Note: Descriptions are shown in the official language in which they were submitted.
2~ 7~
HOECHST AKTIENGESELLSCH~FT HOE 89/F 138 Dr.D/fe
Description
Substituted biphenylcarboxylic acids, a process for their
preparation and novel intermediates
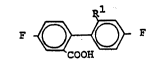
The invention relates to substituted biphenylcarboxylic
acid6 of tho formula I
F ~ F
COOH
in which R1 is hydrogen or methoxy, and to a process for
I their preparation.
The substituted biphenylcarboxylic acids of the formula
I can be converted by cyclization into the corresponding
2,7-difluorofluorenones of the formula IX
IX
F O F
2,7-Difluorospiro~9H-fluorenyl-9,4~-imidazolidine]-2',5'-
~ 15 dione (also called AL 1576, HOE 843 and imirestate), and
,~ 2,7-difluoro-4-methoxyspiro[9H-fluorenyl-9,4'-imidazoli-
dine]-2~,5~-dione, which can used a~ pharmaceuticals for
the treatment of the late effects of diabetes (cf. US
~atent 4,438,272), can be prepared from these in a single
reaction step.
The 2,7-difluorofluorenone, which can easily be prepared
from 4,4'-difluorobiphenyl-2-carboxylic acid, can be
prepared from fluorene in accordance with the prior art
in a 9-stage synthesis, in particular by nitration in the
2-position, reduction to the amine, diazotization and
~' Schiemann reaction to give 2-fluorofluorenone, then ~-
nitration in the 7-position and repetition of the ~aid
reaction sequence to give 2,7-difluorofluorene ~nd final
,,~
~ .. . .
~.~' '' ' '' ,
2- 2~ 8
oxidation with sodium dichromate to give 2,7-difluoro-
9-fluorenone IX (cf., for example, Chem. Ind. 1961, 179,
J. Ned. Chem. ~, 491 (1965), Isr. J. Chem. 1,1 (1963) and
Chem. Ber. 112, 3490 (1979)).
S Thi6 preparation process has many ~tages and the total
yield is 10%, partly on account of isomer separations
necessary in the nitro compound~ stage.
A process for the preparation of 2,7-difluoro-9-fluore-
none described in ~S Patent 4,438,272 starts from 2,7-
diaminofluorene, which is converted into the correspond-
$ng salt of fluoroboric ac~d by treatment with fluoro-
boric acid. The bis-diazonium salt i8 obtained by ~ubse-
quent diazotization, from which 2,7-difluorofluorine is
liberated by warming in xylene. Oxidation to 2,7-
difluoro-9-fluorenone is carried out with RMnO~ in
pyridine.
The 2,7-diaminofluorene used as a starting material in
this synthesis, however, can only be prep~red with
difficulty and in unsatisfactory yields by nitration of
fluorene to 2,7-dinitrofluorene and subsequent reduction,
sinceiSomer mixtures are formed in nitration which have to
be separated ~nd sLnce the 2,7-diaminofluorene is very
sensitive to light and oxidation. Even in this process,
the total yield~ based on fluorene are only about 10%.
A partieul~rly increasingly aerious disadvantage of the
known syntheses of the fluorenones of the formula IX lies
in the fact that amino and nitrofluorene deriv~tives have
carcinogenic properties (cf. J. Natl. Cancer In~t. 10,
- 1201 ~1950), Cancer Res. ~, 1002 (1962), Brit. J. Cancer
4, 235 (1950), Brit. J. Exptl. Pnth. 25, 1 (1944) and
Progressus Med. ~, 79 (1940)).
2,7-Difluoro-4-methoxy-9-fluorenone is prepared ~ia 4 or
5 further reaction steps from 2,7-difluoro-9-fluorenone.
I .:
- 3 ~
The ob~ect of the present invention is to make available
a process for the preparation of 2,7-difluoro-9-fluore-
nones of the formula IX which i8 economical and can be
carried out on a large ~cale, nnd in which the disad-
vantages which occur in the preparation according to theprior art are avoided. This means that the total yield is
high, the product i8 obtained in high purity and that no
reputedly carcinogenic intermediates occur.
Thi6 ob~ect is achieved according to the invention in
that novel biphenylcarboxylic acids of the formula I are
made available, which are converted by cyclization into
the corresponding fluorenone~ in a simple manner, the
latter being obtained in high yields. The disadvantages
described do not occur in the preparation of the bi-
phenylcarboxylic acids of the formula I according to theinvention and in their further processing to give the
fluorenones IX.
