Note: Descriptions are shown in the official language in which they were submitted.
y2011G~~
1,7-FUSED 1H-INDOLE-2-CARBOXYLIC ACID
N-(1 4-BENZODIAZEPIN-3-YL)AMIDES
Backctround of the Invention
The present invention relates to novel amides of 3-
amino-1,4-benzodiazepine derivatives with 1,7-fused 1H-
indole-2-carboxylic acid derivatives and the salts thereof,
and pharmaceutical compositions containing these compounds
and to.a process for the preparation of these compounds.
Published European Patent Application No. 0,167,919
discloses 1,4-benzodiazepine derivatives which are substituted
in the 3-position and have CCR-antagonistic effects.
Cholecystokinin (= c:CK) is a peptide which occurs in
gastrointestinal tissue a.nd in the central nervous system,
has a widely diverse specarum of effects and which exerts,
inter olio, stimulating effects on colon motility,
gallbladder contraction and exocrine pancreas secretion and
inhibitory effects on emptying of the stomach, and also
influences appetite regulation. CCK antagonists are
pharmacologically active substances which are able to bind
to CCK receptors and thus can inhibit CCK-induced processes.
Summary of the Invention
The present invention provides novel compounds having CCK-
ant:agonistic activity and having an impoved activity profile.
y
201165
The invention furthermore prepares novel derivatives of 1, 7-
fused 1H-indole-2-carboxylic acids ~rith valuable pharmacological
properties.
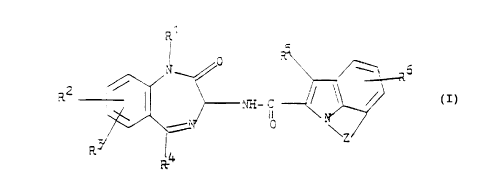
More particularly, the invention provides a compound
corresponding to the formula I
cc ,
i
~ n
V_ i
_ ~ ~~ //~ (1)
r- .r~n i
0 .,\
Z
wherein
R' represents hydrogen, lower alkyl or cycloalkylalkyl
with 4-7 carbon atoms,
Rz represents hydrogen, halogen, lower alkyl, lower alkoxy
or trifluoromethyl,, and
R~ represents hydrogen, halogen, lower alkyl or lower
alkoxy, or
R' and R3 are bonded to two adjacent carbon atoms and
together denote an alkylenedioxy group with 1-2 carbon
atoms,
R4 represents cycloal.kyl with 5 to 6 carbon atoms,
thiophene or an opt:ionaily substituted phenyl group a
(a)
~8
in which
R~ represents hydrogen, halogen, lower alkyl, lower
alkoxy or trifluoromethyl, and
R8 represents hydrogen, halogen, lower alkyl or lower
alkoxy,
- 2 -
~.H
201 ~
RS represents hydrogen or halogen,
R6 represents hydrogen, halogen, lower alkyl, lower alkoxy
or trifluoromethyl, and
Z represents an alkylene chain with 2-4 carbon atoms,
which can optionally be mono- or disubstituted by lower
alkyl, or onto which a 5-6-membered carbocyclic ring
can optionally be fused, or represents a -X-CH2-CHz
chain in which X is bonded to the phenyl ring of the
indole structure and represents oxygen or sulfur,
and the acid addition salts thereof.
It has now been found that the 1,7-fused 1H-indole-
2-carboxylic acid-N-(1,4-benzodiazepin-3-yl)-amides
according to the invention have CCR-antagonistic properties
and are distinguished by a novel type of pharmacological
activity profile with a pronounced component of the effect
promoting emptying of the stomach, along with a good
therapeutic index and low toxicity.
Detailed Description of Preferred Embodiments
The present invention therefore relates to novel 1,7-
fused 1H-indole-2-carboxylic acid-N-(1,4-benzodiazepin-3-
yl)amide compounds corresponding to the formula I
-a
s y
~/ v
;~ ~ /~ (I>
~;~_ ~; ~
I ,~
~o ~4
in which
R~ represents hydrogen, lower alkyl or cycloalkylalkyl
with 4-7 carbon atoms,
:35 R~ represents hydrogen, halogen, lower alkyl, lower alkoxy
or trifluoromethyl, and
- 3 -
2~11~~8
R3 represents hydrogen, halogen, ~eower alkyl or lower
alkoxy, or
RZ and R3 are bonds:d to two adjacent carbon atoms and
together denote an alkylenedioxy group with 1-2 carbon
atoms,
R4 represents cycioalkyl with 5 to 6 carbon atoms,
thiophene or an optionally substituted phenyl group a
R7
l0
R8
in which
R7 represents. hydrogen, halogen, lower alkyl, lower
alkoxy or trifluoromethyl, and
R8 represent; hydrogen, halogen, lower alkyl or lower
alkoxy,
RS represents hydrogen or halogen,
R6 represents hydrogen, halogen, lower alkyl, lower alkoxy
or trifluoromethyl, and
Z represents an alkylene chain with 2-4~ carbon atoms,
which can optionally be mono- or disubstituted by lower
alkyl, or onto which a 5-6-membered carbocyclic ring
can optionally be fused, or represents an -X-CHz-CHZ-
chain in which X is bonded to the phenyl ring of the
indole structure and represents oxygen or sulfur,
and the acid addition salts thereof.
R~ in the compounds of formula I preferably represents
a lower alkyl group. This can be straight chain or branched
and preferably contain 1-4 carbon atoms. Cyclopropylmethyl
may be mentioned a.s example of a cycloalkylalkyl group.
Particularly suitab:Le R' radicals have proved to be straight-
chain and branched alkyl groups with 1-3 carbon atoms,
especially methyl. _
Where the substituents R2, R3 and R4 in the compounds of
formula I represent or contain a lower alkyl group, this can
- 4 -
~0~~6~~
represent a straight or branched alkyl group with 1-5,
preferably 1-4, carbon atoms, especially methyl or ethyl.
Thus, lower alkyl substituents preferably represent methyl,
and lower alkoxy subst;ituents preferably represent methoxy.
Halogen substituents R~, RZ, R3, R5 and R6 represent, in
particular, fluorine, chlorine or bromine.
The substituents R2 and R3 are preferably located in the
7- and 8-positions of the benzodiazepine structure and
preferably represent hydrogen, lower alkoxy, especially
methoxy or lower alkyl, especially methyl, or else chlorine.
A methoxy substituent: in the 8-position has proved to be
particularly advantageous.
The substituent :R4 preferably represents an optionally
substituted phenyl group. The substituents R7 and R8 of the
5-phenyl group preferably represent hydrogen, lower alkyl,
especially methyl, or halogen, especially fluorine or
chlorine, or else lower alkoxy with 1-5 carbon atoms, for
example isopentyloxy. A phenyl group R4 which is
unsubstituted or substituted by fluorine is particularly
suitable.
If the substitue:nt R4 represents a cycloalkyl group, it
is preferably cyclohexyl.
The substituent R6 preferably represents hydrogen or
else halogen, especially fluorine, or lower alkoxy,
especially methoxy. T:he substituent R5 preferably represents
hydrogen. If R5 represents halogen, it is preferably
chlorine.
Z represents a chain with 2-4 chain members, preferably
an alkylene chain with 2-4 carbon atoms. Thus Z forms with
the aminoethylene group to which it is bonded a 5- to 7
membered heterocycle. Z preferably represents a propylene
chain and thus forms together with the indole structure to
which it is bonded, a 4H-pyrrolo[3,2,1-ij]-5,6-dihydro-
quinoline structure. Where the alkylene chain Z is
substituted by lower alkyl, this can contain 1-4 carbon
atoms and represents, in particular, methyl. Where a
- 5 -
20~~.~~~
carboxylic -._-ing is fused onto the alkylene chain Z, this
ring can ire unsaturated or saturated and preferably
represents a benzene: ring.
The compounds of formula I contain an asymmetric carbon
atom in the 3-position of the benzodiazepine structure and
can exist in the D and the L forms or as racemate. The
present invention embraces both the racemic mixtures and the
pure optical isomer: of the compounds of formula I.
The novel amides of formula I and the acid addition
salts thereof are obtained according to the invention by
acylating, in a known manner, amino compounds corresponding
to the formula II
..,
~I ~j
:~2 / v ~TH ( I I )
y
R
in which R' , R2, R3 and R4 have the above meanings, with acids
or reactive acid derivatives corresponding to formula III
~J
a =_ ~_y (III)
in which R5, R6 and Z have the above meanings, and Y
represents hydroxyl. or a reactive group, and, optionally,
converting free compounds of formula I into the acid
addition salts thereof, or converting the acid addition
salts into the free: compounds of formula I.
- 6 -
~4~~~~~~
The acylation of the amino compounds of formula II can
be carried out by c:ustomary methods for the formation of
amide groups by amin.oacylation. Acylating agents which may
be used include acids of formula IIIa
~~/ ~,
'i -,~~ (IIIa)
_.~~,: 4~
in which Rb, R7 and Z have the above meanings, or the
reactive derivatives thereof. Particularly suitable
reactive derivatives are mixed anhydrides, esters and acid
halides. Thus, reactive groups '~ can represent, for
example, lower alkoxy, halogens such as chlorine or bromine
or, preferably, organic sulfonic acid residues, for example
residues of lower alkanesulfonic acids such as, for example,
methanesulfonic acid or of aromatic sulfonic acids such as
benzenesulfonic acid, or benzenesulfonic acids substituted
by lower alkyl or halogen, for example toluenesulfonic acids
or bromobenzenesulfonic acids. The acylation can be carried
out in an organic solvent which is inert under the reaction
conditions, preferably at temperatures between -20 °C and
room temperature. Particularly suitable solvents include
halogenated hydrocarbons such as dichloromethane or aromatic
hydrocarbons such as benzene or toluene or cyclic ethers
such as tetrahydrofuran or dioxane or mixtures of these
solvents.
The acylation can advantageously be carried out,
especially when a mixed anhydride of the acids of formula
IIIa with a sulfonic acid is used as acylating agent, in the
presence of an acid-binding reagent. Suitable acid-binding
agents are bases soluble in the reaction mixture, especially
organic bases such as tert.-lower alkylamines and pyridines
such as, for example, triethylamine, tripropylamine,
~01~~
pyridine, 4-dimethylaminopyridine, 4-diethylaminopyridine or
4-pyrrolidinopyridine. It is also possible for organic
bases employed in excess to act simultaneously as a solvent.
It is possible and advantageous for mixed anhydrides of
the acids of formula :LIIa with organic sulfonic acids to be
obtained in situ by reacting the acids IIIa with an acid
halide, especially then acid chloride of the organic sulfonic
acid, and, without isolation, immediately further reacting
with the amino compound of formula II.
