Note: Descriptions are shown in the official language in which they were submitted.
201.0636
RAN 4070/77
The present invention relates to substituted pyrroles.
More particularly, the invention is concerned with
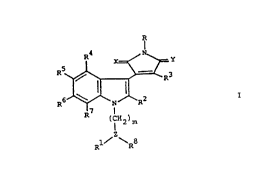
compounds of the general formula
Y
R 3
R
R~ ( iH2)m
2
Rl/ ~R8
I
wherein R represents hydrogen or hydroxy, R1 and
RZ together represent a group of the formula
-(CHZ)n- and R7 represents hydrogen or Rl and
R7 together represent a group of the formula
-(CH2)n- and R2 represents hydrogen; R3
represents an aryl or heteroaryl group: R4, R5 and
R6 each independently represent hydrogen, halogen,
alkyl, hydroxy, alkoxy, haloalkyl, vitro, amino,
acylamino, alkylthio, alkylsulphinyl or alkyl-
sulphonyl; Re represents a group of the formula
-(CEI2)p-R9 or -(CHZ)q_R10~ R9 represents
~ hydrogen, alkylcarbonyl, aminoalkylcarbonyl, cyano,
amidino, alkoxycarbonyl, aryloxycarbonyl, alkyl-
sulphonyl, aminocarbonyl or aminothiocarbonyl; R10
represents hydroxy, alkoxy, halogen, amino, monoalkyl-
amino, dialkylamino, trialkylamino, azido, acylamino,
Me/8.1.90
201.0636
2 -
alkylsulphonylamino, arylsulphonylamino, alkylthio.
alkoxycarbonylamino, aminoacylamino, aminocarbonyl-
amino, isothiocyanato, alkylcarbonyloxy, alkyl-
sulphonyloxy or arylsulphonyloxy, a 5- or 6-membered
saturated nitrogen-containing heterocycle attached via
the nitrogen atom or a group of the formula -U-C(V)-W;
U represents S or NEI; V represents NEI. NN02, NCN,
CHN02; W represents amino, monoalkylamino or
dialkylamino: one of X and Y represents O and the
1p other represents O or (II,H): Z represents CH or N: m
stands for 0-5, with the proviso that m stands for 2-5
when Z represents N; n stands for 1-5; p stands for
0-5; and q stands for 0-5, with the proviso that q
stands for 2-5 when Z stands for N;
as well as pharmaceutically acceptable salts of acidic
compounds of formula I with bases and of basic compounds
of formula I with acids.
Objects of the present invention are the compounds of
formula I and their aforementioned salts per se and as
therapeutically active substances; a process for the
manufacture of said compounds and salts and novel inter-
mediates useful in said process; medicaments containing
said compounds and salts and the manufacture of these
medicaments: and the use of said compounds and salts in
the control or prevention of illnesses, especially in the
control or prevention of inflammatory, immunological.
ontological, bronchopulmonary and cardiovascular disorders
or in the treatment of asthma or AIDS, or for the
3p manufacture of a medicament against inflammatory, immuno-
logical, ontological, bronchopulmonary and cardiovascular
disorders or against asthma or AIDS.
As used herein, the term "alkyl", alone or in
3,5 combination, means a straight-chain or branched-chain
alkyl group containing a maximum of 7, preferably a
-~ 3 -
201,0636
maximum of 4, carbon atoms such as methyl, ethyl, propyl,
isopropyl, butyl, sec.butyl, tert.butyl, pentyl and the
like. The term "alkoxy", alone or in combinations, means
an alkyl group as defined earlier which is attached via an
oxygen atom, examples of alkoxy groups being methoxy.
ethoxy, propoxy, isopropoxy, butoxy, tert.butoxy and the
like. A haloalkyl group can carry one or more halogen
atoms, with examples of such groups being chloromethyl,
trifluoromethyl etc. The term "aryl" means an acyl group
derived from an alkanoic acid containing a maximum of 7,
preferably a maximum of 4, carbon atoms (e. g. formyl
acetyl, propionyl, butyryl etc) or from an aromatic
carboxylic acid (e. g. benzoyl etc). The term "aryl", alone
or in combination, means a monocyclic or polycyclic group.
preferably a monocyclic or bicyclic group, i.e. phenyl or
naphthyl, which can be substituted or unsubstituted, for
example with one or more, preferably one to three,
substituents, selected from halogen, alkyl, hydroxy.
alkoxy, haloalkyl, vitro, amino. acylamino, alkylthio,
pp alkylsulphinyl and alkylsulphonyl. Examples of such aryl
groups are phenyl. 2-chlorophenyl, 3-chlorophenyl,
4-chlorophenyl. 3-bromophenyl, 2-methylphenyl, 3-methyl-
phenyl. 2,5-dimethylphenyl. 4-methoxyphenyl, 2-trifluoro-
methylphenyl, 3-trifluoromethylphenyl, 2-nitrophenyl,
3-nitrophenyl. 4-nitrophenyl, 3-aminophenyl. 4-amino-
phenyl, 4-methylthiophenyl, 4-methylsulphinylphenyl.
4-methylsulphonylphenyl. 1-naphthyl. 2-naphthyl and the
like. The term "heteroaryl" means a 5-membered ar
6-membered heterocyclic aromatic group which can
optionally carry a fused benzene ring and which can be
substituted or unsubstituted, for example with one or
more, preferably one to three, substituents selected from
halogen, alkyl, hydroxy, alkoxy, haloalkyl, vitro, amino,
acylamino, alkylthio, alkylsulphinyl and alkylsulphonyl.
3,5 Examples of such heteroaryl groups are 2-thienyl.
3-thienyl. 3-benzothienyl. 3-benzofuranyl, 2-pyrrolyl.
-Q_
2010636
3-indolyl and the like which can be unsubstituted or
substituted in the manner indicated. The 5- or 6-~membered
saturated nitrogen-containing heterocycle attached via the
nitrogen atom can contain a further nitrogen atom or an
oxygen or a sulphur atom, examples of such heterocycles
being pyrrolidino, piperidino, piperazino, morpholino and
thiamorpholino. The term "halogen" means fluorine,
chlorine, bromine or iodine.
The compounds of formula I in which Z represents CH
and R8 represents a group of the formula -(CEIZ)p-R9
in which p stands for 1-5 or -(CH2)q-R1~ contain an
asymmetric carbon atom and can therefore exist in racemic
or optically active form. The present invention includes
~5 within its scope not only the racemic compounds, but also
the optically active isomers.
In preferred classes of compounds of formula I, R1
and R2 together represent -CHZ- and R~ represents
hydrogen, m stands for 1 or 2 and Z represents CH: or R1
and Rz together represent -(CHZ)Z- and R~
represents hydrogen, m stands for 1 and Z represents CH:
or R1 and R2 together represent -CH2- and R~
represents hydrogen, m stands for 2 and Z represents N: or
R1 and R7 together represent -CHZ- and R2
represents hydrogen, m stands for 1 and Z represents CEI:
or R1 and R~ together represent -(CH2)2- and R2
represents hydrogen, m stands for O and Z represents CH.
R3 preferably represents phenyl, naphthyl, 3-benzo-
thienyl, 3-benzofuranyl or 3-indolyl which is optionally
substituted as defined earlier, especially 1-methyl-3-
-indolyl. Preferably, R4, R5 and R6 each represent
hydrogen. RB preferably represents a group of the
formula -(CH2)q-R10. Preferably, q stands for 1 or
2. Rl~ preferably represents hydroxy, amino, monoalkyl-
amino, dialkylamino, trialkylamino, azido, acylamino,
- 2010636
alkylcarbonyloxy or alkylsulphonyloxy or a group of the
formula -U-C(V)-W. Preferably, U represents S. V
represents NFI and W represents amino.
Especially preferred compounds provided by the
invention are:
3-[e-(Aminomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]-
indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione,
3-[7-(amidinothiomethyl)-6,7.8,9-tetrahydropyrido-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-lI3-pyrrole-
-2,5-dione and
~-[6,7,8,9-tetrahydro-8-[(dimethylamino)methyl]pyrido
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5
~5 -dione
and their pharmaceutically acceptable acid addition salts.
According to the process provided by the present
invention, the compounds of formula I as well as
20 pharmaceutically acceptable salts of acidic compounds of
formula I with bases and of basic compounds of formula I
with acids are manufactured by
(a) for the manufacture of a compound of formula I in
25 which X and Y both represent O, reacting a compound of the
general formula
Rd . O
RS
30 ~~ r~' . Ra
R- ~ ~Rs ~Rs
R~
(~2)m II
35 Rl/Z\kg
- 6 - 2010636
wherein R1. Rz. R3, R4, R5, R6. R~,
R8, Z and m have the significance given earlier.
with ammonia under pressure or with hexamethyldisilazane
and methanol to give a compound of formula I in which R
represents hydrogen or with hydroxylamine.to give a
compound of formula I in which R represents hydroxy, or
(b) for the manufacture of a compound of formula I in
which one of X and Y represents O and the other represents
(H~fi)~ reducing a compound of formula I in which X and Y
both represent O with lithium aluminium hydride, or
(c) if desired, functionally modifying a reactive centre
present in a compound of formula I obtained, and
(d) also if desired, converting an acidic compound of
formula I into a pharmaceutically acceptable salt with a
base or converting a basic compound of formula I into a
pharmaceutically acceptable salt with an acid.
The reaction of a compound of formula II with ammonia
under pressure in accordance with embodiment (a) of the
process is conveniently carried out using aqueous ammonia
(preferably 33% aqueous ammonia) and in the presence of a
water-miscible inert organic solvent such as dimethyl-
formamide or the like. The reaction is preferably carried
out at an elevated temperature, for example a temperature
in the range of about 100°C to about 150°C.
The reaction of a compound of formula.II with hexa-
methyldisilazane and methanol, also in accordance with
embodiment (a) of the process, is conveniently carried out
in an inert organic solvent such as a halogenated hydro-
carbon (e. g. chloroform, carbon tetrachloride or chloro-
3,5 benzene) or an aromatic hydrocarbon (e. g. benzene, toluene
~osos3s
or a xylene) and at an elevated temperature (e.g. a
temperature between about 40°C and 110°C.
The reaction of a compound of formula II with
hydroxylamine, also in accordance with embodiment (a) of
the process, is conveniently carried out in an inert
organic solvent such as dimethylformamide or the like and
at room temperature or an elevated temperature, preferably
at an elevated temperature (e. g. about 100°C).
Expediently, the hydroxylamine is used in the form of a
salt such as the hydrochloride and the reaction is carried
out in the presence of a base such as an alkali metal
carbonate (e. g. sodium or potassium carbonate).
The reduction of a compound of formula I in which X
and Y both represent O with lithium aluminium hydride in
accordance with embodiment (b) of the process is
expediently carried out in an inert organic solvent such
as an aliphatic or cyclic ether (e. g. diethyl ether,
tetrahydrofuran etc) at a temperature between about 0°C
and the reflux temperature of the reaction mixture.
A reactive centre present in a compound of formula I
can be modified,~if desired, in accordance with embodiment
(c) of the process. All of these modifications can be
carried out according to methods known per se. For
example, when Re represents a group of the formula
-(CH2)p-R9 in which R9 represents alkoxycarbonyl
and p stands for O, this group can be converted into a
corresponding group in which R9 represents hydrogen by
treatment with an acid. Aqain, for example, a group of the
formula -(CHZ)g-R10 in which R10 represents alkyl-
carbonyloxy can be converted into a corresponding group in
which R10 represents hydroxy by appropriate base
3,5 treatment. A group of the formula -(CHZ)q-R10 in
which R10 represents hydroxy can be converted into a
2osos3s
_8_
corresponding group in which R10 represents amino, mono-
alkylamino, dialkylamino, trialkylamino or a 5- or
6-membered saturated nitrogen-containing heterocycle
attached via the nitrogen atom by treatment firstly with
trifluoromethanesulphonic anhydride and subsequently with
ammonia, a monoalkylamine, a dialkylamine, a trialkylamine
or an appropriate heterocycle, respectively. A group of
the formula -(CH2)q-R10 in which R10 represents
hydroxy can be reacted with an alkanesulphonic anhydride
to give a corresponding group in which R10 represents
alkylsulphonyloxy. A group of the formula -(CHZ)q-R10
in which R10 represents alkylsulphonyloxy can be
converted into a corresponding group in which R10
represents formamido by reaction with ammonia in dimethyl-
formamide or in Which R10 represents azido by reaction
with an alkali metal azide or in which R10 represents a
group of the formula -U-C(V)-W in which U represents S, V
represents NH and W represents amino by reaction with
thiourea. Further, a group of the formula -(CH2)q-R
in which R10 represents azido can be converted by
catalytic hydrogenation into a corresponding group in
which R10 represents amino. A group of the formula
-(GEI2)q-R10 in which R10 represents alkoxy-
carbonylamino can be converted into a corresponding group
in which R10 represents amino by treatment with an acid.
A group of the formula -(CHZ)q-R10 in which R10
represents amino can be acylated to give a corresponding
group in which R10 represents acylamino or can be
reacted with 3,5-dimethyl-NZ-vitro-1-pyrazole-1-carbox-
amide to give a corresponding group in which R10
represents a group of the formula -U-(C(V)-W wherein U
represents NH, V represents NH and W represents NN02.
Further, a group of the formula -(CH2)q-R10 in which
R10 represents amino can be converted into a
corresponding group in which R10 represents isothio-
cyanato by reaction with 1.1-thiocarbonyldiimidazole. A
201036
- 9 -
group of the formula -(CFi2)p-R9 in which R9
represents cyano can be treated with hydrogen chloride and
subsequently with ammonia to give a corresponding group in
which R9 represents amidino. Again, for example, a
compound of formula I in which Z represents N and Ra
represents a group of the formula -(CHZ)p-R9 wherein
p stands for O and R9 represents hydrogen can be
converted into a corresponding compound in Which R9
represents alkylcarbonyl, alkoxycarbonyl or aralkoxy-
1p carbonyl by appropriate acylation, into a corresponding
compound in which R9 represents alkylsulphonyl by
reaction with an alkanesulphonyl chloride, into a
corresponding compound in which R9 represents amino-
alkylcarbonyl by treatment with a trifluoroacetamido-
alkanoyl chloride and subsequent reaction with ammonia,
into a corresponding compound in which. R9 represents
aminocarbonyl by treatment with 1,1-carbonyldiimidazole
and subsequent reaction with ammonia or into a
corresponding compound in which R9 represents aminothio-
carbonyl by treatment with 1,1-thiocarbonyldiimidazole and
subsequent reaction with ammonia. It will be appreciated
that the foregoing modifications are given by way of
example only and that other modifications within the
purview of a person skilled in the art are also possible.
The conversion of an acidic compound of formula T into
a pharmaceutically acceptable salt in accordance with
embodiment (d) of the .process can be carried out by
treatment with a suitable base in a manner known per se.
Suitable salts are those derived not only from inorganic
bases, for example, sodium salts, potassium salts, calcium
salts and the like, but also from organic bases such as
ethylenediamine, monoethanolamine, diethanolamine and the
like. The conversion of a basic compound of formula I into
a pharmaceutically acceptable salt, also in accordance
with embodiment (d) of the process, can be carried out by
- to -
2010636
treatment with a suitable acid in a manner known per se.
Suitable salts are those derived not only from inorganic
acids, for example, hydrochlorides, hydrobromides, ,
phosphates, sulphates and the like, but also from organic
acids, for example acetates, citrates, fumarates,
tartrates, maleates, methanesulphonates, p-toluene--
sulphonates and the like.
The compounds of formula II which are used as starting
materials in embodiment (a) of the process are novel and
form a further object of the present invention. They can
be prepared by reacting a compound of the general formula
-
a
R'
R6
III
Z
R1/ \R8
wherein R, R, R4, R5, R6, R~, Re, Z and m
have the significance given earlier,
with a compound of the general formula
HOOC-CHZ-R3 IV
wherein R3 has the significance given earlier,
and, Where required, functionally modifying a reactive
centre presont in a compound of formula II obtained.
2010636
- 11 -
The reaction of a compound of formula III with a
compound of formula IV is preferably carried out in the
presence of an acid-binding agent, expediently a tertiary
amine such as a trialkylamine (e. g. triethylamine,
diisopropylethylamine etc), and in an inert organic
solvent such as a halogenated aliphatic hydrocarbon
(dichloromethane etc) at about room temperature.
The optional functional modification of a reactive
substituent present in a compound of formula II can be
carried out in the same manner as described earlier in
connection with the functional modification of a reactive
centre present in a compound of formula I.
