Note: Descriptions are shown in the official language in which they were submitted.
~, ~ /a f1, f ; f~~
~~~..~_~,as.ai~
O.Z. 0050/40948
The stereoselective preparation of
Z-1.2-diarylallyl chlorides and the conversion
thereof into azolylmethyloxiranes and novel
intermediates
The present invention relates to the stereo-
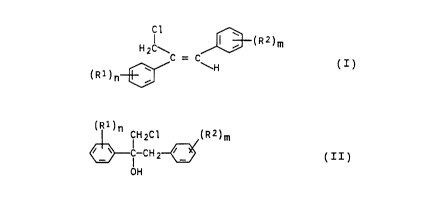
selective preparation of Z-1,2-diarylallyl chlorides of
the general formula I
H C\ ~ ~ (RZ)m
2 _
( R i ) n_~C _ C\H ( I ) .
in which R1 and R2, independently of one another, are
hydrogen, halogen, alkyl, haloalkyl, alkoxy, haloalkoxy
or an unsubstituted or substituted aromatic radical, and
n and m are 1, 2 or 3.
The present invention furthermore relates to the
conversion of Z-1,2-diarylallyl chlorides into the azolyl
methyloxiranes of the formula IV
x~~
HZC~ / ~ ~ '' (~2)m ( IV) ~
(R1)n ~ C _ C\H
in which ( Rl ) ~, ( RZ ) ~ are as defined above, and X is CH or
N.
The present invention also relates to the novel
intermediates I and to the epoxidation products V which
are precursors thereto.
German Laid-Open Applications DE-OS 3,218,129 and
3,218,130, EP-A 196,038 and US-A-3,422,153 indicate that
campounds of the structural type I are useful intermediates
for the preparation of pharmacological, fungicidal and anti-
mycotic active compounds. They have hitherto been obtained
by a free-radical halogenation of corresponding diaryl-
propene compounds (DE-A 3,218,129 or EP-A 196,038) or by
~ .i Gyp C'~ P~)
(uf ;.! ~ ~ :/ ;rd L.
- 2 - O.Z. 0050/40948
oxidation and subsequent substitution (DE-A 3,218,130).
Disadvantages in the methods of the prior art are the use
of expensive reagents, for example expensive halogenating
reagents such as N-bromosuccinimide for the free-radical
bromination, the number of synthetic steps and, in
particular, the low stereoselectivity.
It is generally known that molecules which have
a specific biological or pharmacological action must in
many cases have defined geometrical arrangements of
certain functional groups. In the case of the fungicidal
active compounds of the general formulae III and IV (see
DE-A 2,652,313), it is in particular the Z-configured
compounds (cf. the Cahn, Ingold and Prelog sequence
rule), ie. the compounds in which the substituted or
unsubstituted phenyl radicals are in the trans position
to one another, which have particularly high activity as
crop protection agents.
It is therefore an object of the present inven
tion to find a process by which the intermediates I can
be prepared in high isomeric purity, i:e. with a high
preference for the E- or traps-configuration of the
phenyl radicals on the double bond, and in high yield. It
is a further object of the present invention to find a
preparation process for the fungicidal azolylmethyl-
oxirane IV which uses advantageous intermediates and is
distinguished by high overall yields and by a number of
reaction steps which is low compared with the processes
described in DE-A 3,218,129 and 3,218,130.
Adcording to the prior art, aryl-substituted
alcohols can be converted into the corresponding aryl
substituted olefins or styrenes under acid reaction
conditions, for example using sulfuric acid in an organic
phase (see, for example, Houben-Weyl, Methoden der
organischen Chemie, 4th edition, Volume 5/lb - alkenes,
cycloalkenes, arylalkenes, Georg Thieme Verlag,
Stuttgart, 1972, pp. 62 ff., in particular pp. 70 and 71;
Tetrahedron, 26, (1970) 4277ff.
J.~~enL~l.1
- 3 - O.Z. 0050/40948
It is also known that reactions of this type can
be carried out with the aid of water-absorbing reagents,
for example acetic anhydride. However, relatively high
reaction temperatures are generally necessary for these
elimination reactions which are described in the litera-
ture. Under such reaction conditions, only inadequate E/Z
isomer ratios with respect to the aryl/aryl arrangement
are obtained.