If the acid itself, or an ester, is employed as
acylating agent, the: reaction of the amino compound of
formula II with the acid of formula IIIa or the ester
thereof may also advantageously be carried out in the
presence of a coupling reagent known from peptide chemistry
as suitable for amide formation. Examples of coupling
reagents which promote amide formation with the free acids
by reacting with the acid in situ to form a reactive acid
derivative, include, in particular, alkylcarbodiimides, for
instance, cycloalkylcarbodiimides such as dicyclohexyl-
carbodiimide, or 1-ethyl-3-[3-(dimethylamino)-propyl]-
carbodiimide, carbonyldiimidazole and N-lower-alkyl-2-
halopyridinium salt;, especially halides or tosylates,
preferably N-methyl-~2-chloropyridinium iodide (see, for
example, Mukaiyama in Angew. Chemie 91 789-812). The
reaction in the presence of a coupling reagent can
advantageously be carried out at temperatures from -30 °C to
+50 °C using solvents such as halogenated hydrocarbons and/or
aromatic solvents, optionally in the presence of an acid-
binding amine.
The compounds of formula I can be isolated from the
reaction mixture by known techniques and purified in a known
manner. Acid addition salts can be converted in a customary
manner into the free bases, and the latter can, if desired,
be converted in a known manner into pharmaceutically
acceptable acid addition salts. Examples of suitable
pharmaceutically acceptable acid addition salts of the
g _
201~~~~
compounds of ~ormula I include the salts thereof with
inorganic acids, for example hydrogen halide acids,
particularly hydrochloric acid, sulfuric acid or phosphoric
acids or with organic acids, for example lower aliphatic
mono- or dicarboxylic acids such as lactic acid, malefic
acid, fumaric acid, tartaric acid or acetic acid or sulfonic
acids, for examples lower alkylsulfonic acids such as
methanesulfonic acid or benzenesulfonic acids which are
optionally substituted in the benzene ring by halogen or
lower alkyl, such as p-toluenesulfonic acid or
cyclohexylaminosulfonic acid.
If racemates of the compounds of formula II are
employed in the synthesis, the compounds of formula I are
obtained in the form of racemates. It is possible to obtain
optically active compounds of formula I starting from
optically active foams of the compounds of formula II. The
optically active compounds of formula I can be obtained from
the racemic mixtures in a known manner, for example by
separation by chromatography on chiral separating materials
or by reaction with suitable optically active acids, such as
tartaric acid or J.0-camphorsulfonic acid, and subsequent
fractionation into the optically active antipodes thereof by
fractional crystallization of the resulting salts.
The amine compounds of formula II are known or can be
2~~ prepared by known processes or analogously to known
processes. Thus, t:he amine compounds of formula II can be
obtained, for example, in a known manner by reducing an
oxime compound corresponding to formula IV
3 CI
~~ ~;-=,r_,J~ ( IV )
i
7
..
_ g _
~o~~~~ ~a
in which R' , R2, R3 and R' have the above meanings . The
oximes of formula IV <:an be reduced to the amines of formula
II by customary methods, for example catalytic
hydrogenation, preferably in the presence of a Raney nickel
catalyst or with zinc,iglacial acetic acid as reducing agent.
It may prove advantageous in the reduction with zinc/glacial
acetic acid to add a halogenated organic carboxylic acid to
activate the zinc.
The compounds of formula II contain an asymmetric
carbon atom in the 3-position of the benzodiazepine
structure. They are obtained in the form of racemates in
the synthesis. Then optically active compounds can be
obtained from the racemic mixtures in a known manner, for
example by chromatographic separation on chiral separating
materials or by reaction with suitable optically active
acids, for example tartaric acid or 10-camphorsulfonic acid,
and subsequent fractionation into the optically active
antipodes thereof by fractional crystallization of the
resulting salts. Racemic mixtures of the amines of formula
II can, for the fractionation, also first be reacted with an
optically active amino acid, for example phenylalanine, by
methods customary in peptide chemistry to give the
corresponding amides of the optically active amino acid.
Thus, the racemic compounds of formula II can, for example,
be reacted with an amino acid whose NHz group is protected by
a protective group known from peptide chemistry, for example
tert.butoxycarbonyl (= BOC group), and the protective group
can subsequently be eliminated again in a known manner. The
pair of diastereomE:ric amides produced thereby can be
fractionated in a known manner, for example by fractional
crystallization or chromatography, and subsequently the
amino compound of formula II can be liberated again from the
amides in a known manner.
The oxime compounds of formula I'J can be prepared in a
known manner by nitrosation of compounds of formula V
- 10 -
~~~~~~~)
J
~;~i. ( V
Ft '-
,~i' ~~; r~
r, JI .. 'T
in which R~ , RZ, R3, and R4 have the above meaning. It is
advantageous for i~he compounds of formula V which are
10~ unsubstituted in the 3-position first to be treated with a
strong base, for example an alkali metal alcoholate such as
potassium tert.butylate, in an organic solvent which is
inert under the reaction conditions, for example an aromatic
hydrocarbon such as benzene or toluene or a cyclic ether
1_°°> such as tetrahydrofuran, and then reacted with a
nitrosating
agent, for example with a lower alkyl nitrite such as
isoamyl nitrite or tert.butyl nitrite.
Compounds corresponding to the formula Va
-,
2 ()
~~r-r/
~2 ~j \ (Va)
i
.,
in which R'~ has the meaning given for R' with the exception
of hydrogen, and R2, R3 and R4 have the above meaning, can be
obtained in a known manner by oxidizing 2-chloromethyl-1,4-
benzodiazepine compounds corresponding to the formula VI
3 0 1'
r-r ~
C a~.. L
a
(VI)
Iy,
3 5 ~ ~!,
- 11 -
in which R~' ~ , RZ, R-' and R'' have the above meanings, in a
known manner known. The oxidation can be carried out, for
example, by treating the compounds of the formula VI with a
suitable oxidizing agent in the presence of a solvent which
is inert under tree reaction conditions. Examples of
oxidizing agents which can be employed include potassium
permanganate, chromium trioxide or dichromate salts.
Examples of suitable solvents which are inert to these
oxidizing agents include halogenated hydrocarbons such as
10 dichloromethane, water or acetic acid, or mixtures thereof.
The starting compounds of formula VI are known or can
be prepared by known methods or analogously to known
methods.
The compounds can be obtained, for example, in a known
manner by the methods described in German published
application Nos. DE 22 21 558 or DE 25 20 937 starting from
2-hydroxy-1,3-diaminopropane compounds corresponding to the
formula VIII
~' Pv-C'.-:'-CH-CH--N::-~-~ (VIII)
j
2 ~; R 3,
in which R' ~ , Rz, R3 and R4 have the above meanings . The.
compounds of formula VIII are cyclized by treatment with.
phosphorus oxychloride. For this purpose, the compounds of
formula VIII or the acid addition salts thereof are
advantageously treated with phosphorus oxychloride at a
temperature between 100 and 150 °C, preferably at the boiling
point of the reaction mixture. This results in a mixture of
a 2-chloromethyl-1,4-benzodiazepine compound of formula VI:
3°_i with the 3-chloro-1,5-benzodiazocine compound of formula
VII, which is isomeric thereto,
- 12 -
~~~~~z.~
~,~~ ~- (VII)
R_ I i
i4
R
l0
in which R' ~ , R2, R3' and R4 have the above meanings. The
benzodiazocine compound of formula VII in the mixture can be
rearranged into the. isomeric compound of formula VI in a
known manner, for example by heating the mixture in an
organic solvent which is inert under the reaction
conditions, for example a high-boiling halogenated
hydrocarbon such as tetrachloroethane.
Preparation of 2-hydroxy-1,3-diaminopropane compounds
of formula VIII can start from anilines of formula IX
/~.
FZG ~ (IX)
v
2 5 3;'
in which RZ and R3 have the above meanings. The amino group
in these anilines i~~ first monosubstituted by an R~~ group in
a known manner and subsequently reacted analogously to the
method described in German published application No. DE 28
10 349 with 1,2-epoxypropylphthalimide, or first with
epichlorohydrin and thereafter with phthalimide. The
phthalimide group is subsequ~ tly cleaved in a known manner,
and the resulting compounds of formula X
- 13 -
~0~ ~~'~~
~I-c. ...~-hri,, (X)
-_. , _. _
:/
in which R~~, R~ and R3 have the above meaning, are acylated
with acid halides corresponding to the formula XI
~4-~,0-C~ ( XI )
in which R" has the above meaning.
Compounds of :Formula V can also be obtained starting
from ketones corresponding to the formula XII
R
2 0 Vii.
(XII)
-v
q4
2 _'i
in which R' , Rz, R3 and R~ have the above meanings, in a known
manner, by reacting a ketone of formula XII with a
haloacetyl halide to give a compound corresponding to the
formula XIII
30 F.
-~ ~-CH.,~:al
., c
., _ ~~i (XIII)
/ ~=v
i
x
305 aJ ~=i
- 14 -
~~~~_a'~~;
in which R' , R', R3 and R' have the above meanings, and Hal
represents chlorine or bromine, and subsequently condensing
the compound of formula XIII with ammonia to give a compound
of formula V.
The ketones of formula XII are known or can be prepared
by known methods or analogously to known methods, for
example by reaction of p-substituted anilines corresponding
to formula IX with .acid halides of formula XI, for example
in a Friedel Crafts reaction with subsequent hydrolysis, or
starting from anthranilic acids, by first condensing the
acid with acetic anhydride, reacting the condensation
product in a Grignard reaction with a compound corresponding
to the formula XIV
(XIV)
in which R4 has the above meaning, to give the N-acetyl
derivatives of the compounds of formula XII, and hydrolyzing
the latter.
Compounds corresponding to the formula V in which R'
represents hydrogen can be alkylated in a known manner to
give other compounds of formula V, for example by reaction
with a compound corresponding to the formula XV
1 ~ (XV)
-gal
in which R~~ has t:he above meaning, and Hal represents
chlorine, bromine or iodine.
The esters of formula IIIb
/ v ..
a o I ' -,W'
;j ( IIIb)
- 15 -
~0~~~
in which R~ and Z have the above meanings, and R9 represents
lower alkyl, can be obtained in a known manner starting from
compounds corresponding to the formula XVI
~c- ~I
(~I)
~i
in which R5 and Z have the above meanings, by treating a
1G compound of formula XVI with sodium nitrite to convert it.
into the corresponding N-nitroso compound, and reducing the
nitroso compound to a hydrazine compound corresponding to
the formula XVII
1 ~~ j
/:Vfi ( XVI I )
~~~~ Y
in which R6 and Z have the above meanings, and reacting the
20 hydrazine compound of formula XVII further with a lower
alkyl pyruvate corresponding to the formula XVIII
1
1~
C::3-~~-~~(7-~ (XVIII)
-,
in which R9 has the above meaning, in a known manner under
the conditions of the Fischer indole synthesis, so that a
hydrazone compound corresponding to the formula XIX
3 ()
~Ci~3
~z- nr-c-;
G (XIX)
i:)
- 16 -
2011 ~~S
in which R6, R9 and Z have the above meanings, is formed as
an intermediate which condenses further to give an ester of
formula IIIb. Examples of reducing agents which can be used
to reduce the nitroso compounds include lithium aluminum
hydride in tetrahydrofuran or metallic zinc powder in the
presence of acid. Catalytic hydrogenation of the nitroso
compounds to the hydrazines of formula XVII is also
possible. It is possible and advantageous to prepare the
esters of formula ILIb starting from compounds of formula
XVI in a one-pot process without isolating the individual
intermediates. This entails adding hydrochloric acid to the
reaction mixture containing zinc salts and the hydrazine
compound of formula XVII obtained after reducing the N-
nitroso compound, to further acidify the mixture, and then
adding the pyruvic ester of formula XVIII to the reaction
mixture. Upon addition of the pyruvic ester of formula
XVIII to the react ion mixture, the hydrazone compound of
formula XIX is formed as an intermediate which further
condenses under the reaction conditions to give the ester of
formula IIIb.