The compounds of formula III can be prepared, in turn.
by reacting a compound of the general formula
R4
RS
R6 ~ N ~RZ
R7 ' V
(CH2)m
Rl/ Z~R8
wherein Rl, R2. R4. R5~ R6, R~. Ra, Z
and m have the significance given earlier.
with oxalyl chloride, conveniently in an inert organic
solvent such as a halogenated aliphatic hydrocarbon (e. g.
dichloromethane etc) at a temperature from about 0°C to
the reflex temperature of the solvent. The resulting
compound of formula III can be reacted in situ with the
- 12 - X09"0636
compound of formula IV or can be isolated and purified
(e. g. by concentration followed by crystallization) prior
to the reaction with the compound of formula IV.
The compounds of formula V hereinbefore are known
compounds or analogues of known compounds which can be
prepared in a similar manner to the known compounds.
Further, certain of the Examples hereinafter contain
detailed information containing the preparation of the
1p respective starting materials.
The compounds of formula I and their pharmaceutically
acceptable salts are protein kinase inhibitors; they
inhibit cellular processes, for example cell prolifera-
15 lion, and can be used in the control or prevention of
illnesses, for example in the control or prevention of
inflammatory disorders such as arthritis, immune diseases,
in conjunction with organ transplants and also in
oncology. They inhibit infection of cells with human
20 immunodeficiency virus and are thus useful in the
treatment of AIDS. The compounds and salts of the present
invention also inhibit smooth muscle contraction and can
therefore be used against cardiovascular and broncho-
pulmonary disorders. Further, they are also useful in
25 asthma therapy.
The activity of the present compounds in inhibiting
protein kinase C can be demonstrated by means of the in
vitro assay system described e.g. in BBRC 19 (1979) 1218.
The IC50 figures in the following Table, represent
that concentration of test compound which reduces by 50%
the protein kinase-induced incorporation of 32P from
[Y-32P]ATP into histone.
- 13 -
Table
2010636
Compound IC50
3-[8-(Aminomethyl)-6,7,8,9-tetrahydro-
pyrido[1,2-a]indol-10-yl)-4-(1-methyl-3-
-indolyl)-iH-pyrrolet2,5-dione hydro-
chloride 8 nM
3-[7-(Amidinothiomethyl)-6.7.8.9-tetra-
hydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-
-3-indolyl)-1H-pyrrole-2,5-dione methane-
sulphonate 15 nM
3-[2-(Aminoacetyl)-1,2,3,4-~tetrahydro--
pyrazino[1,2-a]indol-10-yl]-4-(1-methyl-
-3-indolyl)-1H-pycrole-2.5-dione hydro-
20 Chloride 50 nM
3-[7-(2-Aminoethyl)-6,7,8,9-tetrahydro-
pyrido[1,2-a]-indol-10-yl]-4-(1-methyl-
-3-indolyl)-1H-pyrrole-2,5-dione hydro-
25 chloride ZO nM
3-[6,7.8.9-Tetrahydro-8-[(1-piperidino)--
methyl]pyrido[1,2-a]indol-10-yl]-4-(1-
-methyl-3-indolyl)-1H-pyrrole-2.5-dione 30 nM
3-[2,3-Dihydro-2-(dimethylaminomethyl)-
-1H-pyrrolo[1,2-a]indol-9-yl]--4-(1-methyl-
-3-indolyl)-7-H-pyrrole-2,5-dione trifluoro-
methanesulphonate ~ 20 nM
- 1 ~ - 20~.0~36
3-[e-Amidino-6,7,8,9-tetrahydropyrido-
[1,2-a]indol-10-yl]-4-(1-methyl-3-
-indolyl)-1H-pyrrole-2,5-dione hydro-
chloride 60 nM
10
3-[7-(Amidinothiomethyl)-6,7,8,9-tetra-
hydropyrido[1,2-a]indol-10-yl]-4-
-(1-methyl-3-indolyl)-1EI-pyrrole-2,5-dione
methanesulphonate 10 nM
The compounds of formula I and their aforementioned
salts can be used as medicaments, for example, in the form
of pharmaceutical preparations. The pharmaceutical
preparations can be administered orally, for example in
the form of tablets, coated tablets, dragees, hard and
soft gelatine capsules, solutions, emulsions or
suspensions. However, they can also be administered
rectally (e.g. in the form of suppositories) or
parenterally (e.g. in the form of injection solutions)
For the manufacture of pharmaceutical preparations the
compounds of formula I and their aforementioned salts can
be formulated with therapeutically inert, inorganic or
organic carriers. Lactose, maize starch or derivatives
thereof. talc, stearic acid or its salts and tha like can
be used, for example, as such carriers for tablets, coated
tablets, dragees and hard gelatine capsules. Suitable
carriers for soft gelatine capsules are, for example,
vegetable oils, waxes, fats, semi-solid and liquid polyols
and the like. Depending on the nature of the active
substance no carriers are, however, generally required in
the case of soft gelatine capsules. Suitable carriers for
the manufacture of solutions and syrups are, for example.
water, polyols, saccharose, invert sugar, glucose and the
like. Suitable carriers for injection solutions are, for
- 15 - 2030636
example, water, alcohols, polyols, glycerine, vegetable
oils and the like. Suitable carriers for suppositories
are, for example, natural or hardened oils, waxes, fats,
semi-liquid polyols and the like.
The pharmaceutical preparations can also contain
preserving agents, solubilizing agents, stabilizing
agents, wetting agents, emulsifying agents, sweetening
agents, colouring agents, flavouring agents, salts for
1p varying the osmotic pressure, buffers, coating agents or
antioxidants. They can also contain still other thera-
peutically valuable substances. Medicaments containing a
compound of formula I or a salt thereof as defined above
and a therapeutically inert carrier as well as a process
for the manufacture of such medicaments are also objects
of the present invention. This process comprises bringing
a compound of formula I or a salt thereof as defined above
into a galenical administration form together with a
therapeutically inert carrier material and, if desired,
2p one or more other therapeutically active substances.
As mentioned above, the compounds of formula I and
their aforementioned salts can be used in the control or
prevention of illnesses, especially in the control or
prevention of inflammatory, immunological, broncho--
pulmonary and cardiovascular disorders or for the
treatment of asthma or of AIDS. The dosage can vary within
wide .limits and will. of course, be adjusted to the
individual requirements in each particular case. In
gp general, in the case of oral administration to adults, a
daily dosage of about 5 mg to about 500 mg should be
appropriate, although the upper limit may be exceeded when
this is found to be expedient. The daily dosage can be
administered as a single dose or in divided doses.
The following Examples illustrate the present
invention:
- 16 - 20.0636
Example 1
A solution of 2.90 g of 3-[8-(acetoxymethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-
-indolyl)furan-2,5-dione in 30 ml of DMF and 23 ml of 33%
aqueous ammonia was heated to 140°C for 7 hours. The
mixture was extracted with ethyl acetate and the combined
organic extracts were washed with water, dried over
anhydrous sodium sulphate and evaporated to dryness.
Crystallization of the residue from ethyl acetate gave
1.87 g of 3-[6,7,8,9-tetrahydro-8-(hydroxymethyl)pyrido-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-
-dione in the form of a red solid of melting point
262-263°C.
The furandione starting material was prepared as
follows:
a) A solution of 25 g of ethyl indole-2-carboxylate in
400 ml of DMF was added to a stirred solution of 5.5 g of
a 60% dispersion of sodium hydride in mineral oil in 40 ml
of DMF under a nitrogen atmosphere. 30.9 g of ethyl
bromobutyrate were then added dropwise to the mixture at
0°C and the resulting mixture was stirred at room
temperature for 18 hours. The reaction was quenched with
100 ml of water and 30 ml of 2M hydrochloric acid and the
mixture was extracted with dichloromethane. The combined
organic extracts were washed with water, dried over
anhydrous sodium sulphate and evaporated to give 49 g of
3p an oil. This oil was dissolved in ethyl acetate and the
solution was washed with water, dried over anhydrous
sodium sulphate and evaporated to give 39 g of an oil.
This oil was added dropwise to a stirred suspension of
20.5 g of potassium t-butoxide in 750 ml of THF under a
nitrogen atmosphere. After 1 hour Z00 ml of water and then
92 ml of 2M hydrochloric acid were added. The mixture was
concentrated and the resulting precipitate was filtered
- 17 - 2010636
off and dried to give 25.3 g of ethyl 6,7-dihydro-9-
-hydroxypyrido[1,2-a]indole-8-carboxylate. A sample was
crystallized from methanol and gave crystals of melting
point 101-103°C.
b) A suspension of 19.4 g of the carboxylate of a) and
16 spoon spatula measures of Raney nickel in 480 ml of
ethanol and 240 ml of water was heated to reflux for
3.5 hours. A further 4 spoon spatula measures of Raney
nickel were then added and the mixture was heated to
reflux for a further 1.5 hours. The supernatant was
decanted off and the catalyst was washed with ethyl
acetate. The combined organic phases were concentrated and
the precipitate was~filtered off and dried to give 16.3 g
of ethyl 6.7,8,9--tetrahydropyrido[1,2-a]indole-8-
-carboxylate.. A sample was r_rystallized from methanol to
give a solid of melting point 70-72°C.
c) 16.2 g of the carboxylate of b) in 200 ml of THF were
added to a suspension of 2.00 g of lithium aluminium
hydride in 600 ml of THF at 0°C under a nitrogen
atmosphere. After 0.5 hour the reaction was quenched by
the successive additions of ethyl acetate, Water and 2M
hydrochloric acid and the mixture was extracted with
diethyl ether. The combined organic extracts were dried
and evaporated. Crystallization of the residue from
diethyl ether/n-hexane gave 11.5 g of 6,7.8,9-tetra-
hydro-8-(hydroxymethyl)pyrido[1,2-a]indole of melting
point 11.0-111°C.
d) 11.4 g of acetic anhydride were added to a solution of
11.0 g of the pyridoindole from c) in 100 ml of pyridine
and the resulting solution was stirred under a nitrogen
atmosphere for 18 hours. The majority of the pyridine was
3,5 removed by evaporation and the residue was acidified with
2M hydrochloric acid. The mixture was extracted with
diethyl ether and the combined extracts were washed with
- 18 - 201036
sodium bicarbonate solution and with water. The extracts
were dried and evaporated to dryness to give 11.25 g of
8-(acetoxymethyl)-6,7,8,9-tetrahydropyrido(1,2-a]indole of
melting point 63-64°C.
e) 4.13 g of oxalyl chloride were added dropwise to a
solution of 8.2 g of the tetrahydropyridoindole of d) in
160 ml of diethyl ether under a nitrogen atmosphere. After
minutes the solvent was removed under reduced pressure
10 and the residue was dissolved in 330 ml of dichloro-
methane. 6.34 g of 1-methyl-3-indolylacetic acid and
9.20 ml of triethylamine were added to this solution and
the mixture was stirred overnight. A further 4.60 ml of
triethylamine were added. After 4B hours, the solvent was
removed under reduced pressure and the residue was
purified by chromatography on silica gel with ethyl
acetate/petroleum ether (1:2). Crystallization from ethyl
acetate gave 4.02 g of 3-[e-(acet.oxymethyl)-6,7,8,9-tetra-
hydropyrido[1,2-a]indol-10--yl]-4-(1-methyl-3--indolyl)furan--
--2.5-dione of melting point 174-178°C.
Example 2
2.50 g of trifluoromethanesulphonic anhydride in
330 ml of dichloromethane were treated at 0°C under a
nitrogen atmosphere with a suspension of 1.87 g of
pyrroledione product of Example 1 and 0.94 q of collidine
in 280 ml of dichloromethane. After 2.5 hours, the mixture
was allowed to warm to 10°C. 37 ml of 33% aqueous ammonia
were then added and the mixture was allowed to warm to
room temperature overnight. The mixture was washed with
water, dried and evaporated. The residue was subjected to
chromatography on silica gel with dichloromethane/
methanol/acetic acid/water (90:18:3:2). The combined
3,5 product-containing fractions were treated with 2M
hydrochloric acid and evaporated to give 930 mg of
3-[8-(aminomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-
- 19 _ 2010636
-10-yl]-4-(1-methyl-3-indolyl)--1H-pyrrole-2,5-dione
hydrochloride of melting point 310-313°C.
Example 3
265 mg of trifluoromethanesulphonic anhydride in 40 ml
of dichloromethane were treated at 0°C under a nitrogen
atmosphere with a suspension of 200 mg of the pyrroledione
product of Example 1 and 100 mg of collidine in 30 ml of
dichloromethane. After 5 houcs 0.5 ml of a 33% solution of
trimethylamine in ethanol was added and the mixture was
stirred for 18 hours. The resulting precipitate was
filtered off and dried to give 237 mg of 3-[6,7,8,9-tetra-
hydro-8-[(trimethylammonio)methyl]pyrido[1,2-a]indol-10-yl]-
4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione trifluoro-
methanesulphonate of melting point 320-324°C.
Example 4
265 mg of trifluoromethanesulphonic anhydride in 40 ml
of dichloromethane were treated at 0°C under a nitrogen
atmosphere with a suspension of 200 mg of the pyrroledione
product of Example 1 and 100 mg of collidine in 30 ml of
dichloromethane. After 5 hours 0.75 mI of a 33% solution
of methylamine in methylated spirit was added and the
mixture was stirred for 18 hours. A further 0,5 ml of the
aforementioned methylamine solution was thon added. After
4 hours, the solvent was removed by evaporation and the
precipitate was filtered off and purified. by chromato-
graphy on silica gel with dichloromethane/methanol/acetic
acid/water (90:18:3:2). The solid product was stirred with
ethyl acetate saturated with hydrogen chloride for
2 hours. The resulting solid was filtered off and dried to
give 55 mg of 3-[6,7,8.9-tetrahydro-8-[(methylamino)--
'methyl]pyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-
-pyrrole-2,5-dione hydrochloride of melting point
337-340°C.
- 2 0 - 2010636
Example 5
185 mg of trifluoromethanesulphonic anhydride in 30 ml
of dichloromethane were treated at 0°C under a nitrogen
atmosphere with a suspension of 140 mg of the pyrroledione
product of Example 1 and 70 mg of collidine in 25 ml of
dichloromethane. After 1.5 hour 0.8 ml of a 33% solution
of dimethylamine in ethanol was added and the mixture was
stirred for 2.5 hours. The solvent was removed under
reduced pressure and the residue was triturated with
methanol to give a solid which was stirred with ethyl
acetate saturated with hydrogen chloride. The solid was
filtered off and dried to give 70 mg of 3-[6,7,8,9-tetra-
hydro-8-[(dimethylamino)methyl]pyrido[1,2-a]indol-10-yl]-
-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione hydrochloride
of melting point 335-336°C.
Example.6
A solution of 170 mg of the pyrroledione product of
Example 1 in 55 ml of dichloromethane was treated with
87 mg of methanesulphonic anhydride and 1 ml of pyridine.
The resulting solution was stirred under nitrogen for
1 hour. A further 30 mg of methanesulphonic anhydride were
then added. After 1 hour, the mixture was washed with
water, dried and evaporated. Crystallization of the
residue from ethyl acetate/n-hexane gave 150 mg of
3-[6,7,8.9-tetrahydro-8-(methylsulphonyloxymethyl)pyrido-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-
-dione of melting point 259-261°C.
Example 7
A solution of 120 mg of the pyrroledione product of
3,5 Example 6 in 6 ml of DMf and 6 ml of 33% aqueous ammonia
was heated to 140°C for 6 hours. The cooled mixture was
poured into water and the precipitate was filtered off.
- 21 _ 2010F36
The product was purified by chromatography on silica gel
with dichloromethane/acetic acid/methanol/water
(60:18:2:3). Trituration with ethyl acetate gave 50 mg of
3-[8--(formamidomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-
ZO-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5--dione of
melting point 332-334°C.
Example 8
A solution of 100 mg of the pyrroledione product of
Example 6 and 75 mg of thiourea in 5 ml of DMF was heated
to 80°C under a nitrogen atmosphere for 18 hours. The
solvent Was removed by evaporation and the residue was
purified by chromatography on silica gel with dichloro--
methane/methanol/acetic acid/water (90:18:3:2). The
residue was triturated with ethyl acetate to give 80 mg of
3-[8-[(amidinothio)methyl]-6,7,8,9-tetrahydropyrido[1,2-a]-
indol-10-yl]-4--(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione
methanesulphonate of melting point 200-205°C.
Example 9
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[7-(acetoxymethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-
-indolyl)furan-2,5-dione there was prepared 3-[6,7,8,9-
-tetrahydro-7-(hydroxymethyl)pyrido[1,2-a]indol-10-yl]-4-
-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione of melting
point 239-242°C.
The furandione starting material was prepared as
follows:
a) 6.6 ml of a 1.6M solution of n-butyllithium in
3,5 n-hexane were added to a stirred solution of 1.11 g of
diisopropylamine in 150 ml of THF at -78°C undez nitrogen.
