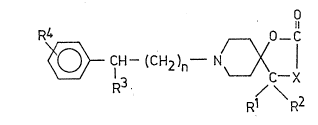Note: Descriptions are shown in the official language in which they were submitted.
~ 2 ~ 2 ~ ~ ~
NOVEL l-OXA-2-OXO-8-AZASPIRO/ 4,57DECANE DERIVATIVES,
PHARMACEUTICAL COMPOSITIONS CONTAINING THEM AND PROCESS
FOR PREPARING SAME
The invention re'ates to novel, therapeutically
active l-oxa-2-oxo-8-azaspiro/ 4,57decane derivatives
of the formula (I),
~4 0 - C (I)
H--(CH2)n--- N~X l
R3 1 \ 2
R R
wherein
X means oxygen or an ~NR group, wherein
R stands for hydrogen, a Cl_l2alkyl, C3 6cyclo-
alkyl, carbocyclic C6 1Oaryl or carbocyclic
C6 1Oaryl-Cl 4alkyl group, the two latter are
optionally substituted on their aromatic part
by one or more, same or different halogen(s),
one or more Cl 4alkyl or Cl 4alkoxy group(s);
Rl and R2 together represent a methylene group or, when
X stands for an ~NR group, wherein R is as defined
above, one of Rl and R2 may represent a hydroxyl
group whereas the other is a methyl group;
A 4586-67 MR/Gi
- 2 ~
R3 stands for hydrogen or a phenyl group optionally
substituted by one or more halogen(s), one or
more Cl_~alkyl or Cl 4alkoxy or hydroxyl group(s);
R4 means hydrogen, one or more halogen(s), Cl 4alkyl,
Cl 4alkoxy, hydroxyl or -trihalomethyl group(s); and
n i5 1, 2 or 3
as well as their acid addition and quaternary ammonium
salts and pharmaceutical compositions containing these
compounds.
The invention also relates to a process ~or the
preparation of the above compounds and compositions as
well as to a method of treatment. The latter comprises
introducing a therapeutically effective amount of a compound
of formula (I) or a pharmaceutically acceptable acid addition
or quaternary ammonium salt thereof into the organism of
a patient.
The compounds of the formula (I) may exist in various
stereoisomeric forms such as geometrical isomers as well
as racemates, individual optical isomers and their mixtures,
all of which may occur in the form of various solvates and
` hydrates. All these compounds and mixtures are within the
scope of the invention.
A number of therapeutically useful l-oxa-2-oxo-3,8-
-diazaspiro/~4,57decane derivatives have been described
in the literature. Such compounds are reported e.g. in the
following publications: C.A. 71, 91359d (1969); C.A. 78,
719668t (1973); C.A. 78, 23876q (1973); C.A. 81, 33153c
and 105368b (1974); C.A. 95, 161765e (1981); as well as
~r;~
in the DE patent specifications Nos. 2,013,729, 2,013,668
and 2,163,000; in the Belgian patent specifications Nos.
775,984, 774,170, 786,631 and 825,444; in the British
patent specification No. 1,100,281; in the published Dutch
patent application No. 7,214,6E19 as well as in the United
States patent specifications Nos. 3,555,033, 3,594,386,
4,244,961 and 4,255,432.
A substantial difference between the compounds of
formula (I) according to the invention and the similar
derivatives known up to the present appears in the nature
of the substituents bound in position 4 and optionally in
position 3 of the spirodecane skeleton.
According to an other aspect of the invention,
there is provided a process for the preparation of compounds
of the formula (I) as well as their acid addition and
quaternary ammonium salts, which comprises
a) reacting a 2-oxo-3,8-diazaspiro/ 4,57decane
derivative of the formula (II),
:
Il (II)
2s ~C l'i--R
R~ 2
- 4 - ~ ~ r~
wherein R, Rl and R2 are as defined above, with a phenyl-
alkane derivative of the formula (III),
F~4
\~--~H -(CH2)n y (III)
R3
wherein R3, R4 as well as n are as defined above and Y
means halogen, a Cl 4alkylsulfonyloxy or arylsulfonyloxy
group,
to obtain compounds of the formula (I), wherein X stands
for an ~NR group and R, Rl, R2, R3, R4 as well as n are
as defined above;
5 or
b) reacting a 4-ethynyl-4-hydroxypiperidine derivative
of the formula (IV),
~4
H--(C112 )n--N~ (IV)
R3 C 3 CH
wherein R3, R4 and n are as defined above, with an iso-
cyanate of the formula R-NC0, wherein R is as defined above,
2 ~ 2 ~ ~
and
0~) cyclizing in an acidic medium the obtained 4-carbamoyl-
oxy-4-ethynylpiperidine derivative of the formula (V),
5 '
O-C- NHR
CH--(C~7 ~ N~
R3 C--C~
wherein R, R3, R4 and n are as defined above and
reacting with water the obtained salt of the 2-imino-
-1,3-dioxolane derivative o:E the formula (VI),
.
11
\~ CH--(CH2 )n--N~X I (VI)
R3 11
CH2
wherein R, R3, R4 and n are as defined above,
to obtain compounds of the formula (I), wherein Rl and
R together represent a methylene group, X means oxygen
and R, R3, R4 as well as n are as
defined above, or
~,
' ~.: ; ,
~) cyclizing in a basic medium the obtained compound of
the formula (V), wherein R, R3, R4 and n are as
defined above,
to obtain compounds of the formula (I), wherein X
means an ~NR group, Rl and R2 together represent a
methylene group and R, R3, R4 as well as n are as
defined above;
or
c) cyclizing in an acidic medium a 4-carbamoyloxy-4-
-ethynylpiperidine derivative of the formula (V) above,
wherein R, R3, R4 and n are as defined above, and reacting
with water the obtained salt of the 2-imino-1,3-dioxolane
derivative of the formula (VI), wherein R, R3, R4 and n
are as defined above,
to obtain compounds of the formula (I), wherein X means
oxygen, R3, R4 as well as n are as defined above and
together with R stands for a methylene group;
or
d) cyclizing in the presence of a base a 4-carbamoyl-
oxy-4-ethynylpiperidine derivative of the formula (V),
wherein R, R3, R4 and n are as defined above,
to ob-tain compounds of the formula (I), wherein X means an
~NR group, Rl together with R2 stands for a methylene
group and R, R3, R4 as well as.n are as defined above;
or
e) reacting a 4-acetyl-4-hydroxypiperidine derivative
of the formula (VII),
R4 OH
~ r ~ n--N~X (VII )
R3
~3
wherein R3, R4 and n are as defined above, with an iso-
cyanate of the formula R-NC0, wherein R is as defined above,
and cyclizing the thus formed 4-acetyl 4-carbamoyloxy-
piperidine derivative of the formula (VIII)
R4 Q C--NHR
~\r CH~(CH2 )n ` N~ . (VIII )
F~3 ll :
wherein R, R3, R4 and n are as defined above,
to obtain compounds of the formula (I), wherein X means
an ~NR sroup, one of Rl and R2 s-tands for a hydroxyl
group and the other is a methyl group, and R, R3, R4 as well
as n are as defined above;
or
f) cyclizing a 4-acetyl-4-carbamoyloxypiperidine
derivative of the formula (VIII), wherein R, R3, R4 and
n are as defined above,
to obtain compounds of the formula (I), wherein X means an
.,
,
. , , ~
-- . ~
- 8 ~
~ NR group, one of Rl and R2 stands for a hydroxyl group,
the other is methyl group and R, R3, R4 as well as n are
as defined above,
then, if desired,
reacting a thus prepared compound of the ~ormula
(I), wherein X means oxygen, R1 and R2 together stand for
a methylene group, R3, R4 and n are as defined above, with
an amine of the formula R-NH2, wherein R is as defined
above, to prepare a compound of the formula ~I), wherein
X means an ~NR group, one of Rl and R2 stands for a
hydroxyl group, the other is methyl group and R, R3, R4
as well as n are as defined above;
and/or
transforming a thus prepared compound of the
formula (I), wherein X, R, Rl, R2, R3, R4 and n are as
defined in the preamble, to an ot;her compound of the formula
(I) falling within the scope of the formula (I);
and/or
reacting with an acid a thus prepared compound of
the formula (I), wherein X, R, Rl, R2, R3, R4 and n are
as defined above to give its acid addition salt and/or
treating with a base a compound of the formula (I), where-
in X, R, Rl, R2, R3, R4 and n are as defined above obtained
as a salt, to liberate the base form thereof and/or convert-
ing a thus prepared compound of the formula (I), whereinX, R, Rl, R2, R3, R4 and n are as defined above, to its
quaternary ammonium salt.
