Note: Descriptions are shown in the official language in which they were submitted.
32122CA
2 ~ Q ~
CARBIDE PRODUCTS AND METHOD AND
APPARATUS FOR THEIR PRODUCTION
~ack~round of the Invention
This invention relates to fl method and apparatus for producing
carbide products, such as silicon carbide. In another aspect, the
invention relates to the composition of such carbide products.
Various carbide powders, such as silicon carbide, are useful
as advanced ceramic materials in the fabrication of highly stressed,
wear resistant ceramic parts, such as those employed in heat engines,
turbo-charger rotors and heat exchangers. Powders which are used to
make such parts must meet stringent particle si~e ~i.e. submicron) and
purity requirements. New synthesis methods currently being researched,
involving plasma and laser heating of gaseous reactants, for example,
are effective in producing submicron, high purity carbide powders, but
employ expensive equipment with high energy demands. Thus, these
methods may not be practical for economlcal, large scale synthesis. A
more conventional method involves electrically heating a mixture of
solid carbon and silicon dioxide. Large chunks of silicon carbide are
produced which must be reduced in size by mechanical grinding. Such
grinding adds undesirable metal impurities and requires a significant
amount of additional energy.
32122CA
Summary of the Invention
It 1SJ therefore, an object of the invention to provide a
method and apparatus whlch are economical in producing a highly pure
carbide product characterized by submicron particles.
The above object is realized in a method which comprises:
providing a reactor having a chamber defined therein which has
longitudinally separated upstream and downstream ends, wherein the
chamber comprises a combustion zone and a reaction zone such that the
combustion zone extends from the upstream end to a boundary between the
zones and such that the reaction zone extends from the boundary to the
downstream end; establishing a flow of a combustible mixture in the
combustion zone so as to generally flow in a direction toward the
reaction zone, wherein the combustible mixture comprises a mixture of a
fuel and an oxidant; combusting the combustible mixture in the
combustion zone to produce hot combustion products; injecting at the
boundary betwesn the zones at least one reactant such that the hot
combustion products carry the reactant(s) toward said downstream end,
wherein the temperature in at least a portion of the reaction zone is at
least about 1400~C and wherein the elemental molar ratio of carbon to
oxygen for the combination of the combustible mixture and reactant(s) is
at least about 0.~:1, wherein the reactant(s) is capable of reacting in
the reaction zone to form a carbide compound; whereby a product powder
comprising the carbide compound is produced in the reaction zone.
According to another aspect of the invention, an apparatus is
provided which comprises: a reactor having a chamber defined therein
which has an upstream end and a downstream end; a ~irst nozzle which has
an outlet end which communicates with the chamber at a position
intermediate the upstream and downstream ends and which comprises first
and second tubular members, whereln the first tubular member is
generally coaxially positioned withln the second tubular member such
that a generally annular space is defined between the interior surface
of the second tubular member and the exterior surface of the first
tubular member; means for passing at least one reactant through the
first tubular member so as to exit the first tubular member into the
chamber, wherein the reactant(s) is capable of reacting in the reactor
chamber to form a carbide product; means for passing a gas through the
32122CA
3 2 ~3 ~
generally annulnr space so as to exit the first nozzle and generally
surround the reactant(s) flowing from the outlet end of the first
nozzle; a second nozzle having an outlet end which communicates with the
chamber at a position closely ad~acent to the upstream end; and means
for passing a combustible mixture through the second nozzle so as to
exit its outlet end into the chamber.
According to yet another aspect of the invention, there is
provided a raw product as collected directly from the above-mentioned
reactor (where a reactant includas a sllicon component) which comprises
silicon carbide and which is characterized by the following weight
percentages: silicon in the amount of about 30 to about 75 weight
percent; carbon in the amount of about 15 to about 50 weight percent;
and oxygen in the amount of about 1 to about 30 weight percent. Such
raw product having a relatively high oxygen content of about 3 weight
percent to about lO weight percent is sinterable to a ceramic part
having a high density of about 2.8 g/cc. Purification of the raw
product by subsequent processing produces an extremely pure silicon
carbide product. The product in accordance with the invention is
composed of submicron particles containing a very low level of
lmpurities as will be discussed in more detail in the Detailed
Description.
The method and apparatus of the inventlon are economical in
requiring the use of inexpensive combustible fuels as the heating
sources and in requiring a minimal investment for construction of the
reactor. Therefore, the invention is particularly well suited to large
scale synthesis of high quality carbide products.
Brief Description of the Drawings
FIG. 1 is a cross-sectional view of a reactor in accordance
with a preferred embodiment of the invention.
FIGS. 2 and 3 are enlarged cross-sectional views of nozzles
whlch are shown ln FIG. 1.
F~G. 4 shows X-ray dlffractlon pàtterns for samples produced
in Example I.
~ ~? ~ 32122CA
FIG. 5 shows infrflred spectral patterns for the samples
produced in Example I.
FIG. 6 is an X-ray diffraction pattern for a sample produced
in Example II using silane as the sllicon-containiDg reactant.
FIG. 7 is a reference X-ray diffraction pattern produced by a
pure sample of crystalline ~-silicon carbide.
FIG. 8 is a graphical representation of the particle size
distribution of a raw product as collected directly from the reactor in
Example VI.
FIG. 9 is a graphical representation of the particle size
distribution of a purified product produced in Example VI.
FIG. 10 is an NMR spectral pattern for a sample produced in
Example VIII.
Detailed Description of the Invention
A preferred embodiment of the invention will now be described
with reference to the drawings.
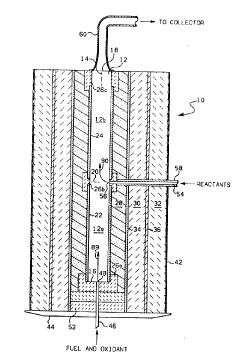
Referring to FIG. 1, there is shown a cross-sectional view of
a reactor 10 having defined therein a chamber 12 which has a
longitudinal axis 14 and longitudinally separated upstream and
downstream ends 16 and 18, respectively. Chamber 12 includes a
combustion zone 12a and a reaction zone 12b situated such that
combustion zone 12a extends from upstream end 16 to an imaginary
boundary 20 between the zones and such that the reaction zone 12b
extends from boundary 20 to downstream end 1~.
Chamber 12 is defined by refractory tubes 22 and 24 and also
inserts 26a, b and c. Such tubes and inserts are preferably composed of
a refractory materia~ resistant to temperatures of at least 2000~C, such
as zirconia, which is commercially available from Zircoa Products of
Solon, OH.
As shown, there is provided several additional coaxially
positioned layers of refractory material which are generally annular in
shape and which surround tubes 22 and 24, including: layer 28,
preferably comprising zirconia powder insulation, available from Zircar
Products of Florida, NY, which allows for contraction and expansion of
32122C~
S ~ 3 ~
this layer; l~yer 30, which preferably comprises alumlna-silica blanket
insulfltion, commercially available under the trademark Fiberfrax~ from
Carborundum of Niagara Falls, NY; and layer 32, which may be of the same
composition as layer 30. A refractory cyclinder 34, preferably low
density thermal insulating alumina available from Zircar Produc*s of
Florida, NY, is illustrated as separating layers 28 and 30, and a metal
cylinder 36 most preferably composed of stainless steel separates layers
30 and 32. Cylinders 34 and 36 assist in providing structural support
for the reactor.
The outermost reEractory layer 32 is held in place by a cloth
material 42, such as fiberglass, which wraps around the exterior surface
of layer 32. The bottom end of the various layers are supported by a
metal plate 44. The reactor is preferably oriented vertically as shown
for the sake of operating convenlence. If any of the refractory
material breaks or cracks it tends to stay in position if the various
layers and tubes are vertically positloned. Therefore, operation can
sometimes continue despite such structural defects. Other reactor
orientations are within the scope of the invention.
Nozzle 46 is connected to a source of fuel and oxidant and has
an outlet end 48 which communicates with the combustion zone 12a of
chamber 12 at a position closely ad~acent to upstream end 16 of chamber
12. As shown, nozzle 46 is surrounded by refractory inserts 52
positioned near upstream end 16. Nozzle 54 is connected to a source of
reactants, discussed later ln detail, and extends through the various
refractory layers to an outlet end 56 which communicates with chamber 12
at boundary 20 intermediate upstream and downstream ends 16 and 18.
Nozzle 54 is surrounded by a refractory tube 58.
Proper positioning of the nozzles with respect to each other
is an important consideration in optimizing operating efficiency and
quality of the product. It ls deslrable for example to positlon nozzle
54 far enough downstream so that substantlally all of the free oxygen
has reacted with the fuel to form combustlon products. Such posltioning
of the nozzles means that there is substantia]ly no free oxygen (~2 in
its free g~seous state, uncombined with any other component) at boundary
20, thus avoiding the undesirable oxldation of one of the reactants, as
will be discussed further in connection with operatlon of the apparatus.
32122CA
It is furthermore desirable to position nozzle 54 sufficiently
downstream from nozzle 46 to avoid the ~et pump effect on gases flowing
from nozzle 46. Thls effect tends to pull the reactants upstream rather
than the intended downstreflm flow. However, in addition to the above
considorations, nozzle 54 should be positioned sufficiently upstream to
ensure that tempcratures to which the reactants are exposed are
conducive to the formation of carbide product.
