Note: Descriptions are shown in the official language in which they were submitted.
Professor Dr. Cerd Dannhard
Konigsbergstr. 26a
D-6236 Eschborn 2
Federal Republic of German
10
Merckle GmbH
Graf-Arco-StraLie 3
7900 Ulm-Donautal
SUBSTITUTED PYRROLE COMPOUNDS AND
TI-IEIR APPLICATIONS IN PHARMACY
t5
1
The invention concerns substituted pyrrole compounds and their
applications to pharmacy and to drugs containing these compounds.
It is known that arachidonic acid metabolizes two different ways. In the
cyclo-oxygenase way the cyclo-oxygenase enzyme metabolizes the arachidonic
acid into
xo prostaglandins. In the lipoxygenase way, the arachidonic acid affected by
the
lipoxygenases is metabolized into the so-called leucotrienes.
The prostaglandins take part in the generation of inflammation, fever and
pain, whereas the leucotrienes are significant in the generation of asthma,
inflammations
and allergies. Frequently non-steroidal antiphlogisncs such as derivatives of
arylacetic-
x5 acid, 2-aryl-propionic-acid and anthranilic acid to fight those symptoms.
These derivatives
inhibit cyclo-oxygenase and thereby prevent the formation of prostaglandins
from
arachidonic acid. However the use of such derivatives is questionable on
account of their
side effects. On the other hand, drugs inhibiting lipoxygenase are not
commercially
available.
Now it was found in surprising manner that certain substituted pyrrole
compounds arc potent cyclo-oxygenase and/or lipoxygenase inhibitors and
therefore are
suitable to prevent allergically induced maladies and to treat the set of
rheumatic
illnesses.
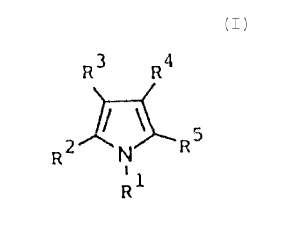
Accordingly the object of the invention are substituted pyrrole compounds
of the general formula I
R3 R4
R N
~o R
~1
where
R~ denotes a Cl - C~2 alkyl group,
RZ is a hydrogen atom or a C~ - C~2 alkyl group, or
R' and RZ together with the carbon atom and the nitrogen atom to which
~s they are bound form a 5- to $- link ring which also may contain a sulfur
heteroatom or
a carbonyl group and which where called for may be substituted with one to two
C, - C4
alkyl groups,
each time, two of the residues R3, R4 and RS independently of each other
are a hydrogen atom, a Cs-C8 cycloalkyl group, a Cl - C~Z alkyl group or an
aryl group
zo which may be substituted by one or two residues selected form a halogen
atom, a vitro-,
a C, - Ca alkoxy, hydroxy, a C,-C4 alkyl or phenoxy group, and
the third of the residues R', R" and RS denotes -CI-IO, -COZI-I, -COSC,-C,,-
alkyl or A-X, with A being a straight chain or branched C, - C8 alkylene group
which may
be interrupted by a hydrogen heteroatom or a carbonyl group, or a CZ - C8
alkenyl group,
2s and with X being COZH, S03I-I, CI-IO, OH or SH.
as well as their pharmaceutically compatible salts and esters, for application
in pharmacy.
In the present case the pharmaceutically compatible salts may be salts of
acid or of base addition. For acid addition :;alts, inorganic acids such as
hydrochloric
acid, sulfuric acid or phosphoric acid are used, or such organic acids as
tartaric acid,
lactic acid, citric acid, malic acid, mandelic acid, ascorbic acid, malefic
acid, fumaric acid,
s gluconic acid and the like.
The base salts include the salts of the compounds of formula I with
inorganic bases such as sodium or potassium hydroxide or with organic bases
such as
mono-, di- or tri-ethanolamine.
The esters of the compounds of formula I include in particular physiologi-
cally easily hydrolyzed esters such as alkyl-, pivaloyloxymethyl-,
acetoxymethyl-,
phthalidyl-, indanyl- and methoxymethyl-ester.
The term "alkyl, alkoxy etc." covers straight-chain or branched alkyl groups
such as methyl, ethyl, n- and i-propyl, n-, i- or t-butyl, n-pentyl,
neopentyl, n-hexyl etc.
Unless otherwise stated, "alkyl" preferably shall denote Cl - C8 alkyl and in
95 particular Ct - C6 alkyl.
Preferably aryl shall denote naphthyl and in particular phenyl.
The expression "halogen atom" covers an atom of fluorine, chlorine,
bromine or iodine, especially a fluorine or chlorine atom.
The cyclo-alkyl residue preferably denotes a cyclopentyl or cyclohexyl
Zo residue.
A preferred embodiment mode are compounds of the formula Ia:
R3
R~
R5 N ~ R4
R5
o s A 4
~'~ .~.. ) ~r
where R3, R' and 1R5 arc the carne as stated above in relation to formula I
and where R~'
and R7 independently from one another denote a hydrogen atom or a Cr - C~
alkyl group.
Especially preferred compounds of formula la are those wherein two of the
residues R', R' and RS denote a phenyl group which is possibly substituted by
one or two
s residues selected from a halogen atom, in particular a fluorine or chlorine
atom, from a
Cr - C~ alkyl-, Cr - Cn alkoxy-, Irydroxy- and phenoxy-group and where the
third residue
denotes A - X, with A being a Cr - C$ alkylene group or a C, - Cg alkenylene
group, and
with X being CO21-I.
In particular AX denotes CZ - C6-alkylene-COZI-I or CZ - C6 alkenylene-
,o COZI-I and in an especially preferred instance it stands for (CI-IZ)ZCO21-
I, C1-IZCOzI-I or
CH = CH COzH.
Preferably R6 and R' are in the 2-position of the pyrrolizine structure and
in particular they designate both a hydrogen atom or a methyl group.
Especially preferred embodiment modes are the compounds of the formulas
~s R3 R3
7 7
R s 4 R
R6 N~ R R6 N~AX
dUC . _~ R4
R~
3
2o R6 N ~ R
R4
where RG and R' denote a hydrogen atom or a CI - C4 alkyl group and R3 and
R~ denote n phenyl group which may be substituted by one or two residues which
are
selected from a halogen atom, a nitro-, CI - Ca alkoxy-, C~ - Ca alkyl- or a
phenoxy- group.
A-X has the same mewing as above in relation to Lhe formula I.
Preferably the substituents of the phenyl group are selected from a halogen
s atom, in particular a fluorine or chlorine atom, from a C1 - C,~ alkyl-, C~ -
C4 alkoxy- and
phenoxy-group. Preferably the substituents shall be in the m- and/or in the p-
position.
Preferably A denotes a C~ - C$ alkylene group or a C~-C$ alkenyiene group
in particular, or a CZ - CG alkylene- or C2 - C6 alkenylene group and X
preferably denotes
COzI-I.
Another embodiment mode are the compounds of formula I
where R~ and RZ independently of one another denote a Cl - C~2 group,
with two of the residues R3, R4 and RS being a phenyl group possibly
substituted by one
or two residues selected from a halogen atom, a Cl - C4 alkyl-, C, - C4 alkoxy-
, hydroxy-
and phenoxy-group, and
,s where the third of the residues R3, R4 and RS is a hydrogen atom or A-X,
with A being a Cl - C6 alkylene- or a CZ - C6 alkenylene-group and X being
COZH or
S03H.
Rl preferably is a Cl - Cs alkyl group, in particular a C~-C6 allyl group and
especially preferred a C4-C6 alkyl group.
zo R' preferably is a Cl-Ca alkyl group and especially a methyl group.
R3 and R'' on one hand and R'~ and RS on the other preferably are a phenyl
group and RS and R3 resp. are A-X, with A being preferably a C~-C,~ alkylene
group or
a Ci C6 alkenylene group.
6
Especially preferred compounds are:
(2,2-dimethyl-6,7-Biphenyl-2,3-dihydro-l I-I-pyrrolizine-5-yl)-acetic acid,
and
3-(2,2-dimethyl-G-(4-phenoxyphenyl}-7-phenyl-2,3-dihydro-II-I-pyrrolizine-S-
yl) propionic acid.
s Further the invention concerns the compounds of the general formula I and
also the corresponding above stated preferred embodiment modes per se, where
R1 and
RZ together denote <~ S to 8 link ring possibly containing a sulfur hetero-
atom or a
carbonyl group and further possibly substituted with one to two Cr-C4 alkyl
groups, and
where R', R4 and R5 assr:me the above meanings but with A being a straight-
chain or a
,o branched C3 C,~ alkylene group which may be interrupted by an oxygen atom
or a
carbonyl group, or denoting a straight-chain or a branched C3 C8 alkenyl
group, or
where Rr through R$ assume the meanings stated in claim 4.
Synthesis of the compounds of formula I wherein R' and RZ together form
a S to 8 link ring, takes place in a first step described by the equation (1)
below:
R~
CHZR3
+ RS - CH - C - R~
ICHZIn N
R6~ X= C1, Br
n = 1 - 3
R3.
R~
(CH ~ n~ / R 9
RS~N
RS
~ ~. ~i ~ ~9 _~. '
This synthesizing; step is elucidated together with the following ones by
means of the illustrative Biphenyl-substituted I>yrroiizine compounds. ~I~hc
first reaction
stage is explained in further detail below:
Reaction diagram (1)
/ \ __
Ethanol,
C\ _ NaHCOy
N + ~C~
BrH2C
/ ~ / \
,o
Ethanol,
NaHCO~,
II + C
N 1 H ' ~5~ N
Br~'~
ri
Ethanol,
CN3 ~ NaHCOy
N + \C Hue, -°-~
C1-- ' \ /' 20.30 °~
/.
The con~~o4~v~~s of r~:vtion are known and described in CI-IEM~KEId-
ZEITUNG, 110, (1968) # 7/$, pp 267, 271 and in ARCH. PHARM. 321, pp 159-162
( 1988).
4
do a second stage, the formyl or methylol group is introduced into the
pyrrole ring and further reaction takes place into the corresponding
derivatives of acetic
acid and ethanol rep. as given by the reaction diagram (2) below:
Reaction diagra~r~ (2)
POCi~, DMF,
er~~z~r!r.~
CHO
UAIH4, THF
-Y-
CHzOH
~ CHO ~ / CHzOH
/' /\
a
CH~OH
~~1~~~~
Pl2CHCOOC~H~,
Cu°, , n
-.~.~ --.~,-
TULUENE
CH2 COOGzHs
_ ~~HF
CH2-CH2OH
r~
CH2-COOC2H5 -~'
/ CH2-CHzOH
/~
NHS ~ CH~-CH..nH
~~~~ 1 ~.~ 10
'These reactions and their conditions are known and arc described in
CI-IEMIKEIZ-LE1TUNG I10 (1968), # 7/8, pp 267-271 and in ARCI-I. PI-IAItM.
321, pp
159-162 (1988).
The preparation of the corresponding derivatives of formic acid, propionic
s acid and acrylic acid is described in ARCM. I'I-IARM. 321, pp 545-549. IL
takes place
according to the reaction diagrams (3) and (4).
'the preparation of the corresponding butyric-acid derivatives takes place
according to the reaction diagram (5). First, using the Friedels-Crafts
acylation with
succinic acid anhydride (E. Berlina, ORG. REAKT. 1949, 5, pp 229-289), Lhe 4-
oxo-
1o butyric acids are prepared which then are reduced by means of the I-Iuang-
Minion
variation of the Wolff-Kishner reduction with hydrazine/KOH in
diethyleneglycol. The
conditions of reaction are known to the expert.
The derivatives of valeric acid can be prepared by means of the 5-oxo-
valeric acids which in turn can be made from the pyrrolizine base bodies by
the Friedel-
1s Crafts acyiation with glutaric acid anhydride in a manner similar to the
preparation of the
butyric acid derivatives. In similar manner the caproic acid derivatives are
obtained by
the Friedel-Crafts acylation of diphenylpyrrolizine with methyl-5-
(chloroformyl-
valerate/AICI~) and saponification of the 6-oxocaproic-acid-methylester
derivatives and
subsequent reduction of the oxovaleric acid with. hydrazine/KOI-I.
zo The preparation of compounds substituted at the phenyl group takes place
similarly. The hydroxy-substituted derivatives are prepared by ether splitting
with BBr3
from the corresponding alkoxy derivatives ('rETRAI-IEDRON, 1968, 24, pp 2289-
2292).