_ 9 _ ~ ~c,~
In the process a) according to the invention, a
2-oxo-3,8-diazaspiroL 4,57decane derivative of the formula
(II) is reacted with a phenylalkane derivative of the
formula (III), wherein Y means a leaving group, e.g. a
mesyloxy or tosyloxy group, or a halogen, preferably
chlorine or bromine. This reaction is preferably carried
out in an inert organic solvent, in the presence of a base
being capable to bind the acid liberated in the reaction.
Various solvents, such as aliphatic alkanols, e.g. ethanol,
isopropanol, butanol; aromatic hydrocarbons, e.g. chloro-
benzene, toluene; ethers, e.g. dibutyl ether or dioxane;
tertiary aliphatic acid amides such as dimethylformamide
or dimethylacetamide; ketones, e.g. acetone, methyl ethyl
ketone or methyl isobutyl ketone or the mixtures of the
above solvents may be used; as acid binding agents inorganic
or tertiary organic bases, e.g. alkaline or earth alkaline
metal carbonates or hydrogen carbonates as well as triethyl-
amine, dimethylaniline or pyridine may be employed -though
an excess of the compound of formula (II) can also be used
for the same purpose. This reaction is accomplished at a
temperature between room temperatue and the boiling point
of the reaction mixture, optionally in the presence of a
catalyst. Suitable catalysts are e.g. the alkaline metal
iodides. The reaction is preferably performed under an
inert gas such as nitrogen or argon.
In the first step of process b) according to the
invention a 4-ethynyl-4-hydroxypiperidine derivative of
the formula (IV) is brought into reaction with an isocyanate
,
,
.
. ~
- 10~
of the formula R-NCO in a manner known per se / Houben-
-Weyl: Methoden der Organischen Chemie ~ol. VIII/3, pages
137 to 147 (1952)7 to give a 4-carbamoyloxy-~-ethynyl-
piperidine derivative of the formula (V). For prepariny
compounds of the formula (I) containing oxygen as X, the
compound of the formula (V) is cyclized in an acidic medium
according to step c~), then the thus-formed 2-imino-1,3-
-dioxolane derivative of the formula (VI) obtained as a
salt is reacted with water; or, for preparing compounds
of the formula (I) containing ~NR group as X, -the compound
of formula (V) is cyclized in a basic medium according -to
step ~).
The cyclization of step ~) is carried out in an
inert organic solvent (i.e. in- a solvent which is inert
under the reaction conditions), in the presence of a suitable
acid, preferably in the presence of a dry hydrogen halide.
Aliphatic or alicyclic ethers such as diethyl ether, di-
propyl ether, diisopropyl ether, dibutyl ether, tetrahydro-
furan or dioxane as well as lower aliphatic carboxylic acids,
e.g. acetic or propionic acid, may be employed.
As a hydrogen halide hydrogen chloride, bromide,
iodide or fluoride, preferably hydrogen chloride or bromide,
are used. After treating with water the thus formed 2-imino-
-1,3-dioxolane hydrohalide salt, the 1-oxa-2-oxo-8-aza-
spiroL~4,57decane derivative of the formula (I) is obtainedas an acid addition salt from which, if desired, the base
can be liberated in a manner known per se.
` The cyc]ization of step ~) is realized in the
presence of a base. Alkaline metal acetates, carbonates,
alkoxides, hydroxides and/or tertiary organic bases, e.g.
pyridine, tripropylamine or picoline, may be used as basic
catalysts in the cyclization; the organic bases may also
serve as solvents for the reaction. Further sui-table
solvents are aliphatic alcohols, e.g. methanol, ethanol,
propanol or butanol; aliphatic, alicyclic or aromatic hydro-
carbons, e.g. hexane, cyclohexane, benzene, toluene or
xylene; acid amides, e.g. dimethylformamide or N-methyl-
-2-pyrrolidone; ethers such as dibutyl ether or dioxane;
nitriles such as acetonitrile; sulfoxides, e.g. dimethyl-
sulfoxide; etc. as well as the mlxtures of the above
solvents. The reaction may be carried out without any solvent,
too, e.g. in a molten state. In order to accelerate the
cyclization the temperature is suitably increased: the
reaction i9 preferably accomplished between 40 C and the
boiling point of the reac-tion mixture. It is suitable to
work under an inert gas such as argon or nitrogen. Accord-
ing to a preferred embodiment the 4-carbamoyloxy-4-ethynyl-
piperidine derivative of the formula (V), formed in the
reaction of the 4-e-thynyl-4-hydroxypiperidine derivative
of the formula (IV) with the isocyanate of the formula
R-NC0, is directly cyclized, without isolation, in the same
reaction mixture, in the presence of a suitable base.
In the processes c) and d) of the invention the
procedures discussed under steps d) and ~) are followed.
`:
- 12 ~
In the process e) of the invention a 4-acetyl-4-
-hydroxypiperidine derivative of the formula (VII) is
reacted with an isocyanate of the formula R-NCO and the
obtained 4-acetyl-4-carbamoyloxypiperidine derivative of
the formula (VIII) is cyclized. The condensation reaction
according to the first step is realized in a manner known
per se / Houben-Weyl: Methoden der Organischen Chemie
Vol. VIII/3, pages 137 to 147 (1952)7. The obtained 4-
-acetyl-4-carbamoyloxypiperidine derivative of the formula
(VIII) is preferably cyclized in the presence of a base.
The cyclization may be carried out under the reaction
conditions described for the step ~) of process b).
Alternatively, according to a preferred embodiment of this
process, the 4-acetyl-4-carbamoyloxypiperidine derivative
of the formula (~'III), obtained in the reaction of the
4-acetyl-4-hydroxypiperidine derivative of the formula (VII)
with the isocyanate of formula R-NCO, is directly cyclized,
without isolation, in the same reaction mixture, in the
presence of a suitable base.
By using the process f) of the invention, the second
step of the process e) is in principle followed.
If desired, the compounds of the formula (I) obtained
by using the processes a) to f) can be transformed to other
compounds being within the scope of the formula (I) in a
manner known ~ se.
Thus, on reacting a compound of the formula (I),
wherein X means oxygen and Rl together with R2 represents
- 13 -
a methylene group, with an amine of the formula R-NH2,
compounds of the formula (I) are obtained, wherein X means
an ~NR group and one of Rl and R2 is a hydroxyl group
whereas the other one means a methyl group. This reaction
may be carried out in a suitable solvent or without any
solvent. Convenient solvents are e.g. aliphatic, alicyclic
or araliphatic alcohols such as e-thanol, butanol, cyclo-
hexanol, benzyl alcohol; aliphatic or aromatic hydro-
carbons such as hexane, heptane~ xylene, chlorobenzene or
nitrobenzene; ethers, e.g. dioxane; ketones, e.g. di-n-
-butyl ketone; tertiary organic bases, e.g. picoline, tri-
ethylamine or pyridine, though an excess of the R-NH2 amine
may also serve as a solvent for the reaction. This procedure -"
may be carried out at a temperature between room temperature
and the boiling point of the reaction mixture, preferably
under an inert gas, e.g. argon or nitrogen.
If desired, the compounds of the formula (I) contain-
ing a hydroxyl and a methyl group, respectively as Rl and
R , can be dehydrated to compounds of the formula (I),
2û wherein Rl and R2 together represent a methylene group.
The dehydration may be achieved under normal or reduced
pressure by using commonly known procedures. Isocyanates,
aliphatic carboxylic acids, aliphatic or aromatic carboxylic
acid anhydrides, Lewis acids, sulfuric acid or aromatic
sulfonic acids can be employed for the dehydration. This
reaction is preferably performed in an organic solvent.
Suitable solvents are e.g. aromatic hydrocarbons such as
benzene, toluene or xylene; ethers such as dioxane, di-n-
~ ~ ~ c~
- 14 -
-butyl ether; or aliphatic carboxylic acids such as acetic
acid. Optionally, the water formed in the reaction may
be removed by azeotropic distillation.
If desired, a water molecule can be introduced in .an
addition reaction into the compounds of formula (I), where-
in Rl and R2 together stand for a methylene group to give
compounds of the formula (I) containing a hydroxyl and a
methyl group, respectively as Rl and R2. This hydration
reaction is accomplished in an aqueous medium, in the
presence of mineral and/or organic acids. As acids e.g.
hydrogen halides, sulfuric, phosphoric, formic acid,
aromatic sulfonic acids, oxalic or trifluoroacetic acid
and the like may be employed. This reaction is carried out
between 5 C and the boiling point of the reaction mixture.
