Note: Descriptions are shown in the official language in which they were submitted.
23305~1163
NO\!~L 2-0~0-1-QYA-8-AZAsPIRO/4,57~ECA~IE DERIVATIVES, PHARI~A-
CEUTICAL COMPOSITIONS CONTAINING THEIl AND PROCESS FOR
PREPQRI~!~ SA!-lE
The invention relates to novel, therapeutically active
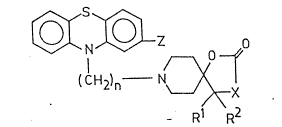
2-oxo-1-oxa-3,8-diazaspiro[4-5]decane derivatives of the
formula (I),
7 0
0 ~
'~ (CH2)n ~ N~X (I )
Rl R2
wherein
X means oxygen or an NR group, ~herein
R stands for hydrogen, a Cl_l2alkyl, C3_6cycloall<yl,
carbocyclic C6_10aryl or carbocyclic C6_10aryl-
C1_4alkyl group, the two latter ones optionall~
being substituted on -their aromatic part by one or
more, same or different halogen(s), one or more
Gl_4alkyl or Cl_4alkoxy group(s);
R1 and R2 tooether represent a methylene group or, when X
stands for an ~NR group, wherein R is as defined above,
one of Rl and R2 may represent a hydroxyl group, where-
as the other is a methyl ~roup;
Z stands for hydrogen, halogen, trihalomethyl or a
C2 4alkanoyl group; and
n is 2 or 3,
as well as their acid addition and quaternary ammonium salts
A 4629-67 MR
- 2 _ 23305-1163 ~
and pharmaceutical compositions containing these co~ounds.
The invention also relates to a process for the prepa-
ration of the above compounds and compositions as well as to
a method of treat~ent. The latter one comprises administer-
ing a therapeutically effective amount of a compound of the
formula (I) or a pharmaceutically acceptable acid addition
or quaternary ammonium salt thereof into the organism of a
patient for influencing the psychic functions or treating
allergic sy~p-to~s.
The compounds of the formula (I) may exist in various
stereoiso~eric forms such as geometrical isomers as well as
racemates, individual optical isomers and their mixtures,
all of which may occur in the form of various solvates and
hydrates. All these compounds and mixtures are within the
scope of the invention.
A number of therapeutically useful 2-oxo-1-oxa-3,8-di-
azaspiro/4-5/decane derivative have been describe~ in the
literature. Such compounds are reported e.g. in the follow-
ing publi~ations: C.A. 71, 913S9d (1969); C.A. 78, 719668t
~û (1973); C.A. 78, 2387q (1973); C.A. 81, 33153c and 105368b
(1974); C.A. 95, 161765e (1981); as well as in the DE patent
specifications Nos. 2,013,729, 2,013,668 and 2,163,000, in BE
patent specifications Nos. 775,984, 774,170, 786,631 and
825,444; in the GB patent speci~ication No. 1,100,218; in the
published N~ patent application No. 7,214,689 as well as in the
US patent specifications Nos. 3,555,033, 3,594,386, 4,244,961
and 4,255,432.
A substantial difference between the compounds of the
formula (I) according to the invention and similar derivati-
ves kr,own up to the present appears in the nature of the
substi-tuents bound in position 4 and optionally in position
3 of the spirodecane skeleton.
According to an other aspect of the invention, -there is
provided a process for the preparation of compounds of the
formula (I) as well as their acid addition and quaternary
ammonium salts, which comprises
a) reacting a 2-oxo-3,8-diazaspiro/4-5/decane derivative
of the formula (II),
. ~0_
HN~
~ `~ -R
/ \ 2 (Il)
wherein R, Rl and R2 are as defined above, with a phe-
nothiazine derivative of the formula (III),
. .
~ [~ ~--
(CH2)" y ( 111)
wherein n and Z are as defined above and Y means halo-
gen, Cl_4alkylsulfonyloxy or arylsulfonyloxy group,
to obtain compounds of the formula (I) "~lherein X
2 ~
-- 4
stands for an NR group and R, n, Rl, R2 as well as Z
are as defined above;
or
b) reacting a 2-oxo-3,8-diazaspiror4,5~decane derivative
of the formula (IV),
Y--(C H2~ N~
Rl R~ :
IG (IV)
wherein R, n, R1 and R2 are as defined above and Y
means halogen, C1_4alkylsulfonyloxy or arylsulfonyloxy
group, with a phenothiazine derivative of the formula
(V),
~ ~
(V) ;' :
wherein Z is as defined above,
to obtain compounds of the formula (I), wherein X
stands for an NR group and R, n, Rl, R2 as well as Z
are as defined above;
or
c) reacting a 4-ethynyl-4-hydroxypiperidine derivative of
. the formula (VI),
. . .
,
~ ~ 2 ~
~ N '~Z OH
r~ ~
(Cl~)n~N~
C _CH ( Vl
wherein n and Z are as defined above, with an isocya-
nate of the formula R-NC0, wherein R is as defined
above, to obtain a 4-carbamoyloxy-4-ethynylpiperidine
derivative of the formula (VII),
~N~Z o--CO--NHR
( CH~)n --- N~
C--CH (Vll )
wherein R, n and Z are as defined above and then
i) cyclizing in an acidic medium the obtained compound
of the formula (VII~, wherein R, n and Z are as
defined abov~ and reacting with water the obtained
salt of 2-imino-1,3-dioxolane derivative of the
formula (VIII),
z NR
( CH~)n--N j~_o
CH2 (Vlll
-- 6
wherein R, n and Z are as defined above, in
order to prepare compounds of the formula tI),
wherein Rl together with R2 stands for a methylene
group, X stands for an oxygen atom and R, n and Z
are as defined above, or
ii) cyclizing in a basic medium the obtained compound
of the formula (VII), wherein R, n and Z are as
defined above,
in order to prepare compounds of the formula (I), wherein
R1 together with R2 stands for a methylene group, X
stands for a NR group, and R, n as well as Z are as
defined above,
or
d) cyclizing in an acidic medium a 4-carbamoyloxy-4-ethy-
nylpiperidine of the formula (VII), wherein R, n and
Z are as defined above, and reacting with water the
obtained salt of 2-imino-1,3-dioxolane derivative of
the formula (VIII), wherein R, n and Z are as defined
above,
to obtain compounds of the formula (I), wherein Rl
together with R2 stands for a methylene group, X means
oxygen and n as well as Z are as defined above;
or
e) cyclizing in a basic medium a 4-carbamoyloxy-4-ethy-
nylpiperidine derivative of the formula (VII), wherein
. R, n and Z are as defined above,
to obtain compounds of the formula ~I), wherein
, : : . : .. :
'; ' '~ '
- 7
together with R2 stands for a methylene group, X means
an -NR group and R, n and Z are as defined above;
or
f) reacting a 4-acetyl-4-hydroxypiperidine derivative of
the formula (X),
$~N~Z OH
(CH2)n C-C1~3 (X)
O
wherein n and Z are as defined above, with an isocya-
nate of the formula R-NCO, wherein R is as defined
above, to obtain a 4-acetyl-4-carbamoyloxypiperidine
derivative of the formula (IX)
~N~ r O-CO--NHR
n - ~ N~X
11 (IX~
wherein R, n and Z are as defined above,
and then cyclizing the obtained compound of the for-
mula (IX),
to obtain compounds of the formula (I), wherein X
means an ~,NR group, one of Rl and R2 stands for a
hydroxyl group and the other is a methyl group, and R,
.
