Note: Descriptions are shown in the official language in which they were submitted.
~(~~I'~'93
, ,...
O.Z. 0050/40553
Aromatic keto compounds, the preparation thereof,
and drugs and cosmetics containing these
The present invention relates to novel aromatic
keto compounds, to processes for the preparation thereof
and to agents prepared therefrom for controlling and
preventing diseases.
US 4,326,055 and CA 1,183,541 disclose the
pharmacological effects of retinoidal benzoic acid
derivatives on topical and systemic therapy of neoplasms
and dermatoses, eg. acne or psoriasis. The disadvantage
of these compounds is that they have a low therapeutic
index because of the side effects comprised by the term
hypervitaminosis A.
We have now found that compounds of the general
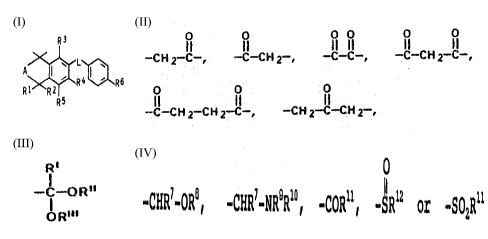
formula I
R3
A I ~ R4 ! ~ - R6 Ii
R1 ~R2I
RS
where
A is ethylene or methylene which can be substituted by
methyl, hydroxyl or oxo,
L is one of the following
II I I! II I~I il
-C H Z C-, -C-CH Z-, --C-C-, -C-C H 1-C-,
0 0 0
-CI-CH1-CHI-C-, -CHI-C-CHZ-,
R1 and R2 are hydrogen or methyl,
R3 is hydrogen, hydroxyl or C1-C6-alkoxy,
R'' is hydrogen, C1-C4-alkyl, halogen or methoxy,
RS is hydrogen or methoxy, and
R6 is hydrogen, methyl, cyano, a CZ-Clo group of the
formula
.~. ~ 20079 35 .~
2
R'
-C-oR"
OR"'
where R',R" and R "' are each alkyl of 1 to 9 carbons, the
total of carbons in R',R",R "' is 2 to 10, and R' can also
be hydrogen, or the following
0
-CHR~-ORB, -CHRT-NR9R1°, -CORM, -SRlZ or -SOZRii
where:
R~ is hydrogen or C1-C4-alkyl,
R8 is hydrogen, C1-C4-alkyl, C1-C20-alkanoyl, or benzoyl
which can be substituted by methoxy, nitro, methyl or
halogen,
R9 and R10 are hydrogen, C1-C4-alkyl, C1-C6-alkanoyl, or
benzoyl which can be substituted by methoxy, nitro, methyl
or halogen, or form together with the nitrogen to which
they are bonded, a 5- or 6-membered saturated heterocyclic
radical which can also contain an oxygen in addition to the
nitrogen,
. R11 is hydrogen, C1-C4-alkyl, or -OR13 or -NR14R15 where
R13 is hydrogen, C1-Cg-alkyl which can be substituted by 1
or 2 hydroxyls, or aryl which can be substituted by methyl,
methoxy or cyano, or aralkyl which can be substituted in
the aryl moiety by methyl, methoxy or halogen, and where
R14 and R15 are hydrogen, or aryl which can be substituted
by methyl, methoxy or cyano, or aralkyl which can be
substituted in the aryl moiety by methyl, methoxy or
halogen, or R14 and R15 form, together with the nitrogen to
which they are bonded, a 5- or 6-membered saturated
~ 20078 3~
2a
heterocyclic radical which can also contain an oxygen in
addition to the nitrogen, and
R12 is Cl-C4-alkyl,
and the physiologically tolerated salts thereof have an
improved profile of action, in particular with regard to
side effects.
Particularly suitable heterocyclic radicals
-NR9R1° and -NR1°R15 are pyrrolidinyl, piperidinyl and
morpholinyl. Preferred substituents on the benzoyl (R°,R°
' 2~~'~935
°~ - 3 - O.Z. 0050/40553
and Rl° ) are methoxy, vitro, methyl or halogen, especially
chlorine or bromine . The preferred aryl ( R'3, Rl°, Rls ) is
phenyl which can be substituted by methyl, methoxy or
cyano. The preferred aralkyl (Rl3,Rl4,Rls) is benzyl, which
can be substituted in the aryl moiety by, in particular,
methyl, methoxy or halogen, preferably chlorine or
bromine.
The preferred halogen for R' is fluorine.
The compounds of the formula I according to the
invention can be prepared by conventional processes. A
review of the various methods for preparing ketones is to
be found in "Methoden der Organischen Chemie" (Houben
Weyl-Miiller), Volume 7/2a-c, Georg Thieme Verlag
Stuttgart.