We have found that the abovementioned objects are
achieved by a process for the stereoselective preparation
of Z-1,2-diarylallyl chlorides of the general formula I
H C\ I. ~ (RZ)m
2 C ; C\H
(I)r
(R1)n
in which R1 and R2, independently of one another, are
hydrogen, halogen, alkyl, haloalkyl, alkoxy, haloalkoxy
or a substituted aromatic radical, and n and m are l, 2
or 3, which comprises dehydrating chlorohydrins of the
formula II
(R1)nCH2C1 (R2)m
C-CHy~ (II),
I
OH
in which the radicals are as defined above, at up to 50°C
in an inert ether or carboxylic acid ester as solvent and
in the presence of carboxylic anhydride and an organic or
inorganic acid.
The process according to the invention gives Z
configured 1,2-diarylallyl chlorides in high stereo
selectivity. In general and in particular in accordance
with the preferred embodiments of the process, the Z:E
ratios are from 8:1 to 15:1, The high regioselectivity
with which the elimination of water occurs is also
surprising, since elimination of water in the direction
Cat3,~~'~\E',!~
~ ~.~ ;.~ .~. ~ E;~ iO
- 4 ° O.Z. 0050/40948
of the chloromethyl side chain to give chlorovinyldiaryl
compounds would have been expected to occur to a greater
extent as a side reaction. In addition, expected compe-
ting reactions, such as substitution instead of elimina-
tion, can successfully be suppressed. The expected
acylation of the alcohol function is also virtually
absent.
The chlorohydrins of the general formula II are
generally known and can be prepared, for example, in
accordance with DE-A 2,851,086, EP-A 47,594 or
EP-A 15,757 in good yields by the addition reaction of
benzyl-Grignard compounds VI with w-chloroacetophenones
VII in accordance with the reaction scheme below:
~R2)m
-I~-Cti2-CI + XMg-CH2~ --~ II
0
VII YI
(X = CI, B~)
In the process for the preparation of the Z-allyl
~ chlorides, it is likewise advantageous to first prepare
the chlorohydrins in diethyl ether and to carry out the
dehydration as a one-pot process by adding inorganic
acid, for example concentrated sulfuric acid, and car
boxylic anhydride to the diethyl ether solution at from
about -10 to 0°C.
It is also possible, instead of an aqueous work-
up in the chlorohydrin synthesis, to liberate the chloro-
hydrin from the magnesium alkoxylate precursor by adding
an equimolar amount of an acid, for example sulfuric
acid, and subsequently to carry out the dehydration.
It is advantageous and according to the invention
here to gradually meter in the carboxylic anhydride, in
which case the 0-acylation of chlorohydrin can be sub-
stantially suppressed compared with the dehydration.
The dehydration according to the invention of the
chlorohydrins II is carried out in an ether or ester as
solvent. In the case of open-chain ethers, those having
('1 ~ ~ A f1, ,(7 =..y
~s~ .s ~l i~~d
- 5 - O.Z. 0050/40948
at least two oxygen atoms, such as ethers of glycols and
low-molecular-weight aliphatic alcohols, for example
ethylene glycol dimethyl ether or ethylene glycol diethyl
ether, are preferred. Cyclic ethers, such as tetrahydro-
furan (THF) and in particular dioxane, are particularly
advantageous. Small amounts of aprotic solvents, such as
ethyl acetate, halogenated hydrocarbons, such as methy-
lene chloride, or THF, can be added, for example, to
dioxane as solvent in order to produce better solvolysis
at low temperatures, for example below about 10°C.
Particularly suitable esters for the process
according to the invention are those made from low-
molecular-weight aliphatic carboxylic acids, in parti-
cular monocarboxylic acids, and low-molecular-weight
aliphatic alcohols, where the term low-molecular-weight
in each case means containing from about 1 to 6 carbon
atoms. The following are specific examples of esters:
ethyl acetate,_ethyl formate, methyl propionate, methyl
butyrate, and methyl or ethyl isobutyrate, ethyl acetate
being preferred.
The amount of solvent is not particularly crucial
and can be varied within broad limits . It is generally
from about 1 to 50~ by weight, in particular from 2.5 to
10~ by weight, based on the chlorohydrin II. A larger
excess of solvent is entirely possible, and mixtures of
the solvents mentioned, for example, in claims 1 to 5 can
also be used for the dehydration, it being possible to
vary the mixing ratios in a broad range of from about
10:1 to 1:10. Addition of from 5 to 20$ by weight, based
on dioxane, have been successful in order to achieve
relatively high space-time yields and high Z-product
proportions.