.- . :
-- 8
n as well as Z are as defined above;
or
g) cyclizing a 4-acetyl-4-carbamoyloxypiperidine deriva-
tive of the formula (IX)g wherein R, n and Z are as
defined above,
to obtain compounds of the formula (I), wherein X
means an NR group, one of Rl and R2 stands for a
hydroxyl group and the other is a methyl group, and R,
n as well as Z are as defined above,
theng if desired,
reacting a thus prepared compound of the formula (I),
wherein X means oxygen, R1 and R2 toge-ther stand for a
methylene group and n as well as z are as defined above,
with an amine of the form~lla R-NH2, wherein R is as
defined above, to prepare a compound of the formula (I),
wherein X means an ~ NR group, one of Rl and R2 stands
for a hydroxyl group, the other is methyl group, and R, n
as well as Z are as defined above;
and/or
transforming a functional group of a thus prepared com-
pound of the formula (I), wherein X, R, n, Rl, R2 and Z
are as defined for the formula (I), to an other one in a
known manner;
and/or
reacting with an acid a thus prepared compound of the
formula (I), wherein X, R, n, Rl, R2 and Z are as defined
above, to give an acid addition salt and/or treating with
a base a compound of the formula (I), wherein X, R, n,
R1, R2 and Z are as defined above, obtained as a salt, to
liberate the free basic form thereof and/or converting a
thus prepared compound of the formula (I), wherein X, R,
n, Rl, R2 and Z are as defined above, to its quaternary
ammonium salt.
In the case of processes a) and b) according to the
invention the condensation reaction is conveniently carried
out in an inert oryanic solvent, in the presence of a base
suitably to bind the acid liberated in the reaction. Useful
solvents for this purpose are e.g. aliphatic alkanols such
as ethanol, isopropanol or butanol; aromatic hydrocarbons
such as toluene, xylene or benzene; ethers such as dibutyl
ether or dioxane; tertiary aliphatic acid amides such as di-
methylformamide or dimethylacetamide; ketones such as ace-
tone, methyl ethyl ketone or methyl isobutyl ketone; or a
mixture of the above solvents.
Useful acid binding agents are e.g. inorganic or
tertiary organic bases such as alkaline metal or alkaline
earth metal carbonates or hydrogen carbonates, alkaline
metal hydroxides, triethyl amine, dimethyl aniline or
pyridine; though an excess of the compounds of the formula
(II) or (V) may also be used as acid binding agent. This
reaction may be realized at a temperature between room
temperature and the boiling point of the reaction mixture,
optionally in the presence of a catalyst. Useful catalysts
are e.g. alkaline metal iodides. It is suitable to work
2 ~ D!~
73305-1163
- 10 -
under an inert gas such as nitrogen or argon during this
reaction.
In the first s-tep of process c) according to the inven-
tion a 4-ethynyl-4 hydroxypiperidine derivative of the for-
mula ~VI) is brought into reaction with an isocyanate of the
formula R-NCO in a manner known per se /Houben-l~leyl: Metho-
den der Organischen Chemie Vol. VIII/39 pages 137 to 147
tl952)/ to give a 4-carbamoyloxy-4-ethynylpiperidine deriva-
tive of the formula (VII), which is then cycli7ed in an
acidic or basic medium, respectively according to step i) or
ii) .
According to step i) a thus obtained 4-carbamoyloxy-4-
ethynylpiperidine of the formula (VII) is cyclized in an
acidic medium and the obtained salt of the 2-imino-1,3-di-
oxolane derivative of the formula (VIII) obtained is reacted
with water. The cyclization is carried out in an anhydrous
solvent being inert to the reaction conditions, in the
presence of a suitable acid, preferably in the presence of a
dry hydrogen halide. Suitable solvent~ for this reaction are
e.g. aliphatic or alicyclic ethers such as diethyl ether,
dipropyl ether, diisopropyl ether, dibutyl ether, tetrahyd-
rofuran or dioxane; or lower aliphatic carboxylic acids such
as acetic or propionic acid.
As a hydrogen halide hydrogen chioride, bromide, iodide
or fluoride, preferably hydrogen chloride or bromide, are
used~ After treating with water the thus formed 2-imino-l,3-
dioxolane hydrohalide salt, the 2-oxo-1-oxa-3,8~diazaspiro-
[4-5]-
2 ~
decane derivative of the formula (I) is obtained as an acidaddition salt, from which, if desired, the free base can be
liberated in a manner known per se.
The cyclization of 4-carbamoyloxy-4-ethynylpiperidine
derivative of the formula (VII) according to step ii) is
realized in the presence of a base. Alkaline rnetal acetates,
carbonates, alkoxides, hydroxides and/or tertiary organic
bases, e.g. pyridine, tripropylamine or picoline, may be
used as basic catalysts in the cyclization. The organic
bases may also serve as solvents for the reac-tion. Further
suitable solvents are aliphatic alcohols, e.g. methanol,
ethanol, propanol or butanol; aliphatic, alicyclic or aroma-
tic hydrocarbons, e.g. hexane, cyclohexane, benzene, toluene
or xylene; acid amides, e.g. dimethyl formamide or N-methyl-
-2-pyrrolidone, ethers such as dibutyl ether or dioxane;
nitriles such as acetonitrile; sulfoxides, e.g. dimethyl
sulfoxide; as well as the mixtures of the above solvents.
The reaction may be carried out without any solvent, too,
i.e. in a molten state. In order to accelerate the cycliza-
?.O tion the tempera-ture is suitably increased: the reaction is
preferably accomplished between 40 C and the boiling point
of the reaction mixture. It is suitable to ~ork under an
inert gas such as argon or nitrogen. According -to a pre-
ferred embodiment the 4-carbamoyloxy-4-ethynylpiperidine
~5 derivative of the formula (VII)~formed in the reaction of a
4-ethynyl 4 hydroxypiperidine derivative of the formula
2 ~ ~ e~
23305-1163
- 12 -
(VI) with an isoc~anate of the formula R-NCO, is directly
cycli~ed without isolation, in the same reaction mixture, in
the presence of a suitable base.
In the processes d~ and e) of the invention the pro-
cedur~s discussed under steps i) and ii) are followed.
In the process f) of the invention a 4-acetyl-4-hydr-
oxypiperidine derivative of the formula (X) is reacted with
an isocyanate of the formula R-NCO and the obtained 4-ace-
tyl 4-carbamoyloxypiperidine deriva-tive of the formula (IX)
is cyclized. The condensation reaction according to the
first step is realized in a manner known per se /Houben-
~leil: Methoden der Organischen Chemie Vol. VIII/3, pages 137
to 147 (1952)/. The obtained 4-acetyl-4-carbamoyloxypiperi-
dine derivative of the formula (IX) is preferably cyclized
in the presence of a base. This cyclization may be carried
out under the reaction conditions described for the step ii)
of process c). Alternatively, acc:ording to a preferred
embodiment of this process, the 4-acetyl-4-carbamoyloxypipe-
ridine derivative of the the formula (IX), obtained in the
reaction of a 4-acetyl-4-hydroxypiperidine derivative of
formula (X) with an isocyanate of formula R-NCO, is directly
cyclized without isolation, in the same reaction mixture, in
the presence of a suitable base.
By using the process g) of the invention, the second
step of process f) is in principle followed.
If desired, the compounds of the formula (I~ obtained
- ~ ~ 2 ~ L~
- 13 -
by using the processes a) to g) can be transformed in a
known way to other compounds being within the scope of the
formula (I).
Thus, on reacting a compound of the formula (I),
wherein X means oxygen and Rl together with R2 represents a
methylene group 9 with an amine of the formula R-NH2, com-
pounds of the formula (I) are obtained, wherein X means an
NR group and one of Rl and R2 is a hydroxyl group whereas
the other one means a methyl group. This reaction may be
carried out in a suitable solvent or without any solvent.
Convenient solvents are e.g. aliphtic, alicyclic or arylali-
phatic alcohols such as ethanol, butanol, cyclohexanol or
benzyl alcohol; aliphatic or aromatic hydrocarbons such as
hexane, heptane, xylene, chlorobenzene or nitrobenzene;
ethers, e.g. di-n-butyl ether or dioxane; tertiary organic
bases, e.g. picoline, triethylamine or pyridine, though an
excess of the R-NH2 amine may also serve as a solvent for
the reaction. This procedure may be carried out at a tem-
perature between room temperature and the boiling point of
~0 the reaction mixture, preferably under an inert gas, e.g.
argon or nitrogen.