It is possible to hydrolyze the esters of formula IIIb
in a known manner to the corresponding acids and/or to
convert the esters :into reactive acid derivatives of such
acids.
Upon conversion into an acid halide, a halogen
substituent R5 is al:~o introduced into the compounds at the
same time.
The 1,7-fused 1H-indole-2-carboxylic acid-N-(1,4
benzodiazepin-3-yl)amide derivatives according to the
invention and the pharmacologically acceptable acid addition
salts thereof have valuable pharmacological properties, in
particular CCK-antagonistic effects and are distinguished by
a novel, favorable a~~tivity profile. Thus, the compounds of
formula I having CCK-antagonizing activity according to the
invention exhibit a pronounced activity of promoting
emptying of the stomach as well as inhibitory influences on
- 17 -
241~~~~~
CCK-induced exocrine pancreas secretion. In the dose range
effective to promote emptying of the stomach, they only
exhibit to a slight extent the side effect of inhibiting
gallbladder contraction and are distinguished by a low
toxicity and a large therapeutic index. Due to their
favorable activity profile, the compounds are suitable for
treating CCK-related disturbances of the emptying of the
stomach.
CCK comprises peptides of various chain lengths and
acts as a hormone and as a neuropeptide. Among the CCK
peptides, the octapeptide CCK-8 is the smallest unit with
the complete spectrum of CCK effects. Therefore, the
following pharmacological tests were performed with CCK-8.
Description of Pharmacoloctical Test Methods
1. Determination of the peripheral CCK receptor binding
affinity of the test substances.
The affinity of the compounds of formula I for
peripheral CCK receptors was measured in vitro on rat
pancreas homogenate. The inhibition of the binding of the
physiologically relevant octopeptide CCK-8 to peripheral CCK
receptors by the test substances was determined.
The receptor binding studies were carried out by a
modification of the method of van Dijk et al. (J.
Neuroscience 4 (1984), 1021-1033) using soya bean trypsin
inhibitor (= SBTI) as protease inhibitor. 3[H]-CCK-8, code
TRK 775, from Amersham Int., having a specific activity of
60-90 Ci/mmol, was used as tritium-labelled CCK-8.
Whole pancreas glands from male Wistar rats with a body
weight of 150-300 g, which had been sacrificed by
decapitation, were freed of fatty tissue and homogenized for
15 seconds in a 50-fold volume of an ice-cold test buffer
solution (10 mmol 2-[4-(2-hydroxyethyl)-piperazin-1-yl]
ethanesulfonic acid (= HEPES~, 130 mmol sodium chloride, 5
mmol magnesium chloride, 0.02 % bacitracins, 0.01 % SBTI, pH
7.4) with a Kinematics Polytron type homogenizer. The
- 18 -
~o~~.~~~
homogenates were then centrifuged at 48,000 g for 10
minutes. This washing method was repeated. After the last
centrifugation, the residue which was obtained each time in
the form of a pellet was suspended in a 550-fold volume of
the test buffer solution and immediately used for the
measurement.
For the binding test, 500 y~l of the tissue homogenate
were incubated with 50 ul of test buffer solution or 50 ~,1
of a solution of the compound to be investigated and with 50
~1 of a 3[H]-CCK-8 solution (final concentration 0.3 nM).
The non-specific binding was assumed to be 0.1 ~mol CCK-8.
The incubation lasted 90 minutes at 25 °C. All the compounds
were measured 3 times at each of several concentrations.
Aqueous solutions were employed as test substance
solutions and were prepared by appropriate dilution of 60 x
10~ molar aqueous stock solutions. Test substances which
are sparingly soluble in water were first dissolved in 96
ethanol, and this solution was diluted with sufficient water
for the ethanol concentration not to exceed 1.6 % by volume
in the solution to be tested.
Bound and free 3[H]-CCK-8 were separated by filtration
through glass fiber filters. The filters were washed twice
with 3 ml of HEPES solution each time and then placed in the
scintillation solution (SAVE scintillation solution from
Packard) in the dark overnight and counted in a Packard Tri-
Carb 1500CT" liquid scintillation counter. The ICSO of the
respective test substance was determined to be that
concentration which caused a 50 o inhibition of the specific
binding of tritiated CCK-8 to the receptors. From this, the
corresponding pKi value was calculated using the Cheng-
Prusoff equation.
The following Table A shows the pKi values for the
affinity of the test substances for peripheral CCK receptors
obtained by the test method described above. The example
numbers listed for the compounds of formula I refer to the
following preparative examples.
- 19 -
2. Determination of the minimum toxic dose.
Male mice weighing 20-25 g were given maximum oral
doses of 300 mg/kg of the test substance. The animals were
observed carefully for three hours for symptoms of toxicity.
In addition, all symptoms and deaths were recorded for a
period of 24 hours after administration. Concomitant
symptoms were likewise observed and recorded. If death or
serious symptoms of toxicity were observed, further mice
were given increasingly lower doses. The lowest dose which
caused death or serious symptoms of toxicity is indicated in
the following Table A as the minimum toxic dose.
- 20 -
20~~.~
Table A
Ex. In vitro binding to peripheral ' Minimum
' toxic
~
No. CCK receptors (pancreas) dose mg/kg mouse
i
~Ki values ; oral
1 i 8.87 ' > 300
i
2a ~ 9.59 '
2b i 7.71 '
i
4 i 8.60 i > 300
5 ~ 8.84 i > 300
i
6 i 8,49
i
g i 8.04 '
i
g t g,10 i > 300
i
10 i 8.75 ' 300
i
11 i 8.62 ' 300
i
13 i 9.00 ' > 300
i
15 i 8.91
i
17 i 8.00 '
i
lg i 8.93 ' > 300
i
~ 9.07 ' > 300
i
20 22 i 9.04 ' > 300
i
23 i 8.00 i > 300
i 7.62 i > 300
26 ~ 8.21
i
i 8.63 ' 300
i
25 33 i 8.43 ; > 300
37 i 9.19
i
3g i 9.01 '
i
3. Investigation of the effect of the test substances on
30 CCK-induced disturbances of emptying of the stomach.
Doses of CCK lead to substantial blockade of the
transpyloric transport of chyme from the stomach into the
duodenum. The ability of the test substances to eliminate
this blocking effect of CCK was investigated.
Female NMRI mice having body weights of 20-25 g were
used in groups of ten animals each. After withdrawal of
- 21 -
2~~ ~~
feed for 24 hours (drinking water ad libitum) the animals
were given an oral dose of the test substance suspended in
a volume of 10 ml/kg of body weight of a 1 % strength
Tylose~ solution (= methylcellulose) or in a volume of 10
ml/kg of body weight of a solubilizer solution which
contains 5 % glycerol, 87 % polyethylene glycol 400, and 8
% water. A control group received only the Tylosee solution
or the solution containing the solubilizer polyethylene
glycol in each case. 60 minutes later the animals were
given a subcutaneous injection of 80 Ig/kg CCK-8. After a
further 5 minutes the animals were each orally administered
0.3 ml of a charcoal suspension (5 % charcoal in 2 ~
strength Tylose solution). The mice were sacrificed 5
minutes later and subsequently dissected. They were
examined to find whether charcoal suspension had advanced
into the duodenum.
Whereas under control conditions, i.e. without a dose
of CCK, charcoal suspension was found in the duodenum of all
the mice, transport of the chyme into the duodenum was
prevented in the groups of control animals which received a
dose of CCK, and traces of charcoal suspension were found in
the duodenum of not more than 5 % of the animals. The
percentage of animals in which the CCK effect was eliminated
and charcoal suspension was found in the duodenum after
administration of the test substances was determined.
The following Table B lists the results obtained in the
foregoing tests for dosages of the test substances which
inhibit the CCK effect in at least 40 % of the animals. It
is evident from the table that the activity of the
substances can often be considerably enhanced by
administration in a solution containing PEG 400 solubilizer.
- 22 -
201 ~~~
Table B
Ex. ' CCK-antagonistic Effect on CCK-induced
No. i Disturbance of Stomach Emptying
Dose ~ % of Animals in
i umol/kg Mouse i which the CCK Effect
oral ~ was Eliminated
1 i 0.215 ; 50
2a i 0.1 i 100
2b i 10 ~ 70
i
4 i 1.0 ~ 45
i
5 ~ 10 i 75
6 i 0.681 ~ 40
i
9 i 0.215 ~ 50
i
10 i 0.1 ~ 40
i
13 i 10 ~ 83
i
i 1.0* i 100
15 i 1.0 ~ 57
i
0.10* ~ 90
i i
17 i 1.0* ~ 50
i
20 i 1.0 ~ 50
i
27 i 1.0 ~ 40
i
33 ~ 1.0 ; 50
* = administered in solution containing solubilizer.
4. Determining the inhibitory effect of Test Substances on
CCK-Induced Gallbladder Emptying.
CCK causes a contraction of the muscles of the
gallbladder and thus an emptying of the gallbladder. This
results in the weight of the gallbladder decreasing compared
with control animals not treated with CCK. Intraperitoneal
doses of 0.1 ~g/kg CCK-8 may result in the weight of the
gallbladders decreasing to about 1./10 of the original
weight.
Female NMRI mice having a body weights of 20-25 g were
used in groups of 10 animals each. After withdrawal of feed
for 24 hours (drinking water ad libitum) the animals
received an oral dose of the test substance suspended in a
- 23 -
201~.~ ~'3
volume of 10 ml/kg of body weight of a 1 % strength Tylose~
solution (= methylcellulose) or in a volume of 10 ml/kg of
body weight of a solubilizer solution which contained 5 %
glycerol, 87 % polyethylene glycol 400, and 8 % water. Two
control groups each received only the Tylose~ solution or
the solution containing the solubilizer polyethylene glycol.
One of the control groups and all the animals treated with
test substance received 0.1 Ig/kg CCK-8 injected i.p. 60
minutes later.
The mice were sacrificed 5 minutes after the CCK-8
administration, and the gallbladders were excised and
weighed.
The following Table C lists the percentage inhibition
of the CCK-8 effect on gallbladder weights caused by the
test substances.
Table C
Ex. ~ CCK-Antagonistic Effect on the Gallbladder
No. ~ Dose ~ % Inhibition of
~
i ~,mol/kg Mouse CCK-induced
oral ~ Gallbladder EmptvinQ
1 i 10 i 66
i 1 i < 25
i
2a i 0.46 ~ 16
2b i 46 ~ 8.3
i
6 ~ 10 i 73
i
9 ~ 100 ~ 99
i
i
4.6 i < 25
15 ~ 100 ~ 100
i
~ 10 i < 25
i
i
0.1* ~ < 25
20 ~ 10 i 25
33 i 10 ~ 19
i
Comparison of the data 'in Tables B and C shows that
CCK-antagonistic effects on the gallbladder were not
observed until the doses of substances were a multiple of
- 24 -
C:
the dosage ranges effective to prevent CCK-induced
disturbances of stomach emptying.
