Note: Descriptions are shown in the official language in which they were submitted.
--1--
IMPROVEMENTS IN OR RELATING TO POLYMERIC
COMPOUNDS
This invention relates to novel polymeric
compounds and more particularly, but not exclusively,
is concerned with such compounds for use in radiation-
sensitive compositions for printing plate or
photoresist production.
In use, whether for printing plate or
photoresist production~ radiation-sensitive
compositions are coated on to a suitable substrate to
form a radiation sensitive plate. The coating is then
image-wise exposed to radiation so that parts of the
coating are struck by the radiation and parts are not.
The radiation-struck and non-radiation struck parts
have differing solubilities in developer liquids and
thus the more soluble parts can be selectively removed
by application of such a liquid to leave an image on
the substrate constituted by the less soluble parts.
~s is well known, radiation-sensitive ~
compounds are considered to be either positive-woxking
or negative-working depending upon whether the effect
of the radiation is to increase or decrease the
solubility of the compounds. Positive-working
compounds are commonly based on quinone-diazides and
negative-working compounds are commonly based on photo-
crosslinkable compounds (e.g. cinnamates), photo-
polymerisable compounds (e.g. (meth)acrylates) or diazo
compounds (e.g. the so-called diazo resins).
Currently, the negative-working compositions
most widely used for lithographic printing plates are
based on diazo compounds in conjunction with a suitable
binder or support resin.
Such compositions have reasonable sensitivity
to radiation and can be developed with aqueous-based
solutions, unlike compositions based on photo-
crosslinkable compounds which require a solvent-based
developer. However, their toughness (which affects the
number of prints that can be obtained from a printing
plate) is substantially less and moreover, unlike other
negative-working compositions and positive-working
compositions based on quinone-diaz;des, they do not
have the capability, after exposure and development, of
being baked (i.e. heated to temperatures of 180~C and
above for a period of a few minutes as described in GB
Patent Specification No.1513368) to increase their
toughness.
It is an object of the present invention to
provide a polymeric compound which is usable as a
binder or support resin for a radiation-sensitive
compound in a radiation-sensitive composition and which
when baked increases the toughness of the composition.
Such polymeric compounds will hereinaiter be referred
to as bakeable polymers.
According to one aspect of the present
invention, there is provided, a bakeable polymer
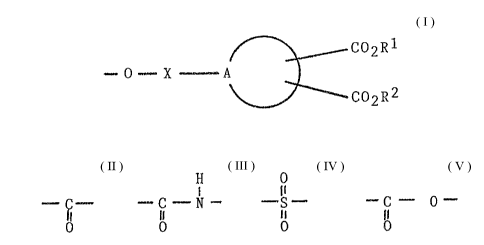
comprising groups of the formula
C02R
~ 0 ~ X - A
~ C02R2
attached to carbon atoms which are part of the backbone
of a polymer containing hydroxyl and optionally epoxide
groups where, X is
H
--C - , - C - N ~ , - ~ - or ~ C - 0-
8 o I o
A is a moiety made up of sufficient carbon atoms to
form a ring or fused ring system, and Rl and R2 are H
or lower alkyl, provided that at least one of R1 and R2
is H, the groups containing Rl and R2 being oriented
relative to one another on the ring system such that
intramolecular anhydride formation can occur through
heating.
The mechanism or chemlcal process which gives
rise to the improvement on baking is unknown but it may
be postulated to occur through two possible routes of
which both or either may occur.
In the first route 9 the groups containing Rl
and R2, cyclise upon heating to form an intramolecular
(cyclic) anhydride. This anhydride will then very
rapidly react with any aYailable hydroxyl or other
suitable functional groups. The conditions of the
baking process are such that the bulk of these groups
will be provided by the backbone polymer and the
product so produced will, therefore, be a crosslinked
polymer.
In the second route, an intermolecular
anhydride could form which gives rise directly to a
crosslinlced system. This intermolecular anhydride
could also further react with hydroxyl groups present
in the backbone polymer but, of course, no further
crosslinking would occur as the initial inter-polymer
link would perforce be broken during this reaction.