The invention therefore relates to the biphenylcarboxylic
acids of the formula I and to a process for their pre-
paration.
The process for the preparation of substituted biphenyl-
carboxylic acids of the formula I comprises
a) reacting a 2-methoxybenzoic acid of the formula II
OCH3
~ CO2H II
or a reactive derivative of this acid with an
ethanolamine of the formula III
~:J'' ' ~ . :
~; . . ' ,
R2 R3
H ~ ~ OH III
~2 R3
in which RZ and R3 are hydrogen or ( Cl-~3 )-alkyl~
preferably methyl~ but where R2 and ~3 are not
simultaneously alkyl, to give an amide of the
genersl formula IV
OCH3 R2 R3
--C- NH - C - C - OH
~ o 12 13 IV
.. . .
in which R2 and R3 have the meanings indicated for
formula III, then cyclizing the amide of the formula -
IV to a dihydrooxazole of the formula V
OCH R2
lD ~ N ~R3 RFj23
in which R2 and R3 have the meanings indicated for
formula III, then reacting the dihydrooxazole of the
formula V with an organometallic reagent of the
formula VI :
Rl ~
F ~ ~
in which Rl has the meaning indicated for formula I
and ~ i8 lithium or Zn-Hal or, preferably, Mg-Hal
and Hal i~ chlorine, brsmine or iodine, to give a
biphenyl derivative of the general formula VII
-- 5 --
~ 7
Rl
F ~ F
~ VII
N O
R2 ~ R3
in which R1 h~s the meaning indicated for formul~ I
and R2 and R3 have the meaning~ indicated for formuls
- III, and hydrolyzing the biphenyl derivative of the
formula VII obt~ined, optionally after converQion
into the corre~ponding alkylated compound of the
formula VIII
F ~ F
¦ VIII
~ R3 _
in which Rl ha~ the meaning indicated for formula I
and R2 and R3 have the meanings indic~ted for formula
III, R4 is (Cl-C3)-alkyl, preferably methyl, and X~ ~8
chloride, bromide, iodide, methylsulfate or othyl-
~ulfate, to the carboxylic acid of the formula I
F ~ F
COOH
in which R1 ha~ the meaning indicated for formula I.
The biphenylcarboxylic acids of the formula I can then be
cyclized directly, or via the corre~ponding acid chloride
or the amide to give the ~ubstituted fluorenone of the
%~ i7~3
formula IX in a manner known per ~e.
The process according to the invention ~as the advantage
that it starts from appropriately 6ubstituted starting
materials and leads in a specific manner to pure bi-
phenylcarboxylic acids of the formula I, in which thepositions of the substituents are unequivocally fixed by
means of the synthesi~ omer separations which ~re
complicated and high in los~es are not nece6sary and the
total yield (about 65%) i~ clearly higher than in the
known processes (about 10%). Only two isolated inter-
mediates (compounds V and VII) have to be passed through,
for example, for the ynthe~is of 4,4'-difluorobiphenyl-
2-carboxylic acid starting from 5-fluoro-2-methoxybenzoic
acid by the process accordin~ to the invention.
The 4,4'-difluorobiphenylcarboxylic acids of the formula
I are obtained in very pure form by the process according
to the invention. This also applies to the fluorenones
which can be prepared therefrom.
It is particularly important that the known carcinogenic
nitro- and aminofluorine derivatives are not passed
through.
In detail, the preparation of the benzamide IV from the
benzoic acid II using the amino alcohol III i~ carried
out in a manner known per ~e by the process according to
the invent$on, for example, the reaction of a (C1-C~)-
alkyl ester of II with the amino alcohol III can be
carried out by heating the components in the presence or
absence of a ~olvent. Acids of the formula II can also
be converted by reaction of the acid II with an acid
chloride, such as ethyl chlorofo~mate or toluenesulfonyl
chloride, in the pre~ence of a base such as, for example,
triethylamine, into a mixed anhydride which then reacts
with an amine of the formula III to give the smides IV.
In a preferred embodiment of the process according to the
invention, the acid II is converted, for example with PC15
~ :J.'` . . ' : ` .- . . ,, , . :
"' : .
- 7 - 2~.55~
or, preferably, thionyl chloride, into the corresponding
acid chloride, which yields the corre~ponding ~mlde IV in
nearly quan~itative yield with an amine III.