Also shown in FIG. 1 is conduit 60 which is connected at one
end to reactor 10 so as to communicate with the downstream end 18 of
chamber 12. Conduit 60 receives carbide produc-t powder therethrough
which then passes to a suitable collector, di~cussed further below.
Conduit 60 in the illustrated embodiment not only functions to transport
the product to the collector, but also functions as a heat exchanger.
The outside of conduit 60 is exposed to a cooling means such as ambient
air which allows heat transfer via both natural convection and
radiation. Such heat transfer effects cooling of the product powder as
it flows through conduit 60, which is highly desirable in order to
prevent undesirable reactions involving, for example, oxidation of the
carbide product to form unwanted oxides. In addition, such cooling of
the product powder is desirable to prevent damage to the collector from
excessively hot product. In instances where a cloth filter bag is used
as the collector, conduit 60 should be of sufficient length to cool the
product powder to a desired temperature, typically below about 100~C,
before it enters the collector. Other types of collectors require less
cooling. If desired, the cooling effect can be further enhanced by
suLlounding conduit 60 with a cooling coil or jacket having coolant
fluid flowing therethrough.
With respect to materials for conduit 60, it is preferable
that a non-metallic material be employed which will not add any
undesirable metal contaminants to the product. If the desired product
ls silicon carbide for example, quartz (silicon dioxide) is preferred
since molecular structures characterized by silicon-oxygen bonds are
already present in the reactor product such that essentially no
Qddltional contaminants will enter the product stream. Quartz is also a
particularly preferred material because of its high emissivity and
excellent thermal shock resistance. However, other heat exchange
32122CA
materials, including metals, are within the scope of certain aspects of
the invention.
The collector can be any suitablo means of collecting the
product powder. One suitable collector, as discussed above, comprises a
cloth filter bag connected to the downstream end of conduit 60. Other
suitable collectors include metal filters, electrostatic precipitators
and cyclone separators~ Of course, regardless of what type of collector
is used a pressure differential skould preferably be established, by a
suitable pump, across the collector to draw the product powder through
conduit 60 and into the collector.
Referring to FIG. 2, there is shown a cross-sectional view of
a portion of nozzle 46 having outlet end 48. Nozzle 46 comprises a
tubular member 62, preferably constructed of a metal such as stainless
steel, which has an inner sidewall 62a and an outer sidewall 62b. Such
sidewalls define a generally annular space 64 therebetween which is
connected to a source of coolant fluid such as water or ethylene glycol
or a combination thereof, which could also include minor amounts of
additives such as corrosion inhibitors, etc. if desired. A tubular
member 66 is positioned within annular space 64 so as to generally
divide the SpQCe into entrance and exit passageways for the coolant
fluid. As shown, coolant fluid flows toward the tip of nozzle 46 as
indicated at 68, and flows away from the tip in the opposite direction
as indicated at 70. The direction of coolant fluid flow may be reversed
if desired. The flow of coolant fluid through space 64 assists in
preventing melting of the metallic tubular members, and also assists in
preventing the flame from burning back (flashback) into nozzle 46 by
keeping the interior of nozzle 46 below the autoignition temperature of
the fuel/oxidant mixture. The interior of nozzle 46 is connected to a
source of fuel and oxidant such that a flow of the fuel/oxidant mixture
is established through nozzle 46 as indicated at 72.
Referring to FIG. 3, there is shown a cross-sectional view of
nozzle 54 havlng outlet end 56. Nozzle 54 is preferably constructed of
the same or similar metallic material as that used for nozzle 46, and
includes tubular members 74 and 76. As shown, tubular member 74 is
positioned generally coaxially within tubular member 76 such that a
generally annular space 78 is defined between the interior surface of
32122CA
8 2 ~
member 76 and the exterior surface of member 74. The interior of
tubular member 74 is connected to a source of reactants to provlde a
flow of reactants therethrough as indicated at 79. Tubular member 76 is
generally of the same design as member 62 in FIG. 2, and includes
respective inner and outer sidewalls 76a and 76b between which there is
defined a generally annular space 80. A tubular member 82 is positioned
within anmJlar space 80 so as to divide it into entrance and exit
passageways. Space 80 is connected to a source of coolant fluid so as
to establish respective entrance and exit flow paths 84 and 86. The
reverse direction of coolant fluid flow can be employed if desired. The
flow of coolant fluid not only assists in preventing melting of the
metallic tubular members, but also helps prevent the formation of
carbide deposits within nozzle 54 by maintaining the temperature of the
nozzle below temperature limits conducive to carbide formation. This
avoids the need to periodically clean carbide deposits from nozzle
surfaces.
Annular space 78 is connected to a purge gas source to
establish a flow of such purge gas through annular space 78 in the
direction of outlet end 56, as indicated at 88. Thus, this flow of
purge gas exits outlet end 56 in a generally annular stream so as to
surround the reactants as they exit the nozzle. This annular gas stream
forms a sheath around the reactants so as to prevent contact of the hot
combustion gases in chamber 12 (see FIG. 1) with the reactants
immediately after their exit from nozzle 54, thereby preventing the
formation of carbide deposits on the tip of nozzle 54. Such deposits,
if not prevented, can eventually lead to blockage of reactant flow from
nozzle 54 and consequent reactor shutdown. Of course, the insulative
sheath of purge gas disperses after only a few milliseconds, but this is
sufficient time to allow the reactants to flow far enough away from the
nozzle tip to prevent formation of undesirable deposits. The choice of
purge gas is not critical, and can be, for example, an inert gas (i.e.
helium or argon), a cooled waste gas as discharged from the reactor, or
a reactive carbon-containing gas (i.e. hydrocarbon) which can contribute
carbon to the reactlve stream for formation of carbides. If a
carbon-containing hydrocarbon is used AS the purge gas, it typically
32122CA
9 2 ~
will not decompose quickly enough to result is any undeslrable carbon
deposits on the tip of no~zle 54.
The various gas flows are preferably established and
controlled by conventional equipment not shown in the drawings. Gfls can
be supplied by, for example, pressurlzed gas bottles. The gas can pass
from such a pressurized container and through an orifice plate whose
orifice is sized to achieve sonic velocity of the gas. Such a sonic
velocity prevents pressure disturbances from traveling upstream, so that
whatever happens downstream near the reactor will not affect the desired
flow rate of gas. A pressure regulator can be employed to control the
rate of flow of the gas.
Turning now to another aspect of the invention, there is
provided a method of making a carbide compound using the above described
apparatus. Reference will be made to the drawings in describing a
preferred embodiment of this method.
As used herein and in the appended claims, a carbide compound
is defined as a compound of a first elemental component and a second,
carbon component. Generally speaking, a carbide compound is produced in
accordance with the illustrated embodiment by reacting two reactants.
The first reactant contains the first component whereas the second
reactant contains the second, carbon component.
According to certain broad aspects of the invention, the first
component as contained in the first reactant may be any element capable
of combining with carbon to form a carbide compound. For example, the
first component may be a metal such as tungsten, chromium, titanium,
zirconium, molybdenum or iron. Halides of such metals are particularly
suitable as the first reactant. Or, the first component may be a
metalloid such as boron or silicon. As stated previously, silicon
carbide is a very useful carbide compound. Ceramic parts can be made
from silicon carbide powder which have excellent mechanical strength and
heat resistance. Therefore, reactants having silicon as the first
component are of particular interest in connection with the present
invention.
Preferred silicon-containing reactants which are compounds of
silicon include silane (SiH4) and substituted silanes. As used herein
and in the appended claims, a substituted silane can be generally
32122CA
expressed by the formula SiABCD where each of A, B, C flnd D can be any
element or combination of elements as long QS at least one of A, B, C
and D is not hydrogen, and where A, B, C and D can be the same or
different. For example, any one of A, B, C and D can be selected from
hydrogen, a halogen, an oxygen-containing group (i.e. OSi(CH3~3), a
nitrogen-containing group (i.e. NHSi(CH3)3), an alkyl group, an aryl
group, a silyl group, or a group containing multiple sillcon atoms.
Examples of such substituted silanes include: alkyl silanes such as
methylsilane ((CH3)SiH3), dimethylsilane ((CH3)2SiH2), trimethylsilflne
((CH3)3SiH) and tetramethylsilane (Si(CH3)4); halogenated silanes such
as dichlorosilane (H2SiCl2~; halogenated methylsilanes such as trimethyl
silicon bromide ((CH3)3SiBr) and dichlorodimethylsilane ((CH3)2SiCl2);
siloxanes such as hexamethyldisiloxans ((CH3)3SiOSi(CH3)3); silazanes
such as hexamethyldisilazane ((CH3)3SiNHSi(CH3)3); and silicon halides
such as silicon tetrachloride (SiCl4). Cyclic and polymeric silicon
compounds are also within the scope of the invention. If desired,
mixtures of any of the preceding silicon-containing compounds can be
employed. Silane is the presently preferred silicon-containing reactant
in view of the quality of the product.