The doses to be used may vary between individuals and,
naturally, vary depending on the type of condition to be
treated, the substance used and the form of administration.
For example, parenteral formulations will generally contain
less active substance than oral products. However, drug
forms with an active substance content of 5-50 mg per single
dose are generally suitable for administration to larger
mammals, in particular humans.
Pharmaceutical compositions may contain the compounds
of formula I or physiologically acceptable acid addition
salts thereof together with conventional pharmaceutical
adjuvants in pharmaceutical forms such as, for example,
tablets, capsules, suppositories or solutions. The
compounds of formula I are distinguished by being readily
soluble in solutions containing customary pharmaceutical
adjuvants and by being highly absorbable. The pharmaceutical
formulations can be prepared by known methods using
customary solid vehicles such as, for example, lactose,
starch or talc or liquid diluents such as, for example,
water, fatty oils or liquid paraffins and using conventional
pharmaceutical adjuvants, for example tablet disintegrating
agents or preservatives.
The following examples are intended to illustrate the
invention in further detail without Limiting its scope in
any way.
Example 1:
3-[(4H-Pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-
amino]-8-methoxy-1-methyl-2-oxo-5-phenyl-1H-2,3-dihydro-1,4-
benzodiazepine.
A) 43 g of Ni-methyl-N~- ( 3-methoxyphenyl ) -2-hydroxy-1, 3
diaminopropane and 31 ml of triethylamine were dissolved in
280 ml of dichloromethane. While cooling in ice, a solution
of 24.4 ml of benzoyl chloride in 20 ml of dichloromethane
- 25 -
was slowly added dropwise to the solution. The reaction
mixture was stirred at room temperature for 1.5 hours. The
reaction solution was subsequently worked up by washing with
water and sodium chloride solution and evaporating the
solvent. The remaining residue was 70.0 g of crude N~-
methyl-N~-(3-methoxyphenyl)-N2-benzoyl-2-hydroxy-1,3-
diaminopropane. The pure product obtained by
recrystallization from toluene/i.sopropanol had a melting
point of 87-89 °C.
B) 64 g of the product obtained above were reacted with
64 ml of phosphorus oxychloride in an oil bath at a bath
temperature of 130 °C for 1.5 hours. The mixture was then
cooled and diluted with dichloromethane, and ice-water was
added to the solution. The organic phase was separated,
washed several times with water and then treated with dilute
sodium hydroxide solution, washed again with water, dried
over sodium sulfate, and evaporated. The residue obtained
was 56.7 g of an oily crude product which contained a
mixture of about 60 % 2-chloromethyl-1-methyl-8-methoxy-5-
phenyl-2,3-dihydro-1H-1,4-benzodiazepine and about 40 % 3-
chloro-1-methyl-9-methoxy-6-phenyl-1,2,3,4-tetrahydro-1,5-
benzodiazocine. To isomerize the benzodiazocine fraction,
the crude mixture was heated in 222 ml of tetrachloroethane
under reflux for 30 minutes. The tetrachloroethane was
subsequently evaporated, and the remaining 2-chloromethyl-
1-methyl-8-methoxy-5-phenyl-2,3-dihydro-1H-1,4-benzo-
diazepine was further processed in the next reaction stage
without purification.
C) 56.7 g of the product obtained above were dissolved
in 285 ml of dichloromethane. To the solution were added
322 ml of 32 % strength aqueous hydrochloric acid, 2481 ml
of water and 255 ml of dichloromethane. A solution of 32.4
g of potassium permanganate in 660 ml of water was
subsequently added dropwise, maintaining the internal
temperature below 15 °C by cooling in ice. The reaction
mixture was then stirred at room temperature for half an
- 26 -
Cs
hour. The reaction mixture was worked up by adding solid
sodium bicarbonate in portions until the reaction mixture
was neutral, filtering out the precipitate which formed from
the solution through asbestos slurry (commercial product
Theorit~) with suction, separating the dichloromethane
phase, and extracting the aqueous phase again with
dichloromethane. The combined dichloromethane extracts were
washed with dilute sodium hydroxide solution and then with
water, dried over magnesium sulfate and evaporated. 55 g of
crude product were obtained. The crude product was
subjected to column chromatography on silica gel under
slightly elevated pressure (flash chromatography) using
cyclohexane/ethyl acetate 4:6 as the eluent to isolate pure
1-methyl-8-methoxy-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-
benzodiazepine. 15.5 g of oily pure product were obtained.
D) 15.5 g of the above product were suspended in 293 ml
of toluene. The suspension was cooled to -20 °C and then
16.4 g of potassium tert.-butylate were added, while
stirring, and the mixture was stirred for a further 15
minutes. Then 9.4 ml of isoamyl nitrite were added, while
cooling, at such a rate that the temperature of the reaction
mixture remained below O °C. It was then stirred at O °C for
minutes. The reaction mixture was then worked up by
stirring it into a mixture of 586 ml of ice-cold water, 29
25 ml of glacial acetic acid and 586 ml of ethyl acetate. After
vigorous mixing the organic phase was separated and the
aqueous phase was extracted once more with ethyl acetate.
The combined organic phases were washed with water and
evaporated. 21 g of crude product were obtained. This was
30 crystallized from toluene/ethanol. The crystals were
separated out, and the mother liquor was again purified by
flash chromatography on 100 g of silica gel using
cyclohexane/ethyl acetate 4:6 as the eluent and evaporated.
The residue was crystallized from ethanol and combined with
the aforementioned crystals. A total of 9.5 g of 3-
hydroxyimino-1-methyl-8-methoxy-2-oxo-5-phenyl-2,3-dihydro-
_ 2~ -
1H-1, 4-benzodiazepine having a melting point of 206-207 °C
was obtained.
E) 9.5 g of the product obtained above were added to a
mixture of 700 ml of glacial acetic acid and 80 ml of
trifluoroacetic acid. The reaction mixture was heated to 40
°C (internal temperature) and a total of 6.9 g of zinc dust
was added in portions while stirring, the mixture was
stirred at 40 °C for a further 2 hours and then 1 g of zinc
dust was again added and the mixture was stirred at 40 °C for
a further 1.5 hours. The mixture was worked up by diluting
with toluene, allowing it to cool and evaporating the
solvent. The remaining residue was taken up in
dichloromethane, and the solution was washed with aqueous
sodium carbonate solution and water. Solids were filtered
out with suction through Theoritm, and the solution was
dried and evaporated. 8.1 g of crude 3-amino-1-methyl-8-
methoxy-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepine
were obtained and were further reacted in the subsequent
reaction stage H without purification.
F) 150 g of 1,2,3,4-tetrahydroquinoline were dissolved
in 1.25 1 of glacial acetic acid. A solution of 80 g of
sodium nitrite in 300 ml of water was added to the solution
while cooling in an ice bath to an internal temperature of
about 15 °C, and the reaction mixture was stirred for a
further 45 minutes. 300 g of zinc dust were added in
portions over the course of 1.5 hours to the reaction
solution which contained the N-nitroso-1,2,3,4-
tetrahydroquinoline formed, during which the reaction
mixture was maintained at an internal temperature of 15-20
°C by cooling in an ice bath. Subsequently 1.75 1 of water
and 1.25 1 of 32 % strength aqueous hydrochloric acid were
added to the mixture, which was stirred for a further 1.5
hours. 130 g of ethyl pyruvate were added to the acidic
reaction mixture which contained the resulting N-amino-
1,2,3,4-tetrahydroquinoline and the zinc salt, and the
mixture was heated to reflux for 1.5 hours and subsequently
_ 28 _
201~.~~~
allowed to stand for a further 16 hours. This involved in
situ condensation of the hydrazone formed as an intermediate
to give ethyl 4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-
carboxylate. The reaction mixture was worked up by
extracting twice with a total of 5 1 of dichloromethane, and
the dichloromethane extracts were combined, washed twice
with a total of 1 1 of water, dried over sodium sulfate and
reduced in volume. 280 g of crude ethyl 4H-pyrrolo[3,2,1-
ij]-5,6-dihydroquinoline-2-carboxylate were obtained and
were purified by chromatography on silica gel using
dichloromethane as the eluent. 151.8 g of purified product
having a melting point of 70-72 °C were obtained.
G) 39 g of ethyl 4H-pyrrolo[3,2,1-ij]-5,6-dihy
droquinoline-2-carboxylate were dissolved in 40 ml of
ethanol, and this solution was added at room temperature to
a solution of 11.3 g of potassium hydroxide in a mixture of
ml of water and 145 ml of ethanol. The reaction mixture
was stirred at room temperature for 90 minutes and then
cooled to 10 °C. The precipitated solid was filtered out
20 with suction and washed three times with 30 ml of ethanol
each time. The mother liquor was reduced to half volume,
and the solid which thereby precipitated was likewise
separated out and washed with ethanol.
All the solid was then dissolved in 150 ml of water,
and the acid was precipitated by acidifying the solution
with concentrated hydrochloric acid to pH 1 to 2. The
precipitated acid was separated out, washed three times with
40 ml of water each time and dried at 60 °C. 32.4 g of 4H
pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carboxylic acid
having a melting point of 212-21.3 °C (decomposition) were
obtained.
H) 5.4 g of 4H-pyrrolo[3,2,1--ij]-5,6-dihydroquinoline-
2-carboxylic acid and 3.67 ml of triethylamine were
dissolved in 119 ml of dichl_oromethane. The solution was
cooled to -20 °C and, while stirring, 2.08 ml of
methanesulfonyl chloride were slowly added dropwise, and the
- 29 -
~~1~~~~
reaction mixture was stirred at -20 °C for a further 15
minutes. To the reaction solution containing the mixed
anhydride from methanesulfonic acid and 4H-pyrrolo[3,2,1-
ij]-5,6-dihydroquinoline-2-carboxylic acid was added
dropwise, while stirring, a solution of 8 g of 3-amino-1-
methyl-8-methoxy-2-oxo-5-phenyl-2,3-dihydro-1H-1,4-
benzodiazepine obtained in stage E and 3.67 ml of
triethylamine in 100 ml of dichloromethane at a temperature
between -15 and -20 °C, and the reaction mixture was stirred
at -15 °C for a further 30 minutes and allowed to warm slowly
(within one hour) to room temperature. The reaction mixture
was worked up by diluting with water, separating the
dichloromethane phase, washing the dichloromethane phase
with sodium bicarbonate solution and then with water, drying
over sodium sulfate, filtering and evaporating the solvent.
14.3 g of the crude title compound were obtained as a
residue. The crude product was purified by column chrom-
atography on 700 g of silica gel under slightly elevated
pressure (flash chromatography) using cyclohexane/ethyl
acetate 1:1 as the eluent. 6.1 g of purified product were
obtained. This was crystallized from 25 ml of ethanol with
addition of a small amount of dichloromethane, and the
crystals were dried at 80 °C for 2 days. 3.3 g of racemic 3-
[(4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-
amino]-8-methoxy-1-methyl-2-oxo-5-phenyl-1H-2,3-dihydro-1,4-
benzodiazepine having a melting point of 175-178 °C were
obtained.