The mixture was allowed to warm to -20°C for 5 minutes and
- 2 2 - 2010636
was then again cooled to -78°C. 1.85 g of 6,7,8,9-tetra-
hydropyrido[1,2-a]indol-6-one in 10 ml of THF were then
added dropwise. After stirring at -78°C for 0.5 hour
1.19 g of ethyl chloroformate were added and the mixture
was allowed to warm to room temperature. The solvent was
removed by evaporation and the residue was partitioned
between diethyl ether and 2M hydrochloric acid. The
ethereal extracts were washed with saturated sodium
bicarbonate solution, dried and concentrated to give an
oil. This oil was purified by chromatography on silica gel
with dichloromethane. Crystallization of the product from
methanol gave 1.35 g of ethyl 6,7,8,9-tetrahydro-6-oxo-
pyrido[1,2-a]indole-7-carboxylate of melting point 82-84°C.
b) 30 ml of a 1M solution of borane in TEIF were added to
a stirred solution of 1.25 g of the carboxylate of a) and
the resulting solution was heated to reflux for 2 hours
under a nitrogen atmosphere. 6 spoon spatula measures of
silica qel were added to the cooled solution and the
solvent was removed by evaporation. The residue was
purified by chromatography on silica gel with ethyl
acetate/n-hexane (1:1) to give an oil. This oil was
dissolved in 60 ml of dichloromethane containing 8 ml of
pyridine and 2 ml of acetic anhydride. After 1B hours, the
solution was washed with 60 ml of 2M hydrochloric acid and
20 ml of saturated sodium bicarbonate solution, dried and
evaporated to give an oil. A solution of this oil in 60 ml
of diethyl ether was treated with 630 mg oxalyl chloride
under a nitrogen atmosphere. Then the solvent was removed
under reduced pressure and the residue was dissolved in
100 ml of dichloromethane. 920 mg of 1-methyl-3-indolyl-
acetic acid and 975 mg of triethylamine were added to this
solution. After 72 hours, the solvent was removed by
evaporation and the residue was purified by chromatography
3,5 on silica gel with ethyl acetate/n-hexane (1:1), Crystal-
lization from ethyl acetate gave 390 mg of 3-[7-(acetoxy-
methyl)-6,7,8,9-tetrahydropyrido(1,2-a]indol-10-yl]-4-
__ 2 3 - iii~~~sa,~s
--(1-methyl-3'-~indolyl)furan-2.5-dione of melting point
190-193°C.
Example 10
200 mg of trifluoromethanesulphonic anhydride in 50 ml
of dichloromethane were treated at 0°C under a nitrogen
atmosphere with a suspension of 150 mg of the pyrroledione
product of Example 9 and 75 mg of collidine in 50 ml of
1p dichloromethane. After 2 hours 4 ml of 33% aqueous ammonia
were added and the mixture was left to warm to room
tempecature overnight. The mixture was washed with water,
dried and evaporated to dryness. The residue was purified
by chromatography on silica gel with dichloromethanel
methanol/acetone/water (90:18:3:2). Crystallization from
dichloromethane/n-hexane gave 85 mg of 3-[7-(aminomethyl)-
-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-
-3-indolyl)-1H-.pyrrole-2,5-dione of melting point
160-165°C.
Example 11
A solution of 120 mg of the pyrroledione product of
Example 9 in 80 ml of'dichloromethane was treated with
2 ml of pyridine and 100 mg of methanesulphonic anhydride
under a nitrogen atmosphere. After stirring for 18 hours
the mixture was washed with 2M hydrochloric acid and
saturated sodium bicarbonate solution, dried and
evaporated to give 130 mg of a gum. This gum was dissolved
in 40 ml of ethanol containing 200 mg of thiourea and tha
mixture was heated to reflux for 72 hours. The solvent was
removed by evaporation and the residue was purified by
chromatography on silica gel with dichloromethane/
methanol/acetone/water (90:18:3:2). Crystallization from
methanol/dichloromethane gave 30 mg of 3-(7-(amidinothio-
methyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-4-
-(1-methyl-3-indolyl)-1H-pyrrole-2.5-dione methanesul-
2 4 - 201.Ofi36
phonate of melting point 195-198°C.
Example 12
A solution of 72 mg of 3-(6,7,8,9-tetrahydropyrido-
(1,2-a]indol-10-yl)-4-(1-methyl-3-indolyl)furan-2,5-dione
in 5 ml of DMF and 5 ml of 33% aqueous ammonia was heated
to 140°C for 4 hours. The resulting crystals were filtered
off and dried to give 50 mg of 3-(6,7,8,9-tetrahydro-
pYrido[1,2-a]indol-10-yl)-4-(1-methyl-3-indolyl)-lEI-pyrrole-
2,5-dione of melting point 286-289°C.
The furandione starting material was prepared as
follows:
a) A solution of 1.03 g of ethyl 6,7-dihydro-9-
-hydroxypyrido[1,2-a]indole-8-carboxylate in 20 ml of
. ethanol, 10 ml of water and 10 ml of concentrated hydro-
chloric acid was heated at 80°C for 3 hours. The solvents
were evaporated to give 740 mg of 7,8-dihydropyrido-
[1,2-a]indol-9(6FI)-one of melting point 138-140°C.
b) A solution of 740 mg of the product of a), 600 mg of
hydrazine hydrate and 440 mg of potassium hydroxide in
2 ml of ethanol and 4 ml of diethylene glycol was heated
at 100°C under reflux for 1.5 hours. Then the mixture was
heated at 180°C for 2 hours. 50 ml of dichloromethane were
added and the organic phase was washed with 2M hydro-
chloric acid and water. The solvent was removed by
evaporation to give 405 mg of 6,7,8,9-tetrahydropyrido-
[1,2-a]i.ndole.
c) 350 mg of oxalyl chloride were added dropwise to a
solution of 450 mg of the product of b) in 13 ml of
3,5 dichloromethane at 0°C. After stirring for'2 hours the
solvent was removed by evaporation and the residue was
dissolved in dichloromethane. 497 mg of 1-methyl-3-
- 2 5 - 2p~,0636
-indolylacetic acid and 0.73 ml of triethylamine were
added to this solution and the mixture was stirred at room
temperature for 60 hours. The solvent was evaporated and
the residue was purified by chromatography on silica gel
with dichloromethane. Trituration of the product with
ethyl acetate gave 100 mg of 3-(6,7,8,9-tetrahydro-
pyrido[1,2-a]indol-10-yl)-4-_(1-methyl-3-indolyl)furan-2,5-
-dione in the form of a red solid of melting point
276-278°C.
Example 13
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[e-(2-acetoxyethyl)-
-6,7,8,9-tetrahydropyrido[1,2-a]indol-10--yl]-4--(1-methyl--
-3-indolyl)furan-2,5-dione there was prepared 3-[6,7,8,9-
-tetrahydro-8-(2-hydcoxyethyl)pyrido[1,2-a]indol-10-yl]-4-
-(1-methyl-3-indolyl)-lEI-pyrrole-2,5-dione of melting
point 261-263°C.
'fhe furandione starting material was prepared as
follows:
a) A solution of 6.52 g of 8-(2-acetoxyethyl)-6,7,8,9-
-tetrahydro-9-oxopyrido[i,2-a]indole in 48 ml of
dichloromethane was treated with 2.5 ml of ethanedithiol
and 3.13 ml of titanium tetrachloride. The resulting
solution was heated at refl:ux under nitrogen for 18 hours.
A further 4 ml of ethanedithiol and 9 ml of titanium
tetrachloride were added and heating was continued for
4.5 hours. The mixture was washed with water, dried over
and evaporated. The residue was purified by chromatography
on silica gel with ethyl acetate/petroleum ether (1:3) to
give 7.7 g of B'-(2-acetoxyethyl)-7~,8~-dihydrospiro-
3~ [1,3-dithiolane-2',9'(6~H)-pyrido[1,2-a]indole].
- 2 6 - 20a.0~36
b) A solution of 5 g of the product of a) in 200 ml of
ethanol was shaken with 8 spoon spatula measures of Raney
nickel for 3.5 hours. The mixture was filtered and the
filter residue was washed with ethanol. The combined
filtrate and washings were evaporated to dryness and the
residue was purified by chromatography on silica gel with
ethyl acetate! petroleum ether (1:2) to give 620 mg of
8-(2-acetoxyethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indole.
c) 1.19 g of oxalyl chloride were added dropwise to a
solution of 2.29 g of the product of b) in 50 ml of
diethyl ether at 0°C. After 2.5 hours the solvent was
removed by evaporation and the residue was dissolved in
dichloromethane. 1.68 g of 1-methyl-3-indolylacetic acid
and 2.45 ml of triethylamine were added to this solution
and the mixture was heated to reflux under nitrogen for
18 hours. The solvent was evaporated and the residue Was
purified by chromatography on silica gel with ethyl
acetate/petroleum ether (1:2). Crystallization from ethyl
acetate gave 625 mg of 3--[8-(2-acetoxyethyl)--6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-
furan-2,5-dione of melting point 159-161°C.
Examvle 14
A solution of 115 mg of 3-[8-.(2-acetoxyethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-
furan-2,5-dione in 1 ml of DMF arid 2 ml of 33% aqueous
ammonia was heated to 140°C for 4 hours. The cooled
mixture was evaporated and the residue was purified by
chromatography on silica gel with ethyl acetate/petroleum
ether (2:1). CrysCallization from ethyl acetate/petroleum
ether gave 13 mg of 3-(8-(2-acetoxyethyl)-6,7,8,9-tetra-
hydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-
-PYrrole-2,5-dione of melting point 272-274°C.
2o~.os3s
- 27 -
Example 15
A solution of 500 mg of the pyrroledione product of
Example 13 in 50 ml of dichloromethane was treated with
218 mg of methanesulphonic anhydride and 1 ml of pyridine.
The resulting solution was stirred at room temperature
under a nitrogen atmosphere for 1 hour. A further 20 mg of
methanesulphonic anhydride were then added and stirring
was continued for 0.5 hour. The mixture was washed with
water, dried and evaporated. Crystallization of the
residue from ethyl acetate/pettoleum ether gave 540 mg of
3-[6,7,8,9-tetrahydto-8-(2-methylsulphonyloxy-ethyl)pyrido-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-lfl-pyrrole-2,5-
-dione of melting point 244-245°C.
Example l6
A solution of 500 mg of the pyrroledione product of
Example 15 and 250 mg of sodium azide in 10 ml of DMF was
heated at 70°C for 3 hours. The solvent was removed by
evaporation and the solid was partitioned between ethyl
acetate and water. The insoluble material was filtered off
and dried to give 425 mg of 3-[B-(2-azidoethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-
1H-pyrrole-2,5-dione of melting point 262-264°C.
Example 17
200 mg of the pyrroledione product of Example 16 in
7-0 ml of methanol containing 40 mg of l0% Pd/C were shaken
under a hydrogen atmosphere at a pressure of 3 atmospheres
for 48 hours. The supernatant was decanted off and
evaporated. The residue was treated with 50 ml of a
saturated solution of hydrogen chloride in ethyl acetate
3,5 and was then purified by chromatography on silica gel with
dichloromethane/methanol/acetic acid/water (60:18:2:3).
Crystallization from ethyl acetate gave 20 mg of
- 2 s - 20.0636
3-[8-(2-aminoethyl)-6,7,8.9-tetrahydropyrido[1,2-a]-
indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione
of melting point 160-165°C.
Example 18
In a manner analogous to that described in the first
paragraph of Example 12, from 3-[2.3-dihydro-lEI-pyrrolo-
[1.2-a]indol-9-yl]-4-(1-methyl-3--indolyl)furan-2,5-dione
there was obtained 3-[2.3-dihyaro-lEI-pyrrolo[1,2-a]indol-
-9-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2.5-dione of
melting point 260-270°C.
The furandione starting material was prepared as
follows:
1'75 mg. of oxalyl chloride were added dropwise to a
solution of 200 mg of 2,3-dihydro.-lEI-pyrrolo[1,2-a]indole
in 7 ml of diethyl ether at 0°C under a~nitrogen
atmosphere. After 1 hour the solvent was removed under
reduced pressure and the residue was dissolved in 14 ml of
dichloromethane. 245 mg of 1-methyl-3-indolylacetic acid
and 265 mg of triethylamine were added to this solution
and the mixture was stirred at room temperature for
72 hours. The solvent was removed under reduced pressure
and the residue was purified by chromatography on silica
gel with ethyl acetate/petroleum ether (1:2). Crystalli-
zation from ethyl acetate gave 70 mg of 3-[2.3-dihydto-
-1H-pyrrolo[1,2-a]indol-9-yl]-4-(1-methyl-3-indolyl)furan-
-2,5-dione of melting point 125-130°C,
Example 19
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[2-(acetoxymethyl)-2,3-
-dihydro-1H-pyrrolo[1,2-a]indol-9-yl]-4-(1-methyl-3-
--indolyl)furan-2,5-dione there was prepared 3-[2.3-
- 29 -
201.0636
-dihydro-2-(hydroxymethyl)-lEI-pyrrolo[1,2-a]indol-9-yl]-4-
-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione of melting
point 238-240°C.
The furandione starting material was prepared as
follows:
a) 6 spoon spatula measures of Raney nickel were added to
a solution of 5.08 g of ethyl 2.3~-dihydro-1-oxo-1H-
-pY~rolo(1,2-a]indole-2-carboxylate in 180 ml of ethanol
and 90 ml of water. The mixture was heated to reflux for
10 hours and then a further 3 spoon spatula measures of
Raney nickel were added. Heating was continued for
5.5 hours, whereupon the mixture was cooled and filtered.
Z'he filter residue was washed with ethyl acetate and
dichloromethane. The combined filtrate and washings were
evaporated and the residue was purified by chromatography
on silica gel with diethyl etherl.petroleum ether (1:2).
Crystallization from methanol gave 635 ing of ethyl
2,3-dihydro-lEI--pyrrolo[1,2-a]indole-2-carboxylate of
melting point 55-57°C.
b) 4 ml of a 1M solution of lithium aluminium hydride in
TEIF were added to a solution of 750 mg of ethyl
2,3-dihydro-1EI-~pyrrolo(1,2-a]indole-2-carboxylate in 30 ml
of TEIF. After 1 hour 30 ml of saturated ammonium chloride
solution were added and the mixture was evaporated. The
residue was extracted with dichloromethane and the organic
extract was dried and evaporated. Crystallization of tho
residue from diethyl ether/petroleum ether gave 355 mg of
2,3-dihydro-2-(hydroxymethyl)-lEI-pyrrolo[1,2-a]indole of
melting point 76-78°C.
c) A solution of 355 mg of the product of b) in 20 ml of
dichloromethane containing 2 ml of acetic anhydride and
2 ml of pyridine was stirred for 2 hours. The solvents
were evaporated and the residue was partitioned between
- 30 -
2osos3s
dichloromethane and water. The organic phase was dried and
evaporated to give 420 mg of 2-(acetoxymethyl)-2,3-
-dihydro-lEI-pyrrolo[1,2-a]indole.
d) 290 mg of oxalyl chloride were added dropwise to a
solution of 420 mg of the product of c) in 14 ml of-
diethyl ether under a nitrogen atmosphere. After 1 hour
the solvent was removed under reduced pressure and the
residue was dissolved in dichloromethane. 420 mg of
1-methyl-3-indolylacetic acid and 485 mg of triethylamine
were added to this solution and the mixture was stirred
for 72 hours. The solvent was evaporated and the residue
was purified by chromatography on silica gel with ethyl
acetate/petroleum ether (1:1). Crystallization from ethyl
acetate gave 90 mg of 3-[2-(acetoxymethyl)--2,3-dihydro-
-lEI-pyrrolo(1,2-a]indol-9-yl]-4-(1-methyl-3-indolyl)furan--
-2,5-dione of melting point 208-211°C.
Example 20
A solution of 150 mg of 3-[2-t-butoxycarbonyl-1.2,3.4-
-tetrahydropyrazino[1,2-a]indol-10--yl]-4-(1-methyl-3-
-indolyl)furan-2,5-dione in 4 mI of DMF and a ml of 33%
aqueous ammonia was heated to 140°C for 4 hours. The
mixture was extracted with ethyl acetate and the organic
extract was washed with water, dried and evaporated to
give a gum. Purification was effected by chromatography on
silica gel with dichloromethane/methanol/acetic acid/
water. The resulting amide was dissolved in 30 ml of
ethanol and 5 ml of 2M hydrochloric acid and the resulting
solution was heated to reflux for 2 hours. Removal of the
solvent by evaporation and trituration of the residue with
ethyl acetate gave 35 mg of 3-[1.2,3,4-tetrahydropyrazino-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-
-2.5-dione hydrochloride of melting point 268-270°C.