The second, carbon-containing reactant is preferably a C~-Cg
carbon compound such as an alcohol or a hydrocarbon. Suitable alcohols
include ethanol and propanol. A hydrocarbon is presently most preferred
and can be selected, by way of example, from the following group:
methane, ethane, propane, butane, pentane, hexane, heptane, octane,
nonane, ethylene, propylene, acetylene, benzene, toluene, cyclopropane,
cyclobutane, cyclopentane, cyclohexane, and mixtures thereof. Although
such Cl-Cg carbon compounds are preferred, any carbon-containing
reactant capable of reacting with the first reactant to form carbide
products is within the scope of certain aspects of the invention.
The fuel, which is in~ected through nozzle 46, is preferably
an unsaturated hydrocarbon (having at least one double or triple bond
between carbon atoms), such as, for example, ethylene, propylene,
butene, propadiene, butadiene, acetylene, propyne, butyne and mixtures
thereof, ~nd can be the same as or different than the hydrocarbon second
reactant. Another preferred group of hydrocarbon fuels are cyclic
hydrocarbons such as cyclopropane, cyclobutane, and mixtures thereof.
3212ZCA
tl 2~
Other types of fuels, such as solld fuels substanttally comprlsing purecarbon, and fuel blends are within the scope of certain aspects of the
invention as long as the desired temperature conditions and carbon to
oxygen ratio, later discussed, are achieved in the reactor.
The oxidant employed should be capable of accepting electrons
from the fuel and is preferably an oxygen-containing gas, most
preferably pure oxygen. Gaseous mixtures which include oxygen as a
single component, such as air, are wlthin the scope of the invention.
In accordance with a preferred procedure for operating the
illustrated apparatus, flow of coolant fluid is started with respect to
nozzles 46 and 54, followed by gradual heating of the reactor to normal
operating temperatures. This is done to avoid thermal shock and
possible breakage to the various refractory materials. One method for
this preheating stage involves initial electrical heating of the
refractory layers with electrical rod heaters (not shown) and heating of
chamber 12 with a coiled wire electrical heater (not shown) inserted
into chamber 12, followed by establishment of a combustion flame in
combustion zone 12a.
In any event, the combustion flame is established in
combustion zone 12a by initiating a flow of gaseous fuel through nozzle
46. If the reactor has been preheated electrically, the fuel should
spontaneously establish a flame by reacting with ambient air at
downstream end 18 of chamber 12. If the combustion flame does not form,
the fuel may be ignited with an appropriate ignition device. After the
flame is established, a flow of air is initiated through nozzle 46 so as
to produce a fuel/air mixture. This causes the flame to propagate
upstream so that the flame establishes itself in combustion zone 12a.
Propagation of the flame ln this manner can be hazardous to an operator
implementing the method such that adequate safety precautions are taken.
The reactor is typically operQted with this fuel/air mixture for a
predetermined period, usually ~ hour to 1 hour. Operation of the
reactor with air as the oxidant is part of the preliminary start-up of
the reflctor to gradually heat the reactor.
A flow of pure oxygen is now commenced through nozzle 46 to
replace the air. The flow of such oxygen is gradually increased and the
flow of air gradually decreased until a fuel/oxygen combustible mixture
32122CA
12
is obtained. The combustion flame should be monitored visually through
downstream end 18 to make sure that the flame does not flash back
upstream so as to enter the noz~le 46 and cause a potentially dangerous
condition. Flashback can be prevented by providing a sufficiently high
velocity of fuel and oxygen exiting nozzle 46.
A flow of the fuel/oxygen mixture is thus established in a
direction generally parallel to axis 14 as indicated at 89, and the fuel
and oxygen flow rates are set to be relatively fuel-rich in preparation
for carbide production. The elemental molar ratio of carbon to oxygen
for the fuel/oxygen mixture is preferably at least about 0.7:1, more
preferably in the range of about 0.8:1 to about 1.2:1, and most
preferably in the range of about 0.9:1 to about 1.1:1. As used herein,
the elemental molar ratio of carbon to oxygen means the molar ratio of
carbon atoms to oxygen atoms. The residence time of the combustible
mixture and hot combustion products formed therefrom in combustion zone
12a is typically about 5 to about 20 milliseconds, which is sufficient
time to consume substantially all of the oxygen before reaching boundary
20. As discussed previously, this is desirable to avo~d the production
of unwanted oxides rather than carbides. Temperature conditions in
combustion zone 12a are typically about 1700~C to about 2000~C.
The substantially gaseous reactants are now in~ected into
chamber 12 at boundary 20, as indicated at 90, in a direction generally
perpendicular to the chamber axis 14 such that the hot combustion
products formed from combustion of the fuel carry the reactants toward
downstream end 18. In the illustrated embodiment, the first and second
reactants are premixed to give a desired molar ratio of silicon to
carbon in the reactants of typically about 1:2 to about 1:4 and passed
in admixture through nozzle 54 so as to exit outlet end 56 into chamber
12. If the first reactant employed is normally a liquid, such first
reactant is placed in vapor form most conveniently by placing it in a
temperature controlled bubbler and passing a purge gas therethrough.
The temperature of the coolant fluid flowing through nozzle 54 can be
elevated to the necessary extent to help prevent condensation of the
first reactant as lt passes through nozzle 54.
Flow rates are ad~usted so that the elemental molar ratio of
carbon to oxygen for the combination of the reactants and fuel/oxygen
2 ~ 3 ~
13
mixture i9 at least about 0.8:1, but i9 preferably in the range of flbout
0.9:1 to about 1.5:1 and most preferflbly in the rango of about 1:1 to
about 1.3:1. Although the reactions occurring in reaction ~one 12b are
numerous and not completely understood, it ls believed that the above
cited carbon to oxygen ratios minimize the productlon of undesirable
oxidizing species such as carbon dioxide and water, and produce partial
pressures of reducing gases like carbon monoxide and hydrogen which flre
favorable to the production of carbides. The preferred carbon to oxygen
ratios for the fuel/oxygen mixture previously discussed (preferably at
least about 0.7:1, more preferably about 0.8:1 to about 1.2:1, and most
preferably about 0.9:1 to about 1.1:1) particularly enhance these
conditions favorable to the production of carbides. In addition,
reactor temperature is somewhat dependent on the carbon to oxygen ratio,
and temperatures conducive to carbide formation are achievable using the
above-discussed carbon to oxygen ratios.
If desired, it is within the scope of the invention to inject
into chamber 12 in admixture with the reactants a boron-containing
compound which will provide boron in the product collected from reaction
zone 12b. Suitable boron-containing c~.poul.ds include boranes, such as
diborane ~B2H6), other boron hydrides, and boron alkyls. As will be
discussed in more detail in the examples, boron is a sintering aid.
Temperature conditions for at least a portion of reaction zone
12b are at least about 1400~C, preferably in the range of about 1400~C
to about 1700~C, most preferably in the range of about 1500~C to about
1600~C. If temperatures at the upper end of these ranges are desired, a
preferred fuel is acetylene or a mixture of acetylene and ethylene.
This is particularly desirable where the first reactant is, for example,
a chlorinated silane such as dichlorodimethylsilane, which requires a
higher temperature than some other reactants to achieve a desirable
reaction rate to form silicon carbide and other products. The
temperature conditions in the reactor can most conveniently be monitored
by means of a thermocouple (not shown) positioned in one of the
refractory layers. The temperature detected by the thermocouple can be
correlated to actual temperature condltions in the reactor. Of course,
a thermosouple can be positioned directly in the chamber 12, but this
requires use of expensive materials such as plfltinum and/or rhodium
32122CA
14 2 ~
which are still subject to deterioration due to the high temperatures in
chamber 12.
Pressure conditions in reaction zons 12b are preferably at or
near atmospheric pressure. Other operating prossures are within the
scope of the invention.
In reaction zone 12b, a product powder is formed from the
reactants which lncludes the desired carbide compound and other
components as is discussed further below. The product powder exits the
reactor through downstream end 18 and passes into and through conduit 60
to the collector. After the desired amount of product powder is
collected, the reactor is shut down by first switching to air as the
oxidant and then gradually decreasing the fuel/oxidant flow rates to
provide gradual cooling of the reactor. It is sometimes desirable to
run the reactor before shutdown for a period of time, i.e. 15 minutes,
at full flow rates to burn out carbon deposits. After shutdown, the
reactor is typically allowed to cool for several hours before the supply
of coolant fluid to the noz~les is terminated.
In the following description of products produced in
accordance with the invention and in claims appended hereto, it is to be
understood that the term "weight percent" as applied to a component of a
composition is based on the total weight of the composition.