The methods mainly used for synthesizing the
compounds according to the invention are described
hereinafters
1. Deoxybenzoins of the formula I (L = -CHZ-CO- or
-CO-CHZ-) can be obtained, for example, by
a) reacting a phosphoric ester of the formula II
R3 X
A I ~ II(ORIBI z
R4 0 II,
R1 R2
R5
where A and Rl-Rs have the abovementioned meanings,
and X is -OR16 or -NR16R1' where R16 and R1' are C1-C4-
alkyl, it being possible for R3 and RlB together to
form a C1-C3-alkylene chain, and R18 is Cl-C4-alkyl,
with an aldehyde of the formula III
I ~ R6 III,
OCH
where R6 has the abovementioned meaning, in the
presence of a base in a Wittig-Horner reaction, and
subjecting the resulting enamine (X = NR16R") or
enol ether (X = OR16) to acid hydrolysis, or by
~C~3'~935
' ~- - 4 - O.Z. 0050/40553
b) reacting a carbonyl chloride of the formula IV or V
R3
COC1 I ~ R6
A~ %~
R4
R1 R2R5 C1C0
IV V,
where A and R1-R6 have the abovementioned meanings,
with a benzyl halide of the formula VI or VII
R3
A ~ ~ Y ~ ~ R6
R 4 Y
R1 ~R21
RS
VI VII.
where A and R1-R6 have the abovementioned meanings,
and Y is chlorine or bromine, in the presence of a
suitable metal, with or without the addition of
catalytic amounts of a transition metal catalyst,
the reaction being between either Iv and VII or V
and VI.
2 . 1, 3-diketo compounds of the formula I according to the
invention, with L = -C(0)CHzC(0)- or the tautomeric
enol compounds, can be prepared, for example, by
a) reacting a ketone of the formula VIII
R3 0
CH3
A
R~ VIII,
R1 R2
RS
where A and R1-R3 have the abovementioned meanings,
with a terephthalic ester of the formula IX
~COOR~B
R1800C
~~~"~935
- 5 - O.Z. 0050/40553
where R18 is C1-C4-alkyl, in the presence of a base,
or by
b) subjecting a chalcone of the formula X
R3 0
A ( ~
~ R4 ~ R6 X~
R1 'RZR
S
where A and R1-R6 have the abovementioned meanings,
to addition of bromine, then eliminating hydrogen
bromide with a suitable base, and finally carrying
out acid hydrolysis.
3. The 1,4-diketo compounds of the formula I according to
the invent ion ( L = -C ( O ) CHZCH2C ( 0 ) - ) c an be prepared by
reacting a chloroethyl ketone of the formula XI,
R3 0
A I ~ Cl XI
R1 R2 ~ R4 i
R5
where A and R1-RS have the abovementioned meanings,
with a suitable base to give a vinyl ketone of the
formula XII
R3 0
R~
R1 R2 XII,
RS
where A and R1-RS have the abovementioned meanings, and
then reacting the latter with a benzaldehyde of the
formula XIII
~ R6
~ XIII,
OHC
where R6 has the abovementioned meaning, in the pres-
ence of an "umpolung" catalyst {see, for example,
H. Stetter, and J. Krasselt, J. Heterocyclic Chem. 14,
2~~~'935
- 6 - O.Z. 0050/40553
573 (1977).
4. It is also possible to convert the compounds of the
formula I which have been prepared by the processes
described under 1. -3 . or by other processes into other
compounds of the formula I according to the invention
by modifying the radical R6.
The Wittig-Horner reaction in l.a) is carried out
in the solvents conventional for this purpose, such as
tetrahydrofuran, diethyl ether, 1,2-dimethoxyethane,
n-hexane, petroleum ether, toluene, dimethyl sulfoxide,
dimethylformamide or mixtures thereof at from -78 to
+60°C, the temperature being greatly dependent on the
base which is employed.
The bases which are used are sodium hydride, the
sodium salt of dimethyl sulfoxide, alcoholates such as
sodium methanolate or potassium tert-butanolate, or
organolithiums such as n-butyllithium. Preferably used
for the metalation of the phosphoric esters of the
formula II is n-butyllithium at from -78 to -60°C, and
this is followed by addition of the benzaldehyde and
allowing the reaction to go to completion at up to 25°C.
The reaction in l.b) requires at least equimolar
amounts of a metal and/or metal salt. Produced as inter
mediates are the corresponding organometallic compounds,
eg. of magnesium, cadmium, zinc or lithium. Zinc is
particularly advantageous, preferably zinc which has
previously been activated with copper or silver. Cata-
lytic amounts of a transition metal catalyst can be
added, preferably containing palladium or nickel, eg.
bis(triphenylphosphine)palladium(II) chloride or
tetrakis(triphenylphosphine)palladium. It is particularly
beneficial to add such catalysts when zinc is used as
reductive metal.
Mainly used as solvents are ethers such as
diethyl ether, tetrahydrofuran, dioxane or 1,2-dimethoxy
ethane, and possibly also aromatic hydrocarbons such as
benzene or toluene. The reactions are carried out at from
~t~~'~935
- 7 - O.Z. 0050/40553
0 to 60°C, preferably at 20-40°C.