Example 2:
Preparation of the optical isomers of 3-[(4H-pyrrolo[3,2,1-
ij]-5,6-dihydroquinoline-2-carbonyl)-amino]-8-methoxy-1-
methyl-2-oxo-5-phenyl-1H-2,3-dihydro-1,4-benzodiazepine.
2a: (-)-isomer, optical rotation [a]p° - -88.2° (c = 0.5 in
dichloromethane).
- 30 -
~~~~.~~~3
A) 30.5 g of racemic 3-amino-1-methyl-8-methoxy-2-oxo-
5-phenyl-2,3-dihydro-1H-1,4-benzodiazepine (prepared
analogously to Example 1 E) were dissolved in 190 ml of
dimethylformamide. To the solution were successively added,
under exclusion of moisture, 28.8 g of N-tert.-
butoxycarbonyl-D-phenylalanine (= BOC-D-phenylalanine), 15
g of 1-hydroxybenzotriazole, 20.7 g of 1-ethyl-3-[3-
(dimethylamino)-propyl-carbodiimide hydrochloride and 15 ml
of triethylamine. The reaction mixture was stirred at room
temperature for 30 minutes. To work up the reaction
mixture, the dimethylformamide was removed by distillation
under reduced pressure, and the residue was dissolved in
ethyl acetate. The solution was shaken with 10 ~ strength
aqueous citric acid solution, the phases were separated from
one another, the aqueous phase was extracted once more with
ethyl acetate, and then the combined organic phases were
washed with 10 ~ strength aqueous sodium hydroxide solution,
water and aqueous sodium chloride solution, dried over
sodium sulfate and filtered. Removal of the solvent by
distillation resulted in 66 g of crude product which was
purified again by flash chromatography on 700 g of silica
gel using cyclohexane/ethyl acetate 1:1 as the eluent.
After evaporation of the solvent, 60 g of 1,1-dimethylethyl-
N-((R)-2-[(2,3-dihydro-1-methyl-2-oxo-5-phenyl-8-methoxy-1H-
1,4-benzodiazepin-3-yl)amino]-2-oxo-1-benzylethyl)-carbamate
were obtained as a 1:1 mixture of diastereomers.
B) 60 g of the mixture of diastereomeric amides
obtained above were dissolved in 480 ml of ethyl acetate.
To eliminate the BOC protective group from the amides, the
solution was saturated with gaseous hydrogen chloride, and
the reaction mixture was stirred for 30 minutes. This
resulted in crystals of the hydrochloride of the liberated
amines in which the diastereomer with (-)-rotation was
enriched. The crystals were filtered out with suction.
Three recrystallizations from ethanol yielded a
diastereomerically pure hydrochloride of 3-phenyl-2-amino-
- 31 -
20116~~~
N-(2,3-dihydro-1-methyl-2-oxo-5-phenyl-8-methoxy-1H-1,4-
benzodiazepin-3-yl)-propionamide with (-)-rotation in
dichloromethane solution. To liberate the base, 10
strength sodium hydroxide solution was added to the
hydrochloride, and the base was extracted from the aqueous
phase with ethyl acetate. The organic phase was washed,
dried and evaporated to result in 18.4 g of
diastereomerically pure 3-phenyl-2-amino-N-(2,3-dihydro-1-
methyl-2-oxo-5-phenyl-8-methoxy-1H-1,4-benzodiazepin-3-yl)-
propionamide. Rotation [a]p~ of -30.6° (c = 0.5 in
dichloromethane).
C) 18.4 g of the diastereomerically pure amide with
(-)-rotation in dichloromethane solution obtained above were
dissolved in 100 ml of dichloromethane. 5.4 ml of phenyl
isothiocyanate were added to the solution under exclusion of
moisture, and the reaction mixture was stirred at room
temperature for 10 minutes. The dichloromethane was then
removed by distillation under reduced pressure, and the
remaining residue was purified by flash chromatography on
500 g of silica gel using cyclohexane/ethyl acetate 1:1 as
the eluent. Evaporation of the solvent resulted in a
resinous foam which was crystallized from ethanol. 20.1 g
of N~-phenyl-NZ-(2-[(2,3-dihydro-1-methyl-2-oxo-5-phenyl-8-
methoxy-1H-1,4-benzodiazepin-3-yl)amino]-2-oxo-1-(benzyl)-
ethyl)-thiourea having a melting point of 138-160 °C were
obtained. Optical rotation: [a]p° - -11.2° (c = 0.5 in
methanol).
D) 30.7 ml of trifluoroacetic acid were added to 20 g
of the thiourea product obtained above, and the reaction
mixture was heated at 50 °C for 20 minutes. The
trifluoroacetic acid was then removed by distillation under
reduced pressure, and the remaining residue was evaporated
twice with dichloromethane, redissolved in dichloromethane
and purified by flash chromatography on 500 g of silica gel,
initially using as the eluent a 1:1 cyclohexane/ethyl
acetate mixture and then a 90:10:1:1 mixture of
- 32 -
~0~~6~~
dichloromethane/methanol/acetic acid/water. The resulting
hydrotrifluoroacetate of (-)-3-amino-2,3-dihydro-1-methyl-
2-oxo-5-phenyl-8-methoxy-1H-1,4-benzodiazepine was dissolved
in dichloromethane, aqueous sodium bicarbonate solution was
added to the solution to liberate the amine, and the
reaction mixture was extracted with dichloromethane. The
dichloromethane phase was separated, dried over sodium
sulfate and filtered: the solvent was removed by
distillation under reduced pressure, and the remaining base
was dried. 8.5 g of (-)3-amino-2,3-dihydro-1-methyl-2-oxo-
5-phenyl-8-methoxy-1H-1,4-benzodiazepine were obtained as a
foam. Optical rotation [a ]p° - -171. 2° (c = 0. 5 in
dichloromethane).
E) In accordance with the method described in Example
1 H), 7.0 g of the (-)-3-amino-1-methyl-8-methoxy-2-oxo-5
phenyl-2,3-dihydro-1H-1,4-benzodiazepine obtained above were
reacted with a reaction solution containing the mixed
anhydride from 4.9 g of 4H-pyrrolo[3,2,1-ij]-5,6
dihydroquinoline-2-carboxylic acid and 1.85 ml of
methanesulfonyl chloride in dichloromethane. The reaction
mixture was worked up as described in Example 1 H).
Purification by flash chromatography resulted in 9.0 g of
purified crystalline product. This was recrystallized twice
more from methanol and once from ethanol to remove any
enantiomeric impurities. 6.7 g of enantiomerically pure
(-)-3-[(4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-
carbonyl)-amino]-8-methoxy-1-methyl-2-oxo-5-phenyl-1H-2,3-
dihydro-1,4-benzodiazepine having a melting point of 201-205
°C and an optical rotation [a]zo of -88.2° (c = 0.5 in
dichloromethane) were obtained.
2 b: 3-[(4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2
carbonyl)-amino]-8-methoxy-1-methyl-2-oxo-5-phenyl-1H-2,3
dihydro-1,4-benzodiazepine with (+)-rotation in dichloro
methane.
- 33 -
201168
A) The mother liquors produced in the preparation of
the diastereomerically pure hydrochloride under 2a B) were
evaporated to obtain the other diastereomeric hydrochloride.
The precipitated hydrochloride was recrystallized four times
from a mixture of acetonitrile and isopropyl acetate. From
the hydrochloride were obtained, analogously to the method
described above, 14.5 g of diastereomerically pure 3-phenyl-
2-amino-N-(2,3-dihydro-1-methyl-2-oxo-5-phenyl-8-methoxy-1H-
1,4-benzodiazepin-3-yl)-propionamide with (+)-rotation in
methanolic solution, having an optical rotation [a]po -
+162.6° (c = 0.5 in methanol).
B) 14.5 g of the amide with (+)-rotation in methanolic
solution obtained above were reacted with 4.3 ml of phenyl
isothiocyanate in dichloromethane analogously to Example 2a
C). The reaction mixture was worked up as described under
Example 2a C). 18.6 g of N~-phenyl-NZ-{2-[(2,3-dihydro-1-
methyl-2-oxo-5-phenyl-8-methoxy-1H-1,4-benzodiazepin-3-yl)-
amino]-2-oxo-1-(benzyl)-ethyl)-thiourea were obtained.
Optical rotation [a]p° - +60.2° (c = 0.5 in methanol).
C) 18.5 g of the thiourea product obtained above were
reacted with 28.4 ml of trifluoroacetic acid analogously to
Example 2a D). The amine was liberated from the resulting
hydrotrifluoroacetate of (+)-3-amino-2,3-dihydro-1-methyl-
2-oxo-5-phenyl-8-methoxy-1H-1,4-benzodiazepine analogously
to Example 2a D). 7.7 g of (+)-3-amino-2,3-dihydro-1-
methyl-2-oxo-5-phenyl-8-methoxy-1H-1,4-benzodiazepine were
obtained. Optical rotation [a]p° - +143.8° (c = 0.5 in
dichloromethane).
D) Analogously to the method described in Example 1 H),
5.0 g of the (+)-3-amino-1-methyl-8-methoxy-2-oxo-5-phenyl
2,3-dihydro-1H-1,4-benzodiazepine obtained above were
reacted with a reaction solution containing the mixed
anhydride from 3.49 g of 3-[(4H-pyrrolo[3,2,1-ij]-5,6
dihydroquinoline-2-carboxylic acid and 1.32 ml of
methanesulfonyl chloride in dichloromethane. The reaction
mixture was worked up as described in Example 1 H). 10 g of
- 34 -
20~~0~~
crude product were obtained. After purification by flash
chromatography, three recrystallizations from methanol were
carried out to remove any enantiomeric impurities. 3.4 g of
enantiomerically pure (+)-3-[(4H-pyrrolo[3,2,1-ij]-5,6-
dihydroquinoline-2-carbonyl)-amino)-8-methoxy-1-methyl-2-
oxo-5-phenyl-1H-2,3-dihydro-1,4-benzodiazepine having a
melting point of 201-205 °C were obtained. Optical rotation
[a)p° - +88.4° (c = 0.5 in dichloromethane).
Example 3:
3-[(4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-
amino]-1-n-pentyl-2-oxo-1H-2,3-dihydro-1,4-benzodiazepine.
