen are known. They can be
prepared, for example, in a manner known per se from L-serine or L-threonine or the
D-isomers thereof by diazotizing the amino group with a nitrite salt, for example
potassium nitrite, in the presence of a hydrohalic acid.
If L-serine is used and compounds of the formula VI are the starting material, it is
possible to obtain compounds of the formula I in which the carbon in the 3-position of the
azetidinone ring has the R-configuration. If L-threonine is used, compounds of the formula
I in which the C atom in the 3-position of the azetidinone ring has the R-configuration and
the 1'-C atom of the hydroxyethyl side chain (Rl is methyl) has the R-configuration. If it
is intended to prepare compounds of the formula I in which the 3-C atom has the R-con-
figuration and the 1'-C atom, for example, of a 1-hydroxyethyl side chain has the S-con-
figuration, allothreonine is used as the starting material. In all the reactions described the
configuration of the C atom which in compounds of the formula I corresponds to the 1'-C
atom of a 1-OR2-lower alkyl side chain having more than one carbon atom in the lower
alkyl moiety remains unchanged.
Amines of the formula IX, in particular the amines having symmetrical substituents, are
known or, if novel, can be prepared in a manner known per se. Thus, for example, a
compound of the formula IX in which R3 and R4' are tert-butoxy can be prepared by the
action of isobutylene on the corresponding acid, usually in dioxane in the presence of
95-98 % sulfuric acid at room temperature. A preferred process for the preparation of
compounds of the formula IX in which Ri and R4' represent phenyl consists in reacting a
phenacylamine hydrochloride with a phenacyl chloride or phenacyl bromide in the
presence of a suitable base, for example triethylamine, and in methylene chloride.
2i~021~4~
The present invention also embraces embodiments of the process in which the starting
material is a compound obtainable at any preliminary seage of the process and the sub-
sequent process stages are carried out. It also embraces a combination of process steps, for
example starting from one of the compounds of the formula VI to X to give a compound
of the formula I or starting from one of the compounds of the formulae IX or X or Xa to
give a compound of the formula VI or IVa, respectively.
The process according to the invention has the advantage that the compounds of the
forrnula I can be prepared stereospecifically in a high yield and in an economical manner,
and that it is possible to use the readily accessible and cheap symmetrically substituted
amines of the formula IX, whose substituents serve on the one hand for the cyclization to
give the ~-lactam compounds and on the other hand as a temporary N-protective group
which can be removed under mild conclitions.
The following examples serve to illustrate the invention. Temperatures are in degrees
centigrade. The specific angles of rotation are [ a ] 20 values. Columns packed with Merck
silica gel 60 can be used for the separation by chromatography in preparative work-up.
Example 1
1.1. A solution of 0.866 g of (3S,4S,l'R)-4-benzoyl-3-(1'-hydroxyethyl)-1-
phenacylazetidin- 2-one and 5.7 g of 3-chloroperbenzoic acid in 30 ml of methylene
chloride is stirred for 8 hours at room temperature. The white suspension is diluted with
methylene chloride and washed successively with a 5 % aqueous solution of sodium thio-
sulfate/potassium iodide, water, aqueous sodium bicarbonate solution and water, dried and
evaporated under a water pump vacuum. The crude product is purified by means of
medium pressure chromatography (solvent: a 2:3 mixture of hexane and ethyl acetate).
After the main fraction has been recrystallized from methylene chloride/diethyl ether,
(3R,4R,l'R)-4-benzoyloxy-1-benzoyloxymethyl-3-(1'-hydroxy-ethyl)-azetidin-2-one is
obtained, melting point 136- 138.
1.2. 0.06 g of imidazole and 0.088 g of tert-butyldimethylchlorosilane are added to a solu-
tion of 0.18 g of (3R,4R,l'R)-4-benzoyloxy-1-benzoyloxymethyl-3-(1'-hydroxyethyl)-
azetidin-2-one in 1 ml of dimethylformamide and the mixture is stirred for 24 hours at
room temperature. A further 0.015 g of imidazole and 0.022 g of tert-butyldimethylchloro-
- 18 -
silane are added to the reaction mixture, which is stirred for a further 6 hours at room tem-
perature and then discharged onto ice and a saturated aqueous solution of sodium bicarbo-
nate, the product is taken up in diethyl ether, and the organic phase is washed once each
with aqueous sodium bicarbonate solution, 1 % aqueous citric acid, water, aqueous
sodium bicarbonate solution and water. The aqueous phases are washed again with diethyl
ether; the combined organic extracts are dried with sodium sulfate and evaporated in a
water pump vacuum. The crude product is dissolved in a 3:2 mixture of hexane and ethyl
acetate and is filtered through silica gel 60. Recrystallization from methylene
chloride/diethyl ether gives pure (3R,4R,l'R)-4-benzoyloxy-1-benzoyloxymethyl-3-[1'-
(tert-butyl dimethylsilyloxy)-ethyl]-azetidin-2-one, melting point 109- 110;
[ ] D = -14.1 (c = 0.545 % in chloroform).