2010636
-- 31 -
The furandione starting material was prepared as
follows:
a) A solution of 450 mg of 1,2,3,4-tetrahydropyrazino--
[1,2-a)indole in 30 ml of dichloromethane was treated at
0°C under a nitrogen atmosphere with 303 mg of triethyl-
amine and 615 mg of di(t-butyl) dicarbonate. The mixture
was stirred at 0°C for 4 hours and then washed with
saturated sodium bicarbonate solution, dried and
evaporated to give an oil. Crystallization from methanol
gave 580 mg of t-butyl 1,2,3,4-tetrahydropyrazino[1,2-a]-
indole-2-carboxylate of melting point 103-105°C.
b) 230 mg of oxalyl chloride were added dropwise to a
~5 stirred solution of 450 mg of the product of a) in 30 ml
of diethyl ether at 0°C. After stirring, the solution was
evaporated and the residue was dissolved in 50 ml of
dichloromethane. 360 mg of 1-methyl-3-indolylacetic acid
and 350 mg of triethylamine were added and the mixture was
20 stirred for 90 hours. The solvent was evaporated and the
residue was purified by chromatography on silica gel with
ethyl acetate/petroleum ether (2:3)to give 180 mg of a
gum. A sample was crystallized from ethyl acetate/n-hexane
to give 3-[2-t-butoxycarbonyl-1,2,3,4-tetrahydropyra-
25 zino[1,2-a]indol-10-yl)-4-(1-methyl-3-indolyl)furan-2,5-
-dione of melting point 125-127°C.
Examvle 21
30 In a manner analogous to that described in the first
paragraph of Example 12, from 3-(S,6-dihydro-4H-pyrrolo-
[3,2,1-ij]quinolin-1-yl)-4-(1-methyl-3-indolyl)furan-2,5-
-dione there was prepared 3-(5,6-dihydro-4II-pyrcolo-
[3,2,1-ij)quinolin-1-yl)-4-(1-methyl-3-indolyl)-1H-pyrrole-
3,5 -2,5-dione of melting point 285-288°C.
2010636
- 32 -
The furandione starting material was prepared as
follows:
1.22 g of oxalyl chloride were added dropwise to a
solution of 1.5 g of 5.6-dihydro-4H-pyrrolo[3,2,1-ij]-
quinoline in 60 ml of dichloromethane under a nitrogen
atmosphere. After 1 hour the solvent was removed under -
reduced pressure and the residue was dissolved in 120 ml
of dichloromethane. 1.9 g of 1-methyl-3-~indolylacetic acid
and 2.02 g of triethylamine were added to this solution
and the mixture was stirred for 18 hours. The solvent was
removed under reduced pressure and the residue was
purified by chromatography on silica gel with ethyl
acetate/petroleum ether (1:2). Further purification by
chromatography was effected with dichloromethane.
Crystallization from ethyl acetate gave 690 mg of
3-(5,6-dihydro-4H-pyrrolo[3,2.1-ij]quinolin-1-yl)-4-
-(1-methyl-3-indolyl)furan-2,5-dione of melting point
217-219°C.
Example 22
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[5-(acetoxymethyl)-5,6-
-dihydro-4H-pyrrolo[3.2,1-ij]quinolin-1-yl]-4-(1-methyl-3-
-indolyl)furan-2,5-dione there Was prepared 3-[5,6-
-dihydco-5-(hydroxymethyl)-4H-pyrrolo[3.2,1-ij]quinolin-1-
-yl]-4-(1-methyl-3-indolyl)-lEI-pyrrole-2,5-dione of
melting point 223-225°C.
The furandione starting material was prepared as
follows:
a) 33.4 ml of a 1.6M solution of n-butyllithium in hexane
were added to a solution of 8.13 ml of diisopropylamine in
420 ml of TEIF at -78°C under a nitrogen atmosphere. After
0.5 hour 4.6 g of 1,2.5,6-tetrahydro-4-oxo-~4FI-pyrrolo-
3 3 - 2o~.os3s
[3,2,1-ij)quinoline were added and the mixture was stirred
at -78°C for 0.5 hour. 2.77 ml of ethyl chloroformate were
added and stirring was continued for 1 hour. The reaction
was quenched with water and the mixture was evaporated.
The residue was purified by chromatography on silica gel
with ethyl acetate/petroleum ether (1:2). Crystallization
from diethyl ether gave 2.8 g of ethyl 1,2,5,6-
-tetrahydro-4-oxo-4FI-pyrrolo[3,2,1-ij]quinoline-5-
-carboxylate of melting point 88-90°C.
b) 15 ml of a 1M solution of borane in TEIF were added to
a solution of 2.8 g of the product of a) in 100 ml of TEIF
and the resulting solution was heated to reflux for
2 hours. A further 55 mh of borane were added and heating
was continued for 12 hours. The solvent was removed under
reduced pressure, water and 2M hydrochloric acid were
added and the mixture was extracted with dichloromethane.
The solvent was evaporated and the residue was dissolved
in diethyl ether. The solution obtained~was treated with
12 ml of a 1M solution of lithium aluminium hydride in
diethyl ether and the mixture was stirred under a nitrogen
atmosphere for 18 hours. Water was added and the mixture
was extracted with dichloromethane. Removal of the solvent
under reduced pressure gave 1.4 g of 1,2,5,6-tetrahydro-
-4H-pYrrolo[3.2.1-ij]quinoline-5-methanol.
c) A solution of 1.4 g of the product of b) in 50 ml of
dichloromethane was treated with 9 ml of acetic anhydride
and 2 ml of pyridine. After 4 hours a further 4 ml of
acetic anhydride were added and the mixture was stirred
for 18 hours. Ths solvent was removed under reduced
pressure and the residue was partitioned between water and
dichloromethane. The organic phase was evaporated and the
residue was dissolved in toluene and heated to reflux in
3,5 the presence of 250 mg of 10% Pd/C for 18 hours. A further
250 mg of 10% Pd/C were then added and heating was
continued for a further 20 hours. The mixture was filtered
- 3 4 - '~ro9..~~a,~~
and the filtrate was evaporated. The residue obtained was
purified by chromatography on silica gel with ethyl
acetate/petroleum ether (1:2) to give 350 mg of 5-(ace-
toxymethyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinoline.
d) 315 mg of oxalyl chloride were added to a solution of
570 mg of the product of c) in 15 ml of dichloromethane
under a nitrogen atmosphere. The solvent was removed under
reduced pressure and the residue was dissolved in
dichloromethane. 472 mg of 1-methyl-3-indolylacetic acid
and 505 mg of triethylamine were added and the mixture was
stirred for 72 hours. The solvent was removed under
reduced pressure and the residue was purified by chromato-
graphy on silica gel with dichloromethane. Crystallization
y5 from ethyl acetate/n-hexane gave 140. mg of 3-[5-(acetoxy-
methyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]quinolin-1-yl]-4-
-(1-methyl-3-indolyl)furan-2,5-dione solid of melting
point 198-200°C.
20 Example 23
In a manner analogous to that described in Example 11,
from the pyrroledione product of Example 22 there was
prepared 3-[5-(amidinothiomethyl)-5,6-dihydro-4H-pyrrolo-
25 (3,2,1-ij]quinolin-1-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole
-2,5-dione methanesulphonate of molting point 190-195°C.
Example 24
30 In a manner analogous to that described in Example 2,
from the pyrroledione product of Example 22 there was
prepared 3-(5-(aminomethyl)-5,6-dihydro-4H-pyrrolo-
(3,2,1-ij]quinolin-1-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-
-2,5-dione hydrochloride of melting point 248-250°C.
- 35 -
Example 25
20~,0~3f
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[8-(acetoxymethyl)-6.7.8,9-
-tetrahydropyrido[1.2-a]indol-10-yl]-4-phenylfuran-2,5-
-dione (obtained as described in the last paragraph of
Example 1 by using phenylacetic acid in place of 1-methyl-
-3-indolylacetic acid) there was prepared 3-[6.7,8,9-
-tetcahydro-8-(hydroxymethyl)pyrido[1,2-a]indol-10-yl]-4-
-phenyl-1H-pyrrole-2.5-dione of melting point 276-278°C.
Examine 26
In a manner analogous to that described in the first
Paragraph of Example 1, from 4-[8-(acetoxymethyl)-6,7.8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-3-(3-benzo[b]thienyl)-
furan-2,5-dione (obtained as described in the last
paragraph of Example 1 by using 3-benzo[b]thienylacetic
acid in place of 1-methyl-3-indolylacetic acid) there was
prepared 3-(3-benzo[b]thienyl)-4-[6.7,8,9-tetrahydro-8--
-(hydroxymethyl)pyrido[1,2-a]indol-10-yl]-1H-pyrrole-2.5-
-dione of melting point 226-7.27°C.
Example 27
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[8-(acetoxymethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-naphthyl)furan-
-2,5-dione (obtained as described in the last paragraph of
Example 1 by using 1-naphthylacetic acid in place of
1-methyl-3-indolylacetic acid) there was prepared
3-[6,7,8,9-tetrahydro-e-(hydroxymethyl)pyrido[1,2-a]indol-
-10-yl]-4-(1-naphthyl)-1H-pyrrole-2.5-dione of melting
point 221-222°C.
36 -
Example 28
201.0636
In a manner analogous to that described in Example 10,
from the pyrroledione product of Example 19. there was
prepared 3-[2-(aminomethyl)-2,3-dihydro-1H-pyrrolo[1,2-a]-
indol-9-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione of
melting point 208-211°C.
Example 29
In an analogous manner to that described in
Example 10, from the pyrroledione product of Example 25,
there was prepared 3-(8-(aminomethyl)-6,7,8,9-tetrahydro--
pyrido[1,2-a]indol-10-yl]-4-phenyl-1H-pyrrole-2,5-dione of
melting point 249-250°C.
Example 30
A suspension of 100 mg of the pyrro2edione product of
Example 20 in 10 ml of dichloromethane was treated under
nitrogen with 0.08 ml of triethylamine and 86 mg of phenyl
chloroformate. The mixture was stirred for 2 hours and
then the solvent was evaporated. Chromatography of the
residue on silica gel with ethyl acetate/n-hexane (1:1)
gave a gum which was dissolved in a mixture of 5 ml of
isopropanol and 10 ml of 33~ aqueous ammonia. The mixture
was diluted with water and extracted with dichloromethane.
The combined dichloromethane extracts were dried and,
evaporated. Crystallization of the residue from ethyl
acetate/n-hexane gave 45 mg of 3-[1:2,3,4-tetrahydro-2-
-(phenoxycarbonyl)pyrazino[1,2-a]indol-10-yl]-4-(1-methyl-
-3-indolyl)-1H-pyrrole-2,5-dione of melting point
160-165°C.
3 7 20~.0~36
Example 31
a) A solution of 80 mg of the pyrroledione product of
Example 20 in 20 ml of dichloromethane was treated with
10 ml of 5% aqueous sodium hydrogen carbonate. The stirred
mixture was treated with a solution of 125 mg of
trifluoroacetamidoacetyl chloride in 5 ml of dichloro-
methane. After 17 hours, the phases were separated and the
organic phase was dried and evaporated. Chromatography of
the residue on silica gel with ethyl acetate/n-hexane
(2:1) and crystallization from ethyl acetate/n-hexane gave
70 mq of 3-[2-[(trifluoroacetamido)acetyl]-1,2,3,4-tetra--
hydropyrazino[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-
-1H-pyrrole-2,5-dione of melting point 170-172°C.
b) A solution of 65 mg of the product of a) in 10 ml of
methanol was treated with 5 ml of 33% aqueous ammonia.
After 4 hours the solvent was cemoved by evaporation and
the residue was partitioned between dichloromethane and
water. The organic phase was washed with water, dried and
evaporated. Chromatography of the residue on silica gel
- with chloroform/methanol/acetic acid/water (60:18:2:3)
gave a gum which was dissolved in glacial acetic acid and
treated with 20 ml of lM hydrochloric acid. Evaporation of
the solvent and trituration of the residue with diethyl
ether gave 35 mg of 3-[2-(aminoacetyl)-1.2,3,4-tetrahydro-
pyrazino[1,2-a]indol-10-yl]-9-(1-methyl-3-indolyl)-1H-
-pyrrole-2,5-dione hydrochloride of melting point 235°C
(decomposition).
Example 32
a) A solution of 100 mg of the pyrroledione product of
Example 20 in 40 ml of dichloromethane was treated under a
nitrogen atmosphere with 125 mg of 1,1-carbonyldiimidazole
and the mixture was stirred for 24 hours. The solution was
washed with water, dried and evaporated. Trituration of
- 38 -
20.0636
the residue with ethyl acetate gave 84 mg of 3-[1,2,3,4-
-tetrahydro-2-(1-imidazolylcarbonyl)pyrazino[1,2-a)indol-10-
yl]-4-(1-methyl-3-indolyl)-lI-I-pyrrole-2,5-dione of melting
point 295°C (decomposition).
b) 80 mg of the product of a) were dissolved in a mixture
of ZO ml of DMF and 20 ml of 33~ aqueous ammonia. The
mixture was stirred for 17 hours and the solvent was
evaporated. Chromatography of the residue on silica gel
with methanol/ethyl acetate (1:9) gave a solid which was
crystallized from methanol. There were obtained 45 mg of
3-[2-carbamoyl--1,2,3,4-tetrahydropyrazino(1.2-a]indol-
-10-yl]-4-(1-methyl-3-indolyl)-lI-I-pyrrole-2,5-dione of
melting point 295°C (decomposition).
Example 33
A solution of 505 mg of the pyrroledione product of
Example 2 in 20 ml of DMF was treated with a solution of
222 mg of 1,1-thiocarbonyldiimidazole in 5 ml of THF.
After 17 hours the solvent was evaporated and the residue
was purified by chromatography on silica gel with
methanol/dichloromethane (1:99). Trituration with n-hexane
gave 297 mg of 3-[6,7.8,9-tetrahydro-8-isothiocyanato-
pYrido[1.2-a]indol-10-yl]-4-(1-methyl-~3-indolyl)-1PI-pyrrole-
2,5-dione in the form of a red solid of melting point
285-287°C.
Examule 34
250 mg of the pyrroledione product of Example 2 were
stirred in a mixture of 25 ml of dichloromethane and 15 ml
of 5% aqueous sodium hydrogen carbonate. The mixture was
treated with 1 ml of benzoyl chloride and stirred foc
17 hours. The phases were separated and the organic phase
was dried and evaporated. Chromatography of the residue on
silica gel with methanol/dichloromethane (7:93) followed
- 39 -
201.0636
by trituration with n-hexane gave 220 mg of 3-[8-(benz-
amidomethyl)-6,7.8.9-tetrahydropyrido[1,2-a]indol-10-yl]-
-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione of melting
point 297-303°C.
Example 35
A solution of 150 mg of 3-[7-acetoxy-6.7,8,9-tetra
hydropyrido[1.2-a]indol-10-yl]-4-(1-methyl-3-indolyl)furan
-2.5-dione in 6 ml of DMF and 6 ml of 33% aqueous ammonia
was heated to 150°C for 6 hours. The mixture was extracted
with ethyl acetate and the organic extracts were washed
with water, dried and evaporated. Crystallization of the
residue from ethyl acetate gave 120 mg of 3-[6,7,8,9-
wtetrahydro-7-hydroxypyrido[1,2-a]indol-10--y1]-4-(1-methyl-
-3-indolyl)-1H-pyrrole-2.5-dione of melting point
252-255°C.
The furandione starting material was prepared as
2~ follows:
a) A solution of 14.0 g of indole-2-methanol in 500 ml of
dichloromethane was stirred with 76.4 g of activated
manganese-IV oxide. After 1 hour the solid was filtered
off and washed with dichloromethane. The combined washings
were concentrated and 33 g of (carbethoxymethylene)-
triphenylphosphorane Were added. The resulting solution
was heated to reflux under a nitrogen atmosphere. The
solvent was evaporated to give an oil which was purified
3p by chromatography on silica gel with ethyl acetate!
n-hexane (1:3). The product was obtained as a 20:1 mixture
of E!Z isomers. Crystallization from methanol gave 11.3 g
of ethyl (E)-2-indolyl-2-propenoate of melting point
120-122°C.
b) A solution of 7.2 g of ethyl (E)-2-indolyl-2-
-propenoate in 120 ml of DMF was treated with 1.47 g of a
- 40 -
2010636
60% dispersion of sodium hydride in mineral oil. The
resulting solution was cooled to 0°C and 7.17 g of t-butyl
bromoacetate were added under an atmosphere of nitrogen.