The water-absorbing agent added to the reaction
mixture is a carboxylic anhydride, in particular an
anhydride of an aliphatic low-molecular-weight mono
carboxylic acid, such as acetic anhydride, propionic
anhydride, butyric anhydride or isobutyric anhydride.
:~ .9 f~ f, ~. i
nd '.l ~ .w >_ s :~n (_.I
- 6 - O.Z. 0050/40948
However, it is also possible for anhydrides of aliphatic
or aromatic dicarboxylic acids, such as malonic anhy-
dride, malefic anhydride, succinic anhydride or phthalic
anhydride to be present.
The dehydration is generally carried out using
from 0.5 to 3 mole equivalents, in particular from 1 to
2 mole equivalents, of anhydride, based on the chloro-
hydrin TI. Larger amounts are possible, but bring no
further advantages.
Particularly advantageous results are obtained
using a combination of dioxane and/or THF as solvent with
acetic anhydride and sulfuric acid or using ethyl acetate
as solvent with isobutyric anhydride and sulfuric acid.
The dehydration is carried out under acidic
reaction conditions, using acids which are conventional
for this purpose, for example organic sulfonic acids,
such as trifluoromethanesulfonic acid, methanesulfonic
acid, pare-toluenesulfonic acid or naphthalenesulfonic
acid and in particular concentrated mineral acids, such
as perchloric acid, phosphoric acid and in particular 30
to 99.9, preferably 50 to 99~, sulfuric acid, or oleum.
More carboxylic anhydride is generally used in the case
of stronger aqueous acids.
The acid can be employed in a catalytic amount,
a stoichiometric amount or in excess, based on II.
Amounts of from about 0.01 to 4 mole equivalents, based
on II, are preferred. When oleum is used, smaller amounts
of from 0.05 to 1 mole equivalents, based on II, are
advantageous.
Any advantageous variant of the process according
to the invention comprises using ketene instead of the
carboxylic anhydride as the water-absorbing agent, if
desired in combination with a stoichiometric or catalytic
amount, based on II, of an aliphatic carboxylic acid. In
this case, it is advantageous to initially introduce the
carboxylic acid, for example one of the abovementioned
low-molecular-weight aliphatic carboxylic acids, and to
CA 02021328 2000-04-17
7
add the gaseous ketene to the reaction mixture, or to add
the gaseous ketene to the chlorohydrin II dissolved in
the solvent without addition of carboxylic acid. The
amount of ketene here corresponds to the abovementioned
amounts for the carboxylic anhydride.
In order to produce a large proportion of the Z
isomer, the dehydration should be carried out at the
lowest possible temperature, ie. at up to about 50°C,
advantageously at from -25 to +40°C, in particular from
-25 to +30°C.
In general, the dehydration is carried out under
l0 atmospheric pressure. It is also possible to carry out the
reaction under reduced pressure or under super-atmospheric
pressure, and in some cases increasing the pressure can
result in an increase in the space-time yield.
The Z-1,2-diarylallyl chlorides of the formula I~
II
(R2)m
C = C~ (I~,
fR1)n
in which R1 and R2, independently of one another, are
20 halogen, C1-C~-alkyl, C1-C6-haloalkyl, C1-C5-alkoxy, C1-C5-
haloalkoxy or an aromatic radical which is unsubstituted or
monosubstituted to trisubstituted by the radicals mentioned
for R1 and R2, and m and n are 1, 2 or 3, which can be
prepared by the process according to the invention, are
likewise the subject-matter of the invention.