What is known, is that for effective cross-
linking to occur, groups containing Rl and R2 must be
oriented relative to one another on the ring system
such that intramolecular (cyclic) anhydride formation
could occur through heating. Thus, for example, in the
case o~ A being a benzene or alicyclic ring system, the
groups containing Rl and R2 must be adjacent each other
whilst in the case of A being a naphthalene ring system
the groups could also be located peri to each other.
The polymer containing hydroxyl, and
optionally epoxide, groups (hereinafter referred to as
the "polymeric material") may be, for example:-
1 Poly (vinyl acetate) or a copolymer
of vinyl acetate with another vinyl
monomer and which has been at least
partially saponified, or esters or
acetal derivatives of such saponifi~d
materials. Examples of such polymers
are poly (vinyl alcohols) having
between 80% and 100~ (by weight)
vinyl alcohol units and molecular
weights of approximately 50,000 and
poly ~vinyl butyrals) and other poly
(vinyl acetals) having at least 5% by
weight of vinyl alcohol units and
molecular weights in the range 20,000
to 80,000.
2 An epoxy resin which is the
condensation product of
epichlorhydrin with an aromatic
hydroxy compound such as bisphenol A
and which has a molecular weight in
the range of 900 to SO~O.
3 A poly (meth) acrylate ester and in
particular one derived ~rom 2-hydroxy
propyl methacrylate or 2-hydroxy
ethyl methacrylate.
~ A copolymer containing free hydroxyl groups
such as a styrene-allyl alcohol copolymer,
A novolak resin which is the condensation
product of a phenol or cresol with
formaldehyde.
6 A polymer derived from a vinyl phenol.
Optionally, the polymeric material may also
contain ester groups derived from aliphatic or aromatic
carboxylic acids such as octanoic acid, lauric acid or
benzoic acid.
According ~o a further aspect of the present
invention, there is provided a process for the
production of a bakeable polymer which comprises
(i) providing a polymer containing a plurality of
hydroxyl and optionally epoxide groups,
(ii) reacting some of the hydroxyl groups (and/or
epoxide groups if present) with a reagent containing
both a cyclic anhydride group and a further functional
group, which further function~l group is capable of
reacting with said hydroxyl groups (and/or epoxide
groups if present) preferentially relative to the
anhydride group, and
(iii) changing the anhydride groups in a conversion
step to carboxylic acid groups or to a carbox~lic acid
group and an ester group.
The further functional group may be an
isocyanate, a carboxylic acid or suitable derivative
thereof, a sulphonic acid or suitable derivative
thereof or a chloroEormate.
Particular reagents containing such a further
functional group are:
3-isocyanato phthalic acid anhydride;
4-isocyanato phthalic acid anhydride;
trimellitic acid anhydride chloride;
4-chlorosulphonyl 1,8~ naphthalic acid
anhydride; and
the Diels-Alder reaction product of sorbic
acid and maleic acid anhydride.
The conversion step eliminates premature crosslinking
and may be carried out by adding water or a primary
alcohol to the reaction mixture. The use of the
alcohol does, of course, produce an ester group which
confers additional oleophilicity to the polymer in the
unbaked state.
According to a further aspect of the
invention, there is provided a radiation-sensitive
2~
composition comprising a radiation-sensitive compound
and a bakeable polymer as defined above~
Whilst any radiation-sensi~ive compound may be
used in the composition, a diazo compound is
particularly suitable because, as has been mentioned
above, baking after exposure and development has no
effect on such compounds when used conventionally.
Suitable diazo compounds are those such as are
described in European Patent Specification No~308O2,
those described in GB Patent Specifications NosO1312925
and 1312926 and those produced by the condensation of
4-diazonium diphenylamine salts with formaldehyde i~e.
conventional diazo resins.
