Note: Descriptions are shown in the official language in which they were submitted.
2 ~ 3 ~
Merck Patent Gesellschaft
mit beschr~nkter Haftung
6100 D a r m s t a d t
Indole derivatives
The invention relates to novel indole derivatives of
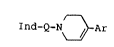
formula I
Ind-Q- ~ -Ar
wherein
Ind is an indol-3-yl radical substituted by -O-CH2-CO-R1,
-NXR2, -NO2, -Co-NR3R4 or -CSNH2,
R1 is OH, OA, NH2, NHA, NA2, NH-CH2COOA or NHCH(CH20H)COOA,
R2 is H, Ac, CONH2, CONHA, CONA2 or SO2A,
R3 is H, A or hydroxyalkyl,
R4 is hydroxyalkyl, AO-alkyl, i~cO-alkyl, ANH-CO-O-al~yl,
AOOC-alkyl, H2NCO-alkyl, HSO3-alkyl, A2N-alkyl, Ar, Ar-
alkyl or Het-alkyl,
R3 and R4 ~ogether are also an alkylene group having 3-7
C atoms, which can be interrupted by O or NR5 and/or sub-
stituted by NA2, NHAc, COOA, CONH2, Ar or Het, and/or can
contain an additional double bond,
R5 is H, A, Ar, Het, Ac, COOA, CH2CONH2, CH2CONHA, CH2CONA2
or CH2CONR6,
R6 is alkylene having 3-7 C atoms,
Q is ~ ( CH2 ) n~ r -CH2-S-CH2CH2-r -CH2-SO-cH2cH2- or
-CH2-SO2-cH2cH2-r
n is 2, 3, 4 or 5,
A is alkyl having 1-4 C atoms,
-alkyl- is alkylene having 1-4 C atoms,
Ar is a phenyl group which is unsubstituted, monosub-
stituted or disubstituted by F, Cl, Br, OA and~or OH or
substituted by a methylenedioxy group, or a thien-2-yl or
thien-3-yl group,
Ac is A-CO- or Ar-CO- and
2 ~ 3 fl
-- 2 --
Het is a saturated or unsaturated 5~membered or 6-
membered heterocyclic radical having 1-4 N, O and/or S
atoms, which can be fused with a benzene ring and/or
monosubstituted or disubstituted by A,
and to their salts.
The ob~ect of the invention was to find novel
compounds capable of being used for the preparation of
drugs.
It has been found that the compounds of formula
I and their biocompatible acid addition salts possess
valuable pharmacological proper~ies. Thus, in particu-
lar, they are active on the central nerYous system,
especially as dopamine stimulants (parkinsonism inhibi
tors) and as ~erotonin agonists and antagonists. Speci-
fically, the compounds of formula I induce contralateral
rotational behaviour in rats suffering from parkinsonism
on one side (this can be established by the method of
Ungerstedt et al., Brain Res. 24 (1970), 485-493). They
inhibit the binding of tritiated dopamine agonists and
antagonists to extrapyramidal receptors (t~is can be
established by the method of Schwarcz et al., J. Neuro-
chemistry 34 (1980), 772-778, and Creese et al., European
J. Pharmacol. 46 (1977), 377-381) and the binding of
tritiated serotonin ligands to hippocampal receptors
(Cossery et al., European J. Pharmacol. 140 (1987), 143-
155). They also modify the accumulation of DOPA in the
corpus striatum and the accumulation of 5-HTP in the
nuclei raphes (Seyfried et al., European J. Pharmacol.
160 (1989), 31-41). In addition, the compounds inhibit
the linguomandibular reflex in narcotized rats (this can
be established on the basis of the methods of Barnett et
al., European J. Pharmacol. 21 (1973), 173-182, and von
Ilhan et al., European J. Pharmacol. 33 (1975), 61-64~.
They also have analgesic and hypotensive effects; thus,
in catheterized, conscious, spontaneously hypertensive
rats (strain: SHR/Okamoto~NIH-MO-CHB-Kisslegg; method:
q.v. Weeks and Jones, Proc. Soc. Exptl. Biol. Med. 104
(1950), 646-648), the directly measured blood pressure is
lowered after oral administration of the compounds.
2 ~ t 1
- 3
Compounds of formula I and their biocompatible
acid addition salts can therefore be uRed as active in-
gredients for anxiolytics, antidepre~sants, neuroleptics,
parkinsonism inhibitors and/or antihypertensives, and
also as intermediates for the preparation of other
pharmaceutical active ingredients.
The invention relates to the indole derivatives
of formula I and to their biocompatible acid addition
salts.
The radical A i5 alkyl having 1, 2, 3 or 4 C
atoms, especially 1 or 2 C atoms, preferably methyl and
also ethyl, n-propyl, isopropyl, n~butyl, isobutyl, sec-
butyl or tert-butyl. OA is preferably methoxy and also
ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, 6ec-
butoxy or tert-butoxy. NHA i preferably methylamino and
also ethylamino, n-propylamino, isopropylamino, n-butyl-
amino, isobutylamino, sec-butylamino or tert-butylamino.
NA2 is preferably dLmethylamino and also N-ethyl-N-
methylamino, diethylamino, di-n-propylamino, diisopropyl-
amino or di-n-butylamino.
Analogously, NH-CH2COOA i8 preferably methoxy-
carbonylmethylamino or ethoxycarbonylmethylamino;
NHCH(CH20~)COOA is preferably l-methoxycarbonyl-2-
hydroxyethylamino or l-ethoxycarbonyl-2-hydroxyethyl-
amino; CONHA is preferably N-m~thylcarbamoyl or N-ethyl-
carbamoyl; CONA2 is preferably N,N-dimethylcarb~moyl or
N,N-diethylcarbamoyl; CS-NH-COOA is pr~ferably methoxy-
carbonylaminothioxo or etho~ycarbonylaminothioxo; S02A is
preferably methylsulphonyl or ethylsulphonyl; COOA is
preferably me~hoxycarbonyl or ethoxycarbonyl; CH2CONHA is
preferably N methylcarbamoylmethyl or N-ethylcarbamoyl-
methyl; and CH2CONA2 is preferably N,N-dimethylcarbamoyl-
methyl or N,N-diethylcarbamoylmethyl.
The group -alkyl- is a linear or branched alky-
lene group having especially 1 or 2 C atoms, preferably-CH2- or -CH2CH2-, hut also e.g. ~CH(CH3)~ CH2)3-,
-CH(CH3)CH2-, -CH2CH(CH3)-, -C(CH3)2- or -(CH2)b-.
Accordingly, hydroxyalkyl is preferably hydroxy-
methyl or 1- or 2-hydroxyethyl, other preferred meanings
2 ,~
- 4
being 3-hydroxypropyl, 4-hydroxybutyl or 2-hydroxy-1,1-
dimethylethyl; AO-alkyl is preferably metho~ymethyl,
ethoxymethyl, 1- or 2-methoxyethyl or 1- or 2~ethoxy-
ethyl; ANH-CO-O-alkyl is preferably N-methylcarbamoyloxy-
methyl, N-ethylcarbamoyloxymethyl, 1- or 2-(N-methylcar-
bamoyloxy)ethyl or 1- or 2-(N-ethylcarbamoyloxy)ethyl;
AOOC-alkyl is preferably methoxycarbonylmethyl, ethoxy-
carbonylmethyl, 1- or 2-methoxycarbonylethyl or 1- or 2-
ethoxycarbonylethyl; HzN-CO-alkyl is preferably carba-
moylmethyl or l- or 2-carbamoylethyl; HSO3-alkyl i8
preferably sulphomethyl or l- or 2-~ulphoethyl; and P~N-
alkyl is preferably dLmethylaminomethyl, diethylamino-
methyl, 1- or 2-dimethylaminoethyl or 1- or 2-diethyl-
aminoethyl.
The radical Ar is preferably unsubstituted
phenyl. If Ar is a substituted phenyl group, it is pre-
ferably monosubstituted. However, it can also be di-
substituted, i~ being possible for the substituents to be
identical or different. Preferred substituents on the
phenyl group are F, Cl, methoxy or OH. Specifically, Ar
is preferably phenyl, o-, m- or p-fluorophenyl, o-, m- or
p-chlorophenyl, o-, m- or p-methoxyphenyl or o-, m- or p-
hydroxyphenyl, and al60 0-, m- or p-bromophenyl, o-, m-
or p-ethoxyphenyl, 2,3-, 2,4-, 2,5-, 2,6-,- 3,4- or 3,5-
dimethoxyphenyl, 3-hydroxy-4-methoxyphenyl, 3-methoxy-4-
hydroxyphenyl, 2,3-, 2,4-, 2,5-, 2,6-, 3,4- or 3,5-di-
hydroxyphenyl, 2,3- or 3,4-methylenedioxyphenyl, thien-2-
yl or thien-3-yl.