1.3. 0.03 g (30 U) of cholesterol esterase (Sigma C 9530, from porcine pancreas) is
dissolved in 33 ml of phosphate buffer pH 7 in a S0 ml round-bottomed flask controlled by
thermostat at 30, and 0.05 g of (3R,4R,l'R)-4-benzoyloxy-1-benzoyloxymethyl-3-Ll'-
(tert-butyldimethylsilyloxy)-ethyl]-azetidin-2-one, dissolved in 6 ml of acetone, is added
in portions in the course of 10 hours. The reaction is followed by thin layer chromato-
graphy (solvent: a 1: 1 mixture of ethyl acetate and hexane) and by high pressure liquid
chromatography (solvent: an 85:15 mixture of acetonitrile and water). After 20 hours, the
reaction mixture is extracted with six times 100 ml each of chloroform. The organic ex-
tract gives a slightly yellow oil which, according to high pressure liquid chromatography,
still contains about 10% of starting material. Chromatography over silica gel (mobile
phase: a 4:1 mixture of methylene chloride and ethyl acetate) gives (3R,4R,l'R)-4-
benzoyloxy- 1-hydroxy-methyl-3-[1 '-(tert-butyldimethylsilyloxy)-ethyl]-azeti~in-2-one,
which, after recrystallization from methylene chloride/diethyl ether/hexane, melts at
83-85; [ a ] D = +68.5 (c = 0.672 % in chloroform).
1.4. 0.21 ml of 8 N chromosulfuric acid (Killiani mixture) is added to a solution of 0.16 g
of (3R,4R,l'R)-4-benzoyloxy-1-hydroxymcthyl-3-[1'-(tert-blltyldimethylsilyloxy)-cthyll-
azetidin-2-one in 10 ml of methylene chloride, and the mixture is stirred for S hours at
room temperature. Excess oxidizing agent is destroyed by adding 1 ml of isopropanol, and
the reaction mixture is applied directly to a columll of 80 g of aluminium oxide (Alox II,
acid) and eluted with ethyl acetate. This gives virtually pure (3R,4R,l'R)-4-benzoyloxy-
3-[1 '-(tert-butyldimethylsilyloxy)-ethyl]-azetidin-2-one, melting point 125- 126; [ a ] D =
71.1 (c = 0.85 % in chloroform), after recrystallization from diethyl ether/hexane.
4~
- 19-
~3R,4R,l'R)-4-Benzoyloxy-3-(1'-hydroxyethyl)-azetidin-2-one, melting point 145-147
~from methylene chloride/diethyl ether) can be obtained from the (3R,4R,l'R)-4-benzoyl-
oxy-3-[1'-(tert-butylsimethylsilyloxy)-ethyl]-azetidin-2-one by treatment with tetrabutyl-
ammonium fluoride trihydrate in tetrahydrofuran and acetic acid.
The starting material can be prepared as follows:
1.5. A solution of 2.7 ml of oxalyl chloride in 8.5 ml of methylene chloride is added
dropwise, in the course of 15 minutes, to a mixture of 6.86 g of dimethylformamide and 40
ml of methylene chloride, cooled to -25. The white suspension is stirred for a further 20
minutes at -25, and a solution of 9.87 g of the dicyclohexylammonium salt of (2R,3R)-
2,3-epoxybutyric acid in 40 ml of methylene chloride is then added in the course of 10
minutes. The reaction mixture is stirred for a further 20 minutes at the same temperature
and 8.7 ml of triethylamine are then added, followed, in portions, by 7.29 g of diphenacyl-
amine hydrochloride. The cooling bath is removed and the reaction mixture is stirred for
21 hours at room temperature. The yellow suspension is then filtered through a sintered
glass suction filter and the residue is washed exhaustively with methylene chloride. The
filtrate is diluted with methylene chloride and washed successively with water,5 %
aqueous citric acid, saturated aqueous sodium bicarbonate solution and water. The
aqueous phases are subsequently extracted twice with methylene chloride, and the com-
bined organic phases are dried over sodium sulfate and evaporated under a water pump
vacuum. The resulting crude product is purified by flash chromatography over 750 g of
silica gel (Merck 9385; mobile ph.lse: a 2: l mixture of hexane and ethyl acetate), and
(2R,3R)-2,3-epoxy-N,N-bis-phenacylbutyramide is obtained in the form of a yellowish oil.
