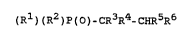Note: Descriptions are shown in the official language in which they were submitted.
2~1277~
HOECHS~ ARTIENGESE~LSCHAFT HOE 89/F 100 Dr. WE/PP
D~rip~ion
Process for ~he preparation of phosphino compound~
The invention relates to a proce&s for the preparation o~
S phosphino compounds of the formula (I)
R3 ~5
~ _ C - H (I)
in which
Rl and R2 independently of one a~other are ~lkyl, alkoxy
or optionally substituted phenyl,
10 R3 and R5 independently of one another are hydrogen,
alkyl, unsubstituted phenyl or phenyl mono- or
multisub titu~d by halogen or mono- or di~ub-
stituted by alkoxy; or are alkoxycarbo~yl,
alkoxycarbonylalkoxy, halogen, cyano, alkoxy,
alkoxyalkoxy, alkylcarbonyl, ~lko~ycarbonyl-
alkyl, carbamoyl, al~ylaminocarbonyl or
dialkylaminocarbonyl,
R4 and R6 independently of one ~nother are h~drogen,
alkyl, un~ub~tituted phenyl or phe~yl mono- or
multi.ubstituted by halogen or mono- or disub-
~tituted by alkoxy; ~r are halo~en, alkoxy
carbonyl, ~lkoxycarbonylalkoxy, cy~no, alkosy,
alkoxyalXoxy, alkylcarbonyl, ~l~oxycarbo~yl-
alkyl, caxbamoyl, al~ylaminocarbonyl or di-
alkyl~minoc~rbonyl; or ~ointly sre a divalent
radical of the formula
-- CO -- ~ -- CO --,
in which
R7 i~ oxygenl a radical of the formula NR*, in
which R* represent~ hydrogen, C~-C6-alkyl,
` :
20~277~
unsubs-ti-tuted phenyl or phenyl mono~ or mul-ti-
substituted by halogen; or is sulfur,
hich cor.prises reacting a compound of the formula (II)
R1-p (II)
in which
R1 and R2 have the meanings defined above and
R3 is alkyl or optionally substituted phenyl,
toge-ther with a compound of the formula (III)
R3 / R5
C = C ~III)
in which R3, R', R5, R6 and R7 have the meanings
defined above,
and
with at least an equimolar amount of a protic organic
substance except carboxylic acids.
The term 'alkyl' denotes, for example, a straight-chain,
branched or cyclic alkyl such as methyl, ethyl, n- and i-
propyl, n-, i-, t- and 2-butyl, pentyl isomers, hexyl
isomers, cyclopentyl, cyclohexyl, heptyl and octyl-
isomers. The term 'alkoxy' denotes an alkyloxy raclical
comprising in the alkyl part the ~eanings cited above as
examples of alkyl. The term 'optionally substituted
phenyl' denotes an unsubstituted phenyl or a phenyl
substituted, for example, by halogen, lower alkoxy or
lower alkyl. The term 'halogen' denotes fluorine, chlor-
ine, bromine ancl/or iodine, preferably chlorine.
,
,,
2~ 2~79
.