If desired, the compounds o~ the formula (I) containing
a hydroxyl and a methyl group, respectively as R1 and R2 can
be dehydrated to compounds of the formula (I), wherein Rl
and R2 together represent a methylene group. The dehydration
may be achieved by using methods generally known from the
, ~ "
- 14 -
literature, e.g. under normal or reduced pressure. Isocya-
nates, aliphatic carboxylic acids, aliphatic or aromatic
carboxylic acid anhydrides, Lewis acids, sulfuric acid or
aromatic sulfonic acids can be employed for the dehydration.
This reaction i5 preferably performed in an organic solvent.
Suitable solvents are e.g. aromatic hydrocarbons such as
benzene, toluene or xylene; ethers such as dioxane or di-n-
-butyl ether; aliphatic carboxylic acids such as acetic
acid. Optionally, the water formed in the reaction may be
lû removed by azeotropic distillation.
If desired, a water molecule can be introduced in an
addition reaction into compounds of the formula (I), wherein
R1 and R2 together stand for a methylene group, to give com-
pounds of the formula (I) containing a hydroxyl and a methyl
group, respectively, as Rl and R2.
This hydration reaction is accomplished in an aqueous
medium, in the presence of mineral and/or organic acids. As
acids, e.g. hydrogen halides, sulfuric, phosphoric or formic
acid, aromatic sulfonic acids, oxalic or trifluoroacetic
2û acid and the like may be employed. This reaction is carried
out between 5 C and the boiling point of the reaction
mixture.
If desired, the compounds of the formula (I) may be
converted into their acid addition salts or quaternary ammo-
nium salts by using known methods. For the preparation ofacid addition salts inorganic or organic acids such as
2 ~ L L~
- 15 -
hydro~qen halides, e.g. hydrochloric acid and hydrobromic
acid, su]furic acid, phosphoric acid as well as formic,
acetic, propionic, oxalic, glycolic, maleic, fumaric,
succinic, tartaric, ascorbinic, citric, malic, salicylic,
lactic, benzoic, cinnamic, aspartic, glutamic, N-ace-tyl-
aspartic or N-acetylglutamic acid as well as alkanesulfonic
acids such as methanesulfonic acid, or arenesulfonic acids,
e.o. p-toluenesulfonic acid and the like, may be used.
The salt formation can be carried out e.g. in such a
way that the corresponding acid is added to the solution of
the compound of the formula (I) in an inert solvent, e.g.
ethanol, and the salt formed is precipitated by adding pre-
ferably a water-immiscible organic solvent, e.g. diethyl
e-ther.
For the preparation of quaternary ammonium salts a
lower alkyl, alkenyl or benzyl halide or an alkyl sulfate
may preferably be employed. The q~aternization is suitably
performed in an organic solvent, such as acetone, aceto-
nitrile, ethanol or their rnixtures, at a temperature range
from room temperature up to the boiling point of the sol-
vent. The acid addition or quaternary ammonium salt obtained
may be isolated e.g. by filtration and, when necessary,
purified by recrystallization.
Conversely, the corresponding free bases can be
liberated from their salts by an alkaline treatment.
Among the starting substances, the compounds of the
' .~' '' ,,
- 16 -
formulae (III) and ~V), as well as R-NCO and R-NH2 are known
or can be prepared analogously to methods known from the
literature.
The starting compounds of the formula (VI), (VII), (IX)
and (X) are new and biologically active, too.
The compounds of the formulae (III) and (IV) may be
prepared e.g. according to ~. Heterocycl. Chem. 21, 613
(19~4) or J. Med. Chem. 11, 622 (196B) or, by using well-
known processes for the alkylation of secondary amines.
The prepara-tion of compounds of the formula (II) is
described in the Hungarian patent application No. 4092/89,
filed simultaneously with the basic Hungarian application
No. 4093/89.
The compounds of the formula (VI) can be prepared e.g.
by the ethynylation reaction of suitably substituted 4-pipe-
ridone derivatives as described e. 9. in the Hungarian
patent specification No. 166,769 or in Farmaco (Pavia) Ed.
Sci. 12, 34 (1957).
The carbamates of the formulae (VII) and (IX), respec-
tively, are obtained e.g. by reacting a compound of the for-
mula (VI) or (X), respectively, with an isocyanate of the
formula R-NCO under conditions described above.
The 4-acetyl-4-hydroxypiperidine derivatives of the
formula (X) can be prepared e.g. by hydrating the cor-
responding 4-ethynyl-4-hydroxypiperidine derivatives of the
formula (VI) /Houben-Weyl: Methoden der Organischen Chemie
- 17 -
Vol. VII/2a, pages 826 to 835 (1973)/ or by the alkaline
treatment of the corresponding 4-methylene-2-oxo-1,3-dioxa-
-8-azaspiror4,5/decane derivatives of the formula (I).
The compounds of the formula (I) according to the
invention show psychotropic and antiallergic actions and
have a broad spectrum of therapeutical possibilities. Thus,
they may be useful e.g. for the treatment of the so-called
functional psychoses and organic psychiatric diseases such
as dementia, delirium, stimulants-induced psychoses and the
like, as well as diskinesias, anxiety, pruritus, nausea,
vomitus and intolerable singultation.
Male CFLP mice with a body-weight of 18 to 25 9 and
male Hannover-Wistar rats with a body-weight of 160 to 180 9
were used for the pharmacological investigations. The test
compounds were orally administered in a 2% Tween-80 suspen-
sion. The pharmacological investigating methods used are
described hereinafter.
1.Protective effect against the amphetamine group toxicity
on a~gregated mice
The method described in Arch. Int. Pharmacodyn. 163, 79
(196~) was used.
The mice were treated with various doses of the test
substances. By one hour following the treatment the animals
were intraperitoneally given 21 mg/kg of d-amphetamine and
then the groups consisting of 10 animals each were tightly
closed together in plexiglass boses of 15x15xlO cm in size.
~3305-1163
- 18 -
A-t the 24th hour following the amphetamine treatment the
surviving animals were counted. The ED50 values of the sub-
stances were calculated from the percentage of the surviving
animals. The ED50 value is the dose protecting 50% of the
animals from perishment. The results are summarized in Table
1.
2. Inhibition of the conditioned _avoidance resQonse (CAR
inhibition) on rats
The method of D. Bovet et al. (Neuropsychopharmacology,
ed. R. Rothlin Vol. 2, p. 142, Elsevier Publishing Co. N.Y.
1961) was used.
Male rats weighing 160 to 180 9 were conditioned in an
au-tomated 6-channel 5huttle box for 10 days. Each one daily
session consisted of 50 cycles and each one cycle lasted for
40 seconds. ~ithin a single cycle an intermitten-t light
stimulus for 15 seconds as conditioned stimulus, and an
electric stimulus of 0.8 mA for lû seconds as unconditioned
stimulus, were used with an intersignal time of 15 seconds.
When an animal changed its half-court during the conditioned
stimulus period, the stimulus was abolished and the foot-
shock was avoided (conditioned avoidance response;
abbreviated: CAR). Animals with a performance of at least
80% on the 8th to 10th days of conditioning were considered
to be useful for the study. ~y 3 hours before the experiment
the animals were treated with various doses of the test sub-
stances and the ED5U value, i.e. the dose causing an 50%
2 ~
- 19 -
inhibition of the conditioned avoidance response of the
animals, was determined. Groups consisting of 10 animals
each were used for each dose. The results are summarized in
Table 1.
3. Measurement of the spontaneous body -temperature
Groups consisting of ln mice each were orally treated
with 30 mg/kg doses of the test substances. After treatment
the rectal temperature of the male mice was determined in
each hour for 5 hours. The average values of temperature
decrease related to the body temperature of the untreated
control animals (37.1+0.8 C) at -the 5th hour after treat-
ment are given in Table 1.
4. Inhibition of the aggressive behaviour of the mice
The method described in J. Pharmacol. Expt. Therap.
1 , 28 tl959) was used.
Pairs of male mice weighing 20 to 25 each were treated
with the test compounds and then placed in cages. By 3 hours
following the treatment an aggressive behaviour was induced
on the animals by using an electric stimulation of 1 mA
current intensity. Each group consisted of 5 pairs of mice.