2fl~.~~~~
Reaction diagr:rrn (3)
l~
._
CtCOSC2H~,
AlCta CH,aCi2
,. N
iC2H5
KOH,
s Ethanol, D ~,
° N s cooH
s ~
~2
Itca~aion diogratia (4) ~
3.
~ ' \
-- ~.. H
I
_ '~~Ig- C-COOG2H5 Ko
CHO arr,cTiotv ~ ~.- Ethanol, DD
N ~ ~ ~ \
H
/ 1
i
H~IPtO~,
.-COOH Ethanol,
- ~~ -
N ~, (CHz)2 COOH
/
w
COOCZHS COOH
H~~ .. --C
H C-H (CH2)~-CG
N.'
13
reaction diagram (S)
agcy cHach_
(CH2)2-COOH
HzN-NH2, NaOH,
DIETHYLENEGLYCOL
(CH2)~ COON
~H2)2 COOH
.OO H (C H2 )3~ COO H
,4
The prclrrration of the compounds of formula I is described below, wherein
Rr and RZ denote a C,-Crz alkyl group. The initial material for the synthesis
of these
compounds are the corresponding 2,3,4- and 2,3,5-substituted pyrrole compounds
of which
the production is described in AUST. J. CI-IEM. 19G<, 19, pp 1971-1885. The
further
s synthesizing steps are elucidated illustratively below starting with 2-
methyl-2,3-diphenyl-
pyrrole and 5-methyl-2,3-diphenylpyrrole. These reactions are shown in the
diagrams (G)
and (7).
The first step is an alkylation of the pyrrole nitrogen atom. This reaction
is carried out in conventional manner, for instance using the corresponding
alkyl halides
,o in the presence of a birse, for instance alkali-metal aicoholates such as
sodium mcthylate,
sodium ethylate or potassium-t-butylate, in an inert solvent such as DMSO,
ethylenegiycol
methylether and the like. The alkylation also can be carried out
heterogeneously with
the corresponding toluene sulfonic-acid alkylesters or alkyl halides using
phase transfer
catalysts, in conventional manner (see for instance CAN. J. CI-IEM. 1977, 55,
pp 4112-
~s 4116). The introduction of acid lateral chains then takes place similarly
to the synthesises
of the corresponding pyrrolizinyl carboxylic acids as described above.
The compounds of the invention are potent inhibitors of cyclo-oxygenase
and/or lipoxygenase. Accordingly they are useful in treating the set of
rheumatic illnesses
and in preventing allergically induced ailments. Therefore the compounds of
the
2o invention represent effective antiphlogistics, analgetics, antipyretics,
anti-allergies and
broncholytics and may be used in thrombosis prophylaxis and in the prophylaxis
of
anaphylactic shock and to treat skin diseases such as psoriasis, urticaria,
acute and chronic
exanthemas of allergic and non-allergic origins.
Reaction diagram (G)
TOLUEfJE SULFONIC _
nCID ME'I'JIYI,ESTEI2
OR
AL(;YLI3ROMIDE
H3 N~ H3C N
H R
CH2~Ct-ICOOCH3,
BFI, CHzCICH2Cl
COMPOUtJD ( R
H
CH3 !~
NHS H3 N COOCH3
i
R
n-~H.r
n-C4Hg KOH,
Ethanol, D
f n-CsH9~
Si ~'CsH~3
~I iso-C4H~
i Neoperrtyl
COON
R
os
~'~.1~ ~~~~
Reaction diagrs~m (7)
'I'OGUEIJE SUI,FONIC
ACID ME'PF1YLESTEt2
OH
Alkyibromld a
H
NzCHCOOCzHy
COMPOUND R CUB, toluened
H
CH3
H5C200C
C2H5
F /
n-C3H7 H3C N
e_ n_CaHs R
f_ n-CSH, ~
KOH,
g n-CsH,3 Elhanoi, D
h~ n-CaH»
j ISO-C4H9
Neopenfyl HOOC
H3
The crampounds of the invention may be administered either as individual
therapeutic substances or as mixtures with other active substances. They may
be
administered alone, but as a rule they may be administered in the form of
drugs, that is
as rnixtures of the active substances with suitable ;pharmaceutical carriers
or diluents. The
s compounds or drugs may be administered orally or parentcrally but preferably
they shall
be dosed orally.
The kind of drug and of pharmaceutical carrier or diluent depends on the
desired method of administration. Oral drugs for instance may be in the form
of tablets
or capsules and may contain conventional excipienis such as binders (for
instance syrup,
,o acacia, gelatin, sorbite, traganth gum or polyvinylpyrrolidane), fillers
(for instance lactose,
sugar, maize starch, calcium phosphate, sorbite or glycine), lubricants (for
instance
magnesium stearate, talcum, polyethyleneglycol or silicon dioxide),
disintegrants (for
instance starch) or wetting agents (for instance sodium lauryl sulfate). Oral
liquid
preparations may be in the form of aqueous or oily suspensions, solutions,
emulsions,
,s syrups, elixirs or sprays etc. or they may be in the form of dry powders to
be reconstituted
with water or another suitable carrier. Such liquid preparations may contain
conventional
additives such as suspending agents, flavorings, diluents or emulsifiers. As
regards
parenteral administration, solutions or suspensions with conventional
pharmaceutical
carriers may be used.
2o The compounds or drugs of the invention can be administered to mammals
(human and animal) in doses of about 0.5 mg to about 100 mg per kg of body
weight per
day. They can be administered in single or multiple doses.
~~ ~~ ~4~:~ ,o
The effectiveness of the compounds of the invention can be determined by
means of the inhibition of S-lipoxygenase or of c:yclo-oxygenase. The research
was carried
out as follows:
TESTING TO DETERMINE THE INI-IIBITION OF 5-LII'OXYGENASE
The source for 5-lipoxygenase were bovine granulocytes which can form
leucotrienes just as human granulocytes do. By stimulation with calcium-
ionophore (see
BIOCI-IEM. BIOPI-IYS. ACTA 1984, 795, pp 499 - 503), mainly LTC4 (leucotriene
C4)
and LTB4 (leucotriene B4) are formed from endagenous arachidonic acid. The
isolation
of the granulocytes and the implementation of the enzyme reaction are
conventional (see
,o BIOCHEM. BIOPI-IYS. ACTH 1984, 795 pp 499 - 503). The blood protected
against
clotting by EDTA first is centrifuged for that purposed and the thrombotic
excess is
removed. Following lysis of the erythrocytes with water the lymphocytes and
monocytes
are separated from the granulocytes by means of a Ficoll gradient. The
granulocytes are
set to a specific cell number. The enzyme reaction is then begun in the
presence or
,s absence of the test substances following the addition of Caz+ with calcium
ionophore. The
synthesis of the leucotrienes is stopped after S minutes by adding a mixture
of methanol
and acetonitrile containing PGB2 as the internal standard and NDGA as the anti-
oxidant.
Next the samples are diluted in water and processed in the manner described in
J.
CHROMATOGR. 1896, 378, pp 208 - 214. LTB4 is measured at the absorption
2o maximum at 270 nm. The arachidonic acid metabolites are observed ,present
in this
research in approximately quantitative manner.
~~~.~~~0~
19
TEST1NCG 'r0 DF,'r>rIRMINIE 'TI IE INHIII3IT10N OF CYCLO-OXYGENASE
In this test the 12-IIH'r amount of 12-hydroxyheptadecatriene acid or
prostaglandin EZ amount formed by bovine thrombocytes following addition of
calcium
ionophore is determined by uv detection following HPLC separation. Following
s centrifuging of the bovine blood, the thrombocytes are isolated from the
surnatant. The
enzyme reaction and the isolation of the formed metabolites take place as when
determining the 5-lipoxygenase inhibitiun, however the incubation time was one
minute.
The detection of 12 I-II-IT following I-IPLC separation takes place at 232 nm.
Tables 1 and 2 list the test results for the compounds of the invention, The
1o test substances were at concentrations of 10 p M.
The Examples below illustrate the invention. All temperatures are
uncorrected. The it spectra -- unless noted otherwise -- are from KBr molded
articles.
The NMR spectra, unless noted otherwise, are 90 Mhz spectra, recorded in CDCI3
with
tetramethylsilane (TMS) as the internal standard.
1s The preparation and the properties of the compounds used as initial
materials used in part for the reactions below and listed in Tables 3, 4 and 5
are
described in the following literature:
ARCH. PHARM. 312, pp 896 - 907 (1979)
ARCH. PHARM. 319, pp 500 - 505 (1986)
zo ARCH. PHARM. 321, pp 159 - 162 (1988)
ARCH. PI-IARM. 321, pp 545 - 549 (1988)
CHEMIKER ZEITUNG, 110, (1986), 7/8, pp 267 - 271.
'rAI3L~ 1: INI-IIBITION OF 5-I_.,IPOXYGENASE AND OF
CYCLO-OXYGI;NASC BY PYRR,OLIZINE COMPOUNDS.
R3
R~
R 4 ( 10 y1~5 )
R 6 Pl
RS
COMPOUND P4 R5 R6 R7 L1
R3 INfIIBITIO~ClppXy-
Nr. ge~
a
genase
Phenyl (CH2)4C02H Phenyl H H 82
Phenyl (CH2)5CO2tiPhenyl H H 80
( ~2 ) Phenyl Phenyl H ti 86
4CC2H
(CH2)5C~2r~Phenyl Phenyl H H 86
Phenyl Phenyl CH2~2H ~3 ~3 94 99
Phenyl Phenyl ( ta-12 CH3 CH.~91 97
) 2C02H
( ~2 ) Phenyl Phenyl CIi3Cli393
2~2H
( ~Z ) Phenyl Phenyl CH CH 96
5~2H 3 3
Phenyl Phenyl CHZCOZH H H 91
Phenyl Phenyl CH=C~1-C02HH H 80
CH=CHC02HPhenyl Phenyl CH3 CH3 91
Phenyl C6H13 (~(=(3iC~p2HH H 82
Phenyl m-C'hl.osphenyl( (~~2 H H 89
) 2Cp2H
Phenyl p-(hlozphenyl( ~2 ) H H 98
zCp2H
Phenyl p-Tolyl ( C7-12 H H 92
) 2Cp2H
Phenyl p-Methoxy- (CH2)2Cp2HH H 91
phenyl
Phenyl p-Phenoxy- (CHZ)ZOpzHH H 98 70
i phenyl
i Phenyl a-naphthyl ( p-(2 H 11 94
) 2pp2H
Phenyl p-Fluorphenyl(Di2)zCp2H~3 ~3 93 (3,3 yM)
Phenyl p-Tolyl. (Cfi2)2C02HCH3 CN3 93 (3.3 pM)
Phenyl p-Ffienoxy-( C1~2 C?-i3C~-1398
) 2Cp2H
phenyl (
3,
3
~;M
2,
T ABLE 2: INI-iIBITION OI~ 5-L,IPOXYGENASE
BY 2-ME'rI-IYL-DIPLIENYLPYRROL1C-ACETIC-ACIDS
R3 R4
R2 ~ ~ RS ( 10 uM )
N
R1
ir~tmamoN(
R1 R2 R3 R4 RS ~Lipoxygenase~
n-CQH~ CN3 PhenylPhenyl (C7-IZ)2COZH
94
n-C5~111 ~3 ~enylPhenyl ( CIi2 ) 2COZH
99
n-C6H13 CH3 PhenylPhenyl (C7-12)ZC02H
99
tVeo-
pentyl CH3 PhenylPhenyl (CH2)ZC02H 97
TABLE 3
EXAMP~T
H CHs
i H ~ 1~
CHO
CH20H
CH2COOC2H~
(CH2)20H
6 COOH
CH2COOH
(CH2)2COOCH3
(CH2)ZCOOH 27a 27~
io CH=CHCOOCZHS (E) ,~
11 CH~HCOOH (E)
23
TABLE 4
EXAMPLE
..._ _ ._ _.._ .~.. ~ _.. H ~ -~.c"3
12 H
13 CHO 7~ 7~1
14 CHaOH ~ lQtz
15 CH2COOC2H5
16 (CH2)20H 1~ 1 ~
1~ COSC2H~ 1~ 1~
1s COOH 21~ 21~
1~ CH2COOH 24a 24b
2o CH~HCOOC2H5 (E) 2$
21 CH~HCOOH (E) ,~Q
22 (CH2)zCOOH ~2
za
TABLE 5
EXeIMPLE
Nr. ~ ~ H I CH3
2 3 hi ,~ ~.