Ester condensations as in 2.a) can in principle
take place with either basic or acid catalysis, but bases
are mostly used, eg. alkali metal hydrides such as sodium
hydride, alkali metal alcoholates such as sodium methano-
late or potassium tart-butanolate, or alkali metal amides
such as those of lithium, sodium or potassium.
Solvents which are expediently used are alcohols
such as methanol or ethanol, ethers such as diethyl
ether, diisopropyl ether, tetrahydrofuran, dioxane or
1,2-dimethoxyethane, aromatic hydrocarbons such as
toluene or xylene, or else dimethylformamide. The reac-
tions are carried out at from 20 to 140°C, preferably
from 80 to 120°C.
Examples of suitable bases in 2.b) are alcohol-
ates such as sodium methanolate or potassium tert-
butanolate. The hydrolysis is brought about with dilute
or concentrated mineral acids such as hydrochloric acid
or sulfuric acid.
Suitable for the elimination of hydrogen chloride
in 3. are, in particular, organic nitrogen bases such as
triethylamine or pyridine. The vinyl ketones of the
formula XII can be isolated, but further reaction in situ
is beneficial. The coupling with the benzaldehyde deriv-
ative of the formula XIII in an umpolung reaction is
catalyzed by cyanide ions or, even better, by thiazolium
salts such as 3-benzyl-5-(2-hydroxyethyl)-4-methyl-
thiazolium chloride. Dimethylformamide is preferably used
as solvent.
Examples of methods 4. are:
Benzoic esters or benzonitriles of the general
formula I where Rg is carboalkoxy or cyano can be con-
verted into the free carboxylic acids and the physio-
logically tolerated salts thereof by hydrolysis. The
hydrolysis is preferably carried out in a mixture of a
lower aliphatic alcohol such as methanol, ethanol,
propanol, isopropanol or n-butanol with water in the
~~3"~935
"' - 8 - O.Z. 0050/40553
presence of an excess of an alkali metal hydroxide,
preferably sodium or potassium hydroxide, at the boiling
point of the reaction mixture.
The amides according to the invention can be
prepared in a conventional manner by converting the
corresponding benzoic acids into more active derivatives,
eg. into the carbonyl halides, azides, imidazolides or
anhydrides, and treating the latter with amines HNRI4Ris.
A carboxylic acid or ester or amide thereof or a
nitrile of the formula I can be reduced in a conventional
manner to the corresponding alcohols or amines. The
reaction is advantageously carried out with a metal
hydride or alkali metal hydride in the presence of a
suitable solvent. The metal hydrides which are preferably
employed are complex metal hydrides such as lithium
aluminum hydride or lithium borohydride or diisobutyl
aluminum hydride. Preferred solvents are ethers such as
diethyl ether, tetrahydrofuran, dioxane or 1,2-dimethoxy-
ethane.
An alcohol or amine of the formula I can be
acylated in a conventional manner with alkanoyl or aroyl
chloride or anhydride to give the corresponding ester or
amide, or alkylated with an alkyl halide, preferably an
alkyl bromide or iodide, to give the corresponding ether
or more highly alkylated amine, or oxidized with suitable
oxidizing agents such as manganese(IV) oxide to give the
corresponding aldehyde.
An aldehyde of the formula I can also be obtained
by reducing the corresponding nitrile of the formula I
with diisobutylaluminum hydride in a solvent, preferably
toluene, hexane or tetrahydrofuran, at from -40°C to room
temperature.
The compounds according to the invention and
their physiologically tolerated salts can, by reason of
their pharmacological properties, be used for the topical
and systemic therapy and prophylaxis of precanceroses and
carcinomas of the skin, the mucous membranes and internal
2~~'~~35
- 9 - O.Z. 0050/40553
organs and for the topical and systemic therapy of acne,
psoriasis and other dermatological disorders associated
with pathological keratinization, especially ichthyosis,
Darier's disease, herpes and leukoplakia but also eczema,
vitiligo, warts, phototoxis (premature ageing) of the
skin, and dry eyes and other corneopathies and for the
treatment of rheumatic disorders, especially those of an
inflammatory or degenerative nature and which affect
joints, muscles, tendons and other parts of the locomotor
system. Preferred indications are: the therapy of dermat-
ological disorders, of skin damage caused by sunlight,
and of iatrogenic skin damage, eg. atrophy induced by
corticosteroids, and the prophylactic treatment of
precanceroses and tumors.
The pharmacological effects can be shown, for
example, in the following tests: the compounds according
to the invention abolish the keratinization which starts
in hamster tracheal tissue in vitro after vitamin A
deficiency. The keratinization is part of the early phase
of carcinogenesis, which is inhibited by the compounds of
the formula I according to the invention in a similar
test in vivo after initiation by chemical compounds, by
energetic radiation or after viral cell transformation.
These methods are described in Cancer Res. 36 (1972) 964-
972 and Nature 250 (1974) 64-66 and 253, (1975) 47-50.
In addition, the compounds according to the
invention inhibit the proliferation of certain malignant
cells. This method is described in J. Natl. Cancer Inst.