After 2 hours the mixture was poured into 100 ml of 2M
hydrochloric acid and extracted with ethyl acetate. The
combined organic extracts were washed with water, dried
and evaporated to give an oil. This oil was purified by
chromatography on silica gel with diethyl ether/petroleum
ether (1:3). Crystallization from diethyl ether/n-hexane
gave 8.1 g of ethyl (E)-3-(1-t-butoxycarbonylmethyl)
-2-indolyl]-2-propenoate of melting point 66-68°C.
c) A solution of 8.0 g of the product of b) in 300 ml of
ethanol was shaken with 800 mg of 10% Pd/C under a
hydrogen atmosphere. The catalyst was filtered off and
washed with ethyl acetate. The combined filtrate and
washings were evaporated to give an oil which was
dissolved in TFIF. The solution was added to a solution of
2.8 g of potassium t-butoxide in THF under a nitrogen
atmosphere. Then the mixture was left tc stir for 1 hour
and the solvent was evaporated. The residue was
pactitioned between ethyl acetate and 2M hydrochloric
acid. The organic phase was washed with water, dried and
evaporated. The residue was purified by chromatography on
silica gel with diethyl ether/n-hexane (1:4). There are
obtained 4.55 g of t-butyl 6,7,8,9-tetrahydro-7-oxo-
-pyrido[I,2-a]indole-6-carboxylate.
d) A solution of 4.5 g of the product of c) in 200 ml of
toluene was treated with four spoon spatula measures of
silica gel and the mixture was heated to reflux for
3 hours under a nitrogen atmosphere. The solid was
filtered off and washed with toluene. The combined
filtrate and washings were evaporated to give a solid.
Crystallization from diethyl ether/n-hexane gave 2.5 g of
8.9-dihydropyrido[1,2-a]indol-7(6H)-one of melting point
126-128°C.
- 41 - 2010636
e) 190 mg of sodium borohydride were added to a stirred
solution of 650 mg of 8,9-dihydropyrido[1.2-a]indol--
-7(6EI)-one in 50 ml of methanol under a nitrogen
atmosphere. The mixture was stirred and then poured into
100 ml of saturated ammonium chloride solution. The
mixture was extracted with ethyl acetate and the combined
extracts were dried and evaporated to give a solid. This
was crystallized from diethyl ether/n-hexane and gave
500 mg of 6.7,8,9-tetrahydro-7-hydroxypyrido[1,2-a]indole
of melting point 99-100°C.
f) A solution of 500 mg of the product of e) in 5 ml of
pyridine and 2 ml of acetic anhydride was stirred for
8 hours. The mixture was poured into 50 ml of 2M
hydrochloric acid and extracted with ethyl acetate. The
combined orgahic extracts were washed with 5% sodium
bicarbonate solution and water, dried and evaporated to
' give 520 mg of an oil. A sample was crystallized from
diethyl ether/n-hexane. and there was obtained
7-acetoxy-6.7.8,9-tetrahydropyrido[1,2-a]indole of melting
point 90-95°C.
g) 320 mg of oxalyl chloride were added to a solution of
500 mg of the product of f) in 50 ml of diethyl ether
under a nitrogen atmosphere. Then the solvent was removed
under reduced pressure and the residue was dissolved in
50 ml of dichloromethane. 378 mg of 1-methyl-3-indolyl-
acetic acid and 505 mg of triethylamine were added to this
solution and the mixture was stirred for 72 hours. The
solvent was removed under reduced pressure and the residue
was purified by chromatography on silica gel with ethyl
acetate/n-hexane (1:1). Crystallization from ethyl acetate
gave 160 mg of 3-[7-acetoxy-6,7,8,9-tetrahydropyrido-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)furan-2.5-dione
of melting point 272-275°C.
- 4 2 - 2010636
Example 36
A solution of 85 mg of 3-[7-t-butoxyfocmamido-6,7,8.9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-
-indolyl)furan-2.5-dione in 5 ml of DMF and 5 ml of 33%
aqueous ammonia was heated to 100°C for 1 hour. The cooled
mixture was pactitioned between ethyl acetate and water.
The organic phase was washed with water, dried and
evaporated. Crystallization from ethyl acetate/n-hexane
gave 70 mg of 3-[7-t-butoxyformamido-6,7,8.9-tetrahydro-
pyrido[1.2-a]indol-.10-yl]-4-(1-methyl-3-indolyl)-1FI-pyrrole-
2.5-dione of melting point 159-163°C.
The furandione starting material was prepared as
15 follows:
a) A suspension of 555 mg of 8.9-dihydropyrido[1.2-a]-
indol-?(6FI)-one and 4.62 g of ammonium acetate in 15 ml of
methanol was treated with 250 mg of sodium cyanoboro-
20 hydride. The mixture was stirred and then partitioned
between ethyl acetate and water. The organic phase was
dried and the solvent was removed under reduced pressure.
The residual oil was subjected to chromatography on silica
gel with 10% methanol in dichloromethane. The indoline
2b obtained was dissolved in toluene and heated to reflux
with 50 mg of 10% Pd/C for 4 hours. The catalyst was
filtered off and washed with toluAne. The combined
filtrate and washings were evaporated to give 170 mg of
7-amino-6,7,8,9-tetrahydropyrido[1,2-a]indole.
b) 225 mg of di-t-butyl dicarbonate were added to a
stirred solution of 175 mg of the product of a) and 112 mg
of triethylamine in 20 ml of diehloromethane at 0°C under
a nitrogen atmosphere. After 18 hours the solution was
3,5 washed with saturated sodium bicarbonate solution, dried
and evaporated to give an oil. Crystallization from
diethyl ether gave 240 mg of 7-t-butoxyformamido--6,7.8,9-
- 43 -
2010636
-tetrahydropyrido[1,2-a]indole of melting point 137-139°C.
c) 127 mg of oxalyl chloride were added to a solution of
240 mg of the product of b) in 30 ml of diethyl ether
under a nitrogen atmosphere. After 10 minutes the solvent
was removed under reduced pressure and the residue was
dissolved in 30 ml of dichloromethane. 170 mg of 1-methyl-
-3-indolylacetic acid and 200 mg of triethylamine were
added to the resulting solution and the mixture was
stirred for 72 hours. The solvent was removed under
reduced pressure and the residue was purified by chromato-
graphy on silica gel with ethyl acetate/n-hexane (1:2).
Crystallization from ethyl acetate/n-hexane gave 100 mg of
3-(7-t-butoxyformamido-6,7.8,9-tetrahydropyrido[1,2-a]indol-
10-yl]-4-(1-methyl-3-indolyl)furan-2,5-dione of melting
point 141-145°C.
Example 37
A saturated solution of hydrogen chloride in 30 ml of
ethyl acetate was added to a stirred suspension of 60 mg
of the pyrroledione product of Example 36 in 50 ml of
ethyl acetate and the mixture was stirred for 18 hours.
The solvent was removed under reduced pressure and the
z5 residue was triturated with ethyl acetate to give 35 mg of
3-[7-amino-6,7,8.9-tetrahydropyrido[1.2-a]indol-l0-yl]-4-(1-
methyl-3-indolyl)-1H-pycrole-2,5-dione hydrochloride of
melting point 260-265°C.
30 Example 38
A solution of 80 mg of 3-(8-t-butoxyformamido-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-tl-methyl-3-indolyl)-
fucan-2,5-dione in 2 ml of DMF and 2 ml of 33~ aqueous
35 ammonia was heated to 100°C for 1 hour. The solution was
cooled and gave 60 mg of 3-[8-t-butoxyformamido-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-
- 4 4 - 2010636
1H-pyrrole-2,5-dione of melting point 153-155°C.
The furandione starting material was prepared as
f o 11 ows
a) A solution of 300 mg of sodium hydroxide in 5 ml of
water was added to a stirred solution of 1.35 g of the
carboxylate product of Example 1b) in 25 ml of ethanol and
the mixture was heated to reflux for 15 minutes. 2 ml of
2M hydrochloric acid and 10 ml of water were added and the
precipitate obtained was filtered off and dried to give
1.14 g of 6,7,8,9-tetrahydropyrido[1,2-a)indole-8-car-
boxylic acid of melting point 244-246°C.
b) A suspension of 900 mg of the product of a) in 1 ml of
water and 20 ml of acetone was cooled to 0°C and treated
With 490 mg of triethylamine followed by 5?6 mg of ethyl
chloroformate. After 0.5 hour 345 mg of sodium azide in
1 ml of water were added and the mixture was stirred at
0°C for 1 hour. The solvent was removed under reduced
pressure and the residue was extracted with dichloro-
methane. The extracts were evaporated and the residue was
purified by chromatography on silica gel with dichloro-
methane. The obtained solid was dissolved in 10 ml of
toluene and heated to 100°C for 4 hours under a nitrogen
atmosphere. The solvent was evaporated to give 700 mg of
6,7,8,9-tetrahydropyrido(1,2-a]indole-e-isocyanate of
melting point 87-89°C.
c) 4 ml of a 2M sodium hydroxide solution were added to a
solution of 700 mg of the product of b) in 50 ml of THF
and the solution obtained was stirred overnight. The
solvent was removed under reduced pressure and the residue
was extracted with dichloromethane. The dichloromethane
3,5 extract was evaporated to give an amine which was
redissolved in dichloromethane. 645 mg of di-t-butyl
dicarbonate and 300 mg of triethylamine were added at 0°C
20.0636
- 95 -
and the mixture was allowed to warm to room temperature
while stirring for 72 hours. The mixture Was washed with
sodium bicarbonate solution and the organic phase was
dried. The solvent was removed under reduced pressure and
the residue was extracted with diethyl ether. The ethereal
extracts were evaporated and the solid obtained was
triturated with petroleum ether to give 550 mg 8-t-butoxy-
formamido-6,7,8,9-tetrahydropyrido[1.2-a]indole of melting
point 155-157°C.
d) 256 mg of oxalyl chloride were added to a solution of
550 mg of the product of c) in 10 ml of diethyl ether at
0°C under a nitrogen atmosphere. After 1 hour the solvent
was removed under reduced pressure and the residue was
dissolved in dichloromethane. 363 mg of 1-methyl-3-
-indolylacetic acid and 390 mg of triethylamine were added
and the mixture was stirred for 40 hours. The solvent was
removed under reduced pressure and the residue was
purified by chromatography on silica gel with ethyl
acetate/petroleum ether (1:2). Crystallization from
diethyl ether/petroleum ether gave 200 mg of
3-[8-t-butoxyformamido-6,7,8,9-tetrahydropyrido[1,2-a]indol-
10-yl]-4-(1-methyl-3-indolyl)furan-2,5-dione of melting
point 155-160°C.
Examule 39
In a manner analogous to that described in Example 37.
from the pyrroledione product of Example 38, there was
Prepared 3-[8-amino-6,7.8,9-tetrahydropyrido[1.2-a]indol-
-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2.5-dione hydro-
chloride of melting point 310-315°C.
Example 40
A solution of 320 mg of 3-[4-(Z-acetoxyethyl)-5.6-
-dihydro-9H-pyrrolo[3.2,1-ij]quinolin-1-yl]-4-(1-methyl-3-
-. 4 6 _ 2o~.os~s
-indolyl)furan-.2,5-dione in 2 ml of DMFand 2 ml of 33%
aqueous ammonia was heated to 140°C for 12 hours. Water
was added to the cooled mixture which was filtered to give
210 mg of a solid. A sample was crystallized from ethyl
acetate to give 3-[4-(2-hydroxyethyl)-5,6-dihydro-
-4H-pyrrolo[3,2,1-ij]quinolin-1-yl)-4-(1-methyl-3-
-indolyl)-1H-pyrrole-2,5-dione of melting point 214-215°C.
The furandione starting material was prepared as
follows:
a) 25 ml of a 1.6M solution of n-butyllithium in n-hexane
were added to a solution of 4.04 g of diisopropylamine in
ml of THF at 0°C under nitrogen. After 10 minutes the
15 stirred solution was cooled to -78°C and a solution of
9.28 g of t-butyl acetate in 20 ml of THF was added. After
10 minutes 3.46 g of 1.2,5,6-tetrahydro-4H-pyrrolo-
[3,2,1-ij]quinolin-9-one in 20 ml of THF. was added
followed by 8 ml of boron trifluoride diethyl etherate.
20 The mixture was stirred at -78°C and then 20 ml of
pyrrolidine were added. The mixture was partitioned
between ethyl acetate and Water and the organic extracts
were washed with water and sodium chloride solution, dried
and evaporated. The residue was purified by chromatography
on silica gel with ethyl acetate/petroleum ether (1:3).
There were obtained 4.1 g of t-butyl (E)-(1,2,5,6-tetra-
hydro-4H-pyrrolo[3.2.1-ij]quinolin--4-ylidene)acetate of
melting point 105-107°C.
b) A solution of 4 g of the product of a) in 400 ml of
methanol was shaken with 280 mg of 10% Pd/C under a
hydrogen atmosphere for 18 hours. The catalyst was
filtered off and the filtrate was evaporated to an oil.
1.99 g of this oil in 100 ml of diethyl ether were treated
with 5 ml of a 1M solution of lithium aluminium hydride in
diethyl ether and the mixture was stirred for 2 hours.
Water was added and the product was extracted with ethyl
- 47 - zo~.os3s
acetate. The ethyl acetate extracts were dried and
concentrated under reduced pressure to give 1.44 g of
1,2,5,6-tetrahydro-4H-pyrrolo[3,2,1-ij]quinoline-4-ethanol.
c} 1.44 g of the product of b) in 40 ml of dichloro-
methane were treated with 10 ml of acetic anhydride and
5 ml of pyridine. The solution obtained was stirred and
then evaporated. The residue was dissolved in dichloro-
methane, the solution was washed with water, the organic
phase was separated, dried and concentrated to give 1.65 g
of 4-(2-acetoxyethyl)-1,2.5,6-tetrahydro-4H-pyrrolo-
[3,2,1-ij]quinoline.
d) A solution of 1.6 g of the product of c) in 50 ml of
xylene and 100 mg of 10% Pd/C was heated to reflux for
12 hours. The catalyst was filtered off and the filtrate
was evaporated to give 1.7 g of 4-(2-acetoxyethyl)-5,6-
-dihydro-4H-pyrrolo[3,2,1-ij]quinoline.
20 e) 935 mg of oxalyl chloride were added to a stirred
solution of 1.7 g of the product of c) in 45 ml of
dichloromethane under a nitrogen atmosphere. After 1 hour
the solvent was removed under reduced pressure and the
residue was dissolved in 90 ml of dichloromethane. 1.38 g
25 of 1-methyl-3-indolylacetic acid and 1.48 g of triethyl-
amine were added to this solution and the mixture obtained
was stirred for 18 hours. The solvent Was removed under
reduced pressure and the residue was purified by chromato-
graphy on silica gel with ethyl acetate/petroleum ether
30 (1:2). Crystallization from methanol/water gave 280 mg of
3-[4-(2-acetoxyethyl)-5,6-dihydro-4H-pyrrolo[3.2.1-ij]-
quinolin-1-yl]-4-(1-methyl-3-indolyl)furan-2,5-dione of
melting point 143-146°C.
- 48 -
Example 41
20.0636
A solution of 400 mg of 3-(8-[(t-butoxyformamido)-
methyl]-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-4-
-(1-methyl-3-indolyl)furan-2,5-dione in 50 ml of DMF and
50 ml of water was treated with 2.5 g of hydroxylamine
hydrochloride and 2.5 g of potassium carbonate and the
solution obtained was heated to 100°C. The solvents were
evaporated and the residue was dissolved in dichloro-
methane, washed with water and dried. The solvent was
removed under reduced pressure and the residue was
crystallized from ethyl acetate/petroleum ether to give
190 mg of 3-[8-[(t-butoxyformamido)methyl]-6.7,8,9-,
-tetrahydropyrido(1,2-a]indol-10-yl]-1-hydroxy-4-(1-methyl-
-3-indolyl)pyrrole-2,5-dione of melting point 238-240°C.
The furandione starting material was prepared as
f o 11 ows
a) 2.4 g of methanesulphonic anhydride and 2 ml of
triethylamine were added to a stirred solution of 2.01 g
of 6,7,8,9-tetrahydropyrido[1,2-a]indole-8-methanol in
40 ml of dichloromethane under a nitrogen atmosphere.
After 18 hours the mixture was washed with saturated
sodium bicarbonate solution, dried and evaporated to an
oil. 1.8 g of this oil were dissolved in 10 ml of
isopropanol and 5 ml of 33% aqueous ammonia and the
mixture was heated to 80°C for 10 hours. The solvent was
removed under reduced pressure and the residue was
partitioned between dichloromethane and saturated sodium
bicarbonate solution. The organic phase was dried and
evaporated to give 1.3 g of 8-aminomethyl-6.7.8.9-tetra-
hydropyrido[1,2-a]indole of melting point 85-90°C.