The invention, as has already been stated, is
not limited to the use of radiation sensitive
compositions based solely on diazo resins. Indeed
performance advantages are also observed, for example,
for light sensitive formulations based on o:Ligomeric
acrylates and/or combinations of oligomeric acrylates
and diazo resins.
In the following examples which illustrate the
invention, Examples 1-9 describe the preparation of
various bakeable po]ymers, Example 10 describes the
deleterious effect of allowing the anhydride function
to react in the absence of the conversion step, whilst
Example 11 describes one, prior art, unbakeable
polymer. Examples 12-20 describe the use of such
polymers in lithogr~phic prin-ting plate production.
EXAMPLE l
22 g of a polyvinyl butyral having a molecular
weight of 30,000 to 34,000 and comprising approximately
80% vinyl butyral units, up to 2% vinyl acetate units
and the remainder vinyl alcohol units, were dissolved
in 150 cm3 N-methyl-2-pyrrolidone (NMP) and cooled to
-10~C. 7.5 g pyridine were then added followed by 17 g
trimellitic anhydride chloride dissolved in 50 cm3 cold
NMP. The acid chloride addition was carried out
dropwise at such a rate as to keep the temperature in
the range -10~C to 0~C. After completing the addition,
the stirred reaction mixture was kept at -5~C to 0~C
and under anhydrous conditions for a fur-ther 16 hours.
15 g of water were then added to the reaction mixture,
keeping the temperature in the range 0~C to +10~C, and
this mixture was allowed to warm to ambient temperature
over six hours. The mixture was then heated to 45~C
and held at 45~C for a further 18 hours. The product
was isolated by drowning out into water to give a white
fibrous precipitate which was filtered, thoroughly,
water-washed in a commercial food processor, filtered
again and finally dried in a hot air oven at 32~C.
The product which weighed 25 g was readily
soluble in tetrahydrofuran ~THF) and titration of the
THF solution with 0.1 M aqueous NaOH gave an acid value
of 120. Analysis by gas phase chromatography (gpc)
showed a smooth molecular weight (mw) profile with no
evidence of crosslinking.
EXAMPLE 2
In a similar manner to that of Example 1,
reaction was effected between a polyvinyl butyral
containing 71% vinyl butyral units, 2% vinyl acetate
units and 27% vinyl alcohol units, and 4-
chlorosulphonyl-1,8-naphthalic anhydride. 20 g of the
polymeric material, 30 g of the anhydride and 8 g of
pyridine were reacted together in 150 cm3 acetonitrile,
at a temperature of 0~C for eight hours. After the
addition of 15 g water at 0~C, the reaction was
continued ror a further 16 hours at 20~C and then for
12 hours at 45~C. Isolation as in Example 1 gave 27 g
of a product having an acid value of 162. The product
was readily soluble in THF, and demonstrated a smooth
--8--
mw profile on gpc analysis.
EXAMPLE 3
The first stage of ~xample 1 was repeated but
after the ~eaction between the acid chloride and the
polymer had been completed (i.e. 16 hours at 0~C), 50
cm3 of methanol were added to the reaction mixture,
keeping the temperature in the range 0~C to tlO~C
during the addition. The temperature was then raised
to 40~C over one hour and held at 40~C for a further
six hours. Isolation was achieved by drowning out the
reaction solution into 2 litres of water, to give a
coarse fibrous white precipitate. Further processing
as before gave 25 g of a product having an acid value
of 73.
The product was again readily soluble in T~F
and analysis by gpc showed a smooth, non-crosslinked,
mw profile.
EXAMPLE 3A
In a similar manner, solketal (2,2-dimethyl-
1,3-dioxolane-4-methanol) tSO cm3) was used in place of
methanol. The product was isolated into a mixture of
150 isopropylalcohol (IPA) and water (1:4), and had an
acid value of 65.
~XAMPLE ~
22 g of the polyvinyl butyral used in Example
1, was dissolved ~n 150 cm3 of NMP at 40-50~C.