In the formula I, the indices m and n are pre-
ferably 1, and the substituents R1 and RZ, independently
of one another, are in particular:
30 hydrogen;
halogen, such as fluorine, chlorine, bromine or iodine,
preferably chlorine or fluorine;
t~1 ,f~?~ ~ ~ s~ ~v, ~ l
!~s i y oa :. ?.p F~ L.:~
8 - O.Z. 0050/40948
linear or branched C1-C~-alkyl, such as methyl, ethyl,
propyl, 1-methylethyl, butyl, 1-methylpropyl, 2-methyl-
propyl, 1,1-dimethylethyl, pentyl, 1-methylbutyl, 2-
methylbutyl, 3-methylbutyl, 1,1-dimethylpropyl, 1,2-
dimethylpropyl, 2,2-dimethylpropyl, 1-ethylpropyl, hexyl,
1-methylpentyl, 2-methylpentyl, 3-methylpentyl, 4-methyl-
pentyl, 1,1-dimethylbutyl, 1,2-dimethylbutyl, 1,3-
dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 3,3-
dimethylbutyl, 1-ethylbutyl, 2-ethylbutyl, 1,1,2-tri-
methylpropyl, 1,2,2-trimethylpropyl, 1-ethyl-1-methyl-
propyl or 1-ethyl-2-methylpropyl;
C1-Cs-haloalkyl, such as fluoromethyl, difluoromethyl,
trifluoromethyl, chlorodifluoromethyl, dichlorofluoro-
methyl, trichloromethyl, 1-fluoroethyl, 2-fluoroethyl,
2,2-difluoroethyl, 2,2,2-trifluoroethyl, 2-chloro-2,2-
difluoroethyl, 2,2-dichloro-2-fluoroethyl, 2,2,2-tri-
chloroethyl or pentafluoroethyl, preferably trifluoro-
methyl;
C1-C5-alkoxy, such as methoxy, ethoxy, propoxy, 1-methyl-
ethoxy, butoxy, 1-methylpropoxy, 2-methylpropoxy or 1,1-
dimethylethoxy, preferably methoxy, ethoxy or propoxy;
C1-C5-haloalkoxy, such as difluoromethoxy, trifluoro-
methoxy, chlorodifluoromethoxy, dichlorofluoromethoxy, 1-
fluoroethoxy, 2-fluoroethoxy, 2,2-difluoroethoxy,
1,1,2,2-tetrafluoroethoxy, 2,2,2-trifluoroethoxy, 2-
chloro-1,1,2-trifluoroethoxy or pentafluoroethoxy,
preferably trifluoromethoxy;
an aromatic radical, for example phenyl whit-h is unsub-
stituted or monosubstituted to trisubstituted by a
radical R3 which has the preferred meaning given for R1 or
RZ, ie. is hydrogen, halogen, linear or branched C1-C,-
alkyl, ~C1-C6-haloalkyl, C1-C$-alkoxy or C1-CS-haloalkoxy.
r., ~ , ~ r
~r l r .r.. s_: F.r i~
- 9 - O.Z. 0050/4094E
R1 is preferably 4-F and RZ is preferably 2-C1.
Z-1,2-diarylallyl chlorides of the general
formula I have unexpected advantages over the 1,2-diaryl
allyl bromides disclosed in DE-A 3,218,129. Besides very
simple epoxidation to give the diaryloxiranes of the
general formula V, it is particularly advantageous that
stereoselective epoxidation means that isomer mixtures of
the oxiranes are not obtained, which is the case starting
from the known Z-1,2-diarylallyl bromides, but instead
oxiranes in which the aryl radicals are transoid are
obtained.
Examples of possible substitution patterns are
shown in Table 1 belowe
'~ !1 .E ~T~ ,-~. f',
~ 2d (-J l
- 10 - O.Z. 0050/40948
Table ~.
~1 ( ~ ~R2)
H2C~ .~ m
C = C
--~ ~H
~R1)n~ (I) r
Compound No . ( Rl ) n ( R2 ) m Melting pt . [ ° C ]
1H ~ L PPm l
1.1 3-C1 3-C1
1.2 4-C1 2,4-diCl
1.3 4-F 2-CH3
1.4 4-F 2-CF3
1.5 H 2-OCF3
1.6 4-F 2-C1 66
1.7 4-OCH3 2-C1
1.8 4-Br 2,4-diCl
1.9 4-C6H5-CH20 3-CH3
1.10 4-p-C1C6H,, 2-Cl
1.11 n-C4H9 2-C1
1. 12 4-CsH$ 2, 4-diCl
1.13 4-F 3-CF3
1.14 4,5-diCl 2-CH3
1.15 4-C6H50 2-C1
1.16 4-C1 2-C1 79-82
In the diarylallyl chlorides I, the Z:E isomer
ratio can be determined in a known manner, for example by
HPhC (high-pressure liquid chromatography), by gas
chromatography or by 'H NMR using the pure Z- and E-
isomers as comparison and standardizing the corresponding
mixing ratios.
