Note: Descriptions are shown in the official language in which they were submitted.
20n~s
BACRGROUND OF T~E INVENTION
The present invention relates to novel
phosphonates which can be employed as precursors to a
variety of biologically-active materials; including 13-
cis-retinoic acid (accutane), retin-A and beta
carotene. The phosphonates of the present invention can
be synthesized by the reaction of a cyclohexenyl-group-
containing C-14 through C-16 aldehyde, such as 2-methyl-
4-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2-butenal, with a
phosphonic acid ester, such as methylenebisphosphonic
acid, tetraethyl ester.
A procedure for producing vitamin A acetate
from beta-ionone has been described by Rei~ and Grassner
[Chemie-Inq.Techn., 45, 646-652 (1973)]:
OH
1 H ~ C ~ ~ HX
2 H2/cataly~t ~
r~ 1 , ~ ~ x ~
~ ~tr ph nyl -
o
~ 2 3 ~ 2 3 t--~ormyl-
~ crotyl ~c-tate
vitamin A ~c-t~t-
2n~4l2s
Similarly, Pommer and Kuhn [Angew.Chem., 72,
911 (1960)~ have described a procedure for preparing
beta-carotene from the same beta-ionone-derived
triphenylphosphonium salt:
~ ~ X ~
formed in the course of the ~eif, et al. synthesis. The
disadvantages of these procedures include the fact that
the triphenylphosphine reactant required for the
syntheses is relatively expensive and that the byproduct
of the reactions, Ph3PO, is not water soluble, thus
making it difficult to isolate the desired product.
Surmatis and Thommen have described a process
for preparing beta-carotene utilizing a phosphonate in a
Wittig-type reaction lJ.Org.Chem., 34, 559 (1969)]. An
essential step of this procedure invoi~es the reaction
of a C-20 dibromo compound with a trialkyl phosphite:
~ CH29r
o
~ c~c ~ cH2~lo~2
^~t-iyrt >
~ o ~ N-OCJ3, pyr~d~n-- >
~ ~, Ç~2Pl01l~2 2- ~X~Ho
r-eLnyL pbo~phon-t- ~
~-t~n~l
~t--e-rot-n-
Z004125
Although the C-20 dibromo compound of
Surmatis, et al. can be reacted with trialkyl
phosphites, the literature does not report similar
reactions for structurally related C-15 halides.
Indeed, the literature shows that the compound l-bromo-
3-methyl-5-(2~6~6-trimethyl-l-cyclohexen-l-yl)-2~4
pentadiene
~ CH2Br
is not stable at room temperature. (aull.Soc.Chim.Fr.,
15 Part II, 746-50 (1973)].
Other procedures for preparing retinoid
intermediates and beta-carotene are shown in the prior
art, e.g., Babler U.S. Patent 4,175,204; F. Frickel,
"The Retinoids", edited by M.B. Sporn, A.B. ~oberts and
D.S. Goodman, Academic Press (Orlando, Florida, 1984),
pp. 77-145; and R.S.H. Liu and A.E. Asato, Tetrahedron,
40, 1931-196g (1984).
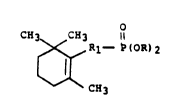
SUMMARY OF THE INVENTION
The novel phosphonate compounds of the present
invention have the structural formula:
C~3 CH3
~ R1- P~OR)2
c~3
20n4l2~
in which R is an alkyl group having up to four carbon
atoms, and Rl is a 3-alkyl pentadienyl group wherein the
alkyl group at the 3 position is methyl, ethyl or
propyl. The two double bonds in the 3-alkyl pentadienyl
group, Rl, can be in the 1,3 or 2,4 positions
(conjugated) or in the 1,4 positions (non-conjugated).
The compounds of the present invention are
systematically named as esters of an alkenylphosphonic
acid~ Thus, for example, when Rl is:
CH3
I
-CH=CH-C=CH-CH2-
and R is ethyl, the compound is named 3-methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-2,4-pentadienylphosphonic
acid, diethyl ester. When Rl is:
-CH=CH-CH-CH=CH-
and R is isopropyl, the compound is named 3-methyl-5-
(2,6,6-trimethyl-1-cyclohexen-1-yl)-1,4-
pentadienylphosphonic acid, diisopropyl ester.