1.6. A solution of 3.48 g of (2R,3R)-2,3-epoxy-N,N-bis-phenacylbutyramide in 100 ml of
tetrahydrofuran is cooled with stirring to -15, and 20.6 ml of a 0.5-molar solution of
lithium hexamethyldisil~zide in tetrallydrofuran is adde(l at a temperature between -10
and -15; the reaction is complete after stirring for about 4 hours within the same tempe-
rature range. The dark red reaction mixture is poured onto 100 ml of an ice cold buffer
solution (pH 6.0) and the product is extracted three times with methylene chloride. The
organic extracts are washed with saturated aqueous sodium chloride solution, dried with
sodium sulfate and evaporated under a water pump vacuum. The crude product obtained is
separated into three constituents by medium pressure chromatography over a 100-fold
amount of a silica gel preparation [LiChroprep(~) ~i 60 made by Merck AG, Darmstadt
s
- 20 -
(West Gerrnany); particle size 25-40 ~-m; mobile phase: a 4:1 mixture of hexane and ethyl
acetate]~ This gives~ as Ihe least polar *action, (3S,4S,1 'R)-4-benzoyl-3-(1 '-trimethylsilyl-
oxyethyl)-1-phenacylazetidin-2-one, as the product of medium polarity, (3S,4R,l'R)-4-
benzoyl-3-(1'-hydroxyethyl)-1-phenacylazetidin-2-one ("cis-compound") of melting point
128-129 (from methylene chloride/diethyl ether/hexane) and, finally, amorphous
(3S,4S,1 'R)-4-benzoyl-3-(1 '-hydroxyethyl)-1-phenacylazetidin-2-one~
Example 2:
2.1. 8~95 g of lead tetraacetate are added under an argon atmosphere and with stirring at
room temperature to a solution of 2~9 g of (3S,4S,l'R)-1-carboxymethyl-3-[1'-(tert-butyl-
dimethylsilyloxy )-ethyl]-2-oxoazetidin-4-carboxylic acid in 90 ml of tetrahydrofuran and
13 ml of dimethylformamide~ After 75 minutes at 25, the excess lead tetraacetate is de-
stroyed by adding 2 2 ml of ethylene glycol dropwise, the resulting white suspension is
filtered through a silica gel preparation (Hyflo), the residue on the filter is subsequently
washed thoroughly with tetrahydrofuran, and the filtrate is evaporated under a water pump
vacuum~ The oily residwe is purified by flash chromatography over 250 ml of silica gel
(silica gel 60; 0~040-0.63 mm; mobile phase: a 60:4 mixture of hexane and ethyl acetate)~
This gives (3R,4R,l'R)-4-acetoxy-1-acetoxymethyl-3-[1'-(tert-butyldi-methylsilyloxy)-
ethyl]-azetidin-2-one in the form of an oil, [ ~ ] D = +9`4 (c = 0 287 % in chloroform).
(3R,4R,1 'R)-4-Acetoxy- 1 -acetoxymethyl-3-[1 '-(dimethyl-(2,3-dimethyl-2-butyl)-silyl-
oxy)-ethyl]-azetidin-2-one is obtained analogously in amorphous form, [ ] D = +3~3
(c = 2.1 % in chloroform), starting from (3S,4S,l'R)-N-carboxymethyl-3-[1'-(dimethyl-
(2,3-dimethyl-2-b utyl)-silyloxy)-ethyll-2-oxoazetidine-4-carboxylic acid~
2.2. 3 ml of a solution of acetyl esterase (aqlleous solution containing 723 U/ml) from
Nocardia mediterranei [see: H.P~ Sch,ir, D~ Gygax, G~M~ Ramos Tombo and O~ Ghisalba,
Appl. Microbiol. Biotechnol~, volume 27, page 451 (1988)l are added to 0~92 g of(3R,4R,1 'R)-4-acetoxy- 1 acetoxymethyl-3-[1 '-(tert-butyldimethylsilyloxy)-ethyl]-
azetidin-2-one in 70 ml of phosphate buffer of pH 7~ The solution is stirred for 24 hours at
room temperature, then a further 2 ml of the above enzymc solution are added and after
two further days the mixture is extracted with methylene chloride~ This gives a crude pro-
duct which contains only small amounts of the starting materiah Purification by chromato-
graphy (silica gel; mobile phase: a 96:4 mixture of methylene chloride and methanol)
gives (3R,4R,1 'R)-4-acetoxy- 1 -hydroxymethyl-3-[1 '-(tert-butyldimethylsilyloxy)-ethyl]-
2~45
- 21 -
azetidin-2-one, which crystalli~es spontaneously and melts at 51-53.