Accordingly, Ar-alkyl is preferably benzyl, 1- or
2-phenylethyl, o-, m- or p-fluorobenzyl, o-, m- or p-
chlorobenzyl, o-, m- or p-methoxybenzyl, 1- or 2-o-, -m-
or -p-methoxyphenylethyl, o-, m- or p-hydroxybenzyl or 1-
or 2-o-, -m- or -p-hydroxyphenylethyl.
Ac is preferably acetyl or benzoyl, other pre-
ferred meanings being propionyl, butyryl, isobutyryl, o-,
m- or p-fluorobenzoyl, o-, m- or p-chlorobenzoyl, o-, m-
or p-hydroxybenzoyl or o-, m- or p-methoxybenzoyl.
Accordingly, Ac~-alkyl is preferably acetoxymethyl, 1- or
2-acetoxyethyl, benzoyloxymethyl or 1- or 2-benzoyloxy-
- 5 - 2~
ethyl; NH~c i8 preferably acetamido or benzamido.
Het i8 preferably furan--2-yl or furan-3-yl,
thien-2-yl or ~hien-3-yl, pyrrol-1-, -2- or -3-yl,
imidazol-l-, -2-, -4- or -5-yl, pyrazol-1-, -3-, -4- or
-5-yl, oxazol-2-, -4- or -5-yl, isoxazol-3-, -4- or -5-
yl, thiazol-2-, -4- or -5-yl, i60thiazol-3-, -4- or -S-
yl, pyrid-2-, -3- or 4-yl or pyrimidin-2-, -4-, -5- or
-6-yl, other preferred meanings being 1,2,3-triazol-1-,
-4- or -5-yl, 1,2,4-triaæol-1-, -3 or -5-yl, tetrazol-l-
or -5-yl, 1,2,3-oxadiazol-4- or -5-yl, 1,2,4-oxadiazol-3-
or -5-yl, 1,3,4-thiadiazol-2- or -S-yl, 1,2,4-thiadiazol-
3- or -5-yl, 1,2,3-thiadiazol-4- or -S-yl, 2H-thiopyran-
2-, -3-, -4-, -5- or -6-yl, 4H-thiopyran-2-, -3- or -4-
yl, pyridazin-3- or -4-yl, pyrazinyl, benzofuran-2-, -3-,
-4-, -5-, -6- or -7-yl, benzothien-2-, -3-, -4-, -5-, -6-
or -7-yl, indol-1-, -2-, -3-, -4-, -5-, -6- or -7-yl,
isoindol-l-, -2-, -3-, -4-, -5-, -6- or -7-yl, benz-
imidazol-1-, -2-, -4- or -5-yl, benzopyrazol-1-, -3~,
-4-, -5-, -6- or -7-yl, benzoxazol-2-, -4-, -5-, -6- or
-7-yl, benzisoxazol-3-, -4-, -5-, -6- or -7-yl, benz-
thiazol-2-, -4-, -5-, -6- or -7-yl, benzisothiazol-2-,
-4-, -5-, -6- or -7-yl, benz-2,1,3-oxadiazol-4-, -5-l -6-
or -7-yl, quinol-2-, -3-, -4-, -5-, -6-, -7- or -8-yl,
isoquinol~ 3-, -4-, -5-, -6-, -7- or -8-yl, car-
bazol-l-, -2-, -3-, -4- or -9-yl, acridin-l-, -2-, -3-,
-4-, -5-, -6-, -7-, -8- or -9-yl, cinnol-3-, -4-, -5-,
-6-l -7- or -8-yl or quinazol-2-l -4-, -5-, -6-, -7- or
-8-yl. The heterocyclic radi~al~ can also be partially
or completely hydrogenated. Het can therefore al~o be
e.g. 2,3-dihydrofuran-2 , -3-, -4- or -5-yl, 2,5-dihydro-
furan-2-, -3-, -4- or -S-yl, tetrahydrofuran-2- or -3-yl,
tetrahydrothien-2- or -3-yl, 2,3-dihydropyrrol-1-, -2-,
-3-, -4- or -5-yl, 2,5-dihydropyrrol-1-, -2-, -3-l -4- or
-5-yl/ pyrrolidin-l-l -2- or -3-yl,
tetrahydroLmidazol-1-l -2- or -4-yl, 2,3-dihydrop-
yrazol-1-, -2-, -3-, -4- or -5-yl, tetrahydropyrazol-l-,
-3- or -4-yl, 1,4-dihydropyrid-1-, -2-, -3- or -4-yl,
1,2,3,4-tetrahydropyrid-1-, -2-, -3-, -4-, -5- or -6-yl,
~ 3,6-tetrahydropyrid-1-, -2-, -3-, -4-, -5- or~-6-yl,
2a~3 ~
piperidin-l-, -2-, -3- or -4-yl, morpholin-2-, -3- or
-4-yl, tetrahydropyran-2-, -3- or -4-yl, 1,4-dioxanyl,
1,3-dioxan-2-, -4- or -5-yl, hexahydropyridazin-l-, -3-
or -4-yl, hexahydropyrimidin-1-, -2-, -4- or -5~yl,
piperazin-l-, -2- or -3-yl, 1,2,3,4-tetrahydroquinol-1-,
~2-, -3-, -4-, -5-, -6-, -7- or -8-yl or 1,2,3,4-tetra-
hydroisoquinol-l-, -2-, -3-, -4-, -5-, -6-, -7- or -8-yl.
The heterocyclic radicals can al~o be substituted
as indicated. Het can preferably al~o be e.g. 4- or 5-
methylthiazol-2-yl~ 4-, 5- or 6-methylpyrimidin-2-yl,
4,5-dimethylthiaæol-2-yl, 3-, 4- or 5-methylfuran-2-yl,
2-, 4- or 5-methylfuran-3-yl, 2,4-dimethylfuran-3-yl, 3-,
4- or 5-methylthien-2-yl, 2-, 4- or 5-methylthien-3-yl or
3-methyl-5-tert-butylthien-2-yl.
The radical Ind is an indol-3-yl radical monosub-
stituted by one of the radicals indicated. It is pre-
ferably substituted in the 5-position or else in the 4-,
6- or 7-position. Substitution in the 1- or 2-position
is a further possibility. Preferred substituents on the
indol-3-yl radical are -NHR2, -NO2 and -CoNR3R4.
Rl is preferably OH, NH2, NA2, NHCH2COOA or
NHCH(CH2OH)COOA.
R2 is preferably CONH2, other preferred meanings
being H, Ac or SO2A.
R3 is preferably H.
R4 i6 preferably hydroxyalkyl, other preferred
meanings being AOOC-alkyl, H2NCO-alkyl, A2N-alkyl, Ar or
HSO3-alkyl.
The group -NR3R4 is preferably also pyrrolidino,
piperidino, morpholino, 4-R5-piperazino or 4-R7-piperi-
dino, R5 preferably being H, A, Ar, pyrLmidin-2-yl, 5-
methylthiazolyl, Ar-CO, COO~, CH2CONHA or pyrrolidino-
carbonylmethyl and R7 preferably being NA2, NHCO~r, COOA,
CONH2, piperidino or morpholino, or else NHCOA, Ar or
another Het group.
The parameter n is preferably 4 and the radical
Q is preferably -tCH2)~- or -CH2S-CH2CH2-, or else
-CH2-SO-CHzCH2-, -CH2-SO2-CH2CH2-, -(CH2)z-, -(CH2) 3- or
- (CH2)5- -
7~
-
Accordingly, the invention relates particularly
to those compounds of formula I in which at least one of
said radicals has one of the meanings indicated above,
especially one of the preferred meanings indicated above.
Some preferred groups of compounds can be expressed by
the following partial formulae Ia to If, which correspond
to formula I and in which the radicals and parameters not
described in greater detail are as defined for formula I,
but in which:
in Ia, Ind is an indol-3-yl radical fiubstituted in the 5-
position by -O-CH2-CO-R1;
in Ib, Ind is an indol-3-yl radical substituted in the 5-
position by -NHR3;
in Ic, Ind i8 a 5-nitroindol-3-yl radical;
in Id, Ind is an indol-3-yl radical substituted in the 5-
position by -CoNR3R4;
in Ie, Ind is an indol-3-yl radical substituted in the 5-
position by -CSNH2; and
in If, Ind is a 5-ureidoindol-3-yl radical.
Especially preferred compounds are those of par-
tial formulae Ig and Iag to Ifg, which correspond to par-
tial formulae I and Ia to If, but in which additionally:
Q is -(CH2~4--
Other especially preferred compounds are those of
partial formulae Ih, Iah to Ifh, Igh and Iagh to Ifgh,
which correspond to partial formulae I, Ia to If, Ig and
Iag to Ifg, but in which additionally-
Ar is phenyl.