3,,5 b) 1.09 g of di-t-butyl dicarbonate were added to a
stirred solution of 890 mg of 8-aminomethyl-6,7,8,9-tetra-
. hydropyrido[1,2-a]indole and 920 mg of triethylamine in
- 4 9 _ ~0~.0636
60 ml of dichloromethane at 0°C under a nitrogen
atmosphere. Aftec 72 hours the organic phase was washed
with saturated sodium bicarbonate solution, dried and
evaporated. The residue was crystallized from petroleum
ether to give 1.03 g of 8-[(t-butoxyformamido)methyl]-
-6,7,8,9-tetrahydropyrido[1,2-a]indole of melting point
80-85°C.
c) 445 mg of oxalyl chloride were added dropwise to a
solution of 1 g of the product of b) in 20 ml of diethyl
ether under a nitrogen atmosphere at 0°C. After 1 hour the
solvent was removed under reduced pressure and the residue
was dissolved in dichloromethane. 630 mg of 1-methyl-3-
-indolylacetic acid and 920 of of triethylamine were
added to this solution and the mixture was stirred for
72 hours. The solvent was removed under reduced pressure
and the residue was purified by chromatography on silica
gel With ethyl aaetate/petroleum.ether (1:2). The
resulting solid was crystallized from diethyl ether and
there were obtained 315 mg of 3-[8-[(t-butoxyformamido)-
methyl]-6,7.8.9-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-
-methyl-3-indolyl)furan-2,5-dione of melting point
124-126°C.
Example 42
In a manner analogous to that described in t;xamplo 37,
fcom the product of Example 41 these was prepared
3-[e-(aminomethyl)-6,7.8,9--tetrahydropyrido[1.2-a]indol-
-10-Y1]-1-hydr,oxy-4-(1-methyl-3-indolyl)pyrrole-2,5-dione
hydrochloride of melting point 280-282°C.
Example 43
In a manner analogous to that described in Example 11.
from the product of Example 40 there was prepared
3-[4-[2-(amidinothio)ethyl]-5,6-dihydro-4H-pyrrolo-
- 5 0 - 209.0636
[3.2,1-ij]quinolin-1-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-
-2,5-dione methanesulphonate of melting point 185-19o°C.
Example 44
In a manner analogous to that described in Example 2,
from the product of Example 40 there was prepared
3-[4-(2-aminoethyl)-5,6-dihydro-4H-pyrrolo[3,2,1-ij]-
quinolin-1-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione
hydrochloride of melting point 193-195°C.
Example 45
In a manner analogous to that described in Example 2,
from the product of Example 26 there was obtained
3-[8-(aminomethyl)-6.7,8,9-tetrahydropyrido[1,2--a]indol-
-10-yl]-4-(3-benzo[b]thienyl)-1H-pyrrole-2,5-dione hydro-
chloride of melting~point 285-287°C.
Example 46
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[e-(acetoxymethyl)-
-6,7,8.9-tetrahydropyrido[1,2-a]indol-10-yl]-4-(2-
-naphthyl)furan-2,5-dione (obtained as described in the
Last paragraph of Example 1 by using Z-naphthylacetic acid
in place of 1-methyl-3-indolylacetic acid) there was
prepared 3-[6.7,8,9-tetcahydro-e-(hydroxymethyl)pyrido-
[1,2-a]indol-10-yl]-4-(2-naphthyl)-1H-pyrrole-2,5-dione of
melting point 260-263°C.
Example 47
In a manner analogous to that described in Example 2,
3,5 from the product of Example 46 there was obtained 3-[8-
-(aminomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-
-4-(2-naphthyl)-1H-pyrrole-2,5-dione hydrochloride of
201.0636
- 51 -
melting point >300°C.
Example 48
20
5 In an analogous manner to that described in
Example 10. from the product of Example 27 there was
prepared 3-[8-(aminomethyl)-6,7,8,9-tetrahydropyrido--
[1,2-a]indol--1-yl]-4-(1-naphthyl)-lEI-pyrrole--2,5-dione of
melting point 167-169°C.
Example 49
In a manner analogous to that described in the first
paragraph of Example 1, from 1.3 g of 3-[9-(acetoxy-
methyl)-7,8,9,10-tetrahydro-6H-azepino[1,2-a]indol-11-yl]-4
(1-methyl-3-indolyl)furan-2,5-dione there were obtained
520 mg of 3-[7.8,9,10-tetrahydro-9-(hydroxymethyl)-6H-
-azepino[1.2-a]indol-11-yl]-4-(1-.methyl-3-indolyl)-lI-I-
-pyrrole-2.5-dione of melting point 268=270°C.
The furandione starting material was prepared as
follows:
a) A solution of 18.9 g of ethyl indole-2-carboxylate in
100 ml of DMF Was added to a suspension of 2.64 g of
sodium hydride in 50 ml of DMF. After 1 hour a solution of
20.9 q of ethyl 5-bromovalerate in 100 ml of DMF was added
dropwise. After 48 hours the mixture was poured into
water, extracted with dichloromethane and the combined
dichloromethane extracts were washed with water, dried and
concentrated to give 26.2 g of ethyl 1-(4-ethoxycarbonyl-
butyl)indole-2-carboxylate.
b) This oil was dissolved in 50 ml of THF and the
solution was added to a stirred suspension of 11.2 g of
potassium t-butoxide in 150 ml of THF. After 36 hours the
mixture was concentrated and the residue was poured into a
52 -
2o~os3s
mixture of water and diethyl ether. The organic phase was
dried and concentrated. Chromatography of the residue on
silica gel with dichloromethane/methanol (9:1) gave a
solid which was recrystallized from ethyl acetate/
n-hexane, there being obtained 6.1 g of ethyl 7,8-dihydro-
-10-hydroxy-6H-azepino(1,2-a]indole-9-carboxylate of
melting point 74-81°C.
c) 5.5 g of this solid were dissolved in 200 ml of
ethanol and treated with 11 spoon spatula measures of
Raney nickel and 400 ml of water. The mixture was heated
at reflux for 4 hours. The cooled mixture was filtered and
the residue was washed with ethyl acetate. The filtrate
was extracted with ethyl acetate. The combined extracts
and washings were dried and concentrated to give an oil
which was purified by chromatography on silica gel with
dichloromethane. there being obtained 2.5 g of ethyl
7,8.9.10-tetrahydro-6H-azepino[1,.2-a]indole-9-carboxylate
of melting point 69-70°C.
d) This solid was dissolved in 50 ml of THF and added
dropwise to a mixture of 0.45 g of lithium aluminium
hydride in 2o ml of THF. The mixture was stirred for
2 hours and then water was added. The resulting mixture
was extracted with diethyl ether and the combined extracts
were dried and concentrated. Chromatography of the residue
on silica gel with dichloromethane gave 1.9o g of
7,8,9,10-tetrahydro-9-(hydroxymethyl)-6H-azepino(1.2-a]-
indole of melting point 109-111°C.
e) 1.8 g of this solid were dissolved in 100 ml of
diethyl ether at 0°C and treated with 1.70 g of acetic
anhydride and 0.66 g of pyridine. After a hours a further
5 g of pyridine were added and the mixture was stirred foe
76 hours. The solvents were removed under reduced pressure
and the residue was chromatographed on silica gel with
dichloromethane. there being obtained 1.98 g of
_ 5 3 _.
20.0636
9-(acetoxymethyl)-7,8,9,10-tetcahydro-6EI-azepino[1.2-a]-
indole of melting point 65°C.
f) 1.90 g of this solid were dissolved in 50 ml of
dichloromethane, the solution was cooled to 0°C and
treated with 1.03 g of oxalyl chloride. After 2 hours the
solvent was removed by evaporation and the residue was
dissolved in dichloromethane and added dropwise to a
solution of 1.5 g of 1-methylindole-3-acetic acid and
1.86 g of triethylamine in dichloromethane. The mixture
was concentrated and the residue was chromatographed on
silica gel with dichloromethane containing 5% methanol by
volume. The solid obtained was recrystallized from ethyl
acetate/n-hexane to give 1.55 g of 3-[9-(acetoxymethyl)-
-~~8~9~10-tetrahydro-6I-I-azepino[1:2-a]indol-11-yl]-4-
-(1-methyl-3-indolyl)furan-2.5-dione of melting point
164-166°C.
Example 50
in a manner analogous to that described in Example 12.
from 0.50 g of 3-[7.8.9.10-tetrahydro-6H-azepino[1:2-a]-
indol-11-yl]-3-(1-methyl-3-indolyl)furan-2,5-dione there
was obtained 0.43 g of 3-[7~8~9,10-tetrahydro-6H-azepino-
[1.2-a]indol-11-yl]-3-(1-methyl-3-indolyl)-1H-pyrrole-2,5-
-dione of melting point >300°C.
The furandione starting matecial was prepared as
follows:
1.5 g of oxalyl chloride were added dropwise to an
ice-cold solution of 2.0 g of 7.8~9~10-tetrahydro-6H-
-azepino[1,2-a]indole (J. Org. Chem. 33, 1968, 4286) in
50 ml of dichloromethane. The mixture was stirred for
2 hours. The solvent was removed in a vacuum and the
residue was dissolved in dichloromethane. The solution
'obtained was added to a solution of 2.2 g of 1-methyl-3-
- 5 4 - ~olos3s
-indolylacetic acid and 2.73 g of triethylamine in 50 ml
of dichloromethane. The mixture was stirred and then
concentrated. The residue was chromatographed on silica
gel with dichloromethane and there was obtained 1.0 g of
3-[7,8,9,10-tetrahydro-6H-azepino[1,2-a]indol-11-yl]-3-
(1-methyl-3-indolyl)furan-2,5-dione of melting point
257-259°C.
Example 51
A solution of 150 mg of the product of Example 49 and
146 mg of 2,6-lutidine in 15 ml of dichloromethane was
added to a solution of 290 mg of trifluoromethanesulphonic
anhydride at 0°C. After 3 hours 25 ml of 33% aqueous
y5 ammonia were added and the mixture was stirred for
16 hours. The mixture was extracted with dichloromethane
and the combined extracts were dried and concentrated.
Chromatography of the residue on silica gel with
dichloromethane/methanol/acetic acid/water (90:18:3:2)
gave 50 mg of 3-[9-(aminomethyl)-7,8,9,10-tetrahydro-
-6H-azepino[1,2-a]indol-11-yl]-4-(1-methyl--3-indolyl)-1H-
-pyrrole-2,5-dione acetate of melting point 215°C
(decomposition).
Example 52
A mixture of 40 mg of the product of Example 51, 20 mq
of sodium bicarbonate and 25 mg of 3,5-dimethyl--N2-
-nitro-1-pyrazole-1-ca=boxamide in 10 ml of ethanol was
heated at reflux for 16 hours. The mixture was
concentrated and the residue was chromatographed on silica
gel with dichloromethane/methanol (9:1). There were
obtained 15 mg of 3-(1-methyl-3-indolyl)-4-[7,8.9,10--
-tetrahydro-9-[(2-nitroguanidino)methyl]-6H-azepino[1,2-a]-
3,5 indol-11-yl]-1H-pyrrole-Z,5-dione of melting point
177-178°C.
- 55 -
~p~.o636
Example 53
In a manner analogous to that described in the first
paragraph of Example 1, from 0.20 g of 3-[8-(acetoxy-
5 methyl)-7.8,9,10-tetrahydro-6H-azepino[1,2-a]indol-11-yl]-4
(1-methyl-3-indolyl)furan-2,5-dione there were obtained
60 mg of 3-[7,8,9,10-tetrahydro-8-(hydroxymethyl)-6H-
-azepino[1,2-a]indol-11-yl]-4-(1-methyl-3-indolyl)-1H-
-pyrrole-2,5-dione of melting point 109-111°C.
The furandione starting material was prepared as
follows:
a) A solution of 5 g of ethyl 6,7-dihydro-9-hydroxy-
pYrido[1,2-a]indole-8-carboxylate (prepared as described
in Example 1) in 200 ml of DMF was treated with 550 mg of
sodium hydride. The mixture was stirred under a nitrogen
atmosphere and then a solution of. 3.6 g of ethyl
bromoacetate in 50 ml of DMF was added.~After 16 hours the
mixture was poured into water and extracted with diethyl
ether. The combined extracts were washed with water, dried
and concentrated to give 4.4 g of ethyl 8-(ethoxycar-
bonyl)-6.7,8,9-tetrahydro-9-oxopyrido[1,2-a]indole-8-
-acetate. '
b) A solution of 5.0 g of the product of a) in 200 ml of
THF was added dropwise to a stirred solution of 2.0 g of
potassium t-butoxide in 50 ml of THF. The mixture was
stirred and then 1 ml of glacial acetic acid was added.
The mixture was poured into water and extracted with
dichloromethane. The combined extracts were dried and
concentrated. The residue was chromatographed on silica
gel with dichloromethanelmethanol (95:5) to give 3.0 g of
diethyl 7,8-dihydro-10-hydroxy-6H-azepino[1.2-a]indole-
-8,9-dicarboxylate.
- 56 -
2~~.~636
c) A mixture of 2.8 g of the product of b) and 0.5 g of
boric acid was heated at 150°C for 1 hour and at 170°C for
3 hours. Ice-water was added to the cooled mixture and the
whole was extracted with dichloromethane. The combined
dichloromethane extracts were dried and concentrated. The
residue was chromatographed on silica gel with dichloro-
methanelmethanol (95:5). There were obtained 2.1 g of
ethyl 7,8.9.10-~tetrahydro-10-oxo-6H-azepino[1.2-a]-
indole-B-carboxylate.
d) 2.1 g of the product of c) were dissolved in 80 m1 of
ethanol and treated with 4 spoon spatula measures of Itaney
nickel and 50 ml of water. The mixture was heated at
reflux for 4 hours, cooled and filtered, and the residue
was washed with ethyl acetate. The filtrate was extracted
with ethyl acetate. The combined extracts and washings
were dried and concentrated. Chromatography of the residue
on silica gel with dichloromethane gave 0.89 g of ethyl
7.8,9,10-tetrahydro-6H-azepino[1,2-a]indole-e-carboxylate.
e) 0.85 g of the product of d) Was dissolved in 50 ml of
THF and added dropwise to a stirred suspension of 140 mq
of lithium aluminium hydride in 50 ml of THF. After the
addition of water. the mixture was extracted with diethyl
ether. The combined extracts were dried and concentrated.
Chromatography of the residue on silica gel with dichloro-
methane/methanol (95:5) gave 0.70 q of 7.8.9,10-tetra-
hydro-e-(hydroxymethyl)-6H-azepino[1.2-a]indole of melting
point 90-91°C.
f) 0.70 g of the product of e) was treated with 0.66 g of
acetic anhydride and 0.39 g of pyridine in 50 ml of
diethyl ether. A further 1 g of pyridine and a further 1 g
of acetic acid were added and the mixture was stirred for
16 hours. Then, the mixture was concentrated and the .
residue was chromatographed an silica gel with dichloro-
methane, there being obtained 0.60 g of 8-(acetoxymethyl)-
- 57 -
209.0636
-7,8.9,10-tetrahydro-6H-azepino[1,2-a]indole of melting
point 77-79°C.
g) A solution of 0.60 g of the product of f) in 50 ml of
dichloromethane was treated dropwise with 0.33 g of oxalyl
chloride. After leaving to stand at 10°C for 2 hours the
solution was concentrated and the residue was dissolved in
dichloromethane. The solution was added to a solution of
0.49 g of 1-methylindole-3-acetic acid and 0.59 g of
y0 triethylamine in dichloromethane. After 16 hours the
mixture was concentrated and the residue was chromato-
graphed on silica gel with dichloromethane/methanol
(95:5). There was obtained 0.51 g of 3-[8-(acetoxymethyl)-
-7,8,9.10-tetrahydro-6H-azepino[1,2-a]indol-11-yl]-4-
y5 -(1-methyl--3-indolyl)furan-2.5-dione of melting point 70°C.
Example 54
A solution of 60 mg of the product 'of Example 53 and
20 60 mg of 2,6-lutidine in 25 ml of dichloromethane was
added dropwise to a solution.of 116 mg of trifluoro-
methanesulphonic anhydride in 25 ml of dichloromethane at
0°C. After 3 hours. 25 ml of aqueous ammonia were added to
the solution. The organic phase was dried and concentra-
25 ted. Chromatography of the residue on silica gel gave
30 mg of 3-[8-(aminomethyl)-7,8,9,10-tetrahydro-6H-
-azepino[1,2-a]indol-11-yl]-4-(1-methyl-3-indolyl)-1H-
-pyrrole-2,5-dione acetate of melting,point 162-163°C.
30 Example 55
A solution of 0.64 g of the product of Example 1 and
0.4 ml of 2,4,6-collidine in 20 ml of dichloromethane was
added dropwise to a solution of 0.75 g of trifluoro-
methanesulphonic anhydride in 10 ml of dichloromethane at
0°C. After 2.5 hours the mixture was treated with 3 ml of
piperidine and stirred for 16 hours. Concentration and
a -- 20x.0636
chromatography of the residue on silica gel with dichloro-
methane/methanol (gradient from 98:2 to 50:50) gave 340 mg
of 3-(6,7,8,9-tetrahydro-8-[(1-piperidino)methyl]pyrido-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-
5 -dione. This was converted into the hydrochloride of
melting point 294°C (decomposition), by treatment with a
saturated solution of hydrogen chloride in ethyl acetate.