24 CHO $.~ $~2
25 CH~OH 11~, 11~
26 CH2COOC2H5 L4,~ ~i4
a~ (CHz)20H 1?~
2 8 COSC~-IS ~ 2_Qt~
2 ~ COOH
3o CHaC00H ,~ 2~,
3i CH~HCOOC2H5 (E) 2~ ,2~
32 CH~HCOOC2H5 (Z) 29b'
33 CH~HCOOH (E) ~ i~
34 CH=CHCOOH (Z) ~11~'
35 (CH2)2COOH
25
Gl=:NERAI_ l'h.OCEDUIZI: hOR I'IZEE'ARING 'I'IIE 4-(DIPI-IENYL-2,3-DII-IYDRO-
lI-I-I'YRROLIZINYL)-4-OXOI3UTYRIC ACIDS
With ice cooling and stirring, 8.8 mmoles (1.17 g) of A1C13 are added in
batches over five minutes to a solution of 4 mmoles of Biphenyl-2,3-dihydro-1H
s pyrrolizine and 4 m~xr (0.40 g) of succinic acid anhydride. Then agitation
continues for
4S minutes at room temperature. Thereupon the batch is poured into 1S0 ml ice
water.
After adding 4 ml of 8 % I-I~PO,,, extraction is performed three times with CI-
ICh, the
organic phases are dried with Na2S04 and the solvent is distilled. Product
isolation is by
means of column chromatography (silica gel, 1st ether, 2nd ether/THF 1+1). The
,o product fractions are concentrated, the residue is dissolved in a little CI-
ICl3. After
adding n-hexane and new concentration, the product is precipitated.
GENERAL PROCEDURE FOR PREPARING TI-IE 4-(DIPI-IENYL-2,3-DIHYDRO-
1H-PYRROLIZINYL)-IiUTRYIC ACIDS
15 O.S mmoles of 4-(Biphenyl-2,3-Bihydro-1H-pyrrolizinyl)-4-oxobutyric acid
are mixed with 10 ml of diethyleneglycol, SO mmoles (2.8 g) of KOH and 30
mmoles (1.S
g) of hydrazine hydrate. The batch is heated 1 hour till reflux (bath
temperature 170 ° C).
Next the reflux condenser is replaced by a distillation bridge and the
temperature is
raised until the inside is at about 210 ° C. This temperature is then
maintained for 2 h.
2o Following cooling, the batch is poured into 1S0 ml I-I20, acidified with 8
% I-I3P0,, and
extraction is carried out three times with ether. The extracts are washed with
HzO, dried
and concentrated. The product is isolated with column chromatography (silica
gel,
ether/TI-iF 4 + 1 in 39, diisopropylether in 4Q, ether in 41a and ether/n-
hexane 4 + 1
in 4~) and is precipitateB using n-hexane.
~~~~~3~~
2s
GENERAL 1'ItOCEDURE rOR PREPARING TI-IE S-(DII'I-IENYL-2,3-DIi-IYDRO-
li-I-PYRROI_IZINYL)-5-OXOVALER1C ACIDS
4 mmoles of Biphenyl-2,3-dihydro-lI-I-pyrrolizine are reacted in lf~ ml of
absolute CIIZCI2 with 4 mmoles (O.~IG g) of glutaric acid anhydride and 8.8
mmoles (1.17
s g) of AlCl3 similarly to the procedure for rnaking the 4-(Biphenyl-2,3-
dihydro-1H-
pyrrolizinyl)-4-oxobutyric acid ("s.S." 222). In deviation, the batch is
poured into ice
water immediately after the addition of AICI~ and then is processed further.
Product
purification takes place by column chromatography (silica gel/ether).
GENERAL PROCEDtJRE FOR PREPARING TI-IE 5-(DIPI-IENYL-2,3-DIHYDRO-
,0 1H-PYRROLIZINYL)-S-VALERIC ACIDS
0.5 mmoles of 5-(Biphenyl-2,3-dihydro-lI-I-pyrrolizinyl)-S-oxovaleric acids
are reacted similarly to the procedure for making the 4-(Biphenyl-2,3-dihydro-
1H-
pyrrolizinyl)-4-oxobutyric acids. The product is isolated by column
chromatography (silica
gel, ether) and precipitated by means of n-hexane.
,s GENERAL PROCEDURE FOR PREPARING THE 6-(DIPI-IENYL-2,3-DII-IYDRO-
lI-I-PYRROLIZINYL)-6-OXOCAPROIC-ACID METHYLESTERS
With ice Gaoling and while stirring, 22 mmoles (2.93 g) of A1CI3 are added
aver 5 minutes and in batches to the solution of 10 mmoles of Biphenyl-2,3-
dihydro-1H-
pyrrolizine and 10 mmoles (1.79 g) of methyl-5-(chloroformyl)-valerate in 50
ml of
2o absolute CHZC12. Immediately thereafter the batch is poured into 150 ml of
ice-cooled
% NaCI solution. Upon addition of 4 ml of 8 % h13P04, extraction with ether is
carried out three times, the organic phases are dried with NazS04 and the
solvent is
distilled. The product is isolated by means of column chromatography (silica
gel, 1st n-
hexane/ether 3-+~2, 2nd n-hexane/ether 1 + 4 in 48, 50a and SOb, or 1st
toluene, 2nd
2s hexane/ether 1+4 in 49). First the eluates are concentrated to about 100
ml, then they
are washed twice with 0.05 n NaOI-I and twice with 10 % NaCI solution and
lastly they
are dried with Na2S0,,. When the solution is concentrated, the products
precipitate or,
following distillation of the solvent, crystallize.
~~ ~ ~'.v~~
27
GENERAI_ PROCEDURE FOR PREPARING 'I'I-iE 6-(DII'IIENYL-2,3-DII-IYDRO-
lI-I-PYRROLIZINYL)-G-CAI'ROIC ACIDS
The solution of 1 mmole of 6-(Biphenyl-2,3-dihydro-lI-I-pyrrolizinyl)-6-
oxocaproic-acid methylester in 10 ml of ethanol is heated to boiling. S ml of
10 % KOH
s previous degassed by boiling are dripped into that solution and heating
continues another
1S minutes with reflex. Following cooling the batch is poured into 100 ml of S
°lo NaCI
and then is acidified with 8 % H3P0,~ and extraction is carried out three
times with ether.
The organic phases are dried by means of Na2S04 and concentrated. The residue
is
reacted with SO mmoles (2.8 g) of KOI-I and 30 mmoles (1.S g) of hydrazine
hydrate in
,0 10 ml of diethyiene glycol similarly to the preparation procedure for the 4-
(Biphenyl-2,3-
dihydro-1H-pyrrolizinyl)-butyric acids and is purified by column
chromatography with
silica gel.
The compounds so prepared and several physical properties of theirs are
listed in the Tables 6 through 8 below.
,s GENERAL PROCEDURE FOR PREPARING THE 2-(DIPI-IENYL-2,3-DIHYDRO-
lI-I-PYRROLIZINYL)-PROPIONIC-ACID ETHYLESTERS
S mmoles of Biphenyl-2,3-dihydro-1H-pyrrolizine dissolved in 4 ml of
absolute toluene are reacted with 7.5 mmoles (0.96 g) of 2-diazopropionic-acid
ethylester
dissolved in 2 ml of absolute toluene similarly to the procedure for preparing
the
2o diphenyl-2,3-dihydro-1H-pyrrolinyl acetic-acid ethylesters. I-Iowever the
time of reaction
is 2 h. Purification is column chromatography (A1ZO3, n-hexane/ether 9+1). The
products are precipiated with ethanol.
GENERALPROCEDURE FOR SAPONIFYING THE 2-(DIPHENYL-2,3-DIHYDRO-
1H-PYRROLIZINYL) PROPIONIC-AICD ETHYLESTERS
2s S mmoles of 5~i or O.S mmoles of 55a and SSb, dissolved in 12 ml or 9 ml
resp. of ethanol are reacted with 8 mI or 2.S ml resp. of 10 °~o
aqueous KOI-I similarly to
the procedure for saponifying the Biphenyl-2,3-dihydro-lI-l:-pyrrolizinyl-
acetic-acid
ethylesters. The time of saponification is 1S min in 54, 60 min in 55a, and
55b.
Purification is by column chromatography (silica gel, 1st n-hexane/ether 1+ 1,
2nd ether).
ao After the eluates have been concentrated, the products are precipitated
with n-hexane.
The compounds so prepared together with some of their properties are
listed in Table 6 and the Table 9.
28
Tabie 6 % ~ ~
. .. . ,y. .
EXAMPLE-. ,. , MEL'I'IC~GjRl J
ppyNZ~
tdr. ~ C
6 COOH ,~ 187-188 1710, 1635
)
CO(CH
3 2
g
37 (CH2)gCOOH ~ 166-167 1705
CO(CH2)3COOH 42 142-143 1710, 1635
3 $
3~ (CH2)aCOOH 4~ 146 1710
40 CO(CH2)4COOH ~ 9~ 1740, 1630
41 (CH2)~COOH ~.~.
CH (CH3)COOC2H5 ,;~_4, - 17 30
4 2
43 CH(CHg)COOH ~ 173 1705
1, (C=O) hand
~H-NMR: b (ppr'~) = 1.06-1.78 (m, 6H, -(~)3-CHz-CO-), 2. i0-2.40 (rn, 2H,
...CH~p-), 2.40-2.76 (rn, 4H, f'yr-CH2 ~a :.-2), 3.02 (t, 2H, J=7 Hz,
C-1), 3.94 (t, 2H, J=7 Hz, C-3), 6.97-7.31 (m, 10H. Arom.)
Table 7
G:,AMPLG MELTING 1 )
' NZ,. R POINT IR
.
e~
44 CO(CH2)2COOH s~ 231-234 1715, 1680
45 (CH2)3COOH ~ 176-178 1715
46 CO(CHZ)3COOH ~ 128 1710, 1660
47 (CH2)4COOH ~ 126-127 1?00
48 CO(CHa)4COOCH3 ~ 112 17d5, 1658
49 (CHp)gCOOH ~ 63 1710
1~ (C=O) Band
G~ '~ a i"' 4
Table 8
I
EXAMPLE ~~ ~ MCi.TING
N_r. H CT.
R - C
~ I
_ R1
a ~
b_
a
b
, ~ '~15, 166 1715.-
CO(CH2)2COOH ~ 190-191187-1881 1645
1645
51 (CH2)3COOH 41 a 41 146-148192-194 17051U
b 7 '
52 CO(CH2)3COOH 44a 44~' 109 167 172 715,
0 .
1660 1660
53 (CH2)4COOH 47a 47~ 111-1121705 129-1301715
54 CO(CH2)4COOCHg ~ ~ 92 1735, 1740,
1655
1645
55 (CH2)SCOOH ~ ~b 123 1710 122 1710
v.
~0
1~
ai
'I'ablc 9
2p~.~ ~ fly.
10 ~- - I - -_ _
EXnMPLE R'= MELTING FT. lf~ (C-=O) I
Nr. R H CH.t a b a b
56 CH(CH3)COOC2H5 55a 55b X33 136 1730 1740
57 CH(CH3)COOH ~ 57a I 57b i 190 1705 ~ 226 1710
is
EXAMPLE 58
5-( 1-(DIPf-IENYL-2,3-DI I-IYDRO-1 ~I-PYRROLIZINE-7-YL)-ETI-IYL)-2,2-DIMETI-
IYLr
1,3-DIOXAN-4,6-DIONE (59a)
The solution of 4 mmoles (1.04 g) of 3a, 4 mmoles (0.58 g) of 2,2-dimethyl-
zo 1,3-dioxan-4,6-dione (Meldrum's acid) and 8 mmoles (0.35 g) of freshly
distilled
acetaldehyde in 40 ml of acetonitrile is made to stand for 24 h at 30 °
C. Following
cooling in the ice bath, the precipitated product is evacuated.