A) l0 g of 2-oxo-5-phenyl-2,3-dihydro-1H-1,4
benzodiazepine were dissolved in 100 ml of tetrahydrofuran
under a nitrogen atmosphere. 1.4 g of sodium hydride in the
form of an 80 % oily suspension were added in portions to
the solution under the nitrogen atmosphere, and the reaction
mixture was heated to reflux for 30 minutes. 9.2 g (= 5.5
ml) of iodopentane were slowly added dropwise, the mixture
was heated to reflux for a further 1.5 hours and then
another 0.3 g of sodium hydride in the form of an oily
suspension was added and, after a further 10 minutes,
another 5.5 ml of iodopentane were added dropwise, and the
mixture was heated to reflux for a further hour. To work up
the reaction mixture, ice-water was added to the reaction
mixture and it was diluted with dichloromethane, the aqueous
phase was separated, and the organic phase was washed to
neutrality with water, dried over sodium sulfate, filtered
and evaporated. 13.5 g of crude product were obtained and
were purified by flash chromatography on about 300 g of
silica gel using cyclohexane/ethyl acetate 1:1 as the
eluent. The purified product was crystallized from
cyclohexane and dried. 6.2 g of pure 2-oxo-1-n-pentyl-5-
phenyl-2,3-dihydro-1H-1,4-benzodiazepine having a melting
point of 93-95 °C were obtained.
- 35 -
20:~1~
B) 6.5 g of the above product were suspended in 122 ml
of toluene. The suspension was cooled to -20 °C, and then
5.88 g of potassium tert.-butylate were added, while
stirring, and the mixture was reacted with 2.84 ml of tert.-
butyl nitrite as described in Example 1D. The reaction
mixture was worked up as in Example 1D. This resulted in
4.7 g of 3-hydroxyimino-2-oxo-1-n-pentyl-5-phenyl-2,3-
dihydro-1H-1,4-benzodiazepine having a melting point of 188-
191 °C.
C) 4.6 g of the product obtained above were dissolved
in a mixture of 328 ml glacial acetic acid and 37.6 ml
trifluoroacetic acid and reduced with a total of 3.1 g of
zinc dust as described in Example lE. The reaction mixture
was worked up as in Example lE. 4.2 g of crude 3-amino-2-
oxo-1-n-pentyl-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepine
were obtained and were further processed without
purification.
D) By the process described in Example 1H), a solution
of 4.2 g of the product obtained above and 1.77 ml of
triethylamine in 50 ml of dichloromethane was added dropwise
at a temperature between -15 °C and -20 °C to a reaction
solution containing the mixed anhydride from methanesulfonic
acid and 2.6 g of 4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-
2-carboxylic acid in 60 ml of dichloromethane. The reaction
mixture was worked up as described in Example 1H). 3.9 g of
3-[(4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-
amino]-2-oxo-1-n-pentyl-5-phenyl-1.H-2,3-dihydro-1,4-
benzodiazepine were obtained as a white resinous foam.
IR spectrum: 1682 cm~~, 1662 cm ~, 1524 cm ~, 1499 cm ~.
Example 4:
3-[(4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-
amino]-1-methyl-2-oxo-5-cyclohexyl-1H-2,3-dihydro-1,4-
benzodiazepine. ,
A) 75.6 g of N~-methyl-N~-phenyl-2-hydroxy-1,3-
diaminopropane and 65 ml of triethylamine were dissolved in
- 36 -
2~~~.~~
600 ml of dichloromethane. The solution was cooled in ice
while a solution of 61 ml of cyclohexylcarbonyl chloride in
50 ml of dichloromethane was slowly added dropwise. The
reaction mixture was stirred at room temperature for 1.5
hours. The reaction solution was subsequently worked up by
washing with water and with sodium chloride solution, and
the solvent was evaporated. The remaining residue was 131
g of crude N~-methyl-N~-phenyl-N2-cyclohexylcarbonyl-2-
hydroxy-1,3-diaminopropane. The crude product was
recrystallized from toluene, washed with ether and dried.
113.8 g of pure product having a melting point of 86-88 °C
were obtained.
B) 87 g of the product obtained above were reacted with
174 ml of phosphorus oxychloride in an oil bath at a bath
temperature of 130 °C for 2 hours. The mixture was then
cooled and diluted with dichloromethane, and ice-water was
added to the solution. The organic phase was separated,
washed several times with water, then treated with dilute
sodium hydroxide solution, again washed with water, dried
over sodium sulfate and evaporated. The residue obtained
was 76.1 g of an oily crude product which contained a
mixture of about 40 % 2-chloromethyl-1-methyl-5-cyclohexyl-
2,3-dihydro-1H-1,4-benzodiazepine and about 60 % 3-chloro-
1-methyl-6-cyclohexyl-1,2,3,4-tetrahydro-1,5-benzodiazocine.
To isomerize the benzodiazocine fraction, the crude mixture
was heated under reflux in 300 ml of tetrachloroethane for
minutes. The tetrachloroethane was then evaporated, and
the remaining 2-chloromethyl-1-methyl-5-cyclohexyl-2,3-
dihydro-1H-benzodiazepine was further processed without
30 purification in the next reaction stage.
C) 19.0 g of the product obtained above were dissolved
in 103 ml of dichloromethane. 116 ml of 32 % strength
aqueous hydrochloric acid, 882 ml of water and 91 ml of
dichloromethane were added to the solution. Subsequently a
solution of 11.65 g of potassium permanganate in 238 ml of
water was added dropwise, maintaining the internal
- 37 -
~0116~~
temperature below 15 °C by cooling with ice. The reaction
mixture was stirred at room temperature for 1.5 hours. Then
another 2 g of potassium permanganate dissolved in 50 ml of
water were added dropwise, and the mixture was stirred at
room temperature for a further hour.. To work up the
reaction mixture, solid sodium bicarbonate was added in
portions to the reaction mixture until neutrality. The
dichloromethane phase was then separated, and the aqueous
phase was again extracted with dichloromethane. The
combined dichloromethane extracts were washed with dilute
sodium hydroxide solution and then with water, dried over
magnesium sulfate and evaporated. 55 g of crude product
were obtained. The crude product was subjected to column
chromatography on 1 kg of silica gel under slightly elevated
pressure (flash chromatography) using cyclohexane/ethyl
acetate 1:1 as the eluent to isolate pure 1-methyl-2-oxo-5-
cyclohexyl-2,3-dihydro-1H-1,4-benzodiazepine, which was
crystallized from ether and dried. 2.3 g of pure product
having a melting point of 98-100 °C were obtained.
D) 8.9 g of the above product were suspended in 201 ml
of toluene. The suspension was cooled to -20 °C and then
9.63 g of potassium tert.-butylate were added while
stirring, and the mixture was stirred for a further 15
minutes. Then 5.5 ml of isoamyl nitrite were added, while
cooling, at such a rate that the temperature of the reaction
mixture remained below 0 °C. It was then stirred at 0 °C for
minutes. The reaction solution was then worked up by
adding it to a stirred mixture of 347 ml of ice-cold water,
16.7 ml of glacial acetic acid, and 347 ml of ethyl acetate.
30 After vigorous mixing the organic phase was separated, and
the aqueous phase was extracted again with ethyl acetate.
The combined organic phases were washed with water and
evaporated. The residue was taken up in toluene and
recrystallized from toluenel ethanol. 12.4 g of crude
product were obtained. This was purified by flash
chromatography on 500 g silica ge:l, using cyclohexane/ethyl
- 38 -
20~i6~y
acetate as the eluent, first in the ratio 1:1 then in the
ratio 4:6. Crystallization of the resulting product from
ethanol resulted in 4.4 g of 3-hydroxyimino-1-methyl-2-oxo
5-cyclohexyl-2,3-dihydro-1H-1,4-benzodiazepine having a
melting point of 205-210 °C.
E) 4.4 g of the product obtained above were added to a
mixture of 375 ml of glacial acetic acid and 42.3 ml of
trifluoroacetic acid. The reaction mixture was heated to 40
°C (internal temperature), and a total of 2.36 g of zinc dust
was added in portions while stirring. The mixture was
stirred at 40 °C for a further 2 hours, and then another 1.1
g of zinc dust were added, and the mixture was stirred at 40
°C for an additional 1.5 hours. The mixture was worked up by
diluting with toluene, allowing it to cool and evaporating
the solvent. The remaining residue was taken up in
dichloromethane, washed with aqueous sodium carbonate
solution and water, dried and evaporated. 4.0 g of crude 3-
amino-1-methyl-2-oxo-5-cyclohexyl-2,3-dihydro-1H-1,4-
benzodiazepine were obtained and were further processed
without purification in the subsequent reaction stage.
F) 2.92 g of 4H-pyrrolo[3,2,1-:ij]-5,6-dihydroquinoline-
2-carboxylic acid (obtained as in Example 1F to 1G) and 1.99
ml of triethylamine were dissolved in 64 ml of
dichloromethane. The solution was cooled to -20 °C and,
while stirring, 1.13 ml of methanesulfonyl chloride were
slowly added dropwise, and the reaction mixture was stirred
at -20 °C for a further 15 minutes. A solution of 4.0 g of
the 3-amino-1-methyl-2-oxo-5-cyclohexyl-2,3-dihydro-1H-1,4-
benzodiazepine obtained in stage E and 1.99 ml of
triethylamine in 54 ml of dichloromethane was added dropwise
at a temperature between -15 and -20 °C to the stirred
reaction solution containing the mixed anhydride from
methanesulfonic acid and 4H-pyrrolo[3,2,1-ij]-5,6-
dihydroquinoline-2-carboxylic acid, and the reaction mixture
was stirred at -15 °C for a further 30 minutes and allowed to
warm slowly (within one hour) to room temperature. The
- 39 -
2011~~«
reaction mixture was worked up by diluting with water,
separating the dichloromethane phase, washing with sodium
bicarbonate and then with water, drying over sodium sulfate,
filtering, and evaporating the solvent. 7.6 g of the crude
title compound were obtained as a residue. The crude
product was purified by column chromatography on 400 g of
silica gel under slightly elevated pressure (flash
chromatography) using cyclohexane/ethyl acetate 1:1 as the
eluent. The purified product was crystallized from ethanol
and dried. 1.5 g of 3-[(4H-pyrrolo[3,2,1-ij]-5,6-
dihydroquinoline-2-carbonyl)-amino]-1-methyl-2-oxo-5-
cyclohexyl-1H-2,3-dihydro-1,4-benzodiazepine having a
melting point of 147 -152 °C were obtained.
Examgle 5:
3-[(8-Fluoro-4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-
carbonyl)-amino-2-oxo-1-methyl-5-phenyl-1H-2,3-dihydro-1,4-
benzodiazepine.
0.99 g of 8-fluoro-4H-pyrrolo[3,2,1-ij]-5,6-dihydro
quinoline-2-carboxylic acid, 1.4 g of 2-chloro-1-methyl
pyridinium iodide, 1.1 ml of triethylamine and 1.2 g of 3
amino-2-oxo-1-methyl-5-phenyl-2,3-dihydro-1H-1,4
benzodiazepine were dissolved in 120 ml of dichloromethane,
and the reaction mixture was boiled to reflux for 1 hour.
The reaction mixture was worked up by allowing it to cool,
adding 5 % strength sodium bicarbonate solution, separating
the organic phase, and extracting the aqueous phase again
with dichloromethane. The combined dichloromethane extracts
were dried over sodium sulfate, and the solvent was
distilled off under reduced pressure. The crude title
compound remaining as a residue was purified by column
chromatography on silica gel using dichloromethane/methanol
99:1. 1.5 g of 3-((8-fluoro-4H-pyrrolo[3,2,1-ij]-5,6-
dihydroquinoline-2-carbonyl)-amino]-2-oxo-1-methyl-5-phenyl-
1H-2,3-dihydro-1,4-benzodiazepine having a melting point of
181-182 °C were obtained.