Other compounds within the scope o~ the
present invention include:
3-methyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-1,3-
pentadienylphosphonic acid, diethyl ester;
3-ethyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid, diethyl ester;
XO(~
-- 5
3-methyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid, dimethyl ester;
3-methyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid, dipropyl ester;
3-methyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid, dibutyl ester;
3-propyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-1,3-
pentadienylphosphonic acid, diethyl ester; and
3-methyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid, diisopropyl ester.
Because of their ability to form known biologically-
active compounds, dialkyl esters of 3-methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-2,4-pentadienylphosphonic
acid, are especially preferred.
The compounds of thè present invention can be
formed by the base-promoted reaction of a C-14 through
C-16 aldehyde having the structure
CH3 CH3
~ R2CHO
CH3
IR3 IR3
wherein R2 is -CH=CH-CH- or -CH2-CH=C- and R3 is
methyl, ethyl or propyl, with a methylenebisphosphonic
acid ester having the structure:
O O
Il 11
(R0)2PcH2p(oR)2
2~)04125
wherein each R, which can be the same or different, is
selected from the class consisting of alkyl groups
having up to four carbon atoms. The preferred aldehydes
are C-14 materials which are derived from beta-ionone.
At room temperature, reaction of the aldehyde and
bisphosphonate ester proceeds rapidly (< 30 minutes) in
an organic solvent containir.g one equivalent of a base
(e.g., a Group I metal alkoxide or sodium hydr ~e):
o o
X~ Q2C~ = O + (RO) 2PCH2P(OR~ 2 + base or~anic solvent
r 1 ~ room temp .
15 ~
11
~<,~ Rl -- P(OR~ 2
11
`~' ~
wherein Rl is a 3~alkyl pentadienyl group, as defined
above, and R is a C-l to C-4 alkyl group. The
phosphonate ester product is soluble in a variety of
organic solvents and can be isolated from the reaction
mixture by convention~l techniques. Yields are
typically in excess of 90%.
A variety of organic solvents, both polar and
nonpolar, can be employed in the foregoing reaction,
3Q including hydrocarbons such as benzene, hexane,
cyclohexane, and toluene; ethers such as tetrahydrofuran
and diethyl ether; ethyl alcohol; polar solvents such as
dimethylformamide and dimethyl sulfoxide; or mixtures of
such organic solvents. Suitable bases include sodium
hydride, Group I metal alkoxides, and alkali metal
carbonates.
Z0041Z~
As noted previously, the double bonds in the
pentadienyl moiety can be in the 1,3-; 1,4- or 2,4-
positions. The 1,3- and 1,4- compounds can be
isomerized to the preferred 2,4- pentadienyl
phosphonates by employing a base catalyst such as an
alkoxide of a Group I metal, i.e., KOC(CH3)3, NaOCH3, or
NaOCH2CH3 with an organic solvent such as dimethyl
sulfoxide (DMSO).
10 ~CH = CHPIOR) 2 O
OR alk~xid~ ~ fX~. CH2P ~OR) 2
/ C~3 DMSo
X' CH2CH = C
~J~ ~ CH = CHP(OR) 2
In general, any orqanic solvent in which the
reactants are soluble may be employed in the practice of
the above two steps ~i.e., preparation of the
phosphonate ester and its subsequent isomerization).
Any base whose conjuqate acid has a PKa of approximately
8 or above can be utilized to promote these reactions.
The aldehyde reactant ~e.q., 2-methyl-4-
(2',6',6'-tri-methyl-1'-cyclohexen-1'-yl)-3-~utenal]
used to synthesize the phosphonate esters can be
prepared in accordance with known procedures. Processes
for synthesi2ing such aldehydes from ~eta-ionone are
shown, for example, in O. Isler, et al.,
Helv.Chim.Acta., 30, 1911 (1947); V. Ramamurthy, et al.,
Tetrahedron, 31, 193 (1975), or M~ ~osenberqer, et al.,
Helv.Chim.Acta, 63, 1665 (1980).