60 (1978) 1035-1041, Experimental Cell Research 117
(1978) 15-22 and Proc. Natl. Acad. Sci. USA 77 (1980)
2937-2940.
The antiarthritic effect of the compounds accord-
ing to the invention can be determined in a conventional
manner in animal experiments using the adjuvant arthritis
or streptococci cell wall induced arthritis model. The
dermatological activity, for example for the treatment of
acne, can be demonstrated, inter alia, by the comedolytic
~~~''~~35
- 10 - O.Z. 0050/40553
activity and the ability to reduce the number of cysts in
the rhino mouse model.
The latter method is described by L . H . Kligman et
al. in the Journal of Investigative Dermatology 73 (1978)
354-358.
The dermatological activity can also be measured
by the reduction in sebaceous glands and the associated
diminution in sebum production by the flank organ of the
hamster . This method is described by E . C . Gomez in J . Am .
Dermatol. 6 (1982) 746-750.
Furthermore, it is possible to determine the
reversal which can be achieved with compounds according
to the invention of skin damage caused by UV light in
animal models. This method is described by L.H. Kligman
et al. in Connect. Tissue Res. 12, (1984) 139-150 and in
the Journal of the American Academy of Dermatology 15
(1986) 779-785.
Accordingly, the invention furthermore relates to
therapeutic agents for topical and systemic administra
tion and to cosmetic agents which contain a compound of
the formula I as active substance in addition to con-
ventional carriers or diluents.
The agents can accordingly be administered
orally, parenterally or topically. Examples of suitable
formulations are uncoated or (film-coated tablets,
capsules, pills, powders, solutions or suspensions,
infusion or injection solutions and pastes, ointments,
gels, creams, lotions, dusting powders, solutions or
emulsions and sprays.
The therapeutic or cosmetic agents can contain
the compounds to be used according to the invention in a
concentration of 0.0001 to 1 %, preferably 0.001 to
0.1 %, for local use, and preferably in a single dose of
0.1 to 50 mg for systemic use, and are administered in
one or more doses each day depending on the nature and
severity of the disorders.
The drugs and cosmetics of the invention are
2C~~~~935
°" - 11 - O.Z. 0050/40553
produced in a conventional manner using the conventional
solid or liquid carriers or diluents and the auxiliaries
which are conventionally used in pharmaceutical tech-
nology to accord with the desired mode of administration
and with a suitable dosage. Tablets can be obtained, for
example, by mixing the active substance with known
auxiliaries, for example inert diluents such as dextrose,
sugar, sorbitol, mannitol or polyvinylpyrrolidone,
disintegrants, such as corn starch or alginic acid,
binders such as starch or gelatin, lubricants such as
magnesium stearate or talc and/or agents to achieve a
depot effect, such as carboxypolymethylene, carboxy
methylcellulose, cellulose acetate phthalate or polyvinyl
acetate. The tablets can also be composed of several
layers.
Appropriate coated tablets can be produced by
coating cores, which have been produced in a similar
manner to the tablets, with conventional coating agents,
for example polyvinylpyrrolidone or shellac, gum arabic,
talc, titanium dioxide or sugar. The coating can also be
composed of several layers, it being possible to use the
auxiliaries mentioned above for tablets.
Solutions or suspensions containing the active
substance according to the invention can additionally
contain taste corrigents such as saccharin, cyclamate or
sugar as well as, for example, flavorings such as vanil-
lin or orange extract. They can moreover contain suspend-
ing auxiliaries such as sodium carboxymethylcellulose or
preservatives such as p-hydroxybenzoates. Capsules
containing active substances can be produced, for ex-
ample, by the active substance being mixed with an inert
carrier such as lactose or sorbitol and encapsulated in
gelatin capsules.
Examples of conventional ingredients of cosmetic
and pharmaceutical formulations for topical use are:
anionic, cationic and nonionic emulsifiers and emulsion
stabilizers which can simultaneously act as bodying
~(~~'~~35
- 12 - O.Z. 0050/40553
agents or gel formers, such as polyvinylpyrrolidone,
fatty alcohols, glycerol monostearate, polyacrylic acids,
cellulose derivatives and ethylene oxide/propylene oxide
block polymers, solid or liquid oily components or fats
of mineral, vegetable or animal origin, synthetic oily
esters such as glycerol triesters and isopropyl myri-
state, hydrophilic components such as glycerol, poly-
ethylene glycol and propylene glycol.
Examples of further ingredients of cosmetics are
sunscreen agents, suntan agents, preservatives, anti
oxidants, pigments, colorants, essential oils and perfume
oils, vitamins, plant extracts, collagen etc. These
substances are described, for example, in the CTFA
Cosmetic Ingredient Dictionary, 3rd edition, Washington
1982.
Some of the compounds according to the invention
have an acidic hydrogen and can therefore be converted
with bases in a conventional manner into a physiologi-
cally tolerated salt which is readily soluble in water.