Yield: 0.44 g (2G %)
melting point: 169 ° C (with dissociation)
zs IR: v ",~x = 1790 (C=O), 1605 (C=C)~cm
EXAMPLE 59
S-(1-(2,2-DIMETIIYL-5,6-DIPIIINYI; 2,3-DIE-IYDRO-lI-I-PYRROLIZINE-7-YL)-
ETIIYL)-2,2-DIME~I'IfYL-1,3-DIOXAN-4,6-D~fONE (59b)
4 mmoles ( 1. IS g) of 3b are reacted in the manner described in relation to
s 59a with acetaldehyde and Meldrum's acid.
Yield: 1.35 g (74 °lo)
Ir: v max = 1700 (C =O), 1755 (C=O), 1605(C=C)/cm
EXAMPLE 60
3-(5,6-DIPHENYL-2,3-DII-IYDRO-lI-I-PYRROLIZINE-7-YL) BUTYRIC-ACID
,o E'1'I-IYLESTERS (GOa)
1 mmole of 59a is dissolved in a mixture of S ml of absolute pyridine and
1.0 ml of absolute ethanol. After adding some copper powder, the batch is
heated to
reflex for 3 h. The copper power is evacuated and the solvent is distilled in
vacuum. The
product is isolated from the residue by column chromatography (silica gel, n-
hexane/
,s CI-IZC1Z 1 + 2).
Yield: 96.5 mg (25 °lo); IR: v,nax- 1735 (C=O), 1605 (C=C)/cm.
EXAMPLE 61
3-(2,2-DIMETHYL-S,6-DIPI-IENYL-2,3-DIHYDRO-1I-I-PYRROLIZINE-7-YL)-
BUTYRIC-ACID ETI-IYLESTER (GOb)
20 2.5 mmoles of 59b are dissolved in a mixture of 20 ml absolute pyridine and
2 ml of absolute ethanol and upon addition of some copper powder the batch is
reacted
as described for 60a. Purification is by column chromatography (silica gel, n-
hexane/-
CHzCIZ 2+ 1) in isolation. ,
Yield: 0.68 g (68 %) IR: vmax = 1745 (C=O), 1605 (C=C)/Cm
2s EXAMPLE 62 -
3-(5,6-DIPI-iENYL-2,3-DII-IYDRO-lI-I-PYRROLIZINE-7-YL)-BUTYRIC ACID (61a)
The eluates are concentrated. The product is made to crystallize using a
little ether and then is made to precipitate extensively by aclding n-hexane.
so Yield: 63 mg (36 %); melting point: 157 ° C (with dissociation)
IR v max = 3300 - 2400 (OI-I), 1710 (C=O), 1605 (C=C)/cm
33
EXAMPLE G3
3-(2,2-DIMETi-lYL-5,G-DIP1-iENYL-2,3-DIi-IYDRO-1 i-I-PYRROLiZINE-7-YL)
13U'I'1'RIC-ACID (61b)
The product precipitates when the eluates are concentrated.
s Yield: 0.26 g (46 %); melting point: 202 - 208 ° C (with
dissociation)
GENERAL PROCEDURE POR PREPARING TIE TItIMETI-IYLPiIENYL-2,3-
DIHYDRO-1-i-I-PYRROLIZINES
0.05 moles (5.6 g) of 2,4,4-trimethyl-0lpyrroline, dissolved in 25 ml of
absolute ethanol, are reacted with 0.05 moles (10.7 g) of 1-bromo-1-
phenylacetone (in G3)
io or a-bromo-propiophenone (in G4) in a manner analogous to the synthesis of
3a. In
deviation from that reference, here, following addition of the NaI-IC03
solution (0.06
motes in 40 ml of i-Iz0) are heated only 4 h to reflex. Purification is
carried out by
means of column chromatography (A1203, n-hexane/ether 9+ 1). Oil remains
following
concentration of the eluate.
is . GENERAL PROCEDURE FOR PREPARING TI-IE TRIMETI-IYLPHENYL-2,3-
Dii-IYDRO-1I-I-PYRROLIZINYL-ACETIC-ACID ETi-IYLESTERS
mmoles (1.13 g) of G3 or of 64, dissolved in S ml of absolute toluene, are
reacted with 7.5 mmoles (0.86 g) of diazo-acetic-acid ethylester dissolved in
4 ml of
absolute toluene similarly Lo the procedure for preparing the Biphenyl-1,2,3-
dihydro-1I-I-
2o pyrrolizinyl acetic-acid ethylesters. The products are obtained as oils
following column
chromatography (A1203, 1st n-hexane/ether 9+ I, 2nd n-hexane/ether 3+2).
SAPONIFICATION OF THE TRIMETI-IYLPI-IENYL-2,3-DII-IYDRO-1I-I-
PYRROZINYL-ACETIC-ACID ETI-IYLESTERS
1.5 mmoles of 65 or 0.5 mmoles of GG, dissolved in 4m1 or 12 ml resp. of
is ethanol are reacted with 2.5 ml or 7.5 mi resp. of 10 % aqueous KOi-I
similarly io the
procedure for saponifying the Biphenyl-2,3-dihydro-lI-I-pyrrolinyl-acetic-acid
ethylesters.
Purification is by column chromatography.
The compounds and several of their properties are listed in the Tables 10
and 11 below.
'('able 10.
H
Ha
H
10 EXAMPLE 1 I
R MELTING P'r. IR
64 ti 63 -- 1605
65 CH2COOC2H5 65 -- 1735
66 CH2CO0rt 67 168 1715
Table 11
R
H3C
H3C
CHI
as
1)
R MELTING ~ I R
PT.
67 H 64 58-6U 1595 (C=C)
68 CHZCOOCZFiS 66 1740
69 CH2COOH 68 141 1710
as
1) (C=0~ band, unless stated otherwise
35
EXAMPLE 70
5-(n-IIL:XYL)-7-I'I-IENYL-2,3-DII-IYDRO-lI-I-PYRIZOLIZ(NE (G9)
In order to dissolve 35 mmoles of n-octanal in 15 ml of ether and S ml of
dioxan, a solution of 35 mmoles (5.6 g) of bromine in 5 ml of CI-1ZC12 is
slawly added
s while stirring. This batch then is mixed with 50 rnl ether and to neutralize
I-IBr, the
mixture is washed carefully twice with a 5 % NaI-IC03 solution. The organic
phase is
dried by means of NazSO,, and concentrated. The residue is dissolved in SO ml
ethanol
and added to a mixture of 35 mmoles of 2 benzyl-Q 1-pyrroline~, SO ml of
ethanol and 50
ml of 10 % NaLICO~ solution. After stirring for 24 at room temperature, the
batch is
,o poured into 500 ml of 10 % NaCI and extraction is carried out twice with
ether. The
organic phases are dried by means NazSO~, the solvent is distilled and the
pyrrolizines so
obtained, 69 and 70, are isolated by column chromatography (A.IzO~, n-
hexane/ether
9+1).
The two isomeric pyrroiizines then are dissolved in SO ml of dichloroethane
,s and are mixed a total of four times, 15 minutes apart, each time with 17
mmoles of
acrylic-acid methylester and 1.0 ml of BF3 etherate. Following addition of 200
ml of 5
NaCI solution, the batch is extracted twice with ether. Following drying by
means of
Na2S04, the solvent is distilled and the unconverted G9 is isolated by means
of column
chromatography (A1203, n-hexane/ether 9+ 1). The eluates are concentrated. The
2o remaining oil solidifies after some time.
Yield: 1.23 g (16 %); Melting point: 46-47 ° C
C»H~N (224.1) IR: vn,ax= 1610 (C=C)/cm
tH-NMR: 8 (ppm) ~ 0.73-1.06 (m, 3H, -CFA), l.Ofri.76 (m, 8H, -CHz-), 2.33-
2.72 (m, 4H, C-2 and Pyr-CHr), 3.04 (t, 2H, J=7 Hz, C-1), 3.83 (t,
2H, J=7 Hz, C-3), 6.20 (s, 1H, C-5), 6.~5-7.56 (m, 5H, Aran.)
36
EXAMPLE 71
5-(S-(n-I-II~XYL)-7-PI-IENYL-2,3-DIHIYDRO-11-I-PYRROLIZINE-6YL)-5-
OXOVALERIC ACID (71)
s 5 mmoles of 5-(n-hexyl)-7-phenyl-2,3-dihydro-11-I-pyrrolizine G9 are reacted
in 20 ml of ChIZCI2 with 5 mmoles (0.5 g) of glutaric acid anhydride and 11
mmoles (1.47
g) of A1CL~ similarly to the procedure far preparing the 5-(Biphenyl-2,3-
Bihydro-lI-I-
pyrrolizinyl)-5-oxovaleric-acids. The product is purified by column
chromatography (silica
gel, 1st n-hexane/ether 1+ 1, 2nd ether). After the eluates are concentrated,
71 remains
1o as an oil.
Yield 0.40 g (21 %)
C2aH3tN03 (381.5)
tH-NMR: a (ppm) = 0.69-1.07 (m. 3H, -CH3), 1.07-1.98 (m, 10H, -CHr), 1.98-
2.65 (m, 6H, C-2 and -CO-~1- -CHz-~-CO-), 2.65-2.95 (m, 4H,
G1 t.uid Pyr-CH2-), 3.91 (t, 2H, J=7 Ha, C-3), 7.07-7.47 (m, 5H,
Arom. )
EXAMPLE 72
5-(5-(n-HEXYL)-7-PHENYL-2,3-DII-IYDRO-1I-I-PYRROLIZINE-6YL)-VALERIC
ACID (72)
1 mmole of 71 is reacted in 10 ml of Biethyleneglycol with 50 mmoles (2.8
2o g) of KOI-I and 30 mmoles (1.5 g) of hydrazine hydrate similarly to the
procedure for
reducing the 4-(Biphenyl-2,3-Bihydro-lII-pyrrolizinyl)-4-oxobutyric acids.
Product
purification is by means of column chromatography (silica gel /
Biisopropylether). After
concentrating the eluates, 72 remains as an oil.
Yield 0.20 g {54 %)
C2aH33N02 (367.5)
IR: vrt,~ = 3600-2400 (OH), 1710 (C~). 1605 (C~) cm-t
tH-NMR: b (pprn) a 0.72-1.12 (m, 3H, -Cl-~), 1.12-1.78 (m, 12H, -CHr), 2.08-
2.70 (m, 8H, C-2, -CHz-CO- tmd 2x pyr..CH2-), 2.90 (t, 2H. J=7 Ha,
61 ),3.87 (t, 2H, J=7 Hz, C-3), 6.95-7.44 (m, 5H, Arum. )
CXAMI'I_ES 73, 74
5-(n-I1~;XYL.)-7-I'IIENYL-2,3-DII-IYDRO-ILI-PYRROLIZINE-6-YL-CARBAL-
DGI iYDE (73) AND b-(n-I-IEXYL)-7-P~ICNYL-2,3-DII-IYDRO-11-i-PYRROLIZ1NE-5-
YL-CARBALDRI-IYDE (74)
s 35 mmoles of octanal, bromine and 2-benzyl-D 1-pyrroline arc reacted each
time similarly to the preparation for 69 and 70. The isolated pyrrolizines
then are
absorbed in 35 ml of absolute benzene and reacted with 105 mmolcs (7.7 g) of
absolute
DMF and 35 mrnoles (5.4 g) of POCK similarly to the procedure of the Vilsmeier
formylation of the Biphenyl-2,3-dihydro-lI-I-pyrrolizines. The two products
are separated
,o by column chromatography with silica gel, first elution taking place with n-
hexane/diiso-
propylether 73 and then with diisopropylether 74. Both products will be in the
form of
oils after the eluates have been concentrated.
Proof of structure for 73: the same product is obtained in the Vilsmeier
formylation of 69.
15 73 Yield 1.74 g
C2oH2sN~ (295.4.