- 40 -
201~~
Example 6:
3-[(4H-Pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-
amino]-1-methyl-2-oxo-5-phenyl-1H-2,3-dihydro-1,4-
benzodiazepine.
A) 98.6 g of 2-aminobenzophenone were dissolved in a
mixture of 650 ml of dichloromethane and 50 ml of water. A
solution of 116.1 g of bromoacetyl bromide in 150 ml of
dichloromethane was added dropwise to this mixture at a
temperature of -10 °C. The reaction mixture was then stirred
at room temperature for a further 2 hours. The reaction
mixture was worked up by adding water, separating the
organic phase, washing again with water, drying and
evaporating under reduced pressure. The crude product
remaining as a residue was crystallized from ether/petroleum
ether. 142 g of 2-[(2-bromoacetyl)-amino]-benzophenone
having a melting point of 96-98 °C were obtained.
B) 71 g of the 2-[(2-bromoacetyl)-amino]-benzophenone
obtained above were dissolved in 500 ml of methanol. A
solution of 75 g of ammonia in 1.2 1 of methanol was added
dropwise to this solution at a temperature of 10 °C. Then
the reaction mixture was first stirred at room temperature
for 1.5 hours and then heated to reflux for 2 hours. The
reaction mixture was worked up by distilling off the
methanol under reduced pressure, dissolving the residue in
dichloromethane, washing the solution with 10 % strength
aqueous sodium hydroxide solution and with water, drying and
concentrating under reduced pressure. The crude product
remaining as residue was crystallized from methanol. 20 g
of 2-oxo-5-phenyl-1H-2,3-dihydro-1,4-benzodiazepine having
a melting point of 178-180 °C were obtained.
C) 60 g of the product obtained above were dissolved in
1.2 1 of dried tetrahydrofuran. While excluding moisture,
34.2 g of potassium tert.-butylate were added to the
solution. A solution of 20.6 ml of methyl iodide in 75 ml
of tetrahydrofuran was then added dropwise, and the reaction
mixture was stirred at room temperature for a further hour.
- 41 -
2~11~~°
To work up the reaction mixture, ice-cold sodium chloride
solution was added; the reaction mixture was diluted with
dichloromethane: the aqueous phase was separated, and the
organic phase was washed with water to neutrality, dried
over sodium sulfate, filtered and evaporated. The crude
product obtained as a residue was recrystallized from
ethanol. 56 g of 1-methyl-2-oxo-5-phenyl-1H-2,3-dihydro-
1,4-benzodiazepine having a melting point of 154-155 °C were
obtained.
D) 50.4 g of the product obtained above were added to
987 ml of toluene, cooled to -20 °C and reacted with 56.4 g
of potassium tert.-butylate and 32.3 ml of isoamyl nitrite
by the method described in Example 1 D). The reaction
mixture was worked up as described in Example 1 D), and the
resulting crude product was crystallized from ethanol. 47.2
g of 3-hydroxyimino-1-methyl-2-oxo-5-phenyl-2,3-dihydro-1H-
1,4-benzodiazepine having a melting point of 239-242 °C were
obtained.
E) 6.8 g of the product obtained above were dissolved
in 500 ml of methanol. 12 g of Raney nickel were added to
the solution which was then hydrogenated at room temperature
under a hydrogen pressure of 6 bar for 12 hours. To work up
the reaction solution, the catalyst was filtered out, and
the solvent was removed by distillation under reduced
pressure. 6 g of crude racemic 3-amino-1-methyl-2-oxo-5-
phenyl-2,3-dihydro-1H-1,4-benzodiazepine were obtained.
This product was dissolved in 50 ml of acetonitrile for
further purification by conversion into its benzenesulfonate
salt. A solution of 3.6 g of benzenesulfonic acid in 22 ml
of acetonitrile was added to the solution, and the reaction
mixture was stirred at room temperature for one hour. The
crystalline precipitate which formed was filtered out with
suction, washed with acetonitrile and then with hexane, and
then dried under reduced pressure. 5.9 g of the
benzenesulfonate of the 3-amino compound having a melting
point of 224-227 °C were obtained. To liberate the amine, 5
- 42 -
~01~_5
g of the benzenesulfonate salt obtained above were dissolved
in dichloromethane, and the solution was shaken with aqueous
sodium carbonate solution. The dichloromethane phase
containing the liberated amine compound was separated, dried
and evaporated. 3.1 g of 3-amino-1-methyl-2-oxo-5-phenyl-
2,3-dihydro-1H-1,4-benzodiazepine were obtained.
F) 0.38 g of 4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-
2-carboxylic acid, 0.50 g of 3-amino-1-methyl-2-oxo-5-
phenyl-2,3-dihydro-1H-1,4-benzodiazepine and 0.46 g of
triethylamine were dissolved in 50 ml of dichloromethane.
0.58 g of 2-chloro-1-methylpyridinium iodide was added to
the solution, and the reaction mixture was boiled to reflux
for one hour. The reaction mixture was then worked up as
described in Example 5. 0.69 g of 3-[(4H-pyrrolo[3,2,1-ij]-
5,6-dihydroquinoline-2-carbonyl)-amino]-1-methyl-2-oxo-5-
phenyl-1H-2,3-dihydro-1,4-benzodiazepine was obtained as a
resinous foam. ~3C NMR spectrum: [data in ppm, (signal
multiplicity)]
167.88 (s), 167.51 (s), 162.32 (s), 142.91 (s),
138.17 (s), 136.40 (s), 131.93 (d), 130.72 (d),
130.69 (d), 130.23 (s), 129.83 (d), 129.83 (d),
129.22 (s), 128.28 (d), 128.28 (d), 124.58 (d),
124.40 (s), 123.09 (s), 121.60 (d), 120.90 (d),
120.66 (d), 119.34 (d), 104.24 (d), 67.40 (d),
44.38 (t), 35.36 (q), 24.89 (t), 23.13 (t).
Example 7:
Preparation of the optical isomers of 3-[(4H-pyrrolo[3,2,1
ij]-5,6-dihydroquinoline-2-carbonyl)-amino]-1-methyl-2-oxo
5-phenyl-1H-2,3-dihydro-1,4-benzodiazepine.
7a: (-)-(3S)-3-[(4H-Pyrrolo[3,2,1-ij]-5,6-dihydro-
quinoline-2-carbonyl)-amino]-1-methyl-2-oxo-5-phenyl-1H-2,3-
dihydro-1,4-benzodiazepine.
A) Analogously to Example 2a A-D, (-)-(3S)-3-amino-1
methyl-2-oxo-5-phenyl-1H-2,3-dihydro-1,4-benzodiazepine
having an optical rotation [a]p~ of -230.8° (c = 1 in
- 43 -
2~1~~~~
acetonitrile) was produced from racemic 3-amino-1-methyl-2-
oxo-5-phenyl-2,3-dihydro-1H-1,4-benzodiazepine (for
preparation, see Example 6 E).
B) Analogously to the method described in Example 1H,
1.7 g of the (-)-(3S)-3-amino-1-methyl-2-oxo-5-phenyl-2,3
dihydro-1H-1,4-benzodiazepine obtained above were reacted
with a reaction solution containing the mixed anhydride from
1.36 g of 4H-pyrrolo-[3,2,1-ij]-5,6-dihydroquinoline-2
carboxylic acid and 0.52 ml of methanesulfonyl chloride in
dichloromethane. The reaction mixture was worked up as
described in Example 1 H). 2.6 g of crystalline product
purified by flash chromatography were obtained. This was
recrystallized from methanol to remove enantiomeric
impurities. 1.4 g of enantiomerically pure (-)-(3S)-3-[(4H-
pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-amino]-
1-methyl-2-oxo-5-phenyl-1H-2,3-dihydro-1,4-benzodiazepine
having a melting point of 151-158 °C and an optical rotation
[a ]pD of -61. 6° (c = 0.5 in methanol) were obtained.
7b: (+)-(3R)-3-[(4H-Pyrrolo[3,2,1-ij]-5,6-dihydro-
quinoline-2-carbonyl)-amino]-1-methyl-2-oxo-5-phenyl-1H-2,3-
dihydro-1,4-benzodiazepine.
A) (+)-(3R)-3-amino-1-methyl-2-oxo-5-phenyl-2,3
dihydro-1H-1,4-benzodiazepine with an optical rotation [a]2D
D
of +259.1° (c = 1 in acetonitrile) was produced in a manner
analagous to Example 2b A-C.
B) Analogous to the method described in Example 1 H),
0.65 g of the (+)-(3R)-3-amino-1-methyl-2-oxo-5-phenyl-2,3-
dihydro-1H-1,4-benzodiazepine obtained above was reacted
with a reaction solution containing the mixed anhydride from
0.52 g of 4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-
carboxylic acid and 0.2 ml of methanesulfonyl chloride in
dichloromethane. The reaction mixture was worked up as
described in Example 1 H)_ Flash chromatography and
crystallization from ethanol resulted in 596 mg of
enantiomerically pure(+)-(3R)-3-[(4H-pyrrolo[3,2,1-ij]-5,6-
- 44 -
2011~'~
dihydroquinoline-2-carbonyl)-amino;-1-methyl-2-oxo-5-phenyl-
1H-2,3-dihydro-1,4-benzodiazepine having a melting point of
147-156 °C.
Optical rotation [ a ] p° - +61. 0° ( c ~- 0 . .~ in
methanol ) .
Example 8:
3-[(4H-Pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-carbonyl)-
amino]-1,7-dimethyl-2-oxo-5-(2-thienyl)-1H-2,3-dihydro-1,4-
benzodiazepine.
A) 30 g of N~-methyl-N~-(4-methylphenyl)-2-hydroxy-1,3-
diaminopropane and 24.2 ml of triethylamine were dissolved
in 225 ml of dichloromethane. A solution of 16.5 ml of
thiophene-2-carbonyl chloride in 50 ml of dichloromethane
was slowly added dropwise to the solution while cooling in
ice. The reaction mixture was allowed to stand at room
temperature for 12 hours. The reaction solution was then
worked up by washing with water and aqueous sodium chloride
solution, separating and drying the organic phase, and
removing the solvent by distillation under reduced pressure.
The residue obtained was 52.2 g of crude oily N~-methyl-N~-
(4-methylphenyl)-NZ-(2-thienoyl)-2-hydroxy-1,3-
diaminopropane, which was employed without further
purification in the next reaction stage.