Examples of suitable salts are ammonium and alkali metal
salts, especially of sodium, potassium and lithium, and
alkaline earth metal salts, especially of calcium or
magnesium, as well as salts with suitable organic bases,
such as C1-C6-alkylamines, eg. methylamine, ethylamine or
cyclohexylamine, or with substiauted C~-C6-alkylamines,
especially hydroxyl-substituted alkylamines such as
diethanolamine, triethanolamine and tris(hydroxymethyl)-
aminomethane, and with piperidine or morpholine.
The amines of the formula I according to the
invention which have been obtained can be converted by
conventional procedures into the acid addition salt of a
physiologically tolerated acid. Examples of suitable
physiologically tolerated inorganic acids are hydro
chloric acid, hydrobromic acid, phosphoric acid or
sulfuric acid, and of organic acids are oxalic acid,
malefic acid, fumaric acid, lactic acid, tartaric acid,
malic acid, citric acid, salicylic acid, adipic acid or
~C~~'7'~35
- 13 - O.Z. 0050/40553
benzoic acid. Others can be found in "Fortschritte der
Arzneimittelforschung" volume 10, pages 224-225,
Birkhauser Verlag, Basle and Stuttgart, 1966.
EXAMPLE 1
1-(4-Carbomethoxyphenyl)-3-(5,6,7,8-tetrahydro-5,5,8,8,-
tetramethyl-2-naphthyl)-1,3-propanedione
A solution of 19.4 g (,0.1 mol) of dimethyl
terephthalate and 23 g (0.1 mol) of 6-acetyl-1,2,3,4
tetrahydro-1,1,4,4-tetramethylnaphthalene in 80 ml of
toluene and 20 ml of dimethoxyethane was added dropwise
under nitrogen to a suspension of 3.6 g (0.15 mol) of
sodium hydride in 21 ml of toluene at 100°C. The mixture
was refluxed for 5 h and, after cooling, 50 ml of water
were added, and the mixture was acidified with 50$
concentrated hydrochloric acid, poured into 500 ml of
water and extracted with chloroform. The precipitated
product was filtered off with suction, and the chloroform
phase was dried over sodium sulfate and concentrated. The
residue and crude product were mixed and washed several
times with hot methanol. Drying of the crystals resulted
in 22.1 g of the title compound of melting point 128°C.
EXAMPLE 2
1-(4-Carbomethoxyphenyl)-3-(2,3-dihydro-1,1,2,3,3-penta-
methyl-5-(1H)-indenyl)-1,3-propanedione
In a similar manner to Example 1, 18.1 g of the
title compound of melting point 120-125°C were obtained
from 23 g (0.1 mol) of 5-acetyl-2,3-dihydro-1,1,2,3,3-
pentamethyl-(1H)-indene and 19.4 g (0.1 mol) of dimethyl
terephthalate.
EXAMPLE 3
1-(4-Carbomethoxyphenyl)-3-(5,6,7,8-tetrahydro-3,8,8-
trimethyl-2-naphthyl)-1,3-propanedione
In a similar manner to Example 1, 4.8 g of the
title compound of melting point 91-92°C were obtained
from 10 g (46 mmol) of 7-acetyl-1,2,3,4-tetrahydro-1,1,6
trimethylnaphthalene and 9 g (46 mmol) of dimethyl
terephthalate.
2(1~~"i ~3S
""' - 14 - O.Z. 0050/40553
EXAMPLE 4
1-(4-Carboxyphenyl)-3-(5,6,7,8-tetrahydro-5,5,8,8-tetra-
methyl-2-naphthyl)-1,3-propanedione
20 g (87 mmol) of 6-acetyl-1,2,3,4-tetrahydro
1,1,4,4-tetramethylnaphthalene, 13 g (87 mmol) of
4-formylbenzoic acid and 8 g of sodium hydroxide pellets
were stirred in 100 ml of methanol at room temperature
overnight. The mixture was then poured into ice/water and
extracted with ether. Remaining ether in the aqueous
phase was removed by blowing nitrogen through. It was
acidified with concentrated hydrochloric acid, the
crystalline precipitate was filtered off with suction,
and drying resulted in 24.9 g of 3-(4-carboxyphenyl)-1-
(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)-2-
propen-1-one of melting point 199-201°C.
A solution of 8.5 g (53 mmol) of bromine in
100 ml of methylene chloride was added dropwise to a
solution of 18.1 g (50 mmol) of this chalconecarboxylic
acid in 100 ml of methylene chloride at -5 to 0°C. The
mixture was stirred for 1 h and then the solvent was
removed under reduced pressure.
The residue was taken up in 85 ml of methanol and
added to the 29.2 g of a 30$ strength methanolic sodium
methanolate solution. The reaction mixture was stirred at
65°C for 3 h, allowed to cool to 25°C and acidified with
9 ml of concentrated hydrochloric acid. The mixture was
again stirred at 65°C for 3 h and then left to stand at
room temperature overnight. The precipitate was filtered
off with suction and dried.