The invention further relates to a process for
the preparation of indole derivatives of formula I and
~heir salts, characterized in that a compound of formula
II
Ind-Q-Xl II
wherein
X1 is X or NH2,
X is Cl, Br, I, OH or an OH group functionally modified
to form a reactive group, and
~ ~ 2 ~
Ind and Q are as defined,
is reacted with a compound of formula III
X2-CH2CH2C~r=CH-CH2X3 III
wherein
X2 and X3 can be identical or different and are each X if
Xl = NH2 or are together NH in other cases, and
Ar is as defined,
or in that a compound which has formula I except that one
or more hydrogen atoms have been replaced by one or more
reducible groups and/or one or more additional C-C and/or
C-N bonds is treated with a reducing agent,
or in that a compound which has formula I except tha~ one
or more hydrogen atoms have been replaced by one or more
solvolyzable groups is trea~ed with a solvolyzing agent,
or in that, to prepare thioethers of formula I in which
Q is -CH2-S-CH2CH2-, a compound of formula IV
I~d-CH2N~R)2 IV
wherein
R is alkyl having 1-4 C atoms or else both radicals R
together are -(CH2)p- or -CH2CH20CH2CH2-,
p is 4 or 5.and
Ind is as defined,
ix reacted with a thiol of formula V
V ~
HS - C~ CH -N ~-Ar
wherein
Ar is as defined,
or with one of it reactive derivatives,
or in that a compound of formula VI
E
Ind-Q-N ~ -Ar
~ VI
9 ~ 3 r~
wherein
one of the radicals E is X, CN or NH2,
the other radical E is H and
Ind, Q, Ar and X sre as defined,
is treated with an agent which eliminates HE,
or in that, to prepare a compound of formula I in which
Ind is an indol-3-yl radical substituted by -O~CH2-CO-R1,
a hydroxyindole which has formula I except that Ind has
been replaced by an indol-3-yl xadical substituted by an
OH group, or one of it reactive derivatives, i6 reacted
with a compound of the iormula X-CHz-CO-Rl (wherein X and
Rl are as defined),
or in that, to prepare a compound of formula I in which
Ind is an indol-3-yl radical substituted by -Co-NR3R4, an
indolecarboxylic acid which has formula I except that Ind
has been replaced by an indol~3-yl radical substituted by
a COOH group, or one of its reacti~e derivatives, is
reacted with a compound of the formula HNR3R4 (wherein R3
and R4 are a~ defined~,
or in that, to prepare a compound of ormula I in which
Ind is an indol-3-yl radical substituted by -CS-NH2, a
cyanoindole which has formula I except that Ind has been
replaced by an indol-3-yl xadical substituted by a CN
group is reacted with H2S or an agent which releases H2S,
and/or in that, if desired, in a compound of formula I,
a thioether group is oxidized to an SO group or SO2 group
or an SO group is oxidized to an SO2 group, and/or an OA
group is cleaved to form an OH group, and/or an Ind group
is converted into another Ind group, and/or in that a
resulting base of formula I is converted into one of its
salts by treatment with an acid or base.
The compounds of formula I are otherwise prepared
by methods known per se, such as those described in the
literature (e.g. in the standard works such as Houben-
Weyl, Methoden der Organischen Chemie ~Methods of OrganicChemistry), Georg-Thieme-Verlag, Stuttgart; Organic ~e~
actions, John Wiley & Sons, Inc., New York; German Offen-
2 ~ J ~
-- 10 --
legungsschrift 33 42 632), namely under reaction con-
ditions such as those which are known and suitable for
said reactions. It i8 also po~sible to make use of
variants known per se, which are not mentioned in greater
detail here.
If deired, the starting materials for the
claimed proce~s can also be formed in situ in such a way
that they are not isolated from the reaction mixture but
are immediately reacted further to give the compounds of
formula I.
In the indole derivatives of formula II, X1 is
preferably X; accordingly, in the compounds of formula
III, x2 and X3 are together preferably NH. The radical X
is preferably Cl or Br, but it can also be I, OH or an OH
qroup functionally modified to form a reactive group,
especially alkylsulphonyloxy having 1-6 C atoms (e.g.
methanesulphonyloxy~ or arylsulphonyloxy having 6-10 C
atoms (e.g. benzenesulphonyloxy, p-toluenesulphonyloxy,
naphthalene-l- or -2-sulphonyloxy).
Accordingly, the indole derivati~es of formula I
can be obtained especially by reacting compounds of the
formula Ind-Q-Cl or Ind-Q-Br with tetrahydropyridine
derivatives of formula III in which x2 and X3 together are
an NH group (designated as IIIa hereafter).
Some of the compounds of formulae II and, in par-
ticular, III are known; the unknown compounds of formulae
II and III can easily be prepared analogously to the
known compounds. Compounds of formula II ~Q
-CH2-S-CH2CH2-) can be prepared e.g. from Mannich bases of
formula IV and thiols of the formula HS-CH2CH2-~, e.g.
HS-CH2CH20H. The sulphoxides and sulphones of formula II
lQ = -CH2-SO-CH2CH2- or -CH2-SO2-CH2CH2-) are accessible by
oxidation of the thioethers (II, Q = -CH2-S-CH2CH2-).
Primary alcohols of the formula Ind-Q- OH can be obtained
e.g. by reducing the appropriate carboxylic acids or
their esters. Treatment with thionyl chloride~ hydrogen
bromide, phosphorus tribromide or sLmilar halogen com-
pounds yields the corresponding halides of the formula
Ind-Q-Hal. The corresponding sulphonyloxy compounds can
~ 3
be obtained from the alcohols Ind-Q-OH by reaction with
the appropriate sulphonyl chlorides.
The iodine compounds of the formula Ind-Q-I can
be obtained e.g. by reacting potassium iodide with the
appropriate p-toluenesulphonic acid esters. The amines
of the formula Ind-Q-NHz can be prepared e.g. from the
halides with potassium phthalimide or by reducing the
appropriate nitriles.
Most of the piperidine derivatives IIIa are known
(q.v. German Offenlegung6schrift 20 60 816) and can be
obtained e.g. by reacting piperid-4-one with organo-
metallic compounds of the formula M-Ar (wherein M i8 an
Li atom or MgHal), this being followed by hydrolysis to
give the corresponding 4-Ar-4-hydro~ypiperidines and, if
desired, by dehydration to give 4-Ar-3,4-dehydropiperi-
dines. Compounds of formula III (X2 and X3 = X in each
case) can be prepared e.g. by redu~ing diesters of the
formula alkylOOC-CH2-CAr=CH-COOalkyl to give diols of the
formula HO-CH2CH2-CAr=CH-CH20H (III, X2 = X3 = OH), this
being followed, if desired, by reaction with SOC12 or
PBr3 .
The reaction of the compounds II and III proceeds
according to methods such as those known from the litera-
ture for the alkylation of amines. The components can be
melted together in the absence of a solvent, in a sealed
tube or an autoclave if necessary. It is also possible,
however, to react the compounds in the presence of an
inert solvent. Examples of suitable solvents are hydro-
earbons such as benzene, toluene or xylene; ketones such
as acetone or butanone; alcohols such as methanol,
ethanol, isopropanol or n-butanol; ethers such as tetra-
hydrofuran (THF) or dioxane; amides such as dimethyl-
formamide (DMF) or N-methylpyrrolidone; or nitriles such
as acetonitrile, or else, if desired, mixtures of these
solvents with one another or mixtures with water. It can
be favourable to add an acid-binding agent, for example
an alkali metal or alkaline earth metal hydroxide, car-
bonate or bicarbonate or another aika ~metal or alkaline
earth metal salt of a weak acid, preferably a potassium,
2 ~ ~ ~. ,` fi~
- 12 -
sodium or calcium salt, or to add an organic base such as
triethylamine, dimethylaniline, pyridine or quinoline, or
an excess of the amine component Ind-Q-NH2 or of the
piperidine derivative of formula IIIa. The reaction tLme
is between a few minut~s and 14 days, depending on the
conditions used, and the reac~ion temperature is between
about 0 and 150, normally between 20 and 130.
A compound of formula I can alss be obtained by
treating a precursor, in which hydrogen atoms have been
replaced by one or more reducible groups and/or one or
more additional C-C and/or C-N bonds, with a reducing
agent, preferably at temperatures of between -80 and
~250, in the presence of at least one inert solvent.
Reducible groups (groups replaceable by hydrogen)
are, in particular, oxygen in a carbonyl group, hydroxyl,
arylsulphonyloxy (e.g. p-toluenesulphonyloxy), N-benzene-
sulphonyl, N-benzyl or O-benzyl.
In principle, compounds containing only one of
the above-mentioned groups or additional bonds, or com-
pounds containing two or more of the above-mentioned
groups or additional bonds adjacent to one another, can
be converted into a compound of formula I by reduction,
it being possible simultaneously to reduce substituents
in the Ind group which are present in the starting com-
pound. This is preferably carried out using nascenthydrogen or complex metal hydrides or by means of a
Wolff-Rishner reduction.