20~ 2~
The methylenebisphosphonic acid ester reactant
can be prepared by reacting methylene bromide with a
trialkyl phosphite [P(OR)3] in accordance with the
procedure shown in B. Costisella, J. fur prakt. Chemie,
324, 537 (1982), e.g.,
2Br2 + 2 P[OCH(C~ ) ] heat
O O
Il 11
[(CH3)2CHO]2 PCH2P[OCH(CH3)2]2
methylenebisphosphonic acid, tetraisopropyl ester, 73
yield.
The compounds of the present invention can be
used in the synthesis of retinoids or beta-carotene.
Illustrative examples of three such syntheses employing
the novel phosphonate compounds are as follows:
~0~)4125
SYnthesis of all-trans-retinoic acid (retin-A~
CH3 O O organic solvent,
~ CHCH = O ~(cH3cH2o)2pcH2p~ocH2cH3)2 2base >
2-methyl-4-l2',6',6'-methyieneb~ho~phonic
trim-thyl-l'-cyclo-acld/tetraet yl eJter
hexen-l '-yl)-3-butenal
CH3 o
~ CHCH = CHp(ocH2cH3)2 ba-e ~c g , KoclcH3)~ ~r
~ 90~ yield
Il
~ CH2P~OCH2CH3)2 CH3 C / COOCH3 ~OC(CH3)3
~ CH3 CH ~ \ H
~solc product)
COCH3
methyl e~t-r of reeinoic acit
200~1~5
-- 10 --
Synthesis of 13-cis-retinoic acid ~accutane)
o
CH2P~OCH2CH3)2 CH3
+ 2 equiv. KOC(CH3)3 + OH ~ ~O
5-hydroxy-4-methyl
2-(5H) furanone
THF/DMSO ~ ~
COOH
13~cls-retinoic acid
SYnthesis of beta-carotene
o
2 equiv ~ ~ CH2P~OCH2CH3)2 + 2 equlv KOC(CH3)3 +
o
H\ / CH = ~/
CH3 C 2 C CH3
/ ~ THF/DMSO ~
~C = CH H 20-C ~'
o
2,7-di~ethyl-2,4,6-octatrie~edial
~ ~ ~ _
3-c~roten- 1>60- yield)
20n4~25
DETAILED DESCRIPTION OF THE INVENTION
The following examples illustrate in greater
detail the practice of the present invention,
S specifically: (i) the preparation of intermediates which
can be utilized to form the phosphonate compounds of the
present invention (Examples I-V); (ii) the preparation
of representative novel phosphonate compounds (Examples
VI-XI); (iii) the preparation of intermediates which can
be reacted with the compounds of the present invention
to form biologically-active materials (Examples XII-
XIV); and, (iv) the preparation of biologically-active
compounds utilizing the novel phosphonate compounds of
the invention (Examples XV-XVII).
EXAMPLE I
Preparation of Methylenebisphosphonic
Acid, TetraethY1 Ester
In accordance with a procedure suggested by
H. Gross, et al., Journal fur prakt. Chemie, 324, 537
(1982~, a mixture of 4.00 ml (57.0 mmoles) of
dibromomethane and 30 mL (175 mmoles) of triethyl
phosphite was gradually warmed to 90C over a period of
15 minutes. After maintaining the temperature at 90C
for an additional 10 minutes, the solution ~as warmed to
140C and kept at that temperature for 2 hours. At that
point, the mixture was warmed to 160C ~external bath
temperature) and heated at that temperature for an addi-
tional 15 hours, during which time ethyl bromide was
slowly distilled out of the reaction mixture. Next,
excess triethyl phosphite was distilled from the
reaction flask, followed by distillative removal of
minor amounts of ethylphosphonic acid, diethyl ester.
The desired product was then obtained by distillation
under reduced pressure, affording 9.03g (55% yield) of
2(~4125
- 12 -
bisphosphonate: bp 145-160C (bath temperature, 0.25
mm). H. Gross, et. al., reported a 70% yield of the
same compound, prepared on a larger scale (150 mmoles of
dibromomethane).
EXAMPLE II
Preparation of Methylenebisphosphonic
Acid, Tetraisopropyl Ester
A mixture of 1.00 ml (14.25 mmoles) of
dibromomethane and 11.0 mL (44.5 mmoles) of triisopropyl
phosphite was heated in the same manner as described in
the procedure of Example I. Removal of excess
triisopropyl phosphite, followed by a minor amount of
isopropylphosphonic acid, diisopropyl ester, by
distillation at reduced pressure, and subsequent
evaporative distillation [bath temperature: 138-152C
(0.25 mm)] afforded 3.57 g (73% yieid) of the desired
bisphosphonate.