- 3 -
Of particular intere~t i~ a proc~ accordin~ to the
invention, in which
R1 and R2 independently of one another are Cl-Ct-alkyl,
phenyl or Cl-C8-alko~y, preferably methyl,
ethyl, phenyl, ~etho~y, ethoxy, propo~y or
bu~oxy, in particular methyl, e~hyl, phenyl,
methoxy or ethoxy,
R3 and R 5 independently of one another are hydrogen,
Cl-C~-al~yl, unsubst~tuted phenyl or phenyl
mono- or multisub~tituted by halogen,o or ~re
C2-C~-alkoxycarbonyl, ~2-co-a~cylcarbonyl,
(Cl-C4-alkyl)~arbonyl-C1-C10 alkyl, halogen,
c:y~no~ C1-CB-a1kOXY~ (C1-C4-a1kOXY)-C1~ a1kOXY~
carbamoyl, N-(Cl C~-alkyl3aminooarbonyl, N,N-
di(Cl-C4-alkyl)aminocarbonyl, 1-(Cl-C~-alko~y)-
l-hydroxymethyl or l,l-bis(C1-C~-alkoxy)~ethyl,
R~ and R6 independently of one another ~re hydrogen,
Cl-C~-alkyl, unsub~titut~d phenyl or phenyl
mono- or multi~ubstituted by halogen; or are
C2-C6-alkoxycarbonyl, C2-CB-alkylcarb~nyl~
( Cl-C4-alkyl ) carbonyl-Cl-C1O-alkyl, halogen,
cyano, Cl-~6-alkoxy, ( Cl-C"~-alkoxy3 -Cl-C4-alkoxy,
carbamoyl, N- ~ Cl-C4-alkyl ) aminocarbo~yl, N, N-
di ( Cl-C4-alkyl ) aminocarbonyl; 1- ( Cl-C4-~lkoxy3 -
l-hydroxymethyl or l,l-bis(C1-C4-alko~r)methyl;
or ~ointly are a divalent radical of the
formula
- CO - R7 - CO -,
in which
R7 i8 oxygen, a radical of the fo~mul~ NR*, ln
whi~h R* represent~ hydrogen, tCl-C6)-alkyl,
un~ub~tituted ph~nyl or phenyl mono- to tri~ub-
3titllted by halogen or i8 ~ulfur.
The process acc:ording to the ~nvention, in which
35 Rl and R2 independently of one ano~her are methyl, ethylt
methoxy, e~hoxy or phenyl,
R3 is hydrogen,
2~127~
",
R~ i~ hydrogen or (Cl-C~-alkoxy~carbonyl,
Rs i~ hydrogen,
R6 i~ hydrogen, halogen, cyano, (Cl-C4~alko~y)~ar-
bo~yl or carbamoyl, preferably (C,-Ca-~lkoxy)-
carbonyl or cyano
is particularly preferred.
The compounds of the fon~ula (I),~re useful intermediate~
in the preparation o~ plant protection ~gents ( see for
example EP-A-30,4~4, US-~-4,399,2B7) and fixe retardants
(Angewand~e Nakromolekulare Chemie 105, pp. 203-215
(1982) and literature cited therein~. It i~ known that a
few representa~ives of the compounds of the formula (I)
are obtainable, for example, by reacting the compounds of
the formula (II) with un~aturated fatty acid$ ~see
Houben-Weyl, ~ethoden der org. Chemie, volume 12tl,
pp. 259-260 (1963) and literature cited therein~. ~ow-
ever, this proce~ gives yield~ of only 10 - 50 % which,
becau~e of the high amounts of efflu~nt ga~e~ ~nd waste
product~ formed, represents a serious drawback both
technically and ecologically. The unreacted phosphorus
component (I) must undergo a c08tly process of di~po6al,
since these compounds repre~ent a fire and toxic hazard
and have an ob~ectionable smell (~ee Houben-~eyl, Method-
en der org. Chemie, Vol. 12~1, p. 14~. In addition,
hecause of ~he po3r yield~, eo~tly purification oper~
ations are needed at the end of the reaction.
In compari60n with the k~own proces~e~, the pre~ent
in~ention relate6 to a highly 6el~ctive, inexpensivs and
~imple pxoce~s which furn~hes the phosphino ~ompound~ of
~he formula (I~ in almo~t quantitative yields a~d in high
puri~y.
The crude products obtained in an almost guantit~tive
yield are usually B0 pure that they can be directly used
for further ch~mical reactions.
For a number of rea~on6 the proce6~ according to the
.