To purify the crude product it was dissolved in
2 N sodium hydroxide solution and extracted 3 x with
ether. Remaining ether was driven out of the aqueous
phase with nitrogen, and it was then acidified with
concentrated hydrochloric acid. The crystals were
filtered off with suction and dried, resulting in 10.3 g
of the title compound of melting point 200-202°C.
~~3~935
'""'" - 15 - 0. Z . 0050/40553
EXAMPLE 5
1-(4-Carbomethoxyphenyl)-4-(5,6,7,8-tetrahydro-5,5,8,8-
tetramethyl-2-naphthyl)-1,4-butaredione
73.5 g (0.55 mol) of anhydrous aluminum chloride
were added a little at a time to a solution of 70 g
(0.55 mol) of chloropropionyl chloride in 200 ml of
methylene chloride at 0 to 5°C and then, at the same
temperature, a solution of 94 g (0.5 mol) of 1,2,3,4
tetrahydro-1,1,4,4-tetramethylnaphthalene in 200 ml of
methylene chloride was added dropwise. The mixture was
stirred at room temperature overnight, poured into 1 1 of
ice/water and extracted 3x with 300 ml of methylene
chloride each time. The combined organic phases were
washed with saturated sodium bicarbonate solution and
water, dried over magnesium sulfate and concentrated.
138 g of 6-(3-chloropropionyl)-1,2,3,4-tetrahydro-
1,1,4,4-tetramethylnaphthalene remained as an oil. The
structure was confirmed by H NMR spectroscopy.
5.6 g (0.02 mol) of 6-(3-chloropropionyl)
1,2,3,4-tetrahydro-1,1,4,4-tetramethylnaphthalene and
3.3 ml (0.024 mol) of triethylamine were stirred in 30 ml
of dimethylformamide at room temperature for 1 h. Then a
solution of 3.6 g (0.025 mol) of methyl 4-formylbenzoate
and 1 g of 3-benzyl-5-(2-hydroxyethyl)-4-methylthiazolium
chloride in 10 ml of dimethylformamide was added drop-
wise. The mixture was stirred for 1 h and then poured in
to ice/water and extracted 2x with ethyl acetate, and the
combined organic extracts were washed several times with
water, dried over magnesium sulfate and concentrated.
Recrystallization of the residue (6.4 g) from ethanol
resulted in 3.7 g of the title compound of melting
139-141°C.
EXAMPLE 6
1-(4-Carbomethoxyphenyl)-2-(5,6,7,8-tetrahydro-5,5,8,8
tetramethyl-2-naphthyl)ethanedione
7.9 g (33 mmol) of potassium tert-butanolate were
added a little at a time to a suspension of 34.5 g
2(~'~'~~35
- 16 - O.Z. 0050/40553
(66 mmol) of 4-carbomethoxybenzyltriphenylphosphonium
bromide were added in 200 ml of dry toluene at room
temperature. The mixture was stirred for 10 min and then
heated to reflux, and 7.5 g (33 mmol) of 5,6,7,8-tetra-
hydro-5,5,8,8-tetramethylnaphthalene-2-carbonyl chloride
were added dropwise. After the solution had become
colorless again it was allowed to cool and solids were
filtered off. The filtrate was concentrated, and the
remaining oil was recrystallized from methanol (11.2 g of
the ylide intermediate of melting point 101-203°C). 3.2 g
of the ylide and 3.1 g of sodium periodate were refluxed
in a mixture of 40 ml of ethanol and 10 ml of water for
1 h. Saturated brine was added to the cooled reaction
mixture, which was then extracted with methylene chlor-
ide. The organic phase was dried over magnesium sulfate
and concentrated. The residue was triturated in
n-heptane, the solids were filtered off, and the filtrate
was concentrated. Recrystallization from ethanol yielded
1.5 g of the title compound of melting point 93-96°C.
EXAMPLE 7
2-(4-Carboxyphenyl)-1-(5,6,7,8-tetrahydro-3-methoxy-
5,5,8,8-tetramethyl-2-naphthyl)ethanone
14.6 g (29 mmol) of 1,2-dibromo-1-(4-cyano
phenyl)-2-(5,6,7,8-tetrahydro-3-methoxy-5,5,8,8-tetra
methyl-2-naphthyl)ethane, which had been obtained by
bromination of (E)-4-[2-(5,6,7,8-tetrahydro-3-methoxy-
5,5,8,8-tetramethyl-2-naphthyl)-1-ethenyl]benzonitrile
with elemental bromine in chloroform, and 15.3 g of
potassium hydroxide pellets were refluxed in 38 ml of
n-butanol for 1 h. The mixture was then poured into
ice/water and the mixture was acidified with 2 N hydro-
chloric acid and extracted 3x with ether. The combined
ether extracts were washed to neutrality with water,
dried over sodium sulfate and concentrated. The residue,
which contained n-butanol, had produced crystals after 3
days, and these were filtered off with suction and washed
with a little ethanol. This resulted in 2.9 g of the
""' - 17 - O.Z. 0050/40553
title compound of melting point 164-165°C.