The cyclization of amides of the formula IV to the
dihydrooxazoles V can be carried out in a manner known
per se using dehydrating agents, such as concentrated
~ulfuric acid, polyphosphoric acid or phosphorus pen-
toxide. Cyclization with thionyl chloride in an inert
solvent such as, for example, diethyl ether, methylene
chloride or toluene is preferred. It can be carried out
at temperatures between -10 and 50~C, preferably at 0 to
- 30C. The free dihydrooxazole derivative V i~ prepared
from the initially resulting hydrochloride using base~
such as, for example, sodium hydroxide solution.
In a particularly preferred embodiment of the process
according to the invention, the dihydrooxazoles V are
prepared from the benzoic acid II without isolating the
amides of the formula IV. In detail, the benzoic acid II
is in this case heated with a small excess of thionyl
chloride in the absence or presence of an inert solvent
such as me~hylene chloride or toluene and the solution of
the resulting acid chloride iB added dropwi~e to the
amino alcohol III. As the amino alcohol alio function6 as
ian wid scavenger (accep~or), it is emploved in twice the m~lar
amount. An alternative to this i8 the use of one mole
each of amino alcohol III and of an inert base such a8
triethylamine. Ring closure to give the dihydrooxazole V
takes place as a result of ~ubsequent addition of the
molar amount of thionyl chloride to the reaction mixture.
After rendering the reaction mixture alkaline with sodium
hydroxide solution and evaporating the organic phase, the
dihydrooxazole V is obtained in nearly quantitative
yield. The product can be purified by di~till~tion.
Surprisingly, the reaction of the dihydrooxazole V with
an organometallic reagent VI proceeds in a si~ple manner
with the substitution of the methoxy group, biphenyl
. ~ .
~ , ~ . ' .' .~
!..-.
- 8 - ZQ~S~8
derivatives of the formula VII being obtained. Signifi-
cant side rea~tions as a re~ult of nucleophilic exchange,
for example of a fluorene atom in the compounds V or VII,
do not occur. Solvents which ~an be usedfor thi~ reaction
are ethers, ~uch as diethyl ether, methyl t-butyl ether
or dibutyl ether; tetrahydrofuran iB preferred. With
organomagne6ium or organozinc compounds, the reaction
takes place at temperatures between 0C and the boiling
point of the solvent, with 4-fluorophenyllithium the
reaction mixture i8 allowed to react at between -60-C and
room temperature. The organometallic reagents VI are
preferably employed in excess to attain a satisfactory
yield, in particular 1.2 to 3.0 moles, preferably 2-3
moles, per mole of dihydrooxazole V. The reaction can be
carried out in the presence or absence of 8 catalytic
amount of palladium compounds such as, for example,
Pd(PPh3)4-
The formation of the biphenyl derivatives VII from the
dihydrooxazoles V and the Grignard reagent ~I (M ~ MgBr)
is preferred. In thi~ case, the Grignard reagent can be
added to the dihydrooxazole or the dihydrooxazole V to
the Grignard reagent.
In a particularly preferred embodiment of the process
according to the invention, for example in the pre-
paration of 4,4'-difluorobiphenyl-2-carboxylic acid, the
magnesium i~ initially introduced with the total amount
of THF, and Vitride (NaAlHz (OCH2CH20CH3)2, 70% strength in
toluene) is added until the evolution of hydrogen iB
complete. The 4-bromofluorobenzene i8 then allowed to run
into the boiling T~, the Grignard reaction starting
immediately. After completion of the formation of the d~
Grignard reagent, the dihydrooxazole V is added with, or
preferably without, solvent. The reaction is complete
after a short time (about 10 minute~). The mixture i~
hydrolyzed with water or saturated ammonium chloride
solution and ad~usted to pH 4-7 with hydrochloric acid in
order to achieve a good phase ~eparation. The product can
~-.. ~. . , . .: , . . . . .... .. ..
2~
- 9 -
be purified by crystallization and/or distillation.
The hydroly~is of the dihydro-1,3-oxazole VII to the
biphenylcarboxylic acid I i6 carried out by hydroly~is
with aqueous acids, for example ~y heating with hydro-
chloric acid, ~ulfuric acid or methanesulfonic acid.