2.3. 2 ml of 8 N chromosulfuric acid (Killiani mixture) are added to a solution of 0.317 g
(3R,4R,1 'R)-4-acetoxy-1-hydroxymethyl-3-[1 '-(tert-butyldimethylsilyloxy)-ethyl]-
a~etidin-2-one in 2 ml of methylene chloride, and the mixture is stirred for 4 hours at room
temperature. Stirring is continued for a further 24 hours after adding a further 0.25 ml of
chromosulfuric acid. Excess oxidizing agent is destroyed by adding 1 ml of isopropanol,
and the reaction mixture is applied directly to a column of 10 g of aluminium oxide
(Alox II, acid) and eluted with a 1: 1 mixture of hexane and ethyl acetate. This gives
(3R,4R,1 'R)-4-acetoxy-3-[1 '-(tert-butyldimethylsilyloxy)-ethyl]-azetidin-2-one, melting
point 107-109 (from low-boiling petroleum ether).
2.4. 0.3 g of (3R,4R,l'R)-4-acetoxy-3-[1'-(tert-butyldimethylsilyloxy)-ethyl]-
azetidin-2-one is added with stirring to a solution of 0.63 g of tetrabutylammonium
fluoride trihydrate in 10 ml of tetrahydrofuran and 0.8 ml of acetic acid, and the rnixture is
stirred for 16 hours at room temperature. The reaction mixture is discharged onto ice water
and extracted three times with methylene chloride. The combined organic extracts are
washed successively with a saturated aqueous solution of sodium bicarbonate and a satu-
rated aqueous solution of sodium chloride, dried over sodium sulfate and evaporated under
a water pump vacuum at a bath temperature of 35. The residue is recrystallized from
methylene chloride/diethyl ether/hexane and affords (3R,4R,l'R)-4-acetoxy-3-(1-hydroxy-
ethyl)-azetidin-2-one, melting point 110- 112.
The starting material can be obtained as follows:
2.5. A solution of 11.6 g of oxalyl chloride in 25 ml of methylene chloride is added drop-
wise, in the course of 15 minutes, to a mixture, coolcd to -25, of 20 g of dimethyl-
formamide and 115 ml of methylcne chloride. The white suspension is stirred for a furthcr
20 minutes at -25, and a solution of 28.5 g of the dicyclohexylammonium salt of(2R,3R,)-2,3-epoxybutyric acid in l lS ml of methylene chloride is then added in the
course of 10 minutes. I`he reaction mixture is stirred for a further 20 minutes at the same
temperature, and a solution of 17 87 g of di-tert-butyl iminodiacetate in 90 ml of methyl-
ene chloride and 9 22 ml of N-methylmorpholine are then added dropwise simultaneously
in the course of lS minutes. The cooling bath is removed and the reaction mixture is
stirred for 3 hours at room temperature. The white suspension is then filtered through a
sintered glass suction filer, the residue is washed with methylene chloride, and the filtrate
04
- 22 -
is diluted with methylene chloride and washed successively with water, 5 % aqueous citric
acid, saturated aqueous sodium bicarbonate solution and water. The aqueous phases are
subsequently extracted twice with methylene chloride, and the combined organic phases
are dried and evaporated under a water pump vacuum. The yellow-brown crude product is
purified by flash chromatography over silica gel (Merck 9385) using a 9: 1 mixture of
methylene chloride and ethyl acetate as the mobile phase. (2R,3R)-N,N-Bis-(tert-butoxy-
carbonylmethyl)-2,3-epoxybutyramide is obtained in the form of a pale yellow oil,
[ a ] D= +52 (c = 1.250 % in chloroform).