The compounds of the formula (I) may be converted
to their acid addition salts or quaternary ammonium salts
by using methods known ~ se. For the preparation of acid
addition salts inorganic or organic acids such as hydrogen
halides, e.g. hydrochloric acid and hydrobromic acid,
sulfuric acid, phosphoric acids as well as formic, acetic,
propionic, oxalic, glycolic, maleic, fumaric, succinic,
- tartaric, ascorbinic, citric, malic, salicylic, lactic,
benzoic, cinnamic, aspartic, glutamic, N-acetil-aspartic
or N-acetylglutamic acid as well as alkanesulfonic acids
such as methanesulfonic acid or arenesulfonic acids, e.g.
p-toluenesulfonic acid and the like may be used.
The salt formation can be carried out e.g. in such
a way that the corresponding acid is added to the solu-tion
.
- 15 ~
of the compound of formula (I) in an inert solvent, e.g.
ethanol, and the salt formed is precipitated by addiny
preferably a water-immiscible organic solvent, e.g. ethyl
ether. For the preparation of quaternary ammonium salts
a lower alkyl, alkenyl or benzyl halide or an alkyl sulfate
may preferably be employed. The quaternization is suitably
performed in an organic solvent such as acetone, aceto-
nitrile, ethanol or their mixtures, at a temperature range
from room temperature up to the boiling point of the solvent.
The acid addition or quaternary ammonium salt obtained may
be isolated e.g. by filtration and, when necessary, purified
by recrystallization.
Conversely, the corresponding bases can be liberated
from their salts ~y an alkaline treatment.
A part of the starting substances used in the process
of the invention are known or can be prepared by using
known methods.
The compounds of the formula (III) can be prepared
e.g. according to Collection Czechoslov. Chem. Commun. 38,
3879 (1973); as well as Chim. Ther. 3, 185 (1969).
The preparation of the compounds of formula (II) is
described in our Hungarian patent application No. 4092/89.
The substances of formula (IV) may be synthetized
e.g. by the ethynylation reaction of suitably substituted
4-piperidone derivatives as described e.g. in the Hungarian
patent specification No. 166,769 or in Farmaco (Pavia) Ed.
Sci. 12, 34 (1957).
- 16 -
The carbamates of the formulae (V) and (VIII),
respectively are obtained e.g. by reacting a compound of
the formula (IV) or (VII), respectively, with an iso-
cyanate of the formula R-NCO under conditions commonly
known in the literature L Houben-Weyl: Methoden der
Organischen Chemie Vol. VIII/3, pages 137 to 147 (1952)7.
The 4-acetyl-4-hydroxypiperidine derivatives of
the formula (VII) can be prepared e.g. by hydrating the
corresponding 4-ethynyl-4-hydroxypiperidine derivatives
of formula (IV) L see e.g. in: Houben-Weyl: Methoden der
Organischen Chemie Vol. VII/2 a, pages B26 to 835 (1973)7
or by the alkaline treatment of the corresponding 1,3-dioxa-
-2-oxo-4-methylene-8-azaspiroL 4,57decane derivatives of
formula (I~
The compounds of formulae (IV), (V), (VII) and ;
(VIII) are novel compounds, up to the present not described
in the literature, which are valuable intermediates in the
synthesis of the novel compounds according to the invention
and in addition, they are biologically active, too.
The compounds of formula (I) as well as their
stereoisomers and salts exhibit valuable pharmacological
properties such as calcium uptake-inhibiting, antihypoxic
and antianoxic effects. Thus they are useful for the
systemic (i.e. oral, rectal or parenteral) treatment of
varm-blooded mammals (including man). They can favourably
be employed for the prevention or therapeutic treatment
of hypoxic brain damages of various origin such as senile
dementia, Alzheimer's disease, ischemic lesions, dis-
-
- 1 7 - ~ ?J
turbances of the cognitive function, multi-infarctual
dementia, hypoxia following atheroschlerosis and the
like.
The calcium uptake-inhibiting action of the novel
compounds of formula (I) was studied on a rat brain
synaptosomal p~eparation by using the method of P. H. Wu
et al. / J. Neurochem. 39, 700 (1982)7.
Wistar rats weighing 180 to 200 9 were decapitated,
their brains were collected in an ice-cold physiological
saline solution, the cortex was removed and purified from
the white substance. The tissue was homogenized in lD
volumes of 0.32 M sucrose solution by using a glass-teflon
potter. Qfter centrifuging the homogenate at a rate of
1000 x 9 at 4 C for 10 minutes, the supernatant was further
centrifuged at 10000 x 9 for 20 rninutes. The sediment was
taken up in a 0.32 M sucrose solution in such a way that
the protein content of the preparation was adjus-ted to
20 mg/ml.
The medium (containing 112 mM of sodium chloride,
5 mM of potassium chloride, 1.3 mM of magnesium chloride,
1.2 mM of sodium dihydrogen phosphate, 1.2 mM of calcium
chloride, 10 mM of glucose, 20 mM of TRIS buffer) used
for the incubation was saturated with carbogen, consisting
of 95% by volume of oxygen and 5% by volume of carbon
dioxide, up to a pH of 7.4. After adding the test substances
to the medium, the synaptosomal preparation was added in an
amount corresponding to 1 mg of protein. The incubation was
carried out in a final volume of 1 ml. The samples were
- 18 - ~2~
pre-incubated at 34 C for 20 minutes. The calciurn uptake
was begun by using a 45CaC12 solution of 75 nCi activity.
Potassium chloride used for investigating the potassium-
-induced 45Ca uptake was employed in 60 mM concentration
whereas sodium chloride was added in the same concentra-tion
to the control samples. The incubation lasted 20 seconds.
The reaction was stopped by 5 ml of a stopping solution
containing 120 mM of sodium chloride,5 mM of potassium
chloride, 5 mM of EGTA and 20 mM of TRIS at pH 7.4. The
samples were filtered through a Wha-tman GF/C filter paper
and twice washed with 5 ml of a washing solution each,
containing 132 mM of sodium chloride, 5 mM of potassium
chloride, 1.3 mM of magnesium chlorid~e, 1.2 mM of calcium
chloride and 20 mM of TRIS at p~ 7.4. (Abbreviations used
above: TRIS means tris(hydroxyme-thyl)aminomethane; EGTA
means ethylene glycol bis( ~-aminoethyl) ether-N,N,N',N'-
-tetraacetic acid).
The filter papers were put into glass cuvets, 10 ml
of a scintillation cocktail each were added, then the
radioactivity of the samples was measured in an 1219
Rackbeta (LKB Wallace) liquid scintillatinn spectrophoto-
meter.
The IC50 values obtained by examination of the
concentration/effect correla-tions are shown in Table I.
IC50 value means the molar concentra-tion of the tes-t compounds
which causes 50% inhibition of the stimulated 45Ca uptake.
1 9
Table I
._
Compound No. I~50
~uM
_
1 5.8
2 5.9
3 2.1
4 5.3
6.1
Numbers for the chemical names and abbreviations used in
the Tables:
1 1-Oxa-2-oxo-3-butyl-4-methylene-a-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane
2 1-Oxa-2-oxo-3-tert-butyl-4-methylene-8-/ 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiroL 4,57decane
3 1-Oxa-2-oxo-3-phenyl-4-methylene-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane
4 1-Oxa-2-oxo-3-cyclohexyl-4-methylene-8-L 4,4-bis(4- -
-fluoroFhenyl)bu~yl7-3,8-diazaspiroL 4,57decane
1-Oxa-2-oxo-3-butyl-4-hydroxy-4-methyl-8-L 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiroL 4,57deeane hydro-
chloride
6 1-Oxa-2-oxo-3,4-dimethyl-4-hydroxy-8-L 2-(4-fluoro-
phenyl)ethyl7-3,~-diazaspiroL 4,57decane
7 1-Oxa-2-oxo-3-propyl-4-hydroxy-4-methyl-8-L 2-(4-fluoro-
phenyl)ethyl7-3,B-diazaspiroL 4,57decane
- 20 - ~ d~ ~
B l-Oxa-2-oxo-3-cyclohexyl-4-hydroxy-4-methyl-8-L 2-(4-
-fluorophenyl)ethyl7-3,8-diazaspiroL 4,57decane
9 1-Oxa-2-oxo-3-ethyl-4-hydroxy-4-methyl-8-t 2-(4-fluoro-
phenyl)ethyl7-3,8-diazaspiro/ 4,57decane.