In a preferred variant of the process according to the
invention, compounds of the general formula VII are
reacted with alkylating agents such as, for example,
(Ca-C3)-alkyl halides, ( C~-C3 )alkyl methanesulfonates,
(Cl-C3)-alkyl toluenesulfonates or di(Cl-C3)alkyl sulfates
to give compounds of the general formula VIII. Dimethyl
sulfate, methyl iodide, methyl bromide and methyl
chloride in solvents such a~ toluene, acetone, 2-butanone
or low molecular weight alcohols are particularly prefer-
red as alkylating agents. The alkylation reaction can becarried out in the presence or, preferably, in the
absence of a base such as ~odium hydroxide solution,
potas~ium carbonate or triethylamine, if appropriate ~lso
in the presence of phase transfer catalysts such as
tetrabutylammonium sulfate. The resulting dihydro-1,3-
oxazolium salt VIII is then hydrolyzed to the biphenyl-
carboxylic acid I. This can be carried out by heating
with aqueous alkali metal hydroxide solutions, such as
sodium hydroxide solution or potassium hydroxide
solution, preferably in the sen~e of a one-pot reaction,
without isolating the salt VIII.
The ring closure of the carboxylic acid I to the fluore-
none IX is carried out in a manner known per ~e, for
example with dehydrating agent such as, for ex~mple,
polyphosphoric acid, at temperatures between about 80 nnd
120C
The compounds of the formulae V, VII and VIII are novel
and repre~ent useful intermediates, for example for the
preparation of biphenylcarboxylic acids of the formula I.
The invention therefore also relates to these
!: .
; ; . , ..
_ 10 -
intermediatec and to processes for their preparation.
Example I
4,4'-Difluorobiphenyl~2-carbo~ylic acid
1) 5-Fluoro-2-metho~ybenzoic ~cid-2-(1-hydrosy-2-
methyl)propyl~ide (IV)
23.0 g (0.135 mol) of 5-fluoro-2-~ethoxybenzoic acid are
dissolved in 50 ml of methylene chloride. 17.6 ml
(O.2 mol) of thionyl chloride and 2 drops of DNF are
added at room temperature and the mixture i8 BlOWly
- 10 heated to boiling. After reaction is complete (th~n layer
checking by addition of methanol to a sample of the
reaction mixture), the mixture is evaporated in vacuo and
the excess of thionyl chloride is removed in vacuo after
adding further methylene chloride twice. The residue
(28.7 g) is di~solved in 100 ml of methylene chloride and
the solution obtained iB added dropwise at 0-5-C to ~ -
solution of 24.06 g (0.27 mol) of 2-amino-2-methyl-1-
propanol in 50 ml of me~hylene chloride. The mixture is
subsequently stirred for 1 hour without ice-coolinq ~nd
washed twice with 100 ml of portion~ of water, then with
50 ml of dil. HCl, dried over anhydrous sodium ~ulfate
and concentrated in vacuo. The crystalline residue
(30.5 g, 93.5% of theory) is sufficiently pure for the
further reactions.
m.p. 97 - 98C (from isopropanol)
'H-NMR: ~ = 4.8 ppm (t, lH), 3.95 ppm ( 8, 3H),
3.65 ppm (d,2H), 1.4 ppm ( 8, 6H)
2) 2-(5-Fluoro-2-~etho~yphenyl)-4,4-dimethyl-4,5-
dihydro-1,3-o~azole (V)
a) 20 g (0.083 mol) of N-2-(1-hydroxy-2-methyl)propyl 5-
fluoro-2-methoxybenzamide are added in portions at 20-
25C to 14.4 ml (0.17 mol) of thionyl chloride, the
reaction mixture having to be cooled periodically with
:-
.. ! , , ~ , . , , :
t . ''
~. ':' , . . . .
5~3
-- 11 --
ice water. The mixture i8 sub~equently stirred for onehour at room temperature, then 100 ml portion~ of ether
are added twice, stirred and in each case the ether pha~e
is then decanted off. The ~emi-crystalline residue i~
taken up in 100 ml of methylene chloride. The ~olution i~
added dropwise with ice cooling at about 15-C to 150 ml
of half-concentrated sodium hydroxide solution. At the
end of the dropwise addition, the pH of the agueous phhse
must be above 10. The aqueous phase is ~eparated off and
again extracted with methylene chloride. The combined
organic phaces are washed with saturated sodium chloride
solution, dried over anhydrous sodium sulfate and evapor-
ated in vacuo. 18.7 g of an oil which gradually crystal-
lizes are obtained. After distillation, the product melts
lS at 35 - 36C. b.p. 0.5 98C
Yield: Crude product 93 - 100~, after distillation
85 - 95%.