The product powder as collected directly from the reactor,
hereafter denoted as "raw" powder, is generally black in appearance, and
in the case of silicon as the first elemental component, contains
silicon carbide, silicon and carbon in addition to that in the silicon
carbide, and oxygen. Such a raw product powder is characterized by the
following wei~ht percentages: silicon in the amount of about 30 to
about 75 weight percent, preferably in the amount of about 50 to about
70 weight percent, and most preferably in the amount of about 55 weight
percent to about 70 weight percent; carbon in the amount of about 15 to
about 50 weight percent, preferably in the amount of about 20 to about
45 weight percent, and most preferably in the amount of about 30 to
about 40 weight percent; and oxygen in the amount of about 1 to about 30
welght percent, preferably ln the amount of about 1 to about 20 weight
percent, and most preferably in the amount of about 1 to about 10 weight
percent. Hydrogen can also be present in the raw product in minor but
32122CA
2 ~, ~
detectable amounts of between abou-t 0 and about 1 weight percent. NMR
analysis, as will be di~cussed further in a subsequent example, is also
taken to indicate that at least some of the silicon atoms in raw product
powder are bonded to both carbon and oxygen atoms. In other words, at
least some of the silicon in the product is simultaneously bonded to
both carbon and oxygen.
The raw product powder in accordance with the invention cfln be
further characterized insofar as a sample of such powder having a
relatively high oxygen content in the range of about 3 to about 10
weight percent is sinterable to a sintered ceramic part having a density
of at least about 2.8 g/cc, or about 85% of the density of pure
crystalline silicon carbide, by a process comprising: pressing the raw
product at a temperature of less than about 100~C to a pressed part
having a density of no more than about 1 g/cc; heating the pressed part
to a temperature of about 1700~C to about 2400~C without application of
compaction force so as to produca the sintered part having the density
of at least about 2.8 g/cc; wherein no steps are performed prior to the
heating step for removal of any appreciable amounts of oxygen from the
raw product or pressed part produced therefrom. As used herein and in
the appended claims, the term "prassing" refers to any technique for
fabricating a self-supporting shape from ceramic particles. Also as
used herein and in the appended claims, the application of a "compaction
force" to a ceramic part means the application of a force to the part by
means of a solid member in contact with the part which mechanically
compacts the part to thereby increase its density.
With respect to particle size, the raw product powder
comprises particles having diameters in the range of about 0.01 to about
0.3 micron.
The raw product powder can be further purified by additional
processing to yield a purified product. This purification process
typically involves two stages carried out in a conventional furnace.
First, the raw powder is heated in an inert gas (i.e. argon) atmosphere
at a temperature of about 1300~C to about 2400~C, most preferably about
1400~C to about 1800~C, for at least about 15 minutes and most
preferably in the range of about 1 hour to about 2 hours. This serves
to react molecular structures having silicon-oxygen bonds with carbon to
32122CA
16 ~ 3 ~J ~ ~
thereby remove oxygen as carbon monoxide and make the silicon avflilable
for reacting with free carbon to form additional silicon carbide. In
certain instances, the raw powder will have insufficient carbon to
remove a substsntial portion of the oxygen, thus necessitating the
addition of carbon to the raw powder before heating in the inert
atmosphere. Second, the powder resulting from the first purification
stage is heated in an oxygen-contalning atmosphere to a temperature of
about 600~C to about 900~C, most preferably about 600~C to about 700~C,
over a period of at least about 15 minutes and most preferably for about
30 minutes to about 2 hours. This stage burns off remaining carbon in
the form of carbon oxides to yield the purified product.
X-ray fluorescence analysis of the purified product indicates
that the product has less than about 1000 ppm of elemental impurities,
wherein such elemental impurities include aluminum and those elements of
higher atomic numbers, except silicon, up to and including uranium.
Most preferably, the product has less than about 600 ppm of such
impurities. As discussed previously, many impurities undesirably
decreàse the strength of sintered carbide parts made from product
powder.
Indlvidual particles of the purified product in the form of a
powder are highly uniform and have diameters which range from about 0.05
micron to about 0.50 micron. As discussed previously, submicron and
uniform particles are vital characteristics in the production of
fine-grained, high strength parts from a carbide powder. Crystallite
size (size of individual crystals) range from about 30 to about 100
angstroms.
Either the raw or purified product can be sintered into heat
resistant, high strength parts in a conventional manner. For example,
appropriate amounts of additives such as boron and carbon or yttrium
oxide and aluminum oxide can be added to such product, followed by
pressing to a desired shape and heating at a temperature of about 1700~C
to about 2400~C.
It i8 to be understood that the above descriptlon pertains to
a preferred embodiment of the invention, but that many variations and
modifications are within the scope of certain aspects of the invention.
For example, it is possible to use excess carbon from the fuel and/or
32122CA
17 2 ~v ~
the first reactant as the source of carbon for producing the carbide
compound, in which case the second, carbon-containing reactant can be
omitted. It is desirable in such an embodiment to utilize a carrier
gas, such as nitrogen, helium, argonJ hydrogen, carbon monoxide or
mixtures thereof in admixture with the flrst reactant to carry the first
reactant into the reactor chamber. Since a mixture of carbon monoxide
and hydrogen is produced as a waste gas by the reactor, the reactor can
serve as a convenient source of such carrier gas. Another possible
variation could involve employing a fuel which includes a preferred
unsaturated hydrocarbon as well as amounts of other types of
hydrocarbons such as saturated hydrocarbons. However, this will
generally decrease the heat produced by the combustion reaction so as to
possibly require a supplemental heat source (i.e. electric, plasma,
microwave, combustion zones exterior to chamber 12 but in heat exchange
relationship with chamber 12J etc.) to obtain the desired temperature
conditions in the reaction zone. In any event it is preferable that the
hot combustion products as produced by combustion in the combustion zone
provide at least about 15% of the energy needed to maintain desired
temperature conditions of at least about 1400~C in the reaction zone.
Examples
Specific examples will now be described to further illustrate
the invention. These examples should not be construed to limit the
invention in any manner.
In each of the following examples, various gaseous flow rates
are given in gram moles/minute (abbreviated to gmol/min hereafter).
Actual measurements of flow rate were taken volumetrically at room
temperature and atmospheric pressure in units of liters/minute. These
volumetric measurements were converted to gmol/min by assuming there are
24.45 liters/mole for any gas at 25~C (room temperature) and at
atmospheric pressure.
All flow rates for gases below are undiluted with any other gases (i.e.
carrier gases) unless specified otherwise.
Wlth respect to elemental analysis results given in various
tables, the carbon and hydrogen weight percentages were obtained by
32122CA
18
means o~ CHNS combustion analysls. The Si percentages were obtalned in
most cases using neutron activation anfllysis. In each example
hereafter, silicon percentages shall be assumed to have been obtained
using neutron activation unless indicated otherwise. In several
examples, as will be indicated, X-ray fluorescence analysis was employed
to determine silicon weight percentages. The oxygen percentages were
obtained using only neutron activation. We~ght percentage results which
are provided in this regard are not normalized to 100% unless specified
otherwise.
In several examples~ the weight percentages obtained from
elemental analysis sum to a total percentage of greater than 100% which
might be considered an unreasonably high value. It was found in this
regard that at least some of this error may have been contributed by the
results of neutron activation analysis for silicon and oxygen. The
neutron activation instrument was calibrated with an analytical standard
sample of silicon dioxide (Puratronic grade, Johnson Matthey Chemical
Ltd., Herts, England). The results of such analysis favorably compared
to the actual weight percentages of silicon and oxygen in the standard
sample. Therefore, every possible effort was made to produce accurate
neutron activation analysis results for silicon and oxygen. After
noting consistently high (i.e. greater than 100%) total weight
percentage results in analyzing products of the invention, a series of
samples were analyzed for silicon by both neutron activation and X-ray
fluorescence. The neutron activation analysis always yielded a weight
percentage of silicon slightly greater than that weight percentage
obtained by X-ray fluorescence analysis of the same sample.
In each example where an elemental analysis was performed,
CHNS analysis revealed detectable amounts of hydrogen. However,
hydrogen weight percentages of less than 1 weight percent are not
reported in the followlng examples.
Wlth respect to termlnology and notations used hereafter, it
will be understood that all degree readings obtained by X-ray
diffraction are for an angle of 2~. In addltion, the notation Si-0
means slllcon bonded to oxygen but denotes no particular molecular
structure.
32122CA
19
Example I
The purpose of this example, at least in part, is to show the
formatlon of silicon carbide over a range of temperatures and carbon to
oxygen ratios.
The apparatus used in this example was substantially similar
to the apparatus shown in FIG. 1. However, the nozzle used for
sidestream injection of reactants was simllar in structure to the nozzle
shown in FIG. 2 and was not adapted to receive a purge gas therethrough.
A flow of water was employed in conjunctlon with each nozzle to serve as
a coolant fluid. A relatively planar Teflon fllter was used to collect
a sample of powder exiting from the reactor. In operation, a
differential pressure was established across the filter so that powder
collected on one side of the filter to form a filter cake. Important
dimensions for this apparatus are given in Table IA.
TABLE IA
I _ Dimenslon
Diameter of chamber 12 2.54cm
Overall length of chamber 12 44.4 cm
Length of combustlon zone 12a 12.0 cm
Length of reaction zone 12b 32.4 cm
Overall diameter of reactor 10 22.0 cm
I.D. of fuel nozzle 0.26cm
I.D. of reactant nozzle 0.22cm
~ sing the above described apparatus, five runs were made using
ethylene (C2H4) as the fuel, pure oxygen as the oxidant,
tetramethylsilane (Sl(CH3)4) as the first, silicon-containing reactant,
and ethylene as the second, carbon-containing reactant. In each run,
the oxygen flow rate was 0.61 gmol/min, and powder was collected for a
perlod of three minutes.