Abbreviations:
n: number of animals
p.o.: oral administration '
i.v.: intravenous injection
S.E.: standard error
The antihypoxic effect was studied by using two
methods. According to the method of C. Caillard et al.
/ Life Sci. 16, 1607 (1975)7 the asphyxial action was
. determined after starvation for 16 hours on CFLP mice
of both sexes weighiny 24 to 26 g. Each snimal was pla-ced
in a separate well-closed glass cylindre. The interval
passing from closing the cylindre until the cessation of
the last visible respiratory movement was registered as
survival time. Animals, surviving longer by 30% than the
average survival time of the control group, were considered
to be protected. The test substances were administered in
an oral dose of 50 mg/kg by 60 minutes before starting
the examination. The results are summarized in Table II.
,
:. . .
- 21 -
Table II
Compound No.Protected%animals n
_
6 60 10
7 40 10
8 - 60 10
9 60 10
The average survival time of the control group was 23.5 + 2.51
sec (X+S.E.) `~
The potassium cyanide test method was used for
determination of the p~otective effect against the histotoxic
hypoxia. A hypoxia af this type ~an be developed by a rapid;
intravenous injection of 5.0 my/kg of potassium cyanide
inducing abdominal contractions and clonic convulsions on
the animals and leading to death of the animals within 2
minutes.
Male Hannover-Wistar rats weighing 160 to 170 9 were
used in this experiment. Animals surviving longer by 30%
than the average survival time of the control group were
considered to be protected. The test substances were orally
administered in various doses 60 minutes before starting
the examination. The ED50 values, i.e. the dose protecting
half of the treated animals from the hypoxia, were calculated
from percentage of the protected animals relating to various
doses (i.e. from the dose-response curve) by using probit
.
:
- 22 -
analysis. The results are summarized in Table III.
Table III
5 Compound No. ED50 P
mg/ky
1 40.9
2 39.9
~ 37.9
: .,
The compounds according to the invention showed a
strong calcium-antagon1zing and antihypoxic activity and
had a Iow toxicity. Their oraI LU50 values (i.e, the dose
lS causing death of 50% of the animals) were found to be
higher than 1000 mg/kg; thus, their therapeutic indices
are favourable.
Thus, the invention relates to a method for the
calcium uptake-inhibiting as well as antihypoxic and anti-
anoxic treatment of mammals. This method comprises administer-
ing a prophylactically or therapeutically effective amount
of a compound of the invention or a pharmaceutically
acceptable acid addition or quaternary salt thereof to a
patient suffering from anoxia connected with e.g. senile
dementia, Alzheimer's disease, ischemic lesions, disturbances
of the cognitive function, multi-infarctual dementia;
hypoxia following atherosclerosis and the like. Depending
from the severity of the disease and the condition of the
; , , :
. ~ ~
~3~
- 23 -
patient, the daily dose amounts to 0.1 to 40 mg/kg which
may be given once or in divided subdoses in oral, rectal
or parenteral route.
The compounds according to the invention can be
converted into pharmaceutical composi-tions in a known
manner. The pharmaceutical compositions may be administered
in oral, rectal and/or parenteral route. For oral administra-
tion, the composition may be formulated e.g. as a table-t,
dragée or capsule. In order -to prepare oral compositions,
e.g. lactose or starch may be used as carriers. Gelatine,
carboxymethylcellulose sodium, methylcellulose, polyvinyl-
pyrrolidone or starch gum are suitable binding or granulat-
ing agents. As disintegrating agents, mainly potato starch
or microcrystalline cellulose may be added thcugh ultra-
amylopectin or formaldehyde-casein and the like are also
useful. Talc, colloidal silicic acid, stearin, calcium
or magnesium stearate and the like are suitable anti-
-adhesive and sliding agents. The liquid oral compositions
of the invention can be prepared in the form of e.g. a
suspension, syrup or elixir which may contain water, glycols,
oils, alcohols as well as colouring and flavouring additives.
Tablets may be prepared e.g. by compression follow-
ing the wet granulation. The mix-ture of the active
ingredient with the carriers and optionally with a part
of the disintegrating additive is granulated wi-th an aqueous,
alcoholic or aqueous-alcoholic solution of the binding
agents in a suitable equipment, then the granulate is dried.
- 24 ~ $~
Subsequently, after mixing the other disintegra-ting,
sliding and anti-adhesive additives to the dried granulate,
the mixture is compressed to table-ts. If desired, the
tablets may be provided with a groove in order to facilitate
the administration. Tablets may also directly be prepared
from a mixture containing the active ingredient and suitable
additives. The tablets may optionally be converted to
dragées by employing the commonly used pharmaceutical
additives, e.g. protective, flavouring or colouring agents
such as sugar, cellulose derivatives (methyl- or ethyl-
cellulose, carboxymethylcellulose sodium and the like),
polyvinylpyrrolidone, calcium phosphate, calcium carbonate,
food dyes, dyeing lacquers, aromatizing agents, iron oxide
pigments and the llke. Capsulatecl compositions are prepared
by filling a mixture of the active ingredient with the
additives into capsules. -~
For rectal administration, the composition of the
invention is formulated as a suppository containing a
carrier mass, the so-called "adeps pro suppositorio" in
addition to the active ingredient. As carriers, vegetable
fats such as hardened vegetable oils, or triglycerides of
C12 18 fatty acids (preferably the carriers bearing the
trade name Witepsol) may be used. The active ingredient
is uniformly distributed in the molten carrier mass, then
suppositories are prepared by moulding.
For parenteral administration, the composition of
the invention is formulated as an injectable solution. For
- 25 -
preparing these injectable solu-tions, the active ingredients
are dissolved in distilled water and/or various organic
solvents, e.g. glycol ethers, if desired, in -the presence
of solubilizing agents such as polyoxyethylene sorbitan
monolaurate or monooleate or monostearate (Tween 20,
Tween 60, Tween 80), respectively. The injectable solution
may further contain various additives (auxiliary agents),
e.g. preservatives such as ethylenediamine tetraacetate
as well as pH-modifying and buffering substances or, if
desired, a local anaesthetic agent such as lidocaine.
Before filling into the ampouls, the injectable solution
containing the composition of the invention is filtered
and after filling.in, it is subjected to sterilization.
On using the pharmaceutical composition of the
invention, the patient is treated with a dose needed to
ensure the desired effect. This dose depends upon several
factors like the severity of the disease, the body-weight
of the patient and the route of administration. The dose
to be used is in every case to be defined by the physician.
The invention also relates to a method for treating
hypoxic brain damages of various origin such as the senile
dementia, Alzheimer's disease hypoxia following athero-
sclerosis, multi-infarctual dementia and disturbances of
the cognitive function. This method comprises administering
a therapeutically effective amount of an active ingredient
of the formula (I) or a pharmaceutically acceptable acid
addition salt thereof to the patient.
- 26 -
The invention is illustrated in detail by the aid
of the following non-limiting Examples.
Example 1
Preparation of l-oxa-2-oxo-3-propyl-4-methylene-8-
L 4,4-bis(4-fluorophenyl)butyl7-3,8-diazaspiroL 4,57-
decane
A mixture containing 8.4 9 of 1-oxa-2-oxo-3-propyl-
-4-methylene-3,B-diazasoiroL 4,57decane, 22.4 9 of 4,4-bis(4-
-fluorophenyl)butyl chloride, 16.6 9 of anhydrous potassium
carbonate and 0.3 9 of potassium iodide in 90 ml of methy]
isobutyl ketone is boiled under reflux and stirring for
8.hours, then the solvent is distilled off under reduced
pressure. After adding benzene and water to the residue,
the organic phase is separated, washed with water to neutral,
dried over anhydrous magnesium sulfate and evaporated under
reduced pressure. The crude product obtained i5 purified
by chromatography on a silica gel column by using ethyl
acetate for elution. The eluates are combined, evaporated
and the residue is recrystallized from diisopropyl ether
to give the title compound in 89% yield, m.p.: 107-108 C.
Analysis:
Calculated for C27H32F2N202
C 71.34; H 7.10; F 8.36; N 6.16~;
found: C 71.50; H 7.23; F 8.28; N 6.07%.
-
.