The ED50 values were calculated from the percentage of
animal pairs showing no aggressive behaviour owing to the
treatment. (En50 value is the dose inhibiting the aggressive
behaviour on 50% of the animals.)
The results are illustrated in Table 2.
~ '~
- 20 -
5. Acute toxicity
Rats were treated with various doses of the tes-t com-
pounds. The perishment of the animals were observed for 14
days. The LD~U value, i.e. the dose causing the death of
half of the animals, was determined from the percentage of
the dead animals.
The results are shown in Table 2.
Chlorpromazine /chemically 2-chloro-10-(3-dimethylami-
nopropyl)phenothiazine/ was used as reference drug in these
investigations.
The abbreviations listed hereinafter are used in the
Tables:
CPZ = chlorpromazine;
A = 5-r3-(2-chloro-lOH-phenothiazin-lû-yl)propyl7-3-methyl-
-4-methylene-2-oxo-1-oxa~3,8-diazaspiro/4,5/decane;
B = 8-/3-(2-chloro-lOH-phenothiazin-10-yl)propyl7-3-e-thyl-4-
-methylene-2-oxo-1-oxa-3,8-diazaspiroL4,57decane;
C = 3-methyl-4-methylene-2-oxo-8-/3-(2 trifluoromethyl-lOH-
-phenothiazin-10-yl)propyl7-1-oxa-3,8-diazaspiro/4,57-
decane;
D = 3-ethyl~4-methylene-2-oxo-8-L3-(2-trifluoromethyl-lOH-
-
-phenothiazin-10-yl)propyl/-1-oxa-3,8-diazaspiro/4,5/-
decane;
E = 8-/3-(2-chloro-lOH-phenothiazin-10-yl)propyl/-4-methyle-
ne-2-oxo-3-propyl-1-oxa-3,8-diazaspiror4,57decane;
- 21 - 2~2~
F = B-,r3-(2-chloro-lOH-phenothiazin-10-yl)propyl7-3,4-dime-
thyl-4-hydroxy-2-oxo-1-oxa-3,8-diazaspiro/4,57decane;
G = 8-r3-(2-chloro-lOH-phenothiazin-10-yl)propyl7-3-ethyl-4-
-hydroxy-4-methyl-2-oxo-1-oxa-3,8-diazaspiro/4,5/decane;
H = 3,4-dimethyl-4-hydroxy-2-oxo-8-/3-(2-trifluoromethyl-
-lOH-phenothiazin-10-yl)propyl7-1-oxa-3,8-diazaspiro-
/4,5/decane;
I = 4-hydroxy-4-methyl-2-oxo-3-propyl-8-/3-(2-tri~luorome-
thyl-lOH-phenothiazin-10-yl)propyl7-1-oxa-3,8-diaza-
spiro/4,5/decane;
p.o. = orally
amph. = amphetamine (d,l-l-phenyl-2-aminopropane)
tox. = toxicity
CAR = conditioned avoidance response
Table 1
Compound Protection CAR Decrease in the
against amph. ED50 spontaneous
ED50mg/kg p.o. temperature
m~/k~ p.o. C
A 0.6 6.0 -B.8
B 1.6 1.6 -7.4
C 0.7 1.9 -9.9
D 4.1 6.8 -8.2
E 1.8 10.6 -5.6
F 0.7 4.6 9.1
G 2.4 6.9 -8.1
H 0.9 3.1 -9.6
I 4.2 5.6 -7.0
CPZ 7.2 17.1 -7.9
.
~ ~ 2 ~
- 22 -
Table 2
Compound Antiaggressive L50
effect~ ED50 mg/kg p.o.
_ mg/kg p.o.
A 1.7 353
B 10.9 ~ 500
C 1.2 - 235
D 2.5 500
E 2.0
CPZ 4.3 225 _
It is obvious from the above Tables that the novel com-
poùnds of the formula (I) according to the invention possess
a significantly stronger effect than that of the reference
drug and their therapeutic range is more advantageous than
- that of CPZ; simultaneously, their effective doses do not
induce any sedative, muscle relaxant or anticholinergic
action and, even in the case of a chronic treatment, they do
not cause the hypersensitization of the dopaminergic system
of the central nervous system. Thus, it is likely that their
side effects will be diminished in rela-tion to the reference
drug in the clinical practice. Being effective antipsychotic
agents, the novel compounds of formula (I) according to the
invention are useful for the treatment of psycho-tic condi-
tions of mammals. The effective doses of course depend on
the condition of the patient, on the severi-ty of the dis-
order to be trea-ted and may be varied from 0.01 mg/kg to 5
mo/kg of body-weight in the case of oral administration.
. : ~ ; .; ~.'; ., .
~ . . .
~2~
- - 23 -
If desired, the compounds according to the invention
can be converted into pharmaceutical compositions. These
compositions may be adrninistered in oral, rectal and/or
parenteral route. For oral administration, the composition
may be formulated e.g. as a tablet, dragée or capsule. In
order to prepare oral compositions, e.g. lactose or starch
may be used as carriers. Gelatine, carboxymethylcellulose
sodium, mehylcellulose, polyvinylpryrrolidone or starch gum
are suitable binding or granulating agents. As disintegrat-
ing agen-ts mainly potato starch or microcrystalline
cellulose may be added though ultraamylopectin or formalde-
hyde-casein and the like are also useful.
Talc, colloidal silicic acid, stearin, calcium or mag-
nesium stearate and the like are suitable an-ti-adhesive and
sliding agents.
Tablets may be prepared e.g. by compression following
the wet granulation. The mixture of the active ingredient
with the carriers and optionally with a part of the dis-
integrating additive is granulated with an aqueous, alco-
2û holic or aqueous-alcoholic solution of the binding agents in
a suitable equipment, then the granulate is dried. Sub-
sequently, after mixing the other disintegrating, sliding
and anti-adhesive additives to the dried granulate, the mix-
ture is compressed to tablets. If desired, the tablets may
be provided with a groove in order to facilitate the admin-
istra-tion. Tablets may also directly be prepared from a mix-
- 24 - 2~
ture cqntaining the active ingredient and suitable
additives. The tablets may optionally be converted to
dragées by employing the commonly used pharmaceutical
additives, e.g. protective, flavouring or colouring agents
such as sugar, cellulose derivatives (methyl- cr ethyl-
cellulose, carboxymethylcellulose sodium and the like),
polyvinylpyrrolidone, calcium phospha-te, calcium carbonate,
food dyes 3 dyeing lacquers, aromatizing agents, iron oxide
pigments and the like. Capsulated compositions are prepared
by filling a mixture of the active ingredient with the
additives into capsules.
For rectal administration, the composition of the
invention is formulated as a suppository containing a
carrier mass, the so-called "adeps pro suppositorio" in
addition -to the active ingredien~t. As carriers, vegetable
fats such as hardened vegetable oils, or triglycerides of
C12_18 fatty acids (preferably -the carriers bearing the
trade name ~itepsol) may be used. The active ingredient is
uniformly distributed in the molten carrier mass, then sup-
positories are prepared by moulding.
For parenteral administration, the composition of theinvention is formulated as injectable solu-tion. For prepar-
ing these injectable solutions, the active ingredients are
dissolved in distilled water and/or various organic sol-
vents, e.g. glycol ethers, if desired, in -the presence of
solubilizing agents such as polyoxyethylene sorbitan mono-
; . ,. ; ;
2~2~
~5
23305~ 3
laurate or monooleate or monostearate (Tween 20, Tween 60 orTween 80), respectively. The injectable solution may further
contain various additives (auxiliary agents), e.g. preservatives
such as benzylalcohol, methyl p-oxybenzoate, propyl
p-oxybenzoate, benzalkonium chloride or phenyl mercury borate;
antioxidants such as ascorbinic acid, tokoferole, sodium
pyrosulfate; and optionally a complexing agent being capable of
binding metal traces, such as ethylenediamine tetraacetate as
well as pH-modifying and buffering substanc~s or, if desired, a
local anaesthetic agent such as lidocaine. Before filling into
the ampoules, the injectable solution containing the composition
of the invention is filtered and after filling in, it is subjected
to sterilization.