The preparation of the fungicidal active com
pounds III and IV, starting from the diarylallyl chlorides
I or the chlorohydrins II, is Shawn in the reaction
scheme below:
-- 11 -- 0. Z . 0050/40948
IH2C1 (R2)m
C-CHZ~ II
(Ri)n ~ O ~H
-Hg0
jl (RZ)m
H2C~ ~ I
C = C
(Ri )n~ \H
X~I
Route a) + H lepoxidation Route b)
-HC1
XII 11 C1 (R2)m
H fCV~\ I s (RZ)m HZC\ ~
2 _
C - C\li (III) . (Ri a C C\H (v)
lRi)n~~
) n'-
Permaleic + X
dIihy'dr'l.de
H
-HC1
X (R2)m
I
H2C\ /0~
C - C (IV)
(R1)n ~ \H
Gtr ~ cj A c1 r: ;, .~
a
~ ,.. ;.a .S. :a ~=.~ a
- 12 .- O.Z. 0050/40948
Route b) can be carried out in a conventional
manner, for example as described in principle in
DE-A 3,218,129. The substitution of the chlorine atom by
the azole or imidazole group in compound V is usually
carried out in an inert solvent, such as dimethyl-
formamide or N-methylpyrrolidone, in the presence of an
inorganic or organic base, for example sodium hydroxide,
potassium hydroxide, sodium carbonate, potassium car-
bonate, dicylohexylamine or dimethylcyclohexylamine.
The intermediates V are novel. With respect to
the preferred radicals R1 and RZ and the indices n and m,
the definitions given for the compounds I apply analo-.
gously. Examples of possible substitution patterns are
given in Table 2 below:
/Y, S') C3 I ~
r
~~d .z_ ':4 ~a
- 13 - O.Z. 0050/40948
Table 2
~ ~RZ)m
'~H ~V)r
(R1)T1 0
Compound No . ( Rl ) n ( RZ ) ~ Melting pt . [ ° C ]
. 1H NMR [ppm]
2.1 3-C1 3-C1
2.2 4-Cl 2,4-diC1
2.3 4-F 2-CH3
2.4 4-F 2-CF3
2.5 H 2-OCF3
2.6 4-F 2-Cl 68-70
2.7 4-OCH3 2-C1
2.8 4-Br 2,4-diCl
2.9 4-C6H5-CHZO 3-CH3
2.10 4-p-C1CBH,, 2-C1
2.11 n-C4H9 2-C1
2 .12 4-C6H5 2, 4-diCl
2.13 4-F 3-CFA
2.14 4,5-diCl 2-CH3
2.15 4-C6H50 2-C1
2.16 4-C1 2-C1
In route a), the first step, ie, the substitu-
tion, proceeds analogously to the final step of route b).
The dehydration and the subsequent substitution can
advantageously be carried out in a one-pot process with-
out isolation and purification of the intermediate II.
The epoxidation of the compounds III is carried
out according to the invention in the presence of a large
excess of permaleic acid, the permaleic acid being
prepared in situ by reacting from 5 to 30 mole equiva
lents,- in particular from 5 to 10 mole equivalents, of
malefic anhydride, based on III, with less than the
c~,~~~ ys,~-y
~.?~ t r .~. -i i..~ i.~
- 14 - O.Z. 0050/40948
stoichiometric amount of hydrogen peroxide solution,
based on the malefic anhydride. In general, anhydride: HZp2
molar ratios of from 1.5 to 10, in particular from 2 to
4, are employed. A 30 to 50~ strength aqueous solution of
hydrogen peroxide may advantageously be used.
The reaction temperature for the epoxidation can
be from 0 to 100°C, in particular from 20 to 80°C.
The epoxidation is carried out in the presence of
an aprotic, polar solvent. Examples of suitable solvents
are halogenated hydrocarbons, such as dichloromethane,
dichloroethane, chlorobenzene or chlorotoluene, or
aromatic hydrocarbons, such as benzene, toluene or
xylene. The amount of solvent is not particularly crucial
and is generally from 5 to 50~ by weight, in particular
from 10 to 20~ by weight, based on the olefin.
Using this epoxidation method, the azolylmethyl-
oxiranes IV can be obtained in considerably higher yields
than in the processes described in DE-A 3,218,129.
The individual synthesis steps are described in
the experimental examples below.
EXAMPLE 1
Preparation of the starting materials II
1-Chloro-2-(4-chlorophenyl)-3-(2-chlorophenyl)propan-2-
ol
5.0 g (0.031 mol) of 2-chlorobenzyl chloride are
added within 5 minutes at from 24 to 36°C to 9.7 g
(0.404 mol) of magnesium turnings in 20 ml of absolute
ether. After the reaction has commenced, a solution of
200 ml of absolute ether and 50.2 g (0.31 mot) of 2-
chlorobenzyl chloride is added dropwise. The mixture is
subsequently refluxed for about a further 10 minutes, the
excess_magnesium is decanted off under nitrogen, and the
Grignard solution is cooled to 0°C. 55.7 g (0.3 mol) of
y r=. c, .~ >
d ~. °,A l"r !