Reactlon zone temperature for each run was measured at a
locatlon along the reactor chamber axls and 20cm upstream from the
downstream end of the chamber. A thermocouple comprislng bare wlres of
32122CA
~ ~ 6,i~
different composltions was employed to measure these temperatures. The
wires was made up of Type B alloys; that i9, 94% plfltinum and 6% rhodium
for one wire, and 70% platinum and 30% rhodium for the other wire. Tbe
two wires were run through a two hole electrlcal insulator made of 99.8%
alumina, and the insulator and wires were encased in a 0.64 cm O.D.
closed end 99.8% alumina tubo to protect the wires from attack by the
silicon reactant. A thermocouple junction was formed by extending the
wires about 0.5 cm beyond the alumina tube and spot weldlng the wires
together. This junction was located on the longitudinal axis of the
reactor chamber. Since the reactor walls are insulated and hence
operate close to the same temperature as the gases in the chamber, the
thermocouple readings were not corrected for radiation error.
Table IB summarizes the various operating conditions for the
five runs. The following parameters are set forth for each run: flow
rates for the ethylene injected as a fuel with the oxygen, the ethylene
reactant injected at the boundary between the combustion and reaction
zones, and the tetramethylsilane gas reactant; the elemental molar ratio
of carbon to oxygen for the combustible mixture only (combustion C:O
ratio); the elemental molar ratio of carbon to oxygen for the
combination of the combustible mixture and reactants which are injected
into the chamber (overall C:O ratio~; and the measured reaction ~-one
temperature for each run.
21 2~#3~
. ~ ~ ~ o~ ~ o
C~ oo ~ o
e o U~
E~
~,, ~ C'l o C~l ,,
o ~ ~ ~
K o ~ ~ ,~ ,i
o
o
~ ~ o
- ~C .. . .
K oo o~1
m e OCo'l o
C~ ~ .. . .
~_ o OO O O O
,1 e
~'-- .
E~ ~
o O O O
o oo o o o
m u
r~~
tL~ e~Inu~
~ oo o o o
3:~ o
O ~C~
~ z
32122US
22
For each run, a small sample (between 0.1 and 0.2 grams) was
taken from the collected powder. Each sample, hereafter designated as
samples 1-5 correspondlng to runs 1-5 respectively, was sub~ected to
both powder X-ray diffraction analysis and infrared analysis.
The resulting powder X-ray diffraction patterns for each of
samples 1, 2, 3, 4 and 5 are shown ln Fig. 4 at reference characters IX,
2X, 3X, 4X and 5X respectlvely. Silicon carbide peaks are at 35.6~ and
C/Si-0 peaks are between 15~ and 30~. Pattern IX, corresponding to the
highest reaction zone temperature but the lowest C:0 ratios, has a very
prominent C/Si-0 peak as indicated at 90. Pattern 5X, corresponding to
the lowest reaction zone temperature but highest C:0 ratios, is very
similar to pattern lX. In contrast, patterns 2X, 3X and 4X have more
prominent silicon carbide peaks, as shown for example at 92 for p~ttern
3X. Patterns 2X, 3X and 4X correspond to those samples produced with
intermediate temperatures and C:O ratios.
Samples 1-5 were also subjected to infrared analysis.
Referring now to Fig. 5, there are shown infrared spectral patterns
produced by transmittance analysis of each of the samples. Patterns lI,
2I, 3I, 4I and 5I correspond to samples 1, 2, 3, 4 and 5 respectively.
Pattern lI has very prominent silicon-oxygen bond absorptions at a
wavenumber (cm~1) of about 1100 and between wavenumbers 400 and 500, as
indicated by reference characters 94 and 96. Pattern 5I is similar but
with slightly less pr3minent silicon-oxygen bond absorptions. Patterns
2I, 3I and 4I can be seen to have more prominent silicon carbide
absorptions at a wavenumber of between 800 and 900 as is shown, for
example, at reference character 98 for pattern 3I.
Example II
The purpose of this example is to show the formation of
silicon carbide using silane (SiH4) as the silicon-containing reactant,
ethylene (C2H4) as the hydrocarbon reactant and two different fuels.
Two runs used an ethylene fuel and the remaining two runs used an
ethylene/acetylene (C2H4/CzH2) mlxture as the fuel. The
ethylene/acetylene mixture was a 67%/33% mixture where the percentages
23 32122US
2 ~
indicated are volume percentages. Each of the four runs were carried
out employtng the apparatus described in Example I. Table IIA
summarizes operating conditions for each of the runs and also data on
the products collected. Pure oxygen was employed as the oxidant at a
flow rate of 0.61 gmol/min.
TABLE IIA
RunC2H4 FuelC2H4+C2Hz Fuel C2H4 Reactant SiH4 Combustion Overall Run Time Product
No.(gmol/min)(gmol/min) (gmol/min)(~mol/min)C:O Ratio C:O Ratio (mins.) (g)
6 0.56 ---- 0.052 0.026 0.92 1.00 4 1.4
7 0.59 ---- 0.055 0.027 0.97 1.06 4 1.2
8 ---- 0.59 0.056 0.027 0.97 1.06 3 1.4
9 ---- 0.62 0.060 0.026 1.03 1.11 ND* 1.6
* ND means no data recorded.
~, ~
32122CA
~ 3 ~
Runs 7 and 9 both produced products visually characterized as
blue-gray flakes~ while run 6 produced gray flakes and run 8 produced
gray particles.
The results of product analysis corresponding to each run are
set forth in Table II B.
Table II B
Run C ~i 0
(wt.%) (wt.%) (wt.%)
6 22.8 61.6 22.3
7 22.3 60.9 24.3
8 31.0 57.5 15.6
9 36.6 58.0 9.9
The X-ray powder diffraction pattern of representative raw
reactor product is preseDted in FIG. 6 produced under the conditions of
run 9. By comparlson to the reference x-ray powder diffraction pattern
of beta crystalline silicon carbide in FIG. 7, it can be seen that the
agreement between the reference diffraction pattern and the pattern of
the product synthesized confirms that the product contains beta
crystalline silicon carbide. However, the existence of beta crystalline
silicon carbide in the raw product does not preclude the presence of
alpha phase and/or amorphous silicon carbide.
Example III
The purpose of this example is to show the formation of
sllicon carbide-containing products using sources of silicon other than
silane and tetramethylsilane. These runs utilized ethylene/acetylene
mixtures (67 vol.%/33 vol.%) as the fuel and either
dichlorodimethylsilane (SiCl2(CH3)z) or hexamethyldisilazane
32122CA
26 2~2 ~ v ~ h~
((CH3)3SiNHSi(CH3)3) as the silicon-containing reactants. The reactor
used in the experiments of this example was the reactor described in
Example I. The run conditions and product characterizations for runs
using dichlorodlmethylsllane are presentod in Table IIIA and those for
hexamethyldisllazane are presented in Table IIIB. The oxygen flow rate
in each run of Tables IIIA and IIIB was 0.61 gmol/mln.
Table IIIA
Silicon Carbide Synthesized from Dichlorodimethylsilane
RunCombustion Overall C2H4+C2H2Fuel siC12(cH3~2 C2H4 Reactant Product Analysis
C:O Ratio C:O Ratio gmol/min gmol/min gmol/min Wt.% Compst.
C H Si O
0.97 1.08 0.59 0.012 0.056 26.8 1.1 47.7 25.6
11 1.02 1.14 0.62 0.016 0.060 27.6 1.1 56.2 15.0
Table IIIB
Silicon Carbide Synthesized from Hexamethyldisilazane
RunCombustion Overall C2H4+C2H2 Fuel(CH3)3SiNHSi(CH3)3C2H~ Reactant Product Analysis
C:O Ratio C:O Ratio gmol/min gmol/min gmol/min wt.% Compst.
12 0.97 1.16 0.59 0.019 0.056 none performed
~.~
~ ~
32122CA
28 2 ~
Wlth respect to product produced by run 10 with
dichlorodimethylsilane, an X-ray powdar diffraction pattern of such
product has a fairly prominent C/Si-O peak between 15~ and 30~, a strong
silicon carbide peak at 35~ and less prominent silicon carbide peaks at
60~ and 72~. An X-ray powder diffractlon pattern for run 11 shows
similar s~licon carbide peaks but a C/Si-O peak far less promiment than
for the run 10 product.
An X-ray powder diffraction pattern was additionally obtained
for the product resulting from run 12 using hexamethyldisilazane. A
broad peak centered at 35~ and a broad peak batween 60~ and 70~ indicate
the presence of beta silicon carbide. The 35~ peak is the most intense
peak for beta silicon carbide while the repsonse at the higher angles
results from the combination of two strong beta silicon carbide peaks at
approximately 60~ and 72~.