2~)~ 2779
- 5 -
invention i8 to be regarded as surprising. Thu~, for
example, the reaction~ of the phosphorus ~mponents (II)
with the alkenes (IlI) do not ~iV9 ri~e, according to
examples in the literature, to t:he products (I) without
reQorting to a protic organic ~ubstance. Only highly
vi~cous polymeric produ~t~ are obtain~d ~ee Anionische
Polymerisation, B. Vollmert, Grundrlss der Nakromoleku-
laren Chemie, Springer-Verlag 1!962, p. 107 ff ~nd lS9,
~ee Compari~on ~xample I). The low yields of up to a
maxLmum of 50 ~ in the examples d2scribed in the liter-
ature are likewise due ~o competing polymerization
reactions as ~econdary reaction~. The desired reac ions
are in some ca~es ~o ~low that the polymerization becomes
the main reaction. (Houben-Weyl, Methoden der org.
Chemie, vol. 12/1, pp. 259 - 260). Surpri~ingly, the
addition according to the lnvention of protic organic
6ubstance~ succeeds in ~ubstantially suppres~ing the
polymerization and in forming the 1s1 adducts of the
formula (I) in almost qua~titative yields.
~he process according to the invention i~ ~urthermore to
be regarded as surprising ina~much as it i8 precisely by
the addition of protic organic substances to the reac-
tions described in the literature in which phosphorus
compounds of the formula (II) are react~d with unsaturat-
ed fatty acids that yields of le~s than 30 ~ ~nd henceeven worse yield~ of the desired 1:1 addu~tE are obtained
~ee Compari~on ~xample II).
The preferred protic organic ~ubstance~ are alcohol~, in
particular Cl-C6-alcohol~, for example, methanol, ethanol,
propanol, i~opropanol, n~ , t- and 2-butanol, further
polyhydric alcohol~ ~uch as ethanediol and glycerol,
Cl-C6-mercaptan~, for example ~ethyl~ercaptan, e~hylmer
captan, propylmercaptan and 1,2 ethanedithiol, amine~, in
parti~ular mono- or di(Cl-C6-alkyl)amines, for e~ample
methyl~mine, ethylamine, dLmethylamine and propylamine,
phenols, thiophenol~, anilines and ~imilar compoundR.
Because the above sub~tances can be generally regarded as
20:L~7~9
-- 6
~'derivatives~' of water on account of their protic proper-
ties, the process according to the invention cannot be
carried out in the presence of water, in contrast -to the
organic protic compounds described, since water rapidly
hydrolyzes the phosphorus components of the formula (II)
(Sander, Chem. Ber. 93 (1960) ].223). Since it is quite
impossible to ob~ain products of the formula (I) with
water as the protic substance because of hydrolytic
reactions taking place (see also Comparison Example III),
it was unexpected for the protic organic substances to be
used according to the invention to be suitable for
achieving high selectivity and yields.
The protic organic substances R-H employed in the process
according to the invention in at least equimolar amounts
serve as reactants and, if appropriate, as solvents, and
are converted, depending on the course of the reaction, at
least in part, for example, to ethers, thioethers,
amines, phene-toles, thiophenetoles or substituted ani-
lines of the formula R-R3, Ra having the meaning defined
above. The products R-R8 are preferably removed from the
reaction mixture during or at the end of the reaction,
for example by distillation. ~xcess reactant or solvent
R-H is preferably removed at the end of the reaction by
vacuum distillation.
Carboxylic acids are likewise excluded from the process
according to the invention. When in the process according
to the invention organic acids, for example formic acid
or acetic acid, are used as protic substances, virtually
no products of the formula (I) are obtained (see
Comparison Example IV). It follows that the protic
organic substance must not possess too acidic character-
istics. Whether a substance is suitable in the sense of
the invention can be readily established in a preliminary
experiment.
The procedure of the process according to the invention
is for example such that the compounds of the
2~277~
- 7 -
formula (II) are dis~olved in the protic organic ~ub-
stance and ~he alkenes (III~ are added ~o the reaction
mix~ure at temperature~ be~ween -20C and 150C, prefer-
ably be~ween 0 and 100C. I~ also poe~ible to add the
components (II) and (III) to the protic orga~ic substance
at the ~ame time.