Preferred starting materials for the reduction
have formula VII
Ind'-L-W VII
wherein
Ind' is an Ind radical which can additionally be substi-
tuted in the 1-position by an arylsulphonyl group or a
benzyl group,
L is Q or a chain which corresponds to the radical Q
except that one or more -CH2- groups have been replaced by
-CO- groups and/or one or more hydrogen atoms have been
2 ~
- 13 -
replaced by OH groups,
W is -N ~ -Ar' or -N ~ Ar' An ;
An~ is an anion of a 6trong acid and
Ar' is a phenyl group which is unsubstituted, monosub-
stituted or difiubstituted by F, Cl, Br, OA, O~ and/or O-
benzyl or substituted by a methylenedioxy group,
but wherein the following meanings canno$ apply simul-
taneously: Ind' = Ind, L - Q and
W = -N ~ Ar
In the compounds of formula VII, L is preferably
-CO-(CH2)~2-CO- [specifically -COCO-, -CO~H2CO-,
-CO-(CH232-CO-, -CO-(CH2) 3-CO- ] ~ ~ ( CH2 ) n l~CO- [ specifically
-CH2CO- ~ -CH2CHz-CO- ~ - ( CH2 ) 3-CO- or -(CH2) 4-CO- ],
-CH2-S-CH2-CO-, -CH2-SO-CH2-CO- or -CH2-SO2-CH2-CO-, further
examples being -CO-CH2CH2-, -CH2-CO-CH2-, -CO-(CH2) 3-,
-CH2-CO-CH2cH2- ~ -CH2CH2-CO-cH2- ~ -CO- ( CH2 ) 4
-CH2-CO- ( ~H2 ) 3-, -CH2CH2-CO-CH2CH2- or -(CH2)3-CO-CH2-.
Compounds of formula VII can be prepared e.g. by
reacting . 4-Ar'-1,2,3,6-tetrahydropyridine or 4-Ar'-
pyridine with a compound of formula VIII
Ind'-L-Xl VIII
wherein
- Ar', Ind', L and Xl are as defined above,
under the conditions indicated above for the re~ction of
II with III.
If nascent hydrogen is used as the reducing
agent, this can be produced e.g. by treating metals with
weak acids or with bases. Thus it is possible e.g. to
use a mixture of zinc with an alkali metal hydroxide
solution or a mixture of iron with acetic acid. It is
also appropriate to use sodium or another alkali metal in
an alcohol such as ethanol-,~3~opropanol, butanol, amyl or
isoamyl alcohol or pnenol. It is also possible to use an
2 ~
- 14 -
aluminium-nickel alloy in aqueous-alkaline solution,
ethanol being added if necessary. Sodium amalgam or
aluminium amalgam in aqueous-alcoholic or aqueous solu-
tion is also suitable for producing the nascent hydrogen.
S The reaction can also be carried out in the heterogeneous
phase, in which case it is convenient to use an aqueous
phase and a benzene or toluene phase.
Other reducing agents which can be used to par~
ticular advantage are complex metal hydrides su~h as
10 LiAlH4, NaBH4, diisobutylaluminium hydride or
NaAl(OCH2CH20CH3)2H2, and diborane, catalysts such as BF3,
AlCl3 or LiBr being added if desired. Solvents which are
suitable for this purpose are, in particular, ethers such
as diethyl ether, di-n-butyl ether, THF, dioxane, diglyme
15 or 1,2-dimethoxyethane, and hydrocarbons such as benzene.
Solvents which are suitable for reduction with NaBH4 are
primarily alcohols such as methanol or ethanol, as well
as water and aqueous alcohols. Reduction by these
methods is preferably carried out at temperatures of
20 between -80 and +150, especially of between about 0 and
about 100.
The reduction of -CO- groups in acid amides (e.g.
those of formula VII in which L is a -(CH2)D1-CO- or
-CHz-S-CH2-CO- group) to -CH2- groups can be carried out
25 to particular advantage with LiAlH4 in THF at temperatures
of between about 0 and 66. Arylsulphonyl protecting
groups located in the l-position of the indole ring can
be sLmultaneously eliminated by reduction.
Reduction of the pyridinium salts of formula VII
30 (wherein W is -N ~ -Ar'An and An is preferably Cl, Br
or CH3S03) to compounds of formula I is carried out e.g.
with NaBH4 in water, methanol or ethanol or in mixtures of
these solvents, a base such as NaOH being added if
desired, at temperatures of between about 0 and 80.
3S N-Benzyl groups can be eliminated by reduction
with sodium in liquid ammonia.
It is also possible to reduce one or mor-e~_ar-
- 15 -
bonyl groups to CH2 groups according to the Wolff-Rishner
method, e.g. by treatment with anhydrous hydrazine in
absolute ethanol, under pressure, at temperatures of
between about 150 and 250. A Rodium alcoholate i8 ad-
vantageously used as the catalyst. The reduction can
also be varied according to the Huang~Minlon method by
carrying out the reaction with hydrazine hydrate in a
high-boiling water-miscible solvent such as diethylene
glycol or triethylene glycol, in the presence of an
alkali such as fiodium hydroxide. ~he reaction mixture is
normally boiled for about 3-4 hours. The water i8 then
distilled off and the hydrazone formed is decomposed at
temperatures of up to ~bout 200. The ~olff-~ishner
reduction can also be carried out with hydrazine in
dimethyl sulphoxide at room temperature.
Compounds which have formula I except that one or
more H atoms have been replaced by one or more solvoly-
zable groups can be solvolyzed, especially hydrolyzed, to
give the compounds of formula I.
The starting materials for the solvolysis can be
obtained for example by reacting IIIa with compounds
which have formula II (X1 = X) except that one or more H
atoms hav~ been replaced by one or more solvolyzable
groups. Thus, in particular, 1 acylindole derivatives
~which have formula I except that, in the 1-position of
the Ind radical, they contain an acyl group, preferably
an alkanoyl, alkylsulphonyl or arylsulphonyl group having
up to 10 C atoms in each case, such as methanesulphonyl,
benzenesulphonyl or p-toluenesulphonyl3 can be hydrolyzed
to give the corresponding indole derivatives unsubstitu-
ted in the l-position of the indole ring, e.g. in an
acidic or, preferably, neutral or alkaline medium at
temperatures of between 0 and 200. Sodium, potassium or
calcium hydroxide, sodium or potassium carbonate, or
ammonia, is conveniently used as the base. The chosen
solvents are preferably water; lower alcohols ~uch as
methanol or ethanol; ethers such as THF or dio~ane;
sulphones such as tetramethylenesulph~e; or mixtures
thereof, especially mix~ures containing water. Hydroly-
- 16 - ~ 3-
sis can also be carried out simply by treatment with
water alone, especially at the boiling point.
Indole derivatives of formula I (Q
-CH2-S-CHzCH2-) can also be obtained by reacting ~annich
bases of formula IV with thiols of formula V or their
salts or reactive derivatives.
Some of the starting materials of formulae IV and
V are known; those which are not known can easily be
prepared analogously to the known compounds. Thus the
Mannich bases of formula IV can be obtained e.g. from
indoles of the formula Ind-H, formaldehyde and amines of
the formula HN(R)2 and the thiols of formula V can be
obtained from the bases of formula IIIa and thiol ~eri-
vatives of the formula HS-CHzCH2-Xl (it also being pos-
sible for the HS group to be protected in an intermediate
step).
Specifically, the reaction of IV with V takes
place in the presence or absence of an inert solvent, at
temperatures of between about -20 and 250, preferably of
between 60 and 150. Examples of suitable solvents are
hydrocarbons such as benzene, toluene, xylenes or mesit-
ylene; tertiary ~ases such as triethylamine, pyridine or
picoline; alcohols such as me~hanol, ethanol or butanol;
glycols and glycol ethers such as ethylene glycol, di-
~thylene glycol or 2-methoxyethanol; ketones such as
acetone; ethers such as THF or dioxane; amides such as
DMF; and sulphoxides such as dimethyl sulphoxide. Mix-
tures of these solvents are also suitable. The thiols of
formula V are conveniently converted first into the cor-
responding mercaptides, preferably by reaction with
sodium or potassium hydroxide or sodium or potassium
ethylate to give the corresponding sodium or potassium
mercaptides. Reactive derivatives of the thiols of
formula V which can be used are preferably the corres-
ponding isothioureas (which have formula V except that
HS- has been replaced by H2N(=NH)-S-) or their salts (e.g.
the isothiuronium chlorides); these are more stable and
easier to handle. Their reaction with the bases IV is
conveniently carried out in a basic medium, e;g. in
- 17 _ 2 ~
aqueous sodium hydroxide solution, at temperatures of
between O and 100.
Compounds of formula I are also obtained by
eliminating HE from compounds of formula VI to form a
double bond. According to the definition of E, the
molecule eliminated can be e.g. a hydrogen halide, water
(dehydration), a carboxylic acid or other acid, ammonia
or HCN. ~he starting materials of formula VI can be
obtained e.g. by reacting II (~1 = X) with a compound of
formula IX
E
HN ~ -Ar IX
wherein E and Ar are as defined.