Pyridine (8 cm3) was added followed by lauroyl chloride
(16 cm3) and the reaction was stirred under anhydrous
conditions and at a temperature of 50~C, for four
hours. This mixture was then cooled to -10~C and a
further aliquot of pyridine (8 cm3) was introducedJ
followed by trimellitic anhydride chloride (15 g)
dissolved in NMP (10 cm3). The reaction was continued
at -5~C for 24 hours after which time water (20 g) and
NMP (20 cm3) were added dropwise, keeping the
temperature below 0~C. The reaction mixture was held
at 0~C for two hours, then at 20~C for a further four
hours and then at 45~C for a further 12 hours.
Isolation was achieved by drowning out the hazy
reaction solution into 2 litres of a mixture of water
(4 parts by volume) and isopropanol (1 part by volume)
the solid from which was then filtered water washed in
a food processor 9 filtered, and dried at 32~C in a hot
air oven. The product, which was obtained as a fine,
white, slightly waxy solid, was readily soluble in THF,
had an acid value of 104 and showed a smooth mw profile
on analysis by gpc.
EXAMPLE 5
30 g of styrene/allyl alcohol copolymer
containing 5.5% to 6.0% by weight hydroxyl units and
having a molecular weight of 2500 was dissolved in 200
cm3 of T~F at room temperature. A catalyst in the form
of dibutyl tin di laurate (0.1 g) was added followed by
10 g of 4-isocyanato phthalic anhydride (obtained by
Curtius rearrangement of the carbonyl azide derived
from trimellitic anhydride chloride; H. Ulrich, R.
~ichter, J. Org. Chem. 38(14), 2557-8, 1973). The
mildly exothermic reaction was held at 20~C for six
hours during which time all the isocyanate (as
determined by IR spectroscopy) had reacted. A mixture
of water (20 cm3) and THF (20 cm3) was then added and
the reaction was left a further four hours at 20~C
before warming to 50~C for 12 hours. The product was
isolated as before into IPA/water (2 litres of a 1:4
mixture). The washed and dried product weighed 36 g
and had an acid value of 118. Analysis by gpc showed a
smooth mw profile and the dried product readily
redissolved in THF.
Measurement of the anhydride content of the
reaction mixture by quantitative IR showed that the
--10--
anhydride function did not decrease signi-Eicantly prior
to addition of the water. Addition of water resulted
in a complete loss of the anhydride function.
EXAMPLE 6
21.6 g of cresol-novolak resin were dissolved
in 100 cm3 NMP and cooled to -10~C. 13.1 g pyridine
were then added, followed by 31.6 g trimellitic
anhydride chloride dissolved in 100 cm3 cold NMP. The
acid chloride was added dropwise at such a rate as to
keep the temperature in the range -5~C to 0~C. After
completing the addition, the stirred reaction mixture
was kept at -2~C to +2~C and under anhydrous conditio~s
for a further 16 hours. 20 g of methanol were then
added to the reaction mixture, keeping the temperature
lS in the range of 0~C to +10~C~ and this mixture was then
allowed to warm to ambient over six hours. The
reaction mixture was then heated to 45~C and held at
45~C for a further 18 hours. The product was isolated
by drowning out into water to give a white fibrous
precipitate which was filtered, thoroughly water washed
in a commercial food processor, filtered again and
finally dried in a hot air oven at 32~C.
The product which wei~hed 22 g was readily
soluble in THF and was found to have an acid value of
93.
EXAMPLE 7
20 g of an epoxy resin obtained by condensing
epichlorohydrin and bisphenol A and having a molecular
weight of 1400 were dissolved in 100 cm3 NMP and cooled
to -10~C. 7.2 g pyridine were then added, followed by
17.3 g trimellitic anhydride chloride dissolved in 50
cm3 cold NMP. The acid chloride solution was added
dropwise at such a rate as to keep the temperature in
the range -10~C to 0~C. After completing the addition,
the stirred reaction mixture was kept at -5~C to 0~C
and under anhydrous conditions for a further 16 hours.