It is equally pos~ible to sta~: with a ~i~ture of the
alkene componen~ ( III) and the protic organic substance
~nd add the phosphorus comp~nent (II) to ~h~a ~olutio~.
The protic organic ~ubstances ~re u~ed i~ at least
equimolar ~mount6. In amount~ greater than eguimolar they
can be additionally employed in the sen~e of an or~anic
solvent.
The proces~ may be carried out without solYent or with
excess protic organic substance ~ 801vent and/or in the
presence of ~u~tomary organic ~olvent~ whieh are inert
under the reaction conditions. ~xample~ of the last-named
~olvents are 601vents ~uch as op~ionally halogena~ed,
aliphatic, cycloaliphatic, aromatic or araliphatic
hydrocarbons, aliphati~ or cycloaliphatic ether~, for
example poly~lycol dialkyl ethers, as well as ketones and
esters. It iB expedient to perform the proce~s in an
inert gas ~tmosphers, for example under n~trog~n, in
order to prevent oxygen inter~ering with the reaction.
The process according to ~he invention may be ~ontinuous
or di~continuous.
The process according ~o the invention i5 elucidated in
greater detail by the examples below.
, : ,.
:, , ' ' ' . ~ , :
.
2 ~ 7 ~ ~
EXAMPLE 1:
Methyl 3-(methoxy-methylphos~hin~l)propionate
80 g of methanol are mixed at room temperature under
nitrogen with 216 g of dimethyl me-thanephosphonate and
the mixture is treated dropwise at room temperature with
172 g of methyl acrylate, the temperature rising to 70C.
A-t the end of the dropwise addition the mixture is
further stirred for 1 hour, at the end of which period
about 90 g of dimethyl ether have separated in a fitted
cooling trap. Excess methanol used as solvent is then
removed by vacuum distillation. The crude product is
obtained in a yield of 365 g with a 96.4 % purity, which
represents a theoretical yield of 97.7 %. The boiling
point of a distilled sample is 96C at 0.027 mbar. The
lH-NMR spectrum and the CHP analysis of the product
correspond to those of a comparison sample, synthesized
by an independent route.
EXAMPLE 2:
Ethyl 3-(methoxy-methylphosphinyl)propionate
A flask filled with nitrogen and fitted with a cooling
trap is charged with 216 g of dimethyl methanephosphon-
ate; the contents are heated to 60C and a mixture of
200 g of ethyl acrylate and lO0 g of methanol is added
dropwise in the course of 1 hour, the temperature rising
to 65C. The reaction mixture is further stirred for l
hour, during which period a total of 91 g of dimethyl
ether are collected in the cooling trap. Excess methanol
is removed by vacuum distillation. The crude product is
obtained in a yield of 362 g with a 97.3 % purity, which
corresponds to a theoretical yield of 97.9 %. The boiling
point of a sample is 100 - 102C at 0.013 mbar.
,
20~L2t7r~
- 9
EXAMPLE 3:
Methyl 3-(methoxy-methYlPhosPhin~l)propiona-te
A mixture of 86 g of methyl acrylate and 80 g of ethanol
is added dropwise in the course of 30 minutes at 60C
under nitrogen to a solution o~ 108 g of dimethyl meth
anephosphonate. The reaction mixture is then stirred at
60C for one hour. A total of 55 g of methyl ethyl ether
are collected in the cooling trap. Excess e-thanol is -then
removed by vacuum distillation. The crude product is
obtained in a yield of 184 g with a 95.4 ~ purity, which
corresponds to a theoretical yield of 97.5 %.
EXAMPLE 4:
3-(Methoxy-ethylphospinyl)prooionamide
71 g of acrylamide and 120 g of n-propylamine are mixed
at room temperature under nitrogen and the mixture is
heated to 50C. 122 g of dimethyl ethanephosphonate are
added dropwise to the reaction solution in the course of
30 minutes. The reaction mixture is then stirred for 5
hours at reflux temperature and the solven-t (low-boiling
solvent such as excess propylamine ~ N-methyl-N-propyl-
amine) is removed by vacuum distillation. The crude
product is obtained in a yield of 175 g with a 94.4 %
purity, which corresponds to a theoretical yield of
92.3 %.