The invention also relates to a method for treating
psychiatric and allergic diseases, e~g. functional psychoses and
organic psychiatric diseases such as dementia, delirium,
stimulants-induced psychoses and the like, as well as dyskinesias,
anxiety, pruritus, nausea, vomitus and intolerable singultation.
This method comprises administering a therapeutically effective
amcunt of an active ingredient of the formula (I) or a pharma-
ceutically acceptable acid addition salt or quaternary ammonium
salt thereof to the patient.
The invention is illustrated in detail by the aid of
the following non-limiting Examples.
Example 1
Preparation of 3-m~thyl-4-methylene-2-oxo-8-[3-(2-
trifluoromethyl-10~-phenothiazin-10-yl)propyl]-1-oxa-3,8-
2~2~
- 25a -
~3305-1163
diazaspiro-[4,5]decane
A mixture containing 11.0 g of 3-methyl~4-mathylene-2~
~, .,
- 26 -
-oxo-l-oxa-3,8-diazaspiro/4,5/decane, 41.3 9 of 3-(2-tri-
fluoromethyl-lOH-phenothiazin-10-yl)propyl chloride, 16.6 9
of anhydrous potassium carbonate and 0.6 9 of potassium
iodide in 110 ml of methyl isobutyl ketone is refluxed under
nitrogen while stirring for 6 hours. After evaporating the
solvent under reduced pressure benzene and water are added
to the evaporation residue, -the organic phase is separated,
washed with water to neutral, dried over anhydrous sodium
sulfate, then the solution is evaporated under reduced
pressure. The solid residue is boiled with hexane, after
cooling down the precipitate is filtered off and recrys-
tallized from ethanol to give the title compound in a yield
of 75.3%; m.p.: 116-117.5 C.
Analysis:
Calculated for C25H26F302S
C 61.33; H 5.35; F 11.64; N B.58; S 6.55%;
found: C 61.50; H 5.38; F 11.38; N 8.34; S 6.34%.
Example 2
Preparation of 8-/3-(2-chloro-lOH-pheno-thiazin-10-yl)pro-
pyl7-4-hydroxy-4-methyl-2-oxo-1-oxa-3,8-diazaspiro/4,5/de-
cane
9.7 9 of 8-/3-(2-chloro-lOH-phenothiazin-10-yl)propyl/-
-4-methylene-2-oxo-3-propyl-1-oxa-3,8-diazaspiro/4,5/decane
are stirred with 12û ml of a 0.5 mol/litre hydrochloric acid
solution at 5 to lû C for 3 hours, then the reaction mix-
ture is made alkaline by adding sodium hydroxide solution
'
- 27 -
and extracted with chloroform. The chloroform phase is
washed with water to neutral, dried over anhydrous magnesium
sulfate and evaporated under reduced pressure. After recrys-
tallization of the evaporation residue from benzene the
tltle compound is obtained in 88% yield, m.p.: 174-176 C.
Analysis:
calculated for C26H32ClN303S:
C 62.19; H 6.42; Cl 7.06; N 8.37; S 6.39%;
found: C 62.10; H 6.58; Cl 6.91; N 8.40; S 6.60%;
On treating an ethanolic solution of the base with
ethereal hydrogen chloride solution the hydrochloride of the
above base is obtained, m.p.: 157-160 C (with decomposi-
tion).
Example 3
Preparation of 8-/3-(2-chloro-lOH-pheno-thiazin-10-yl)pro-
pyl/-3-cyclohexyl-4-hydroxy-4-methyl-2-oxo-1 oxa-3,8-diaza-
spiro/4,5/decane hydrochloride
5.3 g of 8-/3-(2-chloro-lOH-phenothiazin-10-yl)propyl/-
3-cyclohexyl-4-methylene-2-oxo-1-oxa-3,8-diazaspiro/4,5/-
~o decane are dissolved in 5.3 ml of 98% formic acid at room
temperature and 53 ml of 3 mol/litre hydrochloric acid solu-
tion are dropwise added under stirring during 20 minutes.
The precipitate is filtered off, washed with water and dried
to give the title hydrochloride in 98% yield, m.p.: 246-248
C (with decomposition). The base can be liberated from the
hydrochloride by adding aqueous ammonium hydroxide solu-
tion.
~2~
Analysis of the base:
calculated for C29H36ClN303S:
C 64.25; H 6.69; Cl 6.54; N 7.75; S 5.91%;
found: C 64.28; H 6.82; Cl 6.63; N 7.57; S 6.12%;
By using the appropriate starting substances the
following compounds are prepared in an analo~ous manner as
described in Examples 2 or 3 or Example 12 to be described
hereinafter.
3-/3-(2-Acetyl-lûH-phenothiazin-1û-yl)propyl7-3-decyl-4-
-hydroxy-4-me-thyl-2-oxo-1-oxa-3,8-diazaspiro/4,5/decane
hydrochloride, m.p. 154-157 C (with decomposition);
8- r-(2-Chloro-lûH-phenothiazin-lû-yl)propyl7-3,4-dimethyl-
-4-hydroxy-2-oxo-1-oxa-3,8-diazaspiro/4,5/decane, m.p.:
172-173 C; the hydrochloride decomposes at 232-235 C;
8-/3-(2-Chloro-lOH-phenothiazin-10-yl)propyl/-4-hydroxy-3-
-isopropyl-4-methyl-2-oxo-1-oxa-3,8-diazaspiro/4,5/decane,
m.p.: 108-110 C; the hydrochloride decomposes at 127-130
C;
4-Hydroxy-4-methyl-2-oxo-3-phenyl-8-L3-(2-trifluoromethyl-
-lOH-phenothiazin-10-yl)propyl/-1-cxa-3,B-diazaspiro/4,5/-
decane hydrochloride, m.p.: 147-150 C (with decomposi-
tion);
3-Ethyl-4-hydroxy-4-methyl-2-oxo-8-/3-(2-trifluoromethyl-
-lOH-phenothiazin-10-yl)propyl7-1-oxa-3,8-diazaspiro/4,5/-
decane, m.p.: 148-149 C; the hydrochloride decomposes at
186-189 C; and
~ ~ ~ S~ f~
- 29 -
4-Hydroxy-4-methyl-2-oxo-8-/3-(lOH-phenothiazin-10-yl)pro-
pyl7-3-tert-butyl-1-oxa-3,8-diazaspiro/4,5~decane, m.p.:
16g-171 C.
Example 4
Preparation of 8-/3-(2-acetyl-lOH-phenothiazin-10-yl)pro-
pyl7-3-methyl-4-methylene-2-oxo-1-oxa-3,8-diazaspiroL4,5/-
decane
A mixture containing 7.3 9 of 3-methyl-4-methylene-2-
-oxo-l-oxa-3,B-diazaspiroL4,5/decane, 9 ml of anhydrous tri-
ethylamine, 22.0 9 of 3-(2-acetyl-lOH-phenothiazin-10-yl)-
propyl bromide and 80 ml of methyl isobutyl ketone is re-
fluxed under argon while stirring for 6 hours. After cooling
down the organic phase is washed with water, dried over an-
hydrous magnesium sulfate and evaporated under reduced
pressure. The crude product obtained is purified by chroma-
tography on a silica gel column by using first chloroform
and then ethyl acetate as eluating agent. After combining
the ethyl acetate eluate is evaporated under reduced
pressure and the residue is recrystallized from ethanol to
give the title compound in 84.5% yield, m.p.: 105-106 C.
Analysis:
calculated for C26H29N303S:
C 67.36; H 6.30; N 9.06; S 6.92%;
found: C 67.48; H 6.51; N 9.01; S 7.11%.
8y using the appropriate starting substances the
following substances are prepared in an analogous way as
- 30 -
described in Examples 1 or 4 or in Example 14 to be
described hereinafter.