,. ;:
- 27 -
Example 2
Preparation of l-oxa-2-oxo-3-benzyl-4-methylene-8-
/ 2-(4-fluorophenyl)7-3,8-diazaspiro/ 4.57decane
A mixture containing 10.3 9 of 1-oxa-2-oxo-3-benzyl-
-4-methylene-3,8-diazaspiro/ 4,57decane, 16.2 9 of 2-(4-
-fluorophenyl)ethyl bromi-de and 11.2 ml of triethylamine
in 100 ml of methyl isobutyl ketone is refluxed under
argon while stirring for 2.5 hours. After cooling down
and adding water to the reaction mixture, the organic
phase is separated, washPd with saturated sodium chloride
solution, dried over anhydrous sodium sulfate and evaporated
under reduced pressure. The residue is recrystallized under
. clarifying by activated carbon from hexane and then from
diisopropyl ether to obtain the title compound in 31.8%
yield, m.p.: 100-101 C.
Analysis:
Calculated for C23~l25~FN22
C 72.61; H 6.62; F 4.99; N 7.36%;
found: C 72.8; H 6.54; F 5.22; N 7.53%.
~0 :~
_ample 3
Preparation of l-oxa-2-oxo-3-ethyl-4-methylene-8-
(3,3-diphenyl~ropyl)-3,B-diazaspiro/ 4,57decane
hydrogen maleate
A mixture containing 7.9 9 of 1-oxa-2-oxo-3-ethyl-
~4-methylene-3,8-diazaspiroL 4,57decane, 26 9 of 3,3-di-
phenyl-l-tosyloxypropane and 7.4 9 of anhydrous sodium
- 28 -
carbonate in 100 ml of methyl isopropyl ketone is refluxed
under nitrogen while stirring for 5 hours. After evaporating
the reaction mixture under reduced pressure, water is
added to the residue and extracted with chloroform. The
organic phase is washed with water, dried over anhydrous
magnesium sulfate and evaporated under reduced pressure.
The crude produc-t is dissolved in acetone and the title
salt is precipitated by adding an ethereal solution of
maleic acid, m.p.: 168-170 C.
The base can be liberated from the above sal-t by
adding aqueous sodium hydroxide solution.
Analysis of the base:
Calculated for C25H30N202
C 76.89; H 7.74; N 7.17%;
found: C 76.95; H 7.89, N 7.24%.
Example 4
Preparation of l-oxa-2-oxo-3-decyl-4-hydroxy-4-methyl-
-8-L 4,4-bis(4-fluorophenyl)butyl7-3,8-diazaspiro-
_ 4,57decane hydrochloride
5.5 9 of 1-oxa-2-oxo-3-decyl-4-me-thylene-8-L 4,4-
bis(4-fluorophenyl)butyl7-3,8-diazaspiroL 4,57decane are
dissolved in 11 ml of formic acid and after dropping 100 ml
of a 3 mol/litre hydrochloric acid to the solution under
stirring, the reaction mixture is stirred for additional
30 minutes. The crystals precipitated are filtered, washed
with water and dried to give the title hydrochloride in
97.2% yield, m.p.: 109-111 C. The base is liberated by
:~ .
,
adding aquenus sodium carbonate solution to the hydro-
chloride, extracted into chloroform, the organic phase
is washed with water to neutral, dried over anhydrous
sodium sulfate and evaporated under reduced pressure.
After recrystallization from diisopropyl ether the base
melts at 89-90 C.
Analysis:
Calculated for C34H48F2N203
C 71.54; H 8.48; F 6.66; N 4.91%;
~ound: C 71.54; H 8.31; F 6.73; N 4.99%.
Example 5
Preparation of l-oxa-2-oxo=3-cyclohexyl-1-methylene-
-8-L 2-t4-chlorophenyl)ethyl7-3,8-diazaspiro/ 4,57-
- 15 decane
A mixture containing 8.14 9 of 1-oxa-2-oxo-3-cyclo-
hexyl-4-hidroxy-4-methyl-8-/ 2-t4-chlorophenyl)ethyl7-
-3,8-diazaspiro/ 4,57decane and 0.8 9 of p-toluenesulfonic
acid monohydrate in 100 ml of xylene is boiled under stirring
while the water formed in the reaction is azeotropically
distilled out. The reaction is followed by using thin-
-layer chromatography. After termination of the reaction
the mixture is cooled down, made alkaline by adding 5% by
weight aqueous sodium hydroxide solution, then the organic
phase is washed with water to neutral, dried over anhydrous
sodium sulfate and evaporated under reduced pressure. After
recrystallizing the crude product from ethanol the pure
title product is obtained in 92.4% yield, m.p.: 134-135 C.
` - 30 - 2~2~
Analysis:
Calculated for C22H29ClN202
C 67.93; H 7.52; Cl 9.12; N 7.20~,;
found: C 67.88; H 7.65; Cl 9.25; N 7.11%.
Example 6
Preparation of l-oxa-2-oxo-3-methyl-4-methylene-8-
(3-phenvlpropyl)-3,8-diazaspiro/ 4.57decane
A solution of 7.5 9 of 4-ethynyl-4-methylcarbamoyl-
oxy-1-(3-phenylpropyl)piperidine in 100 ml of an ethanolic
sodium ethoxide solution of 0.09 mol/litre concentration
is refluxed under argon for 3 to 4 hours. After cooling
down and adding aqueous ammonium chloride solu-tio~. to the
reaction mixture, the major part of alcohol is distilled
off under reduced pressure. The residue is diluted with
water and extracted with benzene. The benzene phase is
washed with water to neutral, dried over anhydrous magnesium
sulfate and evaporated under reduced pressure. After re-
crystallizing the residue from hexane, the title compound
is obtained in 68% yield, m.p.: 35-36 C.
Analysis:
Calculated for C18H24N202
C 71.97; H 8.05; N 9.33%;
found: C 71.88; H 8.19; N 9.52%.
.
, ............ . . . . .
.
. .
,
:, . ,.:
: , , : ~ ,
': . ' '
- 31 - ~ d~
Example 7
PrEparation of l-oxa-2-oxo-4-methylene-3-phenyl-
-8-(2-phenylethyl)-3,8-diazaspiro/ 4,57decane
A mixture containing 6.2 9 of 4-acetyl-4-hydroxy-
-1-(2-phenylethyl)-piperidine, 2 ml of triethylamine and
11 ml of phenylisocyanate in 20 ml of xylene is refluxed
under argon for 6 hours. After cooling down and diluting
with xylene the reaction mixture is filtered and the filt-
rate is evaporated under reduced pressure. After re-
crystallizing the crude product from ethanol under clarify-
ing by ac-tivated carbon, the title product is obtained in
49.4% yield, m.p.: 137-138 C.
Analysis:
Calculated for C22~z4N202
~5 C 75.83; H 6.94; N 8.04%;
found: C 76.00; H 6.97; N 8.15%.
Example 8
Preparation of l-oxa-2-oxo-3-cvclohexyl-4-hydroxy-
-4-methyl-B-/ 2-(4-chlorophenvl)ethyl7-3,8-diaza-
splro/ 4,57decane hydrochloride
10.7 9 of 1-oxa-2-oxo-3-cyclohexyl-4-hydroxy-4-
-methyl-3,8-diazaspiro/ 4,57decane are refluxed with
13.2 9 of 2-(4-chlorophenyl)ethyl bromide, 8.2 9 of anhydrous
powdered po-tassium carbonate and 0.7 9 of potassium iodide
in 110 ml of methyl isobutyl ketone under nitrogen while
stirring for 6 hours. After evaporating the solven-t under
- 32 ~
reduced pressure and adding water to the residue, the
mixture is extracted with benzene. The combined benzene
solution is washed with water to neutral, dried over
anhydrous sodium sulfate, then the benzene solution is
filtered through an aluminum oxide layer and evaporated
under reduced pressure. After recrystallization of the
residue from hexane, the base is converted to the hydro-
chloride by adding hydrogen chloride in diisopropyl ether
solution. Thus, the title hydrochloride is obtained in
58.4% yield with a decomposition point of 310-315 C.
Analysis of the base:
Calculated for C22H31ClN203
- C 63.93, H 7.68; Cl 8.71; N 6.88%;
fou~d: C 65.10; H 7.53; Cl B.60, N 7.00~.
By using the appropriate starting substances the
following compounds are prepared in an analogous manner
as described in Examples 1 to 3 or 8.
l-Oxa-2-oxo-3-methyl-4-methylene-8-(2-phenylethyl)-3,8-
-diazaspiro/ 4,57decane, m.p.: 119-120 C;
1-Oxa-2-oxo-4-methylene-3-phenyl-8-/ 2-(4-chlorophenyl)-
ethyl7-3,8-diazaspiro/ 4,57decane, m.p.: 134-135 C;
l-Oxa-2-oxo-3-ethyl-4-methylene-8-L 2-(4-methylphenyl)-
; ethyl7-3,8-diazaspiroL 4,57decane hydrochloride, decomp.
at 280-282 C;
1-Oxa-2-oxo-3-cyclohexyl-4-methylene-8-/ 2-(4-fluorophenyl)-
ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 125-126 C;
l-Oxa-2-oxo-4-methylene-3-phenyl~8-/ 2-(4-fluorophenyl)-
ethyl7-3,B-diazaspiroL 4,57decane, m.p.: 145-147 C;
.