15 g of water were then added to the reaction mixture,
keeping the temperature in the range 0~C to +10~C and
this mixture was then allowed to warm to ambient over
six hours. The reaction mixture was then heated to
45~C and held at 45~C for a further 1~ hours. The
product was isolated by drowning out into water to give
a white fibrous precipitate which was filtered,
thoroughly water washed in a commercial food processor,
filtered again and finally dried in a hot air oven at
32~C.
The product which weighed 22 g was readily
soluble in THF and was found to have an acid value of
180.
lS EXAMPLE 8
After evaporation of xylene/butyl acetate
dilution solvent, 25 g of Macrynal S~548, a hydroxy
acrylic copolymer of hydroxy value 66, were dissolved
in 200 cm3 THF at room temperature. A catalyst in the
form of dibutyl tin dilaurate (0.1 g) was added,
followed by 5.6 g 4-isocyanato phthalic anhydride. The
mildly exothermic reaction was held at 20~C for six
hours during which time all the isocyanate (as
determined by IR spectroscopy) had reacted. 20cm3
methanol were then added and the reaction mixture was
left a further four hours at 20~C before warming to
50~C for 12 hours. The product was isolated as in
Example 4 into IPA/water (2 litres of a 1:4 mixture).
The washed and dried product weighed 28g and had an
acid value of 43. Analysis by gpc showed a smooth mw
profile and the dried product readily redissolved in
THF.
EXAMPLE 9
25 g of DP6-3095, a hydroxy acrylic polymer
obtained from Allied Colloids with a hydroxy value of
3 ~
-12-
155, were dissolved in 200 cm3 acetonitrile and cooled
to 0~C. 5.5 g of pyridine were then added followed by
18.6 g of 4-chlorosulphonyl-1,8-naphthalic anhydride
dissolved in 50 cm3 cold acetonitrile. The acid
chloride addition was carried out dropwise at such a
rate as to keep the temperature in the range -2~C to
0~C. After completing the addition, the stirred
reaction mixture was kept at -2~C to 0~C ~or eight
hours. 20 g of water were then added to the reaction
mixture, keeping the temperature in the range 0~C to
+10~C and this mixture was allowed to warm to ambient
over six hoursO The reaction mixture was then heated
to 45~C and held at 45~C for a further 18 hours.
Isolation, as in Example 1, gave 29 g of a
product having an acid value of 190~ The product was
readily soluble in THF, and demonstrated a smooth mw
profile on gpc analysis.
EXAMPLE 10
Example 5 was repeated but no water was added
at the end of the isocyanate reaction. The reaction
was again held at 20~C for four hours and for a further
121 hours at 50~C. The slight]y gelatinous reaction
mixture was isolated as before but this time the dried
product would not fully redissolve in THF indicating
that considerable crosslinking had taken place. In
addition analysis by gpc of the soluble material showed
an uneven profile with a high molecular weight
(excluded) portion.
EXAMPLE 11
- 30 Comparative Example.
20 g of the polyvinylbutyral used in Example 2
were dissolved in 200 cm3 NMP at 70~C. 1.2 g o~ sodium
carbonate were added followed by 14.8 g of trimellitic
acid anhydride. The reaction mixture was warmed to
100~C for four hours before cooling and isolating intO
-13-
2 litres of water. The so]id product was filtered
thoroughly, water washed in a commercial food processor
and re-filtered before drying at 40~C in a hot air
oven.
The product, a white fibrous powder, was
readily soluble in THF, showed a smooth mw profile by
gpc analysis and had an acid value of 110. The yield
amounted to 21 g.