IR: vr"~ a 1660 (C~7), 1610 (C~) c;m-~
~H-NMR: b (ppm) = 0.73-1.07 {m, 3H, -Ct-~), 1.07-1.86 (m, 8H, -CHr), 2.30-
2.70 (m, 2H, C-2), 2.74-3.05 (m, 4H, C-1 a~a Pyr-CHZ-), 3.91 (t, 2H,
J=7 Hz, C-3), 7.05-7.46 (m, 5H, Aran.), 9.87 {s, 1H, -CHO)
20 74 Yield 2.13 g
C2ort25N0 (295.4)
IR: v~ = 1655 {C~), 1610 (C~) cm-t
~H-NMR: 8 (ppm) = 0.68-1.01 (m, 3H, -CH3), 1.01-1.74 (m, 8H, -CHr), 2.30-
2.67 (m, 2H, C-2), 2.67-3.00 (m, 4H, C-1 ~d Pyr-CHr), 4.32 (t, 2H,
J=7 Hz, C-3), 7.19-7.4& (m, 5H, Aran.), 9.67 (s, 1H, -CHO)
38
CXAMPLI'75
3-(fi(n-I iEXYL)-7-1'f-IENYI: 2,3-DII-IYDRO-lI-I-PYRROLIZINE-5-YL)-ACRYLIC-
ACID EThIYLES'hCRs (75)
mrnoles ( I.48 g) of 74 are reacted similarly to the procedure for preparing
s the Biphenyl-2,3-dihydro-I HI-pyrrolizinyl-acrylic-acid ethylesters.
Purification is by column
chromatograpi~y (silica gel, petroleum ether SO-70/acetic-ester 7+'l).
Following
concentration of the eluate, the product is obtained as an oil
Yield 0.45 g (25 °lo)
C2aN3tN02 (365.5)
IR: vr"~ x 1705 (C~), 1610 (C~) Cm-t
tH-NMR: 8 Cpprn) = 0.62-1.02 (m, 3H, -CH3), 1.02-1.76 (m, 11 H, -CH2- and -
O-CH~t- ), 2.23-3.01 (m, 6H, C-2, Pyr-CH2- ~a C-1), 3.93-4.39
(m, 4H, G3 and -0-CHr), 5.89 (AB, 1 H, J=16.1 Hz. =CH-CO-),
7.04-7.48 (m, 5H, Arum.), 7.68 (AB, 1H, J=16.1 Hz, Pyr-CH=)
is EXAMPLE 76
3-(6-(n-HEXYL)-7-PI-IENYL-2,3-DIHYDRO-1I-I-PYRROLIZINE-5-YL)-ACRYLIC
ACID (75)
1.0 mmole (0.37 g) of 75 is reacted similarly to the procedure for
saponifying the Biphenyl-2,3-dihydro-lI-I-pyrrolizine-acrylic-acid
ethylesters.
2o Yield 0.20 g (59 %)
melting point 140 ° C (with dissociation)
C22H27N02 (337.5)
iR: vr"~ = 3300-2200 (OH), 1670 (C~), 1595 (C~) cart
tH-NMR (c!u-DM50): 5 (ppm) = 0.58-0.96 (m, 3H, -CH3), 0.96-1.60 (m, SH,
-CHz-), 2.18-2.73 (m, 4H, C-2 ana Pyr-CHr), 2.86 (t, 2H, J=7 Hz,
C-1), 4.15 (t, 2H, J=7 Hz, C-3), 5.85 (AB, 1H, J=15.8 Hz, ~H-CO-),
7.02-7.53 (m, 5H, Arum.), 7.50 (AB, 1H, J=15.8 Hz, Pyr-CH=)
39
EXAMPLE 77
6-CYCLOI-II;XYL-7-I'1IENYL-2,3-DIIIYDRO-11I-PYRROLIZINE (77)
SO mmoles of I3rz, dissolved in 10 ml of CCI,,, are slowly dripped into the
solution of 50 mrnolcs (G.3 g) of cyclohexylmethylketone in 40 ml of CCI4 with
stirring and
s shielding from light. Tl2is batch is then mixed with 50 ml of CI-lzClz and
carefully washed
twice with a S % NaI-ICO~ solution to neutralize the hIbr. The organic phase
is dried by
means of NazSOa and concentrated. The residue is dissolved in 30 ml of ethanol
and
reacted with the solution of 50 mrnoles of 2-benzyl-~ 1-pyrroline in 20 ml of
ethanol.The
batch is stirred for 24 h at room temperature. After adding 25 ml of a
saturated NaI-IC03
io solution, stirring proceeds for another 24 h. Then 500 ml of 10 % NaCi are
added and
extraction with ether is carried out twice. The organic phases are dried
(Na2S04). The
solvent is distilled and the product is isolated by column chromatography
(A1203,
n-hexane/ether 9+1). The oil (3.0 g) remaining after the eluate was
concentrated is
impure. It is further reacted without any purification.
,s EXAMPLE 78
6-CYCLOI-IEXYL-7-PI-IENYL-2,3-DII-IYDRO-1H-PYRROLIZINE-SYL-CARBAL-
DEI-IYDE (78)
3.0 g of impure 77 are dissolved in S ml of absolute benzene and reacted
zo with 12 mmoles of absolute DMF (0.88 g) and 4 mmoles (0.61 g) of POC13
similarly to
the procedure for the Vilsmeier formylation of the Biphenyl-2,3-dihydro-lI-I-
pyrrolizines.
Purification is by column chromatography (silica gel, ether/n-hexane 3+ 1).
Then the
product is precipitated with ethanol
Yield O.C6 g (4.S % referred to the cyclohexylmethyl ketone)
2s Melting point 135 ° C
C2pHg3NO2 (293.4)
IR: vr,,,e"~ = 1645 (C~), 1610 (C~C) cm-j
~H-NMR: g (ppm) = 1.01-2.13 (m, 10H, -CH~-of the cyclohexyl ring), 2.24-
3.03 (m, 5H, C-1, C-2 and Pyr-CH<), 4.35 (t, 2H, J=7 Hz. C-3), 7.07-
7.51 (m, 5H, Arcxn.), 9.93 (s, 1H, CHO)
40
EXAM I'L.L: 79
3-(G-CYCLOI-iEXYL-7-PI-iENYL-2,3-DII-iYDRO-lI-I-PYRROLIZINE-5-YL)-ACRYLIC-
ACID ETI-IYLES'I'I?RS (79)
s 2 mmoles of 78, dissolved in 4 ml of absolute CI-IZC12, are reacted with a
solution of 2 mmolcs (0.86 g) of ethoxycarbonyl triphenyl phosphoniurn bromide
in 3.5
ml of absolute ethanol and with a solution of Na-ethanolate prepared from b
mmoles
(0.14 g) of sodium and 3 ml of absolute ethanol in a manner similar to the
procedure for
preparing the Biphenyl-2,3-dihydro-lI-I-pyrrolizinyl acrylic-acid ethylesters.
The
~o purification takes place by column chromatography (silica gel, petroleum
ether 50-
70/acetic-ester 7+2). The oil remaining after concentration solidifies after
some time.
Yield: 80 mg (I1 %); melting point: 190 ° C
C2~H~N02 (363.5)
IR: v~ ~ 1715 (0),1605 (C~) crn-I
~H-NMR: 8 (ppm) ~ 0.99-1.99 (m, 10H, -CHr of the cyclohpxyl ring), . 1.31 (t,
,s 3ti, J= 7 Hz, -O-CHI), 2.27-2.93 (m, 5H, C-1, C-2 and
Pyr-CH<), 4.17 (t, 2H, J=7 Hz, C-3), 4.23 (q, 2H, J=7 Hz, -O-~CHr),
5.85 (AB, 1H, J=16.2 Hz, ~H-CG), 7.04-7.48 (m, 5H, Arom.), 7.91
(AB, 1 H, J=16.2 Hz, Pyr-CH=)
EXAMPLE 80
3-(6-CYCLOI-IEXYIr7-PHENYL-2,3-DII-IYDRO-lI-I-PYRROLIZINE -S-YL)-ACRYLIC
2o ACID (80)
0.22 mmoles (80 mg) of 79 dissolved in S ml of ethanol are reacted with 3
ml of 10 % adueous KOH similarly to the procedure for saponifying the Biphenyl-
2,3-
dihydro-lI-I-pyrrolizine acrylic-acid ethylesters (see p 219).
Yield 37 mg (SO %)
2s melting point 201 ° C (with dissociation)
C22H25N02 (335.4)
IR: vm~ = 3200-2400 (OH), 1660 (C~), 15$5 (C~) crtrt
~H-NMR: 5 (ppm) = 0.98-2.05 (m, tOH, -CH2- of the cycloheuyl rmp, 2.29-
2.98 (m, 5H, G1, C-2 ur~d Pyr--CHo), 4.21 (t, 2H, J=7 Hz, C-3), 5.87
(AB, 1H, J=16.0 Hz, ~H-CO-), 7.02-7.55 (m, 5H, Arorn.), 8.01 (AB,
1H, J=16.0 Hz, Pyr-CH=)
6'S n'~7 r A,
. / ~ .4, ~' .,) ~ . s 41
GENERAL PROCEDURE FOR 'I'I-IE N-ALKYLATION OF 2-METHYL-3,4-
DIPI-iENYLPYRROLE
2 I11I11(lleS (0.47 g) of 2-methyl-3,4-Biphenyl-pyrro(e (AUS'r.,d. CI-IEM.
1966,
19, pp 1871-1885) (and] 2.2 mmoles of p-toluene sulfonic-acid rnethylester (in
81b) or
s alkylbromide (for the remaining compounds) and 1 mmole (0.32) g) of
tetrabutyl-
ammonium bromide are mixed with 10 ml ether and 5 m1 of 50 °lo aqueous
NaOI-I.
While agitating strongly, the mixture is heated for 8 h to slow boiling.
Thereupon the
batch is poured into 100 ml of I-IZO acrd extracted twice with ether. The
ether phases are
washed with 1 °lo II~i'O,, and I-1Z0, dried by means of Na2S04 and
concentrated. The
1o product isolation takes place by column chromatography (A1203, n-
hexane/ether 9+ 1).
The compounds so obtained and their physical properties are listed in Table
12 below.
EXAMPLE 88
1-NEOPENTYL-2-METHYL-3,4-DII'I-IENYLPYRROLE (81i)
The above compound cannot be synthesized. I-lowever it may be prepared
as follows:
2 mmoies (0.46 g) of 2-methyl-3,4-Biphenyl-pyrrole, 2.6 mmoles of
potassium-t-butylate and 3 mmoles (0.45 g) of neopentyl bromide in 6 ml of
absolute
zo DMSO are heated for 45 minutes to 130 - 140 ° C. Following cooling,
100 ml Hz0 are
added, acidification with dilute I-I3P04 and next double extraction with ether
are carried
out. The ether phases are washed with I-h0, dried by means NazSO4, and
concentrated.
The product is isolated from the residue by column chromatagraphy (A12O3, n-
hexane/ether 9+ 1) and precipitated with ethanol (similarly to the case for
81h).
zs YielB: 0.31 g (51 °~o); melting point: 132 ° C.
Cz2H~N (303.4) COMPUTED C 87.1 I-I 8.30 N 4.6
MEASURED C 86.9 H 8.21 N 4.5
IR: vr"~= 1605 (C~) cart
MS: m/z (rel.lnt.) = 303 (89°/°, M+~), 288 (9%, M+-CH3, °
273.74), 247 (58°I°,
M+-C4H8, ' 201.34), 246 (100%, 247-H)
tH-NMR: b (ppm) = 1.02 (s, 9H, -CH3), 2.19 (s, 3H, Pyr-CHI), 3.66 (s, 2H,
>N-CHz-), 6.75 (s, iH, C-5), 7.06-7.33 (m, 10H, Arom.)
~0~.~~0~.