The lH-NMR spectrum and the CHP analysis of the product
are in agreement with the corresponding da-ta of a com-
parison sample, synthesized by an independent route.
EXAMPLE 5:
3-(Ethoxy-propylphosphinyl)propionitrile
53 g of acrylonitrile are added dropwise under nitrogen
2 7 7 9
-- 10 --
at a reaction temperature of 70C to a mixture of 160 g
of die-thyl propanephosphonate in 100 g of n-propylmer-
captan in -the course of 2 hours. Af-ter a further 3 hours
the solvent (low-boilin~ solvent such as C3H7SC2H5 and
excess C3H7SH) is removed by vacuum distillation; the
crude product is obtained in a yield of 173 g with a
95.4 % purity, which corresponds to a theoretical yield
of 94.4 %. The boiling point of the product is 117 -
118C at 0.027 mbar.
EXAMPLE 6:
Ethyl 3-(ethoxycarbonyl)-3-(e-thoxy-ethylphosphin~l)-
propionate
172 g of diethyl fumarate are slowly added at 65 - 70C
under nitrogen to a mixture of 140 g of diethyl ethane-
phosphonate in 46 g of ethanol. After a fur-ther 3 hours
the solvent (excess ethanol and diethyl ether) is removed
by vacuum distillation; the crude product is obtained in
a yield of 285 g with a 95.7 % purity, which corresponds
to a theoretical yield of 90.9 ~.
The lH-NMR spectrum and the CHP analysis are in agreement
with the corresponding data of a comparison sample,
synthesized by an independent route.
EXAMPLE 7:
Methyl 3-(dimethoxyphosphinyl)propionate
86 g of methylacryla-te and 100 g of isobutanol are mixed
under nitrogen at room temperature and heated to 80C,
124 g of trimethylphosphite are added dropwise to -the
reaction solution in the course of 1 hour. The reaction
mixture is stirred for 5 hours at reflux temperature and
the solvent (remainder of isobutanol and isobutyl methyl
ether) is removed by vacuum distillation. The crude
product is obtained in a yield of 173 g with a 94.1 %
~0~277~
- 11
purity, which corre~pond~ to a theoretical yield of
83.0 ~. The bo~ling point i~ 104 - 106C at 0.93 mbar.
E~AMPL~ 8:
3-(DLmethylphosphinyl)propionitrile
135 g of isobutyl dimethylpho~p]hinate ~re ~lo~ly added
under nitrogen at 20C to a mixture of 53 g of ~rylo-
nitrile and 32 g of me~hanol, the temperature rising
sl~wly to about S0C. The rea~tion mixture is heated for
about 3 hours and the ~olvent (remainder of methanol and
i~obutyl methyl ether~ i8 remo~ed by diatillation. The
crude product i8 obtained in a yield of 125 g with a
95.3 % purity, which corresponds to a theoretical yield
of 30.9 %. The boiling point of a distilled sample
98 -100C at 0.013 mbar.
EXAMP~E 9:
Methyl 3-~dimethylphosphinyl)-3-metho~ycarbonyl)
propionate
135 g of isobutyl dLmethylpho~phinate and 144 g of
dLmethyl maleate are added under nitrogen simult~neously
dropwise from two ~eparate dkoppin~ funnels at about 70DC
to a solution of 67 g of ethylene glycol. After a further
hour the reaction mixturQ i8 di~tilled under reduced
pre3~ure, i.e. volatile constituent~ Are ~arefully
removed by vacuum distillat~on. The crude product i8
ob~ained in a yield of 215 g with a 95.2 ~ purity, which
corresponds to a theoreti~al yield of 91.8 %.