EXAMPLE 12
A coating solution was prepared having the
following composition:
8 parts by weight (pbw) of a diazo compound as
described in Example 1 of EP-A-0 030 862,
16 pbw of the polymer produced in Example 1,
0.~ pbw of Victoria Pure Blue B0 dye,
0.6 pbw of 85% phosphoric acid, and
1000 pbw of Ethylene glycol monomethyl ether
After filtering to remove any solids, the
solution was whirler coated onto a sheet of
electrochemically grained and anodised aluminium to
give a dry coating weight of 0.82 gm~2. The resultant
radiation sensitive plate was exposed through a
continuous tone Stouffer step-wedge to ultra violet
light in a Berkey-Ascor printing down frame and
developed with an aqueous solution containing sodium
propionate, sodium benzoate and a surfactant. The
de~eloped image of the so produced lithographic
printing plate had a step-wedge reading of solid 4 tail
9. After gumming, this plate was fitted to a rotary
web offset press and found to produce 150,000
acceptable impressions after which the step-wedge had
lost nearly 2 full steps.
A printing plate prepared and developed under
the same conditions was further treated by baking at
200~C for 10 minutes in acco~dance with the treatment
~3
~14-
described in GB 1513368. When used on a rotary web
offset press, 230,000 good impressions were obtained
with a loss of only 1 full step on the step-wedge
reading.
EXAMPLE 13
A coating solution was prepared as in Example
12, but using 16 pbw of the polymer produced in Example
2. Development, exposure (and baking as appropriate)
were effected in a likewise manner to give a finished
printing plate which gave 130,000 good copies on a
rotary web offset press without baking and 200,000 good
copies with baking, i.e. the run length of the printing
plate was increased by about 50% through the baking
process.
EXAMPLE 14
A coating solution having the following
composition was prepared:
35 pbw of the polymer prepared in Example 4,
10 pbw of a diazonium salt condensation
product which was the reaction product of 4-diazonium
diphenylamine and formaldehyde and obtained as the 2-
hydroxy-4-methoxy benzophenone-5-sulphonic acid salt,
2 pbw 85% phosphoric acid,
1.5 pbw of Victoria Pure Blue B0 dye, and
2000 pbw of Ethylene glycol monomethyl ether.
The filtered solution was applied to an
aluminium sheet as in ~xample 12 to give a dry coating
weight of 0.93 gm~2. After treatment as in Example 12
(exposure, development and baking as appropriate)
printing plates were obtained which showed excellent
ink recepti~ity during a proof test. When used on a
rotary weh offset press, 160,000 good impressions were
obtained, which increased by 40% with baking.
EXAMPLE 15
A coating solution having the following
~a~63~fi~3
composition was prepared:
35 pbw of the polymer prepared in Example 3A,
10 pbw of a diazo compound as described in
Example 1 of EP-A-0 030 ~62,
2 pbw of 85% phosphoric acid,
11.5 pbw of Victoria Pure Blue B0 dye, and
2000 pbw of Ethylene glycol monomethyl ether
After filtering, the solution was whirler
coated onto an aluminium substrate which had been
electrochemically grained and anodised and further
treated with an aqueous solution of polyvinyl
phosphonic acid before drying and the application of
the radiation sensitive coating. After exposure as in
Example 12, development was achieved by treating the
plate with an aqueous solution of a surfactant
containing 7% benzyl alcohol. Development was very
rapid to give a step-wedge reading, after inking-in, of
solid 5 tail 9. This rapid development was maintained
even for plates which had been subjected to rapid
ageing by being stored at 30~C and 90% relative
humidity in a cabinet for five-weeks.
When used on a rotary web offset press/ these
plates were found capable of producing up ~o 150,000
good clean impressions. Plates which had been baked,
according to the previously mentioned process, were
found to have an increased performance such that up to
200,000 copies could be obtained.
EXAMPLE 16
A coating solution was prepared having the
following composition:
12 pbw of the polymer produced in Example 5,
12 pbw of a diazonium salt condensation
product which was the reaction product
of 4-diazonium diphenylamine and
formaldehyde and obtained as the 2-
2~
-16-
hydroxy-4-methoxy ben~ophenone-5-
sulphonic acid salt,
1 pbw of Victoria Pure Blue B0 dye,
1 pbw of 85% phosphoric acid, and
1000 pbw of Ethylene glycol monomethyl ether
The filtered solution was applied to an
aluminium sheet as in Example 12 to give a dry coating
weight of 0.87 gm-2. After treatment as in Example 12
(exposure, development and baking as appropriate)
printing plates were obtained. When used on a rotary
web offset press, 80,000 good impressions were obtained
which increased by 55% with baking.