If one of the radicals E = Hal, this substituent can
easily be eliminated under basic reaction conditions.
The following bases can be used: alkali metal hydroxides,
alkali metal carbonates, alcoholate6 such as potassium
tert-butylate, and amine such as dimethylaniline,
pyridine, collidine or quinoline; examples of solvents
used are benzene, toluene, cyclohexane, ~HF or tert-
butanol. The amines serviny as bases can also be used inexcess as solvents. If one of the radicals E is an OH
group, acids such as acetic acid, hydrochloric acid or
mixtures of both are preferably used as water-eliminating
agents. It may be advantageous to add a solvent (e.g.
water or ethanol). The elLmination of acyloxy, alkyl-
sulphonyloxy and alkox~ulphonyloxy radicals or amino
radicals can be carried out under similar conditions.
The elimination of sulphonic acid radicals, e.g. those of
mesylates or to~ylates, is carried out under mild con~
ditions by boiling in DMF or dimethyl sulphoxide with
alkali metal carbonates, e.g. Li2CO3, or with potassium
acetate. Ammonia can be eliminated cimply by heating the
salts of the corresponding amino compounds (especially
th~ 4-amino derivatives). Similarly, HCN can be elimi-
nated from compounds of formula VI (one group E = CN) by
2 ~ 3 ':~ ~
- 18 -
heating. In general, HE i8 eliminated from VI at tem-
peratures of between 0 and about 250~, preferably of
between 50 and 200.
Compounds of formula I in which Ind is an indol-
3-yl radical substituted by -O-CH2 CO-R1 can be obtained
by etherifying appropriate hydroxyindol-3-yl compounds
(q.v. European patent 7399) or their reactive deriva-
tives, in particular their 6alts, e.g. their alkali metal
salts such as their Na or K salts, with compounds of the
formula X-CH2-CO-R1 (e.g. chloroacetic or bromoacetic
acid, methyl or ethyl chloroacetate or bromoacetate,
chloroacetamide or bromoacetamide, chloro-N-methylaceta-
mide or bromo-N-methylacetamide, chloro-N,N-dimethyl-
acetamide or bromo-N,N dimethylacetamide). The etheri-
fication reaction is conveniently carried out in one of
the solvents indicated (e.g. DMF), the hydroxyl compound
first being converted into one of its salts with the aid
of a base, e.g. an alkali metal hydride such as NaH or
KH, or an alkali metal alcoholate such as sodium or
potassium methylate or ethylate, after which the compound
of the formula X-CH2-CO-R1 is added and the mixture is
stirred for a few hours at temperatures of between 0 and
50! preferably of between 20 and 80.
Compounds of formula I in which Ind is an indol-
3-yl radical substituted by -Co-NR3R4 can be obtained by
amidating appropriate carboxyindol-3-yl compounds (q.v.
German Offenlegungsschrift 33 42 632) or their reactive
derivatives, e.g. their acid halides, esters or anhyd-
rides, with amines of the formula HNR3R4. It is preferred
to react the free carboxylic acid with the amine under
the conditions of a peptide synthesis. This reaction i6
preferably carried out in the presence of a dehydrating
agent, e.g. a carbodiimide such as dicyclohexylcarbodi-
imide or N-(3-dimethylaminopropyl)-N-ethylcarbodiimide,
or propanephosphonic anhydride (q.v. Angew. Chem. 92, 129
(1980)), diphenylphosphorylazide or 2- ethoxy-N-ethoxy-
carbonyl-1,2-dihydroquinoline, in an inert solvent, e.g.
a halogenated hydrocarbon such as methylen~:~chloride, an
ether such as THF or dioxane, an amide such as DMF or
2 0 ~
-- 19 --
dimethylacetamide, or a nitrile such as acetonitrile, at
temperatures of between about -10 and 40, preferably of
between 0 and 30D. Instead of the acid or amide, it i8
also possible to use reactive derivatives of these
substances in the reaction, e.g. those in which reactive
groups are blocked by protecting groups in an inter-
mediate step. The acids can also be used in the form of
their activated esters, which are conveniently formed in
situ, e.g. by the addition of l-hydroxybenztriazole or
N-hydroxysuccinimide.
Compounds of formula I in which Ind i8 an indol-
3-yl radical substituted by -CS-NHz can be obtained by
adding H2S on to appropria~e cyanoindol-3-yl compounds.
Agents which release ~2S, e.g. thioacetamide, can also be
used instead of H2S. The reaction is conveniently carried
out in one of the solvents indicated, e.g. DMF, in the
presence of an acid, e.g. HCl, at temperatures of between
O and 200, preferably of between 20 and 160.
Furthermore, one compound of formula I can be
converted into another compound of formula I by methods
known per se.
Thus the thioether group in a thioether of for-
mula I (Q = -CH2-5-C~2CH2-) can be oxidized to an S0 group
or to an SOz group, or the 50 group in a sulphoxide of
formula I (Q = -CH2-SO-CH2CH2-) can be oxidized to an SO2
group. If it is desired to obtain the sulphoxides,
oxidation is carried out for example with hydrogen per-
oxide, peracids such as m-chloroperbenzoic acid, Cr(VI)
co~pounds such as chromic acid~ KMnO4 ~ l-chlorobenztri-
zzole, Ce(IV) compounds such as (NH4)2Ce~NO3)6, or nega-
tively substituted aroma~ic diazonium salts such as o- or
p-nitrophenyldiazonium chloride, or by electrolysis under
relatively mild conditions and at relatively low tempera-
tures (about -80 to +100). If, on the other hand, it is
desired to obtain the sulphones (from the thioethers or
the sulphoxides), the s~me oxidizing agents are used
under more vigorous conditions and/or in excess, and nor-
mally at higher te~eratures. The customary inert sol-
vents may be present or absent in these reactions.
2 01 .1 ~
- 20 -
Examples of suitable inert solvents are water, aqueous
mineral acids, aqueous alkali metal hydroxide ~olutions,
lower alcohols such as methanol or ethanol, esters such
as ethyl acetate, ketones such as acetone, lower car-
boxylic acids such as acetic acid, nitriles such as
acetonitrile, hydrocarbons such as benzene, and chlori-
nated hydrocarbons such as chloroform or CCl~. A pre-
ferred oxidizing agent is 30~ aqueous hydr~gen peroxide.
This yields the sulphoxides if the calculated amount is
used in solvents such as acetic acid, acetone, methanol,
ethanol or aqueous sodium hydroxide solution, at tempera-
tures of between -20 and 100, and to the sulphones if an
excess is used at higher temperatures, preferably in
acetic acid or in a mixture of acetic acid and acetic
anhydride.
Ethers of formula I in which the radical Ar is
monosubstituted or disubstituted by O-alkyl can be
cleaved to form the corresponding hydroxyl deri~atives.
For example, the ethers can be cleaved by treatment with
dimethyl sulphide/boron tribromide complex, e.g. in
toluene, ethers such as THF, or dimethyl sulphoxide, or
by melting with pyridine hydrohalides or aniline hydro-
halides, preferably pyridine hydrochloride, at about 150-
250.
Furthermore, Ind groups can be converted into
other Ind groups by methods known per se. Carboxyl
groups can ba esterified, e.g. by treatment with alcohols
in the presence of acid catalysts, or by reaction with
diazoalkanes. Conversion of the carboxylic acids into
their chlorides, e.g. with SOC12, and ~ubsequent reaction
with NH3 or amines yields the corresponding carboxamides,
which can also be obtained by treating the carboxylic
acid esters with ammonia or amines. Sol~olysis, pre-
ferably hydrolysis under the conditions indicated above,
yields the carboxylic acids from the esters or amides;
in particular, carboxylic acids can be obtained from the
esters by treating the latter with NaOH or ROH in aqueous
alcohols, convenienJ~-ly at temperatures of between about
50 and abo~t 200. The reduction of nitro groups to
2 1 2 ~ r r
-
amino groups is carried out e.g. with metals such as Fe,
Sn or Zn, or with SnCl2, conveniently in aqueous- al-
coholic acids, e.g. in aqueou~-ethanolic hydrochloric
acid, at temperatures of between 20 and 100. Hydroxyl
groups or amino groups can be acylated, e.g. with acid
chlorides such as acetyl chloride, benzoyl chloride or
methanesulphonyl chloride, in the presence of a base such
as triethylamine or pyridine, and in the presence or
absence of one of the sol~ents indicated. Reactions of
hydroxyl compounds with alkyl isocyanates, e.g. in the
presence of a base such as pyridine, yield the corres-
ponding urethanes.