H-NMR: ~ = 6.6 - 7.6 ppm, (m, 3H), 4.05 ppm t~, 2H)
3.8 ppm (s, 3H), 1.35 ppm (8, 6H)
b) (One-pot reaction) `
230 g of 5-fluoro-2-methoxybenzoic acid are dissolved in
100 ml of methylene chloride and 1 ml of DMF. lOB ml of
thionyl chloride are added to this. The mixture iB heated
under reflux until the formation of acid chloride is
complete (about 1 hour). A solution of 293.5 ml of 2-
amino-2-methyl-1-propanol in 200 ml of methylene chloride
is then allowed to run into the clear reaction solution
at 10C. After ~ddition is complete, the mixture is sub-
seguently stirred at room temperature for 30 minutes, and
then 98 ml of thionyl chloride are added dropwise at
20C. The mixture is subseguently stirred for 15 minutes
and about 250 ml of conc. sodium hydroxide solution are
added dropwise until a pH of ~10 is achieved. The organic
phase iB washed once with dilute ~odium hydroxide solu-
tion, dried over sodium ~ulfate and distilled in vacuo.
Yield 276 g (92.9~).
.. . . .
.
, ~ . , .
:, - . , ~ . : .
- 12 - ~ 7~
3) 2-(4,4'-Difluorobiphenyl-2-yl)-4,4-dimethyl-4,5-
dihydro-l,3-osazole YII
1.18 kg of magnesium c~ips are initially introduced in
25 1 of THF and the mixture i8 heated to 50-60C. Vitride
solution in toluene (70% strength) is added until the
evolution of hydrogen iB complete (about 150 ml). The
mixture is heated under reflux and 500 g of 4-bromo-
fluorobenzene are added with ~igorous stirring. The
exothermic reaction ~tarts almo~t in6tantansously. A
further 7.99 kg of 4-bromofluorobenzene are allowed to
run in in such a way that the mixture always boils under
reflux. After completion of the addition, the mixture i~
heated for a further 1 hour. 3.6 k~ of 2-(5-fluoro-2-
methoxyphenyl)-4,4-dimethyl-4,5-dihydro-1,3-oxazole in 5
1 of dry THF are then metered in at reflux temperature
and the mixture i8 heated for a further 15 minutes. The
mixture is c~oled to 10C. 20 1 of saturated nmmonium
chloride solution are metered in at 10-20C with good
cooling and the pH is ad~usted to 6 using about 3.5 1 of
conc. hydrochloric acid. The precipitated salts are
dissolved with water and the phases are separated. The
organic phase is washed with 9 1 of saturated sodium
chloride solution, ~tirred with 200 g of active carbon,
dried with sodium sulfate and filtered off with ~uction.
After evaporating the solvent and drying in vacuo, 4.5 kg
of yellow crystalline product are obtained, which is
sufficiently pure for the further reaction~ (97% pure
after GC analysis, yield 94.2%). It can be purified by
crystallization from isopropanol or diisopropyl ether and
distillation of the mother liquor in a thin film evapora-
tor.
b.p. 126-130C (0.1 mm)
m.p. 83 - 84C
lH-NMRs ~ ~ 6.4 - 7.5 ppm (m~ 7H), 3.85 ppm (8, 2H),
1.28 ppm (8, 6H)
s~ ` . . .
~, ;, . :
r : .
.. . . . .
- 13 - '~ 578
4. a) 2-(4,4'-Difluorobiphenyl-2-yl)~3,4,4-trim~thyl-
4,5-dihydro-1,3-o~azoliu~ iodide (VII, Rl ~ ~, R2,
R~ = C~3, R3 - H, ~
8.6 g of 2-(4,4'-difluorobiphenyl-2-yl)-4,4-dimethyl-4,5-
dihydro-1,3-oxazole are stirred under reflux for 2 days
in 60 ml of acetone and 30 ml of methyl iodide. The
mixture is concentrated in vacuo snd the oily re~idue is
crystallized by adding a little ~cetone. After filtering
off with suction and washing with a little ~cetone, 5.8 g
of cry~tals of m.p. 179C are obtained.