Table 12;
H3 N
R
EXAMPLE R ; I R ( C=C
MELTING )
POINT
IJr. C
811 CH3 81b
82 C2H5 81c I 1600
83 n-C3H7 81d 80 1605
I
i
84 n-C4H9 81e~ 1610
~
852 n-C5t111 81f
) 1
862 n-C6H13 81g
87 iso-C4H9 8lta; 1605
116
1) Tetrahedron Lett. 1969, 55, 4875-4878
Chem. Pharm. Bull. 1974, 22, 61-69
2) mixture with alkyl bromide; it is reacted withwt further purification
43
GENERAL I'ItOCEDURE FOIZ PREPARING 'I'I-IE 3-(5-ME'rI-iYL-3,~-DIPI-IENYL-
I'YRROLE-2-YL)-PROPIONIC-ACID METI-IYLES'1'ERS
1 mmole of 2-methyl-3,4-diphenylpyrrole or of 1-alkyl-2-methyl-3,4
diphenylpyrrole 82b-i and 1.5 mmoles (0.13 g) of acrylic-acid methylester are
dissolved
s in 4 ml of dichlorocthane. Following addition of O.OG mi of BF3 ethylether
complex, the
batch is stirred for 1 h at room temperature and after 15 minutes in each case
again the
same amounts of acrylic-acid methylcster and BF3 ethylester complex are added.
Then
the batch is mixed with 50 ml of I-IZO and extraction with ether is performed
twice. The
ether phases are washed with I-IzO, dried by means of NazS04 and concentrated.
The
~o product is isolated from the residue by column chromatography (A1203, n-
hexane/ether
1+4 (82a) or 1+1 (82b,c) or 3+2 (remaining compounds)). After the eluate has
been
concentrated, an oil remains.
The compounds so obtained and several of their physical properties are
listed in Table 13 below.
is GENERAL PROCEDURE FOR THE SAPONIFICATION OF TI-IE 3-(5-METHYLr3,4-
DIPI-IENYLPYRROLE-2-YL)-PROPIONIC-ACID METIIYLESTERS
The solution of 0.4 mmoles of 3-(2-methyl-3,4-diphenylpyrrole-5-yl)-
propionic-acid methylester 82a or 3-(1-alkyl-2-methyl-3,4-diphenylpyrrole-5-
yl)-propionic-
2o acid methylester 82b-i in 3 ml of ethanol is heated to boiling. 2 m1 of 10
% aqueous
KOH previously degassed by boiling are dripped into the batch which then is
heated with
reflux for another 5 minutes. Following cooling, the batch is poured into 100
ml of 5 %
NaCI solution and is washed twice with 50 ml of ether. The aqueous phase is
acidified
with dilute I-I3P04 and extracted twice with 50 ml of ether. The ether phases
are washed
is with 100 ml of I-Iz0 and dried by means of NazS04. The solvent is
distilled, and where
called for the product is precipitated with hexane.
'the compounds so obtained and several of their physical properties are
shown in Table 14.
44
Table 13
M3
I
R
cnnMYLE R
Nr.
IR(C=0)
89 H 82a
1730
90
CH3 82b 1790
91
C2H5 82c 1740
92
n-C3H 82d
7 1740
93
n-C4H9 82e 1745
,
94
n-C5H11 82f 1740
95 n-C6H13 82g 1740
~
96 i.so-C4 82h 1740
H9 j
Neopentyl 82i. 1740
I
Table 14
COOH
i
R
-.-
ExnMPLE R
Nr.
MELTIMG POINT
IR (C=O
C
98 H . .
CH 60 17 0
3
100 C H 108 1710
2
101 157 1715
n-C H
3 7
102 128 1705
n-C H
4 9
>4~CctiSS~c
lU4 n-C H ..,l 1715
5 11 _
105 n-C H 1715
6 13 _
1715
106 iso-C
H
4 50
107 9 1715
Neopentyl
48
1710
~Q1~~ ~~-
GENERAL PROCEDURE FOR Tf-1E N-ALKYLATION OF 5-METHYL-2,3-
DIPI-IENYLPYRROLE
S mmoles ( 1.17 g) of 5-methyl-2,3-diphenylpyrrole (AUST. J. CI-1EM. 1966,
s 19, pp 1871-1885), S.S mmoles of toluene sulfonic-acid methylester or of
alkyl bromide
and 1 mmole (032 g) of tetrabutyl ammonium bromide are mixed with 10 ml ether
and
S ml of 10 % aqueous NaOI-I. The reaction henceforth is carried out in the
manner of
the procedure for the N-alkylation of 2-methyl-3,4-diphenylpyrrole.
The compounds so obtained and some of their physical properties are
~o shown in Table 15.
GENERAL PROCEDURE FOR PREPARING 'r1-IE (5-METI-IYL-4,5-DIPI-IENYL-
PYRROLE-3-YL)-ACETIC-ACID ETF-IYLESTERS.
2.5 mmoles of methyl-2,3-diphenylpyrrole (AUST. J. CI-IEM. 1966, 19, pp
1871-1885) or of 1-alkyl-5-methyl-2,3-diphenylpyrrole 84b-j, dissolved in 2 ml
of absolute
15 toluene, are reacted similarly to the procedure for preparing the Biphenyl-
2,3-dihydro-1H-
pyrrolizine-acetic-acid ethylesters. Following column chromatography (silica
gel, n-
hexane/ether 1 + 1 in Example 117 or AIZO3, n-hexane-ether 9+ 1 in the
remaining
compounds), the products are obtained in the form of oils
The compounds so obtained and the infra-red Bata relating to them are
zo shown in Table 16.
Table IS
H
R MELTING P'r.1R(C=C)
EXAMPLC C
~o
Nr.
108 CH3 84b 149 1605
109 84c 68-69 1605
C2H5
110 n-C3H~ 84d - 1605
H 84e _ 1605
n-C
111 9
4
84f 1605
112 n-C5H11
1s 84g 1605
113 n-C6H13
114 1) 84h
n-C8H17
115 iso-C4H9
84i 107 1600
116 Neopentyl 84J 89 1605
20 1) mixture with n-CBH~~Br, reacted without further purification
~fl.~ ~~~.~
T:~ble IG
H~C20
EXAMPLE R ~ I R ( C=O
Nr.
117 H 85a 1730
118 CH3 85b ~ 1740
119 C2H5 85c 1735
120 n-C3H7 B5d 1740
121 n-C4H9 85e 1735
122 n-C5H11 85f 1740
123 n-C6H13 85g 1740
124 n-C8H17 85h 1740
125 iso-C4H9 85i 1735
126 Neopentyl 85j 1735
GENERAL PROCEDURE FOR SAPONIFYING THE (2-METHYL-4,5-DIPHENYL-
PYRROLE-3-YL)-ACETIC-ACID ETHYLESTERS
The solution of 0.6 mmoles of (2-methyl-4,S-diphenylpyrrole-3-yl)-acetic-
acid ethylester or of (1-alkyl-2-methyl-4,5-diphenylpyrrole-3-yl)-acetic-acid
ethylester in
zo 5 ml of ethanol is heated to boiling. Then 3 ml of 10 % aqueous KOI-I
previously
degassed by boiling are dripped into the batch which is further heated for 1 h
with reflux.
Following cooling the batch is poured into 100 ml of S °Jo NaCI
solution and the mixture
is then acidified with 8 % I-I3P04 and extracted three times with ether. The
organic
phases are dried by means of NaZS04 and concentrated. Product purification
takes place
zs by column chromatography (silica gel, 1st n-hexane/ether 1+1 in the
Examples 127-131,
fl16~~~
or 2-H 1 in Examples 132-136, 2nd ether). The eluates are concentrated down to
a few
ml. After adding n-hexane and concentrating again, the product precipitates.
The compounds so obtained and their physical data are shown in Table 17.
Table 17
HOC
EXAMPLE R MELTING POINT IR(C=O)
Nr. C
127 H 86a 110 1710
128 CH3 86b 161 1710
129 C2H5 86c 177 1705
130 n-C3H7 86d 172 1700
131 n-C4H9 86e 129 1705
132 n-C5H11 86f 121 1710
133 n-C6H13 86g 127 1710
134 n-C8H17 86h 45 1715
135 iso-C4H9 86i 168 1710
136 Neopentyl 867 163 1710
~fli~~~Q~
GENERAL PROCEDURE FOR PREI'ArIING 1'I-IE 6-ARYL-7-PI-IENYL-2,3-
DII-IYDRO-IN-PYRROLIZINES OR TI-IE 6-PI-IENYL-7-ARYL-2,3-DIHYDRO-1H-
PYRROLIZINES
20 mmoles of aromatics-substituted' or unsubstituted a-bromo-acetophe-
s none dissolved in 25 ml of CI-IzClz are mixed with the solution of 20 mmoles
of
unsubstituted or aromatics-substituted" resp. 2-benzyl-b, l-pyrroline in 50 mI
of ethanol
and are stirred for 24 h aL room temperature. lfien 20 ml of saturated aqueous
NaI-IC03
solution are added and the batch is stirred for another 24 h at roam
temperature. The
batch is poured into 500 ml of 5 % NaCI solution and is extracted three times
each time
io with 100 ml of ether/CI-IzClz 3+ 1. 'Ifie organic phases are dried by means
of NaZS04
and processed in the manner discussed below.
* a-bromo-3-chloroacetophenone, a-bromo-3,4-dimethoxyacetophenone, a-bromo-3,4-
d9chloroaceto
~s phenone and a-bromo-4-phenoxyacetophenone are not available commercially.
They are prepared as
follows:
A solution of 20 mmoles (3.2 g) of bromine In 10 ml of CI-hCta is slowly
dripped into the
solution of 20 mmoles of 3-chloroacetophenone, 3,4-dimethoxyacetophenone, 3,4-
dichloroacetophenone
or 4-phenoxyacetophenone in 15 ml of CHtCt~ and 20 ml of dioxan. Thereupon the
batch is mixed with
20 50 ml of CI-tlCtZ and carefully washed twice with 5 °,6 NaHCO,
solution to remove the Hbr. The organic
phase is dried by means of NaZ SO, and concentrated. The residue is reacted
directly as described above
with 20 mmoles of 2-berrzyl-D 1-pyrroline.
** 2(4-chlorobenzyl}-A1-pyrrollne
The preparation Is similar to that for 2-benzyl-D 1-pyrroline (J. AMER. CHEM.
SOC. 1932,
2s 54, pp 3971-3976)
** 2-(4-methylb~nzyl}-d1-pyrrollne
A Grignard reagent is prepared from O.i5 moles (3.6 g) of Mg and 0.15 moles
(21.1 g)
of methylbenzyl chloride in 150 ml of absolute ether. After dripping 0.15
moles (15.5 g) of 4-
chlorobutyronitrile dissolved in 100 ml absolute ether into the batch, same is
heated for 2 h to reBux. Next
so the ether is distilled and 200 ml of absolute xylene are added. After
further boiling with reflux for 2 h, the
batch is mixed with 100 ml of H~ 0 while being ice-cooled and is acidified
with dilute H, PO,. The aqueous
phase is made alkaline with concentrated NH, and ice-cooling and the generated
precipitate is evacuated.
The filtrate is extracted three times with 50 ml of Cf-~Ct~ and the
precipitate is washed with 100 ml of
~~~~~~i~ ~ 51
CFi~ Ct~. The CFis Cll solutions are combined, dried by means of Na; SO, and
concentrated. The product
Is Isolated from the last residue by distillation (boiling point 117 °
C at 0.1 torr).
Yield 3.7 g (14 96)
C,iH,sN (173.3)
Ir: vmax = 1640 (C=N), 1605 (C=C)/cm
** 2-(4-melhoxybenzyl~-G1-pyrrollne
4-methoxybenzyl magnesium chloride Is prepared from 3.0 moles (72.9 g) of Mg
and 0.15
moles (23.5 g) of 4-methoxybenzylchlorlde fn 500 ml of absolute ether. The
product is reacted further
with 0.'15 moles (15.5 g) of 4-chlorobutyronitrile and is purified by
distillation (boiling point 142 C at 0.1
1o torr).