EXAMPLE 8
2-(4-Carbomethoxyphenyl)-1-(5,6,7,8-tetrahydro-5,5,8,B-
tetramethyl-2-naphthyl)ethanone
A solution of 4 g (16 mmol) of 5,6,7,8-tetra-
hydro-5,5,8,8-tetramethylnaphthalene-2-carbonyl chloride
and 4 g (16 mmol) of methyl 4-bromomethylbenzoate in
20 ml of dimethoxyethane was added dropwise to a solution
or suspension of 0.56 g of bis(triphenylphosphine)pal-
ladium(II) dichloride and 2.1 g of zinc powder in 40 ml
of dimethoxyethane at room temperature. The reaction
mixture was then refluxed for 6 h. After cooling, solids
were filtered off and the filtrate was heated with active
carbon and filtered hot. The filtrate was concentrated
and yielded after recrystallization 3x from methanol and
finally from isopropanol 0.9 g of the title compound of
melting point 103-104°C.
EXAMPLE 9
2-(4-Carbomethoxyphenyl)-1-(5,6,7,8-tetrahydro-4-methoxy-
5,5,8,8-tetramethyl-2-naphthyl)ethanone
5,6,7,8-tetrahydro-4-methoxy-5,5,8,8-tetramethylnaphtha-
lene-2-carbonyl chloride:
A mixture of 20 g (86 mmol) of 1,2,3,4-tetra
hydro-5-methoxy-1,1,4,4,7-pentamethylnaphthalene, 4.9 g
of sodium hydroxide, 58 ml of pyridine and 30 ml of water
was heated to 95°C. Then 32.5 g of potassium permanganate
were added a little at a time at this temperature. The
mixture was stirred at 95°C for 2 h and then cooled, and
6 ml of ethanol were slowly added dropwise. Next day the
manganese dioxide was filtered and washed with hot 2 N
sodium hydroxide solution. The filtrate (2 phases) was
mixed with ether, resulting in 3 phases . The middle phase
was separated out, extracted 2x with petroleum ether and
acidified with concentrated hydrochloric acid. The white
precipitate was filtered off with suction and dried:
9.2 g of 5,6,7,8-tetrahydro-4-methoxy-5,5,8,8-tetra-
methylnaphthalene-2-carboxylic acid.
2(~~'~93 a
- 18 - O.Z. 0050/40553
g (19 mmol) of this were dissolved in 30 ml of
toluene. 3 drops of dimethylformamide were added and then
2.9 g (23 mmol) of oxalyl chloride were slowly added
dropwise . The mixture was stirred at room temperature for
5 30 min and then heated to 70°C and stirred at this
temperature until there was no more evolution of gas. The
mixture was cooled and the solvent was removed under
reduced pressure. The residue was 6.1 g of crude 5,6,7,8-
tetrahydro-4-methoxy-5,5,8.8-tetramethylnaphthalene-2-
carbonyl chloride, whose structure was confirmed by
H NMR.
2-(4-Carbomethoxyphenyl)-1-(5,6,7,8-tetrahydro-4-methoxy-
5,5,8,8-tetramethyl-2-naphthyl)ethanone
2.1 g of zinc powder and 0.21 g of copper(II)
acetate were stirred with 7.3 ml of acetic acid at 20°C,
with cooling, for 30 min. The zinc/copper couple was
filtered off with suction and washed 2 x with dry ether
and 1 x with dry dimethoxyethane. A solution of 4.5 g
(16 mmol) of 5,6,7,8-tetrahydro-4-methoxy-5,5,8,8-tetra-
methylnaphthalene-2-carbonyl chloride and 3.7 g (16 mmol)
of methyl 4-bromomethylbenzoate in 60 ml of dimethoxy-
ethane was added dropwise to a suspension of the zinc/-
copper couple prepared in this way and of 0.56 g of
bis(triphenylphosphine)palladium(II) chloride in 15 ml of
dimethoxyethane at room temperature. The mixture was
stirred for 75 min and then filtered through Celite, and
the filtrate was concentrated. Three recrystallizations
of the residue from methanol yielded 2.0 g of the title
compound of melting point 143-144°C.
EXAMPLE 10
1-(4-Carbomethoxyphenyl)-2-(5,6,7,8-tetrahydro-5,5,8,8-
tetramethyl-2-naphthyl)ethanone
6-Bromomethyl-1,2,3,4-tetrahydro-1,1,4,4-tetramethyl-
naphthalene:
A mixture of 117.5 g (0.66 mol) of N-bromo-
succinimide and 2 g of 2,2'-azodiisobutyronitrile was
added a little at a time to a solution of 121 g (0.6 mol)
~~~D"'~''93a
'""' - 19 - O. Z . 0050/40553
of 1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalene
and 1 spatula-tip of 2,2'-azodiisobutyronitrile in 1 1 of
1,2-dichloroethane at 80°C. After the addition was
complete, the mixture was stirred at about 80°C for
75 min. It was then allowed to cool, the solvent was
removed under reduced pressure, and the residue was
triturated with n-heptane. The precipitate was filtered
off, and the filtrate was concentrated. The residue was
distilled, resulting in 109 g of 6-bromomethyl-1,2,3,4-
tetrahydro-1,1,4,4-tetramethylnaphthalene of boiling
point 122-124°C (0.3 mbar).