- 15 - O.Z. 0050/40948
para-chloro-w-chloroacetophenone dissolved in 350 ml of
toluene are then added dropwise, and the reaction mixture
is stirred at 0°C for 1.5 hours and added dropwise at
from about 2 to 6°C to 1.5 1 of concentrated ammonium
chloride solution. Extraction with methyl tart.-butyl
ether and subsequent conventional work-up gives 92.9 g
(yield 99~, purity according to HPLC: 68.2 0 of 1-chloro-
2-(4-chlorophenyl)-3-(2-chlorophenyi)propan-2-of as a
crude oil, which can be further reacted directly. For
characterization, the product was recrystallized from n-
hexane.
Melting point: 64 to 69°C.
EXAMPLES 2 TO 5 AND COMPARISON EXAMPLES I TO IV
Dehydration of the chlorohydrins II
Z-3-Chloro-2-(4-chlorophenyl)-1-(2-chlorophenyl)propene
(Compound No. 1.16 in Table 1)
24.5 g (0.24 mol) of acetic anhydride are addad
at -2°C to 60 g (0.2 mol) of the chlorinated alcohol
described in Example 1 in 230 ml of dioxane and 23 ml of
tetrahydrofuran, and 2.36 g (0.024 mol) of concentrated
sulfuric acid are subsequently added to the mixture.
After the mixture has been stirred at 0°C for 3 hours,
HPLC analysis shows that virtually all the starting
material has reacted.
A mixture of half-saturated sodium chloride
solution and 50~ strength sodium hydroxide solution is
subsequently added at 0°C over the course of 30 minutes
until the pH is from 8 to 9.
Finally, the organic phase is dried and evapora
tad under reduced pressure and can be used for subsequent
reactions without further purification.
Yield 55.7 g (Z/E=9.1/1), crude oil, recrystal-
lization from n-hexane to get the pure Z-isomer of
melting paint 79 to 82°C.
s3 ~ fh c~. r~
a a ._. r ~,~ J
- 16 - O.Z. 0050/40948
The Z-1,2-diarylallyl chlorides in Table 1 can be
grepared in a similar manner. _
Z-3-chloro-2-(4-fluorophenyl)-1-(2-chlorophenyl)-
propane
(Example No. 1.6 in Table 1)
1-Chloro-2-(4-fluorophenyl)-3-(2-chlorophenyl)-
propan-2-ol, prepared by Grignard addition of 2-chloro-
benzylmagnesium chloride to pare-fluoro-~-chloroaceto-
phenone and employed as the crude material having an HPLC
purity of 78-87~, was reacted as described in Example 2
under the reaction conditions given in Table 2. The
proportions of Z- and E-isomers were determined by HPLC
(high-pressure liquid chromatography) analysis (uncor-
rected relative area percentages).
C~ ~~ : ~ c~ c, .
~rf~~.s~~~1.~~
- 17 - O.Z. 0050/40948
_ r
M
N
t0 M
v E
.r'-~
c ..a
~o
L
y aM
L U
N ~
v
y. H
d C
Y~
Q
O,fD
O_
dd
N 4!7n
of ~c ,~ a cc
N W
L
v
C
O
U
L O
C
M ~1
~
u~ M
i N
> N
0
.,..I
f E so M M ~ m
I
F'
I
N ~
_
( ~n
U v N N N v
I
a
0
v
I w ~_ m m mw a ao
=
'Jy/ ~ 0 0 o c o
V ~ w o 0 o c o
II .n
.~ v .~ ~
0
vs
i~
0
a ~ ~ d
1 s s
o os o o
V! ~
C
N Vf N C N
ro C .- L
O L
w = = ~ N b NU
' x s
1 H : ~
.~'
. .