Example IV
The purpose of this example is to demonstrate the production
of silicon carbide products using tetramethylsilane (TMS) as the
silicon-containing reactant and an ethylene/acetylene mixture (67 vol.%
C2H4, 33 vol.% C2H2) as the fuel instead of ~ust ethylene as in Example
I. Equipment utili~ed in this demonstration was the same as for Example
I. The oxygen flow rate in each run was 0.61 gmol/min. Table IV
presents other process conditions and results of product analysis.
Table IV
~ TMS and Ethylene/Acetylene Fuel Mixture in Silicon Carbide Synthesis
RunCombustionOverallC2H4+C2H2 FuelTMS ~Si(CH3)4)C2H4 ReactantProduct Analysis
C:O RatioC:O Ratiogmol/min gmol/min gmol/~inWt.% Composition
C si o
13 0.97 1.17 0.59 0.033 0.056 38.1 60.8 1.7
14 1.02 1.23 0.62 0.035 0.060 38.0 63.1 1.2
i.,~
32122CA
2~J~$~
X-ray powder dlffraction patterns for the products from runs
13 and 14 oach reveal a strong, sharp peak at about 35~ and weaker but
still well deflned peaks at about 60~ and 72~, thus indicflting silicon
carbide. With respect to C/Si-0, each pattern reveals a broad peak
between 15~ and 30~ which is much weaker than either of the 60~ and 72~
silicon carbide peaks.
Example V
The purpose of this example ls to demonstrate that sidestream
compositions that are incomplete with respect to the necessary carbon to
form the desired silicon carbide compound may be utilized by relying on
carbon from the fuel and/or silicon-containing reactant to supply the
carbon component. This example also shows the formation of silicon
carbide using low overall C/0 ratios well below 1Ø Thus, as has been
explained previously, the sidestream composition may include only a
silicon-containing reactant and one or more of various carrier gases.
Table VA sets forth process conditions for runs in which
hydrogen and helium were used as the carrier gas. The oxygen flow rate
in each run was 0.61 gmol/min. The apparatus of Example I was used to
carry out these runs.
Table VA
Carrier Gss Sidestream Synthesis of Silicon Carbide
Run Combustion Overall C2H4 Fuel Carrier GasCarrier Gas Si~CH~)4
C:O Ratio C:O Ratio gmol/min gmol/min compositiongmol/min
0.85 0.94 0.52 0.048 H2 0.027
16 0.g2 1.02 0.56 0.052 H2 O.C29
17 0.85 0.94 0.52 0.048 He 0.027
18 0.92 1.02 0.56 0.052 He 0.029
;~i
~,.
~ ~ r~ 32122CA
32
X-ray powder diffraction analysis was performed on the product
resulting from each of runs 15-18. Each pattern revealed a promiment
peak at about 35 degrees, thus indicatlng the presence of sillcon
carblde.
Several additional runs, 19-22, were made with hydrogen and
nitrogen as carrier gases, but using a different and larger reactor than
that reactor used for the runs in Table VA. The reactor used for runs
19-22 was substantially similar to that used in Example I, but included
two sidestream reactant nozzles adapted to receive a flow of purge gas
therethrough. The reactant nozzles were located on opposite sides of
the reactor chamber, and each such nozzle was substantlally similar to
nozzle 54 shown in FIG. 3. A Dacron~ bag filter was utilized to collect
product powder exiting from a quartz conduit having one end in
communication with the downstream end of the reactor. Important
dimensions of the reactor are given in Table VB, including dimensions of
tubular members 74 and 76 of nozzle 54.
Table VB
Item Dimension
Diameter of Chamber 12 5.08 cm
Overall length of Chamber 12 53.3 cm
Length of Combustion Zone 12a 27.9 cm
Length of Reaction Zone 12b 25.4 cm
Overall O.D. of Reactor 10 33.0 cm
O.D. of Tubular Member 76 0.952 cm
I.D. of Tubular Member 76 0.394 cm
O.D. of Tubular Member 74 0.317 cm
I.D. of Tubular Member 74 0.175 cm
The structure of the fuel nozzle was similar to that shown in
FIG. 2, wherein this nozzle is denoted as nozzle 46. Dimensions of
nozzle 46 are identical to those of nozzle 54, except with respect to
tubular member 74. Of course, nozzle 46 does not have such an inner
tubular member.
32122CA
33 2 3 ~
Table VC sets forth process conditions and product analysis
for runs 19-22~ In each of these runs, water WAS in~ected as a coolant
fluid into and through annular spaces 64 and 80 defined within
respective nozzles 46 and 54 (see FIGS. 2 and 3). In runs 19-21, no
purge gas was ln~ected into the sidestream reactant nozzles. However,
in run 22, helium was utilized as a purge gas in each such nozzle so as
to flow through annular space 78 of nozzle 54 at a flow rate of 0.15
gmol/min per nozzle. The oxygen flow in each of the runs was 1.09
gmol/min. Ethylene and acetylene were used as the fuel, and two
different silicon-containing reactants (abbreviated as Rct. in Table VC)
were employed. The flow rates indicated for the silicon reactant and
carrier gas are total flow rates from both sidestream reactant nozzles
into the chamber. Flow rates given in subsequent examples for gases
other than purge gas flowing through opposing sidestream reactant
nozzles will similarly be understood to be total flow ra~e from both
such nozzles.
Table VC
Carrier Gas Sidestream Synthesis of Silicon Carbide
Run Combustion Overall C2H~ C2H2 Carrier Gas Carrier Gas Silicon Rct. Silicon Rct. Product Analysis
C:O Ratio C:O Ratio gmol/min gmol/min gmol/min Compostn. gmol/min Compostn. Wt.% Composition
C si o
19 0.83 0.94 0.72 0.180 0.124 H2 0.0634 Si(CH3)4none performed
0.79 0.91 0.69 0.172 0.124 H2 0.0634 Si(CH3~4none performed
21 0.77 0.89 0.67 0.168 0.124 H2 0.0634 Si(CH3)4none performed
22 1.04 1.04 0.84 0.280 0~077 N2 0.0600 SiH419.0 65.8 16.8
~J
.~
32122CA
2 ~ ,3 ;:~
Powder X-ray diffraction patterns obtained for products
resulting from runs 19-21 have prominent, broad peaks between 15~ and
30~ indicativo of carbon and silicon-oxygen bonds. Rach pattern can be
interpreted to have a shoulder on the C/Si-O peak at about 35~. The
patterns, however, do not strongly indicate the presence of crystalline
silicon carbide. It should be noted that X-ray diffraction analysis is
sensitive only to crystalline materials so that the X-ray diffraction
patterns corresponding to runs 19-21 do not necessarily rule out the
presence of amorphous or poorly crystallized silicon carbide. Infrared
analysis was also performed on the products of runs 19-21. Each
resulting spectral pattern shows a clearly defined absorption at a
wavenumber (cm~l) between about 800 and 900, which indicates the
presence of silicon carbide. Prominent absorptions are also located at
wavenumbers of about 1100 and 450. These absorptions indicate the
presence of silicon-oxygen bonds.
In view of the above data, it can be concluded that the
products obtained from runs 19-21 contain silicon carbide which is
poorly crystallized if not amorphous.
As to the product from run 22, an X-ray powder diffraction
pattern revealed sharp, very prominent peaks at about 35~, 60~ and 72~,
indicating the presence of beta silicon carbide, and also peaks at about
28~, 47~ and 56~, indicating the presence of elemental silicon. The
pattern reveals little C/Si-0.
Example VI
The purpose of this example is to demonstrate a representative
particle size distribution obtainable from products produced by the
invention. This example also demonstrates the production of a silicon
carbide product using acetylene as the carbon-containing reactant and
also using a mixture of silicon-containing reactants.
A reactor as described in Example V was utilized to produce
the product of this example. For the instant example, the oxygen flow
rate was 1.09 gmol/min. No purge gas was employed. The fuel was a
mixture of ethylene and acetylene flowing at rates of 0.84 gmol/min and
0.28 gmol/min respectively. The combustion carbon to oxygen ratio was
36 32122CA
1.03. The ceramic formlng reactants were acetylen~ ~a~ gmol/min,
silane at 0.059 gmol/min, and tetramethylsilane at 0.058 gmol/min. This
procedure resulted in a fine black product powder containing 42.8 wt.%
carbon, 48.9 wt.% silicon, and 3.7 wt.% oxygen. An X-ray powder
diffraction pattern shows prominent, well defined peaks at about 35~,
60~ and 72~, thus indicating the presence of beta silicon carbide.
A sample of the raw product powder was analyzed in a Horiba
CAPA-700 Particle Analyzer after the sample had been ultrasonically
dispersed in a dispersant comprising equal parts of a 0.07 weight
percent solution of Triton~X100 (Rohm ~ Haas Company3 in deionized water
and a 0.02 weight percent solution of sodium pyrophosphate in deionized
water. The resulting particle size distribution was as follows wherein
each percentage value is the weight percentage of partlcles examined
falling in the particle diameter range indicated: 0.00 to 0.04
micron-10.5%; 0.04 to 0.05 micron-6.8%; 0.05 to 0.06 micron-8.5%; 0.06
to 0.07 micron-8.6%; 0.07 to 0.08 micron-9.1%; 0.08 to 0.09
micron-10.0%; 0.09 to 0.10 micron-8.9%; 0.10 to 0.20 micron-36.9%; 8 to
9 microns-0.7%. The last figure for the range of 8 to 9 microns is
considered an anomaly in the data. These results are illustrated in the
bar graph of FIG. 8 in which each bar represents one of the
above-mentioned particle diameter ranges. Each bar is positioned at the
particle diameter value which is the upper limit of a particular range.