1-(4-Carbomethoxyphenyl)-2-(5,6,7,8-tetrahydro-5,5,8,8-
tetramethyl-2-naphthyl)ethanone
In a similar manner to Example 9, 0.3 g of the
title compound of melting point 96-97°C was obtained from
10 g (50 mmol) of monomethyl terephthalate and 14 g
(50 mmol) of 6-bromomethyl-1,2,3,4-tetrahydro-1,1,4,4
tetramethylnaphthalene.
EXAMPLE 11
2-(4-Carboxyphenyl)-1-(5,6,7,8-tetrahydro-5,5,8,8-tetra-
methyl-2-naphthyl)ethanone
65.4 g (0.3 mol) of 5,6,7,8-tetrahydro-5,5,8,8-
tetramethyl-2-naphthaldehyde, 56 g (0.38 mol) of ethyl
orthoformate and 0.3 g of 4-toluenesulfonic acid in 75 ml
of absolute ethanol were stirred at room temperature for
16 h. Then 7.6 g of anhydrous sodium carbonate were
added, the mixture was stirred for about 15 min, the
solids were filtered off, and the filtrate was con-
centrated. 92 g of diethyl acetal remained. 73.5 g
(0.25 mol) of this diethyl acetal and 42.2 g (0.25 mol)
of triethyl phosphite were dissolved in 240 ml of dry
methylene chloride and, at -20°C under nitrogen, 38 g
(0.265 mol) of boron trifluoride diethyl etherate were
added dropwise. The mixture was allowed to reach room
temperature overnight, then 80 ml of water were added,
the phases were separated, and the organic phase was
dried over sodium sulfate and concentrated. Purification
~~x'7935
""" - 20 - O.Z. 0050/40553
by column chromatography (silica gel; n-heptane/ethyl
acetate 1:0 ~ 1) resulted in 33.3 g of slightly impure
diethyl 1-ethoxy-1-(5,6,7,8-tetrahydro-5,5,8,8-tetra-
methyl-2-naphthyl)methylphosphonate as an oil whose
structure was confirmed by H and 13C NMR.
28 ml (44 mmol) of a 1.6 M solution of n-butyl-
lithium in n-hexane were slowly added dropwise to a
solution of 7.6 g (20 mmol) of this phosphonate in 30 ml
of absolute tetrahydrofuran at -78°C under nitrogen. The
mixture was stirred for 30 min and then, likewise at
-78°C, a solution of 3.3 g (20 mmol) of methyl 4-formyl-
benzoate in 20 ml of tetrahydrofuran was added dropwise.
The mixture was allowed to warm to room temperature and
was then stirred for 1 h. It was poured into water, the
mixture was extracted with ether, and the ether phase was
washed with water, dried over sodium sulfate and con-
centrated. The residue was purified by column chroma-
tography (silica gel; n-heptane/ethyl acetate 95:5),
resulting in 6.7 g of slightly impure 2-(4-carbomethoxy-
phenyl)-1-ethoxy-1-(5,6,7,8-tetrahydro-5,5,8,8-tetra-
methyl-2-naphthyl)ethene. 2 g (5 mmol) of this and 0.5 g
of potassium hydroxide in a mixture of 15 ml of methanol
and 10 ml of water were refluxed for 3 h. After the
mixture had been cooled it was poured into water and
extracted with ether. The aqueous phase was acidified
with 2 N hydrochloric acid, and the crude product which
separated out as an oil was extracted with ether. The
ether extract was concentrated, and the residue was
dissolved in 16 ml of tetrahydrofuran. 4 ml of 2 N
hydrochloric acid were added, and the reaction mixture
was refluxed for 3 h. It was allowed to cool, water was
added, the phases were separated, and the organic phase
was concentrated. The residue was 0.5 g of the title
compound of melting point 204-206°C, RF=0.39 (TLC: silica
gel; methylene chloride/methanol 9:1).
~(~~'~935
- 21 - O.Z. 0050/40553
EXAMPLE 12
1-(4-Carboxyphenyl)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetra-
methyl-2-naphthyl)ethanedione
3.9 g (10 mmol) of 1-(4-Carbomethoxyphenyl)-2
(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)
ethanedione (Example 6) were refluxed in a mixture of
5 ml of concentrated sulfuric acid, 80 ml of glacial
acetic acid and 40 ml of water for 8 h. The mixture was
then poured into 500 ml of water, and the crystalline
precipitate was filtered off with suction and washed with
water until the washings were neutral. The dried crude
product was recrystallized from cyclohexane, yielding
3.1 g of the title compound of melting point 148-151°C.