. " ~ " >,
cv ca co c ~'"
r
O O O O O U O
U U N U
U
i0
O U U U iCU
i0 O
~
O O O O O
o (~ O O O O
O
Of OI QI 01 OIOI
O1 OI Of'O OI
V N N N N M N
M M C M
U a O O O L N O
1 N N M O N
tp O
rl
W
0
O N
~. m A
ro
p v
z
ro
ro
=
~
.7~t nr ro c
C
~0
m
" o o s
u. ~ ou. sv_
~(7~ ~~ -o~v v~ v~
O O D O
O O p
C O
O
d _
> E E E E E
E E E E
O O O
N N N O
N N
N N N N
N
x
,..9 m W
M ~B In
ro
_ =a
- 18 - O.Z. 0050/40948
.W
'
ro v Nv
v n
' o w o
v ~
L N N L
U t W N
J '
. N p
C N LA .Q
O
Lr
L ~ 3~
d M t0 7 L
Ln ~
U 01 au .N
O O p~ p~
d~
~7NvN Z~ F-ar
_O
su
m W ,~
L
M ~ O
41 _
N m
L
N
7
C
O
U
O
O C
_ M 1A
,D
~ 07 t0
V
> N
P O
~ r
X
d U O 7-
h v.. O , tOfo .
N
d'
_C
L
'D
I
H O O O
.....
d O L
f0 .
tG
C ~ O O N t
7 ~ O O U
\ DJ
W t
N Y
C
C
_U _O
U C ~r
_ O U
C 4- C
O
~ 7 4.
F-
_ N
G7 2
N C O
H
C ~ y
N _ L
C ~ O ~
N _ d-
O O I~ C m
m Y d C7 ~-
I
~ 7 7
d k.
.
U O 01 O
_ 4. ~ N
O'
fl ~ Y
C1 U
Q N 1~. d
m m
L O
dw
W
d N
LY
G; ~ T
~ U
C ~ L
M'
U m ~- N cn
rt1 m d ~ ~ r,
r~ U a~ .-
C
_C
41 C d
O
7~
i~ U Y X
U
m L
O O
O
C p
E OI
_
E t7~OI N
E
U .1.~
~n ., m m
W
00 ~ N
N o
U ~e-
U O
m
r-~ ,~ C
O_ ~ ".y,.,7
c
X
E
w
~~~ J.. ~,~,,-,
l;a.:..~.>r,~.:
- 19 - O.Z. 0050/40948
EXAMPLE 6
Preparation of the chlorohydrin and in situ dehydration
1-Chloro-2-(4-fluorophenyl)-3-{2-chlorophenyl)propan-2-
ol
170 g (1.0 mol) of 2-chlorobenzyl chloride
dissolved in 400 ml of diethyl ether were added to 36.0 g
(1.5 mol) of magnesium turnings in 200 ml of diethyl
ether. 155 g (0.9 mol) of para-fluoro-c~-chloroaceto-
phenone, dissolved in 450 ml of diethyl ether, were
subsequently added dropwise at -10°C, and the mixture is
then stirred for a further 2 hours at 25°C.
49 . 0 g ( 0. 5 mol ) of concentrated sulfuric acid in
300 ml of diethyl ether are then added dropwise at -10°C.
The mixture is allowed to warm to 25°C, and the precipi-
tated salt is filtered off with suction. The crude ether
solution of the chlorohydrin is then employed further.
Z-3-chloro-2-(4-fluorophenyl)-1-(2-chlorophenyl)propene
8.0 g (0.08 mol) of concentrated sulfuric acid
are added at -10°C to 525 ml of the above-described crude
solution, containing about 134.5 g of chlorohydrin
(corresponding to 0.45 mol), and 57.1 g (0.56 mol) of
acetic anhydride are subsequently added dropwise over the
course of 2 hours. A small amount of precipitated salt is
then again filtered off. The solvent is evaporated from
the filtrate, and the crude a11y1 chloride can be used
further for the triazole substitution or for the epoxida-
Lion.
w sa .r~: r;
~~ f i .. :J ~..:
- 20 - O.Z. 0050/40948
EXAMPLE 7
Ketene variant
Z-3-chloro-2-(4-fluorophenyl)-1-(2-chlorophenyl)propene
250 ml of dioxane, 25 ml of tetrahydrofuran,
12.4 g of acetic acid (0.2 mol) and 69 g (0.23 mol) of
crude 1-chloro-2-(4-fluorophenyl)-3-(2-chlorophenyl)-
propan-2-of obtained from the Grignard reaction as per
Example 1 are mixed at 0°C, and 43 g (1.02 mol) of ketene
in gaseous farm are introduced within about 1 hour. After
customary work-up, a virtually identical yield as when
acetic anhydride is used in the above-described Example
2 is achieved according to HPLC analysis. The Z:E isomer
ratio when the reaction is carried out in this manner is
about 11:1.