-NMR: 6 = 8.3 - 8.6 ~m, lH), 7.1 - 7.6 (m, 6H), 5.25
(B, 2H), 2.85 (~, 3H), 1.6 (s, 6H).
b) 2-(4,4'-Difluorobiphenyl-2-yl)-3,4,4-trimethyl-4,5-
dihydro-1,3-osazolium methylsulfate (VIII, R1 = ~,
R2, R~ = CH3, R3 = H, X = So42)
4.3 g of the dihydrooxazole (from 3) are stirred under
reflux for two hours in 30 ml of toluene and 1.7 ml of
dimethyl sulfate. The salt cry6tallizes out overnight. It
is filtered off with suction and washed with a little
acetone, and 5.65 g (92.6%) of product are obtained. Thi6
i8 sufficiently pure for the further reactions. After
recrystallization from isopropanol, it melts at 167-
16g C.
lH-NMR: 6 ~ 8.05 - 8.35 (m, 1~), 7.0 - 7.6 (m, 6H),
5.05 (8, 2H), 3.7 (8, 3H), 2.75 (8~ 3H),
1.5 (8, 6H).
5. 4,4'-Difluorobiphenyl-2-carbo ylic acid (I)
a) 7.18 g of 2-(4,4'-difluorobiphenyl-2-yl)-4,4'-
dimethyl-4,5-dihydro-1,3-oxazole (compound VII) are
hydrolyzed for 3 days under reflux in 75 ml of
dioxane and 80 ml of half-concentrated hydrochloric
acid. The solvent is removed in vacuo, the re~idue
is taken up in a little half-concentrated sodium
hydroxide solution, and the solution i~ wa~hed twice
with S0 ml portions of ether, clarified with active
carbon and acidified with dilute hydrochloric acid.
57~3
The precipitate i6 filtered off with suction and
washed with water. Colorless crystal6 of m.p.
117-119C are obtained. After recrystallization from
isopropanol/water, the product melt6 at 124C. Yield
61%
b) 4.0 g of 2-(4,4'-difluorobiphenyl-2-yl)-3,4,4-
trimethyl-4,5-dihydro-1,3-oxazolium methylsulfate
(compound VIII) and 30 ml of methanol are added to
a 601ution of 1.6 g of NaOH in 30 ml of water. After
2 hours at 65C, the reaction solution i~ acidified
with 2 N HCl and evaporated in vacuo. ~he residue i8
recrystallized in isopropanol/H20 1:4 and dried.
Yield: 2.25 g (99%) of m.p. 124C
c) The carboxylic acid I i8 obtained from the cor~
responding iodide VIII (from 4.a)) in a yield of
92.8% after recrystallization in an analogous
manner. -~
d) One-pot reaction:
100 g (0.348 mol) of 2-(4,4'-difluorobiphenyl-2-yl)-
4,4-dimethyl-4,5-dihydro-1,3-oxazole (compound VII)
are heated under reflux for 1.5 hours with 40 ml
(0.417 mol) of dimethyl 6ulfate in 100 ml of
acetone. During the reaction, a colorles6 precipi-
tate deposit~. 342 ml (1.54 mol) of 184 strength
~odium hydroxide solution are then added dropwi~e to
the su6pension and the acetone i~ ~imultaneously
removed by distillation. The mixture i8 then he~ted
under reflux nt an internal temperature of about
90~C until the hydrolysi6 i6 complete (about 1
hour). The mixture i~ cooled to 20C, the reaction
mixture separating into two pha~es. The lower pha~e
i8 re~ected, and the upper i~ washed twice with
100 ml portions of toluene an~ then clarified with
active carbon. It iB then acidified with concen-
trated hydrochloric acid with cooling, the crystal-
line, colorles6 4,4'-difluorobiphenyl-2-carboxylic
;. ~ - : . . .. - :
i8
- 15 -
acid precipitating. ~his i8 filtered off with
suction, washed with water and dried in vacuo at
30C.
Yield 70.1 g (86.0%)
The carboxylic acid obtained according to ~x~mple I c~n
be cyclized to 2,7-difluoro-9-fluorenone (IX) ~8 follows:
2.34 g of 4,4~-difluorobiphenyl-2-carboxylic acid are
added at 40-50C to 35 g of polyphosphoric acid. The
mixture i~ heated to 100C with stirring for 1 hour. The
yellow suspension is then poured into lS0 ml of ice
water. ~he yellow precipitate is filtered off with
suction and washed thoroughly with water.
Yield: 2.11 g (98%3 of compound IX
m.p. 208 - 210C