EXAMPLE III
Preparation of 2-Methyl-2-[2-l2,6,6-
trimethYl-l-cvclohexen-l-yl)ethenYlloxirane
A mixture of 762 mg (19.1 mmoles) of sodium
hydride (60% dispersion in mineral oil, which was
removed ~y washing with hexane prior to the addition of
DMSO) and 6.0 m~ of anhydrous dimethyl sulfoxide (DMSO)
was heated, protected from atmospheric moisture, at a
bath temperature of 65C for approximately 45 minutes --
until evolution of hydrogen had ceased. After cooling
this mixture to room temperature, it was added dropwise
over a period of 10 minutes to a stirred slurry of
3.9549 (19.38 mmoles) of trimethylsulfonium iodide in
12.0 mL of 1:1 ~v/v) anhydrous DMSO: tetrahydrofuran,
protected from atmospheric moisture and kept cold in an
ice-brine bath at approximately -5C. The resulting
2()~)412~
gray suspension was stirred for an additional 5 minutes,
after which a solution of 1.42g (7.38 mmoles) of beta-
ionone in 3.00 mL of anhydrous tetrahydrofuran was added
dropwise rapidly. This mixture was subsequently stirred
at approximately O~C for 2 hours, after which it was
allowed to warm to room temperature. The product was
isolated, after addition of 1 mL of water to quench the
reaction, by dilution of the mixture with 50 mL of
pentane and 100 mL of 10% aqueous sodium chloride.
Separation of the layers was followed by washing the
organic layer with 10% aqueous sodium chloride (2 x 100
mL), water (1 x 100 mL), and saturated brine
(1 x 100 mL) in successive order. The organic extracts
were then dried over anhydrous sodium sulfate and
subsequently filtered. Removal of the pentane and
tetrahydrofuran by evaporation at reduced pressure
afforded 1.52 g (100~ yield) of the desired epoxide.
EXAMPLE IV
Preparation of 2-Methyl-4-~2,6,6-
trimethyl-l-cyclohexen-l-yl)-3-butenal
~ solution of 1.502 9 (7.28 mmoles) of the
epoxide, prepared as described in Example III, in 6.00
mL of anhydrous ether was added dropwise over 5 minutes
to a stirred suspension of magnesium bromide [prepared
_ situ from 355 mg (1.88 mmoles) of 1,2-dibromoethane
and 48 mg (1.98 milli-g-atoms) of magnesium turnings] in
3.00 mL of anhydrous ether, protected from atmospheric
moisture, at -10C. The resulting mixture was stirred
at -10C for an additional 5 minutes, after which it was
diluted with 20 mL of solvent ether~ The organic layer
was washed in successive order with 15 mL portions of
water and saturated brine, after which it was dried over
anhydrous sodium sulfate and subsequently filtered.
Removal of the ether by evaporation at reduced pressure
20(~1XS
- 14 -
afforded 1.40 g (93% yield) of the desired aldehyde,
whose structure was verified by NMR analysis [~ 9.69,
doublet, J = 1.8 Hz, CHO; ~ 1.25, doublet, J = 7 Hz,
CHCH3]. The procedure used in Examples III a~d IV was
developed by M. Rosenberger, et al. [Helv. Chim. Acta.,
_ , 1665 (1980)]. An alternate route to this same
aldehyde can be found in O. Isler, et al., Helv. Chim.
Acta., 30, 1911 (1947), subsequently modified by R.S.H.
Liu, et al., Tetrahedron, 31, 193 (1975).