For example, the bar at 0.1 micron represents the 0.09 to 0.1 micron
range.
The raw product powder was purified by placing a 1.81 gram
sample of the powder in an open graphite crucible and heating it in an
argon atmosphere at 1600~C for 30 min., with heatup from room
temperature to 1600~C at a rate of 25~C/min. The temperature was
increased to 1610~C and held for 20 minutes and then cooled to room
temperature in the argon atmosphere. Subsequently the sample was heated
in an aluminum oxide crucible in air at 600~C. When the effluent gas
showed a carbon monoxide level below 150ppm the oxidation was
terminated. Such puriflcatlon steps serve to remove molecules
characterized by silicon-oxygen bonds and also carbon from the raw
product. The resulting purified product powder was light green in
color.
2~ 32122CA
37
A sample of this product powder was viewed under a scanning
electron microscope and a photograph taken with a si~e standard bar
imposed on the photograph. The photograph was then mànually flnalyzed by
comparing the particle size of photographed partlcles with the
standardized size bar imposed on the photograph. The particle size data
presented in FIG. 9 is a plot of the particle diameter versus number of
particles for the 218 examined particles.
Example VII
The purpose of this example is to demonstrate the weight
percentage ranges of carbon, silicon and oxygen in raw reactor product
produced in accordance with previous examples.
32122CA
38 2
Table VIIA
Range of Carbon ln Product
Ru~ Wt.% Carbon Table
22 19.0 VC
7 22.3 IIB
6 22.8 IIB
11 26.7 IIIA
26.8 IIIA
8 31.0 IIB
14 38.0 IV
13 38.1 IV
Table VIIB
Range of Silicon in Product
Run Wt.% Silicon Table
47.4 IIIA
11 56.2 IIIA
8 57.5 IIB
9 58.0 IIB
13 60.8 IV
7 60.9 IIB
6 61.6 IIB
14 63.1 IV
22 65.8 VC
Table VIIC
Range of Oxygen ln Product
Run Wt.% Oxygen Table
14 1.2 IV
13 1.7 IV
9 9.9 IIB
8 15.6 IIB
11 15.0 IIIA
22 16.8 VC
6 22.3 IIB
7 24.3 IIB
25.6 IIIA
32122CA
39 2 ~
Example VIII
The purpose of this example ls to demonstrate that at least
some silicon in product powder produced in accordance with the invcntion
is simultaneously bonded to both carbon and oxygen.
A reactor as described in Example I was utilized to produce
the product of this example. The oxygen flow rate was 0.61 gmol/min.,
and the fuel was C2H4 which flowed at a rate of 0.56 gmol/min. The
combustion carbon to oxygen ratio was 0.92. The reactants employed were
tetramethylsilane at 0.033 gmol/min. and ethylene at 0.052 gmol/min.
The resulting raw product powder had the following composition:21.2 wt.%
carbon; 56.6 wt.% silicon; and 25.ô wt.% oxygen.
A sample of the resulting product was analyzed by silicon-29
nuclear magnetic resonance. The nuclear magnetic resonance spectrometer
used was a model WPSY-200 available from Bruker Instruments. Since the
materials examined were solids, the experimental determination utilized
cross polarization magic angle spinning. The resulting spectral
pattern, shown in FIG. 10, displays a large Si-O signal centered around
-llOppm and a silicon carbide signal near -15ppm. The relaxation time
of silicon carbide was measured flnd determined to be approximately 300
seconds. This requires a pulse delay of approximately 1000 seconds,
meaning that a spectral scan can be made about once every 15 minutes.
Since the spectral pattern is time averaged, to improve the signal to
noise ratio, the number of scans required for time averaging in one
twenty-four hour period does not exceed 100 scans. This results in a
low signal to noise ratio.
It should be noted in particular that the pattern of FIG. 10
shows some tailing of the silicon carbide signal toward higher field
strength. This is taken to indicate a structure other than simple
sillcon-carbon bonds, namely simultaneous bonding of some of the silicon
separately to both carbon and oxygen.
Example IX
The purpose of this example is to demonstrate the lower level
of impurities present in silicon carbide-containing product produced by
the invention in comparison to commercially available materials.
32122CA
A sample of product powder from run 13 of Example IV was
sub~ected to a two step purification procedure substantially like that
procedure described in Example VI. A sample of the resulting purified
product was analyzed using X-ray fluorescence. This sample as well as a
sample of commercially available silicon carbide whiskers were scanned
for elemental impurities, where such impurities included aluminum and
those elements of higher atomic numbers up to and including uranium.
The comparison in Table IX is for the purified product in accordance
with the invcntion and the commercially available silicon carbide
whiskers. Only those contaminant elements are shown in Table IX which
were detected for either the commercial whiskers or the purified
product. All other elements were below detectable limits and are
assumed to be ~ero for calculation of total impurities. As discussed
previously, the strength of sintered ceramic parts is adversely affected
by the presence of impurities. Therefore, a lower level of impurities
can lead to greater strength parts produced from the purer product.
Table IX
X-Ray Fluorescence Determination of Impurities
in Purified Final Product and Commercial Whiskers
Element (in ppm)Commercial Whiskers Purified Final Product
Al 1300 300
P 50 25
Cl ND* 50
Ca 2300 20
K 50 ND
Ba 10 ND
Ti 5 2
Nn 1500 ND
Cr 70 ND
Fe 1000 100
Zn 50 30
Pb 20 50
*ND means not detected.
It can be seen from Table IX that the total levels of
elemental impurities in the commercial product e~ceed that of the
purified product of the lnventlon.
32122CA
41
Example X
The purpose of the following example ls to demonstrate that
product produced in accordance with the invention may be sintered to at
least 90% of theoretical density and that such sintering may be
accomplished even when such product contains oxygen as an impurity.
This example also demonstrates the use of propylene (C3H6) as the
carbon-containing reactant and the use of a mixture (C2H4,C2H2) of
carbon~containing reactants.
Densities obtained for various pressed and sintered parts
discussed below were determined by either determining the volume of the
part and weighing the part or using ASTM procedure C 373-72 which
employs Archimedes principle and water.
A. Sintering of Raw Product: Y203 and Al~03 Sintering Aids
The reactor described in Example V was used to prepare a
quantity of reactor product under the conditions set forth below for
further processing and sintering studies. Helium was passed as a purge
gas flt a flow rate of 0.15 gmol/min. through each sidestream reactant
nozzle.
Combustion Fuel Flow Oxygen Sidestream Flows Product Analysis
C:O Ratio 80 vol.% C2H4 Flow SiH4 C3H~ Wt.% Composition
20 vol.% C2H2 gmol/min. gmol/min. gmol/min. C Si O
gmol/min.
1.04 1.12 1.09 0.06 0.11 33.6 5~.5 7.8
The run produced 218 gms of product. The weight percentage for silicon
was obtained by X-ray fluorescence analysis. X-ray powder diffraction
analysis of the product revealed peaks at about 35~, 60~ and 72~,
definitely indicating beta silicon carbide.
15.47g of the raw reactor product produced under the foregoing
conditions was milled with 0.46g yttrium oxide (Y203) and 0.16g aluminum
oxide (Al203) in the presence of 200ml ethanol and 2ml polyethylene
glycol (Carbowax PEG 400, Union Carbide, Danbury, CT) for 4 hours in a
high density polyethylene jar with nylon coated s-teel milling elements.
32122CA
42
The slurry was dried overnight to remove ethanol, crushed and screened
through a number 40 sieve~ and pressed at room temperature into a dlsc
3.34cm in diameterl 0.155cm thick, and weighing 1.25gm using a Carver
laboratory press and 70,000 lb force. At this point, the disc was
determined to have a density of 0.92 g/cc or 29% of pure crystalline
sllicon carbide density, hereinafter referred to as "theoretical
density". The disc was then sintered in a controlled atmosphere furnace
by first evacuating the furnace to less than 200 millitorr and rapidly
heating the sample from room temperature to 1000~C in 15 min. At 1000~C
the temperature was increased to 1500~C using a heating rate of 20~C/min
and was held at 1500~C for 30 minutes. The furnace was brought to
atmospheric pressure using argon and the temperature was raised to
1800~C using a heating rate of 10~C/min and subsquently ralsed to 2150~C
using a heating rate of 2~C/min. The sample was held at 2150~C for 1
hour and then cooled to room temperature. The density of the disc was
determined to be 2.92 g/cc or 91% of theoretical density. It is
important to note that this high density disc was successfully sintered
employing a raw reactor product with 7.8 wt.% oxygen without any oxygen
removal steps prior to sintering and without applying any compaction
force during sintering. It should also be noted that this high density
disc was sintered from a pressed disc having a density of only 29% of
theoretical density.