Example 56
A solution of 0.8 g of the product of Example 1 and
0.44 g of 2,4,6-collidine in 30 ml of dichloromethane was
added to a solution of 0.9 g of trifluoromethanesulphonic
anhydride in 10 ml of dichloromethane at 0°C. After 1,5
hour the mixture was treated with 3.64 g of diisopropyl-
amine and stirred for 16 hours. The mixture was washed
with water and then With saturated aqueous sodium
bicarbonate solution, dried and concentrated. The solid
obtained was dissolved in ethyl acetate~and treated with a
saturated solution of hydrogen chloride in ethyl acetate.
Removal of the solvent in vacuo gave 26o mg of 3-[6,7,8,9-
-tetcahydro-8-((diisopcopylamino)methyl]pytido[1,2-a]-
indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione
hydrochloride of melting point 187°C (decomposition).
Example 57
A solution of 1.0 g of 3-(e-(acetoxymethyl)-6,7,8,9-
-tetrahydropyrido(1,2-a]indol-10-yl]-4-(3-benzofuranyl)-
furan-2,5-dione in 100 ml of chloroform was treated with
13.8 ml of hexamethyldisilazane and 2.73 ml of methanol
and the solution obtained was heated to 50°C while
stirring under a nitrogen atmosphere for 6 hours. A
fucther 13.8 ml of hexamethyldisilazane and 2.73 ml of
methanol were added and the heating was continued for
16 hours. Two fucther additions of the same quantities of
hexamethyldisilazane and methanol were effected and the
- 5 9 - 20.0636
temperature of the mixture was held at 50°C for a further
24 hours. 20 ml of methanol were added and the mixture was
heated to reflux for 15 minutes, cooled and concentrated.
The precipitate was filtered off and triturated in
succession with ethyl acetate and methanol. There were
obtained 630 mg of 3-[8-(acetoxymethyl)-6,7,8.9-tetra-
hydropyrido[1,2-a]indol-10-yl]-4-(3-benzofuranyl)-11I--
pyrrole-2,5-dione of melting point 234-237°C.
The furandione starting material was prepared as
follows:
1.7 g of oxalyl chloride were added dropwise to a
solution of 3.3 g of 8-(acetoxymethyl)-6,7,8,9-tetra-
i5 hYdropyrido[1,2-a]indole in 200 ml of diethyl ether under
a nitrogen atmosphere. After 15 minutes the solvent was
removed under reduced pressure and the residue was
dissolved in dichloromethane. 2.4. g of 3-ben2ofuranyl-
acetic acid and 5.6 ml of triethylamine~were added to this
solution and the mixture was stirred overnight. The
solvent was removed under reduced pressure and the residue
Was pucified by chromatography on silica gel with ethyl
acetate/petcoleum ether (1:2). Crystallization of the
residue from ethyl acetate/petroleum ether gave 1.62 g of
25 3-[8-(acetoxymethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-
-10-yl]-4-(3-benzofuranyl)furan-2,5-dione of melting point
214-215°C.
Examvle 58
A solution of 300 mg of the product of Example 57 in
ml of methanol was treated with 5 ml of 2M sodium
hydroxide. After 10 minutes the mixture was acidified with
5 ml of 2M hydrochloric acid and the methanol was removed
3,5 under reduced pressure. The residue was partitioned
between ethyl acetate and water. The phases were separated
and the organic phase was washed with sodium bicarbonate
- 6 0 - 201.0636
solution and dried. The solution was concentrated and the
precipitate was filtered off to give 190 mg of 3-(3-benzo-
furanyl)-4-[6,7,8,9-tetrahydro--8-(hydroxymethyl)pyrido-
[1,2-a]indol-10-yl]-1H-pyrrole-2,5-dione of melting point
246-248°C.
Example 59
In a manner analogous to that described in Example 2,
from the product of Example 58 there was prepared
3-[8-(aminomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-
-10-yl]-4-(3-benzofuranyl)-1H-pyrrole-2,5-dione hydro-
chloride of melting point 210-212°C.
Examule 60
118 mg of trifluoromethanesulphonic anhydride in 20 ml
of dichloromethane were treated at 0°C under a nitrogen
atmosphere with a suspension of 90 mg of the product of
Example 26 and 45 mg of collidine in 20 ml of dichloro-
methane. After 45 minutes 0.41 ml of a 40% solution of
dimethylamine in water was added and the mixture was
stirred for 1.5 hours. The solution obtained was washed
with water and sodium bicarbonate solution, and then
dried. The solution was concentrated and the resulting
crystals were filtered off and dried to give 60 mg of
3-(3-benzo[b]thienyl)-4-[6,7,8,9-tetrahydro-8-(dimethyl-
aminomethyl)pyrido[1,2-a]indol-10-yl]-1H-pyrrole-2,5-dione
of melting point 285-286°C.
Examuie 61
546 mg of trifluoromethanesulphonic anhydride in e0 ml
of dichloromethane were treated at 0°C under a nitrogen
atmosphere with a suspension of 400 mg of the product of
Example 19 and 208 mg of collidine in 120 ml of dichloro-
methane. After 1 hour 1.9 ml of 40% aqueous dimethylamine
- 61 -
20.0636
were added and the mixture was stirred for 3 hours. The
solvent was removed and the residue was subjected to
chromatography on silica gel with dichloromethane/
methanol/acetone (88:10:2). Trituration with ethyl acetate
followed by recrystallization from methanol gave 295 mg of
3-[2,3-dihydro-2-(dimethylaminomethyl)-1PI-pyrrolo[1,2-a]-
indol-9-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5--dione
trifluoromethanesulphonate of melting point 323-325°C.
Example 62
A solution of 400 mg of 3-[8-cyano-6.7,8,9-tetra--
hydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)furan-
-2,5-dione in 12 ml of DMF and 12 ml of 33% aqueous
ammonia was heated to 140°C for 3 hours. The mixture was
cooled and the resulting solid was filtered off and dried
to give 275 mg of 3-[8-cyano-6.7,8,9-tetrahydropyrido-
[1-,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-
-dione of melting point 312-313°C.
The furandione starting material was prepared as
f o 11 ows
a) A suspension of 4.0 g of the product of Example 38a)
in 4.4 ml of water and 84 ml of acetone was cooled to 0°C
and 2.18 g of triethylamine were added. 2.56 g of ethyl
chloroformate were then added and the resulting solution
was stirred under a nitrogen atmosphere. 0.9 ml of 33%
aqueous ammonia were added and the mixture was allowed to
warm to room temperature. A further 0.5 ml of 33% aqueous
ammonia was added and stirring was continued. The solvent
was evaporated and the residue was extracted with of
dichloromethane. The organic phase was washed with of
water, dried and concentrated to give 2.8 g of
6,7,8,9-tetrahydropyrido[1,2-a]indole-8-carboxamide of
melting point 179-181°C.
zosos3s
- 62 -
b) 991 mg of trifluoroacetic anhydride were added
dropwise to a suspension of 1.0 g of 6,7,8,9-tetrahydro-
pyrido[1,2-a]indole-8-carboxamide in 15 ml of dioxan at
10°C. The mixture was partitioned between dichloromethane
and water and the organic phase was dried. The solvent was
removed under reduced pressure to give an oil which was
subjected to chromatography on silica gel with ethyl
acetate/petroleum ether (l:i). There were obtained 740 mg
of 6,7,8,9-tetrahydropyrido[1,2-a]indole-8-carbonitrile of
melting point 116-118°C.
c) 518 mg of oxalyl chloride were added to a solution of
800 mg of 6,7,8,9-tetrahydropyrido[1,2-a]indole-8-cacbo-
nitrile in 100 ml of diethyl ether under a nitrogen
~5 atmosphere. The solvent was evaporation and the residue
was dissolved in of dichloromethane. 771 mg of 1-methyl-3-
-indolylacetic acid and 1.24 g of triethylamine were added
to this solution and the mixture'was stirred overnight.
The solvent was removed under reduced pressure and the
20 residue was purified by chromatography on silica gel with
10% methanol in dichloromethane. The fractions containing
the desired product were concentrated and the crystals
obtained were filtered off and dried to give 560 mg of
3-[B-cyano-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-4-
25 -(1-methyl-3-indolyl)furan-2,5-dione of melting point
309-31I°C.
Example 63
30 Hydrogen chloride gas was bubbled through a solution
of 200 mg of the product of Example 62 in 250 ml of
methanol at 0°C. The solvent was then removed under
reduced pressure and the residue was dissolved in 50 ml of
dichloromethane and 250 ml of ethanol. Ammonia was bubbled
35 through the solution and the solvent was then evaporated.
The residue was purified by chromatography on silica gel
with dichloromethane/methanol/acetic acid/water
2010636
- 63 -
(90:18:3:2). Trituration with ethyl acetate gave 75 mg of
3-[8-amidino-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-
-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione hydrochloride
of melting point 237-239°C.
example 64
A solution of 50 mg of 3-[8-carbamoyl-6,7.8,9-tetra
hydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)furan
-2,5-dione in 4 ml of nMF and 4 ml of 33% aqueous ammonia
was heated to 140°C. The mixture Was extracted with of
ethyl acetate and the organic phase was washed with water
and then dried. The majority of the solvent was evaporated
and the precipitate obtained was filtered off and dried.
There were obtained 20 mg of 3-[8--carbamoyl-6,7,8.9-tetra-
hydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-
-pyrrole-2,5-dione of melting point 315-316°C.
The furandione starting material was prepared as
Zp follows:
178 mg of oxalyl chloride were added to a solution of
300 mg of the product of Example 62a) in 40 ml of
dichloromethane under a nitrogen atmosphere. Then the
solvent was removed under reduced pressure and the residue
was dissolved in dichloromethane. 265 mg of 1-methyl-3-
-indolylacetic acid and 424 mg of triethylamine were added
and the mixture was stirred for about 60 hours. The
solvent was removed under reduced pressure and the residue
was purified by chromatography on silica gel with 10%
methanol in dichloromethane. Crystallization from ethyl
acetate gave 70 mg of 3-(8-carbamoyl-6,7,8,9-tetra-
hydropyrido(1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-
furan-2,5-dione of melting point 307-309°C.
- 6 4 - 2p9.0636
Example 65
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[e-(acetoxymethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(5-methoxy-1-
-methyl-3-indolyl)furan-2,5-dione there was prepared
3-[6.7,8,9-tetrahydro-8-(hydroxymethyl)pyrido[1.2-a]indol-
-10-yl]-4-(5-methoxy-1-methyl-3-indolyl)-1H-pyrrole-2,5-
-dione of melting point 300-303°C.
The furandione starting material was prepared as
follows:
0.4 ml of oxalyl chloride were added to a solution of
906 mg of 8-(acetoxymethyl)-6,7,8:9-tetrahydropyrido-
[1,2-a]indole in 35 ml of diethyl ether under a nitrogen
atmosphere. Then the solvent was removed under reduced
pressure and the residue was dissolved in of dichloro-
methane. 940 mg of 5-methoxy-1-methyl-3=indolylacetic acid
and 1.16 ml of triethylamine were added and the mixture
was stirred for 40 hours. The solvent was removed under
reduced pressure and the residue was purified by chromato-
graphy on silica gel with ethyl acetate/n-hexane (1:2).
Crystallization from ethyl acetate/petroleum ether gave
250 mg of 3-[8-(acetoxymethyl)-6.7.8.9-tetrahydropyrido-
[1.2-a]indol-10-yl]-4-(5-methoxy-1-methyl-3-indolyl)furan-
-2.5-dione of melting point 259-261°C.
Examvle 66
In a manner analogous to that described in Example 2,
from the product of Example 65 there was prepared
3-[e-(aminomethyl)-6.7.8.9-tetrahydropyrido[1,2-a]indol-l0-
-yl]-4-(5-methoxy-1-methyl-3-indolyl)-1FT-pyrrole-2,5-dione
3,5 hydrochloride of melting point 268-270°C.
- 6 5 - 2010636
Example 67
In a manner analogous to that described in the first
paragraph of Example 1, from 3-[8-(acetoxymethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(5-bromo-1-methyl-
-3-indolyl)furan-2,5-dione there was prepared 3-[6,7,8,9-
-tetrahydro-8-(hydroxymethyl)pyrido[1.2-a]indol-10-yl]-4-
-(5-bromo-1-methyl-~3-indolyl)-1H-pyrcole-2,5-dione of
melting point 316-31B°C.
The furandione starting material was prepared as
f o 11 ows
a) 500 mg of a 60% dispersion of sodium hydride in
~5 mineral oil were added to solution of 1 g of 5-bcomo-
indole-3-acetic acid in 50 ml of TEIF and the mixture was
stirred under a nitrogen atmosphere for 1 hour. 820 mg
(5.8 mmol) of methyl iodide were .then added and the
mixture was stirred under a nitrogen atmosphere for
24 hours. 5 ml of water were added and the solvent was
removed under reduced pressure. The residue was treated
with 2M hydrochloric acid and the precipitate formed was
filtered off, washed with n-hexane and dried. The obtained
solid was recrystallized from diethyl ether to give
5-btomo-1-methyl-3-indolylacetic acid of melting point
192-194°C.
b) 500 mq of oxalyl chloride were added to a solution of
900 mg of the product of a) in 100 ml of diethyl ether
g0 under a nitrogen atmosphere. Then the solvent was
evaporated and the residue was dissolved in dichloro-
methane. 880 mg of 5-bromo-1-methyl-3-indolylacetic acid
and 810 mg of triethylamine were added and the mixture was
stirred for 48 hours. The solvent was removed under
reduced pressure and the residue was purified by chromato-
graphy on silica gel with ethyl acetate/n-hexane (1:1) to
give 400 mg of a solid. A sample was recrystallized from
- 6 6 - I~i~~.~~~6
ethyl acetate to give 3-[8-(acetoxymethyl)-6,7,8,9-tetra--
hydropyrido[1,2-a]indol-10-yl]-4-(5-bromo-1-methyl-3-
-indolyl)furan-2,5-dione of melting point 215-220°C.
Example 68
In a manner analogous to that desceibed in Example 2,
from the product of Example 67 there was prepared
3-[8-(aminomethyl)-6,7,8.9-~tetrahydropyrido[1,2-a]indol-10-
-yl]-4-(5-bromo-1-methyl-3-indolyl)-lt-I-.pyrrole-2,5-dione
hydrochloride of melting point >310°C.
Example 69
~1 solution of 200 mg of 3-[7-(2-acetoxyethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)--
-furan-2,5-dione in 2 ml of DMF and 1 ml of 33% aqueous
ammonia was heated to 140°C. Then 1 ml of a 2M solution of
sodium hydroxide was added to the cooled solution and the
mixture Was stirred for 2 hours. The mixture was acidified
with 2M hydrochloric acid and evaporated. 'The residue was
partitioned between ethyl acetate and of water and the
organic phase was dried. The solvent was evaporated and
the solid obtained was triturated with ethyl acetate to
give 115 mg of 3-[6.7,8,9-tetrahydro-7-(2-hydroxyethyl)-
pyrido[1,2-a]indol--10-y1]-4-(1-methyl-3-indolyl)-1H-pyrrole-
2,5-dione of melting point 236-238°C.
The furandione starting material was prepared as
follows:
a) 400 mg of a 60% dispersion of sodium hydride in
mineral oil were added to a solution of 2.24 g of triethyl
phosphonoacetate in 40 ml of dimethoxyethane under a
nitrogen atmosphere. Then the solution was cooled to 0°C
and 1.85 g of the product of Example 35d) in l0 ml of
dimethoxyethane were added. The mixture was stirred
67 - zo~.os3s
overnight and then evaporated. The residue was dissolved
in dichloromethane and the solution was washed with water,
dried and concentrated. The residue was purified by
chromatography on silica gel with diethyl ether/petroleum
ether (1:3), there being obtained 1.55 g of a mixture of
ethyl (E) and (Z)-(6,7,8,9-tetrahydropyrido[1,2-a]-
indol-7-ylidene)acetate. 1.4 g were dissolved in ethanol
and the solution was shaken with 280 mg of 10% Pd/C under
a hydrogen atmosphere. The catalyst was then filtered off
and the filtrate was evaporated to give 1.2 g of ethyl
6,7.8,9-tetrahydropyrido[1,2-a]indole-7-acetate of melting
point 66-68°C after crystalli2ation from diethyl ether/
petroleum ether.
b) A solution of 1.2 g of the product of a) in 100 ml of
diethyl ether was treated with 3.5 ml of a 1M solution of
lithium aluminium hydride in diethyl ether. After stirring
for 1 hour the mixture was quenched with 50 ml of aqueous
ammonium chloride. The mixture was extracted with of
diethyl ether and the organic phase was dried and
evaporated to give 1.01 g of 6,7,8,9-tetrahydro-7-
-(2-hydroxyethyl)pyrido[1,2-a]indo2e of melting point
70-72°C after crystallization from diethyl ether/petroleum
ether.
c) A solution of 1.04 g of the product of b), in 30 ml of
dichloromethane was treated with 6 ml of acetic anhydride
and 3 ml of pyridine and the solution was stirred under a
nitrogen atmosphere. The mixture was then evaporated to
dryness and the residue was dissolved in dichloromethane.