EXAMPLE V
Preparation of 2-Methyl-4-(2,6,6-
trimethyl-l-cYclohexen-l-yl)-2-butenal
A mixture of 920 mg (4.46 mmoles) of the
aldehyde prepared as described in Example IV and 45 mg
of potassium hydroxide pellets in 3.0 mL of methyl
alcohol containing 0.05 mL of water ;~as stirred,
protected from atmospheric moisture, at 20C for 35
minutes. The product was isolated after dilution of the
mixture with 30mL of 1:1 (v/v) pentane: ether and
subsequent washing of the organic layer with 25 mL
portions of 10% aqueous sodium chloride and saturated
brine. Drying of the organic extracts over anhydrous
magnesium sulfate, followed by filtration and removal of
the pentane and ether at reduced pressure, afforded 916
mg (99.6% yield) of the isomerized aldehyde, whose
structural identity was confirmed by NMR analysis
(~ 9.45, singlet, CHO~.
EXAMPLE VI
Preparation of 3-Methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-1,3-
~ntadienylphosphonic acid, Diethyl E~ter
A solution of 508 mg (1.76 mmoles) of
methylenebisphosphonic acid, tetraethyl ester, prepared
Z0~12~
as described in Example I, in 2.5 mL of benzene and 1.5
mL of anhydrous tetrahydrofuran (THF) was added dropwise
slowly over 5 minutes to a stirred mixture of 69 mg (1.7
mmoles) of sodium hydride (60~ dispersion in mineral
oil, which was removed prior to the reaction by washing
with hexane) and 1.0 mL of benzene, protected from
atmospheric moisture and maintained at a temperature o~
15-20C by use of an external cold water bath. This
mixture was stirred for an additional 15 minutes, after
which a solution of 20~ mg (1.01 mmole) of aldehyde
(prepared as described in Example V) in 2.5 mL of
benzene was added dropwise rapidly. After stirring this
mixture at room temperature for 25 minutes, it was
diluted with 20 mL of 1:1 ~v/v) pentane: ether and
washed in successive order with 7:3 (v/v) lM aqueous
sodium hydroxide: methyl alcohol (2 x 40 mL) to remove
excess bisphosphonate and then with saturated brine (20
mL). The organic layer was then dried over anhydrous
magnesium sulfate and subsequently filtered. Removal of
the pentane, ether, and benzene by evaporation at
reduced pressure afforded 320 mg (93~ yield) of the
desired vinyl phosphonate.
EXAMPLE VII
Preparation of 3-Methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-1,3-
pentadienylphosphonic Acid, Diisopropyl Ester
The ylide was prepared in the manner described
in the procedure of Example VI by reaction of 605 mg
(1.76 mmoles) of methylenebisphosphonic acid,
tetraisopropyl ester (produced in accordance with
Example II), with 69 mg (1.7 mmoles) of 60~ sodium
hydride. Subsequent addition of 195 mg (0.95 mmole) of
the unsaturated aldehyde produced in accordance with
Example V and stirring of the mixture at 20C for 25
ZO(~4125
- 16 -
minutes completed the reaction. The product was
isolated after dilution of the mixture with 20 mL of 1:1
(v/v) pentane: ether and washing in successive order
with 1:1 (v/v) lM aqueous sodium hydroxide: methyl
S alcohol (2 x 40 mL) to remove excess bisphosphonate and
then with saturated brine (20 mL). The organic layer
was then dried over anhydrous magnesium sulfate and
subsequently filtered. Removal of the pentane, ether,
and benzene by evaporation at reduced pressure afforded
313 mg (90% yield) of the desired vinyl phosphonate.
EXAMPLE VIII
Preparation of 3-Methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl~-1,4-
pentadienylphosphonic Acid, DiethYl Ester
The reaction was conducted in the mannerdescribed in the procedure of Example VI using the
following reagents: 2.96 g (10.25 mmoles) of
methylenebisphosphonic acid, tetraethyl ester (produced
in accordance with Example I), in 20 mL of 3:2 (v/v)
benzene: anhydrous tetrahydrofuran; 413 mg (10.3 mmoles)
of 60% sodium hydride in 8.0 mL of benzene; and 1.204 g
(5.85 mmoles) of unsaturated aldehyde (produced in
accordance with Example IV) in 12.0 mL of benzene.
Isolation of the product as described in the procedure
of Example VI afforded 1.901 g (95.5% yield) of the
desired vinyl phosphonate.