Yield 2.4 g (8 °~6)
C,aFi,sNO (189.3)
Ir: v,°d" = 164~ (C=N), 1610 (C=C)/Cm
GENERAL PROCEDURE FOR PREPARING 3-(6-CHLOROPHENYL- AND 6-
NITROPHENYL-7-PHENYL,~2,3-DIl-IYDRO-1H-PYRROLIZINE-S-YL)-PROPIONIC-
ACID METHYLESTERS
4 mmoles of 6 chloro- or 6-nitrophenyl-7-phenyl-2,3-dihydro-1H-pyrrolizine
2o and 6 mmoles (O.S2 g) of acrylic-acid methylester are dissolved in 16 ml of
absolute
dichloroethane. After adding 0.24 ml of BF3 ethylene complex, the batch is
stirred for 1
h at room temperature, and the same amounts of acrylic-acid methylester and
BF3
ethylether complex are added in each case again after 15 minutes. Thereupon
the batch
is poured into 100 mi of 10 % NaCI solution and is extracted twice with
ether/CHzCI2
is 3+1. The organic phases are dried by means of Na2S04 and concentrated. The
residue
then is processed in the manner discussed below.
GENERAL PROCEDURE FOR SAPONIFYING THE 3-(6-CHLOROPI-IENYL-
AND 6-NITROPHENYL-7-PHENYL-2,3-DII-iYDRO-1H-PYRROLIZINE-6-YL)-
PROPIONIC-ACID METHYLESTERS
ao The solution of 1 mmole of the corresponding pyrrolizinyl-propionic-acid
methylester in 10 ml of ethanol is heated to boiling. S ml of 10 % aqueous KOI-
I solution
previous degassed is dripped into the batch which is then heated another 5
minutes with
5z
reflux. Following cooling the batch is poured unto 100 ml of 5 % NaCI solution
and
acidified with 8 % I-I~P04 and extracted three times with ether/CI-IZC12 3+1.
The
organic phases are dried by means of Na2S04, concentrated, and processed in
the manner
described above.
s GENERAL PROCEDURE FOR THE VILSMEIER FORMYLATION OF
AROMATICS-SUBSTITUTED G,7-D1PHENYL-2,3-DIHYDRO-1H-PYRROLIZINES
6 mmoles of the corresponding pyrrolizine dissolved in 6 ml of absolute
benzene are reacted with 18 mmoles (1.32 g) of absolute DMF and 6 mmoles (0.92
g) of
POCl3 similarly to the procedure far the Vilsmeier formylatian of the Biphenyl-
2,3-
,o dihydro-lI-I-pyrrolizines.
GENERAL PROCEDURE FOR PREPARING AROMATICS-SUBSTITUTED 6,7-
DIPHENYL-2,3-DII-IYDRO-lI-i-PYROLLIZINYL-ACRYLIC-ACID ETHYLESTERS
2.5 mmoles of the corresponding carbaldehyde dissolved in 5 rnl of absolute
CI-i2C12 are reacted with the solution of 2.5 mmoles (1.08 g) of
ethoxycarbonyl-
,a methyltriphenyl-phosphonium bromide in 4 ml of absolute ethanol and with a
solution of
Na-ethanolate prepared from 7.5 mmoles (0.I7 g) of sodium and 3 ml of absolute
ethanol
in a manner similar to the procedure for preparing the Biphenyl-2,3-dihydro-lI-
I-
pyrrolizinyl-acrylic-acid ethylesters.
GENERAL PROCEDURE FOR SAPONIFYING AROMATICS-SUBSTITUTED 3-(6
zo 7-DIPHENYL-2,3-DII-IYDRO-1H-PYRROLIZINE-5-YL)-ACRYLIC-ACID ETHYL
ESTERS.
1.2 mmoles of the corresponding pyrrolizinyl-acrylic-acid ethylesters
dissolved in 50 ml of ethanol are reacted with 10 ml of aqueous KOH similarly
to the
procedure for saponifying the 3-(Biphenyl-2,3-dihydra-lI-I-pyrrolizine-5-yl)-
acrylic-acid
zs ethylesters.
GGNf~RAL. I'ROC1-~DURC FOR HYDROGENATING AROMATICS-SUBSTITUTED
3-(6,7-DII'f IENYfd 2,3-DII~IYDRO-Ifd-PYRROLIZINE-5-YL) ACRYLIC ACIDS
0.9 mmoles of the corresponding pyrrolizinyl acrylic acid are dissolved in
30 ml of absolute TI-IF. After adding 10 ml of absolute ethanol and a spatula
tip-ful of
s PtOz (alternatively, palladium also may be used), hydrogenation is carried
out in the
autoclave for about 4 h at 15 bars. Several times a spatula tip-ful of fresh
Pt02 (or
palladium) is added. Upon complete reaction (thin-layer chromatography: silica
gel,
TI-IF), the catalyst is evacuated, the solvent is distilled and the product is
isolated.
GENERAL PROCEDURE FOR SPLi'I~I'ING THE ARYLME'I'f-IYLETHERS
~0 0.5 mmoles of the corresponding methyl compound dissolved in 5 ml of
absolute CHZCIz are dripped into the solution of 0.20 ml of BBr3 in 3 ml of
absolute
CHzCIz at -80 ° C. The mixture is allowed to rise to room temperature
in about 8 h.
After adding 30 ml of HZO, extraction is carried out three times with ether.
The organic
phases are washed twice with a saturated NaCI solution, dried by means of
NazS04 and
is are processed.
The compounds so obtained and their physical properties are shown in the
Tables 18 through 27 below.
2f~~.~ ~~~
Table 18
Jtd
~p8(8
H
FXAt4PLE ' I R ( C=C
Nr. Rmeta ~para vLo~. )
R para P.r.
138 C1 H H 1600
1391) H C1 H
140 N02 H H 117 1605
141 H N02 H 132 1595
142 H H CI 156 1605
1~ Chemiker-Zeitung 1986, 110, 267-271
~0~~~~~~
Table 19
~I nars
~t8
af8
EXAMPLER R R ~ MELTING j R ( C=O
Nr. meta para para E'T )
C
143 C1 H H 99 1735
144 H C1 H 98 1730
145 NOZ H H 1740
146 H NOZ H 1740
147 H H C1 111 1730
56
Table 2Q~
Sara
R~~
N
H2~2-CQOH
GXAMPL!: Rmeta Rpara R~para M~;T. IR(C=O)
Nr. QT.
C
143 C1 H H 99 1735
144 H C1 ti 98 1730
145 NOZ H H 1740
146 H NOZ H I 1740
147 H H C1 111 1730
57
Table Z1
EXAMPLE MELT. IR
Nr. R PT. (C=O)
C
153 H 1605
(C=C)
154 CHO >84 1650
155 CFi=CtiCOOC2H5 122 1715
( E )
156 CH=CHCOOf-I (E) 229 1660
157 (CH2)z OOH 157 1710
Table 22
~o
r ~~a
~,
io ~xNr ~'c Rmeta Rpara R~ para MeLm. OR( C=C
p~r. )
'C
158 CH3 H H 1605
159 H CH3 H 1605
160 OCH3 H N 1605
1611) H OCH3 H
1621) OCH3 OCH3 H
1632) H O-C6H5 H
164 C1 C1 H 119 1605
165 H H CH3 101 1610
166 H H OCH3 1605
20
1)
Chemiker-Zeitung
1986
110,
267-271
2) the product is impure; it is made to react without further purification
Table 23
?~
EXN~PLERmeta Rpara R~para M~oC' IR(C=O
~~w
167 CH3 H H 128 1645
168 H CH3 H 142 1645
169 OCH3 H H 117 1635
1701 H OCH3 H
1711 OCH3 OCH3 H
172 H O-C6H5 H 135 1635
173 Cl CZ H 162 1640
174 H H CFi3 102 1645
59
l) Chemiker-teitung 1986 110, 267-271
Table 24
~para
E?(APIPLERmeta H ara R~ ara MELT, IR(C=O)
Nr. p p eT.
C
175 CH3 N H 142 1710
176 H CH3 H 158 1710
177 OCH3 Fi H 119 1710
178 H OCH3 H 164 1705
179 OCH3 OCH3 H 133 1705
180 H O-C6H5 H 173 1705
1B1 C1 C1 H 122 1715
182 H H CH3 222 1705
COOCZHS
~rlr~ee Zs
EXAMPLLE2 t'. R' MELTING IR(C=O)
((~~td ~uI'u ~7dra PT.
Nr. C
183 CH3 H H 216 1660
184 H CH3 H 221 1680
185 OCH3 H H 205 1670
186 H OCH3 H 216 1675
187 OCH3 OCN3 H 216 1660
188 H O-C6H5 H 221 1660
189 C1 C1 H 229 1660
190 H ti CH3 118 1665
,..
H~~~
~GOH
sz
1'f1Af11
'1('able 2G
AW
a
cx.~MPLrRmeta Rpara ~~para M~L~r. I~(C=O)
Nr. ~T.
C
191 CH3 H H 125 1710
192 H CH3 H 122 1710
193 OCH3 H H 111 1710
194 H OCH3 H 73 1710
195 OCH3 OCH3 H 152 1725
196 OH H H 87 1710
197 H OH H 187 1700
198 OH OH H 76 1710
199 H 0-C6H5 H 178 1705
200 C1 C1 H 144 1710
201 H H CH3 182 1705
(CH2)2-COOH
Table 27
a
GXAt~iPLER R MELT I R (
N.r. p~r~ . PT C=O )
'C
io 202 tl C1 118 1605 (
C=C:
)
203 (Ctf2)2COOCH C1 113 1735
204 (CH2)2COOH C1 207 1710
3
sa
~s EXAMPLE 205
2,2-DIMETI-IYL-6-(4-FLUORO)-7-PHENYL-2,3-DII-IYDRO-1H-PYRROLIZINE (lv)
20 mmoles of a -chlorine-4-fluoroacetophenone dissolved in 25 rnl of CI-IZCIz
are mixed with the solution of 20 mmoles of 2-benzyl-4,4-dimethyl-D 1-
pyrroline [inj 25 ml
zo of ethanol and the batch is stirred for 24 h at a bath temperature of 70
° C. Thereupon
20 ml of saturated aqueous NAI-IC03 solution is added and agitation continues
for
another 24 h at the same temperature. The batch is poured into S00 ml of S
°~o NaCI
solution and is then extracted three times each time with 100 ml of ether/CI-
IZC12 3-h 1.
The organic phases are dried by means of Na2SOa. The product is precipitated
by column
zs chromatography (AhO~, n-hexane/ether 9+1) in the form of an oil
Yield 3.1 g (S1 %)
C2~H2oFN (305.x)
IFi: v~ = 1605 (CaC) cm-~
1H-PlMR: b (p~m) = 1.28 (s, 6H, -CHI), 2.78 (s, 2H, C-1), 3.71 (s, 2H, C-3),
6.64 (s, 1H, C-5), 6.75-7.60 (tn, 9H, ~.~rom.)
ExAMPI-.E 20(~
2,2-D1 ME'I'I-lYL-G-(~1-IvLL10It0)-7-PI IENYL-2,3-DII-IYDRO-II-I-
I'YltROI.OZINE-5-YI,-
CAItt3ALDEIIYDE (Gv)
19 mmolcs of iv dissolved in 9 ml of absolute benzene are reacted with 27
mmoles (1.97
s g) of absolute DMP and 9 mmoles (1.38 g) of I'UCh in a manner similar to the
Vilsmeier
forrnylation of the Biphenyl-2,3-dihydro-11-I-pyrrolizines. Purification is by
means of
column chromatography (silica gel, n-hexane/ether 2-H 1). When the eluates are
concentrateB, the product precipitates.
YielB: 0.9 g (30 %) melting point: 186 ° C
C22H2pFN0 (333.4)
,o
1R: vr,.,~ = 1645 (C~), 1610 (C~) cm-1
1H-NMR: a (ppm) = 1.33 (s, 6H, -CH3), 2.83 (s, 2H, C-1 ), 4.18 (s, 2H. C-3),
6.90"7.44 (m, 9H, Arum.), 9.38 (s, 1H, -CHO)
,s EXAMPLE 207
3-(2,2-DIMETI-IYL-6-(4-PLUORO)-7-PI-IENYL-2,3-DII-IYDR~-lI I-PYRROLIZINE-5-
YL)-ACRYLIC-ACID ETI-IYLESTERS (34v)
2 mmoles of Gv Bissolved in 15 ml of absolute CI-IzClz are reacted with the
solution of 2 mmoles (0.8G g) of ethoxycarbonyl methyltriphenylphosphonium
bromide in
Zo 4 ml of absolute ethanol and a solution of Na-ethanolate prepared from 6
mmoles (0.14
g) sodium and 3 ml absolute ethanol in a manner similar to the procedure for
preparing
the Biphenyl-2,3-Bihydro-lI-I-pyrrolizinyl acrylic-acid ethylesters.