The compounds of formula I can possess one or
more centres of asymmetry. When prepared, they can
lS therefore be obtained as racemates or else in the opti-
cally active form if optically active starting materials
are used. When synthesized, compounds possessing ~wo or
more centres of asymmetry are generally obtained as mix-
tures of racemates, from which the individual racemates
can be isolated in the pure form, for example by recry~-
tallization from inert solvents. If desired, the race-
mates obtained can be mechanically or chemically re~olved
into their optical antipodes by methods known per se.
Preferably, diastereoi60mers are formed from the racemate
by reaction with an optically active resolving agent.
Examples of suitable resolving agents are optically
active aci s such as the D and L forms of tartaric acid,
dibenzoyltartaric acid, diacetyltartaric acid, camphor-
sulphonic acids, mandelic acid, malic acid or lactic
acid.- The different forms of the diastereoisomers can be
resolved in a manner known per se, e.g. by fractional
crystallization, and the optically active compounds of
formula I can be liberated from the diastereoisomers in
a manner known per se.
A base of formula I can be converted with an acid
into the corresponding acid addition salt. Acids which
produce biocompatible salts ar~ suitable for this re-
action. Thus it is possible to use inorganic acids, e.g.
sulph~r-.c acid, hydrohalic acids such as hydrochloric
2 R ~
- 22 -
acid or hydrobromic acid, phosphoric acid6 such as ortho-
phosphoric acid, nitric acid and sulphamic acid, a~ well
as organic acids, i.e. specifically aliphatic, alicyclic,
araliphatic, aromatic or heterocyclic monobasic or poly-
basic carboxylic, sulphonic or sulphuric acids, such as
formic acid, acetic acid, propionic acid, pivalic acid,
diethylacetic acid, malonic acid, succinic acid, pimelic
acid, fumaric acid, maleic acid, lactic acid, tartaric
acid, malic acid, benzoic acid, salicylic acid, 2-phenyl-
propionic acid, citric acid, gluconic acid, ascorbic
acid, nicotinic acid, isonicotinic acid, methanesulphonic
or ethanesulphonic acid, ethanedisulphonic acid, 2-
hydroxyethanesulphonic acid, benzenesulphonic acid, p-
toluenesulphonic acid, naphthalenemonosulphonic and
naphthalenedisulphonic acids and laurylsulphuric acid.
If desired, the free bases of formula I can be
liberated from their salts by treatment with strong bases
such as sodium or potassium hydroxide or sodium or potas-
sium carbonate.
The invention further relates to the use of the
compounds of formula I and their biocompatible salts for
the manufacture of pharmaceutical preparations, espe-
cially by a non-chemical route. For this purpose, they
can be converted into a suitable dosage form together
with at least one excipient or ad~unct and, if approp-
riate, in combination with one or more additional active
ingredients.
The invention further relates to compositions,
especially pharmaceutical preparations, containing at
least one compound of formula I and/or one of their bio-
compatible salts. These preparations can be used as
drugs in human or veterinary medicine. Possible exci
pients are organic or inorganic substances which are
suitable for enteral le.g. oral), parenteral or topical
administration and which do not react with the novel
compounds, examples of such excipients being water,
vegetable oils, benzyl alcohols, polyethylene glycols,
gelatin, carhohydrates such as lactose or starch, mag-
nesium stearate, talc and petroleum jelly. Tablets,
23 ~
coated tablets, capsules, syrups, ~uices, drops or sup-
positories are used in particular for enteral adminis-
tration, solutions, preferzbly oily or aqueous solutions,
as well as suspen6ions, emulsions or implants are used
for parenteral administration, and ointments, creams or
powders are used for topical administration. The novel
compounds can also be lyophilized and the resulting
lyophilizates used e.g. to manufacture in~ectable prepa-
rations.
The preparations indicated can be sterilized
and/or can contain ad~uncts such as lubricants, preser-
vatives, stabilizers and/or wetting agents, emulsifiers,
salts for influencing the osmotic pressure, buffer sub-
stances, colourants, taste correctors and/or flavourings.
If desired, they can also contain one or more additional
active ingredients, e.g. one or more vitamins.
The compounds of formula I and their biocompa-
tible salts can be used for the therapeutic treatment of
the human or animal body and for controlling diseases,
especially parkinsonism, extrapyTamidal disorders in
neuroleptic therapy, depressions and/or psycho~es, and
side-effects in the $reatment of hypertension (e.g. with
~-methyldopa). The compounds can also be used in endo-
crinology and gynaecology, e.g. for the therapeutic
treatment of acromegaly, hypogonadism, secondary amenor-
rhoea, premenstrual syndrome and undesired puerperal
lactation and in general as prolactin inhibitors, and
also for the therapeutic treatment of cerebral disorders
(e.g. migraines), especially in geriatrics in a manner
similar to certain ergot alkaloids.
In these treatments, the substances of the inven-
tion are normally administered analogou~ly to known,
commercially available preparations (e.g. bromocriptine,
dihydroergocornin), preferably in dosages of between
abGut 0.2 and 500 mg, especially of between 0.2 and S0 mg
per dosage unit. The daily dosage is preferably between
about 0.001 and 10 mg/kg of body weight. The low dosages
(about 0.2 to 1 mg per dosage unit; about 0.001 to 0.005
mg/kg of body weight) are particularly suitable for use
2 ~ J? ~ ~f
- 24 -
as anti-migraine preparations; dosages of between 10 and
50 mg per dosage unit are preferred for the other indi-
cation~. However, the partic~lar dose for each indivi-
dual patient depends on a very wide variety of factors,
5 for example the activity of the particular compound used,
age, body weight, general state of health, sex, diet,
time and method of administration, rate of excretion,
drug combination and 6everity of the particular disease
to which the therapy i8 applied. Oral administration is
preferred.
In the following Examples, "working-up in con-
ventional manner" means: Water is added if necessary,
extraction is carried out with methylene chloride, the
prganic phase is separated off, dried over sodium sul-
phate and filtered, the filtrate is evaporated and the
residue is purified by chromatography on silica gel
and/or by crystallization. Temperatures are given in C.
IR = principal bands in the IR spectrum (RBr). MS =
peaks in the mass spectrum. Rf values were obtained by
thin layer chromatography on silica gel.
Example 1
A solution of 26.6 g of 3-(4-chlorobutyl)indol-5-
yl-urea [obtainable by reacting 5-nitroindole with 4-
chlorobutyryl chloride to give 3-(4-chlorobutyryl)-5-
nitroindole, reduction with diborane to 3-(4-chloro-
butyl)-5-nitroindole, hydrogenation to 3-l4-chlorobutyl)-
5-aminoindole and reaction with RCNO] and 16 g of 4-
phenyl-1,2,3,6-ketrahydropyridine in 200 ml of aceto-
nitrile is stirred for 12 hours at 20 and worked up in
conventional manner to give 3-[4-(4-pheny~ 2~3~6
tetrahydropyrid-1-yl)butyl]indol-5-yl-urea dihydrate,
m.p. 158 (decomposition).
The following are obtained analogously from the
appropriate starting materials of formulae II and III:
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)butyl]-5-
nitroindole, m.p. 148-150,
3-[4-(4-p-methoxyphenyl-1,2,3,6-tetrahydropyrid-1-
- 25 - 2 a ~
yl)hutyl]indol-5-yl-urea,
3-[4-(4-(3,4-dimethoxyphenyl)-1,2,3,6-tetrahydropyrid-1-
yl)butyl]indol-5-yl-urea,
3-[4-(4-(3,4-methylenedioxyphenyl)-1,2,3,6-tetrahydro-
pyrid-1-yl)butyl]indol 5-yl-urea,
3-[4-(4-(thien-2-yl)-1,2,3,6-tetrahydrop~rid-1-yl)butyl]-
indol-5-yl-urea,
3-[4-(4-(thien-3-yl)-1,2,3,6-tetrahydropyrid-1-yl)butyl]-
indol-5-yl-urea,
3-[2-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)ethyl]indol-
5-yl-urea,
3~ r 3-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)propyl]indol-
S-yl-urea and
3-~5-(phenyl-1,2,3,6-tetrahydropyrid-1-yl)pentyl]indol-5-
yl-urea.
Example 2
A mixture of 2.46 g of 3-(4-aminobutyl)indol-S-
yl-urea [obtainable from indol-5-yl-urea via 3-(4-chloro-
butyryl)indol-5-yl-urea, 3-(4-chlorobutyl)indol-5-yl-urea
and 3-(4-phthalimidobutyl)indol-5-yl-urea] and 2.15 g of
1,5-dichloro-3-phenylpent-2-ene in 40 ml of acetone and
40 ml of ~ater is boiled for 24 hours and worked up in
conventional manner to give 3-[4-(4-phenyl-1,2,3,6-tetra-
hydropyrid-l-yl)butyl]indol-5-yl-urea ~ihydrate, m.p.
158 (decomposition).