~he lH-N~R 6pectrum and the CHP analy~is correspond to a
compari on ~ample synthecized by an independent route.
The boiling point of the product i8 128 - 130C at
0.013 mbar.
' ' ,
,' ' '~
.
~012~79
- 12 -
EXAMPLE 10:
Methyl ~-(pnenyl-metnox~phosphlnyl)-2-chloroproplona~e
138 g of dimethyl phenylphosphonate are slowly added at
20C under nitrogen to a solution of 120.5 g of methyl 2-
chloroacryla-te and 94 g of phenol. After a reaction time
of 3 hours at 70C phenetole is removed by vacuum distil-
lation. The residue represents 267 g of crude produc-t
with a 93.0 ~ purity, which corresponds to a theoretical
yield of 90.1 %.
The 1H-NMR spectrum and the CHP analysis of the product
correspond to those of a comparison sample, synthesized
by an independent route.
COMPARISON EXAMPLE I: (without protic organic substance)
86 g of methyl acrylate are added to 108 g of dimethyl
methanephosphonate at 70C and the reaction mixture is
heated at 70C for a further 4 hours. The reaction
mixture is worked up by vacuum distillation, yielding
194 g of a highly viscous oil which according to the
lH-NMR and 31P-NMR spectra does not contain the desired
methyl 3-(dimethylphosphinyl)propionate. If the reaction
mixture is worked up by distillation, no distillable
product is obtained, which points to polymerization
occurring in the experiment.
COMPARISON EXAMPLE II: in accordance with examples from
the literature (Houben-Weyl, Methoden der org. Chemie,
volume 12/1, pp. 259-260 and literature cited therein)
108 g of dimethyl methanephosphonate are added dropwise
at 70C to a solution of 86 g of methacrylic acid and
32 g of methanol in the course of 30 minutes. After a
reaction time of 3 hours the reaction mixture is worked
up by vacuum distillation.
. ' ~ '
2~27~9
- 13
59 g of 3-(dimethylpho~phinyl~-2~methylpropionic ~id axe
obtained with a 91.1 % purity, which corresponds to a
theoretical yield of 29.5 %.
COMPARISON ~XAMPhE IIIs (in the presence of water as
protic sub~tance)
108 g of dLmethyl ~ethanephosphonate are added dro~ e
at an lnitial t~mpera~ure of 25C ~o a ~ollltion o~ 86
of methylacrylate and 20 g of wa1:er i~ the course o~ 30
minutes. Th~ temperature ri~e~ to 65C during the addl-
tion. The reac~ion mixture i~ then heated ~or 3 hours at
70C.
After working up of the reaction mixture by vacuum
distillation, no methyl 3-(dimethylphosphinyl)propionate
is ob~ained. On the other hand the hydroly~i~ product
methyl methanephosphonate~ which could be i~ola~ed in an
amount of 91 g by distillation, i~ obtained in a yield of
91.4 % with a 94.4 % purity. ~he boiling poin~ of the
di6tilled ~ample is 55 58C at 10 - 15 torr. ~he ~ ~MR
~pectrum and the CHP analysi~ corre~pond to the compound
obtained from a compari~on sample, ~ynthe~ized by an
independent route.
COMPARISON ~AMPLE IY: (in the presence of formic ~cid as
protic polar substance)
108 g of dLmethyl methsnephosphonat~ are ~dded dropwise
at 70C to a solution o 86 g of ~ethylacryl~te and 50 g
of formic acid ln the course of 2 hour~ And the reac~
mixture i~ heated ~t 70C ~or 2 hours. After ~orking up
the xeaction mi~tur~ by vacuum distillation, 123 g of a
crude produc~ are obtained ~hich fro~ it~ lH-~R and
31P-NMR spectra and gas chromatographic a~alysis cont~ins
none of the desired product. The reaction mixture i~
composed of several ~ubstan es, the bulk of them ~eing
acid compoundR of pho~phoru~ i~ the oxidation ~tage V.
.