8-/3-(2-Acetyl-lOH-phenothiazin-10-yl)propyl~-3,4-dimethyl- -
-4-hydroxy-2-oxo-1-oxa-3,8-diazaspiro/4,5/deGane hydro-
chloride, m.p.: 165-168 C (with decomposition);
8-r3-(2-Chloro-lOH-phenothiazin-10-yl)propyl7-3-methyl-4-me-
thylene-2-oxo-1-oxa-3,8-diazaspiro/4,5/decane, m.p.: 142-
143 C;
4-Methylene-2-oxo-8-/3-(lOH-phenothiazin-10-yl)propyl7-3-n-
-propyl-1-oxa-3,8-diazaspiro/4,5/decane, m.p.: 98-99 C;
3-Cyclohexyl-4-methylene-2-oxo-8-/3-(lOH-phenothiazin-10-
-yl)propyl/-l-oxa-3,8-diazaspiroL~,57decane, m.p.: 159-160
C;
4-Methylene-2-oxo-8-/3-(lOH-phenothiazin-10-yl)propyl/-2-
-oxo-3-tert-butyl-1-oxa-3,8-diazaspiror4,5/decane, m.p.:
96-97 C;
8-L3-(2-Chloro-lOH-phenothiazin-10-yl)propyl/-3-isopropyl-4-
-methylene-2-oxo-1-oxa-3,8-diazaspiro/4,5/decane, m.p.:
162-163 C;
4-Methylene-2-oxo-3-propyl-8-/3-(2-trifluoromethyl-lOH-phe-
nothiazin-10-yl)propyl/-1-oxa-3,8-diazaspiro/4,5/decane,
m.p.: 119-121 C;
8-/3-(2-Acetyl-lOH-phenothiazin-10-yl)propyl7-3-n-decyl-4-
-methylene-2-oxo-1-oxa-3,8-diazaspiro/4,57decane hydrogen
maleate, m.p.: 108-110 C;
3-Methyl-4-methylene-2-oxo-8-/3-(lOH-phenothiazin-10-yl)pro-
- 31 -
pyl/-l-oxa-3,8-diazaspiro/4,5/decane, m.p.: 132-133 C
obtained by reacting 3-methyl-4-methylene-2-oxo-1-oxa-3,8-
-diazaspiro/4,5/decane with 3-(10-phenothiazin-10-yl)pro-
pyl p--toluene-sulfonate;
8-/3-(2-Chloro-lOH-phenothiazin-10-yl)propyl/-3-n-butyl-4-
-methylene-2-oxo-1-oxa-3,8-diazaspiro/4,5/decane, m.p.:
100-101 C;
8-/3-(2-Chloro-lOH-phenothiazin-10-yl)propyl/-3-cyclohexyl-
-4-methylene-2-oxo-1-oxa-3 J 8-diazaspiro/4,5/decane, m.p.:
177-178 C;
8-r3-(2-Chloro-lOH-phenothiazin-10-yl)propyl7-3-ethyl-4-hyd-
roxy-4-methyl-2-oxo-1-oxa-3,8-diazaspiro/4,57decane, m.p.:
155-156 C; the hydrochloride decomposes at 220-222 C;
8-/3-(2-Chloro-lOH-phenothiazin-10-yl)propyl~-4-methylene-2-
-oxo-3-n-propyl-1-oxa-3,8-diazaspiroL4,5/decane, m.p.:
118-119 C;
3-Ethyl-4-methylene-2-oxo-8-/3-(2-trifluoromethyl-lOH-phe-
nothiazin-10-yl)propyl~ oxa-3 ? 8-diazaspiro/4,5/decane,
m.p.: 117-119 C;
3-n-Butyl-4-methylene-2-oxo-8-/3-(2-tri~luoromethyl-lOH-phe-
nothiazin-10-yl)propyl/-1-oxa-3,~-diazaspiroL4,57decane,
m.p.: 104-105 C;
4-Methylene-2-oxo-3-tert-butyl-8-/3-(2-trifluoromethyl-lOH-
phenothiazin-10-yl)propyl/-1-oxa-3,8-diazaspiro/4,5/-
decane, m.p.: 12Y-130 C; and
8-/2-(2-Chloro-lOH-phenothiazin-10-yl)ethyl/-3-n-butyl-4-me-
- 32 -
thylene-2-oxo-1-oxa~3,8-diazaspiro/4,5/decane, m.p.: 108-
1 10 C .
Example 5
Preparation of 4-methylene-2-oxo-8-/3-(lOH-phenothiazin-10-
-yl)propyl7-3-phenyl-1-oxa-3,8-diazaspiroL4,5~decane
12.0 9 of 4-ethynyl-L3-(lOH-phenothiazin-10-yl)propyl/-
-4-phenylcarbamoyloxypiperidine are refluxed in 120 ml of
0.05 mol/litre methanolic sodium methoxide solu-tion under
argon for 3 to 4 hours. After cooling down the sodium meth-
oxide is decomposed by adding aqueous ammonium chloride
solution, then the solution is evaporated to the tenth of
its original volume under reduced pressure, the residue is
diluted with water and extracted with benzene. After washing
the benzene phase with water to neutral and drying over an-
hydrous sodium sulfate the solvent is distilled off and the
evaporation residue is recrystallized from ethyl acetate to
give the title compound in 85.4% yield, m.p.: 98-99 C.
Analysis.
calculated for C29H29N303S:
C 72.02; l1 6.04; N ~.69; S 6.63%;
found: C 72.12; H 6.19; N 8.48; S 6.70%.
8y using the appropriate starting substances the
following compound is prepared in an analogous manner as
described in the preceding Example:
3-Isopropyl-4-methylene-2-oxo-8-L3-(lOH-phenothiazin-10-yl)-
propyl7-1-oxa-3,8-dlazaspiror4,57decane, m.p.: 113-114 C.
. -, ' , . ;
: ' , " ,. ~ ..: ''~
':', " ' ' . :. ~ ` '
' ' ; ~' .,
- 33 -
Example 6
Prepara-tion of 4-methylene-2-oxo-3--phenyl-8-/3-(2-trifluoro-
methyl-lOH-phenothiazin-10-yl)propyl7-1-oxa-3,8-diazaspiro-
L4,5/decane
A mixture containing 10.8 9 of 4-ethynyl-4-hydroxy-1-
-L3-(2-trifluoromethyl-lOH-phenothiazin-10-yl)propyl7piperi-
dine, 0.12 9 of potassium acetate, 3.6 9 of phenyl isocya-
nate and 35 ml of 2-picoline is refluxed under argon while
s-tirring for 4.5 hours. After evaporatlng the solvent under
ln reduced pressure water is added to the residue and the mix-
ture is extracted with Ghloroform. The chloroform phase is
washed with water, dried over anhydrous sodium sulfate and
evaporated under reduced pressure. After recrystallizing the
crude product from ethyl acetate under clarifying with
activated carbon the title compound is obtained in 74.6%
yield, m.p.: 154-155 C.
Analysis:
calculated for C30H28F3N302S
C 65.32; H 5.12; F 10.33; N 7.62; S 5.81%;
~0 found: C 65.40; H 5.31; F 10.12; N 7.57; S 5.8B%.
By using the suitable starting compounds the following
substances are preparedin an analogous way as described in
the preceding Example.
8-/3-(2-Chloro-lOH-phenothiazin-10-yl)propyl7-4-methylene-2-
-oxo-3-phenyl-1-oxa-3,8-diazaspiro/4,5/decane, m.p.: 145-
146 C;
- 34 -
3-Cyclohexyl-4-methylene-2-oxo-8-/3-(2-trifluoromethyl-lOH-
-phenothiazin-10-yl)propyl/-1-oxa-3,8-diazaspiroL4,57de-
cane, m.p.: 175-176 C.