Purification is by column
chromatography (silica gel, CI-IzCl2}. After the eluates have been
concentrateB, the
product remains in the form of a foam.
2s Yield 0.33 g (41 %)
melting point 152 ° C
C26H26FN02 (403.5)
IR: vm~ = 1715 (C~), 1620 (C~) an-1
~H-NMR: b (ppm) = 1.27 (t, 3H, J=7 Hz, -O-CHI), 1.33 (s, 6H. -CH3), 2.86
(s, 2H, C-1 ), 4.00 (s, 2H, C-3), 4.19 (q, 2H, J=7 Hz, -0-~H~-CH3),
5.90 (AB, 1H, J=16.4 Hz, ~H-GU-), 6.87-7.34 (m, 9H, Arom.), 7.48
(AB, 1 H, J=16.4 Hz, Pyr-CH=)
ss
EXAMI'I_I? 208
3-(2,2-DIML'I'11YL-6(4-PLUORO)-7-PI-IENYL-2,3-DIHIYDRO-lI-I-I'YRROLIZINE-S-
YL) ACRYLIC-ACID (35v)
0.6 mmoles c~f 34v dissolved in 30 mt of ethanol are reacted with 6 ml of
s 10 °lo aqueous KOI-d in a manner similar to the saponification of the
Biphenyl-2,3-dihydra-
lI-d-pyrrolizinyl acrylic-acid esters.
Yield 0.20 g (89 %; melting point: 242 ° C
IR: vrr,~ ~- 3300-2200 (OH), 1685, 1670 (C~), 1600 (C~) ar-t
1H-NMR (ds-DMSO): b (ppm) = 1.27 (s, 6H, -CH3), 2.84 (s, 2H, C-1 ), 4.06 (s,
2H, C-3), 5.92 (AB, 1 H, J=16.4 Hz, =CH-GO-), 6.87-7.41 (m, 9H,
,o Arum. ~~ Pyr-~H=)
EXAMPLE 20~
3-(2,2-DI M ETI-IY1J6-(PLUORO)--PI-IENYLr2,3-DII-IYDRO-lI-I-PYRROLIZINE-5-YL)-
~s PROPIONIC ACID (27v)
0.3 mmoles of 27v are reacted in the manner of the procedure for
hydrogenating the aromatics-substituteB 3-(6,7-Biphenyl-2,3-dihydro-lI-I-
pyrrolizine-5-yl)-
acrylic-acids. Palladium is used as the catalyst. The product is precipitated
by means of
hexane.
zo Yield: 0.08 g (71 %) melting point: 182 ° C
C~,HZ4FNOZ (377.5) COMPUTED: C 76.4 I-I 6.41 N 3.7
MEASURED: C 75.8 H 6.56 N 3.3
IR: v",~ = 3300-2400 (OH), i 710 (C~), 1605 (G=C) crrr-t
~H-NMR: b (pprn) = 1.30 (s, 6H, -CH3), 2.28-2.58 (m, 2H, -CHI-CO-), 2.73-
3.03 (m, 2H, Pyr-CHr), 2.81 (s, 2H, C-1 ), 3.68 (s, 2H, C-3), 6.76-
7.28 (m, 9H, Arom.)
se
EXAMPLE 210
2,2-DIMETI-IYL-C-(4-I'IIENOXYPI-IENOL)-7-PHENYL-2,3-DIHYDRO-1H-
PYRROLIZINE (lw)
20 mmoles of 2 bcnzyl-4,4-dimcthyl-d I-[pyrroline lb~ are reacted with 20
s mmoles of a -bromo-4-chloroacetophenone in 50 ml of ethanol similarly to the
procedure
for preparing the f-aryl-7-phenyl-2,3-dihydro-11-I-pyrrolizines. Purification
is by column
chromatography (AI20~, n-hexane/ether 9+ 1). Following concentration of the
eluates,
Iw remains in the form of oil
Yield 2.7 g (36 °lo)
io C27H25NO (379.5)
IFt: v~ - 1605 urxi 1595 (C~) cm's
'H-NMR: b (ppm) = 1.28 (s, 6H, -rCH3), 2.78 (s, 2H, C-1 ), 3.71 (s, 2H, C-3),
6.65 (s, 1 H, C-5), 6.78-7.57 (m, 14H, Arom.
,s EXAMPLE 211
2,2-DIMETI-IYL: 6-(4-PI-IENOXYPHENYL)-7-PI-IENYL-2,3-DII-1YDR0-II-I-
PYRROLIZINE-S-YL-CAItBALDEHYDE (Gw)
S mmoles of lw dissolved in 6 ml of absolute benzene are reacted with 18
mmoles (1.32 g) of absolute DMF and 6 mmoles (0.92 g) of POC13 similarly to
the
Zo procedure for the Vilsmeier formylation of the Biphenyl-2,3-dihydro-lI-i-
pyrrolizines.
Purification is by column chromatography (silica gel, CI-IZCIz). The product
is
precipitated by ethanol.
Yield: 0.94 g (38 %) melting point: 139 ° C
C2eH25N02 (~7.5)
IR: v,n~ = 1645 (C~), 1605 and 1590 (C~) curl
1H-NMR: 8 (ppm) a 1.30 (s, 6H, -CH3). 2.81 (s, 2H, C-1). 4.17 (s, 2H, C-3),
6.84-7.47 (m, 14H, Arum.), 9.40 (s, 1H, -CHO)
61
I:xnMl'I_r 212
3-(2,2-D I M E'r1-I Y I_-li-(4-PI-I ENOXYPI-IENYL)-7-PI-I ENYL-2,3-DII-IYDRO-
1H-
PYItI~,OLIZINC.-5-YL.)-ACRYLIC-nCID ETI-I'YLESTERS (34w)
2 mmolcs of Gw dissolved in S ml of absolute CI-I3C12 are reacted with the
solution of 2 mmoles (0.86 g) of ethoxycarbonyltrimethyltriphenylphosphonium
bromide
in 4 ml of absolute ethanol and a solution of Na-ethanolate prepared from 6
rnmoles
(0.14 g) of sodium and 3 ml of absolute ethanol in a manner similar to the
procedure for
preparing the Biphenyl-2,3-dihydro-11-I-pyrrolizinyl acrylic -acid esters.
Purification is by
column chromatography (silica gel, CI-IZCIZ). The product remains in the form
of foam
,o after the eluates have been concentrated.
Yield: 0.33 g (35 %) melting point: from 69 ° C
C32H3~N03 (477.6)
iR: v,r,~ = 1705 (C~), 1610 (C~) crrr-t
1H-NMR: a (ppm) = 1.28 (t, 3H, J=7 Hz, -O-CHI), 1.33 (s, 6H, -CH3). 2.86
(s, 2H, C-1 ), 4.00 (s, 2H, C-3), 4.19 (q, 2H, J=7 Hz. -0~H3).
5.92 (AB, 1H, ,k~16 Hz, ~CH-CO-), 6.83-7.44 (m, 14H, Arum.), 7.56
'S (AB, 1H, J=16 Hz, Pyr-CH=)
EXAMPLE 213
3-(2,2-DIMETHYL-6-(4-PI-IENOXYPI-IENYL)-7-PHENYL-2,3-DIHYDRO-1I-I-
PYRROLIZINE-5-YL) ACRYLIC ACID (35w)
zo 0.6 mmoles of 34w dissolved in 30'ml of ethanol are reacted with 6 ml of
% aqueous KOI-I in a manner similar to the procedure for saponifying the
diphenyl-
2,3-dihydro-1H-pyrrolizine-5-yl acrylic-acid ethylesters.
Yield: 0.23 g (85 %) . Illeltlrlg polllt: 198 ° C
C~hZ~N03 (449.5) COMPUTED C 80.2 I-I 6.05 N 3.1
2s MEASURED C 79.9 I-I G.07 N 2.7
IR: vr"~ = 3304-2200 (OH), 1675 (C~), 1590 (C=-C) atr~
~H-NMR (ds-DMSO): b (ppm) ~ 1.27 (s. 6H, -CH3), 2.83 (s, 2H, C-1), 4.06 (s,
ao 2H, C-3)> 5.92 (AB, 1H, J=16.2 Hz, ~H-CO-), 6.86-7.57 (m, 15H,
Arum. ~,a Pyr-CH=)
EXAMPLE 214
3-(2,2-DIME'i'IIYL-fi-(4-I'I-IENOXYPIIENYL)-7-PHENYL-2,3-DII-IYDRO-lI-I-
I'YRROLIZINE-5-YL) I'ROPIONIC ACID (2fw)
0.3 mmoles of 35w are reacted in the manner of the procedure for
s hydrogenating the aromatics-suUstituted 3-(6,7-Biphenyl-2,3-dihydro-lI-i-
pyrrolizine-S-yl)
acrylic acids. Palladium is used as the catalyst. The product is precipitated
by n-hexane.
Yield: 0.10 g (74 %) melting point: 149 ° C
CSI i~,,NO~ (451.6) COMPUTED C 79.8 I-I 6.47 N 3.1
MEASURED C 79.5 H 6.61 N 2.7
,o IR: v",~ = 3300-2400 (OH), 1710 (C=O), 1605 ,r,a 1595 (C=C) crrt-t
~H-NPvIR: b (ppm) = 1.29 (s. 6H, -CH3), 2.34-2.63 (m, 2H, -CH2-CO-), 2.77-
3.06 (m, 2H, Pyr-CH2-), 2.83 (s, 2H, C-1), 3.69 (s, 2H, C-3), 6.85-
7.47 (m, 14H, Arom.)
EXAMPLE 21S
~s 2-(2,2-DIMETI-IYL-6-(4-PI-IENOXYPF-IENYL)-7-PI-IENYL-2,3-DII-I1'DRO-lI-I-
PYRROLIZINE-5-YL) PROPIONdC-ACID ETI-IYLESTERS (54a)
2.5 mmoles of 6-(4-phenoxyphenyl)-7-phenyl-2,3-Bihydro-lI-I-pyrrolizine 1bv
dissoiveB in 3 ml of absolute toluene are reacted with 4 mmoles (0.51 g) of 2-
Biazopropionic-acid ethylesters BissolveB in 3 ml of absolute toluene in a
manner similar
zo to the procedure for preparing the Biphenyl-2,3-Bihydro-li-I-pyrrolizinyl
acetic-aciB
ethylesters [ "s.S."201]. Isolation is by means of column chromatography
(A1203, n-hexane-
/ether 9+ 1). The oily product (0.45 g) remaining after concentration of the
eluates is
impure. It is reacteB further without aBBitionai purification.
~~3~ ~~ ~
CXAMPLL 216
2-(2,2-D1M~T1-IYL-6-(4-PI ICNOXYI'FICNYL)-7-PI-ICNYL-2,3-DII-IYDRO-lI-I-
PYRROLIZINE-5-YL) PROPIONIC ACID (5Ga)
s The impure S4a (0.45 g) dissolved in 10 ml of ethanol is reacted with 5 ml
of 10 °lo aqueous KOI-I in a manner similar to the saponification
procedure for the
tliphenyl-2,3-dihydro-ll-I-pyrrolizinyl acetic-acid ethylesters. The time of
saponification
is 90 minutes. Purification is by column chromatography (silica gel,
diisopropylether).
Yield: 90 mg melting point: 186 ° C
C3pH2gN03 (451.6)
IR: v".,~,r = 3300-2400 (OH), 1710 (C~), 1605 ,~,~ 1595 (C=C) cm-t
1H-NMR: b (ppm) s 1.23 (s, 3H, -Chi), 1.32 (s, 3H, -CH3), 1.48 (d, 3H, J=7
Hz, CH3-CH<), 2.72, 2.91 (A8, 2H, J=15.5 Ha, C-1 ), 3.81 (s, 2H,
C-3), 3.96 (q, 1H, J=7 Hz, -CH<), 6.81-7.50 (m, 10H, Arom.)