2.6. A solution of 13.18 g of (2R,3R)-N,N-bis-(tert-butoxycarbonylmethyl)-2,3-epoxy-
butyramide in 100 ml of tetrahydrofuran is cooled with stirring to -15, and 100 ml of a
0.5-molar solution of lithium hexamethyldisilazide in tetrahydrofuran is added at tem-
peratures between -15 and -10. The progress of the reaction is followed by thin layer
chromatography (solvent: a 1:1 mixture of hexane and ethyl acetate); the reaction is com-
plete after stirring for about 4 hours at the temperature indicated above. The reaction mix-
ture is diluted with methylene chloride and poured onto ice, and the organic layer is
washed successively with water, twice with 0. I N hydrochloric acid, water, saturated
aqueous sodium bicarbonate solution and water. The wash water is extracted twice with
methylene chloride, and the combined organic phases are dried over sodium sulfate and
evaporated under a water pump vacuum. After flash chromatography over 750 g of silica
gel 60 (0.040-0.063 mm, Merck; elution with a 6:4 mixture of hexane and ethyl acetate),
the crude product obtained gives (3S ,4S,1 'R)-4-tert-butoxycarbonyl- 1 -tert-butoxycar-
bonylmethyl-3-(1'-hydroxyethyl)-azetidin-2-one, which melts at 79-81;
[ ~ ] D -22.6 (c = 0.574 % in chloroform).
2.7. 2.45 g of imidazole and 3.61 g of tert-butyldimethylchlorosilane are added to a
solution of 6.59 g of (3S,4S,l'R)-4-tert-butoxyc.llbonyl-1-tert-butoxycarbonylmethyl-3-
(I'-hydroxyethyl)-azetidin-2-one in 25 ml of dimethylformamide, and the mixture is
stirred for 6 hours at room temperature. The reaction mixture is discharged onto ice and a
saturated aqueous solution of sodium bicarbon.lte and the product is taken up in diethyl
ether. I he organic phase is washed once each with aqueous sodium bicarbonate solution,
1 % aqueous citric acid, water, aqueous sodium bicarbon.lte solution and water. The
aqueous phases are extracted with diethyl ether, and the combined organic phases are
dried over sodium sulfate and evaporated under a water pump vacuum. The crude product
is dissolved in an 80:20 mixture of hexane and ethyl acetate, and the solution is filtered
through 200 g of silica gel (silica gel 60) to give (3S,4S,l'R)-4-tert-butoyxcarbonyl-1-
2~ 0~5
- 23 -
tert-butoxycarbonylmethyl-3-[1'-(tert-butyldimethylsilyloxy)-ethyl]-azetidin-2-one in the
form of an oily product, [ a ] D = -14.6 (C = 0.553 % in chloroform).
(3S,3S,l'R)-4-tert-Butoxycarbonyl-l-tert-butoxycarbonylmethyl -3-[1'-(dimethyl-
(2,3-dimethyl-2-butyl)-silyloxyethyl]-azetidin-2-one, [ a ] D = -16.8 (C = 2.5 in
chloroforrn), is obtained analogously using dimethyl-(2,3-dimethyl-2-butyl)-chlorosilane.
2.8. 29.3 ml of 1 N aqueous sodium hydroxide solution are added dropwise, with ice
cooling, to a solution, cooled to -5, of 5.4 g of (3S,4S,l'R)-4-tert-butoxycarbonyl-1-tert-
butoxycarbonylmethyl -3-[1'-(tert-butyldimethylsilyloxy)-ethyl]-azetidin-2-one in 120 ml
of ethanol. The reaction mixture is then stirred for 14 hours at 0 to 5, then concentrated to
a volume of about 30 ml under a water pump vacuum on a rotary evaporator (bath tem-
perature 30) and extracted twice with ethyl acetate. Ethyl acetate is added to the aqueous
phase which has been separated off, and the mixture is again cooled to about 0 to 5 and
adjusted to pH 2.0 by dropwise addition of orthophosphoric acid, with stirring and cooling
in ice. The phases are separated, the aqueous layer is extracted three times with ethyl
acetate, and the combined organic phases are dried with sodium sulfate and evaporated in
a water pump vacuum. The crude product obtained is crystallized from ethyl
acetate/hexane and gives (3S,4S,l'R)-1-carboxymethyl-3-[1'-(tert-butyldimethylsilyl-
oxy)-ethyl]-2-oxoazetidine-4-carboxylic acid, which melts at 134-135;
[ (~ ] D = -32.9 (c = 1.058 % in chloroform).
Thecorresponding (3S,4S,l'R)-1-carboxymethyl-3-[1'-(dimethyl-(2,3-dimethyl-2-butyl)-
silyloxy)-ethyl]-2-oxoazetidine-4-carboxylic acid is obtained analogously, starting from
(3S,4S,1 'R)-4-tert-butoxycarbonyl-1-tert-butoxycarbonylmethy -3-[1 '-(dimethyl-(2,3-di-
methyl-2-butyl)-silyloxv)-ethyl] -azetidin-2-one.