Example 3
A suspension of 40.7 g of 3-[4-(4-phenyl-1,2,3,6-
tetrahydropyrid-l-yl)-4-oxo-2-thiabutyl~-5-nitroindole
[obtainable from 4-(5-nitroindol-3-yl)-3-thiabutyric acid
and 4-phenyl-1,2,3,6-tetrahydropyridine] in 3 1 of
absolute THF is added dropwise, with stirring, to a sus-
pension of 11.7 g of LiAlH4 in 1000 ml of absolute THF and
the mixture is decomposed with water and sodium hydroxide
solution and worked up in conventional manner to give
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)-2- thia-
2Q~
- 26 -
butyl]-5-nitroindole hydrochloride, m.p. 157-158. (The
nitro group is not attacked by the reducing agent~)
Example 4
1 g of NaBH4 in 20 ml of water is added, with
stirring, to a solution of 4.65 g of 1-t4-(5-ureidoindol-
3-yl)butyl]-4-phenylpyridinium bromide ~obtainable from
3-(4-bromobutyl)indol-5-yl-urea and 4-phenylpyridine~ in
50 ml of 1 N NaO~ and the mixture is then stirred for a
further 3 hours at 60. After working-up in conventional
manner, 3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)-
butyl~indol-5~yl-urea dihydrate, m.p. 158 (decomposi-
tion), is obtained.
Example 5
4.68 g of l-benzenesulphonyl-3-[4-(4-phenyl-1,2,
3,6-tetrahydropyrid-1-yl)butyl]indol-5-yl-urea [ob~ain-
ablefroml-benzenesulphonyl-3-(4-chlorobutyl)indol-5-yl-
urea and 4-phenyl-1,2,3,6-tetrahydropyridine] are boiled
with l g of KOH in 7 ml of water and 14 ml of ethanol for
16 hours and the mixture is worked up in conventional
manner to give 3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-
yl)butyl]indol-5-yl-urea dihydrate, m.p. 158 (decompo-
sition).
Example 6
2.76 g of Na are dissolved in 180 ml of ethanol,
21.g g of 1-(2-mercap~oethyl)-4-phenyl-1,2,3,6-tetra-
hydropyridine and 23.2 g of 5-ureidogramine are added,
the mixture is boiled for 16 hours and evaporated and the
residue is worked up in conventional manner to give 3-[4-
(4-phenyl-1,2,3,6-tetrahydropyrid-l-yl~-2-thiabutyl]-
indol-5-yl-urea, m.p. 158 (decomposition).
Example 7
2 ~
- 27 -
A mixture oi 26.9 g of 5-ureidogramine hydro-
chlorid~, 29.8 g of S-[2-(4-phenyl-1,2,3,6-tetrahydro-
pyrid-l-yl)e~hyl]isothiuronium chloride and 200 ml of 1.5
N aqueous sodium hydroxide solution is stirred for 4
hours at 40. After working-up in conventional mann~r,
3-[4-(4-phenyl-1,2,3,6-tetrahydxopyrid-1-yl)-2-thia-
butyl]indol-5-yl-urea, m.p. 158 tdecomposition), is
obtained.
3-[4-~4-Phenyl-1,2,3,6-tetrahydropyrid-1-yl3-
2-thiabutyl]-5~aminoindole, Rf 0.18 in toluene/
methanol/triethylamine (8:2:1), i6 obtained analogou~ly
with 5- aminogramine dihydrochloride.
E~ample 8
4.07 g of 3-[4-t4-hydroxy-4-phenylpiperid-1-yl)-
lS butyl]-5-nitroindole [obtainable by reacting 3-t4-bromo-
butyl~-5-nitroindole with piperid-4-one, followed by re-
action with C6H5Li and hydrolysis] are stirred with 40 ml
of 2 N hydrochloric acid for 2 hours at 50 G and tho mix-
ture is worked up in conventional manner to give 3-t4-(4-
phenyl-l,2,3,6-tetrahydropyrid-1-yl)butyl]-S-nitroindole,
m.p. 148-i50.
Example 9
0.24 g of NaH (80~ suspension in mineral oil) is
added to a solution of 3.46 g of 3-[4-(4-phenyl-1,2,3,6-
tetrahydropyrid-1-yl)butyl]-S-hydroxyindole in 35 ml of
DMF and the mixture is &tirred for 1 hour at 20. A
solution of 0.94 g of chloroacetamide in 10 ml of DMF is
then added, the mixture is heated for 2 hour~ at 50,
with stirring, and evaporated and the residue is worked
up in conventional manner to give 3-14-(4-phenyl-1,2,3,6-
tetrahydropyrid-l-yl)butyl]-S-carbamoylmethoxyindole,
m.p. 94-96. Methanesulphonate, m.p. 193-195~.
The following are obtained analogously:
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)butyl]-5-
2 ~ r
-- 28 --
methylcarbamoylmethoxyindole,
3-[4-(4-phenyl-1,2,3,6-tetrahydrop1yrid-l-yl)butyl]-5-di-
methylcarbamoylmethoxyindole hydrochloride, m.p. 211
(decompo~ition), and
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid~1-yl)butyl]-5-
ethoxycarbonylmethoxyindole, Rf 0.56 (CH2C12/CH30H, 9:1).
Example 10
1.01 g of N-methylmorpholine are added to a solu-
tion of 3.74 g of 3-[4-(4-phenyl-1,2,3 6-tetrahydropyrid-
1-yl)butyl]indole-5-carboxylic acid in 50 ml of DMF. A
solution of 0.61 g of 2-aminoethanol in 5 ml of DMF, 1.35
g of l-hydroxybenz~riazole and a ~olution of 1.92 g of
N-(3-dimethylaminopropyl)-N'-ethylcarbodiLmide hydro-
chloride in 20 ml of DMF are added, with stirring. The
mixture is stirred for 16 hours at 20 and the filtrate
is evaporated. After working-up in conventional manner,
3-[4-(4-phenyl-1,2,3,6~tetrahydropyrid-1-yl)butyl]-
indole-5-carboxylic acid N-(2-hydroxyethyl)amide, m.p.
168-170, is obtained.
The following derivative~ of 3-[4-(4-phenyl-
1,2,3,6-tetrahydropyrid-1-yl)butyl]indole-5-carboxylic
acid are obtained analogously with the appropriate
amines:
N-(3-hydroxypropyl)amide, m.p. 154-158,
N-(4-hydroxybutyl)amide,
N-(1,1-dimethyl-2-hydroxyethyl)amide, IR: 3419, 3265,
2927, 1638, 1620, 1579, 1522, 1471, 1448, 1368, 1308,
1129, 1061, 395, 963, 907, 857, 747, 693j
N-(2-hydroxyethyl)-N-methylamide, m.p. 142-145,
N,N-bis(2-hydroxyethyl)amide, MS: 462, 444, 399, 375,
357, 327, 285, 2S7, 227, 19~, 164, 158, 129, 105, 88, 74,
N-(2-methoxyethyl)amide, m.p. 143-144,
N-(2-acetoxyethyl)amide,
N-(2-benzoyloxyethyl)amide, MS: 399, 341, 241, 213, 173,
158, 122, 105, 91, 77,
N-(methoxycarbonylmethyl)amide, m.p. 145-147,
N-(ethoxycarbonylmethyl)amide, m.p. 169,
2 ~ 3 '-~
- 29 -
N-(carbamoylmethyl)amide hydrate, m.p. lB3-185,
N-(2-~ulphoethyl)amide, m.p. 159-160,
N-(2-dimethylaminoethyl)amide, IR 3417, 3240, 1632,
1578, 1535, 1495, 1466, 1445, 1375, 1304, 1244, 1185,
1128, 1097, 1034, 961, 908, ~17, 747, S93,
anilide,
p-fluoroanilide, m.p. 167-168,
N-benzylamide, m.p. 155-156,
N-benzyl-N-methylamide, IR: 3420, 3238, 3028, 2930, 2713,
1620, 1608, 1579, 1495, 1446, 1401, 1358, 1~50, 1193,
1094, lQ76, 1027, 1001, 817/ 747, 697,
N-(2-morpholinoethyl)amide, m.p. 153-154,
N-[2-(pyrid-2-yl)ethyl]amide, m.p. 148-149,
pyrrolidide dihydrochloride, m.p. 188-190 ~decomposi-
tion),
piperidide, m.p. 94-35,
morpholide dihydrochloride, IR: 3415, 3227, 2925, 2858,
2710, 2588, 1621, 1495, 1440, 13B6, 1362, 1329, 1273,
1248, 1192, 1156, 1113, 1068, 1~23, 942, 920, 840, 819,
749, 606,
4-methylpiperazide, IR: 3419, 3226, 3031, 2933, 2856,
2792, 1625, 1604, 1494, 1451, 1440, 1366, 1294, 1255,
1234, 1172, 1139, 1071, 1050, 1023, 964, 925, ~92, 747,
693,
4-phenylpiperazide, IR: 3412, 3231, 3038, 3002, 2919,
2854, 2765, 1626, 1593, 1565, 1479, 1457, 1434, 1377,
1311, 1281, 1238, 1160, 1132, 1098, 1052, 1014, 980, 945,
864, 813, 772, 747, 694,
4-(pyrimidin-2-yl)piperazide dihydrochloride, m.p. 235-
236 (decomposition),
4-t5-methylthiazol-2-yl)piperazide dihydrochloride, m.p.