EXAMPLE IX
Preparation of 3-Methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-1,4-
pentadienYlphosphonic Acid, DiisoPropyl Ester
The reaction was conducted in the manner
described in the procedure of Example VII using the
following reagents: 303 mg (0.88 mmole) of
X0~4~2~j
methylenebisphosphonic acid, tetraisopropyl ester
(produced in accordance with Example II), in 2.5 mL of
3:2 (v/v~ benzene: anhydrous tetrahydrofuran; 36 mg
(0.90 mmole) of 60% sodium hydride in 1.0 mL of benzene;
and 98 mg (0.47 mmole) of unsaturated aldehyde (produced
in accordance with Example IV) in 1.5 mL of benzene.
Isolation of the product as described in the procedure
of Example VII afforded 104 mg (60% yield) of the
desired vinyl phosphonate.
EXAMPLE X
Preparation of 3-Methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-2,4-
pentadienylPhosphonic Acid, Diethyl E ter
A mixture of the vinyl phosphonate produced in
accordance with Example VIII (943 mg, 2.77 mmoles) and
99 mg (0.88 mmoles) of potassium tert-butoxide in 12 mL
of anhydrous dimethyl sulfoxide (DMSO) was stirred,
protected from atmospheric moisture, at 20C for 80
minutes. The prGduct was isolated by dilution of the
reaction mixture with 100 mL of ether and subsequent
washing with 120 mL portions of 10% aqueous sodium
chloride (4 x 120 mL). The organic layer was then dried
over anhydrous magnesium sulfate and filtered. Removal
of the e~her by evaporation at reduced pressure afforded
718 mg (76% yield) of the desired allylic phosphonate,
whose structural integrity was confirmed by NMR analysis
[~ 2.75, doublet of doublets, J = 8 Hz and 22 Hz,
CH2P]. In a similar manner, this allylic phosphonate
could be prepared by isomerization of 3-methyl-5-~2,6,6-
trimethyl-l-cyclohexen-l-yl)-1,3-pentadienylphosphonic
a~id, diethyl ester, produced in accordance with Example
VI.
;~O~lZ5
- 18 -
EXAMPLE XI
Preparation of 3-Methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-2,4-
p~n~adienylphosphonic Acid, Diisopropyl Ester
A mixture of vinyl phosphonate produced in
accordance with Example VII (308 mg, 0.84 mmole) and 88
mg. (0.78 mmole) of potassium tert-butoxide in 4 mL of
anhydrous dimethyl sulfoxide was stirred, protected from
atmospheric moisture, at 20C for 30 minutes. Isolation
of the product in the manner described in the procedure
of Example X afforded 238 mg (77% yield) of the desired
allylic phosphonate. This latter compound could also be
prepared by isomerization of 3-methyl-5-(2,6,6-
trimethyl-l-cyclohexen-l-yl)-1,4-pentadienylphosphonic
acid, diisopropyl ester, produced in accordance with
Example IX.
EXAMPLE XII
Preparation of 2-Butenyl-1,4-
bi~phosphonic Acid, Tetraethvl Ester
A solution of 2.00 mL (18.9 mmoles) of trans-
1,4-dichloro-2-butene in 3.00 mL (17.5 mmoles) of
triethyl phosphite was added dropwise slowly over 25
minutes to a flask containing 5.00 mL (29.2 r~moles) of
triethyl phosphite, maintained at a temperature of
approximately 140C (external oil bath temperature).
This mixture was subsequently heated at 140C for an
additional 12 hours, during which time ethyl chloride
was continuously distilled out of the reaction flask.
At that point, the external oil bath temperature was
raised to 180C to distill over as much of the remaining
triethyl phosphite as possible. The desired product was
then obtained by fractional distillation under reduced
pressure, affording 5.40 9 (87.5% yield) of bis-
phosphonate: bp 161-184C ~bath temperature, 0.25 mm).