EXAMPLES 8 AND 9
Preparation of the azolylmethyloxiranes IV by route a)
Z-3-(1,2,4-triazol-1-yl)-2-(4-chlorophenyl)-1-(2-chloro-
phenyl)propene
6.6 g of sodium hydroxide are added to a solution
of 11.5 g (0.17 mol) of triazole in 150 ml of dimethyl-
formamide, and the mixture is warmed at about 70°C until,
with stirring, a clear solution has been formed. The
mixture is subsequently cooled to 10°C, and 49.5 g of the
Z-3-chloro-2-(4-chlorophenyl)-1-(2-chlorophenyl)propene
as the crude product, prepared as in Example 2, dissolved
in 50 ml of dimethylformamide, are added dropwise within
1 hour, and the mixture is then stirred at room tempera-
ture for a further 4 hours.
200 ml of water are then added, and the mixture
is extracted several times with methyl tert.-butyl ether.
s
G'd .4 c3 c3 r.i
/ y~ 4
FJ !f ~ ,1 t~ end ~.,1
- 21 - O.Z. 0050/40948
The combined organic phases are washed, dried and evapo-
rated at a reduced pressure. Recrystallization from
methyl tert.-butyl ether and n-hexane gives 24.4 g of Z-
3-(1,2,4-triazol-1-yl)-2-(4-chlorophenyl)-1-(2-chloro-
phenyl)propene of melting point 106-110°C.
cis-2-(1,2,4-Triazol-1-ylmethyl)-2-(4-fluorophenyl)-3-(2-
chlorophenyl)oxirane
84 g (0.9 mol) of malefic anhydride and 6 drops of
concentrated sulfuric acid in 90 ml of dichloroethane are _
warmed to 50°C with 22 g of 50% strength hydrogen
peroxide. 28 g (0.089 mol) of Z-3-(1,2,4-triazol-1-yl)-
2-(4-fluorophenyl)-1-(Z-chlorophenyl)propene in 75 ml of
dichloroethane are added dropwise. The mixture is stirred
at this temperature for a further 3 hours and sub-
sequently at 70°C for a further 2.5 hours.
The reaction mixture is cooled, the precipitated
malefic acid is filtered off with suction, and the fil-
trate is washed by shaking with thiosulfate solutian and
dilute sodium hydroxide solution. The organic phase is
dried, substantially evaporated at about 50°C under
reduced pressure, cooled and re-evaporated to give 14 g
of useful product (~ 50% yield).
EXAMPLES 10 AND 11
Preparation of the azolylmethyloxiranes I'V by route b)
cis-1-Chloromethyl-2-(2-chlorophenyl)-1-(4-fluorophenyl)-
oxirane
(Compound No. 2.6 in Table 2)
56.2 g (0.2 mol) . of Z-3-chloro-2-(4-fluoro-
phenyl)-1-(2-chlorophenyl)propene in 530 ml of glacial
acetic_ acid are mixed with 196 g (2 mol) of malefic
anhydride, and 68 g (1 mol) of 50% strength hydrogen
J lJ
- 22 .- O.Z. 0050/40948
peroxide solution are added at 25°C within 1 hour. The
mixture is stirred at 40°C for a further 3 to 4 hours and
subsequently at 25°C for a further 10 hours.
Finally, the reaction mixture is stirred into
3 liters of water and 50 ml of 10~ strength sodium
thiosulfate solution, and a further small amount of
thiosulfate solution is added if necessary until peroxide
is no longer detectable. The colorless precipitate
produced is filtered off with suction and dried. The
crude material is employed without further purification.
(Recrystallization of n-hexane; m.p. 68 to 70°C).
cis-2-(1,2,4-Triazol-1-ylmethyl)-2-(4-fluorophenyl)-3-(2-
chlorophenyl)oxirane
1.5 g (S mmol) of cis-1-chloromethyl-2-(2-chloro-
phenyl)-1-(4-fluorophenyl)oxirane and 0.69 g (7.5 mmol)
of sodium 1,2,4-triazolide are stirred at 75°C for 5
hours in 7 ml of dimethylformamide. After cooling, the
mixture is neutralized by adding a little acetic acid,
and a little water (about 10 ml) is added, a crystalline
product precipitating (yield: 1.4 g). The product is
filtered off with suction, washed with water and dried
under reduced pressure.