' . . . '
.
..
,:, ~ .
l-Oxa-2-oxo-3-ethyl-4-methylene-8-(2-pherlylethyl)-3,8-
-diazaspiroL 4,57decane, m.p.: 121-122 C;
l-Oxa-2-oxo-3-tert-butyl-4-methylene-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane, m.p.: 90-92 C;
1-Oxa-2-oxo-3-isopropyl-4-methylene-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane, m.p.: llB-119 C;
l-Oxa-2-oxo-3-methyl-4-methylene-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane, m.p.: 90-91 C.;
l-Oxa-2-oxo-3-tert-butyl-4-methylene-8-(2-phenylethyl)-3,8-
-diazaspiroL 4,57decane, m.p.: 106-107 C;
l-Oxa-2-oxo-3-isopropyl-4-methylene-8-L 2-(4-chlorophenyl)-
ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 101-102 C;
Oxa-2-oxo-3-methyl-4-methylene-8-L 2-(4-fluorophenyl)-
ethyl7-3,8-diazaspiro~ 4,57dec~ne, m.p.: 74-75 C;
1-Oxa-2-oxo-3-ethyl-4-methylene-8-L 4,4-bis(4-fluorophenyl)-
butyl7-3,8-diazaspiroL 4,57decane, m.p.: 111-112 0;
l-Oxa-2-oxo-3-isopropyl-4-methylene-8-L 2-(4-fluorophenyl)-
ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 103-104 C;
l-Oxa-2-oxo-4-methylene-3-phenyl-8-L 4,4-bis(4-fluorophenyl)-
butyl7-3,8-dia7aspiroL 4,57decane, m.p.: 125-126 C;
l-Oxa-2-oxo-4-methylene-3-propyl-8-L 2-(4-fluorophenyl)-
ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 78-79 C;
l-Oxa-2-oxo-3-L 2-(3,4-dimethoxyphenyl)ethyl7-4-methylene-
-8-(2-phenylethyl)-3,8-diazaspiroL 4,57decane, m.p.: 114-
-115 C,
l-Oxa-2-oxo-3-benzyl-4-methylene-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane, m.p.: 81-82 C;
.
.
:- .
, , , . ' , : ~ . -~ . . .. .
- 34 - ~2~
l-Oxa-2-oxo-3-decyl-4-methylene-8-/ 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane hydrogen maleate,
m.p.: 106-107 C;
l-Oxa-2-oxo-3-cyclohexyl-4-methylene-8-/ 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiro/ 4,57decane, m.p.: 121-122 C;
l-Oxa-2-oxo-3-butyl-4-methylene-8-(2-phenylethyl)-3,8-
-diazaspiroL 4,57decane, m.p.: 70-71 C;
l-Oxa-2-oxo-3-me-thyl-4-methylene-8-/ 2-(4-chlorophenyl)-
ethyl7-3,8-diazaspiro/ 4,57decane, m.p.: 118-119 C;
1-Oxa-2-oxo-3--tert-bu-tyl-4-methylene-8-/ 2-(4-chlorophenyl)-
ethyl7-3,8-diazaspiro/ 4,57decane, m.p.: 104-105 C;
l-Oxa-2-oxo-3-ethyl-4-methylene-8-L 2-(4-fluorophenyl)-
ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 83-84 C;
l-Oxa-2-oxo-3-methyl-4-methylene-8-L 2-(3,4-dimethoxy-
phenyl)ethyl7-3,8-diazaspiro/ 4,57decane hydrochloride,
m.p.: 278-280 C; and ;~
l-Oxa-2-oxo-3-tert-butyl-4-methylene-8-/ 2-(4-fluorophenyl)-
ethyl7-3,8-diazaspiro/ 4,57decane, m.p.: 93-94 C.
~'.
Example 9
:
Preparation of l-oxa-2-oxo-4-methylene-3-(1-naphthyl)-
-8-L 2-(4-fluorophenyl)ethyl7-3,8-diazaspiro/ 4,57-
decane
9.9 9 of 4-e-thynyl-4-hydroxy-1-/ 2-(4-fluorophenyl)-
ethyl7piperidine are heated with 7.0 ml of l-naphthyl iso-
cyanate under argon. A violent exothermic reaction occurs.
The temperature is maintained between 170 C and 180 C
by external cooling for one hour. After cooling down and
.
,
:,"
,
- 35 -
dissolving -the solid residue in chloroform, the solution
is filtered through an aluminum oxide layer and the
filtra-te is evaporated under reduced pressure. Af-ter
recrystallizing the residue from ethanol the title compound
is obtained in 65% yield, m.p.: 160-161 C.
Analysis:
Calculated for C26H25FN202
C 74.98; H 6.05; F 4.56; N 6.73%;
found: C 75.10; H 6.25; F 4.37; N 6.55%.
Example 10
.
Preparation of l-oxa-2-oxo-3-n-butyl-4-methylene-
8-/ 4,4-bis(4-fluorophenyl)butyl7-3,8-diazaspiro-
/ 4,57decane
3.3 9 of n-butyl isocyanate dissolved in 11 ml of
benzene are portionwise added to a gently boiling suspension
containing 11 9 of 4-ethynyl-4-hydroxy-1-/ 4,4-bis(4-fluoro-
phenyl)butyl7piperidine and 0.09 9 of sodium methoxide
in 45 ml of benzene under argon while stirring, then the
mix-ture is refluxed for additional 3 to 4 hours. After
cooling down the benzene solution is washed with water,
dried over anhydrous sodium sulfate, filtered and the
solvent is distilled off under reduced pressure. After
recrystallizing the residue from diisopropyl ether the
title compound is obtained in 79.5% yield, m.p.: 94-95 C
~nalysis
Calculated for C2~H34F2N202
~ . .
- 36 -
C 71.77; H 7.31; F 8.11; N S.98%;
found: C 71.98; H 7.40; F 8.24; N 6.13%.
Example 11
Preparation of l-oxa-2-oxo-3-butyl-4-methylene-8-
/ 2-(4-chlorophenyl)ethyl7-3,8-diazaspiro/ 4,57-
decane
13.2 9 of 4-ethynyl-4-hydroxy-1-/ 2-(4-chloro-
phenyl)ethyl7piperidine are refluxed with 6.5 9 of n-butyl
isocyanate in 40 ml of 2-picoline in the presence of 0.2 9
of anhydrous potassium acetate under argon ~or 6 hours.
After evaporating the solvent under reduced pressure and
dissolving the residue in benzene, the organic phase is
washed with water and dried-over a-nhydrous sodium-sul~fate.
The benzene solution is filtered through an aluminum oxide
layer and evaporated under reduced pressure. After re-
crystallizing the crude product from hexane the title
compound is obtained in 74.5% yield, m.p.: 86-87 C.
Analysis:
Calculated for C20H27C1N22
C 66.19; H 7.50; Cl 9.77; N 7.72%;
found: C 66.23; H 7.57; Cl 9.90; N 7.64%.
The following compounds are prepared similarly as
described in Examples 9, 10 or 11 by using the appropriate
starting substances:
l-Oxa-2-oxo-4-methylene-3-propyl-8-/ 2-(4-chlorophenyl)-
ethyl7-3,a-diazaspiroL 4,57decane, m.p.: 82-8~ C;
. .
3 ~. ~
- 37 -
1-Oxa-2-oxo-3-ethyl-4-methylene-8~L 2-(4-chlorophenyl)-
ethyl7-3,8-diazaspiro/ 4,57decane, m.p.: 106-107 C;
1-Oxa-2-oxo-4-methylene-3-(1-naphthyl)-8-L 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiroL 4,57decane, m.p.:
127-128 C;
1-ûxa-2-oxo-3-tert-butyl-4-methylene-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane, m.p.: 90-92 C;
1-Oxa-2-oxo-3-heptyl-4-methylene-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane hydrogen maleate,
m.p.: 121-122 C;
l-Oxa-2-oxo-3-(3,4-dichlorophenyl)-4-methylene-8-L 2-(4-
-chlorophenyl)ethyl7-3,8-diazaspiro/ 4,57decane hydro-
chloride, decomp. at 292-295 C;
l-Oxa-2-oxo-3-cyclahexyl-4-methylene-8-(2-phenylethyl)-3,8-
-diazaspiroL 4,57decane, m.p.: 152-153 C;
1-Oxa-2-oxo-4-methylene-3-propyl-8-(2-phenylethyl)-3,8-
-diazaspirL 4,57decane, m.p.: 97-98 C; and
l-Oxa-2-oxo-3-butyl-4-methylene-8-L 2-(4-fluorophenyl)ethyl7-
-3,8-diazaspiroL 4,57decane, m.p.: 91-92 C.