ZO~
-- 19 --
EXAMPLE XIII
Preparation of 1,1,8,8-
Tetramethoxy-2,7-dimethyl-2,4,6-octatriene
To a solution of 312 mg (0.95 mmole) of 2-
butenyl-1,4-bisphosphonic acid, tetraethyl ester
(produced in accordance with Example XII), and 0.25 mL
(2.07 mmoles) of pyruvic aldehyde dimethyl acetal
(available from Aldrich Chemical Co.) in 3.25 mL of 12:1
(v/v) anhydrous tetrahydrofuran: dimethyl sulfoxide,
protected from atmospheric moisture and maintained at a
temperature of approximately 5C by use of an external
ice water bath, was added 211 mg (1.88 mmoles) of
potassium tert-butoxide. This mixture was subsequently
stirred in the cold for 15 minutes and then at room
temperature for 7 hours. The product was isolated by
dilution of the mixture with 30 mL of 1:1 (v/v) ether:
pentane and subsequent washing of the organic layer with
10% aqueous sodium chloride (3 x 30 ml). The organic
layer was then dried over anhydrous sodium sulfate and
filtered. Removal of the volatile organic solvents by
evaporation at reduced pressure afforded 151 mq (62
yield) of bisacetal.
EXAMPLE XIV
Preparation of 2,7-Dimethvl-2,4,6-octatrienedial
A solution of 150 mg (0.585 mmole) of 1,1,8,8-
tetramethoxy-2,7-dimethyl-2,4,6-octatriene, produced in
accordance with Example XIII, in 3.5 mL of 4:2:1 (v/v/v)
glacial acetic acid: tetrahydrofuran: water was heated
at 45~C ~external oil bath temperature) for 3 hours.
After cooling the solution to room temperature, the
product was isolated by dilution of the mixture with 25
mL of 4:1 (v/v) ether: dichloromethane and washing the
organic layer in successive order with saturated brine
2()~)412~ri
- 20 -
(2 x 25 mL), 4:1 (v/v) saturated brine: lM aqueous
sodium hydroxide (2 x 25 mL), and saturated brine (~5
mL). The organic layer was then dried over anhydrous
magnesium sulfate and filtered. Removal of the volatile
organic solvents by evaporation at reduced pressure
afforded 86 mg (90% yield) of the desired bisaldehyde,
previously prepared in a similar manner by H. Pommer,
et al., Anqew. Chem., 72, 911 (1960).
EXAMPLE XV
Preparation of Beta-Carotene
To a solution of 192 mg (0.564 mmole) of 3-
methyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid, diethyl ester (produced in
accordance with Example X) and 41 mg (0.25 mmole) of
2,7-dimethyl-2,4,6-octatrienedial (produced in
accordance with Example XIV) in 2.25 mL of 8:1 (v/v)
anhydrous tetrahydrofuran: dimethyl sulfoxide, protected
from atmospheric moisture and maintained at a
temperature of approximately 5C by use of an external
ice water bath, was added 59 mg ~0.526 mmole) of
potassium tert-butoxide. This mixture was subsequently
stirred in the cold for 15 minutes and then at room
temperature for 3~5 hours. The product was isolated by
dilution of the mixture with 25 mL of 4:1 (v/v) ether:
dichloromethane and subsequent washinq of the organic
layer with 25 mL portions of 10% aqueous sodium chloride
(3 x 25 mL). The organic layer was then dried over
anhydrous magnesium sulfate and filtered. Removal of
the volatile organic solvents by evaporation at reduced
pressure, followed by filtration through a small column
of silica gel (10 mL, 60-200 mesh, elution with 40 mL of
3:1 (v/vj hexane: benzene~ to remove any unreacted
starting materlals afforded 82 mg (61% yield) of deep-
purple crystals, identifi~d by NMR analysis as beta-
carotene: mp 183-185C.
lZ.~
- 21 -
EXAMPLE XVI
Preparation of all-trans Retinoic Acid, Ethyl Ester
.