B) 50.2 g of the product obtained above were added to
150 ml of phosphorus oxychloride, and the reaction mixture
was heated at reflux for 90 minutes. It was then cooled and
worked up as described in Example 1 B). 48.5 g of an oily
crude product were obtained which contained a mixture of 2
chloromethyl-1,7-dimethyl-5-(2-thienyl)-2,3-dihydro-1H-1,4
benzodiazepine and 3-chloro-1,8-dimethyl-6-(2-thienyl)-
1,2,3,4-tetrahydro-1,5-benzodiazocine. To isomerize the
benzodiazocine fraction, the crude mixture was dissolved in
280 ml of tetrachloroethane and heated at reflux for one
hour. The tetrachloroethane was then evaporated off, the
residue was dissolved in dichloromethane, and the solution
was washed with 10 ~ strength sodium hydroxide solution and
._ 45 -
2~~~6~~~
then with water and with aqueous sodium chloride solution,
dried and evaporated under reduced pressure. The remaining
residue was dissolved in a mixture of methylene chloride and
methanol, and the solution was filtered through magnesium
silicate (= Florisil~) and reduced in volume. 32.4 g of 2-
chloromethyl-1,7-dimethyl-5-(2-thienyl)-2,3-dihydro-1H-1,4-
benzodiazepine were obtained as an oily substance which was
further processed without further purification in the next
reaction stage.
C) 13.6 g of the substance obtained above were oxidized
with 8.2 g of potassium permanganate by the method described
in Example 1C. The reaction mixture was worked up as
described in Example 1 C). The resulting crude product was
dissolved in dichloromethane and purified by flash
chromatography on silica gel using dichloromethane/methanol
95:5 as the eluent. 3.8 g of oily 1,7-dimethyl-2-oxo-5-(2-
thienyl)-2,3-dihydro-1H-1,4-benzodiazepine were obtained.
D) 7.7 g of the product obtained above were dissolved
in 21o ml of toluene. After cooling to -20 °C, 8.1 g of
potassium tert.-butylate were added under an NZ atmosphere,
and the mixture was stirred for a further 15 minutes. Then
4.6 ml of isoamyl nitrite were added, while cooling, at such
a rate that the temperature of the reaction mixture remained
below 0 °C. The mixture was then stirred at 0 °C for a
further 30 minutes. For working up, the reaction mixture
was added to a stirred mixture of 300 ml of ice-cold water,
300 ml of ethyl acetate and 15 m1 of glacial acetic acid.
The organic phase was separated, and the aqueous phase was
extracted again with ethyl acetate. The organic phases were
combined, washed with water and with aqueous sodium chloride
solution, dried over sodium sulfate and concentrated under
reduced pressure. 6.5 g of 3-hydroxyimino-1,7-dimethyl-2-
oxo-5-(2-thienyl)-2,3-dihydro-1H-1,4-benzodiazepine were
obtained as a resinous foam.
E) 2.0 g of the product~obtained above were dissolved
in 150 ml of methanol. 8 g of Raney nickel were added, and
- 46 -
24~~.4v~y
the mixture was then hydrogenated with a hydrogen pressure
of 4 bar for 7.5 hours. The reaction was worked up by
filtering out the catalyst and distilling off the solvent
under reduced pressure. 1.4 g of crude product remained as
a residue. This was taken up in dichloromethane and
extracted with dilute aqueous hydrochloric acid solution.
The hydrochloric acid phase was separated, made alkaline by
addition of dilute aqueous sodium hydroxide solution and
extracted with dichloromethane. Evaporation of the
dichloromethane extract resulted in 0.7 g of 3-amino-1,7-
dimethyl-2-oxo-5-(2-thienyl)-2,3-dihydro-1H-1,4-
benzodiazepine as a resinous foam, which was further
processed without purification in the following reaction
stage.
F) Analogously to the method described in Example 1 H),
1.15 g of the 3-amino-1,7-dimethyl-2-oxo-5-(2-thienyl)-2,3-
dihydro-1H-1,4-benzodiazepine obtained above were reacted
with a reaction solution containing the mixed anhydride from
0.81 g of 4H-pyrrolo[3,2,1-ij]-5,6-dihydroquinoline-2-
carboxylic acid and 0.32 ml of methanesulfonyl chloride in
dichloromethane. The reaction mixture was worked up as
described in Example 1 H). 2.3 g of crude product were
obtained and were dissolved in dichloromethane and purified
by flash chromatography on silica gel using
dichloromethane/methanol 96:4 as the eluent. Removal of the
solvent by distillation resulted in 0,7 g of residue which
was crystallized from a mixture of cyclohexane and ethyl
acetate. 0.4 g of crystalline 3-[(4H-pyrrolo[3,2,1-ij]-5,6-
dihydroquinoline-2-carbonyl)-amino]-1,7-dimethyl-2-oxo-5-(2-
thienyl)-1H-2,3-dihydro-1,4-benzodiazepine having a melting
point of 204-207 °C were obtained.
The compounds of formula I listed in the following
table were also obtained by the processes described in the
foregoing examples by acylation of the corresponding 3-
aminobenzodiazepine derivatives of formula II.
- 47 -
~o~~o
1~
f~ I~ d' f~ O N If7 01 CD CO I~ h lfl r1 f~ N d' ..
O d' O ri CO N ~D 01 l~ O 01 01
O tf7 M O r"1 Ol N O l~ M l~ ~ O tIl d' 01 d' r-1 U
~O O 00 CO to lf7 I~ ~ O M to
W rl N W N r-1 N N '-i N N N N N rl ~ rl N N rl N
i-1 rl N e-I rl N N N
1 1 I 1 1 I I i I I I I I I I 1 I I I I I I I I
I I I
l M d' rl lp In G1 lfl M M CO d' CO N ~O M 10 M
10 LI1
~ l0
l
'
U1 r rl r-1
r I!7
Ul dl 00 N l
U1 d
'~ 111 M ,~~ O 00 N O l~ M t~ to O d' d' CO M
~D l~ I~ CO d' 111 I~ 01 01 M
QI O r"1 N O N rl N N rl N N N N N r-1 ri r-I N ri N
rl '-i rl N r1 v-I rl N N
G
O .,..i . .,..i
z cn wwm a~s.~n,s.~~wwwa~si~wwwn,a~a~an~sia,a~ri,a.~~si.n,~o
~ I~ >~ ~ >~ >~ ~ >~ I~ I~ >~ >~ ~ ~ >~ ~ ~ >~ ~ s~
a~ >~ ~ ~ a~ ~ ~ ~ ~ o I~
>s
M
x
U rl +I
Lr. O U
I 11 v
~x xxxxxxxxxxxxxxxxx~oxxxx~oo~xxxx xx I~
ix ~ xxxuxxxxxxxxxxxxxxxxxxxxxxxxxx r~
v
I
N
M rl
x ~,
U
1 1 1 ~7
U "~ O U1
1 I I I I I I I I 1 I I I I I I I I i I I I 1 I .N I
I 1
I I 1 N
MMMMMMMMMMMMMMMMMMMMMMMMNN~t~ NN
~~~~~~~~~~~. ~.-.
-
.~~~ 1
~~~~~~~~.-.~~.~.
N N N N N N N N N N N N N N N N N N N N N N N N N
N N ~i N N
xxxxxxxxxxxxxxxxxxxxxxxxa~xx
xxx ?~
U U U U U U U U U U U U U U U U U U U U U U U U U
U U ,~ U U
vvvv~r~rv~rvvvvvv~rvvvvvvv~rvvvv avv I
I 1 1 1 1 I I 1 1 1 1 I 1 1 1 I 1 1 1 1 1 1 1 I 1
1 1 1 1 1
I
b
Grr Lzr fs, fsr U !ir U ~. fJr U fsr w
1 I I I I I I 1 I 1 I i II
xx~x~~~~~x~~~oxxxxx~~o~xxxxxxxxx
cn c ~ ~ ~ ~ >; o
'~
w
a, a. ~ ~ a. a
')y .~ .,.i .~ .~ .,..j .~ M
1~ I '-1 1 r-1 1 1 r1 1 1 ~ w
~
~ fs.~ Gr, fi, Cza O w U O U tsr O Gr. O U O O U
t2
pG
, I 1 1 x 1 I 1 I 1 x 1 1 1 1 1 I 1 1 I
Oa N N N N N N M N M N N N x N x x ~"~ N N d' x x x x o
x x x x
I
s
x >
0
c>~ xxxxxxxxxxxxxxxxxxxxxxxxxxxxxx
U
M M M M M M II
M M M
~xxx
U x U U U U U x x
oUxxoooooUxxxxxUxUUUUxxxxxxxxx n
N I 1 1 1 1 I 1 I 1 1 1 1 1 U
(x
~ ~ ~ h V
I ,.
N 1 ?~ >;T
O S-I
~ N ~
I I I I I I I 1 1 I I I I I O'' M I 1 1 1 1 1 1 1 1 1 1 1 1
M M M M M M M M M M M M M MU Mx M M M M M M M M M M M M M -
ix xxxxxxxxxxxxxx~,xUxxxxxxxxxxxxx
U U U U U U U U U U U U U U U U ~-U U U U U U U U U U U U U II ~~
t~
N II
k' O 0lOriNM V'III~Ot~0001OrlNMd'1f~10hCO01Oe-INMd'InlOI~CO C~
-1 ,-1 ~--1 rl e-i rl e-i ri e-1 rl N N N N N N N N N N M M M M M M M M M wl
U7
- 48 -
2~~~~~~
Example I:
Tablets containing 3-[(4H-pyrrolo[3,2,1-ij)-5,6-dihydro-
quinoline-2-carbonyl)-amino]-8-methoxy-1-methyl-2-oxo-5-
phenyl-1H-2,3-dihydro-1,4-benzodiazepine.
Tablets were prepared containing, per tablet:
3-[(4H-pyrrolo[3,2,1-ij)-5,6-dihydroquinoline-2-
carbonyl)-amino]-8-methoxy-1-methyl-2-oxo-5-phenyl-1H-
2,3-dihydro-1,4-benzodiazepine 20 mg
maize starch 60 mg
lactose 135 mg
gelatin (as 10 % strength solution) 6 mg
The active substance, the maize starch and the lactose were
thickened with the 10 % strength gelatin solution. The
paste was comminuted, and the resulting granules were placed
on a suitable metal sheet and dried at 45 °C. The dried
granules were passed through a comminuting machine and mixed
with the following additional adjuvants in a mixer:
talc 5 ~
magnesium stearate 5 m9
maize starch
and then compressed to form 240 mg tablets.
Example II:
Tablets containing 3-[(4H-pyrrolo[3,2,1-ij)-5,6-dihydro
quinoline-2-carbonyl)-amino]-8-methoxy-1-methyl-2-oxo-5
phenyl-1H-2,3-dihydro-1,4-benzodiazepine.
Tablets with the following composition per tablet were
prepared in analogy to Example I:
3-[(4H-pyrrolo[3,2,1-ij)-5,6-dihydroquinoline-2-
carbonyl)-amino]-8-methoxy-1-methyl-2-oxo-5-phenyl-1H-
2,3-dihydro-1,4-benzodiazepine. 20 mg
maize starch 60 mg
lactose 135 mg
gelatin (as 10 % strength solution) 6 mg
talc 5 ~
magnesium stearate 5 ~
maize starch
- 49 -
~0~ ~_6'~~
The foregoing description and examples have been set
forth merely to illustrate the invention and are not
intended to be limiting. Since modifications of the
described embodiments incorporating the spirit and substance
of the invention may occur to persons skilled in the art,
the scope of the invention should be construed to include
all variations falling within the ambit of the appended
claims and equivalents thereof.
- 50 -