Example 12
Preparation of l-oxa-2-oxo-3-butyl-4-hydroxy-4-methyl-
-8-/ 4,4-bis(4-fluorophenvl)butvl7-3,8-diazaspiro-
~ 4,57decane hydrochloride
3 ml of butylamine dissolved in 3 ml of benzene are
dropped to a solution of 8.3 y of 1,3-dioxa-2-oxo-~-
-methylene-8-L 4,4-bis(4-fluorophenyl)bu-tyl7-8-azaspiro-
~2~
- 38 -
L ~i, 57decane in 17 ml of anhydrous benzene under stirring.
Mean~Jhile the temperature of the reaction mixture raises
` to 3a to 45 C. Thereafter thè reaction mix-ture is stirred
- at room temperature for additional 16 hours, then evaporated
under reduced pressure. After take up of -the residue in
anhydrous ether the title hydrochloride salt is precipitated
by adding ethereal hydrogen chloride solution. The title
salt i5 obtained in 87% yield, m.p.: 218-221 C.
The base is obtained from the hydrochloride by adding
ammonium hydroxide solution.
Analysis of the base:
Calculated for C28H36F2N203
. . . C 69.11; H 7.46; F 7.81; N 9.86%;
~ound: C 69.20; ~ 7.5~; F 7.64, N g.72%.
Example_13
Preparation of l-oxa-2-oxo-3-methyl-4-methylene-8, a-
-bis(3-phenylpropyl)-3 ? 8-diazaspiro! 4,57decan-8-
ium iodide
, :
6.0 9 of 1-oxa-2-oxo-3-methyl-4-methylene-8-(3-phenyl-
~; propyl)-3,B-diazaspiroL 4,57decane are refluxed with 5.4 9
of 3-phenylpropyl iodide in 60 ml of methyl isobutyl ketone
under nitrogen for 4 hours, then the solvent is distilled
~ off under reduced pressure. After adding hexane to the
; 25 residue the solid precipitate is filtered and recrystallized
from ethanol to give the title compound in 86% yield, m.p.:
219-220 C.
.,
;
~ 3
- 39 -
Example 14
Preparation of 1,3-dioxa 2-oxo-4-methylene-8-
/ 4,4-bis(4-fluorophenyl)butvl7-8-azaspiro/ 4,57-
decane hvdrogen maleate
A solution of 16.0 9 of 1-/ 4,4-bis(4-fluorophenyl)-
butyl7-4-butyl-carbamoyloxy-4-ethynylpiperidine in 90 ml
of anhydrous dioxane is saturated by dry gaseous hydrogen
chloride at 15 to 20 C, then the reaction mixture is left
to stand overnight. The solvent is evaporated at 40 to 50 C
under reduced pressure. After adding water to the residue,
the base is liberated with sodium hydrogen carbonate and
extracted into benzene. The benzene solution is dried
over anhydrous ~agnesium sulfate, `then the solvent is
evaporated under reduced pressure. After dissolving the
residue in ethyl acetate, the solution is filtered through
a silica gel column and the solution thus obtained is
evaporated under reduced pressure. Af-ter taking up the
residue in acetone, the title hydrogen maleate salt is
precipitated by maleic acid. The title salt is ob-tained
in 55% yield, m.p.: 149-150 C.
Analysis for the base:
Calculated for c24H2sF2No3
C 69.72; H 6.09; F 9.19; N 3.39%;
found: C 69.85; H 6.17; F 9.35; N 3.12%.
- 40 - ~ f
Example 15
reparation of l-oxa-2-oxo-3,4-dimethyl-4-hydroxy-
-8-(2-phenylethyl)-3,B-diazaspiro/ 4,57decane
A solution containing 9.1 9 of 4-acetyl-4-methyl-
carbamoyloxy-1-(2-phenylethyl)piperidine and 0.5 9 sodium
methoxide in 100 ml of methanol is refluxed under argon
for 4 for 5 hours. Qfter cooling down the sodium methoxide
is decomposed by adding aqueous ammonium chloride solution,
the mixture is diluted with water and methanol is distilled
off under reduced pressure. After filtration the precipitate
obtained is recrystallized from ethanol to give the title
product in 72.1% yield, m.p.: 184-185 C.
", ~nalysis:
Calculated for C17~l24N203
C 67.08; H 7.95; N 9.20%;
found: C 67.11; H 7.90; N 9.03%.
Example 16
Preparation of l-oxa-2-oxo-3-benzyl-4-hydroxy-4-
-methyl-8-/ 2-(4-fluorophenyl)ethyl7-3,8-dlaza-
spiro/ 4~57decane hydrochloride
A solution of 6.1 9 of 1-oxa-2-oxo-3-benzyl-4-
-methylene-8-L 2-(4-fluorophenyl)ethyl7-3,8-diazaspiro-
/ 4,57decane in 120 ml of 0.2 molar hydrochloric acid is
refluxed for 10 minutes and then evaporated to a volume
of 70 ml under reduced pressure. The mixture is cooled
at 1 to 5 C for 30 minutes, then the crystalline precipitate
- 41 -
is filtered and dried to give the title hydrochloride in
98% yield, decomp. at 295 C.
The base is liberated from the hydrochloride by
adding aqueous ammonium hydroxide solution.
Analysis for the base:
Calculated for C23H27FN203
C 69.32; H 6.83; F 4.77; N 7.03%;
found: C 69.40; H 6.64; F 4.85; N 7.16%.
The following compounds are prepared similarly as
described in the Example 12 or 16 by using the appropriate
starting substances:
l-Oxa-2-oxo-3,4-dimethyl-4-hydroxy-8-L 2-(4-fluorophenyl)-
ethyl7-3,8-diazaspiro/ 4,57decane, m.p.: 197-198 C;
l-nxa-2-oxo-3-cyclohexyl-4-hydroxy-4-methyl-8-/ 2-(4-fluoro-
phenyl)ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 189-190 C;
l-Oxa-2-oxo-3-tert-butyl-4-hydroxy-4-methyl-8-L 2-(4-fluoro-
phenyl)ethyl7-3,8-diazaspiroL 4,57decane hydrochloride,
decomp. at 286-288 C;
l-Oxa-2-oxo-3,4-dimethyl-4-hydroxy-8-/ 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane hydrochloride,
decomp. at 220-223 C;
l-Oxa-2-oxo-3-benzyl-4-hydroxy-4-methyl-8-L 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiro/ 4,57decane hydrochloride,
decomp. at 177-179 C;
1-Oxa-2-oxo-4-hydroxy-3-isopropyl-4-methyl-8-L 2-(4-fluoro-
phenyl)ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 157-158 C;
l-Oxa-2-oxo-4-hydroxy-4-methyl-3-phenyl-8-L 4,4-bis(4-
fluorophenyl)butyl7-3,8-diazaspiroL 4,57decane hydrochloride,
- 42 - ~2
decomp. at 274-276 C;
l-Oxa-2-oxo-3-butyl-4-hydroxy-4-methyl-B-L 2-(4-fluoro-
phenyl)ethyl7-3,8-diazaspiroL 4,57decane, m.p.: 155-156 C;
l-Oxa-2-oxo-4-hydroxy-4-methyl-3-propyl-8-/ 2-(4-fluoro-
phenyl)ethyl7-3,8-diazaspiroL 4,57decane hydrochloride,
decomp. at 248 C; the base melts at 138-139 C;
l-Oxa-2-oxo-3-ethyl-4-hydroxy-4-methyl-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane hydrochloride,
decomp. a-t 235 C;
1-Oxa-2-oxo-4-hydroxy-4-methyl-3-(1-naphthyl)-8-L 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiroL 4,57decane hydro-
chloride, decomp. at 180-182 C;
l-Oxa-2-oxo-3-heptyl-4 hydroxy-4-methyl-8-L 4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiro/ 4,57decane, m.p.: 128-129 C;
1-Oxa-2-oxo-4-hydroxy-4-methyl-3-(1-naphthyl)-8-L 2-(4-
-fluorophenyl)ethyl7-3,8-diazasp:iroL 4,57decane hydro-
chloride, decomp. at 288-290 C;
l-Oxa-2-oxo-3-ethyl-4-hydroxy-4-methyl-8-L 2-(4-~luoro-
phenyl)ethyl7-3,8-diazaspiro/ 4,57decane, m.p.: 139-140 C;
1-Oxa-2-oxo-3-cyclohexyl-4-hydroxy-4-methyl-8-/ 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiroL 4,57decane hydro-
chloride, decomp. at 258 260 C;
l-Oxa-2-oxo-4-hydroxy-3-isopropyl-4-methyl-8-/ 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiroL~4,57decane hydro-
chloride, decomp. at 251-253 C;
l-Oxa-2-oxo-3-butyl-4-hydroxy-4-methyl-8-L 4,4-bis(4-fluoro-
phenyl)butyl7-3,8-diazaspiroL 4,57decane hydrochloride,
decomp. at 218-220 C;
- 43 - ~ .J~
l-Oxa-2-oxo~4-hydroxy-4-methyl-3-propyl-8-/ 4,4-bis(4-
-fluorophenyl)butyl7-3,8-diazaspiro/ 4,57decane hydro-
chloride, decomp. at 134-136 C;
; l-Oxa-2-oxo-4-hydroxy-4-methyl-3-phenyl-8-L 2-(4-fluoro-
phenyl)ethyl7-3,8-diazaspiro/ 4,57decane hydrochloride,
decomp. at 346-350 C;
l-Oxa-2-oxo-3-(3,4-dichlorobenzyl)-4-hydroxy-4-methyl-8-
-L 2-(4-chlorophenyl)ethyl7-3,8-diazaspiroL 4,57decane
hydrochloride, decomp. at 310-315 C; and
1-Oxa-2-oxo-4-hydroxy-4-methyl-8-/ 4,4-bis(4-fluorophenyl)-
butyl7-3,8-diazaspiro/ 4,57decane hydrochloride, m.p.:
130-132 C.