The organic phase was washed with 2M hydrochloric acid and
with water, dried and evaporated. The residue was purified
by chromatography on silica gel with diethyl ether/
petroleum ether (1:4) to give 670 mg of 7-(2-acetoxy-
ethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indole.
2010636
- 68 -
d) 250 ul of oxalyl chloride were added to a solution
of 670 mg of the proeuct of c) in 12 ml of dichloromethane
under a nitrogen atmosphere. Then the solvent was removed
under reduced pressure and the residue was dissolved in
dichloromethane. 493 mg of 1-methyl-3-indolylacetic acid
and 527 mg of triethylamine were added to this solution
and the mixture was stirred. Then the solvent was removed
under reduced pressure and the residue was purified by
chromatography on silica gel with ethyl acetate/petcoleum
ether (1:2) to give 350 mg of 3-[7-(2-acetoxy-
ethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indole-10-yl]-4-(1-
-methyl-3-indolyl)furan-2,5-dione of melting point
182-184°C after crystallization from ethyl acetate.
Example 70
In a manner analogous to that described in Example 2,
from the product of Example 69 there was prepared
3-(7-(2-aminoethyl)-6.7.8.9-tetrahydropyrido--
f1,2-a]indol-10-yl]-4-(1-.methyl-3-indolyl)--1H-pyrrole
-2.5-dione hydrochloride of melting point 240-242°C.
Example 71
in a manner analogous to that described in the first
paragraph of Example 1, from 3-(8-(acetoxymethyl)-6,7,8,9-
-tetrahydro-2-methoxy-pyrido(1,2-a]indol-10-yl]-4-(1-
-methyl-3-indolyl)furan-2,5-dione thore was preparod
3-(6,7,8,9-tetrahydro-8-(hydroxymethyl)-2-methoxypyrido-
~0 [1.2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-lEI-pyrrole-2,5-
-dione of melting point 195-197°C.
The furandione starting material was prepared as
follows:
a) 2 g of a 60% sodium hydride dispersion in mineral oil
was washed with n-hexane by decantation and suspended in
20.0636
- 69 -
100 ml of DMF under a nitrogen atmosphere. A solution of
g of ethyl 5-methoxyindole-2-carboxylate in 100 ml of
DMF was added and the mixture was stirred. Then 9.8 g of
ethyl 4-bromobutyrate were added and the mixture was
5 stirred for 2 hours. The mixture was cooled and treated
with 50 ml of 1M hydrochloric acid and 400 ml of water.
The mixture was extracted with diethyl ether and the
combined extracts were washed with sodium chloride
solution. The organic phase was dried and evaporated. The
obtained oil was dissolved in TEIF and added to a mixture
of 5.2 g of potassium t-butoxide in 200 ml of THF under a
nitrogen atmosphere. Then the mixture was cooled and
neutralized with 1M hydrochloric acid. Water was added and
the mixture was extracted with diethyl ether. The combined
extracts were washed with water and sodium chloride
solution and then dried. Evaporation of the solvent and
crystallization of the cesidue fcom,ethyl acetate gave
6.7 g of ethyl 6,7-dihydro-9-~hydtoxy-2-methoxypyrido-
[1.2-a]indole-e-carboxylate of melting point 157-160°C.
b) 5 g of the product of a) in 200 ml of ethanol were .
treated under a nitrogen atmosphere with 10 spoon spatula
measures of Raney nickel and 100 ml of water. The
suspension was heated at reflux, then cooled and filtered.
The solid was washed with ethyl acetate and volatile
constituents were removed in a vacuum from the combined
filtrate and washings. The aqueous suspension was
extracted with ethyl acetate and the combined extracts
were washed with sodium chloride solution and dried.
Evaporation of the solvent and crystallization of the
residue from methanol gave 2.41 g of ethyl 6,7,8,9-tetra-
hydro-2-methoxypyrido(1,2-a]indole-e-carboxylate of
melting point 104-105°C.
c) A solution of 2.3 g of the product of b) in 25 ml of
THF was added to a suspension of 260 mg of lithium
aluminium hydride in 20 ml of THF under a nitrogen
70 _ zo~.os3s
atmosphere. Then the mixture was treated with 10 ml of
ethyl acetate followed by 20 ml of water. The mixture was
acidified to pH 3 with 1M hydrochloric acid and extracted
with diethyl ether. The combined extracts were washed with
water and dried. Removal of the solvent by evaporation
gave 1.85 g of 6,7,8,9-tetrahydro-2-methoxypyrido-
[1,2-a]indole-8-methanol. A sample crystallized from ethyl
acetate/n-hexane melted at 95-96°C.
d) 1 g of the product of c) in 10 ml of pyridine was
treated with 1.5 g of acetic anhydride. Then the solvent
was evaporated and the residue was partitioned between
diethyl ether and 5% aqueous ammonium chloride. The
organic phase was washed with sodium chloride solution,
dried and evaporated. Crystallization of the residue from
diethyl ether/n-hexane gave O.B4 g of 8-acetoxymethyl-
-6.7.8.9-tetrahydro-2-methoxypyrido[1,2-a]indole of
melting point 98-100°C.
e) A suspension of 800 mg of the product of d) in 25 ml
of diethyl ether was treated with 0.27 ml of oxalyl
chloride under a nitrogen atmosphere. Then the solvent was
evaporated, the residue was dissolved in 20 ml of
dichloromethane and treated with 555 mg of N-methylindole-
-3-acetic acid and 0.8 ml of triethylamine. The mixture
Was stirred for 65 hours and then the solvent was removed
under ceduced pressure. Chromatography of the cesidue on
silica gel with ethyl acetate/n-hexane (1:1) gave 380 mg
of 3-[e-(acetoxymethyl)-6,7,8,9-tetrahydro-2-methoxy-
~0 pyrido[1,Z-a]indol-10-yl]-4-(1-methyl-3-indolyl)furan-2,5-
-dione. A sample crystallized from toluene/n-hexane melted
at 131-133°C (decomposition).
Example 72
3~5
In a manner analogous to that described in Example 2,
from the product of Example 71 there was prepared
2010636
- 71 -
3-[8-(aminomethyl)-6.7.8,9-tetrahydro-2-methoxypyrido-
[1,2-a]indol-10-yl]]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-
-dione hydrochloride of melting point 235-238°C
(decomposition).
Example 73
a) A solution of 150 mg of the product of Example 20 in
dichloromethane under a nitrogen atmosphere was treated
with 135 mg of 1.1~-thiocarbonyldiimidazole, After
17 hours, the solution was washed with water and dcied.
The solvent was evaporated and the residue was
crystallized fcom ethyl acetate to give 150 mg of
3-[1,2,3,4-tetrahydro-2-(1-imidazolylthiocarbonyl)-
-pYrazino[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-lI-I-
-pyrrole-2,5-dione of melting point 244-247°C.
b) A solution of 140 mg of the product of a) in 10 ml of
DME' was treated with 20 ml of 33~ aqueous ammonia. After
17 hours, the suspension was filtered and the solid was
washed with water. The solid was dried to give 95 mg of
3-[1.2,3,4-tetrahydro-2-thiocarbamoylpyrazino[1.2-a]indol-
-10-yl]-4-(1-methyl-3-indolyl)-lI-I-pyrrole-2,5-dione of
melting point 278°C (decomposition).
Examv_le 74
A solution of 150 mg of the product of Example 20 in
50 ml of dichloromethane was treated with 3 ml of acetic
anhydride and 3 ml of triethylamine. After 17 hours, the
solution was washed with water. The organic phase was
dried and evaporated. The residue was dissolved in
dichloromethane and treated with 0.08 ml of diethylamine,
After 17 hours the solution was evaporated. Crystalli-
zation of the residue from dichloromethane/n-hexane gave
80 mg of 3-[2-acetyl-1,2.3,4-tetrahydro-pyrazino-
[1,2-a]indol-10-yl]-4-(1-methyl-3-indolyl)-1H-pyrrole-2,5-
20~.0~36
-. 7 2 _
-dione of melting point 308-310°C.
Example 75
In a manner analogous to that described in Example 74,
from the product of Example 2 there was prepared
3-[8-(acetamidomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]-
indol-10-yl ]-4-~ ( 1-methyl- 3-~ indolyl )-lEI- pyrrole--2, 5-~dione
of melting point 270-273°C.
Examine 76
A solution of 150 mg of the product of Example 20 in
40 ml of dichloromethane was treated with 40 mg of
triethylamine and 44 mg of methanesulphonyl chloride.
After 17 hours the solution was washed with water. The
organic phase was dried and evaporated. Chromatography of
the residue on silica gel with ethyl acetate/n-hexane
(2:1) and ethyl acetate gave 95 mg of 3-[1.2,3,4--tetra-
hydro-2-methanesulphonylpyrazino[1.2-a]indol-10-yl]-4-
-(1-methyl-3-indolyl)-1H-pyrrole-2,5-dione of melting
point 298-301°C (decomposition).
Examyle 77
2b
3.0 g of the product of Example 1 were dissolved in
100 ml of THF and the solution was added to a suspension
of 1.8 g of lithium aluminium hydride in 50 ml of THF at
0°C. Then the mixture was heated at reflux for 16 hours.
The mixture was then cooled, treated with 10 ml of water
and extracted with dichloromethane. The combined
dichloromethane extracts were washed with aqueous sodium
bicarbonate solution, dried and concentrated to give a
solid. Chromatography on silica gel with dichloromethane/
methanol (95:5) gave a solid which was purified further by
chromatography to give
20.0636
_. ~3 _
a) 400 mg of 1,5-dihydro-3-[6,7,8,9--tetrahydro--8-
-(hydroxymethyl)pyrido[1,2-a]indol-10-yl]-4-(1-methyl-
-3-indolyl)-2H-pyrrole-2-one of melting point 205-207°C.
There were also obtained
b) 160 mg of 1,5-dihydro-4-~[6,7,8,9-tetrahydro-8-
-(hydroxymethyl)pyrido[1,2-a)indol-10-yl]-3-(1-methyl-3-
-indolyl)-2H-pyrrole-2-one of melting point 201-203°C.
Example 78
In a manner analogous to that described in the first
paragraph of Example 1. from 0.5 g of 3-[8-(acetoxy-
methyl)-6,7,8,9-tetrahydropyrido[1,2-a)indol-10-yl]-4-
-(3-trifluoromethylphenyl)furan-2,5-dione there were
obtained 110 mg of 3-[6,7,8,9-tetrahydro--8-(hydroxy-
methyl)pyrido[1,2-a]indol-10-yl]-4-(3-trifluoromethyl-
phenyl)-1fI-pyrrole-2,5-dione solid of, melting point
77-79°C. .
The furandione starting material was prepared as
follows:
1.7 g of oxalyl chloride were added to a cold (0-4°C)
solution of 3.0 g of B-(acetoxymethyl)-6,7,8,9-tetra-
hydropyrido[1,2-a]indole in 50 ml of dichloromethane.
After 2 hours the solvent was evaporated and the residue
was dissolved in dichlocomethane. The solution was added
to a solution of 2.7 g of (a,a,a-trifluoro-m-tolyl)-
acetic acid and 3.2 g of triethylamine in 70 ml of
dichloromethane. The mixture was stirred for 16 hours and
then concentrated. The residue was chromatographed on
silica gel with dichloromethane/methanol (95:5). There
were obtained 700 mg of 3-[8-(acetoxymethyl)-6,7,8,9-
-tetcahydropyrido[1,2-a]indol-10-yl]-4-(3-trifluoromethyl-
phenyl)furan-2,5-dione of melting point 176-177°C.
20.0636
- 74 -
Example 79
In a manner analogous to that described in the first
paragraph of Example 1, from 1.0 g of 3-[8-(acetoxy-
methyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-4--
-(4-methoxyphenyl)furan-2,5-dione there were obtained
150 mg of 3-[6,7,8,9-~tetrahydto-8-(hydroxymethyl)--
pyrido[1,2-a]indol--10-yl]-4-(4-methoxyphenyl)-lEI-
-pyrrole-2,5-dione of melting point 228°C (decomposition).
The furandione starting material was prepared as
follows:
1.7 g of oxalyl chloride were added to a cold (0-4°C)
solution of 3.0 g of 8-(acetoxymethyl)-6,7,8,9-tetrahydro-
pyrido[1.2-a]indole in 50 ml of dichloromethane. After
2 hours the solvent was evaporated and the residue was
dissolved in dichloromethane. This solution was added to a
solution of 2.24 g of p-methoxyphenylacetic acid and 3.2 g
of triethylamine in 70 ml of dichloromethane. The mixture
was stirred for 16 hours and then concentrated. Chromato-
graphy of the residue on silica gel With dichloromethane/
methanol (95:5) gave 2 g of 3-[8-(acetoxymethyl)-6,7,8,9-
-tetrahydropyrido[1,2-a]indol-10--yl]-4-(4-methoxyphenyl)--
furan-2.5-dione of melting point 79-82°C.
Examele 80
In a manner analogous to that described in the first
paragraph of Example 1, from 0.8 g of 3-[8-(acetoxy-
methyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-
-10-yl]-4-(2-chlorophenyl)furan-2.5-dione there were
obtained 120 mg of 3-(2-chlorophenyl)-4-[6,7.8,9-tetra-
hydro-8-(hydroxymethyl)pyrido[1,2-a]indol-10-yl]-lEI-
-pyrrole-2,5-dione of melting point 232-233°C.
2010636
_ 75 -
The furandione starting material was pcepared as
follows:
2.2 g of oxalyl chloride were added to an ice-cold
solution of 4 g of 8-(acetoxymethyl)-6,7,8.9-tetcahydro-
pycido[1.2-a]indole in 50 ml of dichloromethane. After
2 hours the solvent was evaporated and the residue was
dissolved in diahloromethane. This solution was added to a
solution of 3.0 g of 2-chlocophenylacetic acid and 4.0 g
of triethylamine in dichloromethane. The mixture was
stirred for 16 hours and then concentrated. Chromatography
of the residue on silica gel with dichloromethane/methanol
(95:5) yielded 0.9 g of 3-[8-(acetoxymethyl)-6.7.8.9-
-tetrahydropyrido[1,2-a]indol-10-yl]-4-(2-chlorophenyl)-
fucan-2.5-dione of melting point 168-171°C.
Examule 81
In a manner analogous to that described in Example 51,
from 80 mg of the product of Example 78 there were
obtained 30 mg of 3-[8-(aminomethyl)-6,7.8,9-tetra--
hydropyrido[1.2-a]indol-10-yl]-4-(3-trifluoromethylphenyl)-
-1H-pyrrole-2.5-dione of melting point 202--204°C.
z5 Examule 82
In a manner analogous to that described in Example 51,
fcam 100 mg of the product of Example 79 there were
obtained 88 mg of 3-[B-(aminomethyl)-6.7.8.9-tetta-
hydropyrido[1,2-a]indol-10-yl]-4-(4-methoxyphenyl)-1H-
-pyrrole-2,5-dione of melting point 195-19S°C.
Examvle 83
3,5 In a manner analogous to that described in Example 53.
from 80 mg of the product of Example 80 there were
obtained 57 mg of 3-[8-(aminomethyl)-6.7.8.9-tetrahydro-
2010636
- 76 -
pyrido[1,2-a]indol-10-yl]-4-(2-chlorophenyl)-1H-pyrrole-2,5-
dione of melting point 206-208°C (decomposition).
The following Examples illustrate typical
pharmaceutical preparations containing compounds provided
by the present invention:
Example A
1p Tablets containing the following ingredients may be
produced in a conventional manner:
Incrredient Per tablet
Compound of formula I 5.0 mg
Lactose 125.0 mg
Maize starch 75.0 mg
Talc 4.0 mg
Magnesium stearate ~ 1.0 ma
Tablet weight 210.0 mc~
Example B
Capsules containing the following ingredients may be
produced in a conventional manner:
Inaredient Per capsule
Compound of formula I 10.0 mg
Lactose 165.0 mg
Maize starch 20.0 mg
Talc 5.0 ma
Capsule fill weight 200.0 and