EXAMPLE 17
A coating solution was prepared, having the
following composition:
16 pbw of a diazo compound as described in
Example 1 of EP-A-30862,
8 pbw of the polymer produced in Example 7,
0.8 pbw of Victoria Pure Blue B0 dye,
0.6 pbw of 85% phosphoric acid, and
1000 pbw of Ethylene glycol monomethyl ether.
The filtered solution was applied to an
aluminium sheet as in Example 12 to give a dry coating
weight of 0.82 gm~2. After treatment, as in Example 12
(exposure, development and baking as appropriate)
printing plates were obtained. When used on a rotary
web offset press, 70,000 good impressions were
obtained, which increased by 40~ with baking.
EXAMPLE 18
A printing plate was prepared according to the
recipe and method given in Example 12, but the polymer
(of Example 1) was replaced by an equal weight of the
polymer from Example 11. A dried coating weight of
0.84 gm~2 was observed.
The development and step-wedge characteristics
were essentially indistinguishable from those of
Example 12 and on a printing press 150,000 good copies
were obtained.
On baking an identical plate by the method
previously described and testing under the same process
conditions, the same number of good copies were
obtained, i.e. there was no improvement achieved
through the baking process.
EXAMPLE 19
~ coating solution, having the following
composition was prepared:
12 pbw of a urethane acrylate disclosed as
prepolymer A in Example 1 of
EP-A-0260823
4 pbw of the polymer produced in Example 1
0.6 pbw of 2-(4'-trichloromethylphenacylidene)-
1,3,3-trimethyl 5-chloroindoline
2 pbw of the diazo compourld (41) described in
EP-A 0030862
0.4 pbw of Sudan Yellow and
1000 pbw of ethylene glycol monomethyl ether
The filtered solution was applied to an
aluminium sheet as in Example 12 to give a dry coating
weight of 0.85 gm~2. ~Eter treatment as in Example 12
(exposure, development and baking as appropriate)
printing plates were obtained. When used on a rotary
~eb offset press, 100,000 good impressions were
obtained for the unbaked plate. The baked plate
demonstrated significantly improved performance and
gave a run length of 200,000 good impressions.
The whole experiment was repeated using, in
place of the polymer produced in Example 1, 4 pbw of
the polymer (non-bakeable) produced in Example 11. In
this instance an improvement in performance on baking
was again noted (due presumably to the use of the
3$~
photopolymerisable urethane acrylate) but the degree of
improvement was not as great as when the bakeable
polymer of Rxample 1 was used.
EXAMPLE 20
A coating solution in methyl ethyl ketone
comprising:
12 pbw of the dimethacrylate ester of the
diglycidyl ether of Bisphenol A;
4 pbw of the polymer produced in Example l;
0.6 pbw of 2(4' chlorophenyl)-4,6-bis
(trichloromethyl)-s-triazine; and
0,6 pbw of ethyl Michler's ketone,
was whirler coa~ed onto a sheet of electrochemica]ly
grained and anodised aluminium and dried to form a
radiation sensitive plate. The coating weight was 1.0
gm~2. The dried coating was overcoated with poly(vinyl
alcohol) to prevent oxygen inhibition.
After treatment as in Example 12 (exposure,
development and baking as appropriate) printing plates
were obtained. When used on a rotary web offset press,
lS0,000 good impressions were obtained for the unbaked
plate. The baked plate demonstrated significantly
improved performance and gave a run length of 250,000
good impressions.
The whole experiment was repeated using, in
place of the polymer produced in Example 1, 4 pbw of
the polymer (non-bakeable) produced in Example 11. In
this instance an improvement in performance on baking
was again noted but the degree of improvement was not
as great as when the bakeable polymer of Example 1 was
used.