Example 17
Preparation of 4-ethynyl-4-hydroxy-1-/ 4,4-b1s(4-
-fluorophenyl)butyl7piperidine hydrochloride
Acetylene is introduced into a solution containing
38.4 9 of potassium -tert-butoxide in 25n ml of tetra-
hydrofuran at a temperature between O C and -5 C under
stirring for 30 minutes, then 78.0 9 of l-L 4,4-bis(4-
-fluorophenyl)butyl7-4-piperidone dissolved in 200 ml of
tetrahydrofuran are dropwise added and acetylene is
introduced for additional one hour. Thereafter the reaction
mixture is cooled to -10 C and saturated aqueous ammonium
chloride solution is added under nitrogen. Af-ter evaporat-
ing tetrahydrofuran under reduced pressure, the residue
is extracted with benzene. The benzene solution is washed
- 44 ~
with water, dried over anhydrous magnesium sulfate and
evaporated under reduced pressure. After taking up the
residue in acetone, the hydrochloride is precipitated
by adding hydrogen chloride in diisopropyl ether solution.
The title hydrochloride is obtained in 91.0% yield, m.p.:
166-168 C.
Analysis for the base:
Calculated for C23H25F2N0
C 74.77; H 6.82; F 10.28; N 3.79%;
found: C 74.B5; H 6.66; F 10.15; N 4.00%.
Example 18
Preparation of 4-butylcarbamoyloxy-4-ethynyl-1-
. .
/ 4,4-bis(4-fluorophenyl)butyl7piperidine hydro-
chloride
6.4 ml of butyl isocyanate dissolved in 19 ml of
benzene are dropwise added to a mixture of 18.5 9 of
4-ethynyl-4-hydroxy-1-/ 4,4-bis(4-fluorophenyl)butyl7-
piperidine, 0.35 9 of anhydrous powdered potassium carbonate
and 74 ml of benzene in a nitrogen atmosphere under reflux
and stirring. The mixture is refluxed for additional one
hour, then cooled down and water is added. After separating
the phases the benzene solution is washed with water to
neutral, dried over anhydrous magnesium sulfate, the solu-
tion is filtered through a silica gel column and evaporatedunder reduced pressure. After taking up the residue in
diisopropyl ether, the hydrochloride sal-t is precipitated
- 45 -
by adding hydrogen chloride in diisopropyl ether solution.
The title hydrochloride is obtained in B7.5%, m.p.: 84-89 C.
Analysis of the base:
Calculated for C28H34F2Nn2
C 71.77; H 7.31; F 8.11; N 5.98%;
found: C 71.88; H 7.50; F 8.28; N 5.83%.
Example 19
Preparation of 4-acetyl-4-hydroxy-1-/ 4,4-bis(4-
-fluoro~henyl)butyl7piperidine hydrochloride
A solution of 1,3-dioxa-2-oxo-4-methylene-8-L 4,4-
-bis(4-fluorophenyl)butyl7-8-azaspiro/ 4,57decane in 100 ml
of a 2.8 molar sodium hydroxide solution is stirred at 80
to 90 C under argon. After cooling down the reaction
mixture is extracted with benzene, the benzene layer is
washed with water to neutral, dried over anhydrous sodium
sulfate, then the solvent is distilled off under reduced
pressure. The evaporation residue is dissolved in diiso-
propyl ether and the hydrochloride is precipitated by adding
hydrogen chloride in diisopropyl ether solution. The title
hydrochioride is obtained in 61% yield, m.p.: 62-67 C.
Analysis of the base:
Calculated for C23H27F2N02
C 71.29; H 7.02; F 9.81; N 3.61%;
found: C 71.27; H 7.18; F 9.63; N 3.80%.
;
,
- 46 ~ 3~
Example 20
Pharmaceutical compDsitions containing e g. the
following components ~ingredients) can be prepared from
the compounds according to the invention.
a) Preparation of tablets
50.0 9 of active ingredient are mixed together with
92 9 of lactose, 40 9 of potato s-tarch, 4 9 of polyvinyl-
pyrrolidone, 6 9 of talc, 1 9 of magnesium stearate, 1 9
of colloidal silicon dioxide (Aerosil) and 6 9 of ultra-
amylopectin and, after wet granulation, the product obtained
is compressed to tablets containing 50 mg of the active
ingredient each.
; b) Preparation of dragées
The tablets prep~red as d~escribed ab~ve are covered
in a manner known per se with a coating consisting of sugar
and talc. The dragées are polished by using a mixture of
bee wax and carnaube wax. Each dragée weighes 250 mg.
c) Preparation of capsules
100 mg of active ingredient, 30 9 of sodium lauryl
sulfate, 280 9 of starch, 280 9 of lactose, 4 9 of colloidal
silicon dioxide (Aerosil) and 6 9 of magnesium stearate are
thoroughly mixed together and after sieving, the mixture
obtained is filled into hard gelatine capsules containing
20 mg of the active ingredient each.
d) Preparation of suppositories
30.0 mg of active ingredient are thoroughly mixed
with 60.0 mg of lactose. Simultaneously, 1910.0 mg of
suppository base (e.g. Witepsol 4) are molten (all weights
- 47 -
are calculated for one suppository), cooled to 35 C and
the mixture of the active ingredient and lactose are
mixed thereto by using a homogenizer. The product obtained
is poured into cooled conic moulds. Each suppository
weighes 20ûO mg.
e) Preparation of a suspension
Components in 100 ml of the suspension:
Active ingredient l.ûO g
Sodium hydroxide 0.26 g
10 Citric acid 0.30 9
Nipagin (methyl 4-hydroxybenzoate
sodium salt) 0.10 9
Carbopol 940 (polyacrylic acid)0.3û g
9~ Ethanol 1.0~ 9
15 Raspberry flavour 0.60 g
Sorbitol (aqueous solution of 70%) 71.00 9
Distilled water for injection purpose
up to 100.00 ml
After adding carbopol in little portions to the
solution of nipagin and citric acid in 20 ml of distilled
water under vigorous stirring, the solution obtained is
left to stand for 10 to 12 hours. Subsequently, -the amount
given above of sodium hydroxide dissolved in 1 ml of distilled
water, the aqueous solution of sorbitol and finally the
ethanolic solution of the raspberry flavour are dropped
in under stirring. The active ingredien-t is added in small
portions to this mixture and suspended by using a submerged
homogenizer. Finally, the suspension is supplemented to
;. ' , ' ,~ ,
.. , , i
- 48 -
100 ml by adding dis-tilled water and the syrupy suspension
is led through a colloid mill.
2 0
~'