Examele 7
Preparation of 4-hydroxy-3-isopropyl-4-methyl-2-oxo-8-/3-(2-
-trifluoromethyl-lOH-phenothiazin-10-yl)propyl/-1-oxa-3,8-
-diazaspiro/4,5/decane
A solution containing 5.4 9 of 4-acetyl-4-isopropylcar-
bamoyloxy-l-/3-(2-trifluoromethyl-lOH-phenothiazin-10-yl)-
propyl/piperidine in 54 ml of 0.55 mol/litre ethanolic
sodium ethoxide solution is refluxed under nitrogen for 6
hours. After cooling down and decomposing the reaction mix-
ture with aqueous acetic acid solution the most part of
ethanol is distilled off under reduced pressure. The
evaporation residue is diluted with water and extracted with
methylene chloride. The organic phase is washed wi-th water
to neutral, dried over anhydrous magnesium sulfate, then the
solvent is evaporated under reduced pressure. After
recrystallizing the crude product from benzene the title
compound is obtained in 85.7% yield, m.p.: 187-189 C.
Analysis:
calculated for C27H32F3N303S:
C 60.54; H 6.02; F 10.64; N 7.84; S 5.99%;
found: C 60.65; H 6.22; F 10.45; N 7.63; S 6.07%.
On adding ethereal hydrogen chloride solution to the
solution of the base in ethanol the hydrochloride is
, ' :
2~
precipitated in crystalline form. It is filtered and dried,
m.p.: 212-214 C.
Example 8
Preparation of 3-n-outyl-4-methylene-2-oxo-8-L3-(lOH-pheno-
thiazin-10-yl)propyl/-1-oxa-3,8-diazaspiro/4,5/decane
A mixture of 7.6 9 of 4-acetyl-4-hydroxy-l-/3-tloH-
-phenothiazin-10-yl)propyl/piperidine, 1.5 ml of triethyl-
amine and 11.5 ml of n-butyl isocyanate is refluxed under
argon for 6 to 7 hours. After cooling down and adding n-
-hexane to the reaction mixture the precipitate is filtered
off, dissolved in ethyl acetate and led through a silica gel
layer. After evaporating the solvent under reduced pressure
and recrystallizing the evaporation residue from ethanol the
title product is obtained in 67.4% yield, m.p.: B2-83 C.
Analysis:
calculated for C27H33N302S:
C 69.94; H 7.17; N 9.06; S 6.92%;
found: C 70.17; H 7.28; N 7.28; S 6.77%.
Example 9
Preparation of 8-/3-(2-chloro-lOH-phenothiazin-10-yl)pro-
pyl/-3-ethyl-4-methylene-2-oxo-1-oxa-3,8-diazaspiro/4,57-
decane
9.8 9 of 8-~-(2-chloro-lOH-phenothiazin-10-yl)propyl7-
3-ethyl-4-hydroxy-4-methyl-2-oxo-1-oxa-3,8-diazaspiror4,57-
decane are boiled with 0.8 9 of p-toluenesulfonic acid mono-
hydrate in 150 ml of xylene in a flask equipped with a
- 36 - ?~2^~
water-trap while azeotropically distilling off the water
formed in the reaction. After termination of the reaction (2
to 3 hours) the reaction mixture is cooled down, made
alkaline by adding aqueous sodium hydroxide solutiong the
organic phase is separated, washed with water to neutral,
dried over anhydrous sodium sulfate and evaporated under
reduced pressure. After recrystallization of the evaporation
residue from ethyl acetate the title compound is obtained in
87.5% yield, m.p.: 145-146 C.
Analysis:
calculated for C25H28ClN302S:
C 63.88; H 6.00; Cl 7.54; N 8.94; S 6.82%;
found: C 64.02; H 5.86; Cl 7.37; N 8.B3; S 6.60%.
By using the appropriate starting substances the
following compounds can be prepared in an analogous way as
described in the preceding Example.
8-/3-(2-Chloro-lOH-phenothiazin-10-yl)propyl7-4-methylene-2-
-oxo-3-tert-butyl-l-oxa-3,8-dlazaspiro/4,57decane, m.p.:
79-81 C.
3-Isopropyl-4-methylene-2-oxo-8-L3-(2-trifluoromethyl-lOH-
-phenothiazin-lû-yl)propyl7-l-oxa-3,8-diazaspiroL4,5/-
decane, m.p. 129-130 C.
Example 10
Preparation,of 4-methylene-2-oxo-8-L3-(2-trifluoromethyl-
-lOH-phenothiazin-10-yl)propyl/-1,3-dioxa-8-azaspiro/4, s7-
decane hydrochloride
37 2~2~
Into a solution containing 10.0 9 of 4-butylcarbamoyl-
oxy-4-ethynyl-1-/3-(2-trifluoromethyl-lOH-phenothiazin-10-
-yl)propyl/piperidine in 50 ml of anhydrous dioxane gaseous
dry hydrogen chloride is introduced until saturation at 15
to 20 C, then the reaction mixture is left to stand over-
night. After evaporating the solvent under reduced pressure
water is added to the evaporation residue, the precipi-tate
is filtered off, washed with water and dried to obtain the
title hydrochloride salt in 88.5% yield, m.p.: 230-233 C
(with decomposition).
The base can be liberated from its hydrochloride by
adding sodium hydrogen carbona-te.
Analysis_of the base:
calculated for C24H23F3N203S
C 60.49; H 4.86; F 11.96; N 5.88; S 6.73%;
found: C 60.60; H 4.99; F 11.78; N 6.03; S 6.70%.
Example 11
Preparation of 8-methyl-4-methylene-2-oxo-8-~-(lOH-pheno-
thiazin-10-yl)propyl/-3-propyl-1-oxa-3,8-diazaspiro_4,57de-
can-8-ium iodide
6.0 9 of 4-methylene-2-oxo-8-/3-(lOH-phenothiazin-10-
-yl)propyl/-3-propyl-1-o~a-3,8-diazaspiro/4,57decane are
boiled under reflux with 2.9 9 of methyl iodide in 60 ml of
methyl isobutyl ketone for 2 hours. After cooling down the
crystalline precipitate is filtered off and washed with di-
isopropyl ether previously cooled to 0C. After drying the
. . . , ~ .... .. .
- 38 -
title quaternary ammonium salt is obtained in 98.0% yield,
m.p.: 192-193 C.
The following compound is prepared analogously to the
process described in the preceding Example:
8-Allyl-3-butyl-4-methylene-2-oxo-8-/3-(lOH-phenothiazin-10-
-yl)propyl7-1-oxa-3,8-diazaspiro/4,5/decan-8-ium bromide,
rn.p.: 200-202 C.
Example 12
Preparation of 4-hydroxy-4-methyl-2-oxo-3-propyl-8-/3-(2-
-trifluoromethyl-lOH-phenothiazin-10-yl)propyl/-1-oxa-3,8-
-diazaspiro/4,57decane
4.8 g of 4-me-thylene-2-oxo-8-/3-(2-trifluoromethyl-lOH-
-phenothiazin-10-yl)propyl7-1,3-dioxa-8-azaspiro/4,5/decane
are stirred with 50 ml of n-propylamine overnight, then the -
excess of amine is distilled off under reduced pressure.
After recrystallizing the evaporation residue from benzene
-the title compound is obtained in 89.8% yield, m.p.: 170.5-
172 C.
Analysis:
:
calculated for C27H32F3N303S:
C 60.54; H 6.02; F 10.64; N 7.84; S 5.99~;
found: C 60.47; H 6.11; F 10.71; N 7.89; S 6.15%.
The hydrochloride of the title compound is prepared by
treating an e-thanolic solution of the base with ethereal
hydrogen chloride solu-tion, m.p.: 129-133 C.
By using the appropriate starting substances the
. ~ I . ~; . .. ...
- 39 -
following compound can be prepared analogously as described
in the preceding Example:
4-Hydroxy-4-methyl-2-oxo-8-L3-(2-triflunromethyl-lOH-pheno-
thiazin-10-yl)propyl7-1-oxa-3,8-diazaspiro/4,57decane,
m.p.: 102-103 5.