To a solution of 57 mg (0.40 mmole) of ethyl
3-methyl-4-oxobutenoate (prepared according to a
procedure described by R.W. Curley, Jr.v et al.,
J.Org.Chem., 51, 256 (1986); an alternate synthesis has
been described by A. Guingant, et al., J.Org.Chem., 52,
4788 (1987); the compound is commercially available from
Fluka Chemical Corp., Ron Kon Koma, New York 11779.] and
132 mg (0.388 mmole) of 3-methyl-5-(2,6,6-trimethyl~l-
cyclohexen-l-yl)-2,4-pentadienylphosphonic acid diethyl
ester (produced in accordance with Example X) in 3.5 mL
of 6:1 (v/v) anhydrous tetrahydrofuran: dimethyl
sulfoxide, protected from atmospheric moisture and
maintained at a temperature of approximately 5C by use
of an ice water bath, was added 43 mg (O.38 mmole) of
potassium tert-butoxide. This mixture was subsequently
stirred in the cold for 10 minutes and then at room
temperature for 6 hours. The product was isolated by
dilution of the mixture with 30 mL of 1:1 (v/v) pentane:
ether and subseqùent washing of the organic layer with
30 mL portions of 10% aqueous sodium chloride (3 x 30
mL). The organic layer was then dried over anhydrous
magnesium sulfate and filtered. Removal of the volatile
organic solvents by evaporation at reduced pressure,
followed by filtration through a small column of silica
gel (15 mL, 60-200 mesh, elution with 75 mL of hexane -
4~ ether) to remove any unreacted starting materials,
afforded 76 mg (61% yield) of ethyl retinoate shown by
high-field (300 MHz) NMR analysis to be predominantly
the all trans stereoisomer. The product was
characteri~ed by three broad singlets of equal intensity
at 6 2.37, 2.02, and 1.73 (3 vinyl methyls). For tables
listing spectroscopic properties of retinoids, see:
412S
R.S.H. Liu, et al. Tetrahedron, 40, 1931-1969 (1984).
Ethyl retinoate was also prepared in a similar manner
from ethyl 3-methyl-4-oxobutenoate and 3-methyl-5-
(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid diisopropyl ester (producedin accordance with Example XI).
EXAMPLE XVII
Preparation of 13-cis-Retinoic Acid
To a solution of 88 mg (0.258 mmole) of 3-
methyl-5-(2,6,6-trimethyl-1-cyclohexen-1-yl)-2,4-
pentadienylphosphonic acid diethyl ester (produced in
accordance with Example X) and 37 mg (0.324 mmole) of 5-
hydroxy-4-methyl-2-(SH) furanone [prepared according to
a procedure described by G. Pattenden, et al.,
J.Chem.Soc.(C), 1984 (1968). An alternate synthesis has
been described by C.G. Wermuth, et al., J.Org.Chem., 46,
4889 (1981).] in 2.25 mL of 8:1 (v/v) anhydrous
tetrahydrofuran: dimethyl sulfoxide, protected from
atmospheric moisture and maintained at a temperature of
approximately 5C by use of an ice water bath, was added
64 mg (0.57 mmole) of potassium tert-butoxide. This
mixture was subsequently stirred in the cold for 15
minutes and then at room temperature for 3.5 hours.
After aci'difying the mixture by addition of 0.50 mL of
2M aqueous hydrochloric acid, it was diluted wi~h 25 mL
of 4:1 (v/v) ether: dichloromethane. The organic layer
was washed in successive order with 10% aqueous sodium
chloride (2 x 25 mL), water (1 x 25 mL), and saturated
brine (1 x 25 mL), dried over anhydrous magnesium
sulfate, and filtered. Removal of the volatile organic
solvents by evaporation at reduced pressure, followed by
filtration throu~h a small column of silica gel (6 mL,
40-140 mesh, elution with 25 mL of pentane - 20% ether~
to remove any unreacted phosphonate afforded 44 mg (57
~o~
- 23 -
yield) of orange crystals, shown by NMR analysis (in
CDC13 solution) to be a mixture of stereoisomers. The
predominate stereoisomer (comprising approximately 75-
80% of the mixture) was characterized by a doublet (J =
15 Hz) at ~ 7.81 (vinyl hydrogen bonded to C-12), a
broad singlet at ~ 5.~8 (vinyl hydrogen bonded to C-14),
and a broad singlet at ~ 2.11 (CH3 bonded to C-13). By
comparison with the NMR data reported (in "tau values",
where "tau" = 10 -~) for various stereoisomers of
retinoic acid by Pattenden, et al., [J.Chem.Soc.,C.,
1984-1997 (1968)], this major component was shown to be
13-cis-retinoic acid. The other (minor) component in
the product exhibited broad singlets at ~ 5.82 (vinyl
hydrogen bonded to C-14) and ~ 2.37 (CH3 bonded to C-
13), absorptions characteristic of all-trans retinoic
acid.
Although the foregoing invention has been
described in some detail by way of example, various
changes and modifications to the specific procedures
which have been illustrated may be practiced within the
scope of the appended claims.