B. Sintering of Purified Product: Y2O3 and Al2O3 Sintering Aids
llOg of the raw reactor product as produced in part A. of this
example was mixed with llg carbon black. 114.9g of the resulting mixture
was placed in a graphite box, and the box was placed in a controlled
atmosphere furnace purged with argon. The furnace was heated to 1550~C
at 25~C/min. and held at 1550~C for 2 hours. The resulting powder
contained 36.7 wt.% C, 60.8 wt.% Si, and 1.38 wt.% O. Silicon weight
percentage was obtained by X-ray fluorescence analysis. The powder was
heated in air at 600~C to remove free carbon to thereby produce a
purified product powder.
A 14.90g sample of this purified product was milled as in part
A of this example using 0.33g yttrium oxide, 0.33g aluminum oxide, 60ml
32122CA
43 ~ t
ethanol, and 2ml of polyethylene glycol (Carbowax PEG 400). After
milling, drying, crushing, and sievlng, a dlsc 1.245cm ln diameter and
O.t37cm thick was pre~sed to a denslty of 1.23 g/cc. After placing the
disc in a furnace, the dlsc was sintered by raising the furnance
temperature from room temperature to 1000~C ln 15 mlnutes, lncreaslng
the temperature to 1800~C at a rate of 10~C/mln., and then further
heatlng up to 2200~C at a rate of 2~C/min. Upon reaching 2200~C, the
temperature was held at 2200~C for 90 minutes followed by cooling to
room temperature. This procedure produced a disc having a density of
3.08 g/cc, 96% of theoretical density.
C. Sintering of Raw Product: B Sinterin~ Aid
Another batch ~198 g) of raw product was produced uslng the
same apparatus and procedure used ln part A of thls example. Analysis
of the raw product ylelded weight percentages as follows: 34.8 wt.%
carbon; 57.0 weight% sllicon, as obtained by X-ray fluorescence; and 9.0
wt.% oxygen. X-ray powder diffraction analysis revealed peaks at 35~,
60~ and 72~ indicative of the presence of silicon carbide.
10.55 g of the above product was added to 200ml of a mixture
comprising 48 wt.% ethanol and 52 wt.% heptane. This mixture was milled
until the product powder was wet and formed a slurry. 0.1355 g of
elemental boron (B), available from Callery Chemical Co., Callery, PA,
was added to the slurry and the resulting mixture was milled for 4 hours
in a polyethylene jar with nylon coated steel milling elements. The
solvent, comprising ethanol and heptane, was evaporated and the
resulting dried cake was crushed and screened through a 40 mesh screen.
A disc was pressed from the screened powder. The disc density was 0.887
g/cc which is 28% of theoretical density. After pressing, the disc was
placed in a carbon element furnace and slntered using the following
procedure. The furnace was first evacuated to less than 200 millitorr,
and then heated from room temperature to 1000~C in 15 minutes. The
temperature was then further raised from 1000~C to 1500~C, again over a
period of 15 minutes. The 1500~C temperature was held for 30 minutes,
followed by pressurization of the furnace to 1 atmosphere with argon and
further elevation of the temperature to 2100~C in 15 minutes. The power
32122CA
44
to the furnace was then shut off and allowed to cool to room
temperature. The resulting disc was determined to have a density of
2.84 g/cc, 88% of theoretical density, which WflS obtained employing a
raw product with 9.0 wt.% oxygen without oxygen removal steps prior to
sintering and without application of compsction force during sintering.
It should also be noted that this high density disc was sintered from a
pressed disc having a density of only 28% of theoretical density.
D. Sintering of Purified Product: B Sintering Ald
The reactor described in Example V was utilized to produce
275g of raw product powder under the conditions set forth below. A flow
of helium purge gas at a flow rate of 0.15 gmol/min per reactant no~zle
was also employed in this run.
Combustion Fuel FlowOxygen Sidestream Flows Product AnalysisC:O Ratio80 vol.% C2H4 Flow SiH4 C2H4 C2H2 Wt.% Composition
20 vol.% C2H2 gmol/mingmol/min C Si O
~mol/min
1.04 1.12 1.090.059 0.052 0.07232.8 69.4 10.1
The product was analyzed by X-ray powder diffraction and revealed peflks
at about 35~, 60~ and 72~, indlcstive of the presence of beta silicon
carbide. 73g of the powder obtained in the foregoing synthesis was
placed in a graphite box purged with ~rgon and the box placed in a
controlled atmosphere furnace purged with nitrogen. The furnance was
heated to 1550~C and held at 1550~C for 2 hours. The resulting powder
contained 68.4 wt.% Si, 32.3 wt.% C, 1.2 wt.% O, and 1.1 wt.% N. The
powder was heated in air at 600~C until it turned a light green color.
The powder WAS subsquently washed with 12 wt.% aqueous
hydrofluoric acid, rinsed several times with water and alcohol and
vacuum dried.
23.8 g of the washed and vacuum dried product powder was mixed
with 0.31 g elemental boron (Callery Chemical), l.S9g phenolic resin,
(Plyophen 23-169, BLT Corp., Warren, NJ), 1.27g polyethylene glycol
(Csrbowax~ PEG 400), and 100 ml 8cetone snd milled for 15 hours. After
milling, drying, crushing, snd sieving, a disc 1.30 cm in diameter and
32122CA
2 ~
0.48 cm thlck was pxessed to a density of 1.97 g/cc, or 61% of
theoretica] density. After placing the disc in a controlled atmosphere
furnace, the disc was sintered by evacuating the furnace to less than
200 millitorr and heating from room temperature to 1000~C ln 15 minutes.
At 1000~C the temperature was increased to 1550~C at a rate of 40~C/min.
whereupon the furnace was brought to atmospheric pressure using argon.
Subsquently the furnace was heated to 2200~C at a rate of 40~C/min and
held at 2200~C for 30 minutes, followed by cooling to room temperature.
This produced a disc having a density of 3.10 g/cc, 97% of theoretical
density.
E. Sintering of Raw Product: Introduction of B Sintering
Aid in Synthesis
The reactor described in Example V was utilized to produce
60.2 g of raw product powder under the conditions set forth below. Note
that a mixture (available from Alphagaz, Inc., Walnut Creck, CA) of 1.9
vol.% diborane (BzH6) and silane is employed rather than pure silane in
order to introduce boron sintering aid into the raw product by means of
the synthesis reaction. Ilelium was used as a purge gas through each
reactant nozzle at a flow rate of 0.15 gmol/min.
Combustion Fuel Flow Oxygen Sidestream Flows Product Analysis
C:O Ratio80 vol.% C2H4Flow SiH4+B2H6 C3H6 Wt.% Composition
20 vol.% C2H2 gmol/min gmol/min C Si O
gmol/min
1.04 1.12 1.09 0.06 0.1131.3 59.6 8.23
In the above product analysis results, the silicon weight
percentage was obtained by X~ray fluorescence. The product was also
analyzed for boron content via an inductively coupled plasma system.
The measured mass ratio of B to Si was found to be 0.0091. An X-ray
diffraction pattern of the product shows peaks at 35~, 60~ and 72~ which
are clearly indicative of the presence of beta silicon carbide.
2.47 g of the above synthesized raw product was mixed with
isopropanol, whlch was then sub~ected to about 100 watts of ultrasonic
energy for 5 minutes using a Model W-380 Sonicator manufactured by Heat
Systems Ultrasonics, Inc. of Farmingdale, NY. Another mixture of
2 ~/ ~'J ~
46
isopropanol ~nd 0.12 g of carbon black (type FW18 from Degussa Corp.,
Teterboro, NJ) was prepared using the same ultrasonic procedure. The
two mix-tures were then combined and sub~ected to the ultrasonic
treatment again, followed by air drying on aluminum foil to remove the
isopropanol. 2.50 g of the resulting dried material was added to more
lsopropanol and 0.27 g of oleic acid was also added. The mixture was
then subjected to the above described ultrasonic procedure which
produced a slurry which was dried on aluminum foil, crushed through a 40
mesh screen, flnd pressed at room temperature into a disc. The organic
pressing aid, oleic acid, was burned out in accordance with the
following procedure. The disc was placed on a graphite tray and
inserted into a controlled atmosphere furnace which was heated to a
temperature of 500~C at a rate of 2~C/min. The temperature was then
raised to 1000~C at a rate of 5~C/min., and the 1000~C temperature was
maintained for 60 minutes. The resulting disc was 3.21 cm in diameter,
0.343 cm thick, and weighed 0.85 g giving a presintered density of 27%
of theoretical density.
The disc was then sintered, without application of compaction
force to the disc, using a high temperature furnace as follows: the
furnace was evacuated to 200 millitorr; heated to a temperature of
1000~C in 15 minutes; further elevated in temperature to 1450~C at a
rate of 20~C/min.; pressuri~ed to 1 atmosphere with purlfied argon;
heated from 1450~C to 2100~C at 60~C/min.; held at 2100~C for 25
minutes; and finally cooled to room temperature. The resulting disc
weighed 1.99 grams, was 2.08 cm in diameter and 0.208 cm thick. The
density was determined to be about 2.8 g/cc or 88% of theoretical
density.