211-213 Idecomposition),
4-p-fluorobenzoylpiperazide, m.p. 143-145,
4-ethoxycarbonylpiperazide, m.p. 115-119 (decomposi-
~ion),
4-(carbamoylmethyl)piperazide,
4-(N-isopropylcarbamoylmethyl)piperazide dihydrochloride,
m.p. 156 (decomposition),
4-(N,N-dimethylcarbamoylmethyl)piperazide,
2 ~ 3 ~
- 30 -
4-pyrrolidinocarbonylmethylpiperazide, MS- 553, 457, 441,
394, 357, 327, 296, 227, l9B, 155, 151, 129, 115, 91, 70,
56, 42,
4-dimethylaminopiperidide dihydrochloride dihydrate, de-
composition above 105,
4-p-fluorobenzamidopiperidide, m.p. 110(de~omposition),
4-ethoxycarbonylpiperidide, m.p. 117-119,
4-carbamoylpiperidide, m.p. 93-98,
4-p-chlorophenylpiperidide, IR: 3422, 3236, 3020, 292~,
2854, 1643, 1602, 1493, 1445, 1369, 1320, 1297, 1276,
1231, 1130, 1090, 1033, 1014, 826, 747, 694,
4-piperidinopiperidide dihydrochloride, Rf 0.15 (methyl-
ene chloride/methanol/ethyl acetate 7:2:1),
4-morpholinopiperidide dihydrochloride hydrate, m.p. 90
(decomposition), and
4-phenyl-1,2,3,6-tetrahydropyridide hydrochloride, m.p.
88-89.
The following derivatives of 3-t4-(4-phenyl-
1,2,3,6-tetrahydropyrid-1-yl)butyl~indol-S-oxyacetic acid
are obtained analogously:
N-(ethoxycarbonyl)amide hydrochloride, m.p. 191-193, and
N-~1-ethoxycarbonyl-2-hydroxyethyl)amide hydrochloride,
m.p. 118-120.
- Example 11
HCl is passed into a solutiQn of 3.55 g of 3- [ 4-
(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)butyl]-5-cyano-
indole and l.S g of thioacetamide in 40 ml of DMF until
saturation is reached, and the mixture is boiled for 30
minutes. It is evaporated and the residue is worked up
in conventional manner to give 3-[4-(4-phenyl-1,2,3,6-
tetrahydropyrid-l-yl)butyl]indole-5-thiocarboxamide, de-
compocition at 118.
Example 12
6 ml of 30% H2O2 are added to a boiling solution
of 3.93 g of 3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-
2~ 3
- 3I -
yl)-2-thiabutyl~-5-nitroindole in 50 ml of ethanol and
the mixture is then boiled for 3 hours. After the
addition of a further 4 ml of 30% H2O2, the mixture is
boiled for another 9 hours, cooled and worked up in con-
ventional manner to give 3-[4-(4-phenyl-1,2,3,6-tetra-
hydropyrid~ yl)-2-thiabutyl] 5-nitroindole S-oxlde.
Example 13
9 ml of 30% H2O2 are added to a solution of 3~93
g of 3-~4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)-2-
thiabutyl]-5-nitroindole in 20 ml of acetic acid and the
mixture is boiled for 90 minutes. After working-up in
conventional manner, 3-t4-(4-phenyl-1,2,3,6-tetrahydro-
pyrid-1-yl-)-2-thiabutyl]-5-nitroindole S,S-dioxide, Rf
0.7 ~acetone), is obtained.
Example 14
A mixture of 4.05 g of 3-t4-(4-p-methoxyphenyl-
1,2,3,6-tetrahydropyrid-1-yl)butyl]-5-nitroindole, 3.5 g
of pyridine hydrochloride and 80 ml of pyridine is boiled
for 3 hours. It is cooled and evaporated and the residue
is worked up in conventional manner to give 3-~4-(4-p-
hydroxyphenyl-1,2,3,6-tetrahydropyrid-1-yl)butyl]-5-
nitroindole.
Example 15
9.3 g of SnCl2 are added, with stirring, to a
suspension of 3.75 g of 3-~4-(4-phenyl-1,2,3,6-tetra-
hydropyrid-l-yl)butyl]-5-nitroindole in 45 ml of concen-
trated hydrochloric acid and 30 ml of e$hanol and the
mixture is then boiled for 0.5 hour. It is poured on to
ice and worked up in conventional manner to give 3-[4-(4-
phenyl-1,2,3,6-tetrahydropyrid-l-yl~butyl]-5-Aminoindole,
~.p. 113~.
2 ~
- 32 -
Example 16
A mixture of 2 g of 3--[4-(4-phenyl-1,2,3,6-
tetrahydropyrid-l-yl)-2-thiabutyl]--5 aminoindole, 2mlof
acetyl chloride and 15 ml of triethylamine is stirred for
16 hours a~ 20. After working-up in conventional
manner, 3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)-2-
thiabutyl]-5-acetamidoindole, m.p. 172-173, is obtained.
The following are obtained analogously by acyla-
tion:
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)butyl]-5-
acetamidoindole,
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)butyl~-5-
benzamidoindole,
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl~butyl]-5-
methylsulphonamidoindole,
3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)-2-thia-
butyl]-5-benzamidoindole and
3-t4-(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)-2~thia-
butyl]-5-methylsulphonamidoindole, m.p. 181-183.
Example 17
A mixture of 4.17 g of 3-[4-(4-phenyl-1,2,3,6-
- tetrahydropyrid-1-yl)butyl]indole-5-carboxylicacidN-(2-
hydroxyethyl)amide, 1.41 g of benzoyl chloride and 50 ml
of triethylamine is stirred for 2 hours at 20. After
evaporation and working-up in conventional manner, 3-[4-
(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl~butyl]indole-5-
carboxylic acid N-(2-benzoyloxyethyl)amide is obtained.
Example 18
A mixture of 4.17 g of 3-[4-(4-phenyl-1,2,3,6-
tetrahydropyrid-1-yl)butyl]îndole-5-carboxylicacidN-(2-
hydroxye~hyl)amide, 0.65 g of methyl isocyanate and 40 ml
of pyridine is stirred for 16 hours at 20. After
evaporation and working-up in conventionai manner, 3-[4-
(4-phenyl-1,2,3,6-tetrahydropyrid-1-yl)butyl]indole-5-
2 ~ 1 ~ 3 3 L~
~ 33 -
carboxylic acid N-(2-N-methylcarbamoyloxyethyl)amide,
m.p. 117-118, is obtained.
Example 19
4.32 gof3-[4-(4-phenyl-1,2,3,6-tetrahydropyrid-
1-yl)butyl~-5-ethoxycarbonylmethoxyindole are boiled for
2 hours with 20 ml of water and 100 ml of 2 N ethanolic
KOH and the mixture is worked up in conventional manner
to give 3-[4-(4 phenyl l,2,3,6-tetrahydropyrid-1-yl)-
butyl]indol-5-oxyacetic acid hydrochloride, decomposition
above 175 (sinters at 132).
The following Examples relate to pharmaceutical
preparations containing amines of formula I or their acid
addition salts:
Example A: Tablets
A mixture of 1 kg of 3-[4~(4-phenyl-1,2,3,6-
tetrahydropyrid-1-~l)butyl]indole-5-carboxylicacidN-(2-
hydroxyethyl)amide, 4 kg of lactose/ 1.2 kg of potato
starch, 0.2 kg of talc and 0.1 kg of magnesium stearate
is compressed to tablets in conventional manner so that
each tablet contains 10 mg of active ingredient.
Example B: Coated tablets
Tablets are formed by compression analogously to
Example A and then covered in co~ventional manner with a
coating of sucrose, potato starch, talc, tragacanth and
colourant.
Example C: Capsules
2 kg of3-[4-~4-phenyl-1,2,3,6-tetrahydropyrid-1-
yl)-2-thiabutyl]indol-5-yl-urea are filled into hard
gelatin capsules in conventional manner so that each cap-
sule contains 20 mg of the active ingredient.
2 ~ 3 ~
- 34 -
Example D: Ampoules
A solution of 1 kg of 3-[4 (4-phenyl-1,2,3~6-
tetrahydropyrid-1-yl)butyl]indol-5-yl-urea dihydrate in
60 1 of double-distilled water is filtered under sterile
conditions, filled into ampoules and lyophilized under
sterile conditions and the ampoules are sealed under
~terile conditions. Each ampoule contains 10 mg of
active ingredient.
Tablets, coated tablets, capsules and ampoules
containing one or more of the other active ingredients of
formula I and/or their biocompatible acid addition salts
can be obtained analogously.