Example 13
Preparation of 3,4-dimethyl-4-hydroxy-2-oxo-8-/3-(2-tri-
fluoromethyl-lOH-phenothiazin-10-yl)propy_7-1-oxa-3,8-diaza-
spiror4,5~decane
After adding a solution of 0.4 9 of methylamine in 10
ml of xylene (previously cooled to O C) to the solution of
4.~ 9 of 4-methylene-2-oxo-8-/3-(2-trifluoromethyl-lOH-phe-
nothiazin-10-yl)propy_ 7-1, 3-dioxa-8-azaspiro/4,5/decane in
20 ml of xylene while s-tirring, the reaction mixture is
heated at 70 to 80 C for 60 minutes, then the solvent is
distilled off under reduced pressure. After recrystallizing
the evaporation residue from a mixture of ethanol and n-
-hexane the title product is obtained in 81.5% yield, m.p.:
164-165 C.
Analysis:
calculated for C25~l28F3N303S
C 59.15; H 5,56; F 11.23; N 8.28; S 6.32%;
found: C 59.32; H 5.39; F 11.14; N 8.43; S 6.50%.
The hydrochloride of the title base melts at 194-197 C
with decomposition.
, -
",. .,~ .
~2~
- 40 -
Exarnple 14
Preparation of 3-ethyl-4-methylene-2-oxo-8-r3-(lOH-phe-
nothiazin-10-yl)propyl7-1-oxa-3,8-diazaspiroL4,5/decane
0.6 9 of sodium hydride (60 % oily dispersion) is added
to a solution of 3.0 9 of phenothiazine in 20 ml of anhyd-
rous dimethylformamide under argon, then the reaction mix-
ture is sti~red at 50 to 60 C for 2 hours. Thereafter, 3.9
g of 8-(3-chloropropyl)-3-ethyl-4-methylene-2-oxo-1-oxa-3,8-
-diazaspiro/4,5/decane dissolved in 20 ml of dimethylform-
amide are dropwise added and the mixture is stirred at 40 to
50 C for additional 6 to 7 hours. After cooling down
saturated arnmonium chloride solution is added to the reac-
tion rnixture under argon and the solvent is evaporated under
reduced pressure. After taking up the evaporation residue in
benzene and washing the benzene solution with water the
solution is dried over anhydrous sodium sulfate and then
evaporated under reduced pressure. The residue is
recrystallized from ethanol to give the title product in
57.6% yield, m.p.: 122-123 C.
Analysis:
calculated for C25H29N32:
C 68.93; H 6.71; N 9.65; S 7.36%;
found: C 62.08; H 6.77; N 9.78; S 7,23%.
Example 15
Preparation of 4-ethynyl-4-hydroxy-1-~-(2-trifluoromethyl-
-lOH-phenothiazin-10-yl)propyl7piperidine
-- ?J ~ 2 ~
Gaseous acetylene is bubbled through a solution of 7.7
g of potassium tertiary butoxide in 46 ml of tetrahydrofuran
at O to -5 C for 30 minutes under stirring. Thereafter, the
solution containing 18.4 g of 1-L3-(2-trifluorome-thyl-lOH-
-phenothiazin-10-yl)propyl7-4-piperidone in 40 ml of tetra-
hydrofuran is drop~ise added and acetylene ls introduced for
additional one hour. Then, the reaction mixture is decompos-
ed by adding aqueous saturated ammonium chloride solution
under nitrogen at û C and the solvent is evaporated under
reduced pressure. The evaporation residue is extracted with
benzene, the benzene solution is washed with water to
neutral, dried over anhydrous sodium sulfate and evaporated
under reduced pressure. The crude title product ootained as
evaporation residue is recrystallized from diisopropyl ether
under clarifying by activated carbon to give the crystalline
-title compound in 89% yield, m.p.: 94-96 C.
Analysis:
calculated for C23H23F3N20S
C 63.87; H 5.36; F 13.1B; N 6~48; S 7.41%;
found: C 63.84; H 5.40; F 13.31; N 6.50; S 7.58%.
By using the appropriate starting substances the
following compounds can be prepared in an analogous way as
described in the preceding Example.
4-Ethynyl-4-hydroxy-/3-(2-chloro-lOH-phenothiazin-10-yl3pro-
pyl~pipridine, m.p.: 113-114 C; and
4-Ethynyl-4-hydroxy-1-~-(lOH-phenothiazin-10-yl)propyl7pi-
peridine, m.p.: 118-120 C.
;
. : ~: , . ''
~3~ ~
- 42 -
Example 16
Preparation of 4-butylcarbamoyloxy-4-ethynyl-1-/3-(2-tri-
fluoromethyl-lOH-phenothiazin-10-yl)propyl~piperidine
A mixture containing 21.6 9 of 4-ethynyl-4-hydroxy-1-
-/3-(2-trifluoromethyl-lOH-phenothiazin-10-yl)propyl7piperi-
dine, 7.4 ml of butyl isocyanate and 100 ml of triethylamine
is gently refluxed under nitrogen while stirring for 7 to 8
hours, then the reaction mixture is evaporated under reduced
pressure. After clarifying the evaporation residue by
activated carbon in methanol, the solution is evaporated
under reduced pressure. The crude product obtained as
evaporation residue is recrystallized from diisopropyl ether
under clarifying by activated carbon to give the crystalline
title compound in 74% yield, m.p.: 109-110 C.
A_alysis:
calculated for C28H32F3N202S
C 63.25; H 6.07; F 10,72; N 7.90; S 6.303;
found: C 63.11; H 6.26; F 10.66; N 7.71; S 6.07%.
By using the appropriate starting substances the
following compound can be prepared in an analogous manner as
described in the preceding Example:
4-Acetyl-4-phenylcarbamoyloxy-1-/3-(2-trifluoromethyl-lOH-
-phenothiazin-10-yl)propyl/piperidine, m.p.: 156-157 C.
Example 17
Preparation of 4-acetyl-4-hydroxy-1-/3-(2-trifluoromethyl-
-lOH-phenothiazin-10-yl)propyl7piperidine
- 2 ~ ~ v ~
23305-1163
- 43 -
A solution containing 9.5 9 of 4-methylene-2-oxo-~
-(2-trifluoromethyl-lOH-phenothiazin-10-yl)propyl7-1,3-di-
oxa-~-azaspiro/4,5/decane in 50 ml of benzene is vigorously
stirred with 100 ml o~ 10% aqueous sodium hydroxide solution
at 70 to ~0 C under argon. After termination of the
reaction (which is observed by using thin layer chromato-
graphy) the heterogeneous reaction mixture is cooled down.
~ter separation the benzene layer is washed ~Jith water to
neutral, dried over anhydrous magnesium sulfate and
evaparated under reduced pressure. The evaporation resiude
is recrystallized ~rom isopropyl ether to give the title
compound in 56% yield, m.p.: 75-76 C.
Analysis:
ca1culat~d for C23H25-3N2 2
C 61.31; H 5.59; F 12.65; N 6.22; S 7.12;
found: C 61.33; ~ 5.70; F 12.60; N 6.11; S 7.00.
Example 18
Preparation of tablets with a weight o~ 100 mg containing 10
m~ of active ingredient each
50.0 g of active ingredient are mixed together with
285.0 9 o~ lactose, 100.0 9 of potato starch, 2.5 9 oi
sodium dodecyl sul~ate, 5.0 g o~ polyvinylpyrrolidone
(Kollidon-K 90R), 50.0 9 of microcrystalline cellulose
tAvicelR) and 7.5 9 o~ vegetable oil (SterotexR) and, after
wet granulation, the product obtained ls compressed to
tablets weighing 100 mg each. Each oi the tablets contain~
. ~ .
- 44 -
10 mg of the active ingredient.
Example 19
Preparation of dragées with a weight of 125 mg containing 10
mg of active ingredient each
After coating the tablets prepared as described above
in a known manner with a layer consisting of sugar and talc,
the dragées obtained are polished with a mixture of bee wax
and carnauba wax.
Example 20
Preparation of capsules containing 20 mg of active ingre-
dient each
40.0 9 of active ingredient, 12.0 9 of sodium lauryl
sulfate, 102.0 9 of lactose, 102.0 0 of potato starch, 2.4 9
of magnesium stearate and 1.6 9 of colloidal silicon dioxide
are thoroughly mixed together and the mixture obtained is
filled into hard gelatine capsules containing 20 mg of
active ingredient each.